

Abstract
The t(4;14) translocation in multiple myeloma (MM) simultaneously dysregulates two apparent oncogenes: fibroblast growth factor receptor 3 (FGFR3) controlled by the 3′ immunoglobulin heavy chain enhancer on der(14) and MMSET controlled by the intronic Eμ enhancer on der(4). Although all MM tumors and cell lines with a t(4;14) translocation have dysregulated MMSET, about 25% do not express FGFR3. Therefore, the function of dysregulated wild-type (WT) FGFR3 in the pathogenesis of MM remains unclear. We developed a murine transgenic (TG) model in which WT FGFR3 is over-expressed in B lymphoid cells. Although high levels of FGFR3 resulted in lymphoid hyperplasia in about one-third of older mice, no increase in tumorigenesis was observed. However, double TG FGFR3/Myc mice develop mature B lymphoma tumors that occur with a higher penetrance and shorter latency than in single TG Myc mice (P = 0.006). We conclude that expression of high levels of WT FGFR3 can be oncogenic and cooperate with MYC to generate B lymphoid tumors. This suggests that dysregulated FGFR3 expression is likely to be essential at least for the early stages of pathogenesis of MM tumors that have a t(4;14) translocation.
Keywords: FGFR3, Myc, lymphoma, multiple myeloma, oncogene
Introduction
Multiple myeloma (MM), a mostly incurable neoplasm of terminally differentiated B cells, is the second most common hematologic malignancy, with nearly 20 000 new diagnoses made annually in the United States.1 Translocation of oncogenes into the immunoglobulin heavy chain (IgH) locus at 14q32 seems to be an initiating event in about 40% of MM tumors.2–4 Among the various molecular subtypes of MM, the prognosis of the t(4,14)-positive subset is particularly unfavorable. 5–7 This translocation results in the simultaneous ectopic expression of fibroblast growth factor receptor 3 (FGFR3), encoded by FGFR3, and MMSET, a SET domain-containing protein encoded by MMSET (now officially renamed WHSCI).8
FGFR3 is one of the four high-affinity receptors for a large family of fibroblast growth factors. All FGF receptors share the same general structure in being comprised of three distinct domains: an extracellular domain, a transmembrane domain, and a tyrosine kinase domain. The extracellular domain contains the ligand-binding site and is characterized by two or three Ig-like motifs (IgD1-3). In addition, FGFR1, -2, and -3 have two major alternatively spliced isoforms, IIIb and IIIc. The IIIb isoform is expressed in epithelial cells, whereas the IIIc isoform is present in mesenchymal cells.9,10 Engagement of FGFR3 by ligand in concert with heparin sulfate proteoglycan results in dimerization and autophosphorylation at specific tyrosine residues. This initiates recruitment of adaptor proteins with subsequent activation of multiple signaling cascades, including the ERK/MAPK, PLCγ/PKC, PI3K, and STAT pathways.11–13 Consequently, FGFR3 and its ligand regulate many cellular processes, including proliferation, differentiation, and survival in a specific cell context-dependent manner.
Germline mutations of FGFR3 result in constitutive activation of the receptor in the absence of ligand. Aberrant signaling is associated with a broad spectrum of human skeletal dysplasias ranging from hypochronroplasia to achondroplasia to the more severe thanatophoric dysplasia, depending on the location of the mutation and the degree of receptor activation.14 The same mutations also have been identified in some MM cell lines and primary MM tumors that have t(4;14) translocations.15–17 Earlier studies showed that FGFR3 with activating mutations transforms fibroblasts in vitro, and hematopoietic cells in bone marrow transplantation models in vivo.18 Furthermore, expression of FGFR3 is dysregulated not only in MM, but also in other hematological malignancies, including some acute myeloid leukemia, B-cell and T-cell non-Hodgkin lymphomas, and some Hodgkin lymphoma.19 It also is dysregulated in a variety of solid tumors, such as glioblastoma and carcinomas of the bladder, prostate, and breast.20–22 However, although the effects down-stream from mutations of FGFR3 have been extensively studied, little is known about the effect of ectopic expression of wild-type (WT) FGFR3 in vivo, and specifically in MM.
The manner in which FGFR3 might be involved in the initiation of MM cases bearing the t(4,14) translocation is controversial. As ~25% of t(4;14)-positive tumors do not express FGFR3, some have hypothesized that FGFR3 may not be important in tumorigenesis.23 Others interpret this finding to suggest that dysregulated expression of FGFR3 is required for disease initiation, but not for progression.24 The development of a murine model of t(4;14) MM might help to discriminate between these alternative possibilities. The need for such a model is highlighted further by proposals that FGFR3 is a candidate for targeted therapeutic intervention in MM.25 Although several small molecule inhibitors targeting the tyrosine kinase domain of FGFR3, as well as specific short-hairpin RNAs and ribozymes, are cytotoxic for t(4;14) MM cell lines with mutated FGFR3, the function of unmutated FGFR3 in t(4;14) tumors is unclear.26–30
Here, we have developed a murine model in which a WT hFGFR3 transgene is over-expressed in B lymphoid cells. Although high levels of FGFR3 result in an increased prevalence of lymphoid hyperplasia in these mice, there is not a significant increase in the incidence of tumors. However, we show that FGFR3 cooperates with MYC in double transgenic (TG) mice to cause B lymphomas that occur with a higher penetrance and shorter latency than in single TG Myc mice.
Materials and methods
Expression construct and generation of FGFR3 TG mice
A 3.4 kb Hind III/XbaI fragment of WT hFGFR3 (IIIc isoform) complementary DNA (cDNA) was cloned into pBluescript KS, and the GC-rich segment of the 5′ untranslated region of FGFR3 was replaced with a PCR fragment containing a Kozak consensus sequence and a BamHI restriction site. The modified FGFR3 fragment was subsequently cloned into the BamHI restriction site of the p1026X vector (Figure 1).31 The NotI linearized construct was microinjected into FVB/N zygotes. TG founders were detected by Southern blots of BamH digested tail DNA. Founders were then crossed with WT FVB/N mice to generate stable lines of heterozyous FGFR3 TG mice. All mouse studies were performed under protocols approved by the Institutional Animal Care and Use Committees of NCI and NIAID.
Figure 1.

Expression of hFGFR3 in TG mice. (a) Schematic diagram of p1026X-hFGFR3 TG vector. The vector includes a 9.7 kb NotI fragment that contains 3.2 kb of Lck proximal promoter sequences (thick line), a 1 kb Eμ enhancer fragment (red box) inserted into these sequences, a 3.4 kb human FGFR3 cDNA (green box is coding) cloned into a BamHI site located 37 bp from the lck promoter, and a 2.1 kb human growth hormone (hGH) genomic fragment with five exons (boxes), and a polyadenylation site. FGFR3 and the fourth exon of hGH contain stop codons, as indicated by the asterisks. The spliced hFGFR3-hGH transcript is 4.4 kb. (b) Immunoblot of hFGFR3 in plasma cells. Plasma cells were generated in vitro by 5 days LPS stimulation of spleen cells from four independent F1 TG mice (founders H1, D3, A5, K1) and a non-TG littermate (WT). Proteins from total cell lysates were immunoprecipitated with a polyclonal anti-hFGFR3, fractionated by electrophoresis on a 7.5% polyacrylamide gel in SDS, transferred to nitrocellulose filters, and reacted with a monoclonal anti-hFGFR3 antibody. H929 MM cell line expresses FGFR3 as a result of a t(4;14) translocation. The FGFR3 doublet, which is present in the H929 myeloma cell line and TG splenocytes, probably reflects differential glycosylation as described for other tyrosine kinase receptors.26 (c–f) Expression of hFGFR3 in spleen cells by immunohistochemistry. Sections of normal spleens from hFGFR3 TG mice and normal littermates were immunostained with anti-hFGFR3 antibody (brown) and counterstained with Meyer’s hematoxylin (Ventana) (blue); hFGFR3 TG at × 100 (c) and × 400 magnification (d); non-TG littermate at × 100 (e) and × 400 magnification (f).
Generation of FGFR3/MYC double TG mice
Double TG mice were generated by breeding heterozygous FGFR3 TG mice with 129SvJ/C57BL/6 heterozyous Myc knock-in mice (iMyc).32 For survival studies, double TG (F+/M+), both genotypes of single TG (F+/M−, F−/M+), and WT (F−/M−) mice were aged until they developed tumors or showed discomfort, and then were necropsied. Statistical analysis was performed with Prism software (GraphPad Software Inc.).
Genotyping of TG mice
Genotyping of the mice was performed by PCR from tail DNA. For FGFR3 TG detection, we used primers and cycling condition as described below. Detection of the c-Myc TG was performed using the following primers: forward primer 5′ TCA CGC AGG GCA AAA AAG C 3′ and reverse primer 5′ GAC GAC GAG ACC TTC ATC AAG AAC 3′ complementary to the second and third exon, respectively.
Immunohistochemistry staining: histopathology and flow cytometric immunophenotyping
Spleen sections of TG FGFR3 and WT littermates were immunostained with a rabbit anti-hFGFR3 antibody sc-13121 (Santa Cruz Biothecnology, Santa Cruz, CA, USA). Detection of FGFR3 protein was performed according to standard procedures. Histological diagnoses were made using the criteria in the Bethesda proposals.33,34 Single cell suspensions prepared from bone marrow, spleen, and lymph nodes were subjected to Ficoll gradient separation to isolate mononuclear cells. Cells were stained with monoclonal antibodies specific for CD45R/B220, CD19, CD138, and CD3 (Pharmingen, BD Biosciences, San Diego, CA, USA). For each cell suspension, 10 000 events were collected with an FACScan cytometer (Cytek, CA, USA) and analyzed using Cell-Quest software.
Immunoprecipitation and immunoblot analysis
Plasma cells were induced in vitro by stimulating spleen cells with 30 μg/ml LPS for 5 days. Immunoprecipitates, obtained as elsewhere described,27 were fractionated by 7.5% SDS–PAGE and transferred to a Potran nitrocellulose membrane (Schleicher & Shuell, Keene, NH, USA). Membranes were probed with a monoclonal antibodies to human FGFR3 antibody (sc-B9; Santa Cruz Biotechology), and FGFR3 was detected using a horse-radish peroxidase-conjugated secondary Ab using the enhanced chemiluminescence kit (GE Healthcare, Pittsburgh, PA, USA). H929 MM cell line expressing FGFR3 from t(4;14) translocations was used as control.
RT–PCR analysis
Total RNA from tumor tissues was isolated using TRIZOL (Invitrogen, CA, USA). cDNA was synthesized using Superscript II (Nitrogen). FGFR3 transcripts from the TG were detected by PCR using a reverse primer (5′-AGC TGG TGC AGA CGA TGG GCG CGG AGC-3′) complementary to the second exon of human growth factor genomic sequence and a forward primer in the 3′ untranslated region of hFGFR3 IIIc (5′-TTG TTC CCA CAC CCC AAC ACT TCC-3′).
qRT–PCR
First-strand cDNA synthesis from total RNA was performed by using High Capacity cDNA RT kit (Applied Biosystems, Foster City, CA, USA). TaqMan probes Hs00231528_m1 USF2, Hs00997397_m1 FGFR3, Mm01152664_m1 USF2, and the TaqMan Fast Universal PCR Master Mix Reagents kit (Applied Biosystems) were used for experiments. The comparative CT method (ΔΔCT) was used for relative quantification of gene expression.
Southern blot analysis of tumor samples for detection of clonotypic VDJ rearrangement
Genomic DNA obtained from tumor tissues was digested with EcoRI for analyses of IgH gene organization, and EcoRI plus BamHI for analysis of Igκ light chain organization, fractionated by electrophoresis on 0.7% agarose gels, transferred onto a nylon filter, and hybridized to a [32P] dCTP-labeled 1.2-kb HindII/EcoRI IgH probe spanning JH3 and Eμ or a 1.1 Kb Ck probe as described earlier.32
Generation and culture of tumor cell lines
Tumors cells were obtained from bone marrow by flushing both femurs with PBS, and by mechanical disruption of spleens and lymph nodes. Red cells were removed by Ficoll gradient separation. Cells were then plated at a cell density of 5 × 105 cells/ml in RPMI medium supplemented with 10% fetal bovine serum, 100 μg/ml penicillin, 100 μg/ml streptomycin (Nitrogen), 30 ng/ml aFGF (R&D Systems, MN, USA), and 100 μg/ml heparin (Sigma, MO, USA). Established cell lines were maintained in culture in the presence and absence of ligand.
Detection of FGFR3 mutations
The FGFR3 tyrosine kinase domain was amplified by PCR using primers and cycling conditions described elsewhere,15 and then directly sequenced.
Microarray analyses
Amplified RNAs from six tumors of F+/M+ mice, seven tumors of F−/M+ mice, and reference RNA (Universal Mouse Reference RNA, Stratagene, Cedar Creek, TX, USA) were labeled, respectively, with Cy3 and Cy5 (Quick-Amp labeling kit, Agilent Technologies, Santa Clara, CA, USA). Hybridizations were performed using a mouse NIAID array comprising ~36 000 genes. Image analysis and normalization were performed using Feature Extraction Software (Agilent Technologies). Differentially expressed genes in tumors of F+/M+ mice compared with tumors of F−/M+ mice and cell lines were identified with SAM.35 Subsequently, hierarchical clustering was applied with Genesis.36 Myc expression levels were obtained from 559 nearly diagnosed, untreated tumors (GSE 2658) plus 22 healthy plasma cells and 44 MGUS (GSE 5900).37,38
Results
TG murine model recapitulates overexpression of FGFR3 present in human MM cell lines
We cloned hFGFR3 cDNA into the p1026X vector containing a Lck promoter, the Eμ intronic enhancer and the human growth hormone gene (Figure 1), as this vector had been used by others to achieve high-level expression of a TG in B and T lymphocytes.39,40 We obtained six founder mice on the FVB/N background, which were bred with WT mice from the same strain to obtain F1 heterozygous FGFR3 TG mice. Spleen cells from F1 progeny mice were stimulated in vitro with LPS to drive plasma cell differentiation and total cell extracts were examined for levels of FGFR3 by RT–PCR (data not shown) and by immunoblotting (Figure 1b). The results showed that the levels of WT FGFR3 expression in activated splenic B cells from four different TG mice (founders H1, D3, A5, and K1) were comparable with the levels expressed by the H929 HMCL, which has a t(4;14) translocation with upregulation of WT FGFR3 transcripts, whereas the extracts from the spleen of a WT mouse was completely negative. To further examine the levels of FGFR3 expression in TG mice, we performed IHC analysis of spleen sections from WT and TG mice using a monoclonal antibodies specific for human FGFR3. The receptor was highly expressed in spleens of mice bearing the TG (Figures 1c and d), whereas no staining for human FGFR3 was detected in the spleens of WT mice (Figures 1e and f).
FGFR3 accelerates tumorigenesis in the iMYC mouse model of B-cell malignancies
The MYC oncogene is dysregulated in a variety of human hematological diseases, including lymphomas, plasmacytomas, and MM. Recently, Park et al.32 developed a mouse model of MYC-driven B-cell malignancies by replicating the structure of the t(8;14) translocations associated with endemic human Burkitt lymphoma and some mouse plasmacytomas. This was performed by inserting a mouse Myc cDNA containing a histidine tag into the Igh locus upstream of the intronic Eμ enhancer, yielding iMycEμ mice exhibiting constitutive expression of the knock-in Myc TG. In earlier studies, these mice were shown to develop a spectrum of B-cell malignancies, including diffuse high-grade lymphomas, diffuse large B-cell lymphomas, and plasmacytomas.32,41 Most of these mice died between 6 and 21 months of age.32 Given the long latencies for tumor development in iMycEμ mice, we hypothesized that they could provide a novel platform to evaluate the potential for complementary genetic events to accelerate disease. To test this concept, we crossed FGFR3 TG lines D3 and A5, with the highest transgene expression, and iMycEμ mice. These crosses yielded mice of four genotypes on a new hybrid genetic background: non-TG (F−/M−), single TG FGFR3 (F+/M−), single TG iMycEμ (F−/M+), and double TG FGFR3/iMycEμ (F+/M+). Although F+/M− mice expressing only the FGFR3 TG developed tumors at an advanced age and with a frequency that was not significantly different than that of F−/M− mice (Figure 2), necropsies of non-tumor bearing mice revealed lymphoid hyperplasia in 10 of 31 (32%) F+/M− mice compared with 1 of 27 (3.7%) F−/M− mice. Of 44 mice expressing only the iMycEμ transgene (F−/M+), 75% developed tumors at a median age of 281 days. By comparison, 28 of 30 (95%) mice that expressed both the hFGFR3 and iMycEμ transgenes (F+/M+) developed tumors at a median age of 156 days. A log-rank test showed that the difference between the F−/M+ and F+ M+ survival curves was statistically significant (P=0.006). We obtained similar results in studies of both FGFR3 TG mouse lines (data not shown). These data indicate that WT FGFR3 can accelerate tumorigenesis and, therefore, has oncogenic activity.
Figure 2.

Survival curves for FGFR3 (F) and iMyc (M) TG mice. Tumor incidence is similarly low in F+/M− and F−/M− mice, but is higher in F+/M+ mice versus F−/M+ mice (P=0.006).
Tumors of FGFR3/iMycEμ mice are mature B-cell lymphomas
Analyses of histologic sections from most tumors of both F+/M+ and F−/M+ mice stained with hematoxylin and eosin were consistent with the diagnosis of diffuse high-grade blastic B-cell lymphoma,34 a category that had earlier been misclassified as Burkitt-like lymphoma.33,42 Two of the F+/M+ tumors were readily classified as more mature immunoblastic diffuse large B-cell lymphomas with one showing plasmacytoid differentiation as seen in ~25% of the F−/M+ cases. In contrast, the original iMyc mice in a 129SvJ/C57BL/6 background develop both B-cell lymphomas (50%) and plasmacytomas (~20%). In keeping with the histologic findings, flow cytometric analysis of the F+/M+ and F−/M+ tumors showed them to be uniformly CD3− B220+ CD19+, with some of the tumors also expressing moderate levels of CD138 in a fraction (up to 40%) of cells (data not shown). To further confirm the B-cell origin of these tumors and to study their clonality, we performed Southern blot analysis of DNA prepared from the tumors using probes specific for the Igh locus and the Igk locus (Figure 3). All tumors showed VDJ and/or VKJK rearrangement, with one or two major rearranged fragments in each case. We did not detect significant oligoclonality in any tumor even with tumor samples taken from several different locations in the same mouse, including lymph nodes, spleen, and bone marrow (data not shown). Taken together, the histologic, flow cytometric, and clonality analyses of the F+/M+ tumors indicated that they were clonal mature B-cell lineage lymphoma that generally had features somewhat less mature than those from F−/M+ mice.
Figure 3.

Southern blots of representative B lymphoma tumors from FGFR3/iMyc TG mice. (a) Tumor DNA was digested with EcoRI, subjected to electrophoresis, blotted, and hybridized with a 1.2 kb JH probe. Size markers are to the left, arrows to right indicate the 6.4 kb normal germline locus, and the 5.5 kb locus containing the knocked-in c-myc gene. Rearranged fragments are indicated by an R. (b) Tumor DNA was digested with BamHI and EcoRI, subjected to electrophoresis, blotted, and hybridized with a 1 kB Ck probe. The normal germline locus is indicated by an arrow.
FGFR3/iMyc B lymphomas express high levels of unmutated hFGFR3
To assess the level of TG expression in the tumors from F+/M+ mice, we developed a semi-quantitative RT–PCR using a reverse primer specific for the hGH that allowed us to discriminate the exogenously expressed FGFR3. Our results indicated that the hFGFR3 was expressed at high levels in most tumor cells (Figure 4a). We also assessed hFGFR3 transcript levels by realtime RT–PCR for some tumors and, in three cases, cell lines derived from them (Figures 4b and c). The results confirmed that the F+M+ tumors express variable but high levels of hFGFR3 that generally are as high or higher than for the H929 HMCL that has a t(4;14) translocation. Sequencing of the tyrosine kinase domain of FGFR3 in these tumors did not detect any of the most frequent mutations observed in other malignancies.14,15 These data further support the hypothesis that overexpression of WT FGFR3 contributes to tumorigenesis in mature B-cell malignancies.
Figure 4.

RT–PCR and qRT–PCR to detect hFGFR3 expression in primary tumors and cell lines. (a) Detection of expression of hFGFR3 transgene in tumors derived from F+/M+ mice by RT–PCR. (b) qRT–PCR in F+/M+ tumors (white bar), and in the corresponding cell lines (CL, gray bar) derived from primary tumors #228 and #229 and cultured continuously with FGF. (c) qRT–PCR in three F+/M+ cell lines cultured in the presence (F1, gray bar) or in the absence (F0, white bar) of FGF. hFGFR3 expression is −ΔΔCt, normalized to USF2 and to the H929 human myeloma cell line.
FGFR3 enhances the generation of in vitro tumor cell lines
We next attempted to generate cell lines from F+/M+ and F−/M+ tumors cultured in vitro. Although cell lines were readily generated from 7 of 10 (70%) tumors derived from F+/M+ mice, only two were generated from 12 tumors (17%) from F−/M+ mice. FGF-1 was added to the growth medium initially used to culture the F+ M+ tumors, but subsequently, the lines were easily propagated in the presence or absence of the factor. For all F+ M+ tumor cell lines, the inclusion of FGF-1 had no obvious effect on growth rate. RT–PCR analysis showed continued expression of the hFGFR3 construct in all of these cell lines, although usually at substantially lower levels than in the primary tumors from which they were derived (Figure 4c). For three F+ M+ cell lines grown with or without FGF-1, the levels of hFGFR3 expression were determined by qRT–PCR. In two of the three cell lines, the level of hFGFR3 RNA was substantially higher for the cultures grown with FGF-1 (Figure 4c), suggesting that the presence of FGF-1 provides at least some selection for cells expressing FGFR3. Although the immunophenotypes of the F+ M+ tumor-derived cell lines were similar to the primary tumors (CD3-B220 + CD19+), they differed from the tumors in that all of the cells expressed high levels of CD138 (data not shown).
F+/M+ tumors are transplantable
We injected 106 cells from two different tumor-derived cell lines, 228 and 229, subcutaneously in F−/M− WT mice. After 2 weeks, the recipient mice developed large subcutaneous tumors that later spread to the lymph nodes. Flow cytometric analyses showed that transplanted tumors maintained the same phenotype as the cell lines (data not shown).
Gene expression profiling
We next compared the gene expression profiles of six primary tumors of F+/M+ mice with those of seven primary tumors of F−/M+ mice. Two-dimensional hierarchical clustering showed that the subsets of primary tumors grouped tightly in distinct clusters, indicating that they are significantly different from each other (Figure 5). Among 201 genes differently expressed between lymphomas of F+/M+ and F−/M+ mice, only 16 were upregulated in F+/M+ tumors, with FGFR3 as the third most elevated gene.
Figure 5.
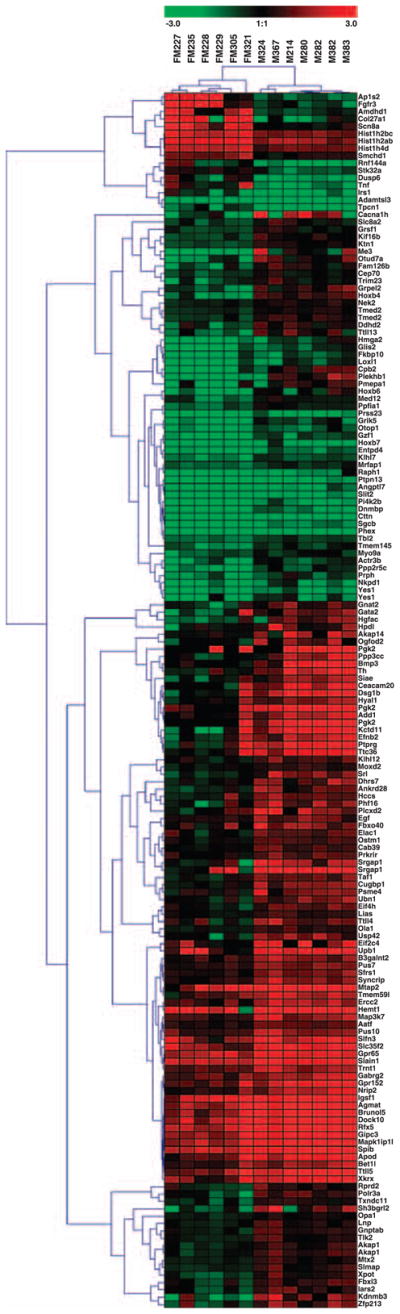
Expression profiling by microarrays of FGFR3 + iMyc TG mice and iMyc TG mice. Two-dimensional hierarchical clustering of gene expression profiles, using distance matrix of Pearson’s correlation, for lymphomas from F+/M+ mice (labeled FM with case number) and F−/M+ mice (labeled M plus case number). Scale bar shown at the top is in log2 scale.
Expression profiles indicate that F+/M+ tumors mice are less differentiated than F−/M+ tumors
As noted above, histologic studies suggested that the tumors of F+/M+ mice were less mature than those from F−/M+ mice. To determine whether this conclusion is confirmed by gene expression, we correlated the gene expression patterns of the two tumor subsets to two master regulators of B-cell lineage differentiation: Pax5 for B cells and Prdm1 for plasma cells. The results of these studies (Table 1) showed that Pax5 was expressed at higher levels and Prdm1 at lower levels in the tumors from F+/M+ mice than those of F−/M+ mice. In keeping with this observation, there was generally increased expression of genes that are activated by Pax5. We conclude that based on histologic features and patterns of gene expression, the tumors of the double TG mice have a somewhat less mature B-cell phenotype than the tumors of iMyc-only mice.
Table 1.
Pax5 targets
| FGFR3iMyc versus iMyc | Ratio | |
|---|---|---|
| Normally induced | ||
| Cd19 | Increased | 1.351 |
| Cd79a | Increased | 1.351 |
| Id3 | Increased | 1.562 |
| Lef1 | Increased | 4.270 |
| Ebf1 | Increased | 1.535 |
| Smarca4 Increased | 1.253 | |
| Rb1 | Decreased | −1.047 |
| Normally repressed | ||
| Prdm1 | Decreased | −1.205 |
The level of expression of Pax5 target genes is compared between F+/M+ and F−/M+ tumor subsets. F+/M+ tumors have generally increased expression of genes that are activated by Pax5 as indicated by the ratio > 1.0. In contrast, Prdm1 is expressed at lower level in the F+/M+ tumors (ratio < 1.0). Values are expressed as log2.
Discussion
The t(4;14) translocation, which is found in ~15% of MM tumors and 30% of HMCL, is the first example of a primary IgH translocation that results in the simultaneous dysregulated expression of two potential oncogenes.8 Ectopic, high expression of FGFR3, which is not expressed in normal B cells or plasma cells, is driven by the 3′ IgH regulatory sequences on the der(14) chromosome. The marked increase of MMSET expression compared with normal B cells or plasma cells is driven by the intronic Eμ enhancer on the der(4)chromosome. The function of these two oncogenes in the transformation process is not well understood. The dysregulation of MMSET seems to be required at all stages of tumor development and progression as all MM tumors and HMCL with t(4;14) retain the der(4) chromosome and express high levels of MMSET. In contrast, ~25% of MM tumors and HMCL with t(4;14) do not express high levels of FGFR3, mostly because the der(14) chromosome is absent, but sometimes because FGFR3 is deleted or epigenetically silenced.23,24 FGFR3 mutations that cause constitutive activation of FGFR3 in patients with achondroplasia and thanataphoric dysplasia have been identified in about 5% of MM tumors and 20% of HMCL that have a t(4;14) and express FGFR3.15,16
Small molecule tyrosine kinase inhibitors targeting FGFR3, as well as short-hairpin RNA and ribozymes that target FGFR3, inhibit proliferation and increase apoptosis of t(4;14) HMCL that express FGFR3 with a kinase-activating mutation. However, these agents do not inhibit in vitro growth of t(4;14) HMCL that express WT FGFR3, even though they can inhibit the enhanced growth that results from addition of exogenous FGF.26–29 The effect of tyrosine kinase inhibitors on intramedullary MM tumors in vivo has not been fully investigated. In one study, however, treatment of primary cultures of cells from t(4;14) MM with the multi-targeted inhibitor of receptor tyrosine kinases, CHIR-258, induced increased apoptosis in four of five tumors that expressed FGFR3, but had no effect on five tumors that were FGFR3 negative.28
Several groups have generated monoclonal antibodies that target the kinase domain of FGFR3. These antibodies inhibit the in vitro growth of HMCL with a subset of the kinase-activating mutations, but have no effect on cells with WT FGFR3 or other activating mutations. The cytotoxic effects of these antibodies on MM tumors or cell lines in vivo or co-cultured in vitro with peripheral blood or bone marrow mononuclear cells seem to be mediated by an antibody-dependent cellular cytotoxicity immune reaction and not by inhibition of FGFR3 function.43
It is clear that the enhanced expression of FGFR3 resulting from the t(4;14) translocation provides a target for kinase-activating mutations that can contribute to tumor progression and can cause at least some MM tumors and HMCL to be fully dependent on the mutant gene. Although the expression of WT FGFR3 is not important in HMCL, it remains to be determined whether ectopic expression of WT FGFR3 is critical early in pathogenesis, and perhaps in most intramedullary t(4;14) cases. However, as activating K- or N-RAS mutations stimulate mostly the same pathways as FGFR3, we suspect that FGFR3 is no longer required in the 20–30% of t(4;14) MM tumors that have K- or N-RAS mutations.15 Significantly, although K- and N-RAS mutations seem to be mutually exclusive with FGFR3 mutations, most t(4;14) MM tumors and HMCL have neither RAS nor FGFR3 mutations. Although cells in the bone marrow or spleen microenvironments express some FGFs, it is not clear if the types of FGF required to stimulate FGFR3 are present in a sufficient quantity for effective stimulation.
Others have shown that FGFR3 containing a kinase-activating mutation can transform 3T3 cells, whereas WT FGFR3 cannot.15,18 In addition, it has been shown that infection of murine bone marrow cells with a retroviral vector containing FGFR3/K650E resulted in a marked leukocytosis and a high frequency of pre-B-cell lymphoid, T lymphoid, or myeloid tumors within 1 month after infection.18 In contrast, parallel experiments in a limited number of mice with a retroviral vector-containing WT FGFR3 resulted in the onset of leukocytosis consistent with acute lymphoblastic leukemia about 1 year after infection.
To validate the apparent oncogenic effect of WT FGFR3 and its cooperation with other oncogenes, we developed a TG mouse (F+/M−) expressing WT hFGFR3 (isoform IIIc) under the control of the LCK promoter and Eμ enhancer to ensure expression in B lymphoid cells. Unlike their control littermates, about one-third of 2-year-old F+/M− mice developed lymphoid hyperplasia. However, although the F+/M− mice occasionally developed lymphoid tumors by 2 years of age, the incidence of lymphomas was not significantly different from control littermates. The F+/M− mice were crossed with iMyc knock-in mice (F−/M+) that develop B lymphomas or plasmacytomas after a relatively long latency. The double TG mice F+/M+ mice developed monoclonal mature B lymphomas with a shorter latency and higher incidence than F−/M+ mice. The absence of plasmacytoma tumors in both the F+/M+ and F−/M+ mice presumably reflects a difference in genetic background from the iMyc mice. As the hFGFR3 TG had no kinase-activating mutations and was expressed at high levels in the F+/M+ lymphomas, these results show that WT FGFR3 is weakly oncogenic and can cooperate with constitutively expressed MYC to enhance B-cell lymphomagenesis. Although MYC rearrangements resulting in enhanced expression is thought to be a late progression event in MM, it is significant that MYC expression is significantly higher in cells from MGUS compared with normal bone marrow plasma cells (P=0.003) and at even higher levels in MM tumors compared with MGUS (P<0.0001)44 (and M Kuehl, unpublished). Therefore, the cooperation of Myc and WT FGFR3 in our lymphoma model may mimic the cooperation that might occur in the early stages of MM with t(4;14). Similar to human MM, the F+/M+ B lymphomas often yielded cell lines that express lower levels of FGFR3 than the corresponding tumor. It thus seems that both the mouse B lymphoma cell lines and human MM cell lines usually do not require FGFR3 for survival and proliferation. As the initial site of origin of the B-cell lymphomas in the F+/M+ mice might be bone marrow, spleen, or some other lymphoid site, we can draw no inferences about the origin of the presumptive ligands that activate the FGFR3 receptor at early stages of tumorigenesis.
In conclusion, the oncogenic effect of WT FGFR3 seems likely to be an important early contribution to the pathogenesis of t(4;14) MM. Significantly, the t(4;14) translocation is the only primary translocation in MM that exclusively targets IgH switch regions, which results in dissociation of the Eμ and 3′ IgH enhancers, thereby enabling the simultaneous dysregulation of FGFR3 and MMSET. Increasing autonomy from the microenvironment is a recurrent theme of tumor progression, and may explain why FGFR3 expression often is lost as MM tumors become independent of extrinsic FGF ligands capable of activating the kinase.
Acknowledgments
We thank Marta Chesi (Mayo Clinic, Scottsdale, AZ) for providing immunohistochemistry (Figures 1c–f). This work was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research (WMK), and National Institute of Allergy and Infectious Diseases (HCM), and Award Number P50CA097274 from the National Cancer Institute (SJ).
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer J Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 2.Bergsagel PL, Chesi M, Nardini E, Brents LA, Kirby SL, Kuehl WM. Promiscuous translocations into immunoglobulin heavy chain switch regions in multiple myeloma. Proc Natl Acad Sci USA. 1996;93:13931–13936. doi: 10.1073/pnas.93.24.13931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuehl WM, Bergsagel PL. Multiple myeloma: evolving genetic events and host interactions. Nat Rev Cancer. 2002;2:175–187. doi: 10.1038/nrc746. [DOI] [PubMed] [Google Scholar]
- 4.Bergsagel PL, Kuehl WM. Molecular pathogenesis and a consequent classification of multiple myeloma. J Clin Oncol. 2005;23:6333–6338. doi: 10.1200/JCO.2005.05.021. [DOI] [PubMed] [Google Scholar]
- 5.Fonseca R, Blood E, Rue M, Harrington D, Oken MM, Kyle RA, et al. Clinical and biologic implications of recurrent genomic aberrations in myeloma. Blood. 2003;101:4569–4575. doi: 10.1182/blood-2002-10-3017. [DOI] [PubMed] [Google Scholar]
- 6.Barlogie B, Shaughnessy J, Tricot G, Jacobson J, Zangari M, Anaissie E, et al. Treatment of multiple myeloma. Blood. 2004;103:20–32. doi: 10.1182/blood-2003-04-1045. [DOI] [PubMed] [Google Scholar]
- 7.Chng WJ, Glebov O, Bergsagel PL, Kuehl WM. Genetic events in the pathogenesis of multiple myeloma. Best Pract Res Clin Haematol. 2007;20:571–596. doi: 10.1016/j.beha.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chesi M, Nardini E, Lim RS, Smith KD, Kuehl WM, Bergsagel PL. The t(4;14) translocation in myeloma dysregulates both FGFR3 and a novel gene, MMSET, resulting in IgH/MMSET hybrid transcripts. Blood. 1998;92:3025–3034. [PubMed] [Google Scholar]
- 9.Eswarakumar VP, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005;16:139–149. doi: 10.1016/j.cytogfr.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 10.Dailey L, Ambrosetti D, Mansukhani A, Basilico C. Mechanisms underlying differential responses to FGF signaling. Cytokine Growth Factor Rev. 2005;16:233–247. doi: 10.1016/j.cytogfr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 11.L’Hote CG, Knowles MA. Cell responses to FGFR3 signalling: growth, differentiation and apoptosis. Exp Cell Res. 2005;304:417–431. doi: 10.1016/j.yexcr.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 12.Onwuazor ON, Wen XY, Wang DY, Zhuang L, Masih-Khan E, Claudio J, et al. Mutation, SNP, and isoform analysis of fibroblast growth factor receptor 3 (FGFR3) in 150 newly diagnosed multiple myeloma patients. Blood. 2003;102:772–773. doi: 10.1182/blood-2003-04-1204. [DOI] [PubMed] [Google Scholar]
- 13.Salazar L, Kashiwada T, Krejci P, Muchowski P, Donoghue D, Wilcox WR, et al. A novel interaction between fibroblast growth factor receptor 3 and the p85 subunit of phosphoinositide 3-kinase: activation-dependent regulation of ERK by p85 in multiple myeloma cells. Hum Mol Genet. 2009;18:1951–1961. doi: 10.1093/hmg/ddp116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tavormina PL, Shiang R, Thompson LM, Zhu YZ, Wilkin DJ, Lachman RS, et al. Thanatophoric dysplasia (types I and II) caused by distinct mutations in fibroblast growth factor receptor 3. Nat Genet. 1995;9:321–328. doi: 10.1038/ng0395-321. [DOI] [PubMed] [Google Scholar]
- 15.Chesi M, Brents LA, Ely SA, Bais C, Robbiani DF, Mesri EA, et al. Activated fibroblast growth factor receptor 3 is an oncogene that contributes to tumor progression in multiple myeloma. Blood. 2001;97:729–736. doi: 10.1182/blood.v97.3.729. [DOI] [PubMed] [Google Scholar]
- 16.Chesi M, Nardini E, Brents LA, Schrock E, Ried T, Kuehl WM, et al. Frequent translocation t(4;14)(p16.3;q32.3) in multiple myeloma is associated with increased expression and activating mutations of fibroblast growth factor receptor 3. Nat Genet. 1997;16:260–264. doi: 10.1038/ng0797-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen J, Williams IR, Lee BH, Duclos N, Huntly BJ, Donoghue DJ, et al. Constitutively activated FGFR3 mutants signal through PLCgamma-dependent and -independent pathways for hematopoietic transformation. Blood. 2005;106:328–337. doi: 10.1182/blood-2004-09-3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Z, Zhu YX, Plowright EE, Bergsagel PL, Chesi M, Patterson B, et al. The myeloma-associated oncogene fibroblast growth factor receptor 3 is transforming in hematopoietic cells. Blood. 2001;97:2413–2419. doi: 10.1182/blood.v97.8.2413. [DOI] [PubMed] [Google Scholar]
- 19.Khnykin D, Troen G, Berner JM, Delabie J. The expression of fibroblast growth factors and their receptors in Hodgkin’s lymphoma. J Pathol. 2006;208:431–438. doi: 10.1002/path.1900. [DOI] [PubMed] [Google Scholar]
- 20.Gomez-Roman JJ, Saenz P, Molina M, Cuevas Gonzalez J, Escuredo K, Santa Cruz S, et al. Fibroblast growth factor receptor 3 is overexpressed in urinary tract carcinomas and modulates the neoplastic cell growth. Clin Cancer Res. 2005;11 (2 Pt 1):459–465. [PubMed] [Google Scholar]
- 21.Tomlinson DC, Baldo O, Harnden P, Knowles MA. FGFR3 protein expression and its relationship to mutation status and prognostic variables in bladder cancer. J Pathol. 2007;213:91–98. doi: 10.1002/path.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grose R, Dickson C. Fibroblast growth factor signaling in tumorigenesis. Cytokine Growth Factor Rev. 2005;16:179–186. doi: 10.1016/j.cytogfr.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 23.Keats JJ, Reiman T, Belch AR, Pilarski LM. Ten years and counting: so what do we know about t(4;14)(p16; q32) multiple myeloma. Leuk Lymphoma. 2006;47:2289–2300. doi: 10.1080/10428190600822128. [DOI] [PubMed] [Google Scholar]
- 24.Chesi M, Bergsagel PL, Kuehl WM. The enigma of ectopic expression of FGFR3 in multiple myeloma: a critical initiating event or just a target for mutational activation during tumor progression. Curr Opin Hematol. 2002;9:288–293. doi: 10.1097/00062752-200207000-00005. [DOI] [PubMed] [Google Scholar]
- 25.Paterson JL, Li Z, Wen XY, Masih-Khan E, Chang H, Pollett JB, et al. Preclinical studies of fibroblast growth factor receptor 3 as a therapeutic target in multiple myeloma. Br J Haematol. 2004;124:595–603. doi: 10.1111/j.1365-2141.2004.04814.x. [DOI] [PubMed] [Google Scholar]
- 26.Trudel S, Ely S, Farooqi Y, Affer M, Robbiani DF, Chesi M, et al. Inhibition of fibroblast growth factor receptor 3 induces differentiation and apoptosis in t(4;14) myeloma. Blood. 2004;103:3521–3528. doi: 10.1182/blood-2003-10-3650. [DOI] [PubMed] [Google Scholar]
- 27.Trudel S, Stewart AK, Rom E, Wei E, Li ZH, Kotzer S, et al. The inhibitory anti-FGFR3 antibody, PRO-001, is cytotoxic to t(4;14) multiple myeloma cells. Blood. 2006;107:4039–4046. doi: 10.1182/blood-2005-10-4179. [DOI] [PubMed] [Google Scholar]
- 28.Trudel S, Li ZH, Wei E, Wiesmann M, Chang H, Chen C, et al. CHIR-258, a novel, multitargeted tyrosine kinase inhibitor for the potential treatment of t(4;14) multiple myeloma. Blood. 2005;105:2941–2948. doi: 10.1182/blood-2004-10-3913. [DOI] [PubMed] [Google Scholar]
- 29.Masih-Khan E, Trudel S, Heise C, Li Z, Paterson J, Nadeem V, et al. MIP-1alpha (CCL3) is a downstream target of FGFR3 and RAS-MAPK signaling in multiple myeloma. Blood. 2006;108:3465–3471. doi: 10.1182/blood-2006-04-017087. [DOI] [PubMed] [Google Scholar]
- 30.Qian S, Somlo G, Zhou B, Zhu L, Mi S, Mo X, et al. Ribozyme cleavage leads to decreased expression of fibroblast growth factor receptor 3 in human multiple myeloma cells, which is associated with apoptosis and downregulation of vascular endothelial growth factor. Oligonucleotides. 2005;15:1–11. doi: 10.1089/oli.2005.15.1. [DOI] [PubMed] [Google Scholar]
- 31.Iritani BM, Forbush KA, Farrar MA, Perlmutter RM. Control of B cell development by Ras-mediated activation of Raf. EMBO J. 1997;16:7019–7031. doi: 10.1093/emboj/16.23.7019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park SS, Kim JS, Tessarollo L, Owens JD, Peng L, Han SS, et al. Insertion of c-Myc into Igh induces B-cell and plasma-cell neoplasms in mice. Cancer Res. 2005;65:1306–1315. doi: 10.1158/0008-5472.CAN-04-0268. [DOI] [PubMed] [Google Scholar]
- 33.Morse HC, III, Anver MR, Fredrickson TN, Haines DC, Harris AW, Harris NL, et al. Bethesda proposals for classification of lymphoid neoplasms in mice. Blood. 2002;100:246–258. doi: 10.1182/blood.v100.1.246. [DOI] [PubMed] [Google Scholar]
- 34.Janz S, Morse H, Teitell MA. Mouse models of mature B-cell and plasma cell neoplasm. In: Li S, editor. Mouse Models of Human Blood Cancers: Basic Research and Pre-clinical Application. Springer; 2008. pp. 180–200. [Google Scholar]
- 35.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sturn A, Quackenbush J, Trajanoski Z. Genesis: cluster analysis of microarray data. Bioinformatics. 2002;18:207–208. doi: 10.1093/bioinformatics/18.1.207. [DOI] [PubMed] [Google Scholar]
- 37.Zhan F, Barlogie B, Arzoumanian V, Huang Y, Williams DR, Hollmig K, et al. Gene-expression signature of benign monoclonal gammopathy evident in multiple myeloma is linked to good prognosis. Blood. 2007;109:1692–1700. doi: 10.1182/blood-2006-07-037077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhan F, Huang Y, Colla S, Stewart JP, Hanamura I, Gupta S, et al. The molecular classification of multiple myeloma. Blood. 2006;108:2020–2028. doi: 10.1182/blood-2005-11-013458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cariappa A, Chen L, Haider K, Tang M, Nebelitskiy E, Moran ST, et al. A catalytically inactive form of protein kinase C-associated kinase/receptor interacting protein 4, a protein kinase C beta-associated kinase that mediates NF-kappa B activation, interferes with early B cell development. J Immunol. 2003;171:1875–1880. doi: 10.4049/jimmunol.171.4.1875. [DOI] [PubMed] [Google Scholar]
- 40.Iseki M, Kubo-Akashi C, Kwon SM, Yamaguchi A, Takatsu K, Takaki S. APS, an adaptor molecule containing PH and SH2 domains, has a negative regulatory role in B cell proliferation. Biochem Biophys Res Commun. 2005;330:1005–1013. doi: 10.1016/j.bbrc.2005.03.073. [DOI] [PubMed] [Google Scholar]
- 41.Kim JS, Han SS, Park SS, McNeil N, Janz S. Plasma cell tumour progression in iMycEmu gene-insertion mice. J Pathol. 2006;209:44–55. doi: 10.1002/path.1940. [DOI] [PubMed] [Google Scholar]
- 42.Zhu D, Qi CF, Morse HC, III, Janz S, Stevenson FK. Deregulated expression of the Myc cellular oncogene drives development of mouse ‘Burkitt-like’ lymphomas from naive B cells. Blood. 2005;105:2135–2137. doi: 10.1182/blood-2004-07-2573. [DOI] [PubMed] [Google Scholar]
- 43.Qing J, Du X, Chen Y, Chan P, Li H, Wu P, et al. Antibody-based targeting of FGFR3 in bladder carcinoma and t(4;14)-positive multiple myeloma in mice. J Clin Invest. 2009;119:1216–1229. doi: 10.1172/JCI38017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chesi M, Robbiani DF, Sebag M, Chng WJ, Affer M, Tiedemann R, et al. AID-dependent activation of a MYC transgene induces multiple myeloma in a conditional mouse model of post-germinal center malignancies. Cancer Cell. 2008;13:167–180. doi: 10.1016/j.ccr.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]