

Abstract
The importance of a protein-protein interaction to a signaling pathway can be established by showing that amino acid mutations that weaken the interaction disrupt signaling, and that additional mutations that rescue the interaction recover signaling. Identifying rescue mutations, often referred to as second-site suppressor mutations, controls against scenarios in which the initial deleterious mutation inactivates the protein or disrupts alternative protein-protein interactions. Here, we test a structure-based protocol for identifying second-site suppressor mutations that is based on a strategy previously described by Kortemme and Baker. The molecular modeling software Rosetta is used to scan an interface for point mutations that are predicted to weaken binding but can be rescued by mutations on the partner protein. The protocol typically identifies three types of specificity switches: knob-in-to-hole redesigns, switching hydrophobic interactions to hydrogen bond interactions, and replacing polar interactions with non-polar interactions. Computational predictions were tested with two separate protein complexes; the G-protein Gαi1 bound to the RGS14 GoLoco motif, and UbcH7 bound to the ubiquitin ligase E6AP. Eight designs were experimentally tested. Swapping a buried hydrophobic residue with a polar residue dramatically weakened binding affinities. In none of these cases were we able to identify compensating mutations that returned binding to wild type affinity, highlighting the challenges inherent in designing buried hydrogen bond networks. The strongest specificity switches were a knob-in-to-hole design (20-fold) and the replacement of a charge-charge interaction with non-polar interactions (55-fold). In two cases, specificity was further tuned by including mutations distant from the initial design.
Keywords: Computational Protein Design, Protein-Protein Interactions, Protein Binding Specificity, Rosetta Molecular Modeling Software
Introduction
Defining the role of a protein in a signaling pathway can be challenging because many proteins interact with multiple upstream and downstream binding partners. Amino acid mutations that disrupt interactions with one partner often disrupt interactions with several partners. If there are changes in signaling that accompany a mutation, it is not clear which protein-protein interaction is responsible. One strategy for identifying the key interactions is to use a genetic screen to search for suppressor mutations on the binding partners1. Random mutations are introduced into a genome, a set of proteins or a single protein and screened to identify mutations that suppress the initial deleterious mutation. An alternative approach is to use structure-based molecular modeling. In this case, high resolution structures of the protein-protein interfaces are used to predict mutations that will weaken binding but then can be compensated by further mutations on the partner protein2. The potential advantages of this approach are that it can be quick if the computational predictions are accurate and it does not require the development of a genetic screen.
Several computational approaches have been used to redesign protein binding specificities3; 4. The methods vary by how much explicit negative design is performed. For instance, consider a scenario where protein A naturally binds protein B and protein C with equal affinity and the goal is to redesign protein A so that it binds more tightly to B. One approach is to perform a sequence optimization simulation that only explicitly optimizes the binding energy between protein A and B5. This is referred to as positive design, and does not include explicit calculations of the binding energy between protein A and C. This approach has been used to successfully redesign the binding specificity of calmodulin for target peptides6; 7. The redesigned calmodulin maintains wild-type affinity for the target sequence from smooth muscle myosin light chain kinase, but binds less tightly to a group of other native binding partners. One advantage of using only positive design is that it is not necessary to have an accurate structure or model of the undesired interactions. Another strategy is to simultaneously include both positive and negative design in the design process, and explicitly optimize the energy gap between the target and undesired interactions8; 9; 10. This is often referred to as multi-state design. Multi-state design has been used to redesign binding specificities between coiled-coils and create a heterodimer from the SspB adopter protein homodimer11. Recently, Grigoryan et al. used large-scale multi-state design simulations to create sequences that recognize specific members of the Bzip family of coiled-coils 12. Designs based entirely on positive design did not attain the desired specificities.
A third strategy for redesigning binding specificities is to separate negative and positive design into different steps. Kortemme et al. used this approach to design second-site suppressor mutations2. First, they scanned through a protein-protein interface for mutations predicted to destabilize the interaction. For each deleterious mutation, a design simulation was run to determine if compensating mutations could be identified by redesigning surrounding residues (residues on both sides of the interface were allowed to vary). Using the interaction between colicin E7 and the immunity protein Im7 as a model system, they created two designs that exhibited a ~20-fold change in binding specifity. For instance, in one case wild type E7 bound to mutated Im7 with an affinity of ~300 nM, while redesigned E7 bound to mutated Im7 with an affinity ~12 nM. Because the affinity of the wild type pair was too strong to measure, they were not able to determine how the affinity of the redesigned pair compared to the wild type pair.
Here, we provide further testing of the second-site suppressor strategy for redesigning protein-protein binding specificities. In particular, we are interested if specificity switches can be achieved with only one or two mutations on each side of the interface. In general, redesigns that minimize the number of mutations required for changing binding specificities will be more attractive for in cell studies as they are less likely to perturb other features of the protein that may be critical for function. Additionally, redesigns focused on just a few residues allow more specific feedback as to which types of interactions, for instance hydrogen bonding versus van der Waals, can be modeled more reliably. We focus on three types of switches: knob-in-to-hole redesigns, switching hydrophobic interactions to hydrogen bond interactions, and replacing polar interactions with non-polar interactions. Predictions were tested with two separate complexes; Gαi1 from the heterotrimeric G-protein system bound to the RGS14 GoLoco peptide, and the E2, UbcH7, bound to the E3, E6AP from the ubiquitin pathway 13; 14. Both interfaces are large (> 2000 Å2), and contain a mixture of hydrophobic and polar interactions.
Results
Despite having large interfaces, a relatively small number of second-site suppressor designs were identified for both model systems. For the interaction between Gαi1 and the GoLoco peptide 84 interface positions were screened, and for the interaction between UbcH7 and E6AP 44 interface positions were screened. At each residue position, all possible point mutations, except to cysteine, were tested. For mutations that were predicted to destabilize binding by more than 0.5 kcal/mol, a second round of simulations was performed to identify compensating mutations. All residues within 5.5 Å of the point mutation were allowed to vary, including residues on the same chain as the mutation. The results from the second round of simulations were filtered to select for rescue mutations that brought binding within 0.3 kcal/mol of the wild type interaction yet filter out mutations that destabilize the individual protein chains by more than 1 kcal/mol based on the predicted ΔΔGobind, ΔΔGochainA and ΔGochainB. For UbcH7 and E6AP 16 designs passed the filters (from 360 destabilizing mutations) and for Gαi1 and the GoLoco peptide 24 designs passed the filters (from 440 destabilizing mutations). From the 40 designs (Tables S.1 and S.2), 4 designs were selected from each model system (Table 1). Emphasis was placed on selecting design that represent the three types of specificity switches, designs that represent different regions of each protein: protein interface, and designs that involved fewer numbers of mutations. Additionally for the Gαi1/GoLoco complex, designs that mutated residues near the GDP binding pocket and designs that varied GoLoco Q508 were not considered for experimental testing. In a previous study to identify affinity enhancing mutations at the Gαi1/GoLoco interface we found that Rosetta underpredicts favorable interactions made by Q508 15.
Table 1.
Computational and Experimental Summary of Designs
| Protein 1 | Protein 2 | Kd (exp) (μM) | Fold change | LJRatr | LJRrep | HbR | SolR | ElecR | Tot.R | type | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gαi1 | GoLoco | ||||||||||
| WT | WT | 0.10 ± .02 | −75* | 39* | −17* | 42* | −3.6* | −15* | - | ||
| #1 | WT | V507M | 1.04 ± .06 | −0.5 | 11.9 | 0.3 | 0.9 | 0.1 | 12.6 | knob-in-hole | |
| E245L, L249A | V507M | 0.05 ± .01 | 21 | 0.0 | −3.2 | 0.0 | 0.8 | 0.0 | −2.4 | ||
| #2 | K248E, S252L | WT | 0.32 ± .06 | −1.3 | 25.7 | 0.3 | 0.8 | −1.0 | 24.5 | knob-in-hole | |
| K248E, S252L | L503K | 0.08 ± .01 | 4 | 0.8 | −2.0 | −0.1 | 0.2 | −0.6 | −1.7 | ||
| #3 | WT | L504A | 2.4 ± 1.0 | 2.5 | −2.5 | 0.0 | 0.4 | 0.0 | 0.4 | knob-in-hole | |
| F223V, L249F | L504A | > 5 | - | 1.8 | −2.6 | 0.0 | 0.6 | 0.0 | −0.3 | ||
| #3a | WT | L504A, F529W | > 5 | distant site | |||||||
| F223V, L249F, Q147L, E245L | L504A, F529W | 0.04 ± 0.1 | >125 | ||||||||
| #4 | WT | L519T | 16 ± 5 | 2.0 | −1.0 | −1.1 | 0.8 | −0.1 | 0.6 | add hbond | |
| I78S, A111Q | L519T | > 20 | - | 1.1 | −1.0 | −1.6 | 1.7 | −0.2 | −0.1 | ||
| UbcH7 | E6AP | ||||||||||
| WT | WT | 5.0 ± 0.5 | −25* | 0.7* | −4.8* | 15* | −5* | −19* | |||
| #5 | F63H | WT | > 100 | 1.2 | −0.2 | 0.0 | 0.9 | 0.0 | 1.9 | add hbond | |
| F63H | F690Y | > 100 | - | −0.2 | −0.1 | −1.4 | 1.8 | 0.1 | 0.2 | ||
| #6 | WT | V634R | 11 ± 2 | 1.3 | −0.1 | 0.0 | 0.2 | −0.3 | 1.0 | add hbond | |
| K64E | V634R | 28 ± 8 | - | −1.6 | 0.1 | −0.7 | 2.9 | −0.4 | 0.2 | ||
| #7 | E60L | WT | 14 ± 1 | −0.9 | 0.2 | 0.3 | 0.4 | 0.6 | 0.6 | polar to nonpolar | |
| E60L | T662F, Q661W | 7.9 ± 4.4 | 2 | −2.5 | 0.2 | 0.4 | 0.0 | 1.1 | −0.9 | ||
| #8 | K96S | WT | 5.5 ± 0.5 | 0.8 | −0.1 | 0.1 | −1.2 | 1.1 | 0.7 | polar to nonpolar | |
| K96S | D641K | 0.1 ± 0.01 | 55 | 0.4 | 0.0 | 0.0 | −1.5 | 1.4 | 0.3 | ||
| #8a | K96S F63H A98W |
WT | 100 | distant site | |||||||
| K96S F63H A98W |
D641K | 2.5 | 40 | ||||||||
Kd(exp): Experimentally determined dissociation constant
Fold change: Kd(WT-Mut)/Kd(Mut-Mut)
R : columns with the R superscript are changes in energy of binding as calculated with Rosetta
for the wt-wt pairs the total binding energies for each term are shown
LJatr: portion of Lennard Jones curve where E < 035
LJrep: portion of Lennard Jones curve where E > 035
Hbond: hydrogen bonding energy16
Sol: solvation energy29
Elec: knowledge-based electrostatics – only pertains to side chains with a non-zero net charge35
Tot.: total binding energy calculated with Rosetta
Type: type of specificity switch (Knob-in-hole – suppressor mutation based on relieving a steric clash, hbond – hydrogen bonds used to accommodate buried polar groups, polar to nonpolar – designs based on the addition of van der Waals interactions and more favorable desolvation energies, distant site, uses mutations that are not near the destabilizing mutation)
Description and Experimental Characterization of Second-Site Suppressor Designs
Knob-in-to-hole designs
Three designs selected for experimental characterization were based on the introduction of a bump or hole at the interface that could be accommodated by pruning back or filling space with neighboring residues. In the first design (design #1 in Table 1) valine 507 on the GoLoco peptide is mutated to a methionine. The methionine is predicted to clash with the β-carbon of glutamate 245 from Gαi1 (Figure 1). To accommodate the methionine, two mutations were made to Gαi1, E245L and L249A. Pruning back residue 249 creates an open space that can be filled by an alternative rotamer of methionine 507. Switching glutamate 245 to a leucine is primarily isosteric and it does not relieve any clashes, however, this mutation is predicted to recoup favorable desolvation energy that is lost by mutating leucine 249 to alanine. Experimentally, this is one of our best performing designs (Figure 1, Figure S.1, Table 1). V507M weakens binding affinity from 100 nM to just over 1 μM, but when combined with E245L and L249A binding affinity returns to 50 nM.
Figure 1.

Models of a steric switch designed at the Gαi1 (green) and GoLoco (blue) interface (design #1). A) Wild type interface. B) The mutation V507M is predicted to create a clash at the interface that is accommodated by L249A (panel C). D) Binding curves as measured by fluorescence polarization, Gαi1 was titrated into GoLoco labeled with fluorescein.
The initial destabilizing mutation for the second knob-in-to-hole design (design #2) was S252L in Gαi1. The new leucine is predicted to clash with L503 from GoLoco. Rosetta compensated for this mutation with two mutations: L503K on GoLoco and K248E on Gαi1. K503 is predicted to open up space for L252, and E248 is predicted to form favorable interactions with K503 (Figure 2). It is interesting that net changes in charge-charge interactions are not predicted to be the main determinant of altered specificity in this design (Table 1). This is because R506 on the GoLoco peptide adopts an alternative rotamer in the redesign (to accommodate K503) and is further from several glutamates on Gαi1. Experiments indicate that L503K GoLoco rescues S252L, K248E Gαi1 as predicted, although the initial destabilization with S252L, K248E is modest. S252L, K248E weakens Gαi1 binding for GoLoco from 100 nM to 320 nM, and the compensating mutation, L503K, restores binding to 80 nM (Figure S.2). In these experiments we choose to characterize the double mutant of Gαi1 (S252L, K248E) with WT GoLoco, and not the single mutant (S252L), because this is how these molecules would be best used in in vivo suppressor studies, i.e. destabilizing mutations are made to one partner and then the protein with the suppressor mutation(s) is tested to determine if it rescues function.
Figure 2.

Models of a steric switch designed at the Gαi1 – GoLoco interface (design #2). A) Wild type interface. B) The mutations S252L and K248E are predicted to create a clash at the interface between L503 and L252 that is accommodated by L503K (panel C). The predicted contribution of electrostatics to binding energy is reduced in the final design by rearrangement of R506 to accommodate K503.
In design #3, L504 on GoLoco is mutated to an alanine creating a hole in the middle of the interface. Rosetta compensated with L249F and F223V on Gαi1. Experiments indicated that L504A GoLoco significantly reduced affinity for Gαi1 (Kd = 2.4 μM) but that the compensating mutations were not successful (Table 1). Because the initial deleterious mutation was a promising beginning for creating an altered specificity interface, we tested if we could suppress GoLoco L504A by including additional mutations that are not adjacent to the initial mutation. In a previous study, we identified several mutations (Q147L, E245L on Gαi1, and F529W on GoLoco) that enhance the affinity of the GoLoco/Gαi1 interaction 15. We added three of them to our initial design, creating a designed pair with the sequences L504A, F529W GoLoco and F223V, L249F, Q147L, E245L Gαi1. None of these mutations are immediately adjacent to the initial set of mutations (Figure 3a). F529W was expected to enhance the affinity of L504A GoLoco for wild type Gαi1, but binding remained weak (Kd > 5 μM). The affinity increasing mutations to Gαi1 however, had the desired effect; F223V, L249F, Q147L, E245L Gαi1 bound L504A, F529W GoLoco with a dissociation constant of 40 nM, thus completely rescuing L504A, F529W GoLoco (Figure 3b). This result indicates that making use of residues distant from the initial perturbation is an effective way to rationally design a suppressor. One advantage of this approach is that it is possible to make use of previous structure-activity studies.
Figure 3.

Using residues distant from the initial mutation to create a second-site suppressor. A) Mutations in the initial design (design #3) F223V, L249F Gαi1: L504A GoLoco are shown in red. This design pair has reduced affinity compared to the wild type pair (~5 μM compared to 0.1 μM). Addition of affinity enhancing mutations, Q147L-Gαi1, E245L-Gαi1 and F529W-GoLoco (shown in blue) creates a suppressible system. B) L504A, F529W GoLoco bind WT Gαi1 weakly (Kd ~ 5 μM), but has strong affinity for F223V, L249F, Q147L, E245L Gαi1 (Kd = 0.04 μM).
Designs with new hydrogen bonds
Three designs were based on the introduction of new hydrogen bonds. In each case the initial destabilizing mutation replaced a hydrophobic amino acid with a polar amino acid. These mutations were predicted to create less favorable desolvation energies for binding (column labeled Solv – Table 1). In an attempt to recover from these mutations, Rosetta introduced additional polar amino acids that were predicted to satisfy the hydrogen bond potential of the first mutation. The rescue mutations were also predicted to create less favorable desolvation energies for binding, but the prediction was that the new hydrogen bonds and more favorable Lennard-Jones attractive scores would counterbalance desolvation penalties. Design #4 starts with the replacement of leucine 519 from GoLoco with a threonine. Despite being within hydrogen bond distance of serine 75 on Gαi1, this mutation is predicted to be destabilizing because of desolvation costs and less favorable packing. To suppress L519T Rosetta introduces two mutations: I78S and A111Q. The complete design results in three new predicted hydrogen bonds: Gαi1 Q111 epsilon nitrogen with GoLoco T519 gamma oxygen, Gαi1 S78 gamma hydrogen with Gαi1 Q111 epsilon oxygen, and GoLoco T519 gamma oxygen with Gαi1 S75 gamma oxygen. The geometric parameters for these bonds are within the ranges seen in naturally occurring proteins 16, however, they are not optimal (Figure 4). Particularly sub-optimal is the donor-hydrogen-acceptor angle of 115° for Q111 epsilon nitrogen with T519 gamma oxygen. The experimental results show that the initial mutation is destabilizing, Kd rises from 100 nM to 16 μM, but the rescue mutations do not recover any of the loss in binding (Figure S.3).
Figure 4.
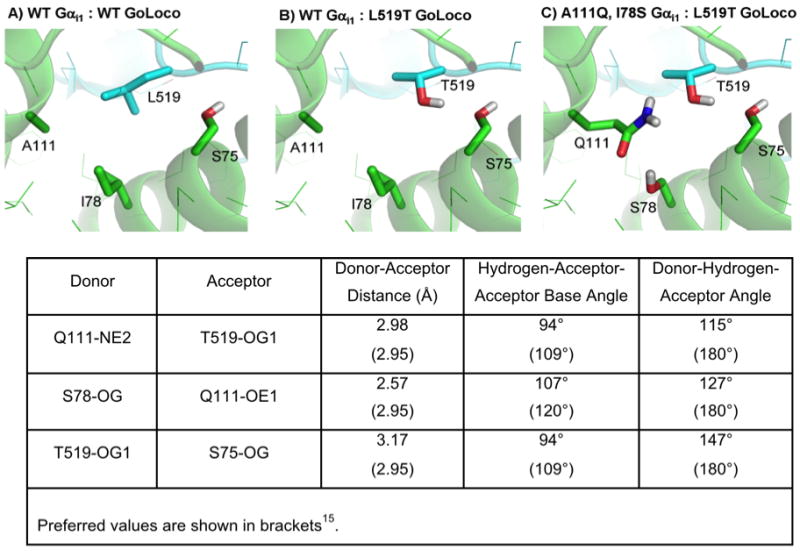
Design of a non-polar to polar switch at the GoLoco (blue) and Gαi1 (green) interface (design #4). A) Wild type interface. B) Destabilizing mutation L519T. C) Mutations designed to compensate for L519T, A111Q and I78S. D) Hydrogen bond parameters for the redesigned residues.
In design #5, phenylalanine 63 on UbcH7 was mutated to a histidine and phenylalanine 690 on E6AP was mutated to a tyrosine forming a putative hydrogen bond between the hydroxyl oxygen on the tyrosine and the epsilon nitrogen of histidine (Figure 5). The new hydrogen bond was predicted to have favorable geometric parameters (typical values in protein crystal structures are shown in brackets16): the distance between the hydrogen and oxygen is 1.95 Å (1.7°–2.1°), the angle defined by the donor nitrogen, hydrogen, and acceptor oxygen is 147° (140°–180°), and the angle defined by the hydrogen, acceptor oxygen and oxygen base is 127°(100–160). A potential weakness of this design is the lack of a hydrogen partner for the delta nitrogen of the histidine, which appears to be inaccessible to water in the bound state. The experimental results for this design are similar to the other designs based on hydrogen bonding. The initial mutation is destabilizing, the dissociation constant changes from 5 μM to greater than 50 μM with the F63H mutation, however, the designed rescue mutation, F690Y, does not recover any binding affinity.
Figure 5.

Design of a non-polar to polar switch at the UbcH7 (purple) and E6AP interface (yellow) (design #5). A) Wild type interface. B) F63H is predicted to create a buried polar atom that can be accommodated by forming a hydrogen bond with a tyrosine introduced at residue 690 of E6AP (panel C). D) Binding experiments indicate that F690Y does not rescue binding.
The two previously described hydrogen bond designs (design 4 and 5) make use of polar, but uncharged side chains. Design #6 is based on the creation of a solvent exposed salt bridge: V634R E6AP paired with K64E UbcH7. Unlike the previous two designs based on new hydrogen bonds, the initial mutation in this case, V634R, does not dramatically destabilize binding (Kd = 11 μM, Kd(wt) = 5 μM). This is probably because R634 is solvent exposed and therefore there is not a large desolvation penalty for making the mutation. The suppressor mutation, K64E, does not recover wild type affinity (Kd = 28 μM) (Figure S.4). This may be because only one of the glutamate oxygens is predicted to hydrogen bond with 634R, while the other is partially removed from solvent.
Replacing polar interactions with hydrophobic interactions
Two designs made to the UbcH7-E6AP interface make use of polar to non-polar swaps (Figure 6). In both cases the residues are on the periphery of the interface and are only partially buried. In design #7, E60L UbcH7 is predicted to weaken binding because of less favorable hydrogen bonding, solvation energies and electrostatic interactions. T662F and Q661W E6AP compensate by forming favorable van der Waals contacts as reflected in changes to the Lennard-Jones score. Experimentally the mutations behave as predicted, although the changes are modest. E60L UbcH7 reduces binding affinity 3-fold (14 μM) and T662F, Q661W E6AP brings affinity back to 7 μM (Figure S.5).
Figure 6.

Design of two polar to non-polar switches at the interface of E6AP (yellow) and UbcH7 (purple). Panels A–C: the mutation E60L is compensated with T662F and Q661W (design #7). Panels D–F: the mutation K96S is compensated with D641K (design #8). Despite only involving polar amino acids, design #8 is classified as polar to non-polar because a salt bridge interaction in the WT (K96 with D641) is replaced with hydrophobic interactions between the methylene groups on K641, the beta carbon of S96 and the side chain of proline 97.
In design #8, K96S UbcH7 removes a salt bridge interaction with D641 E6AP. Rosetta compensates K96S by introducing a lysine at position at 641. We classify this as a polar to hydrophobic switch because the hydrophobic methylene groups on K641 are predicted to pack against the methlylene carbon on serine 96 as well as the side chain of proline 97. The Rosetta solvation score for K96S UbcH7, D641K E6AP is 1.5 kcal/mol more favorable than the wild type interaction. Experimentally, K96S has a modest effect on binding affinity, but when combined with D641K there is a large increase in binding affinity, Kd = 100 nM, providing a 55-fold change in binding specificity (Figure S.6). Given the large change in specificity, we examined if we could use additional mutations to tune this design to create a system in which the mutant-wild type pair was destabilized relative to the wild type pair. Two mutations on the UbcH7 protein that are structurally removed from design #8 were selected. The first mutation, F63H, from design #5, and the second, A98W, from previous work14. Adding these two further mutations, F63H and A98W, to K96S weakens binding with WT E6AP to 100 μM. The rescue mutation, D641K E6AP, brings binding back to 2.5 μM, a 40-fold change in binding specificity (design #8a).
Discussion
Two of the more successful second-site suppressor designs (design #1: 20-fold change in binding specificity, design #3a: >125-fold change in binding specificity) were based on knob-in-to-hole designs and used primarily hydrophobic amino acids. In design #3a, we also used affinity-enhancing mutations distant from the initial destabilization to suppress the mutant GoLoco sequence. Similar changes in binding specificity have been observed for other computer-based interface designs that make use of hydrophobic packing10. Sauer and co-workers redesigned packing at 8 hydrophobic residues at the center of the SspB homodimer interface to create a heterodimer that was >100-fold more favorable than the respective homodimers; although, the heterodimer was significantly destabilized relative to the wild type homodimer11. Shifman and colleagues have created several calmodulin variants that switch binding specificity by 20- to 900-fold for target peptides6; 7. These designs rely primarily on hydrophobic repacking and a small number of charge swaps of partially exposed charged amino acids.
Our designs that made use of polar to hydrophobic swaps on the periphery of the interface bound stronger or as tightly as the wild type pair, but the initial ‘perturbing’ mutation did not dramatically destabilize binding. Serrano and co-workers obtained similar results when redesigning a PDZ domain to recognize new peptide substrates17. In one case, they swapped two partially exposed polar residues on the target peptide from polar to hydrophobic. This had a modest effect on binding to the wild type PDZ domain (5-fold weaker), however, when they compensated with new hydrophobic residues on the PDZ domain they were able to increase binding affinity 20-fold. These results are consistent with our previous study that found that increasing buried hydrophobic surface area at protein-protein interfaces is an effective strategy for strengthening protein binding affinities15.
Our third set of designs centered on the creation of new hydrogen bonds. A variety of studies have indicated that buried hydrogen bonds at naturally occurring protein interfaces can be a strong source of specificity 18; 19. A well-known example of this is a pair of hydrogen bonding asparagines from the homodimeric coiled-coil GCN420. When the asparagine is mutated to a leucine the protein no longer forms a specific dimer, but rather there is an equilibrium between dimers and trimers. Interestingly, however, the thermal unfolding midpoint for the mutant is 45° higher than the wild type GCN4. It appears that the main role of the asparagine is to destabilize alternative conformations, not increase the free energy gap between the folded state and the unfolded state. Is this a general result? Are buried hydrogen bonds inherently less stabilizing than buried hydrophobic interactions21; 22; 23; 24? This could be one conclusion drawn from our studies. In both cases in which we attempted to design buried hydrogen bonds, we saw a significant loss in binding affinity. Similar results have been observed in other design studies that included the creation of buried hydrogen bonds. Baker and coworkers redesigned the interface between colicin E7 and the immunity protein Im7 to have four new hydrogen bonds25. The binding affinity of the redesigned pair was several orders of magnitude weaker than the wild type pair. A crystal structure of the E7-Im7 redesign showed that the hydrogen bonds were forming as designed, however, their geometric parameters were not ideal. One of the designed hydrogen bonds had a hydrogen acceptor distance of 3.0 Å (the preferred distance is 2 Å), and all of the donor-hydrogen-acceptor angles were significantly non-linear (< 150°). Similar deviations from ideality are present in the models of our designs. In design #4, all three hydrogen bonds have donor-hydrogen-acceptor angles below 150°. The new hydrogen bond between a tyrosine and a histidine in design #5 has close to ideal parameters, but there is only a hydrogen bond partner for one of the histidine nitrogens. When the protein is in the unbound state, both nitrogens will probably form hydrogen bonds with solvent. These limitations in the designed hydrogen bonds indicate that the designs may have reduced binding affinity, not because hydrogen bonds are intrinsically less favorable than hydrophobic interactions, but rather, because they are sub-optimal hydrogen bonds. There are many naturally occurring protein-protein interactions in which the key residues for binding (hot-spot residues) are polar amino acids26.
Creating optimal hydrogen bonds is challenging because not only are hydrogen bonds dependent on distance and orientation, but most polar amino acids prefer to form multiple hydrogen bonds. This means that optimizing one hydrogen bond often conflicts with optimizing others. To examine the sensitivity of design #4 to small backbone perturbations we decided to look at the other GoLoco-Gαi1 complex in the asymmetric unit of the crystal structure. Despite being very similar in structure (the RMSD between residues 78,111, and 519 is 0.2 Å) the redesign has a predicted binding energy that is 1 kcal/mol less favorable than that obtained with the structure pair used in these studies.
The correlation between the computational and experimental changes in binding energies for all of the mutations studied here is poor (R2 < 0.1, Table 1, Figure S.7). Contributing to the poor correlation is the over prediction of steric repulsion and the failure to accurately predict the energies of buried hydrogen bond networks. Additionally, the creation of an altered specificity interface may be inherently difficult, i.e. randomly chosen mutations at an interface are unlikely to have wild type binding affinities. From this perspective, it is exciting that four of the eight designs maintained wild type or better binding affinities. Of the ~3000 point mutations computationally tested at the two interfaces, only 40 of the mutations were predicted to be destabilizing and recoverable via neighboring mutations on the partner protein. Similar results have been observed when computationally redesigning protein cores, suggesting there is a restricted set of low energy sequences for a fixed backbone27. One solution to this constraint from a design and protein evolution perspective is to also make use of mutations that are non-local to the original specificity changing mutation. As demonstrated here, these mutations can be used to set baseline affinity, and therefore, create altered specificity designs with affinities similar to the wild type proteins25.
In summary, we find that we can most reliably design second-site suppressor mutations that rely on sterics or the swap of hydrophobic for polar interactions. The design of new hydrogen bond networks holds promise for creating even more dramatic changes in specificity, but this may require extensive side chain and backbone optimization to find conformations that can simultaneously satisfy multiple bonds.
Methods
Rosetta
Second-site suppressor mutations were designed using the molecular modeling program Rosetta28. Rosetta’s core full atom energy function is a linear sum of molecular mechanics and knowledge-based terms: a 6–12 Lennard-Jones potential, the Lazaridis-Karplus implicit solvation model29, an empirically based hydrogen bonding potential16, backbone dependent rotamer probabilities30, a knowledge-based electrostatics energy potential, amino acid probabilities based on particular regions of φ/ψ space, and reference energies that approximate the energies of amino acids in the unfolded state27. Within Rosetta, there are several variations on this core energy function. For the studies described here, we used a version of Rosetta with a dampened repulsion potential (command line option, -soft_rep_design, Rosetta v 2.1). See supplementary material of Dantas et al. for a complete description of this version of the Rosetta energy function31. It is referred to as Rosetta_DampRep.
Side chain flexibility is modeled by allowing amino acids to adopt different rotamers. We use Dunbrack’s backbone dependent rotamer library supplemented with rotamers that vary chi 1 and chi 2 one standard deviation away from their most probable values30. Low energy combinations of side chain conformations are identified using Monte Carlo optimization with simulated annealing.
Computational screening for suppressor mutations
The protein-protein interface was scanned for amino acid point mutations predicted to destabilize binding (ΔΔGobinding > 0.5 kcal/mol). Residues adjacent to the site of a mutation (within 10 Å Cα-Cα distance) were repacked with Rosetta’s side chain optimization protocol to best accommodate the mutation27. Binding energies were calculated by subtracting the energy of the complex from the energies of the individual chains. Any residues that were repacked in the mutant complex were also repacked in the wild type complex.
For each destabilizing point mutation a sequence optimization simulation was performed to search for compensating mutations, residues with any atom within 5.5 Å of the point mutant were allowed to switch identity 27. Residues on both sides of the interface were allowed to change identity during this process. A bonus energy of −0.6 kcal/mol was given to the wild type neighbors during these simulations in order to favor designs that had small numbers of mutations. Residues within 10 Å of any of the mutated residues were repacked during the binding energy calculations. The resulting second-site suppressor designs were then evaluated by comparing the predicted binding energy for the design with the predicted binding energy of the wild-type complex. Designs with predicted binding energies equal to or less than 0.3 kcal/mol were considered, indicating the designs were predicted to achieve binding affinities similar to that of the wild-type complexes. Designs were not considered for experimental studies if the mutations destabilized either of the individual proteins by more than 1 kcal/mol.
Designs
The atomic coordinates for Gαi1 complexed with the GoLoco motif from RGS14 are taken from the crystal structure PDB 2OM214. Designs containing mutations on sequence positions of Gαi1 or the GoLoco motif identified as important for binding GDP or Magnesium were not considered12, 32. These sequence positions for Gαi1 were the following: K46, S47, T181, V179, D200, G203. These sequence positions for the GoLoco motif were the following: D514, Q515 and R516. Additionally, designs containing mutations in the Switch II region of Gαi1 and interfaced with the GoLoco motif were not excluded but were avoided. These sequence positions were the following: D200-R208. The atomic coordinates for UbcH7 complexed with E6AP are taken from the crystal structure PDB 1C4Z13.
Construction and cloning of protein designs
We used the GoLoco motif of RGS14 (residues 496–531) expressed as a fusion to Tenascin as previously described15. We used the N-terminal-truncated, hexahistidine-tagged expression construct of human Gαi1 with the first 25 codons of the Gα open reading frame removed, as previously described13. The UbcH7 and E6AP expression plasmids have been previously described33. Point mutations were introduced using the QuickChange® site-directed mutagenesis protocol (Stratagene) and all vectors were verified by DNA sequencing.
Protein Production
The GoLoco motif peptide and Gαi1 were expressed as previously described15. E6AP and UbcH7 were expressed and purified as previously described33. For both systems, gel filtration was used to make sure the proteins remained monomeric after redesign. Protein concentrations were determined by measuring absorbance at 280 nm. Extinction coefficients were calculated using the method described by Gill and von Hipple34.
Fluorescence polarization binding analysis
The thiol-reactive fluorescent probe 6-IAF (Molecular Probes) was conjugated to the unique cysteine on the GoLoco motif as previously described15. Fluorescence polarization assays were carried out on a Jobin Yvon Horiba Spec FluoroLog-3 instrument (Jobin Yvon Inc.) performed in L-format with the excitation wavelength set at 495 nm and the emission wavelength set at 520 nm. Binding assays were performed with the GoLoco motif diluted to between 50 nM and 100 nM and the excitation and emission slit widths adjusted to give a fluorescence intensity >100,000 counts per second. Data were analyzed using a model for single site binding according to equation (1), which was incorporated into equation (2) to account for the observed polarization:
| (1) |
| (2) |
Where [A:B] is the concentration of fluorescein-GoLoco motif protein and Gαi1 complex formed, [At} is the total concentration of fluorescein-GoLoco motif protein, [Bt] is the concentration of Gαi1, Pmax is the maximum polarization observed when all fluorescein-GoLoco motif protein is bound to Gαi1, and Pobs is the measured polarization at a given concentration of Gαi1. The data were fit according to equation (2) using non-linear regression with SigmaPlot software to obtain fitted parameters for Kd, Pmax, and Po.
The fluorophore bodipy (507/545)-iodoacetamide (Molecular Probes) was conjugated to UbcH7 as previously described33. Binding assays were performed as previously described33.
Supplementary Material
Acknowledgments
We thank F.S. Willard and C.R. McCudden for aid in protein expression and purification. This research was supported by an award from the W.M. Keck foundation and the grant GM073960 from the National Institutes of Health.
References
- 1.Prelich G. Suppression mechanisms: themes from variations. Trends Genet. 1999;15:261–6. doi: 10.1016/s0168-9525(99)01749-7. [DOI] [PubMed] [Google Scholar]
- 2.Kortemme T, Joachimiak LA, Bullock AN, Schuler AD, Stoddard BL, Baker D. Computational redesign of protein-protein interaction specificity. Nat Struct Mol Biol. 2004;11:371–9. doi: 10.1038/nsmb749. [DOI] [PubMed] [Google Scholar]
- 3.Kortemme T, Baker D. Computational design of protein-protein interactions. Curr Opin Chem Biol. 2004;8:91–7. doi: 10.1016/j.cbpa.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 4.Karanicolas J, Kuhlman B. Computational design of affinity and specificity at protein-protein interfaces. Curr Opin Struct Biol. 2009;19:458–63. doi: 10.1016/j.sbi.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Potapov V, Reichmann D, Abramovich R, Filchtinski D, Zohar N, Ben Halevy D, Edelman M, Sobolev V, Schreiber G. Computational redesign of a protein-protein interface for high affinity and binding specificity using modular architecture and naturally occurring template fragments. J Mol Biol. 2008;384:109–19. doi: 10.1016/j.jmb.2008.08.078. [DOI] [PubMed] [Google Scholar]
- 6.Yosef E, Politi R, Choi MH, Shifman JM. Computational Design of Calmodulin Mutants with up to 900-Fold Increase in Binding Specificity. J Mol Biol. 2008 doi: 10.1016/j.jmb.2008.09.053. [DOI] [PubMed] [Google Scholar]
- 7.Shifman JM, Mayo SL. Exploring the origins of binding specificity through the computational redesign of calmodulin. Proc Natl Acad Sci U S A. 2003;100:13274–9. doi: 10.1073/pnas.2234277100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Havranek JJ, Harbury PB. Automated design of specificity in molecular recognition. Nat Struct Biol. 2003;10:45–52. doi: 10.1038/nsb877. [DOI] [PubMed] [Google Scholar]
- 9.Humphris EL, Kortemme T. Design of multi-specificity in protein interfaces. PLoS Comput Biol. 2007;3:e164. doi: 10.1371/journal.pcbi.0030164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ali MH, Taylor CM, Grigoryan G, Allen KN, Imperiali B, Keating AE. Design of a heterospecific, tetrameric, 21-residue miniprotein with mixed alpha/beta structure. Structure. 2005;13:225–34. doi: 10.1016/j.str.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 11.Bolon DN, Grant RA, Baker TA, Sauer RT. Specificity versus stability in computational protein design. Proc Natl Acad Sci U S A. 2005;102:12724–9. doi: 10.1073/pnas.0506124102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grigoryan G, Reinke AW, Keating AE. Design of protein-interaction specificity gives selective bZIP-binding peptides. Nature. 2009;458:859–64. doi: 10.1038/nature07885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kimple RJ, Kimple ME, Betts L, Sondek J, Siderovski DP. Structural determinants for GoLoco-induced inhibition of nucleotide release by Galpha subunits. Nature. 2002;416:878–81. doi: 10.1038/416878a. [DOI] [PubMed] [Google Scholar]
- 14.Huang L, Kinnucan E, Wang G, Beaudenon S, Howley PM, Huibregtse JM, Pavletich NP. Structure of an E6AP-UbcH7 complex: insights into ubiquitination by the E2-E3 enzyme cascade. Science. 1999;286:1321–6. doi: 10.1126/science.286.5443.1321. [DOI] [PubMed] [Google Scholar]
- 15.Sammond DW, Eletr ZM, Purbeck C, Kimple RJ, Siderovski DP, Kuhlman B. Structure-based protocol for identifying mutations that enhance protein-protein binding affinities. J Mol Biol. 2007;371:1392–404. doi: 10.1016/j.jmb.2007.05.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kortemme T, Morozov AV, Baker D. An orientation-dependent hydrogen bonding potential improves prediction of specificity and structure for proteins and protein-protein complexes. J Mol Biol. 2003;326:1239–59. doi: 10.1016/s0022-2836(03)00021-4. [DOI] [PubMed] [Google Scholar]
- 17.Reina J, Lacroix E, Hobson SD, Fernandez-Ballester G, Rybin V, Schwab MS, Serrano L, Gonzalez C. Computer-aided design of a PDZ domain to recognize new target sequences. Nat Struct Biol. 2002;9:621–7. doi: 10.1038/nsb815. [DOI] [PubMed] [Google Scholar]
- 18.Fersht AR, Shi JP, Knill-Jones J, Lowe DM, Wilkinson AJ, Blow DM, Brick P, Carter P, Waye MM, Winter G. Hydrogen bonding and biological specificity analysed by protein engineering. Nature. 1985;314:235–8. doi: 10.1038/314235a0. [DOI] [PubMed] [Google Scholar]
- 19.Xu D, Tsai CJ, Nussinov R. Hydrogen bonds and salt bridges across protein-protein interfaces. Protein Eng. 1997;10:999–1012. doi: 10.1093/protein/10.9.999. [DOI] [PubMed] [Google Scholar]
- 20.Harbury PB, Zhang T, Kim PS, Alber T. A switch between two-, three-, and four-stranded coiled coils in GCN4 leucine zipper mutants. Science. 1993;262:1401–7. doi: 10.1126/science.8248779. [DOI] [PubMed] [Google Scholar]
- 21.Honig B, Yang AS. Free energy balance in protein folding. Adv Protein Chem. 1995;46:27–58. doi: 10.1016/s0065-3233(08)60331-9. [DOI] [PubMed] [Google Scholar]
- 22.Myers JK, Pace CN. Hydrogen bonding stabilizes globular proteins. Biophys J. 1996;71:2033–9. doi: 10.1016/S0006-3495(96)79401-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dill KA. Dominant forces in protein folding. Biochemistry. 1990;29:7133–55. doi: 10.1021/bi00483a001. [DOI] [PubMed] [Google Scholar]
- 24.Rose GD, Wolfenden R. Hydrogen bonding, hydrophobicity, packing, and protein folding. Annu Rev Biophys Biomol Struct. 1993;22:381–415. doi: 10.1146/annurev.bb.22.060193.002121. [DOI] [PubMed] [Google Scholar]
- 25.Joachimiak LA, Kortemme T, Stoddard BL, Baker D. Computational design of a new hydrogen bond network and at least a 300-fold specificity switch at a protein-protein interface. J Mol Biol. 2006;361:195–208. doi: 10.1016/j.jmb.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 26.Moreira IS, Fernandes PA, Ramos MJ. Hot spots--a review of the protein-protein interface determinant amino-acid residues. Proteins. 2007;68:803–12. doi: 10.1002/prot.21396. [DOI] [PubMed] [Google Scholar]
- 27.Kuhlman B, Baker D. Native protein sequences are close to optimal for their structures. Proc Natl Acad Sci U S A. 2000;97:10383–8. doi: 10.1073/pnas.97.19.10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rohl CA, Strauss CE, Misura KM, Baker D. Protein structure prediction using Rosetta. Methods Enzymol. 2004;383:66–93. doi: 10.1016/S0076-6879(04)83004-0. [DOI] [PubMed] [Google Scholar]
- 29.Lazaridis T, Karplus M. Effective energy function for proteins in solution. Proteins: Struct Func Genet. 1999;35:132–152. doi: 10.1002/(sici)1097-0134(19990501)35:2<133::aid-prot1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 30.Dunbrack RL, Cohen FE. Bayesian statistical analysis of protein side-chain rotamer preferences. Protein Sci. 1997;6:1661–1681. doi: 10.1002/pro.5560060807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dantas G, Corrent C, Reichow SL, Havranek JJ, Eletr ZM, Isern NG, Kuhlman B, Varani G, Merritt EA, Baker D. High-resolution Structural and Thermodynamic Analysis of Extreme Stabilization of Human Procarboxypeptidase by Computational Protein Design. J Mol Biol. 2006 doi: 10.1016/j.jmb.2006.11.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coleman DE, Sprang SR. Crystal structures of the G protein Gi alpha 1 complexed with GDP and Mg2+: a crystallographic titration experiment. Biochemistry. 1998;37:14376–85. doi: 10.1021/bi9810306. [DOI] [PubMed] [Google Scholar]
- 33.Eletr ZM, Huang DT, Duda DM, Schulman BA, Kuhlman B. E2 conjugating enzymes must disengage from their E1 enzymes before E3-dependent ubiquitin and ubiquitin-like transfer. Nat Struct Mol Biol. 2005;12:933–4. doi: 10.1038/nsmb984. [DOI] [PubMed] [Google Scholar]
- 34.Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem. 1989;182:319–26. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 35.Kuhlman B, Dantas G, Ireton GC, Varani G, Stoddard BL, Baker D. Design of a novel globular protein fold with atomic-level accuracy. Science. 2003;302:1364–8. doi: 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.