

Abstract
The discovery of superoxide dismutases (SODs), which convert superoxide radicals to molecular oxygen and hydrogen peroxide, has been termed the most important discovery of modern biology never to win a Nobel Prize. Here, we review the reasons this discovery has been underappreciated, as well as discuss the robust results supporting its premier biological importance and utility for current research. We highlight our understanding of SOD function gained through structural biology analyses, which reveal important hydrogen-bonding schemes and metal-binding motifs. These structural features create remarkable enzymes that promote catalysis at faster than diffusion-limited rates by using electrostatic guidance. These architectures additionally alter the redox potential of the active site metal center to a range suitable for the superoxide disproportionation reaction and protect against inhibition of catalysis by molecules such as phosphate. SOD structures may also control their enzymatic activity through product inhibition; manipulation of these product inhibition levels has the potential to generate therapeutic forms of SOD. Markedly, structural destabilization of the SOD architecture can lead to disease, as mutations in Cu,ZnSOD may result in familial amyotrophic lateral sclerosis, a relatively common, rapidly progressing and fatal neurodegenerative disorder. We describe our current understanding of how these Cu,ZnSOD mutations may lead to aggregation/fibril formation, as a detailed understanding of these mechanisms provides new avenues for the development of therapeutics against this so far untreatable neurodegenerative pathology.
Keywords: Superoxide dismutase, Reactive oxygen species, Amyotrophic lateral sclerosis, Lou Gehrig’s disease, Protein crystallography
1. Introduction
The ubiquitous superoxide dismutases (SODs) catalyze the disproportionation of superoxide to molecular oxygen and peroxide and thus are critical for protecting the cell against the toxic products of aerobic respiration [1,2]. As noted by Nick Lane in his book Oxygen: The Molecule that Made the World [3], the discovery and naming of superoxide dismutase activity [1] is “… in the opinion of many, the most important discovery of modern biology never to win a Nobel prize.” This discovery and its implications transformed research on oxygen free radicals and anti-oxidants and their biological implications: There are greater than 60,000 scientific papers published on the superoxide free radical and its functions in more than 100 human pathologies.
This review is based on the following tenets of the biology, chemistry and biochemistry of reactive oxygen species. (a) Superoxide is generated by many life processes, which include aerobic metabolism, oxidative phosphorylation and photosynthesis, in addition to the respiratory burst in the immune response of stimulated macrophages and neutrophils [4–6]. Early proposals that cells lack superoxide [7,8] have failed the test of time. (b) Superoxide and superoxide-dependent formation of hydroxyl radicals are important in oxygen toxicity [9–11]. If unchecked, reactive oxygen species (ROS) including the superoxide radical can result in inflammation [12–15] and inflict cell injury that includes DNA damage mediated by Fenton chemistry [16,17]. This ROS-mediated cellular damage is implicated in many human pathologies, including ischemic reperfusion injury, cardiovascular disease, cancer, aging and neurodegenerative disease [18–29]. (c) SODs are enzymes that disproportionate superoxide anion radicals at some of the fastest enzyme rates known. Furthermore, SODs function as master keys controlling cellular ROS levels, and SOD and SOD mimetics may have potential uses as therapeutic agents in oxidative stress-related diseases [28–37]. Also, upregulation of SOD expression can suppress the malignant phenotype of human melanoma cells [38]. Early proposals suggesting that superoxide is non-toxic and that superoxide removal is not the biological role of SOD were unsupported by subsequent research [7,8,39]. Likewise, the proposed phosphate inhibition of SOD was shown to be incorrect [40], agreeing with the structural analyses [41,42]; the initial study reporting inhibition had mistakenly adjusted the ionic strength with sodium fluoride [43], (d) Reduced structural integrity and stability of mutant human SOD is a causative factor in the disease familial amyotrophic lateral sclerosis (FALS, discussed in detail in our Cu,ZnSOD section), as initially proposed in 1993 [44], and supported by subsequent research [45–63]. This FALS hypothesis is preferred over alternative proposals that FALS mutations cause increased stability [64] or a gain of a toxic peroxidation activity [65]. In this review we describe the seminal discoveries on SODs, with particular emphasis on the contributions of structural biochemistry to our understanding of SOD function and dysfunction.
2. Distinct classes of SOD
Three classes of SOD have evolved with distinct protein folds and different catalytic metal ions: the Cu,ZnSODs, MnSOD/FeSODs and NiSODs. Cu,ZnSOD (also known as SOD1 and SOD3 in humans) occurs in eukaryotes and some prokaryotes, and point mutations in human Cu,ZnSOD are linked to the fatal neurodegenerative disease amyotrophic lateral sclerosis (ALS, also known as Lou Gehrig’s disease) [48,63,64,66,67]. FeSOD and MnSOD (also referred to as SOD2 in humans) appear to have evolved from a common ancestral gene, with the FeSOD gene observed in primitive eukaryotes, the plastids of plants and in bacteria [68]. Phylogenetic analysis of MnSOD indicates that it occurs in all the major domains of life, in the mitochondria of eukaryotes and the cytoplasm of many bacteria [68]. FeSOD and MnSOD have diverged significantly from each other, so that the two metals cannot functionally substitute for each other in Mn/FeSODs from most species. The more recently discovered NiSOD has been found only in bacteria. Common to all three classes of SOD is the disproportionation reaction, occurring through alternate oxidation and reduction of their catalytic metal ions, and rather remarkably, SOD catalysis takes place at rates close to diffusion limits. Although the protein architectures of the three SOD classes are distinct, all crucially provide electrostatic guidance for the superoxide substrate and alter the metal ion redox potential to a range suitable for superoxide disproportionation. These structures also provide for a suitable proton source and may control enzymatic activity through product inhibition. Extensive interest in the SODs lead to tens of thousands of related publications. Here, we have focused our discussions on the structure-based breakthroughs that lead to our current insights into the molecular mechanisms of the SODs, particularly the human enzymes. We also highlight key mechanistic issues, describing where they are generally agreed upon or in debate.
3. Cu,ZnSOD
In 1938, Mann and Keilin [69] isolated a bovine erythrocyte protein with a blue color contributed by bound copper. They termed the ~31.5 kDa protein hemocuprein. In the 1950s, human homologues of erythrocuprein were isolated from erythrocytes [70,71] and perhaps more pertinent to ALS, cerebrocuprein from the brain [72]. In 1969, 30 years after the initial isolation, McCord and Fridovich appropriately renamed the protein superoxide dismutase, after discovering that the enzyme catalyzed the dismutation of superoxide radicals (O2·−) to molecular oxygen (O2) and hydrogen peroxide (H2O2) [1]. In 1982, the first complete three-dimensional structure of Cu,ZnSOD, the product of the SOD1 gene from bovine erythrocytes (BtCu,ZnSOD, PDB code 2SOD) [73], allowed researchers to establish the structural basis for Cu,ZnSOD’s enzymatic mechanism [41] and rapid catalysis [74]. In 1992, the structure of human Cu,ZnSOD (HsCu,ZnSOD, PDB code 1SOS) [75] was solved revealing that the enzyme’s fold and domain organization were highly conserved in eukaryotes (Fig. 1) and later providing insights into ALS [44]. Since then, a variety of Cu,ZnSOD structures have been determined from diverse eukaryotic species. These include X-ray crystal structures of Cu,ZnSODs from budding yeast [76–78], trematode [79], frog [80], spinach [81], and flower P. atrosanguina [82] and NMR structures of the human [83] Cu,ZnSOD. Also, a recent combined sub 1 Å crystal and solution small-angle X-ray scattering structure of a Cu,ZnSOD has been determined from a thermophilic deep-sea hydrothermal vent worm Alvinella pompejana (ApCu, ZnSOD) [59]. This structure allowed further insights into the mechanism and stability of Cu,ZnSODs (Fig. 2).
Fig. 1.

Cu,ZnSOD sequence conservation, fold, and structural and functional regions. (A) Structure-based alignment of eukaryotic Cu,ZnSOD proteins with solved structures: HsSOD, H. sapiens; BtSOD, B. taurus; ApSOD, A. pompejana; XlSOD, X. laevis; SoSOD, S. oleracea; PaSOD, P. atrosanguina; SmSOD, S. mansoni; Sc, S. cerevisiae. Structural elements and secondary structure of HsSOD are noted above the alignment. Asterisks mark ALS sites in HsSOD. Letters below alignment: C, copper-binding ligand; D, disulfide cysteine; B, bridging histidine; Z, zinc-binding ligand; H, H2O2 liganding residue in ApSOD. (B) HsSOD structure 1PU0. Key structural elements in (A) are color coded with abbreviations (V-loop, Variable loop; GK1 and GK2, Greek key loops 1 and 2; S-S, Disulfide region). N and C denote termini. Metal-liganding residues are shown as sticks with the bridging histidine His61 in white.
Fig. 2.

(A) Electrostatic potential mapped onto the Alvinella pompejana surface from the ApSOD-H2O2 complex crystal structure. Electrostatic potential (blue positive, red negative) shows active site electrostatic attraction for superoxide anion. The H2O2 molecule (red spheres) occupies the position of the substrate/intermediate/product of the reaction. The spheres representing reduced copper (left) and zinc (right) are brought forward to show their relative positions. (B) Sliced version of the surface. From this view, the steric restrictions on the substrate/intermediate/product are readily recognized. Within the crystals, the copper ions (gold) were found in both the oxidized and reduced forms and show the short distance required for copper ion movement during SOD catalysis.
As shown from the first BtCu,ZnSOD and HsCu,ZnSOD structures [73,74], eukaryotic Cu,ZnSODs are highly conserved from the primary to quaternary structure. Cu,ZnSODs are composed of two identical subunits related by a two-fold symmetry axis. Each subunit consists of a β-barrel composed of eight antiparallel β-strands arranged in a Greek key motif [84]. Tightly packed hydrophobic residues form the core of the barrel, and loops β3/β4 and β6/β7 form the +3 β-strand Greek key connections (GK1 and GK2) [73,85]. Two conserved Leu residues within these Greek key loops fill the ends of the β-barrel and are termed the cork residues. Two other major external loops form the active site channel. The first, the β4/β5 loop, tethers the dimer interface with the active site zinc and contains a stabilizing intervening disulfide bond that aids stability [42,73,74,85,86]. The disulfide stabilizes both the subunit fold and the dimer interface. The second, β7/β8 or electrostatic loop (EL), guides and accelerates the substrate O2·− into the active site [42,74]. In fact, the evolution of Cu,ZnSOD dismutase and the Greek key β-barrel as well as functional roles of the various residue positions was defined by structural analyses allowing functional equivalences to be identified [85]. Furthermore, identification of electrostatic guidance in Cu,ZnSOD [42,74] opened the door to the examination of electrostatic guidance in other transient interactions involving electron transfer, as illustrated by computational analyses of plastocyanin with cytochrome c [87]. Overall, the stable Greek key scaffold supports elements for electrostatic guidance, dimer formation, and active site metallochemistry.
The Cu and Zn sites are positioned outside the β-barrel in the active site channel; the metal ions tie structural elements together, as well as provide catalytic roles. Furthermore, hydrophobic anchors tie the β-barrel to the active site Cu ion. The active site in each Cu,ZnSOD subunit contains one Cu ion ligated by three histidines when in the reduced state and one Zn ion ligated by one aspartic acid and three histidines, whose side chains all reside outside of the β-barrel. One of the histidine ligands of the Zn ion ligands also ligates the Cu ion when in the oxidized state and thus has been termed the bridging histidine. The exposed Cu ion is additionally coordinated by a water molecule and the substrate/product/reaction intermediate/water depending upon the particular reaction step. Importantly, steric selection allows superoxide, but not larger anions such as phosphate, into the active site. Of interest, while the bridging histidine has been shown to have different conformations depending on the redox state of the active site, more recent advances have revealed that the copper ion can be found occupying two positions in a single crystal [59,79,88,89].
To accomplish its in vivo functions, Cu,ZnSOD requires high stability and optimally fast catalysis. Both the β-barrel fold and a tight hydrophobic dimer interface provide structural stability to Cu, ZnSOD. The hydrophobic β-barrel core and main-chain β-sheet hydrogen bonds [85,90] were shown to be key in promoting not only structural integrity, but also folding, as a set of circular permutation mutants with swapped connections of the inter-β-strand loops and N- and C-termini yielded active enzymes [91]. Both the binding of the active site metal ions and formation of the conserved disulfide bond in each subunit also contribute to the framework stability and specificity of the protein fold and dimer assembly.
3.1. Cu,ZnSOD mechanism
Superoxide dismutases protect cells from reactive oxygen species by catalyzing the disproportionation of superoxide anion radicals into molecular oxygen and hydrogen peroxide. As the difference between molecular oxygen and the superoxide radical is a single electron, the enzyme must have extreme specificity and be finely tuned to perform its catalytic role. Furthermore, the substrate must be distinguished from other important diatomic regulatory species that are roughly the same size, such as nitric oxide [92,93]. In 1983, a structure-based mechanism was proposed [41], but due to the resolution limits at the time, the binding modes of the substrate/intermediate/product were estimated. Subsequent structures and analyses of the enzyme [42,74,94–98] in both the oxidized [99] and reduced [78] states, including inhibitor complexes [99,100], agreed with the general mechanism.
Recent high resolution structures of ApCu,ZnSOD, alone and in complex with H2O2 [59], the first SOD structure with product bound, aided a more unified general mechanism for Cu,ZnSOD that took into account steric restrictions at the Cu(I) binding site and how they relate to the observation of copper at two sites [79,88,89] (Fig. 2). These structures support an inner sphere mechanism [101], as geometrical restraints did not support an outer sphere proposal [99]. Following electrostatic recognition [42] and guidance [74] of the substrate into the active site by positively charged residues such as HsCu,ZnSOD Lys136 [94] and Arg143 [95], the first half reaction begins with the O2·− substrate binding to Cu(II). Cu(II) is then reduced to Cu(I), and O2·− is oxidized to molecular oxygen O2. The Cu ion to bridging histidine (HsCu,ZnSOD His63) bond is broken, leaving His63 Nε1 protonated. In the second half reaction, a proton from His63 Nε1 and an electron from Cu(I) are donated to O2·−, whereupon Cu(I) is oxidized to Cu(II), and O2·− is reduced to hydrogen peroxide or [59]. The copper bind to the bridging histidine is then restored [41].
From the new ApCu,ZnSOD structures, it was proposed that the positively charged Cu(I) ion would be attracted to the negatively charged O2·− substrate rather than shuttling an negatively charged electron to the likewise negatively charged substrate by an outer sphere mechanism. The binding modes of thiocyanate [100] and azide [99] inhibitors in other SOD structures are different than that observed in the H2O2 complex structure. However, the Cu ion appears to transition through coordination geometry similar to that observed in inhibitor bound structures; since the H2O2 oxygen proximal to the Cu ion is bound in nearly the same position as observed for the nearest inhibitor atoms to the copper. Another observation was that the hydrogen-bonding network formed by structurally conserved water molecules would align the reaction intermediate orbitals to promote coupled proton and electron transfer, then realign them for proper stabilization in solution [59].
Recently, the structure of the human extracellular Cu,ZnSOD (SOD3) protein was determined [102]. The crystal structure revealed a tetramer composed of dimers that are similar to the human SOD1 dimers. However, SOD3 does not appear to play a major role in ALS, perhaps due to stabilizing features. This glycoprotein contains Cu and Zn ions and catalyzes the same reaction as the SOD1 encoded enzyme [103,104]. Compared to SOD1 proteins, each SOD3 subunit has additional disordered N-terminal residues, longer loops at the β1/β2 and GK1 positions and an extra C-terminal α-helix. Within the tetramer, these longer loops interleave between dimers. SOD3 is stabilized by an additional intrasubunit disulfide bond not found in SOD1, one half of which is contributed by the SOD3 residue equivalent to Cys6 of human SOD1. Superposition of the SOD3 structure and the ApCu,ZnSOD-H2O2 complex [59] indicates a similar active site and that the mechanism of catalysis for SOD3 is likely conserved with that of the SOD1 enzymes.
3.2. Bacterial pathogenesis
Cu,ZnSODs encoded by the sodC gene are found in many prokaryotes, including Gram-negative pathogens, in which the enzyme has been located in the periplasm or attached to the outer membrane [105–111]. Mutation of the sodC gene was shown to attenuate virulence [112–114]. One hypothesis for Cu,ZnSOD’s role in pathogenesis is to protect bacteria from the actions of host phagocytes that expose cells to free radicals through a respiratory burst, where NADPH oxidase and nitric oxide synthase may play roles [112,114–116]. Thus, the recognition of superoxide as a defensive cytotoxin made the determination of bacterial SOD structures important for understanding pathogenesis. The first prokaryotic Cu,ZnSOD structures solved [117,118] revealed conservation of the subunit fold and electrostatic recognition mechanism [42] for attracting the substrate, but a unique dimer interface. Comparison of Cu,ZnSOD structures [119–122] from a variety of species determined that whereas the subunit fold is conserved between prokaryotic and eukaryotic Cu,ZnSODs, their quaternary structures differ, suggesting that these differences may be exploited for therapeutic interventions of infection.
4. Amyotrophic lateral sclerosis
ALS is one of the most common human neurological disorders, affecting 1 in 200,000 people [123]. First described by Jean-Martin Charcot in 1869 [124], ALS usually strikes in mid-life and selectively kills motor neurons, leading to progressive paralysis and death, typically in 1–5 years. Cu,ZnSOD constitutes 1% total cytosolic protein in neurons [125] and mutations in Cu,ZnSOD cause about 20% of inherited or familial ALS (FALS), which is inherited in an autosomal dominant manner [44,64]. Remarkably, over 100 single-residue mutations at >70 Cu,ZnSOD sites occur in the 153 amino acid protein [126–128] (Fig. 3). The underlying molecular mechanism by which >100 distinct mutations can lead to the same FALS pathology remains controversial [123,129–132]. Initial research emphasis was directed toward hypotheses that included gain of functions [133,134], involving reactions such as peroxidation and increased tyrosine nitration [135–137]; glutamate-based excito-toxic death [138]; neurofilament disorganization leading to axonal strangulation [139]; reduced or altered Cu,ZnSOD activity arising from imperfectly folded proteins and leading to increased oxidative damage [44]; and toxicity of intracellular Cu,ZnSOD aggregates resulting from protein misfolding or impaired protein degradation [44,46]. Some hypotheses do not account for all the mutations, for example, neither peroxidation nor nitration activities are shared by all FALS mutants, especially those missing the Cu site (e.g., H46R, H48Q). In 1993, a unified framework destabilization hypothesis was proposed for FALS [44], and 10 years later FALS Cu,ZnSOD mutants were shown to have substantially increased propensity to form fibrous aggregates [48].
Fig. 3.

Distribution of ALS mutations within the HsSOD Structure. (A) SOD residues known to be mutated in ALS patients, shown with colored side chains and spheres for glycine Cα atoms. One subunit is yellow, the other orange. The majority of the β-barrel core and edge residues, and the dimer interface residues are ALS SOD mutation sites. These residues are important for maintaining β-barrel stability and quaternary structure. Many outer surface residues that are mutated in ALS patients contain side chain oxygen atoms potentially important for maintaining surface electrostatic potential or for stabilizing hydrogen bonding and salt-bridge interactions. (B) SOD residues not implicated in ALS, colored and shown from the same view as in (A). The active site (lower left) and the outer surface contain many residues not linked to ALS.
Many neurodegenerative diseases (Alzheimer’s, Huntington’s, and Parkinson’s diseases and ALS) involve protein aggregates in the brain [126,140,141]. These aggregates also occur in both mouse and cell culture models of ALS and are immunoreactive to Cu,ZnSOD antibodies [46,142]. In cell culture models of ALS, detergents or reducing agents do not dissociate these cytoplasmic aggregates. Furthermore, these aggregates are detected biochemically in transgenic mice months before symptoms [143], as Lewy body-like hyaline inclusions [144]. The framework destabilization mechanism for FALS is consistent with proposals that FALS mutant Cu, ZnSOD-mediated toxicity may occur through co-precipitation of destabilized Cu,ZnSOD with key cellular components [46,123,143]. Cellular components including the copper chaperone for SOD (CCS) [145], nitric oxide synthase (NOS) and phosphorylated neurofilaments [146], co-aggregate with mutant Cu,ZnSOD, and the resulting aggregates react with antibodies to ubiquitin and nitrotyrosine residues [144,146]. The post-mitotic motor neurons affected by ALS are more than a meter long in humans, and their neurofilament-based transport system involves extended protein lifetimes and vulnerability to stress. A role for mitochondrial damage in neurodegenerative diseases [147] suggests defective mitochondrial transport and/or mitochondrial damage may act in FALS [148,149]. Mass spectrometry based-proteomic data indicate that Cu,ZnSOD is present in purified human mitochondria. Thus, Cu,ZnSOD is positioned where aggregation can logically be proposed to result in mitochondrial defects leading to apoptosis through chronic caspase activation [150], consistent with motor neuron pathophysiology in FALS [142,151–154].
Exciting new results link specific non-wild-type interactions of FALS mutant Cu,ZnSODs with proteins from two stress-response pathways [155]: Derlin-1 in the endoplasmic-reticulum-associated degradation (ERAD) pathway for misfolded proteins [156] and Rac1 in the NADPH oxidase complex for oxidative burst in immune cells [157]. Derlin-1 is a transmembrane protein complex component that recognizes and retro-translocates misfolded proteins from the endoplasmic reticulum to the cytosol for ubiquitination and proteosomal degradation [158]. FALS mutant (A4V, G85R, and G93A), but not wild-type human Cu,ZnSODs, interact with Derlin-1, as shown by in vitro and in vivo pull-down binding assays [156]. This protein –protein interaction produced endoplasmic reticulum stress, as assayed by apoptosis signal-regulating kinase 1 (ASK1) activation in cell culture. Furthermore, FALS mutant Cu,ZnSODs co-immunoprecipitated a C-terminal cytoplasmic tail construct from Derlin-1 (but not from homologues Derlin-2 and Derlin-3). The construct containing the 12-residue Derlin-1 sequence 105-FLYRWLPSRRGG-116 was competent to make or block this interaction, to block ASK1 activation and to attenuate the mutant Cu,ZnSOD-induced death of motor neurons in spinal cord cultures from mouse embryos [156]. These findings link aberrant binding of Derlin-1 by FALS mutant Cu,ZnSOD proteins to endoplasmic reticulum stress, ERAD defects, and motor neuron death.
Rac1, a member of the Rho GTPase family, is a key activating component of the NADPH oxidase complex that generates extracellular O2·− in immune cells [159], including the microglia associated with neurons. Wild-type Cu,ZnSOD regulates NADPH oxidase activation via redox-regulated binding to Rac1, whereas FALS mutant Cu,ZnSODs remain bound to Rac1, leading to deregulated O2·− production [157]. The binding of wild-type Cu, ZnSOD to Rac1 was favored by bound GDP analogs and disfavored by GTP analogs or the absence of bound nucleotide. Rac1 reduction with DTT switched this nucleotide preference, presumably by triggering the well-known conformational switch for Rho GTPases. H2O2 (~50 pM) inhibited the binding of Rac1 to Cu,ZnSOD, and this inhibition was reversed by DTT treatment of Rac1. Pull-down assays with Rac1 constructs showed that Cu,ZnSOD most efficiently bound the region of Rac1 (residues 35–70) that includes the switch I, switch II; and nucleotide-binding motifs involved in Rac1 conformational changes associated with nucleotide binding and hydrolysis, and Rac1 interactions with protein effectors. Cu,ZnSOD, but not de-metallated Cu,ZnSOD, bound Rac1 and inhibited its GTPase activity. FALS mutant Cu,ZnSODs (L8Q and G10V) showed enhanced Rac1 binding that was redox insensitive. Treatment of FALS mutant Cu,ZnSOD (G93A) mice with the NADPH oxidase inhibitor apocyanin slowed disease progression and increased lifespan [157]. These findings link aberrant binding of Rac1 by mutant Cu,ZnSOD to excessive superoxide production and resultant inflammation.
FALS mutants have also been implicated in aberrant interactions with the chromogranin proteins of neurosecretory vesicles [160], the inducible cytosolic chaperone Hsc70 [161], and the heavy chain of the dynein complex responsible for retrograde transport in neurons [162,163]. Aberrant and deregulated binding of FALS mutant Cu, ZnSODs to protein partners are predictable consequences from the framework destabilization hypothesis [48,59] and likely contribute to the many different oxidative and inflammatory symptoms characteristic of ALS pathology. The formation of stable rather than transient SOD binding to protein partners can be expected to alter biological outcomes, as shown directly for DNA damage responses [164]. Fortunately, the basis for the assemblies of β barrel domains has been analyzed in some detail [165]. Furthermore, new facilities and methods for small angle X-ray scattering (SAXS) provide promising techniques to examine these framework and assembly aberrations in solution [166,167]. SAXS may prove especially valuable for examining SOD mutant dynamics as emerging NMR data on the dynamics of ALS SOD mutants [168] supports the destabilization hypothesis proposed from analyses of crystal structures [44,48,59]. Moreover, structure-based design of small-molecule ligands that can induce local protein conformational changes may provide a strategy to stabilize the native SOD fold and assembly [169].
5. Role of Cu,ZnSOD stability in ALS
Superoxide dismutases are highly stable. Purification can involve heat denaturation of cellular extracts [59] and organic solvent extraction [1]. BtCu,ZnSOD is one of the most stable enzymes from a mesophile: this enzyme has a melting temperature of 89–104 °C depending on conditions [170,171] and remains active in 8 M urea [172], 4% sodium dodecyl sulfate [173] or at elevated temperatures [86].
Part of the stability of Cu,ZnSOD is imparted by the Greek key β-barrel [84], a fold utilized by stable extracellular proteins, such as immunoglobins [174]. The β-barrel’s integrity is maintained by main-chain hydrogen bonds, a tightly packed hydrophobic core and the Leu cork residues [73]. Many of the loops between strands are composed of short turns. The β2/β3 loop, or variable loop, contains insertions depending on species. The more stable BtCu,ZnSOD and ApCu,ZnSOD contain the shortest variable loops, as well as a greater number of conspicuous proline caps that may restrict movement at the apex of some of their loops [59,175].
The two longest Cu,ZnSOD loops (β4/β5 and β7/β8) that form the active site channel exhibit other stabilizing features. The electrostatic β7/β8 loop contains a short helix and is additionally stabilized both by hydrogen-bonding interactions and by the tightly packed and hydrogen-bonded ordered water molecules filling the active site channel. The extended loop is divided into several regions. The first makes a connection to the dimer interface where it is further stabilized by a hydrogen bond to GK2 and a disulfide bond to the β-barrel (Fig. 1). The disulfide bond will remain intact under many reducing conditions, including large doses of synchrotron X-ray radiation [59]. The metal bridging His at the beginning of the Zn binding region further couples this loop to the remainder of the protein. Therefore, the dimer interface, disulfide bond and metal binding anchor the protein’s largest loop.
HsCu,ZnSOD has two cysteines that do not form a disulfide bond. The first, Cys6 is relatively conserved in many species; among those with solved structures, spinach and yeast Cu,ZnSODs have an Ala residue at this position. The second, Cys111, is rare and is usually occupied by Ser, as is the case for all other eukaryotic Cu,ZnSODs with solved structures to date. These additional cysteines are implicated in the irreversibility of folding when tested in vitro. In fact, a more stable Cu,ZnSOD was engineered by mutating these residues to their counterparts found in other organisms [74,86,176]. This C6A/C111S mutant has been very useful as an in vitro tool for characterizing ALS mutations, as the fold and enzymatic function of the protein is conserved, but complex interpretation of ALS mutations are decreased by limiting irreversible aggregation and free Cys oxidation effects [48,54,56,86,94,177–179].
What do these structural features mean in terms of ALS? Examination of the ALS mutations mapped onto the primary structure of HsCu,ZnSOD shows that the mutations are dispersed throughout the amino acid sequence (Fig. 1). However, when the mutations are mapped upon the tertiary or quaternary structure of the protein, insights are revealed into how they may function in a unified sense to give rise to ALS, as they are dispersed about the structure (Fig. 3). Rather than each mutation site or clusters of mutations in different functional sections of the protein, giving rise to a separate mechanism, the structural destabilization hypothesis would encompass the widespread distribution of ALS mutation sites. Thus far, it has been shown that amyloid-like fibers may be formed by Cu,ZnSOD under different conditions, and that ALS mutations accelerate this process [48]. Staining the fibers with dyes such as Congo red or thiofiavin T indicates that β-sheet interactions are present. Reduction of the disulfide bond and loss of active site metal ions also accelerate fiber formation [180,181].
Our early proposal linked ALS to structural defects in HsCu,ZnSOD and hypothesized that FALS mutations reduced the structural integrity of the dimeric enzyme [44]. Moreover, this analysis pointed out possible structural weaknesses occurring in the human enzyme that let the majority of the β-sheet or β-barrel remain intact [59]. Computational analysis also supported destabilization of FALS mutants and its correlation with disease severity [63]. The evidence mounts that SOD fibrils containing partially folded Cu,ZnSOD subunits are a likely common denominator in causing ALS pathology. Aberrant interactions of ALS mutant Cu,ZnSODs with other proteins important in cellular machinery, likely also stem from framework destabilization stemming again from misfolding rather than different chemical reactions of Cu,ZnSOD. Side reactions and loss of superoxide dismutase activity may occur after misfolding, nucleation and growth of aggregates. However, when we take into account the positions of ALS sites, their roles in maintaining structure, and the observations made by many laboratories, the likely initiator of defective superoxide dismutase pathologies in FALS is a common loss of structural integrity resulting from destabilization of the Cu,ZnSOD framework. Listed below are examples of structural and biochemical results that support the Cu,ZnSOD framework destabilzation mechanism for FALS mutants, categorized by structural and functional elements of the protein.
5.1. The β-barrel
Packing distortions imparted by ALS mutations in the cork residues and other interior side chains key to hydrophobic packing are predicted to locally perturb the fold of the β-barrel. Perturbation of the β-barrel, in turn, may destabilize important loop and dimer interface interactions. When the ALS mutations are mapped onto the HsCu,ZnSOD structure (Fig. 3), it is easily visualized that the majority of hydrophobic packing residues are ALS mutations, including the cork residues Leu38 and Leu106 that are held in place by the two Greek key loops and are represented by the L38R, L38V, and L106V ALS mutations.
His43 is a β-barrel residue that makes van der Waals contacts with core residue Leu38. The H43R Cu,ZnSOD structure showed that the Arg43 substitution results in loss of a hydrogen bond and a steric clash with Thr39, which influenced packing with the surrounding residues including significant loss of contact with the Leu38 cork residue [48]. Even when metallated and in the more stable C6A/C111S background, the H43R mutant loses roughly 40% of activity. Moreover, the mutant protein was shown by electron microscopy (EM) and atomic force microscopy (AFM) to rapidly form fibril aggregates with dimensions suggestive of loss of dimerization and partial loss of the β-barrel fold. The mutant protein did, however, bind dyes that associate with amyloid-like fibers.
Ile113 technically resides within GK2 but contributes to the cork region of the β-barrel along with Leu106. Crystallographic structural analysis of the I113T mutant showed that, although the β-barrel fold was not significantly changed, packing interactions were decreased with Ala4, a key component of the dimer interface. The major structural consequence of the I113T mutation was the change in subunit orientation about the dimer interface. This change was more pronounced in small-angle X-ray scattering experiments in solution [53].
5.2. β-barrel loops
The ALS mutation sites in the β-barrel loops are positioned to either destabilize the hydrophobic packing of the barrel or weaken electrostatic interactions that tether and stabilize the loops and and/or the β-strands with which they associate. As stated above, the loops appear to be key for maintaining the proper fold of the β-barrel [59]. For instance, the G37R and G41D mutations, are located at opposite β-strand/loop junctions of the 1st Greek key connection (GK1). Gly41 has torsion angles disfavored for other residues. Both Gly residues also contribute hydrogen bonds to the β5/β6 turn that opens toward the opposite end of the dimer interface, making the β-strands more likely than others to form new β-strand interactions. The G37R mutant structure showed changes in hydrogen-bonding networks important for stability and different copper binding in the two subunits of the dimer [182].
In mice, the G85R mutation results in motor neuron degeneration and paralytic symptoms, associated with insoluble aggregates [183,184]. Four G85R Cu,ZnSOD structures [47] showed that this mutation results in the loss of metal binding or the introduction of zinc into the copper site. Superposition of the structures also revealed a perturbed dimer interface, and movements of loops away from the mutation site. One G85R structure, which had a Cys111 modification to reduce unwanted disulfide bond formation, resulted in a protein with disordered loops. Moreover, some of the subunits bound a water molecule within the β-barrel, weakening β-strand interactions and resulting in perturbations to the fold.
5.3. The dimer interface
The dimer interface is directly connected to many other structural features of Cu,ZnSOD: It forms the edge of the β-barrel, and through the β4/β5 loop, is immediately linked to the disulfide bond and metal binding ligands. Some of the most severe ALS mutations map not to the active site but to the dimer interface (Fig. 4), which supports the framework destabilization hypothesis, as opposed to altered catalytic activities, as the predominant cause of FALS.
Fig. 4.

Disease onset and severity mapped onto ALS mutation sites in HsSOD structure. (A) FALS mutation sites (spheres) are found throughout the SOD sequence at sites expected to reduce the integrity of the β-barrel, especially the packing within the β-barrel core, “cork” regions, the dimer interface (center) and other framework features. (B) 90° rotation from (A) around horizontal axis. (C) Color codes for the spheres in (A) and (B) show the severity and age of onset linked to each mutation. Mutations linked to the most rapid and severe mutations are located within the dimer interface or in the β-barrel at positions likely to destabilize the dimer interface. The active site Cu and Zn ions are also shown as spheres within cages. Data from Wang et al. [63] were used to generate the figure.
The A4V mutant HsCu,ZnSOD structure revealed that changes in the small side chain within the β-barrel propagate to the nearby dimer interface [177]. Superposition onto the wild-type structure shows no significant backbone changes, but instead subtle changes from the A4V mutation that propagate from Val4 through Ile113 and Ile151 to the dimer interface, and from Val4 to Leu106 to Phe20. Furthermore, buried surface area differences calculated using the program Tiny Probe [185] indicate that these differences are due to an increase in the volume of residue 4 in the A4V structure and a decrease in buried surface area (increase in solvent accessibility) of ~11 Å2 for cork residue Leu106 and Ile113. The increased solvent accessibility of Leu106 together with subtle changes in dimer interface residues caused by the A4V mutation are predicted to compromise the architectural stability and assembly specificity of Cu,ZnSOD. Like the H43R mutant, the A4V mutant was shown to rapidly form aggregates by EM and AFM with dimensions and dye affinity suggestive of a mostly intact β-barrel within the fibrils [48]. Destabilization of the mutant protein was also shown by hydrogen/deuterium exchange and mass spectrometry [58]. The A4V mutation is also associated with the most rapid disease progression, suggesting that structural integrity of the Cu,ZnSOD dimer interface is of high importance [186].
5.4. The disulfide bond and metal-binding ligands
Reduction of the disulfide bond promotes Cu,ZnSOD aggregation [181]. The conserved disulfide bond anchors key elements forming the active site channel and is also directly tethered to the dimer interface and metal ligands, as discussed previously. Both disulfide cysteines are ALS mutation sites. A C57A/C146A double mutant (in the context of the stable C6A/C111S construct) was engineered to show the effects of loss of the disulfide bond [52]. The crystal structure revealed movement of the β4/β5 loop, which tethers the dimer interface to the disulfide bond and metal-binding ligands. In solution, the apo form of this mutant protein was found to consist of individual subunits that reverted back to dimers in the presence of Cu and Zn ions. Molecular dynamics simulations support the importance of the metal ions and the disulfide bond as stabilizing features [49].
ALS patients with mutations in metal-binding residues have been identified (Fig. 1). Through the Cu ion, both His46 and His48 tether β4 to the junction of β7 and the electrostatic loop via His120. In the oxidized state, the Cu ion is also linked via bridging His63 to the Zn ion and its associated loop regions outside of the β-barrel. Therefore, these Cu ion ligands have structural, as well as, catalytic roles. The H46R Cu,ZnSOD structure, as well as that of the S134N mutant, which is mutated near Zn binding ligand His71, showed that the proteins exist in amyloid-like filament structures within crystals [50]. Another H46R structure had disordered loops encompassing the zinc-binding region and electrostatic loops [45].
Two zinc ligands, His80 and Asp83, were mutated to serine to preclude zinc binding [56]. These mutations perturbed the dimer interface, imparting a 9° rotational twist between the two subunits. The active site β4/β5 and β7/β8 loops, which house the disulfide bond and residues important for electrostatic attraction of superoxide, were partially disordered. The disulfide bond was found in two conformational states. Although the mutant protein was more subject to aggregation in the presence of a reducing agent, it formed heterodimers with wild-type Cu,ZnSOD, suggesting that the β-barrel folds remained substantially intact in solution.
5.5. The outer surface
Many side chains at ALS mutation sites are surface-exposed away from the active site and the dimer interface and would therefore apparently play seemingly little or no role in altering active site chemistry (Figs. 3 and 4). However, as seen in Fig. 3, many of these side chains are polar or charged (mainly negative). Mutations at these sites may change the electrostatic potential of the surface (Fig. 5) to promote misfolding and/or nucleate aggregation. The E21K ALS mutation, which lies on the outer β-barrel surface surrounded (Fig. 5) by positively charged Lys3, Lys23, and Lys30, likely serves to neutralize these positive charges. Nearby ALS mutation site Glu100, adjacent to Lys30, participates in the same charge network as Glu21; modeling of the E100G structure suggests loop changes that propagate toward the dimer interface. Mapping the electrostatic potential onto the molecular surface (Fig. 5) reveals that these changes in surface charge may promote aberrant interactions to nucleate aggregation.
Fig. 5.
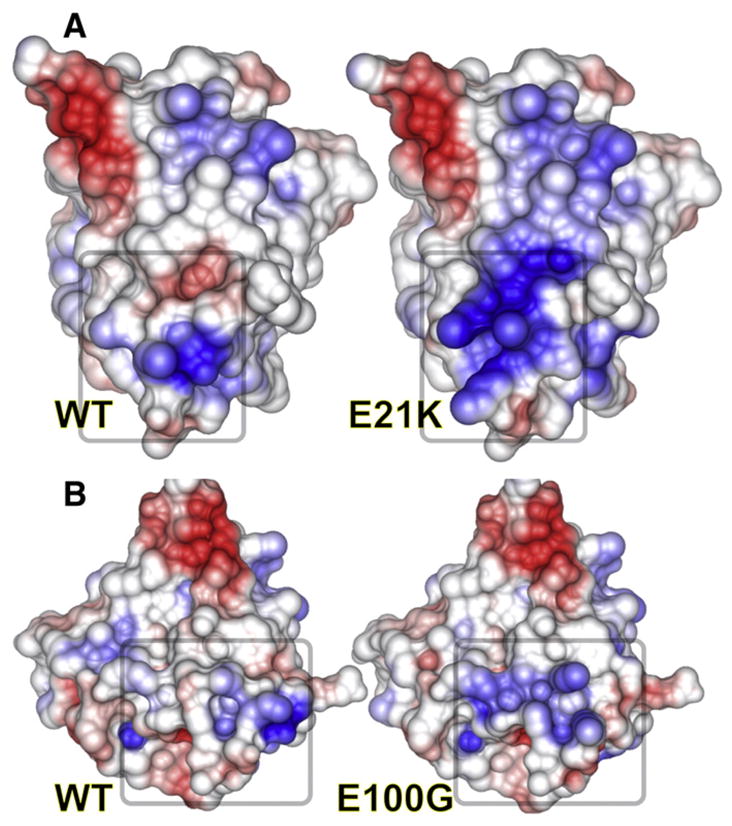
Effects of single-point mutations in HsSOD on electrostatic potential. Electrostatic potential was mapped onto the surfaces of wild-type (left) vs. mutant (right) HsSOD models for the E21K and E100G HsSOD mutants. Loss of charge complementarity can dramatically change the electrostatic potential and reduce stability, both of which may result in aberrant SOD interactions.
6. Manganese and iron superoxide dismutases
In bacteria the cytoplasmic Mn and FeSODs have defensive functions in protecting the cell from ROS, and there are indications that in pathogenic bacteria they may have additional functions in infecting and colonizing their host (reviewed in ref. [187]). The eukaryotic mitochondrial MnSOD is critically required because as much as 90% of cellular ROS can be generated in this organelle. Also, mitochondrial DNA is particularly susceptible to oxidative damage, due to the extreme levels of oxygen metabolism, its relatively in-efficient DNA repair and because of a lack of histones [188]. The important cellular function for mitochondrial MnSOD was highlighted by a study on mice that were nullizygous for the MnSOD gene (sod2). A lack of MnSOD resulted in dilated cardiomyopathy, hepatic lipid accumulation, mitochondrial defects and early neonatal death [189,190]. Markedly, the sod2 nullizygous mice were treated with a SOD mimetic expected to temper these pathologies and enhance their survival [191]. However, surviving mice acquired a spongiform neurodegenerative disorder, with a similar phenotype to certain mitochondrial abnormality disorders and lethality at 3 weeks of age [191]. This neuropathology highlighted roles for MnSOD in controlling ROS levels in the brain, due to insufficient SOD activity across the blood–brain barrier [191]. A later study determined that superoxide-catalase mimetics that are porous to the blood–brain barrier abrogated the spongiform encephalopathy [192]. This result suggests that MnSOD and MnSOD mimetics may have therapeutic potential in combating neurodegenerative diseases associated with oxidative stress, including the spongiform encephalopathies, Alzheimer’s and Parkinson’s diseases. Moreover, combining MnSOD with catalase or creating superoxide-catalase mimetics, in order to remove the hydrogen peroxide product, may have additional therapeutic benefit. For example the median and maximum life spans of transgenic mice were significantly increased when overexpressing catalase in the mitochondria and peroxisome [193]. This was noted to delay cardiac pathology, cataract development and mitochondrial deletions [193].
6.1. MnSOD and FeSOD structural determination
Structural studies helping define the molecular mechanisms behind catalysis and product inhibition of MnSOD and FeSOD first achieved success in the late 1980s and early 1990s [194–199]. In the Mn/FeSOD structures the polypetide chain is divided into N-terminal helices and a C-terminal α/β domain. The N-terminal domain mediates multimerization, with most of the bacterial structures forming dimers. Human MnSOD (hMnSOD) differs by assembling into a tetramer, through forming a dimer of dimers that creates two symmetrical four-helix bundles [198,200], (Fig. 6A). Notably, the I58T polymorphism in hMnSOD modifies Ile58, which is the largest contributor of buried surface area in the tetramerization interface. Structural biochemistry studies revealed that the I58T polymorphism causes packing defects and it significantly reduces the thermal stability of hMnSOD, resulting in rapid inactivation at temperatures elevated above normal that are associated with fever or inflammation [201]. However, epidemiology studies have focused not on the I58T, but on the V16A and A9V polymorphisms of the MnSOD precusor that disrupts targeting of MnSOD to mitochondrial. Results on V16A and A9V have proved mixed, with some groups indicating links to cancer, diabetic nephropathy or tardive dyskinesia in patients with schizophrenia [202–211].
Fig. 6.

Human MnSOD crystal structure. (A) The wild-type homotetrameric MnSOD structure (1LUV.pdb), containing two symmetrical four-helix bundles and four C-terminal α/β domains, is depicted with the four separate polypeptide chains colored in cyan, blue, green and yellow, and active site manganese ions depicted as magenta spheres. (B) MnSOD active site and hydrogen-bonding scheme. The side chains of active site residues His26, His74, His163, and Asp159 bind the Mn ion, in conjunction with a solvent molecule. A hydrogen-bonding network, depicted in orange spheres, is observed in the MnSOD active site, extending from the metal-bound solvent. The solvent forms a hydrogen bond to Gln143, and the network is continued with a hydrogen bond to Tyr34. A conserved water molecule then mediates the hydrogen bond between Tyr34 and His30, and His30 also forms a hydrogen bond with Tyr166 from an adjacent subunit.
The active site of Mn/FeSOD is located between the N and C-terminal domains, and it differs from that of Cu,ZnSOD by containing a single metal ion. The metal ion is coordinated in a strained trigonal bipyramidal geometry by four amino acid side chains, His26, His74, Asp159 and His163 (human sequence), and by one solvent molecule, as observed in the human structure [198](Fig. 6B). The superoxide reaches the active site through a funnel that uses electrostatics for guidance and has a narrow entrance to the active site, limiting access to only small ions. Also at this funnel vertex are residues His30 and Tyr34, which function as gatekeeper residues, specifically blocking access to the metal ion; with computational analysis suggesting that Tyr34 moves to allow passage of the superoxide to the metal center [212].
6.2. Proposed MnSOD and FeSOD molecular mechanisms
The Mn/FeSOD catalytic reaction requires cycling between the Mn/Fe2+ and Mn/Fe3+ states; the midpoint potential for these two states, as measured in the human MnSOD, falls within a suitable range for the disproportionation of superoxide at 393 ± 29 mV [213]. A few different mechanisms have been proposed for the catalytic reaction. One is known as the 5-6-5-reaction mechanism, where the metal is five-coordinated in its resting state and six-coordinated when the anion is bound [196,199,214]. In this model, superoxide is thought to bind in the same manner as anionic inhibitors, such as fluoride, hydroxide and azide. Crystal structures of E. coli FeSOD and T. thermophilus MnSOD have revealed that the azide inhibitor forms a sixth ligand, creating a distorted octahedral manganese ion [199]. X-ray absorption studies further supported this 5-6-5 mechanism, revealing that FeSOD at neutral pH is five-coordinated, at pH 10.5 it is six-coordinated and at pH 9.4 is in a 1:1 mixture, with the sixth ligand presumed to be hydroxide [214]. A second proposed mechanism, associative displacement [215] was based on temperature-dependent absorption or thermochromism of anion complexes of MnSOD. In this mechanism the inactive inhibitor-bound enzyme is six-coordinated, while the active form remains five-coordinated, with substrate binding causing the displacement of one of the manganese ligands. A third proposed mechanism suggested that the catalytic reaction occurs through an outer sphere mechanism and instead utilizes an alternate anion-binding site in the active site, possibly at the base of the funnel [216–218]. This proposal is based upon various anions inhibiting the activity of FeSOD, but without directly coordinating the metal center. Clearly, structures of Mn/FeSOD in complex with superoxide are very much warranted to help delineate which of these proposed mechanisms holds true for this class.
Also important to the catalytic mechanism is that mitochondrial MnSOD has been characterized by increased product inhibition when compared with bacterial forms of the enzyme. This product inhibition also distinguishes MnSOD from FeSOD, as deactivation of the active site of FeSOD occurs instead through Fenton chemistry [4]. This MnSOD product inhibition is most likely important for preventing the overproduction of toxic hydrogen peroxide in eukaryotic cells. Studies on hMnSOD catalysis revealed an initial burst, which is then inhibited at the micromolar level by the peroxide reaction product, consistent with the formation of a reversible, dead-end complex. The structure of this peroxide inhibited form of hMnSOD has not been directly observed, but computational and spectroscopic methods also support a direct complex with the metal ion [219,220].
6.3. Mn/FeSOD active site hydrogen bond network
The active site of Mn/FeSOD contains a hydrogen bond network that extends from the metal bound solvent molecule to solvent-exposed residues at the interface between subunits [198,221] (Fig. 6B). This hydrogen bond network has been suggested to support proton transfer in catalysis; this would provide a means for both delivering protons to the active site and regulating the pKa of the active site water [221]. In human MnSOD the hydrogen bond network is created through the metal bound solvent forming a hydrogen bond with Gln143 Nε. The oxygen of the carboxamide Gln143 side chain continues the network by forming a hydrogen bond to the Tyr34 hydroxyl group. A water molecule situated between Tyr34 and the His30 side chains mediates the hydrogen bonding between these two residues. This His30 side chain, which is near the protein surface, completes the network with a bond with the Tyr166 hydroxyl group from an adjacent subunit. Results defining how these network side chains control the activity of hMnSOD have undergone extensive analysis, using both structure-based mutagenesis [222–226] and chemical modification approaches [227–229]. These studies revealed that any modification of residues in the hydrogen bond network affects both the activity and the stability of the enzyme, even though these alterations cause only minimal structural changes to the active site [222–225,227,230]. Mutation of hydrogen bond network residues Tyr34, His30 or Tyr166 results in a 10- to 40-fold decrease in k(cat), likely due to less efficient proton transfer to the product peroxide [225,231]. Interestingly, substitution of these network amino acids may permit water molecules to replace the endogenous side chain if space allows and perform the necessary interactions to maintain the hydrogen bond network, though at these lower rates [224,227,230].
Interestingly, the domain architecture contributing the active site Gln143 (human sequence) is observed to correlate with metal ion specificity [194,198,232–234]. In hMnSOD Gln143 comes from the C-terminal domain, compared to the N-terminal equivalent Gln in FeSOD (Gln69 in E. coli FeSOD). Substitution of Mn with Fe in the active site of MnSOD markedly reduces the activity of the enzyme, especially at high pH [235,236]; this occurs even though the active sites are 100% conserved in sequence between Mn and FeSOD. The pH sensitivity suggested the binding of a hydroxide to the Fe ion, which was later confirmed in the crystal structure of Fe-substituted E. coli MnSOD [237]. Inhibition occurs through the hydroxide blocking the substrate access funnel and hydrogen bonding to Tyr34 [237] and likely affecting the redox potential of the bound metal [238]. However, certain cannibalistic SODs can function with either metal, and this gain of function may be in part due to the replacement of the strictly conserved Gln by His in this group of enzymes [239,240].
6.4. Structure-based mutational analyses
In addition to its role in metal ion specificity, Gln143 is also critical for in promoting efficient catalysis and maintaining appropriate levels of product inhibition. Mutation of Gln143 reduces activity by two to three orders of magnitude, as determined by stopped-flow spectrophotometry and pulse radiolysis [223,241]. Interestingly, the Gln143 mutants differ from the wild-type protein by having little to no product inhibition even at micromolar concentrations of peroxide. This is of interest because the development of hMnSOD mutants exhibiting limited product inhibition, while retaining a reasonable kinetic rate, may have a therapeutic value. One study aiming to develop such a therapeutic hMnSOD used a directed evolution approach, revealing that two mutations, C140S and N73S, result in a five-fold increase in activity of the Q143A mutant that maintains limited product inhibition [242]. Certain mutations of the hydrogen bond network partners Tyr34, His30 and Tyr166 may also be useful for generating therapeutic SOD, as they are observed to increase the dissociation of the product-inhibited complex [225,231]. Furthermore, studies on one of these mutants with reduced product inhibition, H30N, have revealed that this mutant has both anti-proliferative effects in vitro and anti-tumor effects in vivo, when it is overexpressed [243].
Extensive studies have been conducted on Tyr34 due to its role as an active site gatekeeper residue, controlling access to the metal ion, in addition to having key functions in the hydrogen bond network. The initial structural biochemistry analysis of an Y34F mutant determined that the steady-state turnover of the mutant resembled that of the wild-type enzyme, but at high superoxide levels the rate decreased by 10-fold [222,227]. Markedly, this finding suggested that conservation of Tyr34 is actually due to constraints from extreme conditions affecting cell survival, instead of normal cellular conditions. This result was followed by a study defining Tyr34 function through using Fourier transform infrared (FTIR) spectroscopy and X-ray crystallography analyses on a form of hMnSOD that had the tyrosine side chains replaced with 3-fluorotyrosine [227]. Comparison of the FTIR spectra for 3-fluorotyrosine hMnSOD and a 3-fluorotyrosine hMnSOD Y34F mutant experimentally confirmed that the Tyr34 phenolic hydroxyl is hydrogen bonded, and that it is a proton donor to an adjacent water molecule. A recent structural biochemistry report characterized four further Tyr34 substitutions, in addition to Y34F. This set of experiments revealed that Tyr34 functions to control the gating ratio between catalysis and product inhibition, where substitutions produced a more product-inhibited form of the enzyme [231]. Encouragingly, this recent report also identified in three of the mutations a previously unknown intermediate in catalysis and characterizing this intermediate should help to elucidate a more detailed understanding of enzymatic mechanism. Another area of biological importance is the inactivation of hMnSOD through nitration of Tyr34. Nitration can occur through the reaction of superoxide and nitric oxide in the cell, which generates peroxynitrite [244], a moiety with known roles in cardiovascular disease [245,246]. The crystal structure of nitrated Tyr34 hMnSOD revealed that inhibition of catalysis is likely due to both the steric effect of 3-nitrotyrosine34 perturbing both substrate access and binding and due to the alteration to the hydrogen bond network [228].
Important roles for residues that are not part of the hydrogen bond network, but are close to the active site have been revealed. Trp123 forms a hydrogen bond to the carboxamide of Gln143 and replacement of Trp123 inhibits both catalysis as much as fifty fold and the gating between catalysis and peroxide inhibition, to yield a more product inhibited enzyme. Similar results are observed with Trp161, which resides near the metal chelating Asp159 and the Mn bound solvent, and with Glu162 that forms a hydrogen bond to the metal chelating residue His163 of an adjacent subunit; mutation of these residues lowers enzymatic activity and significantly increases peroxide inhibition. Structural data on the Trp123, Trp161 and Glu162 mutants further support the idea that the defects are due to more subtle electronic effects to the hydrogen bond network rather than gross structural changes. Strikingly, the mutation of Phe66 produces hMnSOD with lower product inhibition and its altered mechanism resembles E. coli MnSOD, through decreasing the rate constant for the oxidative addition of superoxide to Mn2+. Phe66 is adjacent to Tyr34 and it resides at the dimer-packing interface that differs between human and E. coli MnSOD, suggesting that this region plays a role in the increased product inhibition observed in the human enzyme.
Overall, mutation of the well-conserved residues in and around the Mn/FeSOD active site generally results in lower catalytic activity and altered product inhibition; this indicates that Mn/FeSOD have been highly tuned to remove superoxide at near diffusion-limited rates and in a controlled manner. Hopefully, future structure-based analyses will reveal further key insights into the mechanisms of mitochondrial MnSOD and its bacterial counterparts, and will include structure determinations of MnSOD complexes with superoxide or peroxide.
7. NiSOD
The sodN gene, originally identified in Streptomyces [247,248] and cyanobacteria [249], encodes a third, more recently characterized class of SOD with a Ni metal center. Data mining suggests that NiSOD occurs not only in actinobacterial species common to soil and marine environments, but also in proteobacteria that include notable pathogenic species, the obligate intracellular pathogens chlamydiae and green algae eukarya [250]. NiSOD is a protein of ~120 amino acids, containing an acidic 14-residue N-terminal pro-sequence that is removed in the mature enzyme [247]. Initial characterization of NiSOD indicated that it differed from the other SOD classes in its sequence, spectroscopic properties and immunological cross-reactivity. In the absence of Ni it was suggested that NiSOD is a monomer [251], while the presence of Ni promotes formation of a tetrameric species [247]. X-ray and electron paramagnetic resonance spectroscopy studies indicated that Streptomyces seoulensis NiSOD contained an oxidized Ni3+ ion with an axial nitrogen ligand [251]. Also, extended X-ray absorption fine structure (EXAFS) data suggested a dinuclear Ni–Ni active site and thiolate ligation that is not observed in the other classes of SOD [251].
7.1. NiSOD crystal structure
Our laboratory reported the crystal structure of S. coelicolor NiSOD [252] and Kristina Djinović-Carugo and colleagues defined the S. seoulensis homologue [253]; together these studies provided an increased understanding of NiSOD architecture, metal coordination and molecular mechanism. The two NiSOD crystal structures are similar, containing a fold, assembly and active site unique among the SODs. NiSOD forms a right-handed 4-helix bundle, which assembles into a spherical homohexameric structure with an outside diameter of approximately 60 Å and a 20 Å deep central cavity (Fig. 7A). Each subunit contains a N-terminal hook-like structure that projects from the four-helix bundle and chelates a single Ni ion; the closest distance between two Ni ions is 23 Å, ruling out a previously suggested dinuclear Ni–Ni active site. A nine-residue motif, His-Cys-X-X-Pro-Cys-Gly-X-Tyr, forms the novel hook structure (Fig. 7B) and this sequence provides nearly all the key interactions for both chelating the Ni ion and for catalysis. Incubation with cyanide removes Ni from the structure, disordering the hook motif, but not affecting the hexameric structure [252]. These results, together with previous biochemical data [254], indicate a mechanism for NiSOD maturation; hexameric assembly occurs first and is followed by Ni mediated proteolytic cleavage of the 14-residue N-terminal extension. Interestingly, both the oxidized Ni3+ state and reduced Ni2+ state were observed in the structural studies [252,253]. Ni2+ has four-coordinate square planar geometry [252,253], with ligation by the backbone nitrogen atoms of His1 and Cys2, and thiolate side chains (as suggested by EXAFS data) of Cys2 and Cys6. Ni3+ has five-coordinate pyramidal geometry, with axial ligation by the His1 side chain. Ligation by the main-chain nitrogen of His1 explains why proteolytic maturation to free His1 is crucial for activity.
Fig. 7.

S. coelicolor NiSOD crystal structure. (A) NiSOD forms a hexameric assembly (1T6U.pdb) of right-handed four-helix bundles that create a ~60 Å diameter structure with a ~20 Å deep central cavity. Extending from each four-helix bundle are N-terminal Ni-hooks, with the Ni ions displayed as orange spheres. (B) The nine-residue NiSOD metal-binding hook contains the conserved His-Cys-X-X-Pro-Cys-Gly-X-Tyr motif, which chelates a single metal ion and provides key residues for catalysis.
Despite its independent evolution, NiSOD employs strategies similar to those of the other SOD classes for restricting access to the active site. Superoxide access in other SODs is controlled using a combination of size constraint [41,198] and electrostatic steering [42,74,198,255], in addition to the gatekeeper tyrosine for Mn/FeSOD [198,256–258]. In NiSOD, a narrow active site channel also limits access to the catalytic metal site, a nearby group of conserved lysine residues likely aids electrostatic steering, and Tyr9 functions as a gatekeeper. As in other SODs, the protein environment in NiSOD adjusts the midpoint potential of the metal ion into a range suitable for superoxide dismutation (−0.16 to 0.89 V vs. NHE) [259]. However, the metal ligation differs in NiSOD, as the Ni ion does not have a bound solvent molecule associated with proton supply and redox tuning as in MnSOD/FeSOD [260]. Instead, the axial His ligand favors the Ni3+ state, and the cysteine ligands that are also observed in the other characterized redox-active Ni enzymes [261], likely function to reduce the redox potential [262,263].
7.2. Proposed NiSOD molecular mechanisms
Substrate binding in NiSOD has yet to be observed through enzyme-based methods. In NiSOD, two mechanisms have been proposed for superoxide dismutation. First, an inner sphere mechanism was put forward from the discovery of the active site topography and the metal ligand geometry of the S. coelicolor NiSOD structure [252]. The superoxide was proposed to bind the open axial Ni coordination position opposite to the His1 side chain and at the base of the active site channel. Electron transfer from Ni2+ to the superoxide substrate is coupled with protein transfer, which likely comes from the backbone amides of Asp3 or Cys6, or the side chain hydroxyl of Tyr9. Upon Ni oxidation the active site converts to square pyramidal geometry and superoxide again binds axially to the Ni ion. Electron transfer from the superoxide reduces Ni3+ to Ni2+ and generates molecular oxygen, completing the catalytic cycle. The second, recently proposed mechanism has the same general reaction cycle, but an outer sphere, rather than inner sphere, mechanism. This outer sphere mechanism is based on the crystal structure of an Y9F NiSOD mutant with a bound chloride ion, which is suggested to mimic the binding of superoxide substrate or azide inhibitor. The chloride ion is situated between the backbone amides of Asp3 and Cys6 and it does not directly ligate the Ni ion [264]. Recent characterization of a cyanide adduct of the Ni-hook peptide showed direct binding of cyanide to the Ni ion, supporting an inner sphere mechanism for NiSOD [265].
8. Discussion
Structural biochemistry studies have provided striking insights into the functions of the superoxide dismutases. Crystallographic studies confirmed that the FeSODs and MnSODs share a common fold, suggesting a common ancestor. The evolution of similar SODs with different catalytic metal ions has been linked to the availability of these transition metals in the atmosphere during different geological eras. FeSOD may be the more ancestral of the two, evolving when Fe2+ was more abundant; as O2 levels increased, selective pressure shifted towards the more available Mn3+ [266]. Cu,ZnSODs have a distinct fold from Fe/MnSODs, indicating a distinct ancestor, yet the evolutionary origin of Cu,ZnSODs may also be linked to increased O2 levels, which converted insoluble Cu1+ to soluble Cu2+. Despite different evolutionary origins, all the SOD structures have evolved to react at diffusion-limited rates with the aid of electrostatic guidance, alter the redox potential of the transition metal into a range suitable for disproportionation of superoxide, and sequester the active site metal ion to protect against inhibition by other anions such as phosphate.
SOD enzymatic function provides a protective effect to the cell, as ROS mediated damage is linked to many disease states. If left unchecked, superoxide can convert to the hydroxyl radical, which is especially damaging and can cause alterations to DNA, damage proteins and lipids. Damage to DNA can include base mutations right up to chromosomal rearrangements and thus can promote genomic instability and tumorigenesis (reviewed in ref. [21]). Oxidative damage to DNA-processing enzymes, such as DNA polymerases or DNA repair proteins, can also lead to mutagenesis. Interestingly, oxidative damage has been noted in the key DNA repair protein WRN [267], a DNA exonuclease/helicase [268,269] whose dysfunction results in a rapid-aging phenotype (reviewed in refs. [270,271]). Peroxynitrite, generated by ROS reacting with NO, can also attack key cellular proteins [272–274], such as those controlling signal transduction processes or proteins modulating ROS levels, such as MnSOD [246,275]. A mutagenic effect also occurs through lipid peroxidation, as these peroxides can breakdown into mutagenic carbonyl products, such as genotoxic 4-hydroxylnonenal [276]. ROS may additionally damage mitochondrial DNA, a process which is likely relevant to aging and to human disease, including neurodegenerative disorders [277,278].
FALS-inducing mutations in Cu,ZnSOD provide a direct link to neurodegenerative disease. The recognition that SOD1-linked FALS resulted from structural destabilization [44,48] rather than increased stability and activity [64] or a gain of new toxic enzyme activity [65] provides a unified understanding of how mutations frequently cause disease. Other examples of disease-causing protein structural destabilization include mutations in the DNA repair proteins, XPD, Mre11, and Nbs1 that can similarly cause cancer, aging, and neurodegenerative diseases [279–281]. Structural destabilization in SOD1 results in an amyloid-like of fibril aggregate, resembling those found in Alzheimer’s, Huntington’s, Parkinson’s and prion diseases. A single mutation is sufficient to cause FALS, as SOD1 has a core β-sheet structure that appears to be relatively susceptible to aggregation and fibril formation. Interestingly, the increased aggregation propensity of certain SOD1 mutants directly relates to decreasing patient survival [63,282]. Thus, it is likely that the differing SOD mutations result in a general structural destabilization mechanism [44,48] and promote aggregation to a greater or lesser extent. Many of the SOD1 mutations result in a decreased net charge of the enzyme, a property suggested to increase aggregation propensity of proteins [283]. Thus, the likelihood that the 100 plus mutations all contribute to the disease via different mechanisms, as suggested in refs. [132,284] is low. Therefore, this detailed understanding of SOD1 molecular mechanisms and aggregation propensities provides a new avenue of research to generate therapeutics to FALS, for which currently there is no effective treatment. Molecules targeting SOD1 could function at different levels, by down-regulating transcription [285], by helping maintain SOD1 structural stability, preventing aggregation and/or disrupting fibril formation.
Interestingly, MnSOD and its small molecule mimetics may form useful therapeutics by limiting the free radical mediated damage in amyloid-forming neurodegenerative diseases. Also, MnSOD mimetics may help reduce the cellular load of ROS species to help alleviate or reduce the symptoms of neuropathologies generated from inherited DNA-repair defects, including Cockayne syndrome. Interestingly, studies on MnSOD and/or catalase and their mimetics in mice have proved fruitful, revealing potential functions in protecting against neurodegenerative disease and in slowing aging phenotypes [191,192]. Moreover, mouse studies have also revealed other potentially important roles, for example a polymer-conjugated BtCu,ZnSOD protected against death from influenza virus infection [286]. This protective role of BtCu,ZnSOD appears to counter the pathogenicity of influenza virus infection caused, at least in part, by an overreaction of the immune response. Early structure-based genetic engineering of HsCu,ZnSOD sought to increase in vivo stability for biotechnology and medical applications [287]. With the increased knowledge from many SOD structures, including the ultra-stable ApSOD, substantial improvements to these strategies can be envisioned. Also, the more recent discovery that in vivo SOD expression levels can be modified through the Nrf2 transcription factor and its regulator Keap1 [288] may provide a new approach to SOD-mediated therapies.
To date, the structural biochemistry of Cu,ZnSOD has defined the stable subunit and dimer assembly that braces the entire side of subunit, while acting to double the activity per molecule, and provide the efficient bi-directional electrostatic recognition and consequent faster-than-diffusion rate. The stable native Cu,ZnSOD fold excludes non-substrate anions and provides a substantial barrier to aberrant interactions. All known experimental data support the proposal that structural defects cause Lou Gehrig’s disease. The framework destabilization hypothesis whereby loss of native SOD fold-assembly is the molecular basis for the subsequent death of motor neurons provides both a unified general mechanism and a strategy for intervention. Although the discovery of SODs never won the Nobel Prize, it has certainly had enormous impacts on our understanding of cell biology and the medical implications of reactive oxygen species including nitric oxide. The unified framework destabilization hypothesis [44,48,59] proposed from structures and biochemistry best matches all relevant available data. Collectively, the results on the structural biology of SODs, as outlined here, have provided profound insights into molecular mechanisms for ROS control. Moreover, such SOD structural analyses are now providing detailed mechanistic understanding relevant to developing the tools and therapeutics for treating degenerative disease states involving ROS and protein destabilization.
Besides the direct link of SOD destabilization to ALS, the major roles of superoxide and of SODs in biology and medicine are becoming increasingly evident as outlined above. The discovery of superoxide dismutase activity by Fridovich and McCord [1] had divided the opinion of researchers into two camps. One agreed with and built upon on this initial finding. The second thought that superoxide was unlikely to exist in cells, or to interact with cellular substances, or that superoxide dismutase did not function to remove superoxide, and that SOD activity was inhibited by phosphate [7,8,39,43,289]. However, due to the many breakthroughs in SOD research, initiated by Fridovich and McCord and which we have outlined in this review, there is now overwhelming evidence for critical roles of superoxide and of SODs in controlling ROS to alleviate pathogenesis, aging, degenerative diseases, and cancer.
Acknowledgments
We dedicate this review to all patients and their families, and the pioneering researchers in the SOD field, especially Irwin Fridovich and Joseph McCord, who discovered SOD activity; David and Jane Richardson, who tackled the three-dimensional structure of SOD; Stefan Marklund, who discovered SOD3; James Lepock, who characterized SOD stability; Robert Hallewell, who cloned human SOD1; Joan Valentine who characterized the Cu and Zn ion binding; Barry Halliwell, who characterized superoxide toxicity; Joseph Beckman and Bruce Freeman, who identified the cytotoxicity resulting from interactions of superoxide with nitric oxide; Danielle Touati, who used genetics to show the role of SOD in cells; Bernard Babior, who discovered superoxide in the oxidative burst of macrophages; Martha Ludwig, who characterized the first FeSOD structures; Teepu Siddique and Robert Brown, who identified SOD mutations in ALS patients; Don Cleveland, who identified aggregates containing SOD1 as common to FALS disease; and Larry Oberley who defined SOD roles in cancer. We thank Michael Pique for 30+ years of insightful SOD computer graphics, and members of the Tainer and the Getzoff laboratories at TSRI for their critical comments on this manuscript. Work on SOD in the authors’ laboratories was funded by the National Institutes of Health (GM39345 and GM37684).
Abbreviations
- SOD
superoxide dismutase
- O2·−
superoxide
- ROS
reactive oxygen species
- ALS
amyotrophic lateral sclerosis
- FALS
familial ALS
References
- 1.McCord JM, Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein) J Biol Chem. 1969;244:6049–6055. [PubMed] [Google Scholar]
- 2.Fridovich I. Superoxide anion radical (O2-.), superoxide dismutases, and related matters. J Biol Chem. 1997;272:18515–18517. doi: 10.1074/jbc.272.30.18515. [DOI] [PubMed] [Google Scholar]
- 3.Lane N. Oxygen: The Molecule that Made the World. Oxford University Press; USA: 2003. [Google Scholar]
- 4.Fridovich I. Superoxide dismutases. Adv Enzymol Relat Areas Mol Biol. 1986;58:61–97. doi: 10.1002/9780470123041.ch2. [DOI] [PubMed] [Google Scholar]
- 5.Babior BM, Kipnes RS, Curnutte JT. Biological defense mechanisms. The production by leukocytes of superoxide, a potential bactericidal agent. J Clin Invest. 1973;52:741–744. doi: 10.1172/JCI107236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnston RB, Jr, Godzik CA, Cohn ZA. Increased superoxide anion production by immunologically activated and chemically elicited macrophages. J Exp Med. 1978;148:115–127. doi: 10.1084/jem.148.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sawyer DTV, Valentine JS. How super is superoxide. Acc Chem Res. 1981;14:393–400. [Google Scholar]
- 8.Sawyer DTV, Valentine JS. How innocuous is superoxide? Reply to comments. Acc Chem Res. 1982;15:200. [Google Scholar]
- 9.McCord JM, Keele BB, Jr, Fridovich I. An enzyme-based theory of obligate anaerobiosis: the physiological function of superoxide dismutase. Proc Natl Acad Sci U S A. 1971;68:1024–1027. doi: 10.1073/pnas.68.5.1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Halliwell BJMC. Superoxide and superoxide-dependent formation of hyrdoxyl radicals are important in oxygen-toxicity. Trends Biochem Sci. 1982;7:270–272. [Google Scholar]
- 11.Carlioz A, Touati D. Isolation of superoxide dismutase mutants in Escherichia coli: is superoxide dismutase necessary for aerobic life? EMBO J. 1986;5:623–630. doi: 10.1002/j.1460-2075.1986.tb04256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCord JM. Free radicals and inflammation: protection of synovial fluid by superoxide dismutase. Science. 1974;185:529–531. doi: 10.1126/science.185.4150.529. [DOI] [PubMed] [Google Scholar]
- 13.Salin ML, McCord JM. Free radicals and inflammation. Protection of phagocytosine leukocytes by superoxide dismutase. J Clin Invest. 1975;56:1319–1323. doi: 10.1172/JCI108208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Petrone WF, English DK, Wong K, McCord JM. Free radicals and inflammation: superoxide-dependent activation of a neutrophil chemotactic factor in plasma. Proc Natl Acad Sci U S A. 1980;77:1159–1163. doi: 10.1073/pnas.77.2.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Turner SR, Tainer JA, Lynn WS. Biogenesis of chemotactic molecules by the arachidonate lipoxygenase system of platelets. Nature. 1975;257:680–681. doi: 10.1038/257680a0. [DOI] [PubMed] [Google Scholar]
- 16.Imlay JA, Chin SM, Linn S. Toxic DNA damage by hydrogen peroxide through the Fenton reaction in vivo and in vitro. Science. 1988;240:640–642. doi: 10.1126/science.2834821. [DOI] [PubMed] [Google Scholar]
- 17.Imlay JA, Linn S. DNA damage and oxygen radical toxicity. Science. 1988;240:1302–1309. doi: 10.1126/science.3287616. [DOI] [PubMed] [Google Scholar]
- 18.Perry JJ, Fan L, Tainer JA. Developing master keys to brain pathology, cancer and aging from the structural biology of proteins controlling reactive oxygen species and DNA repair. Neuroscience. 2007;145:1280–1299. doi: 10.1016/j.neuroscience.2006.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Halliwell B. Reactive oxygen species and the central nervous system. J Neurochem. 1992;59:1609–1623. doi: 10.1111/j.1471-4159.1992.tb10990.x. [DOI] [PubMed] [Google Scholar]
- 20.Darley-Usmar V, Halliwell B. Blood radicals: reactive nitrogen species, reactive oxygen species, transition metal ions, and the vascular system. Pharm Res. 1996;13:649–662. doi: 10.1023/a:1016079012214. [DOI] [PubMed] [Google Scholar]
- 21.Wiseman H, Halliwell B. Damage to DNA by reactive oxygen and nitrogen species: role in inflammatory disease and progression to cancer. Biochem J. 1996;313(Pt. 1):17–29. doi: 10.1042/bj3130017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stadtman ER, Berlett BS. Reactive oxygen-mediated protein oxidation in aging and disease. Chem Res Toxicol. 1997;10:485–494. doi: 10.1021/tx960133r. [DOI] [PubMed] [Google Scholar]
- 23.Halliwell B. Free radicals, reactive oxygen species and human disease: a critical evaluation with special reference to atherosclerosis. Br J Exp Pathol. 1989;70:737–757. [PMC free article] [PubMed] [Google Scholar]
- 24.Oberley LW, Buettner GR. Role of superoxide dismutase in cancer: a review. Cancer Res. 1979;39:1141–1149. [PubMed] [Google Scholar]
- 25.Granger DN, Rutili G, McCord JM. Superoxide radicals in feline intestinal ischemia. Gastroenterology. 1981;81:22–29. [PubMed] [Google Scholar]
- 26.Parks DA, Bulkley GB, Granger DN, Hamilton SR, McCord JM. Ischemic injury in the cat small intestine: role of superoxide radicals. Gastroenterology. 1982;82:9–15. [PubMed] [Google Scholar]
- 27.Tolmasoff JM, Ono T, Cutler RG. Superoxide dismutase: correlation with lifespan and specific metabolic rate in primate species. Proc Natl Acad Sci U S A. 1980;77:2777–2781. doi: 10.1073/pnas.77.5.2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Orr WC, Sohal RS. Extension of life-span by overexpression of superoxide dismutase and catalase in Drosophila melanogaster. Science. 1994;263:1128–1130. doi: 10.1126/science.8108730. [DOI] [PubMed] [Google Scholar]
- 29.Naslund U, Haggmark S, Johansson G, Marklund SL, Reiz S, Oberg A. Superoxide dismutase and catalase reduce infarct size in a porcine myocardial occlusion-reperfusion model. J Mol Cell Cardiol. 1986;18:1077–1084. doi: 10.1016/s0022-2828(86)80294-2. [DOI] [PubMed] [Google Scholar]
- 30.Costa CA, Amaral TA, Carvalho LC, Ognibene DT, da Silva AF, Moss MB, Valenca SS, de Moura RS, Resende AC. Antioxidant treatment with tempol and apocynin prevents endothelial dysfunction and development of renovascular hypertension. Am J Hypertens. 2009;22(12):1242–1249. doi: 10.1038/ajh.2009.186. [Electronic publication ahead of print 2009 Sep 24] [DOI] [PubMed] [Google Scholar]
- 31.Rahman I. Antioxidant therapeutic advances in COPD. Ther Adv Respir Dis. 2008;2:351–374. doi: 10.1177/1753465808098224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kawakami S, Matsuda A, Sunagawa T, Noda Y, Kaneko T, Tahara S, Hiraumi Y, Adachi S, Matsui H, Ando K, Fujita T, Maruyama N, Shirasawa T, Shimizu T. Antioxidant, EUK-8, prevents murine dilated cardiomyopathy. Circ J. 2009;73:2125–2134. doi: 10.1253/circj.cj-09-0204. [DOI] [PubMed] [Google Scholar]
- 33.Kim BS, Cha HN, Kim YW, Kim JY, Dan JM, Park SY. Inhibition of lipid infusion-induced skeletal muscle insulin resistance by cotreatment with tempol and glutathione in mice. J Pharmacol Sci. 2009;110:370–380. doi: 10.1254/jphs.09046fp. [DOI] [PubMed] [Google Scholar]
- 34.Shopova VL, Dancheva VY, Salovsky PT, Stoyanova AM. Protective effects of a superoxide dismutase/catalase mimetic compound against paraquat pneumotoxicity in rat lung. Respirology. 2009;14:504–510. doi: 10.1111/j.1440-1843.2009.01531.x. [DOI] [PubMed] [Google Scholar]
- 35.Kawakami T, Urakami S, Hirata H, Tanaka Y, Nakajima K, Enokida H, Shiina H, Ogishima T, Tokizane T, Kawamoto K, Miura K, Ishii N, Dahiya R. Superoxide dismutase analog (Tempol: 4-hydroxy-2, 2, 6, 6-tetramethylpiperidine 1-oxyl) treatment restores erectile function in diabetes-induced impotence. Int J Impot Res. 2009;21:348–355. doi: 10.1038/ijir.2009.28. [Electronic publication ahead of print 25 June 2009] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peixoto EB, Pessoa BS, Biswas SK, Lopes de Faria JB. Antioxidant SOD mimetic prevents NADPH oxidase-induced oxidative stress and renal damage in the early stage of experimental diabetes and hypertension. Am J Nephrol. 2009;29:309–318. doi: 10.1159/000163767. [DOI] [PubMed] [Google Scholar]
- 37.Abdallah DM, El-Abhar HS, Abdel-Aziz DH. TEMPOL, a membrane-permeable radical scavenger, attenuates gastric mucosal damage induced by ischemia/reperfusion: a key role for superoxide anion. Eur J Pharmacol. 2009;603:93–97. doi: 10.1016/j.ejphar.2008.11.057. [DOI] [PubMed] [Google Scholar]
- 38.Church SL, Grant JW, Ridnour LA, Oberley LW, Swanson PE, Meltzer PS, Trent JM. Increased manganese superoxide dismutase expression suppresses the malignant phenotype of human melanoma cells. Proc Natl Acad Sci U S A. 1993;90:3113–3117. doi: 10.1073/pnas.90.7.3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paschen WUW. Problems concerning the biochemical action of superoxide dismutase (erthrocuperin) Hoppe Seylers Z Physiol Chem. 1975;356:727–737. doi: 10.1515/bchm2.1975.356.s1.727. [DOI] [PubMed] [Google Scholar]
- 40.Beyer WF, Jr, Wang Y, Fridovich I. Phosphate inhibition of the copper-and zinc-containing superoxide dismutase: a reexamination. Biochemistry. 1986;25:6084–6088. doi: 10.1021/bi00368a037. [DOI] [PubMed] [Google Scholar]
- 41.Tainer JA, Getzoff ED, Richardson JS, Richardson DC. Structure and mechanism of copper, zinc superoxide dismutase. Nature. 1983;306:284–287. doi: 10.1038/306284a0. [DOI] [PubMed] [Google Scholar]
- 42.Getzoff ED, Tainer JA, Weiner PK, Kollman PA, Richardson JS, Richardson DC. Electrostatic recognition between superoxide and copper, zinc superoxide dismutase. Nature. 1983;306:287–290. doi: 10.1038/306287a0. [DOI] [PubMed] [Google Scholar]
- 43.Mota de Freitas D, Valentine JS. Phosphate is an inhibitor of copper-zinc superoxide dismutase. Biochemistry. 1984;23:2079–2082. doi: 10.1021/bi00304a031. [DOI] [PubMed] [Google Scholar]
- 44.Deng HX, Hentati A, Tainer JA, Iqbal Z, Cayabyab A, Hung WY, Getzoff ED, Hu P, Herzfeldt B, Roos RP, Warner C, Deng G, Soriano E, Smyth C, Parge HE, Ahmed A, Roses AD, Hallewell RA, Pericack-Vance MA, Siddique T. Amyotrophic lateral sclerosis and structural defects in Cu,Zn superoxide dismutase. Science. 1993;261:1047–1051. doi: 10.1126/science.8351519. [DOI] [PubMed] [Google Scholar]
- 45.Antonyuk S, Elam JS, Hough MA, Strange RW, Doucette PA, Rodriguez JA, Hayward LJ, Valentine JS, Hart PJ, Hasnain SS. Structural consequences of the familial amyotrophic lateral sclerosis SOD1 mutant His46Arg. Protein Sci. 2005;14:1201–1213. doi: 10.1110/ps.041256705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, Reaume AG, Scott RW, Cleveland DW. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science. 1998;281:1851–1854. doi: 10.1126/science.281.5384.1851. [DOI] [PubMed] [Google Scholar]
- 47.Cao X, Antonyuk SV, Seetharaman SV, Whitson LJ, Taylor AB, Holloway SP, Strange RW, Doucette PA, Valentine JS, Tiwari A, Hayward LJ, Padua S, Cohlberg JA, Hasnain SS, Hart PJ. Structures of the G85R variant of SOD1 in familial amyotrophic lateral sclerosis. J Biol Chem. 2008;283:16169–16177. doi: 10.1074/jbc.M801522200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.DiDonato M, Craig L, Huff ME, Thayer MM, Cardoso RM, Kassmann CJ, Lo TP, Bruns CK, Powers ET, Kelly JW, Getzoff ED, Tainer JA. ALS mutants of human superoxide dismutase form fibrous aggregates via framework destabilization. J Mol Biol. 2003;332:601–615. doi: 10.1016/s0022-2836(03)00889-1. [DOI] [PubMed] [Google Scholar]
- 49.Ding F, Dokholyan NV. Dynamical roles of metal ions and the disulfide bond in Cu, Zn superoxide dismutase folding and aggregation. Proc Natl Acad Sci U S A. 2008;105:19696–19701. doi: 10.1073/pnas.0803266105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Elam JS, Taylor AB, Strange R, Antonyuk S, Doucette PA, Rodriguez JA, Hasnain SS, Hayward LJ, Valentine JS, Yeates TO, Hart PJ. Amyloid-like filaments and water-filled nanotubes formed by SOD1 mutant proteins linked to familial ALS. Nat Struct Biol. 2003;10:461–467. doi: 10.1038/nsb935. [DOI] [PubMed] [Google Scholar]
- 51.Gidalevitz T, Krupinski T, Garcia S, Morimoto RI. Destabilizing protein polymorphisms in the genetic background direct phenotypic expression of mutant SOD1 toxicity. PLoS Genet. 2009;5:e1000399. doi: 10.1371/journal.pgen.1000399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hornberg A, Logan DT, Marklund SL, Oliveberg M. The coupling between disulphide status, metallation and dimer interface strength in Cu/Zn superoxide dismutase. J Mol Biol. 2007;365:333–342. doi: 10.1016/j.jmb.2006.09.048. [DOI] [PubMed] [Google Scholar]
- 53.Hough MA, Grossmann JG, Antonyuk SV, Strange RW, Doucette PA, Rodriguez JA, Whitson LJ, Hart PJ, Hayward LJ, Valentine JS, Hasnain SS. Dimer destabilization in superoxide dismutase may result in disease-causing properties: structures of motor neuron disease mutants. Proc Natl Acad Sci U S A. 2004;101:5976–5981. doi: 10.1073/pnas.0305143101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lindberg MJ, Bystrom R, Boknas N, Andersen PM, Oliveberg M. Systematically perturbed folding patterns of amyotrophic lateral sclerosis (ALS)-associated SOD1 mutants. Proc Natl Acad Sci U S A. 2005;102:9754–9759. doi: 10.1073/pnas.0501957102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nordlund A, Oliveberg M. SOD1-associated ALS: a promising system for elucidating the origin of protein-misfolding disease. HFSP J. 2008;2:354–364. doi: 10.2976/1.2995726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roberts BR, Tainer JA, Getzoff ED, Malencik DA, Anderson SR, Bomben VC, Meyers KR, Karplus PA, Beckman JS. Structural characterization of zinc-deficient human superoxide dismutase and implications for ALS. J Mol Biol. 2007;373:877–890. doi: 10.1016/j.jmb.2007.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rumfeldt JA, Lepock JR, Meiering EM. Unfolding and folding kinetics of amyotrophic lateral sclerosis-associated mutant Cu,Zn superoxide dismutases. J Mol Biol. 2009;385:278–298. doi: 10.1016/j.jmb.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 58.Shaw BF, Durazo A, Nersissian AM, Whitelegge JP, Faull KF, Valentine JS. Local unfolding in a destabilized, pathogenic variant of superoxide dismutase 1 observed with H/D exchange and mass spectrometry. J Biol Chem. 2006;281:18167–18176. doi: 10.1074/jbc.M600623200. [DOI] [PubMed] [Google Scholar]
- 59.Shin DS, Didonato M, Barondeau DP, Hura GL, Hitomi C, Berglund JA, Getzoff ED, Cary SC, Tainer JA. Superoxide dismutase from the eukaryotic thermophile Alvinella pompejana: structures, stability, mechanism, and insights into amyotrophic lateral sclerosis. J Mol Biol. 2009;385:1534–1555. doi: 10.1016/j.jmb.2008.11.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Strange RW, Yong CW, Smith W, Hasnain SS. Molecular dynamics using atomic-resolution structure reveal structural fluctuations that may lead to polymerization of human Cu-Zn superoxide dismutase. Proc Natl Acad Sci U S A. 2007;104:10040–10044. doi: 10.1073/pnas.0703857104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tiwari A, Liba A, Sohn SH, Seetharaman SV, Bilsel O, Matthews CR, Hart PJ, Valentine JS, Hayward LJ. Metal deficiency increases aberrant hydrophobicity of mutant superoxide dismutases that cause amyotrophic lateral sclerosis. J Biol Chem. 2009;284(40):27746–27758. doi: 10.1074/jbc.M109.043729. [Electronic publication ahead of print 2009 Aug 3] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang J, Farr GW, Hall DH, Li F, Furtak K, Dreier L, Horwich AL. An ALS-linked mutant SOD1 produces a locomotor defect associated with aggregation and synaptic dysfunction when expressed in neurons of Caenorhabditis elegans. PLoS Genet. 2009;5:e1000350. doi: 10.1371/journal.pgen.1000350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang Q, Johnson JL, Agar NY, Agar JN. Protein aggregation and protein instability govern familial amyotrophic lateral sclerosis patient survival. PLoS Biol. 2008;6:e170. doi: 10.1371/journal.pbio.0060170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng H-X, Rahmani Z, Krizus A, McKenna-Yasek D, Cayabyab A, Gaston SM, Berger R, Tanzi RE, Halperin JJ, Herzfeldt B, Van den Bergh R, Hung W-Y, Bird T, Deng G, Mulder DW, Smyth C, Laing NG, Soriano E, Pericak–Vance MA, Haines J, Rouleau GA, Gusella JS, Robert Horvitz H, Brown RH., Jr Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 65.Wiedau-Pazos M, Goto JJ, Rabizadeh S, Gralla EB, Roe JA, Lee MK, Valentine JS, Bredesen DE. Altered reactivity of superoxide dismutase in familial amyotrophic lateral sclerosis. Science. 1996;271:515–518. doi: 10.1126/science.271.5248.515. [DOI] [PubMed] [Google Scholar]
- 66.Majoor-Krakauer D, Willems PJ, Hofman A. Genetic epidemiology of amyotrophic lateral sclerosis. Clin Genet. 2003;63:83–101. doi: 10.1046/j.0009-9163.2002.00001.x. [DOI] [PubMed] [Google Scholar]
- 67.Andersen PM. Amyotrophic lateral sclerosis associated with mutations in the CuZn superoxide dismutase gene. Curr Neurol Neurosci Rep. 2006;6:37–46. doi: 10.1007/s11910-996-0008-9. [DOI] [PubMed] [Google Scholar]
- 68.Smith MW, Doolittle RF. A comparison of evolutionary rates of the two major kinds of superoxide dismutase. J Mol Evol. 1992;34:175–184. doi: 10.1007/BF00182394. [DOI] [PubMed] [Google Scholar]
- 69.Mann T, Keilin D. Haemocuprein and hepatocuprein, copper-protein compounds of blood and liver in mammals. Proc Roy Soc Ser B-Biol Sci. 1938;126:303–315. [Google Scholar]
- 70.Kimmel JR, Markowitz H, Brown DM. Some chemical and physical properties of erythrocuprein. J Biol Chem. 1959;234:46–50. [PubMed] [Google Scholar]
- 71.Markowitz H, Cartwright GE, Wintrobe MM. Studies on copper metabolism. XXVII. The isolation and properties of an erythrocyte cuproprotein (erythrocuprein) J Biol Chem. 1959;234:40–45. [PubMed] [Google Scholar]
- 72.Porter H, Folch J. CerebrocupreinI, A copper-containing protein isolated from brain. J Neurochem. 1957;1:260–271. doi: 10.1111/j.1471-4159.1957.tb12081.x. [DOI] [PubMed] [Google Scholar]
- 73.Tainer JA, Getzoff ED, Beem KM, Richardson JS, Richardson DC. Determination and analysis of the 2 A-structure of copper, zinc superoxide dismutase. J Mol Biol. 1982;160:181–217. doi: 10.1016/0022-2836(82)90174-7. [DOI] [PubMed] [Google Scholar]
- 74.Getzoff ED, Cabelli DE, Fisher CL, Parge HE, Viezzoli MS, Banci L, Hallewell RA. Faster superoxide dismutase mutants designed by enhancing electrostatic guidance. Nature. 1992;358:347–351. doi: 10.1038/358347a0. [DOI] [PubMed] [Google Scholar]
- 75.Parge HE, Hallewell RA, Tainer JA. Atomic structures of wild-type and thermostable mutant recombinant human Cu,Zn superoxide dismutase. Proc Natl Acad Sci U S A. 1992;89:6109–6113. doi: 10.1073/pnas.89.13.6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Djinovic K, Gatti G, Coda A, Antolini L, Pelosi G, Desideri A, Falconi M, Marmocchi F, Rolilio G, Bolognesi M. Structure solution and molecular dynamics refinement of the yeast Cu,Zn enzyme superoxide dismutase. Acta Crystallogr B. 1991;47(Pt 6):918–927. doi: 10.1107/s0108768191004949. [DOI] [PubMed] [Google Scholar]
- 77.Djinovic K, Gatti G, Coda A, Antolini L, Pelosi G, Desideri A, Falconi M, Marmocchi F, Rotilio G, Bolognesi M. Crystal structure of yeast Cu,Zn superoxide dismutase. Crystallographic refinement at 2.5 A resolution. J Mol Biol. 1992;225:791–809. doi: 10.1016/0022-2836(92)90401-5. [DOI] [PubMed] [Google Scholar]
- 78.Ogihara NL, Parge HE, Hart PJ, Weiss MS, Goto JJ, Crane BR, Tsang J, Slater K, Roe JA, Valentine JS, Eisenberg D, Tainer JA. Unusual trigonal-planar copper configuration revealed in the atomic structure of yeast copper-zinc superoxide dismutase. Biochemistry. 1996;35:2316–2321. doi: 10.1021/bi951930b. [DOI] [PubMed] [Google Scholar]
- 79.Cardoso RM, Silva CH, Ulian de Araujo AP, Tanaka T, Tanaka M, Garratt RC. Structure of the cytosolic Cu,Zn superoxide dismutase from Schistosoma mansoni. Acta Crystallogr D Biol Crystallogr. 2004;60:1569–1578. doi: 10.1107/S0907444904016798. [DOI] [PubMed] [Google Scholar]
- 80.Carugo KD, Battistoni A, Carri MT, Polticelli F, Desideri A, Rotilio G, Coda A, Wilson KS, Bolognesi M. Three-dimensional structure of Xenopus laevis Cu,Zn superoxide dismutase b determined by X-ray crystallography at 1.5 A resolution. Acta Crystallogr D Biol Crystallogr. 1996;52:176–188. doi: 10.1107/S0907444995007608. [DOI] [PubMed] [Google Scholar]
- 81.Kitagawa Y, Tanaka N, Hata Y, Kusunoki M, Lee GP, Katsube Y, Asada K, Aibara S, Morita Y. Three-dimensional structure of Cu,Zn-superoxide dismutase from spinach at 2.0 A resolution. J Biochem (Tokyo) 1991;109:477–485. doi: 10.1093/oxfordjournals.jbchem.a123407. [DOI] [PubMed] [Google Scholar]
- 82.Yogavel M, Mishra PC, Gill J, Bhardwaj PK, Dutt S, Kumar S, Ahuja PS, Sharma A. Structure of a superoxide dismutase and implications for copper-ion chelation. Acta Crystallogr D Biol Crystallogr. 2008;D64:892–901. doi: 10.1107/S0907444908019069. [DOI] [PubMed] [Google Scholar]
- 83.Banci L, Bertini I, Cramaro F, Del Conte R, Viezzoli MS. The solution structure of reduced dimeric copper zinc superoxide dismutase. The structural effects of dimerization. Eur J Biochem. 2002;269:1905–1915. doi: 10.1046/j.1432-1033.2002.02840.x. [DOI] [PubMed] [Google Scholar]
- 84.Richardson JS. β-Sheet topology and the relatedness of proteins. Nature. 1977;268:495–500. doi: 10.1038/268495a0. [DOI] [PubMed] [Google Scholar]
- 85.Getzoff ED, Tainer JA, Stempien MM, Bell GI, Hallewell RA. Evolution of CuZn superoxide dismutase and the Greek key beta-barrel structural motif. Proteins. 1989;5:322–336. doi: 10.1002/prot.340050408. [DOI] [PubMed] [Google Scholar]
- 86.Hallewell RA, Imlay KC, Lee P, Fong NM, Gallegos C, Getzoff ED, Tainer JA, Cabelli DE, Tekamp-Olson P, Mullenbach GT, Cousens LS. Thermostabilization of recombinant human and bovine CuZn superoxide dismutases by replacement of free cysteines. Biochem Biophys Res Commun. 1991;181:474–480. doi: 10.1016/s0006-291x(05)81443-3. [DOI] [PubMed] [Google Scholar]
- 87.Roberts VA, Freeman HC, Olson AJ, Tainer JA, Getzoff ED. Electrostatic orientation of the electron-transfer complex between plastocyanin and cytochrome c. J Biol Chem. 1991;266:13431–13441. [PubMed] [Google Scholar]
- 88.Hough MA, Hasnain SS. Structure of fully reduced bovine copper zinc superoxide dismutase at 1.15 A. Structure. 2003;11:937–946. doi: 10.1016/s0969-2126(03)00155-2. [DOI] [PubMed] [Google Scholar]
- 89.Strange RW, Antonyuk SV, Hough MA, Doucette PA, Valentine JS, Hasnain SS. Variable metallation of human superoxide dismutase: atomic resolution crystal structures of Cu-Zn, Zn-Zn and As-isolated wild-type enzymes. J Mol Biol. 2006;356:1152–1162. doi: 10.1016/j.jmb.2005.11.081. [DOI] [PubMed] [Google Scholar]
- 90.Bordo D, Djinovic K, Bolognesi M. Conserved patterns in the Cu,Zn superoxide dismutase family. J Mol Biol. 1994;238:366–386. doi: 10.1006/jmbi.1994.1298. [DOI] [PubMed] [Google Scholar]
- 91.Boissinot M, Karnas S, Lepock JR, Cabelli DE, Tainer JA, Getzoff ED, Hallewell RA. Function of the Greek key connection analysed using circular permutants of superoxide dismutase. EMBO J. 1997;16:2171–2178. doi: 10.1093/emboj/16.9.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Crane BR, Arvai AS, Gachhui R, Wu C, Ghosh DK, Getzoff ED, Stuehr DJ, Tainer JA. The structure of nitric oxide synthase oxygenase domain and inhibitor complexes. Science. 1997;278:425–431. doi: 10.1126/science.278.5337.425. [DOI] [PubMed] [Google Scholar]
- 93.Crane BR, Arvai AS, Ghosh DK, Wu C, Getzoff ED, Stuehr DJ, Tainer JA. Structure of nitric oxide synthase oxygenase dimer with pterin and substrate. Science. 1998;279:2121–2126. doi: 10.1126/science.279.5359.2121. [DOI] [PubMed] [Google Scholar]
- 94.Fisher CL, Cabelli DE, Hallewell RA, Beroza P, Lo TP, Getzoff ED, Tainer JA. Computational, pulse-radiolytic, and structural investigations of lysine-136 and its role in the electrostatic triad of human Cu,Zn superoxide dismutase. Proteins. 1997;29:103–112. doi: 10.1002/(sici)1097-0134(199709)29:1<103::aid-prot8>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 95.Fisher CL, Cabelli DE, Tainer JA, Hallewell RA, Getzoff ED. The role of arginine 143 in the electrostatics and mechanism of Cu,Zn superoxide dismutase: computational and experimental evaluation by mutational analysis. Proteins. 1994;19:24–34. doi: 10.1002/prot.340190105. [DOI] [PubMed] [Google Scholar]
- 96.Fisher CL, Hallewell RA, Roberts VA, Tainer JA, Getzoff ED. Probing the structural basis for enzyme-substrate recognition in Cu,Zn superoxide dismutase. Free Radic Res Commun. 1991;12-13(Pt. 1):287–296. doi: 10.3109/10715769109145797. [DOI] [PubMed] [Google Scholar]
- 97.Roberts VA, Fisher CL, Redford SM, McRee DE, Parge HE, Getzoff ED, Tainer JA. Mechanism and atomic structure of superoxide dismutase. Free Radic Res Commun. 1991;12-13(Pt 1):269–278. doi: 10.3109/10715769109145795. [DOI] [PubMed] [Google Scholar]
- 98.Tainer JA, Hallewell RA, Roberts VA, Parge HE, Getzoff ED. Probing enzyme-substrate recognition and catalytic mechanism in Cu,Zn superoxide dismutase. Basic Life Sci. 1988;49:635–640. doi: 10.1007/978-1-4684-5568-7_100. [DOI] [PubMed] [Google Scholar]
- 99.Hart PJ, Balbirnie MM, Ogihara NL, Nersissian AM, Weiss MS, Valentine JS, Eisenberg D. A structure-based mechanism for copper-zinc superoxide dismutase. Biochemistry. 1999;38:2167–2178. doi: 10.1021/bi982284u. [DOI] [PubMed] [Google Scholar]
- 100.Ferraroni M, Rypniewski WR, Bruni B, Orioli P, Mangani S. Crystallographic determination of reduced bovine superoxide dismutase at pH 5.0 and of anion binding to its active site. J Biol Inorg Chem. 1998;3:411–422. [Google Scholar]
- 101.Smirnov VV, Roth JP. Mechanisms of electron transfer in catalysis by copper zinc superoxide dismutase. J Am Chem Soc. 2006;128:16424–16425. doi: 10.1021/ja066369r. [DOI] [PubMed] [Google Scholar]
- 102.Antonyuk SV, Strange RW, Marklund SL, Hasnain SS. The structure of human extracellular copper-zinc superoxide dismutase at 1.7 A resolution: insights into heparin and collagen binding. J Mol Biol. 2009;388:310–326. doi: 10.1016/j.jmb.2009.03.026. [DOI] [PubMed] [Google Scholar]
- 103.Marklund SL. Extracellular superoxide dismutase in human tissues and human cell lines. J Clin Invest. 1984;74:1398–1403. doi: 10.1172/JCI111550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Marklund SL. Product of extracellular-superoxide dismutase catalysis. FEBS Lett. 1985;184:237–239. doi: 10.1016/0014-5793(85)80613-x. [DOI] [PubMed] [Google Scholar]
- 105.Benov L, Chang LY, Day B, Fridovich I. Copper, zinc superoxide dismutase in Escherichia coli: periplasmic localization. Arch Biochem Biophys. 1995;319:508–511. doi: 10.1006/abbi.1995.1324. [DOI] [PubMed] [Google Scholar]
- 106.Canvin J, Langford PR, Wilks KE, Kroll JS. Identification of sodC encoding periplasmic [Cu,Zn]-superoxide dismutase in Salmonella. FEMS Microbiol Lett. 1996;136:215–220. doi: 10.1111/j.1574-6968.1996.tb08052.x. [DOI] [PubMed] [Google Scholar]
- 107.Kroll JS, Langford PR, Loynds BM. Copper-zinc superoxide dismutase of Haemophilus influenzae and H. parainfluenzae. J Bacteriol. 1991;173:7449–7457. doi: 10.1128/jb.173.23.7449-7457.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kroll JS, Langford PR, Wilks KE, Keil AD. Bacterial [Cu,Zn]-superoxide dismutase: phylogenetically distinct from the eukaryotic enzyme. and not so rare after all! Microbiology. 1995;141(Pt. 9):2271–2279. doi: 10.1099/13500872-141-9-2271. [DOI] [PubMed] [Google Scholar]
- 109.Lainson FA, Thomson N, Rowe HA, Langford PR, Aitchison KD, Donachie W, Kroll JS. Occurrence of [copper, zinc]-cofactored superoxide dismutase in Pasteurella haemolytica and its serotype distribution. FEMS Microbiol Lett. 1996;142:11–17. doi: 10.1111/j.1574-6968.1996.tb08400.x. [DOI] [PubMed] [Google Scholar]
- 110.Langford PR, Loynds BM, Kroll JS. Cloning and molecular characterization of Cu,Zn superoxide dismutase from Actinobacillus pleuropneumoniae. Infect Immun. 1996;64:5035–5041. doi: 10.1128/iai.64.12.5035-5041.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Stabel TJ, Sha Z, Mayfield JE. Periplasmic location of Brucella abortus Cu/Zn superoxide dismutase. Vet Microbiol. 1994;38:307–314. doi: 10.1016/0378-1135(94)90149-x. [DOI] [PubMed] [Google Scholar]
- 112.Farrant JL, Sansone A, Canvin JR, Pallen MJ, Langford PR, Wallis TS, Dougan G, Kroll JS. Bacterial copper-and zinc-cofactored superoxide dismutase contributes to the pathogenesis of systemic salmonellosis. Mol Microbiol. 1997;25:785–796. doi: 10.1046/j.1365-2958.1997.5151877.x. [DOI] [PubMed] [Google Scholar]
- 113.Tatum FM, Detilleux PG, Sacks JM, Halling SM. Construction of Cu-Zn superoxide dismutase deletion mutants of Brucella abortus: analysis of survival in vitro in epithelial and phagocytic cells and in vivo in mice. Infect Immun. 1992;60:2863–2869. doi: 10.1128/iai.60.7.2863-2869.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wilks KE, Dunn KL, Farrant JL, Reddin KM, Gorringe AR, Langford PR, Kroll JS. Periplasmic superoxide dismutase in meningococcal pathogenicity. Infect Immun. 1998;66:213–217. doi: 10.1128/iai.66.1.213-217.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Battistoni A. Role of prokaryotic Cu,Zn superoxide dismutase in pathogenesis. Biochem Soc Trans. 2003;31:1326–1329. doi: 10.1042/bst0311326. [DOI] [PubMed] [Google Scholar]
- 116.De Groote MA, Ochsner UA, Shiloh MU, Nathan C, McCord JM, Dinauer MC, Libby SJ, Vazquez-Torres A, Xu Y, Fang FC. Periplasmic superoxide dismutase protects Salmonella from products of phagocyte NADPH-oxidase and nitric oxide synthase. Proc Natl Acad Sci U S A. 1997;94:13997–14001. doi: 10.1073/pnas.94.25.13997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bourne Y, Redford SM, Steinman HM, Lepock JR, Tainer JA, Getzoff ED. Novel dimeric interface and electrostatic recognition in bacterial Cu,Zn superoxide dismutase. Proc Natl Acad Sci U S A. 1996;93:12774–12779. doi: 10.1073/pnas.93.23.12774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Redford SM, McRee DE, Getzoff ED, Steinman HM, Tainer JA. Crystallographic characterization of a Cu,Zn superoxide dismutase from Photobacterium leiognathi. J Mol Biol. 1990;212:449–451. doi: 10.1016/0022-2836(90)90323-E. [DOI] [PubMed] [Google Scholar]
- 119.Bordo D, Matak D, Djinovic-Carugo K, Rosano C, Pesce A, Bolognesi M, Stroppolo ME, Falconi M, Battistoni A, Desideri A. Evolutionary constraints for dimer formation in prokaryotic Cu,Zn superoxide dismutase. J Mol Biol. 1999;285:283–296. doi: 10.1006/jmbi.1998.2267. [DOI] [PubMed] [Google Scholar]
- 120.Forest KT, Langford PR, Kroll JS, Getzoff ED. Cu,Zn superoxide dismutase structure from a microbial pathogen establishes a class with a conserved dimer interface. J Mol Biol. 2000;296:145–153. doi: 10.1006/jmbi.1999.3448. [DOI] [PubMed] [Google Scholar]
- 121.Pesce A, Battistoni A, Stroppolo ME, Polizio F, Nardini M, Kroll JS, Langford PR, O’Neill P, Sette M, Desideri A, Bolognesi M. Functional and crystallographic characterization of Salmonella typhimurium Cu,Zn superoxide dismutase coded by the sodCI virulence gene. J Mol Biol. 2000;302:465–478. doi: 10.1006/jmbi.2000.4074. [DOI] [PubMed] [Google Scholar]
- 122.Pesce A, Capasso C, Battistoni A, Folcarelli S, Rotilio G, Desideri A, Bolognesi M. Unique structural features of the monomeric Cu,Zn superoxide dismutase from Escherichia coli,revealedZ by X-ray crystallography. J Mol Biol. 1997;274:408–420. doi: 10.1006/jmbi.1997.1400. [DOI] [PubMed] [Google Scholar]
- 123.Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci. 2001;2:806–819. doi: 10.1038/35097565. [DOI] [PubMed] [Google Scholar]
- 124.Charcot JM, Joffory A. Deux cas d’atrophie musculaire progressive avec lesions de la substance grise et des faisceaux antero-lateraux de la moelle epiniere. Arch Physiol Neurol Pathol. 1869;2:744–754. [Google Scholar]
- 125.Siddique T, Deng HX. Genetics of amyotrophic lateral sclerosis. Hum Mol Genet. 1996;5:1465–1470. doi: 10.1093/hmg/5.supplement_1.1465. [DOI] [PubMed] [Google Scholar]
- 126.Dobson CM. Protein folding and its links with human disease. Biochem Soc Symp. 2001:1–26. [PubMed] [Google Scholar]
- 127.Andersen PM. Genetics of sporadic ALS. Amyotroph Lateral Scler Other Motor Neuron Disord. 2001;2(Suppl 1):S37–S41. doi: 10.1080/14660820152415726. [DOI] [PubMed] [Google Scholar]
- 128.Gaudette M, Hirano M, Siddique T. Current status of SOD1 mutations in familial amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:83–89. doi: 10.1080/14660820050515377. [DOI] [PubMed] [Google Scholar]
- 129.Julien JP. ALS: astrocytes move in as deadly neighbors. Nat Neurosci. 2007;10:535–537. doi: 10.1038/nn0507-535. [DOI] [PubMed] [Google Scholar]
- 130.Potter SZ, Valentine JS. The perplexing role of copper-zinc superoxide dismutase in amyotrophic lateral sclerosis (Lou Gehrig’s disease) J Biol Inorg Chem. 2003;8:373–380. doi: 10.1007/s00775-003-0447-6. [DOI] [PubMed] [Google Scholar]
- 131.Rodriguez JA, Shaw BF, Durazo A, Sohn SH, Doucette PA, Nersissian AM, Faull KF, Eggers DK, Tiwari A, Hayward LJ, Valentine JS. Destabilization of apoprotein is insufficient to explain Cu,Zn-superoxide dismutase-linked ALS pathogenesis. Proc Natl Acad Sci U S A. 2005;102:10516–10521. doi: 10.1073/pnas.0502515102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Shaw BF, Valentine JS. How do ALS-associated mutations in superoxide dismutase 1 promote aggregation of the protein? Trends Biochem Sci. 2007;32:78–85. doi: 10.1016/j.tibs.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 133.Wong PC, Borchelt DR. Motor neuron disease caused by mutations in superoxide dismutase 1. Curr Opin Neurol. 1995;8:294–301. doi: 10.1097/00019052-199508000-00008. [DOI] [PubMed] [Google Scholar]
- 134.Brown RH., Jr Amyotrophic lateral sclerosis: recent insights from genetics and transgenic mice. Cell. 1995;80:687–692. doi: 10.1016/0092-8674(95)90346-1. [DOI] [PubMed] [Google Scholar]
- 135.Crow JP, Ye YZ, Strong M, Kirk M, Barnes S, Beckman JS. Superoxide dismutase catalyzes nitration of tyrosines by peroxynitrite in the rod and head domains of neurofilament-L. J Neurochem. 1997;69:1945–1953. doi: 10.1046/j.1471-4159.1997.69051945.x. [DOI] [PubMed] [Google Scholar]
- 136.Beckman JS, Carson M, Smith CD, Koppenol WH. ALS, SOD and peroxynitrite. Nature. 1993;364:584. doi: 10.1038/364584a0. [DOI] [PubMed] [Google Scholar]
- 137.Zhang H, Joseph J, Gurney M, Becker D, Kalyanaraman B. Bicarbonate enhances peroxidase activity of Cu,Zn-superoxide dismutase. Role of carbonate anion radical and scavenging of carbonate anion radical by metalloporphyrin antioxidant enzyme mimetics. J Biol Chem. 2002;277:1013–1020. doi: 10.1074/jbc.M108585200. [DOI] [PubMed] [Google Scholar]
- 138.Cleveland DW, Liu J. Oxidation versus aggregation — how do SOD1 mutants cause ALS? Nat Med. 2000;6:1320–1321. doi: 10.1038/82122. [DOI] [PubMed] [Google Scholar]
- 139.Hirano A, Donnenfeld H, Sasaki S, Nakano I. Fine structural observations of neurofilamentous changes in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 1984;43:461–470. doi: 10.1097/00005072-198409000-00001. [DOI] [PubMed] [Google Scholar]
- 140.Wanker EE. Protein aggregation in Huntington’s and Parkinson’s disease: implications for therapy. Mol Med Today. 2000;6:387–391. doi: 10.1016/s1357-4310(00)01761-5. [DOI] [PubMed] [Google Scholar]
- 141.Soto C. Protein misfolding and disease; protein refolding and therapy. FEBS Lett. 2001;498:204–207. doi: 10.1016/s0014-5793(01)02486-3. [DOI] [PubMed] [Google Scholar]
- 142.Durham HD, Roy J, Dong L, Figlewicz DA. Aggregation of mutant Cu/Zn superoxide dismutase proteins in a culture model of ALS. J Neuropathol Exp Neurol. 1997;56:523–530. doi: 10.1097/00005072-199705000-00008. [DOI] [PubMed] [Google Scholar]
- 143.Johnston JA, Dalton MJ, Gurney ME, Kopito RR. Formation of high molecular weight complexes of mutant Cu, Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2000;97:12571–12576. doi: 10.1073/pnas.220417997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Shibata N, Hirano A, Kobayashi M, Siddique T, Deng HX, Hung WY, Kato T, Asayama K. Intense superoxide dismutase-1 immunoreactivity in intracytoplasmic hyaline inclusions of familial amyotrophic lateral sclerosis with posterior column involvement. J Neuropathol Exp Neurol. 1996;55:481–490. doi: 10.1097/00005072-199604000-00011. [DOI] [PubMed] [Google Scholar]
- 145.Kato S, Sumi-Akamaru H, Fujimura H, Sakoda S, Kato M, Hirano A, Takikawa M, Ohama E. Copper chaperone for superoxide dismutase co-aggregates with superoxide dismutase 1 (SOD1) in neuronal Lewy body-like hyaline inclusions: an immunohistochemical study on familial amyotrophic lateral sclerosis with SOD1 gene mutation. Acta Neuropathol (Berl) 2001;102:233–238. doi: 10.1007/s004010000355. [DOI] [PubMed] [Google Scholar]
- 146.Chou SM, Wang HS, Komai K. Colocalization of NOS and SOD1 in neurofilament accumulation within motor neurons of amyotrophic lateral sclerosis: an immunohistochemical study. J Chem Neuroanat. 1996;10:249–258. doi: 10.1016/0891-0618(96)00137-8. [DOI] [PubMed] [Google Scholar]
- 147.Nicholls DG, Budd SL. Mitochondria and neuronal survival. Physiol Rev. 2000;80:315–360. doi: 10.1152/physrev.2000.80.1.315. [DOI] [PubMed] [Google Scholar]
- 148.Jaarsma D, Rognoni F, van Duijn W, Verspaget HW, Haasdijk ED, Holstege JC. CuZn superoxide dismutase (SOD1) accumulates in vacuolated mitochondria in transgenic mice expressing amyotrophic lateral sclerosis-linked SOD1 mutations. Acta Neuropathol (Berl) 2001;102:293–305. doi: 10.1007/s004010100399. [DOI] [PubMed] [Google Scholar]
- 149.Kong J, Xu Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J Neurosci. 1998;18:3241–3250. doi: 10.1523/JNEUROSCI.18-09-03241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ferri KF, Kroemer G. Mitochondria-the suicide organelles. BioEssays. 2001;23:111–115. doi: 10.1002/1521-1878(200102)23:2<111::AID-BIES1016>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 151.Pasinelli P, Borchelt DR, Houseweart MK, Cleveland DW, Brown RH., Jr Caspase-1 is activated in neural cells and tissue with amyotrophic lateral sclerosis-associated mutations in copper-zinc superoxide dismutase [published erratum appears in Proc Natl Acad Sci U S A 1999 Mar 16;96(6):3330] Proc Natl Acad Sci U S A. 1998;95:15763–15768. doi: 10.1073/pnas.95.26.15763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Rabizadeh S, Gralla EB, Borchelt DR, Gwinn R, Valentine JS, Sisodia S, Wong P, Lee M, Hahn H, Bredesen DE. Mutations associated with amyotrophic lateral sclerosis convert superoxide dismutase from an antiapoptotic gene to a proapoptotic gene: studies in yeast and neural cells. Proc Natl Acad Sci U S A. 1995;92:3024–3028. doi: 10.1073/pnas.92.7.3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Li M, Ona VO, Guegan C, Chen M, Jackson-Lewis V, Andrews LJ, Olszewski AJ, Stieg PE, Lee JP, Przedborski S, Friedlander RM. Functional role of caspase-1 and caspase-3 in an ALS transgenic mouse model. Science. 2000;288:335–339. doi: 10.1126/science.288.5464.335. [DOI] [PubMed] [Google Scholar]
- 154.Pasinelli P, Houseweart MK, Brown RH, Jr, Cleveland DW. Caspase-1 and-3 are sequentially activated in motor neuron death in Cu,Zn superoxide dismutase-mediated familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2000;97:13901–13906. doi: 10.1073/pnas.240305897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Polymenidou M, Cleveland DW. Motor neuron disease: The curious ways of ALS. Nature. 2008;454:284–285. doi: 10.1038/454284a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Nishitoh H, Kadowaki H, Nagai A, Maruyama T, Yokota T, Fukutomi H, Noguchi T, Matsuzawa A, Takeda K, Ichijo H. ALS-linked mutant SOD1 induces ER stress-and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev. 2008;22:1451–1464. doi: 10.1101/gad.1640108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Harraz MM, Marden JJ, Zhou W, Zhang Y, Williams A, Sharov VS, Nelson K, Luo M, Paulson H, Schoneich C, Engelhardt JF. SOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS model. J Clin Invest. 2008;118:659–670. doi: 10.1172/JCI34060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lilley BN, Ploegh HL. A membrane protein required for dislocation of misfolded proteins from the ER. Nature. 2004;429:834–840. doi: 10.1038/nature02592. [DOI] [PubMed] [Google Scholar]
- 159.Babior BM. NADPH oxidase. Curr Opin Immunol. 2004;16:42–47. doi: 10.1016/j.coi.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 160.Urushitani M, Sik A, Sakurai T, Nukina N, Takahashi R, Julien JP. Chromogranin-mediated secretion of mutant superoxide dismutase proteins linked to amyotrophic lateral sclerosis. Nat Neurosci. 2006;9:108–118. doi: 10.1038/nn1603. [DOI] [PubMed] [Google Scholar]
- 161.Wang J, Farr GW, Zeiss CJ, Rodriguez-Gil DJ, Wilson JH, Furtak K, Rutkowski DT, Kaufman RJ, Ruse CI, Yates JR, 3rd, Perrin S, Feany MB, Horwich AL. Progressive aggregation despite chaperone associations of a mutant SOD1-YFP in transgenic mice that develop ALS. Proc Natl Acad Sci U S A. 2009;106:1392–1397. doi: 10.1073/pnas.0813045106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kieran D, Hafezparast M, Bohnert S, Dick JR, Martin J, Schiavo G, Fisher EM, Greensmith L. A mutation in dynein rescues axonal transport defects and extends the life span of ALS mice. J Cell Biol. 2005;169:561–567. doi: 10.1083/jcb.200501085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Zhang F, Strom AL, Fukada K, Lee S, Hayward LJ, Zhu H. Interaction between familial amyotrophic lateral sclerosis (ALS)-linked SOD1 mutants and the dynein complex. J Biol Chem. 2007;282:16691–16699. doi: 10.1074/jbc.M609743200. [DOI] [PubMed] [Google Scholar]
- 164.Tubbs JL, Latypov V, Kanugula S, Butt A, Melikishvili M, Kraehenbuehl R, Fleck O, Marriott A, Watson AJ, Verbeek B, McGown G, Thorncroft M, Santibanez-Koref MF, Millington C, Arvai AS, Kroeger MD, Peterson LA, Williams DM, Fried MG, Margison GP, Pegg AE, Tainer JA. Flipping of alkylated DNA damage bridges base and nucleotide excision repair. Nature. 2009;459:808–813. doi: 10.1038/nature08076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Getzoff ED, Tainer JA, Olson AJ. Recognition and interactions controlling the assemblies of beta barrel domains. Biophys J. 1986;49:191–206. doi: 10.1016/S0006-3495(86)83634-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Hura GL, Menon AL, Hammel M, Rambo RP, Poole FL, 2nd, Tsutakawa SE, Jenney FE, Jr, Classen S, Frankel KA, Hopkins RC, Yang SJ, Scott JW, Dillard BD, Adams MW, Tainer JA. Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS) Nat Methods. 2009;6:606–612. doi: 10.1038/nmeth.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Putnam CD, Hammel M, Hura GL, Tainer JA. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q Rev Biophys. 2007;40:191–285. doi: 10.1017/S0033583507004635. [DOI] [PubMed] [Google Scholar]
- 168.Teilum K, Smith MH, Schulz E, Christensen LC, Solomentsev G, Oliveberg M, Akke M. Transient structural distortion of metal-free Cu/Zn superoxide dismutase triggers aberrant oligomerization. Proc Natl Acad Sci U S A. 2009;106:18273–18278. doi: 10.1073/pnas.0907387106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Garcin ED, Arvai AS, Rosenfeld RJ, Kroeger MD, Crane BR, Andersson G, Andrews G, Hamley PJ, Mallinder PR, Nicholls DJ, St-Gallay SA, Tinker AC, Gensmantel NP, Mete A, Cheshire DR, Connolly S, Stuehr DJ, Aberg A, Wallace AV, Tainer JA, Getzoff ED. Anchored plasticity opens doors for selective inhibitor design in nitric oxide synthase. Nat Chem Biol. 2008;4:700–707. doi: 10.1038/nchembio.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Lepock JR, Arnold LD, Torrie BH, Andrews B, Kruuv J. Structural analyses of various Cu2+, Zn2+-superoxide dismutases by differential scanning calorimetry and Raman spectroscopy. Arch Biochem Biophys. 1985;241:243–251. doi: 10.1016/0003-9861(85)90380-7. [DOI] [PubMed] [Google Scholar]
- 171.Roe JA, Butler A, Scholler DM, Valentine JS, Marky L, Breslauer KJ. Differential scanning calorimetry of Cu,Zn-superoxide dismutase, the apoprotein, and its zinc-substituted derivatives. Biochemistry. 1988;27:950–958. doi: 10.1021/bi00403a017. [DOI] [PubMed] [Google Scholar]
- 172.Malinowski DP, Fridovich I. Subunit association and side-chain reactivities of bovine erythrocyte superoxide dismutase in denaturing solvents. Biochemistry. 1979;18:5055–5060. doi: 10.1021/bi00590a005. [DOI] [PubMed] [Google Scholar]
- 173.Forman HJ, Fridovich I. On the stability of bovine superoxide dismutase. The effects of metals. J Biol Chem. 1973;248:2645–2649. [PubMed] [Google Scholar]
- 174.Richardson JS, Richardson DC, Thomas KA, Silverton EW, Davies DR. Similarity of three-dimensional structure between the immunoglobulin domain and the copper, zinc superoxide dismutase subunit. J Mol Biol. 1976;102:221–235. doi: 10.1016/s0022-2836(76)80050-2. [DOI] [PubMed] [Google Scholar]
- 175.Bhattacharyya R, Chakrabarti P. Stereospecific interactions of proline residues in protein structures and complexes. J Mol Biol. 2003;331:925–940. doi: 10.1016/s0022-2836(03)00759-9. [DOI] [PubMed] [Google Scholar]
- 176.McRee DE, Redford SM, Getzoff ED, Lepock JR, Hallewell RA, Tainer JA. Changes in crystallographic structure and thermostability of a Cu,Zn superoxide dismutase mutant resulting from the removal of a buried cysteine. J Biol Chem. 1990;265:14234–14241. doi: 10.2210/pdb3sod/pdb. [DOI] [PubMed] [Google Scholar]
- 177.Cardoso RM, Thayer MM, DiDonato M, Lo TP, Bruns CK, Getzoff ED, Tainer JA. Insights into Lou Gehrig’s disease from the structure and instability of the A4V mutant of human Cu,Zn superoxide dismutase. J Mol Biol. 2002;324:247–256. doi: 10.1016/s0022-2836(02)01090-2. [DOI] [PubMed] [Google Scholar]
- 178.Khare SD, Dokholyan NV. Common dynamical signatures of familial amyotrophic lateral sclerosis-associated structurally diverse Cu, Zn superoxide dismutase mutants. Proc Natl Acad Sci U S A. 2006;103:3147–3152. doi: 10.1073/pnas.0511266103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Lepock JR, Frey HE, Hallewell RA. Contribution of conformational stability and reversibility of unfolding to the increased thermostability of human and bovine superoxide dismutase mutated at free cysteines. J Biol Chem. 1990;265:21612–21618. [PubMed] [Google Scholar]
- 180.Chattopadhyay M, Durazo A, Sohn SH, Strong CD, Gralla EB, Whitelegge JP, Valentine JS. Initiation and elongation in fibrillation of ALS-linked superoxide dismutase. Proc Natl Acad Sci U S A. 2008;105:18663–18668. doi: 10.1073/pnas.0807058105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Furukawa Y, Kaneko K, Yamanaka K, O’Halloran TV, Nukina N. Complete loss of post-translational modifications triggers fibrillar aggregation of SOD1 in the familial form of amyotrophic lateral sclerosis. J Biol Chem. 2008;283:24167–24176. doi: 10.1074/jbc.M802083200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Hart PJ, Liu H, Pellegrini M, Nersissian AM, Gralla EB, Valentine JS, Eisenberg D. Subunit asymmetry in the three-dimensional structure of a human CuZnSOD mutant found in familial amyotrophic lateral sclerosis. Protein Sci. 1998;7:545–555. doi: 10.1002/pro.5560070302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Jonsson PA, Graffmo KS, Andersen PM, Brannstrom T, Lindberg M, Oliveberg M, Marklund SL. Disulphide-reduced superoxide dismutase-1 in CNS of transgenic amyotrophic lateral sclerosis models. Brain. 2006;129:451–464. doi: 10.1093/brain/awh704. [DOI] [PubMed] [Google Scholar]
- 184.Zetterstrom P, Stewart HG, Bergemalm D, Jonsson PA, Graffmo KS, Andersen PM, Brannstrom T, Oliveberg M, Marklund SL. Soluble misfolded subfractions of mutant superoxide dismutase-1s are enriched in spinal cords throughout life in murine ALS models. Proc Natl Acad Sci U S A. 2007;104:14157–14162. doi: 10.1073/pnas.0700477104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Pellequer JL, Chen S, Roberts VA, Tainer JA, Getzoff ED. Unraveling the effect of changes in conformation and compactness at the antibody V(L)-V(H) interface upon antigen binding. J Mol Recognit. 1999;12:267–275. doi: 10.1002/(SICI)1099-1352(199907/08)12:4<267::AID-JMR465>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 186.Juneja T, Pericak-Vance MA, Laing NG, Dave S, Siddique T. Prognosis in familial amyotrophic lateral sclerosis: progression and survival in patients with Glu100Gly and Ala4Val mutations in Cu,Zn superoxide dismutase. Neurology. 1997;48:55–57. doi: 10.1212/wnl.48.1.55. [DOI] [PubMed] [Google Scholar]
- 187.Lynch M, Kuramitsu H. Expression and role of superoxide dismutases (SOD) in pathogenic bacteria. Microbes Infect. 2000;2:1245–1255. doi: 10.1016/s1286-4579(00)01278-8. [DOI] [PubMed] [Google Scholar]
- 188.Richter C, Park J, Ames BN. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc Natl Acad Sci U S A. 1988;85:6465–6467. doi: 10.1073/pnas.85.17.6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, Wallace DC, Epstein CJ. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet. 1995;11:376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- 190.Melov S, Coskun P, Patel M, Tuinstra R, Cottrell B, Jun AS, Zastawny TH, Dizdaroglu M, Goodman SI, Huang TT, Miziorko H, Epstein CJ, Wallace DC. Mitochondrial disease in superoxide dismutase 2 mutant mice. Proc Natl Acad Sci U S A. 1999;96:846–851. doi: 10.1073/pnas.96.3.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Melov S, Schneider JA, Day BJ, Hinerfeld D, Coskun P, Mirra SS, Crapo JD, Wallace DC. A novel neurological phenotype in mice lacking mitochondrial manganese superoxide dismutase. Nat Genet. 1998;18:159–163. doi: 10.1038/ng0298-159. [DOI] [PubMed] [Google Scholar]
- 192.Melov S, Doctrow SR, Schneider JA, Haberson J, Patel M, Coskun PE, Huffman K, Wallace DC, Malfroy B. Lifespan extension and rescue of spongiform encephalopathy in superoxide dismutase 2 nullizygous mice treated with superoxide dismutase-catalase mimetics. J Neurosci. 2001;21:8348–8353. doi: 10.1523/JNEUROSCI.21-21-08348.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, Wallace DC, Rabinovitch PS. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308:1909–1911. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- 194.Parker MW, Blake CC. Crystal structure of manganese superoxide dismutase from Bacillus stearothermophilus at 2.4 A resolution. J Mol Biol. 1988;199:649–661. doi: 10.1016/0022-2836(88)90308-7. [DOI] [PubMed] [Google Scholar]
- 195.Carlioz A, Ludwig ML, Stallings WC, Fee JA, Steinman HM, Touati D. Iron superoxide dismutase. Nucleotide sequence of the gene from Escherichia coli K12 and correlations with crystal structures. J Biol Chem. 1988;263:1555–1562. [PubMed] [Google Scholar]
- 196.Ludwig ML, Metzger AL, Pattridge KA, Stallings WC. Manganese superoxide dismutase from Thermus thermophilus. A structural model refined at 1.8 A resolution. J Mol Biol. 1991;219:335–358. doi: 10.1016/0022-2836(91)90569-r. [DOI] [PubMed] [Google Scholar]
- 197.Stoddard BL, Howell PL, Ringe D, Petsko GA. The 2.1-A resolution structure of iron superoxide dismutase from Pseudomonas ovalis. Biochemistry. 1990;29:8885–8893. doi: 10.1021/bi00490a002. [DOI] [PubMed] [Google Scholar]
- 198.Borgstahl GE, Parge HE, Hickey MJ, Beyer WF, Jr, Hallewell RA, Tainer JA. The structure of human mitochondrial manganese superoxide dismutase reveals a novel tetrameric interface of two 4-helix bundles. Cell. 1992;71:107–118. doi: 10.1016/0092-8674(92)90270-m. [DOI] [PubMed] [Google Scholar]
- 199.Lah MS, Dixon MM, Pattridge KA, Stallings WC, Fee JA, Ludwig ML. Structure-function in Escherichia coli iron superoxide dismutase: comparisons with the manganese enzyme from Thermus thermophilus. Biochemistry. 1995;34:1646–1660. doi: 10.1021/bi00005a021. [DOI] [PubMed] [Google Scholar]
- 200.Wagner UG, Pattridge KA, Ludwig ML, Stallings WC, Werber MM, Oefner C, Frolow F, Sussman JL. Comparison of the crystal structures of genetically engineered human manganese superoxide dismutase and manganese superoxide dismutase from Thermus thermophilus: differences in dimer-dimer interaction. Protein Sci. 1993;2:814–825. doi: 10.1002/pro.5560020511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Borgstahl GE, Parge HE, Hickey MJ, Johnson MJ, Boissinot M, Hallewell RA, Lepock JR, Cabelli DE, Tainer JA. Human mitochondrial manganese super-oxide dismutase polymorphic variant Ile58Thr reduces activity by destabilizing the tetrameric interface. Biochemistry. 1996;35:4287–4297. doi: 10.1021/bi951892w. [DOI] [PubMed] [Google Scholar]
- 202.Nomiyama T, Tanaka Y, Piao L, Nagasaka K, Sakai K, Ogihara T, Nakajima K, Watada H, Kawamori R. The polymorphism of manganese superoxide dismutase is associated with diabetic nephropathy in Japanese type 2 diabetic patients. J Hum Genet. 2003;48:138–141. doi: 10.1007/s100380300021. [DOI] [PubMed] [Google Scholar]
- 203.Mollsten A, Marklund SL, Wessman M, Svensson M, Forsblom C, Parkkonen M, Brismar K, Groop PH, Dahlquist G. A functional polymorphism in the manganese superoxide dismutase gene and diabetic nephropathy. Diabetes. 2007;56:265–269. doi: 10.2337/db06-0698. [DOI] [PubMed] [Google Scholar]
- 204.Liu L, Zheng T, Wang N, Wang F, Li M, Jiang J, Zhao R, Li L, Zhao W, Zhu Q, Jia W. The manganese superoxide dismutase Val16Ala polymorphism is associated with decreased risk of diabetic nephropathy in Chinese patients with type 2 diabetes. Mol Cell Biochem. 2009;322:87–91. doi: 10.1007/s11010-008-9943-x. [DOI] [PubMed] [Google Scholar]
- 205.Wang S, Wang F, Shi X, Dai J, Peng Y, Guo X, Wang X, Shen H, Hu Z. Association between manganese superoxide dismutase (MnSOD) Val-9Ala polymorphism and cancer risk — a meta-analysis. Eur J Cancer. 2009;48(16):2874–2881. doi: 10.1016/j.ejca.2009.04.024. [DOI] [PubMed] [Google Scholar]
- 206.Iguchi T, Sugita S, Wang CY, Newman NB, Nakatani T, Haas GP. MnSOD genotype and prostate cancer risk as a function of NAT genotype and smoking status. In Vivo. 2009;23:7–12. [PMC free article] [PubMed] [Google Scholar]
- 207.Zejnilovic J, Akev N, Yilmaz H, Isbir T. Association between manganese superoxide dismutase polymorphism and risk of lung cancer. Cancer Genet Cytogenet. 2009;189:1–4. doi: 10.1016/j.cancergencyto.2008.06.017. [DOI] [PubMed] [Google Scholar]
- 208.Pae CU. Additive effect between quinine oxidoreductase gene (NQO1: Pro187Ser) and manganese superoxide dismutase gene (MnSOD: Ala-9Val) polymorphisms on tardive dyskinesia in patients with schizophrenia. Psychiatry Res. 2008;161:336–338. doi: 10.1016/j.psychres.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 209.Mikhak B, Hunter DJ, Spiegelman D, Platz EA, Wu K, Erdman JW, Jr, Giovannucci E. Manganese superoxide dismutase (MnSOD) gene polymorphism, interactions with carotenoid levels and prostate cancer risk. Carcinogenesis. 2008;29:2335–2340. doi: 10.1093/carcin/bgn212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Arsova-Sarafinovska Z, Matevska N, Petrovski D, Banev S, Dzikova S, Georgiev V, Sikole A, Sayal A, Aydin A, Suturkova L, Dimovski AJ. Manganese superoxide dismutase (MnSOD) genetic polymorphism is associated with risk of early-onset prostate cancer. Cell Biochem Funct. 2008;26:771–777. doi: 10.1002/cbf.1504. [DOI] [PubMed] [Google Scholar]
- 211.Bica CG, de Moura da Silva LL, Toscani NV, da Cruz IB, Sa G, Graudenz MS, Zettler CG. MnSOD gene polymorphism association with steroid-dependent cancer. Pathol Oncol Res. 2009;15:19–24. doi: 10.1007/s12253-008-9064-6. [DOI] [PubMed] [Google Scholar]
- 212.Sines J, Allison S, Wierzbicki A, McCammon JA. Brownian dynamics simulation of the superoxide-superoxide dismutase reaction: iron and manganse enzymes. J Phys Chem. 1990;94:959–961. [Google Scholar]
- 213.Leveque VJ, Vance CK, Nick HS, Silverman DN. Redox properties of human manganese superoxide dismutase and active-site mutants. Biochemistry. 2001;40:10586–10591. doi: 10.1021/bi010792p. [DOI] [PubMed] [Google Scholar]
- 214.Tierney DL, Fee JA, Ludwig ML, Penner-Hahn JE. X-ray absorption spectroscopy of the iron site in Escherichia coli Fe(III) superoxide dismutase. Biochemistry. 1995;34:1661–1668. doi: 10.1021/bi00005a022. [DOI] [PubMed] [Google Scholar]
- 215.Whittaker MM, Whittaker JW. Low-temperature thermochromism marks a change in coordination for the metal ion in manganese superoxide dismutase. Biochemistry. 1996;35:6762–6770. doi: 10.1021/bi960088m. [DOI] [PubMed] [Google Scholar]
- 216.Fee JA, McClune GJ, O’Neill P, Fielden EM. Saturation behavior of superoxide dismutation catalyzed by the iron containing superoxide dismutase of E. coli B. Biochem Biophys Res Commun. 1981;100:377–384. doi: 10.1016/s0006-291x(81)80107-6. [DOI] [PubMed] [Google Scholar]
- 217.Benovic J, Tillman T, Cudd A, Fridovich I. Electrostatic facilitation of the reaction catalyzed by the manganese-containing and the iron-containing superoxide dismutases. Arch Biochem Biophys. 1983;221:329–332. doi: 10.1016/0003-9861(83)90151-0. [DOI] [PubMed] [Google Scholar]
- 218.Miller AF, Sorkin DL, Padmakumar K. Anion binding properties of reduced and oxidized iron-containing superoxide dismutase reveal no requirement for tyrosine 34. Biochemistry. 2005;44:5969–5981. doi: 10.1021/bi0476331. [DOI] [PubMed] [Google Scholar]
- 219.Bull CN, Yoshida T, Fee JA. Kinetic-studies of superoxide dismutasese: properties of the manganese-containing protein from Thermus-thermophilus. J Am Chem Soc. 1991;113:4069–4076. [Google Scholar]
- 220.Jackson TA, Karapetian A, Miller AF, Brunold TC. Probing the geometric and electronic structures of the low-temperature azide adduct and the product-inhibited form of oxidized manganese superoxide dismutase. Biochemistry. 2005;44:1504–1520. doi: 10.1021/bi048639t. [DOI] [PubMed] [Google Scholar]
- 221.Silverman DN, Nick HS. Catalytic pathway of manganese superoxide dismutase by direct observation of superoxide. Methods Enzymol. 2002;349:61–74. doi: 10.1016/s0076-6879(02)49321-4. [DOI] [PubMed] [Google Scholar]
- 222.Guan Y, Hickey MJ, Borgstahl GE, Hallewell RA, Lepock JR, O’Connor D, Hsieh Y, Nick HS, Silverman DN, Tainer JA. Crystal structure of Y34F mutant human mitochondrial manganese superoxide dismutase and the functional role of tyrosine 34. Biochemistry. 1998;37:4722–4730. doi: 10.1021/bi972394l. [DOI] [PubMed] [Google Scholar]
- 223.Leveque VJ, Stroupe ME, Lepock JR, Cabelli DE, Tainer JA, Nick HS, Silverman DN. Multiple replacements of glutamine 143 in human manganese superoxide dismutase: effects on structure, stability, and catalysis. Biochemistry. 2000;39:7131–7137. doi: 10.1021/bi9929958. [DOI] [PubMed] [Google Scholar]
- 224.Hearn AS, Stroupe ME, Cabelli DE, Ramilo CA, Luba JP, Tainer JA, Nick HS, Silverman DN. Catalytic and structural effects of amino acid substitution at histidine 30 in human manganese superoxide dismutase: insertion of valine C gamma into the substrate access channel. Biochemistry. 2003;42:2781–2789. doi: 10.1021/bi0266481. [DOI] [PubMed] [Google Scholar]
- 225.Hearn AS, Fan L, Lepock JR, Luba JP, Greenleaf WB, Cabelli DE, Tainer JA, Nick HS, Silverman DN. Amino acid substitution at the dimeric interface of human manganese superoxide dismutase. J Biol Chem. 2004;279:5861–5866. doi: 10.1074/jbc.M311310200. [DOI] [PubMed] [Google Scholar]
- 226.Greenleaf WB, Perry JJ, Hearn AS, Cabelli DE, Lepock JR, Stroupe ME, Tainer JA, Nick HS, Silverman DN. Role of hydrogen bonding in the active site of human manganese superoxide dismutase. Biochemistry. 2004;43:7038–7045. doi: 10.1021/bi049888k. [DOI] [PubMed] [Google Scholar]
- 227.Ayala I, Perry JJ, Szczepanski J, Tainer JA, Vala MT, Nick HS, Silverman DN. Hydrogen bonding in human manganese superoxide dismutase containing 3-fluorotyrosine. Biophys J. 2005;89:4171–4179. doi: 10.1529/biophysj.105.060616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Quint P, Reutzel R, Mikulski R, McKenna R, Silverman DN. Crystal structure of nitrated human manganese superoxide dismutase: mechanism of inactivation. Free Radic Biol Med. 2006;40:453–458. doi: 10.1016/j.freeradbiomed.2005.08.045. [DOI] [PubMed] [Google Scholar]
- 229.Ren X, Tu C, Bhatt D, Perry JJP, Tainer JA, Cabelli DE, Silverman DN. Kinetic and structural characterization of human manganese superoxide dismutase containing 3-fluorotyrosines. J Mol Struct. 2006;790:168–173. [Google Scholar]
- 230.Cabelli DE, Guan Y, Leveque V, Hearn AS, Tainer JA, Nick HS, Silverman DN. Role of tryptophan 161 in catalysis by human manganese superoxide dismutase. Biochemistry. 1999;38:11686–11692. doi: 10.1021/bi9909142. [DOI] [PubMed] [Google Scholar]
- 231.Perry JJ, Hearn AS, Cabelli DE, Nick HS, Tainer JA, Silverman DN. Contribution of human manganese superoxide dismutase tyrosine 34 to structure and catalysis. Biochemistry. 2009;48:3417–3424. doi: 10.1021/bi8023288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Whittaker JW. Prokaryotic manganese superoxide dismutases. Methods Enzymol. 2002;349:80–90. doi: 10.1016/s0076-6879(02)49323-8. [DOI] [PubMed] [Google Scholar]
- 233.Parker MW, Blake CC. Iron-and manganese-containing superoxide dismutases can be distinguished by analysis of their primary structures. FEBS Lett. 1988;229:377–382. doi: 10.1016/0014-5793(88)81160-8. [DOI] [PubMed] [Google Scholar]
- 234.Parker MW, Blake CC, Barra D, Bossa F, Schinina ME, Bannister WH, Bannister JV. Structural identity between the iron-and manganese-containing superoxide dismutases. Protein Eng. 1987;1:393–397. doi: 10.1093/protein/1.5.393. [DOI] [PubMed] [Google Scholar]
- 235.Yamakura F, Kobayashi K, Ue H, Konno M. The pH-dependent changes of the enzymic activity and spectroscopic properties of iron-substituted manganese superoxide dismutase. A study on the metal-specific activity of Mn-containing superoxide dismutase. Eur J Biochem. 1995;227:700–706. doi: 10.1111/j.1432-1033.1995.tb20191.x. [DOI] [PubMed] [Google Scholar]
- 236.Whittaker MM, Whittaker JW. Mutagenesis of a proton linkage pathway in Escherichia coli manganese superoxide dismutase. Biochemistry. 1997;36:8923–8931. doi: 10.1021/bi9704212. [DOI] [PubMed] [Google Scholar]
- 237.Edwards RA, Whittaker MM, Whittaker JW, Jameson GB, Baker EN. Distinct metal environment in Fe-substituted manganese superoxide dismutase provides a structural basis of metal specificity. J Am Chem Soc. 1998;120:9684–9685. [Google Scholar]
- 238.Vance CK, Miller AF. Spectroscopic comparisons of the pH dependencies of Fe-substituted (Mn)superoxide dismutase and Fe-superoxide dismutase. Biochemistry. 1998;37:5518–5527. doi: 10.1021/bi972580r. [DOI] [PubMed] [Google Scholar]
- 239.Martin ME, Byers BR, Olson MO, Salin ML, Arceneaux JE, Tolbert C. A Streptococcus mutans superoxide dismutase that is active with either manganese or iron as a cofactor. J Biol Chem. 1986;261:9361–9367. [PubMed] [Google Scholar]
- 240.Sugio S, Hiraoka BY, Yamakura F. Crystal structure of cambialistic superoxide dismutase from porphyromonas gingivalis. Eur J Biochem. 2000;267:3487–3495. doi: 10.1046/j.1432-1327.2000.01373.x. [DOI] [PubMed] [Google Scholar]
- 241.Hsieh Y, Guan Y, Tu C, Bratt PJ, Angerhofer A, Lepock JR, Hickey MJ, Tainer JA, Nick HS, Silverman DN. Probing the active site of human manganese superoxide dismutase: the role of glutamine 143. Biochemistry. 1998;37:4731–4739. doi: 10.1021/bi972395d. [DOI] [PubMed] [Google Scholar]
- 242.Chockalingam K, Luba J, Nick HS, Silverman DN, Zhao H. Engineering and characterization of human manganese superoxide dismutase mutants with high activity and low product inhibition. FEBS J. 2006;273:4853–4861. doi: 10.1111/j.1742-4658.2006.05484.x. [DOI] [PubMed] [Google Scholar]
- 243.Davis CA, Hearn AS, Fletcher B, Bickford J, Garcia JE, Leveque V, Melendez JA, Silverman DN, Zucali J, Agarwal A, Nick HS. Potent anti-tumor effects of an active site mutant of human manganese-superoxide dismutase. Evolutionary conservation of product inhibition. J Biol Chem. 2004;279:12769–12776. doi: 10.1074/jbc.M310623200. [DOI] [PubMed] [Google Scholar]
- 244.Szabo C, Ischiropoulos H, Radi R. Peroxynitrite: biochemistry pathophysiology and development of therapeutics. Nat Rev Drug Discovery. 2007;6:662–680. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- 245.Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Peluffo G, Radi R. Biochemistry of protein tyrosine nitration in cardiovascular pathology. Cardiovasc Res. 2007;75:291–302. doi: 10.1016/j.cardiores.2007.04.024. [DOI] [PubMed] [Google Scholar]
- 247.Youn HD, Kim EJ, Roe JH, Hah YC, Kang SO. A novel nickel-containing superoxide dismutase from Streptomyces spp. Biochem J. 1996;318(Pt. 3):889–896. doi: 10.1042/bj3180889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Youn HD, Youn H, Lee JW, Yim YI, Lee JK, Hah YC, Kang SO. Unique isozymes of superoxide dismutase in Streptomyces griseus. Arch Biochem Biophys. 1996;334:341–348. doi: 10.1006/abbi.1996.0463. [DOI] [PubMed] [Google Scholar]
- 249.Palenik B, Brahamsha B, Larimer FW, Land M, Hauser L, Chain P, Lamerdin J, Regala W, Allen EE, McCarren J, Paulsen I, Dufresne A, Partensky F, Webb EA, Waterbury J. The genome of a motile marine Synechococcus. Nature. 2003;424:1037–1042. doi: 10.1038/nature01943. [DOI] [PubMed] [Google Scholar]
- 250.Schmidt A, Gube M, Kothe E. In silico analysis of nickel containing superoxide dismutase evolution and regulation. J Basic Microbiol. 2009;49:109–118. doi: 10.1002/jobm.200800293. [DOI] [PubMed] [Google Scholar]
- 251.Choudhury SB, Lee JW, Davidson G, Yim YI, Bose K, Sharma ML, Kang SO, Cabelli DE, Maroney MJ. Examination of the nickel site structure and reaction mechanism in Streptomyces seoulensis superoxide dismutase. Biochemistry. 1999;38:3744–3752. doi: 10.1021/bi982537j. [DOI] [PubMed] [Google Scholar]
- 252.Barondeau DP, Kassmann CJ, Bruns CK, Tainer JA, Getzoff ED. Nickel superoxide dismutase structure and mechanism. Biochemistry. 2004;43:8038–8047. doi: 10.1021/bi0496081. [DOI] [PubMed] [Google Scholar]
- 253.Wuerges J, Lee JW, Yim YI, Yim HS, Kang SO, Djinovic Carugo K. Crystal structure of nickel-containing superoxide dismutase reveals another type of active site. Proc Natl Acad Sci U S A. 2004;101:8569–8574. doi: 10.1073/pnas.0308514101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Kim EJ, Chung HJ, Suh B, Hah YC, Roe JH. Transcriptional and post-transcriptional regulation by nickel of sodN gene encoding nickel-containing superoxide dismutase from Streptomyces coelicolor Muller. Mol Microbiol. 1998;27:187–195. doi: 10.1046/j.1365-2958.1998.00674.x. [DOI] [PubMed] [Google Scholar]
- 255.Banci L, Bertini I, Cabelli DE, Hallewell RA, Luchinat C, Viezzoli MS. Advances in the understanding of the structure-function relationship in Cu,Zn superoxide dismutase. Free Radic Res Commun. 1991;12-13(Pt 1):239–251. doi: 10.3109/10715769109145792. [DOI] [PubMed] [Google Scholar]
- 256.Miller AF, Padmakumar K, Sorkin DL, Karapetian A, Vance CK. Proton-coupled electron transfer in Fe-superoxide dismutase and Mn-superoxide dismutase. J Inorg Biochem. 2003;93:71–83. doi: 10.1016/s0162-0134(02)00621-9. [DOI] [PubMed] [Google Scholar]
- 257.Maliekal J, Karapetian A, Vance C, Yikilmaz E, Wu Q, Jackson T, Brunold TC, Spiro TG, Miller AF. Comparison and contrasts between the active site PKs of Mn-superoxide dismutase and those of Fe-superoxide dismutase. J Am Chem Soc. 2002;124:15064–15075. doi: 10.1021/ja027319z. [DOI] [PubMed] [Google Scholar]
- 258.Hunter T, Ikebukuro K, Bannister WH, Bannister JV, Hunter GJ. The conserved residue tyrosine 34 is essential for maximal activity of iron-superoxide dismutase from Escherichia coli. Biochemistry. 1997;36:4925–4933. doi: 10.1021/bi9629541. [DOI] [PubMed] [Google Scholar]
- 259.Holm RH, Kennepohl P, Solomon EI. structural and functional aspects of metal sites in biology. Chem Rev. 1996;96:2239–2314. doi: 10.1021/cr9500390. [DOI] [PubMed] [Google Scholar]
- 260.Miller AF. Redox tuning over almost 1 V in a structurally conserved active site: lessons from Fe-containing superoxide dismutase. Acc Chem Res. 2008;41:501–510. doi: 10.1021/ar700237u. [DOI] [PubMed] [Google Scholar]
- 261.Maroney MJ. Structure/function relationships in nickel metallobiochemistry. Curr Opin Chem Biol. 1999;3:188–199. doi: 10.1016/S1367-5931(99)80032-5. [DOI] [PubMed] [Google Scholar]
- 262.Fiedler AT, Bryngelson PA, Maroney MJ, Brunold TC. Spectroscopic and computational studies of Ni superoxide dismutase: electronic structure contributions to enzymatic function. J Am Chem Soc. 2005;127:5449–5462. doi: 10.1021/ja042521i. [DOI] [PubMed] [Google Scholar]
- 263.Pelmenschikov V, Siegbahn PE. Nickel superoxide dismutase reaction mechanism studied by hybrid density functional methods. J Am Chem Soc. 2006;128:7466–7475. doi: 10.1021/ja053665f. [DOI] [PubMed] [Google Scholar]
- 264.Herbst RW, Guce A, Bryngelson PA, Higgins KA, Ryan KC, Cabelli DE, Garman SC, Maroney MJ. Role of conserved tyrosine residues in NiSOD catalysis: a case of convergent evolution. Biochemistry. 2009;48:3354–3369. doi: 10.1021/bi802029t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Tietze D, Breitzke H, Imhof D, Kothe E, Weston J, Buntkowsky G. New insight into the mode of action of nickel superoxide dismutase by investigating metallopeptide substrate models. Chemistry. 2009;15:517–523. doi: 10.1002/chem.200800870. [DOI] [PubMed] [Google Scholar]
- 266.Bannister WH, Bannister JV, Barra D, Bond J, Bossa F. Evolutionary aspects of superoxide dismutase: the copper/zinc enzyme. Free Radic Res Commun. 1991;12(Pt. 1):349–361. doi: 10.3109/10715769109145804. [DOI] [PubMed] [Google Scholar]
- 267.Harrigan JA, Piotrowski J, Di Noto L, Levine RL, Bohr VA. Metal-catalyzed oxidation of the Werner syndrome protein causes loss of catalytic activities and impaired protein-protein interactions. J Biol Chem. 2007;282:36403–36411. doi: 10.1074/jbc.M706107200. [DOI] [PubMed] [Google Scholar]
- 268.Perry JJ, Yannone SM, Holden LG, Hitomi C, Asaithamby A, Han S, Cooper PK, Chen DJ, Tainer JA. WRN exonuclease structure and molecular mechanism imply an editing role in DNA end processing. Nat Struct Mol Biol. 2006;13:414–422. doi: 10.1038/nsmb1088. [DOI] [PubMed] [Google Scholar]
- 269.Shen JC, Gray MD, Oshima J, Kamath-Loeb AS, Fry M, Loeb LA. Werner syndrome protein. I. DNA helicase and dna exonuclease reside on the same polypeptide. J Biol Chem. 1998;273:34139–34144. doi: 10.1074/jbc.273.51.34139. [DOI] [PubMed] [Google Scholar]
- 270.Bohr VA. Rising from the RecQ-age: the role of human RecQ helicases in genome maintenance. Trends Biochem Sci. 2008;33:609–620. doi: 10.1016/j.tibs.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Ozgenc A, Loeb LA. Werner Syndrome, aging and cancer. Genome Dyn. 2006;1:206–217. doi: 10.1159/000092509. [DOI] [PubMed] [Google Scholar]
- 272.Beckman JS, Chen J, Ischiropoulos H, Crow JP. Oxidative chemistry of peroxynitrite. Methods Enzymol. 1994;233:229–240. doi: 10.1016/s0076-6879(94)33026-3. [DOI] [PubMed] [Google Scholar]
- 273.Beckmann JS, Ye YZ, Anderson PG, Chen J, Accavitti MA, Tarpey MM, White CR. Extensive nitration of protein tyrosines in human atherosclerosis detected by immunohistochemistry. Biol Chem Hoppe Seyler. 1994;375:81–88. doi: 10.1515/bchm3.1994.375.2.81. [DOI] [PubMed] [Google Scholar]
- 274.Eiserich JP, Vossen V, O’Neill CA, Halliwell B, Cross CE, van der Vliet A. Molecular mechanisms of damage by excess nitrogen oxides: nitration of tyrosine by gas-phase cigarette smoke. FEBS Lett. 1994;353:53–56. doi: 10.1016/0014-5793(94)01011-0. [DOI] [PubMed] [Google Scholar]
- 275.Beckman JS, Chen J, Crow JP, Ye YZ. Reactions of nitric oxide, superoxide and peroxynitrite with superoxide dismutase in neurodegeneration. Prog Brain Res. 1994;103:371–380. doi: 10.1016/s0079-6123(08)61151-6. [DOI] [PubMed] [Google Scholar]
- 276.Esterbauer H. Cytotoxicity and genotoxicity of lipid-oxidation products. Am J Clin Nutr. 1993;57:779S–785S. doi: 10.1093/ajcn/57.5.779S. discussion 785S–786S. [DOI] [PubMed] [Google Scholar]
- 277.Harman D. Free radical theory of aging. Mutat Res. 1992;275:257–266. doi: 10.1016/0921-8734(92)90030-s. [DOI] [PubMed] [Google Scholar]
- 278.Shigenaga MK, Hagen TM, Ames BN. Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci U S A. 1994;91:10771–10778. doi: 10.1073/pnas.91.23.10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Fan L, Fuss JO, Cheng QJ, Arvai AS, Hammel M, Roberts VA, Cooper PK, Tainer JA. XPD helicase structures and activities: insights into the cancer and aging phenotypes from XPD mutations. Cell. 2008;133:789–800. doi: 10.1016/j.cell.2008.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Williams RS, Moncalian G, Williams JS, Yamada Y, Limbo O, Shin DS, Groocock LM, Cahill D, Hitomi C, Guenther G, Moiani D, Carney JP, Russell P, Tainer JA. Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell. 2008;135:97–109. doi: 10.1016/j.cell.2008.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Williams RS, Dodson GE, Limbo O, Yamada Y, Williams JS, Guenther G, Classen S, Glover JN, Iwasaki H, Russell P, Tainer JA. Nbs1 flexibly tethers Ctp1 and Mre11-Rad50 to coordinate DNA double-strand break processing and repair. Cell. 2009;139:87–99. doi: 10.1016/j.cell.2009.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Meiering EM. The threat of instability: neurodegeneration predicted by protein destabilization and aggregation propensity. PLoS Biol. 2008;6:e193. doi: 10.1371/journal.pbio.0060193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Chiti F, Stefani M, Taddei N, Ramponi G, Dobson CM. Rationalization of the effects of mutations on peptide and protein aggregation rates. Nature. 2003;424:805–808. doi: 10.1038/nature01891. [DOI] [PubMed] [Google Scholar]
- 284.Valentine JS, Doucette PA, Zittin Potter S. Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis. Annu Rev Biochem. 2005;74:563–593. doi: 10.1146/annurev.biochem.72.121801.161647. [DOI] [PubMed] [Google Scholar]
- 285.Zhong Z, Ilieva H, Hallagan L, Bell R, Singh I, Paquette N, Thiyagarajan M, Deane R, Fernandez JA, Lane S, Zlokovic AB, Liu T, Griffin JH, Chow N, Castellino FJ, Stojanovic K, Cleveland DW, Zlokovic BV. Activated protein C therapy slows ALS-like disease in mice by transcriptionally inhibiting SOD1 in motor neurons and microglia cells. J Clin Invest. 2009;119(11):3437–3449. doi: 10.1172/JCI38476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Oda T, Akaike T, Hamamoto T, Suzuki F, Hirano T, Maeda H. Oxygen radicals in influenza-induced pathogenesis and treatment with pyran polymer-conjugated SOD. Science. 1989;244:974–976. doi: 10.1126/science.2543070. [DOI] [PubMed] [Google Scholar]
- 287.Hallewell RA, Laria I, Tabrizi A, Carlin G, Getzoff ED, Tainer JA, Cousens LS, Mullenbach GT. Genetically engineered polymers of human CuZn superoxide dismutase. Biochemistry and serum half-lives. J Biol Chem. 1989;264:5260–5268. [PubMed] [Google Scholar]
- 288.Nelson SK, Bose SK, Grunwald GK, Myhill P, McCord JM. The induction of human superoxide dismutase and catalase in vivo: a fundamentally new approach to antioxidant therapy. Free Radic Biol Med. 2006;40:341–347. doi: 10.1016/j.freeradbiomed.2005.08.043. [DOI] [PubMed] [Google Scholar]
- 289.Fee JA. Is superoxide important in oxygen poisoning? Trends Biochem Sci. 1982;7:84–86. [Google Scholar]