

Abstract
Glycosphingolipids (GSLs) and gangliosides are a group of bioactive glycolipids that include cerebrosides, globosides, and gangliosides. These lipids play major roles in signal transduction, cell adhesion, modulating growth factor/hormone receptor, antigen recognition, and protein trafficking. Specific genetic defects in lysosomal hydrolases disrupt normal GSL and ganglioside metabolism leading to their excess accumulation in cellular compartments, particularly in the lysosome, i.e., lysosomal storage diseases (LSDs). The storage diseases of GSLs and gangliosides affect all organ systems, but the central nervous system (CNS) is primarily involved in many. Current treatments can attenuate the visceral disease, but the management of CNS involvement remains an unmet medical need. Early interventions that alter the CNS disease have shown promise in delaying neurologic involvement in several CNS LSDs. Consequently, effective treatment for such devastating inherited diseases requires an understanding of the early developmental and pathological mechanisms of GSL and ganglioside flux (synthesis and degradation) that underlie the CNS diseases. These are the focus of this review.
Keywords: lysosomal storage diseases, pathological mechanism, brain development, neuronal degeneration
Glycosphingolipids (GSLs) consist of ceramide and one or more attached carbohydrates. The nature of the oligosaccharide head groups categorizes the GSLs into neutral or acidic types due to the absence or presence of sialic acid residues, respectively (Fig. 1). Gangliosides are sialic acid containing GSLs. In eukaryotic cells, GSLs and gangliosides compose 10–20% of the total lipids (1), which are important to a variety of cellular functions. GSLs and gangliosides are synthesized at the endoplasmic reticulum (ER) and are remodeled during transit from cis to trans Golgi by a series of glycosyl- and sialyl-transferases. These are then transported to the intracellular compartments and the plasma membrane where they become enriched in microdomains and membrane bilayers. During plasma membrane turnover, GSLs and gangliosides can be internalized and partially or completely degraded in the endosomal/lysosomal system to sphingosine and free fatty acids that are then transported or flipped across late endosomal and lysosomal membranes for recycling or for use as signaling molecules (2, 3).
Fig. 1.

Schematic view of the GSL metabolism pathways. The synthesis of GSLs and gangliosides progress stepwise and are catalyzed by membranous glycosyltransferases in the ER or Golgi apparatus (see text). The degradation reactions are also sequential and occur within the lysosomes by various hydrolases. The black arrows show the synthesis pathway in the ER and Golgi and the green arrows show the degradation pathway in the lysosome. The enzymes in o-, a-, b-, and c-ganglioside series are numbered: 1. GM2/GD2/GA2/GT2-synthase (β-1,4-N-acetyl-galactosaminyltransferase, GalNacT), 2. GA1/GM1a/GD1b/GT1c-synthase (UDP-Gal:βGalNAc β-1,3-galactosyltransferase), 3. sialyltransferase IV, 4. sialyltransferase V, 5. sialyltransferase VII, 6. sialidase. Abbreviations: 3-KSR (3-ketosphinganine reductase), Sk (sphingosine kinase), Sa1P (sphinganine 1-phosphate), S1P (sphingosine 1-phosphate), S1-P Pase (sphingosine 1-phosphate phosphatase), (Sa) N-ACT (sphinganine N-acyltransferase), DHCerS (dihydroceramide desaturase), CerS (ceramide synthase, also called longevity assurance genes), SMS (sphingomyelin synthase), GT3 synthase (α-N-acetyl-neuraminide α-2,8- sialyltransferase). The chemical structures are adapted from (3, 36, 100, 313–315) and http://www.cybercolloids.net/library/sugars/hexoses.php. Nomenclature of the enzymes and protein are from IUBMB (http://www.chem.qmul.ac.uk/iubmb/enzyme) (37, 43, 315).
GSL metabolic pathways
GSL biosynthesis begins with condensation of serine and palmitoyl-CoA catalyzed by serine-palmitoyltransferase (SPT) on the cytoplasmic face of the ER, leading to de novo biosynthesis of ceramide, the core of GSLs (Fig. 1) (3–5). Ceramide consists of a fatty acid acyl chain that varies in length and saturation, and a sphingoid base that differs in the number and position of double bonds and hydroxyl groups (6–8). The fatty acid chain length of ceramide is controlled by tissue- and cell-specific ceramide synthases (also called longevity assurance genes) (9). In addition, ceramide can be generated by acid sphingomyelinase (aSMase) hydrolysis of sphingomyelin in the lysosome or at the plasma membrane and by activities of secreted aSMase at the plasma membrane or associated with lipoproteins (Figs. 1 and 2) (10). Neutral sphingomyelinase (nSMase) also cleaves plasma membrane sphingomyelin to ceramide (11). In the salvage pathway, lysosomally derived sphingosine can be reacylated (Fig. 2) (12). Once formed, ceramide is sorted to three pathways: 1) GalCer synthesis in the ER that is followed by 3-sulfo-GalCer (sulfatide) synthesis in the Golgi (13, 14); 2) GlcCer synthesis on the cytoplasmic face of the Golgi as the precursor of most GSLs; and 3) ceramide transfer protein (CERT) delivery to the mid-Golgi for sphingomyelin synthesis (15, 16). In the trans Golgi lumen, the transfer of a β-galactose onto GlcCer by lactosylceramide (LacCer) synthase forms LacCer (17). Several galactosyl-, N-acetylgalactosaminyl-, N-acetylglucosaminyl-, and sialyltransferases can elongate the oligosaccharide chain of GSLs along the luminal side of Golgi (18, 19), thereby defining the different series of GSLs. Addition of sialic acid to LacCer forms GM3 ganglioside by LacCer α-2,3-sialyltransferase (GM3 synthase) that initiates synthesis of the ganglioside or sialo-GSL series (Fig. 1). The type of sugars in the oligosaccharide backbone depends on the activity of glycosyltransferases in the Golgi, the cell type, and the developmental or disease stage (19–22) (see below).
Fig. 2.

Intracellular topology of GSL biosynthesis and trafficking. Ceramide, formed by condensation of serine and palmitoyl-CoA on the cytoplasmic face of ER, has one of three fates (16): a) conversion to galactosylceramide (GalCer) in the ER lumen, which is subsequently converted to sulfatide in the mid-Golgi (13, 14), b) vesicular transport to the cytoplasmic face of the cis-Golgi where it is a precursor for GlcCer synthesis (26, 316), and c) transport by ceramide transfer protein (CERT) to the mid-Golgi where sphingomyelin is formed within the lumen (319). Once formed, GlcCer also has several fates (27, 317): 1) transport by FAPP2 to the ER and/or to the trans-Golgi lumen where it is converted to lactosylceramide (LacCer) (17, 26, 27), 2) to the cytoplasmic side of the plasma membrane by unknown mechanisms, and 3) to the extracellular matrix by exocytosis. Addition of sialic acid to LacCer initiates synthesis of gangliosides or the sialo-GSL series (318). Ceramide can also be generated through degradation of sphingomyelin in the lysosome or at the plasma membrane by aSMase and nSMase (10). Newly synthesized GSLs can exit the cell by exocytic vesicles while membrane and extracellular GSLs can be transported intracellularly via endocytosis with subsequent degradation sphingosine and free fatty acids by hydrolases in the lysosome (2, 10). Abbreviations: DHCer (dihydroceramide), S1P (sphingosine 1-phosphate), SM (sphingomyelin).
In addition to CERT, several other proteins participate in GSL trafficking in the cells. GlcCer can be flipped into the Golgi lumen or across plasma membrane by the ATP-binding cassette transporter [also called multidrug resistance protein, MDR1, or P-glycoprotein (Pgp)]. Although this has been shown for short-chain GlcCer or neutral GlcCer, transport of naturally occurring GlcCer by Pgp has not been shown in vivo (23–25). Four-phosphate adaptor protein 2 (FAPP2) can transport newly synthesized GlcCer to the trans Golgi and back to the ER (26). It is not clear how FAPP2 transports GlcCer through the cytosol to the plasma membrane (26, 27). Although glycolipid transfer protein has been shown to have binding affinity for GSLs, transport of GSLs by glycolipid transfer protein has not been reported (28). The mechanisms of intracellular transport of GSLs continue to emerge.
The catabolism of complex GSLs also proceeds by stepwise, sequential removal of sugars by lysosomal exohydrolases to the final common products, sphingosine and fatty acids (Fig. 1). Individual defects in GSL hydrolases (Fig. 3) result in excessive accumulation of specific GSLs in lysosomes leading to the various lysosomal storage diseases (LSDs) (see Table 1). Nonenzymatic proteins are essential to GSL degradation either by presenting lipid substrates to their cognate enzymes or by interacting with their specific enzyme (2). Two genes, PSAP (prosaposin) and GM2A (GM2 activator protein), encode five such proteins (Fig. 3) (2, 29). Four saposins (A, B, C, and D) or sphingolipid activator proteins (Sap) are derived from proteolytic cleavage of a single precursor protein, prosaposin, in the late endosome and lysosome (30, 31). Each of these saposins has specificity for a particular GSL hydrolase (Table 1).
Fig. 3.

Disorders of GSL and ganglioside degradation. Inherited diseases (violet) caused by genetic defects of individual hydrolases/proteins (green) in the GSL and ganglioside degradation pathway. Increased levels of lysosphingolipids occur in the GSL LSDs, e.g., glucosylsphingosine in Gaucher disease, Lyso-Gb3 in Fabry disease, and galactosylsphingosine in Krabbe disease. Variant AB is GM2 activator deficiency disease. Tay-Sach disease, Sandhoff disease, and Variant AB are GM2 gangliosidosis. Abbreviations are listed in Fig. 1 legend.
TABLE 1.
Human and mouse disorders of GSL and ganglioside degradation
| Disease (Frequencies) | Gene Symbol, Chr. Location,a Name(s) | Species | Gene Defectb | Major Storage Materials | Phenotype | Onset | Lifespan | References |
|---|---|---|---|---|---|---|---|---|
| Farber disease (lipo granulomatosis) (rare) | ASAH1 Chr 8: p22-p21.3 AC, N-acylsphingosine amidohydrolase 1; N-acylsphingosine deacylase, AC, ASAH1 | Human | >17 Different mutations including point mutations and splice-site mutations | Ceramide, hydroxyl ceramide | Granulomas, lipid-laden macrophages in the CNS and viscera, mild or no neurological involvement, subcutaneous skin nodules near or over joints | 0 days – 1.7 year | 3 days–16 year | (96, 108, 321–323) |
| Asah1 Chr 8: 42425551-42460127(-) Asah1−/− | Mouse | Three copies of targeting constructs (PGK-neo cassette replacing exons 3 through 5) inserted into intron 12 | Ceramide | Embryonic lethal | 2-cell stage (E0–E1) | <E8.5 | (39, 102) | |
| Asah1 Chr 8: 42425551-42460127(-) Asah1+/- | Mouse | Three copies of targeting constructs (PGK-neo cassette replacing exons 3 through 5) inserted into intron 12 | Ceramide | Normal, progressive lipid-laden inclusions in liver Kupffer cells. | 6 months | Normal lifespan | (102) | |
| NPA (0.5 to 1/100,000) | SMPD1 Chr 11: p15.4- p15.1 aSMase, sphingomyelin phosphodiesterase 1, aSMase, ASM, aSMase, Zn-SMase | Human | >100 mutations Single nucleotide deletions, missense mutations | Sphingomyelin, Cholesterol, Bis (monoaclglycero)phosphate, GlcCer, LacCer Gb3, GM2, GM3 | Neurovisceral disease, hepatosplenomegaly, failure to thrive, rapid neurodegeneration, foamy cells in multiple organs | 3–6 months | 2–3 years | (104, 109, 324, 325) |
| NPB (0.5 to 1/100,000) | Same as NPA | Human | A single mutation, a 3-base deletion (ΔR608) | Sphingomyelin, Cholesterol, Bis(monoaclglyc ero)phosphate, GlcCer, LacCer, Gb3, GM2, GM3 | Visceral disease, minor neurological involvement, mental retardation, foamy cells in multiple organs | Early childhood to adulthod | Adolescence to adulthood | (104, 109, 325) |
| NPA/B | Smpd1 Chr 7: 112702939-112706901(+) ASM−/− | Mouse | Insertion of a neomycin resistance cassette into exon 3 | Sphingomyelin | Hepatosplenomegaly, tremors, ataxia, impaired coordination, lethargic, poor feeding, hunched posture, progressive CNS disease, Purkinje cell loss Dysregulated Ca2+ homeostasis | 2–3 months | 4 months | (106, 109) |
| Asm Asm−/− | Mouse | Insertion of a neomycin resistance cassette into exon 2 | Sphingomyelin, Cholesterol | Hepatosplenomegaly, mild tremors, ataxia, lethargic, poor feeding, hunched posture, progressive CNS disease, Purkinje cell loss, atrophy of brain, fertile | 2 months | 6–8 months | (105) | |
| NPC (1/120,000 to 1/150,000) | NPC1 (95% cases) Chr18:q11-q12 NPC1 | Human | Infantile and "classic" (severe) phenotype: Premature stop codon mutations,missense mutations in the sterol-sensing domain (SSD), A1054T in the cysteine-rich luminal loop "Variant" (mild) phenotype: Missense mutations (I943M, V950M, G986S, G992R, P1007A) clustered within the cysteine-rich luminal loop between transmembrane domains 8 and 9 | Cholesterol, Sphingomyelin, GlcCer, LacCer and GM2 | Progressive Purkinje cell loss, ataxia, dystonia, dementia, variable hepa tosplenomegaly, sea blue histiocytes in bone marrow, liver, spleen, and lung | In utero to adult | <6 months to adult | (312, 326–330) |
| Npc1 Chr 18: 12348202-12394909(-)spm NPC1−/− | Mouse | Spontaneous mutation | Cholesterol, GlcCer, LacCer and GM2 | Progressive Purkinje cell loss, demyelination, tremors, hind limb paralysis, poor fee ding, foamy macrophages in viscera, hepatosplenomegaly, enlarge lymph nodes, infertile | 7 weeks | 12–14 weeks | (260, 326, 331) | |
| Npc1 Chr 18: 12348202- 12394909(-)nih NPC1−/− | Mouse | Spontaneous mutation, transposon insertion deleting 11/13 transmembrane domains | Cholesterol, Sphingomyelin, lyso(bis)phosphatidic acid, GlcCer, LacCer, GA2, GM2 and GM3 | Progressive Purkinje cell loss, demyelination, tremors, hind limb paralysis, poor feeding, foamy macrophages in viscera | 5–7 weeks | 11–14 weeks | (218, 260, 326, 332–334) | |
| NPC2/HE1 (5% cases) Chr 14: q24.3 HE1, NP-C2, NPC2, HE1/NPC2 | Human | Severe disease: 27delG leading to early termination of protein, a missense mutation (S67P),E20X, E118X, S67P, and E20X/27delG mutations Milder disease: splice mutation (VS2+5G→A) in the consensus sequence of the 5′ donor site of intron 2 | Cholesterol, Sphingomyelin, GlcCer, LacCer and GM2 | Same as NPC1 | In utero to adult | <6 mos to adult | (326–330) | |
| Npc2 Chr 12: 86097442-86113848(-) | Mouse | Insertion of neo cassette into intron 3 | Cholesterol, Sphingomyelin, lyso(bis)phosphatidic acid, GlcCer, LacCer, GA2, GM2 and GM3 | Weight loss, tremors, ataxia, generalized locomotor dysfunction | 7.9 weeks | 13–18.5 weeks | (200) | |
| Krabbe disease (1/100,000) | GALC Chr 14: q31 Galactocereborside β-galactosidase Galactosylceramide β-galactosidase | Human | >60 mutationsInfantile patients:502C→T polymorphism associated with a 30 kb deletion in intron 10Late onset patients: missense mutations (R63H, G95S, M101L, G268S, G270D, Y298C, and I234T), nonsense mutation (S7X), a one-base deletion (805delG), mutations interfere with the splicing of intron 1and intron 6 | GalCer, Galactosylsphingosine | Progressive CNS and PNS involvement, hypertonicity/ hyperactive reflexes, progressive flaccidity, peripheral neuropathy, severe developmental delay, blindness, spastic paraparesis, dementia and loss of vision in later onset patients, infiltration of characteristic "globoid cells" | 3–6 months or 10– 40 years | < 2 years or adult | (111, 113, 335, 336, 337, 338) |
| Galc Chr 12: 99440510-99497547(-) twitcher | Mouse | Spontaneous mutation; G to A transition at codon 339 | GalCer, Galactosylsphingosine | Generalized tremors, progressive weakness, wasting, dys/demyelination, abnormal multinucleated "globoid" cells infiltration, gray matter unaffected | 3 weeks | 6.5–12 weeks | (116, 209, 210, 339, 340) | |
| Galc Chr 12: 99440510-99497547(-) Transgenic Krabbe | Mouse | Insertion of human mutation (H168C) in exon 5 | GalCer, Galactosylsphingosine | Tremor, hindleg weakness, microphage infiltration in PNS | 25–30 days | 58 days | (120) | |
| MLD (1/40,000) | ARSA Chr 22: q13.31- qter Arylsulphatase A cerebroside-3-sulfate 3-sulfohydrolase, ASA | Human | >40 mutations Deletions, insertions, splice site mutation, and missense mutations | Sulfatide | Difficulty walking after the first year of life, progressive peripheral neuropathies, muscle wasting/weakness, developmental delay, seizures, dementia | 6 months –16+ years | 5–63+ years | (122, 123, 341, 342) |
| Asa Chr 15: 89302959-89306545(-) Asa−/− | Mouse | A neomycin selection cassette inserted into exon 4 | Sulfatide | Mild phenotype compared with humans, normal lifespan, no widespread demyelination | 1 year | Normal lifespan | (129) | |
| Asa + Gal3st1 | Mouse | ASA−/− mice overexpressing sulfatide synthesizing enzyme CST | Sulfatide | More severe phenotype of ASA deficiency alone, more aggressive CNS and PNS demyelination | 1 year | < 2 years | (130) | |
| Fabry disease (1/40,000 to 1/117,000) | GLA Chr X: q22-q21 α-Galactosidase A | Human | >400 mutations in the GLA gene, including missense (76.4%), nonsense (16.4%), frameshift (3.6%), and splice site defects (3.6%) | Gb3, lyso-Gb3 | Males more severely affected, painful acroparesthesias, angiokeratoma, renal disease, cardiomyopathy, stroke, no clear neuronal involvement, ∼50% of heterozygous females affected | Childhood to 30+ years | 20–75.4 years | (137, 295, 343–348) |
| Gla Chr X: 131122688- 131135664(-) Gla−/− | Mouse | A neomycin resistance cassette inserted into fragment containing part of exon 3 and intron 3 | Gb3 | Normal lipid accumulation in kidneys, endothelium, liver, and peripheral nerve | Normal lifespan | (137, 138, 349) | ||
| Gaucher disease Type 1 - nonneuronopathic (1/800 in Ashkenazi Jewish, 1/100,000 in non-Jewish) | GBA1 Chr 1: q21 Acid β-glucosidase, β-glucosidase, glucosylceramide-β-glucosidase, glucosylceramidase, glucocerebrosidase, GCase | Human | Mutation substitution of amino acid asparagine for serine (N370S), N370S/84GG, N370S/L444P associated with Gaucher disease type 1 > 350 mutations associated with all Gaucher disease variants | GlcCer, GM1, GM2, GM3, GD3, Glucosylsphingosine | No neuronal involvement, chronic bone marrow expansion, bony deterioration, hepatosplenomegaly, hyper splenism, hepatic dysfunction, extensive fibrosis and tissue scarring, associated with Parkinson disease | 4–25 years | 6–80+ years | (95, 141, 226) |
| Gba1 Chr 3: 89006850-89012603(+) | Mouse | Human disease point mutations (N370S, D409V, D409H, and V394L) at gba1 locus in exon 9 | GlcCer | Mild phenotype with occasional storage cells in lung, spleen and liver, lipid accumulation in viscera, not in brain | <24 h or 3–7 months | Neonatal lethal (N370S), or normal lifespan (V394L, D409V, D409H) | (350) | |
| Mouse | Conditional gba knockout in hematopoietic and endothelial cells | GlcCer | Modest storage cells in liver and spleen, progressive splenomegaly, no bone marrow involvement | 16 weeks | Normal lifespan | (351) | ||
| Mouse | Conditional gba knockout in hematopoeitic endothelial cells | GlcCer | Splenomegaly and microcytic anemia | 12 months post induction | Normal lifespan | (352) | ||
| Gaucher disease type 2 - neuronopathic (1/500,000) | GBA1 Chr 1: q21 Acid β-glucosidase, β-glucosidase, glucosylceramide-β-glucosidase, glucosylceramidase, glucocerebrosidase, GCase | Human | A mutation causing leucine to proline change (L444P) associated with the neuronopathic forms of Gaucher disease, numerous other mutations | GlcCer, GM1, GM2, GM3, GD3, Glucosylsphingosine | Progressive CNS and lung involvement, neuronal death/drop-out, visceral involvement as type 1 disease | 3 months | <2 years | (95, 141) |
| Gba1 Chr 3: 89006850-89012603(+) Gba−/− | Mouse | A neomycin resistance cassette was inserted into exons 9 and 10 | GlcCer, Glucosylsphingosine | Mice die <24 h, lipid storage in lung, brain, a nd liver, glucosylsphingosine in brain and viscera | Embryonic lethal | (148) | ||
| Mouse | Conditional knockout (skin rescue), a floxed neo cassette was inserted into intron 8 | GlcCer | Rapid motor dysfunction, severe neurodegener ation, apoptotic cell death in brain, seizures | 7 days | 14 days | (149) | ||
| Mouse | Conditional knockout (skin rescue), reactivation of a low activity allele in keratinocytes | GlcCer | GlcCer accumulation brain, spleen, and liver, infiltration of Gaucher cells in sple en and liver. Rapid motor dysfunction, severe neurodegeneration, and cell death in CNS | 10 days | 14 days | (150) | ||
| Gaucher disease type 3 -neuronopathic (1/100,000) | GBA Chr 1: q21 Acid β-glucosidase, glucosylceramide-β-glucosidase, glucosylceramidase, glucocerebrosidase, GCase | Human | Homozygosity for L444P and D409H, numerous other mutations | GlcCer | Slower progression than Type 2 disease, normal intelligence, short stature with splenomegaly, abnormal eye movements, variable seizures | 1–5 years | 20–40 years | (95, 141, 353) |
| Complete Prosaposin/Sap deficiency (rare) | PSAP Chr 10: q21 Prosaposin, Sap precursor, PS, SAP1 | Human | 1 bp deletion (c.803delG) within the SAP-B domain of the prosaposin gene leads to a frameshift and premature stop codon, A to T transversion in the initiation codon | GlcCer, LacCer, Gb3, Ceramide, Sulfatides, Globotetraosylceramide | Rapid neurological disease, neuronal storage, loss of cortical neurons, astrocytosis, demyelination | 3 weeks | 16 weeks to 2 years | (176–178, 181) |
| Psap Chr 10: 59740375-59765342(+) PS−/− | Mouse | Exon 3 disrupted by insertion of a neomycin selection cassette | GlcCer, LacCer, Gb3, Ceramide, Sulfatides, Globotetraosylceramide | Rapidly neurological disease, hypomyelination, infl ammation, multiple GSLs accumulation in various organs | 20 days | Neonatal lethality or 5–7 weeks | (190) | |
| Psap Chr 10: 59740375-59765342(+) PS-NA | Mouse | Expression of transgenic prosaposin in PS−/− | GlcCer, LacCer, Ceramide, Sulfatides, | Ataxia, waddle gait, hypomyelination, inflammation, multi ple GSLs accumulation in various organs, Purkinje cell loss | 10–12 weeks | 7 months | (192) | |
| SapA deficiency late onset Krabbe disease(rare) | PSAP Saposin A, Sap A | Human | A 3 bp deletion in the SapA coding sequence of the prosaposin gene causing deletion of a conserved valine at amino acid number 11 of the SapA protein | GalCer, Galac tosylsphingosine | Normal development, rapid neurologic deterioration, loss of acquired milestones, vegetative state, eye contact and spontaneous movement at end stage | 3.5 months | 8 months | (187) |
| Psap Sap A−/− | Mouse | Amino acid substitution in the SapA domain on exon 4, Cys → Phe | GalCer, Galac tosylsphingosine | Hind limb paralysis, tremors, shaking at end stage, pathology and biochemistry identical but milder than twitcher mice | 2.5 months | 5 months | (191) | |
| SapB deficiency - variant MLD (rare) | PSAP Sap B | Human | Mutations destroy glycosylation sites, in-frame deletion of the first 21 bases of exon 6 | Sulfatide, Gangliosides, LacCer, Gb3, Digalactosylceramide | Variant form of MLD, normal ASA activity in white blood cells, increased lipid in urine | 1–10 years | 5–22 years | (124) |
| Psap Sap B−/− | Mouse | Amino acid substitution was introduced into the cysteine in SapB domain on exon 7, Cys →Phe | Hydroxy and nonhydroxy fatty acid sulfatide, LacCer, Gb3 | Neuromotor deterioration, minor head tremor, closely resembles MLD mice, no demyelination unlike human disease | 15 months | <2 years | (194) | |
| SapC deficiency (rare) | PSAP Sap C | Human | Neuronopathic: mutations on SapC domain (p.C382G, p.C382F, p.C315S) and premature stop codon in the SapD domain (p.Q430X), each on separate allele Non-neuronopathic: missense mutation on the SapC domain and the initiation codon mutation (p.L349P/ p.M1L) | GlcCer | Hepatosplenomegaly and severe mental deterioration, no Gaucher cells in bone marrow, slight retardation, focal seizures then generalized, ataxia, tremors opthalmoplegia, dysarthia, spastic tetraparesis, normal GCase activity | 1–8 years | 14–15.5 years | (124, 188, 189, 354–356) |
| Psap Sap C−/− | Mouse | Amino acid substitution on the SapC domain in exon 11, Cys → Pro | GlcCer, LacCer, LacSph | Progressive ataxia, Purkinje cell loss, cerebellum atrophy, impaired hippocampal LTP, slower disease progression than human, reduced GCase activity | 1 year | <2 years | (193) | |
| Psap Sap D−/− | Mouse | Amino acid substitution on the SapD domain in exon 13, Cys → Ser | Ceramide and hydroxyl ceramide | Renal degeneration, progressive polyuria, progressive, selective loss of cerebellar Purkinje cells | 6 months | 15 months | (98) | |
| Psap Sap CD−/− | Mouse | Amino acid substitutions were introduced into cysteine on SapC, (Cys → Pro) and D domains (Cys → Ser) | GlcCer and α-hydroxy ceramides | Severe neurological phenotype with ataxia, kyphotic posturing, hind limb paralysis, lipid accumulation in brain and kidney | 4 weeks | 8 weeks | (266) | |
| β-glucosidase (Gba) + prosaposin/ Sap deficiency | Gba + Psap V394L/PS-NA D409H/PS-NA | Mouse | Gba point mutants (V394L or D409H) crossed with mice expressing a low level Psap transgene (NA) | GlcCer, LacCer, Gb3 | Engorged macrophages infiltration in liver, lung, spleen, thymus, Purkinje cell loss, ataxia | 12 weeks | 22 weeks | (357) |
| Gba +Psap V394L/Sap C | Mouse | Gba point mutant crossed with SapC- deficiency mice | GlcCer, glucosylsphingosine | Hind limb paresis, axonal degeneration in CNS, hippocampal LTP attenuated | 30 days | 48 days | (205) | |
| GM1 gangliosidosis (1/100,000 to 1/300,000) | GLB1 Chr 3: p2.33 GM1- β-Galactosidase acid- β-galactosidase | Human | 78 missense/ nonsense mutations, 10 splicing mutations, 7 insertions, and 7 deletions Missense/nonsense mutations are primarily located in exon 2, 6, and 15 | GM1, GA1, GM2, GM3, GD1A, lyso-GM1, GlcCer, LacCer, oligosaccharides, keratan sulfate | Hepatosplenomegaly, localized skeletal involvement, CNS deterioration, seizures, mental regression, dystonia, gait disturbances, dysarthria | Birth to 3 years | 1–30 years | (155, 358, 359) |
| Glb1 Chr 9: 114310237-114383495(+) GM1 gangliosidosis mouse | Mouse | A neomycin resistance gene was inserted into exon 6 | GM1, GA1, GM2, GM3, GD1A, lyso-GM1, GlcCer, LacCer, oligosaccharides, keratan sulfate | Progressive spastic diplegia, clinical, pathological, biochemical manifestations similar to human | 4 months | 7–10 months | (156, 360) | |
| GM2 gangliosidosis Tay-Sachs (variant B) (1/4000 in Jewish 1/320,000 in non-Jewish) | HEX A Chr 15: q23-q24 β-hexosaminidase A (αβ) β-hexosaminidase S (αα) Hexosaminidase A | Human | Single nucleotide changes, deletions or insertions of varying size | GM2, GD1aGalNac, GA2, lyso-GM2 | CNS disease, rapid mental and motor deterioration, variable early dementia in adults | 3 months to adult | 2–40 years | (161, 162) |
| Hex A Chr 9: 59387504-59412914(+) Tay-Sachs mouse | Mouse | A neomycin resistance cassette was inserted into and disrupted exon 8 of the gene | GM2, GD1aGalNac, GA2, lyso-GM2 | Mouse normal, rare storage neurons in posterior horn of spinal cord, no obvious storage cells in viscera | Normal | (361, 362) | ||
| GM2 gangliosidosis Sandhoff disease (variant 0) (1/1000,000 in Jewish 1/390,000 in non- Jewish) | HEX B Chr 5: q13 β-hexosaminidases A (αβ) and B (ββ) Hexosaminidase A/B, HexA/HexB | Human | The most common mutation is a deletion of 16 kb including the HEXB promoter, exons 1–5, and part of intron 5 | GM2, GD1aGalNac, globoside, oligosaccharides, lyso-GM2 | Neurologic symptoms begin within first year, facial dysmorphism, skeletal dysplasia, hepatosplenomegaly | 5–6 months | 1.5–5.5 years | (161, 172, 363) |
| Hexb Chr 13: 97946362-97968225(-) Sandhoff mouse | Mouse | A neomycin resistance cassette was inserted into and disrupted exon 13 of the gene | GM2, GD1aGalNac, globoside, oligosaccharides, lyso-GM2 (PND2) | Motor function deterioration, head tremor, ataxia, bradykinesia, impaired balance, hind limb paralysis | 3 months | 5 months | (169) | |
| GM2 gangliosidosis GM2 Activator deficiency (Tay Sachs variant AB), (rare) | GM2A Chr 5: q31. 3-q33.1GM2 Activator GM2 ganglioside activator protein | Human | Single nonsense mutation in exon 2 | GM2, GA2 (minor) | Infantile acute encephalopathic phenot ype, closely resembles Tay-Sachs disease | 1 month | 5 months–5 years | (165, 172, 364) |
| Gm2a Chr 11: 54911617-54924400(+) Gm2a−/− | Mouse | A neomycin resistance cassette replaced exon 3 and exon 4 | GM2, GA2(minor) | Mouse normal, minor storage in cerebellum unlike Tay-Sachs disease mice | Normal | (175) |
Mouse Genome Database at the Mouse Genome Informatics Web site, The Jackson Laboratory, Bar Harbor, ME. World Wide Web (URL: http://www.informatics.jax.org), January, 2010.
Detailed mutation information for each disease can be found in the references.
The interconnection of synthesis and degradation pathway network
Many enzymes in GSL metabolic pathways are regulated in response to extra- and intracellular stimuli leading to modulation of bioactive lipid levels (32). The synthesis/degradation pathways (Fig. 1) of GSL metabolism form a network with the product of one enzyme serving as a substrate for other enzymes; e.g., ceramide formed from sphingomyelin may act directly or serve as a substrate for ceramidase, for sphingomyelin synthase, or for GCS, thereby being "converted" to sphingosine, sphingomyelin, and a by product, diacylglycerol, or GlcCer, respectively (33). Also, many cell stimuli modulate the function of more than one of these enzymes in a cell- or tissue-specific manner; upregulation of GCS by tumor necrosis factor α (TNFα) and interleukin (IL)-1 has been reported in the liver (34). The inhibition of GCS and sphingomyelin synthase activities by TNFα was found in the rhabdomyosarcoma cells (35). Thus, the coordinate regulation of such enzymes in response to cellular stimuli alters the lipid flux through this network (Fig. 1) (32). In addition, cellular compartmentalization topologically restricts enzymes and their products to subcellular organelles in which metabolic fluxes are modulated by enzymatic and GSL endocytotic/exocytotic balances (36, 37). Also, disruption of ER- and Golgi-associated endocytotic/exocytotic systems by unrelated pathways (e.g., mucopolysaccharide degradation) may increase levels of a particular GSL and cause secondary accumulations of other GSLs and gangliosides (38). These secondary storage compounds contribute directly to disease pathogenesis and to complex metabolic events leading to multiple, apparently unrelated substrate storage diseases (See Pathological consequences of disorders in GSL metabolism).
Roles of GSLs during normal development and adult stage
Changes in brain GSL synthesis and metabolism correlate with the stage-specific brain development and function, indicating a coordinated spatial-temporal regulation (Fig. 4). Elucidating the timing of GSL synthesis and the alterations of GSL fluxes in the disease states is essential to understanding the pathogenesis and propagation of the various GSL LSDs.
Fig. 4.

GSLs and gangliosides in developmental stages of the nervous system. The diagram shows GSL profiles during development of the mouse nervous system. Mouse embryonic developmental stages are from single cell (∼ day 1), 16 cells (∼ day 3) to fetal (day 19). The mouse embryonic and neural developmental stages are described (320). Colored (vertical) bars above the timeline illustrate overlapping developmental stages. The horizontal gradient bars show the dynamic changes of GSL and ganglioside levels that occur during the stages of mouse embryo and neural development based upon experimental observations (22, 39, 40, 43, 44) (Sun et al., unpublished observations). The dark green indicates increased levels of the specific lipids and the less dense green implies lower levels. Ceramide could be identified at 2-4 cell stage (39). The hatched bar demonstrates the hypothetical ceramide levels since ceramide synthesis remains active throughout life.
In the mouse embryo, ceramide, the core structure of GSLs, is detected by the 2-4-cell stage following fertilization, before neural tube formation, and is present throughout life (39, 40). The globo-series lipids occur at the 2-cell stage (E0.5) followed by the lacto-series at E1.5, and the ganglio series at E7 (40–42) (Fig. 4). GlcCer is present by E11 during the neuronal stem cell proliferation stage. The concentrations of GlcCer are greater during the neurogenesis and astrocytogenesis at mid-embryonic stages (E12 and E14); these steadily decline at later stages (43, 44). By postnatal day 11, GlcCer continues to decrease coincidently with axonal dendritic arborization during adulthood (43) (Sun et al., unpublished observations). Around E16 during fetal development, the central nervous system (CNS) ganglioside distribution patterns shift sequentially from predominantly GM3 and GD3 to the a- and b-series (GD1a, GD1b, and GT1b) and GM1 (22). GalCer and sulfatide become detectable at E17 and predominate at maturity in the adult mouse brain (43). A steady increase in sulfatide content begins at about postnatal day 7 and continues into adulthood in the rat, correlating with active myelin formation. This is regional, because sulfatide levels in the spinal cord are 5-fold higher than in cerebral cortex (45).
These lipid shifts are primarily modulated by differential expression of specific glycosyltransferases because glycosidases vary little during brain development. GD3-synthase and GM2/GD2-synthase are key enzymes in ganglioside biosynthetic pathways (Fig. 1). GM2/GD2-synthase expression significantly increases during in utero development (46–48) and ex vivo in differentiating primary neural precursor cells (43). In comparison, the key enzymes, GCS, ceramide UDP-galactosyltransferase (CGT), and GM3 synthase are not differentially expressed during development (43).
Defects of GSL biosynthesis in mice and some human patients provided insights into the importance of GSLs and gangliosides during brain development (Table 2). The specific roles of GSLs in survival, proliferation, and differentiation have been demonstrated by knockout mice with embryonic lethality (GCS and SPT) (49, 50) and severe neurodegenerative diseases (CGT) (51, 52). Disrupted gangolioside synthesis in mice with either GM3 synthase (53, 54), GM2/GD2 synthase (55), or α-N-acetyl-neuraminide α-2,8- sialyltransferase (GD3 synthase) (56) knocked out causes differential neurological impairments with normal brain formation. Mice lacking CGT are unable to synthesize galactosylceramide. They showed normal growth but had nerve conduction deficits (51, 52). A compensatory increase in GlcCer was observed. The early embryonic lethality of the GCS-deficient mouse delineates the essential roles of complex GSLs in survival (50). Furthermore, the neurodegeneration in the GCS neuronal knockout showed the importance of GSLs in the maintenance of the CNS (57, 58).
TABLE 2.
Disorders of GSL and ganglioside synthesis
| Affected Gene(s) | Enzyme, Name(s) | Affected GSLs | Species | Phenotype | Lifespan | References |
|---|---|---|---|---|---|---|
| SPTLC1 | Serine palmitoyltransferase, long chain base unit 1, SPTLC1 | Increased de novo glucosyl ceramide, accumulation of neurotoxic 1-deoxy-sphinganine and 1-deoxymethyl- sphingonine | Human | Distal sensory loss, sensorineural hearing loss starting between 20 and 50 years | Normal | (59, 60, 62) |
| Sptlc1 Sptlc2 | Serine palmitoyltransferase, long chain base unit 1 Sptlc1 or 2,SPT Sptlc1−/− and Sptlc2−/− Sptlcl+/− and Sptlc2+/− | Mouse | Embryonic lethal | (49) | ||
| Decrease ceramide and sphingosine in liver and plasma, decreased SIP levels in plasma | ||||||
| SIAT9 | GM3 synthase CMP-NeuAc:LacCer α2,3-sialyltransferase LacCer α-2,3-sialyltransferase | No a- and b-series gangliosides | Human | Developed severe epileptic seizures starting between 2 weeks and 3 months | 2–18+ years | (64) |
| Siat9 | GM3 synthase ST3 β-galactoside α-2,3-sialyltransferase | Mouse | Hearing loss detected 13 days after birth; increased insulin sensitivity detected in 6–8 week old mice | Normal lifespan | (53, 54) | |
| Siat8a | GD3 synthase CMPsialic acid:GM3 -2,8-sialyltransferase α-N-acetyl-neuraminate α-2,8- sialyltransferase | No b-series ganglioside | Mouse | Normal | Normal lifespan | (56) |
| GALGT1 | GM2/GD2 synthase UDP-N-acetyl-D-galactosamine: UDP-GalNAc: GM3 N-acetylgalactosaminyltransferase | GM3 accumulation | Human | Poor physical and motor development, frequent seizures | 2–3 months | (61) |
| Galgt1 | GM2/GD2 synthase UDP-N-acetyl-D-galactosamine: GM3/GM2/GD2 synthase, β-1,4-N-acetylgalactosaminyltransferase, GalNAcT. | Lack all complex gangliosides, express high levels of GD3 and GM3 | Mouse | Slight reduced neural conduction velocity due to axonal and degeneration and demyelination in CNS and PNS, detected in 10–16 week old mice, males infertile | > 1 year | (55, 365, 366) |
| Galgt1 + siat8a | GM2/GD2, and GD3 synthases | GM3 ganglioside only | Mouse | Fatal audiogenic seizures and peripheral nerve degeneration detected in 2–4 month old mice, males infertile | 3–9 mos | (367) |
| Galgt1 + Siat9 | GM2 and GM3 synthases | No higher order gangliosides | Mouse | Severe neurodegeneration hind limb weakness, ataxia, and tremors starting at 2 weeks old | 2–5 mos | (368) |
| Cgt | Galactosyltransferase CGT | Lack galactosylceramide and sulfatide | Mouse | Severe generalized tremor and mild ataxia starting between 1 and 2 weeks | 2.5–4 weeks | (52) |
| Lack galactosylceramide and sulfatide | Mouse | Severe dys/demyelination and loss of nerve conduction velocity starting between 1 and 2 weeks | 3.5–4 weeks | (51) | ||
| Ugcg | Glucosylceramide synthase GCS | No GlcCer-based gangliosides | Mouse | Embryonic lethal | E7.5 | (50) |
| Ugcg knockout in neurons and glial cells | Trace levels of GlcCer- based gangliosides in neural and glial cells | Mouse | Cerebellum and peripheral nerve dysfunction, reduced axon branching of Purkinje cells beginning at 5 days after birth | 3 weeks | (58) | |
| Ugcg knockout in keratinocyte cells | Decreased GlcCer in epidermis | Mouse | Desquamation of the stratum corneum, transdermal water loss leading to death starting from 3 days postnatally | 5 days | (369) |
To date, three human diseases are associated with mutations in enzymes involved in de novo GSL synthesis (59–61). SPT consists of three subunits and catalyzes the first step in sphingolipid synthesis (Fig. 1). Six missense mutations in SPT long-chain subunit 1 (SPTLC1) were reported in 26 families that have autosomal dominant hereditary sensory and autonomic neuropathy type 1 (62, 63). These SPT mutations caused changes in substrate specificity that led to formation of two atypical neurotoxic deoxyl-sphingoid bases by condensation of palmitoyl-CoA and alanine or glycine, instead of serine (62). Thus, hereditary sensory and autonomic neuropathy type 1 is caused by a gain-of-function mutation. GM3 synthase catalyzes the first step of complex gangliosides synthesis (Fig. 1). A loss-of-function mutation in GM3 synthase (SIAT9) was identified in a cohort characterized by autosomal recessive infantile-onset symptomatic epilepsy syndrome (64). A nonsense mutation in exon 8 of the GM3 synthase gene produced premature termination that could abolish enzymatic activity (64). Affected individuals are homozygous for the mutation while heterozygous carriers are unaffected (64). A single case of GM2 synthase deficiency was reported by biochemical characterization only (61). The patient died at 3 months after presenting with abnormal motor function and seizures. GM3 accumulated in the brain and liver (61).
Phenotypic analyses from GSL synthase-deficient mice reveal that lack of all GSL is incompatible with embryonic development (49, 50). Simple to complex GSLs are nonessential for early brain development but are important for brain maturation and maintenance (51–56). Compensatory interchange between various GSLs also signifies additional complexities in their metabolism.
Distribution and functions of GSLs
The sialic acid-containing GSLs are ubiquitously expressed in the outer leaflet of the plasma membranes, e.g., liver (65), and are most abundant in CNS and other nervous tissues (22, 65, 66), but there is significant tissue/ regional variability in distribution. High concentrations of GD1a are present in extraneural tissues, erythrocytes, buffy coat, bone marrow, testis, spleen, and liver, whereas different GSLs are in high amounts in other tissues, e.g., GM4 in kidney, GM2 in bone marrow, GM1 in erythrocytes, and GM3 in intestine (19). GA2, GM2, and GM3 are at very low or nondetectable levels in normal brains from humans, cats, and mice (66). Cellular specificity is exemplified by GD1b localization to small neurons, whereas O-Ac-disialoganglioside localizes to large neurons (67). In liver, GM1 was detected on the canalicular and sinusoidal lining cells, but nearly absent from liver parenchymal cells; GM1 was increased in hepatic sinusoidal membranes during cholestasis (68). In skeletal muscle, the neutral GSLs and gangliosides were mostly in membrane vesicles with GlcCer predominating and only trace levels of LacCer (69, 70). GalCer, sulfatide, and sphingomyelin are structural to myelin sheaths and are major lipids in oligodendrocytes and Schwann cells (71). In skin, ceramide comprises about 50% of total epidermal lipid and is generated from GlcCer in the lamellar bodies (72). These cellular microdifferences in GSLs indicate subtle, yet potentially important, metabolic roles that have not been elucidated. Cellular distribution of GSLs can be altered in pathologic states. In MDCK kidney cells, mitochondrial and peroxisomal GSLs are very low/absent (73), whereas GD1b and GD3 had high contents in mitochondria from malignant hepatomas (74). During the apoptotic process, GD3 is rapidly synthesized and relocated from the cis Golgi to mitochondria, leading to the opening of mitochondrial permeability transition pores and release of apoptogenic factors (75).
GSLs have differentially ordered domains to create selectivity in membrane transport that is important in the spatial organization of cells (76). GSLs are enriched to 30–40 mol% in some cell types, e.g., in the apical membranes of intestinal and urinary tract epithelial cells or in the myelin of axons (77–79). Complex GSLs are generally enriched on the plasma membrane, but GlcCer is mostly localized to intracellular membranes (80). GSLs are found in the vacuoles of the exocytotic and endocytotic systems with low content in the ER (65, 73). Complex GSLs are not translocatable to the cytosolic surface of Golgi (81).
Rafts are ordered structures that participate in cell recognition and signaling. These microdomains or lipid rafts are composed of GSLs, sphingomyelin, and cholesterol (33, 82) that provide environments for enrichment of specific proteins (83) in a variety of membranes. Examples include Src family kinases that are important for signal transduction (84), glycosylation-dependent adhesion/ recognition and signaling (85), and G-protein-coupled receptor-signaling (86).
Additional roles of GSLs, including ceramide, sphingosine, sphingosine-1-phosphate, lysoglycolipids, and lysogangliosides, are their inhibition or activation of apoptosis, proliferation, and stress responses (87). Cellular ceramide is an important second messenger in signal transduction that modulates a variety of these physiologic and stress responses (Fig. 2) (87). Several possible intracellular ceramide-target enzymes and signaling pathways have been proposed, including the activation of Ras/extracellular signal-regulated kinase (ERK)-MAPK cascade (88), protein kinases PKCζ (89), kinase suppressor of Ras/MAPK (90), SAPK/JNK signaling pathway (91), Raf-1 (92), protein phosphatases PP1 and PP2A (93), and JNK (91); all contribute to the control of cell growth, proliferation, and death through various downstream intermediates.
For the field of GSL LSDs, a major challenge remains in the unification of the cell and developmental specificity, biological functions, and pathogenic mechanisms. Given the plethora of diverse and essential cellular functions, the severity of disruptions in GSL metabolism would be anticipated to include more than just accumulation of the GSL in cells and architectural distortions of specific cell types. Such mechanistic understanding of the GSL storage diseases is just beginning to be delineated (See below).
DISORDERS OF GSL AND GANGLIOSIDE DEGRADATION IN HUMANS AND MOUSE MODELS
The catabolic defects in neutral GSL and ganglioside metabolism result in LSDs with complex, multi-system progressive diseases, including neurodegeneration, that can present early in utero or childhood. All the catabolic enzymes for these diseases exist in the lysosomes (Fig. 3). Later onset and more attenuated variants of each disorder have been or are anticipated to be present in humans (Table 1). All these diseases are autosomal recessive except Fabry disease, which is X-linked. The phenotypic variants and the molecular causes have been variably delineated. Attempts have been made to correlate the genotype with phenotype. This is a complex and incomplete task because of the variability in presentation, nonuniform clinical delineation, and lack of complete allele characterization. However, the residual in situ hydrolase activities in patients are a major, but not unique, determinant of the age at onset and/or severity of the disease. Work by Sandhoff et al. (94) clearly established in situ residual activity as a critical correlate of phenotype with a direct, albeit nonlinear, relationship between this activity and the age at onset in Tay-Sachs disease and metachromatic leukodystrophy (MLD). Extensive clinical data show similar correlations in Gaucher disease type 1 (95). Mouse models of several LSDs have been developed (Table 1). Although the mouse phenotypes are qualitatively similar to its corresponding human disease, such models can show substantial differences because of differences in GSL metabolism between humans and mice. Analyses of such mouse models of GSL synthesis and degradation have provided insight into early pathological mechanisms of GSL and ganglioside flux aberrations.
The disorders of ceramide, GSLs, and gangliosides degradation in humans and mice are summarized below. The phenotypes and genotypes of these disorders are summarized in Table 1.
Disorders of ceramide degradation
Disorders of ceramide degradation are caused by acid ceramidase (AC; EC 3.5.1.23) deficiencies that lead to Farber disease, a.k.a. Farber lipogranulomatosis (96, 97). AC catalyzes the hydrolysis of ceramide to free fatty acid and sphingosine, and its function is optimized by saposin D (Figs. 1 and 3) (98). However, AC can also synthesize ceramide from sphingosine and free fatty acids (Fig. 1) (99, 100). This reverse reaction is optimally catalyzed at pH ∼6.0 compared with the hydrolytic activity optimum of pH ∼4.5, suggesting that the two reactions likely occur in different subcellular compartments. There are seven variant forms of Farber disease that are distinguished by severity and tissue involvement (96). Although allogenic hematopoietic stem cell transplantation in Farber disease patients without CNS manifestation showed improvement in joint movement (101), there is currently no effective therapy for this disease. The AC-deficient mouse (Asah1−/−) undergoes apoptotic death at the 2-cell stage (E1; Fig. 4) because of excessive ceramide accumulation (39, 102), demonstrating that AC is expressed in the embryo and that ceramide degradation is essential for survival beyond the 2-cell stage (E1) (102). In addition to increasing ceramide pools, loss of AC activity is thought to concurrently reduce pools of sphingosine and sphingosine-1-phosphate, two lipids known to promote cell growth and differentiation (103).
Disorders of GSL degradation
Mutations in the SMPD1 gene that encodes aSMase (ASM; EC 3.1.4.12) cause two types of Niemann-Pick disease: Types A and B (NPA and B) (Fig. 1, Table 1). NPA presents a neurovisceral phenotype, whereas NPB exhibits visceral manifestation (104). The ASM-deficient mice, developed independently in two laboratories, resemble NPA or NPB phenotypes (Table 1) (105, 106). Neither model has detectable lysosomal ASM activity in any tissue but has reduced levels of plasma membrane-bound nSMase activity, which cleaves nonlysosomal sphingomyelin at neutral pH (Fig. 2) (106, 107). The ASM gene is expressed prior to E11 during neuronal stem cell proliferation, and deficient ASM activity, due to genetic defects, can lead to loss of signaling molecules that regulate their proliferation very early on in brain development (108, 109).
Krabbe disease, globoid leukodystrophy disease, is a rapidly progressive, demyelinating disease that results from insufficient cleavage of galactosylceramide and galactosylsphingosine by galactosylceramide-β-galactosidase (GALC; EC 3.2.1.46) (Table 1, Fig. 3). This enzyme has specificity for both galactosylceramide and galactosylsphingosine, the latter being a specific substrate for this enzyme whereas galactosylceramide can be cleaved by at least two other lysosomal β-galactosidases (Fig. 1) (110), potentially accounting for the lack of large accumulations of galactosylceramide in the CNS (111). GALC has a number of nonsynonymous polymorphisms (missense or nonsense mutations) that affect the expression and function of the enzyme (111, 112). There are four forms of the disease that differ in age of onset (113). Secondary pathological changes including reactive astrocytic gliosis, infiltration of unique and often multinucleated macrophages (globoid cells), and other inflammatory responses accelerate disease progression (111). Bone marrow/stem cell transplantation with histocompatible cells has shown some positive effects on the CNS and peripheral nervous system (PNS) phenotypes in the infantile onset disease when performed prior to the onset of significant neurological involvement. However, the dementia and other aspects of the CNS and PNS disease eventually lead to death (114). GALC-deficient mice (twitcher mouse) were discovered at the Jackson Laboratory in 1976 (115, 116). This naturally occurring model is an excellent model of the infantile human disease (117–119). A transgenic globoid leukodystrophy disease Krabbe disease model was created by ‘knock-in’ of a missense mutation (H168C) (120). The analyses of these models demonstrated that there is a correlation between accumulation of galactosylsphingosine and the neuronal defects. The accumulation of galactosylsphingosine was detected before myelin formation (119). Bone marrow transplantation in twitcher mice stabilized galactosylsphingosine at low levels accompanied with remyelination (121). These findings provide support for galactosylsphingosine as the offending agent responsible for triggering proinflammation, oligodendrocyte depletion, and dysmyelination in Krabbe disease.
MLD is caused by a deficiency of arylsulfatase A (ASA; EC 3.1.6.8) that leads to the accumulation of sulfatide, a major lipid of myelin, in the CNS and PNS (Fig. 3) (122, 123). Currently, five allelic forms of ASA deficiency exist (Table 1). Also, there are two nonallelic variants of ASA deficiency: 1) MLD due to saposin B deficiency (described below) leads to inability to cleave sulfatide and globotrioaosylceramide (Gb3) in vivo as a result of the activator protein deficiency and impaired lipid presentation to the enzyme, even though ASA and α-galactosidase A are normally expressed and without mutations (124); and 2) multiple sulfatase deficiency is a disorder that combines features of a mucopolysaccharidosis with those of MLD (125) and results in a deficiency of all cellular sulfatases due to defects in a posttranslational enzyme essential for modification of an active site cysteine that is present in all sulfatases (122, 126). Details of multiple sulfatase deficiency have been reviewed (125, 127). For the infantile and juvenile MLD variants, bone marrow/stem cell transplantation has been attempted with initial improvement/stabilization followed by later progression of the CNS and PNS diseases (128). ASA-deficient mice (Asa−/−) have a very slowly progressive sulfatide storage resembling the early stages of human cases (129). A more severe phenotype of ASA deficiency was achieved by overexpressing the transgene for the sulfatide-synthesizing enzyme, galactose-3-O-sulfotransferase (CST), encoded by Gal3st1, in oligodendrocytes (130). These models have been used in preclinical trials of MLD (131, 132).
Fabry disease, the only X-linked GSL storage disease, is due to a deficiency of α-galactosidase A (133), the enzyme that cleaves Gb3 and di-galactosylceramide (Fig. 3, Table 1). Uncharacteristic of most X-linked traits, both males and females have significant to major clinical involvement (134). Affected patients accumulate the substrate, Gb3, in most tissues and organs (135), but the progressive deposition of Gb3 in capillary endothelial cells leads to many of the morbid manifestations (136). Increased deacylated Gb3, globotriaosylsphingosine (lyso-Gb3), in patient plasma suggests that it may be involved in pathogenesis of Fabry disease (137). The mouse model of Fabry disease (Gla−/−) has accumulation of Gb3 in peripheral nerves and alterations of sensory nerve function that resembles neuropathic pain in Fabry disease patients (138). Gb3 accumulation in endothelium leads to vascular dysfunction, thereby providing an in vivo model to delineate the basis of cardio- and cerebrovascular complications associated with Fabry disease (139, 140).
Gaucher disease results from insufficient cleavage of GlcCer and glucosylsphingosine by the lysosomal enzyme, acid β-glucosidase or glucocerebrosidase (GCase) (Fig. 3) (95, 141). A homologous pseudogene (ψGBA) located 16 kb downstream from the GBA gene has complicated the molecular diagnosis of Gaucher disease (142). The phenotypes are a continuum of degrees of involvement but can be divided categorically into neuronopathic (types 2 and 3) and nonneuronopathic (type 1) variants (Table 1). A nonlysosomal β-glucosidase (GBA2) has been identified as participating in GlcCer degradation (143). Inhibition or knockout of GBA2 activity is associated with GlcCer accumulation in testis and impairment in spermatogenesis (144). However, the link of GBA2 to Gaucher disease is unclear. Enzyme replacement therapy has been successfully used to treat Gaucher disease type 1 (145), whereas new substrate synthesis inhibition and enzyme enhancement therapies are in clinical trials to treat type 1 and 3 diseases (146, 147). There are no direct treatments available for Gaucher disease type 2. The Gaucher disease mouse models (Table 1) demonstrate that accumulation of GCase substrates, GlcCer and glucosylsphingosine, begin at embryonic day 13 (148). Also, substrate storage does not affect brain formation but does lead to early and severe degeneration of the brain and disruption of the skin permeability barrier (149, 150). The downstream biological consequences of GlcCer and glucosylsphingosine accumulation are currently being investigated (see section III).
Disorders of ganglioside degradation
Two types of ganglioside degradation disorders are characterized. GM1 gangliosidosis results from deficiency of lysosomal GM1-β-galactosidase (EC 3.2.1.23) that cleaves ganglioside GM1 (Fig. 3, Table 1), its asialo derivative, GA1, galactose-containing oligosaccharides, keratin sulfate, and other β-galactose-containing glycoconjugates including galactosylceramide (151). GM1-β-galactosidase is part of a multi-enzyme complex with α-neuraminidase and "protective protein"/cathepsin A (152, 153) that is assembled posttranslationally during transit through the Golgi (154). There is an inverse correlation between disease severity and residual β-galactosidase activity in GM1 gangliosidosis patients (155). GM1 gangliosidosis mice show severe neurological defects and resemble the human disease (156, 157). Preclinical studies using substrate reduction and gene therapies have been tested in animal models of GM1-gangliosidosis (158–160).
GM2 gangliosidoses are a group of severe neurodegenerative disorders unified by the excessive accumulation of GM2 ganglioside mainly in neuronal cells. GM2 is specifically hydrolyzed by β-hexosaminidase A (EC 3.2.1.52) (Fig. 3). Two genes are required for the formation of this enzyme. The HEXA and HEXB genes encode the α- and β-subunits, respectively (161, 162). The subunits assemble in the Golgi forming αβ2 [β-hexosaminidase A (Hex A)] and β3 [β-hexosaminidase B (Hex B)] (161, 163, 164). The nonallelic GM2A gene encodes the GM2 activator protein that complexes with the ganglioside GM2 (165) (see below). Defects in any of these three genes may lead to impaired degradation of GM2 gangliosides and related substrates. Tay-Sachs disease (variant B) is the classic GM2 gangliosidosis and results from the deficiency of the α-subunit that produces the specific loss of Hex A, whereas Hex B is at normal or somewhat increased levels, thereby designated as variant B (161). This specific deficiency in Hex A produces GM2 ganglioside accumulation in brain, because GM2 ganglioside is synthesized in appreciable amounts only in the CNS (Fig. 3). Currently, there is no effective treatment for Tay-Sachs disease, although trials of "molecular chaperone" approaches are underway for the late onset variants with residual Hex A activity (166). Sandhoff disease (variant 0) is caused by mutations of the HEXB gene that leads to loss of the β-subunit, resulting in deficiency of both Hex A and Hex B activities (Fig. 3). Unlike Tay-Sachs patients who store mainly GM2-ganglioside, Sandhoff patients accumulate additional N-acetyl β-galactosyl terminated glycolipids and oligosaccharides in the CNS and viscera (167, 168).
The Tay-Sachs disease mouse model (Hex A−/−) displays a milder phenotype compared with Tay-Sachs patients. This is accounted for by the differences in GM2 catabolism between mouse and human (169). Two pathways have been proposed for GM2 metabolism in mice. One involves GM2 hydrolysis to GM3 mainly by Hex A and GM2 activator, which is the major pathway in humans (Fig. 3) (161). The second one is specific to mice and involves GM2 hydrolysis by sialidase first to GA2 and then to LacCer by Hex B. In Hex A−/− mice with a complete loss of Hex A, GA2 can be degraded by Hex B (169). Unlike Hex A−/− mice that have minor GM2 storage in neurons and no detectable neurological impairment, Sandhoff mice (Hex B−/−) are severely affected. The lack of Hex A and B activities in Sandhoff mice provides an authentic model of acute human Sandhoff disease (169). Substrate reduction therapy with N-butyldeoxygalactonojirimycin (NB-DGJ), an imino sugar that inhibits GCS, has been tested in adult (170) and neonatal (171) Sandhoff mice. NB-DGJ significantly reduced total brain and GM2 ganglioside content when administered from postnatal days 2–5 in Sandhoff mice to a greater extent than in the adult mice. No adverse effects were found with NB-DGJ treatment in either age group. These results indicate that earlier intervention is an effective method for managing GSL LSDs.
Activator protein deficiency
The physiologic importance of sphingolipid activator proteins in GSL degradation has been highlighted in the patients with mutations in the GM2 activator and the prosaposin genes; the latter encodes a precursor of four saposins (A, B, C, and D).
GM2 activator (variant AB) deficiency is caused by mutations that lead to an inability to form GM2/GM2-activator complexes in the presence of normal amounts of Hex A and Hex B (Fig. 3) (165, 172, 173). Mechanistically, the GM2 activator protein is a "liftase" that extracts GM2 from intra-lysosomal membranes making the GM2 terminal N-acetyl-galactosamine accessible to β-hexosaminidase A for cleavage (174). The Gm2a−/− mice store mostly GM2-ganglioside and low amounts of GA2 (175), similar to the Hex A−/− mice, thereby establishing its essential role in the GM2 catabolism (175).
Prosaposin/total saposin deficiency was found in four families and was associated with multiple GSL storage (Table 1) (176–178). Saposin (Sap) A is an in vivo activator for GALC (124). Sap B has lipid transfer properties and participates in degradation of sulfatide by ASA, digalactosylceramide, and Gb3 by α-galactosidase A, and GM1 ganglioside and LacCer by β-galactosidases (Fig. 3) (179, 180). Sap C is required for GCase to achieve maximal activity in vitro and ex vivo and protects GCase from proteolysis (181, 182). In comparison to Sap B, Sap C physically interacts with either phospholipid membrane or acid β-glucosidase to optimize this enzyme's activity (183, 184). Direct binding of Sap C to the substrate, GlcCer, has not been shown as part of this optimization (185, 186). Individual Sap A, Sap B, or Sap C deficiencies in humans are rare (181, 187–189). Sap D activates AC for degradation of ceramide. Heteroallelic mutations were identified in the Sap D domain in a patient with an additional mutation in the Sap C domain on a different allele (189). Prosaposin and all four Sap-deficient mice have been developed (98, 190–194). In general, individual Sap deficiencies in mice results in slowly progressive diseases compared with total Sap-deficient mice or their cognate enzyme knockout mice (Fig. 3, Table 1). These could be due to cross-compensation in GSL degradation by multiple saposins (Fig. 1). Proinflammation is a common pathological finding in the CNS from such models because of GSL accumulation. The clinical, pathological, and biochemical phenotypes of prosaposin knockout mice closely resembles those of the human disease, whereas Sap B−/− and Sap C−/− models are more slowly developing than those in humans.
Lipid trafficking disorders
Niemann Pick disease Type C (NPC) is not a disorder directly related to the metabolism of GSLs and gangliosides but rather has major involvement in the egress of free cholesterol from the lysosome (195). NPC is characterized by the accumulation of unesterified cholesterol and other lipids in endosomal/lysosomal compartments that stem from inherited deficiencies of the NPC1 or NPC2 proteins. NPC1 is a transmembrane protein and NPC2 is a soluble protein. Both are involved in intralysosomal cholesterol trafficking (196). Recently, NPC2 was proposed to act as a donor of cholesterol to NPC1 that facilitates cholesterol transport across the lysosomal membrane (196). Loss-of-function mutations in the NPC1 gene also lead to failure of the calcium-mediated fusion of endosomes with lysosomes, resulting in the accumulation of GlcCer, LacCer, and GM2 in late endosomes and lysosomes (195). The exact mechanism of disease causality by either cholesterol (primarily) or GSLs (secondary or primary) accumulation has not been resolved. Elevation of sphingosine was found before cholesterol and GSL accumulation (195). The importance of sphingosine in NPC1 has been addressed recently (197). NPC1-deficient mouse models (NPC1−/−) show similarities with the human clinical NPC1 phenotypes (198). Lipid profile and disease phenotype in NPC1, NPC2, and NPC1/NPC2 double mutant mice were qualitatively and quantitatively very similar, indicating that both proteins operate cooperatively without compensation for each other in trafficking cholesterol to the lysosomes (196, 198–200).
Most GSL LSDs present with neurological phenotypes that begin early in life. The analyses of the mouse models show that the lipid accumulation in the CNS starts very early on in development. Such storage may subtly alter brain and cellular functions. GSL accumulation has been postulated as a trigger to abnormal cellular responses that potentiate the pathological disease process. Although some of the mouse phenotypes differ from the analogous human disease, the biochemical profiles in the GSL synthesis or degradation pathway have provided insight into the pathological mechanisms and GSL flux aberrations. However, systematic analyses of GSL profiles in relation to early brain development (Fig. 4) and the early molecular events have yet to be elucidated. Such understanding should be useful in defining the initiating and propagating mechanisms that would provide a road map for designing therapeutic strategies.
PATHOLOGICAL CONSEQUENCES OF DISORDERS IN GSL METABOLISM
GSL disorders present complex disease phenotypes and diverse pathological changes. The defects in GSL metabolism affect cellular and molecular functions and lead to abnormalities of the CNS and visceral organs (Fig. 5). The mechanisms leading to this pathology are poorly understood. Are these disorders triggered by accumulated lipid or insults from secondary molecular defects in the cells? Does inflammation potentiate the disease? What causes neuronal drop out? Could lesions result from lack of products as well as from storage of substrate in the GSL pathway? The following section summarizes some of the pathological and molecular findings, which may be preludes to understanding the signature pathways in the GSL diseases.
Fig. 5.
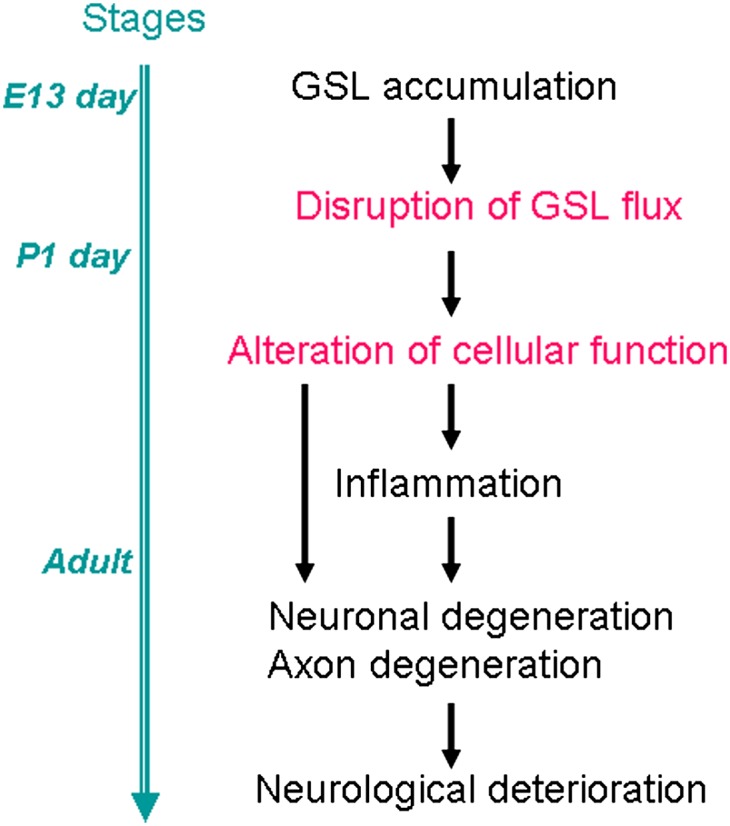
Hypothetical scheme for neuronal disease progression in the nervous system. Accumulation of GSLs causes abnormalities of neuronal cell functions in mouse models and the human diseases. Abnormal degradation of GSL may disrupt the normal flux of GSL in the cell, which can affect a variety of cellular functions. How the GSL accumulation alters GSL flux and cellular function is largely unknown. The changes of the cellular function may cause neuronal degeneration. Inflammation is often detected before the neuronal and axonal defects and it plays a role in potentiating disease progression. The blue line on the left shows the stage of brain development.
Evidence of GSL accumulation correlating with disease progression
Disruption of GSL flux is thought to initiate and potentiate LSD phenotypes. The slow accumulation of sulfatide in Asa−/− mice does not disrupt myelin formation, which occurs in the first 3 weeks postnatally in the mice but does destabilize myelin integrity in adults (201). Although lysosomal accumulation of Gb3 begins in utero, the signs and symptoms of Fabry disease may not be apparent until childhood or adolescence (202). The association of GlcCer, and, especially glucosylsphingosine, with neuronal toxicity is evident in humans and mice with Gaucher disease. GSL composition studies show that glucosylsphingosine and GlcCer in Gaucher type 1 brains are substantially lower (3–10%) than that in Gaucher disease type 2 brain (203), suggesting a relationship between substrate accumulation and severity of brain disease. Galactosylsphingosine levels in twitcher mice brain progressively increase from 20 to 40 days as does the neurological phenotype (119, 204). Unlike galactosylsphingosine in Krabbe disease, glucosylsphingosine has not been proven in vivo as a direct cause of CNS Gaucher disease. However, a recent glucosylsphingosine storage disease mouse model supports this concept (205). The correlations between disease progression and age-dependent GSL levels also occur in NPC disease (206), Tay-Sachs disease (207), and GM1 gangliosidosis mice (156).
GSL accumulation occurs prior to the onset of a disease phenotype, e.g., glucosylsphingosine accumulation was found at the 11th week of pregnancy in human Gaucher disease (208). Elevated glucosylsphingosine can be detected as early as E13 during neurogenesis in Gba knockout mice (208). In V394L/SapC−/− mice, increased glucosylsphingosine is present by 14 days, i.e., before the onset of neurological signs at 30 days (Table 1). Abnormal accumulation of galactosylsphingosine in twitcher mice is detected in the brain and sciatic nerves at postnatal day 7, before the onset of demyelination, and progressively increases to 100-fold normal levels, predominantly in white matter (209, 210). In normal mice, GM2 is not detectable, but GM2 is elevated at postnatal day 2 in Sandhoff mice before any pathological signs (171). Such early lipid accumulation may trigger molecular alterations, e.g., proinflammation, as shown by global gene expression analyses in Sandhoff disease mice in which upregulation of inflammation-related genes preceded neuronal death (211). Similar temporal analyses of CNS tissues from prosaposin-deficient mice reveal the regionally specific gene expression abnormalities including upregulation of numerous proinflammatory genes prior to neuronal degeneration (212). Although low-level excess of GSLs may not lead to early histopathological changes, they do cause cellular alterations that initiate disease processes leading to eventual end stage disease. A final common pathway in the CNS for the GSL LSDs appears to be varying types and degrees of inflammatory and proinflammatory reactions leading to apoptotic or other types of programmed cell death.
Increased lipid levels at early embryonic stages also correlate with reduced expression of genes for neurotrophic factors [brain-derived neurotrophic factor (BDNF) and nerve growth factor (NGF)] and MAPK pathway genes (ERK) in the cortex, brainstem, and cerebellum of neuronopathic Gaucher disease mice at E17.5, E19.5, and P1 (213). The deficiencies in neurotrophic factors (BDNF and NGF) cause sensory neuron degeneration in mice (214, 215). These early abnormalities in lipid and neurotrophic factors correlated with substantial reductions in cellular density, neurodegeneration, and microglial infiltration in Gba knockout mouse brains at E13–14 (149, 150, 208). Injection of the GCase inhibitor (cyclopellitol) into wild-type mice led to a 17- to 31-fold increase of glucosylsphingosine in brain, liver, and spleen with concomitant neurological abnormalities (216). These findings provide further evidence for the relationship of abnormal substrate levels in GCase deficiencies and neuronopathic phenotypes. These data suggest that a toxic stimulus is present long before birth or at least early in postnatal life, and that therapy begun during infancy may not prevent progressive neurologic damage. In this regard, the conditional experiments of Xu et al. (150) are instructive. A chemically induced model of CNS Gaucher disease was produced by administering a covalent inhibitor, conduritol B epoxide (CBE), of GCase to neonatal mice (150). Wild-type and GCase point mutated mice were given daily injections of CBE from postnatal day 5 to day 11. About 1 day after CBE treatment was stopped, the mice developed neuropathic signs. No mice survived if CBE was continued through day 13. Because the wild-type and mutant GCases have t1/2 ∼60 h in cells (217), the neuropathic signs in wild-type mice or those with D409H homozygosity stabilized for the next several months in the absence of CBE. However, with a lower activity mutant, D409V/null, the neuropathic signs progressed to very severe within 2 months; unlike the wild-type and D409H homozygotes, the glucosylceramide levels in brain did not decrease, and there was continuing evidence of neuroinflammation. These data support the concept that specific reconstitution levels of in situ activity must occur at the appropriate developmental time frame to avoid propagation of disease. Furthermore, once established, the neuronopathic disease does not resolve but can be stabilized.
Secondary storage or deficiency of GSLs
Alterations of GSL flux can directly affect membrane composition and disrupt cell homeostasis that may cause secondary storage (38) and potentiate disease processes. Such secondary accumulations of GSLs and gangliosides are common features associated with neuropathology in several LSDs, e.g., Niemann-Pick diseases with GM2 and other GSL accumulation (218–220). Pronounced increases in gangliosides, GlcCer, and LacCer occur in prosaposin deficiency (178, 190) and increases in GM2 and GM3 are evident in the cathepsin D-deficient mouse (221), as they are in the CNS of several mucopolysaccharidoses, MLD, Tay-Sachs disease, and Gaucher disease (222). Importantly, in neurons with secondary gangliosides accumulation, the pathological changes were essentially identical to those in the gangliosidosis (223). Because GM2 and GM3 are precursors of many gangliosides and are the final common metabolites in the ganglioside degradative pathway (224), the discovery of secondary ganglioside accumulation indicates the importance of the endosomal/lysosomal system in the overall regulation of ganglioside expression in neurons.
In the NPC 1 mice, sphingosine accumulation reduces the lysosomal calcium pool and may trigger secondary GSL storage (195). GCase mutations, even in the heterozygous state, are predisposing factors for Parkinson disease (225, 226). It is unknown whether the GSL or other cellular abnormalities determine the association between these two diseases, but increased α-synuclein deposition and Lewy bodies occur in Gaucher disease type 1 brains (227).
A clear connection has yet to be established between the primary lysosomal defect and the accumulation or deficiency of secondary compounds that become involved in the storage process. Signal transduction derangements and altered trafficking of GSLs in the endosomal/lysosomal system would be possible causes. Regardless of the mechanism, secondary GSL storage compounds appear to be actively involved in disease pathogenesis.
Inflammation
Inflammation, constituting a local response to cellular insult, occurs in visceral and CNS tissues as a general pathological event in GSL and ganglioside LSDs. The excess lipid is causally linked to a proinflammatory response, including activation of the microglial/macrophage system to produce inflammatory cytokines leading to CNS damage. In Gaucher disease type 1, GlcCer accumulates mainly in cells of mononuclear phagocyte origin. Serum levels of macrophage-derived cytokines that are pro and antiinflammatory mediators have been variously elevated (228). They include IL-1α, IL-1 receptor antagonist, IL-6, TNFα, pulmonary and activation-regulated chemokine, and soluble IL-2 receptor (229–232). Such mediators clearly could play a role in disease progression (233–235). In ASM knockout mice, the majority of genes with elevated expression in lung and brain were those involved in the immune/inflammatory response (236). Activation of microglial cells and astrocytes in CNS and PNS parallels such cytokine increases. Ex vivo, gangliosides activate microglia to produce the proinflammatory mediators nitric oxide and TNFα (237). Alternatively, gangliosides also suppress toll-like receptor-induced proinflammatory cytokine expression (238). In Sandhoff disease mice and humans, macrophage/microglial activation and subsequent cytokine expression result in neuronal dysfunction (211). Similarly, progressive CNS inflammatory changes were coincident with the onset of clinical signs in Tay-Sachs, Sandhoff, and GM1 gangliosidosis mice (239).
Use of antiinflammatory drugs in several GSL LSD models slowed disease progression (240, 241). This indicates that multiple inflammatory pathways participate. Nonsteroidal cyclooxygenase-dependent antiinflammatory drugs (e.g., indomethacin, aspirin, and ibuprofen) extended the lifespans of Sandhoff disease and NPC1 mice (240, 242). In Sap A−/− mice, such treatment suppressed only early, but not later, proinflammation, indicating involvement of the cyclooxygenase pathway, but it is not the major inflammatory pathogenic pathway (Barnes et al., unpublished observations). Strikingly, continuous high levels of estrogen during the pregnancy period of Sap A−/− mice protected against proinflammation and demyelination even though galactosylceramide and galactosylsphingosine continued to accumulate (191, 243). These studies implicate the proinflammatory reactions in the propagation and potentially initiation of GSL-mediated CNS degeneration.
The underlying molecular mechanisms of GSL- or ganglioside-induced inflammation are not fully elucidated. Several disparate results have suggested signaling pathways. Macrophage inducible nitric oxide synthase can be regulated by GSL-mediated intracellular calcium elevation (244). AMP-activated protein kinase plays a role in regulating astrocytic galactosylsphingosine-mediated inflammatory responses (245). Similarly, sulfatide induces inflammatory cytokine production in microglia and astrocytes in addition to stimulating phosphorylation of p38, ERK, and JNK (246). Whole genome transcriptome analyses of prosaposin-deficient mice implicated CEBPδ as a common mediator in the STAT3 proinflammatory pathway (212). GlcCer-derived ceramide may play an antiinflammatory role by mediating p38δ MAPK activation, p38δ phosphorylation, and regulation of IL-6 (247); the partial deficiency of GlcCer-derived ceramide in Gaucher disease may release, via p38δ MAPK, suppression of IL-6 and the consequent downstream inflammatory effects (247). Greater insights into these basic mechanisms could facilitate developing adjunctive approaches to altering the progression of CNS disease in the GSL LSDs.
Several GSLs have roles as immune modulators through invariant natural killer T (iNKT) cell selection that relies on interaction of glycolipid antigens through the MHC class I-related glycoprotein CD1d in late endosome/lysosomes (248, 249). In addition to α-galactosylceramide, the mammalian lipid, isoglobotrihexosylceramide, was proposed as an endogenous glycolipid antigen (249). This has been questioned since the isoglobotrihexosylceramide synthase knockout mice do not have iNKT cell abnormalities (250). Deficiency of iNKT cells occurs in prosaposin deficiency, Sandhoff disease, and NPC1 disease (248, 249), indicating that several GSLs, gangliosides, and lysophospholipids may participate in iNKT cell selection (251), but the role of these cells and the molecular connection remain unclear.
Neuronal degeneration
The majority of GSL LSDs with CNS involvement result from processes that lead to neuronal death (Table 1, Fig. 5). CNS neuronal cell loss and gliosis in Gaucher disease types 2 and 3 correlate with GlcCer and, particularly, glucosylsphingosine levels (252, 253). Neurological deterioration in human GM1 gangliosidosis can span a period from birth up to 30 years; apoptosis is the major mechanism of neuronal death, but this is not universal, because several other cell death mechanisms have been implicated in different LSDs. Apoptosis is well understood and is linked to an unfolded protein response (UPR) that includes upregulation of BiP and CHOP, and activation of JNK2 and caspase-12 (254). GM1 loading of wild-type neurospheres causes depletion of ER calcium stores and activation of the apoptotic pathway, implicating ER GM1 accumulation as an inducer of UPR (254). However, the UPR is not general for GSL LSDs, because UPR is not evident in the Gaucher disease brain (255). Apoptotic cell death does occur in mouse and human Sandhoff and Tay-Sachs diseases that have CNS accumulation of GM2 and GA2 (169, 256). However, no Hex A or Hex B protein is made in the respective models and, thus, malfolding of a mutant protein cannot be implicated. Apoptotic neuronal death is not evident in several GSL disorders, including prosaposin knockout, Gaucher disease, and individual saposin deficient models. Recently, alterations of autophagy pathways in several LSDs, e.g., NPC1 and Batten disease (257), suggest that impairment of the autophagosome system that can lead to neuron death by as-yet-undetermined mechanisms. Altered calcium homeostasis was described in NPC1 disease (195), ASM knockout mice (102, 109), Sandhoff mice (258) and GM1 gangliosidosis mice (254, 259). A mechanistic link among GSL accumulation, Ca2+ homeostasis, and neuronal cell death may be of significance for delineating the neuronal degeneration in GSL disorder diseases.
Purkinje cells are a unique population of neurons that are very vulnerable to pathogenic insults, and their degeneration occurs in several GSL storage disease mice (98, 192, 193, 260, 261). The losses of Purkinje cells in these mice progress chronologically from lobule I to X. In ASM knockout mice, progressive Purkinje cell loss begins at ∼7 weeks, coincident with the appearance of deficits in motor function (261). Purkinje cell death is detected in 60 day old NPC1 mice that store cholesterol and multiple GSLs (GM2, GM3, GlcCer, and LacCer) (260). Decreased Purkinje cell numbers occur in human Sap C deficiency (262) and Sap C−/− mice (193). Purkinje cell loss is also observed in Sap D-deficient mice (98). LC/MS analyses of cerebella from Sap C−/− or Sap D−/− mice revealed small increases in lactosylsphingosine (LacSph) and LacCer or hydroxy ceramide, respectively (98, 193). By comparison, Sap A−/− and Sap B−/− mice do not exhibit Purkinje cell death, probably due to their selective effects on myelin development (191, 212). Although GM2 storage is detected in Purkinje cells of Sandhoff disease and GM2 activator knockout mice, Purkinje cell death was not reported (169, 175). Thus, some GSLs, e.g., ceramide, GlcCer, and LacCer, or LacSph, may cause Purkinje cell loss, but others may not. Several mouse models unrelated to GSL disorders also present with Purkinje cell loss (263–265), thereby implicating other pathways in degeneration of these cells. The mechanism(s) by which Purkinje cells degenerate are unknown, but negative TUNEL assays in Sap C−/− and Sap D−/− mouse brains suggest that apoptosis is not the major mechanism (243, 266). Recently, an imbalance was proposed between induction and flux through the autophagic pathway in contributing to cell stress and neuronal loss in NPC (267, 268). Neurochemical studies in NPC1 mice demonstrate serotonin and its main metabolite, 5-hydroxyindoleacetic acid, are altered in the cerebellum and cortex of these mice. The levels of the inhibitory amino acid glycine also were 3-fold higher in the cerebellum of NPC1 and increased L-DOPA was detected in the vermis of the cerebellum. These results implicate changes in neurotransmitters that could be a molecular basis of cerebellar neuropathology leading to Purkinje cell death (269).
Axonal degeneration
Axonal degeneration can precede and lead to neuronal death by the inflammatory, myelination defects, and abnormal lipid and protein metabolism (270) that occur in GSL and ganglioside LSDs. In GM1 and GM2 gangliosidosis, axonal spheroids and dendritogenesis correlate with progressive ganglioside accumulation (271). In humans and mice with Sap C deficiency, the soma of neurons and Purkinje cells have normal initial ultrastructure, but inclusion bodies and focal swellings of dendrites develop (193, 262). Dysregulation of autophagosomes in axonal termini has been implicated in axon degeneration (272) but has not been explored in GSL LSDs. The pathological roles of axonal spheroids in several GSL disorders, and possible links to neuronal degeneration, have been discussed in a recent review (273).
Abnormal morphology in the sensory nervous system is a common pathology in the GSL LSDs. Foamy storage material occurs in the dorsal root ganglion of GM1 gangliosidosis, Sandhoff disease, GM2 activator deficiency, Sap B−/−, Sap C−/−, Sap D−/−, prosaposin knockout mice, and NPC1 mice, as well as GM2/GD2 synthase knockouts that lack complex gangliosides (274). The storage of GSLs in dorsal root ganglion causes uncoordinated body movement in those models. GM2/GD2 synthase knockout, twitcher, and Sap A−/− mice exhibit degenerative changes in sciatic nerve, including myelin fragmentation and myelin layer separation with resultant synaptic rearrangement (118, 191, 274). GalCer or sulfatide are components of myelin GSL, and their excess accumulation causes demyelination in ASA, Krabbe disease, Sap B-deficient patients, and mouse models (124) (130, 275). In primary neuronal and neuroblastoma cultures, lysosomal accumulation of sulfatide mediates increases in endosomal ceramide that leads to apoptosis (276), thereby implicating GSL flux alterations due to sulfatide storage in neuronal death.
The psychosine hypothesis proposed that lysosphingolipids are responsible for the neuronal disease pathology and cell death mediated through aberrant modulation of PKC pathways (277). The findings that other lyso-lipids, lyso-sulfatide, LacSph, lyso-GM1, lyso-Gb3, and glucosylsphingosine are present in the GSL disorders extended this hypothesis to the lyso-sphingolipid hypothesis (278, 279). Experimental evidence to support this hypothesis is growing. Galactosylsphinosine (psychosine) is undetectable in normal tissues but is at high levels in the brains of Krabbe (globoid cell leukodystrophy) patients and mouse models (twitcher) (280). The pathology of infantile Krabbe disease includes severe dys/demyelination caused by apoptotic loss of myelin-forming oligodendrocytes and Schwann cells thought to be linked to accumulation of the highly toxic GALC substrate, galactosylsphingosine (277, 281, 282). Importantly, this was the first disease in which a lysoglycolipid was implicated as a major pathogen and led to the postulation that lysoglycolipids were the toxic agents that were responsible for the disease progression in many GSL storage diseases. In the twitcher mouse brain, galactosylsphingosine accumulation leads to alteration in lipid raft structure, which may cause abnormal cellular function in oligodendrocyte and Schwann cells and lead to retrograde axonal degeneration (283). A similar mechanism may be operative in neuronopathic Gaucher disease mice exhibiting increased CNS glucosylsphingosine levels (205, 208). Axonal degeneration has been correlated with elevated GlcCer and glucosylsphingosine levels (205), and glucosylsphingosine toxicity has been demonstrated in neurons by suppression of neural outgrowth (284). Both GlcCer and glucosylsphingosine modulate Ca2+ flux in brain microsomes (285). Defective Ca2+ homeostasis, especially increases of Ca2+release, presumably from the ER, has been suggested to be responsible for neuronal cell death (286). However, the direct effect of in vivo GlcCer and glucosylsphingosine levels on neuron and axon degeneration remains to be clarified. Several other lyso-glycolipids have been detected in GSL LSDs and their role and/or mechanism of toxicity provide fertile ground for developing support for this universal lyso-sphingolipid hypothesis.
Electrophysiologic studies in GSL disorders
The application of long-term potentiation (LTP) analyses has provided insight into the pathogenesis and levels of neural involvement of the GSL and lysosomal disorders (193, 212, 287). For such analyses, a constant level of presynaptic stimulation is converted to a large postsynaptic output, i.e., LTP, a leading experiment to study learning and memory (288). Sap C−/− and V394L/SapC−/− mice have been tested and exhibit reductions of LTP, together with marked accumulation of GlcCer and glucosylsphingosine, suggesting abnormal synaptic plasticity (193, 205). Sap B−/− hippocampi have sulfatide accumulation and normal LTP, showing that although excess sulfatide induced myelin dysfunction, it does not affect LTP (194). Although the molecular basis for LTP alteration association with GSL levels has not been elucidated, calcium dysregulation has been implicated as having a direct effect on LTP (289), which is mediated through glutamate effects on Na+ and Ca2+ flux. Indeed, disrupted calcium homeostasis was found in isolated brain microsomes from a Gaucher disease type 2 patient, the Sandhoff disease mouse (285, 290), and the ASM knockout mouse (291). Thus, selected abnormal GSL levels could alter LTP by disruption of calcium-mediated vesicular release of neurotransmitters or related receptor functions, as suggested recently for galactosylsphingosine interference of lipid raft assembly (283). The alteration of LTP in selected GSL disorders also could result from secondary pathology, such as proinflammation and neurotrophin defect (292, 293). Neurotrophins have been suggested to modulate LTP (292). BDNF, a positive effector, deficiency was found in NPC1 mice (294), whereas NGF deficiency occurs in Gaucher disease and Sandhoff disease (213). Such deficits in the brain could lead to impairment of LTP and form a basis of more global cognitive and learning deficits in patients.
THERAPY
Treatments for the GSL LSDs (Table 3) focus on two primary approaches to decrease the excess accumulated/accumulating GSLs by increasing their degradation rates or decreasing their synthesis rates. The former centers on increasing the hydrolytic capacity of the GSL enzymes by reconstituting target cells to above-threshold amounts of the specific enzyme function via several different approaches: 1) directly supplying the needed enzyme (so called enzyme replacement therapy or ERT) that can be taken up into target cells and delivered as active enzyme to the lysosome by receptor-mediated endocytosis; 2) indirectly providing enzyme by surrogate factories, transplantation of unaffected cells (e.g., hematopoietic stem cells or induced pluripotent stem cells) or addition of genes to cells that can produce wild-type or greater in vivo levels of the needed enzyme for secretion and internalization by the target deficient cells, so called, metabolic cross correction by cell or gene therapy; and 3) use of small molecules that interact directly with the specific mutant enzyme in cells to increase its activity in situ by improving trafficking to the lysosome, enhancing its stability, improving its catalytic rate constant, reestablishing proteostasis by diminishing the UPR that a mutant enzyme creates, or a combination of all these mechanisms. This approach has been called molecular chaperone therapy, but is more appropriately termed enzyme enhancement therapy or EET. The current studies and trials with EET use potent competitive inhibitors of selected lysosomal enzymes as the chaperone agents.
TABLE 3.
Therapy for disorders of GSL and ganglioside metabolism
| Diseases | Therapy | Trade Name | Co. | Pipeline | References |
|---|---|---|---|---|---|
| Gaucher disease type 1 | ERT | Ceredase | Genzyme | FDA approved (Apr 1991) | (145) Genzyme website |
| Cerezyme | Genzyme | FDA approved (May 1994) | |||
| Velaglucerase | TKT Shire | Phase 3 trials completed FDA, fast track status (Aug 2009) | |||
| prGCD | Protalix | Phase 3 trials completed FDA, fast track status (Aug 2009) | |||
| SSIT | Zavesca | Actelion | FDA approved (Jul 2003). EMEA approved (Nov 2002). Phase 4 trial results expected in 2010 | (146, 370) | |
| Genz-112368 | Genzyme | Entering Phase 3 trials | (307) | ||
| EET | Plicera | Amicus | EMEA approved (Oct 2007).Phase 2 trials completed | (310) | |
| EXR-202 (Ambroxol) | ExSAR | Approved by European and other foreign regulatory agencies for other indications. Planning Phase 1 trials in the US | ExSAR Web site | ||
| Gaucher disease type 3 | ERT | Cerezyme | Genzyme | FDA approved visceral components of Type 3 disease (2003) | (147) |
| SSIT | Zavesca | Actelion | Phase 3 trials underway in UK | www.gaucher.org.uk | |
| Fabry disease | ERT | Fabrazyme | Genzyme | EMEA approved (Aug 2001).FDA approved (April 2003).Phase 4 trials | (295, 343, 371) |
| Replagal | TKT Shire | EMEA approved (Aug 2001), seeking FDA approval | |||
| EET | Amigal | Amicus | Phase 3 trials underway | Amicus Web site | |
| Late onset Tay- Sach disease | EET | EXR-101 (Pyrimethamine) | ExSAR | Phase 1 trials temporarily suspended | (166), clinicaltrials.gov |
| SSIT | Zavesca | Actelion | Phase 2 trials ongoing | (372) clinicaltrials.gov | |
| Sandhoff disease | SSIT | Zavesca | Actelion | Phase 3 trials completed | (372, 373) |
| NB-DGJ | Toronto Research Chemicals | Preclinical trials in animal models | (171) | ||
| GM1 gangliosidosis | SSIT | NB-DGJ | Toronto Research Chemicals | Preclinical trials in animal models | (158) |
| NPB | ERT | Recombinant human aSMase (rhASM) | Genzyme | Phase 1 Trials complete April 2009. Phase 3 trials begun for 2010. Preclinical trials in animal models (clearance of lipids in liver, spleen and lung) | Genzyme website (374) |
| NPC | SSIT | Zavesca | Actelion | EMEA approved (2003) Phase 2-3 trials | (305, 375) |
| Krabbe disease | BMT/stem cell transplantation/ gene therapy | Non-FDA approved for Krabbe disease. Ongoing preclinical trials in animal models | (301, 376, 377) | ||
| MLD | BMT/stem cell transplantation/ gene therapy | Non-FDA approved for MLD.Preclinical trials in animal models | (131, 300, 378) | ||
| Farber disease | BMT/stem cell transplantation | Non-FDA approved for non-CNS Farber disease | (101) |
Treatments by increasing GSL degradation
The success of ERT in ameliorating many, but not all, of the visceral manifestations of Gaucher disease type 1 has spurred the development of this approach for other LSDs with major visceral involvement, such as Fabry disease (295). This restriction to visceral disease derives mainly from the inability of intravenously administered enzymes to cross the blood brain barrier in therapeutically significant amounts. Data are now emerging that ligands, other than the typical sugars, i.e., mannose-6-phosphate or mannose, may allow transit of proteins from the circulation across the blood brain barrier, particularly at very high plasma concentrations, as shown in the mouse models of β-glucuronidase or ASA deficiencies (132, 296). Such effects were enhanced by using an amino acid tag to increase the delivery of the enzyme to the brain substance (297). In addition, the use of intraventricular or intrathecal administration of appropriate enzymes could provide for significant therapeutic effects in the CNS. Similar considerations also apply to postneonatal delivery of AAV viral vectors for genetic transduction following intravenous administration. This limitation appears to have been partially overcome by the inclusion of disease-specific peptide ligands into AAV vectors that would then bind to endothelial cells of affected animals and express the needed enzyme, thereby facilitating uptake into affected CNS cells including neurons (298). Such an approach led to widespread distribution of the expressed enzyme in the brain following intravenous administration with resultant biochemical and phenotypic improvement.
Transplantation of hematopoietic, embryonic, neural progenitor, or induced pluripotent stem cells with or without transduced additional genes is being used to deliver the needed therapeutic enzymes (299–303). Such cells can either add to existing cell populations in the brain, i.e., glia or neurons, and/or act as metabolic factories for the enzymes that can then be secreted and taken up by enzyme deficient cells in the CNS and other organs to varying degrees. Such approaches have had varying success in altering the CNS phenotypes because of the timing and nature of the underlying CNS pathology in the GSL LSDs. The degree of reversibility is determined by the time of treatment and/or the overall distribution of the therapeutic cells and/or enzyme to different regions of the brain (see below).
Treatments by decreasing GSL synthesis
The approach of inhibiting the synthesis of GSLs to treat the corresponding diseases was first suggested by Norman Radin some 30 years ago. The general concept is to inhibit GCS, because GlcCer is the precursor of the lipids in the majority of GSL LSDs, but more specific enzymes for the synthesis of higher GSLs and gangliosides could also be targeted. Because the complete deficiency of GCS is lethal in utero, partial inhibition of GlcCer synthesis is the goal with the intent to reestablish a balance between synthesis and degradation. For this approach to work, either residual, albeit low, enzyme activity or an alternative pathway for GSL disposal is needed. This approach has been termed Substrate Reduction Therapy. However, this is a misnomer, beccause the goal of all the therapeutic approaches is to reduce substrate accumulation; a better term would be substrate synthesis inhibition therapy or SSIT.
Oral SSIT with N-butyl-deoxynojirimycin (NB-DNJ) (miglustat or Zavesca; Actelion, Basal Switzerland) has been approved by the EMEA and FDA for treatment of Gaucher disease type 1 patients who cannot tolerate ERT (147, 304). This drug is in Phase 3 trials for Sandhoff disease (158) and for NPC1; miglustat has been approved by EMEA (305). The therapeutic effects of NB-DNJ are less than achieved with intravenous ERT using mannose-terminated GCase (304). Use of NB-DNJ in Gaucher disease type 3 patients with established CNS disease failed to show significant improvement in neurological signs, specifically abnormal eye movements (147). However, sufficient neurological effects were found in NPC1 disease for EMEA approval for that condition (306) (http://nnpdf.org; http://www.emea.europa.eu/pdfs/human/opinion/Zavesca_33596508en.pdf). More recently, a more potent GCS inhibitor (Genz-112638) based on the parent compound, 1-phenyl-2-decanoylamino-3-morpholino-1-propanol, has shown major clinical and biochemical effects in visceral organs from humans and mice with Gaucher disease type 1 variants (307). Importantly, the lungs were substantially cleared of GlcCer storage in the mice, an organ that has poor biochemical response to ERT (307). Genz-112638 is a Pgp substrate and, although it rapidly crossed the blood brain barrier, significant levels of the drug are not achieved due to rapid removal by Pgp (307). Phase II data of Genz-112638 from the human trial show similar safety and initial efficacy at 6–12 months to ERT with imiglucerase (308).
A clear advantage of drugs used for SSIT and EET is their ability to cross the blood brain barrier in potentially therapeutically significant amounts. Furthermore, the EET approach to re-engineer the mutant enzyme in situ for better function has additional advantages of oral administration and avoiding immunological reactions and the necessity of specific ligands for receptor mediated endocytosis. The first generation of these agents for the GSL storage diseases are primarily iminosugars that are either 5- or 1- deoxy or 3,4-dideoxy glycoside derivatives of glucose or galactose, and they are potent reversible competitive inhibitors of specific lysosomal hydrolases. The current agents, isofagomine [3,4-dideoxy-5- (hydroxymethyl) piperidine] and DGJ (1-deoxy galactonojirimycin), have shown significant ability to enhance the activity of a great variety of different mutant GCases or α-galactosidases A, respectively, in tissue culture and in peripheral blood leukocytes of affected patients with Gaucher or Fabry disease, respectively (309–311). Clinical trials in Fabry disease are ongoing with DGJ, but the development of isofagomine has been stopped due to the lack of clinical effect in all but one patient with Gaucher disease type 1 (http://files.shareholder.com/downloads/AMTX/763312895×0×322616/e986f511-4f40-4412-972d-068e6d7dd058/FOLD_News_2009_10_2_General.pdf). Clearly, alternative drugs with different targets on the mutant enzymes might have greater clinical effects.
For CNS LSDs, early therapeutic interventions have been shown to improve diseases outcome in several LSD mouse models. NPC1 mice treated with cyclodextrin at P7 versus P21 showed longer lifespans (312). NB-DGJ treatment started at P2, when GM2 accumulation was detected, significantly reduced total gangliosides in Sandhoff disease mice (171). Isofagomine treatment on V394/SapC−/− mice started in utero (E15) extended lifespan versus treatment started at P21 (Sun et al., unpublished observations). The dosage used in the early intervention at embryonic stage is critical, because they may be toxic in utero effects. Understanding the GSL profile and function during brain development (Figs. 4 and 5) will be important to design the therapy and to define the timing for effective treatment of CNS LSDs without disrupting normal brain development.
The disease stage and pathogenic mechanisms of the CNS GSL storage diseases are of major importance to the development of therapies. Many of the studies of ERT, cell transplantation, and gene therapies have shown promising results in animal models in which the disease processes may be reversible, e.g., GSL storage is present in selected CNS cells, but the neurons remain viable. In such cases, one can envision the reestablishment of GSL flux in such cells might lead to recovery of their function and a positive effect on phenotype. In comparison, if the disease process has neuronal loss as a major pathogenic mechanism, recovery or even stabilization might be impossible; e.g., in neuronopathic Gaucher disease mice, neither complete enzyme reconstitution within neurons nor provision of microglia with wild-type GCase activity led to restoration or rescue of the CNS disease (150). Neuronal death is predominant in these models. Much more detail of the pathogenic mechanisms will be required for development of disease-specific appropriate therapeutic approaches.
CONCLUSIONS AND PERSPECTIVES
Advances in understanding the pathophysiology and treatment of the GSL diseases hinge upon molecular insights into the basic and initial mechanisms of disease causation. Elucidation of the earliest molecular events caused by the subclinical accumulation of GSLs could provide initial targets that are fundamental to interfering with the propagation of the neuronal death pathways. Such insights are critically important as the primary neuronal disease, cell death versus GSL storage, would require highly different intervention strategies and timing of initiation. Recent proposals that retrograde axonal degeneration due to lipid raft aberrations provide additional pathways and targets for therapy and for the understanding of the role of GSLs in cell development and physiology. These rare diseases continue to provide platforms for the general understanding of GSL biochemistry and therapy for many diverse and more common diseases.
Acknowledgments
The authors thank Brandy Morris and Amanda Manning for their clerical expertise.
Footnotes
Abbreviations:
- AC
- acid ceramidase
- ASA
- arylsulfatase A
- aSMase
- acid sphingomyelinase
- BDNF
- brain-derived neurotrophic factor
- CBE
- conduritol B epoxide
- CERT
- ceramide transfer protein
- CGT
- ceramide UDP-galactosyltransferase
- CNS
- central nervous system
- EET
- enzyme enhancement therapy
- ER
- endoplasmic reticulum
- ERK
- extracellular signal-regulated kinase
- FAPP2
- four-phosphate adaptor protein 2
- GALC
- galactosylceramide-β-galactosidase
- Gb3
- globotrioaosylshyceramide
- GCase
- acidβ-glucosidase or glucocerebrosidase
- GCS
- glucosylceramide synthase
- GD3 synthase
- α-N-acetyl-neuraminide α-2,8-sialyltransferase
- GM3 synthase
- LacCer α-2,3-sialyltransferase
- GSL
- glycosphingolipids
- Hex A
- β-hexosaminidase A
- Hex B
- β-hexosaminidase B
- IL
- interleukin
- iNKT
- invariant natural killer T
- LacCer
- lactosylceramide
- LacSph
- lactosylsphingosine
- LSD
- lysosomal storage disease
- LTP
- long-term potentiation
- lyso-Gb3
- globotriaosylsphingosine
- MLD
- metachromatic leukodystrophy
- NB-DGJ
- N-butyldeoxygalactonojirimycin
- NB-DNJ
- N-butyl-deoxynojirimycin
- NGF
- nerve growth factor
- NPA/B
- Niemann-Pick disease Types A and B
- nSMase
- Neutral sphingomyelinase
- Pgp
- P-glycoprotein
- PNS
- peripheral nervous system
- Sap
- saposin
- SPT
- serine-palmitoyltransferase
- SSIT
- substrate synthesis inhibition therapy
- TNFα
- tumor necrosis factor-α
- UPR
- unfolded protein response
This work was supported by National Institutes of Health grants to G.A.G. (NS64352, HD59823, and DK36729). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health.
REFERENCES
- 1.Holthuis J. C., Pomorski T., Raggers R. J., Sprong H., Van Meer G. 2001. The organizing potential of sphingolipids in intracellular membrane transport. Physiol. Rev. 81: 1689–1723. [DOI] [PubMed] [Google Scholar]
- 2.Kolter T., Sandhoff K. 2005. Principles of lysosomal membrane digestion: stimulation of sphingolipid degradation by sphingolipid activator proteins and anionic lysosomal lipids. Annu. Rev. Cell Dev. Biol. 21: 81–103. [DOI] [PubMed] [Google Scholar]
- 3.Lahiri S., Futerman A. H. 2007. The metabolism and function of sphingolipids and glycosphingolipids. Cell. Mol. Life Sci. 64: 2270–2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mandon E. C., Ehses I., Rother J., van Echten G., Sandhoff K. 1992. Subcellular localization and membrane topology of serine palmitoyltransferase, 3-dehydrosphinganine reductase, and sphinganine N-acyltransferase in mouse liver. J. Biol. Chem. 267: 11144–11148. [PubMed] [Google Scholar]
- 5.Michel C., van Echten-Deckert G., Rother J., Sandhoff K., Wang E., Merrill A. H., Jr 1997. Characterization of ceramide synthesis. A dihydroceramide desaturase introduces the 4,5-trans-double bond of sphingosine at the level of dihydroceramide. J. Biol. Chem. 272: 22432–22437. [DOI] [PubMed] [Google Scholar]
- 6.Pruett S. T., Bushnev A., Hagedorn K., Adiga M., Haynes C. A., Sullards M. C., Liotta D. C., Merrill A. H., Jr 2008. Biodiversity of sphingoid bases ("sphingosines") and related amino alcohols. J. Lipid Res. 49: 1621–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosenthal M. D. 1987. Fatty acid metabolism of isolated mammalian cells. Prog. Lipid Res. 26: 87–124. [DOI] [PubMed] [Google Scholar]
- 8.Rother J., van Echten G., Schwarzmann G., Sandhoff K. 1992. Biosynthesis of sphingolipids: dihydroceramide and not sphinganine is desaturated by cultured cells. Biochem. Biophys. Res. Commun. 189: 14–20. [DOI] [PubMed] [Google Scholar]
- 9.Pewzner-Jung Y., Ben-Dor S., Futerman A. H. 2006. When do Lasses (longevity assurance genes) become CerS (ceramide synthases)?: insights into the regulation of ceramide synthesis. J. Biol. Chem. 281: 25001–25005. [DOI] [PubMed] [Google Scholar]
- 10.Jenkins R. W., Canals D., Hannun Y. A. 2009. Roles and regulation of secretory and lysosomal acid sphingomyelinase. Cell. Signal. 21: 836–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marchesini N., Hannun Y. A. 2004. Acid and neutral sphingomyelinases: roles and mechanisms of regulation. Biochem. Cell Biol. 82: 27–44. [DOI] [PubMed] [Google Scholar]
- 12.Kitatani K., Sheldon K., Rajagopalan V., Anelli V., Jenkins R. W., Sun Y., Grabowski G. A., Obeid L. M., Hannun Y. A. 2009. Involvement of acid beta-glucosidase 1 in the salvage pathway of ceramide formation. J. Biol. Chem. 284: 12972–12978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burkart T., Siegrist H. P., Herschkowitz N. N., Wiesmann U. N. 1977. 3′-phosphoadenylylsulfate:galactosylceramide 3′-sulfotransferase. An optimized assay in homogenates of developing brain. Biochim. Biophys. Acta. 483: 303–311. [DOI] [PubMed] [Google Scholar]
- 14.Yaghootfam A., Sorkalla T., Haberlein H., Gieselmann V., Kappler J., Eckhardt M. 2007. Cerebroside sulfotransferase forms homodimers in living cells. Biochemistry. 46: 9260–9269. [DOI] [PubMed] [Google Scholar]
- 15.Futerman A. H., Stieger B., Hubbard A. L., Pagano R. E. 1990. Sphingomyelin synthesis in rat liver occurs predominantly at the cis and medial cisternae of the Golgi apparatus. J. Biol. Chem. 265: 8650–8657. [PubMed] [Google Scholar]
- 16.Hanada K., Kumagai K., Yasuda S., Miura Y., Kawano M., Fukasawa M., Nishijima M. 2003. Molecular machinery for non-vesicular trafficking of ceramide. Nature. 426: 803–809. [DOI] [PubMed] [Google Scholar]
- 17.Lannert H., Bunning C., Jeckel D., Wieland F. T. 1994. Lactosylceramide is synthesized in the lumen of the Golgi apparatus. FEBS Lett. 342: 91–96. [DOI] [PubMed] [Google Scholar]
- 18.Lloyd K. O., Furukawa K. 1998. Biosynthesis and functions of gangliosides: recent advances. Glycoconj. J. 15: 627–636. [DOI] [PubMed] [Google Scholar]
- 19.Iwamori M., Shimomura J., Tsuyuhara S., Nagai Y. 1984. Gangliosides of various rat tissues: distribution of ganglio-N-tetraose-containing gangliosides and tissue-characteristic composition of gangliosides. J Biochem. 95: 761–770. [DOI] [PubMed] [Google Scholar]
- 20.Freischutz B., Saito M., Rahmann H., Yu R. K. 1994. Activities of five different sialyltransferases in fish and rat brains. J. Neurochem. 62: 1965–1973. [DOI] [PubMed] [Google Scholar]
- 21.Senn H. J., Manke C., Dieter P., Tran-Thi T. A., Fitzke E., Gerok W., Decker K. 1990. Ganglioside biosynthesis in rat liver: different distribution of ganglioside synthases in hepatocytes, Kupffer cells, and sinusoidal endothelial cells. Arch. Biochem. Biophys. 278: 161–167. [DOI] [PubMed] [Google Scholar]
- 22.Yu R. K., Nakatani Y., Yanagisawa M. 2009. The role of glycosphingolipid metabolism in the developing brain. J. Lipid Res. 50 (Suppl.): S440–S445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eckford P. D., Sharom F. J. 2005. The reconstituted P-glycoprotein multidrug transporter is a flippase for glucosylceramide and other simple glycosphingolipids. Biochem. J. 389: 517–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Rosa M. F., Sillence D., Ackerley C., Lingwood C. 2004. Role of multiple drug resistance protein 1 in neutral but not acidic glycosphingolipid biosynthesis. J. Biol. Chem. 279: 7867–7876. [DOI] [PubMed] [Google Scholar]
- 25.van Helvoort A., Smith A. J., Sprong H., Fritzsche I., Schinkel A. H., Borst P., van Meer G. 1996. MDR1 P-glycoprotein is a lipid translocase of broad specificity, while MDR3 P-glycoprotein specifically translocates phosphatidylcholine. Cell. 87: 507–517. [DOI] [PubMed] [Google Scholar]
- 26.Halter D., Neumann S., van Dijk S. M., Wolthoorn J., de Maziere A. M., Vieira O. V., Mattjus P., Klumperman J., van Meer G., Sprong H. 2007. Pre- and post-Golgi translocation of glucosylceramide in glycosphingolipid synthesis. J. Cell Biol. 179: 101–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.D'Angelo G., Polishchuk E., Di Tullio G., Santoro M., Di Campli A., Godi A., West G., Bielawski J., Chuang C. C., van der Spoel A. C., et al. 2007. Glycosphingolipid synthesis requires FAPP2 transfer of glucosylceramide. Nature. 449: 62–67. [DOI] [PubMed] [Google Scholar]
- 28.Brown R. E., Mattjus P. 2007. Glycolipid transfer proteins. Biochim. Biophys. Acta. 1771: 746–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rorman E. G., Grabowski G. A. 1989. Molecular cloning of a human co-beta-glucosidase cDNA: evidence that four sphingolipid hydrolase activator proteins are encoded by single genes in humans and rats. Genomics. 5: 486–492. [DOI] [PubMed] [Google Scholar]
- 30.Hiraiwa M., Martin B. M., Kishimoto Y., Conner G. E., Tsuji S., O'Brien J. S. 1997. Lysosomal proteolysis of prosaposin, the precursor of saposins (sphingolipid activator proteins): its mechanism and inhibition by ganglioside. Arch. Biochem. Biophys. 341: 17–24. [DOI] [PubMed] [Google Scholar]
- 31.Leonova T., Qi X., Bencosme A., Ponce E., Sun Y., Grabowski G. A. 1996. Proteolytic processing patterns of prosaposin in insect and mammalian cells. J. Biol. Chem. 271: 17312–17320. [DOI] [PubMed] [Google Scholar]
- 32.Hannun Y. A., Luberto C., Argraves K. M. 2001. Enzymes of sphingolipid metabolism: from modular to integrative signaling. Biochemistry. 40: 4893–4903. [DOI] [PubMed] [Google Scholar]
- 33.Degroote S., Wolthoorn J., van Meer G. 2004. The cell biology of glycosphingolipids. Semin. Cell Dev. Biol. 15: 375–387. [DOI] [PubMed] [Google Scholar]
- 34.Memon R. A., Holleran W. M., Uchida Y., Moser A. H., Ichikawa S., Hirabayashi Y., Grunfeld C., Feingold K. R. 1999. Regulation of glycosphingolipid metabolism in liver during the acute phase response. J. Biol. Chem. 274: 19707–19713. [DOI] [PubMed] [Google Scholar]
- 35.Bourteele S., Hausser A., Doppler H., Horn-Muller J., Ropke C., Schwarzmann G., Pfizenmaier K., Muller G. 1998. Tumor necrosis factor induces ceramide oscillations and negatively controls sphingolipid synthases by caspases in apoptotic Kym-1 cells. J. Biol. Chem. 273: 31245–31251. [DOI] [PubMed] [Google Scholar]
- 36.Huwiler A., Kolter T., Pfeilschifter J., Sandhoff K. 2000. Physiology and pathophysiology of sphingolipid metabolism and signaling. Biochim. Biophys. Acta. 1485: 63–99. [DOI] [PubMed] [Google Scholar]
- 37.Tettamanti G., Bassi R., Viani P., Riboni L. 2003. Salvage pathways in glycosphingolipid metabolism. Biochimie. 85: 423–437. [DOI] [PubMed] [Google Scholar]
- 38.Walkley S. U., Vanier M. T. 2009. Secondary lipid accumulation in lysosomal disease. Biochim. Biophys. Acta. 1793: 726–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eliyahu E., Park J. H., Shtraizent N., He X., Schuchman E. H. 2007. Acid ceramidase is a novel factor required for early embryo survival. FASEB J. 21: 1403–1409. [DOI] [PubMed] [Google Scholar]
- 40.Hakomori S. I. 2000. Cell adhesion/recognition and signal transduction through glycosphingolipid microdomain. Glycoconj. J. 17: 143–151. [DOI] [PubMed] [Google Scholar]
- 41.Fenderson B. A., Andrews P. W., Nudelman E., Clausen H., Hakomori S. 1987. Glycolipid core structure switching from globo- to lacto- and ganglio-series during retinoic acid-induced differentiation of TERA-2-derived human embryonal carcinoma cells. Dev. Biol. 122: 21–34. [DOI] [PubMed] [Google Scholar]
- 42.Kannagi R., Levery S. B., Ishigami F., Hakomori S., Shevinsky L. H., Knowles B. B., Solter D. 1983. New globoseries glycosphingolipids in human teratocarcinoma reactive with the monoclonal antibody directed to a developmentally regulated antigen, stage-specific embryonic antigen 3. J. Biol. Chem. 258: 8934–8942. [PubMed] [Google Scholar]
- 43.Ngamukote S., Yanagisawa M., Ariga T., Ando S., Yu R. K. 2007. Developmental changes of glycosphingolipids and expression of glycogenes in mouse brains. J. Neurochem. 103: 2327–2341. [DOI] [PubMed] [Google Scholar]
- 44.Seyfried T. N., Ariga T. 1992. Neutral glycolipid abnormalities in a t-complex mutant mouse embryo. Biochem. Genet. 30: 557–565. [DOI] [PubMed] [Google Scholar]
- 45.van der Pal R. H., Klein W., van Golde L. M., Lopes-Cardozo M. 1990. Galactosylceramide sulfotransferase, arylsulfatase A and cerebroside sulfatase activity in different regions of developing rat brain. Biochim. Biophys. Acta. 1043: 91–96. [DOI] [PubMed] [Google Scholar]
- 46.Ishii A., Ikeda T., Hitoshi S., Fujimoto I., Torii T., Sakuma K., Nakakita S., Hase S., Ikenaka K. 2007. Developmental changes in the expression of glycogenes and the content of N-glycans in the mouse cerebral cortex. Glycobiology. 17: 261–276. [DOI] [PubMed] [Google Scholar]
- 47.Yamamoto A., Haraguchi M., Yamashiro S., Fukumoto S., Furukawa K., Takamiya K., Atsuta M., Shiku H. 1996. Heterogeneity in the expression pattern of two ganglioside synthase genes during mouse brain development. J. Neurochem. 66: 26–34. [DOI] [PubMed] [Google Scholar]
- 48.Yu R. K., Macala L. J., Taki T., Weinfield H. M., Yu F. S. 1988. Developmental changes in ganglioside composition and synthesis in embryonic rat brain. J. Neurochem. 50: 1825–1829. [DOI] [PubMed] [Google Scholar]
- 49.Hojjati M. R., Li Z., Jiang X. C. 2005. Serine palmitoyl-CoA transferase (SPT) deficiency and sphingolipid levels in mice. Biochim. Biophys. Acta. 1737: 44–51. [DOI] [PubMed] [Google Scholar]
- 50.Yamashita T., Wada R., Sasaki T., Deng C., Bierfreund U., Sandhoff K., Proia R. L. 1999. A vital role for glycosphingolipid synthesis during development and differentiation. Proc. Natl. Acad. Sci. USA. 96: 9142–9147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bosio A., Binczek E., Stoffel W. 1996. Functional breakdown of the lipid bilayer of the myelin membrane in central and peripheral nervous system by disrupted galactocerebroside synthesis. Proc. Natl. Acad. Sci. USA. 93: 13280–13285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Coetzee T., Fujita N., Dupree J., Shi R., Blight A., Suzuki K., Popko B. 1996. Myelination in the absence of galactocerebroside and sulfatide: normal structure with abnormal function and regional instability. Cell. 86: 209–219. [DOI] [PubMed] [Google Scholar]
- 53.Yoshikawa M., Go S., Takasaki K., Kakazu Y., Ohashi M., Nagafuku M., Kabayama K., Sekimoto J., Suzuki S., Takaiwa K., et al. 2009. Mice lacking ganglioside GM3 synthase exhibit complete hearing loss due to selective degeneration of the organ of Corti. Proc. Natl. Acad. Sci. USA. 106: 9483–9488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yamashita T., Hashiramoto A., Haluzik M., Mizukami H., Beck S., Norton A., Kono M., Tsuji S., Daniotti J. L., Werth N., et al. 2003. Enhanced insulin sensitivity in mice lacking ganglioside GM3. Proc. Natl. Acad. Sci. USA. 100: 3445–3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Takamiya K., Yamamoto A., Furukawa K., Yamashiro S., Shin M., Okada M., Fukumoto S., Haraguchi M., Takeda N., Fujimura K., et al. 1996. Mice with disrupted GM2/GD2 synthase gene lack complex gangliosides but exhibit only subtle defects in their nervous system. Proc. Natl. Acad. Sci. USA. 93: 10662–10667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Handa Y., Ozaki N., Honda T., Furukawa K., Tomita Y., Inoue M., Furukawa K., Okada M., Sugiura Y. 2005. GD3 synthase gene knockout mice exhibit thermal hyperalgesia and mechanical allodynia but decreased response to formalin-induced prolonged noxious stimulation. Pain. 117: 271–279. [DOI] [PubMed] [Google Scholar]
- 57.Saadat L., Dupree J. L., Kilkus J., Han X., Traka M., Proia R. L., Dawson G., Popko B. 2010. Absence of oligodendroglial glucosylceramide synthesis does not result in CNS myelin abnormalities or alter the dysmyelinating phenotype of CGT-deficient mice. Glia. 58: 391–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jennemann R., Sandhoff R., Wang S., Kiss E., Gretz N., Zuliani C., Martin-Villalba A., Jager R., Schorle H., Kenzelmann M., et al. 2005. Cell-specific deletion of glucosylceramide synthase in brain leads to severe neural defects after birth. Proc. Natl. Acad. Sci. USA. 102: 12459–12464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Auer-Grumbach M. 2008. Hereditary sensory neuropathy type I. Orphanet J. Rare Dis. 3: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dawkins J. L., Hulme D. J., Brahmbhatt S. B., Auer-Grumbach M., Nicholson G. A. 2001. Mutations in SPTLC1, encoding serine palmitoyltransferase, long chain base subunit-1, cause hereditary sensory neuropathy type I. Nat. Genet. 27: 309–312. [DOI] [PubMed] [Google Scholar]
- 61.Fishman P. H., Max S. R., Tallman J. F., Brady R. O., Maclaren N. K., Cornblath M. 1975. Deficient Ganglioside Biosynthesis: a novel human sphingolipidosis. Science. 187: 68–70. [DOI] [PubMed] [Google Scholar]
- 62.Penno A., Reilly M. M., Houlden H., Laura M., Rentsch K., Niederkofler V., Stoeckli E. T., Nicholson G., Eichler F., Brown R. H., Jr., von Eckardstein A., Hornemann T. Hereditary sensory neuropathy type 1 is caused by the accumulation of two neurotoxic sphingolipids. J Biol Chem. Epub 2010 Jan 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rotthier A., Baets J., De Vriendt E., Jacobs A., Auer-Grumbach M., Levy N., Bonello-Palot N., Kilic S. S., Weis J., Nascimento A., et al. 2009. Genes for hereditary sensory and autonomic neuropathies: a genotype-phenotype correlation. Brain. 132: 2699–2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Simpson M. A., Cross H., Proukakis C., Priestman D. A., Neville D. C., Reinkensmeier G., Wang H., Wiznitzer M., Gurtz K., Verganelaki A., et al. 2004. Infantile-onset symptomatic epilepsy syndrome caused by a homozygous loss-of-function mutation of GM3 synthase. Nat. Genet. 36: 1225–1229. [DOI] [PubMed] [Google Scholar]
- 65.Matyas G. R., Morre D. J. 1987. Subcellular distribution and biosynthesis of rat liver gangliosides. Biochim. Biophys. Acta. 921: 599–614. [DOI] [PubMed] [Google Scholar]
- 66.Baek R. C., Martin D. R., Cox N. R., Seyfried T. N. 2009. Comparative analysis of brain lipids in mice, cats, and humans with Sandhoff disease. Lipids. 44: 197–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kawashima I., Nagata I., Tai T. 1996. Immunocytochemical analysis of gangliosides in rat primary cerebellar cultures using specific monoclonal antibodies. Brain Res. 732: 75–86. [DOI] [PubMed] [Google Scholar]
- 68.Jirkovska M., Majer F., Smidova J., Stritesky J., Shaik G. M., Draber P., Vitek L., Marecek Z., Smid F. 2007. Changes in GM1 ganglioside content and localization in cholestatic rat liver. Glycoconj. J. 24: 231–241. [DOI] [PubMed] [Google Scholar]
- 69.Muthing J., Maurer U., Neumann U., Kniep B., Weber-Schurholz S. 1998. Glycosphingolipids of skeletal muscle: I. Subcellular distribution of neutral glycosphingolipids and gangliosides in rabbit skeletal muscle. Carbohydr. Res. 307: 135–145. [DOI] [PubMed] [Google Scholar]
- 70.Muthing J., Maurer U., Sostaric K., Neumann U., Brandt H., Duvar S., Peter-Katalinic J., Weber-Schurholz S. 1994. Different distributions of glycosphingolipids in mouse and rabbit skeletal muscle demonstrated by biochemical and immunohistological analyses. J Biochem. 115: 248–256. [DOI] [PubMed] [Google Scholar]
- 71.Schaeren-Wiemers N., van der Bijl P., Schwab M. E. 1995. The UDP-galactose:ceramide galactosyltransferase: expression pattern in oligodendrocytes and Schwann cells during myelination and substrate preference for hydroxyceramide. J. Neurochem. 65: 2267–2278. [DOI] [PubMed] [Google Scholar]
- 72.Feingold K. R. 2007. Thematic review series: skin lipids. The role of epidermal lipids in cutaneous permeability barrier homeostasis. J. Lipid Res. 48: 2531–2546. [DOI] [PubMed] [Google Scholar]
- 73.van Genderen I. L., van Meer G., Slot J. W., Geuze H. J., Voorhout W. F. 1991. Subcellular localization of Forssman glycolipid in epithelial MDCK cells by immuno-electronmicroscopy after freeze-substitution. J. Cell Biol. 115: 1009–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dyatlovitskaya E. V., Novikov A. M., Gorkova N. P., Bergelson L. D. 1976. Gangliosides of hepatoma 27, normal and regenerating rat liver. Eur. J. Biochem. 63: 357–364. [DOI] [PubMed] [Google Scholar]
- 75.Malisan F., Testi R. 2002. GD3 ganglioside and apoptosis. Biochim. Biophys. Acta. 1585: 179–187. [DOI] [PubMed] [Google Scholar]
- 76.Simons K., van Meer G. 1988. Lipid sorting in epithelial cells. Biochemistry. 27: 6197–6202. [DOI] [PubMed] [Google Scholar]
- 77.Sprong H., Degroote S., Claessens T., van Drunen J., Oorschot V., Westerink B. H., Hirabayashi Y., Klumperman J., van der Sluijs P., van Meer G. 2001. Glycosphingolipids are required for sorting melanosomal proteins in the Golgi complex. J. Cell Biol. 155: 369–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Brasitus T. A., Schachter D. 1980. Lipid dynamics and lipid-protein interactions in rat enterocyte basolateral and microvillus membranes. Biochemistry. 19: 2763–2769. [DOI] [PubMed] [Google Scholar]
- 79.Forstner G. G., Tanaka K., Isselbacher K. J. 1968. Lipid composition of the isolated rat intestinal microvillus membrane. Biochem. J. 109: 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Warnock D. E., Roberts C., Lutz M. S., Blackburn W. A., Young W. W., Jr., Baenziger J. U. 1993. Determination of plasma membrane lipid mass and composition in cultured Chinese hamster ovary cells using high gradient magnetic affinity chromatography. J. Biol. Chem. 268: 10145–10153. [PubMed] [Google Scholar]
- 81.Buton X., Herve P., Kubelt J., Tannert A., Burger K. N., Fellmann P., Muller P., Herrmann A., Seigneuret M., Devaux P. F. 2002. Transbilayer movement of monohexosylsphingolipids in endoplasmic reticulum and Golgi membranes. Biochemistry. 41: 13106–13115. [DOI] [PubMed] [Google Scholar]
- 82.Simons K., Toomre D. 2000. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 1: 31–39. [DOI] [PubMed] [Google Scholar]
- 83.Rodgers W., Crise B., Rose J. K. 1994. Signals determining protein tyrosine kinase and glycosyl-phosphatidylinositol-anchored protein targeting to a glycolipid-enriched membrane fraction. Mol. Cell. Biol. 14: 5384–5391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hakomori S., Handa K. 2002. Glycosphingolipid-dependent cross-talk between glycosynapses interfacing tumor cells with their host cells: essential basis to define tumor malignancy. FEBS Lett. 531: 88–92. [DOI] [PubMed] [Google Scholar]
- 85.Hakomori S. 2003. Structure, organization, and function of glycosphingolipids in membrane. Curr. Opin. Hematol. 10: 16–24. [DOI] [PubMed] [Google Scholar]
- 86.Patel H. H., Murray F., Insel P. A. 2008. G-protein-coupled receptor-signaling components in membrane raft and caveolae microdomains. Handb. Exp. Pharmacol. 167–184. [DOI] [PubMed] [Google Scholar]
- 87.Hannun Y. A., Obeid L. M. 2002. The ceramide-centric universe of lipid-mediated cell regulation: stress encounters of the lipid kind. J. Biol. Chem. 277: 25847–25850. [DOI] [PubMed] [Google Scholar]
- 88.Brown M. D., Sacks D. B. 2009. Protein scaffolds in MAP kinase signalling. Cell. Signal. 21: 462–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Muller G., Ayoub M., Storz P., Rennecke J., Fabbro D., Pfizenmaier K. 1995. PKC zeta is a molecular switch in signal transduction of TNF-alpha, bifunctionally regulated by ceramide and arachidonic acid. EMBO J. 14: 1961–1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang Y., Yao B., Delikat S., Bayoumy S., Lin X. H., Basu S., McGinley M., Chan-Hui P. Y., Lichenstein H., Kolesnick R. 1997. Kinase suppressor of Ras is ceramide-activated protein kinase. Cell. 89: 63–72. [DOI] [PubMed] [Google Scholar]
- 91.Verheij M., Bose R., Lin X. H., Yao B., Jarvis W. D., Grant S., Birrer M. J., Szabo E., Zon L. I., Kyriakis J. M., et al. 1996. Requirement for ceramide-initiated SAPK/JNK signalling in stress-induced apoptosis. Nature. 380: 75–79. [DOI] [PubMed] [Google Scholar]
- 92.Huwiler A., Brunner J., Hummel R., Vervoordeldonk M., Stabel S., van den Bosch H., Pfeilschifter J. 1996. Ceramide-binding and activation defines protein kinase c-Raf as a ceramide-activated protein kinase. Proc. Natl. Acad. Sci. USA. 93: 6959–6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ruvolo P. P., Clark W., Mumby M., Gao F., May W. S. 2002. A functional role for the B56 alpha-subunit of protein phosphatase 2A in ceramide-mediated regulation of Bcl2 phosphorylation status and function. J. Biol. Chem. 277: 22847–22852. [DOI] [PubMed] [Google Scholar]
- 94.Leinekugel P., Michel S., Conzelmann E., Sandhoff K. 1992. Quantitative correlation between the residual activity of beta-hexosaminidase A and arylsulfatase A and the severity of the resulting lysosomal storage disease. Hum. Genet. 88: 513–523. [DOI] [PubMed] [Google Scholar]
- 95.Grabowski G. A., Kolodny E. H., Weinreb N. J., Rosenbloom B. E., Prakash-Cheng A., Kaplan P., Charrow J., Pastores G. M., Mistry P. K. 2006. Gaucher disease: phenotypic and genetic variation. Chapter 146.1. In The Metabolic and Molecular Bases of Inherited Diseases Scriver C. R., Sly W. S., Beaudet A., Valle D., Childs B., McGraw-Hill, New York. [Google Scholar]
- 96.Moser H., Linke T., Fensom A. H., Levade T., Sandhoff K. 2001. Acid ceramidase deficiency: Farber lipogranulomatosis. In The Metabolic and Molecular Basis of Inherited Disease Scriver C. R., Beaudet A. L., Sly W. S., Valle D., McGraw-Hill, New York: 3573–3588. [Google Scholar]
- 97.Sugita M., Dulaney J. T., Moser H. W. 1972. Ceramidase deficiency in Farber's disease (lipogranulomatosis). Science. 178: 1100–1102. [DOI] [PubMed] [Google Scholar]
- 98.Matsuda J., Kido M., Tadano-Aritomi K., Ishizuka I., Tominaga K., Toida K., Takeda E., Suzuki K., Kuroda Y. 2004. Mutation in saposin D domain of sphingolipid activator protein gene causes urinary system defects and cerebellar Purkinje cell degeneration with accumulation of hydroxy fatty acid-containing ceramide in mouse. Hum. Mol. Genet. 13: 2709–2723. [DOI] [PubMed] [Google Scholar]
- 99.Gatt S. 1963. Enzymic hydrolysis and synthesis of ceramides. J. Biol. Chem. 238: 3131–3133. [PubMed] [Google Scholar]
- 100.Okino N., He X., Gatt S., Sandhoff K., Ito M., Schuchman E. H. 2003. The reverse activity of human acid ceramidase. J. Biol. Chem. 278: 29948–29953. [DOI] [PubMed] [Google Scholar]
- 101.Ehlert K., Frosch M., Fehse N., Zander A., Roth J., Vormoor J. 2007. Farber disease: clinical presentation, pathogenesis and a new approach to treatment. Pediatr. Rheumatol. Online J. 5: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li C. M., Park J. H., Simonaro C. M., He X., Gordon R. E., Friedman A. H., Ehleiter D., Paris F., Manova K., Hepbildikler S., et al. 2002. Insertional mutagenesis of the mouse acid ceramidase gene leads to early embryonic lethality in homozygotes and progressive lipid storage disease in heterozygotes. Genomics. 79: 218–224. [DOI] [PubMed] [Google Scholar]
- 103.Cutler R. G., Mattson M. P. 2001. Sphingomyelin and ceramide as regulators of development and lifespan. Mech. Ageing Dev. 122: 895–908. [DOI] [PubMed] [Google Scholar]
- 104.Schuchman E. H., Desnick R. J. 2001. Niemann-Pick Disease Types A and B: acid sphingomyelinase deficencies. In The Metabolic and Molecular Basis of Inherited Disease Scriver C. R., Beaudet A.L., Sly W.S., Valle D., editor McGraw-Hill; 3589–3610. [Google Scholar]
- 105.Horinouchi K., Erlich S., Perl D. P., Ferlinz K., Bisgaier C. L., Sandhoff K., Desnick R. J., Stewart C. L., Schuchman E. H. 1995. Acid sphingomyelinase deficient mice: a model of types A and B Niemann-Pick disease. Nat. Genet. 10: 288–293. [DOI] [PubMed] [Google Scholar]
- 106.Otterbach B., Stoffel W. 1995. Acid sphingomyelinase-deficient mice mimic the neurovisceral form of human lysosomal storage disease (Niemann-Pick disease). Cell. 81: 1053–1061. [DOI] [PubMed] [Google Scholar]
- 107.Hostetler K. Y., Yazaki P. J. 1979. The subcellular localization of neutral sphingomyelinase in rat liver. J. Lipid Res. 20: 456–463. [PubMed] [Google Scholar]
- 108.Li C. M., Park J. H., He X., Levy B., Chen F., Arai K., Adler D. A., Disteche C. M., Koch J., Sandhoff K., et al. 1999. The human acid ceramidase gene (ASAH): structure, chromosomal location, mutation analysis, and expression. Genomics. 62: 223–231. [DOI] [PubMed] [Google Scholar]
- 109.Schuchman E. H. 2007. The pathogenesis and treatment of acid sphingomyelinase-deficient Niemann-Pick disease. J. Inherit. Metab. Dis. 30: 654–663. [DOI] [PubMed] [Google Scholar]
- 110.Kobayashi T., Shinnoh N., Goto I., Kuroiwa Y. 1985. Hydrolysis of galactosylceramide is catalyzed by two genetically distinct acid beta-galactosidases. J. Biol. Chem. 260: 14982–14987. [PubMed] [Google Scholar]
- 111.Wenger D., Suzuki K., Suzuki Y., Suzuki K. 2001. Galactosylceramide lipodosis: globoid cell leukodystrophy (Krabbe disease). In The Metabolic and Molecular Basis of Inherited Disease Scriver C. R., Beaudet A. L., Sly W. S., Valle D., McGraw-Hill, New York: 3669–3694. [Google Scholar]
- 112.Suzuki A., Furukawa K. 2003. [Glycobiology basics: glycolipids]. Tanpakushitsu Kakusan Koso. 48: 923–928. [PubMed] [Google Scholar]
- 113.Wenger D. A., Rafi M. A., Luzi P., Datto J., Costantino-Ceccarini E. 2000. Krabbe disease: genetic aspects and progress toward therapy. Mol. Genet. Metab. 70: 1–9. [DOI] [PubMed] [Google Scholar]
- 114.Duffner P. K., Caviness V. S., Jr., Erbe R. W., Patterson M. C., Schultz K. R., Wenger D. A., Whitley C. 2009. The long-term outcomes of presymptomatic infants transplanted for Krabbe disease: report of the workshop held on July 11 and 12, 2008, Holiday Valley, New York. Genet. Med. 11: 450–454. [DOI] [PubMed] [Google Scholar]
- 115.Sakai N., Inui K., Tatsumi N., Fukushima H., Nishigaki T., Taniike M., Nishimoto J., Tsukamoto H., Yanagihara I., Ozono K., et al. 1996. Molecular cloning and expression of cDNA for murine galactocerebrosidase and mutation analysis of the twitcher mouse, a model of Krabbe's disease. J. Neurochem. 66: 1118–1124. [DOI] [PubMed] [Google Scholar]
- 116.Kobayashi T., Yamanaka T., Jacobs J. M., Teixeira F., Suzuki K. 1980. The Twitcher mouse: an enzymatically authentic model of human globoid cell leukodystrophy (Krabbe disease). Brain Res. 202: 479–483. [DOI] [PubMed] [Google Scholar]
- 117.Suzuki K., Suzuki K. 1995. The twitcher mouse: a model for Krabbe disease and for experimental therapies. Brain Pathol. 5: 249–258. [DOI] [PubMed] [Google Scholar]
- 118.Suzuki K., Taniike M. 1995. Murine model of genetic demyelinating disease: the twitcher mouse. Microsc. Res. Tech. 32: 204–214. [DOI] [PubMed] [Google Scholar]
- 119.Taniike M., Suzuki K. 1994. Spacio-temporal progression of demyelination in twitcher mouse: with clinico-pathological correlation. Acta Neuropathol. 88: 228–236. [DOI] [PubMed] [Google Scholar]
- 120.Luzi P., Rafi M. A., Zaka M., Curtis M., Vanier M. T., Wenger D. A. 2001. Generation of a mouse with low galactocerebrosidase activity by gene targeting: a new model of globoid cell leukodystrophy (Krabbe disease). Mol. Genet. Metab. 73: 211–223. [DOI] [PubMed] [Google Scholar]
- 121.Hoogerbrugge P. M., Suzuki K., Poorthuis B. J., Kobayashi T., Wagemaker G., van Bekkum D. W. 1988. Donor-derived cells in the central nervous system of twitcher mice after bone marrow transplantation. Science. 239: 1035–1038. [DOI] [PubMed] [Google Scholar]
- 122.Figura K., Gieselmann V., Jaeken J. 2001. Metachromatic leukodystrophy. In The Metabolic and Molecular Basis of Inherited Disease Scriver C. R., Beaudet A. L., Sly W. S., Valle D., McGraw-Hill, New York: 3695–3724. [Google Scholar]
- 123.Wolfe H. J., Pietra G. G. 1964. The visceral lesions of metachromatic leukodystrophy. Am. J. Pathol. 44: 921–930. [PMC free article] [PubMed] [Google Scholar]
- 124.Sandhoff K., Kolter T., Harzer K. 2001. Sphingolipid activator proteins. In The Metabolic and Molecular Basis of Inherited Disease Scriver C. R., Beaudet A. L., Sly W. S., Valle D., McGraw-Hill, New York: 3371–3388. [Google Scholar]
- 125.Dierks T., Schlotawa L., Frese M. A., Radhakrishnan K., von Figura K., Schmidt B. 2009. Molecular basis of multiple sulfatase deficiency, mucolipidosis II/III and Niemann-Pick C1 disease: lysosomal storage disorders caused by defects of non-lysosomal proteins. Biochim. Biophys. Acta. 1793: 710–725. [DOI] [PubMed] [Google Scholar]
- 126.Cosma M. P., Pepe S., Annunziata I., Newbold R. F., Grompe M., Parenti G., Ballabio A. 2003. The multiple sulfatase deficiency gene encodes an essential and limiting factor for the activity of sulfatases. Cell. 113: 445–456. [DOI] [PubMed] [Google Scholar]
- 127.Schlotawa L., Steinfeld R., von Figura K., Dierks T., Gartner J. 2008. Molecular analysis of SUMF1 mutations: stability and residual activity of mutant formylglycine-generating enzyme determine disease severity in multiple sulfatase deficiency. Hum. Mutat. 29: 205. [DOI] [PubMed] [Google Scholar]
- 128.Biffi A., Lucchini G., Rovelli A., Sessa M. 2008. Metachromatic leukodystrophy: an overview of current and prospective treatments. Bone Marrow Transplant. 42 (Suppl. 2): S2–S6. [DOI] [PubMed] [Google Scholar]
- 129.Hess B., Saftig P., Hartmann D., Coenen R., Lullmann-Rauch R., Goebel H. H., Evers M., von Figura K., D'Hooge R., Nagels G., et al. 1996. Phenotype of arylsulfatase A-deficient mice: relationship to human metachromatic leukodystrophy. Proc. Natl. Acad. Sci. USA. 93: 14821–14826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ramakrishnan H., Hedayati K. K., Lullmann-Rauch R., Wessig C., Fewou S. N., Maier H., Goebel H. H., Gieselmann V., Eckhardt M. 2007. Increasing sulfatide synthesis in myelin-forming cells of arylsulfatase A-deficient mice causes demyelination and neurological symptoms reminiscent of human metachromatic leukodystrophy. J. Neurosci. 27: 9482–9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Matzner U., Gieselmann V. 2005. Gene therapy of metachromatic leukodystrophy. Expert Opin. Biol. Ther. 5: 55–65. [DOI] [PubMed] [Google Scholar]
- 132.Matzner U., Lullmann-Rauch R., Stroobants S., Andersson C., Weigelt C., Eistrup C., Fogh J., D'Hooge R., Gieselmann V. 2009. Enzyme replacement improves ataxic gait and central nervous system histopathology in a mouse model of metachromatic leukodystrophy. Mol. Ther. 17: 600–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Brady R. O., Gal A. E., Bradley R. M., Martensson E., Warshaw A. L., Laster L. 1967. Enzymatic defect in Fabry's disease. Ceramidetrihexosidase deficiency. N. Engl. J. Med. 276: 1163–1167. [DOI] [PubMed] [Google Scholar]
- 134.Desnick R. J., Brady R., Barranger J., Collins A. J., Germain D. P., Goldman M., Grabowski G., Packman S., Wilcox W. R. 2003. Fabry disease, an under-recognized multisystemic disorder: expert recommendations for diagnosis, management, and enzyme replacement therapy. Ann. Intern. Med. 138: 338–346. [DOI] [PubMed] [Google Scholar]
- 135.Nance C. S., Klein C. J., Banikazemi M., Dikman S. H., Phelps R. G., McArthur J. C., Rodriguez M., Desnick R. J. 2006. Later-onset Fabry disease: an adult variant presenting with the cramp-fasciculation syndrome. Arch. Neurol. 63: 453–457. [DOI] [PubMed] [Google Scholar]
- 136.Elleder M. 2003. Sequelae of storage in Fabry disease–pathology and comparison with other lysosomal storage diseases. Acta Paediatr. Suppl. 92: 46–53, discussion 45. [DOI] [PubMed] [Google Scholar]
- 137.Aerts J. M., Groener J. E., Kuiper S., Donker-Koopman W. E., Strijland A., Ottenhoff R., van Roomen C., Mirzaian M., Wijburg F. A., Linthorst G. E., et al. 2008. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc. Natl. Acad. Sci. USA. 105: 2812–2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Rodrigues L. G., Ferraz M. J., Rodrigues D., Pais-Vieira M., Lima D., Brady R. O., Sousa M. M., Sa-Miranda M. C. 2009. Neurophysiological, behavioral and morphological abnormalities in the Fabry knockout mice. Neurobiol. Dis. 33: 48–56. [DOI] [PubMed] [Google Scholar]
- 139.Park J. L., Whitesall S. E., D'Alecy L. G., Shu L., Shayman J. A. 2008. Vascular dysfunction in the alpha-galactosidase A-knockout mouse is an endothelial cell-, plasma membrane-based defect. Clin. Exp. Pharmacol. Physiol. 35: 1156–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Shu L., Park J. L., Byun J., Pennathur S., Kollmeyer J., Shayman J. A. 2009. Decreased nitric oxide bioavailability in a mouse model of fabry disease. J. Am. Soc. Nephrol. 20: 1975–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Beutler E., Grabowski G. A. 2001. Gaucher disease. In The Metabolic and Molecular Basis of Inherited Disease Scriver C. R., Beaudet A. L., Sly W. S., Valle D., McGraw-Hill, New York: 3635–3668. [Google Scholar]
- 142.Horowitz M., Wilder S., Horowitz Z., Reiner O., Gelbart T., Beutler E. 1989. The human glucocerebrosidase gene and pseudogene: structure and evolution. Genomics. 4: 87–96. [DOI] [PubMed] [Google Scholar]
- 143.Walden C. M., Sandhoff R., Chuang C. C., Yildiz Y., Butters T. D., Dwek R. A., Platt F. M., van der Spoel A. C. 2007. Accumulation of glucosylceramide in murine testis, caused by inhibition of beta-glucosidase 2: implications for spermatogenesis. J. Biol. Chem. 282: 32655–32664. [DOI] [PubMed] [Google Scholar]
- 144.Yildiz Y., Matern H., Thompson B., Allegood J. C., Warren R. L., Ramirez D. M., Hammer R. E., Hamra F. K., Matern S., Russell D. W. 2006. Mutation of beta-glucosidase 2 causes glycolipid storage disease and impaired male fertility. J. Clin. Invest. 116: 2985–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Burrow T. A., Hopkin R. J., Leslie N. D., Tinkle B. T., Grabowski G. A. 2007. Enzyme reconstitution/replacement therapy for lysosomal storage diseases. Curr. Opin. Pediatr. 19: 628–635. [DOI] [PubMed] [Google Scholar]
- 146.Elstein D., Dweck A., Attias D., Hadas-Halpern I., Zevin S., Altarescu G., Aerts J. F., van Weely S., Zimran A. 2007. Oral maintenance clinical trial with miglustat for type I Gaucher disease: switch from or combination with intravenous enzyme replacement. Blood. 110: 2296–2301. [DOI] [PubMed] [Google Scholar]
- 147.Schiffmann R., Fitzgibbon E. J., Harris C., DeVile C., Davies E. H., Abel L., van Schaik I. N., Benko W., Timmons M., Ries M., et al. 2008. Randomized, controlled trial of miglustat in Gaucher's disease type 3. Ann. Neurol. 64: 514–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Tybulewicz V. L. J., Tremblay M. L., LaMarca M. E., Willemsen R., Stubblefield B. K., Winfield S., Zablocka B., Sidransky E., Martin B. M., Huang S. P., et al. 1992. Animal model of Gaucher's disease from targeted disruption of the mouse glucocerebrosidase gene. Nature. 357: 407–410. [DOI] [PubMed] [Google Scholar]
- 149.Enquist I. B., Lo Bianco C., Ooka A., Nilsson E., Mansson J. E., Ehinger M., Richter J., Brady R. O., Kirik D., Karlsson S. 2007. Murine models of acute neuronopathic Gaucher disease. Proc. Natl. Acad. Sci. USA. 104: 17483–17488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Xu Y. H., Reboulet R., Quinn B., Huelsken J., Witte D., Grabowski G. A. 2008. Dependence of reversibility and progression of mouse neuronopathic Gaucher disease on acid beta-glucosidase residual activity levels. Mol. Genet. Metab. 94: 190–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Suzuki K. 1968. Cerebral GM1-gangliosidosis: chemical pathology of visceral organs. Science. 159: 1471–1472. [DOI] [PubMed] [Google Scholar]
- 152.Hinek A. 1996. Biological roles of the non-integrin elastin/laminin receptor. Biol. Chem. 377: 471–480. [PubMed] [Google Scholar]
- 153.Privitera S., Prody C. A., Callahan J. W., Hinek A. 1998. The 67-kDa enzymatically inactive alternatively spliced variant of beta-galactosidase is identical to the elastin/laminin-binding protein. J. Biol. Chem. 273: 6319–6326. [DOI] [PubMed] [Google Scholar]
- 154.Callahan J. W. 1999. Molecular basis of GM1 gangliosidosis and Morquio disease, type B. Structure-function studies of lysosomal beta-galactosidase and the non-lysosomal beta-galactosidase-like protein. Biochim. Biophys. Acta. 1455: 85–103. [DOI] [PubMed] [Google Scholar]
- 155.Suzuki K., Oshima A., Nanba E. 2001. β−Galactosidase Deficiency (β-Galactosidosis): GM1 gangliosidosis and Morquio B Disease. In The Metabolic and Molecular Basis of Inherited Disease Scriver C. R., Beaudet A. L., Sly W. S., Valle D., McGraw-Hill, New York: 3775–3809. [Google Scholar]
- 156.Hahn C. N., del Pilar Martin M., Schroder M., Vanier M. T., Hara Y., Suzuki K., d'Azzo A. 1997. Generalized CNS disease and massive GM1-ganglioside accumulation in mice defective in lysosomal acid beta-galactosidase. Hum. Mol. Genet. 6: 205–211. [DOI] [PubMed] [Google Scholar]
- 157.Matsuda J., Suzuki O., Oshima A., Ogura A., Naiki M., Suzuki Y. 1997. Neurological manifestations of knockout mice with beta-galactosidase deficiency. Brain Dev. 19: 19–20. [DOI] [PubMed] [Google Scholar]
- 158.Kasperzyk J. L., d'Azzo A., Platt F. M., Alroy J., Seyfried T. N. 2005. Substrate reduction reduces gangliosides in postnatal cerebrum-brainstem and cerebellum in GM1 gangliosidosis mice. J. Lipid Res. 46: 744–751. [DOI] [PubMed] [Google Scholar]
- 159.Broekman M. L., Baek R. C., Comer L. A., Fernandez J. L., Seyfried T. N., Sena-Esteves M. 2007. Complete correction of enzymatic deficiency and neurochemistry in the GM1-gangliosidosis mouse brain by neonatal adeno-associated virus-mediated gene delivery. Mol. Ther. 15: 30–37. [DOI] [PubMed] [Google Scholar]
- 160.Elliot-Smith E., Speak A. O., Lloyd-Evans E., Smith D. A., van der Spoel A. C., Jeyakumar M., Butters T. D., Dwek R. A., d'Azzo A., Platt F. M. 2008. Beneficial effects of substrate reduction therapy in a mouse model of GM1 gangliosidosis. Mol. Genet. Metab. 94: 204–211. [DOI] [PubMed] [Google Scholar]
- 161.Gravel R., Kaback M. M., Proia R. L., Sandhoff K., Suzuki K., Suzuki K. 2001. The GM2 gangliosidoses. In The Metabolic and Molecular Basis of Inherited Disease Scriver C. R., Beaudet A. L., Sly W. S., Valle D., McGraw-Hill, New York: 3827–3876. [Google Scholar]
- 162.Srivastava S. K., Beutler E. 1973. Hexosaminidase-A and hexosaminidase-B: studies in Tay-Sachs’ and Sandhoff's disease. Nature. 241: 463. [DOI] [PubMed] [Google Scholar]
- 163.Proia R. L., d'Azzo A., Neufeld E. F. 1984. Association of alpha- and beta-subunits during the biosynthesis of beta-hexosaminidase in cultured human fibroblasts. J. Biol. Chem. 259: 3350–3354. [PubMed] [Google Scholar]
- 164.Sonderfeld-Fresko S., Proia R. L. 1988. Synthesis and assembly of a catalytically active lysosomal enzyme, beta-hexosaminidase B, in a cell-free system. J. Biol. Chem. 263: 13463–13469. [PubMed] [Google Scholar]
- 165.Conzelmann E., Burg J., Stephan G., Sandhoff K. 1982. Complexing of glycolipids and their transfer between membranes by the activator protein for degradation of lysosomal ganglioside GM2. Eur. J. Biochem. 123: 455–464. [DOI] [PubMed] [Google Scholar]
- 166.Almassian B. 2009. 4. Phase I trial of pyrimethamine to treat LOTS. Mol. Genet. Metab. 96: S12–S13. [Google Scholar]
- 167.Beutler E., Kuhl W., Comings D. 1975. Hexosaminidase isozyme in type O Gm2 gangliosidosis (Sandhoff-Jatzkewitz disease). Am. J. Hum. Genet. 27: 628–638. [PMC free article] [PubMed] [Google Scholar]
- 168.Srivastava S. K., Beutler E. 1974. Studies on human beta-D-N-acetylhexosaminidases. 3. Biochemical genetics of Tay-Sachs and Sandhoff's diseases. J. Biol. Chem. 249: 2054–2057. [PubMed] [Google Scholar]
- 169.Sango K., Yamanaka S., Hoffmann A., Okuda Y., Grinberg A., Westphal H., McDonald M. P., Crawley J. N., Sandhoff K., Suzuki K., et al. 1995. Mouse models of Tay-Sachs and Sandhoff diseases differ in neurologic phenotype and ganglioside metabolism. Nat. Genet. 11: 170–176. [DOI] [PubMed] [Google Scholar]
- 170.Andersson U., Smith D., Jeyakumar M., Butters T. D., Borja M. C., Dwek R. A., Platt F. M. 2004. Improved outcome of N-butyldeoxygalactonojirimycin-mediated substrate reduction therapy in a mouse model of Sandhoff disease. Neurobiol. Dis. 16: 506–515. [DOI] [PubMed] [Google Scholar]
- 171.Baek R. C., Kasperzyk J. L., Platt F. M., Seyfried T. N. 2008. N-butyldeoxygalactonojirimycin reduces brain ganglioside and GM2 content in neonatal Sandhoff disease mice. Neurochem. Int. 52: 1125–1133. [DOI] [PubMed] [Google Scholar]
- 172.Sakuraba H., Itoh K., Shimmoto M., Utsumi K., Kase R., Hashimoto Y., Ozawa T., Ohwada Y., Imataka G., Eguchi M., et al. 1999. GM2 gangliosidosis AB variant: clinical and biochemical studies of a Japanese patient. Neurology. 52: 372–377. [DOI] [PubMed] [Google Scholar]
- 173.Schroder M., Schnabel D., Suzuki K., Sandhoff K. 1991. A mutation in the gene of a glycolipid-binding protein (GM2 activator) that causes GM2-gangliosidosis variant AB. FEBS Lett. 290: 1–3. [DOI] [PubMed] [Google Scholar]
- 174.Schulze H., Kolter T., Sandhoff K. 2009. Principles of lysosomal membrane degradation: cellular topology and biochemistry of lysosomal lipid degradation. Biochim. Biophys. Acta. 1793: 674–683. [DOI] [PubMed] [Google Scholar]
- 175.Liu Y., Hoffmann A., Grinberg A., Westphal H., McDonald M. P., Miller K. M., Crawley J. N., Sandhoff K., Suzuki K., Proia R. L. 1997. Mouse model of GM2 activator deficiency manifests cerebellar pathology and motor impairment. Proc. Natl. Acad. Sci. USA. 94: 8138–8143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Elleder M., Jerabkova M., Befekadu A., Hrebicek M., Berna L., Ledvinova J., Hulkova H., Rosewich H., Schymik N., Paton B. C., et al. 2005. Prosaposin deficiency–a rarely diagnosed, rapidly progressing, neonatal neurovisceral lipid storage disease. Report of a further patient. Neuropediatrics. 36: 171–180. [DOI] [PubMed] [Google Scholar]
- 177.Harzer K., Paton B. C., Poulos A., Kustermann-Kuhn B., Roggendorf W., Grisar T., Popp M. 1989. Sphingolipid activator protein deficiency in a 16-week-old atypical Gaucher disease patient and his fetal sibling: biochemical signs of combined sphingolipidoses. Eur. J. Pediatr. 149: 31–39. [DOI] [PubMed] [Google Scholar]
- 178.Hulkova H., Cervenkova M., Ledvinova J., Tochackova M., Hrebicek M., Poupetova H., Befekadu A., Berna L., Paton B. C., Harzer K., et al. 2001. A novel mutation in the coding region of the prosaposin gene leads to a complete deficiency of prosaposin and saposins, and is associated with a complex sphingolipidosis dominated by lactosylceramide accumulation. Hum. Mol. Genet. 10: 927–940. [DOI] [PubMed] [Google Scholar]
- 179.Ciaffoni F., Tatti M., Boe A., Salvioli R., Fluharty A., Sonnino S., Vaccaro A. M. 2006. Saposin B binds and transfers phospholipids. J. Lipid Res. 47: 1045–1053. [DOI] [PubMed] [Google Scholar]
- 180.Ahn V. E., Faull K. F., Whitelegge J. P., Fluharty A. L., Prive G. G. 2003. Crystal structure of saposin B reveals a dimeric shell for lipid binding. Proc. Natl. Acad. Sci. USA. 100: 38–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Sandhoff K., Kolter T., Harzer K. 2001. Sphingolipid activator proteins. In The Metabolic and Molecular Basis of Inherited Disease Scriver C. R., Beaudet A. L., Sly W. S., Valle D., McGraw-Hill, New York: 3371–3387. [Google Scholar]
- 182.Sun Y., Qi X., Grabowski G. A. 2003. Saposin C is required for normal resistance of acid beta-glucosidase to proteolytic degradation. J. Biol. Chem. 278: 31918–31923. [DOI] [PubMed] [Google Scholar]
- 183.Alattia J. R., Shaw J. E., Yip C. M., Prive G. G. 2007. Molecular imaging of membrane interfaces reveals mode of beta-glucosidase activation by saposin C. Proc. Natl. Acad. Sci. USA. 104: 17394–17399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Qi X., Grabowski G. A. 1998. Acid beta-glucosidase: intrinsic fluorescence and conformational changes induced by phospholipids and saposin C. Biochemistry. 37: 11544–11554. [DOI] [PubMed] [Google Scholar]
- 185.Soeda S., Hiraiwa M., O'Brien J. S., Kishimoto Y. 1993. Binding of cerebrosides and sulfatides to saposins A-D. J. Biol. Chem. 268: 18519–18523. [PubMed] [Google Scholar]
- 186.Vaccaro A. M., Tatti M., Ciaffoni F., Salvioli R., Barca A., Scerch C. 1997. Effect of saposins A and C on the enzymatic hydrolysis of liposomal glucosylceramide. J. Biol. Chem. 272: 16862–16867. [DOI] [PubMed] [Google Scholar]
- 187.Spiegel R., Bach G., Sury V., Mengistu G., Meidan B., Shalev S., Shneor Y., Mandel H., Zeigler M. 2005. A mutation in the saposin A coding region of the prosaposin gene in an infant presenting as Krabbe disease: first report of saposin A deficiency in humans. Mol. Genet. Metab. 84: 160–166. [DOI] [PubMed] [Google Scholar]
- 188.Tylki-Szymanska A., Czartoryska B., Vanier M. T., Poorthuis B. J., Groener J. A., Lugowska A., Millat G., Vaccaro A. M., Jurkiewicz E. 2007. Non-neuronopathic Gaucher disease due to saposin C deficiency. Clin. Genet. 72: 538–542. [DOI] [PubMed] [Google Scholar]
- 189.Diaz-Font A., Cormand B., Santamaria R., Vilageliu L., Grinberg D., Chabas A. 2005. A mutation within the saposin D domain in a Gaucher disease patient with normal glucocerebrosidase activity. Hum. Genet. 117: 275–277. [DOI] [PubMed] [Google Scholar]
- 190.Fujita N., Suzuki K., Vanier M. T., Popko B., Maeda N., Klein A., Henseler M., Sandhoff K., Nakayasu H. 1996. Targeted disruption of the mouse sphingolipid activator protein gene: a complex phenotype, including severe leukodystrophy and wide-spread storage of multiple sphingolipids. Hum. Mol. Genet. 5: 711–725. [DOI] [PubMed] [Google Scholar]
- 191.Matsuda J., Vanier M. T., Saito Y., Tohyama J., Suzuki K., Suzuki K. 2001. A mutation in the saposin A domain of the sphingolipid activator protein (prosaposin) gene results in a late-onset, chronic form of globoid cell leukodystrophy in the mouse. Hum. Mol. Genet. 10: 1191–1199. [DOI] [PubMed] [Google Scholar]
- 192.Sun Y., Qi X., Witte D. P., Ponce E., Kondoh K., Quinn B., Grabowski G. A. 2002. Prosaposin: threshold rescue and analysis of the "neuritogenic" region in transgenic mice. Mol. Genet. Metab. 76: 271–286. [DOI] [PubMed] [Google Scholar]
- 193.Sun Y., Ran H., Zamzow M., Kitatani K., Skelton M. R., Williams M. T., Vorhees C. V., Witte D. P., Hannun Y. A., Grabowski G. A. 2010. Specific saposin C deficiency: CNS impairment and acid {beta}-glucosidase effects in the mouse. Hum Mol Genet. 19: 634–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Sun Y., Witte D. P., Ran H., Zamzow M., Barnes S., Cheng H., Han X., Williams M. T., Skelton M. R., Vorhees C. V., et al. 2008. Neurological deficits and glycosphingolipid accumulation in saposin B deficient mice. Hum. Mol. Genet. 17: 2345–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Lloyd-Evans E., Morgan A. J., He X., Smith D. A., Elliot-Smith E., Sillence D. J., Churchill G. C., Schuchman E. H., Galione A., Platt F. M. 2008. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 14: 1247–1255. [DOI] [PubMed] [Google Scholar]
- 196.Kwon H. J., Abi-Mosleh L., Wang M. L., Deisenhofer J., Goldstein J. L., Brown M. S., Infante R. E. 2009. Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell. 137: 1213–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Lloyd-Evans E., Platt F. M. Lipids on trial: the search for the offending metabolite in Niemann-Pick type C Disease. Traffic. Epub 2010 Jan 6. [DOI] [PubMed] [Google Scholar]
- 198.Loftus S. K., Morris J. A., Carstea E. D., Gu J. Z., Cummings C., Brown A., Ellison J., Ohno K., Rosenfeld M. A., Tagle D. A., et al. 1997. Murine model of Niemann-Pick C disease: mutation in a cholesterol homeostasis gene. Science. 277: 232–235. [DOI] [PubMed] [Google Scholar]
- 199.Infante R. E., Wang M. L., Radhakrishnan A., Kwon H. J., Brown M. S., Goldstein J. L. 2008. NPC2 facilitates bidirectional transfer of cholesterol between NPC1 and lipid bilayers, a step in cholesterol egress from lysosomes. Proc. Natl. Acad. Sci. USA. 105: 15287–15292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Sleat D. E., Wiseman J. A., El-Banna M., Price S. M., Verot L., Shen M. M., Tint G. S., Vanier M. T., Walkley S. U., Lobel P. 2004. Genetic evidence for nonredundant functional cooperativity between NPC1 and NPC2 in lipid transport. Proc. Natl. Acad. Sci. USA. 101: 5886–5891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Hess B., Kafert S., Heinisch U., Wenger D. A., Zlotogora J., Gieselmann V. 1996. Characterization of two arylsulfatase A missense mutations D335V and T274M causing late infantile metachromatic leukodystrophy. Hum. Mutat. 7: 311–317. [DOI] [PubMed] [Google Scholar]
- 202.Pintos-Morell G., Beck M. 2009. Fabry disease in children and the effects of enzyme replacement treatment. Eur. J. Pediatr. 168: 1355–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Nilsson O., Grabowski G. A., Ludman M. D., Desnick R. J., Svennerholm L. 1985. Glycosphingolipid studies of visceral tissues and brain from type 1 Gaucher disease variants. Clin. Genet. 27: 443–450. [DOI] [PubMed] [Google Scholar]
- 204.Tohyama J., Vanier M. T., Suzuki K., Ezoe T., Matsuda J. 2000. Paradoxical influence of acid beta-galactosidase gene dosage on phenotype of the twitcher mouse (genetic galactosylceramidase deficiency). Hum. Mol. Genet. 9: 1699–1707. [DOI] [PubMed] [Google Scholar]
- 205.Sun Y., Liou B., Ran H., Skelton M. R., Williams M. T., Vorhees C. V., Kitatani K., Hannun Y. A., Witte D. P., Xu Y. H., et al. 2010. Neuronopathic Gaucher disease in the mouse: Viable combined selective saposin C deficiency and mutant glucocerebrosidase (V394L) mice with glucosylsphingosine and glucosylceramide accumulation and progressive neurological deficits. Hum Mol Genet. 19: 1088–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Vanier M. T. 1999. Lipid changes in Niemann-Pick disease type C brain: personal experience and review of the literature. Neurochem. Res. 24: 481–489. [DOI] [PubMed] [Google Scholar]
- 207.Frey L. C., Ringel S. P., Filley C. M. 2005. The natural history of cognitive dysfunction in late-onset GM2 gangliosidosis. Arch. Neurol. 62: 989–994. [DOI] [PubMed] [Google Scholar]
- 208.Orvisky E., Sidransky E., McKinney C. E., Lamarca M. E., Samimi R., Krasnewich D., Martin B. M., Ginns E. I. 2000. Glucosylsphingosine accumulation in mice and patients with type 2 Gaucher disease begins early in gestation. Pediatr. Res. 48: 233–237. [DOI] [PubMed] [Google Scholar]
- 209.Tanaka K., Nagara H., Kobayashi T., Goto I. 1988. The twitcher mouse: accumulation of galactosylsphingosine and pathology of the sciatic nerve. Brain Res. 454: 340–346. [DOI] [PubMed] [Google Scholar]
- 210.Tanaka K., Nagara H., Kobayashi T., Goto I. 1989. The twitcher mouse: accumulation of galactosylsphingosine and pathology of the central nervous system. Brain Res. 482: 347–350. [DOI] [PubMed] [Google Scholar]
- 211.Wada R., Tifft C. J., Proia R. L. 2000. Microglial activation precedes acute neurodegeneration in Sandhoff disease and is suppressed by bone marrow transplantation. Proc. Natl. Acad. Sci. USA. 97: 10954–10959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Sun Y., Jia L., Williams M. T., Zamzow M., Ran H., Quinn B., Aronow B. J., Vorhees C. V., Witte D. P., Grabowski G. A. 2008. Temporal gene expression profiling reveals CEBPD as a candidate regulator of brain disease in prosaposin deficient mice. BMC Neurosci. 9: 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Kim E. Y., Hong Y. B., Go S. H., Lee B., Jung S. C. 2006. Downregulation of neurotrophic factors in the brain of a mouse model of Gaucher disease; implications for neuronal loss in Gaucher disease. Exp. Mol. Med. 38: 348–356. [DOI] [PubMed] [Google Scholar]
- 214.Crowley C., Spencer S. D., Nishimura M. C., Chen K. S., Pitts-Meek S., Armanini M. P., Ling L. H., McMahon S. B., Shelton D. L., Levinson A. D., et al. 1994. Mice lacking nerve growth factor display perinatal loss of sensory and sympathetic neurons yet develop basal forebrain cholinergic neurons. Cell. 76: 1001–1011. [DOI] [PubMed] [Google Scholar]
- 215.Ernfors P., Lee K. F., Jaenisch R. 1994. Mice lacking brain-derived neurotrophic factor develop with sensory deficits. Nature. 368: 147–150. [DOI] [PubMed] [Google Scholar]
- 216.Atsumi S., Nosaka C., Iinuma H., Umezawa K. 1993. Accumulation of tissue glucosylsphingosine in Gaucher-like mouse induced by the glucosylceramidase inhibitor cyclophellitol. Arch. Biochem. Biophys. 304: 302–304. [DOI] [PubMed] [Google Scholar]
- 217.Leonova T., Grabowski G. A. 2000. Fate and sorting of acid beta-glucosidase in transgenic mammalian cells. Mol. Genet. Metab. 70: 281–294. [DOI] [PubMed] [Google Scholar]
- 218.Taniguchi M., Shinoda Y., Ninomiya H., Vanier M. T., Ohno K. 2001. Sites and temporal changes of gangliosides GM1/GM2 storage in the Niemann-Pick disease type C mouse brain. Brain Dev. 23: 414–421. [DOI] [PubMed] [Google Scholar]
- 219.Brunngraber E. G., Berra B., Zambotti V. 1973. Altered levels of tissue glycoproteins, gangliosides, glycosaminoglycans and lipids in Niemann-Pick's disease. Clin. Chim. Acta. 48: 173–181. [DOI] [PubMed] [Google Scholar]
- 220.Walkley S. U. 2004. Secondary accumulation of gangliosides in lysosomal storage disorders. Semin. Cell Dev. Biol. 15: 433–444. [DOI] [PubMed] [Google Scholar]
- 221.Jabs S., Quitsch A., Kakela R., Koch B., Tyynela J., Brade H., Glatzel M., Walkley S., Saftig P., Vanier M. T., et al. 2008. Accumulation of bis(monoacylglycero)phosphate and gangliosides in mouse models of neuronal ceroid lipofuscinosis. J. Neurochem. 106: 1415–1425. [DOI] [PubMed] [Google Scholar]
- 222.Rouser G., Galli C., Kritchevsky G. 1965. Lipid class composition of normal human brain and variations in metachromatic leucodystrophy, Tay-Sachs, Niemann-Pick, chronic Gaucher's and Alzheimer's diseases. J. Am. Oil Chem. Soc. 42: 404–410. [DOI] [PubMed] [Google Scholar]
- 223.Walkley S. U. 1998. Cellular pathology of lysosomal storage disorders. Brain Pathol. 8: 175–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Sandhoff K., Kolter T. 1996. Topology of glycosphingolipid degradation. Trends Cell Biol. 6: 98–103. [DOI] [PubMed] [Google Scholar]
- 225.Goker-Alpan O., Lopez G., Vithayathil J., Davis J., Hallett M., Sidransky E. 2008. The spectrum of parkinsonian manifestations associated with glucocerebrosidase mutations. Arch. Neurol. 65: 1353–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Latourelle J. C., Pankratz N., Dumitriu A., Wilk J. B., Goldwurm S., Pezzoli G., Mariani C. B., DeStefano A. L., Halter C., Gusella J. F., et al. 2009. Genomewide association study for onset age in Parkinson disease. BMC Med. Genet. 10: 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Wong K., Sidransky E., Verma A., Mixon T., Sandberg G. D., Wakefield L. K., Morrison A., Lwin A., Colegial C., Allman J. M., et al. 2004. Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol. Genet. Metab. 82: 192–207. [DOI] [PubMed] [Google Scholar]
- 228.Jmoudiak M., Futerman A. H. 2005. Gaucher disease: pathological mechanisms and modern management. Br. J. Haematol. 129: 178–188. [DOI] [PubMed] [Google Scholar]
- 229.Boot R. G., Verhoek M., de Fost M., Hollak C. E., Maas M., Bleijlevens B., van Breemen M. J., van Meurs M., Boven L. A., Laman J. D., et al. 2004. Marked elevation of the chemokine CCL18/PARC in Gaucher disease: a novel surrogate marker for assessing therapeutic intervention. Blood. 103: 33–39. [DOI] [PubMed] [Google Scholar]
- 230.Barak V., Acker M., Nisman B., Kalickman I., Abrahamov A., Zimran A., Yatziv S. 1999. Cytokines in Gaucher's disease. Eur. Cytokine Netw. 10: 205–210. [PubMed] [Google Scholar]
- 231.Boven L. A., van Meurs M., Boot R. G., Mehta A., Boon L., Aerts J. M., Laman J. D. 2004. Gaucher cells demonstrate a distinct macrophage phenotype and resemble alternatively activated macrophages. Am. J. Clin. Pathol. 122: 359–369. [DOI] [PubMed] [Google Scholar]
- 232.Hollak C. E., Evers L., Aerts J. M., van Oers M. H. 1997. Elevated levels of M-CSF, sCD14 and IL8 in type 1 Gaucher disease. Blood Cells Mol. Dis. 23: 201–212. [DOI] [PubMed] [Google Scholar]
- 233.Frings W., Dreier J., Sorg C. 2002. Only the soluble form of the scavenger receptor CD163 acts inhibitory on phorbol ester-activated T-lymphocytes, whereas membrane-bound protein has no effect. FEBS Lett. 526: 93–96. [DOI] [PubMed] [Google Scholar]
- 234.Gordon S. 2003. Alternative activation of macrophages. Nat. Rev. Immunol. 3: 23–35. [DOI] [PubMed] [Google Scholar]
- 235.Gratchev A., Guillot P., Hakiy N., Politz O., Orfanos C. E., Schledzewski K., Goerdt S. 2001. Alternatively activated macrophages differentially express fibronectin and its splice variants and the extracellular matrix protein betaIG-H3. Scand. J. Immunol. 53: 386–392. [DOI] [PubMed] [Google Scholar]
- 236.Dhami R., Passini M. A., Schuchman E. H. 2006. Identification of novel biomarkers for Niemann-Pick disease using gene expression analysis of acid sphingomyelinase knockout mice. Mol. Ther. 13: 556–564. [DOI] [PubMed] [Google Scholar]
- 237.Pyo H., Joe E., Jung S., Lee S. H., Jou I. 1999. Gangliosides activate cultured rat brain microglia. J. Biol. Chem. 274: 34584–34589. [DOI] [PubMed] [Google Scholar]
- 238.Shen W., Stone K., Jales A., Leitenberg D., Ladisch S. 2008. Inhibition of TLR activation and up-regulation of IL-1R-associated kinase-M expression by exogenous gangliosides. J. Immunol. 180: 4425–4432. [DOI] [PubMed] [Google Scholar]
- 239.Jeyakumar M., Thomas R., Elliot-Smith E., Smith D. A., van der Spoel A. C., d'Azzo A., Perry V. H., Butters T. D., Dwek R. A., Platt F. M. 2003. Central nervous system inflammation is a hallmark of pathogenesis in mouse models of GM1 and GM2 gangliosidosis. Brain. 126: 974–987. [DOI] [PubMed] [Google Scholar]
- 240.Jeyakumar M., Smith D. A., Williams I. M., Borja M. C., Neville D. C., Butters T. D., Dwek R. A., Platt F. M. 2004. NSAIDs increase survival in the Sandhoff disease mouse: synergy with N-butyldeoxynojirimycin. Ann. Neurol. 56: 642–649. [DOI] [PubMed] [Google Scholar]
- 241.Wu Y. P., Proia R. L. 2004. Deletion of macrophage-inflammatory protein 1 alpha retards neurodegeneration in Sandhoff disease mice. Proc. Natl. Acad. Sci. USA. 101: 8425–8430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Smith D., Wallom K. L., Williams I. M., Jeyakumar M., Platt F. M. 2009. Beneficial effects of anti-inflammatory therapy in a mouse model of Niemann-Pick disease type C1. Neurobiol Dis. 36: 242–251. [DOI] [PubMed] [Google Scholar]
- 243.Matsuda J., Vanier M. T., Saito Y., Suzuki K. 2001. Dramatic phenotypic improvement during pregnancy in a genetic leukodystrophy: estrogen appears to be a critical factor. Hum. Mol. Genet. 10: 2709–2715. [DOI] [PubMed] [Google Scholar]
- 244.Korhonen R., Kankaanranta H., Lahti A., Lahde M., Knowles R. G., Moilanen E. 2001. Bi-directional effects of the elevation of intracellular calcium on the expression of inducible nitric oxide synthase in J774 macrophages exposed to low and to high concentrations of endotoxin. Biochem. J. 354: 351–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Giri S., Khan M., Nath N., Singh I., Singh A. K. 2008. The role of AMPK in psychosine mediated effects on oligodendrocytes and astrocytes: implication for Krabbe disease. J. Neurochem. 105: 1820–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Jeon S. B., Yoon H. J., Park S. H., Kim I. H., Park E. J. 2008. Sulfatide, a major lipid component of myelin sheath, activates inflammatory responses as an endogenous stimulator in brain-resident immune cells. J. Immunol. 181: 8077–8087. [DOI] [PubMed] [Google Scholar]
- 247.Kitatani K., Sheldon K., Anelli V., Jenkins R. W., Sun Y., Grabowski G. A., Obeid L. M., Hannun Y. A. 2009. Acid beta-glucosidase 1 counteracts p38delta-dependent induction of interleukin-6: possible role for ceramide as an anti-inflammatory lipid. J. Biol. Chem. 284: 12979–12988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Gadola S. D., Silk J. D., Jeans A., Illarionov P. A., Salio M., Besra G. S., Dwek R., Butters T. D., Platt F. M., Cerundolo V. 2006. Impaired selection of invariant natural killer T cells in diverse mouse models of glycosphingolipid lysosomal storage diseases. J. Exp. Med. 203: 2293–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Zhou D., Mattner J., Cantu C., 3rd, Schrantz N., Yin N., Gao Y., Sagiv Y., Hudspeth K., Wu Y. P., Yamashita T., et al. 2004. Lysosomal glycosphingolipid recognition by NKT cells. Science. 306: 1786–1789. [DOI] [PubMed] [Google Scholar]
- 250.Porubsky S., Speak A. O., Luckow B., Cerundolo V., Platt F. M., Grone H. J. 2007. Normal development and function of invariant natural killer T cells in mice with isoglobotrihexosylceramide (iGb3) deficiency. Proc. Natl. Acad. Sci. USA. 104: 5977–5982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Cox D., Fox L., Tian R., Bardet W., Skaley M., Mojsilovic D., Gumperz J., Hildebrand W. 2009. Determination of cellular lipids bound to human CD1d molecules. PLoS One. 4: e5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Conradi N. G., Sourander P., Nilsson O., Svennerholm L., Erikson A. 1984. Neuropathology of the Norrbottnian type of Gaucher disease. Morphological and biochemical studies. Acta Neuropathol. 65: 99–109. [DOI] [PubMed] [Google Scholar]
- 253.Kaye E. M., Ullman M. D., Wilson E. R., Barranger J. A. 1986. Type 2 and type 3 Gaucher disease: a morphological and biochemical study. Ann. Neurol. 20: 223–230. [DOI] [PubMed] [Google Scholar]
- 254.Tessitore A., Martin M. del P., Sano R., Ma Y., Mann L., Ingrassia A., Laywell E. D., Steindler D. A., Hendershot L. M., d'Azzo A. 2004. GM1-ganglioside-mediated activation of the unfolded protein response causes neuronal death in a neurodegenerative gangliosidosis. Mol. Cell. 15: 753–766. [DOI] [PubMed] [Google Scholar]
- 255.Farfel-Becker T., Vitner E., Dekel H., Leshem N., Enquist I. B., Karlsson S., Futerman A. H. 2009. No evidence for activation of the unfolded protein response in neuronopathic models of Gaucher disease. Hum. Mol. Genet. 18: 1482–1488. [DOI] [PubMed] [Google Scholar]
- 256.Huang J. Q., Trasler J. M., Igdoura S., Michaud J., Hanal N., Gravel R. A. 1997. Apoptotic cell death in mouse models of GM2 gangliosidosis and observations on human Tay-Sachs and Sandhoff diseases. Hum. Mol. Genet. 6: 1879–1885. [DOI] [PubMed] [Google Scholar]
- 257.Nixon R. A., Yang D. S., Lee J. H. 2008. Neurodegenerative lysosomal disorders: a continuum from development to late age. Autophagy. 4: 590–599. [DOI] [PubMed] [Google Scholar]
- 258.Pelled D., Lloyd-Evans E., Riebeling C., Jeyakumar M., Platt F. M., Futerman A. H. 2003. Inhibition of calcium uptake via the sarco/endoplasmic reticulum Ca2+-ATPase in a mouse model of Sandhoff disease and prevention by treatment with N-butyldeoxynojirimycin. J. Biol. Chem. 278: 29496–29501. [DOI] [PubMed] [Google Scholar]
- 259.d'Azzo A., Tessitore A., Sano R. 2006. Gangliosides as apoptotic signals in ER stress response. Cell Death Differ. 13: 404–414. [DOI] [PubMed] [Google Scholar]
- 260.Higashi Y., Murayama S., Pentchev P. G., Suzuki K. 1993. Cerebellar degeneration in the Niemann-Pick type C mouse. Acta Neuropathol. 85: 175–184. [DOI] [PubMed] [Google Scholar]
- 261.Macauley S. L., Sidman R. L., Schuchman E. H., Taksir T., Stewart G. R. 2008. Neuropathology of the acid sphingomyelinase knockout mouse model of Niemann-Pick A disease including structure-function studies associated with cerebellar Purkinje cell degeneration. Exp. Neurol. 214: 181–192. [DOI] [PubMed] [Google Scholar]
- 262.Pampols T., Pineda M., Giros M. L., Ferrer I., Cusi V., Chabas A., Sanmarti F. X., Vanier M. T., Christomanou H. 1999. Neuronopathic juvenile glucosylceramidosis due to sap-C deficiency: clinical course, neuropathology and brain lipid composition in this Gaucher disease variant. Acta Neuropathol (Berl). 97: 91–97. [DOI] [PubMed] [Google Scholar]
- 263.Mannan A. U., Roussa E., Kraus C., Rickmann M., Maenner J., Nayernia K., Krieglstein K., Reis A., Engel W. 2004. Mutation in the gene encoding lysosomal acid phosphatase (Acp2) causes cerebellum and skin malformation in mouse. Neurogenetics. 5: 229–238. [DOI] [PubMed] [Google Scholar]
- 264.Rudelius M., Osanger A., Kohlmann S., Augustin M., Piontek G., Heinzmann U., Jennen G., Russ A., Matiasek K., Stumm G., et al. 2006. A missense mutation in the WD40 domain of murine Lyst is linked to severe progressive Purkinje cell degeneration. Acta Neuropathol. 112: 267–276. [DOI] [PubMed] [Google Scholar]
- 265.Weimer J. M., Benedict J. W., Getty A. L., Pontikis C. C., Lim M. J., Cooper J. D., Pearce D. A. 2009. Cerebellar defects in a mouse model of juvenile neuronal ceroid lipofuscinosis. Brain Res. 1266: 93–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Sun Y., Witte D. P., Zamzow M., Ran H., Quinn B., Matsuda J., Grabowski G. A. 2007. Combined saposin C and D deficiencies in mice lead to a neuronopathic phenotype, glucosylceramide and alpha-hydroxy ceramide accumulation, and altered prosaposin trafficking. Hum. Mol. Genet. 16: 957–971. [DOI] [PubMed] [Google Scholar]
- 267.Pacheco C. D., Kunkel R., Lieberman A. P. 2007. Autophagy in Niemann-Pick C disease is dependent upon Beclin-1 and responsive to lipid trafficking defects. Hum. Mol. Genet. 16: 1495–1503. [DOI] [PubMed] [Google Scholar]
- 268.Pacheco C. D., Lieberman A. P. 2008. The pathogenesis of Niemann-Pick type C disease: a role for autophagy? Expert Rev. Mol. Med. 10: e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Yadid G., Sotnik-Barkai I., Tornatore C., Baker-Cairns B., Harvey-White J., Pentchev P. G., Goldin E. 1998. Neurochemical alterations in the cerebellum of a murine model of Niemann-Pick type C disease. Brain Res. 799: 250–256. [DOI] [PubMed] [Google Scholar]
- 270.Coleman M. 2005. Axon degeneration mechanisms: commonality amid diversity. Nat. Rev. Neurosci. 6: 889–898. [DOI] [PubMed] [Google Scholar]
- 271.Walkley S. U., Zervas M., Wiseman S. 2000. Gangliosides as modulators of dendritogenesis in normal and storage disease-affected pyramidal neurons. Cereb. Cortex. 10: 1028–1037. [DOI] [PubMed] [Google Scholar]
- 272.Komatsu M., Wang Q. J., Holstein G. R., Friedrich V. L., Jr., Iwata J., Kominami E., Chait B. T., Tanaka K., Yue Z. 2007. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc. Natl. Acad. Sci. USA. 104: 14489–14494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Walkley S. U. 2009. Pathogenic cascades in lysosomal disease: Why so complex? J. Inherit. Metab. Dis. 32: 181–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Sugiura Y., Furukawa K., Tajima O., Mii S., Honda T., Furukawa K. 2005. Sensory nerve-dominant nerve degeneration and remodeling in the mutant mice lacking complex gangliosides. Neuroscience. 135: 1167–1178. [DOI] [PubMed] [Google Scholar]
- 275.Eckhardt M., Hedayati K. K., Pitsch J., Lullmann-Rauch R., Beck H., Fewou S. N., Gieselmann V. 2007. Sulfatide storage in neurons causes hyperexcitability and axonal degeneration in a mouse model of metachromatic leukodystrophy. J. Neurosci. 27: 9009–9021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Zeng Y., Cheng H., Jiang X., Han X. 2008. Endosomes and lysosomes play distinct roles in sulfatide-induced neuroblastoma apoptosis: potential mechanisms contributing to abnormal sulfatide metabolism in related neuronal diseases. Biochem. J. 410: 81–92. [DOI] [PubMed] [Google Scholar]
- 277.Miyatake T., Suzuki K. 1972. Globoid cell leukodystrophy: additional deficiency of psychosine galactosidase. Biochem. Biophys. Res. Commun. 48: 539–543. [DOI] [PubMed] [Google Scholar]
- 278.Hannun Y. A., Bell R. M. 1987. Lysosphingolipids inhibit protein kinase C: implications for the sphingolipidoses. Science. 235: 670–674. [DOI] [PubMed] [Google Scholar]
- 279.Hannun Y. A., Bell R. M. 1989. Functions of sphingolipids and sphingolipid breakdown products in cellular regulation. Science. 243: 500–507. [DOI] [PubMed] [Google Scholar]
- 280.Igisu H., Suzuki K. 1984. Glycolipids of the spinal cord, sciatic nerve, and systemic organs of the twitcher mouse. J. Neuropathol. Exp. Neurol. 43: 22–36. [DOI] [PubMed] [Google Scholar]
- 281.Igisu H., Suzuki K. 1984. Progressive accumulation of toxic metabolite in a genetic leukodystrophy. Science. 224: 753–755. [DOI] [PubMed] [Google Scholar]
- 282.Svennerholm L., Vanier M. T., Mansson J. E. 1980. Krabbe disease: a galactosylsphingosine (psychosine) lipidosis. J. Lipid Res. 21: 53–64. [PubMed] [Google Scholar]
- 283.White A. B., Givogri M. I., Lopez-Rosas A., Cao H., van Breemen R., Thinakaran G., Bongarzone E. R. 2009. Psychosine accumulates in membrane microdomains in the brain of krabbe patients, disrupting the raft architecture. J. Neurosci. 29: 6068–6077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Schueler U. H., Kolter T., Kaneski C. R., Blusztajn J. K., Herkenham M., Sandhoff K., Brady R. O. 2003. Toxicity of glucosylsphingosine (glucopsychosine) to cultured neuronal cells: a model system for assessing neuronal damage in Gaucher disease type 2 and 3. Neurobiol. Dis. 14: 595–601. [DOI] [PubMed] [Google Scholar]
- 285.Lloyd-Evans E., Pelled D., Riebeling C., Bodennec J., de-Morgan A., Waller H., Schiffmann R., Futerman A. H. 2003. Glucosylceramide and glucosylsphingosine modulate calcium mobilization from brain microsomes via different mechanisms. J. Biol. Chem. 278: 23594–23599. [DOI] [PubMed] [Google Scholar]
- 286.Kacher Y., Futerman A. H. 2006. Genetic diseases of sphingolipid metabolism: pathological mechanisms and therapeutic options. FEBS Lett. 580: 5510–5517. [DOI] [PubMed] [Google Scholar]
- 287.D'Hooge R., Lullmann-Rauch R., Beckers T., Balschun D., Schwake M., Reiss K., von Figura K., Saftig P. 2005. Neurocognitive and psychotiform behavioral alterations and enhanced hippocampal long-term potentiation in transgenic mice displaying neuropathological features of human alpha-mannosidosis. J. Neurosci. 25: 6539–6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Malenka R. C., Nicoll R. A. 1999. Long-term potentiation–a decade of progress? Science. 285: 1870–1874. [DOI] [PubMed] [Google Scholar]
- 289.Foster T. C., Kumar A. 2002. Calcium dysregulation in the aging brain. Neuroscientist. 8: 297–301. [DOI] [PubMed] [Google Scholar]
- 290.Lloyd-Evans E., Pelled D., Riebeling C., Futerman A. H. 2003. Lyso-glycosphingolipids mobilize calcium from brain microsomes via multiple mechanisms. Biochem. J. 375: 561–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Ginzburg L., Futerman A. H. 2005. Defective calcium homeostasis in the cerebellum in a mouse model of Niemann-Pick A disease. J. Neurochem. 95: 1619–1628. [DOI] [PubMed] [Google Scholar]
- 292.Bramham C. R., Messaoudi E. 2005. BDNF function in adult synaptic plasticity: the synaptic consolidation hypothesis. Prog. Neurobiol. 76: 99–125. [DOI] [PubMed] [Google Scholar]
- 293.Nolan Y., Martin D., Campbell V. A., Lynch M. A. 2004. Evidence of a protective effect of phosphatidylserine-containing liposomes on lipopolysaccharide-induced impairment of long-term potentiation in the rat hippocampus. J. Neuroimmunol. 151: 12–23. [DOI] [PubMed] [Google Scholar]
- 294.Henderson L. P., Lin L., Prasad A., Paul C. A., Chang T. Y., Maue R. A. 2000. Embryonic striatal neurons from niemann-pick type C mice exhibit defects in cholesterol metabolism and neurotrophin responsiveness. J. Biol. Chem. 275: 20179–20187. [DOI] [PubMed] [Google Scholar]
- 295.Schiffmann R. 2009. Fabry disease. Pharmacol. Ther. 122: 65–77. [DOI] [PubMed] [Google Scholar]
- 296.Grubb J. H., Vogler C., Levy B., Galvin N., Tan Y., Sly W. S. 2008. Chemically modified beta-glucuronidase crosses blood-brain barrier and clears neuronal storage in murine mucopolysaccharidosis VII. Proc. Natl. Acad. Sci. USA. 105: 2616–2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Montano A. M., Oikawa H., Tomatsu S., Nishioka T., Vogler C., Gutierrez M. A., Oguma T., Tan Y., Grubb J. H., Dung V. C., et al. 2008. Acidic amino acid tag enhances response to enzyme replacement in mucopolysaccharidosis type VII mice. Mol. Genet. Metab. 94: 178–189. [DOI] [PubMed] [Google Scholar]
- 298.Chen Y. H., Chang M., Davidson B. L. 2009. Molecular signatures of disease brain endothelia provide new sites for CNS-directed enzyme therapy. Nat. Med. 15: 1215–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Wang D., Zhang W., Kalfa T. A., Grabowski G., Davies S., Malik P., Pan D. 2009. Reprogramming erythroid cells for lysosomal enzyme production leads to visceral and CNS cross-correction in mice with Hurler syndrome. Proc. Natl. Acad. Sci. USA. 106: 19958–19963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Biffi A., Capotondo A., Fasano S., del Carro U., Marchesini S., Azuma H., Malaguti M. C., Amadio S., Brambilla R., Grompe M., et al. 2006. Gene therapy of metachromatic leukodystrophy reverses neurological damage and deficits in mice. J. Clin. Invest. 116: 3070–3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Galbiati F., Givogri M. I., Cantuti L., Rosas A. L., Cao H., van Breemen R., Bongarzone E. R. 2009. Combined hematopoietic and lentiviral gene-transfer therapies in newborn Twitcher mice reveal contemporaneous neurodegeneration and demyelination in Krabbe disease. J. Neurosci. Res. 87: 1748–1759. [DOI] [PubMed] [Google Scholar]
- 302.Klein D., Schmandt T., Muth-Kohne E., Perez-Bouza A., Segschneider M., Gieselmann V., Brustle O. 2006. Embryonic stem cell-based reduction of central nervous system sulfatide storage in an animal model of metachromatic leukodystrophy. Gene Ther. 13: 1686–1695. [DOI] [PubMed] [Google Scholar]
- 303.Papadeas S. T., Maragakis N. J. 2009. Advances in stem cell research for amyotrophic lateral sclerosis. Curr Opin Biotechnol.. 20: 545–51. [DOI] [PubMed] [Google Scholar]
- 304.Weinreb N. J., Barranger J. A., Charrow J., Grabowski G. A., Mankin H. J., Mistry P. 2005. Guidance on the use of miglustat for treating patients with type 1 Gaucher disease. Am. J. Hematol. 80: 223–229. [DOI] [PubMed] [Google Scholar]
- 305.Wraith J. E., Imrie J. 2009. New therapies in the management of Niemann-Pick type C disease: clinical utility of miglustat. Ther Clin Risk Manag. 5: 877–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Galanaud D., Tourbah A., Lehericy S., Leveque N., Heron B., Billette de Villemeur T., Guffon N., Feillet F., Baumann N., Vanier M. T., et al. 2009. 24 month-treatment with miglustat of three patients with Niemann-Pick disease type C: follow up using brain spectroscopy. Mol. Genet. Metab. 96: 55–58. [DOI] [PubMed] [Google Scholar]
- 307.McEachern K. A., Fung J., Komarnitsky S., Siegel C. S., Chuang W. L., Hutto E., Shayman J. A., Grabowski G. A., Aerts J. M., Cheng S. H., et al. 2007. A specific and potent inhibitor of glucosylceramide synthase for substrate inhibition therapy of Gaucher disease. Mol. Genet. Metab. 91: 259–267. [DOI] [PubMed] [Google Scholar]
- 308.Peterschmitt J., Lukina E., Watman N., Arreguin E. A., Banikazemi M., Pastores G., Lastrebner M., Dragosky M., Rosenbaum H., Phillips M., et al. 2009. Genz-112638 for Gaucher disease type 1: Phase 2 clinical trial results after one year of treatment. In 59th Annual Meeting on The American Society of Human Genetics, Honolulu, Hawaii. Available at http://www.ashg.org/2009meeting/abstracts/fulltext. [Google Scholar]
- 309.Benjamin E. R., Flanagan J. J., Schilling A., Chang H. H., Agarwal L., Katz E., Wu X., Pine C., Wustman B., Desnick R. J., et al. 2009. The pharmacological chaperone 1-deoxygalactonojirimycin increases alpha-galactosidase A levels in Fabry patient cell lines. J. Inherit. Metab. Dis. 32: 424–440. [DOI] [PubMed] [Google Scholar]
- 310.Steet R. A., Chung S., Wustman B., Powe A., Do H., Kornfeld S. A. 2006. The iminosugar isofagomine increases the activity of N370S mutant acid beta-glucosidase in Gaucher fibroblasts by several mechanisms. Proc. Natl. Acad. Sci. USA. 103: 13813–13818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Yu Z., Sawkar A. R., Whalen L. J., Wong C. H., Kelly J. W. 2007. Isofagomine- and 2,5-anhydro-2,5-imino-D-glucitol-based glucocerebrosidase pharmacological chaperones for Gaucher disease intervention. J. Med. Chem. 50: 94–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Davidson C. D., Ali N. F., Micsenyi M. C., Stephney G., Renault S., Dobrenis K., Ory D. S., Vanier M. T., Walkley S. U. 2009. Chronic cyclodextrin treatment of murine Niemann-Pick C disease ameliorates neuronal cholesterol and glycosphingolipid storage and disease progression. PLoS One. 4: e6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Galonic D. P., Gin D. Y. 2007. Chemical glycosylation in the synthesis of glycoconjugate antitumour vaccines. Nature. 446: 1000–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Furukawa K., Tokuda N., Okuda T., Tajima O. 2004. Glycosphingolipids in engineered mice: insights into function. Semin. Cell Dev. Biol. 15: 389–396. [DOI] [PubMed] [Google Scholar]
- 315.Sabourdy F., Kedjouar B., Sorli S. C., Colie S., Milhas D., Salma Y., Levade T. 2008. Functions of sphingolipid metabolism in mammals–lessons from genetic defects. Biochim. Biophys. Acta. 1781: 145–183. [DOI] [PubMed] [Google Scholar]
- 316.Jeckel D., Karrenbauer A., Burger K. N., van Meer G., Wieland F. 1992. Glucosylceramide is synthesized at the cytosolic surface of various Golgi subfractions. J. Cell Biol. 117: 259–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.van Meer G., Wolthoorn J., Degroote S. 2003. The fate and function of glycosphingolipid glucosylceramide. Philos. Trans. R. Soc. Lond. B Biol. Sci. 358: 869–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Kolter T., Proia R. L., Sandhoff K. 2002. Combinatorial ganglioside biosynthesis. J. Biol. Chem. 277: 25859–25862. [DOI] [PubMed] [Google Scholar]
- 319.Fukasawa M., Nishijima M., Hanada K. 1999. Genetic evidence for ATP-dependent endoplasmic reticulum-to-Golgi apparatus trafficking of ceramide for sphingomyelin synthesis in Chinese hamster ovary cells. J. Cell Biol. 144: 673–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Hogan B., Beddington R., Costantini F., Lacy E., 1994. Manipulatiing the mouse embryo: a laboraory manual. Second edition Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York. [Google Scholar]
- 321.Koch J., Gartner S., Li C. M., Quintern L. E., Bernardo K., Levran O., Schnabel D., Desnick R. J., Schuchman E. H., Sandhoff K. 1996. Molecular cloning and characterization of a full-length complementary DNA encoding human acid ceramidase. Identification of the first molecular lesion causing Farber disease. J. Biol. Chem. 271: 33110–33115. [DOI] [PubMed] [Google Scholar]
- 322.Li C. M., Hong S. B., Kopal G., He X., Linke T., Hou W. S., Koch J., Gatt S., Sandhoff K., Schuchman E. H. 1998. Cloning and characterization of the full-length cDNA and genomic sequences encoding murine acid ceramidase. Genomics. 50: 267–274. [DOI] [PubMed] [Google Scholar]
- 323.Park J. H., Schuchman E. H. 2006. Acid ceramidase and human disease. Biochim. Biophys. Acta. 1758: 2133–2138. [DOI] [PubMed] [Google Scholar]
- 324.Simonaro C. M., Park J. H., Eliyahu E., Shtraizent N., McGovern M. M., Schuchman E. H. 2006. Imprinting at the SMPD1 locus: implications for acid sphingomyelinase-deficient Niemann-Pick disease. Am. J. Hum. Genet. 78: 865–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Sogawa H., Horino K., Nakamura F., Kudoh T., Oyanagi K., Yamanouchi T., Minami R., Nakao T., Watanabe A., Matsuura Y. 1978. Chronic Niemann-Pick disease with sphingomyelinase deficiency in two brothers with mental retardation. Eur. J. Pediatr. 128: 235–240. [DOI] [PubMed] [Google Scholar]
- 326.Patterson M. C., Vanier M. T., Suzuki K., Morris J. M., Carstea E., Neufeld E. B., Blanchette-Mackie J. E., Pentchev P. G. 2001. Niemann-Pick Disease Type C: A Lipid Trafficking Disorder.In The Metabolic and Molecular Basis of Inherited Disease Scriver C. R., Beaudet A. L., Sly W. S., Valle D., McGraw-Hill, New York: 3611–3633. [Google Scholar]
- 327.Tang Y., Li H., Liu J. P. 2010. Niemann-Pick Disease Type C: from molecule to clinic. Clin. Exp. Pharmacol. Physiol. 37: 132–140. [DOI] [PubMed] [Google Scholar]
- 328.Vanier M. T., Millat G. 2003. Niemann-Pick disease type C. Clin. Genet. 64: 269–281. [DOI] [PubMed] [Google Scholar]
- 329.Millat G., Chikh K., Naureckiene S., Sleat D. E., Fensom A. H., Higaki K., Elleder M., Lobel P., Vanier M. T. 2001. Niemann-Pick disease type C: spectrum of HE1 mutations and genotype/phenotype correlations in the NPC2 group. Am. J. Hum. Genet. 69: 1013–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Naureckiene S., Sleat D. E., Lackland H., Fensom A., Vanier M. T., Wattiaux R., Jadot M., Lobel P. 2000. Identification of HE1 as the second gene of Niemann-Pick C disease. Science. 290: 2298–2301. [DOI] [PubMed] [Google Scholar]
- 331.Miyawaki S., Mitsuoka S., Sakiyama T., Kitagawa T. 1982. Sphingomyelinosis, a new mutation in the mouse: a model of Niemann-Pick disease in humans. J. Hered. 73: 257–263. [DOI] [PubMed] [Google Scholar]
- 332.Pentchev P. G., Boothe A. D., Kruth H. S., Weintroub H., Stivers J., Brady R. O. 1984. A genetic storage disorder in BALB/C mice with a metabolic block in esterification of exogenous cholesterol. J. Biol. Chem. 259: 5784–5791. [PubMed] [Google Scholar]
- 333.Reid P. C., Sakashita N., Sugii S., Ohno-Iwashita Y., Shimada Y., Hickey W. F., Chang T. Y. 2004. A novel cholesterol stain reveals early neuronal cholesterol accumulation in the Niemann-Pick type C1 mouse brain. J. Lipid Res. 45: 582–591. [DOI] [PubMed] [Google Scholar]
- 334.Treiber-Held S., Distl R., Meske V., Albert F., Ohm T. G. 2003. Spatial and temporal distribution of intracellular free cholesterol in brains of a Niemann-Pick type C mouse model showing hyperphosphorylated tau protein. Implications for Alzheimer's disease. J. Pathol. 200: 95–103. [DOI] [PubMed] [Google Scholar]
- 335.Lissens W., Arena A., Seneca S., Rafi M., Sorge G., Liebaers I., Wenger D., Fiumara A. 2007. A single mutation in the GALC gene is responsible for the majority of late onset Krabbe disease patients in the Catania (Sicily, Italy) region. Hum. Mutat. 28: 742. [DOI] [PubMed] [Google Scholar]
- 336.Xu C., Sakai N., Taniike M., Inui K., Ozono K. 2006. Six novel mutations detected in the GALC gene in 17 Japanese patients with Krabbe disease, and new genotype-phenotype correlation. J. Hum. Genet. 51: 548–554. [DOI] [PubMed] [Google Scholar]
- 337.De Gasperi R., Gama Sosa M. A., Sartorato E. L., Battistini S., MacFarlane H., Gusella J. F., Krivit W., Kolodny E. H. 1996. Molecular heterogeneity of late-onset forms of globoid-cell leukodystrophy. Am. J. Hum. Genet. 59: 1233–1242. [PMC free article] [PubMed] [Google Scholar]
- 338.Furuya H., Kukita Y., Nagano S., Sakai Y., Yamashita Y., Fukuyama H., Inatomi Y., Saito Y., Koike R., Tsuji S., et al. 1997. Adult onset globoid cell leukodystrophy (Krabbe disease): analysis of galactosylceramidase cDNA from four Japanese patients. Hum. Genet. 100: 450–456. [DOI] [PubMed] [Google Scholar]
- 339.Duchen L. W., Eicher E. M., Jacobs J. M., Scaravilli F., Teixeira F. 1980. Hereditary leucodystrophy in the mouse: the new mutant twitcher. Brain. 103: 695–710. [DOI] [PubMed] [Google Scholar]
- 340.Mohri I., Taniike M., Taniguchi H., Kanekiyo T., Aritake K., Inui T., Fukumoto N., Eguchi N., Kushi A., Sasai H., et al. 2006. Prostaglandin D2-mediated microglia/astrocyte interaction enhances astrogliosis and demyelination in twitcher. J. Neurosci. 26: 4383–4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Phelan M. C., Thomas G. R., Saul R. A., Rogers R. C., Taylor H. A., Wenger D. A., McDermid H. E. 1992. Cytogenetic, biochemical, and molecular analyses of a 22q13 deletion. Am. J. Med. Genet. 43: 872–876. [DOI] [PubMed] [Google Scholar]
- 342.Gieselmann V., Polten A., Kreysing J., von Figura K. 1989. Arylsulfatase A pseudodeficiency: loss of a polyadenylylation signal and N-glycosylation site. Proc. Natl. Acad. Sci. USA. 86: 9436–9440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Hoffmann B. 2009. Fabry disease: recent advances in pathology, diagnosis, treatment and monitoring. Orphanet J. Rare Dis. 4: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Desnick R. J., Astrin K. H., Bishop D. F. 1989. Fabry disease: molecular genetics of the inherited nephropathy. Adv. Nephrol. Necker Hosp. 18: 113–127. [PubMed] [Google Scholar]
- 345.Grzeschik K. H., Grzeschik A. M., Banhof S., Romeo G., Siniscalco M., van Someren H., Meera Khan P., Westerveld A., Bootsma D. 1972. X-linkage of human -galactosidase. Nat. New Biol. 240: 48–50. [DOI] [PubMed] [Google Scholar]
- 346.Ramaswami U., Whybra C., Parini R., Pintos-Morell G., Mehta A., Sunder-Plassmann G., Widmer U., Beck M. 2006. Clinical manifestations of Fabry disease in children: data from the Fabry Outcome Survey. Acta Paediatr. 95: 86–92. [DOI] [PubMed] [Google Scholar]
- 347.Meikle P. J., Hopwood J. J., Clague A. E., Carey W. F. 1999. Prevalence of lysosomal storage disorders. JAMA. 281: 249–254. [DOI] [PubMed] [Google Scholar]
- 348.Shabbeer J., Yasuda M., Benson S. D., Desnick R. J. 2006. Fabry disease: identification of 50 novel alpha-galactosidase A mutations causing the classic phenotype and three-dimensional structural analysis of 29 missense mutations. Hum. Genomics. 2: 297–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Ohshima T., Murray G. J., Swaim W. D., Longenecker G., Quirk J. M., Cardarelli C. O., Sugimoto Y., Pastan I., Gottesman M. M., Brady R. O., et al. 1997. alpha-Galactosidase A deficient mice: a model of Fabry disease. Proc. Natl. Acad. Sci. USA. 94: 2540–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Xu Y. H., Quinn B., Witte D., Grabowski G. A. 2003. Viable mouse models of acid beta-glucosidase deficiency: the defect in Gaucher disease. Am. J. Pathol. 163: 2093–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Sinclair G. B., Jevon G., Colobong K. E., Randall D. R., Choy F. Y., Clarke L. A. 2007. Generation of a conditional knockout of murine glucocerebrosidase: utility for the study of Gaucher disease. Mol. Genet. Metab. 90: 148–156. [DOI] [PubMed] [Google Scholar]
- 352.Enquist I. B., Nilsson E., Ooka A., Mansson J. E., Olsson K., Ehinger M., Brady R. O., Richter J., Karlsson S. 2006. Effective cell and gene therapy in a murine model of Gaucher disease. Proc. Natl. Acad. Sci. USA. 103: 13819–13824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Dreborg S., Erikson A., Hagberg B. 1980. Gaucher disease–Norrbottnian type. I. General clinical description. Eur. J. Pediatr. 133: 107–118. [DOI] [PubMed] [Google Scholar]
- 354.Amsallem D., Rodriguez D., Vanier M., al E. 2005. Third case of Gaucher disease with sap-C deficiency and evaluation of twelve months’ therapy by miglustat. J. Inherit. Metab. Dis. 28: 152. [Google Scholar]
- 355.Christomanou H., Chabas A., Pampols T., Guardiola A. 1989. Activator protein deficient Gaucher's disease. A second patient with the newly identified lipid storage disorder. Klin. Wochenschr. 67: 999–1003. [DOI] [PubMed] [Google Scholar]
- 356.Rafi M. A., de Gala G., Zhang X. L., Wenger D. A. 1993. Mutational analysis in a patient with a variant form of Gaucher disease caused by SAP-2 deficiency. Somat. Cell Mol. Genet. 19: 1–7. [DOI] [PubMed] [Google Scholar]
- 357.Sun Y., Quinn B., Witte D. P., Grabowski G. A. 2005. Gaucher disease mouse models: point mutations at the acid {beta}-glucosidase locus combined with low-level prosaposin expression lead to disease variants. J. Lipid Res. 46: 2102–2113. [DOI] [PubMed] [Google Scholar]
- 358.Brunetti-Pierri N., Scaglia F. 2008. GM1 gangliosidosis: review of clinical, molecular, and therapeutic aspects. Mol. Genet. Metab. 94: 391–396. [DOI] [PubMed] [Google Scholar]
- 359.Takano T., Yamanouchi Y. 1993. Assignment of human beta-galactosidase-A gene to 3p21.33 by fluorescence in situ hybridization. Hum. Genet. 92: 403–404. [DOI] [PubMed] [Google Scholar]
- 360.Matsuda J., Suzuki O., Oshima A., Ogura A., Noguchi Y., Yamamoto Y., Asano T., Takimoto K., Sukegawa K., Suzuki Y., et al. 1997. Beta-galactosidase-deficient mouse as an animal model for GM1-gangliosidosis. Glycoconj. J. 14: 729–736. [DOI] [PubMed] [Google Scholar]
- 361.Yamanaka S., Johnson M. D., Grinberg A., Westphal H., Crawley J. N., Taniike M., Suzuki K., Proia R. L. 1994. Targeted disruption of the Hexa gene results in mice with biochemical and pathologic features of Tay-Sachs disease. Proc. Natl. Acad. Sci. USA. 91: 9975–9979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Taniike M., Yamanaka S., Proia R. L., Langaman C., Bone-Turrentine T., Suzuki K. 1995. Neuropathology of mice with targeted disruption of Hexa gene, a model of Tay-Sachs disease. Acta Neuropathol. 89: 296–304. [DOI] [PubMed] [Google Scholar]
- 363.Zampieri S., Filocamo M., Buratti E., Stroppiano M., Vlahovicek K., Rosso N., Bignulin E., Regis S., Carnevale F., Bembi B., et al. 2009. Molecular and functional analysis of the HEXB gene in Italian patients affected with Sandhoff disease: identification of six novel alleles. Neurogenetics. 10: 49–58. [DOI] [PubMed] [Google Scholar]
- 364.Chen B., Rigat B., Curry C., Mahuran D. J. 1999. Structure of the GM2A gene: identification of an exon 2 nonsense mutation and a naturally occurring transcript with an in-frame deletion of exon 2. Am. J. Hum. Genet. 65: 77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Sandhoff R., Geyer R., Jennemann R., Paret C., Kiss E., Yamashita T., Gorgas K., Sijmonsma T. P., Iwamori M., Finaz C., et al. 2005. Novel class of glycosphingolipids involved in male fertility. J. Biol. Chem. 280: 27310–27318. [DOI] [PubMed] [Google Scholar]
- 366.Sheikh K. A., Sun J., Liu Y., Kawai H., Crawford T. O., Proia R. L., Griffin J. W., Schnaar R. L. 1999. Mice lacking complex gangliosides develop Wallerian degeneration and myelination defects. Proc. Natl. Acad. Sci. USA. 96: 7532–7537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Kawai H., Allende M. L., Wada R., Kono M., Sango K., Deng C., Miyakawa T., Crawley J. N., Werth N., Bierfreund U., et al. 2001. Mice expressing only monosialoganglioside GM3 exhibit lethal audiogenic seizures. J. Biol. Chem. 276: 6885–6888. [DOI] [PubMed] [Google Scholar]
- 368.Yamashita T., Wu Y. P., Sandhoff R., Werth N., Mizukami H., Ellis J. M., Dupree J. L., Geyer R., Sandhoff K., Proia R. L. 2005. Interruption of ganglioside synthesis produces central nervous system degeneration and altered axon-glial interactions. Proc. Natl. Acad. Sci. USA. 102: 2725–2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Jennemann R., Sandhoff R., Langbein L., Kaden S., Rothermel U., Gallala H., Sandhoff K., Wiegandt H., Grone H. J. 2007. Integrity and barrier function of the epidermis critically depend on glucosylceramide synthesis. J. Biol. Chem. 282: 3083–3094. [DOI] [PubMed] [Google Scholar]
- 370.Pastores G. M., Elstein D., Hrebicek M., Zimran A. 2007. Effect of miglustat on bone disease in adults with type 1 Gaucher disease: a pooled analysis of three multinational, open-label studies. Clin. Ther. 29: 1645–1654. [DOI] [PubMed] [Google Scholar]
- 371.Lidove O., Joly D., Barbey F., Bekri S., Alexandra J. F., Peigne V., Jaussaud R., Papo T. 2007. Clinical results of enzyme replacement therapy in Fabry disease: a comprehensive review of literature. Int. J. Clin. Pract. 61: 293–302. [DOI] [PubMed] [Google Scholar]
- 372.Aerts J. M., Hollak C. E., Boot R. G., Groener J. E., Maas M. 2006. Substrate reduction therapy of glycosphingolipid storage disorders. J. Inherit. Metab. Dis. 29: 449–456. [DOI] [PubMed] [Google Scholar]
- 373.Wortmann S. B., Lefeber D. J., Dekomien G., Willemsen M. A., Wevers R. A., Morava E. Substrate deprivation therapy in juvenile Sandhoff disease. J Inherit Metab Dis. Epub 2009 Nov 4. [DOI] [PubMed] [Google Scholar]
- 374.Ziegler R. J., Brown C., Barbon C. M., D'Angona A. M., Schuchman E. H., Andrews L., Thurberg B. L., McPherson J. M., Karey K. P., Cheng S. H. 2009. Pulmonary delivery of recombinant acid sphingomyelinase improves clearance of lysosomal sphingomyelin from the lungs of a murine model of Niemann-Pick disease. Mol. Genet. Metab. 97: 35–42. [DOI] [PubMed] [Google Scholar]
- 375.Pineda M., Wraith J. E., Mengel E., Sedel F., Hwu W. L., Rohrbach M., Bembi B., Walterfang M., Korenke G. C., Marquardt T., et al. 2009. Miglustat in patients with Niemann-Pick disease Type C (NP-C): a multicenter observational retrospective cohort study. Mol Genet Metab. 98: 243–249. [DOI] [PubMed] [Google Scholar]
- 376.Escolar M. L., Poe M. D., Provenzale J. M., Richards K. C., Allison J., Wood S., Wenger D. A., Pietryga D., Wall D., Champagne M., et al. 2005. Transplantation of umbilical-cord blood in babies with infantile Krabbe's disease. N. Engl. J. Med. 352: 2069–2081. [DOI] [PubMed] [Google Scholar]
- 377.Strazza M., Luddi A., Carbone M., Rafi M. A., Costantino-Ceccarini E., Wenger D. A. 2009. Significant correction of pathology in brains of twitcher mice following injection of genetically modified mouse neural progenitor cells. Mol. Genet. Metab. 97: 27–34. [DOI] [PubMed] [Google Scholar]
- 378.Matzner U., Herbst E., Hedayati K. K., Lullmann-Rauch R., Wessig C., Schroder S., Eistrup C., Moller C., Fogh J., Gieselmann V. 2005. Enzyme replacement improves nervous system pathology and function in a mouse model for metachromatic leukodystrophy. Hum. Mol. Genet. 14: 1139–1152. [DOI] [PubMed] [Google Scholar]