

Abstract
A wide range of cell stresses, including DNA damage, signal to p53 through post-translational modification of p53. The cytoplasmic functions of p53 are emerging as an important constituent of p53’s role in tumor suppression. Here we report that deletion of the Cul9 (formerly Parc) gene, which encodes an E3 ubiquitin ligase that binds to p53 and localizes in the cytoplasm, resulted in spontaneous tumor development, accelerated Eμ-Myc-induced lymphomagenesis and rendered mice susceptible to carcinogenesis. Cul9-p53 double mutant mice exhibited indistinguishable tumor phenotypes as p53 single mutant mice, indicating that the function of Cul9 in tumor suppression is largely mediated by p53. Deletion of Cul9 had no significant effect on cell cycle progression, but attenuated DNA damage-induced apoptosis. Ectopic expression of wild-type CUL9, but not a point mutant CUL9 deficient in p53 binding, promotes apoptosis. These results demonstrate CUL9 as a potential p53 activating E3 ligase in the cytoplasm.
Keywords: CUL9, p53, apoptosis, tumor suppression
Introduction
Cullins are a family of evolutionarily conserved proteins that bind to the small RING finger protein, ROC1 (also known as RBX1), to constitute a large number of distinct E3 ubiquitin ligases. Mammalian genomes contain six closely related cullin genes (CUL1, 2, 3, 4A, 4B and 5). In addition, three other proteins, CUL7, CUL9 (changed from the former name of PARC following the recommendation by HUGO Nomenclature Committee) [(1, 2), Supplementary Fig. S1A, B] and APC2 (3, 4), contain significant sequence homology to cullins over a ~180 residue region that binds with ROC1 or a homologous small RING protein, APC11. CUL7 and CUL9 share extensive sequence homology and contain several additional functional domains, including an N-terminal p53-binding sequence (2, 5–7), a region similar to HERC2 (hect domain and rcc1 domain protein 2), a centrally located DOC1 domain, and a C-terminal sequence unique to CUL9 that contains two RING fingers separated by a sequence known as IBR (in between the RING fingers).
Both the function and mechanism of CUL9 are poorly defined. Two notable features of CUL9 [(2), Supplementary Fig. S1C], which are also shared by CUL7(5–7), are its exclusive localization in the cytoplasm and binding with p53. The significances of both features are unclear. Although it was initially reported that CUL9/PARC sequesters p53 in the cytoplasm and thereby antagonizes p53-mediated apoptosis (2), this finding does not appear to be reproducible as the deletion of mouse Cul9/Parc had no detectable effect on p53 stability, target gene induction or localization (8), and CUL9 did not sequester p53 in the cytoplasm in most cells, even when CUL9 or both CUL9 and p53 were overexpressed (Supplementary Fig. S1C). In this paper, we report an extensive genetic analysis that has identified CUL9 as a p53-dependent tumor suppressor.
Materials and Methods
Generating Cul9 mutant mice
The targeting construct was generated to delete a 5.7 kb genomic fragment containing exons 2 to 7 encoding the N-terminal 664 amino acid residues of the mouse Cul9 gene. Briefly, a 17.8 kb mouse genomic DNA fragment spanning the Cul9 locus was isolated by PCR from mouse embryonic stem (ES) cell genomic DNA. LoxP sites were introduced flanking exons 2 and 7 of Cul9 by site-directed mutagenesis (Stratagene). A neomycin (neo) resistance gene flanked by frt sites was inserted upstream of the loxP site upstream of mouse exon 2 as a positive selection marker for homologous recombination, and the thymidine kinase gene was inserted upstream of exon 1 to allow for negative selection using gancyclovir against random integration. The linearized targeting construct was electroporated into E14 ES cells and selected with G418 and ganglocyclovir. Doubly-resistant clones were screened for homologous recombination events and confirmed by Southern blot analysis. After removal of the neo marker by transfection of Flp recombinase, three independent Cul9flox/+ ES clones were injected into C57BL/6 blastocysts, and chimeric mice were crossed with C57BL/6 mice to generate Cul9flox/+ heterozygotes. Germline transmitted Cul9flox/+ mice were then crossed with EIIa-Cre transgenic mice [B6.FVB-Tg (EIIa-Cre), Jackson Laboratory] to generate Cul9+/− heterozygotes. Successful deletion of exons 2–7 in Cul9+/− mice was confirmed by PCR and Southern blotting (Supplementary Fig. 2).
Mice, histopathology, immunohistochemistry and carcinogenesis
Cul9 heterozygotes were backcrossed for 10 generations with BL/6 mice. p53 null (p53tm1Tyj) and Eμ-Myc transgenic (B6.Cg-Tg(IghMyc)22Bri/J) mice in BL/6 background were obtained from Jackson Laboratory and bred to the Cul9 mutant mice to create double mutant mice. Most analyses, including body weight and tumor development, on Cul9 and Cul9;p53 double mutant mice were performed with mice backcrossed 4–5 generations to BL/6. Analysis on Cul9;Eμ-Myc mice was performed using Cul9 mutant mice that were backcrossed 10 generations to BL/6. For the determination of spontaneous tumor incidence, Cul9, p53 and Cul9;p53 mutant mice with visible tumors, exhibiting morbidity, weight loss or paralysis were sacrificed. The remaining surviving mice were sacrificed at 27 months of age (for WT, Cul9+/− and Cul9−/−) or at 21 months of age (for p53+/−, Cul9+/−;p53+/−, and Cul9−/−;p53+/−). For the determination of lymphoma development in Eμ-Myc and Eμ-Myc;Cul9 mutants, mice were sacrificed when they developed visible tumors or exhibited morbidity. The remaining surviving mice were sacrificed at five months of age. A full autopsy was performed and tissues were fixed and histologically examined by two pathologists after H&E staining. Immunohistochemistry was performed as described previously (9).
To determine the carcinogenic susceptibility of Cul9 mutants, mice of different genotypes were injected intraperitoneally at 7 weeks of ages with N-Nitrosodiethylamine (DEN, Sigma, St. Louis, MO) at 10mg/kg body weight. All mice were sacrificed and full necropsies were performed at 11 months of age. All tumors were confirmed by two individual pathologists. The numbers of lung tumors were counted based on the H.E. staining confirmed lesions.
ES and MEF cells
Cul9 null ES cells were generated by two rounds of targeting. Briefly, the linearized targeting construct was electroporated into E14 ES cells and selected with G418 and ganglocyclovir. Doubly-resistant clones were screened for homologous recombination events and confirmed by Southern blot analysis (Supplementary Fig. 2). The neo marker was removed by Flp recombinase, and clones confirmed for neo excision were subjected to another round of targeting, selection and screening for homologous recombination. Cul9NEOflox/flox ES clones were expanded and subjected to both plasmid-mediated (CMV-Cre) and retroviral Cre expression. Deletion of exons 2–7 from both alleles was confirmed by genomic PCR, RT-PCR and Western blot (Supplementary Fig. 2). p53 null mice were bred to the Cul9 mutant mice to create double heterozygous mice. Cul9+/− and p53+/−;Cul9+/− mice were intercrossed separately and E13.5-E14.5 embryos were isolated to generate MEFs as described (10). Early passage MEFs (< passage 4) from individual embryos were plated in 100-mm plates and incubated in DMEM plus 10% FBS.
Proliferation and apoptosis assays
For BrdU labeling, ES cells or early passage MEFs (< passage 4) from individual embryos were grown in media containing 10 μM BrdU for 1 hour. After trypsinization and PBS wash, cells were fixed and permeabilized. Incorporated BrdU was exposed to DNase treatment and then stained with FITC-conjugated anti-BrdU antibody. Total DNA was stained by PI (Propidium Iodide). For apoptosis analysis, cells were exposed to the indicated amounts of UV radiation, harvested 24h after UV treatment, stained with Annexin V–FITC and then analyzed by FACS. FACS analysis was performed using the CYAN ADP Analyzer (Dako, Fort Collins, Colorado). Data analysis was performed using Summit v4.3 software (Dako, Fort Collins, Colorado).
To analyze apoptosis in the thymus in vivo, mice were irradiated with 10 gray (Gy) of γ-irradiation and sacrificed after 8 hours. Thymus sections were analyzed for apoptosis using ApoTag kit (Chemicon) according to the manufacturer’s protocol. Thymocytes derived from the irradiated mice were also stained with either JC-1 or Annexin V–FITC and propidium iodide (PI), and then analyzed by FACS. JC-1 staining determines mitochondrial membrane potential (ΔΨm). It enters cells and gives a green fluorescent signal, and is processed to give an additional red fluorescent signal only in mitochondria with preserved ΔΨm. Cells with preservation of ΔΨm areJC-1Ghigh and JC-1Rhigh, and cells with loss of ΔΨm are JC-1Ghigh and JC-1Rlow.
Results
Cul9 mutant mice develop spontaneous tumors
To determine the function of CUL9, we generated Cul9−/− mice by homologous recombination using a targeting construct that deletes a 5.7 kb genomic fragment containing exons 2 to 7 that encodes the N-terminal 664 amino acid residues of Cul9 protein (Supplementary Fig. S2A – F). The Cul9−/− mice, as previosuly reported (8), were produced in Mendelian ratio without significant developmental defect. The body weight of both male and female Cul9−/− mice, however, were decreased (Fig. 1A). After monitoring a cohort of 28 Cul9−/−, 23 Cul9+/−, and 12 wild-type mice for 27 months, we found that 22 (79%) Cul9−/− mice developed tumors in multiple organs or tissues, including lymphoma, sarcoma and tumors in pituitary, lung, liver and ovary (Fig. 1B,C, and Supplementary Fig. S3). Cul9+/− heterozygote mice also developed tumors with a similar spectrum and incidence, 74% (n=23), while only 33% (n=12) WT mice displayed tumors at similar age. Lymphoma and sarcoma developed in Cul9 mutant mice frequently infiltrated into multiple organs, including thymus, spleen, liver, pancreas, lung and skin (Supplementary Fig. S3). Both Cul9−/− and Cul9+/− mice had a decreased life span compared with WT controls. The mean tumor-free survival times for Cul9−/−, Cul9+/− and WT were 19.9, 20.8 and 26.4 months, respectively (Fig. 1C). Only 2 out of 16 (13%) tumors developed in Cul9+/− mice were found to have lost the remaining WT allele (Fig. 1D, and data not shown). These results reveal a haploinsufficient function of Cul9 in tumor suppression.
Figure 1. Cul9 deficient mice develop spontaneous tumors.

A. Loss of Cul9 results in body weight decrease. Male and female mice at the indicated ages were weighed. Averages and standard deviations were calculated from 4 to 5 mice in each time point.
B. Tumor incidence and spectrum in WT and Cul9 mutant mice.
C. Tumor-free survival curve of WT and Cul9 mutant mice.
D. Genomic DNA extracted from the microdissected samples of Cul9 heterozygote mice was amplified by PCR to detect Cul9 alleles.
Cul9 deficiency promotes lymphomagenesis and metastasis in Eμ-Myc mice
Eμ-Myc transgenic mice ectopically express the c-myc oncogene from the immunoglobulin heavy chain enhancer, and reproducibly develop and die from tumors of the B lymphocyte lineage with a mean survival time of 4 months (11, 12). To further corroborate the tumor suppression function of Cul9, we generated Cul9;Eμ-Myc double mutant mice and followed lymphoma development for 5 months. No lymphoma developed in Cul9 mutant mice in the absence of ectopic Myc expression by this age. Loss of Cul9 significantly accelerated the onset of lymphoma development in Eμ-Myc mice. The mean lymphoma-free survival times were reduced from 126.4 days in Cul9+/+;Eμ-Myc mice to 108.9 days in Cul9+/−;Eμ-Myc and 85.1 days in Cul9−/−;Eμ-Myc mice, respectively (Fig. 2A, B). The earliest lymphomas were detected in Cul9−/−;Eμ-Myc mice at 25 days of age. 3 out of 5 Cul9+/− or Cul9−/− mice younger than 2 months of age developed lymphoma, but no tumor developed in Cul9+/+ mice by this age. Overall, 66% (6/9) of the Cul9+/+;Eμ-Myc mice developed lymphoma, whereas 80% of Cul9+/−;Eμ-Myc mice (16/20) and 100% (10/10) Cul9−/−;Eμ-Myc mice had lymphoma in 5 months. All tumors that developed were either B cell lymphoma or pre-B lymphomas regardless of the genotypes (Supplmentary Fig. S4B).
Figure 2. Cul9 deficiency accelerates Eμ-Myc-induced lymphomagenesis and increases the susceptibility to carcinogen.
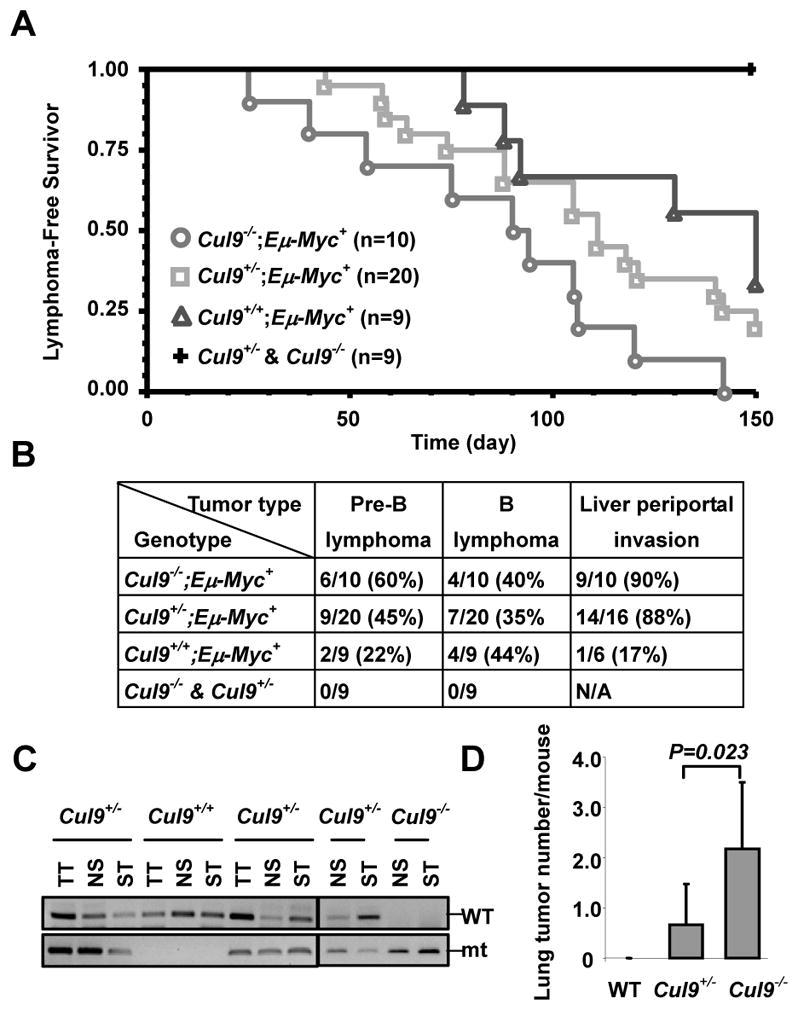
A. Lymphoma-free survival curve of Cul9, Eμ-Myc and Eμ-Myc;Cul9 double mutant mice. All mice were euthanized within 5 months, followed by pathological examination and FACS analysis.
B. Lymphoma incidence, subtype, and lymphoma numbers invaded into liver (periportal invasion) in Cul9, Eμ-Myc and Eμ-Myc;Cul9 double mutant mice. Note, lymphoma was diagnosed and typed by H.E. staining and FACS analysis.
C. Genomic DNA extracted from the representative lymphomas derived from 3 Cul9 heterozygotes mice and from WT or Cul9 homozygote mouse was amplified by PCR to detect Cul9 alleles.
D. Average numbers of lung tumors derived from the mice treated with DEN.
Haplo or complete loss of Cul9 not only accelerated the lymphoma development in Eμ-Myc mice, but also transformed Eμ-Myc–induced lymphoma to become more malignant. All but 3 lymphomas (n=26) developed in Cul9+/−;Eμ-Myc or Cul9−/−;Eμ-Myc mice exhibited extensive periportal invasion with lymphoma cell clusters spreading throughout the liver parenchym and various degrees of lung and pancreas infiltration (Supplmentary Fig. S4A). In contrast, lymphomas developed in Cul9+/+;Eμ-Myc mice displayed little nonlymphoid organ infiltration, with only one Cul9+/+;Eμ-Myc lymphoma (n=6) showing minor periportal invasion in the liver (Fig. 2B, and Supplmentary Fig. S4A). No significant difference was observed in tumor invasion and infiltration between Cul9+/−;Eμ-Myc and Cul9−/−;Eμ-Myc mice, nor did we detect the loss of the remaining WT allele of Cul9 in lymphomas developed in Cul9+/−;Eμ-Myc mice (n=10, Fig. 2C, and data not shown). Together, these results provide further evidence supporting Cul9 as a haploinsufficient tumor suppressor.
Cul9 mutant mice are susceptible to carcinogenesis
Spontaneous tumor development and acceleration of Eμ-Myc-induced lymphomagesis in Cul9 mutant mice led us to examine the susceptibility of Cul9 mutant mice to tumorigenesis after continuous low-dose exposure to a commonly used chemical carcinogen diethylnitrosamine (DEN), which mainly targets the lung and liver. As shown in Supplmentary Fig. S5A, both Cul9+/−and Cul9−/− mice are more susceptible to DEN-induced tumorigenesis. By the age of 11 months, when all mice were scarified for histological and pathological analysis, none of three WT mice had developed detectable tumor lesions. In contrast, 3 out of 6 Cul9+/− heterozygote mice and 5 out of 6 Cul9−/− homozygous mice developed multiple tumors in lung with Cul9−/− mice developing significantly more tumors per lung than the Cul9+/− mice (Fig. 2D and Supplmentary Fig. S5A). Most lung tumors developed in DEN-treated Cul9 mutant mice were relatively well-differentiated adenomas (Supplmentary Fig. S5B). Two lung tumors derived from Cul9−/− mice were adenocarcinomas with high mitotic indices and invasive potential. Two Cul9−/− mice developed lymphoma with multiple organ infiltration, one of which also developed hepatocellular carcinoma with metastasis to spleen, pancreas, and lymph node. In addition, we also observed more severe hepatocyte dysplasia in Cul9 mutant livers than in wild type counterparts. Taken together, these results further corroborate the function of Cul9 in tumor suppression.
Tumor suppression function of Cul9 is largely mediated by p53
To determine the functional interaction of Cul9 and p53, we generated Cul9-p53 double mutant mice. After monitoring a cohort of total 73 mice of various genotypes over a period of 21 months, we found that unlike accelerated lymphoma development of Eμ-Myc mice, neither haplo nor complete loss in Cul9 significantly affected tumor development in either p53+/− heterozygote or p53−/− null mutant mice (Fig. 3A, B). The mean tumor-free survival times were comparable in both p53+/− (14 mon. for p53+/−;Cul9−/− and 15.1 mon. for p53+/−;Cul9+/− vs. 14.6 mon for p53+/− mice) and p53−/− (4.3 mon. for p53−/−;Cul9−/− and 4.6 mon. for p53−/−;Cul9+/− vs. 5.3 mon for p53−/− mice) backgrounds, indicating that combinatorial loss of Cul9 and p53 had little or no effect on tumor-free survival as compared with animals lacking p53 alone. The lack of significant effect on tumor initiation and progression in p53 mutant mice by Cul9 mutation can not be attributed to either a lack of function of Cul9 in suppressing lymphomas, nor early death of p53 mutant mice from lymphomas becoming rapidly lethal since Eμ-Myc mice developed similar types of lymphomas and died even earlier. One interpretation for the collaboration of Cul9 mutation with Myc overexpression, but not with p53 mutation, to promote tumor growth is that Cul9 suppresses tumor growth via a pathway that is distinct from Myc-promoted tumorigenesis, but is largely mediated by p53.
Figure 3. Functional dependency of Cul9 on p53 in tumor suppression.

A. Tumor incidence and spectrum in Cul9, p53 single and p53;Cul9 double mutant mice.
B. Tumor-free survival curve of Cul9, p53 single and p53;Cul9 double mutant mice.
Disruption of Cul9 attenuates DNA damage-induced apoptosis
Given the functional dependency of CUL9 on p53 in tumor suppression, we examined two cellular activities—arresting cell cycle progression and promoting apoptosis—underlying the function of p53 in tumor suppression. Deletion of one or both alleles of Cul9 did not cause any significant alteration in the cell cycle phase distribution in either asynchronously growing embryonic stem (ES, Fig. 4A) or mouse embryonic fibroblasts (MEFs, Fig. 4B). These results indicate that CUL9, a relatively young gene evolved after the emergence of vertebrates, does not have an essential function in cell cycle progression.
Figure 4. Disruption of Cul9 reduced DNA damage-induced apoptosis.
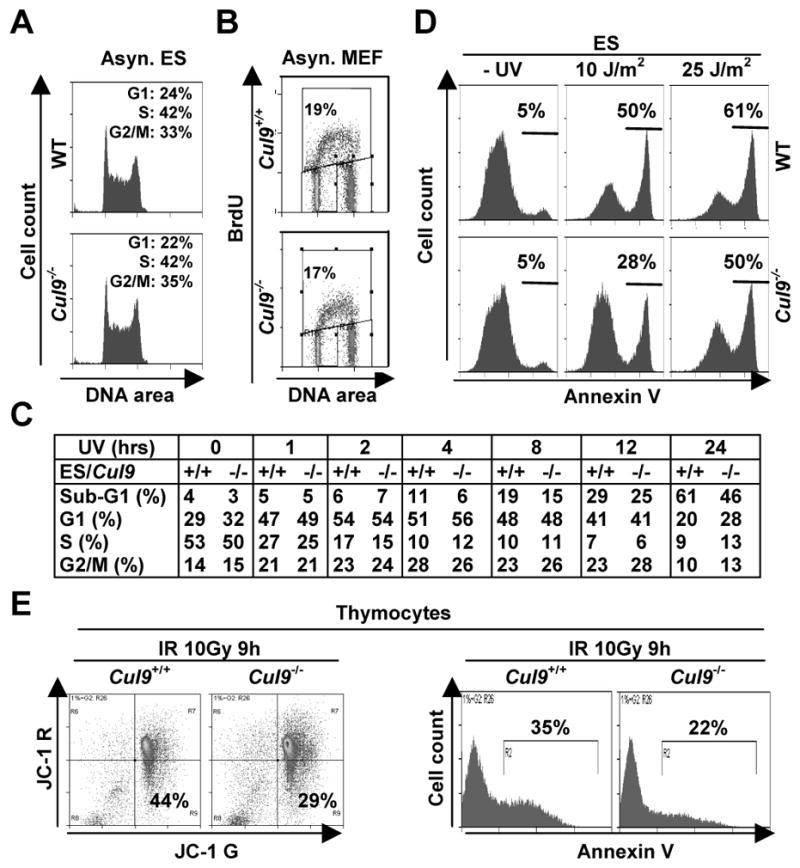
A. Asynchronously growing WT and Cul9−/− ES cells were analyzed by FACS.
B. Asynchronously growing WT and Cul9−/− MEFs cells were pulse labeled with 10 μM BrdU for 1 hour and analyzed by FACS.
C. Cul9f/f and Cul9−/− ES cells were exposed to 50 J/m2 UV irradiation, pulse labeled with 10 μM BrdU for 1 hour, and harvested at indicated time after UV and stained with anti-BrdU-FITC and PI. The distribution of cells in different phases of the cell cycle is determined by flow cytometry and shown in the table.
D. Cul9f/f and Cul9−/− ES cells were exposed to the indicated dosage of UV for 24 hours and then stained for Annexin V. The percentages of Annexin V positive cells are indicated.
E. Littermate mice at 4 months of age were irradiated (10 Gy), 9 hours later freshly isolated thymocytes were stained with JC-1 (left panel) and Annexin V (right panel). The percentages of apoptotic cells, as measured by the mitochondrial membrane potential (ΔΨm or JC-1Ghigh and JC-1Rlow), are indicated. A representative of three independent experiments is shown.
We next examined the function of Cul9 in response to two common types of DNA damage, UV and γ-irradiations, in both in vitro cultured cells and in tissues of mutant mice. Cul9−/− ES cells were exposed to UV irradiation for various lengths of time, pulse labeled with BrdU and analyzed by flow-cytometry. UV irradiation quickly—within an hour—reduced the S-phase (BrdU+) cell population by almost 50% and accumulated both G1 and G2 populations (Fig. 4C and Supplementary Fig. S6A). Although not causing a significant change in the relative distribution of these three cell populations, deletion of Cul9 decreased UV-induced apoptosis. Starting at 4 hours after UV irradiation, there was an evident accumulation of an apoptotic (sub-G1) cell population in WT ES cells that continuously increased throughout the experimental duration of 24 hours. Loss of function of Cul9 attenuated, but did not completely eliminate, the accumulation of apoptotic population (Fig. 4C). To confirm the proapoptotic function of Cul9, we analyzed UV-irradiated ES cells by Annexin V staining which more sensitively and accurately detects early apoptotic cells. We found that deletion of Cul9 reduced Annexin V positive cells in response to both 10 J/m2 and 25 J/m2 of UV doses (Fig. 4D). Together, these results demonstrate that Cul9 plays a significant role in promoting UV-induced apoptosis.
We next examined the function of Cul9 in cellular response to DNA damage in vivo. Littermate mice were γ-irradiated and dissected 9 hours after irradiation, and thymocytes were isolated from thymus and analyzed by flow-cytometry. JC-1 staining, which detects early apoptotic cells by measuring the mitochondrial membrane potential (ΔΨm), demonstrated that deletion of Cul9 attenuated apoptosis, reducing the percentages of thymocytes with loss of ΔΨm from 44% in the WT thymus to 29% in Cul9−/− thymus (Fig. 4E). Parallel Annexin V staining of these irradiated thymocytes confirmed decreased apoptosis from 35% in WT to 22% in Cul9−/−thymus. We also performed a TUNEL assay in γ-irradiated thymus (Supplementary Fig. S6B) and spleen (data not shown) and found that deletion of Cul9 substantially reduced apoptosis in both organs.
Cul9 promotes p53-dependent apoptosis
To determine how Cul9 and p53 functionally interact with each other to promote DNA damage-induced apoptosis, we UV irradiated asynchronously growing MEFs of different genotypes and determined both cell cycle progression by coupled BrdU pulse labeling and flow cytometry and apoptosis by Annexin V staining 24 hours after UV irradiation. Unlike deletion of Cul9, which had little effect on the cell cycle progression of MEF cells as measured by the BrdU incorporation (24% vs. 23%), deletion of p53 increased S-phase (BrdU+) cell population by 54% (24% in WT vs. 37% in p53−/− MEFs, Fig. 5A), consistent with an activity of p53 in constraining G1 progression. Co-deletion of both Cul9 and p53 genes further increased the S-phase cell population to 45%. Annexin V staining showed that singular deletion of either Cul9 or p53 reduced UV-induced apoptosis when compared with the WT MEFs (53%), with p53 disruption (37%) exhibiting a more pronounced effect than Cul9 deletion (44%, Fig. 5B). Co-deletion of both Cul9 and p53 genes resulted in a slight further reduction of apoptosis (32%), indicating the function of Cul9 in promoting apoptosis is mostly, but not entirely, dependent on p53.
Figure 5. CUL9 promotes a p53-dependent apoptosis.

A, B. Cul9 collaborates with p53 in promoting apoptosis. MEFs (passage 1) of different genotypes were pulse labeled with 10 μM BrdU for 1 hour and stained with anti-BrdU-FITC (A) or were irradiated with UV (50 J/m2) for 24 hours and stained with Annexin V (B). DNA synthesis and apoptosis were determined by flow cytometry. The percentages of BrdU+ and apoptotic cells are indicated.
C. Littermate mice at 5 months of age were irradiated (10 Gy). Thymus was isolated and fixed at the indicated time, and stained with an antibody against Bax and DAPI. Thymus not irradiated was used as control.
D. Deletion of Cul9 reduces the accumulation of p53 and p53 target gene, Bax, in MEF cells. Total cell lystes prepared from UV-irradiated MEF cells of different genotypes were separated by SDS-PAGE, followed by direct immunoblotting with indicated antibodies. The quantification is included in Supplementary Fig. 8.
E. Plasmids expressing different human CUL9 mutants were co-transfected with p53 expressing plasmid into U2OS cells and CUL9-p53 binding was determined by IP-western.
F. WT MEF cells were infected for 2 days with the lentivirus indicated, stained with PI, and analyzed by FACS. The percentages of cells with <2N (sub-G1) and >4N DNA (polyploidy) contents were indicated. The expression levels of WT and mutant Cul9 transduced by lentivirus were confirmed by Western blot (left panel).
To further corroborate the p53-mediated function of Cul9 in promoting apoptosis, we examined the expression of Bax, a pro-apoptosis gene and a downstream target of p53, in Cul9-deficient thymus in response to γ-irradiation. The steady state level of Bax protein was nearly undetectable in both un-irradiated WT and Cul9-deficient spleen (Fig. 5C, and data not shown). Bax levels were significantly increased in WT thymus after γ-irradiation for 2 and 8 hours, respectively, but these increases were substantially reduced in Cul9-deficient thymus (Fig. 5C).
We further examined the UV-induced p53 induction in cultured ES and MEFs. Western blotting analyses showed that deletion of Cul9 had little effect on the basal level of p53 in unperturbed asynchronously growing ES and MEFs (Fig. 5D, and Supplementary Fig. S2F, H, S7A, B, C). Cul9 deletion reduced, but did not abolish, the accumulation of p53 following UV irradiation. Accordingly, deletion of Cul9 did not significantly affect the basal level of Bax in unperturbed ES and MEF cells, but it reduced the Bax protein accumulation following UV irradiation. Deletion of p53, on the other hand, slightly decreased Cul9 protein level in embryos at E13. 5, E15.5, and E17.5 (Supplementary Fig. S8A and data not shown), but did not affect Cul9 expression in adult tissues (Supplementary Fig. S8B), indicating that p53 does not play a major role in the regulation of Cul9 gene expression.
Finally, to provide further biochemical evidence supporting a functional dependency on p53 for the proapoptotic activity of CUL9, we carried out a series of deletion and mutation analyses and identified two point mutants of human CUL9, CUL9Q417A and CUL9F419A , that completely disrupted CUL9’s binding with p53 (Fig. 5E for Q417A mutant and data not shown for F419A). We then generated a lentivirus expressing both the WT and Q417A mutant CUL9. Transduction of MEFs with WT Cul9 lentivirus increased the apoptotic (sub-G1) cell population by 71%, from 14.1% to 24.1% (Fig. 5F). Remarkably, transduction of CUL9Q417A, when expressed at a similar level, did not cause any significant increase of apoptotic cells (14.0%). Instead, ectopic expression of CUL9Q417A substantially increased the polyploidy cell population, resulting in an increase of cells with more than 4N DNA content from 16.5% in control cells and 14.2% in cells transduced with WT CUL9 to 24.6% in MEFs transduced with CUL9Q417A. Transduction of U2OS cells with WT CUL9 and CUL9Q417A lentivirus also increased apoptosis and polyploidy, respectively, as evidenced by Annexin V and DAPI staining (data not shown). These results suggest that increased CUL9 level causes a defect leading to polyploidy cells that would normally be eliminated by a p53-mediated apoptosis pathway, but become accumulated when the CUL9-p53 interaction is disrupted.
Discussion
In this paper, we provide compelling genetic evidence that CUL9 (formerly PARC) is a tumor suppressor; deletion of Cul9 in mice resulted in spontaneous tumor development, accelerated Eμ-Myc-induced lymphomagenesis and predisposed mice to carcinogenesis. Consistent with its broad expression (2), tumors spontaneously developed in Cul9 mutant mice cross a wide spectrum of at least 9 different organs or tissues, with pituitary adenomas being more prevalent than others. One notable feature of tumorigenesis in Cul9 mutant mice is that both the penetrance of tumor development and tumor-free survival are very similar between Cul9 heterozygotes and homozygotes, suggesting haploinsufficiency of Cul9 in tumor suppression. This notion is supported by the observation that in both Cul9+/− single and Cul9+/−;Eμ-Myc double mutants mice, the remaining WT allele of Cul9 was retained in most tumors examined. The CUL9 gene has not been reported thus far to be frequently mutated in human tumors. In addition to being a haploinsufficient gene whose decreased expression may be sufficiently reduced by its function in tumor suppression, the functional dependency of CUL9 on p53 in tumor suppression may also explain the lack of wide spread CUL9 mutations in human cancers. Deletion of both alleles of Cul9 did not exhibit any significant synergistic tumorigenic effect in p53 heterozygotes, suggesting that even complete loss of CUL9 function would not offer a significant selection advantage for the growth of human tumor cells with even haploid loss or partial reduction of p53 activity.
CUL9 joins a growing number of p53 regulators that mediate different cellular stresses to activate p53, leading to either cell cycle arrest or apoptosis. There are three unique features of CUL9 in signaling p53 to suppress tumorigenesis. First, CUL9 signals p53 selectively toward apoptosis and does not appear to cause a cell cycle arrest following DNA damage. In several cell types we have examined (ES, MEFs, thymocytes, and spleenocytes), deletion of Cul9 caused only an attenuated apoptosis without significantly affecting the cell cycle phase distribution. Second, CUL9 is an E3 ubiquitin ligase that may function as a p53 activator. More than a dozen E3 ubiquitin ligases have been linked to p53 regulation [for a recent compilation, see ref.(13)], all promoting p53 degradation and thus acting functionally as a negative regulator of p53. In contrast, the functions of CUL9 in tumor suppression (Fig. 3) and in promoting apoptosis (Fig. 5) are both dependent on p53. To the best of our knowledge, CUL9 represents the first E3 ligase that binds directly to and is functionally dependent on p53. Thirdly, CUL9 localizes exclusively in the cytoplasm. In addition to the well- established function of p53 as a nuclear transcription factor, emerging evidence suggests that p53 has an important role in the cytoplasm, especially in mitochondrial-mediated apoptosis (14). The regulation, in particular the activation, of p53 in the cytoplasm remains poorly understood. Given its cytoplasmic localization and functional dependency on p53, we suspect that CUL9 may activate p53 in the cytoplasm.
The biochemical mechanism by which CUL9 regulates p53 remains unclear. Following DNA damage, the induction of p53 was moderately and reproducibly reduced in Cul9 deficient organs and cells. A reduced p53 induction is consistent with and likely underlines the attenuated apoptosis in Cul9 deficient organs in vivo and in both ES and MEF cells cultured in vitro after DNA damage. However, unlike the overexpression or decrease of other p53 E3 ligases, especially MDM2, we did not detect a significant change in the steady state level and half-life of p53 in cells with either Cul9 deletion or overexpression. We have also carried out extensive investigation of subcellular localization and stress-linked phosphorylation at various sites in WT and Cul9 null cells following DNA damage. Thus far, we have not detected a significant change in either subcellular distribution or phosphorylation of p53. Thus, it remains a challenge to elucidate the molecular mechanism by which CUL9, a cytoplasmic localized E3 ligase, signals to p53.
Supplementary Material
Acknowledgments
We thank J. DeCaprio (Dana-Farber Cancer Institute) for providing a CUL9/PARC lentiviral plasmid, and discussions. This study was supported by NIH grants CA065572 and GM067113 to Y.X.
References
- 1.Dias DC, Dolios G, Wang R, Pan ZQ. CUL7: A DOC domain-containing cullin selectively binds Skp1.Fbx29 to form an SCF-like complex. Proc Natl Acad Sci U S A. 2002;99:16601–6. doi: 10.1073/pnas.252646399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nikolaev AY, Li M, Puskas N, Qin J, Gu W. Parc: a cytoplasmic anchor for p53. Cell. 2003;112:29–40. doi: 10.1016/s0092-8674(02)01255-2. [DOI] [PubMed] [Google Scholar]
- 3.Zachariae W, Shevchenko A, Andrews PD, Ciosk R, Galova M, Stark MJR, et al. Mass spectrometric analysis of the anaphase-promoting complex from yeast: identification of a subunit related to cullins. Science. 1998;279:1216–9. doi: 10.1126/science.279.5354.1216. [DOI] [PubMed] [Google Scholar]
- 4.Yu H, Peters J-M, King RW, Page AM, Hieter P, Kirschner MW. Identification of a cullin homology region in a subunit of the anaphase-promoting complex. Science. 1998;279:1219–22. doi: 10.1126/science.279.5354.1219. [DOI] [PubMed] [Google Scholar]
- 5.Andrews P, He YJ, Xiong Y. Cytoplasmic localized ubiquitin ligase cullin 7 binds to p53 and promotes cell growth by antagonizing p53 function. Oncogene. 2006;25:4534–48. doi: 10.1038/sj.onc.1209490. [DOI] [PubMed] [Google Scholar]
- 6.Kaustov L, Lukin J, Lemak A, Duan S, Ho M, Doherty R, et al. The conserved CPH domains of Cul7 and PARC are protein-protein interaction modules that bind the tetramerization domain of p53. J Biol Chem. 2007;282:11300–7. doi: 10.1074/jbc.M611297200. [DOI] [PubMed] [Google Scholar]
- 7.Kasper JS, Arai T, DeCaprio JA. A novel p53-binding domain in CUL7. Biochem Biophys Res Commun. 2006;348:132–8. doi: 10.1016/j.bbrc.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 8.Skaar JR, Arai T, DeCaprio JA. Dimerization of CUL7 and PARC is not required for all CUL7 functions and mouse development. Mol Cell Biol. 2005;25:5579–89. doi: 10.1128/MCB.25.13.5579-5589.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bai F, Pei XH, Godfrey VL, Xiong Y. Haploinsufficiency of p18INK4c sensitizes mice to carcinogen-induced tumorigenesis. Mol Cell Biol. 2003;23:1269–77. doi: 10.1128/MCB.23.4.1269-1277.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pei XH, Bai F, Tsutsui T, Kiyokawa H, Xiong Y. Genetic evidence for functional dependency of p18Ink4c on Cdk4. Mol Cell Biol. 2004;24:6653–64. doi: 10.1128/MCB.24.15.6653-6664.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, et al. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985;318:533–8. doi: 10.1038/318533a0. [DOI] [PubMed] [Google Scholar]
- 12.Langdon WY, Harris AW, Cory S, Adams JM. The c-myc oncogene perturbs B lymphocyte development in E-mu-myc transgenic mice. Cell. 1986;47:11–8. doi: 10.1016/0092-8674(86)90361-2. [DOI] [PubMed] [Google Scholar]
- 13.Jain AK, Barton MC. Regulation of p53: TRIM24 enters the RING. Cell Cycle. 2009;8:3668–74. doi: 10.4161/cc.8.22.9979. [DOI] [PubMed] [Google Scholar]
- 14.Green DR, Kroemer G. Cytoplasmic functions of the tumour suppressor p53. Nature. 2009;458:1127–30. doi: 10.1038/nature07986. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.