

Abstract
ABL-family proteins comprise one of the best conserved branches of the tyrosine kinases. Each ABL protein contains an SH3-SH2-TK (Src homology 3–Src homology 2–tyrosine kinase) domain cassette, which confers autoregulated kinase activity and is common among nonreceptor tyrosine kinases. This cassette is coupled to an actin-binding and -bundling domain, which makes ABL proteins capable of connecting phosphoregulation with actin-filament reorganization. Two vertebrate paralogs, ABL1 and ABL2, have evolved to perform specialized functions. ABL1 includes nuclear localization signals and a DNA binding domain through which it mediates DNA damage-repair functions, whereas ABL2 has additional binding capacity for actin and for microtubules to enhance its cytoskeletal remodeling functions. Several types of posttranslational modifications control ABL catalytic activity, subcellular localization, and stability, with consequences for both cytoplasmic and nuclear ABL functions. Binding partners provide additional regulation of ABL catalytic activity, substrate specificity, and downstream signaling. Information on ABL regulatory mechanisms is being mined to provide new therapeutic strategies against hematopoietic malignancies caused by BCR-ABL1 and related leukemogenic proteins.
Introduction
ABL (1) genes were first encountered in the guise of a tumor gene in the Abelson murine lymphosarcoma virus (2). The product of the virally transduced oncogene, v-abl, showed tyrosine kinase activity (3, 4) and was determined to be an altered form of cellular Abl1 (encoded by the c-Abl gene) (5). The human ortholog of Abl1 was later identified as part of a mutationally activated fusion oncoprotein, BCR-ABL1 (6), common in chronic myeloid leukemia (CML) patients. ABL2 [also known as Abl-related gene or Arg (7)] is a paralog of ABL1 identified by sequence similarity (8). Direct biochemical analyses, cell biology observations, animal experiments, and human leukemia studies have produced working models of ABL function in normal (9, 10) and transformed cells (11).
Central to the biochemical and physiological functions of ABL proteins are their combination of a regulated SH3-SH2-TK (Src homology 3–Src homology 2–tyrosine kinase) domain cassette with cytoskeletal protein– and DNA-binding domains (Fig. 1), a combination that confers unique signaling capabilities. This review focuses on ABL protein evolution, function, and mechanisms of regulation.
Fig. 1.

ABL domain structure and motif conservation. Linear depiction of human ABL1 and ABL2 (long, b isoforms), D. melanogaster Abl, and M. brevicollis Abl1 and Abl2. my, myristoylation site (*site present but modification not verified); G BD, G-actin–binding domain; and MT BD, microtubule-binding domain. Blue triangle, NES; magenta triangle, NLS; and green triangle, proline-rich motif with capacity to bind SH3 or WW domains.
The ABL Subfamily of Tyrosine Kinases
Evolution and domain structure
ABL genes are found in all metazoans, which suggests that their structure and function were fixed relatively early in tyrosine kinase evolution. Vertebrate genomes encode two closely related paralogs, ABL1 and ABL2, with conserved domain structure (Fig. 1) and intron-exon boundaries (www.ensembl.org), which suggests a gene duplication origin. Nonvertebrate metazoans (Strongylocentrotus purpuratus, Caenorhabditis elegans, and Drosophila melanogaster) have a single ABL gene, which shows strong conservation through the SH3-SH2-TK cassette and terminal actin-binding domain. The choanoflagellate Monosiga brevicollis, a unicellular protist and proximal progenitor of metazoans, encodes two nonreceptor tyrosine kinases that align throughout the ABL SH3-SH2-TK domain cassette but terminate soon thereafter (Fig. 1). This suggests an early origin for ABL kinases, with the addition of an extended carboxy terminus during the metazoan radiation.
The primary clade of human nonreceptor tyrosine kinases [see (12) for kinome dendrogram] has 22 members, all but three of which share an SH3-SH2-TK domain structure. The reiteration of this domain cassette implies strong selective pressure, which reflects multiple contributions to function and regulation (13–15). Alignments using the SH3-SH2-TK sequence from human and fly nonreceptor tyrosine kinases show that ABL1 and ABL2 are more closely related to each other, and to their fly ortholog, than any other nonreceptor tyrosine kinase is related to its closest paralog or ortholog (Fig. 2). Human ABL1 and ABL2 are more than 90% identical in this domain cassette (Fig. 3).
Fig. 2.
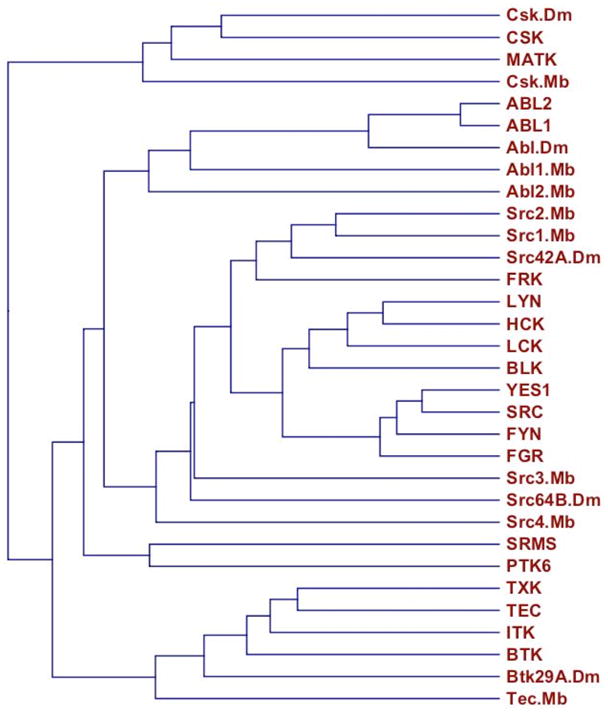
SH3-SH2-TK–family proteins. A dendrogram of the SH3-SH2-TK sequence cassettes from H. sapiens, D. melanogaster (Dm), and the protist M. brevicollis (Mb). The dendrogram was created using CLC Sequence Viewer (www.clcbio.com).
Fig. 3.

Sequence alignment of human ABL1b and ABL2b. Identity (*) and strong or weak sequence conservation [(:)colon for strong conservation; (.) period for weak conservation] are indicated. Domains are indicated with the following underlines: plain, SH3; heavy, SH2; dashed, TK; or wavy, terminal F-actin binding. Shading is used to highlight the following features: sequences deleted in human leukemogenic ABL fusions (gray), phosphorylation sites with known function (red), SH3 and WW domain proline-rich ligands (yellow), K- or R-rich NLS motifs (purple), and NES (blue). Additional reported phosphorylation sites are indicated with a red over-line. The DFG kinase signature is italicized. Alignment was performed using ClustalW (248).
Downstream of the TK domain, vertebrate ABL proteins contain domains for binding cytoskeletal components (F-actin, G-actin, and microtubules) and DNA. Of these, only the carboxy-terminal F-actin–binding domain is identifiable in sea urchin (S. purpuratus) and fruit fly (D. melanogaster) Abl (Fig. 4). In addition, fruit fly Abl is overwhelmingly localized to the cytoplasm and plasma membrane (16–19), which suggests that it lacks the nuclear functions attributed to vertebrate Abl1. Considering proposed mechanisms of gene duplication and evolution [reviewed in (20)], it is tempting to speculate that the fixation of vertebrate Abl paralogs is attributable to the selective value of new functions, rather than to a simple partition of preexisting functions. During the “fate-determination phase” (20) following Abl gene duplication, nuclear-localization and DNA binding properties could have emerged in the nascent Abl1, whereas cytoskeletal remodeling capabilities were expanded and refined in Abl2. These changes might have been subsequently preserved by the complex demands of vertebrate development and differentiation, as well as the requirement for enhanced DNA damage repair capabilities in these longer-lived organisms.
Fig. 4.




Sequence alignment of ABL proteins from representative organisms. Representative Chordates are: Primates (Homo sapiens, Hs); Rodentia (Mus musculus, Mm); Marsupialia (Monodelphis domestica, Md); Aves (Gallus gallus); Amphibia (Xenopus tropicalis, Xt); and Pisces (Danio rerio, Dr). Representative Echinoderm is sea urchin (S. purpuratus, Sp); representative Arthropod is fruit fly (Drosophila melanogaster, Dm). Sequence identity and similarity is denoted as in Fig. 3. Color shading corresponds to the description in Fig. 3. Note that the first line shows only those vertebrate ABL2 sequences for which a long (myristoylated) isoform was identified. A few large insertions (mostly in AblDm) relative to other sequences are indicated. Sequences were obtained from Ensemble and GenBank. Alignment was performed using ClustalW.
Three PxxP (21) motif SH3-binding sites belonging to class 2 [αPxαPx(R or K), reviewed in (22)] are well conserved between ABL1 and ABL2 (Fig. 3). The first is conserved in sea urchin but not fruit fly, the third is apparent in fruit fly but not sea urchin, and the second is absent from both (Fig. 4). ABL1 proteins have an additional class 2 motif that is not found in ABL2 or in nonvertebrates (Fig. 4). Human ABL2 has a single class 1 motif [(R or K)xαPxαP] that is missing from all ABL orthologs except sea urchin Abl (Fig. 4). A PPxY motif implicated in WW domain binding (23) is present in the TK domain, and extended proline-rich sequences with potential for either SH3- or WW-domain interactions are found at variable positions near the carboxy termini of metazoan ABL proteins (Fig. 4). There does not appear to be a simple progressive development of these binding motifs, which suggests a complex evolutionary path.
Three K- and R-rich nuclear localization signal (NLS) motifs have been identified in ABL1 (24), consistent with its partial distribution in the nucleus. The equivalent positions in ABL2 have less K and R content (Fig. 3), consistent with its cytoplasmic localization. Alignments with sea urchin and fruit fly Abl proteins (Fig. 4) suggest that NLS function developed relatively late. An L-, V-, and I-rich nuclear export signal (NES), probably engaged by XPO1 (exportin 1) (25), is found in the ABL1 carboxy terminus and is likely responsible for the ability of ABL1 to shuttle between nucleus and cytoplasm (26). Sequence alignment (Fig. 4) suggests that this NES developed gradually and in concert with the nuclear functions of ABL1.
ABL gene expression
In mice, both Abl genes are expressed widely, but not uniformly [(27) and Biogps.gnf.org]. Relatively little is known, however, about the promoter elements and transcription factors that regulate the expression of ABL genes. ABL1 mRNA translation is silenced by microRNA-203 (miR203) (28), and sequence analysis suggests additional posttranscriptional regulators for ABL1 (miR196) and ABL2 (miR26 and miR1297) (targetscan.org).
Each human ABL gene gives rise to two primary transcripts (a and b) using alternate exons. The first exons of ABL1b and ABL2b encode target sites for cotranslational myristoylation (glycine at position 2). The relative proportions and tissue distribution of each isoform have not been established.
Role in development
There is ample evidence for ABL functions during development. The single Drosophila Abl gene is essential for viability (29, 30) and plays a key role in central nervous system development (29). Abl controls growth cone guidance and synaptogenesis, acting downstream of the Robo receptor (31, 32) and antagonistically to tyrosine phosphatases (33, 34). Abl is also required during dendritic morphogenesis (35). These developmental functions require reorganization of actin (34, 35) and microtubule (36) structures and involve the Abl phosphorylation substrates Enabled (31, 34, 35, 37), Abi (Abl interactor protein) (38), and Cables (32). Abl has a critical role in the developmental morphogenesis of epithelial tissues, including head involution and dorsal closure (39, 40), ventral furrow formation (18), and wing construction (41). These epithelial cell functions reflect, at least in part, the contribution of Abl to actin dynamics (18, 40). C. elegans Abl controls epithelial morphogenesis (42) but, in contrast to Drosophila, a null mutation in the C. elegans Abl gene confers only conditional phenotypes. These include sensitivity of germ cells to various types of stress (43, 44), rescue of apoptotic cell engulfment defects (45), and altered Shigella flexneri pathogenesis (46).
Both vertebrate Abl1 and Abl2 are required for normal development. Mice with a disrupted Abl1 gene show defective hematopoiesis and low viability (47, 48), osteoporosis (49), decreased systolic blood pressure (50), and cardiac hyperplasia (51). Abl1 deficits also impair the development and responsiveness of B cells (52, 53) and T cells (54). Ablation of the Abl2 gene has distinct effects, leading only to relatively subtle neuronal defects (27, 55). An Abl1−/− Abl2−/− double mutation, however, causes embryonic lethality with abnormalities in neuroepithelial cells and defects in neurulation (27). Hence, Abl paralogs make unique contributions to vertebrate development while retaining substantial functional overlap.
Biochemistry and Regulation of ABL Tyrosine Kinases
Tyrosine kinase activity
An essential property of ABL proteins is their tyrosine kinase activity. Tyrosine phosphorylation can modify substrate protein activity, localization, and partnering capacity. Consequently, knowledge of ABL catalytic regulation and substrate specificity is integral to understanding ABL function.
Structural data and biochemical studies have revealed multiple autoinhibitory mechanisms that constrain the enzymatic activity of the ABL-family kinases. A myristoyl group attached to the amino terminal glycine of ABL1 and ABL2 “b” isoform proteins can nestle into a surface pocket in the kinase domain, contributing to an autoinhibitory fold (56). A short amino-terminal “Cap” sequence stabilizes the inactive conformation of ABL1 through additional surface interactions (57). Downstream of the “Cap” peptide are SH3 and SH2 domains that cradle the kinase domain, imposing a “locked” inactive state (58, 59). Tyrosine kinase activity of ABL1 is increased by disrupting these autoinhibitory interactions, as demonstrated with the Cap domain mutation in which lysine at position 7 is replaced by alanine (K7A) (56), the SH3 domain mutation W118A (60), the SH2 domain mutations E157A and Y158D (56) and the SH2-TK linker-region double mutation P242E, P249E (also known as “PP”) (60). These key residues are identical (W118, E157, Y158, P242, and P249) or conserved (K7) in the ABL2 protein (Fig. 3).
Phosphatidylinositol 4,5-bisphosphate (PIP2), a phospholipase C–generated second messenger, inhibits ABL1 and ABL2 tyrosine kinase activity (61). PIP2 binds the ABL1 SH2 domain through residues normally required for phosphotyrosine binding (62), but the mechanism of inhibition has not been determined.
Phosphorylations in the amino-terminal half of ABL proteins, including phosphorylation within the kinase domain, can substantially alter catalytic activity (see below). These amino-terminal phosphorylation sites are conserved between ABL1 and ABL2, and phosphorylation at some sites can affect the activity of oncogenic ABL fusion proteins such as BCR-ABL1.
Covalent modification of ABL proteins
Tyrosine phosphorylation of members of the ABL family of kinases occurs in trans by ABL1 and ABL2 (63, 64), a reaction often referred to as “autophosphorylation.” ABL-family kinases are also phosphorylated by members of the SRC family of tyrosine kinases (63–65) and by PDGFR (61). Tyrosine phosphorylation of ABL1-Y245 (equivalent to ABL2-Y272), which resides in the linker segment between the SH2 and kinase domains, and ABL1-Y412 (ABL2-Y439), which lies in the activation loop of the kinase domain, correlate with increased kinase activity (63, 64). Phosphorylation of ABL1-Y89 (conserved in ABL2) by members of the SRC family of kinases (66, 67) disrupts SH3 domain–based autoinhibitory interactions and intermolecular associations, such as that with ABI1, and also enhances kinase activity (66). Phosphorylation of ABL2-Y261 (conserved in ABL1) promotes ABL function through protein stabilization (68). Tyrosine phosphorylation at these same sites within the oncogenic fusion protein BCR-ABL1 correlates with its ability to transform cells (67), which demonstrates that “constitutively active” ABL mutants still respond to positive regulation. Phosphorylation of ABL1-Y272 in the kinase domain P loop inhibits ABL1 kinase activity (69) and BCR-ABL1 transforming activity (70), whereas phosphorylation of the nearby ABL1-Y276 enhances transformation and appears to promote kinase activity (70). Phosphosite entries for ABL1 and ABL2 include other phosphorylated tyrosines (Fig. 3). Possible contributions of these modifications to ABL function are not clear, although ABL1-Y158 and ABL1-Y331 contribute to intramolecular folding (56, 71), which may be altered by phosphorylation,
Several ABL phosphotyrosines are predicted SH2 domain binding sites (lilab.uwo.ca/SMALI.htm). Phosphorylated ABL1-Y89 (ABL2-Y116) and ABL1-Y276 (ABL2-Y303) match well with the preferred pYDxV binding site for NCK-family SH2 domains (72) and may contribute to the interaction of ABL1 with NCK1 (73). ABL1-pY134 (ABL2-pY161) and ABL1-pY147 (ABL2-pY174) are predicted to associate with the RASA1 [Ras GTPase–activating protein 1] SH2 domain, consistent with RASA1 binding to BCR-ABL1 (74). ABL1-pY251 is predicted to bind the BLNK (B cell linker protein) SH2 domain, whereas ABL1-pY276 (ABL2-pY303) is predicted to bind the HCK SH2 domain, but these interactions have not been experimentally validated.
ABL1 is phosphorylated on T754 (75), which resides at the position normally occupied by phosphoserine in type 1 consensus 14-3-3 binding sites: RSxpSxP (76). Phosphorylation of T754 leads to 14-3-3 binding, which favors the cytoplasmic localization of ABL1 (75). 14-3-3 proteins exist primarily as dimers, which allows them to cross-link multiple sites within a protein or to bridge two target proteins (76), but it is not yet know whether 14-3-3 dimerization contributes to ABL regulation. Phosphorylation of T754 can be carried out by the kinases CLK1, CLK4, MST1, MST2, and TTK in vitro (77) but has not been well studied in vivo. Although ABL2 has a conserved threonine at the same position as ABL1-T754 (Fig. 3), upstream elements of the 14-3-3–binding consensus sequence are absent, and there is no experimental evidence that ABL2 binds 14-3-3 proteins.
Phosphorylation of ABL1 residues S637 and S638 by PAK2 (p21-activated kinase 2) alters the surface charge environment immediately downstream of the third PxxP motif (PTPPKRSS638) and reduces binding to the SH3 domain protein and ABL inhibitor ABI1 (78). This may explain in part the stimulatory effect of PAK2 on ABL kinase activity and the negative feedback resulting from ABL-mediated PAK2 phosphorylation (79). These residues are conserved in ABL2 (Fig. 3), which suggests a similar type of regulation. For reasons less well understood, phosphorylation of these same residues increases ABL1 binding to CRK (78). Additional phosphorylations on ABL1, including that on S588, have been attributed to a CDC2-associated kinase and are coupled to cell division (80).
ABL1 is acetylated at K730 in the second NLS (81). This modification, performed by EP300 (E1A binding protein p300), promotes the cytoplasmic translocation of ABL1.
An amino-terminal myristoyl modification, likely carried out by a myristoyl-CoA:protein N-myristoyltransferase (NMT1 and NMT2 in humans), stabilizes the inactive ABL kinase conformational fold through an intramolecular interaction. Myristoylation of other proteins, including members of the SRC family of tyrosine kinases, has more typically been associated with lipid-membrane interactions that regulate protein localization (82). When the ABL1 myristoyl group is released from the kinase domain by treatment with GNF-2, an allosteric inhibitor compound that binds to the same pocket (83), ABL1 translocates to the endoplasmic reticulum (84). This result raises the possibility that physiological signals may also expose the myristoyl group on ABL proteins and trigger its subcellular relocalization.
Both ABL proteins are subject to polyubiquitination, which leads to their degradation (68, 85). The ubiquitin ligase CBL, itself an ABL substrate (86, 87), has been directly implicated in this modification (83). Some evidence suggests that ubiquitination of ABL2 may itself be regulated by phosphorylation of Y261 in response to oxidative stress (68). ABL1 is also targeted for caspase-mediated cleavage during some types of apoptosis (88).
Substrate specificity
Defining target site specificity is critical for understanding normal and leukemogenic ABL signal transduction pathways. ABL tyrosine phosphorylation can directly influence catalytic activity, SH2 domain binding, and subcellular localization of substrate proteins. Substantial progress has been made in defining ABL target phosphorylation sites, but the ability to accurately predict ABL substrates has remained elusive.
In vitro kinase reactions with peptide libraries suggest a preferred ABL target site: (L- or I- or V-) pY-x-x-P (89, 90). Analysis of 119 separately validated ABL1, ABL2, and BCR-ABL1 substrates (Table 2) confirmed a strong preference for proline at position +3 and for aliphatic amino acids (L, I, or V) at position −1, as well as revealing an enrichment for acidic residues (D or E) at positions −4, −3, and +1 (Fig. 5). Global phosphopeptide analysis of cells expressing BCR-ABL1 is generally consistent with this result (91). These target site preferences presumably reflect structural requirements of the ABL catalytic site, a presumption confirmed, at least in part, by analyses of ABL active site mutations (92).
Table 2.
Proteins reported as substrates of ABL1, ABL2, or BCR-ABL1 (nomenclature as in Table 1).
| PROTEIN | SUBSTRATE | AA | REF | ALIASES | PARALOGS | FUNCTION |
|---|---|---|---|---|---|---|
| ABI1 | PVKPPTVPNDYMTSPARLGSQ | 213 | 18328268 | Abi-1 | ABI2,3 | cytoskeleton, ABL reg |
| ABL1 | ENDPNLFVALYDFVASGDNTL | 89 | 18775435 | c-Abl | ABL2 | tyrosine kinase |
| ABL1 | PAPKRNKPTVYGVSPNYDKWE | 245 | 12748290 | c-Abl | ABL2 | tyrosine kinase |
| ABL1 | GLSRLMTGDTYTAHAGAKFPI | 412 | 12748290 | c-Abl | ABL2 | tyrosine kinase |
| ABL2 | VADGLVTTLHYPAPKCNKPTV | 261 | 15735735 | Arg | ABL1 | tyrosine kinase |
| ABL2 | PAPKCNKPTVYGVSPIHDKWE | 272 | 12748290 | Arg | ABL1 | tyrosine kinase |
| ABL2 | GLSRLMTGDTYTAHAGAKFPI | 439 | 12748290 | Arg | ABL1 | tyrosine kinase |
| ABL2 | RAASSSSVVPYLPRLPILPSK | 568 | 12748290 | Arg | ABL1 | tyrosine kinase |
| ABL2 | REMENQPHKKYELTGNFSSVA | 683 | 12748290 | Arg | ABL1 | tyrosine kinase |
| ANXA1 | AWFIENEEQEYVQTVKSSKGG | 21 | 2457390 | lipocortin | ANXA2-12,13 | signaling, endocytosis |
| APBB1 | NELVQKFQVYYLGNVPVAKPV | 547 | 15031292 | Fe65 | APBB2,3 | transcription adaptor |
| APP | RHLSKMQQNGYENPTYKFFEQ | 682 | 11279131 | amyloid pp | AD1 | unclear (signaling?) |
| BCAR1 | HLLAPGPQDIYDVPPVRGLLP | 267 | 7780740 | p130CAS | NEDD9, EFS | signal adaptor |
| BCR | QPGADAEKPFYVNVEFHHERG | 177 | 8112292 | BCR1 | ABR | GAP for RAC, signaling |
| BCR | PLEYQPYQSIYVGGMMEGEGK | 283 | 8622703 | BCR1 | ABR | GAP for RAC, signaling |
| BCR | SSRVSPSPTTYRMFRDKSRSP | 360 | 8622703 | BCR1 | ABR | GAP for RAC, signaling |
| BTK | TSELKKVVALYDYMPMNANDL | 223 | 12445832 | AGMX1, XLA | BMX | tyrosine kinase |
| CABLES1 | 10896159 | ik3-1 | CABLES2 | signal adaptor, ccyc | ||
| CASP9 | ESLRGNADLAYILSMEPCGHC | 153 | 15657060 | CASP2,8,10 | protease, apoptosis | |
| CAT | KLVNANGEAVYCKFHYKTDQG | 231 | 12777400 | catalase | catalase | |
| CAT | NCPYRARVANYQRDGPMCMQD | 386 | 12777400 | catalase | catalase | |
| CAV1 | GKYVDSEGHLYTVPIREQGNI | 14 | 12531427 | caveolin, CAV | CAV2,3 | microdomains, endocyt. |
| CBL | EQCEGEEDTEYMTPSSRPLRP | 700 | 15556646 | CBL2 | CBLB, C | ubiquitin lig, endocyt. |
| CBL | PEESENEDDGYDVPKPPVPAV | 774 | 8649859 | CBL2 | CBLB, C | ubiquitin lig, endocyt. |
| CD19 | 11120811 | BCR co-receptor | ||||
| CDK5 | EKLEKIGEGTYGTVFKAKNRE | 15 | 10896159 | PSSALRE | CDK2,3, CDC2 | S/T kinase, ccyc, cytosk |
| CDKN1B# | EVEKGSLPEFYYRPPRPPKGA | 88 | 17254966 | KIP1, p27 | CDKN1A, C | kinase inhib, ccyc |
| CDV3* | WKEFEQREVDYSGLRVQAMQI | 120 | 12606058 | H41, TPP36 | unknown | |
| CHRNB1 | 15340048 | nic Ach recpt | CHRND, E, G | ion channel, recpt | ||
| CRK | QPLGGPEPGPYAQPSVNTPLP | 221 | 8194526 | CRKII | CRKL | signal adaptor |
| CRKL | SYGIPEPAHAYAQPQTTTPLP | 207 | 9710592 | CRK | signal adaptor | |
| CTNNB1 | EMAQNAVRLHYGLPVVVKLLH | 489 | 16099633 | Catenin-beta | CTNNBL1 | signaling, transcript. |
| CTNND1 | 11891774 | Catenin-delta | CTNND2, ARVCF | signaling, adhesion | ||
| CTTN* | IEDRPPSSPIYEDAAPFKAEP | 421 | 17306540 | cortactin | HCLS1 | signal adaptor, cytosk. |
| CTTN* | QGLTYTSEPVYETTEAPGHYQ | 466 | 17306540 | cortactin | HCLS1 | signal adaptor, cytosk. |
| CTTN* | PGHYQAEDDTYDGYESDLGIT | 482 | 17306540 | cortactin | HCLS1 | signal adaptor, cytosk. |
| DBN1 | 17892306 | drebrin 1 | cytosk. (postsynaptic) | |||
| DDB1 | 12107171 | XPE | DNA repair, cplx w/DDB2 | |||
| DDB2 | 12107171 | UV-DDB2 | DNA repair, cplx w/DDB1 | |||
| DNM2 | 19833721 | DYN2 | DNM1,3 | GTPase, endocyt. | ||
| DOK1# | QELLDSPPALYAEPLDSLRIA | 296 | 16497976 | p62Dok | DOK2-6 | signal adaptor |
| DOK1# | IAPCPSQDSLYSDPLDSTSAQ | 315 | 16497976 | p62Dok | DOK2-6 | signal adaptor |
| DOK1 | KLTDPKEDPIYDEPEGLAPVP | 362 | 15148308 | p62Dok | DOK2-6 | signal adaptor |
| DOK1# | WCQARVKEEGYELPYNPATDD | 398 | 16497976 | p62Dok | DOK2-6 | signal adaptor |
| DOK1# | ELPYNPATDDYAVPPPRSTKP | 409 | 16497976 | p62Dok | DOK2-6 | signal adaptor |
| DOK1# | SGIKSHNSALYSQVQKSGASG | 449 | 16497976 | p62Dok | DOK2-6 | signal adaptor |
| DOK2# | APRPRGQEGEYAVPFDAVARS | 299 | 16858728 | p56dok-2 | DOK1,3-6 | signal adaptor |
| EGFR | FKGSTAENAEYLRVAPQSSEF | 1173 | 16943190 | ERBB1 | ERBB2-4 | tyrosine kinase (recpt) |
| ENAH | PPPPLPSGPAYASALPPPPGP | 296 | 12672821 | MENA | VASP, EVL | cytosk., actin pol. |
| ENO1 | AVPSGASTGIYEALELRDNDK | 44 | 6330085 | Enolase | ENO2,3 | metabolism |
| EPHB2 | 11494128 | Tyro5 | EPHB1,3,4,6 | tyrosine kinase (recpt) | ||
| ERCC3 | 9874796 | XPB, TFIIH | DNA repair | |||
| ERCC6 | NLTGANRVVIYDPDWNPSTDT | 932 | 17626041 | CKN2, CSP | ERCC6L | DNA repair |
| ESR1 | VYLDSSKPAVYNYPEGAAYEF | 52 | 20101225 | Era | ESR2 | steroid recpt |
| ESR1 | FKRSIQGHNDYMCPATNQCTI | 219 | 20101225 | Era | ESR2 | steroid recpt |
| GNB2L1 | MWKLTRDETNYGIPQRALRGH | 52 | 19423701 | RACK1, HLC-7 | GNB2 | signal scaffold |
| GPX1 | KNEEILNSLKYVRPGGGFEPN | 96 | 12893824 | GPX2-8 | enzyme, antioxidant | |
| GRB2 | HGQTGMFPRNYVTPVNRNV | 299 | 11726515 | NCKAP2 | GRB7,10,14 | signal adaptor |
| GRLF1* | VKPRNEEENIYSVPHDSTQGK | 1105 | 15084284 | p190RhoGAP | ARHGAP5 | GAP for RHO, cytosk. |
| HCLS1 | 18305217 | HS1 | CTTN | signal adaptor, cytosk. | ||
| HSPA1A | 17892306 | HSP70 | HSPA1B,1L | stress response | ||
| INPPL1 | QMAKTLSEVDYAPAGPARSAL | 1135 | 16858728 | SHIP2 | INPP5D | PtdInsP phosphatase |
| IRS1 | 12560071 | HIRS-1 | IRS2,3L,4 | signaling | ||
| JAK2 | LTKVLPQDKEYYKVKEPGESP | 1007 | 11593427 | JTK10 | JAK1,3, TYK2 | tyrosine kinase |
| JUN | SASLHSEPPVYANLSNFNPGA | 170 | 10637231 | c-Jun | JUNB, JUND | transcription |
| LGALS3 | AYPGAPAPGVYPGPPSGPGAY | 79 | 20600357 | GALIG, MAC-2 | LGALS1-14 | signaling lectin |
| LGALS3 | ATGAYPATGPYGAPAGPLIVP | 107 | 20600357 | GALIG, MAC-2 | LGALS1-14 | signaling lectin |
| LGALS3 | GAPAGPLIVPYNLPLPGGVVP | 118 | 20600357 | GALIG, MAC-2 | LGALS1-14 | signaling lectin |
| LASP1 | PHHIPTSAPVYQQPQQQPVAQ | 171 | 15138294 | MLN50 | NEBL | cytoskeleton, migration |
| MAP3K1 | 10866655 | MEKK1 | MAP3K2-15 | S/T kinase | ||
| MAPT | AKTDHGAEIVYKSPVVSGDTS | 305 | 16014719 | tau | cytosk., microtub. | |
| MDM2 | QELSDEDDEVYQVTVYQAGES | 276 | 16702947 | Hdm2 | MDM4 | DNA damage/apop/signal |
| MDM2 | QASQSQESEDYSQPSTSSSII | 394 | 12110584 | Hdm2 | MDM4 | DNA damage/apop/signal |
| MDM4 | EMFTVKEVMHYLGQYIMVKQL | 55 | 19075013 | Mdmx | MDM2 | DNA damage/apop/signal |
| MDM4 | SFSVKNPSPLYDMLRKNLVTL | 99 | 19075013 | Mdmx | MDM2 | DNA damage/apop/signal |
| MSH5 | 16397227 | MutS hom 5 | MSH2-4,6 | DNA repair | ||
| MUC1 | SAGNGGSSLSYTNPAVAATSA | 463 | 16888623 | PUM, Y60 | barrier funct., signal | |
| MYOD1* | LCSFETADDFYDDPCFDSPDL | 30 | 12415271 | MYF3, PUM | transcription | |
| MYOD1* | RSNCSDGMMDYSGPPSGPRRQ | 213 | 12415271 | MYF3, PUM | transcription | |
| MYH10 | 17892306 | MyosinIIB | MYH9,11,14 | motor prot., cytosk. | ||
| NCK1 | 11494134 | NCK2 | signal adaptor | |||
| NCOA3 | MMQHPQAASIYQSSEMKGWPS | 1357 | 18765637 | AIB1, SRC-3 | NCOA1, 2 | steroid recpt co-act. |
| NEDD9 | TPVRTGHGYVYEYPSRYQKDV | 166 | 8668148 | HEF1, Cas-L | BCAR1, EFS | signal adaptor |
| NFKBIA | FTEFTEDELPYDDCVFGGQRL | 305 | 12167702 | IkBa | NFKBIB | signaling, NFKB inhib. |
| NFKBIB | PRRPREAPDTYLAQGPDRTPD | 161 | 16286457 | IkBb | NFKBIA | signaling, NFKB inhib. |
| OGDH | 17892306 | 2oxoglut dhg | OGDHL, DHTKD1 | metabolism | ||
| PAK2 | 11121037 | PAKgamma | PAK1,3 | S/T kinase | ||
| PDGFRB | PIYIITEYCRYGDLVDYLHRN | 686 | 14993293 | PDGFR | PDGFRA | tyrosine kinase (recpt) |
| PDGFRB | AQPAHASDEIYEIMQKCWEEK | 934 | 14993293 | PDGFR | PDGFRA | tyrosine kinase (recpt) |
| PDGFRB | RLLGEGYKKKYQQVDEEFLRS | 970 | 14993293 | PDGFR | PDGFRA | tyrosine kinase (recpt) |
| PIK3AP1 | QCHLGQEEDVYHTVDDDEAFS | 513 | 15893754 | BCAP | signal adaptor (BCR) | |
| PIK3AP1 | GAHQLPDNEPYIFKVFAEKSQ | 553 | 15893754 | BCAP | signal adaptor (BCR) | |
| PIK3AP1 | EKSQERPGNFYVSSESIRKGP | 570 | 15893754 | BCAP | signal adaptor (BCR) | |
| PIK3AP1 | PWRDRPQSSIYDPFAGMKTPG | 594 | 15893754 | BCAP | signal adaptor (BCR) | |
| PIK3AP1 | HLPAKVEFGVYESGPRKSVIP | 694 | 15893754 | BCAP | signal adaptor (BCR) | |
| PLCG1 | LEKIGTAEPDYGALYEGRNPG | 771 | 12652307 | PLCg-1 | PLCG2 | phospholipase |
| PLCG1 | NKAKGKKFLQYNRLQLSRIYP | 1003 | 12652307 | PLCg-1 | PLCG2 | phospholipase |
| PLSCR1 | AGFPVPNQPVYNQPVYNQPVG | 69 | 11390389 | MMTRA1B | PLSCR2-5 | phospholipid scrablase |
| PLSCR1 | PNQPVYNQPVYNQPVGAAGVP | 74 | 11390389 | MMTRA1B | PLSCR2-5 | phospholipid scrablase |
| POLR2A | SPSYSPT repeat | CTD | 7504297 | POLR2 | POLR2B-L | RNA polym., transcript. |
| PRKCD | KLDTTESVGIYQGFEKKPEVS | 311 | 17126298 | PKC-δ | PRKCE, H, Q | S/T kinase |
| PRKD1 | LFQNETGSRYYKEIPLSEILR | 463 | 12637538 | PKD, PKCμ | PRKD2,3 | S/T kinase |
| PRKDC | 9109492 | DNA-PK | S/T kinase, DNA reapir | |||
| PSMA7 | RLYQTDPSGTYHAWKANAIGR | 153 | 16678104 | C6, XAPC7 | PSMA8 | proteasome component |
| PSTPIP1 | TPTPERNEGVYTAIAVQEIQG | 345 | 11163214 | H-PIP, PAPAS | PSTPIP2 | signal adaptor |
| PTPN6 | LQSQKGQESEYGNITYPPAMK | 536 | 8692915 | SHPTP1 | PTPN11 | tyrosine phosphatase |
| PTPN6 | RTSSKHKEDVYENLHTKNKRE | 564 | 8692915 | SHPTP1 | PTPN11 | tyrosine phosphatase |
| PTPN11 | THIKIQNTGDYYDLYGGEKFA | 62 | 8195176 | SHPTP2 | PTPN6 | tyrosine phosphatase |
| PTPN11 | AEMREDSARVYENVGLMQQQK | 580 | 18827006 | SHPTP2 | PTPN6 | tyrosine phosphatase |
| PXN | RPVFLSEETPYSYPTGNHTYQ | 31 | 11695992 | paxillin | cytoskeleton, actin reg | |
| PXN | CSRVGEEEHVYSFPNKQKSAE | 118 | 11695992 | paxillin | cytoskeleton, actin reg | |
| RAD9A | HSLSRIGDELYLEPLEDGLSL | 28 | 11971963 | RAD9 | RAD9B | DNA repair |
| RAD51 | AGFHTVEAVAYAPKKELINIK | 54 | 9461559 | RECA | DNA repair | |
| RAD51 | GRGETRICKIYDSPCLPEAEA | 315 | 11684015 | RECA | DNA repair | |
| RAD52 | DFVDLNNGKFYVGVCAFVRVQ | 104 | 12379650 | DNA repair | ||
| RAN | VFHRKKNLQYYDISAKSYNNF | 147 | 11420673 | TC4 | GTPase, nucl. transport | |
| RAPGEF1 | QSTAPIPSVPYAPFAAILPFQ | 504 | 20581864 | C3G | RAPGEF2-6 | signaling, GEF for RAP |
| RAPH1 | RYFLLRASGIYYVPKGKAKVS | 426 | 20417104 | lamellipodin | cytoskeleton, actin reg | |
| RAPH1 | DHVNVYYGQDYRNKYKAPTDY | 456 | 20417104 | lamellipodin | cytoskeleton, actin reg | |
| RAPH1 | AGYGGSHISGYATLRRGPPPA | 1226 | 20417104 | lamellipodin | cytoskeleton, actin reg | |
| RB1 | SPLRIPGGNIYISPLKSPYKI | 805 | 16158058 | RB | transcript., ccyc | |
| RIN1 | AREKPAQDPLYDVPNASGGQA | 36 | 15886098 | RIN2,3 | RAS eff, RAB & ABL activ | |
| ROBO1 | TNLMLPESTVYGDVDLSNKIN | 1038 | 17618275 | ROBO2-4 | axon guidance recpt | |
| ROBO1 | FVNPSGQPTPYATTQLIQSNL | 1073 | 17618275 | ROBO2-4 | axon guidance recpt | |
| ROBO1 | QQKQEVAPVQYNIVEQNKLNK | 1114 | 17618275 | ROBO2-4 | axon guidance recpt | |
| SHC1 | EEEEPPDHQYYNDFPGKEPPL | 350 | 1623525 | SHC, p66 | SHC2-4 | signal adaptor |
| SHD | 9315092 | signal adaptor | ||||
| SHE | 9315092 | Shg | signal adaptor | |||
| SIVA1 | ELSRGVCAERYSQEVFEKTKR | 34 | 11278261 | CD27BP | signaling, apop. | |
| SORBS1* | RKYRAEPKSIYEYQPGKSSVL | 326 | 19891780 | CAP, ponsin | SORBS2,3 | signal adaptor |
| SORBS1* | DITSEPPGYIYSSNFHAVKRE | 360 | 19891780 | CAP, ponsin | SORBS2,3 | signal adaptor |
| SORBS1* | SLDLCSYQALYSYVPQNDDEL | 632 | 19891780 | CAP, ponsin | SORBS2,3 | signal adaptor |
| SORBS2 | 9211900 | ArgBP2 | SORBS1,3 | signal adaptor | ||
| SORBS3* | DGSLNPDPAWYQTWPGPGSRP | 127 | 16831423 | vinexin | SORBS1,2 | signal adaptor |
| SOS1 | 15039778 | SOS2 | GEF for H/K/NRAS | |||
| STAT1 | 8940193 | STAT91 | STAT2-4 | signaling, transcript. | ||
| STAT3 | PEADPGSAAPYLKTKFICVTP | 705 | 8940193 | APRF | STAT1,2,4 | signaling, transcript. |
| STAT5A | TPVLAKAVDGYVKPQIKQVVP | 694 | 8940193 | MGF | STAT5B,6 | signaling, transcript. |
| STAT5B | GFLLKIKLGHYATQLQNTYDR | 90 | 8940193 | STAT5A,6 | signaling, transcript. | |
| STAT6 | 8940193 | IL-4-STAT | STAT5A,B | signaling, transcript. | ||
| STXBP1 | 17892306 | MUNC18-1 | STXBP2,3 | trafficking, exocytosis | ||
| SYNJ2 | 15659545 | synaptojanin2 | SYNJ1 | PtdInsP phosph, endocyt | ||
| TERT | 10837221 | TRT, TP2 | telomerase RTase | |||
| TOP1 | TFFAKMLDHEYTTKEIFRKNF | 268 | 15448168 | TOP1MT | topoisom, DNA repl/rep | |
| TP73 | HAASVPTHSPYAQPSSTFDTM | 99 | 10391251 | p73 | TP63, TP53 | transcription; apop. |
| TRIP10 | 19631450 | CIP4 | FNBP1,FNBP1L | signal adaptor, endocyt | ||
| TUB | 10455176 | tubby | TULP1-4 | transcript., signaling | ||
| TUBA1 | 17892306 | alpha-tubulin | TUBA1A-C | cytoskeleton, microtub. | ||
| TUBB | 17892306 | beta-tubulin | TUBB1 | cytoskeleton, microtub. | ||
| WASF2 | RDDGKEALKFYTDPSYFFDLW | 150 | 15657136 | WAVE2 | WASF1,3,4 | cytoskeleton, actin reg |
| WASF3 | RDDKKDGLKFYTDPSYFFDLW | 151 | 17623672 | WAVE3 | WASF1,2,4 | cytoskeleton, actin reg |
| WASF3 | TRSHASDVTDYSYPATPNHSL | 248 | 17623672 | WAVE3 | WASF1,2,4 | cytoskeleton, actin reg |
| WASF3 | YGMLPAQIIEYYNPSGPPPPP | 337 | 17623672 | WAVE3 | WASF1,2,4 | cytoskeleton, actin reg |
| WASF3 | TILSRRIAVEYSDSDDDSEFD | 486 | 17623672 | WAVE3 | WASF1,2,4 | cytoskeleton, actin reg |
| WASL | KNPEITTNRFYGPQVNNISHT | 175 | 16199863 | N-WASP | WAS | cytoskeleton, actin reg |
| WASL | LKDRETSKVIYDFIEKTGGVE | 256 | 16199863 | N-WASP | WAS | cytoskeleton, actin reg |
| WRN | 12944467 | RECQ3 | DNA repair | |||
| YAP1 | STDSGLSMSSYSVPRTPDDFL | 357 | 18280240 | YES-AP1 | transcript., apop. | |
| YTHDC1 | 15175272 | YT521 | YTHDC2 | splicing factor | ||
| ZDHHC16 | 12021275 | APH2 | signaling, apoptosis |
“Substrate” shows phosphorylated tyrosine (bold black) and 10 residues flanking each side. Also highlighted: P (+1 to +5, red); D and E (−5 to +5, green) and I, L, and V (−1, blue). No sequence appears if a target has not been mapped. “AA” gives the position of target Y;
indicates targets shown for BCR-ABL1 only;
indicates sites reported for mouse or rat ortholog, but conserved in human.
“Ref” provides literature citations as PMID numbers.
Fig. 5.

ABL target site consensus. (Top) Sequence logo was created from 119 phosphorylation sites in Table 2 using Weblogo (249) at weblogo.berkeley.edu. The line at position zero represents the phosphorylated tyrosine. (Bottom) Amino acid position weight matrix generated using Python. Positions contributing most heavily to the consensus target site, as described in the text, are shown in bold.
There are caveats to identification of phosphorylation targets on the basis of consensus-derived sequences. First, peptide substrates may not accurately mimic protein substrates. Second, protein substrates identified in cells may be indirect targets, in other words, they may be substrates for kinases activated downstream of ABL1, ABL2, or BCR-ABL1. Only a few direct ABL targets have been experimentally determined using mutations that render the kinase dependent on an ATP analog (93) (94).
The SH2 domain contributes to ABL catalytic activity (71) and target site specificity (15). When the ABL SH2 domain is replaced with the SH2 domain from another protein, there is a shift in substrate profile (95). Moreover, SH2 binding preference correlates with target site preference, and juxtaposition of an appropriate SH2 ligand site enhances phosphorylation of an ABL target site. This result suggests a processive phosphorylation mechanism, that is to say, successive phosphorylation of different tyrosines on the same substrate (96, 97). Processivity would also explain why ABL-mediated phosphorylation shows accelerated, autocatalytic kinetics when substrate peptides are surface bound at high density (98). In this model, a newly phosphorylated tyrosine moves from the ABL catalytic site to the SH2 pocket, positioning another tyrosine residue for efficient phosphorylation.
Recent work in the field of histone modification has uncovered proteins such as Clr4 that contain both a catalytic site (histone modification “writer”) and a separate binding site (histone modification “reader”) that coordinate processive histone modification (99, 100). By analogy, the combination of a tyrosine kinase domain (pY “writer”) with an SH2 domain (pY “reader”) in ABL and other nonreceptor tyrosine kinases may have evolved in part to increase phosphorylation efficiency of multitarget proteins or complexes. It may also facilitate the phosphorylation of “poor” target sites by docking them into the catalytic pocket. A corollary of the processivity model is that consensus ABL kinase targets based on a database of known phosphorylation sites, may over-represent amino acids that contribute little to substrate affinity (Km) but facilitate SH2 binding.
Processive phosphorylation is probably integral to ABL function. Multiple target sites exist in many ABL substrates, including BCAR1 (96), CAT (101), CBL (86), DOK1 (102), GAB2 (103), CTTN (104), MDM2 (105), PIK3AP1 (106), PLCG1 (107), PTPN11 (108), PXN (109), POLR2A (RNA polymerase) (110), and RAD51 (111). Indeed, the ABL SH2 domain is essential for efficient phosphorylation of the 52 target sites in the repetitive carboxy-terminal domain of POLR2A (112). In addition, the list of known ABL substrates has many adaptor proteins (including CRK, CRKL, DOK1, GRB2, and NCK1) that, subsequent to phosphorylation, might deliver their associated proteins to the ABL catalytic site after docking at the ABL-SH2 domain.
This simple processive phosphorylation model implies that the ABL catalytic site and SH2 pocket have coevolved to recognize the same sequences (in other words, the best kinase target sequences, once phosphorylated, would also be the best SH2-binding targets). However, some targets within a multitarget substrate appear to require the prior phosphorylation of another target site. RAD51, an established ABL substrate (113, 114) has two ABL target sites: AY54APK (poor consensus) and IY315DSP (good consensus). In transfected cells, a RAD51Y54F mutant was efficiently phosphorylated on Y315, but a RAD51Y315F mutant had no detectable Y54 phosphorylation (115). This suggests that Y315 phosphorylation must occur first, followed by reorientation of the RAD51 substrate so that pY315 is bound to the ABL-SH2 domain and Y54 is positioned at the ABL catalytic site.
The processive phosphorylation data and RAD51 results suggest a “hierarchical processivity” model in which the substrate target site most compatible with ABL kinase domain preferences is phosphorylated with greatest efficiency. If this site is also compatible with the ABL SH2 domain specificity, it will then reposition and dock in the SH2 pocket. This mechanism could enable ABL kinases to phosphorylate otherwise poor targets on the same substrate if they are properly positioned (Fig. 6A). By extension, relatively poor substrate proteins might be recruited to ABL through a complex with strong substrates that can also dock with the SH2 pocket (Fig. 6B). A corollary to the hierarchical processivity model is that consensus ABL phosphorylation sites determined by statistical analysis may actually represent an average of “primary” sites, with high specificity for the catalytic pocket, and “secondary” sites that require the prior SH2 docking of an associated primary site to guide them into the catalytic site.
Fig. 6.

Hierarchical processivity model. (A) The catalytically active conformation of ABL1 is depicted with SH3, SH2, and TK domains labeled (carboxy-terminal domains not shown). A “primary,” consensus, tyrosine target (1) is phosphorylated, then relocated to the SH2 domain. This guides a “secondary,” nonconsensus, tyrosine (2) into the catalytic site. (B) Same steps as in (A) except that the secondary tyrosine is in a separate protein associated with the initial substrate.
Target-site phosphorylation of some ABL substrates requires an escort for delivery to the ABL catalytic site, independent of processive phosphorylation. Enah (also known as Mena) phosphorylation at the target site AY296ASA is enhanced by Abi1, which binds to both Enah and Abl1 (116). Similarly, CABLES enhances ABL-mediated tyrosine phosphorylation of CDK5 (117) and CTNNB1 (β-catenin) (32) by directly linking kinase and substrate. This use of adaptor proteins to recruit targets further extends the reach of ABL kinases beyond the restrictive preferences of its catalytic site and SH2 domain.
In summary, ABL substrate specificity is driven by both target sequence and domain-guided protein-protein interactions. The role of other determinants is suggested by quantitative shifts in substrate phosphorylation patterns among point mutation variants of BCR-ABL1 (70).
Regulation through tyrosine phosphatases
Given the importance of tyrosine phosphorylation in ABL activation and downstream signaling, the regulatory role of protein tyrosine phosphatases (PTPs) is perhaps not surprising. BCR-ABL1 transforming properties are blocked by PTPN1 (118) and by a variant form of PTPRO (119), which suggests that some PTPs function as negative regulators by reversing ABL-mediated tyrosine phosphorylation. In response to ionizing radiation treatment, ABL1 phosphorylates PTPN6 (also known as SHP-1), which binds to the SH3 domain of ABL1, on Tyr536 and Tyr564 (120). PTPN6 is also associated with BCR-ABL1, and PTPN6 overexpression diminishes some transformation-associated phenotypes of the CML-derived cell line K562 (121). PTPN11 (SHP-2) is an ABL1 substrate that facilitates growth factor–induced mitogenic responses (122). However, delivery of the PTPN11 phosphatase domain to BCR-ABL1 through an ABL-binding domain causes a transformation block (123), which indicates that this phosphatase can also inhibit ABL-mediated tyrosine phosphorylation signaling. PTPN12 (PTP-PEST) and PTPN18 (PTP-HSCF) are recruited by the PSTPIP1 adaptor to dephosphorylated ABL and suppress its function (124).
Interaction Partners That Regulate ABL Activity and Function
ABL self-association
Any discussion of ABL protein partners must consider self-association. Wild-type ABL proteins in cultured cells can exist as oligomers, the formation of which requires their kinase activity and an intact amino-terminal domain (125). Chemically induced dimerization of ABL1 increases both its tyrosine kinase activity and transformation capacity (126), and homophilic interaction domains encoded by translocation partners markedly enhance the catalytic activity and leukemogenic potential of ABL fusion proteins such as BCR-ABL1 and ETV6-ABL1 (discussed below). These data suggest that close proximity of partners within a dimer or oligomer enhances transphosphorylation that directly activates ABL kinases. ABL1-ABL2 heterodimers can form in response to oxidative stress and mediate apoptosis (127), which suggests functional cooperativity between ABL-family members.
Cytoskeleton components
The capacity to directly bind cytoskeletal elements is a defining characteristic of ABL proteins (Fig. 1). A conserved calponin homology (CH)–type F-actin–binding domain is located at the C terminus of ABL proteins, and ABL1 also has G-actin binding properties (128). An (I or L)WEQ (talinlike) domain, found in ABL2 only, provides additional F-actin binding properties (129). These domains mediate the formation of F-actin bundles (128, 129), which are critical for membrane protrusions. Binding to filamentous actin inhibits ABL kinase activity (130), which suggests regulatory feedback. The microtubule-binding domain of ABL2 (131) provides an additional capability for remodeling the cytoskeleton through cross-association with both thick and thin filaments.
A common signal transduction theme among ABL partners
Numerous ABL1 and ABL2 partners have been identified (Table 1). Many of these are ABL substrates, but their association with ABL is stable enough to be detected. The ABL SH2 domain may engage phosphotyrosines on a partner or, conversely, a partner SH2 domain may bind a phosphotyrosine on ABL. Similarly, the ABL SH3 domain engages PxxP motifs on partner proteins, whereas some partner SH3 domains bind to ABL PxxP motifs. Other binding motifs are also employed, and in some cases the mode of interaction is unknown.
Table 1.
ABL-interacting proteins. Human protein names (www.genenames.org) are used throughout, although in some cases the murine gene and protein was reported.
| PROTEIN | DOMAIN | REF | SUB | ALIASES | PARALOGS | FUNCTION COMMENTS |
|---|---|---|---|---|---|---|
| ABI1 | SH3, PxxP | 7590237 | yes | NAP1BP | ABI2,3 | cytoskeleton, ABL kinase reg |
| ABI2 | SH3, PxxP | 7590236 | AblBP3 | ABI1,3 | cytoskeleton, ABL kinase reg | |
| ACR | SH3 | 16446784 | acrosin | protease | ||
| APBB1 | PR | 11279131 | yes | Fe65 | APBB2,3 | transcription adaptor |
| ATM | SH3 | 9168117 | TEL1, ATC | ATR | DNA repair, damage response | |
| ATR | unknown | 15050919 | MEC1, SCKL | ATM | DNA repair, damage response | |
| BCR | SH2 | 1383690 | ABR | signaling, GAP for RAC | ||
| BIN1 | C-term | 9356459 | AMPHL, ALP1 | cytoskeleton, endocytosis | ||
| BRCA1 | SH3 | 12024016 | BRCC1 | DNA repair, damage response | ||
| BTK | unknown | 12445832 | yes | AGMX1, XLA | BMX | tyrosine kinase |
| CABLES1 | SH3+ | 10896159 | yes | ik3-1 | CABLES2 | signal adaptor, cell cycle |
| CABLES2 | unknown | 11955625 | ik3-2 | CABLES1 | signal adaptor, cell cycle | |
| CASP9 | SH3 | 15657060 | yes | CASP2,8,10 | protease, apoptosis | |
| CAV1 | unknown | 16151024 | yes | caveolin | CAV2,3 | membrane protein, endocytosis |
| CBL | SH2, SH3 | 11494134 | yes | CBL2 | CBLB, C | ubiquitin ligase, endocytosis |
| CD19 | unknown | 11120811 | yes | BCR co-receptor | ||
| CDON | SH3 | 19470755 | CDO | BOC | Ig superfamily recpt, adhesion | |
| CPSF6 | SH3 | 8943360 | AAP1, CFIM | cleavage & polyA factor | ||
| CREB1 | SH2, TK | 7565761 | CREB | ATF1, CREM | transcription, cAMP response | |
| CRK | PxxP | 7926767 | yes | CRKL | signal adaptor | |
| CRKL | (PxxP) | 8083188 | yes | CRK | signal adaptor | |
| CTNND1 | SH3 | 11891774 | yes | catenin δ | CTNND2, ARVCF | adhesion, signaling |
| DAB1 | SH2 | 9009273 | disabled | DAB2 | signal adaptor | |
| DDB1 | TK | 12107171 | yes | XPE | DNA repair, complex w/DDB2 | |
| DOK1 | pY | 10567556 | yes | p62Dok | DOK2,3-7 | signal adaptor |
| DOK2 | SH3 | 12777393 | yes | p56Dok | DOK2,3-7 | signal adaptor |
| DOK3 | pY | 10567556 | yes | DOKL | DOK1,2,4-7 | signal adaptor |
| EGFR | SH2 | 7678409 | yes | ERBB1 | ERBB2-4 | tyrosine kinase (receptor type) |
| ENAH | SH3 | 16446784 | yes | MENA | VASP, EVL | cytoskeleton, actin polym |
| EPHB2 | SH2+Cter | 11494128 | yes | Tyro5 | EPHB1,3,4,6 | tyrosine kinase (receptor type) |
| ERBB2 | SH2 | 19275932 | neu, HER2 | EGFR, ERBB3,4 | tyrosine kinase (receptor type) | |
| ERCC6 | SH3 | 17626041 | yes | CSP, CKN2 | ERCC6L | DNA repair |
| ESR1 | SH3 | 20101225 | yes | Era | ESR2 | steroid recpt, transcript. |
| EVL | SH3 | 16446784 | RNB6 | ENAH, VASP | cytoskeleton, actin polym | |
| GPX1 | SH3 | 12893824 | yes | GPX2-8 | glutathione peroxidase | |
| GRB2 | PxxP | 7926767 | yes | NCKAP2 | GRB7,10,14 | signal adaptor |
| GRIN2D | SH3 | 10777567 | NR2D | GRIN2A-C | NMDA recpt., ABL kinase inhib | |
| HCLS1 | SH2 | 18305217 | yes | HS1 | CTTN | cytoskeleton, actin regulator |
| HDAC4 | SH3 | 16446784 | HD4 | HDAC5,7,9 | deacetylase | |
| INPPL1 | SH3 | 10194451 | yes | SHIP2 | INPP5D | phosphoinositide phosphatase |
| JAK1 | C-term | 9774693 | JTK3 | JAK2,3, TYK2 | tyrosine kinase | |
| JAK2 | C-term | 11593427 | yes | JTK10 | JAK1,3, TYK2 | tyrosine kinase |
| JUN | SH2 | 10637231 | yes | AP-1 | JUNB, JUND | transcription |
| LASP1 | SH3 | 15138294 | yes | MLN50 | NEBL | cytoskeleton, cell migration |
| LRRN2 | SH3 | 16446784 | LRRN5, GAC1 | LRRN1,3,4 | leucine rich repeat, signaling | |
| MAP3K1 | SH3 | 10866655 | yes | MEKK1 | MAP3K2-15 | MAPK (JNK) activation, signaling |
| MAP4K1 | SH3 | 9003777 | HPK1 | MAP4K2-5 | MAPK (JNK) activation, signaling | |
| MAP4K5 | unknown | 9949177 | KHS, GCKR | MAP4K1-4 | MAPK (JNK) activation, signaling | |
| MAPT | unknown | 16014719 | yes | tau | cytoskeleton, microtubule dynamics | |
| MDM2 | unknown | 12110584 | yes | MDM4 | DNA damage/apoptosis/signaling | |
| MDM4 | unknown | 19075013 | yes | MDMX | MDM2 | DNA damage/apoptosis/signaling |
| MSH5 | SH3 | 16397227 | yes | MutS hom5 | MSH2-4,6 | DNA repair |
| MUC1 | SH2 | 16888623 | yes | PUM, PEM | barrier function, signaling | |
| MUSK | unknown | 12796783 | yes | tyrosine kinase (receptor type) | ||
| NCK1 | PxxP | 7926767 | yes | NCKalpha | NCK2 | signal adaptor |
| NCK2 | unknown | 10887126 | yes | GRB4, NCKβ | NCK1 | signal adaptor |
| NEDD9 | unknown | 8668148 | yes | HEF1, CAS-L | BCAR1, EFS | signal adaptor |
| NFKBIA | SH2+ | 12167702 | yes | IKBA, NFKBI | NFKBIB, D, E,Z | signaling, NFKB inhibitor |
| NTRK1 | unknown | 10679771 | yes | TrkA | NTRK2,3 | neurotrophic RTK (also 10708759) |
| PAK2 | unknown | 11121037 | yes | PAKgamma | PAK1,3 | S/T kinase |
| PDE4D | SH3 | 10571082 | DPDE3 | PDE4A-C | cAMP phosphodiesterase | |
| PDGFRB | SH2 | 19275932 | yes | PDGFR | PDGFRA | tyrosine kinase (receptor type) |
| PIK3R1 | pYXXM | 8781408 | p85 | PIK3R2-5 | phosphoinositide-3-kinase reg | |
| PLCG1 | SH2, SH3 | 12652307 | yes | PLCgamma | PLCG2 | phospholipase |
| PLSCR1 | SH3 | 11390389 | yes | MMTRA1B | PLSCR2-5 | phospholipid scrablase |
| PLXNA1 | unknown | 18660502 | plexin | PLXNA2-4 | signaling, SEMA response | |
| POLR2A | SH2 | 7533294 | yes | RNA pol. 2 | POLR2B-L | transcription, polymerase |
| PRDX1 | SH3 | 9334312 | PAG, MSP23 | PRDX2-6 | peroxiredoxin, ABL kinase inhib | |
| PRKDC | SH3 | 9312071 | yes | DNAPK | S/T kinase, DNA reapir | |
| PSMA7 | SH2, SH3 | 16678104 | yes | C6, XAPC7 | PSMA8 | proteasome component |
| PSTPIP1 | C-term | 11163214 | yes | PSTPIP | PSTPIP2 | signal adaptor |
| PTK2B | SH2 | 14654952 | FAK2, PYK2 | PTK2 | cytoskeleton, adhesion | |
| PTPN6 | SH3 | 8692915 | yes | SHPTP1 | PTPN11 | tyrosine phosphatase |
| PXN | unknown | 9603926 | yes | paxillin | cytoskeleton, actin regulator | |
| RAD9A | SH3 | 11971963 | yes | RAD9 | RAD9B | DNA repair |
| RAD51 | SH3 | 9461559 | yes | RECA | DNA repair | |
| RAD52 | unknown | 12379650 | yes | DNA repair | ||
| RAN | unknown | 11420673 | yes | TC4 | GTPase, nuclear transport | |
| RAPH1 | SH2 | 20417104 | yes | lamellipodin | cytoskeleton, actin reg | |
| RB1 | TK | 8242749 | yes | RB | cell cycle, inhibits ABL kinase | |
| RFX1 | SH3 | 9583676 | RFX2-8 | transcription factor | ||
| RIN1 | SH2, SH3 | 9144171 | yes | RIN2,3 | RAS effector, RAB & ABL activator | |
| ROBO | unknown | 17618275 | ROBO1-4 | axon guidance receptor | ||
| SEM6A | SH3 | 16446784 | semaphorin | SEMA6B-D | receptor-mediated signaling | |
| SEM6D | SH3 | 16446784 | semaphorin | SEMA6A-C | receptor-mediated signaling | |
| SH3BP1 | SH3 | 1379745 | 3BP1 | SH3BP2,4,5 | Rac GAP | |
| SH3BP2 | SH3 | 1379745 | 3BP2 | SH3BP1,4,5 | TNFalpha regulator | |
| SHC1 | SH2 | 8617726 | yes | SHC, p66 | SHC2-4 | signal adaptor |
| SHD | unknown | 9315092 | yes | signal adaptor | ||
| SHE | unknown | 9315092 | Shg | signal adaptor | ||
| SIVA1 | SH2, SH3 | 11278261 | yes | CD27BP | signaling, apoptosis | |
| SLC9A2 | SH3 | 10187839 | NHE2 | SLC9A1,3-11 | Na+/H+ exchanger | |
| SORBS1 | PxxP | 11374898 | yes | ponsin, CAP | SORBS2,3 | signal adaptor |
| SORBS2 | PxxP | 9211900 | yes | ArgBP2 | SORBS1,3 | signal adaptor |
| SORBS3 | PxxP | 16831423 | yes | vinexin | SORBS1,2 | signal adaptor |
| SOS1 | unknown | 15039778 | yes | SOS2 | GEF for H/K/NRAS | |
| ST5 | SH3 | 9632734 | HTS1 | DENND2A, C, D | MAPK signal regulation | |
| TERT | SH3 | 10837221 | yes | TRT, TP2 | telomerase reverse transcript. | |
| TMEM131 | SH3 | 16446784 | RW1 | transmembrane protein | ||
| TLN2 | SH3 | 19234221 | talin2 | TLN1 | cytoskeleton, integrin signaling | |
| TOPBP1 | DBD | 15961388 | yes | TOP2BP1 | DNA repair | |
| TP53 | C-term | 7651743 | yes | p53 | TP63, TP73 | transcription; apoptosis |
| TP73 | SH3 | 10391251 | yes | p73 | TP53, TP63 | transcription; apoptosis |
| TREX1 | SH3 | 16446784 | AGS1, DRN3 | DNA repair, exonuclease | ||
| TUB | SH2 | 10455176 | yes | tubby | TULP1-4 | transcription, signaling |
| VASP | unknown | 12087107 | yes | ENAH, EVL | cytoskeleton, actin regulator | |
| VAV1 | unknown | 11790798 | yes | VAV | VAV2,3 | Rho GEF |
| WAS | TK | 12899629 | WASP | WASL | cytoskeleton, actin polym | |
| WASF2 | SH3 | 16899465 | yes | WAVE2 | WASF1,3,4 | cytoskeleton, actin regulator |
| WASF4 | SH3 | 16446784 | WASF1-3 | cytoskeleton, actin regulator | ||
| WRN | SH3 | 12944467 | yes | RECQ3 | DNA repair | |
| YLPM1 | SH3 | 16446784 | ZAP3 | assoc. w/PP1 phosphatase | ||
| YTHDC1 | unknown | 15175272 | YT521 | YTHDC2 | splicing factor | |
| YWHA- | RSVpTLP | 15696159 | 14-3-3 | ABDEGHQZ | subcellular localization | |
| ZAP70 | SH2 | 7760813 | SRK, STD | SYK, PTK2 | tyrosine kinase | |
| ZDHHC16 | C-term | 12021275 | yes | APH2 | signaling, apoptosis |
“Domain” indicates site of interaction on ABL, if known. “Ref” provides literature citations as PMID numbers. “Sub” identifies proteins that are also known ABL substrates.
The ABL interactor proteins ABI1 and ABI2 were initially identified as proteins that bind to PxxP motifs in the carboxy half of ABL1 through their own SH3 domain, while at the same time interacting with the SH3 domain of ABL1 through their own PxxP motifs (132, 133). ABI proteins influence ABL function, although the mechanism for this is not well established. ABI1 can inhibit the transforming activity of v-Abl (133), and ABI1-derived phosphopeptides can inhibit ABL1 activity through an apparent allosteric effect (134). However, ABI1 also enhances the ABL-mediated phosphorylation of several substrates (116, 135, 136) and may facilitate the oligomerization of ABL proteins (125), normally an activating event. ABI1 and ABI2 participate in a WAVE protein complex that promotes actin remodeling at the leading edge of motile cells (137). A Drosophila ortholog, dAbi, plays an opposing role to Abl in axonogenesis and synaptogenesis (38), but appears to increase Abl kinase activity (138). A third mammalian ABI paralog, ABI3, is not well characterized.
The RAS effector protein RIN1 binds directly to ABL1 and ABL2, causing an increase in tyrosine kinase catalytic efficiency (139). The proposed activation mechanism invokes ABL-SH3–domain attachment to a PxxP motif in RIN1. Subsequent ABL kinase domain–mediated phosphorylation of RIN1-Y36 leads to ABL-SH2–domain binding at that site. By disengaging ABL autoinhibitory domains, RIN1 is thought to stabilize a catalytically active ABL conformation. Activation by RIN1 does not require prior phosphorylation of ABL by SRC-family kinases (139), which suggests that this is an independent activation pathway.
Several interacting proteins appear to function as negative regulators of ABL’s catalytic activity. PRDX1 (PAG) binds to the ABL1-SH3 domain, inhibits ABL’s kinase activity, and overcomes the cytostatic effects of overexpressed ABL1 (140). PSTPIP1 binds to the ABL1-P589xxP and P779xxP motifs and recruits PEST-type PTPs that inhibit ABL’s kinase activity (124). ABL inhibition by TUSC2 (FUS1) involves a short (20 amino acid) peptide through an uncharacterized mechanism (141).
Many of the documented ABL-interacting proteins are adaptors that mediate signal transduction and cytoskeleton dynamics. The CRK and CRKL adaptor proteins bind to ABL1 (142–144), primarily through the interaction of two SH3 domains on CRK (and on CRKL) with the P545xxP and P589xxP motifs of ABL1 (145), and promote tyrosine kinase activity (146). These interactions serve to dock CRK proteins for subsequent tyrosine phosphorylation (145). Several other multi-SH3 domain adaptor proteins also form stable interactions with ABL1. These include NCK1 (145), SORBS1 (ponsin) (145), SORBS2 (ArgBP1) (145, 147), and SORBS3 (vinexin) (148), all of which engage the P631xxP motif of ABL1 (145). All three PxxP motifs mentioned above are well-conserved in ABL2 (Fig. 3), which suggests that it makes similar contacts with CRK, NCK, and SORBS proteins. Other SH3 domain–bearing adaptors implicated as ABL partners are the DOK- family (149, 150), GRB- family (151), and SHC- family (152) proteins. Many of these are mediators of cytoskeleton remodeling.
Cortactin (CTTN), another SH3 domain protein involved in actin dynamics (153), binds to the P573xxP of ABL2 (154). This sequence includes an unusual upstream iteration forming a PxαPxαPxαPxK motif, and mutation of all four prolines is needed to block cortactin binding. The additional upstream prolines are not observed in fish or frog ABL2, nor are they present in any ABL1 sequence (Fig. 4), which suggests that this extended motif evolved relatively late to mediate specialized functions. The fourth PxxP motif (P779xxP) in ABL1 is not conserved in ABL2. Rather, ABL2 encodes a PxxP motif of type 2 (KxαP949xαP) for which no specific binding partners have been identified.
The ABL-SH3 domain has a binding propensity distinct from the type 1 and type 2 categories discussed above, showing a preference for PPx(F,Y,W)xPPP(L,I,V,G,A)P peptides (155). Additional peptide-binding data and modeling were used to develop an ABL-SH3–binding site–motif matrix. WAS-family proteins (WASF1-4), actin reorganization proteins that activate ARP2/3 complexes, were among the strongest ABL-SH3–domain binders predicted with this matrix (156). These results are consistent with experimental evidence that ABL1 binds and signals through WASF proteins (157). Other ABL-SH3 domain–interacting proteins are listed in Table 2.
Both ABL1 and ABL2 have proline-rich sequences, distinct from their SH3-ligand motifs, that conform to established WW domain–ligand motifs of Group 1 (PPxY) and Group 2/3 (PPPPP) (158). The WW-domain protein APBB1 (Fe65) binds to ABL1 (159, 160). From the structure of the APBB1 WW domain (161), it likely binds to PPPPP922. The physiological relevance of this interaction, and potential interactions with APBB2 and APBB3, has not been fully explored.
Cellular Functions of ABL Proteins
Reviews focusing on ABL protein contributions to actin remodeling (162), cell adhesion and motility (9), DNA damage response (163, 164), and microbial pathogen response (165) provide excellent and comprehensive coverage of these areas. Provided below is a brief overview of ABL functions, with an emphasis on ABL partners and substrates.
Actin binding, bundling, and remodeling
ABL-family tyrosine kinases act in the cytoplasm to coordinate actin remodeling in response to appropriate stimuli. This function is mediated by tyrosine phosphorylation of actin cytoskeleton-remodeling proteins and by the ABL carboxy-terminal filamentous actin (F-actin)–binding and –bundling domain (166). ABL-mediated actin remodeling has been studied primarily in the context of cell adhesion and motility (see below), axon guidance (34), and the formation of microspikes (167) and synapses (168–170).
The list of known ABL substrates (Table 1) and binders (Table 2) shows that cytoskeleton-remodeling proteins are highly represented. These include F-actin–binding proteins involved in branch formation (WASF and WASL); depolymerization and severing (CFL1); membrane anchoring (ANXA1 and ANXA2); movement (Myosin IIB); and signaling (DBN1, DBNL, CTTN, RAPH1, ENAH, VASP, and EVL). ABL also promotes the association of proteins within this group, as in the case of RAPH1 and ENAH during dorsal ruffling and axonal morphogenesis (171). Microtubule subunits (TUBA and TUBB) are ABL-kinase substrates, although the target sites and effects on polymerization are unknown. The microtubule-binding proteins MAPT (tau) and PXN (paxillin) are also ABL substrates.
Cell motility and adhesion
Mammalian cells mutated in both ABL genes (Abl1−/−Abl2−/−), or wild-type cells treated with an inhibitor of ABL’s catalytic activity, show increased motility with altered adhesion and spreading (172–174). Conversely, increased ABL activity leads to reduced motility (172, 173). Moreover, ABL proteins localize to sites of actin remodeling (102,131, 154, 175, 176).
Among direct ABL partners and substrates, the CRK-family (CRK and CRKL) and CAS-family (BCAR1, NEDD9, and EFS) proteins are key regulators of cell attachment and motility (177). ABL-mediated phosphorylation of CRK disrupts its binding to CAS and leads to reduced cell migration (178). The CAS-associated proteins CASS4 and CD2AP are also ABL substrates. Of note is the phosphorylation of multiple YxxP sites on BCAR1, NEDD9, and CASS4 [12, 11, and 8 sites, respectively, in Phosphosite (www.phosphosite.org)].
Interaction of ABL1 with the integrin β2–binding and –activating protein TLN2 (179) provides a direct means for ABL proteins to influence attachment to the extracellular matrix. The role of ABL1 in cell migration involves collaboration with SRC-family kinases (SFKs) (180), which likely involves the phosphoregulation of ABL proteins by SFKs (see Covalent modification of ABL proteins). In a Drosophila epithelial cell invasion model, Abl increased SFK activity through a positive-feedback loop (41), which demonstrates the integral relationship of ABL- and SRC-family kinases.
Receptor endocytosis and autophagy
Actin remodeling has been directly implicated in receptor endocytosis (181–183), and ABL proteins likely contribute to this process by coordinating cytoskeleton remodeling with phosphoregulation of receptors and endocytic factors. Multiple receptor tyrosine kinases (EGFR, ERBB2, NTRK1, PDGFRB, ROS, MUSK, and EPHB2) interact with, and are in most cases phosphorylated by, ABL proteins (Tables 1 and 2). ABL interactions promote endocytosis of EGFR (184), facilitate the formation of neuromuscular synapses through MUSK (168), and inhibit PDGFRB-mediated chemotaxis (185). ABL-family tyrosine kinases also modulate the endocytosis of activated B cell receptor complexes (186).
ABL-family tyrosine kinases phosphorylate CAV1 [caveolin, a plasma membrane scaffolding protein that regulates receptor signaling (187)], as well as RIN1 [a RAS effector and RAB5 GEF in endocytosis (188, 189)] and ITSN2 [CDC42 GEF required for caveolae endocytosis (190)]. In addition, ABL proteins functionally engage the CBL family of ubiquitin ligases that drive receptor down-regulation and actin remodeling (191), as well as the CBL-associated protein SORBS1. ABL phosphorylation of CBL leads to increased EGFR stability (184). The membrane invaginations associated with autophagy and the engulfment of apoptotic cells also depend on ABL proteins (45, 192).
The role of ABL1 in DNA damage response and apoptosis
Three NLS motifs (24) and a NES motif (26), together with a regulated 14-3-3 interaction (75), facilitate ABL1 translocation between the cytoplasm and nucleus (193). Its DNA binding domain (194) allows ABL1 to associate directly with DNA in response to damage signals. Many ABL-binding partners and substrates are known mediators of DNA repair (Tables 1 and 2). These include ATM, ATR, DDB1, DDB2, ERCC3, ERCC6, RAD9A, RAD51, RAD52, and WRN. In addition, ABL proteins are found in complexes with MLH1 (195) and other functionally relevant DNA repair proteins. The DNA binding domain and NLS and NES motifs of ABL1 are weak or unrecognizable in fruit fly Abl, sea urchin Abl, and mammalian ABL2 proteins, which suggests that DNA damage-response functions evolved after ABL gene duplication.
ABL1 overexpression causes cell cycle arrest (196) and apoptosis (197) in cultured cells, which suggests that ABL facilitates a repair checkpoint following moderate DNA damage but cell death after severe damage. Several ABL targets (MDM2, MDM4, TP53, and TP73) are primary regulators for this type of damage-induced apoptosis.
Leukemogenic ABL Proteins
Several excellent reviews of ABL oncogenes in hematopoietic malignancies are available (11, 198). This section provides a brief introduction to the topic and then focuses on the current understanding of ABL function in cell transformation and disease.
BCR-ABL1 and related fusion proteins
ABL genes are activated by chromosome translocations in various hematopoietic malignancies. Chronic myeloid leukemia (CML) is characterized in almost all cases by a t(9;22)(q34;q11) translocation (199) that fuses the BCR (breakpoint cluster region) and ABL1 genes (6). Chromatin structural elements (200) and microhomologies and interspersed repeat sequences (201) may contribute to these translocations. The BCR-ABL1 fusion gene product (p210) has constitutive tyrosine kinase activity and is leukemogenic in model systems (11).
Three to 5% of childhood (202) and 20 to 30% of adult acute lymphoblastic leukemia (ALL) cases have a similar translocation (203). The resulting BCR-ABL1 (p190) fusion protein includes less BCR sequence than its CML counterpart. Additional BCR-ABL1 fusion variants have been reported in some leukemias (204, 205). Other leukemogenic ABL1 fusions include NUP214-ABL1 (191) EML1-ABL1 (206), and ETV6-ABL1 (207). In addition, ABL2 is activated by a translocation in some acute myeloid leukemias (AMLs) (208).
Although ABL fusions vary in the amount and identity of upstream partners, and even in which ABL paralog is involved, the ABL breakpoint position consistently leads to removal of the amino-terminal Cap peptide but retention of the SH3 domain (Fig. 7). What remains unclear is why the autoinhibitory SH3 domain is preserved, given that deletion of this domain enhances the catalytic activity of human ABL1. Indeed, the murine Gag-Abl1 (v-Abl) fusion disrupts the SH3 domain (Fig. 7) and is a potent oncogene (209). The presence of autoinhibitory SH3 sequences in human ABL fusion genes might be explained by a requirement for signaling through SH3-interaction partners (Table 2) during leukemia initiation or progression. Notably, murine v-Abl, with a disrupted SH3 domain, produces a phenotypically different disease from that caused by BCR-ABL1 in model systems (210).
Fig. 7.

(Top) Seven translocation-derived ABL1 and ABL2 fusion oncoproteins. The following diseases are associated with each fusion: BCR-ABL1 p190 (also called p185) (ALL), p210 (CML), p230 (CNL), ETV6-ABL1 (CML and AML), NUP214-ABL1 (TALL), EML1-ABL1 (T-ALL), and ETV6-ABL2 (AML). (Bottom) The murine v-Abl oncoprotein results from a retroviral Gag gene fusion that deletes the Abl1 amino terminus and most of the SH3 domain. v-Abl causes B cell lymphomas.
There is a conspicuous absence of ABL-activating mutations in solid tumors, even though ABL fusion genes can transform human fibroblasts in culture (211) and enhanced ABL signaling may contribute to epithelial cell malignancies (212), as well as to the invasive growth of breast cancer cells (213). Furthermore, no other member of the SH3-SH2-TK type nonreceptor tyrosine kinase family is mutationally activated in spontaneous human cancers, even though SRC was first identified as the rodent oncogene in Rous sarcoma virus (214) and cultured human cells are transformed by mutationally activated SRC-family kinases (215) and TEK-family kinases (216). Overexpressed and overactive SRC-family kinases have been detected in many types of tumors [reviewed in (217)], however, and BCR-ABL1 may work in part through SRC activation (218–220).
Kinase activity of ABL fusion proteins
Elimination of the ABL myristoylation site and the amino-terminal Cap domain, both of which participate in stabilizing the inactive conformation of the kinase domain, partly explains the increased and constitutive kinase activity of oncogenic ABL fusion proteins. Homophilic interaction domains in BCR and ETV6 mediate fusion protein oligomerization (221, 222), which promotes transphosphorylation and further increases ABL’s catalytic activity. Reduced nuclear localization (223) may contribute further to transforming activity by ABL1 fusions by interfering with their DNA damage-response functions.
Consistent retention of the SH3 domain implies that the kinase activity of ABL fusion proteins, although constitutive, can be enhanced through derepression. Indeed, RIN1, which activates ABL1 and ABL2 by binding the SH3 and SH2 domains to relieve autoinhibition, increases the transforming ability of BCR in hematopoietic cell lines and primary bone marrow cells (224) and enhances the leukemogenic properties of BCR-ABL1 in a murine model system (224). Like ABL1 and ABL2, BCR-ABL1 proteins are subject to regulation by phosphatases, including PTPN1 (118) and PTPN6 (225). These observations show that constitutively active ABL oncoproteins remain responsive to positive and negative regulation.
ABL inhibitors in leukemia therapy
Transformation by ABL fusion proteins is inextricably tied to their tyrosine kinase activity, which suggests that targeted kinase inhibitors should be therapeutically useful. Imatinib mesylate (also known as STI571 or Gleevec) is an ATP-competitive inhibitor that stabilizes the inactive ABL kinase–domain conformation (226). Imatinib is an effective first-line treatment for CML (227), which validates the signal pathway blockade approach to cancer treatment, and other BCR-ABL1 inhibitors have been added to the CML pharmacopeia (228).
Some people with CML do not respond to imatinib (229), and even individuals with responsive disease, who must remain on the drug indefinitely, can relapse. Drug resistance is typically a consequence of mutations in the BCR-ABL1 kinase domain (230, 231) but may also result from mutations in the SH3 and SH2 domains (232, 233) or BCR-ABL1 amplification (230). BCR-ABL1–positive ALL is refractory to imatinib alone and shows high rates of resistance and relapse to chemotherapy combined with imatinib (234). Dasatinib, which is among the more effective second-generation ABL active-site inhibitors, also inhibits SRC-family kinases (235), and its efficacy in CML likely reflects the cooperation of SFKs with BCR-ABL1 in eliciting cell transformation. When different ABL inhibitors are used to treat disease sequentially, however, resistance can return through the accumulation of multiple kinase-domain mutations (236), which suggests that a combination approach might be more efficacious. One drug-resistant mutation in the kinase gatekeeper residue (237), BCR-ABL1T315I, is refractory to all established inhibitors but may respond to new drugs (238).
The discovery of allosteric (noncompetitive with ATP) ABL inhibitors (83) represents a promising new direction for the treatment of BCR-ABL1–positive leukemias. These compounds (GNF-2 and GNF-5) target the ABL1 myristate-binding pocket (unoccupied in BCR-ABL1) to stabilize an inactive kinase-domain conformation (239). Combining GNF-5 with catalytic site inhibitors suppresses the emergence of resistance mutations. GNF compounds have limited potency against BCR-ABL1T315I but worked additively with an ATP-competitive inhibitor to block this mutation in a bone marrow–transplantation leukemia model (239). Long-term effective therapy would likely benefit from pairing inhibitors of ABL’s kinase activity with drugs that target ABL allosteric sites (239, 240) or drugs that target direct activators of the ABL-family kinases (139). Other combination approaches to therapy of BCR-ABL1–driven leukemias include the pairing of inhibitors of ABL’s catalytic activity with drugs that reduce BCR-ABL1 expression (241) or stability (242), or that disable collaborative pathways (243–246). Targeting functions required for leukemia cell survival might also provide a useful approach [reviewed in (247)].
Acknowledgments
The author thanks Steven Goff and Thomas Graeber for comments and criticisms, and Jiyong Park for performing the position weight matrix analysis. I also acknowledge NIH CA136699 as a source of support.
- GLOSS
ABL, family proteins couple a highly regulated tyrosine kinase domain with an actin-binding and -bundling domain to carry out a set of unique and essential functions. The ABL genes are among the earliest identifiable genes encoding tyrosine kinases, and they show remarkable sequence conservation. Gene duplication produced two vertebrate ABL paralogs with specialized properties. ABL1 evolved nuclear localization signals and a DNA binding domain to mediate damage repair functions. ABL2 developed additional binding domains for actin and microtubules, extending its cytoskeletal remodeling functions. This review surveys the recent literature and available databases with a focus on ABL evolution and the mechanisms regulating ABL’s catalytic activity and substrate specificity. A better understanding of these properties could facilitate the design of new treatments for malignancies driven by ABL fusion proteins
Footnotes
This manuscript has been accepted for publication in Science Signaling. This version has not undergone final editing. Please refer to the complete version of record at http://www.sciencesignaling.org/. The manuscript may not be reproduced or used in any manner that does not fall within the fair use provisions of the Copyright Act without the prior, written permission of AAAS.
References and Notes
- 1.Upper case is used for human genes and proteins (ABL1 and ABL2) as well as for discussion of gene and protein families (ABL). First-letter-only upper case (Abl1 and Abl2) is used when referring to genes and proteins from all other species in this review. Human Genome Organization nomenclature (www.genenames.org) is used. More common gene names are provided in the text, with additional aliases given in Table 1 and Table 2.
- 2.Abelson HT, Rabstein LS. Lymphosarcoma: Virus-induced thymic-independent disease in mice. Cancer Res. 1970;30:2213–2222. [PubMed] [Google Scholar]
- 3.Sefton BM, Hunter T, Raschke WC. Evidence that the Abelson virus protein functions in vivo as a protein kinase that phosphorylates tyrosine. Proc Natl Acad Sci USA. 1981;78:1552–1556. doi: 10.1073/pnas.78.3.1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Witte ON, Dasgupta A, Baltimore D. Abelson murine leukaemia virus protein is phosphorylated in vitro to form phosphotyrosine. Nature. 1980;283:826–831. doi: 10.1038/283826a0. [DOI] [PubMed] [Google Scholar]
- 5.Goff SP, Gilboa E, Witte ON, Baltimore D. Structure of the Abelson murine leukemia virus genome and the homologous cellular gene: Studies with cloned viral DNA. Cell. 1980;22:777–785. doi: 10.1016/0092-8674(80)90554-1. [DOI] [PubMed] [Google Scholar]
- 6.Ben-Neriah Y, Daley GQ, Mes-Masson AM, Witte ON, Baltimore D. The chronic myelogenous leukemia-specific P210 protein is the product of the bcr/abl hybrid gene. Science. 1986;233:212–214. doi: 10.1126/science.3460176. [DOI] [PubMed] [Google Scholar]
- 7.Perego R, Ron D, Kruh GD. Arg encodes a widely expressed 145 kDa protein-tyrosine kinase. Oncogene. 1991;6:1899–1902. [PubMed] [Google Scholar]
- 8.Kruh GD, Perego R, Miki T, Aaronson SA. The complete coding sequence of arg defines the Abelson subfamily of cytoplasmic tyrosine kinases. Proc Natl Acad Sci USA. 1990;87:5802–5806. doi: 10.1073/pnas.87.15.5802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bradley WD, Koleske AJ. Regulation of cell migration and morphogenesis by Abl-family kinases: Emerging mechanisms and physiological contexts. J Cell Sci. 2009;122:3441–3454. doi: 10.1242/jcs.039859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gu JJ, Ryu JR, Pendergast AM. Abl tyrosine kinases in T-cell signaling. Immunol Rev. 2009;228:170–183. doi: 10.1111/j.1600-065X.2008.00751.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wong S, Witte ON. The BCR-ABL story: Bench to bedside and back. Annu Rev Immunol. 2004;22:247–306. doi: 10.1146/annurev.immunol.22.012703.104753. [DOI] [PubMed] [Google Scholar]
- 12.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 13.Pawson T, Kofler M. Kinome signaling through regulated protein-protein interactions in normal and cancer cells. Curr Opin Cell Biol. 2009;21:147–153. doi: 10.1016/j.ceb.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 14.Pincus D, Letunic I, Bork P, Lim WA. Evolution of the phospho-tyrosine signaling machinery in premetazoan lineages. Proc Natl Acad Sci USA. 2008;105:9680–9684. doi: 10.1073/pnas.0803161105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yadav SS, Miller WT. The evolutionarily conserved arrangement of domains in SRC family kinases is important for substrate recognition. Biochemistry. 2008;47:10871–10880. doi: 10.1021/bi800930e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bennett RL, Hoffmann FM. Increased levels of the Drosophila Abelson tyrosine kinase in nerves and muscles: Subcellular localization and mutant phenotypes imply a role in cell-cell interactions. Development. 1992;116:953–966. doi: 10.1242/dev.116.4.953. [DOI] [PubMed] [Google Scholar]
- 17.Bonini NM, Leiserson WM, Benzer S. Multiple roles of the eyes absent gene in Drosophila. Dev Biol. 1998;196:42–57. doi: 10.1006/dbio.1997.8845. [DOI] [PubMed] [Google Scholar]
- 18.Fox DT, Peifer M. Abelson kinase (Abl) and RhoGEF2 regulate actin organization during cell constriction in Drosophila. Development. 2007;134:567–578. doi: 10.1242/dev.02748. [DOI] [PubMed] [Google Scholar]
- 19.Xiong W, Dabbouseh NM, Rebay I. Interactions with the Abelson tyrosine kinase reveal compartmentalization of eyes absent function between nucleus and cytoplasm. Dev Cell. 2009;16:271–279. doi: 10.1016/j.devcel.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Innan H, Kondrashov F. The evolution of gene duplications: Classifying and distinguishing between models. Nat Rev Genet. 2010;11:97–108. doi: 10.1038/nrg2689. [DOI] [PubMed] [Google Scholar]
- 21.Single-letter abbreviations for the amino acid residues used in this review are as follows: A, alanine; C, cysteine; D, aspartic acid; E, glutamic acid; F, phenylalanine; G, glycine; H, histidine; I, isoleucine; K, lysine; L, leucine; M, methionine; N, asparagine; P, proline; Q, glutamine; R, arginine; S, serine; T, threonine; V, valine; W, tryptophan; Y, tyrosine; x, any amino acid; α, hydrophobic amino acid.
- 22.Mayer BJ. SH3 domains: Complexity in moderation. J Cell Sci. 2001;114:1253–1263. doi: 10.1242/jcs.114.7.1253. [DOI] [PubMed] [Google Scholar]
- 23.Macias MJ, Wiesner S, Sudol M. WW and SH3 domains, two different scaffolds to recognize proline-rich ligands. FEBS Lett. 2002;513:30–37. doi: 10.1016/s0014-5793(01)03290-2. [DOI] [PubMed] [Google Scholar]
- 24.Wen ST, Jackson PK, Van Etten RA. The cytostatic function of c-Abl is controlled by multiple nuclear localization signals and requires the p53 and Rb tumor suppressor gene products. EMBO J. 1996;15:1583–1595. [PMC free article] [PubMed] [Google Scholar]
- 25.Henderson BR, Eleftheriou A. A comparison of the activity, sequence specificity, and CRM1-dependence of different nuclear export signals. Exp Cell Res. 2000;256:213–224. doi: 10.1006/excr.2000.4825. [DOI] [PubMed] [Google Scholar]
- 26.Taagepera S, McDonald D, Loeb JE, Whitaker LL, McElroy AK, Wang JY, Hope TJ. Nuclear-cytoplasmic shuttling of C-ABL tyrosine kinase. Proc Natl Acad Sci USA. 1998;95:7457–7462. doi: 10.1073/pnas.95.13.7457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koleske AJ, Gifford AM, Scott ML, Nee M, Bronson RT, Miczek KA, Baltimore D. Essential roles for the Abl and Arg tyrosine kinases in neurulation. Neuron. 1998;21:1259–1272. doi: 10.1016/s0896-6273(00)80646-7. [DOI] [PubMed] [Google Scholar]
- 28.Bueno MJ, Pérez de Castro I, Gómez de Cedrón M, Santos J, Calin GA, Cigudosa JC, Croce CM, Fernández-Piqueras J, Malumbres M. Genetic and epigenetic silencing of microRNA-203 enhances ABL1 and BCR-ABL1 oncogene expression. Cancer Cell. 2008;13:496–506. doi: 10.1016/j.ccr.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 29.Gertler FB, Doctor JS, Hoffmann FM. Genetic suppression of mutations in the Drosophila abl proto-oncogene homolog. Science. 1990;248:857–860. doi: 10.1126/science.2188361. [DOI] [PubMed] [Google Scholar]
- 30.Henkemeyer MJ, Gertler FB, Goodman W, Hoffmann FM. The Drosophila Abelson proto-oncogene homolog: Identification of mutant alleles that have pleiotropic effects late in development. Cell. 1987;51:821–828. doi: 10.1016/0092-8674(87)90105-x. [DOI] [PubMed] [Google Scholar]
- 31.Bashaw GJ, Kidd T, Murray D, Pawson T, Goodman CS. Repulsive axon guidance: Abelson and Enabled play opposing roles downstream of the roundabout receptor. Cell. 2000;101:703–715. doi: 10.1016/s0092-8674(00)80883-1. [DOI] [PubMed] [Google Scholar]
- 32.Rhee J, Buchan T, Zukerberg L, Lilien J, Balsamo J. Cables links Robo-bound Abl kinase to N-cadherin-bound beta-catenin to mediate Slit-induced modulation of adhesion and transcription. Nat Cell Biol. 2007;9:883–892. doi: 10.1038/ncb1614. [DOI] [PubMed] [Google Scholar]
- 33.Song JK, Giniger E, Desai CJ. The receptor protein tyrosine phosphatase PTP69D antagonizes Abl tyrosine kinase to guide axons in Drosophila. Mech Dev. 2008;125:247–256. doi: 10.1016/j.mod.2007.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wills Z, Bateman J, Korey CA, Comer A, Van Vactor D. The tyrosine kinase Abl and its substrate enabled collaborate with the receptor phosphatase Dlar to control motor axon guidance. Neuron. 1999;22:301–312. doi: 10.1016/s0896-6273(00)81091-0. [DOI] [PubMed] [Google Scholar]
- 35.Li W, Li Y, Gao FB. Abelson, enabled, and p120 catenin exert distinct effects on dendritic morphogenesis in Drosophila. Dev Dyn. 2005;234:512–522. doi: 10.1002/dvdy.20496. [DOI] [PubMed] [Google Scholar]
- 36.Lee H, Engel U, Rusch J, Scherrer S, Sheard K, Van Vactor D. The microtubule plus end tracking protein Orbit/MAST/CLASP acts downstream of the tyrosine kinase Abl in mediating axon guidance. Neuron. 2004;42:913–926. doi: 10.1016/j.neuron.2004.05.020. [DOI] [PubMed] [Google Scholar]
- 37.Gertler FB, Comer AR, Juang JL, Ahern SM, Clark MJ, Liebl EC, Hoffmann FM. enabled, a dosage-sensitive suppressor of mutations in the Drosophila Abl tyrosine kinase, encodes an Abl substrate with SH3 domain-binding properties. Genes Dev. 1995;9:521–533. doi: 10.1101/gad.9.5.521. [DOI] [PubMed] [Google Scholar]
- 38.Lin TY, Huang CH, Kao HH, Liou GG, Yeh SR, Cheng CM, Chen MH, Pan RL, Juang JL. Abi plays an opposing role to Abl in Drosophila axonogenesis and synaptogenesis. Development. 2009;136:3099–3107. doi: 10.1242/dev.033324. [DOI] [PubMed] [Google Scholar]
- 39.Grevengoed EE, Loureiro JJ, Jesse TL, Peifer M. Abelson kinase regulates epithelial morphogenesis in Drosophila. J Cell Biol. 2001;155:1185–1198. doi: 10.1083/jcb.200105102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stevens TL, Rogers EM, Koontz LM, Fox DT, Homem CC, Nowotarski SH, Artabazon NB, Peifer M. Using Bcr-Abl to examine mechanisms by which abl kinase regulates morphogenesis in Drosophila. Mol Biol Cell. 2008;19:378–393. doi: 10.1091/mbc.E07-01-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Singh J, Aaronson SA, Mlodzik M. Drosophila Abelson kinase mediates cell invasion and proliferation through two distinct MAPK pathways. Oncogene. 2010;29:4033–4045. doi: 10.1038/onc.2010.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sheffield M, Loveless T, Hardin J, Pettitt J. C. elegans Enabled exhibits novel interactions with N-WASP, Abl, and cell-cell junctions. Curr Biol. 2007;17:1791–1796. doi: 10.1016/j.cub.2007.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deng X, Hofmann ER, Villanueva A, Hobert O, Capodieci P, Veach DR, Yin X, Campodonico L, Glekas A, Cordon-Cardo C, Clarkson B, Bornmann WG, Fuks Z, Hengartner MO, Kolesnick R. Caenorhabditis elegans ABL-1 antagonizes p53-mediated germline apoptosis after ionizing irradiation. Nat Genet. 2004;36:906–912. doi: 10.1038/ng1396. [DOI] [PubMed] [Google Scholar]
- 44.Salinas LS, Maldonado E, Navarro RE. Stress-induced germ cell apoptosis by a p53 independent pathway in Caenorhabditis elegans. Cell Death Differ. 2006;13:2129–2139. doi: 10.1038/sj.cdd.4401976. [DOI] [PubMed] [Google Scholar]
- 45.Hurwitz ME, Vanderzalm PJ, Bloom L, Goldman J, Garriga G, Horvitz HR, Green DR. Abl kinase inhibits the engulfment of apoptotic [corrected] cells in Caenorhabditis elegans. PLoS Biol. 2009;7:e1000099. doi: 10.1371/journal.pbio.1000099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burton EA, Pendergast AM, Aballay A. The Caenorhabditis elegans ABL-1 tyrosine kinase is required for Shigella flexneri pathogenesis. Appl Environ Microbiol. 2006;72:5043–5051. doi: 10.1128/AEM.00558-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schwartzberg PL, Stall AM, Hardin JD, Bowdish KS, Humaran T, Boast S, Harbison ML, Robertson EJ, Goff SP. Mice homozygous for the ablm1 mutation show poor viability and depletion of selected B and T cell populations. Cell. 1991;65:1165–1175. doi: 10.1016/0092-8674(91)90012-n. [DOI] [PubMed] [Google Scholar]
- 48.Tybulewicz VL, Crawford CE, Jackson PK, Bronson RT, Mulligan RC. Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell. 1991;65:1153–1163. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- 49.Li B, Boast S, de los Santos K, Schieren I, Quiroz M, Teitelbaum SL, Tondravi MM, Goff SP. Mice deficient in Abl are osteoporotic and have defects in osteoblast maturation. Nat Genet. 2000;24:304–308. doi: 10.1038/73542. [DOI] [PubMed] [Google Scholar]
- 50.Chen S, Wang R, Li QF, Tang DD. Abl knockout differentially affects p130 Crk-associated substrate, vinculin, and paxillin in blood vessels of mice. Am J Physiol Heart Circ Physiol. 2009;297:H533–H539. doi: 10.1152/ajpheart.00237.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qiu Z, Cang Y, Goff SP. c-Abl tyrosine kinase regulates cardiac growth and development. Proc Natl Acad Sci USA. 2010;107:1136–1141. doi: 10.1073/pnas.0913131107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brightbill H, Schlissel MS. The effects of c-Abl mutation on developing B cell differentiation and survival. Int Immunol. 2009;21:575–585. doi: 10.1093/intimm/dxp027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liberatore RA, Goff SP. c-Abl-deficient mice exhibit reduced numbers of peritoneal B-1 cells and defects in BCR-induced B cell activation. Int Immunol. 2009;21:403–414. doi: 10.1093/intimm/dxp006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Silberman I, Sionov RV, Zuckerman V, Haupt S, Goldberg Z, Strasser A, Ben-Sasson ZS, Baniyash M, Koleske AJ, Haupt Y. T cell survival and function requires the c-Abl tyrosine kinase. Cell Cycle. 2008;7:3847–3857. doi: 10.4161/cc.7.24.7267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gourley SL, Koleske AJ, Taylor JR. Loss of dendrite stabilization by the Abl-related gene (Arg) kinase regulates behavioral flexibility and sensitivity to cocaine. Proc Natl Acad Sci USA. 2009;106:16859–16864. doi: 10.1073/pnas.0902286106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hantschel O, Nagar B, Guettler S, Kretzschmar J, Dorey K, Kuriyan J, Superti-Furga G. A myristoyl/phosphotyrosine switch regulates c-Abl. Cell. 2003;112:845–857. doi: 10.1016/s0092-8674(03)00191-0. [DOI] [PubMed] [Google Scholar]
- 57.Pluk H, Dorey K, Superti-Furga G. Autoinhibition of c-Abl. Cell. 2002;108:247–259. doi: 10.1016/s0092-8674(02)00623-2. [DOI] [PubMed] [Google Scholar]
- 58.Nagar B, Hantschel O, Seeliger M, Davies JM, Weis WI, Superti-Furga G, Kuriyan J. Organization of the SH3-SH2 unit in active and inactive forms of the cAbl tyrosine kinase. Mol Cell. 2006;21:787–798. doi: 10.1016/j.molcel.2006.01.035. [DOI] [PubMed] [Google Scholar]
- 59.Smith KM, Yacobi R, Van Etten RA. Autoinhibition of Bcr-Abl through its SH3 domain. Mol Cell. 2003;12:27–37. doi: 10.1016/S1097-2765(03)00274-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Barilá D, Superti-Furga G. An intramolecular SH3-domain interaction regulates cAbl activity. Nat Genet. 1998;18:280–282. doi: 10.1038/ng0398-280. [DOI] [PubMed] [Google Scholar]
- 61.Plattner R, Koleske AJ, Kazlauskas A, Pendergast AM. Bidirectional signaling links the Abelson kinases to the platelet-derived growth factor receptor. Mol Cell Biol. 2004;24:2573–2583. doi: 10.1128/MCB.24.6.2573-2583.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tokonzaba E, Capelluto DG, Kutateladze TG, Overduin M. Phosphoinositide, phosphopeptide and pyridone interactions of the Abl SH2 domain. Chem Biol Drug Des. 2006;67:230–237. doi: 10.1111/j.1747-0285.2006.00361.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brasher BB, Van Etten RA. c-Abl has high intrinsic tyrosine kinase activity that is stimulated by mutation of the Src homology 3 domain and by autophosphorylation at two distinct regulatory tyrosines. J Biol Chem. 2000;275:35631–35637. doi: 10.1074/jbc.M005401200. [DOI] [PubMed] [Google Scholar]
- 64.Tanis KQ, Veach D, Duewel HS, Bornmann WG, Koleske AJ. Two distinct phosphorylation pathways have additive effects on Abl family kinase activation. Mol Cell Biol. 2003;23:3884–3896. doi: 10.1128/MCB.23.11.3884-3896.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Furstoss O, Dorey K, Simon V, Barilà D, Superti-Furga G, Roche S. c-Abl is an effector of Src for growth factor-induced c-myc expression and DNA synthesis. EMBO J. 2002;21:514–524. doi: 10.1093/emboj/21.4.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen S, O’Reilly LP, Smithgall TE, Engen JR. Tyrosine phosphorylation in the SH3 domain disrupts negative regulatory interactions within the c-Abl kinase core. J Mol Biol. 2008;383:414–423. doi: 10.1016/j.jmb.2008.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Meyn MA, 3rd, Wilson MB, Abdi FA, Fahey N, Schiavone AP, Wu J, Hochrein JM, Engen JR, Smithgall TE. Src family kinases phosphorylate the Bcr-Abl SH3-SH2 region and modulate Bcr-Abl transforming activity. J Biol Chem. 2006;281:30907–30916. doi: 10.1074/jbc.M605902200. [DOI] [PubMed] [Google Scholar]
- 68.Cao C, Li Y, Leng Y, Li P, Ma Q, Kufe D. Ubiquitination and degradation of the Arg tyrosine kinase is regulated by oxidative stress. Oncogene. 2005;24:2433–2440. doi: 10.1038/sj.onc.1208454. [DOI] [PubMed] [Google Scholar]
- 69.Allen PB, Wiedemann LM. An activating mutation in the ATP binding site of the ABL kinase domain. J Biol Chem. 1996;271:19585–19591. doi: 10.1074/jbc.271.32.19585. [DOI] [PubMed] [Google Scholar]
- 70.Skaggs BJ, Gorre ME, Ryvkin A, Burgess MR, Xie Y, Han Y, Komisopoulou E, Brown LM, Loo JA, Landaw EM, Sawyers CL, Graeber TG. Phosphorylation of the ATP-binding loop directs oncogenicity of drug-resistant BCR-ABL mutants. Proc Natl Acad Sci USA. 2006;103:19466–19471. doi: 10.1073/pnas.0609239103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Filippakopoulos P, Kofler M, Hantschel O, Gish GD, Grebien F, Salah E, Neudecker P, Kay LE, Turk BE, Superti-Furga G, Pawson T, Knapp S. Structural coupling of SH2-kinase domains links Fes and Abl substrate recognition and kinase activation. Cell. 2008;134:793–803. doi: 10.1016/j.cell.2008.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Blasutig IM, New LA, Thanabalasuriar A, Dayarathna TK, Goudreault M, Quaggin SE, Li SS, Gruenheid S, Jones N, Pawson T. Phosphorylated YDXV motifs and Nck SH2/SH3 adaptors act cooperatively to induce actin reorganization. Mol Cell Biol. 2008;28:2035–2046. doi: 10.1128/MCB.01770-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Miyoshi-Akiyama T, Aleman LM, Smith JM, Adler CE, Mayer BJ. Regulation of Cbl phosphorylation by the Abl tyrosine kinase and the Nck SH2/SH3 adaptor. Oncogene. 2001;20:4058–4069. doi: 10.1038/sj.onc.1204528. [DOI] [PubMed] [Google Scholar]
- 74.Frackelton AR, Jr, Kumar PS, Kannan B, Clark JW. Tyrosine phosphorylated proteins in chronic myelogenous leukemia. Leuk Lymphoma. 1993;11(Suppl 1):125–129. doi: 10.3109/10428199309047875. [DOI] [PubMed] [Google Scholar]
- 75.Yoshida K, Yamaguchi T, Natsume T, Kufe D, Miki Y. JNK phosphorylation of 14-3-3 proteins regulates nuclear targeting of c-Abl in the apoptotic response to DNA damage. Nat Cell Biol. 2005;7:278–285. doi: 10.1038/ncb1228. [DOI] [PubMed] [Google Scholar]
- 76.Yaffe MB, Rittinger K, Volinia S, Caron PR, Aitken A, Leffers H, Gamblin SJ, Smerdon SJ, Cantley LC. The structural basis for 14-3-3:phosphopeptide binding specificity. Cell. 1997;91:961–971. doi: 10.1016/s0092-8674(00)80487-0. [DOI] [PubMed] [Google Scholar]
- 77.Nihira K, Taira N, Miki Y, Yoshida K. TTK/Mps1 controls nuclear targeting of cAbl by 14-3-3-coupled phosphorylation in response to oxidative stress. Oncogene. 2008;27:7285–7295. doi: 10.1038/onc.2008.334. [DOI] [PubMed] [Google Scholar]
- 78.Jung JH, Pendergast AM, Zipfel PA, Traugh JA. Phosphorylation of c-Abl by protein kinase Pak2 regulates differential binding of ABI2 and CRK. Biochemistry. 2008;47:1094–1104. doi: 10.1021/bi701533j. [DOI] [PubMed] [Google Scholar]
- 79.Roig J, Tuazon PT, Zipfel PA, Pendergast AM, Traugh JA. Functional interaction between c-Abl and the p21-activated protein kinase gamma-PAK. Proc Natl Acad Sci USA. 2000;97:14346–14351. doi: 10.1073/pnas.97.26.14346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kipreos ET, Wang JY. Differential phosphorylation of c-Abl in cell cycle determined by cdc2 kinase and phosphatase activity. Science. 1990;248:217–220. doi: 10.1126/science.2183353. [DOI] [PubMed] [Google Scholar]
- 81.di Bari MG, Ciuffini L, Mingardi M, Testi R, Soddu S, Barilà D. c-Abl acetylation by histone acetyltransferases regulates its nuclear-cytoplasmic localization. EMBO Rep. 2006;7:727–733. doi: 10.1038/sj.embor.7400700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Farazi TA, Waksman G, Gordon JI. The biology and enzymology of protein N-myristoylation. J Biol Chem. 2001;276:39501–39504. doi: 10.1074/jbc.R100042200. [DOI] [PubMed] [Google Scholar]
- 83.Adrián FJ, Ding Q, Sim T, Velentza A, Sloan C, Liu Y, Zhang G, Hur W, Ding S, Manley P, Mestan J, Fabbro D, Gray NS. Allosteric inhibitors of Bcr-abl-dependent cell proliferation. Nat Chem Biol. 2006;2:95–102. doi: 10.1038/nchembio760. [DOI] [PubMed] [Google Scholar]
- 84.Choi Y, Seeliger MA, Panjarian SB, Kim H, Deng X, Sim T, Couch B, Koleske AJ, Smithgall TE, Gray NS. N-myristoylated c-Abl tyrosine kinase localizes to the endoplasmic reticulum upon binding to an allosteric inhibitor. J Biol Chem. 2009;284:29005–29014. doi: 10.1074/jbc.M109.026633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Soubeyran P, Barac A, Szymkiewicz I, Dikic I. Cbl-ArgBP2 complex mediates ubiquitination and degradation of c-Abl. Biochem J. 2003;370:29–34. doi: 10.1042/BJ20021539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Andoniou CE, Thien CB, Langdon WY. The two major sites of cbl tyrosine phosphorylation in abl-transformed cells select the crkL SH2 domain. Oncogene. 1996;12:1981–1989. [PubMed] [Google Scholar]
- 87.Grossmann AH, Kolibaba KS, Willis SG, Corbin AS, Langdon WS, Deininger MW, Druker BJ. Catalytic domains of tyrosine kinases determine the phosphorylation sites within c-Cbl. FEBS Lett. 2004;577:555–562. doi: 10.1016/j.febslet.2004.10.054. [DOI] [PubMed] [Google Scholar]
- 88.Podar K, Raab MS, Tonon G, Sattler M, Barilà D, Zhang J, Tai YT, Yasui H, Raje N, DePinho RA, Hideshima T, Chauhan D, Anderson KC. Up-regulation of c-Jun inhibits proliferation and induces apoptosis via caspase-triggered c-Abl cleavage in human multiple myeloma. Cancer Res. 2007;67:1680–1688. doi: 10.1158/0008-5472.CAN-06-1863. [DOI] [PubMed] [Google Scholar]
- 89.Cujec TP, Medeiros PF, Hammond P, Rise C, Kreider BL. Selection of v-abl tyrosine kinase substrate sequences from randomized peptide and cellular proteomic libraries using mRNA display. Chem Biol. 2002;9:253–264. doi: 10.1016/s1074-5521(02)00098-4. [DOI] [PubMed] [Google Scholar]
- 90.Songyang Z, Carraway KL, 3rd, Eck MJ, Harrison SC, Feldman RA, Mohammadi M, Schlessinger J, Hubbard SR, Smith DP, Eng C, Lorenzo MJ, Ponder BAJ, Mayer BJ, Cantley LC. Catalytic specificity of protein-tyrosine kinases is critical for selective signalling. Nature. 1995;373:536–539. doi: 10.1038/373536a0. [DOI] [PubMed] [Google Scholar]
- 91.Goss VL, Lee KA, Moritz A, Nardone J, Spek EJ, MacNeill J, Rush J, Comb MJ, Polakiewicz RD. A common phosphotyrosine signature for the Bcr-Abl kinase. Blood. 2006;107:4888–4897. doi: 10.1182/blood-2005-08-3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Till JH, Chan PM, Miller WT. Engineering the substrate specificity of the Abl tyrosine kinase. J Biol Chem. 1999;274:4995–5003. doi: 10.1074/jbc.274.8.4995. [DOI] [PubMed] [Google Scholar]
- 93.Shah K, Liu Y, Deirmengian C, Shokat KM. Engineering unnatural nucleotide specificity for Rous sarcoma virus tyrosine kinase to uniquely label its direct substrates. Proc Natl Acad Sci USA. 1997;94:3565–3570. doi: 10.1073/pnas.94.8.3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Boyle SN, Koleske AJ. Use of a chemical genetic technique to identify myosin IIb as a substrate of the Abl-related gene (Arg) tyrosine kinase. Biochemistry. 2007;46:11614–11620. doi: 10.1021/bi701119s. [DOI] [PubMed] [Google Scholar]
- 95.Mayer BJ, Baltimore D. Mutagenic analysis of the roles of SH2 and SH3 domains in regulation of the Abl tyrosine kinase. Mol Cell Biol. 1994;14:2883–2894. doi: 10.1128/mcb.14.5.2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mayer BJ, Hirai H, Sakai R. Evidence that SH2 domains promote processive phosphorylation by protein-tyrosine kinases. Curr Biol. 1995;5:296–305. doi: 10.1016/s0960-9822(95)00060-1. [DOI] [PubMed] [Google Scholar]
- 97.Pellicena P, Stowell KR, Miller WT. Enhanced phosphorylation of Src family kinase substrates containing SH2 domain binding sites. J Biol Chem. 1998;273:15325–15328. doi: 10.1074/jbc.273.25.15325. [DOI] [PubMed] [Google Scholar]
- 98.Liao X, Su J, Mrksich M. An adaptor domain-mediated autocatalytic interfacial kinase reaction. Chemistry (Easton) 2009;15:12303–12309. doi: 10.1002/chem.200901345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Baker LA, Allis CD, Wang GG. PHD fingers in human diseases: Disorders arising from misinterpreting epigenetic marks. Mutat Res. 2008;647:3–12. doi: 10.1016/j.mrfmmm.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhang K, Mosch K, Fischle W, Grewal SI. Roles of the Clr4 methyltransferase complex in nucleation, spreading and maintenance of heterochromatin. Nat Struct Mol Biol. 2008;15:381–388. doi: 10.1038/nsmb.1406. [DOI] [PubMed] [Google Scholar]
- 101.Cao C, Leng Y, Liu X, Yi Y, Li P, Kufe D. Catalase is regulated by ubiquitination and proteosomal degradation. Role of the c-Abl and Arg tyrosine kinases. Biochemistry. 2003;42:10348–10353. doi: 10.1021/bi035023f. [DOI] [PubMed] [Google Scholar]
- 102.Woodring PJ, Meisenhelder J, Johnson SA, Zhou GL, Field J, Shah K, Bladt F, Pawson T, Niki M, Pandolfi PP, Wang JY, Hunter T. c-Abl phosphorylates Dok1 to promote filopodia during cell spreading. J Cell Biol. 2004;165:493–503. doi: 10.1083/jcb.200312171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dorsey JF, Cunnick JM, Mane SM, Wu J. Regulation of the Erk2-Elk1 signaling pathway and megakaryocytic differentiation of Bcr-Abl(+) K562 leukemic cells by Gab2. Blood. 2002;99:1388–1397. doi: 10.1182/blood.v99.4.1388. [DOI] [PubMed] [Google Scholar]
- 104.Boyle SN, Michaud GA, Schweitzer B, Predki PF, Koleske AJ. A critical role for cortactin phosphorylation by Abl-family kinases in PDGF-induced dorsal-wave formation. Curr Biol. 2007;17:445–451. doi: 10.1016/j.cub.2007.01.057. [DOI] [PubMed] [Google Scholar]
- 105.Goldberg Z, Vogt Sionov R, Berger M, Zwang Y, Perets R, Van Etten RA, Oren M, Taya Y, Haupt Y. Tyrosine phosphorylation of Mdm2 by c-Abl: Implications for p53 regulation. EMBO J. 2002;21:3715–3727. doi: 10.1093/emboj/cdf384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Maruoka M, Suzuki J, Kawata S, Yoshida K, Hirao N, Sato S, Goff SP, Takeya T, Tani K, Shishido T. Identification of B cell adaptor for PI3-kinase (BCAP) as an Abl interactor 1-regulated substrate of Abl kinases. FEBS Lett. 2005;579:2986–2990. doi: 10.1016/j.febslet.2005.04.052. [DOI] [PubMed] [Google Scholar]
- 107.Plattner R, Irvin BJ, Guo S, Blackburn K, Kazlauskas A, Abraham RT, York JD, Pendergast AM. A new link between the c-Abl tyrosine kinase and phosphoinositide signalling through PLC-gamma1. Nat Cell Biol. 2003;5:309–319. doi: 10.1038/ncb949. [DOI] [PubMed] [Google Scholar]
- 108.Tauchi T, Feng GS, Shen R, Song HY, Donner D, Pawson T, Broxmeyer HE. SH2-containing phosphotyrosine phosphatase Syp is a target of p210bcr-abl tyrosine kinase. J Biol Chem. 1994;269:15381–15387. [PubMed] [Google Scholar]
- 109.Schaller MD, Schaefer EM. Multiple stimuli induce tyrosine phosphorylation of the Crk-binding sites of paxillin. Biochem J. 2001;360:57–66. doi: 10.1042/0264-6021:3600057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Baskaran R, Dahmus ME, Wang JY. Tyrosine phosphorylation of mammalian RNA polymerase II carboxyl-terminal domain. Proc Natl Acad Sci USA. 1993;90:11167–11171. doi: 10.1073/pnas.90.23.11167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Slupianek A, Schmutte C, Tombline G, Nieborowska-Skorska M, Hoser G, Nowicki MO, Pierce AJ, Fishel R, Skorski T. BCR/ABL regulates mammalian RecA homologs, resulting in drug resistance. Mol Cell. 2001;8:795–806. doi: 10.1016/s1097-2765(01)00357-4. [DOI] [PubMed] [Google Scholar]
- 112.Duyster J, Baskaran R, Wang JY. Src homology 2 domain as a specificity determinant in the c-Abl-mediated tyrosine phosphorylation of the RNA polymerase II carboxyl-terminal repeated domain. Proc Natl Acad Sci USA. 1995;92:1555–1559. doi: 10.1073/pnas.92.5.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chen G, Yuan SS, Liu W, Xu Y, Trujillo K, Song B, Cong F, Goff SP, Wu Y, Arlinghaus R, Baltimore D, Gasser PJ, Park MS, Sung P, Lee EY. Radiation-induced assembly of Rad51 and Rad52 recombination complex requires ATM and c-Abl. J Biol Chem. 1999;274:12748–12752. doi: 10.1074/jbc.274.18.12748. [DOI] [PubMed] [Google Scholar]
- 114.Yuan ZM, Huang Y, Ishiko T, Nakada S, Utsugisawa T, Kharbanda S, Wang R, Sung P, Shinohara A, Weichselbaum R, Kufe D. Regulation of Rad51 function by c-Abl in response to DNA damage. J Biol Chem. 1998;273:3799–3802. doi: 10.1074/jbc.273.7.3799. [DOI] [PubMed] [Google Scholar]
- 115.Popova M, Shimizu H, Yamamoto K, Lebechec M, Takahashi M, Fleury F. Detection of c-Abl kinase-promoted phosphorylation of Rad51 by specific antibodies reveals that Y54 phosphorylation is dependent on that of Y315. FEBS Lett. 2009;583:1867–1872. doi: 10.1016/j.febslet.2009.04.044. [DOI] [PubMed] [Google Scholar]
- 116.Tani K, Sato S, Sukezane T, Kojima H, Hirose H, Hanafusa H, Shishido T. Abl interactor 1 promotes tyrosine 296 phosphorylation of mammalian enabled (Mena) by c-Abl kinase. J Biol Chem. 2003;278:21685–21692. doi: 10.1074/jbc.M301447200. [DOI] [PubMed] [Google Scholar]
- 117.Zukerberg LR, Patrick GN, Nikolic M, Humbert S, Wu CL, Lanier LM, Gertler FB, Vidal M, Van Etten RA, Tsai LH. Cables links Cdk5 and c-Abl and facilitates Cdk5 tyrosine phosphorylation, kinase upregulation, and neurite outgrowth. Neuron. 2000;26:633–646. doi: 10.1016/s0896-6273(00)81200-3. [DOI] [PubMed] [Google Scholar]
- 118.LaMontagne KR, Jr, Flint AJ, Franza BR, Jr, Pandergast AM, Tonks NK. Protein tyrosine phosphatase 1B antagonizes signalling by oncoprotein tyrosine kinase p210 bcr-abl in vivo. Mol Cell Biol. 1998;18:2965–2975. doi: 10.1128/mcb.18.5.2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Motiwala T, Majumder S, Ghoshal K, Kutay H, Datta J, Roy S, Lucas DM, Jacob ST. PTPROt inactivates the oncogenic fusion protein BCR/ABL and suppresses transformation of K562 cells. J Biol Chem. 2009;284:455–464. doi: 10.1074/jbc.M802840200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 120.Kharbanda S, Bharti A, Pei D, Wang J, Pandey P, Ren R, Weichselbaum R, Walsh CT, Kufe D. The stress response to ionizing radiation involoves c-Abl-dependent phosphorylation of SHPTP1. Proc Natl Acad Sci USA. 1996;93:6898–6901. doi: 10.1073/pnas.93.14.6898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bruecher-Encke B, Griffin JD, Neel BG, Lorenz U. Role of the tyrosine phosphatase SHP-1 in K562 cell differentiation. Leukemia. 2001;15:1424–1432. doi: 10.1038/sj.leu.2402214. [DOI] [PubMed] [Google Scholar]
- 122.Mitra S, Beach C, Feng GS, Plattner R. SHP-2 is a novel target of Abl kinases during cell proliferation. J Cell Sci. 2008;121:3335–3346. doi: 10.1242/jcs.035691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lim YM, Wong S, Lau G, Witte ON, Colicelli J. BCR/ABL inhibition by an escort/phosphatase fusion protein. Proc Natl Acad Sci USA. 2000;97:12233–12238. doi: 10.1073/pnas.210253497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cong F, Spencer S, Côté JF, Wu Y, Tremblay ML, Lasky LA, Goff SP. Cytoskeletal protein PSTPIP1 directs the PEST-type protein tyrosine phosphatase to the c-Abl kinase to mediate Abl dephosphorylation. Mol Cell. 2000;6:1413–1423. doi: 10.1016/s1097-2765(00)00138-6. [DOI] [PubMed] [Google Scholar]
- 125.Fan PD, Cong F, Goff SP. Homo- and hetero-oligomerization of the c-Abl kinase and Abelson-interactor-1. Cancer Res. 2003;63:873–877. [PubMed] [Google Scholar]
- 126.Smith KM, Van Etten RA. Activation of c-Abl kinase activity and transformation by a chemical inducer of dimerization. J Biol Chem. 2001;276:24372–24379. doi: 10.1074/jbc.M100786200. [DOI] [PubMed] [Google Scholar]
- 127.Cao C, Leng Y, Li C, Kufe D. Functional interaction between the c-Abl and Arg protein-tyrosine kinases in the oxidative stress response. J Biol Chem. 2003;278:12961–12967. doi: 10.1074/jbc.M300058200. [DOI] [PubMed] [Google Scholar]
- 128.Van Etten RA, Jackson PK, Baltimore D, Sanders MC, Matsudaira PT, Janmey PA. The COOH terminus of the c-Abl tyrosine kinase contains distinct F- and G-actin binding domains with bundling activity. J Cell Biol. 1994;124:325–340. doi: 10.1083/jcb.124.3.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wang Y, Miller AL, Mooseker MS, Koleske AJ. The Abl-related gene (Arg) nonreceptor tyrosine kinase uses two F-actin-binding domains to bundle F-actin. Proc Natl Acad Sci USA. 2001;98:14865–14870. doi: 10.1073/pnas.251249298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Woodring PJ, Hunter T, Wang JY. Inhibition of c-Abl tyrosine kinase activity by filamentous actin. J Biol Chem. 2001;276:27104–27110. doi: 10.1074/jbc.M100559200. [DOI] [PubMed] [Google Scholar]
- 131.Miller AL, Wang Y, Mooseker MS, Koleske AJ. The Abl-related gene (Arg) requires its F-actin-microtubule cross-linking activity to regulate lamellipodial dynamics during fibroblast adhesion. J Cell Biol. 2004;165:407–419. doi: 10.1083/jcb.200308055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Dai Z, Pendergast AM. Abi-2, a novel SH3-containing protein interacts with the c-Abl tyrosine kinase and modulates c-Abl transforming activity. Genes Dev. 1995;9:2569–2582. doi: 10.1101/gad.9.21.2569. [DOI] [PubMed] [Google Scholar]
- 133.Shi Y, Alin K, Goff SP. Abl-interactor-1, a novel SH3 protein binding to the carboxy-terminal portion of the Abl protein, suppresses v-abl transforming activity. Genes Dev. 1995;9:2583–2597. doi: 10.1101/gad.9.21.2583. [DOI] [PubMed] [Google Scholar]
- 134.Xiong X, Cui P, Hossain S, Xu R, Warner B, Guo X, An X, Debnath AK, Cowburn D, Kotula L. Allosteric inhibition of the nonMyristoylated c-Abl tyrosine kinase by phosphopeptides derived from Abi1/Hssh3bp1. Biochim Biophys Acta. 2008;1783:737–747. doi: 10.1016/j.bbamcr.2008.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Leng Y, Zhang J, Badour K, Arpaia E, Freeman S, Cheung P, Siu M, Siminovitch K. Abelson-interactor-1 promotes WAVE2 membrane translocation and Abelson-mediated tyrosine phosphorylation required for WAVE2 activation. Proc Natl Acad Sci USA. 2005;102:1098–1103. doi: 10.1073/pnas.0409120102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lin TY, Huang CH, Chou WG, Juang JL. Abi enhances Abl-mediated CDC2 phosphorylation and inactivation. J Biomed Sci. 2004;11:902–910. doi: 10.1007/BF02254375. [DOI] [PubMed] [Google Scholar]
- 137.Lebensohn AM, Kirschner MW. Activation of the WAVE complex by coincident signals controls actin assembly. Mol Cell. 2009;36:512–524. doi: 10.1016/j.molcel.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Juang JL, Hoffmann FM. Drosophila Abelson interacting protein (dAbi) is a positive regulator of Abelson tyrosine kinase activity. Oncogene. 1999;18:5138–5147. doi: 10.1038/sj.onc.1202911. [DOI] [PubMed] [Google Scholar]
- 139.Cao X, Tanis KQ, Koleske AJ, Colicelli J. Enhancement of ABL kinase catalytic efficiency by a direct binding regulator is independent of other regulatory mechanisms. J Biol Chem. 2008;283:31401–31407. doi: 10.1074/jbc.M804002200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wen ST, Van Etten RA. The PAG gene product, a stress-induced protein with antioxidant properties, is an Abl SH3-binding protein and a physiological inhibitor of c-Abl tyrosine kinase activity. Genes Dev. 1997;11:2456–2467. doi: 10.1101/gad.11.19.2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lin J, Sun T, Ji L, Deng W, Roth J, Minna J, Arlinghaus R. Oncogenic activation of c-Abl in non-small cell lung cancer cells lacking FUS1 expression: Inhibition of c-Abl by the tumor suppressor gene product Fus1. Oncogene. 2007;26:6989–6996. doi: 10.1038/sj.onc.1210500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Feller SM, Knudsen B, Hanafusa H. c-Abl kinase regulates the protein binding activity of c-Crk. EMBO J. 1994;13:2341–2351. doi: 10.1002/j.1460-2075.1994.tb06518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ren R, Ye ZS, Baltimore D. Abl protein-tyrosine kinase selects the Crk adapter as a substrate using SH3-binding sites. Genes Dev. 1994;8:783–795. doi: 10.1101/gad.8.7.783. [DOI] [PubMed] [Google Scholar]
- 144.Wang B, Mysliwiec T, Feller SM, Knudsen B, Hanafusa H, Kruh GD. Proline-rich sequences mediate the interaction of the Arg protein tyrosine kinase with Crk. Oncogene. 1996;13:1379–1385. [PubMed] [Google Scholar]
- 145.Antoku S, Saksela K, Rivera GM, Mayer BJ. A crucial role in cell spreading for the interaction of Abl PxxP motifs with Crk and Nck adaptors. J Cell Sci. 2008;121:3071–3082. doi: 10.1242/jcs.031575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Reichman C, Singh K, Liu Y, Singh S, Li H, Fajardo JE, Fiser A, Birge RB. Transactivation of Abl by the Crk II adapter protein requires a PNAY sequence in the Crk C-terminal SH3 domain. Oncogene. 2005;24:8187–8199. doi: 10.1038/sj.onc.1208988. [DOI] [PubMed] [Google Scholar]
- 147.Wang B, Mysliwiec T, Krainc D, Jensen RA, Sonoda G, Testa JR, Golemis EA, Kruh GD. Identification of ArgBP1, an Arg protein tyrosine kinase binding protein that is the human homologue of a CNS-specific Xenopus gene. Oncogene. 1996;12:1921–1929. [PubMed] [Google Scholar]
- 148.Mitsushima M, Ueda K, Kioka N. Vinexin beta regulates the phosphorylation of epidermal growth factor receptor on the cell surface. Genes Cells. 2006;11:971–982. doi: 10.1111/j.1365-2443.2006.00995.x. [DOI] [PubMed] [Google Scholar]
- 149.Cong F, Yuan B, Goff SP. Characterization of a novel member of the DOK family that binds and modulates Abl signaling. Mol Cell Biol. 1999;19:8314–8325. doi: 10.1128/mcb.19.12.8314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Master Z, Tran J, Bishnoi A, Chen SH, Ebos JM, Van Slyke P, Kerbel RS, Dumont DJ. Dok-R binds c-Abl and regulates Abl kinase activity and mediates cytoskeletal reorganization. J Biol Chem. 2003;278:30170–30179. doi: 10.1074/jbc.M301339200. [DOI] [PubMed] [Google Scholar]
- 151.Renshaw MW, Lewis JM, Schwartz MA. The c-Abl tyrosine kinase contributes to the transient activation of MAP kinase in cells plated on fibronectin. Oncogene. 2000;19:3216–3219. doi: 10.1038/sj.onc.1203667. [DOI] [PubMed] [Google Scholar]
- 152.Raffel GD, Parmar K, Rosenberg N. In vivo association of v-Abl with Shc mediated by a non-phosphotyrosine-dependent SH2 interaction. J Biol Chem. 1996;271:4640–4645. doi: 10.1074/jbc.271.9.4640. [DOI] [PubMed] [Google Scholar]
- 153.Weaver AM, Young ME, Lee WL, Cooper JA. Integration of signals to the Arp2/3 complex. Curr Opin Cell Biol. 2003;15:23–30. doi: 10.1016/s0955-0674(02)00015-7. [DOI] [PubMed] [Google Scholar]
- 154.Lapetina S, Mader CC, Machida K, Mayer BJ, Koleske AJ. Arg interacts with cortactin to promote adhesion-dependent cell edge protrusion. J Cell Biol. 2009;185:503–519. doi: 10.1083/jcb.200809085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Sparks AB, Rider JE, Hoffman NG, Fowlkes DM, Quillam LA, Kay BK. Distinct ligand preferences of Src homology 3 domains from Src, Yes, Abl, Cortactin, p53bp2, PLCgamma, Crk, and Grb2. Proc Natl Acad Sci USA. 1996;93:1540–1544. doi: 10.1073/pnas.93.4.1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hou T, Chen K, McLaughlin WA, Lu B, Wang W. Computational analysis and prediction of the binding motif and protein interacting partners of the Abl SH3 domain. PLOS Comput Biol. 2006;2:e1. doi: 10.1371/journal.pcbi.0020001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Westphal RS, Soderling SH, Alto NM, Langeberg LK, Scott JD. Scar/WAVE-1, a Wiskott-Aldrich syndrome protein, assembles an actin-associated multi-kinase scaffold. EMBO J. 2000;19:4589–4600. doi: 10.1093/emboj/19.17.4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kato Y, Nagata K, Takahashi M, Lian L, Herrero JJ, Sudol M, Tanokura M. Common mechanism of ligand recognition by group II/III WW domains: Redefining their functional classification. J Biol Chem. 2004;279:31833–31841. doi: 10.1074/jbc.M404719200. [DOI] [PubMed] [Google Scholar]
- 159.Perkinton MS, Standen CL, Lau KF, Kesavapany S, Byers HL, Ward M, McLoughlin DM, Miller CC. The c-Abl tyrosine kinase phosphorylates the Fe65 adaptor protein to stimulate Fe65/amyloid precursor protein nuclear signaling. J Biol Chem. 2004;279:22084–22091. doi: 10.1074/jbc.M311479200. [DOI] [PubMed] [Google Scholar]
- 160.Zambrano N, Bruni P, Minopoli G, Mosca R, Molino D, Russo C, Schettini G, Sudol M, Russo T. The beta-amyloid precursor protein APP is tyrosine-phosphorylated in cells expressing a constitutively active form of the Abl protoncogene. J Biol Chem. 2001;276:19787–19792. doi: 10.1074/jbc.M100792200. [DOI] [PubMed] [Google Scholar]
- 161.Meiyappan M, Birrane G, Ladias JA. Structural basis for polyproline recognition by the FE65 WW domain. J Mol Biol. 2007;372:970–980. doi: 10.1016/j.jmb.2007.06.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Woodring PJ, Hunter T, Wang JY. Regulation of F-actin-dependent processes by the Abl family of tyrosine kinases. J Cell Sci. 2003;116:2613–2626. doi: 10.1242/jcs.00622. [DOI] [PubMed] [Google Scholar]
- 163.Shaul Y, Ben-Yehoyada M. Role of c-Abl in the DNA damage stress response. Cell Res. 2005;15:33–35. doi: 10.1038/sj.cr.7290261. [DOI] [PubMed] [Google Scholar]
- 164.Wang JY. Regulation of cell death by the Abl tyrosine kinase. Oncogene. 2000;19:5643–5650. doi: 10.1038/sj.onc.1203878. [DOI] [PubMed] [Google Scholar]
- 165.Backert S, Feller SM, Wessler S. Emerging roles of Abl family tyrosine kinases in microbial pathogenesis. Trends Biochem Sci. 2008;33:80–90. doi: 10.1016/j.tibs.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 166.Hernández SE, Krishnaswami M, Miller AL, Koleske AJ. How do Abl family kinases regulate cell shape and movement? Trends Cell Biol. 2004;14:36–44. doi: 10.1016/j.tcb.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 167.Woodring PJ, Litwack ED, O’Leary DD, Lucero GR, Wang JY, Hunter T. Modulation of the F-actin cytoskeleton by c-Abl tyrosine kinase in cell spreading and neurite extension. J Cell Biol. 2002;156:879–892. doi: 10.1083/jcb.200110014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Finn AJ, Feng G, Pendergast AM. Postsynaptic requirement for Abl kinases in assembly of the neuromuscular junction. Nat Neurosci. 2003;6:717–723. doi: 10.1038/nn1071. [DOI] [PubMed] [Google Scholar]
- 169.Huang Y, Comiskey EO, Dupree RS, Li S, Koleske AJ, Burkhardt JK. The c-Abl tyrosine kinase regulates actin remodeling at the immune synapse. Blood. 2008;112:111–119. doi: 10.1182/blood-2007-10-118232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Sfakianos MK, Eisman A, Gourley SL, Bradley WD, Scheetz AJ, Settleman J, Taylor JR, Greer CA, Williamson A, Koleske AJ. Inhibition of Rho via Arg and p190RhoGAP in the postnatal mouse hippocampus regulates dendritic spine maturation, synapse and dendrite stability, and behavior. J Neurosci. 2007;27:10982–10992. doi: 10.1523/JNEUROSCI.0793-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Michael M, Vehlow A, Navarro C, Krause M. c-Abl, Lamellipodin, and Ena/VASP proteins cooperate in dorsal ruffling of fibroblasts and axonal morphogenesis. Curr Biol. 2010;20:783–791. doi: 10.1016/j.cub.2010.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Hu H, Bliss JM, Wang Y, Colicelli J. RIN1 is an ABL tyrosine kinase activator and a regulator of epithelial-cell adhesion and migration. Curr Biol. 2005;15:815–823. doi: 10.1016/j.cub.2005.03.049. [DOI] [PubMed] [Google Scholar]
- 173.Kain KH, Klemke RL. Inhibition of cell migration by Abl family tyrosine kinases through uncoupling of Crk-CAS complexes. J Biol Chem. 2001;276:16185–16192. doi: 10.1074/jbc.M100095200. [DOI] [PubMed] [Google Scholar]
- 174.Zandy NL, Playford M, Pendergast AM. Abl tyrosine kinases regulate cell-cell adhesion through Rho GTPases. Proc Natl Acad Sci USA. 2007;104:17686–17691. doi: 10.1073/pnas.0703077104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Sossey-Alaoui K, Li X, Cowell JK. c-Abl-mediated phosphorylation of WAVE3 is required for lamellipodia formation and cell migration. J Biol Chem. 2007;282:26257–26265. doi: 10.1074/jbc.M701484200. [DOI] [PubMed] [Google Scholar]
- 176.Nolz JC, Nacusi LP, Segovis CM, Medeiros RB, Mitchell JS, Shimizu Y, Billadeau DD. The WAVE2 complex regulates T cell receptor signaling to integrins via Abl- and CrkL-C3G-mediated activation of Rap1. J Cell Biol. 2008;182:1231–1244. doi: 10.1083/jcb.200801121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Chodniewicz D, Klemke RL. Regulation of integrin-mediated cellular responses through assembly of a CAS/Crk scaffold. Biochim Biophys Acta. 2004;1692:63–76. doi: 10.1016/j.bbamcr.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 178.Takino T, Tamura M, Miyamori H, Araki M, Matsumoto K, Sato H, Yamada KM. Tyrosine phosphorylation of the CrkII adaptor protein modulates cell migration. J Cell Sci. 2003;116:3145–3155. doi: 10.1242/jcs.00632. [DOI] [PubMed] [Google Scholar]
- 179.Cui L, Chen C, Xu T, Zhang J, Shang X, Luo J, Chen L, Ba X, Zeng X. c-Abl kinase is required for beta 2 integrin-mediated neutrophil adhesion. J Immunol. 2009;182:3233–3242. doi: 10.4049/jimmunol.0802621. [DOI] [PubMed] [Google Scholar]
- 180.Baruzzi A, Iacobucci I, Soverini S, Lowell CA, Martinelli G, Berton G. c-Abl and Src-family kinases cross-talk in regulation of myeloid cell migration. FEBS Lett. 2010;584:15–21. doi: 10.1016/j.febslet.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kaksonen M, Toret CP, Drubin DG. A modular design for the clathrin- and actin-mediated endocytosis machinery. Cell. 2005;123:305–320. doi: 10.1016/j.cell.2005.09.024. [DOI] [PubMed] [Google Scholar]
- 182.Saffarian S, Cocucci E, Kirchhausen T, Hughson F. Distinct dynamics of endocytic clathrin-coated pits and coated plaques. PLoS Biol. 2009;7:e1000191. doi: 10.1371/journal.pbio.1000191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Yarar D, Waterman-Storer CM, Schmid SL. A dynamic actin cytoskeleton functions at multiple stages of clathrin-mediated endocytosis. Mol Biol Cell. 2005;16:964–975. doi: 10.1091/mbc.E04-09-0774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Tanos B, Pendergast AM. Abl tyrosine kinase regulates endocytosis of the epidermal growth factor receptor. J Biol Chem. 2006;281:32714–32723. doi: 10.1074/jbc.M603126200. [DOI] [PubMed] [Google Scholar]
- 185.Srinivasan D, Kaetzel DM, Plattner R. Reciprocal regulation of Abl and receptor tyrosine kinases. Cell Signal. 2009;21:1143–1150. doi: 10.1016/j.cellsig.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Jacob M, Todd LA, Majumdar RS, Li Y, Yamamoto K, Puré E. Endogenous cAbl regulates receptor endocytosis. Cell Signal. 2009;21:1308–1316. doi: 10.1016/j.cellsig.2009.03.016. [DOI] [PubMed] [Google Scholar]
- 187.Lajoie P, Goetz JG, Dennis JW, Nabi IR. Lattices, rafts, and scaffolds: Domain regulation of receptor signaling at the plasma membrane. J Cell Biol. 2009;185:381–385. doi: 10.1083/jcb.200811059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Han L, Colicelli J. A human protein selected for interference with Ras function interacts directly with Ras and competes with Raf1. Mol Cell Biol. 1995;15:1318–1323. doi: 10.1128/mcb.15.3.1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Tall GG, Barbieri MA, Stahl PD, Horazdovsky BF. Ras-activated endocytosis is mediated by the Rab5 guanine nucleotide exchange activity of RIN1. Dev Cell. 2001;1:73–82. doi: 10.1016/s1534-5807(01)00008-9. [DOI] [PubMed] [Google Scholar]
- 190.Klein IK, Predescu DN, Sharma T, Knezevic I, Malik AB, Predescu S. Intersectin-2L regulates caveola endocytosis secondary to Cdc42-mediated actin polymerization. J Biol Chem. 2009;284:25953–25961. doi: 10.1074/jbc.M109.035071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Graux C, Cools J, Melotte C, Quentmeier H, Ferrando A, Levine R, Vermeesch JR, Stul M, Dutta B, Boeckx N, Bosly A, Heimann P, Uyttebroeck A, Mentens N, Somers R, MacLeod RA, Drexler HG, Look AT, Gilliland DG, Michaux L, Vandenberghe P, Wlodarska I, Marynen P, Hagemeijer A. Fusion of NUP214 to ABL1 on amplified episomes in T-cell acute lymphoblastic leukemia. Nat Genet. 2004;36:1084–1089. doi: 10.1038/ng1425. [DOI] [PubMed] [Google Scholar]
- 192.Yogalingam G, Pendergast AM. Abl kinases regulate autophagy by promoting the trafficking and function of lysosomal components. J Biol Chem. 2008;283:35941–35953. doi: 10.1074/jbc.M804543200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Lewis JM, Baskaran R, Taagepera S, Schwartz MA, Wang JY. Integrin regulation of c-Abl tyrosine kinase activity and cytoplasmic-nuclear transport. Proc Natl Acad Sci USA. 1996;93:15174–15179. doi: 10.1073/pnas.93.26.15174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Miao YJ, Wang JYJ. Binding of A/T-rich DNA by three high mobility group-like domains in c-Abl tyrosine kinase. J Biol Chem. 1996;271:22823–22830. doi: 10.1074/jbc.271.37.22823. [DOI] [PubMed] [Google Scholar]
- 195.Kim WJ, Rajasekaran B, Brown KD. MLH1- and ATM-dependent MAPK signaling is activated through c-Abl in response to the alkylator N-methyl-N′-nitro-N′-nitrosoguanidine. J Biol Chem. 2007;282:32021–32031. doi: 10.1074/jbc.M701451200. [DOI] [PubMed] [Google Scholar]
- 196.Sawyers CL, McLaughlin J, Goga A, Havlik M, Witte O. The nuclear tyrosine kinase c-Abl negatively regulates cell growth. Cell. 1994;77:121–131. doi: 10.1016/0092-8674(94)90240-2. [DOI] [PubMed] [Google Scholar]
- 197.Yuan ZM, Huang Y, Ishiko T, Kharbanda S, Weichselbaum R, Kufe D. Regulation of DNA damage-induced apoptosis by the c-Abl tyrosine kinase. Proc Natl Acad Sci USA. 1997;94:1437–1440. doi: 10.1073/pnas.94.4.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Van Etten RA. Mechanisms of transformation by the BCR-ABL oncogene: New perspectives in the post-imatinib era. Leuk Res. 2004;28(Suppl 1):S21–S28. doi: 10.1016/j.leukres.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 199.Westbrook CA, Le Beau MM, Diaz MO, Groffen J, Rowley JD. Chromosomal localization and characterization of c-abl in the t(6;9) of acute nonlymphocytic leukemia. Proc Natl Acad Sci USA. 1985;82:8742–8746. doi: 10.1073/pnas.82.24.8742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Strick R, Zhang Y, Emmanuel N, Strissel PL. Common chromatin structures at breakpoint cluster regions may lead to chromosomal translocations found in chronic and acute leukemias. Hum Genet. 2006;119:479–495. doi: 10.1007/s00439-006-0146-9. [DOI] [PubMed] [Google Scholar]
- 201.Mattarucchi E, Guerini V, Rambaldi A, Campiotti L, Venco A, Pasquali F, Lo Curto F, Porta G. Microhomologies and interspersed repeat elements at genomic breakpoints in chronic myeloid leukemia. Genes Chromosomes Cancer. 2008;47:625–632. doi: 10.1002/gcc.20568. [DOI] [PubMed] [Google Scholar]
- 202.Carroll WL, Bhojwani D, Min DJ, Raetz E, Relling M, Davies S, Downing JR, Willman CL, Reed JC. Pediatric acute lymphoblastic leukemia. Hematology Am Soc Hematol Educ Program. 2003;2003:102–131. doi: 10.1182/asheducation-2003.1.102. [DOI] [PubMed] [Google Scholar]
- 203.Faderl S, Kantarjian HM, Talpaz M, Estrov Z. Clinical significance of cytogenetic abnormalities in adult acute lymphoblastic leukemia. Blood. 1998;91:3995–4019. [PubMed] [Google Scholar]
- 204.Demehri S, Paschka P, Schultheis B, Lange T, Koizumi T, Sugimoto T, Branford S, Lim LC, Kegel T, Martinelli G, Hochhaus A, Druker BJ, Deininger MWN. e8a2 BCR-ABL: More frequent than other atypical BCR-ABL variants? Leukemia. 2005;19:681–684. doi: 10.1038/sj.leu.2403604. [DOI] [PubMed] [Google Scholar]
- 205.Melo JV. The diversity of BCR-ABL fusion proteins and their relationship to leukemia phenotype. Blood. 1996;88:2375–2384. [PubMed] [Google Scholar]
- 206.De Keersmaecker K, Graux C, Odero MD, Mentens N, Somers R, Maertens J, Wlodarska I, Vandenberghe P, Hagemeijer A, Marynen P, Cools J. Fusion of EML1 to ABL1 in T-cell acute lymphoblastic leukemia with cryptic t(9;14)(q34;q32) Blood. 2005;105:4849–4852. doi: 10.1182/blood-2004-12-4897. [DOI] [PubMed] [Google Scholar]
- 207.Janssen JW, Ridge SA, Papadopoulos P, Cotter F, Ludwig WD, Fonatsch C, Rieder H, Ostertag W, Bartram CR, Wiedemann LM. The fusion of TEL and ABL in human acute lymphoblastic leukaemia is a rare event. Br J Haematol. 1995;90:222–224. doi: 10.1111/j.1365-2141.1995.tb03407.x. [DOI] [PubMed] [Google Scholar]
- 208.Iijima Y, Ito T, Oikawa T, Eguchi M, Eguchi-Ishimae M, Kamada N, Kishi K, Asano S, Sakaki Y, Sato Y. A new ETV6/TEL partner gene, ARG (ABL-related gene or ABL2), identified in an AML-M3 cell line with a t(1;12)(q25;p13) translocation. Blood. 2000;95:2126–2131. [PubMed] [Google Scholar]
- 209.Kelliher MA, McLaughlin J, Witte ON, Rosenberg N. Induction of a chronic myelogenous leukemia-like syndrome in mice with v-abl and BCR/ABL. Proc Natl Acad Sci USA. 1990;87:6649–6653. doi: 10.1073/pnas.87.17.6649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Clark SS, Chen E, Fizzotti M, Witte ON, Malkovska V. BCR-ABL and v-abl oncogenes induce distinct patterns of thymic lymphoma involving different lymphocyte subsets. J Virol. 1993;67:6033–6046. doi: 10.1128/jvi.67.10.6033-6046.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Lugo TG, Witte ON. The BCR-ABL oncogene transforms Rat-1 cells and cooperates with v-myc. Mol Cell Biol. 1989;9:1263–1270. doi: 10.1128/mcb.9.3.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Lin J, Arlinghaus R. Activated c-Abl tyrosine kinase in malignant solid tumors. Oncogene. 2008;27:4385–4391. doi: 10.1038/onc.2008.86. [DOI] [PubMed] [Google Scholar]
- 213.Srinivasan D, Plattner R. Activation of Abl tyrosine kinases promotes invasion of aggressive breast cancer cells. Cancer Res. 2006;66:5648–5655. doi: 10.1158/0008-5472.CAN-06-0734. [DOI] [PubMed] [Google Scholar]
- 214.Duesberg PH, Wang LH, Beemon K, Kawai S, Hanafusa H. Sequences and functions of Rous sarcoma virus RNA. Hamatol Bluttransfus. 1976;19:327–340. doi: 10.1007/978-3-642-87524-3_34. [DOI] [PubMed] [Google Scholar]
- 215.Levy JB, Iba H, Hanafusa H. Activation of the transforming potential of p60c-src by a single amino acid change. Proc Natl Acad Sci USA. 1986;83:4228–4232. doi: 10.1073/pnas.83.12.4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Li T, Tsukada S, Satterthwaite A, Havlik MH, Park H, Takatsu K, Witte ON. Activation of Bruton’s tyrosine kinase (BTK) by a point mutation in its pleckstrin homology (PH) domain. Immunity. 1995;2:451–460. doi: 10.1016/1074-7613(95)90026-8. [DOI] [PubMed] [Google Scholar]
- 217.Russello SV, Shore SK. SRC in human carcinogenesis. Front Biosci. 2004;9:139–144. doi: 10.2741/1138. [DOI] [PubMed] [Google Scholar]
- 218.Danhauser-Riedl S, Warmuth M, Druker BJ, Emmerich B, Hallek M. Activation of Src kinases p53/56lyn and p59hck by p210bcr/abl in myeloid cells. Cancer Res. 1996;56:3589–3596. [PubMed] [Google Scholar]
- 219.Hu Y, Liu Y, Pelletier S, Buchdunger E, Warmuth M, Fabbro D, Hallek M, Van Etten RA, Li S. Requirement of Src kinases Lyn, Hck and Fgr for BCR-ABL1-induced B-lymphoblastic leukemia but not chronic myeloid leukemia. Nat Genet. 2004;36:453–461. doi: 10.1038/ng1343. [DOI] [PubMed] [Google Scholar]
- 220.Rubbi L, Brown L, Galvan E, Komisopoulou E, Chen SS, Titz B, Skaggs BJ, Pellegrini M, Graeber TG. Kinase subnetwork delineation by global phosphoproteomics reveals Bcr-Abl-mediated overpowering of Src negative feedback mechanisms. 2010 (submitted) [Google Scholar]
- 221.Tognon CE, Mackereth CD, Somasiri AM, McIntosh LP, Sorensen PH. Mutations in the SAM domain of the ETV6-NTRK3 chimeric tyrosine kinase block polymerization and transformation activity. Mol Cell Biol. 2004;24:4636–4650. doi: 10.1128/MCB.24.11.4636-4650.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Zhao X, Ghaffari S, Lodish H, Malashkevich VN, Kim PS. Structure of the Bcr-Abl oncoprotein oligomerization domain. Nat Struct Biol. 2002;9:117–120. doi: 10.1038/nsb747. [DOI] [PubMed] [Google Scholar]
- 223.Van Etten RA, Jackson P, Baltimore D. The mouse type IV c-abl gene product is a nuclear protein, and activation of transforming ability is associated with cytoplasmic localization. Cell. 1989;58:669–678. doi: 10.1016/0092-8674(89)90102-5. [DOI] [PubMed] [Google Scholar]
- 224.Afar DE, Han L, McLaughlin J, Wong S, Dhaka A, Parmar K, Rosenberg N, Witte ON, Colicelli J. Regulation of the oncogenic activity of BCR-ABL by a tightly bound substrate protein RIN1. Immunity. 1997;6:773–782. doi: 10.1016/s1074-7613(00)80452-5. [DOI] [PubMed] [Google Scholar]
- 225.Liedtke M, Pandey P, Kumar S, Kharbanda S, Kufe D. Regulation of Bcr-Abl-induced SAP kinase activity and transformation by the SHPTP1 protein tyrosine phosphatase. Oncogene. 1998;17:1889–1892. doi: 10.1038/sj.onc.1202117. [DOI] [PubMed] [Google Scholar]
- 226.Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J. Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science. 2000;289:1938–1942. doi: 10.1126/science.289.5486.1938. [DOI] [PubMed] [Google Scholar]
- 227.Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R, Ohno-Jones S, Sawyers CL. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–1037. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 228.Quintás-Cardama A, Kantarjian H, Cortes J. Imatinib and beyond—exploring the full potential of targeted therapy for CML. Nat Rev Clin Oncol. 2009;6:535–543. doi: 10.1038/nrclinonc.2009.112. [DOI] [PubMed] [Google Scholar]
- 229.Ramirez P, DiPersio JF. Therapy options in imatinib failures. Oncologist. 2008;13:424–434. doi: 10.1634/theoncologist.2007-0170. [DOI] [PubMed] [Google Scholar]
- 230.Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, Sawyers CL. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–880. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 231.Shah NP, Nicoll JM, Nagar B, Gorre ME, Paquette RL, Kuriyan J, Sawyers CL. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2:117–125. doi: 10.1016/s1535-6108(02)00096-x. [DOI] [PubMed] [Google Scholar]
- 232.Azam M, Latek RR, Daley GQ. Mechanisms of autoinhibition and STI-571/imatinib resistance revealed by mutagenesis of BCR-ABL. Cell. 2003;112:831–843. doi: 10.1016/s0092-8674(03)00190-9. [DOI] [PubMed] [Google Scholar]
- 233.Sherbenou DW, Hantschel O, Kaupe I, Willis S, Bumm T, Turaga LP, Lange T, Dao K-H, Press RD, Druker BJ, Superti-Furga G, Deininger MW. BCR-ABL SH3-SH2 domain mutations in chronic myeloid leukemia patients on imatinib. Blood. 2010 doi: 10.1182/blood-2008-10-183665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Yanada M, Ohno R, Naoe T. Recent advances in the treatment of Philadelphia chromosome-positive acute lymphoblastic leukemia. Int J Hematol. 2009;89:3–13. doi: 10.1007/s12185-008-0223-z. [DOI] [PubMed] [Google Scholar]
- 235.Lombardo LJ, Lee FY, Chen P, Norris D, Barrish JC, Behnia K, Castaneda S, Cornelius LA, Das J, Doweyko AM, Fairchild C, Hunt JT, Inigo I, Johnston K, Kamath A, Kan D, Klei H, Marathe P, Pang S, Peterson R, Pitt S, Schieven GL, Schmidt RJ, Tokarski J, Wen ML, Wityak J, Borzilleri RM. Discovery of N-(2-chloro-6-methyl- phenyl)-2-(6-(4-(2-hydroxyethyl)-piperazin-1-yl)-2-methylpyrimidin-4- ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J Med Chem. 2004;47:6658–6661. doi: 10.1021/jm049486a. [DOI] [PubMed] [Google Scholar]
- 236.Shah NP, Skaggs BJ, Branford S, Hughes TP, Nicoll JM, Paquette RL, Sawyers CL. Sequential ABL kinase inhibitor therapy selects for compound drug-resistant BCR-ABL mutations with altered oncogenic potency. J Clin Invest. 2007;117:2562–2569. doi: 10.1172/JCI30890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Liu Y, Shah K, Yang F, Witucki L, Shokat KM. A molecular gate which controls unnatural ATP analogue recognition by the tyrosine kinase v-Src. Bioorg Med Chem. 1998;6:1219–1226. doi: 10.1016/s0968-0896(98)00099-6. [DOI] [PubMed] [Google Scholar]
- 238.O’Hare T, Shakespeare WC, Zhu X, Eide CA, Rivera VM, Wang F, Adrian LT, Zhou T, Huang WS, Xu Q, Metcalf CA, 3rd, Tyner JW, Loriaux MM, Corbin AS, Wardwell S, Ning Y, Keats JA, Wang Y, Sundaramoorthi R, Thomas M, Zhou D, Snodgrass J, Commodore L, Sawyer TK, Dalgarno DC, Deininger MW, Druker BJ, Clackson T. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell. 2009;16:401–412. doi: 10.1016/j.ccr.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Zhang J, Adrián FJ, Jahnke W, Cowan-Jacob SW, Li AG, Iacob RE, Sim T, Powers J, Dierks C, Sun F, Guo GR, Ding Q, Okram B, Choi Y, Wojciechowski A, Deng X, Liu G, Fendrich G, Strauss A, Vajpai N, Grzesiek S, Tuntland T, Liu Y, Bursulaya B, Azam M, Manley PW, Engen JR, Daley GQ, Warmuth M, Gray NS. Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature. 2010;463:501–506. doi: 10.1038/nature08675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Weisberg E, Deng X, Choi HG, Barrett R, Adamia S, Ray A, Moreno D, Kung AL, Gray N, Griffin JD. Beneficial effects of combining a type II ATP competitive inhibitor with an allosteric competitive inhibitor of BCR-ABL for the treatment of imatinib-sensitive and imatinib-resistant CML. Leukemia. 2010;24:1375–1378. doi: 10.1038/leu.2010.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Chen R, Gandhi V, Plunkett W. A sequential blockade strategy for the design of combination therapies to overcome oncogene addiction in chronic myelogenous leukemia. Cancer Res. 2006;66:10959–10966. doi: 10.1158/0008-5472.CAN-06-1216. [DOI] [PubMed] [Google Scholar]
- 242.Wu LX, Xu JH, Zhang KZ, Lin Q, Huang XW, Wen CX, Chen YZ. Disruption of the Bcr-Abl/Hsp90 protein complex: A possible mechanism to inhibit Bcr-Abl-positive human leukemic blasts by novobiocin. Leukemia. 2008;22:1402–1409. doi: 10.1038/leu.2008.89. [DOI] [PubMed] [Google Scholar]
- 243.Dierks C, Beigi R, Guo GR, Zirlik K, Stegert MR, Manley P, Trussell C, Schmitt-Graeff A, Landwerlin K, Veelken H, Warmuth M. Expansion of Bcr-Abl-positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer Cell. 2008;14:238–249. doi: 10.1016/j.ccr.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 244.Gregory MA, Phang TL, Neviani P, Alvarez-Calderon F, Eide CA, O’Hare T, Zaberezhnyy V, Williams RT, Druker BJ, Perrotti D, Degregori J. Wnt/Ca2+/NFAT signaling maintains survival of Ph+ leukemia cells upon inhibition of Bcr-Abl. Cancer Cell. 2010;18:74–87. doi: 10.1016/j.ccr.2010.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Hess P, Pihan G, Sawyers CL, Flavell RA, Davis RJ. Survival signaling mediated by c-Jun NH(2)-terminal kinase in transformed B lymphoblasts. Nat Genet. 2002;32:201–205. doi: 10.1038/ng946. [DOI] [PubMed] [Google Scholar]
- 246.Zhao C, Chen A, Jamieson CH, Fereshteh M, Abrahamsson A, Blum J, Kwon HY, Kim J, Chute JP, Rizzieri D, Munchhof M, VanArsdale T, Beachy PA, Reya T. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature. 2009;458:776–779. doi: 10.1038/nature07737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Heaney NB, Holyoake TL. Therapeutic targets in chronic myeloid leukaemia. Hematol Oncol. 2007;25:66–75. doi: 10.1002/hon.813. [DOI] [PubMed] [Google Scholar]
- 248.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]