

Abstract
Abnormal dopaminergic transmission is implicated in schizophrenia, attention deficit hyperactivity disorder, and drug addiction. In an attempt to model aspects of these disorders, we have generated hyperdopaminergic mutant mice by reducing expression of the dopamine transporter (DAT) to 10% of wild-type levels (DAT knockdown). Fast-scan cyclic voltammetry and in vivo microdialysis revealed that released dopamine was cleared at a slow rate in knockdown mice, which resulted in a higher extracellular dopamine concentration. Unlike the DAT knockout mice, the DAT knockdown mice do not display a growth retardation phenotype. They have normal home cage activity but display hyperactivity and impaired response habituation in novel environments. In addition, we show that both the indirect dopamine receptor agonist amphetamine and the direct agonists apomorphine and quinpirole inhibit locomotor activity in the DAT knockdown mice, leading to the hypothesis that a shift in the balance between dopamine auto and heteroreceptor function may contribute to the therapeutic effect of psychostimulants in attention deficit hyperactivity disorder.
Abnormal dopaminergic transmission has been implicated in a number of psychiatric and neurological disorders including schizophrenia and attention deficit hyperactivity disorder (ADHD) (1–3). To investigate the potential neurobiological basis of ADHD, we attempted to model aspects of this disorder by genetic manipulation of the gene encoding the dopamine transporter (DAT). DAT is the membrane transporter that recycles released dopamine and is the main mechanism for clearance of released dopamine (4, 5). We chose DAT as our target for a number of reasons. First, it is the major target for amphetamine and methylphenidate that are the treatments for ADHD (3). Second, an association between ADHD and polymorphisms in the DAT gene has been reported (6–8). Third, the DAT knockout displays hyperactivity and its hyperactivity can be reduced by psychostimulants, making it a potential animal model of ADHD (9). However, behavioral analysis in the DAT knockout is complicated by growth retardation phenotype (10). Furthermore, the DAT knockout lacks the dopaminergic target of psychostimulants, so it cannot reveal any potential dopaminergic mechanism in the treatment of ADHD by psychostimulants. Therefore, a mutant mouse with a decreased DAT level rather than a complete knockout may offer advantages as an animal model of ADHD. In the present study, we generated mutant mice that express 10% of wild-type DAT levels. These mice display chronic hyperdopaminergic activity but no apparent developmental defects; they model some of the behavioral and pharmacological characteristics of ADHD.
Materials and Methods
Generation of the DAT Knockdown Mice.
A 7.5-kb HindIII fragment containing the first two exons of the DAT gene was excised from a phage DNA isolated from a mouse 129 Sv/J genomic library (5). “NotI” and “AscI” sites were introduced by PCR in the second exon right upstream of the translational start with the Kozak sequence reconstructed. A NotI–AscI cassette was inserted to generate the targeting construct (see Fig. 1a). The cassette contained the tetracycline-dependent transactivator tTA (11) (from pUHD15–1, gift of Hermann Bujard, Center for Molecular Biology, Heidelberg, Germany), the neomycin-resistance gene (PGK-neo-pA), the tet-operators, and the human cytomegalovirus minimal promoter (tetO, from pUHD10–3, gift of Hermann Bujard). The original purpose of this targeting construct was to generate a transgenic line in which the DAT level could be regulated by tetracycline (12). However, no tTA expression was detected in the mutants either by in situ hybridization or by crossing with a teto-lacZ reporter line. This is most likely due to interference from the PGK promoter (13, 14). The insertion of an extra 4-kb DNA sequence (tTA-neo-tetO) resulted in a reduction in DAT expression levels. A defective transcription caused by extra sequences in the promoter region has been reported (15, 16). W9.5 embryonic stem cells were electroporated (Bio-Rad Gene Pulse; 800 V and 3 μF) with 30 μg of linearized targeting construct. G418-resistant clones were screened by Southern blot for homologous recombination with a 3′ external probe (see Fig. 1a). Positive cells from one clone were injected into C57BL6/J blastocysts to generate chimeras. One of the chimeras was mated with 129 Sv/J females to generate heterozygous mutants on a 129 Sv/J genetic background (17, 18). The resulting heterozygous mice were bred to generate wild-type, heterozygous, and homozygous mutant mice. All animal procedures were approved by Columbia University Animal Care Committee.
Figure 1.

Generation of the DAT knockdown mutant line. (a) The HindIII fragment represents the subcloned genomic DNA from which the targeting construct was made. Filled and open boxes represent coding and untranslated exons, respectively. The targeting construct was made by inserting the “tTA-neo-tetO” cassette right upstream of ATG. The 3′ external probe (0.8 kb) used for Southern analysis distinguished the EcoRI restriction fragment sizes for wild-type (15 kb) and recombinant DNA (8 kb). (b) Genomic Southern blot analysis of tail biopsies. Genomic DNA from offspring obtained after breeding of heterozygote mice was digested with EcoRI and probed with the external probe. Wild-type and recombinant restriction fragments had the expected 15-kb and 8-kb size, respectively. (c) Western blot analysis of DAT in striatum. A strong immunoreactive band (85 kDa) was seen in the wild-type mouse brain that corresponded to DAT. In the DAT knockdown, immunoreactivity was drastically reduced to approximately 10% of the wild-type level as measured by densitometry. The internal control α-tubulin immunoreactivity (55 kDa) did not differ between the two genotypes. (d) Immunocytochemical analysis of DAT in midbrain. Robust immunoreactivity was seen in the ventral tegmental area (VTA), substantia nigra pars compacta (SNc), and substantia nigra pars reticulata (SNr) of the wild-type brain. DAT immunoreactivity was drastically reduced in the DAT knockdown. (Calibration bar = 500 μm.)
Western Blot Analysis of DAT.
Striatal tissue (10 μg) was isolated, suspended in sample buffer (62.5 mM Tris⋅HCl, pH 6.8/20% glycerol/2% SDS/0.01% bromophenol blue/1 mM DTT), and subjected to SDS/PAGE. Proteins were electrophorectically transferred to poly(vinylidene difluoride) membrane, and nonspecific sites were blocked in 5% nonfat dry milk in TBS (135 mM NaCl/2.5 mM KCl/50 mM Tris/0.1% Tween-20, pH 7.4). Membranes then were incubated in the presence of a mAb to the N terminus of DAT (mAb DAT-Nt; Chemicon) (19) in TBS with 2% nonfat dry milk. DAT immunoreactivity was detected by using a sheep anti-rat horseradish peroxidase secondary antibody (ICN) and enhanced chemiluminescence (Pierce). Membranes then were stripped and reprobed with an antibody to α-tubulin to confirm equal loading of samples.
Immunohistochemistry.
Animals were deeply anesthetized with sodium pentobarbital and transcardially perfused with 4% paraformaldehyde. The brains were equilibrated in 30% sucrose overnight. Frozen sections (45 μm) were cut, preblocked with normal goat serum, and then incubated with primary antibody (DAT-Nt) over 2 nights at 4°C. DAT immunoreactivity was visualized with the avidin-biotin-peroxidase method (Vectostatin Elite ABC; Vector Laboratories). The peroxidase reaction was developed in 0.05% diaminobenzidine and 0.01% H2O2.
Cyclic Voltammetry in Striatal Slices.
Mice were decapitated and brains were rapidly removed. Coronal slices (400 μm thick) containing the striatum were placed in a recording chamber and superfused with an artificial cerebrospinal fluid. The recording carbon-fiber electrodes were prepared as described (20). A potentiostat (EI-400, Ensman Instrumentation, Bloomington, IN) was used for fast-scan cyclic voltammetry. The electrode potential was linearly scanned from −400 to 1,000 mV at 300 V/s, repeated every 100 ms. The peak oxidation current for dopamine occurred between 500 and 700 mV. Each electrode was calibrated with 10 μM dopamine at the end of the experiment to convert the oxidation current to dopamine concentration. Dopamine release was evoked by single-pulse stimulations (350 μA, 4 ms) from a bipolar stimulating electrode (MS 303/3, Plastics One, Roanoke, VA). Both electrodes were placed in the dorsolateral portion of the caudate-putamen 100–200 μm away from each other (21). Cyclic voltammograms were constructed by subtracting the background current obtained before release (15–50 nA) from the current measured after release. In every case, dopamine was the substance detected as identified by its characteristic cyclic voltammogram.
HPLC Assessment of Brain Tissue Content of Dopamine and Metabolites.
Dissected striatum of adult mice were homogenized in 0.1 M HClO4 containing 100 ng/ml 3,4-dihydroxybenzylamine as an internal standard. Homogenates were centrifuged for 10 min at 10,000 g. Supernatants were filtered through a 0.22-μm filter and analyzed for levels of dopamine, 3,4-dihydroxyphenylacetic acid (DOPAC), and homovanillic acid (HVA) using HPLC with electrochemical detection. Dopamine and metabolites were separated on a microbore reverse-phase column (C-18, 5 μm, 1 × 150 mm, Unijet, BAS, West Lafayette, IN) with a mobile phase consisting of 0.03 M citrate-phosphate buffer with 2.1 mM octyl sodium sulfate, 0.1 mM EDTA, 10 mM NaCl, and 17% methanol (pH 3.6) at a flow rate of 90 μl/min and detected by a 3-mm glass carbon electrode (Unijet, BAS) set at +0.8 V. The volume of injection was 5 μl.
In Vivo Microdialysis.
Mice were anesthetized with chloral hydrate (400 mg/kg, i.p.) and placed in a stereotaxic frame. Dialysis probes (2-mm membrane length, 0.24 mm o.d., Cuprophane, 6-kDa cutoff, CMA-11, CMA/Microdialysis, Solna, Sweden) with CMA-11 guide cannulae were implanted into the right striatum. The stereotaxic coordinates for implantation of microdialysis probes were: antero-posterior 0.0 mm, daiso-ventral −4.4 mm, lateral 2.5 mm relative to bregma (22). Placement of the probe was verified by histological examination subsequent to the experiments. Twenty four hours after surgery, the dialysis probe was connected to a syringe pump and perfused with artificial cerebrospinal fluid. To measure “true” extracellular concentration of dopamine, low perfusion rate microdialysis (23) was performed (50 nl/min for 6–7 h and collected to a tube containing 2 μl of 0.4 M HClO4 each 90 min). Perfusate samples were assayed for dopamine by using HPLC-electrochemical detection.
Behavioral Studies.
All mice (3–4 months old) were kept on a 06:00–18:00 light cycle. Behavioral tests were performed during the light period except for home cage activity. Independent animals were used for all four behavioral tests.
Basal activity monitoring in home cage.
Mice (n = 6 for each genotype, both males and females) were single-housed and brought to the testing room (06:00–18:00 light cycle) 12 h before testing. Cages were placed onto a Stoelting activity meter connected to a six-channel amplifier and a computer (24). Activity was monitored continuously for 24 h, and the sampling period was 15 min with 0-sec interval between adjacent sampling periods.
Open-field test.
The open-field test was used to assess animals' exploratory activity and reactivity to novel environment (24, 25). Animals (n = 8 for each genotype, all males) were placed in one of the four square open chambers (40 cm long × 40 cm wide × 37 cm high) located in a 200 cm × 200-cm sound-attenuating behavioral test room. Illumination of the test room was the same as the mouse colony room. No background noise was provided. They were monitored by a video-tracking system equipped with infrared beams (PolyTrack, San Diego Instruments, San Diego) that records the animal's location and path (horizontal activity), as well as the number of rearings (vertical activity).
Novel object exploration test.
This test was used to assess animals' exploratory activity and reactivity to discrete novel stimuli (24, 25). Mice (n = 10 for each genotype, all males) were placed into one corner of the open field. Objects (3 × 3 × 3-cm cubes) were secured to the center of the open-field floor with tapes. The computer divided each open field into two separate regions: one 16 × 16-cm square in the center and the surrounding region. Animals were naïve to the object.
Exploratory behavior in the Y maze.
The free-choice exploration paradigm in Y maze was used to assess preference and/or habituation to novelty (25, 26). The Y maze was made of black Plexiglas. The three arms (A, B, and C, converged at 120o of angular deviation from each other) were identical in their shapes (34 × 10 × 18 cm) but were different in stripes that mark the walls. Mice (n = 8–10 for group, all males) naïve to the Y maze were brought to the testing room 1 h before testing. Mice were placed individually in arm A and allowed to explore arms A and B for 10 or 60 min with arm C blocked. They were then allowed to explore all three arms and the time they spent in arm C was recorded during the 5-min test. An arm entry was defined as the body of a mouse (not necessarily the tail) completely entering into an arm compartment.
Drug Treatment.
All drugs were administered i.p. except for apomorphine (s.c.). SKF-81297 [(±)-6-chloro-7,8-dihydroxy-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrobromide] and amphetamine [S(+)-α-methylphenylethylamine sulfate] were dissolved in saline. Apomorphine [R(-)-10,11-dihydroxyaporphine hydrochloride] was dissolved in 0.1% ascorbic acid. Quinpirole {trans-(-)4aR-4,4a,5,6,7,8,8a,9-octahydro-5-propyl-1H-pyrazolo[3,4-g]quinoline hydrochloride} was dissolved in water. All drugs were purchased from Sigma. Animals' locomotor and rearing activities were monitored right after injections (n = 6–8 for each dose and genotype).
Autoradiography.
Coronal fresh-frozen sections were cut at 20 μm and were thaw-mounted onto slides. Wild-type and mutant brain (n = 4 for each genotype) sections of comparable brain regions were mounted to the same slides. Sections were dried at room temperature and preincubated for 30 min in 50 mM Tris buffer containing 120 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2 (pH 7.4). For dopamine D1 receptor binding, sections were incubated for 90 min in buffer supplemented with 2 nM N-methyl-3H-SCH23390 (85.0 Ci/mmol, Amersham Pharmacia) and 100 nM ketanserin (to block 5-hydroxytryptamine 2 receptor binding). Nonspecific binding was determined in the presence of 10 μM flupenthixol. For dopamine D2 receptor binding, sections were incubated for 90 min in buffer supplemented with 50 pM 2′-[125I]-iodospiperone (2,200 Ci/mmol, NEN) and 100 nM ketanserin. Nonspecific binding was determined in the presence of 1 μM spiperone.
Data Analysis.
Data were analyzed by using STATVIEW 4.5 (Abacus Concepts, Berkeley, CA). Unpaired two-tailed Student's t test was used when genotype was the only grouping variable. ANOVA was used when genotype was not the only grouping variable and when data were collected in a single trial of a single session. Repeated measure ANOVA was used when data were collected in multiple trials of a single session. Nested repeated measure ANOVA was used when data were collected in multiple trials in more than one session.
Results
Generation of the DAT Knockdown Mutant Line.
We have generated, by homologous recombination in embryonic stem cells, a strain of mice with a modified DAT locus (see Methods and Fig. 1 a and b). Expression of the DAT protein was assessed by immunohistochemistry (Fig. 1d) and Western blot analysis (Fig. 1c). In wild-type mice, DAT was distributed throughout the ventral tegmental area (VTA), substantia nigra pars compacta (SNc), substantia nigra pars reticulata (SNr), and striatum (not shown). Dopaminergic cell bodies, axons, and dendrites were strongly labeled (Fig. 1d). In the DAT knockdown mice, DAT immunoreactivity was drastically reduced. However, low levels remain in all anatomical areas with discernable cell bodies, axons, and dendrites. Western blot (Fig. 1c) of DAT in the striatum revealed an approximate 90% loss of DAT immunoreactivity in the mutant mice.
Normal Development in DAT Knockdown Mice.
Heterozygous mice were crossed to generate wild-type, heterozygous, and homozygous mutant mice. There was no obvious sex bias in the offspring, and the expected Mendelian ratio was observed. Because the DAT knockout mice have anterior pituitary hypoplasia, dwarfism, lactation deficits, and high mortality (10), we looked for these features in the DAT knockdown mice. The knockdown mice did not differ from the wild-type mice in their body weight, litter size, pituitary weight (see Fig. 6, which is published as supplemental material on the PNAS web site, www.pnas.org), or gross anatomy of the pituitary (not shown); mothers lactated normally (not shown); there was no premature death (animal numbers: +/+/+/−/−/− = 42:78:40 in adults). In summary, the low levels of DAT present in the knockdown appear to be sufficient to rescue the pituitary hypoplasia and growth retardation phenotype seen in the knockout.
The DAT Knockdown Mice Have a Chronic Hyperdopaminergic Tone.
Release and uptake of dopamine were measured in striatal slices with fast-scan cyclic voltammetry. Compared with that in wild-type mice, electrically stimulated dopamine release was cleared at a much slower rate in the knockdown mice (Fig. 2a). The amount of dopamine released in the knockdown mice was approximately 25% of wild-type levels. This decreased release of dopamine is likely due to deficient dopamine recycling and the resulting diminished pool of releasable dopamine (4, 27).
Figure 2.

The DAT knockdown mice have a hyperdopaminergic tone. (a) Cyclic voltammetry was used to record dopamine efflux in the striatum (n = 4 for each genotype). Dopamine release was evoked by a single electrical pulse, applied at the time indicated by the arrow. Extracellular dopamine was sampled every 100 ms. The rising portion of the curve represents mainly release, whereas the falling portion represents clearance via the DAT. Release and clearance parameters were obtained by fitting recordings to a Michaelis–Menten-based kinetic model. (Inset) Representative cyclic voltammograms (current/voltage curves). (b) The basal level of extracellular dopamine concentration was measured by quantitative in vivo microdialysis on freely moving animals in their home cages (n = 4 for each genotype). The extracellular dopamine concentration is significantly higher in knockdown than in wild-type mice (t = 3.0, P = 0.02). (c) Total tissue (n = 4 for each genotype) content of dopamine and its metabolites were measured by HPLC-coupled electrochemical detection. DOPAC/dopamine and HVA/dopamine ratios were used as an index of dopamine turnover. Both DOPAC/dopamine (t = 3.7, P = 0.01) and HVA/dopamine (t = 8.0, P = 0.0002) ratios were significantly higher in knockdown than in wild-type mice. Error bar represents SEM.
To determine whether the decreased clearance of released dopamine influenced in vivo dopamine levels, we performed quantitative microdialysis on freely moving animals in their home cages. We observed a 70% increase in extracellular striatal dopamine in the mutants (Fig. 2b). HPLC-coupled electrochemical analysis of the total tissue content of dopamine and its metabolites revealed increased DOPAC/dopamine and HVA/dopamine ratios in knockdown mice (Fig. 2c), indicating increased turnover of dopamine. Total tissue content of dopamine in the mutants was approximately 45% of wild-type levels (data not shown), consistent with their decreased release.
The DAT Knockdown Mice Display Hyperactivity and Impaired Response Habituation.
Basal activity was monitored continuously in the home cage for 24 h. The mutant and wild-type mice were not different in their home cage activity. Nocturnal activity was significantly higher for both genotypes (wild-type day: 127.5 ± 35.5; wild-type night: 302.8 ± 57.3; knockdown day: 124.6 ± 32.2; knockdown night: 387.7 ± 49.7; P = 0.42 for genotype difference, P = 0.0001 for day-night difference, P = 0.27 for genotype × day-night interaction; also see Fig. 7, which is published as supplemental material).
We next analyzed locomotor and rearing activities in response to a novel environment. Naïve mice were placed in an open field and their locomotor and rearing activities were measured. As shown in Fig. 3 a and b, both the mutant and the wild-type mice displayed similar locomotor and rearing activities during the initial open-field exposure. Both groups reached same baseline activities after habituation. However, the DAT knockdown mice had an overall higher locomotor activity (Fig. 3a) and rearing activity (Fig. 3b). In addition, there was a significant genotype × trial interaction in rearing, suggesting a genotype difference in habituation.
Figure 3.

The DAT knockdown mice are hyperactive and have decreased response habituation. (a) Naïve animals were exposed to the open field and their locomotor activity was monitored for 3 h (5 min per point). Knockdown mice showed an overall hyperactivity [genotype difference, F(1,14) = 5.0, P = 0.04]. (b) Knockdown mice had higher overall rearing activity [genotype difference, F(1,14) = 4.5, P = 0.05]. There was also a significant genotype difference in the shape of the habituation curve, suggesting a deficit in response habituation in the knockdown [genotype × trial interaction, F(35,490) = 1.54, P = 0.026]. (c) Mice that have been exposed to the open field were re-exposed to the same open field 1 h per day for 6 consecutive days. There was a significant genotype difference in overall locomotor activity [genotype difference, F(1,10) = 15.3, P = 0.003] and in habituation [genotype × trial interaction, F(11,110) = 6.9, P < 0.0001]. (d) In rearing activity, there was significant genotype difference in overall activity [genotype difference, F(1,10) = 19.5, P = 0.0013] and in habituation [genotype × trial interaction, F(11,110) = 13.2, P < 0.0001]. (e) Novel object exploration was measured by time spent in the center of the open field where the novel object was placed. There was no genotype difference before the object was placed. With the novel object, the knockdown displayed more exploratory activity toward the object than the wild type. A similar pattern was observed in the second day with the same procedure. There was a significant genotype difference in mice's response to the novel object [object × genotype interaction, F(1,18) = 8.5, P = 0.01]. (f) Similar results were obtained when specific exploratory activity was measured by percent path traveled in the center [object × genotype interaction, F(1,18) = 7.3, P = 0.015]. Error bar represents SEM.
After their initial exposure to the open field, mice were re-exposed to the same open field for 6 consecutive days. The differences between the knockdown and the wild-type mice continued to develop during repeated re-exposures (Fig. 3 c and d). The knockdown mice displayed overall hyperactivity in both path length and rearing activity measures. In addition, there were significant genotype × trial interactions in both of these measures, suggesting a genotype difference in habituation.
In the novel object exploration test, we studied specific exploratory activity directed toward a novel object placed in the center of the open field. Exploratory activity was assessed by the time spent in the center of the open field and by the percent path length traveled in the center. In the absence of a novel object, the wild-type and knockdown mice spent the same time and traveled the same percent path length in the center (Fig. 3 e and f), suggesting that these two measures segregate well from measures of locomotor activity. After the introduction of a novel object into the center of the open field, there was a dramatic increase in the activities in the center for both genotypes. However, this exploratory activity toward the novel object continued for only 5 min in wild-type mice whereas it continued throughout the 30-min session in the knockdown mice (Fig. 3 e and f), indicating an increased novel object exploratory activity in the knockdown mice. On the second day in the same procedure the knockdown mice again displayed increased exploratory activity toward the same object (Fig. 3 e and f).
In the Y-maze test, we studied animals' preference for the novel arm after habituation in other arms. Mice were first allowed to explore two arms for 10 min before a new arm was opened for exploration. Their exploratory activity in the new arm was measured as the time spent in the new arm during the 5-min test. As shown in Fig. 4, the wild-type mice spent most of the 5 min exploring the newly introduced arm whereas the mutants spent no more time in the new arm than in the other two arms. To test whether the knockdown's lack of preference for the new arm was related to insufficient habituation in the first two arms, we increased the habituation time in the first two arms to 60 min before. As a result, both genotypes spent most of their time in the newly introduced arm (Fig. 4).
Figure 4.

The DAT knockdown mice have impaired exploratory preference after short habituation. Mice were first allowed to explore freely in two arms for 10 min and then were allowed to explore freely in all three arms. During the 5-min test, wild-type mice but not knockdown mice spent most of their time exploring the newly introduced arm. However, when mice were first allowed to explore two arms for 60 min, both genotypes spent most of their time exploring the newly introduced arm [genotype × habituation time interaction, F(1,34) = 7.0, P = 0.01]. Error bar represents SEM.
The DAT Knockdown Mice Display Altered Responses to Amphetamine and Dopamine Agonists.
To assess the behavioral output from dopamine receptor activation, we studied locomotor responses to different doses of the indirect dopamine agonist amphetamine, the D2-selective agonist quinpirole, the D1-selective agonist SKF-81297, and the mixed D1/D2 agonist apomorphine.
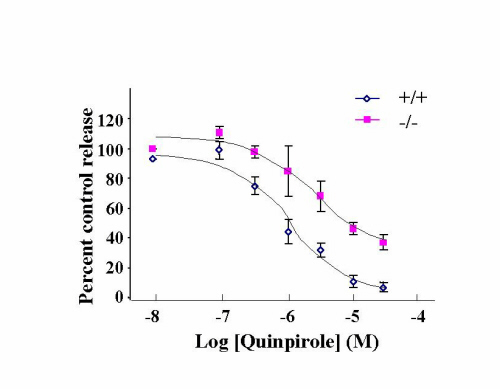
In wild-type mice, amphetamine increased locomotor activities in a dose-dependent manner (Fig. 5 a and a′); apomorphine and quinpirole had an inhibitory effect at low doses, but an excitatory effect at high doses (Fig. 5 b, c, b′, and c′). In contrast, all doses of amphetamine, apomorphine, and quinpirole inhibited locomotor activities in the DAT knockdown mice (Fig. 5 a, b, c, a′, b′, and c′). A biphasic pattern can be seen in response to 2 mg/kg apomorphine (Fig. 5b). Stereotypy was seen only in wild-type mice in response to 2 and 3 mg/kg of amphetamine (data not shown). Inhibition of locomotion by quinpirole and apomorphine at low doses has been reported to be due to activation of D2 autoreceptors that inhibit dopamine synthesis, release, and dopamine neuron firing whereas activation of postsynaptic D2 heteroreceptors results in increased locomotion (28, 29). Therefore, responses to D2 agonists are often biphasic. In summary, inhibition of locomotion by D2 agonists is much more pronounced in the knockdown than in wild-type mice.
Figure 5.

The DAT knockdown mice display altered responses to amphetamine and dopamine agonists. (a) Locomotor activities of knockdown and wild-type mice were monitored for 60 min (5 min per point) after injections of amphetamine. All mice had been habituated to the open field before the test. Amphetamine stimulated the locomotor activity of wild-type mice in a dose-dependent manner. In contrast, it inhibited the locomotor activity of knockdown mice in a dose-dependent manner [genotype × treatment interaction, F(3,64) = 6.8, P = 0.0005]. (b) Apomorphine inhibited locomotion at low doses, but stimulated locomotion at high doses in wild-type mice (b, c, b′, and c′). In contrast, it inhibited locomotion in the knockdown mice at all doses. There was significant genotype × treatment interaction [F(5,95) = 17.9, P < 0.0001]. (c) Quinpirole inhibited locomotor activity of wild-type mice at 2 and 6 mg/kg but stimulated locomotor activity at 20 mg/kg. In contrast, it inhibited locmotor activity in the knockdown mice at all doses [genotype × treatment interaction, F(5,83) = 5.7, P = 0.0001]. (d) SKF-81297 stimulated locomotor activity in both genotypes (main effect of drug treatment, P < 0.0001) but wild-type mice responded more [genotype × treatment interaction, F(2,58) = 6.1, P = 0.004]. (a′–d′) Bar graphs that show average locomotor activity during the 1-h session. Error bar represents SEM.
The locomotor stimulatory effect of SKF-81297 was seen in both wild-type and knockdown mice (Fig. 5 d and d′), but was reduced in knockdown mice, suggesting a decrease in postsynaptic D1 receptor function in the knockdown mice.
Discussion
In the present study, we generated mutant mice that express 10% of wild-type DAT levels (DAT knockdown). Three lines of evidence indicate that the DAT knockdown mice are in a chronic state of hyperdopaminergic activity: a decreased clearance of released dopamine as revealed by fast-scan cyclic voltammetry; a higher extracellular dopamine concentration as revealed by in vivo microdialysis; and a higher dopamine turnover as shown by a higher total tissue metabolite/dopamine ratio.
The DAT knockdown mice do not display apparent developmental impairments as assessed by their body weight, mortality, pituitary weight, pituitary gross anatomy, and maternal behavior. This is very different from the DAT knockout generated previously (10). It is therefore likely that the 10% remaining DAT is sufficient to rescue the growth retardation phenotype seen in the DAT knockout mice.
The DAT knockdown mice had normal home cage activity but they were significantly hyperactive in novel environments (open field), suggesting that hyperactivity may be related to increased responses to novelty (24, 30). In the novel object exploration test, we demonstrated further that the knockdown's hyperactivity occurred specifically in response to novel stimuli. Because the knockdown and wild-type mice have very similar horizontal and vertical activities at the beginning of the first exposure to the open field, it is possible that hyperactivity in the knockdown may be due to a decreased response habituation to novelty. A deficit in habituation often will lead to hyperactivity, even though hyperactivity is not necessarily caused by a habituation deficit (31–33). We therefore analyzed the time course of their activity in the open field. Our statistics support a significant habituation deficit in some but not all behavioral tests. There was a significant genotype × trial interaction in rearing activity during the initial exposure to the open field indicative of an habituation deficit. There were also significant genotype × trial interactions in both locomotor and rearing activities after repeated short exposures to the same open-field environment. These data suggest that hyperactivity in the knockdown may be due at least in part to a decreased response habituation in a novel environment.
In the Y-maze test, we demonstrated that decreased habituation was related to an impaired exploratory preference. It is worth noting that decreased response habituation has been implicated in impaired attention in human studies (34–36). Habituation diminishes unwanted responses as a result of repeated exposures to neutral stimuli and enables the organism to attend selectively to relevant stimuli. Whether there is an attentional component in the genotype difference in the Y-maze test deserves further analysis in the future using specific behavioral paradigms that assess attentional processes.
It is still highly controversial whether ADHD is characterized by a hyperdopaminergic or hypodopaminergic transmission (3). Consistent with the hyperdopamine hypothesis, hyperactivity usually is related to a hyperdopaminergic state (37). Furthermore, recent studies in ADHD patients have found a positive correlation between high dopamine metabolite HVA levels and hyperactivity (38, 39). However, because psychostimulants such as amphetamine and methylphenidate enhance dopaminergic transmission and also appear to improve ADHD symptoms, it also has been proposed that ADHD may be characterized by hypodopaminergic transmission (3). The present study with the DAT knockdown mice and previous studies with the DAT knockout mice (9) support the hyperdopamine hypothesis and provide potential explanations for the calming effect of psychostimulants. In the DAT knockout mice, psychostimulants inhibit locomotor activity and this inhibitory effect may be mediated by an increased release of serotonin (9). Because the DAT knockout lacks the dopaminergic target of psychostimulants, it cannot reveal any potential dopaminergic mechanisms. The present data from the DAT knockdown mice provides a potential dopaminergic mechanism that may contribute to the calming effect of psychostimulants. First, all drugs (amphetamine, apomorphine, and quinpirole) that can activate dopamine autoreceptors have a more pronounced inhibitory effect on locomotor activity in the DAT knockdown mice than in wild-type mice. Second, drugs that do not have an autoreceptor component (SKF-81297) have a less pronounced stimulatory effect on locomotor activity in the DAT knockdown mice than in wild-type mice. These results suggest that the inhibitory effect of amphetamine in the DAT knockdown may be the result of an altered balance between autoreceptor and heteroreceptor functions. We therefore studied dopamine receptor levels by ligand binding autoradiography and studied autoreceptor-mediated inhibition of dopamine release by voltammetry. There was no change in postsynaptic D1 or D2 receptor levels and there was a 50% reduction in D2 autoreceptor levels in the knockdown (see Fig. 8, which is published as supplemental material). In the voltammetry study, our preliminary data indicate no significant difference in autoreceptor-mediated inhibition of dopamine release between the knockdown and wild type (see Fig. 9, which is published as supplemental material). These observations favor the hypothesis that D2 autoreceptor function may be unchanged in the knockdown whereas D1/D2 heteroreceptor function may be decreased.
Taken together, results from both the DAT knockout and knockdown mice support the hyperdopaminergic hypothesis for ADHD. There are two potential mechanisms via which psychostimulants may inhibit hyperactivity: increased serotonergic activity and/or a shift in the balance between dopamine autoreceptor and heteroreceptor function.
The DAT knockdown line appears to be an animal model that exhibits some of the behavioral and pharmacological characteristics of ADHD. ADHD has a substantial genetic component, with a heritability of 0.75–0.91 (40), and recent studies have indicated an association between a polymorphism in the human DAT gene and ADHD (6–8). However, ADHD is a complex psychiatric disorder that is most likely heterogeneous and polygenic. Discrete genetic manipulations such as the DAT knockdown may only mimic a subset of its symptoms. Nevertheless, the study of these mutant mice may provide insights into the mechanisms by which an altered neurotransmitter system may impair a fundamental behavioral process such as response habituation and may point to new potential targets for the treatment of ADHD.
Supplementary Material
Acknowledgments
We thank Monica Mendelsohn and Sylvie Ramboz for help with embryonic stem cell culture and blastocysts injections. This work was supported in part by Solvay Pharmaceuticals and Bristol Meyers Squibb Neuroscience Award (to R.H.) and a National Alliance for Research on Schizophrenia and Depression Young Investigator Award (to X.Z.). M.G.C. is an Investigator of the Howard Hughes Medical Institute.
Abbreviations
- ADHD
attention deficit hyperactivity disorder
- DAT
dopamine transporter
- DOPAC
3,4-dihydroxyphenylacetic acid
- HVA
homovanillic acid
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Cohen J D, Servan-Schreiber D. Psychol Rev. 1992;99:45–77. doi: 10.1037/0033-295x.99.1.45. [DOI] [PubMed] [Google Scholar]
- 2.Self D W, Nestler E J. Annu Rev Neurosci. 1995;18:463–495. doi: 10.1146/annurev.ne.18.030195.002335. [DOI] [PubMed] [Google Scholar]
- 3.Swanson J, Castellanos F X, Murias M, LaHoste G, Kennedy J. Curr Opin Neurobiol. 1998;8:263–271. doi: 10.1016/s0959-4388(98)80150-5. [DOI] [PubMed] [Google Scholar]
- 4.Jones S R, Gainetdinov R R, Jaber M, Giros B, Wightman R M, Caron M G. Proc Natl Acad Sci USA. 1998;95:4029–4034. doi: 10.1073/pnas.95.7.4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giros B, Jaber M, Jones S R, Wightman R M, Caron M G. Nature (London) 1996;379:606–612. doi: 10.1038/379606a0. [DOI] [PubMed] [Google Scholar]
- 6.Cook E H, Jr, Stein M A, Krasowski M D, Cox N J, Olkon D M, Kieffer J E, Leventhal B L. Am J Hum Genet. 1995;56:993–998. [PMC free article] [PubMed] [Google Scholar]
- 7.Gill M, Daly G, Heron S, Hawi Z, Fitzgerald M. Mol Psychiatry. 1997;2:311–313. doi: 10.1038/sj.mp.4000290. [DOI] [PubMed] [Google Scholar]
- 8.Waldman I D, Rowe D C, Abramowitz A, Kozel S T, Mohr J H, Sherman S L, Cleveland H H, Sanders M L, Gard J M, Stever C. Am J Hum Genet. 1998;63:1767–1776. doi: 10.1086/302132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gainetdinov R R, Wetsel W C, Jones S R, Levin E D, Jaber M, Caron M G. Science. 1999;283:397–401. doi: 10.1126/science.283.5400.397. [DOI] [PubMed] [Google Scholar]
- 10.Bosse R, Fumagalli F, Jaber M, Giros B, Gainetdinov R R, Wetsel W C, Missale C, Caron M G. Neuron. 1997;19:127–138. doi: 10.1016/s0896-6273(00)80353-0. [DOI] [PubMed] [Google Scholar]
- 11.Gossen M, Bujard H. Proc Natl Acad Sci USA. 1992;89:5547–5551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bond C T, Sprengel R, Bissonnette J M, Kaufmann W A, Pribnow D, Neelands T, Storck T, Baetscher M, Jerecic J, Maylie J, et al. Science. 2000;289:1942–1946. doi: 10.1126/science.289.5486.1942. [DOI] [PubMed] [Google Scholar]
- 13.Fiering S, Epner E, Robinson K, Zhuang Y, Telling A, Hu M, Martin D I, Enver T, Ley T J, Groudine M. Genes Dev. 1995;9:2203–2213. doi: 10.1101/gad.9.18.2203. [DOI] [PubMed] [Google Scholar]
- 14.Olson E N, Arnold H H, Rigby P W, Wold B J. Cell. 1996;85:1–4. doi: 10.1016/s0092-8674(00)81073-9. [DOI] [PubMed] [Google Scholar]
- 15.Takahashi K, Vigneron M, Matthes H, Wildeman A, Zenke M, Chambon P. Nature (London) 1986;319:121–126. doi: 10.1038/319121a0. [DOI] [PubMed] [Google Scholar]
- 16.Tansey W P, Schaufele F, Heslewood M, Handford C, Reudelhuber T L, Catanzaro D F. J Biol Chem. 1993;268:14906–14911. [PubMed] [Google Scholar]
- 17.Crawley J N E A. Psychopharmacology. 1997;132:107–124. doi: 10.1007/s002130050327. [DOI] [PubMed] [Google Scholar]
- 18.Phillips T J, Hen R, Crabbe J C. Psychopharmacology. 1999;147:5–7. doi: 10.1007/s002130051128. [DOI] [PubMed] [Google Scholar]
- 19.Miller G W, Staley J K, Heilman C J, Perez J T, Mash D C, Rye D B, Levey A I. Ann Neurol. 1997;41:530–539. doi: 10.1002/ana.410410417. [DOI] [PubMed] [Google Scholar]
- 20.Kawagoe K T, Zimmerman J B, Wightman R M. J Neurosci Methods. 1993;48:225–240. doi: 10.1016/0165-0270(93)90094-8. [DOI] [PubMed] [Google Scholar]
- 21.Jones S R, Garris P A, Kilts C D, Wightman R M. J Neurochem. 1995;64:2581–2589. doi: 10.1046/j.1471-4159.1995.64062581.x. [DOI] [PubMed] [Google Scholar]
- 22.Franklin K B J, Paxinos G. The Mouse Brain in Stereotaxic Coordinates. San Diego: Academic; 1997. [Google Scholar]
- 23.Smith A D, Olson R J, Justice J B., Jr J Neurosci Methods. 1992;44:33–41. doi: 10.1016/0165-0270(92)90111-p. [DOI] [PubMed] [Google Scholar]
- 24.Grailhe R, Waeber C, Dulawa S C, Hornung J P, Zhuang X, Brunner D, Geyer M A, Hen R. Neuron. 1999;22:581–591. doi: 10.1016/s0896-6273(00)80712-6. [DOI] [PubMed] [Google Scholar]
- 25.Malleret G, Hen R, Guillou J L, Segu L, Buhot M C. J Neurosci. 1999;19:6157–6168. doi: 10.1523/JNEUROSCI.19-14-06157.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dellu F, Contarino A, Simon H, Koob G F, Gold L H. Neurobiol Learn Mem. 2000;73:31–48. doi: 10.1006/nlme.1999.3919. [DOI] [PubMed] [Google Scholar]
- 27.Gainetdinov R R, Jones S R, Fumagalli F, Wightman R M, Caron M G. Brain Res Brain Res Rev. 1998;26:148–153. doi: 10.1016/s0165-0173(97)00063-5. [DOI] [PubMed] [Google Scholar]
- 28.Van Hartesveldt C. Pharmacol Biochem Behav. 1997;58:955–960. doi: 10.1016/s0091-3057(97)00332-8. [DOI] [PubMed] [Google Scholar]
- 29.Jones S R, Gainetdinov R R, Hu X-T, Cooper D C, Wightman R M, White F J, Caron M G. Nat Neurosci. 1999;2:649–655. doi: 10.1038/10204. [DOI] [PubMed] [Google Scholar]
- 30.Crabbe J C. Pharmacol Biochem Behav. 1986;25:289–292. doi: 10.1016/0091-3057(86)90267-4. [DOI] [PubMed] [Google Scholar]
- 31.Laviola G, Renna G, Bignami G, Cuomo V. Int J Dev Neurosci. 1988;6:431–438. doi: 10.1016/0736-5748(88)90049-4. [DOI] [PubMed] [Google Scholar]
- 32.Hendley E D, Ohlsson W G. Am J Physiol. 1991;261:H583–H589. doi: 10.1152/ajpheart.1991.261.2.H583. [DOI] [PubMed] [Google Scholar]
- 33.Gerlai R, Roder J. Behav Brain Res. 1993;59:119–124. doi: 10.1016/0166-4328(93)90157-l. [DOI] [PubMed] [Google Scholar]
- 34.Cohen R A, Sparling-Cohen Y A. In: The Neuropsychology of Attention. Cohen R A, editor. New York: Plenum; 1993. pp. 75–93. [Google Scholar]
- 35.Tipper S P, Bourque T A, Anderson S H, Brehaut J C. J Exp Child Psychol. 1989;48:353–378. doi: 10.1016/0022-0965(89)90047-7. [DOI] [PubMed] [Google Scholar]
- 36.Walters W F, Wright D C. In: The Orienting Reflex in Humans. Kimmel H D, van Olst E H, Orlebeke J F, editors. Hillsdale, NJ: Lawrence Erlbaum; 1979. pp. 101–121. [Google Scholar]
- 37.Nestler E J. Cell. 1994;79:923–926. doi: 10.1016/0092-8674(94)90022-1. [DOI] [PubMed] [Google Scholar]
- 38.Castellanos F X, Elia J, Kruesi M J, Gulotta C S, Mefford I N, Potter W Z, Ritchie G F, Rapoport J L. Psychiatry Res. 1994;52:305–316. doi: 10.1016/0165-1781(94)90076-0. [DOI] [PubMed] [Google Scholar]
- 39.Castellanos F X, Elia J, Kruesi M J, Marsh W L, Gulotta C S, Potter W Z, Ritchie G F, Hamburger S D, Rapoport J L. Neuropsychopharmacology. 1996;14:125–137. doi: 10.1016/0893-133X(95)00077-Q. [DOI] [PubMed] [Google Scholar]
- 40.Levy F, Hay D A, McStephen M, Wood C, Waldman I. J Am Acad Child Adolesc Psychiatry. 1997;36:737–744. doi: 10.1097/00004583-199706000-00009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}