

Abstract
In the search for bioactive sphingosine 1-phosphate (S1P) receptor ligands, a series of 2-amino-2-heterocyclic-propanols were synthesized. These molecules were discovered to be substrates of human-sphingosine kinases 1 and 2 (SPHK1 and SPHK2). When phosphorylated, the resultant phosphates showed varied activities at the five sphingosine-1-phosphate (S1P) receptors (S1P1–5). Agonism at S1P1 was displayed in vivo by induction of lymphopenia. A stereochemical preference of the quaternary carbon was crucial for phosphorylation by the kinases and alters binding affinities at the S1P receptors. Oxazole and oxadiazole compounds are superior kinase substrates to FTY720, the prototypical prodrug immunomodulator, fingolimod (FTY720). The oxazole-derived structure was the most active for human SPHK2. Imidazole analogues were less active substrates for SPHKs, but more potent and selective agonists of the S1P1 receptor; additionally, the imidazole class of compounds rendered mice lymphopenic.
Introduction
Five membrane-bound sphingosine 1-phosphate (S1P, Figure 1) receptors control physiological processes, including heart rate, tissue permeability,1 wound healing,2 immune cell trafficking3 and oligodendrocyte function.4 Receptor expression and metabolism of sphingolipid signaling molecules enable endogenous S1P to control these diverse functions with specificity while being present at concentrations of 200 to 450 nM in plasma.5,6 Our laboratories have attempted to describe these signaling pathways by investigating the structure-activity-relationship of individual S1P receptors through the synthesis and biological characterization of non-natural S1P receptor ligands. Previously, we reported diverse classes of S1P analogues with various receptor affinities; including S1P4 and S1P1,5 selective agonists,7,8 as well as some of the first S1P1,3 antagonists.9,10
Figure 1.

S1P is active at five S1P receptors (S1P1–5). VPC44152 is a non-hydrolysable phosphonate agonist at S1P1,4,5. Various 2-amino-2-heterocyclic propanols were designed as prodrugs (converted by sphingosine kinases to active phosphates) for targeting S1P receptors.
Important in the successful development of S1P ligands is their incorporation into sphingosine metabolism. In view of natural S1P biosynthesis and degradation, one pathway for ligand inactivation involves lysophospholipid phosphatases.11,12 These enzymes dephosphorylate S1P and related molecules to primary alcohols that are physiologically inactive at the five receptors. We and others illustrated the synthesis of phosphonate mimetics that are more chemically resistant to phosphatase activity (bioactive VPC44152, Figure 1).10,13 This report describes the synthesis and biological characterization of S1P ligands that are prone to phosphorylation by one or both of the known sphingosine kinases (SPHKs).14 Substrates for SPHKs may obtain therapeutically useful equilibriums between their alcohol and phosphate states in vivo, as investigated by S1P1 induced lymphopenia.
A series of 2-amino-2-heterocyclic-propanols were investigated, based on our previous discovery of S1P1 selective agonists that contained N-aryl amide moieties within their linker region (Figure 1). This series was tested for activity at the known mouse (mSPHK) and human (hSPHK) sphingosine kinases. These potential kinase substrates were tested in vitro; and, following chemical phosphorylation, the compounds were evaluated at the five individual S1P receptors; and finally, the substrates were tested in vivo, for the induction of S1P1 mediated lymphopenia.
Imidazole, oxazole, and oxadiazole containing compounds are phosphorylated by SPHKs, with hSPHK2 being the more active species. This activity was dependant on the chirality of the C-2 carbon. One of two oxadiazoles was a better kinase substrate for than FTY720, the prototypical S1P prodrug. The corresponding oxazole showed the highest activity at SPHK2. Imidazole based compounds were comparatively less active substrates at the SPHKs but their phosphorylated congeners were more potent and selective agonists at the S1P1 receptor. Meta-substituted compounds in these series (found to be antagonists of S1P1,3 receptors) were not substrates for SPHKs. This is consistent with our previous model, in which the stereochemical preference for antagonism is opposite to that favored by enzymatic phosphorylation.
Chemistry
The synthesis of 4(5)-phenylimidazoles (Scheme 1) was envisioned through a Davidson-like cyclodehydration.15,16,17,18,19 Compound 1 was attained from the Freidel-Crafts acylation of commercially available 1-phenyloctane and 2-bromoacetyl bromide as previously described.10 N-Boc-α-methylserines were converted to their cesium salts under sonication,20 and alkylated with α-bromoketone 1 to form the desired -acyloxyketones, R- and S-2, in robust yields. α-Acyloxyketones were cyclized to optically active phenylimidazoles R-and S-3 by careful heating with NH4OAc in xylenes. The 2-amino-1-propanols, 3, were deprotected under acidic conditions and neutralized to yield the optically active final compounds VPC44211 and VPC44217. N-Boc protected compounds were also converted to the corresponding phosphates by standard phosphoramidite methodology. Subsequent deprotection provided the bis-ammonium trifluoroacetate salts VPC44218 and VPC44239.
Scheme 1.

Synthesis of chiral 4(5)-phenylimidazoles. Reagents and conditions: a.) AlCl3, neat, 0 °C to rt, 4h. (84%); b.) Cs2CO3, EtOH, sonication, 5 – 10 min.; then α-bromoketone in DMF, rt, overnight (86–94%); c.) NH4OAc, Xylenes, Dean-Stark, 110–120 °C, 1–3h. (50–60%); d.) TFA, CH2Cl2, 0 °C to rt, 4h. (80–84%); e.) N,N-di-iso-propyl-di-tert-butyl-phosphoramidite, 3% tetrazole in acetonitrile, CH2Cl2, 4–8h.; then 30% H2O2(aq) rt, 4h. (33–37%).
A procedure to create the 4-phenyloxazole ring system (Scheme 2) was readily available; however, the acidic conditions necessary for the cyclization meant that new protection schemes for the α-methyl-serine were necessary.21 A tert-butyldiphenylsilyl (TBDPS) ether was used successfully22 with a benzyloxycarbonyl (Cbz) protection for the amine. Literature procedures for the selective hydrolysis of the methyl ester protected acid (6) in the presence of the TBDPS ether further increased the utility of this protection scheme.23 Formation of the corresponding α-acyloxyketone proceeded smoothly, but the cyclization step to form the desired intermediate 9 proved low yielding. The 4-phenyloxazole was converted by standard methods to amino alcohol VPC92153 and amino phosphate VPC92249.
Scheme 2.

Synthesis of 4-phenyloxazoles. Reagents and Conditions: a.) 10% sat. aq. Na2CO3, Dioxanes, N-(Benzyloxycarbonyloxy)succinimide; b.) TMSCHN2, 6:1 benzene/MeOH (79% over 2 steps); c.) TBDPSCl, imidazole, DMAP, CH2Cl2 (82%); d.) NaOH, H2O/i-PrOH (96%); e.) Cs2CO3, EtOH, sonication, 5 – 10 min.; then α-bromoketone in DMF, rt, overnight (94%); f.) NH4OAc, AcOH, 90 °C, 10h (33%); g.) t-Bu4NF, THF (77%); h.) N,N-di-iso-propyl-di-tert-butyl-phosphoramidite, 3% tetrazole in acetonitrile, CH2Cl2, overnight.; then 30% H2O2(aq) rt, 4h. (59%); i.) Pd/C, H2, EtOH, rt, overnight (97%); then TFA, CH2Cl2, 4h (quantitative); j.) t-Bu4NF, THF (77%); then Pd/C, H2, EtOH (quantitative).
1,2,4-oxadiazoles are established peptide bond mimetics and comparable to the 4(5)-phenylimidazoles.24 They are smaller in diameter, considerably less basic than their imidazole counterparts, and allow for hydrogen-bond acceptance, but not donation. Two isomers exist in which the nitrogen atom occupies a similar location compared with the imidazole ring. To approach the two isomers, a common pathway for construction of the oxadiazole ring was desired. Previously, 1,2,4-oxadiazoles were constructed by the condensation of activated carboxylic acids with amidoximes in the presence of strong base. 25,26,27,28,29,30 Several common condensation methods suggested by the literature for coupling carboxylic acids and amidoximes afforded little or no success (DCC,31 EDC,32 and DIC/HOBT33). Following the literature through extensions to mild condensation strategies, a general method for the conjoining of various carboxylic acids and amidoximes remained elusive.34 We found PyBOP, the common and mild condensation reagent, worked well for the coupling of both oxadiazole isomers.
With this strategy in hand, the synthesis of the 1,2,4-oxadiazole isomer commenced with the conversion of commercially available 4-iodobenzonitrile to the para-alkynylaniline 12 through a Verkade-modified Sonogashira reaction (Scheme 3).35 Selective reduction of the arylalkyne was accomplished by hydrogenation over Lindlar’s catalyst to afford para-octylbenzonitrile 13. Using methods pioneered by Tiemann and Kruger,36 and optimized by Eitner and Weitz,37 hydroxylamine heated in ethanol gave reliable yields of the amidoxime 14.38,39
Scheme 3.

Synthesis of benzylamidoxime 14. Reagents and conditions: a.) Pd(OAc)2, Bu4NOAc, 1-octyne, DMF, rt, overnight (85–99%); b.) H2, Pd on BaSO4, EtOH, 45 psi, rt, 1h. (>95%); c.) NH2OH·HCl, Et3N, 95% EtOH(aq), 75 °C, 3h (74%).
Commercially available 2-methyl-(D,L)-serine was converted to acid 15 in two convenient steps (Scheme 4). Carboxylic acid 15 was coupled with amidoxime 14 to form acylamidoxime 16 following our newly established PyBOP coupling strategy. The resulting intermediate was cyclized, providing near quantitative yields of 17. Global deprotection was successful upon the addition of TFA to attain 3-phenyl-1,2,4-oxadiazole VPC45064 following basic workup. Protected amino alcohol 17 was also converted to the ammonium phosphate VPC45070 under standard conditions.
Scheme 4.

Reagents and Conditions. a.) i. 10% Na2CO3 in H2O, rt, 5–10 min.; then Boc2O in Dioxanes, rt, 0.5 – 2 days; ii. 2,2-dimethoxypropane, BF3 OEt2, Acetone, rt, 1–3 h. (>95%, two steps); b.) PyBOP, i-Pr2NEt, CH2Cl2, rt, 4h. (88%); c.) DMF, 110 °C, 3–4 h (71%); d.) TFA, CH2Cl2, rt, 3h.; then NaHCO3, rt, 15 min. (90%). e.) i. TFA, CH2Cl2, rt, 3h.; then NaHCO3, rt, 15 min. (90%); ii. 10% Na2CO3 in H2O, rt, 5–10 min.; then Boc2O in Dioxanes, rt, 4 – 6 h. (67%, two steps); f.) N,N-di-iso-propyl-di-tert-butyl-phosphoramidite, 3% tetrazole in acetonitrile, CH2Cl2, overnight; then 30% H2O2(aq) rt, 4h. (57%); g.) TFA, CH2Cl2, rt, 3h. (>80%).
Inversion of the oxadiazole substitution pattern relied on the proper conversion of α-methyl serine to the amidoxime derivative 20 (Scheme 5). The desired 5-phenyl-1,2,4-oxadiazole more closely approximates the position of the nitrogens in the 4(5)-phenylimidazole compounds. Carboxylic acid 15 was converted to the primary amide 18 through formation of the mixed anhydride followed by addition of either NH3(g) or NH4OH(aq). Convenient and effective dehydration conditions40 were used to convert the amide to the nitrile-serinoid 19. Treatment of this nitrile with hydroxylamine yielded the desired amidoxime analogue of serine 20.
Scheme 5.

Reagents and Conditions: a.) i. iso-butyl chloroformate, Et3N, THF, −10 °C, 30 min.; ii. NH4OH, 0 °C to rt. 1h. (60–80%); b.) Et3N, trifluoroacetic anhydride (TFAA), 0.1M THF 0 °C to rt, 30 min. (85–95%); c.) NH2OH·HCl, Et3N, 95% EtOH(aq), 75 °C, 3–5h. (85%).
The methyl benzoyl ester 21, previously synthesized by esterification, (Scheme 6) provided the methyl para-octynylbenzoyl ester 22 by a modified Sonogashira coupling. Hydrogenation over Pd/C was achieved to yield the alkylbenzoyl ester 23. Saponification of 23 provided the para-octylbenzoic acid 24 efficiently, which was condensed with the sterically congested amidoxime 20 to yield 25 under PyBOP coupling conditions. The purified intermediate was cyclized to the 5-phenyl-1,2,4-oxadiazole, 26, as previously described. The N-Boc and N,O-isopropylidene were deprotected with TFA and treated with basic conditions, providing the desired 2-amino-1-propanol VPC45129.
Scheme 6.

Reactants and Conditions. a.) MeOH, SOCl2, 0 °C to rt, 24h. (81%); b.) 1-octyne, Pd(OAc)2, Bu4NOAc, DMF, rt, overnight (83%); c.) H2, Pd/C, EtOH, rt, 4–6 h. (99%); d.) 20% NaOH(aq), 95% EtOH(aq), rt, 2h., then 1N H2SO4 rt, 15 min. (99%); e.) 20, PyBOP, i-Pr2NEt, CH2Cl2, rt, 4h (43%); f.) DMF, 110 °C, 16h. (60%); g.) TFA, CH2Cl2, rt, 3h.; then NaHCO3, rt, 15 min. (>90%).
Once obtained, VPC45129 was subjected to anhydrous N-Boc protection and subsequent phosphorylation and deprotection to yield the corresponding phosphate (VPC46023) as a white solid (Scheme 7).
Scheme 7.

Synthesis of 5-phenyl-1,2,4-oxadiazole phosphate VPC46032. Reagents and conditions: a.) Boc2O, TEA, CH2Cl2; b.) Tetrazole, MeCN, N,N-diisopropylditertbutylphosphoramidite; 30% H2O2; c.) TMSBr, DCM (32% over 3 steps).
Synthesis of a 4-phenylthiazole derivative began with the serine-derived amide 18, which was next converted to the thioamide 27 with the use of Lawesson’s reagent (Scheme 8). The α-iminothioketone formed by the base-initiated S-alkylation of compound 27 was dehydrated in situ to give a separable mixture of the desired thiazole 28 and the incomplete dihydrothiazole 29.41,42 This one pot reaction was not optimized, but on re-treatment of the dihydrothiazole intermediate 29 with dry lutidine and TFAA, the dehydration was completed in excellent yields. Thiazole 28 was deprotected with TFA and neutralized to provide the desired aminoalcohol VPC45214.
Scheme 8.

Synthesis of thiazole VPC45214. a.) Lawesson’s Reagent, THF, rt, 4h. (46%); b.) i. KHCO3, DME, −15 °C, 15 min; ii. α-bromoketone, −15 °C, 30 min., then rt, 30 min.; iii. TFAA, Lutidine, DME, −15 °C to rt, 12 h. (39%); c.) TFAA, lutidine, DME, −15 °C to rt, overnight, (>95%); d.) TFA, CH2Cl2, rt, 6h.; then NaHCO3, 15 min. (62%)
Biology
The final 2-heterocyclic-2-amino-1-propanols were analyzed as substrates of four SPHKs (h-SPHK1,2 and m-SPHK1,2, as previously described43). Phosphorylation was compared to that of the natural substrate of the kinases, D-erythro-sphingosine.
Most of our compounds (Figure 2) exhibited activity at SPHK2 with the exception of the 4-phenylthiazole (VPC45124). The (S)-stereoisomer of the imidazole (VPC44217) was virtually inactive at the kinases while the R-stereoisomer (VPC44211), having the natural configuration about the quaternary carbon, had approximately 20% the activity of sphingosine at hSPHK2. This stereoselective preference was upheld when comparing the racemic mixture of the 3-phenyl-1,2,4-oxadiazole (VPC45064) and its R- stereoisomer (VPC45080), and recapitulates the observed stereoselectivity of SPHK2 for the methylated FTY720 analogs, AAL149 and AAL151.44 The 5-phenyl-1,2,4-oxadiazole (VPC45129) and the 4-phenyloxazole (VPC92153) performed exceptionally well in the phosphorylation assay. VPC92153 displayed the best activity at SPHK2. While VPC45129 displayed moderate activity at SPHK2, it was the only alcohol in the series to have significant activity at SPHK1. It should be noted that very few synthetic analogs display activity at SPHK1, making this particular oxadiazole-containing compound unusual.
Figure 2.
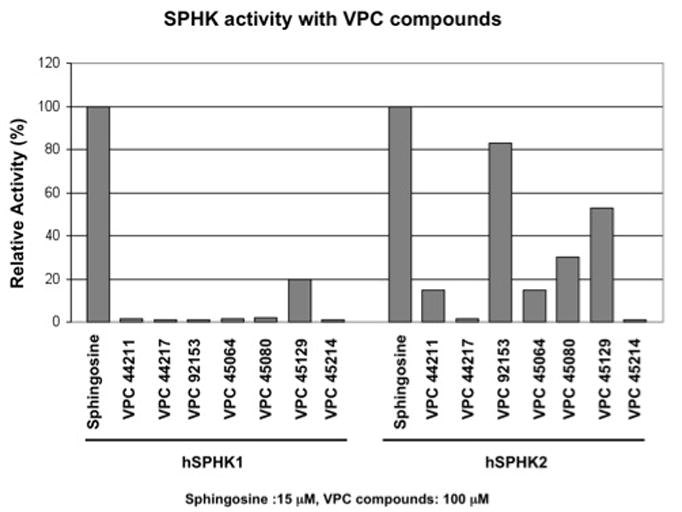
Comparison of imidazole, thiazole, oxadiazole and oxazole 2-amino-alcohols. Compounds were analyzed as possible substrates for the kinases hSPHK1 (hSK1) and hSPHK2 (hSK2).
Due to extensive work by our laboratories and others, it is now well understood that lymphopenia induced by S1P receptor agonists, such as FTY720, is the direct result of potency at the S1P1 receptor after in vivo phosphorylation by SPHK2.43 It has also been demonstrated that the bradycardia evoked by FTY720 is linked to agonism at the S1P3 receptor, at least in rodents.45 Thus it is of interest to determine receptor activity in assessing aminoalcohols as S1P receptor prodrug agonists. Each phosphate was subjected to our standard GTP-[γ-35S] assay as previously described.6–10
On initial examination of the data (Table 1), the selectivity between the S1P1 and S1P3 receptor has been greatly improved relative to FTY720. In most cases a difference of two log orders of selectivity was observed. The only exception was the 3-phenyl-1,2,4-oxadiazole phosphate, (VPC45070) which was considerably less potent at S1P1 and was equipotent at S1P3. These phosphates also appear to be good agonists for the S1P4 receptor, providing some insight into S1P4 agonist SAR. However, while these analogs are approximately equipotent to the natural ligand, S1P itself is a surprisingly poor agonist. Experimental potencies of S1P at S1P4 are in the high nanomolar range according to our assays. The 4-phenylthiazole phosphate was not included in the receptor screening because it was such a poor substrate for the SPHKs. The ligand was, therefore an unlikely candidate as a S1P1 receptor prodrug. Because of the detrimental side effects of FTY720’s potency at the S1P3 receptor, and the ability of this class of heterocycles to discriminate between S1P1 and S1P3, the therapeutic potential of these compounds becomes immediately apparent.
Table 1.
GTP-[γ-35S] ASSAYS
| S1P Receptors | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| S1P1 | S1P2 | S1P3 | S1P4 | S1P5 | ||||||
| EC50 | Emax | EC50 | Emax | EC50 | Emax | EC50 | Emax | EC50 | Emax | |
| S1P | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| FTY720P | 0.28 | 1.00 | NAA | 0.00 | 0.23 | 0.50 | 0.15 | 0.78 | 4.32 | 0.56 |
| VPC44218 | 1.09 | 0.87 | NAA | 0.00 | 50.77 | 0.65 | 0.59 | 0.93 | 3.62 | 0.83 |
| VPC44239 | 8.09 | 0.92 | NAA | 0.00 | 1,000 | 0.61 | 4.13 | 1.19 | 16.15 | 0.83 |
| VPC45070 | 24.40 | 0.89 | NAA | 0.00 | 22.99 | 0.51 | 0.52 | 0.81 | 4.35 | 0.73 |
| VPC46023 | 2.50 | 1.00 | NA | 0.00 | NA | NA | NA | NA | 21.60 | 1.00 |
| VPC92249 | 4.28 | 0.80 | NA | 0.00 | 52.52 | 1.00 | NA | NA | 24.07 | 0.60 |
NAA = No Agonist Activity detected. NA = Not Assessed. a.EC50 and b.EMAX values were normalized to those of S1P. In a typical assay, the EC50 value of S1P was 10 nanoM (100 nanoM at S1P4) and the membrane bound GTP[γ-35S] varied from 1,000 to 3,000 cpm in response to increasing S1P concentrations.
While receptor data provides some insight as to how these compounds should work as a potential therapy, the ultimate test of these heterocyclic sphingosine analogs lies in their ability to induce lymphopenia in vivo. Disappointingly, most of these aminoalcohols were not effective at lowering lymphocyte counts despite their unprecedented activity at the SPHKs and receptor potencies (data not shown). However, the 4-phenylimidazole analogs performed exceptionally well; lowering lymphocyte counts approximately 75% in most cases (Figure 3).
Figure 3.
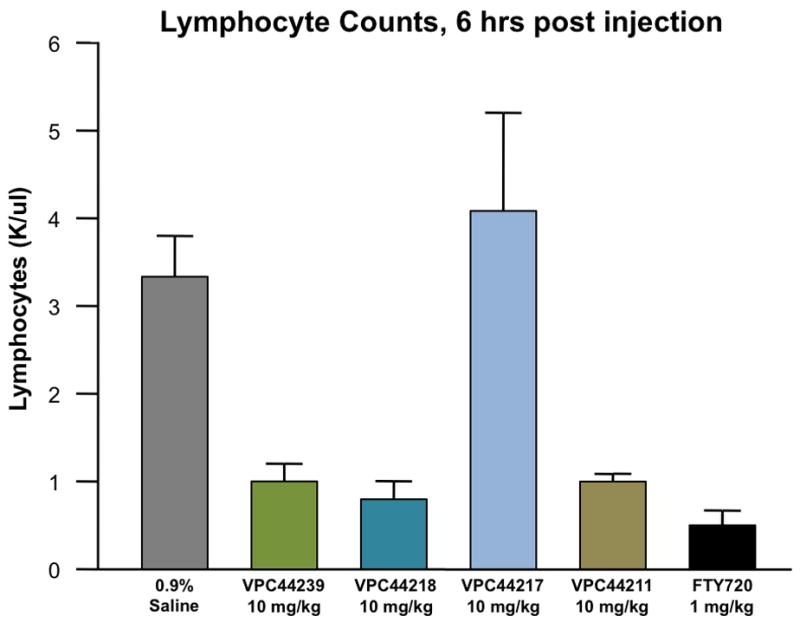
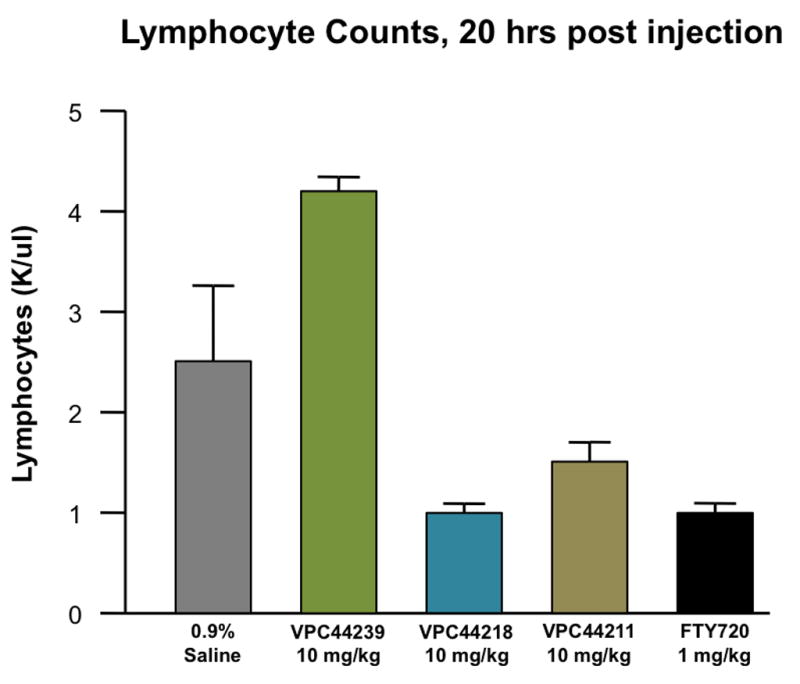
A) Four chiral phenylimidazole compounds VPC44239, VPC44218, VPC44217 and VPC44211 were compared for their stimulation of lymphopenia using our standard in vivo assay.6 B.) Twenty hours post ip injection, the alcohol VPC44211 and phosphate VPC44218 treated mice experienced nearly equivalent levels of lymphocyte depletion from the periphery.46
The imidazole containing compounds that exhibited low nanomolar binding constants effectively induced lymphopenia in mice. S1P analogues containing the natural configuration (VPC44211) induced lymphopenia for more than 20 hours while the unnatural aminoalcohol (VPC44217) did not cause this effect. This finding is consistent with our initial kinase studies, where the analog with the unnatural stereochemistry was a poor substrate for either sphingosine kinase. Although this study produced only a single heterocyclic analog of desirable activity in vivo, we have demonstrated there is a clear structure-activity-relationship for this class of SPHK substrates. Elucidating the elements that make these amino alcohols substrates for SPHK1 and SPHK2 while dialing out S1P3 potency, it becomes possible to deliver immunosuppressants with significantly less detrimental S1P3 related side effects.
Conclusion
At the outset of this study, we sought to not only further our understanding of the elements of SPHK1 and SPHK2 substrate SAR, but also design more metabolically stable S1P1 receptor agonist prodrugs. Additionally, we hoped to improve S1P1/S1P3 receptor selectivity, which would make these compounds more attractive as potential therapeutic agents. While in vitro data was initially quite promising, our newly synthesized series of heterocyclic S1P receptor prodrugs did not prove viable therapeutic candidates after in vivo analysis of lymphocyte levels. A likely cause for this result is the rate of dephosphorylation of these analogs by any number of lysophospholipid phosphatases, which would be revealed by a low agonist (phosphate) : parent (alcohol) drug ratio in plasma. Another possibility is rapid clearance of these compounds in mice. We are currently evaluating these possibilities.
Due to their implication in a number of disease states, such as cancer and tumor growth, inhibitors of the sphingosine kinases are quite desirable. Here, we have presented a body of research that displays some of the most remarkable substrates of SPHK1 and SPHK2 yet reported in the literature; compounds whose rates of phosphorylation are beginning to approach that of the natural ligand. We hope to take this data forward in an effort to design a novel class of SPHK inhibitors that could be used as tools to answer questions about the role S1P in various disease states. Such tools have the potential to validate sphingosine kinases as drug targets.
Supplementary Material
Supplemental Scheme 1
Reagents and conditions. a.) 2,2-dimenthoxypropane, BF3 OEt, Acetone, rt, 4 hours (91%); b.) PyBOP, i-Pr2NEt, CH2Cl2; 14, rt, 4h (82%); c.) DMF, 110 °C, 24h (71%); d.) TFA, CH2Cl2, 3h, (85%).
Acknowledgments
This research was funded by grants from the NIH: R01 GM 067958 (KRL) and T32 GM 007055 (PCK, AHS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sanchez Teresa, Hla Timothy. Structural and functional characteristics of S1P receptors. J Cell Biochem. 2004;92:913–922. doi: 10.1002/jcb.20127. [DOI] [PubMed] [Google Scholar]
- 2.Birgbauer E, Chun J. New developments in the biological functions of lysophospholipids. Cell Molec Life Sci. 2006;63:2695–2701. doi: 10.1007/s00018-006-6155-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosen Hugh, Sanna M Germana, Cahalan Stuart M, Gonzalez-Cabrera Pedro J. Tipping the gatekeeper: S1P regulation of endothelial barrier function. Trends Immunol. 2007;28:102–107. doi: 10.1016/j.it.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 4.Dev Kumlesh K, Mullershausen Florian, Mattes Henri, Kuhn Rainer R, Bilbe Graeme, Hoyer Daniel, Mir Anis. Brain sphingosine-1-phosphate receptors: Implication for FTY720 in the treatment of multiple sclerosis. Pharmacol Therapeutics. 2008;117:77–93. doi: 10.1016/j.pharmthera.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 5.Igarashi Y, Yatomi Y. Sphingosine 1-phosphate is a Blood Constituent Released from Activated Platelets, Possibly Playing a Variety of Physiological and Pathological Roles. Acta Biochim Polon. 1998;45:299–309. [PubMed] [Google Scholar]
- 6.Foss FW, Clemens JJ, Davis MD, Snyder AH, Zigler MA, Lynch KR, Macdonald TL. Synthesis, stability, and implications of phosphothioate agonists of sphingosine-1-phosphate receptors. Bioorganic & Medicinal Chemistry Letters. 2005;15(20):4470–4474. doi: 10.1016/j.bmcl.2005.07.057. [DOI] [PubMed] [Google Scholar]
- 7.Clemens JJ, Davis MD, Lynch KR, Macdonald TL. Synthesis of benzimidazole based analogues of sphingosine-1-phosphate: discovery of potent, subtype-selective S1P4 receptor agonists. Bioorganic & Medicinal Chemistry Letters. 2004;14(19):4903–4906. doi: 10.1016/j.bmcl.2004.07.030. [DOI] [PubMed] [Google Scholar]
- 8.Clemens JJ, Davis MD, Lynch KR, Macdonald TL. Synthesis of para-alkyl aryl amide analogues of sphingosine-1-phosphate: Discovery of potent S1P receptor agonists. Bioorganic & Medicinal Chemistry Letters. 2003;13(20):3401–3404. doi: 10.1016/s0960-894x(03)00812-6. [DOI] [PubMed] [Google Scholar]; Clemens JJ, Davis MD, Lynch KR, Macdonald TL. Synthesis of 4(5)-phenylimidazole-based analogues of sphingosine-1-phosphate and FTY720: Discovery of potent SIP1 receptor agonists. Bioorganic & Medicinal Chemistry Letters. 2005;15(15):3568–3572. doi: 10.1016/j.bmcl.2005.05.097. [DOI] [PubMed] [Google Scholar]
- 9.Davis MD, Clemens JJ, Macdonald TL, Lynch KR. Sphingosine 1-phosphate analogs as receptor antagonists. Journal of Biological Chemistry. 2005;280:9833–9841. doi: 10.1074/jbc.M412356200. [DOI] [PubMed] [Google Scholar]
- 10.Foss FW, Snyder AH, Davis MD, Rouse M, Okusa MD, Lynch KR, Macdonald TL. Synthesis and biological evaluation of γ-aminophosphonates as potent, subtype-selective sphingosine 1-phosphate receptor agonists and antagonists. Bioorganic & Medicinal Chemistry. 2007;15:663–677. doi: 10.1016/j.bmc.2006.10.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pyne S, Pyne NJ. Sphingosine 1-phosphate signalling in mammalian cells. Biochemical Journal. 2000;349:385–402. doi: 10.1042/0264-6021:3490385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spiegel S, Milstien S. Sphingosine-1-phosphate: An enigmatic signalling lipid. Nature Reviews Molecular Cell Biology. 2003;4(5):397–407. doi: 10.1038/nrm1103. [DOI] [PubMed] [Google Scholar]
- 13.Sanna MG, Wang SK, Gonzalez-Cabrera PJ, Don A, Marsolais D, Matheu MP, Wei SH, Parker I, Jo EJ, Cheng WC, Cahalan MD, Wong CH, Rosen H. Enhancement of capillary leakage and restoration of lymphocyte egress by a chiral S1P1 antagonist in vivo. Nature Chemical Biology. 2006;2(8):434–441. doi: 10.1038/nchembio804. [DOI] [PubMed] [Google Scholar]
- 14.Paugh SW, Payne SG, Barbour SE, Milstien S, Spiegel S. The immunosuppressant FTY720 is phosphorylated by sphingosine kinase type 2. Febs Letters. 2003;554(1–2):189–193. doi: 10.1016/s0014-5793(03)01168-2. [DOI] [PubMed] [Google Scholar]
- 15.Davidson D, Weiss M, Jelling M. The Action of Ammonia on Benzil. J Org Chem. 1937;2:319–327. [Google Scholar]
- 16.Clemens JJ, Davis MD, Lynch KR, Macdonald TL. Synthesis of 4(5)-phenylimidazole-based analogues of sphingosine-1-phosphate and FTY720: Discovery of potent SIP1 receptor agonists. Bioorganic & Medicinal Chemistry Letters. 2005;15(15):3568–3572. doi: 10.1016/j.bmcl.2005.05.097. [DOI] [PubMed] [Google Scholar]
- 17.Gordon T, Hansen P, Morgan B, Singh J, Baizman E, Ward S. Peptide Azoles: A New Class of Biologically Active Dipeptide Mimetics. Bioorg Med Chem Lett. 1993;3:915–920. [Google Scholar]
- 18.Gordon TD, Singh J, Hansen PE, Morgan BA. Synthetic Approaches to the ‘Azole’ Peptide Mimetics. Tetrahedron Lett. 1993;34:1901–1904. [Google Scholar]
- 19.Poitout L, Roubert P, Contour-Galcera MO, Moinet C, Lannoy J, Pommier J, Plas P, Big D, Thurieau C. Identification of Potent Non-Peptide Somatostatin Antagonists with sst3 Selectivity. J Med Chem. 2001;44:2990–3000. doi: 10.1021/jm0108449. [DOI] [PubMed] [Google Scholar]
- 20.Without sonication the alcohol solution may be stirred at elevated temperatures (~35 °C) to ensure solvation of the cesium carbonate. Conversion to the cesium carboxylate occurs within one hour under these conditions
- 21.Huang W, Pei J, Chen B, et al. A Novel Improved Procedure for the Synthesis of Oxazoles. Tetrahedron. 1996;52:10131–10136. [Google Scholar]
- 22.Hanessian S, Lavallee P. The Preparation and Synthetic Utility of tert-Butyldiphenylsilyl Ethers. Can J Chem. 1975;53:2975–2977. [Google Scholar]
- 23.Chiang YCP, Yang SS, Heck JV, et al. Total Synthesis of L-659,699, a Novel Inhibitor of Cholesterol Biosynthesis. J Org Chem. 1989;54:5708–5712. [Google Scholar]
- 24.Eicher T, Hauptmann S, Speicher A. In: The Chemistry of Heterocycles: Structure, Reactions, Synthesis, and Applications. 2. Suschitzky H, Suschitzky J, translators. Wiley-VCCH; 2003. pp. 191–196. [Google Scholar]
- 25.Mathvink RJ, Barritta AM, Candelore MR, Cascieri MA, Deng L, Tota L, Strader CD, Wyvratt MJ, Fisher MH, Weber AE. Bioorg Med Chem Lett. 1999;9:1869–1874. doi: 10.1016/s0960-894x(99)00277-2. [DOI] [PubMed] [Google Scholar]
- 26.Swain CJ, Baker R, Kneen C, Moseley J, Saunders J, Seward EM, Stevenson G, Beer M, Stanton J, Watling K. J Med Chem. 1991;34:140–151. doi: 10.1021/jm00105a021. [DOI] [PubMed] [Google Scholar]
- 27.Borg S, Vollinga RC, Labarre M, Payza K, Terenius L, Luthman K. J Med Chem. 1999;42:4331–4342. doi: 10.1021/jm990197+. [DOI] [PubMed] [Google Scholar]
- 28.Orlek BS, Blaney FE, Brown F, Clark MSG, Hadley MS, Hatcher J, Riley GJ, Rosenberg HE, Wadsworth HJ, Wyman P. J Med Chem. 1991;34:2726–2735. doi: 10.1021/jm00113a009. [DOI] [PubMed] [Google Scholar]
- 29.LaMattina JL, Mularski CJ. J Org Chem. 1984;49:4800–4805. [Google Scholar]
- 30.Borg S, Estenne-Bouhtou G, Luthman K, Csoregh I, Hesseling W, Hacksell U. Synthesis of 1,2,4-Oxadiazole-, 1,3,4-Oxadiazole-, and 1,2,4-Triazole-Derived Dipeptidomimetics. J Org Chem. 1995;60:3112–3120. [Google Scholar]
- 31.Liang GB, Feng DD. An Improved Oxadiazole Synthesis Using Peptide Coupling Reagents. Tetrahedron Lett. 1996;37:6627–6630. [Google Scholar]
- 32.Hamze A, Hernandez JF, Fulcrand P, Martinez J. Synthesis of Various 3-Substituted 1,2,4-Oxadiazole-Containing Chiral β3- and α-Amino Acids from Fmoc-Protected Aspartic Acid. J Org Chem. 2003;68:7316–7321. doi: 10.1021/jo0345953. [DOI] [PubMed] [Google Scholar]
- 33.Hamze A, Hernandez JF, Martinez J. Synthesis of (R) and (S) Enantiomers of Fmoc-Protected 1,2,4-Oxadiazole-Containing β3-Amino Acids from Fmoc-(R)-β-Hasp(OtBu)-OH. Tetrahedron Lett. 2003;44:6079–6082. [Google Scholar]
- 34.Poulain RF, Tartar AL, Deprez BP. Parallel Synthesis of 1,2,4-Oxadiazoles from Carboxylic Acids Using an Improved, Uronium-Based, Activatin. Tetrahedron Lett. 2001;42:1495–1498. references 3–11 therein. [Google Scholar]
- 35.Urgaonkar S, Verkade JG. Ligand-, Copper-, and Amine-Free Sonogashira Reaction of Aryl Iodides and Bromides with Terminal Alkynes. J Org Chem. 2004;69:5752–5755. doi: 10.1021/jo049325e. [DOI] [PubMed] [Google Scholar]
- 36.Tiemann F. Effect of Hydroxylamine on Nitriles. Ber. 1884;17:126–129. [Google Scholar]
- 37.Eitner P, Weitz H. Ber. 1893;26:2840. [Google Scholar]
- 38.Eloy F, Lenaers R. The Chemistry of Amidoximes and Related Compounds. Chem Rev. 1962;62:155–183. [Google Scholar]
- 39.Quan C, Kurth M. Solid-Phase Synthesis of 5-Isoxazol-4-yl-[1,2,4]oxadiazoles. J Org Chem. 2004;69:1470–1474. doi: 10.1021/jo0352124. [DOI] [PubMed] [Google Scholar]
- 40.Campagna F, Carotti A, Casini G. A Convenient Synthesis of Nitriles from Primary Amides Under Mild Conditions. Tetrahedron Lett. 1977;21:1813–1816. [Google Scholar]
- 41.Aguliar E, Meyers AI. Reinvestigation of a Modified Hantzsch Thiazole Synthesis. Tetrahedron Lett. 1994;35:2473–2476. [Google Scholar]
- 42.Bredenkamp MW, Holzapfel CW, van Zyl WJ. Synth Commun. 1990;20:2235. [Google Scholar]
- 43.Kharel Y, Lee S, Snyder AH, et al. Sphingosine Kinase 2 is Required for Modulation of Lymphocyte Traffic by FTY720. J Biol Chem. 2005;280:36865–36872. doi: 10.1074/jbc.M506293200. [DOI] [PubMed] [Google Scholar]
- 44.Brinkmann V, Davis MD, Heise CE, Albert R, Cottens W, Hof R, Bruns C, Prieschl E, Baumruker T, Hiestand P, Foster C, Lynch KR. The immune modulator, FTY720, targets sphingosine 1-phosphate receptors. J Biological Chemistry. 2002;277:21453–21457. doi: 10.1074/jbc.C200176200. [DOI] [PubMed] [Google Scholar]
- 45.Sanna MG, Liao J, Euijung J, et al. Sphingosine-1-Phosphate (S1P) Receptor Subtypes S1P1 and S1P3, Respectively, Regulate Lymphocyte Recirculation and Heart Rate. J Biol Chem. 2004;279:13839–13848. doi: 10.1074/jbc.M311743200. [DOI] [PubMed] [Google Scholar]
- 46.Data taken from: Foss, F. W. “Synthesis of Bioavailable Sphingosine-1-Phosphate Receptor Ligands: Structure-Activity-Relationship, Enzymatic Regulation, and Immunosuppression.” University of Virginia, 2006.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Scheme 1
Reagents and conditions. a.) 2,2-dimenthoxypropane, BF3 OEt, Acetone, rt, 4 hours (91%); b.) PyBOP, i-Pr2NEt, CH2Cl2; 14, rt, 4h (82%); c.) DMF, 110 °C, 24h (71%); d.) TFA, CH2Cl2, 3h, (85%).