

Abstract
Apoptosis is a common antiviral defensive mechanism that potentially limits viral reproduction and spread. Many viruses possess apoptosis-suppressing tools. Here, we show that the productive infection of HeLa cells with encephalomyocarditis virus (a cardiovirus) was not accompanied by full-fledged apoptosis (although the activation of caspases was detected late in infection) but rather elicited a strong antiapoptotic state, as evidenced by the resistance of infected cells to viral and nonviral apoptosis inducers. The development of the antiapoptotic state appeared to depend on a function(s) of the viral leader (L) protein, since its mutational inactivation resulted in the efflux of cytochrome c from mitochondria, the early activation of caspases, and the appearance of morphological and biochemical signs of apoptosis in a significant proportion of infected cells. Infection with both wild-type and L-deficient viruses induced the fragmentation of mitochondria, which in the former case was not accompanied with cytochrome c efflux. Although the exact nature of the antiapoptotic function(s) of cardioviruses remains obscure, our results suggested that it includes previously undescribed mechanisms operating upstream and possibly downstream of the mitochondrial level, and that L is involved in the control of these mechanisms. We propose that cardiovirus L belongs to a class of viral proteins, dubbed here security proteins, whose roles consist solely, or largely, in counteracting host antidefenses. Unrelated L proteins of other picornaviruses as well as their highly variable 2A proteins also may be security proteins. These proteins appear to be independent acquisitions in the evolution of picornaviruses, implying multiple cases of functional (though not structural) convergence.
Cells that are infected with a virus recognize the invader's presence by their innate immunity machinery and switch on a variety of defensive mechanisms. The infecting virus, on the other hand, may possess tools capable of interfering with host antiviral responses. The outcome of the infection, both in terms of the efficiency of virus growth and the extent of host pathology, depends on the trade-off between these defensive and counterdefensive measures.
Cellular innate immunity involves multiple pathways, and one powerful defense is apoptosis, or the programmed self-sacrifice of the infected cell, potentially limiting viral reproduction and spread (10). However, many viruses are able to suppress this defensive mechanism (14, 37). Remarkably, virus-elicited pathology may be specific for a given type of cells and a given virus. Unraveling the interplay between pathways leading to the death or survival of the infected cells is an important task that may provide clues to understanding viral pathogenesis and, possibly, may indicate new directions for searching for antiviral drugs.
Picornaviruses are a family of small nonenveloped animal viruses that includes important human and animal pathogens such as polioviruses, rhinoviruses, hepatitis A virus, foot-and-mouth disease viruses, and many others (89). Their genome is represented by a single-stranded 7.2- to 8-kb RNA molecule of positive polarity encoding about a dozen mature proteins (generated by the limited proteolysis of a single polyprotein precursor), nearly all of which are directly involved in the replication of the viral RNA and formation of virions (1).
The first picornavirus demonstrated to interact with the host cell apoptotic machinery by both triggering and suppressing the apoptotic response was poliovirus (95). Since then, a wealth of data has been accumulated that shows that the activation of apoptotic pathways is a widespread, though not universal, response to picornavirus infection. Thus, apoptosis-inducing capacity was reported for coxsackieviruses B3, B4, and B5 (22, 54, 82), enteroviruses 70 and 71 (25, 27, 60, 88), human rhinoviruses 1B, 9, 14, and 16 (32, 92, 100), foot-and-mouth disease virus (53, 76), avian encephalomyelitis virus (62, 63), and hepatitis A virus (16, 43) and was the subject of several recent reviews (15, 102). The antiapoptotic activity of picornaviruses was studied predominantly by using poliovirus (3, 8, 13, 72) and coxsackievirus B3 (21, 36, 85).
The present study is focused on the interaction of cardioviruses, which are representatives of a genus in the picornavirus family, with the apoptotic machinery of infected cells. Our interest in this topic stemmed from the fact that these viruses, e.g., encephalomyocarditis virus (EMCV) and its strain mengovirus (MV), as well as the less-related Theiler's murine encephalomyelitis virus (TMEV), while sharing major features of genome organization and reproductive strategy with other family members, encode a unique protein that is not found in other picornaviruses. Indeed, the leader (L) protein, a derivative of the N-terminal portion of the viral polyprotein (55), appears to be a major player in controlling the virus-host interaction. On the one hand, it is devoid of any known enzymatic activity, and L-lacking mutants are viable, at least in certain cultured cells (19, 57, 106). On the other hand, the L protein appears to inhibit host translation (35, 106), suppresses interferon production (46, 83, 98), and impairs nucleocytoplasmic traffic (11, 30, 61, 80, 81). It has been hypothesized that cardiovirus L protein also is involved in the interaction with defensive apoptotic machinery.
Previous studies have demonstrated that TMEV infection may induce apoptosis, especially in partially restrictive cells (50, 51). EMCV also exerted a similar effect in certain cell lines (87, 103). The reason(s) underlying variability in the apoptosis-inducing effects of cardioviruses remains unexplained. Here, we demonstrate that the productive cardiovirus infection of susceptible HeLa cells resulted in their cytopathic death, which was not accompanied by clear signs of apoptosis. On the contrary, the infected cells acquired an antiapoptotic state, as evidenced by their failure to develop an apoptotic response to viral and nonviral apoptosis inducers. However, the antiapoptotic state failed to develop in cells infected with a mutant virus with inactivated L, and this mutant instead elicited caspase-dependent apoptosis preceded by cytochrome c efflux. These data suggest that the wild-type (wt) L protein is involved, directly or otherwise, in the control of viral antiapoptotic function(s).
MATERIALS AND METHODS
Cells and viruses.
HeLa-B cells (95), human rhabdomyosarcoma RD cells, and baby hamster kidney BHK-21 cells were grown in Dulbecco's modified Eagle medium with 10% fetal bovine serum. wt MV was derived from the plasmid pM16.1 (34). An MV mutant encoding an L protein with a destroyed Zn finger, C19A/C22A, was described previously (46). In control experiments, poliovirus type 1 strain Mahoney was used.
Infection and single-cycle growth experiments.
Cells were detached by EDTA treatment, plated onto 35-mm petri dishes (Corning-Costar), and cultivated overnight at 37°C under 5% CO2 in Eagle medium with 10% fetal bovine serum. At the end of cultivation, the plates contained 7 × 105 to 9 × 105 cells. The growth medium was discarded, and the virus was added in a volume of 1 ml to provide the input multiplicity of infection (MOI) indicated. After a 30-min incubation at room temperature, the cells were washed with 2.5 ml Eagle medium, and 1 ml of serum-free Eagle medium was added. After incubation at 37°C for the indicated time intervals, the cells were detached by EDTA treatment, combined with the supernatant medium, subjected to three cycles of freezing-thawing, and frozen at −80°C until viral plaque titration.
Plaque titration.
For EMCV titration, RD cell monolayers grown on 35-mm petri dishes for 3 days were used. The growth medium was discarded, and sample dilutions were introduced in a volume of 0.1 ml. After virus adsorption for 30 min at room temperature, the cells were overlaid with agarose (Amresco; final concentration of 0.4%), fetal bovine serum (2%), and neutral red (0.002%) in Earle's solution supplemented with 3% sodium bicarbonate and 1 mg/ml kanamycin. Plaque counts were made after a 24-h incubation at 37°C in 5% CO2. For the plaque assay of MV and its mutant, 1-day cultures of BHK-21 cells were used. The overlay in this case contained 0.9% agarose and no neutral red. After 2 or 3 days of incubation at 37°C in 5% CO2, the cultures were fixed with 10% trichloroacetic acid, the overlay was removed, and the cells were stained with 0.15% crystal violet. BHK-21 cells, rather than RD cells, were used for the titration of MV and its mutant, because the former cells are relatively more permissive for the growth of L-deficient MV mutants (106). It should be noted, however, that the titration of MVs on BHK-21 cells produced markedly fewer plaques than on RD cells.
Real-time PCR.
Total RNA was isolated from cell monolayers by TRI reagent (Sigma) according to the manufacturer's protocol, and its concentration was determined spectrophotometrically. Two micrograms of RNA was reverse transcribed with the avian myeloblastosis virus reverse transcriptase (Promega) primed with oligonucleotide MG5 (GAATTCCTCTGCCAATAACC; corresponding to nucleotides [nt] 6997 to 7016). The real-time reverse transcription-PCR assays were performed on a Chromo4 four-color real-time PCR system (Bio-Rad Laboratories) by using a commercially available real-time PCR kit (Syntol, Russia) according to the supplier's protocol with oligonucleotide primers MGVL1 (CGCTAGGAATGCGTAGAACA; nt 6601 to 6620) and MGVR1 (AGCTCGTCCTTGAGGAATGT; nt 6717 to 6736). Oligonucleotide MGVP1 (6-carboxyfluorescein-TGGGAAACCGCCACTCTTATCCC-black hole quencher 1; nt 6633 to 6655) was used as the probe. The standard curve was generated by using serial dilutions (corresponding to 102 to 107 molecules) of a sucrose gradient-purified (40) full-length RNA transcript of wt MV obtained from plasmid pM16.1. The thermal cycling conditions were as follows: 5 min at 95°C for hot activation, followed by 45 cycles of 15 s at 95°C and 30 s at 61°C.
Apoptosis induction.
Protein synthesis inhibitor cycloheximide (CHI; 100 μg/ml) or the transcription inhibitor actinomycin D (ActD; 3 μg/ml) was used as a nonviral inducer of apoptosis. To activate the apoptotic program by poliovirus infection, the viral reproduction was interrupted at 1.5 h postinfection (p.i.) by the addition of guanidine-HCl (100 μg/ml) to the infected cells at either the onset of infection or 1.5 h p.i. A broad-spectrum caspase inhibitor, Q-VD-OPh (23), hereinafter referred to as QVD, was used to suppress apoptosis.
TUNEL assay.
Infected and mock-infected cells were grown on coverslips for the time intervals indicated, stained with Hoechst-33342, and fixed at room temperature with Safe Fix for 30 min. The cells were treated with 96 and 70% ethanol and stored at −20°C. The assay was performed by using the apoptosis detection system fluorescein kit (Promega) according to the manufacturer's instructions. The filter cube I3 was used for the registration of TUNEL (terminal deoxyribonucleotide transferase-mediated dUTP nick end labeling)-positive cells.
DNA fragmentation electrophoretic assay.
DNA fragmentation was assayed essentially as described previously (95). Briefly, the cells were detached from the plastic by EDTA treatment, suspended in a buffer containing 20 mM EDTA and 10 mM Tris-HCl, pH 7.4, and lysed with 0.5% Triton X-100 for 20 min in an ice bath. The suspension was subjected to centrifugation in an Eppendorf minicentifuge (12,000 rpm, 15 min, 4°C), and the nucleus-free supernatant was treated with phenol-sodium dodecyl sulfate (SDS). The nucleic acids were precipitated with ethanol, dissolved in 10 μl of H2O, and treated with RNase A (10 μg/ml, 37°C, 30 min). Samples were subjected to electrophoresis on 1.5% agarose gels.
Caspase activation assay.
The caspase activity in cellular extracts was determined by measuring the cleavage of peptide Ac-DEVD-pNA, a synthetic analog of the natural substrates of caspase-3 and -7. The determination of this DEVDase activity was performed essentially as described previously (3). The cells were lysed with cell lysis buffer from the Clontech CPP-32 colorimetric detection kit. The enzymatic activity was determined in 100-μl reaction mixtures containing 45 μl of cell lysate, 100 μM Ac-DEVD-pNA (Calbiochem), 50 μl of 2× reaction buffer [100 mM piperazine-N,N′-bis(2-ethanesulfonic acid), pH 7.0, 0.2 mM EDTA, 20% glycerol, 2 mM dithiothreitol], and where indicated, 10 μM zVAD-(OMe)-fmk. The optical density was measured at 405 nm. For the identification of activated caspases, extracts of virus-infected and mock-infected cells were prepared by sonication and heating for 10 min at 95°C in lysis buffer (2% SDS, 50 mM 2-mercaptoethanol, 50 mM Tris-HCl [pH 6.8]), and the conversion of procaspases into caspases was investigated by Western blotting by using anti-caspase-3 (585), anti-caspase-8 (3-1-3), and anti-caspase-9 (824) antibodies (all from Santa Cruz). Anti-actin antibodies were from Sigma.
Mitochondria fragmentation and cytochrome c efflux assays.
HeLa-B cells or HeLa-B cells expressing the enhanced yellow fluorescent protein fused with the mitochondria-targeting sequence from subunit VIII of human cytochrome c oxidase (EYFP-mito) were used. The latter was obtained by transformation with pEYFP-mito vector (Clontech). One-day-old subconfluent monolayers on coverslips were infected and/or exposed to inhibitors for the indicated times. The immunostaining was performed as described previously (11). Briefly, infected and mock-infected cells were fixed for 20 min with 3.7% paraformaldehyde in phosphate-buffered saline (PBS), washed, and permeabilized with 0.2% Triton X-100 in PBS for 10 min. Cells were washed three times with PBS for 5 min each and blocked with 1% bovine serum albumin (BSA) for 1 h at 37°C. The incubations with primary (anti-cytochrome c; Pharmingen) and secondary (anti-mouse IgG-Alexa 546 conjugate; Invitrogen) antibodies were performed at 37°C in buffer for immunofluorescence (0.1% BSA, 0.05% Tween-20 in PBS). The cells then were washed three times for 5 min with the same buffer. The second wash contained Hoechst-33342 dye. The coverslips were mounted in Mowiol and inspected with Leica DMLS or Zeiss Axiovert 200M microscopes.
Image processing.
The color images obtained by microscopy were transformed into the grayscale mode by using Adobe Photoshop 7.0. Developed films (in the Western blot assays) were scanned and processed with the same program. When appropriate, the brightness and contrast of the whole row of grayscale pictures in a given panel were equally optimized (to make visible details presented in the color images) by using the Brightness/Contrast tool of Adobe Photoshop 7.0. In the case of Fig. 2, three-dimensional image stacks underwent equal deconvolution for each fluorochrome channel using Axiovision release 3.1 software (Carl Zeiss).
FIG. 2.

Fragmentation of mitochondria without cytochrome c efflux upon productive EMCV infection. Mock-infected and EMCV-infected HeLa cells were incubated for 5 h, immunostained for cytochrome c, and stained for DNA with Hoechst-33342. Alterations in the nuclear shape and structure, typical of cytopathic effect, revealed by Hoechst staining indicated that the cells in the right panel were undergoing viral infection.
RESULTS
Antiapoptotic state and cytopathic (necrotic) death of HeLa cells productively infected with EMCV.
HeLa cells are permissive for EMCV, and the viral yield may approach 104 PFU/cell by 6 to 8 h p.i. (Fig. 1A). By this time, the overwhelming majority of infected cells died, exhibiting typical signs of cytopathic effect, with the rounding up and complete disorganization of the cytoplasmic structure as seen by phase-contrast microscopy, as well as moderate chromatin condensation and nuclear deformation and shrinking as seen upon Hoechst staining (Fig. 1B). No ostensible signs of apoptosis could be detected in the infected cells. Indeed, nuclear fragmentation was not observed and chromatin condensation was not as intense as that seen in typical apoptosis (e.g., induced by restrictive poliovirus infection [Fig. 1B] or the nonviral apoptotic inducer ActD [see Fig. 3A]). When assayed for DNA degradation by TUNEL assay, many infected cells produced only a very faint signal (Fig. 1B). Also, only a slight degradation of cellular DNA to high-molecular-mass species could be observed at late steps of EMCV infection by electrophoretic analysis (Fig. 1C). Neither cytopathic effect nor viral yield was appreciably affected by a pan-caspase inhibitor, QVD (Fig. 1). EMCV infection did not cause any marked cytochrome c release from mitochondria, another hallmark of apoptosis, but unexpectedly, the extensive fragmentation of mitochondria could be observed (Fig. 2). Indeed, a network of thread-like mitochondria was transformed into a set of punctated structures in the infected cell. Of note, mitochondrial fission is known to be a factor involved in the control of apoptotic machinery (24, 77, 104).
FIG. 1.

EMCV reproduction in HeLa cells. (A) One-step growth curve. Cells were infected at an input MOI of ∼500 PFU/cell, and the harvest was plaque assayed in RD cells. Results from a separate experiment performed in the absence (solid symbols) or presence (open symbols) of 20 μM QVD are presented in the inset. (B) Mock-infected cells, EMCV-infected cells, and cells abortively infected with poliovirus (i.e., with 100 μg/ml of guanidine-HCl added at 1.5 h p.i. [polio+gua]), all in the absence or presence of 20 μM QVD, were fixed at 7 h p.i. and stained for the TUNEL assay and with Hoechst-33342. (C) Electrophoretic investigation of DNA from samples similar to those shown in panel B.
FIG. 3.

EMCV inhibition of ActD-triggered apoptosis. (A) HeLa cells were preincubated in serum-free medium for 1.5 h and then mock infected or EMCV infected and incubated at 37°C for 5 h in the absence and presence of 3 μg/ml ActD. The cells were stained for the TUNEL assay and with Hoechst-33342. (B) Quantification of TUNEL-positive cells in the cell populations from the experiment shown in panel A. (C) Electrophoretic investigation of DNA from cell samples shown in panel A. (D) Samples similar to those shown in panel A were immunostained for cytochrome c.
Remarkably, EMCV induced an antiapoptotic state, as evidenced by the resistance of the infected cells to a nonviral apoptotic inducer. The treatment of uninfected cells with 3 μg/ml of ActD for 5 h resulted in the development of apoptosis in a significant proportion of cells, judging by morphological (cytoplasmic blebbing, strong chromatin condensation, and nuclear fragmentation) and biochemical (positive TUNEL signal and DNA fragmentation to oligonucleosome-sized fragments) criteria. All of these alterations could be prevented by QVD, confirming that ActD treatment led to a bona fide caspase-dependent apoptosis. The ActD-induced apoptosis was strongly, if not completely, suppressed in EMCV-infected cells (Fig. 3A to C). EMCV infection also prevented cytochrome c efflux from mitochondria caused by ActD treatment (Fig. 3D). Since EMCV prevented mitochondria damage-mediated cytochrome c efflux, we conclude that the viral antiapoptotic mechanism operated, at least in part, at the mitochondrial level or upstream of mitochondria. The yield of infectious cardioviruses is known to be essentially unaffected by ActD, and in the experiment shown in Fig. 3, ∼1.2 × 104 PFU/cell were produced by 5 h p.i. in the presence and absence of the drug. ActD did not prevent cell destruction caused by EMCV (Fig. 3).
As shown previously (2, 95) (Fig. 1), restrictive poliovirus infection induces apoptosis as a result of the activation of a mitochondrion-dependent and caspase-dependent pathway (13). This apoptosis also was suppressed by coinfection with MV (Fig. 4).
FIG. 4.
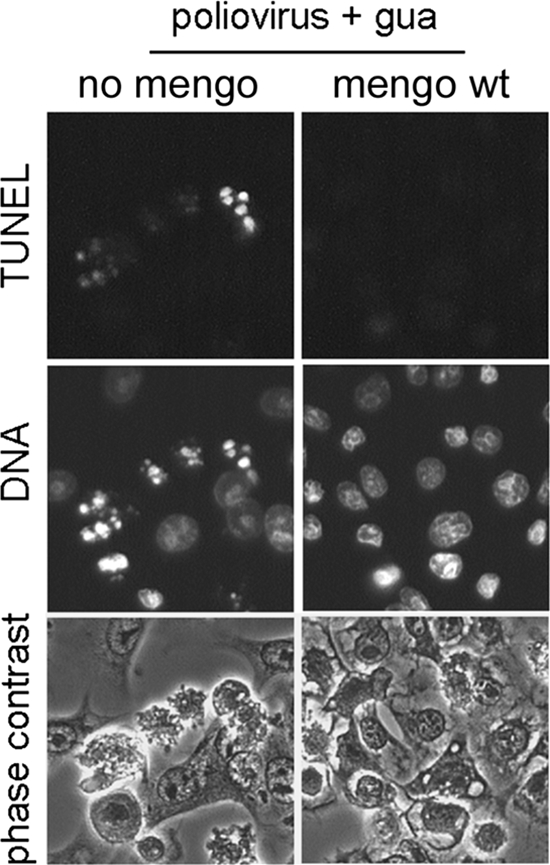
MV (mengo) inhibition of apoptosis triggered by restrictive poliovirus infection. HeLa cells were infected with poliovirus (MOI, ∼800 PFU/cell), and simultaneously portions of samples (the right column) were infected with wt MV (MOI, ∼10 PFU/cell). Guanidine-HCl (100 μg/ml) was added at 1.5 h p.i. to all cultures (poliovirus + gua). The cells were fixed at 7 h p.i. and stained for the TUNEL assay and with Hoechst-33342. The yield of MV was not appreciably changed in the presence of guanidine.
All of these data strongly suggested that EMCV infection switched on an antiapoptotic program in the infected cells.
Mutant cardiovirus with impaired L protein induced apoptosis.
To ascertain the possible involvement of cardiovirus L protein in the control of apoptotic machinery, an L-deficient variant of MV with two mutations, C19A/C22A, destroying the Zn finger motif of the protein (46) was investigated. Previous studies showed that the destruction of this motif resulted in the functional inactivation of L (46, 61).
At 5 h p.i., the C19A/C22A mutant produced usually, but not always, severalfold fewer infectious virus particles than did its wt counterpart, and this difference was somewhat increased by 7 h p.i. (Fig. 5A). However, in some experiments, the mutant and wt MV exhibited more similar growth curves (not shown). The quantification of viral RNA molecules by real-time PCR (Fig. 5B) demonstrated a good parallelism between RNA replication and the generation of infectious progeny, with very close ratios of the number of RNA molecules to PFU for both the wt and C19A/C22A mutant (not shown). As could be expected, this ratio somewhat decreased with time, likely reflecting the maturation of virions. Infection with the mutant ended up with complete cell destruction (Fig. 6A).
FIG. 5.

Effects of mutations in the L protein on MV reproduction in HeLa cells. Accumulation of infectious virus (A) and viral RNA molecules (B) during the one-step growth of wt and C19A/C22A MVs, as determined by plaques assay and real-time PCR, respectively. The input MOI for both viruses was ∼20 PFU/cell. Average data ± standard deviations for seven independent experiments are shown in panel A, and those for two (at 3 h p.i.) and three (at 5 and 7 h p.i.) independent experiments are shown in panel B. At least three replicate cultures were investigated at each time point in each real-time experiment.
FIG. 6.

Apoptosis-triggering activity of L-deficient MV mutant. (A) HeLa cells were mock infected or infected (MOI, ∼20 PFU/ml) with wt MV or its L− mutant for 7 h at 37°C, and then they were stained with Hoechst-33342 and for the TUNEL assay. Appropriate samples were incubated with 20 μM QVD. The virus yields in the absence and presence of the inhibitor were essentially the same, 257 and 285 PFU/cell, respectively. (B) Quantification of TUNEL-positive cells in samples of cells in the experiment shown in panel A. (C) Electrophoretic investigation of DNA from mock-infected and virus-infected cells.
In distinction from the wt MV, the L-deficient mutant elicited a marked apoptosis in HeLa cells, as judged by cytoplasmic blebbing, chromatin condensation, nuclear fragmentation, TUNEL assay (Fig. 6A, B), and DNA laddering (Fig. 6C) criteria. None of these signs of apoptosis could be seen in the mutant-infected cells incubated in the presence of QVD (Fig. 6).
One may argue that the activation of the apoptotic program is due to a somewhat less efficient reproduction of the L-deficient mutant. Indeed, the interruption of ongoing poliovirus reproduction at an early step by the inhibition of translation by a low dose (10 μg/ml) of CHI (by itself not inducing apoptosis in a significant proportion of cells) resulted in the suppression of the viral antiapoptotic program and the development of apoptosis (3). We investigated whether a similar treatment also would activate the apoptotic program in cardiovirus-infected cells. CHI addition at 1.5 h p.i. diminished the yield of infectious virus by ∼103-fold. However, such a treatment did not lead to an increase in the proportion of TUNEL-positive cells in the EMCV-infected population, although the fraction of such cells in the uninfected population slightly increased (Fig. 7). As expected (3), CHI addition to poliovirus-infected cells, which were used as a control, resulted in the development of an appreciable apoptotic response. We concluded that a significant suppression of ongoing cardiovirus reproduction did not result, by itself, in the development of apoptosis.
FIG. 7.

Effect of the interruption of EMCV and poliovirus (polio) reproduction on the development of apoptotic response. HeLa cells were infected with EMCV or poliovirus (both at an MOI of ∼500 PFU/cell), and CHI (final concentration, 10 μg/ml) was added to appropriate samples at 1.5 h p.i. Cells were fixed at 7 h p.i., and the proportion of TUNEL-positive cells was determined.
Thus, in distinction from wt cardioviruses inducing an antiapoptotic state, L-deficient MV activated the apoptotic program, suggesting that L protein was involved, directly or otherwise, in the control of the apoptotic machinery of the infected cells.
The apoptotic pathway activated by L-deficient cardiovirus mutants.
The prevention by QVD of apoptosis triggered by the L− mutant (Fig. 6) suggested the involvement of a caspase(s). This conclusion was supported by measuring the DEVDase activity, i.e., the capacity to cleave DEVD-pNA, a synthetic analog of the natural substrates of caspase-3 and -7. As shown in Fig. 8A, infection with the C19A/C22A mutant resulted in a marked and early (i.e., already at 5 h p.i.) activation of a DEVDase. This activity was only slightly increased by 7 h p.i., and it was practically fully inhibited by QVD. The cells infected with wt MV also demonstrated an increase in DEVDase activity, which, at 5 h p.i., was significantly lower than that of the mutant-infected cells, and approached a similarly high level during the next 2 h. A comparable increase in DEVDase activity also was observed in cells infected with EMCV (not shown). The ability of wt virus to activate DEVDase activity was unexpected, because no obvious signs of apoptosis were observed under such conditions.
FIG. 8.

Effects of infection with wt and C19A/C22A mutant MVs on caspase activation. (A) DEVDase activation. HeLa cells were infected with appropriate viruses for 7 h, and DEVDase activity was assayed in cell extracts prepared at 5 and 7 h p.i. as described in Materials and Methods. (B) Western blot analysis of partial proteolysis of caspase-3, -8, and -9 in virus-infected cells at 5 and 7 h p.i. and in cells treated with 100 μg/ml of CHI for 5 h. The bands corresponding to procaspase-9 and processed caspases are marked by arrowhead and arrows, respectively. A slower-migrating band in the caspase-3 assay in the presence of QVD (asterisk) was reproducible in the inhibitor-containing samples; its nature was not further investigated.
To identify the caspase(s) activated upon infection, immunoblot assays were performed. The results (Fig. 8B) demonstrated the proteolytic cleavage (suggestive of activation) of caspase-9 and -3 and, to a lesser extent, of caspase-8 in the L-deficient mutant-infected cells already at 5 h p.i., with more intense bands of proteolytic products at 7 h p.i. Some activation, albeit less prominent and occurring later (i.e., at 7 h p.i.), was observed in wt-infected cells, in line with the results of the DEVDase assay. Similar proteolytic products also were detected in cells undergoing CHI-elicited apoptosis. QVD prevented the processing of procaspases (in the case of caspase-3, an aberrant proteolytic product was reproducibly observed in samples treated with the inhibitor).
To further characterize the pathway leading to apoptosis in cells infected with the L− mutant, we looked at the status of mitochondria and cytochrome c. To this end, cells expressing the enhanced yellow fluorescent protein fused with the mitochondria-targeting sequence from human cytochrome c oxidase (EYFP-mito) were generated. An advantage of these cells consisted in the possibility to evaluate mitochondrial fragmentation independently of cytochrome efflux. The former could be judged by the altered pattern of fluorescence emitted by EYFP (transition from thread-like to punctated signals), whereas the latter could be seen as diffuse clouds after immunostaining with anti-cytochrome antibodies. In cells infected with the C19A/C22A mutant, mitochondria were fragmented and the extensive exit of cytochrome c from mitochondria could be demonstrated by 7 h p.i. (Fig. 9A). The proportion of the mutant-infected cells exhibiting cytochrome c efflux was significantly greater than that in cells rendered apoptotic after treatment with CHI (Fig. 9B). On the other hand, nearly no wt virus-infected cells demonstrated the exit of cytochrome c from fragmented mitochondria (in line with data in Fig. 2). The anti-caspase drug QVD failed to prevent mitochondrion fission in cells infected with wt and L-deficient MV, indicating that this fission took place upstream of caspase activation; the cytoplasmic exit of cytochrome c in mutant-infected cells also was not prevented (Fig. 9).
FIG. 9.

Fragmentation of mitochondria and cytoplasmic exit of cytochrome c in cells infected with the L mutant. (A) HeLa cells expressing mitochondria-targeted EYFP (EYFP-mito) were infected with wt and C19A/C22A MVs for 7 h or were treated with 100 μg/ml of CHI for 4 h and investigated for the localization of cytochrome c by indirect immunofluorescence or mitochondrial integrity by the direct observation of EYFP. Appropriate samples were incubated in the presence of 20 μM QVD during infection or CHI treatment. DNA was stained with Hoechst 33342. (B) Percentage of cells demonstrating cytochrome c efflux.
Thus, infection with the L-deficient cardiovirus activated a caspase-dependent, cytochrome c-mediated apoptotic pathway. This activation appeared to be due, at least in part, to alterations in the mitochondrial structure, and in particular to the enhanced permeability of its outer membrane, allowing cytochrome c efflux. The mitochondrial injury occurred through a caspase-independent mechanism, since neither fragmentation nor cytochrome c efflux triggered by the mutant were prevented by the caspase inhibitor.
DISCUSSION
Apoptosis-triggering and apoptosis-preventing functions of cardioviruses.
The results presented here demonstrate that EMCV is capable of switching on two opposing pathways in HeLa cells, the host defensive apoptotic response and a viral antiapoptotic counterdefense. As a result of the dominance of the latter, the cells died without obvious signs of apoptosis. In possessing both apoptosis-triggering and apoptosis-suppressing activities, cardioviruses resemble poliovirus (3, 8, 9, 13, 95) and coxsackievirus B3 (22, 36, 85). It is yet to be determined whether other representatives of the picornavirus family also are equipped with these opposing functions.
The competition between the opposing programs may involve multiple players and is influenced by a variety of genetic (viral and cellular), physiological, and environmental factors. The contribution of host factors is illustrated by the ability of cardioviruses to induce apoptosis in some, but not all, types of cells (5, 50, 51, 87, 103). Furthermore, the L-deficient MV, exhibiting a highly apoptogenic activity in HeLa cells, failed to elicit apoptosis in a subline of BHK-21 cells, which proved to be resistant to certain nonviral apoptotic inducers as well (unpublished data). For poliovirus, variable responses of apoptotic machinery of different cells also were reported: this virus, being unable to induce apoptosis in productively infected HeLa cells (2, 95), triggered apoptosis in others (4, 9, 84). On the other hand, abortive poliovirus infection fails to trigger apoptosis in RD cells (84).
One of the possible reasons for the variation of the outcomes of cardiovirus-cell interactions could be the rate of the accumulation of L in the infected cells as well as the biological relevance of cellular targets of L in specific cells. The time course and efficiency of different steps of viral reproduction in these cells also may affect this outcome. The consequences of the trade-off between viral apoptosis-inducing and apoptosis-preventing activities are not expected to be uniform either. In certain picornavirus/cell systems, the inhibition of apoptosis may adversely affect viral reproduction and/or externalization (32, 65). In contrast, the yield of infectious L-deficient MV in HeLa cells was essentially unaffected by QVD, a caspase inhibitor.
Accordingly, one should be cautious in extrapolating observations made in one type of cells to another one, and especially to the events occurring in the infected organism. TMEV (86, 96, 97), porcine EMCV (17), and poliovirus (39) were reported to elicit apoptosis in certain cells of animals. It is not clear if these apoptotic events always developed as a direct consequence of viral reproduction in the target cells or if they were due to some secondary, e.g., immunological, reaction (52). If the former explanation is true, it means that the viral antiapoptotic function was not efficient enough to overcome the apoptotic responses in these cases. However, this should not necessarily be so in some other in vivo settings.
The apoptotic program elicited by cardioviruses.
The activation of the apoptotic program upon cardiovirus infection becomes evident when the viral antiapoptotic function(s) is suppressed, as is the case in infection with L-deficient mutants (another MV mutant with a nearly complete deletion of L protein exhibited apoptosis-inducing activity similar to that of the Zn finger mutant described above; unpublished data). Under these conditions, numerous hallmarks of apoptosis were observed: cytochrome c exit from mitochondria, the activation of caspase-9, caspase-3, and caspase-8, cytoplasmic blebbing, chromatin condensation, nuclear fragmentation, and extensive DNA degradation (as revealed by both TUNEL assay and electrophoretic analysis). Collectively, these alterations pointed to the involvement of a mitochondrion-dependent, cytochrome c-dependent, and caspase-dependent apoptotic pathway(s).
Picornaviruses are known to activate apoptotic pathways through interaction with plasma membrane receptors or intracellular sensors (8, 15, 53, 102). Viral capsid proteins were implicated in triggering apoptosis both from without (9, 53, 76) and from within the cell (47, 62). Other possible players are viral proteases 3C and 2A (18, 26, 42, 60, 105) and viral nonstructural proteins 2B, 3A (64), and 2C (63). Double-stranded RNA also may serve as an apoptosis inducer (49, 99). However, the exact nature of cardioviral activator(s) of the apoptotic pathway(s) and of their immediate cellular targets have yet to be elucidated.
An intriguing observation made in this study was mitochondrial fragmentation in the virus-infected cells. In other systems, mitochondrial fission may take place either upstream or downstream of the mitochondrial membrane permeabilization, a key step in the mitochondrion-dependent apoptotic pathways (24, 77, 104). A possibility that this fission is an upstream event in the cardiovirus-activated apoptotic pathway is in line with the recently reported ability of poliovirus 2B and 3A proteins to induce the fragmentation of the organelle (64). However, further research is needed to ascertain the significance of mitochondrial fission for cardiovirus-triggered apoptosis.
The antiapoptotic program elicited by cardioviruses.
The nature of the cardiovirus antiapoptotic program is even less certain. Only limited information is available on the antiapoptotic tools exploited by other picornaviruses. Coxsackievirus B3 (36) and poliovirus (8) were reported to activate the phosphatidylinositol 3-kinase/Akt signaling pathway, thereby promoting cell survival. 2B protein of the former virus may suppress the apoptotic program by changing intracellular distribution of Ca2+ (21). Other antiapoptotic mechanisms used by enteroviruses may involve the inactivation or degradation of caspases (13, 85) and the depletion of receptors of extracellular proapoptotic ligands (72). In other viruses, a much greater variety of viral antiapoptotic tools are known (6, 20, 28, 68, 73, 74, 93, 94, 101). These tools may operate at transcriptional or posttranscriptional levels, with targets in mitochondria as well as upstream or downstream of them. The antiapoptotic state elicited by cardioviruses may exploit some of these mechanisms.
This study implicates cardiovirus L protein in the viral antiapoptotic activity; indeed, its inactivation led to the development of apoptosis. The mechanism of this antiapoptotic function has yet to be determined. It cannot be ruled out that it is associated, at least in part, with the less efficient growth of L− mutants. Indeed, the balance of viral apoptosis-triggering and apoptosis-suppressing functions, whatever their nature, could well depend on the time course of the synthesis of relevant proteins. This may explain the apoptotic activity of wt cardioviruses under partially restrictive conditions in certain systems (50, 51). However, the somewhat impaired reproduction of L-deficient mutants is hardly a decisive cause of their apoptotic activity. The difference between the infectious particles (or RNA molecules) of wt and L− virus produced by 5 h p.i. (when there already was a marked apoptosis in the mutant-infected cells; not shown) was relatively small. Moreover, the interruption of cardiovirus reproduction by a low dose of CHI did not lead to apoptosis. We propose that L protein has a more direct target(s) among the components of the cellular apoptotic/ antiapoptotic machinery.
As mentioned above, we detected the fragmentation of mitochondria upon infection with wt virus, when the development of apoptosis essentially was suppressed. This fission was not accompanied with the exit of cytochrome c into the cytoplasm. Therefore, one may speculate that the wt EMCV-elicited mitochondrial fission is involved in the viral antiapoptotic function rather than representing an upstream apoptotic event. Indeed, it was reported that mitochondrial fission may suppress apoptosis in some cases, e.g., upon ceramide treatment, oxidative stress, or serum deprivation (77, 91). Under these conditions, a major proapoptotic factor is the redistribution of Ca2+ from endoplasmic reticulum stores to mitochondria. The fragmentation of these organelles would result in impeding the propagation of Ca2+ waves through the mitochondrial network (77, 91). If mitochondrial fission plays a role in the cardiovirus antiapoptotic activity, one may further speculate that an alteration in Ca2+ distribution is an important component of the apoptotic response to EMCV infection. It is noteworthy that the expression of EMCV protein 2B leads to a decrease in Ca2+ content within the endoplasmic reticulum (29).
Interestingly, a viral protein is known that exert its antiapoptotic function via mitochondrial fission. The immediate-early protein vMIA of human cytomegalovirus (41) recruits proapoptotic protein Bax to mitochondria and somehow inactivates it there (7, 79); this process is associated with the disruption of the mitochondrial network (67). The antiapoptotic state elicited by vMIA is effective against nonviral apoptosis inducers (66). However, we were unable to reveal any significant similarity between amino acid sequences of vMIA and cardiovirus L.
Another intriguing observation was the activation of several caspases in nonapoptotic HeLa cells infected with wt cardioviruses. The activation of caspase-8, -9, -3, and -7 late upon EMCV infection (at 7 and 24 h p.i.) also was reported by others (49). A broad group of viral antiapoptotic proteins are known to counteract various caspase activators or caspases themselves (20, 85). However, the interruption of an apoptotic pathway downstream of caspase activation is rather puzzling. It suggests the existence of another cardiovirus antiapoptotic function, operating downstream of caspases and suppressing significant DNA degradation. Further research is needed to clarify this point.
Cardiovirus L exhibits a variety of effects on the innate immunity system (see below). It has yet to be elucidated how and whether these diverse effects are related to each other.
It should be noted that the genome of some, but not all, strains of TMEV contains, within the L-coding gene, an out-of-frame sequence encoding a short polypeptide, called L* (58), which was reported to exhibit antiapoptotic activity (38, 48). However, EMCV does not express L*, and EMCV L has no structural resemblance to L* of TMEV-like viruses.
Cardiovirus L protein as a security protein.
The leader proteins of cardioviruses are not essential for viral reproduction, as evidenced by the viability of L-deleted mutants in certain cells (19, 57, 106). These proteins appeared to target predominantly host metabolism and infrastructure by inhibiting host translation (35, 106), interfering with the interferon system (46, 83, 98, 107), increasing the bidirectional permeability of the nuclear pores and inhibiting active nucleocytoplasmic transport (11, 30, 61, 80, 81), and suppressing apoptosis (this study). We suggest that such nonessential anti-host proteins could be named security proteins.
Bearing the same name but being structurally unrelated, the L protein of aphthoviruses, a protease, also is not essential (78), but it inhibits host translation by the cleavage of translation initiation factors and disables the host interferon system (31, 33, 45). Remarkably, 2A protein of cardioviruses, being nonessential though important (69, 90, 108), also exhibits anti-host functions by inhibiting cell translation (44). 2A protease proteins of enterovirus and rhinovirus (unrelated to 2A proteins of cardioviruses), though apparently essential (70), also perform a set of anti-defensive functions very similar to those exhibited by L proteins of cardioviruses and aphthoviruses, namely the inhibition of host translation (59), the disruption of controllable nucleocytoplasmic traffic (12, 75), the suppression of interferon action (71), and others (56). We propose that L proteins of aphthoviruses as well as 2A proteins of cardioviruses, enteroviruses, and rhinoviruses also be included in the group of security proteins. It seems likely that this group will be joined by other picornavirus proteins.
The fact that the picornavirus security proteins, despite their structural unrelatedness, serve similar functions (largely or even solely neutralizing host antidefenses) strongly suggests that they had been acquired independently, and perhaps relatively recently, in the course of evolution, and that the major driving force behind these acquisitions was positive selection for anti-defensive capabilities.
Obviously, the possession of security proteins is not an exclusive privilege of picornaviruses.
ADDENDUM IN PROOF
After the article was revised, we became aware of a paper by Fan et al. (J. Fan, K.-N. Son, S. Y. Arslan, Z. Liang, and H. L. Lipton, J. Virol., 83:6546-6553, 2009) reporting that ectopic expression of individual Theiler's murine encephalomyelitis virus (BeAn strain) leader protein in BHK-21 cells resulted in the development of apoptosis. It therefore seems that cardiovirus L proteins may, depending on conditions, exihibit either antiapoptotic or proapoptoic activity.
Acknowledgments
This study was supported by grants from the NWO-RFBR (to V.I.A.), Russian Foundation for Basic Research (to P.V.L. and V.I.A.), Scientific School Support Program (to V.I.A.), and the Russian President's Grant for Young Scientists (to P.V.L.). S.V.H. is supported by a mosaic grant from the NWO. F.J.M.V.K. is supported by a VIDI grant from the NWO.
Footnotes
Published ahead of print on 6 May 2009.
REFERENCES
- 1.Agol, V. I. 2002. Picornavirus genome: an overview, p. 127-148. In B. L. Semler and E. Wimmer (ed.), Molecular biology of picornaviruses. ASM Press, Washington, DC.
- 2.Agol, V. I., G. A. Belov, K. Bienz, D. Egger, M. S. Kolesnikova, N. T. Raikhlin, L. I. Romanova, E. A. Smirnova, and E. A. Tolskaya. 1998. Two types of death of poliovirus-infected cells: caspase involvement in the apoptosis but not cytopathic effect. Virology 252343-353. [DOI] [PubMed] [Google Scholar]
- 3.Agol, V. I., G. A. Belov, K. Bienz, D. Egger, M. S. Kolesnikova, L. I. Romanova, L. V. Sladkova, and E. A. Tolskaya. 2000. Competing death programs in poliovirus-infected cells: commitment switch in the middle of infectious cycle. J. Virol. 745534-5541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ammendolia, M. G., A. Tinari, A. Calcabrini, and F. Superti. 1999. Poliovirus infection induces apoptosis in CaCo-2 cells. J. Med. Virol. 59122-129. [PubMed] [Google Scholar]
- 5.Anderson, R., E. Harting, M. S. Frey, J. L. Leibowitz, and R. C. Miranda. 2000. Theiler's murine encephalomyelitis virus induces rapid necrosis and delayed apoptosis in myelinated mouse cerebellar explant cultures. Brain Res. 868259-267. [DOI] [PubMed] [Google Scholar]
- 6.Andoniou, C. E., and M. A. Degli-Esposti. 2006. Insights into the mechanisms of CMV-mediated interference with cellular apoptosis. Immunol. Cell Biol. 8499-106. [DOI] [PubMed] [Google Scholar]
- 7.Arnoult, D., L. M. Bartle, A. Skaletskaya, D. Poncet, N. Zamzami, P. U. Park, J. Sharpe, Ri. J. Youle, and V. S. Goldmacher. 2004. Cytomegalovirus cell death suppressor vMIA blocks Bax- but not Bak-mediated apoptosis by binding and sequestering Bax at mitochondria. Proc. Natl. Acad. Sci. USA 1017988-7993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Autret, A., S. Martin-Latil, C. Brisac, L. Mousson, F. Colbère-Garapin, and B. Blondel. 2008. Early phosphatidylinositol 3-kinase/Akt pathway activation limits poliovirus-induced JNK-mediated cell death. J. Virol. 823796-3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Autret, A., S. Martin-Latil, L. Mousson, A. Wirotius, F. Petit, D. Arnoult, F. Colbère-Garapin, J. Estaquier, and B. Blondel. 2007. Poliovirus induces Bax-dependent cell death mediated by c-Jun NH2-terminal kinase. J. Virol. 817504-7516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barber, G. N. 2001. Host defense, viruses and apoptosis. Cell Death Differ. 8113-126. [DOI] [PubMed] [Google Scholar]
- 11.Bardina, M. V., P. V. Lidsky, E. V. Sheval, K. V. Fominykh, F. J. M. van Kuppeveld, V. Y. Polyakov, and V. I. Agol. 2009. Mengovirus-induced rearrangement of the nuclear pore complex: hijacking cellular phosphorylation machinery. J. Virol. 833150-3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Belov, G. A., P. V. Lidsky, O. V. Mikitas, D. Egger, K. A. Lukyanov, K. Bienz, and V. I. Agol. 2004. Bidirectional increase in permeability of nuclear envelope upon poliovirus infection and accompanying alterations of nuclear pores. J. Virol. 7810166-10177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Belov, G. A., L. I. Romanova, E. A. Tolskaya, M. S. Kolesnikova, Y. A. Lazebnik, and V. I. Agol. 2003. The major apoptotic pathway activated and suppressed by poliovirus. J. Virol. 7745-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Best, S. M. 2008. Viral subversion of apoptotic enzymes: escape from death row. Annu. Rev. Microbiol. 62171-192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blondel, B., A. Autret, C. Brisac, S. Martin-Latil, L. Mousson, I. Pelletier, J. Estaquier, and F. Colbere-Garapin. 2009. Apoptotic signaling cascades operating in poliovirus-infected cells. Front. Biosci. 142181-2192. [DOI] [PubMed] [Google Scholar]
- 16.Brack, K., W. Frings, A. Dotzauer, and A. Vallbracht. 1998. A cytopathogenic, apoptosis-inducing variant of hepatitis A virus. J. Virol. 723370-3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brewer, L. A., H. C. Lwamba, M. P. Murtaugh, A. C. Palmenberg, C. Brown, and M. K. Njenga. 2001. Porcine encephalomyocarditis virus persists in pig myocardium and infects human myocardial cells. J. Virol. 7511621-11629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Calandria, C., A. Irurzun, A. Barco, and L. Carrasco. 2004. Individual expression of poliovirus 2Apro and 3Cpro induces activation of caspase-3 and PARP cleavage in HeLa cells. Virus Res. 10439-49. [DOI] [PubMed] [Google Scholar]
- 19.Calenoff, M. A., C. S. Badshah, M. C. Dal Canto, H. L. Lipton, and M. K. Rundell. 1995. The leader polypeptide of Theiler's virus is essential for neurovirulence but not for virus growth in BHK cells. J. Virol. 695544-5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Callus, B. A., and D. L. Vaux. 2007. Caspase inhibitors: viral, cellular and chemical. Cell Death Differ. 1473-78. [DOI] [PubMed] [Google Scholar]
- 21.Campanella, M., A. S. de Jong, K. W. Lanke, W. J. Melchers, P. H. Willems, P. Pinton, R. Rizzuto, and F. J. M. van Kuppeveld. 2004. The coxsackievirus 2B protein suppresses apoptotic host cell responses by manipulating intracellular Ca2+ homeostasis. J. Biol. Chem. 27918440-18450. [DOI] [PubMed] [Google Scholar]
- 22.Carthy, C. M., D. J. Granville, K. A. Watson, D. R. Anderson, J. E. Wilson, D. Yang, D. W. Hunt, and B. M. McManus. 1998. Caspase activation and specific cleavage of substrates after coxsackievirus B3-induced cytopathic effect in HeLa cells. J. Virol. 727669-7675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Caserta, T. M., A. N. Smith, A. D. Gultice, M. A. Reedy, and T. L. Brown. 2003. Q.-VD-OPh, a broad spectrum caspase inhibitor with potent antiapoptotic properties. Apoptosis 8345-352. [DOI] [PubMed] [Google Scholar]
- 24.Chan, D. C. 2006. Mitochondrial fusion and fission in mammals. Annu. Rev. Cell Dev. Biol. 2279-99. [DOI] [PubMed] [Google Scholar]
- 25.Chang, S.-C., J.-Y. Lin, L. Y.-C. Lo, M.-L. Li, and S.-R. Shih. 2004. Diverse apoptotic pathways in enterovirus 71-infected cells. J. Neurovirol. 10338-349. [DOI] [PubMed] [Google Scholar]
- 26.Chau, D. H., J. Yuan, H. Zhang, P. Cheung, T. Lim, Z. Liu, A. Sall, and D. Yang. 2007. Coxsackievirus B3 proteases 2A and 3C induce apoptotic cell death through mitochondrial injury and cleavage of eIF4GI but not DAP5/p97/NAT1. Apoptosis 12513-524. [DOI] [PubMed] [Google Scholar]
- 27.Chen, D., D. E. Texada, C. Duggan, Y. Deng, T. B. Redens, and M. P. Langford. 2006. Caspase-3 and -7 mediate apoptosis of human Chang's conjunctival cells induced by enterovirus 70. Virology 347307-322. [DOI] [PubMed] [Google Scholar]
- 28.Clemens, M. J. 2006. Epstein-Barr virus: inhibition of apoptosis as a mechanism of cell transformation. Int. J. Biochem. Cell Biol. 38164-169. [DOI] [PubMed] [Google Scholar]
- 29.de Jong, A. S., F. de Mattia, M. M. Van Dommelen, K. Lanke, W. J. G. Melchers, P. H. G. M. Willems, and F. J. M. van Kuppeveld. 2008. Functional analysis of picornavirus 2B proteins: effects on calcium homeostasis and intracellular protein trafficking. J. Virol. 823782-3790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Delhaye, S., V. van Pesch, and T. Michiels. 2004. The leader protein of Theiler's virus interferes with nucleocytoplasmic trafficking of cellular proteins. J. Virol. 784357-4362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de los Santos, T., S. de Avila Botton, R. Weiblen, and M. J. Grubman. 2006. The leader proteinase of foot-and-mouth disease virus inhibits the induction of beta interferon mRNA and blocks the host innate immune response. J. Virol. 801906-1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deszcz, L., E. Gaudernak, E. Kuechler, and J. Seipelt. 2005. Apoptotic events induced by human rhinovirus infection. J. Gen. Virol. 861379-1389. [DOI] [PubMed] [Google Scholar]
- 33.Devaney, M. A., V. N. Vakharia, R. E. Lloyd, E. Ehrenfeld, and M. J. Grubman. 1988. Leader protein of foot-and-mouth disease required for cleavage of the p220 component of the cap binding protein complex. J. Virol. 624407-4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duke, G. M., and A. C. Palmenberg. 1989. Cloning and synthesis of infectious cardiovirus RNAs containing short, discrete poly(C) tracts. J. Virol. 631822-1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dvorak, C. M., D. J. Hall, M. Hill, M. Riddle, A. Pranter, J. Dillman, M. Deibel, and A. C. Palmenberg. 2001. Leader protein of encephalomyocarditis virus binds zinc, is phosphorylated during viral infection, and affects the efficiency of genome translation. Virology 290261-271. [DOI] [PubMed] [Google Scholar]
- 36.Esfandiarei, M., S. Boroomand, A. Suarez, X. Si, M. Rahmani, and B. McManus. 2007. Coxsackievirus B3 activates nuclear factor kappa B transcription factor via a phosphatidylinositol-3 kinase/protein kinase B-dependent pathway to improve host cell viability. Cell Microbiol. 92358-2371. [DOI] [PubMed] [Google Scholar]
- 37.Galluzzi, L., C. Brenner, E. Morselli, Z. Touat, and G. Kroemer. 2008. Viral control of mitochondrial apoptosis. PLoS Pathog. 4e1000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ghadge, G. D., L. Ma. S. Sato, J. Kim, and R. P. Roos. 1998. A protein critical for a Theiler's virus-induced immune system-mediated demyelinating disease has a cell type-specific antiapoptotic effect and a key role in virus persistence. J. Virol. 728605-8612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Girard, S., T. Couderc, J. Destombes, D. Thiesson, F. Delpeyroux, and B. Blondel. 1999. Poliovirus induces apoptosis in the mouse central nervous system. J. Virol. 736066-6072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gmyl, A. P., E. V. Belousov, S. V. Maslova, E. V. Khitrina, A. B. Chetverin, and V. I. Agol. 1999. Nonreplicative RNA recombination in poliovirus. J. Virol. 738958-8965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goldmacher, V. S., L. M. Bartle, A. Skaletskaya, C. A. Dionne, N. L. Kedersha, C. A. Vater, J. W. Han, R. J. Lutz, S. Watanabe, E. D. Cahir McFarland, E. D. Kieff, E. S. Mocarski, and T. Chittenden. 1999. A cytomegalovirus-encoded mitochondria-localized inhibitor of apoptosis structurally unrelated to Bcl-2. Proc. Natl. Acad. Sci. USA 9612536-12541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goldstaub, D., A. Gradi, Z. Bercovitch, Z. Grosmann, Y. Nophar, S. Luria, N. Sonenberg, and C. Kahana. 2000. Poliovirus 2A protease induces apoptotic cell death. Mol. Cell. Biol. 201271-1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gosert, R., D. Egger, and K. Bienz. 2000. A cytopathic and a cell culture adapted hepatitis A virus strain differ in cell killing but not in intracellular membrane rearrangements. Virology 266157-169. [DOI] [PubMed] [Google Scholar]
- 44.Groppo, R., and A. C. Palmenberg. 2007. Cardiovirus 2A protein associates with 40S but not 80S ribosome subunits during infection. J. Virol. 8113067-13074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grubman, M. J., M. P. Moraes, F. Diaz-San Segundo, L. Pena, and T. de Los Santos. 2008. Evading the host immune response: how foot-and-mouth disease virus has become an effective pathogen. FEMS Immunol. Med. Microbiol. 538-17. [DOI] [PubMed] [Google Scholar]
- 46.Hato, S. V., C. Ricour, B. M. Schulte, K. H. Lanke, M. de Bruijni, J. Zoll, W. J. Melchers, T. Michiels, and F. J. M. van Kuppeveld. 2007. The mengovirus leader protein blocks interferon-alpha/beta gene transcription and inhibits activation of interferon regulatory factor 3. Cell. Microbiol. 92921-2930. [DOI] [PubMed] [Google Scholar]
- 47.Henke, A., H. Launhardt, K. Klement, A. Stelzner, R. Zell, and T. Munder. 2000. Apoptosis in coxsackievirus B3-caused diseases: interaction between the capsid protein VP2 and the proapoptotic protein siva. J. Virol. 744284-4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Himeda, T., Y. Ohara, K. Asakura, Y. Kontani, and M. Sawada. 2005. A lentiviral expression system demonstrates that L* protein of Theiler's murine encephalomyelitis virus (TMEV) has an anti-apoptotic effect in a macrophage cell line. Microb. Pathog. 38201-207. [DOI] [PubMed] [Google Scholar]
- 49.Iordanov, M. S., O. P. Ryabinina, P. Schneider, and B. E. Magun. 2005. Two mechanisms of caspase 9 processing in double-stranded RNA- and virus-triggered apoptosis. Apoptosis 10153-166. [DOI] [PubMed] [Google Scholar]
- 50.Jelachich, M. L., and H. L. Lipton. 1996. Theiler's murine encephalomyelitis virus kills restrictive but not permissive cells by apoptosis. J. Virol. 706856-6861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jelachich, M. L., and H. L. Lipton. 1999. Restricted Theiler's murine encephalomyelitis virus infection in murine macrophages induces apoptosis. J. Gen. Virol. 801701-1705. [DOI] [PubMed] [Google Scholar]
- 52.Jelachich, M. L., and H. L. Lipton. 2001. Theiler's murine encephalomyelitis virus induces apoptosis in gamma interferon activated M1 differentiated myelomonocytic cells through a mechanism involving tumor necrosis factor alpha (TNF-α) and TNF-α-related apoptosis-inducing ligand. J. Virol. 755930-5938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jin, H., C. Xiao, G. Zhao, X. Du, Y. Yu, Y. Kang, and B. Wang. 2007. Induction of immature dendritic cell apoptosis by foot and mouth disease virus is an integrin receptor mediated event before viral infection. J. Cell. Biochem. 102980-991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Joo, C. H., Y. K. Kim, H. Lee, H. Hong, S. Y. Yoon, and D. Kim. 2002. Coxsackievirus B4-induced neuronal apoptosis in rat cortical cultures. Neurosci. Lett. 326175-178. [DOI] [PubMed] [Google Scholar]
- 55.Kazachkov, Y. A., T. V. Chernovskaya, E. Y. Siyanova, Y. V. Svitkin, T. Y. Ugarova, and V. I. Agol. 1982. Leader polypeptides encoded in the 5′-region of the encephalomyocarditis virus genome. FEBS Lett. 141153-156. [DOI] [PubMed] [Google Scholar]
- 56.Kempf, B. J., and D. J. Barton. 2008. Poliovirus 2APro increases viral mRNA and polysome stability coordinately in time with cleavage of eIF4G. J. Virol. 825847-5859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kong, W. P., G. D. Ghadge, and R. P. Roos. 1994. Involvement of cardiovirus leader in host cell-restricted virus expression. Proc. Natl. Acad. Sci. USA 911796-1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kong, W. P., and R. P. Roos. 1991. Alternative translation initiation site in the DA strain of Theiler's murine encephalomyelitis virus. J. Virol. 653395-3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kuechler, E., J. Seipelt, H.-D. Liebig, and W. Sommergruber. 2002. Picornavirus proteinase-mediated shutoff of host cell translation: Direct cleavage of a cellular initiation factor, p. 301-311. In B. L. Semler and E. Wimmer (ed.), Molecular biology of picornaviruses. ASM Press, Washington, DC.
- 60.Kuo, R. L., S. H. Kung, Y. Y. Hsu, and W. T. Liu. 2002. Infection with enterovirus 71 or expression of its 2A protease induces apoptotic cell death. J. Gen. Virol. 831367-1376. [DOI] [PubMed] [Google Scholar]
- 61.Lidsky, P. V., S. Hato, M. V. Bardina, A. G. Aminev, A. C. Palmenberg, F. J. M. van Kuppeveld, and V. I. Agol. 2006. Nucleocytoplasmic traffic disorder induced by cardioviruses. J. Virol. 802705-2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu, J., T. Wei, and J. Kwang. 2002. Avian encephalomyelitis virus induces apoptosis via major structural protein VP3. Virology 30039-49. [DOI] [PubMed] [Google Scholar]
- 63.Liu, J., T. Wei, and J. Kwang. 2004. Avian encephalomyelitis virus nonstructural protein 2C induces apoptosis by activating cytochrome c/caspase-9 pathway. Virology 318169-182. [DOI] [PubMed] [Google Scholar]
- 64.Madan, V., A. Castelló, and L. Carrasco. 2008. Viroporins from RNA viruses induce caspase-dependent apoptosis. Cell. Microbiol. 10437-451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Martin, U., N. Jarasch, M. Nestler, A. Rassmann, T. Munder, S. Seitz, R. Zell, P. Wutzler, and A. Henke. 2007. Antiviral effects of pan-caspase inhibitors on the replication of coxsackievirus B3. Apoptosis 12525-533. [DOI] [PubMed] [Google Scholar]
- 66.McCormick, A. L., C. D. Meiering, G. B. Smith, and E. S. Mocarski. 2005. Mitochondrial cell death suppressors carried by human and murine cytomegalovirus confer resistance to proteasome inhibitor-induced apoptosis. J. Virol. 7912205-12217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McCormick, A. L., V. L. Smith, D. Chow, and E. S. Mocarski. 2003. Disruption of mitochondrial networks by the human cytomegalovirus UL37 gene product viral mitochondrion-localized inhibitor of apoptosis. J. Virol. 77631-641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McLean, J. E., A. Ruck, A. Shirazian, F. Pooyaei-Mehr, and Z. F. Zakeri. 2008. Viral manipulation of cell death. Curr. Pharm. Des. 14198-220. [DOI] [PubMed] [Google Scholar]
- 69.Michiels, T., V. Dejong, R. Rodrigus, and C. Shaw-Jacson. 1997. Protein 2A is not required for Theiler's virus replication. J. Virol. 719549-9556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Molla, A., A. V. Paul, M. Schmid, S. K. Jang, and E. Wimmer. 1993. Studies on dicistronic polioviruses implicate viral proteinase 2Apro in RNA replication. Virology 196739-747. [DOI] [PubMed] [Google Scholar]
- 71.Morrison, J. M., and V. R. Racaniello. 2009. Proteinase 2Apro is essential for enterovirus replication in type I interferon-treated cells. J. Virol. 834412-4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Neznanov, N., A. Kondratova, K. M. Chumakov, B. Angres, B. Zhumabayeva, V. I. Agol, and A. V. Gudkov. 2001. Poliovirus protein 3A inhibits tumor necrosis factor (TNF)-induced apoptosis by eliminating the TNF receptor from the cell surface. J. Virol. 7510409-10420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nguyen, M. L., and J. A. Blaho. 2007. Apoptosis during herpes simplex virus infection. Adv. Virus Res. 6967-97. [DOI] [PubMed] [Google Scholar]
- 74.Norris, K. L., and R. J. Youle. 2008. Cytomegalovirus proteins vMIA and m38.5 link mitochondrial morphogenesis to Bcl-2 family proteins. J. Virol. 826232-6243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Park, N., P. Katikaneni, T. Skern, and K. E. Gustin. 2008. Differential targeting of nuclear pore complex proteins in poliovirus-infected cells. J. Virol. 821647-1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Peng, J. M., S. M. Liang, and C. M. Liang. 2004. VP1 of foot-and-mouth disease virus induces apoptosis via the Akt signaling pathway. J. Biol. Chem. 27952168-52174. [DOI] [PubMed] [Google Scholar]
- 77.Perfettini, J. L., T. Roumier, and G. Kroemer. 2005. Mitochondrial fusion and fission in the control of apoptosis. Trends Cell Biol. 15179-183. [DOI] [PubMed] [Google Scholar]
- 78.Piccone, M. E., E. Rieder, P. W. Mason, and M. J. Grubman. 1995. The foot-and-mouth disease virus leader proteinase gene is not required for viral replication. J. Virol. 695376-5382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Poncet, D., N. Larochette, A.-L. Pauleau, P. Boya, A.-A. Jalil, P.-F. Cartron, F. Vallette, C. Schnebelen, L. M. Bartle, A. Skaletskaya, D. Boutolleau, J.-C. Martinou, V. S. Goldmacher, G. Kroemer, and N. Zamzami. 2004. An anti-apoptotic viral protein that recruits Bax to mitochondria. J. Biol. Chem. 27922605-22614. [DOI] [PubMed] [Google Scholar]
- 80.Porter, F. W., Y. A. Bochkov, A. J. Albee, C. Wiese, and A. C. Palmenberg. 2006. A picornavirus protein interacts with Ran-GTPase and disrupts nucleocytoplasmic transport. Proc. Natl. Acad. Sci. USA 10312417-12422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Porter, F. W., and A. C. Palmenberg. 2009. Leader-induced phosphorylation of nucleoporins correlates with nuclear trafficking inhibition by cardioviruses. J. Virol. 831941-1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rasilainen, S., P. Ylipaasto, M. Roivainen, L. Bouwens, R. Lapatto, T. Hovi, and T. Otonkoski. 2004. Mechanisms of beta cell death during restricted and unrestricted enterovirus infection. J. Med. Virol. 72451-461. [DOI] [PubMed] [Google Scholar]
- 83.Ricour, C., S. Delhaye, S. V. Hato, T. D. Olenyik, B. Michel, F. J. M. van Kuppeveld, K. E. Gustin, and T. Michiels. 2009. Inhibition of mRNA export and dimerization of interferon regulatory factor 3 by Theiler's virus leader protein. J. Gen. Virol. 90177-186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Romanova, L. I., G. A. Belov, P. V. Lidsky, E. A. Tolskaya, M. S. Kolesnikova, A. G. Evstafieva, A. B. Vartapetian, D. Egger, K. Bienz, and V. I. Agol. 2005. Variability in apoptotic response to poliovirus infection. Virology 331292-306. [DOI] [PubMed] [Google Scholar]
- 85.Salako, M. A., M. J. Carter, and G. E. N. Kass. 2006. Coxsackievirus protein 2BC blocks host cell apoptosis by inhibiting caspase-3. J. Biol. Chem. 28116296-16304. [DOI] [PubMed] [Google Scholar]
- 86.Schlitt, B. P., M. Felrice, M. L. Jelachich, and H. L. Lipton. 2003. Apoptotic cells, including macrophages, are prominent in Theiler's virus-induced inflammatory, demyelinating lesions. J. Virol. 774383-4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schwarz, E. M., C. Badorff, T. S. Hiura, R. Wessely, A. Badorff, I. M. Verma, and K. U. Knowlton. 1998. NF-κB-mediated inhibition of apoptosis is required for encephalomyocarditis virus virulence: a mechanism of resistance in p50 knockout mice. J. Virol. 725654-5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shih, S. R., K. F. Weng, V. Stollar, and M. L. Li. 2008. Viral protein synthesis is required for enterovirus 71 to induce apoptosis in human glioblastoma cells. J. Neurovirol. 1453-61. [DOI] [PubMed] [Google Scholar]
- 89.Stanway, G., T. Hovi, N. J. Knowles, and T. Hyypiä. 2002. Molecular and biological basis of picornavirus taxonomy. In B. L. Semler and E. Wimmer (ed.), Molecular biology of picornaviruses. ASM Press, Washington, DC.
- 90.Svitkin, Y. V., H. Hahn, A. C. Gingras, A. C. Palmenberg, and N. Sonenberg. 1998. Rapamycin and wortmannin enhance replication of a defective encephalomyocarditis virus. J. Virol. 725811-5819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Szabadkai, G., A. M. Simoni, M. Chami, M. R. Wieckowski, R. J. Youle, and R. Rizzuto. 2004. Drp-1-dependent division of the mitochondrial network blocks intraorganellar Ca2+ waves and protects against Ca2+-mediated apoptosis. Mol. Cell 1659-68. [DOI] [PubMed] [Google Scholar]
- 92.Taimen, P., H. Berghäll, R. Vainionpää, and M. Kallajoki. 2004. NuMA and nuclear lamins are cleaved during viral infection—inhibition of caspase activity prevents cleavage and rescues HeLa cells from measles virus-induced but not from rhinovirus 1B-induced cell death. Virology 32085-98. [DOI] [PubMed] [Google Scholar]
- 93.Tan, Y. J., S. G. Lim, and W. Hong. 2007. Regulation of cell death during infection by the severe acute respiratory syndrome coronavirus and other coronaviruses. Cell. Microbiol. 92552-2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Taylor, J. M., and M. Barry. 2006. Near death experiences: poxvirus regulation of apoptotic death. Virology 344139-150. [DOI] [PubMed] [Google Scholar]
- 95.Tolskaya, E. A., L. I. Romanova, M. S. Kolesnikova, T. A. Ivannikova, E. A. Smirnova, N. T. Raikhlin, and V. I. Agol. 1995. Apoptosis-inducing and apoptosis-preventing functions of poliovirus. J. Virol. 691181-1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tsunoda, I., C. I. Kurtz, and R. S. Fujinami. 1997. Apoptosis in acute and chronic central nervous system disease induced by Theiler's murine encephalomyelitis virus. Virology 228388-393. [DOI] [PubMed] [Google Scholar]
- 97.Tsunoda, I., J. E. Libbey, and R. S. Fujinami. 2007. TGF-β1 suppresses T cell infiltration and VP2 puff B mutation enhances apoptosis in acute polioencephalitis induced by Theiler's virus. J. Neuroimmunol. 19080-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.van Pesch, V., O. van Eyll, and T. Michiels. 2001. The leader protein of Theiler's virus inhibits immediate-early alpha/beta interferon production. J. Virol. 757811-7817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Vercammen, E., J. Staal, and R. Beyaert. 2008. Sensing of viral infection and activation of innate immunity by toll-like receptor 3. Clin. Microbiol. Rev. 2113-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wark, P. A., S. L. Johnston, F. Bucchieri, R. Powell, S. Puddicombe, V. Laza-Stanca, S. T. Holgate, and D. E. Davies. 2005. Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J. Exp. Med. 201937-947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.White, E. 2006. Mechanisms of apoptosis regulation by viral oncogenes in infection and tumorigenesis. Cell Death Differ. 131371-1377. [DOI] [PubMed] [Google Scholar]
- 102.Whitton, J. L., C. T. Cornell, and R. Feuer. 2005. Host and virus determinants of picornavirus pathogenesis and tropism. Nat. Rev. Microbiol. 3765-776. [DOI] [PubMed] [Google Scholar]
- 103.Yeung, M. C., D. L. Chang, R. E. Camantigue, and A. S. Lau. 1999. Inhibitory role of the host apoptogenic gene PKR in the establishment of persistent infection by encephalomyocarditis virus in U937 cells. Proc. Natl. Acad. Sci. USA 9611860-11865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Youle, R. J., and M. Karbowski. 2005. Mitochondrial fission in apoptosis. Nat. Rev. Mol. Cell Biol. 6657-663. [DOI] [PubMed] [Google Scholar]
- 105.Zaragoza, C., M. Saura, E. Y. Padalko, E. Lopez-Rivera, T. R. Lizarbe, S. Lamas, and C. J. Lowenstein. 2006. Viral protease cleavage of inhibitor of kappa-B alpha triggers host cell apoptosis. Proc. Natl. Acad. Sci. USA 10319051-19056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zoll, J., J. M. Galama, F. J. M. van Kuppeveld, and W. J. Melchers. 1996. Mengovirus leader is involved in the inhibition of host cell protein synthesis. J. Virol. 704948-4952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zoll, J., W. J. Melchers, J. M. Galama, and F. J. M. van Kuppeveld. 2002. The mengovirus leader protein suppresses alpha/beta interferon production by inhibition of the iron/ferritin-mediated activation of NF-κB. J. Virol. 769664-9672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zoll, J., F. J. M. van Kuppeveld, J. M. Galama, and W. J. Melchers. 1998. Genetic analysis of mengovirus protein 2A: its function in polyprotein processing and virus reproduction. J. Gen. Virol. 7917-25. [DOI] [PubMed] [Google Scholar]