


Abstract
Designer molecules that can specifically target pre-determined DNA sequences provide a means to modulate endogenous gene function. Different classes of sequence-specific DNA-binding agents have been developed, including triplex-forming molecules, synthetic polyamides and designer zinc finger proteins. These different types of designer molecules with their different principles of engineered sequence specificity are reviewed in this paper. Furthermore, we explore and discuss the potential of these molecules as therapeutic modulators of endogenous gene function, focusing on modulation by stable gene modification and by regulation of gene transcription.
INTRODUCTION
Knowledge of the biochemical and genetic abnormalities underlying disease allows for the rational design and development of therapies and therapeutic agents. A promising rational drug design approach is the development of agents that are designed to specifically modulate endogenous gene function through sequence-specific interaction with DNA. These designer molecules would be invaluable therapeutic tools in those cases where the molecular bases of a given disease would rationally warrant the activation or inhibition of a particular gene, or a set of genes.
DNA-sequence-specific modulation of endogenous gene function, as compared with conventional gene therapy approaches, is an attractive concept. In comparison with strategies that target downstream gene products (mRNA or protein), modulation of endogenous gene function (at the DNA level), be it permanent or transient, is potentially more effective because theoretically fewer ‘copies’ would have to be targeted. Also with respect to ‘gene augmentation’ strategies, the concept of endogenous gene modulation has appealing qualities. First, correction or up-regulation of a mutated or under-expressed endogenous gene ensures the correct stoichiomieric expression of the different splice variants of that gene, whereas an introduced gene usually concerns only a single cDNA variant. Furthermore, corrective endogenous gene targeting, unlike approaches introducing integrating gene copies, is not anticipated to suffer from loss of function due to methylation.
Several distinct mechanisms exist through which sequence-specific binding of duplex DNA can be achieved and on which strategies to make agents with designed DNA-sequence specificities have been based. First of all, there is the phenomenon of triple helix formation. Single-stranded oligonucleotides have been found to be able to bind duplex DNA in the major groove in a sequence-specific manner, thereby forming a triple helical structure. Secondly, there is the mechanism of certain synthetic polyamides that have been engineered by rational design to bind—with sequence specificity—in the minor groove of duplex DNA. Finally, there are different mechanisms by which natural DNA binding proteins are capable of binding duplex DNA in a sequence-specific manner. Several attempts have been described to exploit the DNA binding capacity of these proteins. One particular type of DNA binding protein, the Cys2His2 zinc finger type of protein, has proven to be particularly well-suited as a framework for the development of designer molecules with new sequence specificities.
First, this literature study will review the advances made in the development of agents with engineered DNA-sequence specificities, covering the modalities of triplex-forming oligonucleotides (TFOs), synthetic polyamides and Cys2His2 zinc finger proteins. Next, it explores the application of these different agents as modulators of endogenous gene function, focusing on stable gene modification and on regulation of gene transcription.
TRIPLEX-FORMING MOLECULES
One approach for tailoring molecules that specifically recognize pre-determined sequences has been based on triple helix formation. The phenomenon of triple helix formation is the binding of a third polynucleotide strand to a duplex nucleic acid, thereby forming a triple-helical structure (Fig. 1) (1). It was found that the third nucleotide strand binds in the major groove of the duplex, to one of the two Watson–Crick strands in a sequence-specific manner (2–4). This property of sequence specificity has based the potential of triplex-based applications that aim to target a single-strand DNA (TFO) to a specific target site in duplex DNA.
Figure 1.
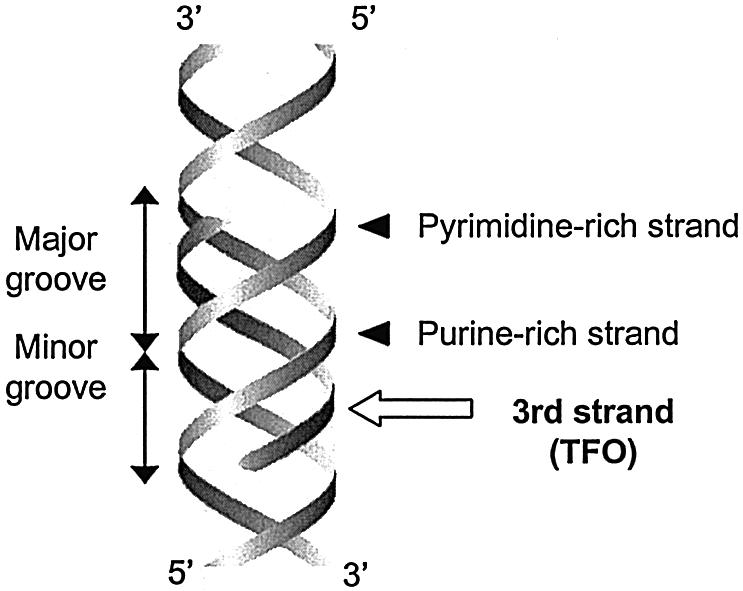
Representation of a TFO binding in the major groove of DNA.
Research on exploiting the sequence specificity of TFO binding, much of which has been reviewed recently by Faria et al. (5,6), Knauert and Glazer (7) and Vasquez and Glazer (8), showed that there are some general principles on how TFOs recognize DNA sequences. It has been established that TFOs bind along the major groove of a duplex DNA target site, to a purine-rich target site strand. Respective to this target strand, TFOs can bind either in a parallel or an anti-parallel fashion (as explained below). The nucleotide bases of the target strand are specifically recognized by TFO bases through consecutive hydrogen interactions. Essentially, each base of the TFO binds to one target strand base, thereby forming—as the targeted base is already half of a Watson–Crick base pair—a base triplet. The possible ‘base-pairing’ combinations that were found to exist between the bases in the TFO and the bases in the purine-rich target strand have given rise to generalized recognition codes for binding of TFOs to DNA (Fig. 2).
Figure 2.
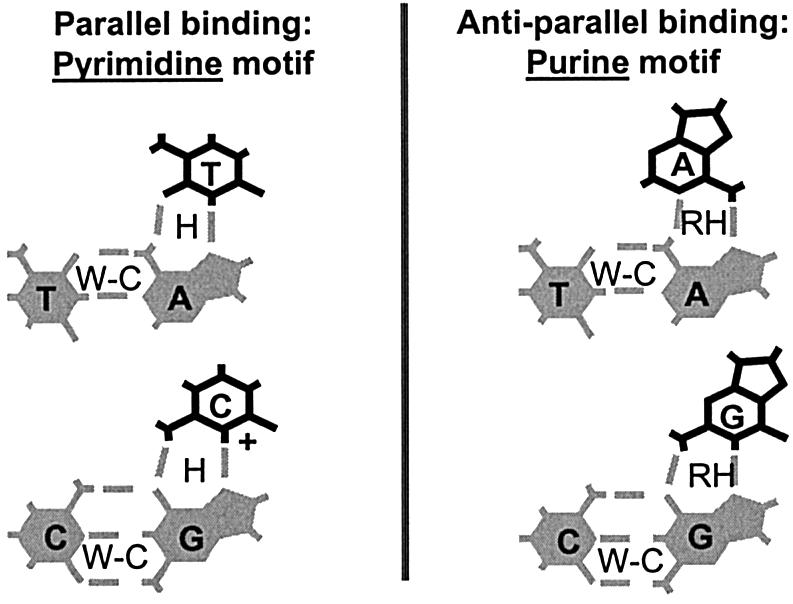
Recognition motifs for TFO-DNA binding. Note the requirement for protonation of cytosine (at N3) in the pyrimidine motif. W-C, Watson–Crick hydrogen bonds; H, Hoogsteen hydrogen bonds; RH, Reverse Hoogsteen hydrogen bonds.
The DNA-recognition codes are different for the parallel and the anti-parallel fashion of TFO binding. When a TFO binds in parallel respective to the purine-rich target strand, it does so according to the so-called pyrimidine motif (Fig. 2) (4,9,10). In this motif the adenine and guanine residues of the purine-rich target strand are, respectively, recognized—through Hoogsteen hydrogen bonds—by thymine and cytosines (hence ‘pyrimidine motif’) of the TFO. A requirement for triplex formation by pyrimidine motif TFOs is that the nitrogen atom at position 3 on the cytosine residue is protonated (11). At physiological pH levels this is not the case and, therefore, this requirement constitutes a limitation for parallel binding by unmodified TFOs in cellular contexts. In case of the anti-parallel fashion of binding, the TFO binds to the target strand according to the so-called purine motif (Fig. 2) (4,9,12). Here the adenines and guanines of the purine-rich target strand are, respectively, recognized—through Reverse Hoogsteen bonds—by adenines and guanines (hence ‘purine motif’) of the TFO.
The general principles for TFO DNA recognition as described above indicate that TFOs can recognize purine-rich target strands of duplex DNA and that they do so according to specific TFO base–target strand base interactions. Due to these characteristics, TFOs are potentially applicable as designer molecules that specifically recognize pre-determined endogenous DNA sequences. However, a major limitation to this application is that TFOs can only bind to purine-rich target strands. This limits the choice of endogenous gene target sites to polypurine-polypyrimidine stretches in duplex DNA. It would be advantageous if, besides the purine bases, the pyrimidine bases could also be specifically recognized by TFOs, as this would enable TFOs to be targeted to any desired genomic site.
As already mentioned, TFOs bind target sites that are rich in purines, implying that at least some pyrimidine bases in the purine-rich target site are apparently tolerated. Different studies have demonstrated that this is the case when those ‘inversion’ sites in the purine-rich target strand are met by certain bases in the TFO molecule (13–15). These matching bases in the TFO are different for the parallel and the anti-parallel fashion of binding. However, although these tolerations of pyrimidines among a majority of purines provide slightly more flexibility in target site choice, target strands—for TFOs consisting of natural nucleotide bases—are still required to consist mostly of purines.
As outlined above, the requirement for a polypurine-polypyrimidine target stretch for triplex formation is a great limitation to the application of TFOs. Apart from this, however, there have been more limitations to the use of TFOs. These have concerned binding affinity and specificity, uptake into cells and tissues, and in vivo stability. Major research efforts have been devoted to addressing these limitations by improving TFO characteristics through chemical modifications. Of course only those applications where TFOs are administered as chemical drugs can benefit from these improving modifications; any gene therapeutic application where TFOs are transferred to cells as genetic information to be expressed in situ cannot make use of such improvements. The many chemical TFO modifications have involved modifications to the phosphodiester backbone (16–19), the ribose (20–24) and the base moiety (25–27), as outlined below.
Modifications to the TFO backbone have often been made in attempts to increase the general affinity of the TFO for duplex DNA (16,18,19,28). This has been done by making neutral or positive backbones, thereby eliminating the electrostatic repulsion between the TFO and the duplex. Backbone modifications often involved the replacement of certain of its residues, such as the non-bridging (negatively charged) oxygen on the phosphodiester phosphate (18,19). However, in case of peptide nucleic acids (PNAs)—which are molecules that can form very stable triplexes—the whole phosphodiester backbone is replaced by an uncharged polyamide backbone (29–31). Modifications made to the ribose moiety in oligonucleotides have also been able to generate derivatives with advantageous properties (20–24). These modifications were mostly made at the 2′ position of ribose and have led to TFO derivatives that were nuclease-resistant, formed more stable triplexes, or were chemically more stable. Modifications made to nucleotide bases—mainly at carbon positions 4 and 5 of pyrimidine bases, and at carbon position 8 of purine bases (8)—have also led to some improvements to TFOs (25–27). As already mentioned, an important limitation of TFOs of the pyrimidine motif is that protonation of the nitrogen at position 3 of cytosine—which does not occur at physiological pH levels—is required for triplex formation (Fig. 2). In order to address this problem, several studies have worked on the derivation of cytosine analogs that allow for pH-independent triplex formation (25–27). Furthermore, base modifications have also been made in attempts to expand the triplex-binding code to recognize all four natural bases instead of only the purines, thereby addressing one of the major problems of TFO applicability. As mentioned above, inversion sites (i.e. sites where a pyrimidine disrupts a homopurine stretch) are tolerated to some extent when matched by certain bases. Various chemical base analogs were shown to have higher affinity at the different inversion sites than these natural bases had (32–34). Thus, the use of TFOs with chemically modified bases has potential to overcome, at least in part, the requirement of polypurine-polypyrimidine target sites.
SYNTHETIC POLYAMIDES
By exploiting the DNA-binding mechanisms of the natural products netropsin and distamycin, different groups, among which those of Dervan, Denny, Goodsell and Lown, have attempted to rationally design a mechanism of sequence-specific minor groove DNA recognition, according to a universal code, by small synthetic polyamides (35–40). Based on these efforts, a certain class of synthetic polyamides has been generated that serves well as a framework for engineered sequence specificity (Fig. 3) (35,38,41–43). These polyamides can bind in the minor groove of DNA through specific recognition of the four Watson–Crick base pairs by cognate pairs of certain amino acids, namely hydroxypyrrole (Hp), imidazole (Im) and pyrrole (Py). The principle for this polyamide mechanism of DNA recognition is a structure containing two side-by-side anti-parallel polyamide stretches. This structure—which is mostly a hairpin—allows for side-by-side amino acid pairings to be formed. In the minor groove of DNA, each of the four Watson–Crick base pairs is specifically bound, through hydrogen bonds, by one of these side-by-side pairs of Hp, Im and Py amino acids. Specifically, the polyamide amino acid pairs Py/Im, Py/Hp, Hp/Py and Im/Py respectively recognize the C–G, A–T, T–A and G–C base pairs. This framework of sequence-specific DNA-binding by synthetic polyamides is exemplified by Figure 4A.
Figure 3.
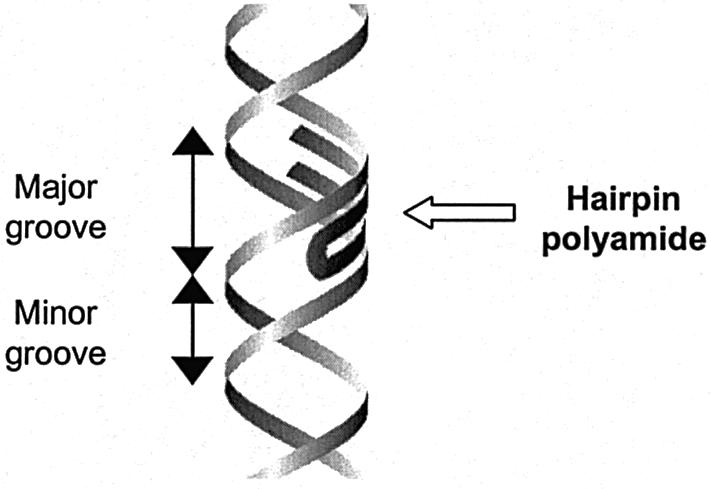
Representation of a hairpin polyamide binding in the minor groove of DNA.
Figure 4.

(A) Exemplification of the sequence-specific binding of a hairpin polyamide. (B) Representation of hairpin polyamides with extended DNA target sites. See text for details.
Being relatively small molecules, the synthetic polyamides described above are interesting candidates for DNA-sequence-specific modulators of gene expression. However, a shortcoming of the first generation of these molecules is the relative shortness of their DNA target sites. These target sites cannot be extended simply by increasing the number of contiguous amino acid pairings in the polyamide molecule, as this is thought to lead to over-bending of the polyamide structure relative to the minor groove of DNA (44). In this regard, the upper structural limit of polyamide length was found to be five amino acid pairings, which can recognize a DNA target site of up to five W-C base pairs.
To extend the DNA binding sites of polyamides beyond five base pairs, several approaches have been explored (38,42). Among these has been the extension of hairpin polyamides by insertion of β-alanine pairs that relax the curvature of the polyamide ligand (Fig. 4B) (43). Another approach for targeting longer DNA sites—of up to 10 or 11 bp—is the generation of covalent dimers of hairpin polyamides. For instance, tandem polyamide hairpins have been made by connecting two hairpins turn-to-tail (Fig. 4B) (44). In this regard, a recent study found several general motifs that can be used to link any two hairpins in the turn-to-tail manner (44). Another recent study also made covalent dimers of polyamide hairpins, but in this case they were connected turn-to-turn (Fig. 4B) (45).
DESIGNER ZINC FINGER PROTEINS
The structural principles of sequence-specific DNA recognition by DNA-binding proteins, especially certain types of transcription factors (TFs), could provide frameworks for the design of tailor-made, site-specific DNA binding proteins. In general, natural TFs have a modular structure with at least two functional domains: a DNA-binding domain (DBD) and a trans-effecting domain. In addition, some TFs contain dimerization domains, and a few, small molecule regulated TFs, harbor ligand-binding domains. The DBDs of most TFs bind to DNA through a structural motif that presents an α-helix to the major groove of DNA. There are four major types of these motifs, namely the helix–turn–helix motif, the zinc finger motif, the leucine zipper motif and the helix–loop–helix motif. As a framework for the design of new DNA binding proteins, the Cys2His2 (or class I) zinc finger motif, the most common DNA-binding motif in eukaryotes, has been used most extensively and has shown great promise (46–49).
Cys2His2 zinc finger proteins are attractive as a framework for novel DNA-binding proteins because of their relatively simple and modular structure. Besides the modularity provided by the separation of DBD and other functional domains, Cys2His2 zinc finger proteins also show modularity in their DBDs. The DBDs of Cys2His2 zinc finger proteins consist of several distinct Cys2His2 zinc finger motifs that function relatively independently from each other. Each of these motifs (Fig. 5) has a ββα structure and consists of a certain consensus sequence, namely X2-C-X2,4-C-X12-H-X3,4,5-H (where X can be any amino acid residue) (47). Its structure is stabilized by hydrophobic interactions and by coordination of a zinc ion by the two cysteine and two histidine residues. Cys2His2 zinc finger motifs typically function by binding DNA—through presentation of their α helix in the major groove—at a 3-bp sub-site. They are commonly arranged as a covalent tandem of two, three or more separate motifs, together comprising the DBD of a TF (46–49).
Figure 5.
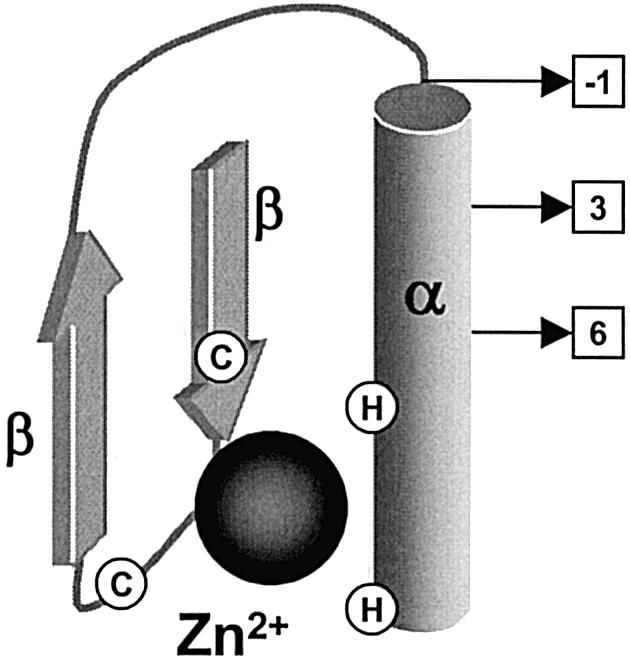
A Cys2His2 zinc finger motif showing secondary structure motifs (α, β) and the localization of the conserved cysteine (C) and histidine (H) residues that coordinate the zinc ion, which is depicted by the black sphere. The residues primarily involved with sequence-specific DNA binding are depicted by the squares. The numbers in the squares represent the amino acid positions relative to the first residue of the α-helix.
The principles of Cys2His2 zinc finger DNA recognition have been elucidated to quite an extent. Based mainly on research on the DBD of a certain natural 3-finger protein, Zif268, it is believed that Cys2His2 class zinc finger proteins primarily bind to one strand of the DNA double helix, and that they do so predominantly via positions –1, 3 and 6 of the α-helix of each of its zinc finger motifs (Fig. 5) (47,49,50). These α-helix amino acid positions would, respectively, contact the 3′-, middle and 5′-nucleotides of the 3-bp sub-site. Interestingly, these amino acids often showed striking conservations for recognition of the same nucleotides in different target sequences. However, attempts to extract from these conservations universal coding rules that relate zinc finger protein sequences and DNA binding site preferences have met with limited success (51–53). The lack of a simple recognition code of one amino acid to one nucleotide is thought to be due to the fact that apart from the primary contact residues (i.e. amino acids at positions –1, 3 and 6 in the zinc finger α-helix) flanking amino acids also play important roles in base specificity (46–49). In this regard, it has been found that the individual fingers of a zinc finger protein, rather than interacting with consecutive 3-bp DNA targets, actually recognize 1 bp overlapping 4-bp DNA targets (54). In this scheme, an additional fourth base—which is in the antisense strand relative to the first three recognized bases—is recognized by amino acid residue 2 of the zinc finger α-helix. Taking into account this fourth critical amino acid residue (at position 2 of the α-helix), a recent attempt to formulate a simple DNA recognition code for Cys2His2 zinc finger binding was relatively successful (55).
As it has proven difficult to devise universal coding rules for high affinity DNA binding, engineering of sequence specificity of Cys2His2 type zinc fingers has generally not been based on rational design alone, but has also relied on procedures that select for target DNA affinity (46–49). Many of these selection procedures used phage display of randomized zinc fingers of the natural 3-fingered protein Zif268 and have involved in vitro selection against target DNA sequences (50,56–59). Other procedures employed in vivo selection using yeast-based screening systems (60,61).
As mentioned above, each Cys2His2 zinc finger motif of a polydactyl zinc finger protein binds to its sub-site relatively independently from its neighboring motifs. The so-called ‘parallel’ selection strategy is based on the assumption that this functional independence is absolute. Ideally it is then possible to separately select a zinc finger for each individual sub-site and subsequently make combinations of these fingers—in any desired order—to design new polydactyl proteins (49). Thus, if for each of the 64 possible DNA triplet sub-sites a specific zinc finger motif were pre-selected, it would be possible to target any DNA sequence just by stitching together combinations of these predefined zinc finger motifs (51,57,58). The group of Barbas III, using phage display selection and optimization through systematic site-directed mutagenesis, was able to develop zinc finger motifs specific for the 5′-GNN-3′ and 5′-ANN-3′ families of DNA triplets, which constitute half of all possible DNA triplet sub-sites (49,57,58,62). Although not every DNA sequence can be targeted by polydactyl proteins assembled from these motifs, it is believed that enough suitable, genome-specific target sites will exist in a given promoter region of interest (49).
Making polydactyl zinc finger proteins using the parallel selection strategy has the advantage that once the zinc finger motifs are selected and optimized, new proteins can easily be constructed, from pre-defined motifs, using standard PCR methods. Ideally, no further design or selection will then be required. However, in practice, functional independence of neighboring zinc finger motifs is not absolute and, therefore, some cases require further optimization of the selected motifs (46–49).
The Pabo group addressed concerns of functional dependence of neighboring fingers by developing a ‘sequential’ selection strategy (59). In this strategy, the individual fingers are sequentially selected in the context of their neighboring fingers, thereby preventing potential problems that could be caused by target site overlap. Since any desired sequence can be targeted, this strategy seems especially useful in those cases where there is no flexibility in target choice. A disadvantage of sequential selection is, however, that it requires the generation of and selection from multiple zinc finger libraries for each new polydactyl protein to be made (46–49,59).
A selection method that combines some of the advantages of the parallel and sequential approach has been developed more recently (50). This ‘bipartite’ strategy makes use of two pre-generated libraries, in each of which one-and-a-half fingers of a three finger protein are partially (affecting only the key residues of sequence recognition) randomized. In one library the N-terminal part of the protein is randomized and in the other the C-terminal. Selection from these two libraries is carried out in parallel against the two halves (respectively the 3′-end and the 5′-end half) of the target sequence of interest. After phage display, the selections of the two libraries are recombined and final selections are performed against the full target sequence to obtain the optimal recombinant protein.
Like the sequential selection strategy, the bipartite system addresses the concerns of target-site overlap and also has the potential to target any desired sequence. The advantage over sequential selection is that, due to the use of pre-generated libraries, construction time would be shortened drastically. For the construction of each new polydactyl protein, the bipartite approach requires, however, multiple rounds of phage display selection, which in case of the parallel selection strategy, with its pre-defined finger motifs, is not necessary (47–50).
Each of the above-mentioned engineering strategies generated 3-fingered zinc finger proteins that were targeted to sites of 9–10 bp. However, proteins targeted to extended sites (of 16–18 bp) should have increased affinity for their targets and would potentially be genome-specific. Therefore, it has been attempted to generate longer proteins by covalently stitching together more than three fingers using canonical linkers, i.e. the short linkers by which the fingers in natural 3-fingered proteins (like Zif268) are linked (Fig. 6A). However, the resulting 6- or 9-fingered proteins exhibited relatively low increases in binding affinity, as compared to their 3-fingered constituents (63). It is assumed that these results are due to the proteins with more than 3 fingers linked canonically being too rigid and thus having to force too much unfolding of the DNA duplex.
Figure 6.

Polydactyl zinc finger proteins with extended DNA-target sites. (A) Two 3-finger units linked by a canonical linker. (B) Two 3-finger units linked by a flexible linker. (C) Two 3-finger units linked by a structured linker. (D) Three 2-finger units linked by slightly-longer-than-canonical linkers. (E) Two 2-finger units linked non-covalently through dimerization domains.
In recognition of the difficulties encountered with canonical linkers, there have been several other approaches of covalently linking together zinc finger units using non-canonical linkers (Fig. 6). In one study, two 3-finger units were linked by flexible linkers that were four or seven residues longer (respectively spanning gaps of up to 1 and 2 bp) than canonical linkers. The resulting 6-fingered proteins showed more than 6000-fold tighter binding than the 3-fingered constituent parts (64). Other studies with multi-fingered proteins containing flexible linkers also showed tighter binding, but of another magnitude, namely 100- to 500-fold (46). Another approach for the covalent linkage of zinc finger units employed structured linkers. TFIIIA finger 4 and a non-sequence-specific zinc finger have been used to link two 3-finger units (65). These structured linkers were shown to be capable of spanning up to 10 bp of non-bound DNA and generally resulted in the binding of target sequences with more specificity and affinity as compared to the use of long flexible linkers. Yet another approach of covalent linking involves slightly longer-than-canonical linkers which have been used to make 6-finger proteins by linking three 2-fingered units together (66). Interestingly, these ‘3 × 2’ 6-fingered proteins were shown to be more specific for their targets then their ‘2 × 3’ 6-finger counterparts.
Non-covalent linkage of 2- or 3-fingered units also provides a means to extend DNA target sites. Zinc finger units can be linked by dimerization if fused to dimerization domains (Fig. 6E). In this regard, homodimerization of 2-fingered proteins has been mediated by genetic fusion to Gal4 dimerization domains (67). In another study, homo-dimerization of 2-fingered proteins was mediated by peptides that had evolved in vitro from random sequences (68). In a third study, homo- and heterodimerization of 2-fingered proteins was accomplished by the leucine zippers of fos and jun (69).
GENE MODIFICATION
In the previous sections, several types of designer molecules with engineered sequence specificities were described. These designer molecules differ in DNA-binding mechanisms and other properties, and consequently have different potencies for different types of gene modulation applications. By linking activators or repressors to the designer molecules, the expression of target genes can be artificially induced or repressed, as discussed—along with other strategies—in the next section, which deals with non-covalent transcription modulation. The present section will explore the potential of designer DNA binding molecules as modulators of endogenous gene function through stable gene modification.
A challenging gene therapy direction is the induction of stable, site-specific gene modifications. In respective disease contexts, it could be advantageous to either inactivate or correct a certain endogenous gene. Sequence-specific designer molecules can potentially contribute in bringing about such gene modifications. Two mechanisms by which designer molecules have been shown to contribute to covalent gene modification are (i) targeted mutagenesis, and (ii) targeted stimulation of homologous recombination and thereby stimulation of gene repair. The status of the research on stable gene modification will be described for each of the designer molecule types.
Gene modification by triplex-forming molecules
There have been various strategies for triplex-directed DNA damage. These have included those where TFOs are covalently linked to DNA-reactive agents (70–80). The rationale of these strategies is that the sequence-specific TFO conjugate mediates co-localization of the tethered mutagenic agent with the site targeted for mutagenesis. However, besides mutagen–TFO conjugates, TFO molecules alone have also been demonstrated to increase mutagenesis (74,75,81,82).
Several types of DNA reactive agents that have been conjugated to TFOs for the purpose of directed mutagenesis are photoinducable crosslinkers (70–76), radioactive agents (77), alkylating groups (78,79) and certain molecules that can recruit DNA-damaging cellular enzymes (80). Psoralen is a photoinducible crosslinker and is the most extensively studied mutagenic agent that has been conjugated to TFOs (70–76). When this mutagen is subjected to UV-A irradiation, it becomes cross-linked to the DNA which results in mutations. Psoralen–TFO conjugates have mediated gene mutagenesis at targeted sites on extrachromosomal reporter plasmids, both in vitro and in vivo (70–72), and on chromosomal sequences in mammalian cells (73–76). In a recent study, a TFO conjugated to a radioactive agent (125I) was used to produce sequence-specific breaks within the human multidrug resistance (mdr1) gene of cultured cells (77). Another recent study made use of TFOs conjugated—at both the 5′ and 3′ ends—to an alkylating agent (phenylacetate mustard) (78). Using a reporter plasmid with a preformed triple helix, this bis-conjugate was able to suppress HER-2/neu promoter activity by 60–70% in cancer cells. Usage of a different type of alkylating agent, 2-amino-6-vinylpurine, was also reported recently (79). It was shown that in vitro formed triplexes between TFOs bearing this new nucleoside derivative and a reporter plasmid were able to deliver—during passage of the TFO treated plasmid in repair-deficient human cells—site-specific modifications to a supF reporter gene. Yet another approach for triplex-mediated gene mutagenesis is the recruitment, by modified TFOs, of cellular enzymes capable of inflicting DNA damage. Recruiters that have been used for this purpose are camptothecin and rebeccamycin, both of which function by recruiting the cellular enzyme topoisomerase I. The feasibility of these procedures was shown in vitro (80).
Interestingly, TFOs that are not conjugated to any mutagen are also capable of directing mutagenesis, albeit to a lesser extent than psoralen-TFOs (74,75,81,82). It has been suggested that this mutagenicity of unconjugated TFOs is due to provocation—by the triplex structures alone—of certain DNA-repair pathways that are not error-proof in their DNA processing (81). An early study that had capitalized on this intrinsic mutagenicity of TFOs was the first work that provided evidence of efficacy of targeted mutagenesis of genomic sequences in live animals (82). In this study systemic administration of TFOs to transgenic mice containing a chromosomal cognate triplex target site resulted in a 5-fold enhancement of mutation frequency at that site. Although the observed efficiency was relatively low, this study showed that triplex-mediated mutagenesis holds some promise in in vivo contexts.
Another mechanism by which TFOs have been shown to stably modify genes is the induction of homologous recombination and thereby the stimulation of gene repair. Strategies that aim to repair genes through homologous recombination are generally limited to the low frequency of homologous events in mammalian cells (83). DNA damage, especially when including double-strand breaks, was shown to enhance the frequency of homologous recombination (84–86). As TFOs have the capability to direct site-specific DNA damage (see above), they are potential tools that can enhance homologous recombination (and thereby gene repair).
The feasibility of TFOs as inducers of recombination was shown both with psoralen conjugated (87–89) and non-conjugated TFOs (90–93). In some of these studies, certain bi-functional molecules were used, namely TFOs that were linked to a short recombination donor DNA fragment (90,93). In this approach the TFO would, besides sensitizing a target site to recombination, also function to direct the homologous donor to that site. Other studies provoked homologous recombination between direct repeats located on either extra- or intrachromosomal reporter constructs. An example of the latter is a study that was able to induce intrachromosomal recombination in mouse cells through intracellular production of ssDNA triplex-forming molecules (93).
Gene modification by synthetic polyamides
In contrast to triplex-forming molecules, synthetic polyamides have not been shown to exert DNA modifying effects by themselves. However, analogous to triplex-forming molecules, polyamides can be coupled to mutagenic agents and, thereby, have applicability for directed mutagenesis and, potentially, for stimulation of gene repair by homologous recombination. Until now, there have only been a few demonstrations of mutagenic polyamide-conjugates (94–96). In these reports, polyamides that were conjugated to DNA alkylating moieties cyclopropylpyrroloindole (95) and chlorambucil (94) were able to damage and crosslink SV40 DNA, predominantly at their designated 6–7-bp target sites. Furthermore, these polyamide conjugates exhibited inhibiting effects on cell cycling and cell growth. Of significance, the chlorambucil-polyamide compound was found to be more potent in this respect than its parent compound chlorambucil, which is routinely used as an anti-tumor drug. Although the above studies demonstrate the feasibility of polyamide-conjugate mediated mutagenesis, it remains to be established whether these types of compounds are capable of directing mutagenesis to specific target genes. In accordance, stimulation of homologous recombination by targeted mutagenesis has also not been demonstrated yet.
Gene modification by designer zinc finger proteins
In principle, designer zinc finger proteins are capable of gene modification by the same two mechanisms as TFOs, namely by directing mutagenesis to specific sites, and based on this, by increasing the occurrence of recombination events at specific sites. Several studies showed that both these mechanisms of gene modification could be mediated by certain designer zinc finger proteins that were composed of a zinc finger DNA binding unit genetically fused to the non-specific cleavage domains of FokI (97–104). These ‘chimeric nucleases’, which have been reviewed by Chandrasegaran and Smith (105), can deliver site-specific double strand breaks (DSBs) to DNA and, as DSBs cause localized increases in recombination rates, have potential as stimulators of gene targeting. Two recent back-to-back publications in Science described enhanced gene targeting with chimeric nucleases (102,104). One study showed stimulated targeting of an integrated GFP reporter construct in human 293 cells (104), while the other demonstrated increased targeting of the yellow gene on the X chromosome in Drosophila (102).
REGULATION OF GENE TRANSCRIPTION
A promising application for designer DNA-binding agents is the therapeutic modulation of endogenous genes at the root of their function, namely at the level of transcription. In various disease contexts, it would be very beneficial to have the capability of transiently turning on or off specific endogenous genes. Therefore, tools capable of gene-specific modulation of endogenous gene transcription could be of great therapeutic value. The different designer DNA-binding agents described in this study—TFOs, polyamides, designer zinc finger proteins—all have potential as such tools. Here their applicability as modulators of gene transcription will be explored.
There are several general mechanisms that designer DNA-binding agents have employed to modulate gene transcription (Fig. 7). These mechanisms of transcription regulation include (i) blocking of transcription elongation by targeting a site within the transcribed region of the gene (Fig. 7A), (ii-a) blocking or (ii-b) promoting transcription initiation by targeting a site within the regulatory region of a gene (Fig. 7B), and (iii-a) repression or (iii-b) activation of gene transcription by fusion of an appropriate trans-effector domain to the DNA-binding agent (Fig. 7C). In this section, it will be investigated to what extent the different designer molecules have employed these distinct modes of transcription modulation.
Figure 7.

Mechanisms by which designer DNA-binding agents can modulate gene transcription. (A) Inhibition of transcription elongation by targeting within the transcribed region of a gene. (B) Inhibition or stimulation of transcription initiation by targeting within the regulatory region of a gene. (C) Activation or repression of gene transcription by conjugation to an appropriate effector domain (ED).
Regulation of gene transcription by triplex-forming molecules
The first reported case of triplex-mediated modulation of transcription employed blocking of transcription initiation (mechanism ii-a). In this mechanism, the DNA-binding agent interferes with the binding of transcriptional activators or with the formation of the transcription initiation complex. In that particular study, triplex formation was targeted to the regulatory region of the c-myc gene and this resulted in inhibition of transcription in vitro (106). Since then, there have been more studies that demonstrate inhibition of transcription by TFOs targeted to regulatory sequences of genes. These studies were performed either in vitro or in vivo, on extrachromosomal (107–112), but also on endogenous targets. Modulation of expression of endogenous gene targets has been successful for a broad range of genes including c-myc (113,114), interleukin-2 receptor (115), aldehyde dehydrogenase (116), GM-CSF (117), HER2/neu (118), monocyte chemoattractant protein-1 (MCP-1) (119) and Ets2 (120).
In addition to transcription inhibition, triplex formation targeted to the regulatory region of a gene could potentially result in transcription activation (mechanism ii-b). This is the case when the triplex would interfere with the binding of a transcriptional repressor. This principle was shown in a study where a TFO—designed to specifically bind at a repressor binding site—was able to up-regulate murine immunoglobulin heavy chain transcription in B cell culture lines (121).
Targeting triplex formation to sites within the transcribed sequence of a gene may inhibit transcription by bringing RNA polymerase to a halt, that is by blocking transcription elongation (mechanism i). This mechanism has been demonstrated, both with (extrachromosomal) reporter constructs (122–124) and with endogenous genes (125–127). In general, it has been found that blocking of elongation with DNA oligonucleotides that have natural (unmodified) phosphodiester backbones is hard to accomplish, probably due to the requirement that triplex stability must be sufficiently high to physically arrest the RNA polymerase. Accordingly, most studies that block transcription elongation by TFO binding do so by covalent crosslinking of the TFO and duplex DNA. However, several studies that use N3′-P5′ phosphoramidate linkages instead of the normal phosphodiester backbone report inhibition of transcription elongation by non-covalent triplexes, both in vitro (17,128) and in vivo (129,130). Endogenous genes that have been down-regulated by triplex-mediated inhibition of transcription elongation have included insulin-like growth factor-1 (IGF-1) (125), IGF-1 receptor (IGF-1R) (126), tumor necrosis factor (127), ICAM-1 (131), and survivin (132). In the above-mentioned studies targeting IGF-1 and IGF-1R genes, the triplex-forming molecules consisted of RNAs that were generated intracellularly. In these experiments, cells that constitutively expressed triplex-forming RNAs showed down-regulation of the respective genes.
By fusing transcriptional activator domains to designer DNA-binding molecules, they can serve as artificial transcription activators (mechanism iii-b). These artificial transcription activators would function, analogous to natural transcription activators, by recruiting the transcriptional machinery to a promoter near the transcription factor binding site. There has been one report demonstrating this mechanism for TFOs (133). A TFO conjugated to VP16, which is a 16 amino acid peptide transcriptional activator, was able to increase transcription from a plasmid construct.
Another mechanism to achieve triplex-mediated activation of transcription involves the unique way of triplex formation by bis-PNAs, which are molecules that consist of two PNA strands covalently linked together (30). Triplex formation by bis-PNA molecules involves invasion of the DNA duplex and displacement of the oligopyrimidine DNA strand. One PNA strand replaces the displaced DNA strand by base-pairing with the oligopurine target strand, while the second PNA strand is folded back to the consequent PNA:DNA duplex to form a PNA*PNA:DNA triplex. This structure, with its single stranded DNA loop resembling a transcription bubble, has been shown to function as an artificial promoter in vitro and in mammalian cells (134,135).
Regulation of gene transcription by synthetic polyamides
Employment of synthetic polyamides as transcription modulators is a relatively new field of research. However, several studies have already reported on polyamide mediated regulation of gene transcription in cellular contexts. In this regard, two studies demonstrated that polyamides targeted to regulatory sequences of genes were able to inhibit gene transcription through mechanism ii-a, that is blocking of transcription initiation. First of all, hairpin polyamides targeted within the binding site of transcription factor TFIIIA inhibited RNA polymerase III mediated transcription of the 5S RNA genes, in vitro and in cultured Xenopus kidney cells (136). In another study, hairpin polyamides were targeted at transcription factor binding sites within the HIV-I enhancer/promoter region (137). This gave inhibition of HIV-I transcription in a cell free assay and—when using two polyamides targeted at different transcription factor binding sites—to 99% inhibition of virus replication in cells. Another interesting example of blocking of transcription initiation—in cell free experiments, however—concerns polyamides designed for targeting HPV-encoded E2 (a transcription and replication factor) DNA binding sites in the HPV genome (138). This study demonstrated that a tandem hairpin polyamide indirectly inhibited binding of E2 to its DNA target sites according to a model in which these target sites are prevented from acquiring their intrinsic bend necessary for E2 protein binding.
Targeting of synthetic polyamides to transcription regulatory regions has not only been pursued for the purpose of blocking, but also for promotion of transcription initiation (mechanism ii-b). This is exemplified by a study by Coull et al. in which a HIV-LTR targeted polyamide specifically blocked binding of a certain host factor, LSF, resulting in de-repression of the LTR (139). Importantly, the increased LTR expression was observed using integrated reporter constructs in cultured cells.
Polyamides have also been used for the activation of transcription by conjugation to transcriptional activator domains (mechanism iii-b). A hairpin polyamide was linked to the 20 amino acid peptide activation domain AH (140). This 4.2 kDa molecule was able to up-regulate transcription in a cell free system. A related study attempted to make a synthetic polyamide transcriptional activator of minimal size (141). A hairpin polyamide was linked to the 16 amino acid peptide activation domain VP16. This molecule, with a total size of 3.2 kDa, was also capable of up-regulating transcription in a cell free system. More recently, Arora et al. linked transcriptional activators to DNA binding polyamides using rigid poly-l-proline linkers (142). Again in cell-free experiments, they found that an optimal linker length (36– 45 Å) exists for transcriptional activation by their polyamide conjugates.
Despite some successes in cultured cells, it remains difficult to echo the results of cell free assays in experiments using cells (143). It has been suggested that this difficulty of intracellular use of polyamides could be due to inefficient polyamide travel to the nucleus (35,144). However, with respect to the intracellular biological activity of polyamides, Laemmli’s group showed significant in vivo results using polyamides targeted to satellite regions of Drosophila chromosomes (145). When fed to Drosophila, these polyamides were able to induce altered gene expression by modifying chromatin structure.
Regulation of gene transcription by designer zinc finger proteins
An early case of designer zinc finger protein-mediated modulation of gene transcription employed a 3-finger protein that was targeted to a site deep within the transcribed region of an integrated reporter construct (51). This molecule inhibited transcription from the reporter construct, by blocking transcription elongation (mechanism i). Several other attempts to inhibit transcription by targeting within the transcribed region of a gene showed no apparent inhibition (146–148). Therefore, it is now generally believed that zinc finger proteins are not very well suited for this particular mechanism of transcription modulation (49).
Interfering with natural transcription factor binding is probably less challenging than placing a steric blockade in the path of RNA polymerase. Therefore, it is conceivable that designer zinc finger proteins will hold more promise for modulating transcription initiation (mechanism ii) than that they were shown to hold for blocking transcription elongation (mechanism i). Indeed, several studies reported that zinc finger proteins targeted within the regulatory sequences of genes were able to block transcription initiation (61,64,146, 148,149). These studies used either transiently transfected or integrated reporter constructs.
The most promising applications of designer zinc finger proteins are probably those that mimic the functional modularity of natural transcription factors. By genetically fusing engineered zinc finger DBDs to natural non-specific transcriptional EDs, it becomes possible to make new, artificial transcription factors. Several different transcriptional effector domains have been reported to be fused to designed zinc finger DBDs. These effector domains have included the activation domains VP16 (150) and p65 (151), and the repression domains KRAB (152), SID (153) and v-ErbA (154). Designer zinc finger units fused to the appropriate effector domains allow for both activation and repression of target gene transcription.
Several endogenous genes have already been modulated by these artificial zinc finger transcription factors. The first study to report on the regulation of an endogenous chromosomal gene employed a 5-fingered protein fused to two KRAB domains, to down-regulate the MDR1 multidrug resistance gene (61). Two other studies reported on 6-fingered zinc finger proteins fused to either KRAB or VP64 (a tetramer of VP16) effector domains, that were able to down- or up-regulate the ErbB-2 and ErbB-3 genes (62,155). Furthermore, a 3-finger protein fused to VP16 was able to activate erythropoietin gene transcription (156). VEGF-A gene transcription was shown to be activated by 3-fingered proteins fused to either VP16 or p65 (157). More recently, the nuclear hormone receptor PPARγ was effectively knocked down by a 6-finger protein fused to KRAB (158). Also recently, a 5-fingered zinc finger protein fused to VP16 was reported to induce bax expression (159). Furthermore, in another recent study, 3-fingered zinc finger proteins were able to specifically modulate the transcription of IGF2 and H19 genes (160). This time, transcriptional activation was performed by protein fusions containing either VP16 or p65, while repression was accomplished using the v-ErbA transcriptional repressor protein. Of significance, these designer transcription factors were able to reactivate transcriptionally silent alleles of the IGF2 and H19 genes, thus demonstrating that regulation of imprinted genes is feasible.
Recently, the therapeutic potency of artificial zinc finger transcription factors was demonstrated for the first time in a living animal model (161). After administration into two different mouse models, artificial transcription factors consisting of VP16 fused to DBDs [containing either 3 (157) or 6 (161) zinc fingers] that were targeted to regulatory sequences of VEGF-A were shown to induce angiogenesis in mouse ears and to accelerate wound healing.
DISCUSSION
Engineering of sequence specificity
Overall, all three different DNA binding modalities described in this review have proven to be suitable as frameworks for the engineering of novel sequence specificity. All three exhibit a degree of modularity in their DNA-recognition mechanisms down to the level of single nucleotide base or base-pair contacts. It is essentially this modularity that yields the amenability of these agents to manipulation of sequence specificity.
For gene modulation purposes, it would be very useful if a given type of designer molecule could be targeted to any desired sequence, so that there would be no limitation in target site choice. In this regard, TFOs have a major limitation as their DNA-recognition code is restricted to recognition of purine-rich target strands. Despite promising attempts to expand the TFO DNA-recognition code by usage of novel, synthetic base analogs, the requirement remains for the greater part of the target strand to consist of purines. In contrast, polyamides have been successfully developed to recognize all four Watson–Crick base pairs and therefore can potentially address any DNA target site. However, it is not conceivable that all possible sites would actually be bound with the necessary affinity and sufficient specificity. Based on already characterized polyamides, it has been estimated that the present generation of polyamides can target—with sufficient affinity and specificity—as much as 50% of the potential target DNA sites in any promoter (35). Compared to TFOs and polyamides, designer Cys2His2 zinc finger proteins probably exhibit the most flexibility in target site choice. It is thought that virtually any sequence can be targeted by zinc finger proteins since their generation can make use of high affinity selection against DNA targets. However, selection procedures are laborious and therefore, in practice, zinc finger proteins with novel specificities are often made by assembly from available repertoires of zinc finger domains with pre-defined specificity. Although somewhat limiting target site choice, this assembly process has proven feasible, as many of the designer zinc finger proteins discussed in this review were constructed this way.
In vivo delivery and functionality
With the ultimate goal of in vivo modulation of endogenous genes in human patients, various related issues like delivery, cellular uptake, nuclear transport and target DNA accessibility must be addressed for each of the different types of designer DNA-binding molecules. In relation to each of these issues, the different types of agents have very diverse characteristics and will therefore have distinct requirements and promises for their employment as gene modulators.
As TFOs are anionic molecules, and, compared to the synthetic polyamides, have a relatively high molecular weight (35), they suffer from poor cellular uptake when administered directly (7,35). In contrast, polyamides, which are uncharged and relatively small, are mostly quite cell-permeable. Strategies to improve direct delivery of TFOs to cells have included cationic lipids (118,162), DNA condensing agents (163), cell permeabilization agents (88,117,164) and coupling of the TFO to hydrophobic residues like cholesterol (165). Furthermore, indirect delivery of TFOs, i.e. intracellular production of TFOs by expression vectors, might also provide means to achieve more efficient delivery to cells, especially in in vivo contexts. In this regard, systems for the intracellular production of RNA (166) and of ssDNA (167) have been developed.
Regarding cellular uptake, designer zinc finger proteins could, in theory, be delivered to cells directly as a protein. However, the different studies reporting on endogenous gene regulation by designer zinc finger proteins (that were described in this study) all delivered these proteins into cells as a transgene. Vectors that have been used for the purpose of introducing zinc finger protein encoding genes to cells have included plasmids, retroviruses and adenoviruses.
In order to reach their DNA targets, designer DNA-binding molecules must, besides penetrating the cellular membrane, be transported to the nucleus. In this regard, it is hypothesized by Sedelnikova et al. that TFOs, upon delivery into cells, become trapped by unknown binding factors that keep TFOs from finding their gene targets (77). Interestingly, TFOs would efficiently be transported into the nucleus by these factors, but would remain bound to them there. This is in contrast to TFOs that were conjugated to the nuclear localization signal (NLS) peptide of SV40 large T antigene (77). These, and other NLS conjugates, are believed to be delivered into the nucleus (by the nuclear pore transport system) and, upon delivery, released by the transport protein, thereby being potentially available to interact with their DNA targets (77,168). In this regard, most designer zinc finger proteins have been genetically fused to NLSs, and therefore also travel to the nucleus via NLS-mediated mechanisms.
The field of DNA-binding polyamides has encountered difficulties with intracellular transport. Although cell-permeable and considerably smaller than some proteins capable of crossing through the nuclear pores (169), polyamides do not appear to travel to the nucleus efficiently. In a variety of cell types polyamides were found to accumulate mainly in the cytoplasm (or cytoplasmic vesicles), not in the nucleus (38,144,170). The group of Dervan has addressed these poorly understood mechanisms of polyamide cellular entry and localization in order to re-engineer polyamides with specific nuclear uptake properties (38,144). In this regard, recent studies have introduced new side-by-side amino acid pairs (benzimidazole/imidazole, imidazopyridine/pyrrole and hydroxybenzimidazole/pyrrole) that can participate in DNA minor groove recognition by synthetic polyamides (171,172). It is thought that novel polyamide classes harboring new types of amino-acid pairs may possess altered, potentially useful properties with respect to cell permeation, cellular distribution and degradation.
Once designer DNA-binding agents are delivered to the nucleus and are available for interaction with their DNA targets, the question remains whether these targets are sufficiently accessible for binding within their chromosomal contexts. In this regard, a general approach, to ensure that inaccessibility due to chromatin structure is not an issue, is to target within DNase-I hypersensitive regions, as was done in a study using designer zinc finger proteins (157). As outlined below, several studies have addressed the important issues of target accessibility and in vivo functionality.
In case of TFOs, Macris and Glazer recently showed that induction of transcriptional activity at a chromosomal target site in mammalian cells resulted in increased TFO binding (173). Their results also showed strong RNA polymerase activity-dependence of this observed enhancement of chromosomal TFO targeting, indicating that the active transcription through a triplex target region is especially favorable for triplex formation. Related to this, Majumdar et al. recently reported that the biology of the cell, specifically the cell cycle status, affects the efficiency of TFO targeting (174). They demonstrated that psoralen-linked TFOs exhibited greatest mutagenic activity in S phase cells.
Polyamides exhibit unique capabilities with respect to target DNA accessibility in the context of chromatin structure. Of importance, polyamides have been shown to be capable of binding efficiently to nucleosomal DNA, even if their DNA targets are partially facing the histone octamer (175). Related to this, polyamides that bind targets within nucleosomal DNA were shown to be able to influence nucleosome translocation (176,177). Thus polyamides display potential for the regulation of endogenous genes by unique mechanisms. In fact, the in vivo feasibility of altering gene expression through modification of chromatin structure using sequence-specific polyamides had already been shown earlier by Laemmli and co-workers (145).
Concerning designer zinc finger proteins, two studies that address the issue of their in vivo functionality were published recently (178,179). First, Bae et al. isolated zinc finger domains from the human genome and tested their sequence-specificities using a reporter gene expression assay in yeast (178). The subsequent usage of these characterized human zinc fingers as modular building blocks for the derivation of novel sequence-specific DNA-binding proteins resulted—upon fusion with appropriate effector domains—in the generation of potent modulators of gene transcription. It is proposed by Bae et al. that these naturally occurring human zinc fingers, when compared with engineered zinc fingers, have favorable DNA-binding properties in vivo and, furthermore, may be preferable in therapeutic settings as they are less likely to provoke host immune responses. Another intriguing approach that selects for in vivo functionality and which exemplifies the flexibility of Cys2His2 zinc finger engineering came from Blancafort et al. (179). They made repertoires of artificial transcriptional activators that contained DBDs consisting of randomized combinations of pre-characterized zinc fingers. These libraries were delivered to cells by a retroviral vector, after which infected cells were selected (by FACS) for the over-expression of certain surface proteins of interest. Several of the individual zinc finger regions that were recovered from selected cells (by PCR) were confirmed to be able to up- or down regulate (by fusion to the appropriate effector) the relevant proteins. Thus, this report demonstrated the feasibility of directly selecting zinc fingers for their in vivo functionality.
SEQUENCE-SPECIFIC MODULATION OF ENDOGENOUS GENE FUNCTION: CONCLUSIONS AND PROSPECTS
Modulation of endogenous gene function through the induction of covalent, site-specific gene modifications represents a new challenge in the field of gene therapy. Counteraction of the effects of abnormal—or abnormally regulated—genes by permanent and specific inactivation or correction of those genes is clearly a very rational and elegant concept. TFOs and designer ZFPs should hold promise in this field of gene modulation as they both have been shown to be able to deliver site-specific damage to genes, as reviewed in this study. Such TFO- or designer ZFP-delivered damage to DNA has utility as it could achieve inactivation of the targeted gene or, quite the opposite, repair of the gene concerned by serving to attract corrective homologous recombination upon co-delivery of recombination donor (gene targeting). Until now, however, targeting ratios for TFO- or designer ZFP-mediated mutagenesis and recombination have been low. Therefore, most utility of the different gene modification technologies probably lies in ex vivo approaches that can select for modified cells, or in the treatment of those types of diseases that do not require the majority of cells to be corrected. Furthermore, there could also be promise for certain diseases in which corrected cells would have an acquired growth advantage.
Modulation of endogenous gene function through regulation of gene transcription is another important and appealing gene therapy endpoint for which designer DNA-binding molecules would have great utility. Each of the types of designer DNA-binding molecules described in this study has potential as a sequence-specific transcriptional modulator of endogenous genes. TFOs have been shown to inhibit several endogenous genes by blocking the initiation or the elongation of transcription, respectively, by interfering with transcription factor binding and arresting RNA polymerase. If delivery issues are addressed, TFOs probably have most promise as transcription inhibitors, as activation of transcription by TFOs would involve the risk of the intrinsic mutagenicity of TFOs. Polyamides have been shown to block the initiation of transcription of at least one endogenous gene (136). The utility of polyamides might lie for a great part in their cellular uptake properties. It is conceivable that, in order not to compromise these properties, they will not be conjugated to too large effector domains. Therefore, if their nuclear transport difficulties could be resolved, they would probably have good potential as transcription inhibitors (or possibly also as activators) by interfering with transcription factor binding. However, polyamides certainly also hold promise as small-molecule transcription activators when conjugated to peptide effector domains of minimal size. Designer ZFPs show significant potential as artificial transcription factors. Fused to transcriptional effector domains, they were shown to be potent transcriptional mediators of several endogenous genes. Moreover, it has recently been established that they can evoke therapeutically relevant levels of effects in mouse models (161). Furthermore, they show the most versatility with respect to the usage of functional domains. Examples of potentially useful domains other than transcriptional effectors are domains with transporter function and ligand binding domains. Approaches fusing zinc finger DBDs with different types of ligand binding domains have already resulted in the construction of several types of small molecule inducible transcription factors (49,180–183).
In conclusion, in the field of therapeutic gene modulation, engineering of DNA-binding agents with designed sequence specificity represents an exciting new approach. Modulation of endogenous genes has clear advantages over conventional gene therapy strategies. However, further research is warranted for this approach to show full potency, especially on expanding the target site choice and on the issues of delivery and DNA accessibility. Nonetheless, the different types of designer DNA-binding agents described here clearly have potential as powerful sequence-specific gene modulators, by exerting their effect either through stable gene modification or through regulation of transcription.
Acknowledgments
ACKNOWLEDGEMENT
We thank Rob Hoeben (Department of Molecular Cell Biology, Leiden University Medical Center, Leiden, The Netherlands) for critical reading of the manuscript.
REFERENCES
- 1.Felsenfeld G., Davies,D.R. and Rich,A. (1957) Formation of a three stranded polynucleotide molecule. J. Am. Chem. Soc., 79, 2023–2024. [Google Scholar]
- 2.Le Doan T., Perrouault,L., Praseuth,D., Habhoub,N., Decout,J.L., Thuong,N.T., Lhomme,J. and Helene,C. (1987) Sequence-specific recognition, photocrosslinking and cleavage of the DNA double helix by an oligo-[alpha]-thymidylate covalently linked to an azidoproflavine derivative. Nucleic Acids Res., 15, 7749–7760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moser H.E. and Dervan,P.B. (1987) Sequence-specific cleavage of double helical DNA by triple helix formation. Science, 238, 645–650. [DOI] [PubMed] [Google Scholar]
- 4.Letai A.G., Palladino,M.A., Fromm,E., Rizzo,V. and Fresco,J.R. (1988) Specificity in formation of triple-stranded nucleic acid helical complexes: studies with agarose-linked polyribonucleotide affinity columns. Biochemistry, 27, 9108–9112. [DOI] [PubMed] [Google Scholar]
- 5.Faria M. and Giovannangeli,C. (2001) Triplex-forming molecules: from concepts to applications. J. Gene Med., 3, 299–310. [DOI] [PubMed] [Google Scholar]
- 6.Faria M. and Ulrich,H. (2002) The use of synthetic oligonucleotides as protein inhibitors and anticode drugs in cancer therapy: accomplishments and limitations. Curr. Cancer Drug Targets, 2, 355–368. [DOI] [PubMed] [Google Scholar]
- 7.Knauert M.P. and Glazer,P.M. (2001) Triplex forming oligonucleotides: sequence-specific tools for gene targeting. Hum. Mol. Genet., 10, 2243–2251. [DOI] [PubMed] [Google Scholar]
- 8.Vasquez K.M. and Glazer,P.M. (2002) Triplex-forming oligonucleotides: principles and applications. Q. Rev. Biophys., 35, 89–107. [DOI] [PubMed] [Google Scholar]
- 9.Vasquez K.M. and Wilson,J.H. (1998) Triplex-directed modification of genes and gene activity. Trends Biochem. Sci., 23, 4–9. [DOI] [PubMed] [Google Scholar]
- 10.Fossella J.A., Kim,Y.J., Shih,H., Richards,E.G. and Fresco,J.R. (1993) Relative specificities in binding of Watson–Crick base pairs by third strand residues in a DNA pyrimidine triplex motif. Nucleic Acids Res., 21, 4511–4515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee J.S., Johnson,D.A. and Morgan,A.R. (1979) Complexes formed by (pyrimidine)n.(purine)n DNAs on lowering the pH are three-stranded. Nucleic Acids Res., 6, 3073–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hardenbol P. and Van Dyke,M.W. (1996) Sequence specificity of triplex DNA formation: analysis by a combinatorial approach, restriction endonuclease protection selection and amplification. Proc. Natl Acad. Sci. USA, 93, 2811–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Griffin L.C. and Dervan,P.B. (1989) Recognition of thymine adenine base pairs by guanine in a pyrimidine triple helix motif. Science, 245, 967–971. [DOI] [PubMed] [Google Scholar]
- 14.Wang E., Malek,S. and Feigon,J. (1992) Structure of a G.T.A triplet in an intramolecular DNA triplex. Biochemistry, 31, 4838–4846. [DOI] [PubMed] [Google Scholar]
- 15.Jiang L. and Russu,I.M. (2001) Proton exchange and local stability in a DNA triple helix containing a G.TA triad. Nucleic Acids Res., 29, 4231–4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arya D.P. and Bruice,T.C. (1999) Triple-helix formation of DNA oligomers with methylthiourea-linked nucleosides (DNmt): a kinetic and thermodynamic analysis. Proc. Natl Acad. Sci. USA, 96, 4384–4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Escude C., Giovannangeli,C., Sun,J.S., Lloyd,D.H., Chen,J.K., Gryaznov,S.M., Garestier,T. and Helene,C. (1996) Stable triple helices formed by oligonucleotide N3′→P5′ phosphoramidates inhibit transcription elongation. Proc. Natl Acad. Sci. USA, 93, 4365–4369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dagle J.M. and Weeks,D.L. (1996) Positively charged oligonucleotides overcome potassium-mediated inhibition of triplex DNA formation. Nucleic Acids Res., 24, 2143–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ehrenmann F., Vasseur,J.J. and Debart,F. (2001) Alpha-oligonucleotides with anionic phosphodiester and cationic phosphoramidate linkages enhanced stability of DNA triple helix. Nucl. Nucl., 20, 797–799. [DOI] [PubMed] [Google Scholar]
- 20.Shimizu M., Konishi,A., Shimada,Y., Inoue,H. and Ohtsuka,E. (1992) Oligo(2′-O-methyl)ribonucleotides. Effective probes for duplex DNA. FEBS Lett., 302, 155–158. [DOI] [PubMed] [Google Scholar]
- 21.Escude C., Sun,J.S., Rougee,M., Garestier,T. and Helene,C. (1992) Stable triple helices are formed upon binding of RNA oligonucleotides and their 2′-O-methyl derivatives to double-helical DNA. C. R. Acad. Sci. III, 315, 521–525. [PubMed] [Google Scholar]
- 22.Torigoe H., Hari,Y., Sekiguchi,M., Obika,S. and Imanishi,T. (2001) 2′-O,4′-C-methylene bridged nucleic acid modification promotes pyrimidine motif triplex DNA formation at physiological pH: thermodynamic and kinetic studies. J. Biol. Chem., 276, 2354–2360. [DOI] [PubMed] [Google Scholar]
- 23.Lacroix L., Arimondo,P.B., Takasugi,M., Helene,C. and Mergny,J.L. (2000) Pyrimidine morpholino oligonucleotides form a stable triple helix in the absence of magnesium ions. Biochem. Biophys. Res. Commun., 270, 363–369. [DOI] [PubMed] [Google Scholar]
- 24.Blommers M.J., Natt,F., Jahnke,W. and Cuenoud,B. (1998) Dual recognition of double-stranded DNA by 2′-aminoethoxy-modified oligonucleotides: the solution structure of an intramolecular triplex obtained by NMR spectroscopy. Biochemistry, 37, 17714–17725. [DOI] [PubMed] [Google Scholar]
- 25.Lee J.S., Woodsworth,M.L., Latimer,L.J. and Morgan,A.R. (1984) Poly(pyrimidine).poly(purine) synthetic DNAs containing 5-methylcytosine form stable triplexes at neutral pH. Nucleic Acids Res., 12, 6603–6614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller P.S., Bi,G., Kipp,S.A., Fok,V. and DeLong,R.K. (1996) Triplex formation by a psoralen-conjugated oligodeoxyribonucleotide containing the base analog 8-oxo-adenine. Nucleic Acids Res., 24, 730–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cassidy S.A., Slickers,P., Trent,J.O., Capaldi,D.C., Roselt,P.D., Reese,C.B., Neidle,S. and Fox,K.R. (1997) Recognition of GC base pairs by triplex forming oligonucleotides containing nucleosides derived from 2-aminopyridine. Nucleic Acids Res., 25, 4891–4898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Escude C., Giovannangeli,C., Sun,J.S., Lloyd,D.H., Chen,J.K., Gryaznov,S.M., Garestier,T. and Helene,C. (1996) Stable triple helices formed by oligonucleotide N3′→P5′ phosphoramidates inhibit transcription elongation. Proc. Natl Acad. Sci. USA, 93, 4365–4369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cherny D.Y., Belotserkovskii,B.P., Frank-Kamenetskii,M.D., Egholm,M., Buchardt,O., Berg,R.H. and Nielsen,P.E. (1993) DNA unwinding upon strand-displacement binding of a thymine-substituted polyamide to double-stranded DNA. Proc. Natl Acad. Sci. USA, 90, 1667–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nielsen P.E. (2001) Targeting double stranded DNA with peptide nucleic acid (PNA). Curr. Med. Chem., 8, 545–550. [DOI] [PubMed] [Google Scholar]
- 31.Ray A. and Norden,B. (2000) Peptide nucleic acid (PNA): its medical and biotechnical applications and promise for the future. FASEB J., 14, 1041–1060. [DOI] [PubMed] [Google Scholar]
- 32.Durland R.H., Rao,T.S., Revankar,G.R., Tinsley,J.H., Myrick,M.A., Seth,D.M., Rayford,J., Singh,P. and Jayaraman,K. (1994) Binding of T and T analogs to CG base pairs in antiparallel triplexes. Nucleic Acids Res., 22, 3233–3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stilz H.U. and Dervan,P.B. (1993) Specific recognition of CG base pairs by 2-deoxynebularine within the purine.purine.pyrimidine triple-helix motif. Biochemistry, 32, 2177–2185. [DOI] [PubMed] [Google Scholar]
- 34.Li J.S., Fan,Y.H., Zhang,Y., Marky,L.A. and Gold,B. (2003) Design of triple helix forming C-glycoside molecules. J. Am. Chem. Soc., 125, 2084–2093. [DOI] [PubMed] [Google Scholar]
- 35.Dervan P.B. (2001) Molecular recognition of DNA by small molecules. Bioorg. Med. Chem., 9, 2215–2235. [DOI] [PubMed] [Google Scholar]
- 36.Denny W.A. (2001) DNA minor groove alkylating agents. Curr. Med. Chem., 8, 533–544. [DOI] [PubMed] [Google Scholar]
- 37.Goodsell D.S. (2001) Sequence recognition of DNA by lexitropsins. Curr. Med. Chem., 8, 509–516. [DOI] [PubMed] [Google Scholar]
- 38.Dervan P.B. and Edelson,B.S. (2003) Recognition of the DNA minor groove by pyrrole-imidazole polyamides. Curr. Opin. Struct. Biol., 13, 284–299. [DOI] [PubMed] [Google Scholar]
- 39.Lown J.W. (1988) Lexitropsins: rational design of DNA sequence reading agents as novel anti-cancer agents and potential cellular probes. Anticancer Drug Des., 3, 25–40. [PubMed] [Google Scholar]
- 40.Reddy B.S., Sharma,S.K. and Lown,J.W. (2001) Recent developments in sequence selective minor groove DNA effectors. Curr. Med. Chem., 8, 475–508. [DOI] [PubMed] [Google Scholar]
- 41.Trauger J.W., Baird,E.E. and Dervan,P.B. (1996) Recognition of DNA by designed ligands at subnanomolar concentrations. Nature, 382, 559–561. [DOI] [PubMed] [Google Scholar]
- 42.Wemmer D.E. (2000) Designed sequence-specific minor groove ligands. Annu. Rev. Biophys. Biomol. Struct., 29, 439–461. [DOI] [PubMed] [Google Scholar]
- 43.White S., Szewczyk,J.W., Turner,J.M., Baird,E.E. and Dervan,P.B. (1998) Recognition of the four Watson–Crick base pairs in the DNA minor groove by synthetic ligands. Nature, 391, 468–471. [DOI] [PubMed] [Google Scholar]
- 44.Kers I. and Dervan,P.B. (2002) Search for the optimal linker in tandem hairpin polyamides. Bioorg. Med. Chem., 10, 3339–3349. [DOI] [PubMed] [Google Scholar]
- 45.Weyermann P. and Dervan,P.B. (2002) Recognition of ten base pairs of DNA by head-to-head hairpin dimers. J. Am. Chem. Soc., 124, 6872–6878. [DOI] [PubMed] [Google Scholar]
- 46.Choo Y. and Isalan,M. (2000) Advances in zinc finger engineering. Curr. Opin. Struct. Biol., 10, 411–416. [DOI] [PubMed] [Google Scholar]
- 47.Pabo C.O., Peisach,E. and Grant,R.A. (2001) Design and selection of novel Cys2His2 zinc finger proteins. Annu. Rev. Biochem., 70, 313–340. [DOI] [PubMed] [Google Scholar]
- 48.Segal D.J. and Barbas,C.F.,III (2001) Custom DNA-binding proteins come of age: polydactyl zinc-finger proteins. Curr. Opin. Biotechnol., 12, 632–637. [DOI] [PubMed] [Google Scholar]
- 49.Beerli R.R. and Barbas,C.F.,III (2002) Engineering polydactyl zinc-finger transcription factors. Nat. Biotechnol., 20, 135–141. [DOI] [PubMed] [Google Scholar]
- 50.Isalan M., Klug,A. and Choo,Y. (2001) A rapid, generally applicable method to engineer zinc fingers illustrated by targeting the HIV-1 promoter. Nat. Biotechnol., 19, 656–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Choo Y., Sanchez-Garcia,I. and Klug,A. (1994) In vivo repression by a site-specific DNA-binding protein designed against an oncogenic sequence. Nature, 372, 642–645. [DOI] [PubMed] [Google Scholar]
- 52.Wolfe S.A., Greisman,H.A., Ramm,E.I. and Pabo,C.O. (1999) Analysis of zinc fingers optimized via phage display: evaluating the utility of a recognition code. J. Mol. Biol., 285, 1917–1934. [DOI] [PubMed] [Google Scholar]
- 53.Suzuki M. and Yagi,N. (1994) DNA recognition code of transcription factors in the helix–turn–helix, probe helix, hormone receptor, and zinc finger families. Proc. Natl Acad. Sci. USA, 91, 12357–12361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim C.A. and Berg,J.M. (1996) A 2.2 Å resolution crystal structure of a designed zinc finger protein bound to DNA. Nature Struct. Biol., 3, 940–945. [DOI] [PubMed] [Google Scholar]
- 55.Sera T. and Uranga,C. (2002) Rational design of artificial zinc-finger proteins using a nondegenerate recognition code table. Biochemistry, 41, 7074–7081. [DOI] [PubMed] [Google Scholar]
- 56.Choo Y. and Klug,A. (1994) Toward a code for the interactions of zinc fingers with DNA: selection of randomized fingers displayed on phage. Proc. Natl Acad. Sci. USA, 91, 11163–11167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dreier B., Segal,D.J. and Barbas,C.F.,III (2000) Insights into the molecular recognition of the 5′-GNN-3′ family of DNA sequences by zinc finger domains. J. Mol. Biol., 303, 489–502. [DOI] [PubMed] [Google Scholar]
- 58.Segal D.J., Dreier,B., Beerli,R.R. and Barbas,C.F.,III (1999) Toward controlling gene expression at will: selection and design of zinc finger domains recognizing each of the 5′-GNN-3′ DNA target sequences. Proc. Natl Acad. Sci. USA, 96, 2758–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Greisman H.A. and Pabo,C.O. (1997) A general strategy for selecting high-affinity zinc finger proteins for diverse DNA target sites. Science, 275, 657–661. [DOI] [PubMed] [Google Scholar]
- 60.Cheng X., Boyer,J.L. and Juliano,R.L. (1997) Selection of peptides that functionally replace a zinc finger in the Sp1 transcription factor by using a yeast combinatorial library. Proc. Natl Acad. Sci. USA, 94, 14120–14125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bartsevich V.V. and Juliano,R.L. (2000) Regulation of the MDR1 gene by transcriptional repressors selected using peptide combinatorial libraries. Mol. Pharmacol., 58, 1–10. [DOI] [PubMed] [Google Scholar]
- 62.Dreier B., Beerli,R.R., Segal,D.J., Flippin,J.D. and Barbas,C.F.,III (2001) Development of zinc finger domains for recognition of the 5′-ANN-3′ family of DNA sequences and their use in the construction of artificial transcription factors. J. Biol. Chem., 276, 29466–29478. [DOI] [PubMed] [Google Scholar]
- 63.Kamiuchi T., Abe,E., Imanishi,M., Kaji,T., Nagaoka,M. and Sugiura,Y. (1998) Artificial nine zinc-finger peptide with 30 base pair binding sites. Biochemistry, 37, 13827–13834. [DOI] [PubMed] [Google Scholar]
- 64.Kim J.S. and Pabo,C.O. (1998) Getting a handhold on DNA: design of poly-zinc finger proteins with femtomolar dissociation constants. Proc. Natl Acad. Sci. USA, 95, 2812–2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Moore M., Choo,Y. and Klug,A. (2001) Design of polyzinc finger peptides with structured linkers. Proc. Natl Acad. Sci. USA, 98, 1432–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Moore M., Klug,A. and Choo,Y. (2001) Improved DNA binding specificity from polyzinc finger peptides by using strings of two-finger units. Proc. Natl Acad. Sci. USA, 98, 1437–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pomerantz J.L., Wolfe,S.A. and Pabo,C.O. (1998) Structure-based design of a dimeric zinc finger protein. Biochemistry, 37, 965–970. [DOI] [PubMed] [Google Scholar]
- 68.Wang B.S. and Pabo,C.O. (1999) Dimerization of zinc fingers mediated by peptides evolved in vitro from random sequences. Proc. Natl Acad. Sci. USA, 96, 9568–9573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wolfe S.A., Ramm,E.I. and Pabo,C.O. (2000) Combining structure-based design with phage display to create new Cys(2)His(2) zinc finger dimers. Structure Fold. Des., 8, 739–750. [DOI] [PubMed] [Google Scholar]
- 70.Havre P.A. and Glazer,P.M. (1993) Targeted mutagenesis of simian virus 40 DNA mediated by a triple helix-forming oligonucleotide. J. Virol., 67, 7324–7331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Havre P.A., Gunther,E.J., Gasparro,F.P. and Glazer,P.M. (1993) Targeted mutagenesis of DNA using triple helix-forming oligonucleotides linked to psoralen. Proc. Natl Acad. Sci. USA, 90, 7879–7883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang G., Levy,D.D., Seidman,M.M. and Glazer,P.M. (1995) Targeted mutagenesis in mammalian cells mediated by intracellular triple helix formation. Mol. Cell. Biol., 15, 1759–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Majumdar A., Khorlin,A., Dyatkina,N., Lin,F.L., Powell,J., Liu,J., Fei,Z., Khripine,Y., Watanabe,K.A., George,J. et al. (1998) Targeted gene knockout mediated by triple helix forming oligonucleotides. Nature Genet., 20, 212–214. [DOI] [PubMed] [Google Scholar]
- 74.Vasquez K.M., Wang,G., Havre,P.A. and Glazer,P.M. (1999) Chromosomal mutations induced by triplex-forming oligonucleotides in mammalian cells. Nucleic Acids Res., 27, 1176–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vasquez K.M., Dagle,J.M., Weeks,D.L. and Glazer,P.M. (2001) Chromosome targeting at short polypurine sites by cationic triplex-forming oligonucleotides. J. Biol. Chem., 276, 38536–38541. [DOI] [PubMed] [Google Scholar]
- 76.Barre F.X., Ait-Si-Ali,S., Giovannangeli,C., Luis,R., Robin,P., Pritchard,L.L., Helene,C. and Harel-Bellan,A. (2000) Unambiguous demonstration of triple-helix-directed gene modification. Proc. Natl Acad. Sci. USA, 97, 3084–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sedelnikova O.A., Karamychev,V.N., Panyutin,I.G. and Neumann,R.D. (2002) Sequence-specific gene cleavage in intact mammalian cells by 125I-labeled triplex-forming oligonucleotides conjugated with nuclear localization signal peptide. Antisense Nucleic Acid Drug Dev., 12, 43–49. [DOI] [PubMed] [Google Scholar]
- 78.Ziemba A.J., Reed,M.W., Raney,K.D., Byrd,A.B. and Ebbinghaus,S.W. (2003) A bis-alkylating triplex forming oligonucleotide inhibits intracellular reporter gene expression and prevents triplex unwinding due to helicase activity. Biochemistry, 42, 5013–5024. [DOI] [PubMed] [Google Scholar]
- 79.Nagatsugi F., Sasaki,S., Miller,P.S. and Seidman,M.M. (2003) Site-specific mutagenesis by triple helix-forming oligonucleotides containing a reactive nucleoside analog. Nucleic Acids Res., 31, e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Arimondo P.B., Bailly,C., Boutorine,A., Sun,J.S., Garestier,T. and Helene,C. (1999) Targeting topoisomerase I cleavage to specific sequences of DNA by triple helix-forming oligonucleotide conjugates. A comparison between a rebeccamycin derivative and camptothecin. C. R. Acad. Sci. III, 322, 785–790. [DOI] [PubMed] [Google Scholar]
- 81.Wang G., Seidman,M.M. and Glazer,P.M. (1996) Mutagenesis in mammalian cells induced by triple helix formation and transcription-coupled repair. Science, 271, 802–805. [DOI] [PubMed] [Google Scholar]
- 82.Vasquez K.M., Narayanan,L. and Glazer,P.M. (2000) Specific mutations induced by triplex-forming oligonucleotides in mice. Science, 290, 530–533. [DOI] [PubMed] [Google Scholar]
- 83.Vasquez K.M., Marburger,K., Intody,Z. and Wilson,J.H. (2001) Manipulating the mammalian genome by homologous recombination. Proc. Natl Acad. Sci. USA, 98, 8403–8410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Brenner D.A., Smigocki,A.C. and Camerini-Otero,R.D. (1985) Effect of insertions, deletions, and double-strand breaks on homologous recombination in mouse L cells. Mol. Cell. Biol., 5, 684–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wake C.T., Vernaleone,F. and Wilson,J.H. (1985) Topological requirements for homologous recombination among DNA molecules transfected into mammalian cells. Mol. Cell. Biol., 5, 2080–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sargent R.G., Rolig,R.L., Kilburn,A.E., Adair,G.M., Wilson,J.H. and Nairn,R.S. (1997) Recombination-dependent deletion formation in mammalian cells deficient in the nucleotide excision repair gene ERCC1. Proc. Natl Acad. Sci. USA, 94, 13122–13127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sandor Z. and Bredberg,A. (1995) Triple helix directed psoralen adducts induce a low frequency of recombination in an SV40 shuttle vector. Biochim. Biophys. Acta, 1263, 235–240. [DOI] [PubMed] [Google Scholar]
- 88.Faruqi A.F., Seidman,M.M., Segal,D.J., Carroll,D. and Glazer,P.M. (1996) Recombination induced by triple-helix-targeted DNA damage in mammalian cells. Mol. Cell. Biol., 16, 6820–6828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Luo Z., Macris,M.A., Faruqi,A.F. and Glazer,P.M. (2000) High-frequency intrachromosomal gene conversion induced by triplex-forming oligonucleotides microinjected into mouse cells. Proc. Natl Acad. Sci. USA, 97, 9003–9008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chan P.P., Lin,M., Faruqi,A.F., Powell,J., Seidman,M.M. and Glazer,P.M. (1999) Targeted correction of an episomal gene in mammalian cells by a short DNA fragment tethered to a triplex-forming oligonucleotide. J. Biol. Chem., 274, 11541–11548. [DOI] [PubMed] [Google Scholar]
- 91.Faruqi A.F., Datta,H.J., Carroll,D., Seidman,M.M. and Glazer,P.M. (2000) Triple-helix formation induces recombination in mammalian cells via a nucleotide excision repair-dependent pathway. Mol. Cell. Biol., 20, 990–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Datta H.J., Chan,P.P., Vasquez,K.M., Gupta,R.C. and Glazer,P.M. (2001) Triplex-induced recombination in human cell-free extracts. Dependence on XPA and HsRad51. J. Biol. Chem., 276, 18018–18023. [DOI] [PubMed] [Google Scholar]
- 93.Datta H.J. and Glazer,P.M. (2001) Intracellular generation of single-stranded DNA for chromosomal triplex formation and induced recombination. Nucleic Acids Res., 29, 5140–5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang Y.D., Dziegielewski,J., Wurtz,N.R., Dziegielewska,B., Dervan,P.B. and Beerman,T.A. (2003) DNA crosslinking and biological activity of a hairpin polyamide-chlorambucil conjugate. Nucleic Acids Res., 31, 1208–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang Y.D., Dziegielewski,J., Chang,A.Y., Dervan,P.B. and Beerman,T.A. (2002) Cell-free and cellular activities of a DNA sequence selective hairpin polyamide-CBI conjugate. J. Biol. Chem., 277, 42431–42437. [DOI] [PubMed] [Google Scholar]
- 96.Bando T., Narita,A., Saito,I. and Sugiyama,H. (2002) Molecular design of a pyrrole-imidazole hairpin polyamides for effective DNA alkylation. Chemistry, 8, 4781–4790. [DOI] [PubMed] [Google Scholar]
- 97.Kim Y.G., Cha,J. and Chandrasegaran,S. (1996) Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc. Natl Acad. Sci. USA, 93, 1156–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Huang B., Schaeffer,C.J., Li,Q. and Tsai,M.D. (1996) Splase: a new class IIS zinc-finger restriction endonuclease with specificity for Sp1 binding sites. J. Protein Chem., 15, 481–489. [DOI] [PubMed] [Google Scholar]
- 99.Smith J., Berg,J.M. and Chandrasegaran,S. (1999) A detailed study of the substrate specificity of a chimeric restriction enzyme. Nucleic Acids Res., 27, 674–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Smith J., Bibikova,M., Whitby,F.G., Reddy,A.R., Chandrasegaran,S. and Carroll,D. (2000) Requirements for double-strand cleavage by chimeric restriction enzymes with zinc finger DNA-recognition domains. Nucleic Acids Res., 28, 3361–3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bibikova M., Carroll,D., Segal,D.J., Trautman,J.K., Smith,J., Kim,Y.G. and Chandrasegaran,S. (2001) Stimulation of homologous recombination through targeted cleavage by chimeric nucleases. Mol. Cell. Biol., 21, 289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bibikova M., Beumer,K., Trautman,J.K. and Carroll,D. (2003) Enhancing gene targeting with designed zinc finger nucleases. Science, 300, 764. [DOI] [PubMed] [Google Scholar]
- 103.Bibikova M., Golic,M., Golic,K.G. and Carroll,D. (2002) Targeted chromosomal cleavage and mutagenesis in Drosophila using zinc-finger nucleases. Genetics, 161, 1169–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Porteus M.H. and Baltimore,D. (2003) Chimeric nucleases stimulate gene targeting in human cells. Science, 300, 763. [DOI] [PubMed] [Google Scholar]
- 105.Chandrasegaran S. and Smith,J. (1999) Chimeric restriction enzymes: what is next? Biol. Chem., 380, 841–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cooney M., Czernuszewicz,G., Postel,E.H., Flint,S.J. and Hogan,M.E. (1988) Site-specific oligonucleotide binding represses transcription of the human c-myc gene in vitro. Science, 241, 456–459. [DOI] [PubMed] [Google Scholar]
- 107.Grigoriev M., Praseuth,D., Robin,P., Hemar,A., Saison-Behmoaras,T., Dautry-Varsat,A., Thuong,N.T., Helene,C. and Harel-Bellan,A. (1992) A triple helix-forming oligonucleotide-intercalator conjugate acts as a transcriptional repressor via inhibition of NF kappa B binding to interleukin-2 receptor alpha-regulatory sequence. J. Biol. Chem., 267, 3389–3395. [PubMed] [Google Scholar]
- 108.Mayfield C., Ebbinghaus,S., Gee,J., Jones,D., Rodu,B., Squibb,M. and Miller,D. (1994) Triplex formation by the human Ha-ras promoter inhibits Sp1 binding and in vitro transcription. J. Biol. Chem., 269, 18232–18238. [PubMed] [Google Scholar]
- 109.Duval-Valentin G., Thuong,N.T. and Helene,C. (1992) Specific inhibition of transcription by triple helix-forming oligonucleotides. Proc. Natl Acad. Sci. USA, 89, 504–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kim H.G. and Miller,D.M. (1995) Inhibition of in vitro transcription by a triplex-forming oligonucleotide targeted to human c-myc P2 promoter. Biochemistry, 34, 8165–8171. [DOI] [PubMed] [Google Scholar]
- 111.Joseph J., Kandala,J.C., Veerapanane,D., Weber,K.T. and Guntaka,R.V. (1997) Antiparallel polypurine phosphorothioate oligonucleotides form stable triplexes with the rat alpha1(I) collagen gene promoter and inhibit transcription in cultured rat fibroblasts. Nucleic Acids Res., 25, 2182–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ritchie S., Boyd,F.M., Wong,J. and Bonham,K. (2000) Transcription of the human c-Src promoter is dependent on Sp1, a novel pyrimidine binding factor SPy, and can be inhibited by triplex-forming oligonucleotides. J. Biol. Chem., 275, 847–854. [DOI] [PubMed] [Google Scholar]
- 113.Postel E.H., Flint,S.J., Kessler,D.J. and Hogan,M.E. (1991) Evidence that a triplex-forming oligodeoxyribonucleotide binds to the c-myc promoter in HeLa cells, thereby reducing c-myc mRNA levels. Proc. Natl Acad. Sci. USA, 88, 8227–8231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Thomas T.J., Faaland,C.A., Gallo,M.A. and Thomas,T. (1995) Suppression of c-myc oncogene expression by a polyamine-complexed triplex forming oligonucleotide in MCF-7 breast cancer cells. Nucleic Acids Res., 23, 3594–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Orson F.M., Thomas,D.W., McShan,W.M., Kessler,D.J. and Hogan,M.E. (1991) Oligonucleotide inhibition of IL2R alpha mRNA transcription by promoter region collinear triplex formation in lymphocytes. Nucleic Acids Res., 19, 3435–3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tu G.C., Cao,Q.N. and Israel,Y. (1995) Inhibition of gene expression by triple helix formation in hepatoma cells. J. Biol. Chem., 270, 28402–28407. [DOI] [PubMed] [Google Scholar]
- 117.Kochetkova M. and Shannon,M.F. (1996) DNA triplex formation selectively inhibits granulocyte-macrophage colony-stimulating factor gene expression in human T cells. J. Biol. Chem., 271, 14438–14444. [DOI] [PubMed] [Google Scholar]
- 118.Porumb H., Gousset,H., Letellier,R., Salle,V., Briane,D., Vassy,J., Amor-Gueret,M., Israel,L. and Taillandier,E. (1996) Temporary ex vivo inhibition of the expression of the human oncogene HER2 (NEU) by a triple helix-forming oligonucleotide. Cancer Res., 56, 515–522. [PubMed] [Google Scholar]
- 119.Marchand P., Resch,K. and Radeke,H.H. (2000) Selective inhibition of monocyte chemoattractant protein-1 gene expression in human embryonal kidney cells by specific triple helix-forming oligonucleotides. J. Immunol., 164, 2070–2076. [DOI] [PubMed] [Google Scholar]
- 120.Carbone G.M., McGuffie,E.M., Collier,A. and Catapano,C.V. (2003) Selective inhibition of transcription of the Ets2 gene in prostate cancer cells by a triplex-forming oligonucleotide. Nucleic Acids Res., 31, 833–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Neurath M.F., Max,E.E. and Strober,W. (1995) Pax5 (BSAP) regulates the murine immunoglobulin 3′ alpha enhancer by suppressing binding of NF-alpha P, a protein that controls heavy chain transcription. Proc. Natl Acad. Sci. USA, 92, 5336–5340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Musso M., Wang,J.C. and Van Dyke,M.W. (1996) In vivo persistence of DNA triple helices containing psoralen-conjugated oligodeoxyribonucleotides. Nucleic Acids Res., 24, 4924–4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bonham M.A., Brown,S., Boyd,A.L., Brown,P.H., Bruckenstein,D.A., Hanvey,J.C., Thomson,S.A., Pipe,A., Hassman,F., Bisi,J.E. et al. (1995) An assessment of the antisense properties of RNase H-competent and steric-blocking oligomers. Nucleic Acids Res., 23, 1197–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Degols G., Clarenc,J.P., Lebleu,B. and Leonetti,J.P. (1994) Reversible inhibition of gene expression by a psoralen functionalized triple helix forming oligonucleotide in intact cells. J. Biol. Chem., 269, 16933–16937. [PubMed] [Google Scholar]
- 125.Shevelev A., Burfeind,P., Schulze,E., Rininsland,F., Johnson,T.R., Trojan,J., Chernicky,C.L., Helene,C., Ilan,J. and Ilan,J. (1997) Potential triple helix-mediated inhibition of IGF-I gene expression significantly reduces tumorigenicity of glioblastoma in an animal model. Cancer Gene Ther., 4, 105–112. [PubMed] [Google Scholar]
- 126.Rininsland F., Johnson,T.R., Chernicky,C.L., Schulze,E., Burfeind,P. and Ilan,J. (1997) Suppression of insulin-like growth factor type I receptor by a triple-helix strategy inhibits IGF-I transcription and tumorigenic potential of rat C6 glioblastoma cells. Proc. Natl Acad. Sci. USA, 94, 5854–5859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Aggarwal B.B., Schwarz,L., Hogan,M.E. and Rando,R.F. (1996) Triple helix-forming oligodeoxyribonucleotides targeted to the human tumor necrosis factor (TNF) gene inhibit TNF production and block the TNF-dependent growth of human glioblastoma tumor cells. Cancer Res., 56, 5156–5164. [PubMed] [Google Scholar]
- 128.Giovannangeli C., Perrouault,L., Escude,C., Gryaznov,S. and Helene,C. (1996) Efficient inhibition of transcription elongation in vitro by oligonucleotide phosphoramidates targeted to proviral HIV DNA. J. Mol. Biol., 261, 386–398. [DOI] [PubMed] [Google Scholar]
- 129.Faria M., Wood,C.D., Perrouault,L., Nelson,J.S., Winter,A., White,M.R., Helene,C. and Giovannangeli,C. (2000) Targeted inhibition of transcription elongation in cells mediated by triplex-forming oligonucleotides. Proc. Natl Acad. Sci. USA, 97, 3862–3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Faria M., Wood,C.D., White,M.R., Helene,C. and Giovannangeli,C. (2001) Transcription inhibition induced by modified triple helix-forming oligonucleotides: a quantitative assay for evaluation in cells. J. Mol. Biol., 306, 15–24. [DOI] [PubMed] [Google Scholar]
- 131.Besch R., Giovannangeli,C., Kammerbauer,C. and Degitz,K. (2002) Specific inhibition of ICAM-1 expression mediated by gene targeting with triplex-forming oligonucleotides. J. Biol. Chem., 277, 32473–32479. [DOI] [PubMed] [Google Scholar]
- 132.Shen C., Buck,A., Polat,B., Schmid-Kotsas,A., Matuschek,C., Gross,H.J., Bachem,M. and Reske,S.N. (2003) Triplex-forming oligodeoxynucleotides targeting survivin inhibit proliferation and induce apoptosis of human lung carcinoma cells. Cancer Gene Ther., 10, 403–410. [DOI] [PubMed] [Google Scholar]
- 133.Kuznetsova S., Ait-Si-Ali,S., Nagibneva,I., Troalen,F., Le Villain,J.P., Harel-Bellan,A. and Svinarchuk,F. (1999) Gene activation by triplex-forming oligonucleotide coupled to the activating domain of protein VP16. Nucleic Acids Res., 27, 3995–4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Mollegaard N.E., Buchardt,O., Egholm,M. and Nielsen,P.E. (1994) Peptide nucleic acid.DNA strand displacement loops as artificial transcription promoters. Proc. Natl Acad. Sci. USA, 91, 3892–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wang G., Xu,X., Pace,B., Dean,D.A., Glazer,P.M., Chan,P., Goodman,S.R. and Shokolenko,I. (1999) Peptide nucleic acid (PNA) binding-mediated induction of human gamma-globin gene expression. Nucleic Acids Res., 27, 2806–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gottesfeld J.M., Neely,L., Trauger,J.W., Baird,E.E. and Dervan,P.B. (1997) Regulation of gene expression by small molecules. Nature, 387, 202–205. [DOI] [PubMed] [Google Scholar]
- 137.Dickinson L.A., Gulizia,R.J., Trauger,J.W., Baird,E.E., Mosier,D.E., Gottesfeld,J.M. and Dervan,P.B. (1998) Inhibition of RNA polymerase II transcription in human cells by synthetic DNA-binding ligands. Proc. Natl Acad. Sci. USA, 95, 12890–12895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Schaal T.D., Mallet,W.G., McMinn,D.L., Nguyen,N.V., Sopko,M.M., John,S. and Parekh,B.S. (2003) Inhibition of human papilloma virus E2 DNA binding protein by covalently linked polyamides. Nucleic Acids Res., 31, 1282–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Coull J.J., He,G., Melander,C., Rucker,V.C., Dervan,P.B. and Margolis,D.M. (2002) Targeted derepression of the human immunodeficiency virus type 1 long terminal repeat by pyrrole-imidazole polyamides. J. Virol., 76, 12349–12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Mapp A.K., Ansari,A.Z., Ptashne,M. and Dervan,P.B. (2000) Activation of gene expression by small molecule transcription factors. Proc. Natl Acad. Sci. USA, 97, 3930–3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ansari A.Z., Mapp,A.K., Nguyen,D.H., Dervan,P.B. and Ptashne,M. (2001) Towards a minimal motif for artificial transcriptional activators. Chem. Biol., 8, 583–592. [DOI] [PubMed] [Google Scholar]
- 142.Arora P.S., Ansari,A.Z., Best,T.P., Ptashne,M. and Dervan,P.B. (2002) Design of artificial transcriptional activators with rigid poly-L-proline linkers. J. Am. Chem. Soc., 124, 13067–13071. [DOI] [PubMed] [Google Scholar]
- 143.Chiang S.Y., Burli,R.W., Benz,C.C., Gawron,L., Scott,G.K., Dervan,P.B. and Beerman,T.A. (2000) Targeting the ets binding site of the HER2/neu promoter with pyrrole-imidazole polyamides. J. Biol. Chem., 275, 24246–24254. [DOI] [PubMed] [Google Scholar]
- 144.Belitsky J.M., Leslie,S.J., Arora,P.S., Beerman,T.A. and Dervan,P.B. (2002) Cellular uptake of N-methylpyrrole/N-methylimidazole polyamide-dye conjugates. Bioorg. Med. Chem., 10, 3313–3318. [DOI] [PubMed] [Google Scholar]
- 145.Janssen S., Cuvier,O., Muller,M. and Laemmli,U.K. (2000) Specific gain- and loss-of-function phenotypes induced by satellite-specific DNA-binding drugs fed to Drosophila melanogaster. Mol. Cell, 6, 1013–1024. [DOI] [PubMed] [Google Scholar]
- 146.Kang J.S. and Kim,J.S. (2000) Zinc finger proteins as designer transcription factors. J. Biol. Chem., 275, 8742–8748. [DOI] [PubMed] [Google Scholar]
- 147.Beerli R.R., Segal,D.J., Dreier,B. and Barbas,C.F.,III (1998) Toward controlling gene expression at will: specific regulation of the erbB-2/HER-2 promoter by using polydactyl zinc finger proteins constructed from modular building blocks. Proc. Natl Acad. Sci. USA, 95, 14628–14633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kim J.S. and Pabo,C.O. (1997) Transcriptional repression by zinc finger peptides. Exploring the potential for applications in gene therapy. J. Biol. Chem., 272, 29795–29800. [DOI] [PubMed] [Google Scholar]
- 149.Kim J.S., Kim,J., Cepek,K.L., Sharp,P.A. and Pabo,C.O. (1997) Design of TATA box-binding protein/zinc finger fusions for targeted regulation of gene expression. Proc. Natl Acad. Sci. USA, 94, 3616–3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Sadowski I., Ma,J., Triezenberg,S. and Ptashne,M. (1988) GAL4-VP16 is an unusually potent transcriptional activator. Nature, 335, 563–564. [DOI] [PubMed] [Google Scholar]
- 151.Fujita T., Nolan,G.P., Ghosh,S. and Baltimore,D. (1992) Independent modes of transcriptional activation by the p50 and p65 subunits of NF-kappa B. Genes Dev., 6, 775–787. [DOI] [PubMed] [Google Scholar]
- 152.Margolin J.F., Friedman,J.R., Meyer,W.K., Vissing,H., Thiesen,H.J. and Rauscher,F.J.,III (1994) Kruppel-associated boxes are potent transcriptional repression domains. Proc. Natl Acad. Sci. USA, 91, 4509–4513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Heinzel T., Lavinsky,R.M., Mullen,T.M., Soderstrom,M., Laherty,C.D., Torchia,J., Yang,W.M., Brard,G., Ngo,S.D., Davie,J.R. et al. (1997) A complex containing N-CoR, mSin3 and histone deacetylase mediates transcriptional repression. Nature, 387, 43–48. [DOI] [PubMed] [Google Scholar]
- 154.Damm K., Thompson,C.C. and Evans,R.M. (1989) Protein encoded by v-erbA functions as a thyroid-hormone receptor antagonist. Nature, 339, 593–597. [DOI] [PubMed] [Google Scholar]
- 155.Beerli R.R., Dreier,B. and Barbas,C.F.,III (2000) Positive and negative regulation of endogenous genes by designed transcription factors. Proc. Natl Acad. Sci. USA, 97, 1495–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Zhang L., Spratt,S.K., Liu,Q., Johnstone,B., Qi,H., Raschke,E.E., Jamieson,A.C., Rebar,E.J., Wolffe,A.P. and Case,C.C. (2000) Synthetic zinc finger transcription factor action at an endogenous chromosomal site. Activation of the human erythropoietin gene. J. Biol. Chem., 275, 33850–33860. [DOI] [PubMed] [Google Scholar]
- 157.Liu P.Q., Rebar,E.J., Zhang,L., Liu,Q., Jamieson,A.C., Liang,Y., Qi,H., Li,P.X., Chen,B., Mendel,M.C. et al. (2001) Regulation of an endogenous locus using a panel of designed zinc finger proteins targeted to accessible chromatin regions. Activation of vascular endothelial growth factor A. J. Biol. Chem., 276, 11323–11334. [DOI] [PubMed] [Google Scholar]
- 158.Ren D., Collingwood,T.N., Rebar,E.J., Wolffe,A.P. and Camp,H.S. (2002) PPARgamma knockdown by engineered transcription factors: exogenous PPARgamma2 but not PPARgamma1 reactivates adipogenesis. Genes Dev., 16, 27–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Falke D., Fisher,M., Ye,D. and Juliano,R.L. (2003) Design of artificial transcription factors to selectively regulate the pro-apoptotic bax gene. Nucleic Acids Res., 31, e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Jouvenot Y., Ginjala,V., Zhang,L., Liu,P.Q., Oshimura,M., Feinberg,A.P., Wolffe,A.P., Ohlsson,R. and Gregory,P.D. (2003) Targeted regulation of imprinted genes by synthetic zinc-finger transcription factors. Gene Ther., 10, 513–522. [DOI] [PubMed] [Google Scholar]
- 161.Rebar E.J., Huang,Y., Hickey,R., Nath,A.K., Meoli,D., Nath,S., Chen,B., Xu,L., Liang,Y., Jamieson,A.C. et al. (2002) Induction of angiogenesis in a mouse model using engineered transcription factors. Nat. Med., 8, 1427–1432. [DOI] [PubMed] [Google Scholar]
- 162.Hobbs C.A. and Yoon,K. (1994) Differential regulation of gene expression in vivo by triple helix-forming oligonucleotides as detected by a reporter enzyme. Antisense Res. Dev., 4, 1–8. [DOI] [PubMed] [Google Scholar]
- 163.Boussif O., Lezoualc’h,F., Zanta,M.A., Mergny,M.D., Scherman,D., Demeneix,B. and Behr,J.P. (1995) A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc. Natl Acad. Sci. USA, 92, 7297–7301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Macaulay V.M., Bates,P.J., McLean,M.J., Rowlands,M.G., Jenkins,T.C., Ashworth,A. and Neidle,S. (1995) Inhibition of aromatase expression by a psoralen-linked triplex-forming oligonucleotide targeted to a coding sequence. FEBS Lett., 372, 222–228. [DOI] [PubMed] [Google Scholar]
- 165.Vlassov V.V., Balakireva,L.A. and Yakubov,L.A. (1994) Transport of oligonucleotides across natural and model membranes. Biochim. Biophys. Acta, 1197, 95–108. [DOI] [PubMed] [Google Scholar]
- 166.Noonberg S.B., Scott,G.K., Garovoy,M.R., Benz,C.C. and Hunt,C.A. (1994) In vivo generation of highly abundant sequence-specific oligonucleotides for antisense and triplex gene regulation. Nucleic Acids Res., 22, 2830–2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Chen Y., Ji,Y.J., Roxby,R. and Conrad,C. (2000) In vivo expression of single-stranded DNA in mammalian cells with DNA enzyme sequences targeted to C-raf. Antisense Nucleic Acid Drug Dev., 10, 415–422. [DOI] [PubMed] [Google Scholar]
- 168.Ragin A., Morgan,R. and Chmielewski,J. (2002) Cellular import mediated by nuclear localization signal Peptide sequences. Chem. Biol., 9, 943. [DOI] [PubMed] [Google Scholar]
- 169.Rout M.P. and Aitchison,J.D. (2001) The nuclear pore complex as a transport machine. J. Biol. Chem., 276, 16593–16596. [DOI] [PubMed] [Google Scholar]
- 170.Crowley K.S., Phillion,D.P., Woodard,S.S., Schweitzer,B.A., Singh,M., Shabany,H., Burnette,B., Hippenmeyer,P., Heitmeier,M. and Bashkin,J.K. (2003) Controlling the intracellular localization of fluorescent polyamide analogues in cultured cells. Bioorg. Med. Chem. Lett., 13, 1565–1570. [DOI] [PubMed] [Google Scholar]
- 171.Briehn C.A., Weyermann,P. and Dervan,P.B. (2003) Alternative heterocycles for DNA recognition: the benzimidazole/imidazole pair. Chemistry, 9, 2110–2122. [DOI] [PubMed] [Google Scholar]
- 172.Renneberg D. and Dervan,P.B. (2003) Imidazopyridine/pyrrole and hydroxybenzimidazole/pyrrole pairs for DNA minor groove recognition. J. Am. Chem. Soc., 125, 5707–5716. [DOI] [PubMed] [Google Scholar]
- 173.Macris M.A. and Glazer,P.M. (2003) Transcription dependence of chromosomal gene targeting by triplex-forming oligonucleotides. J. Biol. Chem., 278, 3357–3362. [DOI] [PubMed] [Google Scholar]
- 174.Majumdar A., Puri,N., Cuenoud,B., Natt,F., Martin,P., Khorlin,A., Dyatkina,N., George,A.J., Miller,P.S. and Seidman,M.M. (2003) Cell cycle modulation of gene targeting by a triple helix-forming oligonucleotide. J. Biol. Chem., 278, 11072–11077. [DOI] [PubMed] [Google Scholar]
- 175.Gottesfeld J.M., Melander,C., Suto,R.K., Raviol,H., Luger,K. and Dervan,P.B. (2001) Sequence-specific recognition of DNA in the nucleosome by pyrrole-imidazole polyamides. J. Mol. Biol., 309, 615–629. [DOI] [PubMed] [Google Scholar]
- 176.Gottesfeld J.M., Belitsky,J.M., Melander,C., Dervan,P.B. and Luger,K. (2002) Blocking transcription through a nucleosome with synthetic DNA ligands. J. Mol. Biol., 321, 249–263. [DOI] [PubMed] [Google Scholar]
- 177.Suto R.K., Edayathumangalam,R.S., White,C.L., Melander,C., Gottesfeld,J.M., Dervan,P.B. and Luger,K. (2003) Crystal structures of nucleosome core particles in complex with minor groove DNA-binding ligands. J. Mol. Biol., 326, 371–380. [DOI] [PubMed] [Google Scholar]
- 178.Bae K.H., Do,K.Y., Shin,H.C., Hwang,M.S., Ryu,E.H., Park,K.S., Yang,H.Y., Lee,D.K., Lee,Y., Park,J. et al. (2003) Human zinc fingers as building blocks in the construction of artificial transcription factors. Nat. Biotechnol., 21, 275–280. [DOI] [PubMed] [Google Scholar]
- 179.Blancafort P., Magnenat,L. and Barbas,C.F. (2003) Scanning the human genome with combinatorial transcription factor libraries. Nat. Biotechnol., 21, 269–274. [DOI] [PubMed] [Google Scholar]
- 180.Clackson T. (2000) Regulated gene expression systems. Gene Ther., 7, 120–125. [DOI] [PubMed] [Google Scholar]
- 181.Auricchio A., Rivera,V., Clackson,T., O’Connor,E., Maguire,A., Tolentino,M., Bennett,J. and Wilson,J. (2002) Pharmacological regulation of protein expression from adeno-associated viral vectors in the eye. Mol. Ther., 6, 238. [DOI] [PubMed] [Google Scholar]
- 182.Beerli R.R., Schopfer,U., Dreier,B. and Barbas,C.F.,III (2000) Chemically regulated zinc finger transcription factors. J. Biol. Chem., 275, 32617–32627. [DOI] [PubMed] [Google Scholar]
- 183.Pollock R., Giel,M., Linher,K. and Clackson,T. (2002) Regulation of endogenous gene expression with a small-molecule dimerizer. Nat. Biotechnol., 20, 729–733. [DOI] [PubMed] [Google Scholar]