

Abstract
The insulin-like growth factor type 1 receptor (IGF-1R) controls aging and cellular stress, both of which play major roles in liver disease. Stimulation of insulin-like growth factor signaling can generate cell death in vitro. Here, we tested whether IGF-1R contributes to stress insult in the liver. Cholestatic liver injury was induced by bile duct ligation in control and liver-specific IGF-1R knockout (LIGFREKO) mice. LIGFREKO mice displayed less bile duct ligation-induced hepatocyte damage than controls, while no differences in bile acid serum levels or better adaptation to cholestasis by efflux transporters were found. We therefore tested whether stress pathways contributed to this phenomenon; oxidative stress, ascertained by both malondialdehyde content and heme oxygenase-1 expression, was similar in knockout and control animals. However, together with a lower level of eukaryotic initiation factor-2 α phosphorylation, the endoplasmic reticulum stress protein CHOP and its downstream pro-apoptotic target Bax were induced to lesser extents in LIGFREKO mice than in controls. Expression levels of cytokeratin 19, transforming growth factor-β1, α-smooth muscle actin, and collagen α1(I) in LIGFREKO mice were all lower than in controls, indicating reduced ductular and fibrogenic responses and increased cholestasis tolerance in these mutants. This stress resistance phenotype was also evidenced by longer post-bile duct ligation survival in mutants than controls. These results indicate that IGF-1R contributes to cholestatic liver injury, and suggests the involvement of both CHOP and Bax in this process.
The family of insulin-like growth factors (IGFs) comprises two ligands, IGF-I and IGF-II, that activate the IGF type 1 receptor (IGF-1R), a transmembrane tyrosine kinase receptor structurally and functionally related to the insulin receptor.1 It was recently discovered that IGF-1R regulates lifespan and response to oxidative stress. Although complete knockout of IGF-1R is not compatible with normal life, heterozygous knockout mice live significantly longer than control littermates.2 IGF-1R-deficient mice also show longer than control survival after exposure to paraquat and increased resistance to oxygen-induced lung injury; also, embryonic fibroblasts from these mutants are more resistant to oxidative stress induced by hydrogen peroxide.2,3 Consistent with these findings, but in apparent contradiction with pro-survival and pro-mitogenic functions of IGF-1R, transfection experiments indicate that IGF-1R can generate cell death signals in vitro.4,5 IGF signaling can also provoke apoptosis in cancer cell lines under low oxygen tension.6 In this setting, IGF-induced cell death is related to enhanced endoplasmic reticulum (ER) stress and requires CCAAT/enhancer binding protein homologous protein (CHOP).6 CHOP, also known as growth arrest- and DNA damage-inducible gene 153, is a key factor in ER stress-mediated apoptosis.7 Recent studies implicate ER stress in neurodegenerative, cardiovascular and liver diseases,8,9 and CHOP-deficient mice display resistance to apoptosis in animal models of these diseases.10,11 In particular, CHOP deficiency attenuates cell death induced by alcohol or cholestasis in the liver.12,13 Various lines of evidence, including the re-expression of fetal IGF-II, and overexpression of IGF-I, IGF-1R and its downstream signaling molecules indicate that the IGF-1R pathway is activated in liver disease.14,15,16,17 Indeed, IGF-1R may control regeneration,18 fibrogenesis15,19 and carcinogenesis20 in the liver. Hepatocellular injury is the major triggering event of the wound healing response that leads to liver fibrosis and cancer, so we investigated whether IGF-1R influences the hepatocellular stress response in the liver. We used a mouse model of liver-specific Igf1r gene inactivation that we established previously,18,21 and examined cellular stress and fibrogenic responses induced by cholestasis in these animals. We found that IGF-1R deletion confers protection against ER stress and cellular injury induced by cholestasis in the liver.
Materials and Methods
Animal Husbandry
A conditional Igf1rflox allele was generated by flanking exon 3 of Igf1r with loxP sites. Igf1rflox/+ mice were backcrossed and maintained in both 129/Sv (129) and C57Bl/6 (B6) genetic backgrounds. The excision of exon 3 inactivates the Igf1rflox allele (giving the Igf1rnull allele), which no longer produces IGF-1R.22 We obtained an efficient liver-specific knockout using mice producing Cre recombinase under control of the albumin promoter and α-fetoprotein enhancer (AlfpCre) in hepatocytes.23 AlfpCre mice were generated by injecting FVB/N zygotes and then backcrossed into B6 genetic background for more than 10 generations. The N10 B6 AlfpCre mice were then crossed with B6 Igf1rflox/flox mice. AlfpCre+/0;Igf1rflox/+ animals were mated with 129 Igf1rflox/flox mice, and Igf1rflox/flox mice identified among offspring. Approximately half the mice were also hemizygous for AlfpCre, and therefore developed Igf1r gene inactivation in hepatocytes. These conditional mutants were named LIGFREKO, for liver-specific IGF-1R knockout. Igf1rflox/flox littermates that had not inherited the AlfpCre transgene served as controls. All were B6/129 first generation (F1) hybrids and therefore of identical genetic background. Experiments were performed in compliance with national guidelines for the care and use of laboratory animals. Animals had free access to standard mouse chow and water, and were housed under specific pathogen-free conditions in temperature-controlled rooms and 12:12-hour light-dark cycles. Genotyping was performed by PCR from skin biopsy samples, as described.21 Cre-lox deletion of exon 3 in the liver was detected by triplex PCR using primers 5′-CCATGGGTGTTAAATGTAATGGC-3′, 5′-ATGAATGCTGGTGAGGGTTGTCTT-3′ and 5′-ATCTTGGAGTGGTGGGTCTGTTTC-3′, as described.21
Animal Surgery
Cholestatic liver injury was induced by double ligation and section of the common bile duct, in LIGFREKO mice and control littermates at 8 to 10 weeks of age, under isoflurane anesthesia. Cholecystectomy was performed after ligation of the cystic duct. Sham operations consisted in laparotomy and careful bile duct exposure without ligation. On postoperative days 3 or 21, livers were removed under isoflurane anesthesia and blood withdrawn from the vena cava. Liver samples were fixed in 10% formalin and embedded in paraffin for immunohistochemistry and histology, or frozen in liquid nitrogen, and stored at −80°C for DNA, RNA, and protein analyses.
Biochemistry
Serum concentrations of bile acids, total bilirubin, and aspartate and alanine aminotransferases were measured using standard methods. The malondialdehyde content of liver tissue (10% liver homogenate in 150 mmol/L KCl) was determined by measuring thiobarbituric acid-reactive substances.24
Histology, Terminal Deoxynucleotidyl Transferase dUTP Nick-End Labeling Assay, and Cytokeratin 19 Immunochemistry
Sections (5 μm thick) of formalin-fixed, paraffin-embedded liver tissue were stained with H&E for histopathological analyses. The areas of bile infarcts (coalescent dead hepatocytes in obstructive cholestasis) were measured by morphometry using digital image analysis (Mercator, Exploranova, La Rochelle, France). Bile infarct areas were measured at ×100 magnification and are expressed relative to the total surface analyzed. The pathologist who performed histological analysis was not informed of the experimental group studied. The terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assay was performed using Apoptag Plus peroxidase in situ apoptosis detection kit (Chemicon, Temecula, CA), following the manufacturer’s instructions. Sections were pretreated with proteinase K (0.02 mg/ml) for 15 minutes. The color was developed using aminoethyl carbazole peroxidase substrate kit (Vector, Burlingame, CA). The sections were counterstained with hematoxylin. Cytokeratin 19 (CK19) immunohistochemistry was performed using an anti-CK19 polyclonal antibody (1/20, Abcam, Cambridge, MA). CK19-positive areas were measured and expressed relative to the total surface analyzed.
Western Blot
Liver tissue was homogenized in radioimmunoprecipitation assay buffer (150 mmol/L NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mmol/L Tris-HCl, pH 7.5) containing a cocktail of phosphatase and protease inhibitors and centrifuged (10,000 × g) at 4°C for 10 minutes. Equal amounts (40 μg) of protein were resolved by SDS-polyacrylamide gel electrophoresis, and transferred to nitrocellulose filters (GE Health care, Buckinghamshire, UK). Blots were probed with antibodies against heme oxygenase (1:1000, US Biological, Swampscott, MA), c-Jun NH2-terminal kinase (JNK) (1:1000, Cell Signaling, Danvers, MA), phospho-JNK (p-JNK) (1:1000, Cell Signaling), eukaryotic initiation factor-2 α (eIF2α) (1:500, Santa Cruz Biotechnology, Santa Cruz, CA), and phospho-eIF2α (p-eIF2α) (1:500, Cell Signaling). Immunosignals were visualized by enhanced chemiluminescence (Pierce, Rockford, IL).
Reverse Transcription-Polymerase Chain Reaction
Total RNA was extracted from liver samples using RNA Plus Reagent (MP Biomedicals, Illkirch, France) and Mixer Mill MM 300 (Qiagen, Hilden, Germany). Complementary DNA was synthesized using random hexamer primers and Moloney murine leukemia virus reverse transcriptase (Promega, Madison, WI). Quantitative real-time PCR (QPCR) was performed using Platinum SyberGreen qPCR superMix-UDG kit (Invitrogen, Carlsbad, CA) and a LightCycler 1.5 (Roche Diagnostics, Mannheim, Germany). Forward and reverse primers used were: 5′-GCACCCAGAGCATGTACTGT-3′ and 5′-GTTATTGCCTCTCCGGATGT-3′ for Igf1r; 5′-CATGCCCAAGACTCAGAAGT-3′ and 5′-GGCTCCTCCTACATTCTGTA-3′ for Igf1; 5′-TTCCAGGTACCAATGGGGAT-3′ and 5′-ACTGAAGCGTGTCAACAAGC-3′ for Igf2; 5′-GAGTGGTGGACAGAAGCAAA-3′ and 5′-TGAGGTAGCCATGTCCAGAA-3′ for Abcb11 that encodes a bile salt export pump (Bsep); 5′-TACAGGAAGGCTCTGGTCAT-3′ and 5′-GGATCTGCCAGAGGAAGTAT-3′ for Abcc3 that encodes multidrug resistance-associated protein 3 (Mrp3); 5′-TTAGATGGGCCTCTGGTTCT-3′ and 5′-GCCCACAATTCCAACCTTT-3′ for Abcc4 that encodes Mrp4; 5′-AGGAGGAAATTACTGCCCTG-3′ and 5′-CTCAATCCGAGCAAGGTAGG-3′ for CK19; 5′-CATCCACGAAACCACCTATA-3′ and 5′-AAGGTAGACAGCGAAGCCAA-3′ for smooth muscle α-actin (α-SMA); 5′-GAAACCCGAGGTATGCTTGA-3′ and 5′-GACCAGGAGGACCAGGAAGT-3′ for collagen α(I) (Col1A1); 5′-GTCAGACATTCGGGAAGCAG-3′ and 5′-GCGTATCAGTGGGGGTCA-3′ for transforming growth factor-α (TGF-α); 5′-GGAAACGAAGAGGAAGAATC-3′ and 5′-CACTCTGTTTCCGTTTCCTA-3′ for Chop; 5′-CATGAGGATCATCAATGAGC-3′ and 5′-TGATGAAGTGTTCCATGACC-3′ for Grp78; 5′-ATGGAGCTGCAGAGGATGAT-3′ and 5′-GAAGTTGCCATCAGCAAACA-3′ for Bax; and 5′-GAGCGAAAGCATTTGCCAAG-3′ and 5′-GGCATCGTTTATGGTCGGAA-3′ for 18S.
Statistics
Data are expressed as means ± SEM. Groups were compared using Student’s t-test. Log-rank analyses were performed to compare Kaplan-Meier curves. P < 0.05 was considered to be statistically significant.
Results
To assess the role of IGF-1R in liver injury we used previously established liver IGF-1R-deficient (LIGFREKO) mice. Mice carrying a floxed Igf1r allele (Igf1rflox)22 were mated with AlfpCre transgenics expressing Cre recombinase in hepatocytes,23 to obtain LIGFREKO mutants (Igf1rflox/floxTg;AlfpCre). Inactivation of the Igf1r gene in LIGFREKO hepatocytes starts shortly before birth and is complete in 8- to 10-week-old animals. The absence of a functional Igf1r gene does not affect postnatal hepatic development or adult liver morphology.18,21 Igf1rflox/flox littermates that had not inherited AlfpCre served as controls.
Up-Regulation of IGF System Components in Cholestatic Liver Injury
Cholestatic liver injury was induced by surgical bile duct ligation (BDL). First, we examined the expression of Igf1r, Igf1, and Igf2 in the liver of sham-operated and BDL Igf1rflox/flox control mice. Quantitative RT-PCR showed a 3.5-fold increase in hepatic levels of Igf1r mRNA 3 days after BDL, and a ninefold increase after 21 days (Figure 1A), while the expression of Igf1 remained stable (Figure 1B). The expression of Igf2 showed a fourfold increase 3 days after BDL and remained at similarly high levels 21 days after BDL (Figure 1C).
Figure 1.

Changes of hepatic Igf1r, Igf1, and Igf2 expression associated with cholestatic liver injury. Liver samples from Sham or BDL control mice were examined 3 or 21 days after surgery by RT-QPCR for expression of Igf1r (A), Igf1 (B), and Igf2 (C). Data were normalized to 18S rRNA and represent means ± SEM for 4 to 6 animals. *P < 0.05. NS, not significant.
Reduction of Cholestatic Liver Injury by IGF-1R Deletion
To examine the effects of IGF-1R on cholestatic liver injury, we compared the pathological changes in control and LIGFREKO mice, 3 days after BDL, when acute hepatocellular injury is maximal.25 Serum bilirubin and bile acids increased equally after BDL in control and LIGFREKO mice (Figure 2, A and B), indicating that cholestasis was induced to similar extent in the two groups. In contrast, serum levels of alanine and aspartate aminotransferases increased to a lesser extent in LIGFREKO than in control mice (Figure 2, C and D). In control BDL mice, there was massive cell death in areas of bile infarcts, which contained TUNEL-positive hepatocytes (Figure 3A); the extent of bile infarct was significantly lower in LIGFREKO mice than controls (Figure 3, A and B). By 21 days post-BDL, only rare bile infarcts were detected (not shown), and serum concentrations of aminotransferases did not differ significantly between genotypes (aspartate aminotransferase, 471 ± 60 vs. 450 ± 43 U/L and alanine aminotransferase, 208 ± 21 vs. 221 ± 35 U/L, in LIGFREKO and controls, respectively). Since endoluminal pressure is a determinant of liver damage, LIGFREKO animals could have developed less damage because of reduced pressure in the biliary tree. Basal bile flow, however, was not different in LIGFREKO and control mice (1.55 ± 0.26 vs. 1.49 ± 0.27 μl/min/g of liver weight; means ± SEM for four animals), which excluded this possibility.
Figure 2.

Cholestasis and liver injury in LIGFREKO mice. Blood samples were collected from Sham or BDL, control and LIGFREKO mice on postoperative day 3. Panels show serum concentrations of (A) bile acids, (B) total bilirubin, (C) aspartate aminotransferase (ASAT), and (D) alanine aminotransferase (ALAT). Data represent means ± SEM for 5 to 10 animals. *P < 0.05; **P < 0.01; ***P < 0.005.
Figure 3.

Bile infarcts in LIGFREKO mice. Liver samples were collected from Sham or BDL, control and LIGFREKO mice on postoperative day 3, stained for H&E and for TUNEL. A: Liver was morphologically normal in control and LIGFREKO sham groups. After BDL, bile infarcts (arrows) were smaller in LIGFREKO than in control mice. Clusters of TUNEL-positive hepatocytes were observed only in bile infarct areas (asterisk). Micrographs are representative of 5 to 10 animals in each group. B: Infarct area was assessed by digital image analysis. Data are expressed as a percentage of whole sections and represent means ± SEM for 10 animals. *P < 0.05.
A Role for Adaptive Transport Mechanisms in Liver Protection
Biochemical and histopathological findings indicated that the absence of IGF-1R confers protection against acute cholestatic liver injury. Cholestasis causes alterations in hepatic expression of ATP-binding cassette transporters, in particular overexpression of Abcc3/Mrp3 and Abcc4/Mrp4 baso-lateral efflux pumps,26,27 whereas expression of the canalicular export pump Abcb11/Bsep is maintained or moderately down-regulated.28 Accordingly, we found that 3 days after BDL, Abcc3/Mrp3 and Abcc4/Mrp4 expression was substantially increased in controls (Figure 4, A and B), whereas Abcb11/Bsep expression tended to be lower (Figure 4C; not significant). In LIGFREKO mice, BDL-induced changes in Abcc3/Mrp3 and Abcb11/Bsep expression were similar to those in controls (Figure 4, A and C), whereas Abcc4/Mrp4 was less up-regulated in LIGFREKO than control mice (Figure 4B). Mrp4 plays an essential protective role in the adaptive response to obstructive cholestatic liver injury.29 Therefore, we could not attribute the better tolerance of cholestasis in LIGFREKO mice to enhanced adaptive transport.
Figure 4.

Post-BDL changes in efflux ABC transporters in LIGFREKO. Liver samples were collected from Sham or BDL, control and LIGFREKO mice on postoperative day 3 and were examined for (A) Mrp3, (B) Mrp4, and (C) bile salt export pump expression by RT-QPCR. Data were normalized to 18S rRNA and represent means ± SEM for 5 to 6 animals. NS, not significant; *P < 0.05; ***P < 0.005.
IGF-1R Induces Cellular Stress Pathways after Bile Duct Ligation
Next, we examined the influence of IGF-1R on the cellular stress pathways that are activated and potentially involved in hepatocellular injury caused by cholestasis. It has been suggested that bile acid-induced cell death is mediated by reactive oxygen species. In our model, the malondialdehyde concentration increased after BDL indicating that lipid peroxidation was induced; however, there was no difference between control and LIGFREKO mice (see supplemental Figure S1A at http://ajp.amjpathol.org). Similarly, hepatic expression of the oxidative stress-responsive heme oxygenase-1 was up-regulated to similar extent in both BDL groups (see supplemental Figure S1B at http://ajp.amjpathol.org). It has also been suggested that ER stress may be involved in hepatocyte injury caused by cholestasis. Two independent factors, JNK and CHOP, are activated in response to ER stress,9 and JNK transduces hepatocyte cell death caused by reactive oxygen species or bile acids.30,31 We found that although JNK activation was induced in the liver of BDL mice, the extent of JNK phosphorylation did not differ between control and LIGFREKO mice (Figure 5A). Likewise, the expression of GRP78, a key molecular chaperone promoting recovery from ER stress, was induced to the same degree in BDL control and LIGFREKO livers (Figure 5B). These findings suggest that none of JNK, GRP78, and oxidative stress is involved in the resistance associated with IGF-1R deletion in BDL-induced liver injury. CHOP is up-regulated by ER stress and is an essential mediator in ER stress-mediated apoptosis. In agreement with a previous report, we found that in the liver of control BDL mice, CHOP and the pro-apoptotic factor Bax, a downstream target in the CHOP-mediated ER stress pathway, were overexpressed (Figure 5, C and D). The induction of CHOP and Bax expression was conspicuously lower in LIGFREKO BDL mice than controls (Figure 5, C and D). In addition, the ER stress transducer eIF2α involved in the regulation of CHOP was less activated in LIGFREKO BDL mice, as indicated by a lower level of phosphorylation, than controls (Figure 5E). Altogether, these results suggested that in acute cholestatic liver injury, a CHOP-mediated ER stress pathway is induced via IGF-1R and contributes to hepatocyte cell death.
Figure 5.

Induction and IGF-1R regulation of major stress pathways associated with cholestatic liver injury. Liver samples were collected from Sham or BDL, control and LIGFREKO mice on postoperative day 3 and were examined. A: JNK activation determined by western blotting for p-JNK and total JNK protein. Blots are representative of groups of 5 animals. B: GRP78 expression measured by RT-QPCR. C: CHOP expression determined by Western blot (upper, representative blot) and RT-QPCR (lower). D: Bax expression measured by RT-QPCR. Data were normalized to 18S rRNA and represent means ± SEM for 5 to 8 animals. *P < 0.05; **P < 0.01; ***P < 0.005. E: eIF2α activation determined by Western blot analysis of p-eIF2α and total eIF2α proteins. Blots are representative of groups of 5 animals.
Effect of IGF-1R on Ductular and Fibrogenic Response in Cholestatic Liver Injury
Fibrogenesis is closely related to the ductular reaction in BDL-induced liver injury. The expression of CK19, a marker of cholangiocytes in liver, was markedly high in control mice, as assessed by quantitative RT-PCR, indicating an increase in bile duct mass as early as 3 days after BDL (Figure 6A). The induction of CK19 expression was lower in LIGFREKO mice than in controls (Figure 6A). In accordance, morphometry of CK19 immunostaining showed reduced bile duct mass in LIGFREKO BDL mice, in comparison with control BDL mice (supplemental Figure S2 at http://ajp.amjpathol.org). Marked overexpression of TGF-β1, a potent profibrogenic factor, α-SMA, a myofibroblast marker, and Col1A1, a marker of liver fibrogenesis, was observed in control mice 3 days after BDL (Figure 6, B–D). In LIGFREKO mice, the mRNAs for CK19, TGF-β1, α-SMA, and Col1A1 were all significantly less abundant than in controls (Figure 6, B–D). These results clearly indicate that the early fibrogenic response to acute cholestatic injury was attenuated in LIGFREKO mice. However, the differences between the two genotypes disappeared after 21 days (Figure 6, E–H), the time by which fibrosis is stabilized.25 Note that CK19 and α-SMA expression levels were lower at 21 days, consistent with reduced proliferation of cholangiocytes and myofibroblasts at this advanced stage of repair.
Figure 6.

Ductular and fibrogenic response to cholestatic liver injury in LIGFREKO mice. Liver samples were collected from Sham or BDL, control and LIGFREKO mice on postoperative day 3 and were examined by RT-QPCR for expression of (A) CK19, (B) TGF-β1, (C) α-SMA, and (D) Col1A1. We performed the same experiments as in (A) to (D) for animals at 21 days post-BDL (E–H). Data were normalized to 18S rRNA and represent means ± SEM for 6 to 8 animals. *P < 0.05; **P < 0.01; ***P < 0.005.
Survival of LIGFREKO Mice with Cholestatic Liver Injury
Survival may be the most relevant criterion to demonstrate protection against liver injury. If IGF-1R is important for BDL-induced liver injury, mortality related to BDL-induced liver injury should be affected by Igf1r inactivation in the liver. Ten days after BDL, seven of nine control mice, but only two of eight LIGFREKO mice, had died (Figure 7). All sham-operated animals survived 21 days after surgery. These findings provided further evidence that IGF-1R makes a significant contribution to the poor tolerance of acute liver injury.
Figure 7.
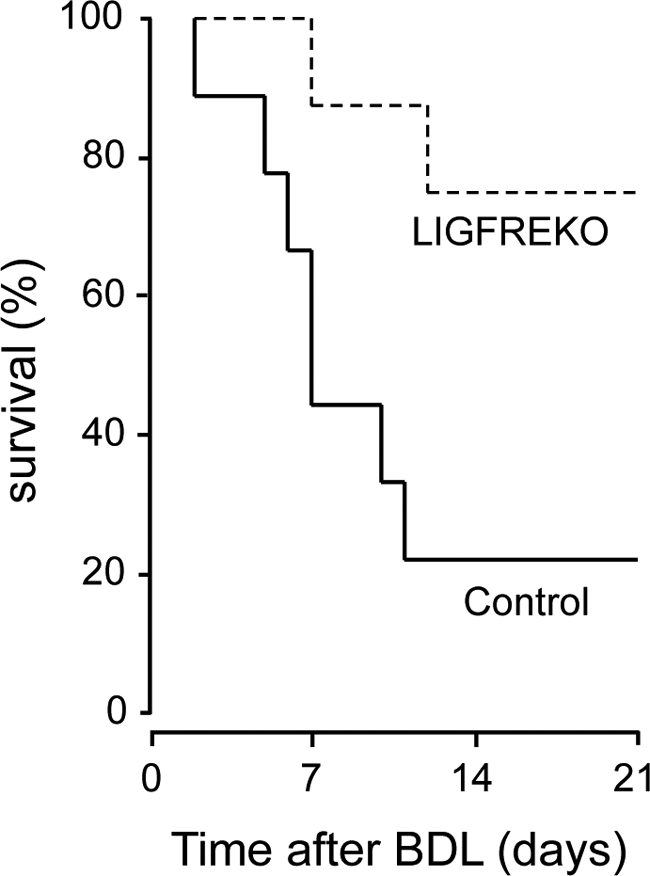
Impact of IGF-1R on survival in BDL mice. Survival after BDL was monitored for 21 days and censored thereafter. Kaplan-Meier survival curves for 9 control and 8 LIGFREKO mice were compared (P < 0.05, log-rank test).
Discussion
Choleostasis, a common feature of many forms of liver disease, is characterized by the retention of bile acids in liver and serum. The accumulation of bile acids damages liver cells through mechanisms that are largely unknown. Here, we demonstrate the pivotal role of an IGF-1R-dependent stress pathway. Hepatocytes express little IGF-1R and receptor abundance in liver is relatively low.32 However, high levels of IGF-I production, together with increased expression of IGF-1R and IGF-II after BDL are sufficient for significant biological effects. To study the consequences of IGF-1R loss-of-function, we inactivated the Igf1r gene in hepatocytes. In our experimental model, the LIGFREKO mice were subjected to BDL at the age of 8 to 10 weeks, by which age the Igf1r gene is completely inactivated in hepatocytes.18 There is little if any possibility that Igf1r null hepatocytes received compensatory IGF signaling through estrogen or insulin receptor cross talk. Estrogen receptor signaling modulates expression and activation of the IGF pathway components. Conversely, signal transduction molecules downstream of IGF-1R enhance estrogen receptor signaling.33,34 Yet, several studies have demonstrated that blockade of the IGF-1R pathway also diminished estrogen receptor-dependent signaling, which is opposite of what one would expect if there was compensatory signaling via estrogen receptors. Since hepatocytes of LIGFREKO mice completely lack IGF-1R, synergistic interaction with insulin receptors via hybrid receptor formation can also be excluded.
The Igf1r deletion from liver cells caused a marked reduction in hepatocellular injury after BDL, as ascertained by the differences in serum levels of aminotransferases and areas of bile infarct. No difference in bile acid serum level or in efflux transporter expression was found that could account for the better tolerance of BDL in LIGFREKO mice. Mrp4 was less induced in the liver of LIGFREKO than control mice, further arguing against a role of adaptive transport in the observed protection. However, this lack of induction may be related to reduced ER stress, because Mrp4 is regulated by ER stress factors, including NFE2-related factor 2.35,36
Both oxidative stress and ER stress are induced in cholestatic liver injury.13,37,38 Malondialdehyde and heme oxygenase-1 expression were both increased in the liver of BDL mice. These changes were not attenuated in LIGFREKO mice, suggesting that the protection conferred by IGF-1R deletion was independent of oxidative stress. Together with previous studies showing that antioxidants do not affect BDL-induced liver injury37 or bile acid-induced hepatocyte apoptosis,39 our findings support the view that reactive oxygen species are not major mediators of hepatocellular injury in cholestasis. A direct causal relationship between the ER stress protein CHOP and hepatocyte apoptosis in cholestasis has been established,13 and we observed that CHOP induction by cholestasis was attenuated in the absence of IGF-1R. CHOP is a transcription factor activated by ER stress that induces apoptosis via the translocation of Bax from the cytosol to mitochondria.7,40 In the BDL experimental model, hepatocyte apoptosis has been shown to be associated with Bax overexpression and translocation.13,41 Here, we report that reduced hepatocyte damage was associated with a decrease in the overexpression of both CHOP and Bax in LIGFREKO mice. ER stress also signals through GRP78 chaperone protein and JNK activation, both of which were similarly induced by BDL, irrespective of whether or not cell death was prevented by the IGF-1R deletion (Figure 5, A and B), or by CHOP deletion.13 Our findings suggest that a CHOP-mediated ER stress pathway is possibly activated by IGF-1R, and that this pathway could make a significant contribution to hepatocyte cell death in cholestasis. CHOP induction by ER stress is nearly completely abolished in PERK−/− cells and in cells that express non-phosphorylatable eIF2α (S51A).42,43 Here, we show that eIF2α was less activated in LIGFREKO BDL mice than in controls, suggesting that the PERK/eIF2α signaling pathway may play a role in the regulation of CHOP by IGF-1R. However, we cannot exclude that the observed changes may also be part of a more global adaptive response, involving other molecular pathways.
IGF-I is a potent inhibitor of apoptosis,44 and it was reported recently that IGF-I could protect MCF-7 breast cancer cells and NIH/3T3 fibroblasts from ER stress-induced apoptosis.45 Nevertheless, IGF-I has also been identified as a positive regulator of CHOP expression,46 and, in hypoxic cancer cells, IGFs induce apoptosis through increased ER stress response by a CHOP-mediated mechanism.6 The cellular response to IGF-1R activation may therefore depend on a balance between survival and apoptosis signaling. Possibly, damage associated with BDL-induced cholestasis is too severe for the cells to recover, and IGF-1R interacts with stress pathways to induce an apoptogenic program. Lethal events in BDL animals occurred within 10 days, ie, the period of acute hepatocellular injury.25 After BDL, significantly more control than LIGFREKO mice died, providing further evidence that IGF-1R is deleterious in acute liver injury.
Cholestasis causes liver fibrosis, a process in which hepatocyte injury activates matrix-producing myofibroblastic cells, primarily by TGF-β-dependent mechanisms. Lower levels of TGF-β1, α-SMA, and Col1A1, 3 days after BDL, clearly indicated that the fibrogenic response was reduced in LIGFREKO mice, which was explained by less hepatocellular injury. In cholestatic disorders, liver fibrosis develops in close correlation with a ductular reaction.47 Cytokeratin 19, a marker of the biliary lineage, was induced to a lesser extent in the liver of LIGFREKO than control mice, consistent with the current view that hepatocyte damage triggers the proliferation of ductular cells, which then leads to scar production.48,49 However, cholangiocytes are also a direct target of IGF-I mitogenic effects in BDL animals50 and it was previously shown that the AlfpCre transgene we used in this study drives Cre recombinase expression also in cholangiocytes (in addition to hepatocytes, but not in any other cell type in the liver).23,51 Therefore, IGF-1R deletion in these cells may have contributed to the lack of cholangiocyte expansion. Three weeks after BDL, there was no longer evidence of the previous protection against fibrosis in LIGFREKO mice. A possible explanation is that there is selection during recovery from BDL, such that only animals with good protection against insult and fibrosis survive. However, we cannot exclude the possibility that the CHOP-mediated stress insult occurring via IGF-1R is restricted to acute injury, as recently proposed in a study involving BDL experiments in CHOP-knockout mice.13
Circulating IGF-I levels are decreased in advanced liver disease. IGF-I replacement has beneficial effects on nutritional status, malabsorption, osteopenia, and hypogonadism in experimental cirrhosis; IGF-I also improves liver function and reduces fibrosis in such conditions.52 Therapy allowing sustained hepatic expression of IGF-I is being considered as a strategy to halt the progression of liver cirrhosis.53 In several reports using rat models of liver injury or aging, exogenous IGF-I rescued liver cells from apoptosis.52,53,54,55,56 In LIGFREKO mice, in contrast, endogenous IGF-I signaling was suppressed before hepatic lesions were induced by BDL. Constitutively reduced IGF signaling can increase cellular stress resistance through adaptive changes in gene expression and cellular physiology 2,3,57,58 In addition, when administered intravenously or by gene therapy, IGF-I reaches all cell types in the liver and may also act on extra-hepatic tissues, which likely generates indirect effects that cannot be distinguished from direct effects on hepatocytes. In LIGFREKO mice, the inactivation of IGF-1R is targeted to liver epithelial cells, allowing us to selectively test the physiological role of IGF signaling in these cells. Effects of IGF on hepatocytes are thus excluded, while in other liver cell populations, eg, endothelial cells, Kupffer cells or stellate cells, IGF signaling is preserved. From our previous results in liver regeneration18 we infer that persistently diminished IGF-1R signaling in LIFREKO mutants can both down-regulate cell renewal and promote resistance to hepatocellular stress. This conclusion is in line with currently proposed pathophysiological functioning of somatotropic signals.59 The underlying mechanism may be IGF-dependent regulation of a large number of target genes, possibly through epigenetic control, as previously observed in similar context.57,60,61 Collectively, our findings indicate that the IGF-I pathway can aggravate liver damage in stressful conditions. This possibility should be taken into account in the design of IGF-I replacement therapy for cirrhotic patients.
Supplementary Material
Acknowledgments
We thank Isabelle Renault for animal care, Elisabeth Lasnier and Nelly Bosselut for biochemistry, Alex Edelman and Associates (SARL) for language revision, and Christéle Desbois-Mouthon for comments.
Footnotes
Address reprint requests to Chantal Housset, M.D., Ph.D., Inserm UMR_S 938, CdR Saint-Antoine, Faculté de Médecine Pierre et Marie Curie, Site Saint-Antoine, 27 rue Chaligny, 75012 Paris, France. E-mail: chantal.housset@inserm.fr.
Supported by ANR grants NT05-3 42491, R06452DS, EU Network of Excellence LifeSpan (036894), and Académie de Médecine.
Supplemental material for this article can be found on http://ajp.amjpathol.org.
References
- Adams TE, Epa VC, Garrett TP, Ward CW. Structure and function of the type 1 insulin-like growth factor receptor. Cell Mol Life Sci. 2000;57:1050–1093. doi: 10.1007/PL00000744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzenberger M, Dupont J, Ducos B, Leneuve P, Geloen A, Even PC, Cervera P, Le Bouc Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature. 2003;421:182–187. doi: 10.1038/nature01298. [DOI] [PubMed] [Google Scholar]
- Ahamed K, Epaud R, Holzenberger M, Bonora M, Flejou JF, Puard J, Clement A, Henrion-Caude A. Deficiency in type 1 insulin-like growth factor receptor in mice protects against oxygen-induced lung injury. Respir Res. 2005;6:31. doi: 10.1186/1465-9921-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Lehar S, Corvi C, Payne G, O'Connor R. Expression of the insulin-like growth factor I receptor C terminus as a myristylated protein leads to induction of apoptosis in tumor cells. Cancer Res. 1998;58:570–576. [PubMed] [Google Scholar]
- Sperandio S, de Belle I, Bredesen DE. An alternative, nonapoptotic form of programmed cell death. Proc Natl Acad Sci USA. 2000;97:14376–14381. doi: 10.1073/pnas.97.26.14376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo H, Murata K, Mukai M, Ishikawa O, Inoue M. Activation of insulin-like growth factor signaling induces apoptotic cell death under prolonged hypoxia by enhancing endoplasmic reticulum stress response. Cancer Res. 2007;67:8095–8103. doi: 10.1158/0008-5472.CAN-06-3389. [DOI] [PubMed] [Google Scholar]
- Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11:381–389. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- Yoshida H. ER stress and diseases. FEBS J. 2007;274:630–658. doi: 10.1111/j.1742-4658.2007.05639.x. [DOI] [PubMed] [Google Scholar]
- Ji C, Kaplowitz N. ER stress: can the liver cope? J Hepatol. 2006;45:321–333. doi: 10.1016/j.jhep.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Oyadomari S, Koizumi A, Takeda K, Gotoh T, Akira S, Araki E, Mori M. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J Clin Invest. 2002;109:525–532. doi: 10.1172/JCI14550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajiri S, Oyadomari S, Yano S, Morioka M, Gotoh T, Hamada JI, Ushio Y, Mori M. Ischemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving CHOP. Cell Death Differ. 2004;11:403–415. doi: 10.1038/sj.cdd.4401365. [DOI] [PubMed] [Google Scholar]
- Ji C, Mehrian-Shai R, Chan C, Hsu YH, Kaplowitz N. Role of CHOP in hepatic apoptosis in the murine model of intragastric ethanol feeding. Alcohol Clin Exp Res. 2005;29:1496–1503. doi: 10.1097/01.alc.0000174691.03751.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamaki N, Hatano E, Taura K, Tada M, Kodama Y, Nitta T, Iwaisako K, Seo S, Nakajima A, Ikai I, Uemoto S. CHOP deficiency attenuates cholestasis-induced liver fibrosis by reduction of hepatocyte injury. Am J Physiol Gastrointest Liver Physiol. 2008;294:G498–G505. doi: 10.1152/ajpgi.00482.2007. [DOI] [PubMed] [Google Scholar]
- Sedlaczek N, Hasilik A, Neuhaus P, Schuppan D, Herbst H. Focal overexpression of insulin-like growth factor 2 by hepatocytes and cholangiocytes in viral liver cirrhosis. Br J Cancer. 2003;88:733–739. doi: 10.1038/sj.bjc.6600777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XZ, Chen ZX, Zhang LJ, Chen YX, Li D, Chen FL, Huang YH. Expression of insulin-like growth factor 1 and insulin-like growth factor 1 receptor and its intervention by interleukin-10 in experimental hepatic fibrosis. World J Gastroenterol. 2003;9:1287–1291. doi: 10.3748/wjg.v9.i6.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffer E, Housset C, Cacheux W, Wendum D, Desbois-Mouthon C, Rey C, Clergue F, Poupon R, Barbu V, Rosmorduc O. Gefitinib, an EGFR inhibitor, prevents hepatocellular carcinoma development in the rat liver with cirrhosis. Hepatology. 2005;41:307–314. doi: 10.1002/hep.20538. [DOI] [PubMed] [Google Scholar]
- Boissan M, Beurel E, Wendum D, Rey C, Lecluse Y, Housset C, Lacombe ML, Desbois-Mouthon C. Overexpression of insulin receptor substrate-2 in human and murine hepatocellular carcinoma. Am J Pathol. 2005;167:869–877. doi: 10.1016/S0002-9440(10)62058-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desbois-Mouthon C, Wendum D, Cadoret A, Rey C, Leneuve P, Blaise A, Housset C, Tronche F, Le Bouc Y, Holzenberger M. Hepatocyte proliferation during liver regeneration is impaired in mice with liver-specific IGF-1R knockout. FASEB J. 2006;20:773–775. doi: 10.1096/fj.05-4704fje. [DOI] [PubMed] [Google Scholar]
- Saile B, DiRocco P, Dudas J, El-Armouche H, Sebb H, Eisenbach C, Neubauer K, Ramadori G. IGF-I induces DNA synthesis and apoptosis in rat liver hepatic stellate cells (HSC) but DNA synthesis and proliferation in rat liver myofibroblasts (rMF). Lab Invest. 2004;84:1037–1049. doi: 10.1038/labinvest.3700116. [DOI] [PubMed] [Google Scholar]
- Scharf JG, Braulke T. The role of the IGF axis in hepatocarcinogenesis. Horm Metab Res. 2003;35:685–693. doi: 10.1055/s-2004-814151. [DOI] [PubMed] [Google Scholar]
- Cadoret A, Desbois-Mouthon C, Wendum D, Leneuve P, Perret C, Tronche F, Housset C, Holzenberger M. c-myc-induced hepatocarcinogenesis in the absence of IGF-I receptor. Int J Cancer. 2005;114:668–672. doi: 10.1002/ijc.20805. [DOI] [PubMed] [Google Scholar]
- Holzenberger M, Leneuve P, Hamard G, Ducos B, Perin L, Binoux M, Le Bouc Y. A targeted partial invalidation of the insulin-like growth factor I receptor gene in mice causes a postnatal growth deficit. Endocrinology. 2000;141:2557–2566. doi: 10.1210/endo.141.7.7550. [DOI] [PubMed] [Google Scholar]
- Kellendonk C, Opherk C, Anlag K, Schutz G, Tronche F. Hepatocyte-specific expression of Cre recombinase. Genesis. 2000;26:151–153. doi: 10.1002/(sici)1526-968x(200002)26:2<151::aid-gene17>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Mihara M, Uchiyama M. Determination of malonaldehyde precursor in tissues by thiobarbituric acid test. Anal Biochem. 1978;86:271–278. doi: 10.1016/0003-2697(78)90342-1. [DOI] [PubMed] [Google Scholar]
- Georgiev P, Jochum W, Heinrich S, Jang JH, Nocito A, Dahm F, Clavien PA. Characterization of time-related changes after experimental bile duct ligation. Br J Surg. 2008;95:646–656. doi: 10.1002/bjs.6050. [DOI] [PubMed] [Google Scholar]
- Wagner M, Fickert P, Zollner G, Fuchsbichler A, Silbert D, Tsybrovskyy O, Zatloukal K, Guo GL, Schuetz JD, Gonzalez FJ, Marschall HU, Denk H, Trauner M. Role of farnesoid X receptor in determining hepatic ABC transporter expression and liver injury in bile duct-ligated mice. Gastroenterology. 2003;125:825–838. doi: 10.1016/s0016-5085(03)01068-0. [DOI] [PubMed] [Google Scholar]
- Denk GU, Soroka CJ, Takeyama Y, Chen WS, Schuetz JD, Boyer JL. Multidrug resistance-associated protein 4 is up-regulated in liver but down-regulated in kidney in obstructive cholestasis in the rat. J Hepatol. 2004;40:585–591. doi: 10.1016/j.jhep.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Lee JM, Trauner M, Soroka CJ, Stieger B, Meier PJ, Boyer JL. Expression of the bile salt export pump is maintained after chronic cholestasis in the rat. Gastroenterology. 2000;118:163–172. doi: 10.1016/s0016-5085(00)70425-2. [DOI] [PubMed] [Google Scholar]
- Mennone A, Soroka CJ, Cai SY, Harry K, Adachi M, Hagey L, Schuetz JD, Boyer JL. Mrp4−/− mice have an impaired cytoprotective response in obstructive cholestasis. Hepatology. 2006;43:1013–1021. doi: 10.1002/hep.21158. [DOI] [PubMed] [Google Scholar]
- Conde de la Rosa L, Schoemaker MH, Vrenken TE, Buist-Homan M, Havinga R, Jansen PL, Moshage H. Superoxide anions and hydrogen peroxide induce hepatocyte death by different mechanisms: involvement of JNK and ERK MAP kinases. J Hepatol. 2006;44:918–929. doi: 10.1016/j.jhep.2005.07.034. [DOI] [PubMed] [Google Scholar]
- Higuchi H, Grambihler A, Canbay A, Bronk SF, Gores GJ. Bile acids up-regulate death receptor 5/TRAIL-receptor 2 expression via a c-Jun N-terminal kinase-dependent pathway involving Sp1. J Biol Chem. 2004;279:51–60. doi: 10.1074/jbc.M309476200. [DOI] [PubMed] [Google Scholar]
- Kim J, Wende AR, Sena S, Theobald HA, Soto J, Sloan C, Wayment BE, Litwin SE, Holzenberger M, Leroith D, Abel ED. IGF-1 receptor signaling is required for exercise-induced cardiac hypertrophy. Mol Endocrinol. 2008;22:2531–2543. doi: 10.1210/me.2008-0265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont J, Le Roith D. Insulin-like growth factor 1 and oestradiol promote cell proliferation of MCF-7 breast cancer cells: new insights into their synergistic effects. Mol Pathol. 2001;54:149–154. doi: 10.1136/mp.54.3.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shupnik MA. Crosstalk between steroid receptors and the c-Src-receptor tyrosine kinase pathways: implications for cell proliferation. Oncogene. 2004;23:7979–7989. doi: 10.1038/sj.onc.1208076. [DOI] [PubMed] [Google Scholar]
- Cullinan SB, Diehl JA. Coordination of ER and oxidative stress signaling: the PERK/Nrf2 signaling pathway. Int J Biochem Cell Biol. 2006;38:317–332. doi: 10.1016/j.biocel.2005.09.018. [DOI] [PubMed] [Google Scholar]
- Aleksunes LM, Slitt AL, Maher JM, Augustine LM, Goedken MJ, Chan JY, Cherrington NJ, Klaassen CD, Manautou JE. Induction of Mrp3 and Mrp4 transporters during acetaminophen hepatotoxicity is dependent on Nrf2. Toxicol Appl Pharmacol. 2008;226:74–83. doi: 10.1016/j.taap.2007.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron V, Muriel P. Role of glutathione, lipid peroxidation and antioxidants on acute bile-duct obstruction in the rat. Biochim Biophys Acta. 1999;1472:173–180. doi: 10.1016/s0304-4165(99)00118-x. [DOI] [PubMed] [Google Scholar]
- Mencin A, Seki E, Osawa Y, Kodama Y, De Minicis S, Knowles M, Brenner DA. Alpha-1 antitrypsin Z protein (PiZ) increases hepatic fibrosis in a murine model of cholestasis. Hepatology. 2007;46:1443–1452. doi: 10.1002/hep.21832. [DOI] [PubMed] [Google Scholar]
- Vrenken TE, Buist-Homan M, Faber KN, Moshage H. reactive oxygen species are not involved in bile acid induced apoptosis of hepatocytes. Hepatology. 2008;48 (S1):408A. doi: 10.1016/j.jhep.2008.07.019. doi: 10.1002/hep.22642. [DOI] [PubMed] [Google Scholar]
- Gotoh T, Terada K, Oyadomari S, Mori M. hsp70-DnaJ chaperone pair prevents nitric oxide- and CHOP-induced apoptosis by inhibiting translocation of Bax to mitochondria. Cell Death Differ. 2004;11:390–402. doi: 10.1038/sj.cdd.4401369. [DOI] [PubMed] [Google Scholar]
- Miyoshi H, Rust C, Roberts PJ, Burgart LJ, Gores GJ. Hepatocyte apoptosis after bile duct ligation in the mouse involves Fas. Gastroenterology. 1999;117:669–677. doi: 10.1016/s0016-5085(99)70461-0. [DOI] [PubMed] [Google Scholar]
- Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, Saunders T, Bonner-Weir S, Kaufman RJ. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell. 2001;7:1165–1176. doi: 10.1016/s1097-2765(01)00265-9. [DOI] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6:1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- Kooijman R. Regulation of apoptosis by insulin-like growth factor (IGF)-I. Cytokine Growth Factor Rev. 2006;17:305–323. doi: 10.1016/j.cytogfr.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Novosyadlyy R, Kurshan N, Lann D, Vijayakumar A, Yakar S, LeRoith D. Insulin-like growth factor-I protects cells from ER stress-induced apoptosis via enhancement of the adaptive capacity of endoplasmic reticulum. Cell Death Differ. 2008;15:1304–1317. doi: 10.1038/cdd.2008.52. [DOI] [PubMed] [Google Scholar]
- Entingh AJ, Law BK, Moses HL. Induction of the C/EBP homologous protein (CHOP) by amino acid deprivation requires insulin-like growth factor I, phosphatidylinositol 3-kinase, and mammalian target of rapamycin signaling. Endocrinology. 2001;142:221–228. doi: 10.1210/endo.142.1.7906. [DOI] [PubMed] [Google Scholar]
- Beaussier M, Wendum D, Fouassier L, Rey C, Barbu V, Lasnier E, Lienhart A, Scoazec JY, Rosmorduc O, Housset C. Adaptative bile duct proliferative response in experimental bile duct ischemia. J Hepatol. 2005;42:257–265. doi: 10.1016/j.jhep.2004.10.025. [DOI] [PubMed] [Google Scholar]
- Clouston AD, Powell EE, Walsh MJ, Richardson MM, Demetris AJ, Jonsson JR. Fibrosis correlates with a ductular reaction in hepatitis C: roles of impaired replication, progenitor cells and steatosis. Hepatology. 2005;41:809–818. doi: 10.1002/hep.20650. [DOI] [PubMed] [Google Scholar]
- Richardson MM, Jonsson JR, Powell EE, Brunt EM, Neuschwander-Tetri BA, Bhathal PS, Dixon JB, Weltman MD, Tilg H, Moschen AR, Purdie DM, Demetris AJ, Clouston AD. Progressive fibrosis in nonalcoholic steatohepatitis: association with altered regeneration and a ductular reaction. Gastroenterology. 2007;133:80–90. doi: 10.1053/j.gastro.2007.05.012. [DOI] [PubMed] [Google Scholar]
- Alvaro D, Metalli VD, Alpini G, Onori P, Franchitto A, Barbaro B, Glaser SS, Francis H, Cantafora A, Blotta I, Attili AF, Gaudio E. The intrahepatic biliary epithelium is a target of the growth hormone/insulin-like growth factor 1 axis. J Hepatol. 2005;43:875–883. doi: 10.1016/j.jhep.2005.04.011. [DOI] [PubMed] [Google Scholar]
- Coffinier C, Gresh L, Fiette L, Tronche F, Schutz G, Babinet C, Pontoglio M, Yaniv M, Barra J. Bile system morphogenesis defects and liver dysfunction upon targeted deletion of HNF1beta. Development. 2002;129:1829–1838. doi: 10.1242/dev.129.8.1829. [DOI] [PubMed] [Google Scholar]
- Castilla-Cortazar I, Garcia M, Muguerza B, Quiroga J, Perez R, Santidrian S, Prieto J. Hepatoprotective effects of insulin-like growth factor I in rats with carbon tetrachloride-induced cirrhosis. Gastroenterology. 1997;113:1682–1691. doi: 10.1053/gast.1997.v113.pm9352873. [DOI] [PubMed] [Google Scholar]
- Vera M, Sobrevals L, Zaratiegui M, Martinez L, Palencia B, Rodriguez CM, Prieto J, Fortes P. Liver transduction with a simian virus 40 vector encoding insulin-like growth factor I reduces hepatic damage and the development of liver cirrhosis. Gene Ther. 2007;14:203–210. doi: 10.1038/sj.gt.3302858. [DOI] [PubMed] [Google Scholar]
- Canturk NZ, Canturk Z, Ozden M, Dalcik H, Yardimoglu M, Tulubas F. Protective effect of IGF-1 on experimental liver cirrhosis-induced common bile duct ligation. Hepatogastroenterology. 2003;50:2061–2066. [PubMed] [Google Scholar]
- Sheen-Chen SM, Ho HT, Chia-Pei L, Hung KS, Eng HL. The effect of insulin-like growth factor-I on hepatocyte apoptosis after bile duct ligation in rat. Dig Dis Sci. 2006;51:2220–2224. doi: 10.1007/s10620-006-9127-z. [DOI] [PubMed] [Google Scholar]
- Puche JE, Garcia-Fernandez M, Muntane J, Rioja J, Gonzalez-Baron S, Castilla Cortazar I. Low doses of insulin-like growth factor-I induce mitochondrial protection in aging rats. Endocrinology. 2008;149:2620–2627. doi: 10.1210/en.2007-1563. [DOI] [PubMed] [Google Scholar]
- Maynard SP, Miller RA. Fibroblasts from long-lived Snell dwarf mice are resistant to oxygen-induced in vitro growth arrest. Aging Cell. 2006;5:89–96. doi: 10.1111/j.1474-9726.2006.00187.x. [DOI] [PubMed] [Google Scholar]
- Harper JM, Salmon AB, Leiser SF, Galecki AT, Miller RA. Skin-derived fibroblasts from long-lived species are resistant to some, but not all, lethal stresses and to the mitochondrial inhibitor rotenone. Aging Cell. 2007;6:1–13. doi: 10.1111/j.1474-9726.2006.00255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedernhofer LJ, Garinis GA, Raams A, Lalai AS, Robinson AR, Appeldoorn E, Odijk H, Oostendorp R, Ahmad A, van Leeuwen W, Theil AF, Vermeulen W, van der Horst GT, Meinecke P, Kleijer WJ, Vijg J, Jaspers NG, Hoeijmakers JH. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature. 2006;444:1038–1043. doi: 10.1038/nature05456. [DOI] [PubMed] [Google Scholar]
- Murakami S, Salmon A, Miller RA. Multiplex stress resistance in cells from long-lived dwarf mice. FASEB J. 2003;17:1565–1566. doi: 10.1096/fj.02-1092fje. [DOI] [PubMed] [Google Scholar]
- Murakami S. Stress resistance in long-lived mouse models. Exp Gerontol. 2006;41:1014–1019. doi: 10.1016/j.exger.2006.06.061. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.