

SUMMARY
Prenatal exposure to ethanol in humans results in a wide range of developmental abnormalities, including growth deficiency, developmental delay, reduced brain size, permanent neurobehavioral abnormalities and fetal death. Here we describe the use of Drosophila melanogaster as a model for exploring the effects of ethanol exposure on development and behavior. We show that developmental ethanol exposure causes reduced viability, developmental delay and reduced adult body size. We find that flies reared on ethanol-containing food have smaller brains and imaginal discs, which is due to reduced cell division rather than increased apoptosis. Additionally, we show that, as in mammals, flies reared on ethanol have altered responses to ethanol vapor exposure as adults, including increased locomotor activation, resistance to the sedating effects of the drug and reduced tolerance development upon repeated ethanol exposure. We have found that the developmental and behavioral defects are largely due to the effects of ethanol on insulin signaling; specifically, a reduction in Drosophila insulin-like peptide (Dilp) and insulin receptor expression. Transgenic expression of Dilp proteins in the larval brain suppressed both the developmental and behavioral abnormalities displayed by ethanol-reared adult flies. Our results thus establish Drosophila as a useful model system to uncover the complex etiology of fetal alcohol syndrome.
INTRODUCTION
The ability of alcohol to cause developmental anomalies has been demonstrated in a broad range of taxa, from insects to mammals. In humans, alcohol consumption during pregnancy can result in fetal alcohol syndrome (FAS), which consists of a persistent growth deficiency, craniofacial dysmorphology and deficient brain growth with associated neurocognitive deficits (Jones and Smith, 1973). FAS is the leading known cause of congenital mental retardation in the Western world (Pulsifer, 1996), and the most severe form of a broad range of disorders known as fetal alcohol spectrum disorder (FASD) (Hoyme et al., 2005). The prevalence of FAS in the world is one to three per 1000 births, indicating a serious medical and societal problem (May and Gossage, 2001). Despite the growing awareness of FAS and FASD (FAS/FASD) and posted warnings on alcoholic beverages, consumption of alcohol during pregnancy continues, highlighting the need for an understanding of the molecular basis of FAS and developing novel treatments to mitigate the complications of gestational ethanol exposure.
The long-lasting neurobehavioral impairments are arguably the most serious consequences of FAS/FASD. Individuals with FAS can exhibit deficits in attention, memory and motor coordination, display hyperactivity, and can suffer from disturbances in food consumption and sleep (Clarren and Smith, 1978; Eckardt et al., 1998). Animal models have shown that adult responses to ethanol are significantly altered by developmental ethanol exposure. For instance, adult mice exposed prenatally to ethanol have been reported to be more sensitive to the locomotor stimulating effects of low doses of ethanol (Becker et al., 1993), be resistant to the disruptive effects of ethanol on operant responding (Middaugh and Ayers, 1988), and be defective in the development of tolerance to the motor incoordinating effects of ethanol (Becker et al., 1996). These behavioral changes are directly linked to the sensitivity of the developing nervous system to the toxic effects of ethanol. Numerous studies have shown that damage to the nervous system is related to the timing, pattern and dose of alcohol exposure during fetal development. For example, a single ethanol exposure in mice administered 7 days after birth (P7; equivalent to the third trimester of pregnancy in humans) causes generalized loss in brain mass (Samson and Diaz, 1981), whereas earlier exposure leads to less deleterious effects (Tran et al., 2000). Conversely, studies in primates have found that earlier exposures to ethanol are as damaging as longer exposures that also included the earlier time window (Clarren et al., 1992; Schneider et al., 2001). This was also observed in zebrafish, in which early ethanol exposure, from midblastula to organogenesis, causes most of the ethanol-induced developmental defects (Reimers et al., 2004). Intriguingly, most of the ethanol sensitivity periods correlate with episodes of intense cellular growth and proliferation (Ikonomidou et al., 2000). Whether these periods of sensitivity are also relevant to the behavioral alterations associated with FAS/FASD has not been examined.
The timing of critical periods of alcohol sensitivity and the mechanisms by which ethanol exposure during these periods affects both development and adult behavior can be investigated using Drosophila melanogaster, the common fruit fly. The use of Drosophila for the study of FAS/FASD-like disorders is sensible for a number of reasons: first, previous studies have shown that flies are susceptible to the developmental toxicity of ethanol; larvae reared on ethanol-containing food exhibit appendage abnormalities (Ranganathan et al., 1987a; Ranganathan et al., 1987b). Second, the external development of flies eliminates the complications of maternal-placenta-fetal interactions seen in mammalian studies. Adult females lay embryos (eggs) that hatch as larvae after 1 day. These larvae grow tremendously over the next 4 days as they voluntarily consume food, and molt twice. During the final larval instar, larvae stop eating, leave the food (wander) and form a puparium, signaling the onset of metamorphosis. The duration of metamorphosis is 4 days, after which the adult fly emerges. Thus, the life cycle of the fly is such that developmental ethanol exposure and the consumption of ethanol-containing food are voluntary, unlike gestational mammals, and occurs mostly during the larval stage of development; involuntary ethanol exposure during metamorphosis can, however, be achieved experimentally. Third, flies allow for mutagenesis screens, which, coupled with gene cloning and genetic pathway analysis, can be used to identify and elucidate mechanisms underlying ethanol teratogenesis. This is important given that genetic factors have been shown to modulate numerous aspects of alcohol-related disorders, including those of FAS (Ducci and Goldman, 2008).
The molecular mechanisms underlying FAS/FASD are complicated owing to the fact that ethanol and its metabolic products interact with many different gene products in a diversity of developing tissues. Studies have attributed the teratogenic effects of alcohol to ethanol metabolism and related oxidative stress (Kotch and Sulik, 1992), to neuronal cell loss and migration defects (Ikonomidou et al., 2000; Rovasio and Battiato, 1995), to changes in DNA methylation patterns (Kaminen-Ahola et al., 2010; Liu et al., 2009), and to a reduction in retinoic acid production (Yelin et al., 2005). Previous work has also shown that developmental ethanol exposure inhibits both trophic and neurotrophic growth factors and/or their signal transduction pathways. For example, developmental ethanol exposure inhibits the expression of insulin, insulin-like growth factor (IGF)-II, and the IGF-I and -II receptors in the rodent brain (de la Monte et al., 2005). In addition, IGF-I is protective against ethanol-related neuronal toxicity in cultured neurons (Barclay et al., 2005) and ameliorates the motor coordination defects caused by developmental alcohol exposure in rats (McGough et al., 2009). The ability of ethanol to interfere with growth factor signaling, and insulin and/or IGF signaling in particular, has numerous effects, including those on cell proliferation, growth, viability, energy metabolism and synapse formation (Luo and Miller, 1998).
In Drosophila, as in mammals, organismal and cellular growth is regulated by the insulin signaling pathway. There are seven Drosophila insulin-like peptides (Dilps), which govern growth, fat and carbohydrate metabolism, reproduction, and longevity (Geminard et al., 2009). The Dilp genes are expressed in a variety of larval and adult tissues, and their products activate a common insulin receptor (InR) (Brogiolo et al., 2001; Chen et al., 1996). Four Dilps, encoded by the dilp1, dilp2, dilp3 and dilp5 genes, are expressed in a small subset of specialized neurosecretory cells, the insulin producing cells (IPCs). These Dilps show overlapping expression patterns and are necessary to drive the extensive growth occurring during larval development (Gronke et al., 2010). Ablation of the IPCs during larval development leads to reduced body size, developmental delay and lethality, phenotypes that are similar to what is observed with mutants in the InR (Broughton et al., 2005; Chen et al., 1996; Ikeya et al., 2002; Rulifson et al., 2002). The expression of dilp6 in the fat body controls growth, specifically during pupal development (Okamoto et al., 2009; Slaidina et al., 2009), whereas dilp4 and dilp7 are not involved in growth regulation, but instead have possible roles in axon guidance and female fecundity, respectively (Gronke et al., 2010; Yang et al., 2008; Song et al., 2003).
Here we present a Drosophila model of alcohol-induced teratogenesis that exhibits several features of FAS, including profound changes in adult behavior. We find that flies reared continuously on ethanol-containing food show a dose-dependent developmental delay, reduced eclosion as adults, a reduction in adult mass and altered behavioral responses to vaporized ethanol as adults. We demonstrate that many of these phenotypes can be attributed to ethanol interfering with the insulin signaling pathway in the brain, specifically with the expression of dilp2 and InR. We demonstrate that Dilp expression in the brain and in the whole larva can ameliorate both the developmental and behavioral alterations associated with developmental ethanol exposure. Our findings validate Drosophila as a useful animal model to uncover the molecular basis of FAS. Thus, studies of ethanol teratogenesis in Drosophila should provide new and complimentary mechanistic insights into the toxicity of developmental ethanol exposure in mammals, including humans.
RESULTS
Ethanol-reared flies display reduced viability and have developmental delay
To establish Drosophila as a model system to investigate FASD, we first determined the optimal ethanol-dosing regimen to induce developmental toxicity in flies. From embryogenesis until adult eclosion, flies of our wild-type strain, white1118 Berlin (wB), were reared on media supplemented with 0, 5, 10 or 12% ethanol. To avoid evaporation of the ethanol in the food and to expose flies during metamorphosis (when they do not feed), the vials in which the flies develop were placed in a water bath (controls) or in a 5% ethanol bath (experimental), and the duration of development and adult eclosion rates were monitored (see Methods for a more detailed protocol). There was a striking decrease in the number of flies undergoing adult eclosion in groups reared on ethanol food compared with control groups, with the effect being more severe at higher ethanol concentrations (Fig. 1A). This reduced eclosion was due to lethality during both the larval and pupal stages; only a fraction of larvae developed to the pupal stage and, of the pupae that formed, only a fraction eclosed as adult flies (supplementary material Fig. S1A).
Fig. 1.

Ethanol-reared flies show reduced viability and developmental delay. (A) The percentage of flies undergoing eclosion (mean + s.e.m.) differs between control (0% ethanol) and ethanol-reared flies (5, 10 and 12% ethanol-food) (Dunnett’s, n=10, *P<0.0001). (B) Duration of development (from egg to adult) is prolonged by ethanol-rearing conditions. Cumulative eclosion rates (percentage of flies undergoing eclosion) of ethanol-reared flies differ from that of control flies (repeated measures ANOVA, n=10, *P<0.0001 between and within groups). Time to 50% total eclosion (at which 50% of flies had eclosed) differs between ethanol-reared and control flies and is indicated with arrowheads (Dunnett’s, n=10, *P<0.0001). Colors correspond to treatment groups shown in A. (C) At eclosion, adult mass is reduced by developmental ethanol exposure (Kruskal-Wallis tests, n=6, *P=0.0032 and 0.0242 for adult females and males, respectively).
In addition to decreasing viability, ethanol exposure during development significantly slowed the rate of egg-to-adult development (Fig. 1B). On regular media, wB flies began eclosion 10 days after egg laying (AEL), with 50% total eclosion (the point at which 50% of the flies have eclosed) occurring by 10.2 AEL (Fig. 1B). By contrast, wB reared on 5%-ethanol food began eclosion with a 1-day delay, 11 days AEL, and 50% total eclosion occurred 11.3 days AEL (Fig. 1B). Flies reared on 10- and 12%-ethanol food were more severely delayed to eclosion, as 50% total eclosion was observed 13.1 and 14.3 days AEL, respectively (Fig. 1B). A detailed analysis of the developmental delay revealed that the duration of only the second and third larval instars was affected by ethanol exposure (supplementary material Fig. S1B), whereas the duration of metamorphosis was normal (data not shown). In contrast to a previous study, our ethanol-rearing protocol did not cause adult patterning defects (at any concentration tested) (Ranganathan et al., 1987a); however, at the time of eclosion, both female and male flies had decreased body weight (Fig. 1C). Importantly, we found that the presence of ethanol (5%) in the food did not significantly alter larval food consumption (supplementary material Fig. S2); the developmental delay, lethality and reduced body size are therefore not due to lack of nutrition. Finally, adult lifespan was not affected by developmental ethanol exposure (5%; supplementary material Fig. S1C). These data indicate that developmental ethanol exposure is detrimental to the correct timing of development and to survival to adulthood.
Ethanol-reared flies exhibit neurobehavioral changes
Because fetal ethanol exposure in mammals leads to altered adult responses to ethanol exposure (Middaugh et al., 1988; Becker et al., 1993) (for a review, see Becker et al., 1996), we tested adult flies reared in 5% ethanol for their responses to the stimulating and sedating effects of ethanol vapor as well as their ability to develop tolerance to ethanol-induced sedation.
We exposed adult flies reared on 5% ethanol to a concentration of ethanol vapor that stimulates locomotor activity and analyzed their behavior using a locomotor tracking assay (Wolf et al., 2002) (Fig. 2A). In control flies, which were not exposed to ethanol during development, this concentration of ethanol vapor caused a transient olfactory startle response (first minute) that was followed by a sustained period of enhanced locomotor activity. As the concentration of internal ethanol increased, flies slowed down and began to sedate. This pattern is very similar to that seen in mice, where low doses of ethanol stimulate locomotion, whereas higher doses lead to motor incoordination and, eventually, sedation (Phillips and Shen, 1996). Flies reared in 5% ethanol displayed significantly increased and sustained locomotor activation when exposed to ethanol vapor as adults, achieving, on average, a velocity 1.6 mm/second faster than that seen in control flies (grown on food without ethanol). Thus, ethanol exposure during development leads to increased sensitivity to the locomotor activating effects of ethanol in adulthood.
Fig. 2.

Flies reared in ethanol display permanent neurobehavioral changes. (A) Ethanol-reared flies display increased locomotor hyperactivation when exposed to a moderate concentration of ethanol vapor. Flies were exposed to a 70:80 ratio of vaporized ethanol:humidified air (E/A) starting at time 0. Ethanol-reared flies achieved a peak velocity of 8.2 mm/second, compared with 5.6 mm/second for control flies (Student’s t-test, n=8, *P<0.001). (B) Ethanol-reared flies are resistant to ethanol-induced sedation. Upon exposure to a high (100:50 E/A) concentration of ethanol, control flies achieved 50% sedation 4.6 minutes sooner than ethanol-reared flies, reflecting a 34% increase in sedation resistance for flies reared on ethanol (Student’s t-test, n=12, *P<0.001). (C,D) Ethanol-reared flies are defective in tolerance development. Flies were exposed to a sedating dose of ethanol (110:40 E/A), allowed to rest for 4 hours and then exposed to a second dose; sedation times were calculated for each exposure. Control flies require an additional 9 minutes to achieve ST50 upon a second exposure, indicating the development of functional ethanol tolerance. Ethanol-reared flies develop only 4.1 minutes of tolerance (Student’s t-test, n=12, *P=0.019). In all experiments, flies were transferred as first-instar larvae to media containing 5% ethanol, collected upon eclosion, and behavioral tests were performed 2 days later.
Next, we tested whether ethanol-reared adult flies displayed altered responses to ethanol-induced sedation. We exposed flies to a sedating concentration of ethanol vapor and examined the flies for loss of the righting reflex (LORR) at defined intervals throughout the exposure. We found that the time needed for 50% of control flies to become sedated (ST50) was 13.6 minutes, whereas flies reared on food containing 5% ethanol had an ST50 of 18.2 minutes (Fig. 2B). Thus, flies reared on 5% ethanol showed a 34% increase in ST50, a marked resistance to the sedating effects of ethanol.
Finally, we tested flies reared on 5% ethanol for their ability to develop tolerance to the sedating and/or motor incoordinating effects of ethanol exposure (Fig. 2C,D). We exposed adult flies to a sedating concentration of ethanol vapor for 35 minutes, calculated the ST50 for the first exposure, then, 4 hours later, exposed them to a second dose of ethanol vapor. Control flies (reared in the absence of ethanol) showed ST50 of 10.4 and 19.1 minutes during the first and second exposures, respectively. The difference in ST50 between the second and first ethanol vapor exposures, 8.7 minutes, is defined as tolerance (Berger et al., 2008). Ethanol-reared flies, as expected, had a higher ST50 (15.7 minutes) during their first ethanol vapor exposure, which increased to 19.8 minutes during the second exposure. Thus, ethanol-reared flies showed an increase of ST50 of only 4.1 minutes, a reduction of approximately 47% relative to control flies. It is possible that the tolerance defect we observed in the ethanol-reared adult flies might be due to a ceiling effect of the assay during the second ethanol exposure. However, we believe that this is not the case because we have found that many ethanol-resistant mutants can develop normal ethanol tolerance (Berger et al., 2008). Furthermore, it is possible to generate ethanol-reared flies with normal sedation sensitivity that nevertheless are defective in the development of ethanol tolerance (see later). Taken together, these results suggest that regions of the nervous system and/or specific signaling mechanisms that are required for establishing normal ethanol sensitivity and tolerance in the adult fly are altered by exposure to ethanol during development.
Ethanol-reared adult flies showed normal ethanol absorption and metabolism (supplementary material Fig. S3). In addition, western blot analysis showed that expression of alcohol dehydrogenase, the principal metabolizing enzyme for ethanol, was not different between adult ethanol-reared and control flies (data not shown). It should be noted that adult flies exposed to ethanol during development have long metabolized the ethanol when tested for adult ethanol behaviors (supplementary material Figs S3 and S4). These data indicate that the ethanol sensitivity phenotype of ethanol-reared flies is not caused by altered drug pharmacokinetics.
Critical periods of ethanol toxicity
Studies in mammals, including humans, have shown that both the timing and duration of ethanol exposure are important factors in the development of FAS (Sulik et al., 1981; Sulik et al., 1986). To assess the critical periods during which developmental ethanol exposure leads to a decline in adult viability and developmental delay, we reared wB flies on 5%-ethanol food during discrete developmental stages. For example, ethanol exposure was limited to embryogenesis, larval or pupal stages, or, alternatively, to a combination of these developmental stages (Table 1).
Table 1.
Critical periods of ethanol toxicity: survival and developmental delay

Exposure to 5% ethanol during embryogenesis did not affect adult viability or time to eclosion, most probably because ethanol does not cross the eggshell. Ethanol exposure that was limited to larval development, by contrast, caused a 1-day delay in eclosion and a reduction in adult viability from 93% in control flies to 63% in ethanol-reared flies. When ethanol exposure was restricted to the pupal stage, there was a small but significant decline in adult viability, yet developmental timing was normal (Table 1). These results confirm that the developmental delay induced by ethanol is solely due to exposure during the larval stages, whereas the reduced viability has both larval and pupal components. The effects of ethanol were also examined during individual larval stages and, interestingly, exposure to ethanol during either the second or third larval instars resulted in a developmental delay, whereas a decline in adult viability was only observed with exposure during the third larval instar (Table 1). Thus, both the second and third larval instars are particularly sensitive to the toxic effects of developmental ethanol exposure. Taken together, these data demonstrate that the critical time period for ethanol toxicity in flies is primarily during larval development, although ethanol exposure during metamorphosis also contributes to the decline in adult viability.
Next, we determined whether the neurobehavioral phenotypes were due to ethanol exposure during distinct developmental time periods (Table 2). Flies were transferred from regular to ethanol-containing food (5%) at specific larval stages and allowed to complete development before being tested for acute ethanol sensitivity and tolerance as adults. As expected, flies grown on ethanol-containing food throughout development displayed increased sedation resistance and reduced tolerance development compared with controls. Flies were similarly affected when transferred to ethanol-containing food as second-instar larvae, indicating that the critical period(s) for both phenotypes (adult resistance and tolerance) was during or after the second instar phase. When flies were transferred to ethanol-containing food as third-instar larvae, they showed sedation profiles indistinguishable from those of control flies (Table 2), indicating that the critical period for development of resistance to ethanol sedation is during the second larval instar. These flies, however, failed to develop normal ethanol tolerance (Table 2), indicating that the critical period for this phenotype is during or after the third larval instar phase. Importantly, we did not see a cumulative effect of longer exposure to ethanol; flies exposed to ethanol from the late-third-instar stage onwards showed the same reduction in tolerance as flies exposed to ethanol throughout development (Table 2). These data show that the critical periods of developmental ethanol exposure that lead to enhanced resistance to sedation (the second larval instar) and a reduced ability to develop tolerance (the third larval instar and pupal stage) are separable, suggesting that these two phenotypes are caused by distinct effects of ethanol on development.
Table 2.
Critical periods of ethanol toxicity: behavior

To determine whether the differences we observed in ethanol toxicity, developmental delay and adult behavior had a pharmacokinetic basis, we measured the concentration of ethanol in animals exposed to developmental ethanol (5% ethanol). Ethanol-reared animals showed a steady increase of internal ethanol concentration during each larval instar, achieving a maximum internal ethanol concentration during metamorphosis (supplementary material Fig. S4). Such observations suggest that the discrete ethanol-sensitivity periods are not simply due to the rise of internal ethanol concentration during larval and metamorphic development, with each phenotype having a different ethanol concentration threshold, but rather that individual tissues and/or cells are differentially sensitive to ethanol at distinct developmental stages.
Effects of developmental ethanol exposure on proliferation and apoptosis in the larval brain and the imaginal discs
In a number of animal models, ethanol treatment causes increased apoptosis in the developing CNS, resulting in reduced brain mass (Ikonomidou et al., 2000; McGee and Riley, 2006). Ethanol exposure also alters the rate of neuronal cell division in the immature brain, reducing the number of new cells as well as deterring neural outgrowth (Guerri, 1998). We observed a reduction in larval brain size upon rearing flies on ethanol-containing food; compared with controls, brain lobes were reduced by 20% and 40% in larvae reared on 5%- and 10%-ethanol food, respectively (Fig. 3A–D). To determine whether the small CNS phenotype was due to increased apoptosis, we examined cell death in the brains of control and ethanol-reared larvae at various developmental stages (early-, mid-and late-third-instar larvae) by TUNEL labeling. The brains of control and ethanol-reared larvae showed nearly equivalent levels of cell death at each developmental stage examined (supplementary material Fig. S5; data not shown), indicating that the reduced size of the CNS was not due to ethanol-induced apoptosis.
Fig. 3.

Developmental ethanol exposure causes defective proliferation in the larval CNS. The number of replicating cells (S phase) in the CNS of control and ethanol-reared larvae was assessed by pulse labeling with BrdU at 24–28 hours post-hatching, with dissection of wandering third-instar larvae.
Representative samples of brains from larvae reared on (A) 0% ethanol, (B) 5% ethanol and (C) 10% ethanol (40×magnification) are shown. Arrows indicate labeling in the mushroom body (MB), optic lobe (OL) and thoracic (T) neurons. (D) The size of brain lobes differs between ethanol-reared (5 and 10% ethanol) and control larvae (Dunnett’s, n=10, *P<0.0047).
To investigate defective proliferation as a possible mechanism for the small brain phenotype, we monitored replicating cells (S phase) by BrdU incorporation in the developing brains of both ethanol-reared and control larvae. Larvae were labeled with BrdU for 4 hours at the transition from the first to second larval instars and dissected for analysis as wandering late-third-instar larvae. All cells in the brain that undergo S phase during the labeling period incorporate BrdU, including neuroblasts, ganglion mother cells and immature neurons; their progenitors also retain BrdU label (Datta, 1995). Control larvae showed high levels of BrdU-labeled neurons in the larval brain lobes as well as in the ventral ganglion (Fig. 3A). By contrast, ethanol-reared larvae showed a drastic reduction of dividing cells in the larval CNS, both in the brain lobes and the ventral ganglion (Fig. 3B,C). In order to determine whether this pattern of defective proliferation also occurred during later larval stages, ethanol-reared and control larvae were labeled with BrdU at the transition from the second to third larval instar. Labeling at this later stage revealed an equivalent number of dividing cells in larvae reared on 0%- and 5%-ethanol food (supplementary material Fig. S6A,B). However, larvae reared on 10%-ethanol food showed a dramatic reduction in replicating cells in the brain (supplementary material Fig. S6C). These results are consistent with the small brain phenotype observed predominantly in larvae grown on 10%-ethanol food. Taken together, these data suggest that the small brain phenotype observed in flies reared on ethanol-containing food is caused by reduced proliferation, not enhanced apoptosis, and that distinct developmental periods are differentially sensitive to the effects of ethanol on proliferating CNS cells.
We did not observe gross morphological defects in the adult CNS upon developmental ethanol exposure to 5%-ethanol food (data not shown); it is likely, however, that subtle defects were present but not easily detectable.
To investigate whether structures that are rapidly growing in larvae are generally affected by developmental exposure to 5% ethanol, we examined the growth of imaginal discs, the larval precursors of the adult fly appendages. As in the larval brain, imaginal discs of ethanol-reared larvae were significantly reduced in size compared with those of control larvae (supplementary material Fig. S7). To assess whether developmental ethanol exposure slowed cell proliferation in the imaginal discs, we used a heat-shock-induced flip-out GAL4 driver to induce permanent, heritable expression of GFP in random clones of cells (Neufeld et al., 1998). At 48 hours after heat shock, we counted the number of GFP-expressing cells per clone to determine in vivo rates of cell division. We found that cell-doubling time in the wing imaginal discs increased from 14.4±0.87 hours in control larvae to 18.5±0.61 hours in ethanol-reared larvae (Student’s t-test, P<0.0001; n=83 and 89 clones in control and 5%-ethanol-reared larvae, respectively). In addition, the small disc phenotype was not caused by abnormal morphogen signaling, because the expression of several morphogens in the wing imaginal discs of ethanol-reared larvae, including that of wingless, decapentapelagic and hedgehog, were normal in pattern and intensity compared with control discs (supplementary material Fig. S7; data not shown). Taken together with the defective proliferation observed in the larval CNS of ethanol-reared larvae, these data indicate that developmental ethanol exposure slows cell proliferation in the developing fly larvae, and this effect probably contributes to the ethanol-induced delay to eclosion and adult lethality.
Developmental ethanol exposure interferes with insulin signaling
Our observations that developmental ethanol exposure leads to reduced larval growth, delayed development, increased stored triglycerides (data not shown), small imaginal discs and small adult flies – all of which are phenotypes that are also seen upon impaired insulin signaling (Garofalo, 2002; Rulifson et al., 2002) – suggested that ethanol rearing might interfere with the insulin signaling pathway. This interference could occur at several levels, such as ligand production and/or secretion, receptor expression, and signal transduction. To investigate whether developmental ethanol exposure interfered with Dilp production, we analyzed dilp2 transcript and protein levels by quantitative real-time (RT)-PCR (qPCR) and immunohistochemistry, respectively, in both ethanol-reared and unexposed control larvae. Expression of the dilp2 gene was downregulated upon developmental ethanol exposure by approximately 50% and 25% in the larval brain and in whole larvae, respectively (Fig. 4A). Additionally, ethanol-reared larvae showed reduced Dilp2 protein in the insulin producing cells (IPCs) of the larval brain (Fig. 4B–D). This reduction in dilp2 transcript and protein was not caused by the loss of Dilp-producing cells (as observed by the number of dilp2-lacZ-expressing cells) in ethanol-reared larvae (data not shown).
Fig. 4.

Developmental ethanol exposure alters insulin levels. (A) Developmental ethanol exposure reduces dilp2 expression in larval brain and whole larvae as quantified by qPCR. mRNA levels are expressed as fold difference relative to control larvae (Kruskal-Wallis test, n=3, *P<0.04). (B,C) Dilp2 is reduced in ethanol-reared wandering larvae. (B,C) Representative images of Dilp2 in control (B) and ethanol-reared (C) larvae. Arrows indicate IPCs. Asterisk shows the esophagus. (D) Quantification of Dilp2 in the IPCs (Student’s t-test, n=10–13, *P<0.001). (E) Larval expression of dilp5 in IPCs of the CNS, using dilp2-GAL4, ameliorates the developmental delay and lethality induced by developmental ethanol exposure. Total eclosion (mean + s.e.m.) of dilp5-overexpressing larvae (dilp2-GAL4/+; UAS-dilp5/+) differs from control larvae (dilp2-GAL4/+and UAS-dilp5/+) (one-way ANOVA, with Tukey HSD post-hoc analysis, n=6, *P=0.045).
(F) Cumulative eclosion rates of dilp5-overexpressing larvae differs from control larvae (repeated measures ANOVA, n=6, between groups *P=0.0134, within groups *P=0.001). Time to 50% total eclosion (indicated by arrowheads) differs between dilp5-overexpressing larvae and control larvae (Dunnett’s, n=6, *P<0.001). n corresponds to the number of vials, containing 100 animals each.
To examine whether the ethanol-induced reduction in dilp2 expression is causally related to ethanol toxicity, we tested whether increasing Dilp expression would rescue the developmental delay and reduced viability caused by developmental ethanol exposure. We used dilp2-GAL4, which drives GAL4 specifically in the IPCs, to induce expression of dilp2 and dilp5, normally expressed in the IPCs, as well as dilp6, the expression of which is found in the fat body and has been shown to act redundantly to the IPC-expressed Dilps (Gronke et al., 2010). Increasing expression of dilp5 and dilp6 ameliorated both the lethality and delay phenotypes associated with developmental ethanol exposure (Fig. 4E,F; supplementary material Fig. S8A,B), whereas overexpression of dilp2 did not rescue either phenotype (data not shown). However, the ethanol-induced developmental phenotypes were improved when dilp2 was expressed ubiquitously using armadillo-GAL4 (arm-GAL4) as a driver (supplementary material Fig. S8C,D). Taken together, these experiments show that increasing Dilp expression, specifically dilp5 and dilp6, in brain IPCs is sufficient to ameliorate the lethality and delay phenotypes induced by developmental ethanol exposure. Interestingly, expression of dilp2 lessened the ethanol-induced developmental phenotypes, but only when expression was driven throughout the animal. It is possible that expression of dilp2, unlike dilp5 or dilp6, is needed in the imaginal discs for efficient rescue of ethanol-induced toxicity (see Discussion). Increasing Dilp expression in flies that were reared on regular media did not alter adult viability or developmental time to eclosion (supplementary material Fig. S9), indicating that the rescue we observed in ethanol-reared animals was not due to general developmental differences. Together, these data show that developmental ethanol exposure impairs larval growth and decreases adult viability by interfering with Dilp production in the developing brain and possibly other larval tissues.
To further explore the idea that ethanol rearing interferes with the insulin signaling pathway, we investigated whether InR expression was also altered by developmental ethanol exposure. We found that InR transcripts were reduced by 70% in the larval brain, but were normal in the whole larva upon ethanol rearing (Fig. 5A), implying that CNS InR expression is particularly sensitive to inhibition by ethanol. If the insulin signaling pathway was compromised by developmental ethanol exposure, then InR mutants should be particularly sensitive to developmental ethanol exposure. To address this possibility, we examined three InR mutant alleles: InR5545, InRGC25 and InREC34. Because these alleles are all homozygous lethal (Chen et al., 1996), we assessed the response of heterozygous flies to developmental ethanol exposure. When reared on 5%-ethanol food, we observed only a slight reduction in adult viability in the InR heterozygotes compared with controls (data not shown). However, rearing flies on 7.5% ethanol food reduced adult viability from 61% in controls to ∼40% in InR heterozygotes (Fig. 5B). Ethanol rearing did not produce developmental delay in the InR mutants (data not shown), suggesting that more pronounced reductions in InR expression are needed for the manifestation of this phenotype. Importantly, InR heterozygotes reared on regular media showed no differences in development time or adult viability compared with controls (supplementary material Fig. S10).
Fig. 5.

Developmental ethanol exposure reduces InR expression and InR mutants are sensitive to developmental ethanol toxicity.
(A) Developmental ethanol exposure reduces InR expression specifically in the larval brain. mRNA levels are expressed as fold difference relative to control larvae (Kruskal-Wallis test, n=3, *P=0.0369). (B) Heterozygous mutants for InR are sensitive to the decrease in eclosion induced by developmental ethanol exposure (Dunnett’s, n=6, *P<0.001).
Taken together, our data leads us to propose that developmental ethanol exposure in flies inhibits the insulin signaling pathway by inhibiting both ligand and receptor expression, and that this inhibition is responsible for the developmental delay and reduced viability seen in ethanol-reared flies. Our data also suggest that the CNS InR is particularly sensitive to inhibition by ethanol.
Altered insulin signaling is responsible for ethanol-induced neurobehavioral changes
Perturbations of insulin signaling in the nervous system can lead to changes in sensitivity to ethanol-induced sedation (Corl et al., 2005). We therefore hypothesized that the reduction in Dilp-InR signaling during development might be responsible for the persistent neurobehavioral changes observed in ethanol-reared flies. To test this, we expressed UAS-dilp2 and UAS-dilp6 under the control of dilp2-GAL4 in larvae grown on 5% ethanol, and tested the resultant adults for sedation resistance and tolerance. Consistent with data obtained with wB flies, this developmental exposure led to increased resistance to sedation and a reduced ability to develop tolerance in the genetic control flies (Fig. 6A). However, expression of either dilp2 or dilp6 restored normal sensitivity to ethanol-induced sedation in flies reared on 5% ethanol, indicating a complete rescue of this phenotype by transgenic Dilp expression (Fig. 6A; supplementary material Fig. S11A). In addition, increasing expression of dilp2 (but not dilp6; supplementary material Fig. S11B) restored normal ethanol tolerance to ethanol-reared flies (Fig. 6B). Thus, the observed reduction in insulin signaling in flies reared on ethanol seems to be responsible for both the sedation resistance and tolerance development phenotypes. Interestingly, as for the rescue of the developmental phenotypes, there are differences in the effectiveness of different Dilps in restoring normal behavior to ethanol-reared flies.
Fig. 6.
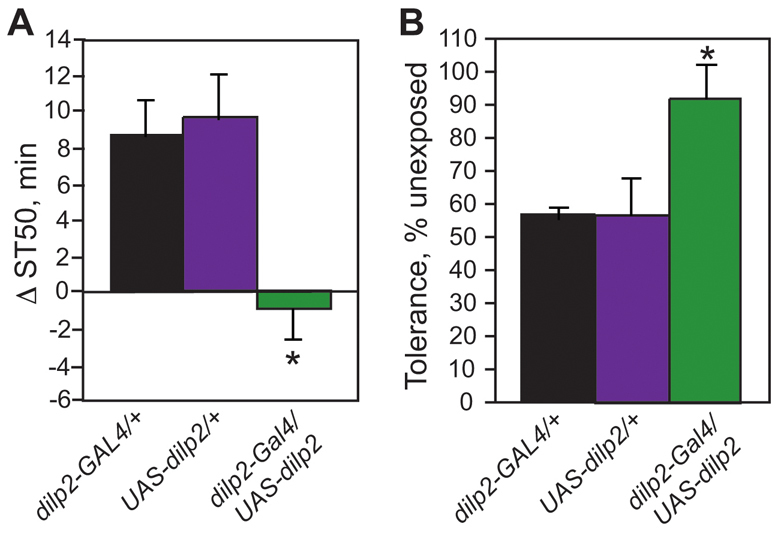
dilp2 expression rescues both sedation resistance and tolerance development. (A) dilp2 expression rescues ethanol-induced sedation resistance. Data are presented as difference in time to 50% sedation (ST50) for ethanol-reared flies as compared with control flies of the same genotype. dilp2-GAL4/UAS-dilp2 flies do not display increased sedation resistance upon ethanol-rearing, whereas both genetic background controls show the expected increase in ST50 (one-way ANOVA with Tukey HSD post-hoc analysis, n=12, *P<0.01). (B) dilp2 expression rescues ethanol-induced tolerance defects. Data are presented as the percent control tolerance (tolerance developed by ethanol-reared flies divided by tolerance developed by control flies of the same genotype multiplied by 100). Although both genetic background controls show a 40–50% reduction in tolerance when reared in ethanol, dilp2-GAL4/UAS-dilp2 flies develop normal levels of ethanol tolerance (one-way ANOVA with Tukey HSD post-hoc analysis, n=6, *P<0.05).
DISCUSSION
A Drosophila model of FAS
Prenatal alcohol exposure can cause FAS, a complex disorder with numerous developmental, morphological and neurological deficits (Jones and Smith, 1973). In this study, we investigated the effects of developmental ethanol exposure in the fruit fly Drosophila melanogaster, and found many features in common with FAS. Flies reared on ethanol-containing food displayed a dose-dependent developmental delay, a small larval CNS, increased developmental mortality and reduced adult size. Developmental ethanol exposure also altered ethanol-responsive behaviors in adult flies. Ethanol-reared flies were hypersensitive to the stimulating effects of ethanol, abnormally resistant to ethanol-induced sedation, and defective in tolerance development, all phenotypes also observed in mammalian FAS models.
The phenotypic similarities between our model and mammalian FAS models extend to the specificity of critical periods for ethanol-induced developmental phenotypes. We found that, with the exception of ethanol-induced lethality, all of the phenotypes examined exhibit discrete critical periods. The critical period for ethanol-induced growth delay is during the second and third larval instars. Increased sedation resistance results from exposure during the second larval instar, whereas the tolerance defect maps to the late-third-instar or early-pupal stage. Of all the phenotypes examined, only the ethanol-induced lethality is cumulative, increasing in severity with longer exposure times. As with developmental ethanol studies in other organisms, these data indicate that the critical periods of ethanol sensitivity vary depending on what is being measured, i.e. viability versus developmental delay (Blader and Strahle, 1998; Oxendine et al., 2006). Such differences undoubtedly reflect the effects of ethanol on specific developmental events or processes. For example, in the case of the ethanol-induced growth delay, the critical period is a time of rapid cell division and growth in the larva. Our investigations show that ethanol exposure during this growth period interferes with cell division in the brain and imaginal discs (Fig. 3; supplementary material Fig. S7). Therefore, one explanation for the observed phenotypes is that insufficient cellular proliferation leads to delayed growth. Indeed, it is known that reduced cell proliferation in the imaginal discs can cause developmental delay (Brogiolo et al., 2001; Stieper et al., 2008). Taken together, these data suggest that reduction in imaginal disc size explains the ethanol-induced developmental delay.
The ethanol-induced defect in tolerance development is particularly interesting, because, unlike the other phenotypes examined, its critical period (late third instar to pupation) is during periods of neuronal differentiation, outgrowth and remodeling, rather than intense cell division. This might indicate that normal development of tolerance is dependent on neurite outgrowth and axon targeting, both of which depend on insulin signaling (Scolnick et al., 2008; Song et al., 2003).
Insulin signaling is reduced in flies reared in ethanol
Our studies show that, as in mammals, developmental ethanol exposure in flies leads to diminished insulin signaling. Expression of dilp2 and InR was reduced in the brains of larvae reared on ethanol, and the growth and viability phenotypes were rescued by transgenic supplementation with several Dilps. Similarly, Dilp overexpression rescued the sedation sensitivity and tolerance defects of adult flies subjected to developmental ethanol exposure.
It is interesting to note that ubiquitous overexpression of dilp2 was required to rescue viability and developmental delay, whereas overexpression of dilp2 in the brain rescued only the adult behavioral phenotypes of ethanol-reared animals. By contrast, expression of dilp6 in the brain (driven by dilp2-GAL4) was sufficient to rescue both the growth and viability defects, as well as sedation resistance. Although it is possible that the differences are a result of lower expression of the UAS-dilp2 transgene relative to UAS-dilp6, we consider this possibility unlikely because previous studies have found that overexpression of UAS-dilp2 is more effective than UAS-dilp6 to drive increased body size (Ikeya et al., 2002). These results might therefore reveal a previously unidentified specificity of function for dilp2 in regulating behavior when expressed in the brain. Alternatively, it is possible that dilp2 is less efficiently transported from the CNS to other tissues or has lower affinity for InR, such that CNS-specific expression is insufficient to rescue the imaginal-disc-mediated growth and viability defects.
Although our results with manipulations of the insulin pathway are satisfying as validation of our model system, they also suggest a potential mechanism for the adult behavioral phenotypes caused by developmental ethanol exposure. In the Drosophila eye, InR is required for proper photoreceptor axon guidance (Song et al., 2003). Similarly, IGF-I is a chemoattractant for axon growth cones in cultured rodent neurons, and IGF signaling is required for proper targeting of axons in the rodent olfactory bulb (Scolnick et al., 2008). It is reasonable to hypothesize that some or all of the behavioral defects caused by developmental ethanol exposure (and that are rescued by Dilp supplementation) are a result of improper neurite outgrowth and/or axon targeting during development. In Drosophila, it is possible to test this hypothesis in a relatively straightforward manner. For example, developmental exposure to ethanol results in increased ethanol-induced locomotion. It is known that a specific pair of dopaminergic neurons in the fly brain mediates ethanol-induced hyperactivity (Kong et al., 2010); similarly, neurons in the pars intercerebralis and central complex have been implicated in sensitivity to ethanol-induced sedation (Rodan et al., 2002). We will be able to examine the general organization of these brain regions using GFP driven by specific GAL4 lines. Analysis of detailed axon pathfinding could then be focused to the relevant neurons using the MARCM technique (Lee and Luo, 2001). We can therefore use this model to investigate the requirement for insulin signaling in the development of the nervous system and how it impacts adult behavior.
Potential significance for FAS
Our results in Drosophila and those of McGough and colleagues in rats (McGough et al., 2009) demonstrate that some of the deleterious effects of developmental ethanol exposure can be ameliorated by replacing lost insulin signaling. These results are important because they illustrate the potential for pharmacological intervention in FAS.
Despite decades of public awareness campaigns and widespread understanding that alcohol consumption during pregnancy can result in birth defects, the rate of alcohol abuse during pregnancy remains unchanged (Sampson et al., 1997). This is probably due in large part to the addictive effects of alcohol, and highlights the need for alternative solutions to the problem of prenatal ethanol exposure.
It is clear from human epidemiological data that genetic factors can modulate the teratogenic effects of alcohol. Monozygotic twins from alcohol-abusing mothers display concordance for FAS defects, whereas dizygotic twins do not (Christoffel and Salafsky, 1975; Streissguth and Dehaene, 1993). Moreover, different inbred mouse and chick strains, in which the timing of alcohol administration and blood alcohol concentration was controlled for, differ in their susceptibilities to ethanol teratogenesis (Boehm et al., 1997; Bupp Becker et al., 1998; Su et al., 2001). It is likely that many genes, in addition to those involved in insulin signaling, confer risk or protection from alcohol injury, yet none have been conclusively identified. Furthermore, insulin supplementation does not rescue the learning and memory defects of the disease, which are the most devastating symptoms of FAS (McGough et al., 2009), indicating the existence of other important developmental targets of ethanol.
The molecular identification of genetic factors that influence developmental ethanol toxicity has been hindered largely by the difficulty of performing unbiased, forward genetic screens in vertebrate systems, and it is here that our model is most useful. Our demonstration of both phenotypic and molecular conservation of developmental ethanol effects between flies and mammals indicates that Drosophila is a good model for further elucidation of the mechanisms of action of developmental ethanol exposure. This, in turn, will allow the identification of novel pharmacological targets to prevent/ameliorate the development of FAS.
METHODS
Drosophila strains and culture
Flies were raised at 25°C and 70% relative humidity on standard cornmeal/molasses medium. All experiments were carried out in a wB genetic background. The UAS-Dilp and dilp2-GAL4 strains were kindly provided by Ernst Hafen (IMSB, Zurich, Switzerland) and Eric Rulifson (UCSF, San Francisco, CA), respectively. The InR alleles were gifts from Marc Tatar (UCD, Davis, CA). All behavioral assays used 20–25 male flies aged 2–4 days after eclosion at the start of the experiment. Flies analyzed for behavior were subjected to brief (<5 minutes) CO2 anesthesia no less than 24 hours before behavioral assays.
Developmental ethanol exposure
Egg collections were taken for 2–3 hours on Petri dishes containing 0, 5, 10 and 12% ethanol food. After 24 hours of development, 100 newly hatched larvae were transferred to vials containing either ethanol food or control food, and placed in a 5% ethanol bath (experimental conditions) or water bath (control conditions). The ethanol bath ensures that developing animals are exposed to ethanol during their entire development, which continues for another 10–16 days. The number of newly eclosed adult flies was counted daily between 9–16 days AEL (or 9–14 days AEL), and these data were used to generate cumulative eclosion rate plots, a direct measurement of egg-to-adult survival, and the time to 50% of total eclosion.
To determine critical periods for ethanol toxicity, larvae were collected from control food plates as they reached the desired developmental stage (first, second, or third larval instar), transferred to 5%-ethanol-containing food (or control food) and grown as described above. Survival and development time were calculated as described above, and newly eclosed adult flies were collected and subjected to behavioral experiments as described below.
Behavioral assays
All behavioral assays were performed in the locomotor tracking system (Wolf et al., 2002). Sedation sensitivity was quantified by exposing groups of 20–25 flies to an ethanol concentration of 100:50 (ethanol vapor: humidified air) (Corl et al., 2009; Rothenfluh et al., 2006). The tubes were spun at 2- to 5-minute intervals, and the number of flies having lost righting ability was counted at each time point. From these data, we calculated ST50 (time to 50% sedation). Ethanol-induced locomotor activation was quantified by exposing groups of 20–25 flies to an ethanol concentration of 70:80 (ethanol vapor:humidified air) and analyzing their activity for a period of 30 minutes in 2- to 10-minute intervals.
qPCR
Control and ethanol-reared brains and whole larvae were snap frozen in liquid nitrogen and stored at −80°C. Total RNA was extracted using TRIzol (GIBCO) according to the manufacturer’s instructions. mRNA in total RNA was reverse transcribed using TaqMan Reverse Transcription Reagents (Applied Biosystems) according to the manufacturer’s specifications. cDNA was analyzed by quantitative, real-time PCR using the ABI PRISM 7700 Sequence Detection System (Applied Biosystems). dilp2, InR and rp49 probe and primers (Dm01822534_g1, Dm02136224_g1 and Dm02151827_g1, respectively) were obtained from Applied Biosystems. rp49 transcript levels were used as an endogenous normalization control for RNA samples, and relative mRNA abundance was calculated using the comparative ΔCt method. Each sample was analyzed in triplicate. As negative controls, we used both no-template and DNAse-treated non-reverse-transcribed mRNA samples; no significant amplification was observed in these samples.
BrdU labeling
Larvae were reared on 0% and 5% ethanol-containing food as described above, then placed on BrdU-containing food (0.7 mg/ml) for 4 hours during the first-to-second larval instar transition (24–28 hours AEL) or second-to-third larval instar transition (68–72 hours AEL). We chose the first-to-second larval instar transition because previous analysis showed that this early developmental stage was not delayed by developmental ethanol exposure and because all three neuroblast populations (mushroom body, optic lobe and thoracic) are actively cycling (Datta, 1995). The second-to-third instar transition can be identified by anterior spiracle morphology. After BrdU labeling, larvae were transferred back to 0%- or 5%-ethanol-containing food and brains were dissected from wandering larvae (larvae with half-empty gut). Brains were fixed for 30 minutes in 4% paraformaldehyde in PBS. Prior to incubation with anti-BrdU antibody, brains were incubated in 2N HCl for 1 hour, then neutralized three times for 5 minutes each in PBS with 0.1% Triton-X.
Immunostaining and imaging
Discs were fixed for 20 minutes in 4% paraformaldehyde in PBS. Larval brains were fixed for 30 minutes on ice in 4% paraformaldehyde in PBS. Rabbit anti-Dilp2 (Eric Rulifson) was used at 1:500, mouse anti-BrdU (Becton Dickinson) was used at 1:100 and rabbit anti-β-galactosidase (Cappel) was used at 1:1000. Images were collected with Leica TCS SP2 confocal laser scanning microscope.
To ensure consistency of Dilp2 staining in controls and ethanol-reared larvae, immunostaining was performed in parallel and aliquots from the same primary and secondary antibody dilutions were used. Dilp2 levels in the IPCs of the CNS were measured using projection confocal images (as seen in Fig. 4B) and ImageJ. Briefly, IPCs from ethanol-reared and control larvae were outlined and then pixel intensity was measured.
The size of brain lobes from ethanol-reared (5% and 10% ethanol) and control larvae were measured using projection confocal images (as seen in Fig. 3A–C) and ImageJ. wB brain lobes were used as a normalization control, and relative brain lobe size in the ethanol-reared larvae was calculated.
Clonal induction and cell doubling time
Because ethanol exposure causes a developmental delay, cell clones were induced at the second-to-third instar transition, a developmental stage distinguished by anterior spiracle morphology. Larvae of the genotype y,w,hs-flp122;Actin5c>CD2>GAL4, UAS-GFP were heat shocked for 15 minutes at 37°C at 72 hours AEL (or its equivalent in ethanol-reared larvae, which was ∼84 hours AEL). Discs were dissected and fixed 48 hours later. The number of cells per clone was counted on a Leica TCS SP2 confocal laser scanning microscope and cell doubling times were calculated with the following formula: (log2)(hour)/logN, where N=cell number per clone and hour=age of clones.
TUNEL assay
TUNEL assay was performed using the In Situ Cell Detection Kit, TMR red (Roche). Larval brains were fixed at 4°C for 30 minutes in 4% paraformaldehyde in PBS. Brains were then permeabilized in 0.1 M sodium citrate for 1 hour, followed by treatment with 5 μg/ml proteinase K in PBS for 2 minutes. Brains were washed twice in PBS then incubated in TUNEL labeling solution for 1 hour at 37°C. Brains were washed three times with PBS then mounted in Fluoromount mounting media.
Ethanol absorption assay
Internal ethanol concentration was measured in extracts from 0%-and 5%-ethanol-reared animals at distinct developmental stages (first-, second- and third-instar larvae, pupae, and adults). Animals were frozen at the indicated developmental stages then assayed for ethanol content using a colorimetric enzymatic kit (Diagnostic Chemicals) (Moore et al., 1998). In order to calculate the internal ethanol concentration (millimolar) of 0%- and 5%-ethanol-reared larvae, pupae and adult flies, their volumes were measured. Animal volume was measured by placing 25–100 animals, of the desired developmental stage, into graduated Eppendorf tubes on ice. Then, 500 μl of 100% ethanol was added to the tubes. The amount of displaced liquid (animal volume) was determined by removing ethanol, by Pipetman, until the meniscus read 500 μl. The calculations of internal ethanol concentration (millimolar) in larvae, pupae and adults, presented in supplementary material Figs S3 and S4, used the average value of these volume measurements (see supplementary material Table S1).
TRANSLATIONAL IMPACT.
Clinical issue
Consumption of alcohol during pregnancy can cause a complex disorder that is commonly known as fetal alcohol syndrome (FAS). Individuals with FAS exhibit a variety of symptoms, including persistent growth deficiencies, birth defects and mental retardation. Ethanol exposure is especially damaging to the developing nervous system and this has long-term consequences on adult behavior. The toxicity of developmental ethanol exposure has been attributed to numerous mechanisms, including ethanol metabolism and related oxidative stress, neuronal cell loss, and inhibition of growth factors and/or their signal transduction pathways. Although human epidemiological data and studies of animal models indicate that genetic factors confer risk for and protection from FAS, no genes that alter susceptibility to the syndrome have been conclusively identified.
Despite growing awareness of the dangers of drinking during pregnancy, the worldwide prevalence of FAS remains steady, at one to three per 1000 births. The high frequency of FAS coupled with the failure of public awareness programs highlights the need to understand more about the molecular basis of FAS, and to develop novel treatments to mitigate the complications of gestational ethanol exposure.
Results
To establish Drosophila as a genetic model for the study of FAS, the authors rear flies in the presence of ethanol and investigate the developmental and behavioral consequences of this exposure. Ethanol-reared flies show several phenotypes that are common to FAS, including reduced viability, developmental delay and small adult body size. Additionally, developmental ethanol exposure significantly impairs cell proliferation in the larval brain and imaginal discs. Finally, ethanol-reared flies exhibit persistent behavioral changes. These developmental and behavioral defects are found to be due largely to a reduction in Drosophila insulin-like peptide and insulin receptor expression.
Implications and future directions
This study demonstrates that Drosophila is a useful animal model for the study of the genetic and molecular mechanisms underlying the toxic effects of developmental ethanol exposure. The finding that, as in mammals, developmental ethanol exposure in flies leads to reduced insulin signaling might have important implications for understanding the cellular mechanisms underlying the behavioral problems associated with FAS in humans. In flies and mammals, insulin and insulin-like growth factors are known to be involved in axon guidance, neurite outgrowth and synaptogenesis. It is therefore possible that some of the behavioral deficits associated with FAS are due to miswiring of the developing brain, rather than just the loss of neurons owing to cell death or reduced cell division.
Drosophila is particularly powerful as a model system given the ability to rapidly identify molecules of interest through forward genetics. Follow-up studies will involve genetic and molecular screens to identify novel mechanisms that contribute to developmental ethanol toxicity. These approaches will aid in the understanding of neurodevelopmental pathways that are altered by exposure to ethanol, which, in turn, might lead to the identification of drug targets for therapeutic intervention of FAS in humans.
Ethanol absorption of control and ethanol-reared adult flies was measured during exposure to vaporized ethanol at 0, 5, 15 and 30 minutes (110:40, vaporized ethanol: humidified air) (Moore et al., 1998).
Larval feeding assay
Larvae were reared on 0%- or 5%-ethanol food as described above. At the second-to-third instar transition (staged by spiracle morphology) or the late-third-instar–wandering-larvae transition (staged by the presence of wandering larvae in the vial), larvae were taken out of the food and washed with distilled water. Approximately 1 ml of the 0%- and 5%-ethanol food was mixed with 0.1% Coomassie Brilliant Blue dye (Bio-Rad) and poured into Petri dishes. Groups of 25 control or ethanol-reared larvae were placed on these Petri dishes for either 15 or 30 minutes. After feeding, larvae were washed in distilled water, and frozen immediately in liquid nitrogen. Larvae were homogenized in 300 μl distilled water, then 100 μl of 50% ethanol was added to the tubes. The samples were centrifuged at 13 g for 10 minutes, and supernatants were placed in new tubes (the last step was repeated three to four times until supernatants were clear). For analysis, 200 μl of supernatant was placed in a 96-well plate and the OD at 633 nm read in a spectrophotometer. Six groups of 25 larvae per treatment were analyzed for each time point.
Lifespan
For lifespan analysis, flies were maintained in vials at a density of 25 flies per vial on standard cornmeal/molasses medium. Flies were transferred to new vials three times per week.
Supplementary Material
Acknowledgments
We thank members of the Heberlein lab for their helpful suggestions regarding experimental design and for critical readings of the manuscript; David Tran and Miri VanHoven for helpful discussions; and Eric Rulifson for generously donating the anti-Dilp2 antibody. This research was supported by grants from the National Institute on Alcohol Abuse and Alcoholism to K.D.M., R.L.F. and U.H.
Footnotes
COMPETING INTERESTS
The authors declare that they do not have any competing or financial interests.
AUTHOR CONTRIBUTIONS
K.D.M. and R.L.F. performed and analyzed experiments describing the developmental and adult behavioral phenotypes induced by ethanol exposure, respectively. U.H. helped with experimental design. All authors contributed to manuscript preparation.
SUPPLEMENTARY MATERIAL
Supplementary material for this article is available at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.006411/-/DC1
REFERENCES
- Barclay D. C., Hallbergson A. F., Montague J. R., Mudd L. M. (2005). Reversal of ethanol toxicity in embryonic neurons with growth factors and estrogen. Brain Res. Bull. 67, 459–465 [DOI] [PubMed] [Google Scholar]
- Becker H. C., Hale R. L., Boggan W. O., Randall C. L. (1993). Effects of prenatal ethanol exposure on later sensitivity to the low-dose stimulant actions of ethanol in mouse offspring: possible role of catecholamines. Alcohol Clin. Exp. Res. 17, 1325–1336 [DOI] [PubMed] [Google Scholar]
- Becker H. C., Diaz-Granados J. L., Randall C. L. (1996). Teratogenic actions of ethanol in the mouse: a minireview. Pharmacol. Biochem. Behav. 55, 501–513 [DOI] [PubMed] [Google Scholar]
- Berger K. H., Kong E. C., Dubnau J., Tully T., Moore M. S., Heberlein U. (2008). Ethanol sensitivity and tolerance in long-term memory mutants of Drosophila melanogaster. Alcohol Clin. Exp. Res. 32, 895–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blader P., Strahle U. (1998). Ethanol impairs migration of the prechordal plate in the zebrafish embryo. Dev. Biol. 201, 185–201 [DOI] [PubMed] [Google Scholar]
- Boehm S. L., II, Lundahl K. R., Caldwell J., Gilliam D. M. (1997). Ethanol teratogenesis in the C57BL/6J, DBA/2J, and A/J inbred mouse strains. Alcohol 14, 389–395 [DOI] [PubMed] [Google Scholar]
- Brogiolo W., Stocker H., Ikeya T., Rintelen F., Fernandez R., Hafen E. (2001). An evolutionarily conserved function of the Drosophila insulin receptor and insulin-like peptides in growth control. Curr. Biol. 11, 213–221 [DOI] [PubMed] [Google Scholar]
- Broughton S. J., Piper M. D., Ikeya T., Bass T. M., Jacobson J., Driege Y., Martinez P., Hafen E., Withers D. J., Leevers S. J., et al. (2005). Longer lifespan, altered metabolism, and stress resistance in Drosophila from ablation of cells making insulin-like ligands. Proc. Natl. Acad. Sci. USA 102, 3105–3110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bupp Becker S. R., Shibley I. A., Jr (1998). Teratogenicity of ethanol in different chicken strains. Alcohol Alcohol. 33, 457–464 [DOI] [PubMed] [Google Scholar]
- Chen C., Jack J., Garofalo R. S. (1996). The Drosophila insulin receptor is required for normal growth. Endocrinology 137, 846–856 [DOI] [PubMed] [Google Scholar]
- Christoffel K. K., Salafsky I. (1975). Fetal alcohol syndrome in dizygotic twins. J. Pediatr. 87, 963–967 [DOI] [PubMed] [Google Scholar]
- Clarren S. K., Smith D. W. (1978). The fetal alcohol syndrome. Lamp 35, 4–7 [PubMed] [Google Scholar]
- Clarren S. K., Astley S. J., Gunderson V. M., Spellman D. (1992). Cognitive and behavioral deficits in nonhuman primates associated with very early embryonic binge exposures to ethanol. J. Pediatr. 121, 789–796 [DOI] [PubMed] [Google Scholar]
- Corl A. B., Rodan A. R., Heberlein U. (2005). Insulin signaling in the nervous system regulates ethanol intoxication in Drosophila melanogaster. Nat. Neurosci. 8, 18–19 [DOI] [PubMed] [Google Scholar]
- Corl A. B., Berger K. H., Ophir-Shohat G., Gesch J., Simms J. A., Bartlett S., Heberlein U. (2009) Happyhour, a Ste20 family kinase, implicates EGFR signaling in ethanol-induced behaviors. Cell 137, 949–960 [DOI] [PubMed] [Google Scholar]
- Datta S. (1995). Control of proliferation activation in quiescent neuroblasts of the Drosophila central nervous system. Development 121, 1173–1182 [DOI] [PubMed] [Google Scholar]
- de la Monte S. M., Xu X. J., Wands J. R. (2005). Ethanol inhibits insulin expression and actions in the developing brain. Cell. Mol. Life Sci. 62, 1131–1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducci F., Goldman D. (2008). Genetic approaches to addiction: genes and alcohol. Addiction 103, 1414–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckardt M. J., File S. E., Gessa G. L., Grant K. A., Guerri C., Hoffman P. L., Kalant H., Koob G. F., Li T. K., Tabakoff B. (1998). Effects of moderate alcohol consumption on the central nervous system. Alcohol. Clin. Exp. Res. 22, 998–1040 [DOI] [PubMed] [Google Scholar]
- Garofalo R. S. (2002). Genetic analysis of insulin signaling in Drosophila. Trends Endocrinol. Metab. 13, 156–162 [DOI] [PubMed] [Google Scholar]
- Geminard C., Rulifson E. J., Leopold P. (2009) Remote control of insulin secretion by fat cells in Drosophila. Cell Metab. 10, 199–207 [DOI] [PubMed] [Google Scholar]
- Gronke S., Clarke D. F., Broughton S., Andrews T. D., Partridge L. (2010). Molecular evolution and functional characterization of Drosophila insulin-like peptides. PLoS Genet. 6, e1000857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerri C. (1998). Neuroanatomical and neurophysiological mechanisms involved in central nervous system dysfunctions induced by prenatal alcohol exposure. Alcohol. Clin. Exp. Res. 22, 304–312 [DOI] [PubMed] [Google Scholar]
- Hoyme H. E., May P. A., Kalberg W. O., Kodituwakku P., Gossage J. P., Trujillo P. M., Buckley D. G., Miller J. H., Aragon A. S., Khaole N., et al. (2005). A practical clinical approach to diagnosis of fetal alcohol spectrum disorders: clarification of the 1996 institute of medicine criteria. Pediatrics 115, 39–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeya T., Galic M., Belawat P., Nairz K., Hafen E. (2002). Nutrient-dependent expression of insulin-like peptides from neuroendocrine cells in the CNS contributes to growth regulation in Drosophila. Curr. Biol. 12, 1293–1300 [DOI] [PubMed] [Google Scholar]
- Ikonomidou C., Bittigau P., Ishimaru M. J., Wozniak D. F., Koch C., Genz K., Price M. T., Stefovska V., Horster F., Tenkova T., et al. (2000). Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science 287, 1056–1060 [DOI] [PubMed] [Google Scholar]
- Jones K. L., Smith D. W. (1973). Recognition of the fetal alcohol syndrome in early infancy. Lancet 302, 999–1001 [DOI] [PubMed] [Google Scholar]
- Kaminen-Ahola N., Ahola A., Maga M., Mallitt K. A., Fahey P., Cox T. C., Whitelaw E., Chong S. (2010). Maternal ethanol consumption alters the epigenotype and the phenotype of offspring in a mouse model. PLoS Genet. 6, e1000811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong E. C., Woo K., Li H., Lebestky T., Mayer N., Sniffen M. R., Heberlein U., Bainton R. J., Hirsh J., Wolf F. W. (2010). A pair of dopamine neurons target the D1-like dopamine receptor DopR in the central complex to promote ethanol-stimulated locomotion in Drosophila. PLoS One 5, e9954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotch L. E., Sulik K. K. (1992). Experimental fetal alcohol syndrome: proposed pathogenic basis for a variety of associated facial and brain anomalies. Am. J. Med. Genet. 44, 168–176 [DOI] [PubMed] [Google Scholar]
- Lee T., Luo L. (2001). Mosaic analysis with a repressible cell marker (MARCM) for Drosophila neural development. Trends Neurosci. 24, 251–254 [DOI] [PubMed] [Google Scholar]
- Liu Y., Balaraman Y., Wang G., Nephew K. P., Zhou F. C. (2009). Alcohol exposure alters DNA methylation profiles in mouse embryos at early neurulation. Epigenetics 4, 500–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J., Miller M. W. (1998). Growth factor-mediated neural proliferation: target of ethanol toxicity. Brain Res. Brain Res. Rev. 27, 157–167 [DOI] [PubMed] [Google Scholar]
- May P. A., Gossage J. P. (2001). Estimating the prevalence of fetal alcohol syndrome. A summary. Alcohol Res. Health. 25, 159–167 [PMC free article] [PubMed] [Google Scholar]
- McGee C. L., Riley E. P. (2006). Brain imaging and fetal alcohol spectrum disorders. Ann. Ist. Super. Sanita 42, 46–52 [PubMed] [Google Scholar]
- McGough N. N., Thomas J. D., Dominguez H. D., Riley E. P. (2009). Insulin-like growth factor-I mitigates motor coordination deficits associated with neonatal alcohol exposure in rats. Neurotoxicol. Teratol. 31, 40–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middaugh L. D., Ayers K. L. (1988). Effects of ethanol on mature offspring of mice given ethanol during pregnancy. Alcohol. Clin. Exp. Res. 12, 388–393 [DOI] [PubMed] [Google Scholar]
- Moore M. S., DeZazzo J., Luk A. Y., Tully T., Singh S. M., Heberlein U. (1998). Ethanol intoxication in Drosophila: genetic and pharmacological evidence for regulation by the cAMP signaling pathway. Cell 93, 997–1007 [DOI] [PubMed] [Google Scholar]
- Neufeld T. P., de la Cruz A. F., Johnston L. A., Edgar B. A. (1998). Coordination of growth and cell division in the Drosophila wing. Cell 93, 1183–1193 [DOI] [PubMed] [Google Scholar]
- Okamoto N., Yamanaka N., Yagi Y., Nishida Y., Kataoka H., O’Connor M. B., Mizoguchi A. (2009). A fat body-derived IGF-like peptide regulates postfeeding growth in Drosophila. Dev. Cell 17, 885–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxendine S. L., Cowden J., Hinton D. E., Padilla S. (2006). Vulnerable windows for developmental ethanol toxicity in the Japanese medaka fish (Oryzias latipes). Aquat. Toxicol. 80, 396–404 [DOI] [PubMed] [Google Scholar]
- Phillips T. J., Shen E. H. (1996). Neurochemical bases of locomotion and ethanol stimulant effects. Int. Rev. Neurobiol. 39, 243–282 [DOI] [PubMed] [Google Scholar]
- Pulsifer M. B. (1996). The neuropsychology of mental retardation. J. Int. Neuropsychol. Soc. 2, 159–176 [DOI] [PubMed] [Google Scholar]
- Ranganathan S., Davis D. G., Hood R. D. (1987a). Developmental toxicity of ethanol in Drosophila melanogaster. Teratology 36, 45–49 [DOI] [PubMed] [Google Scholar]
- Ranganathan S., Davis D. G., Leeper J. D., Hood R. D. (1987b). Effects of differential alcohol dehydrogenase activity on the developmental toxicity of ethanol in Drosophila melanogaster. Teratology 36, 329–334 [DOI] [PubMed] [Google Scholar]
- Reimers M. J., Flockton A. R., Tanguay R. L. (2004) Ethanol- and acetaldehyde-mediated developmental toxicity in zebrafish. Neurotoxicol. Teratol. 26, 769–781 [DOI] [PubMed] [Google Scholar]
- Rodan A. R., Kiger J. A., Jr, Heberlein U. (2002). Functional dissection of neuroanatomical loci regulating ethanol sensitivity in Drosophila. J. Neurosci. 22, 9490–9501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenfluh A., Threlkeld R. J., Bainton R. J., Tsai L. T.-Y., Lasek A. W., Heberlein U. (2006). Distinct behavioral responses to ethanol are regulated by alternate RhoGAP18B isoforms. Cell 127, 199–211 [DOI] [PubMed] [Google Scholar]
- Rovasio R. A., Battiato N. L. (1995). Role of early migratory neural crest cells in developmental anomalies induced by ethanol. Int. J. Dev. Biol. 39, 421–422 [PubMed] [Google Scholar]
- Rulifson E. J., Kim S. K., Nusse R. (2002). Ablation of insulin-producing neurons in flies: growth and diabetic phenotypes. Science 296, 1118–1120 [DOI] [PubMed] [Google Scholar]
- Sampson P. D., Streissguth A. P., Bookstein F. L., Little R. E., Clarren S. K., Dehaene P., Hanson J. W., Graham J. M., Jr (1997). Incidence of fetal alcohol syndrome and prevalence of alcohol-related neurodevelopmental disorder. Teratology 56, 317–326 [DOI] [PubMed] [Google Scholar]
- Samson H. H., Diaz J. (1981). Altered development of brain by neonatal ethanol exposure: zinc levels during and after exposure. Alcohol. Clin. Exp. Res. 5, 563–569 [DOI] [PubMed] [Google Scholar]
- Schneider M. L., Moore C. F., Kraemer G. W. (2001). Moderate alcohol during pregnancy: learning and behavior in adolescent rhesus monkeys. Alcohol. Clin. Exp. Res. 25, 1383–1392 [PubMed] [Google Scholar]
- Scolnick J. A., Cui K., Duggan C. D., Xuan S., Yuan X. B., Efstratiadis A., Ngai J. (2008). Role of IGF signaling in olfactory sensory map formation and axon guidance. Neuron 57, 847–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaidina M., Delanoue R., Gronke S., Partridge L., Leopold P. (2009). A Drosophila insulin-like peptide promotes growth during nonfeeding states. Dev. Cell 17, 874–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J., Wu L., Chen Z., Kohanski R. A., Pick L. (2003). Axons guided by insulin receptor in Drosophila visual system. Science 300, 502–505 [DOI] [PubMed] [Google Scholar]
- Stieper B. C., Kupershtok M., Driscoll M. V., Shingleton A. W. (2008). Imaginal discs regulate developmental timing in Drosophila melanogaster. Dev. Biol. 321, 18–26 [DOI] [PubMed] [Google Scholar]
- Streissguth A. P., Dehaene P. (1993). Fetal alcohol syndrome in twins of alcoholic mothers: concordance of diagnosis and IQ. Am. J. Med. Genet. 47, 857–861 [DOI] [PubMed] [Google Scholar]
- Su B., Debelak K. A., Tessmer L. L., Cartwright M. M., Smith S. M. (2001). Genetic influences on craniofacial outcome in an avian model of prenatal alcohol exposure. Alcohol. Clin. Exp. Res. 25, 60–69 [PubMed] [Google Scholar]
- Sulik K. K., Johnston M. C., Webb M. A. (1981). Fetal alcohol syndrome: embryogenesis in a mouse model. Science 214, 936–938 [DOI] [PubMed] [Google Scholar]
- Sulik K. K., Johnston M. C., Daft P. A., Russell W. E., Dehart D. B. (1986). Fetal alcohol syndrome and DiGeorge anomaly: critical ethanol exposure periods for craniofacial malformations as illustrated in an animal model. Am. J. Med. Genet. Suppl. 2, 97–112 [DOI] [PubMed] [Google Scholar]
- Tran T. D., Cronise K., Marino M. D., Jenkins W. J., Kelly S. J. (2000). Critical periods for the effects of alcohol exposure on brain weight, body weight, activity and investigation. Behav. Brain Res. 116, 99–110 [DOI] [PubMed] [Google Scholar]
- Wolf F. W., Rodan A. R., Tsai L. T., Heberlein U. (2002). High-resolution analysis of ethanol-induced locomotor stimulation in Drosophila. J. Neurosci. 22, 11035–11044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C. H., Belawat P., Hafen E., Jan L. Y., Jan Y. N. (2008). Drosophila egg-laying site selection as a system to study simple decision-making processes. Science 319, 1679–1683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yelin R., Schyr R. B., Kot H., Zins S., Frumkin A., Pillemer G., Fainsod A. (2005). Ethanol exposure affects gene expression in the embryonic organizer and reduces retinoic acid levels. Dev. Biol. 279, 193–204 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.