

Abstract
Autoimmune gastritis is a common autoimmune disorder characterized by chronic inflammatory cell infiltrates, atrophy of the corpus and fundus, and the occurrence of autoantibodies to parietal cell antigen. In CCR7-deficient mice, autoimmune gastritis developed spontaneously and was accompanied by metaplasia of the gastric mucosa and by the formation of tertiary lymphoid organs at gastric mucosal sites. T cells of CCR7-deficient mice showed an activated phenotype in the gastric mucosa, mesenteric lymph nodes, and peripheral blood. In addition, elevated serum IgG levels specific to gastric parietal cell antigen were detected. Because the role of organized lymphocytic aggregates at this inflammatory site is not completely understood, we first analyzed the cellular requirements for the formation of these structures. Autoreactive CD4+ T cells were pivotal for tertiary lymphoid follicle formation, most likely in cooperation with dendritic cells, macrophages, and B cells. Second, we analyzed the necessity of secondary lymph nodes and tertiary lymphoid organs for the development of autoimmune gastritis using CCR7 single- and CCR7/lymphotoxin α double-deficient mice. Strikingly, manifestation of autoimmune gastritis was observed in the absence of secondary lymph nodes and preceded the development of tertiary lymphoid organs. Taken together, these findings identify an inflammatory process where gastric autoreactive T cells independent of organized tertiary lymphoid organs and classic lymph nodes can induce and maintain autoimmune gastritis.
Autoimmunity is often accompanied by the development of tertiary lymphoid organs (TLOs) in the target organ. TLO formation has been reported in inflamed tissues of various autoimmune diseases, including rheumatoid arthritis, Sjögren's syndrome, type 1 diabetes mellitus, and autoimmune gastritis (AIG).1–5 Although TLOs share numerous functional characteristics with secondary lymphoid organs (SLOs), their unique function in autoimmune disease pathogenesis is still unclear.6,7 More specifically, it remains to be elucidated whether these structures are relevant for priming of T cells, development of autoantibody-producing plasma cells, and accumulation of autoantigen-specific T- and B-cell clones.
In a murine AIG model induced by neonatal thymectomy, Katakai and colleagues found ectopic lymphoid organization in the chronic gastric lesions. Number and size of ectopic gastric lymphoid clusters could be correlated with serum autoantibody titers, although development of typical germinal centers (GCs) was rare in this AIG mouse model.5,8 In a nonobese diabetic mouse model of type 1 diabetes, the presence of GCs, a skewed repertoire, and somatic hypermutation of B-cell receptors was described in islet TLOs, which implies that T- and B-cell interactions take place at the inflamed site.4 More recently, a CXCL13-dependent B lymphocyte lodging within islet TLOs was shown. On CXCL13 blockade, B-lymphocyte organization in TLOs was completely abrogated.9 However, induction of a skewed B-cell repertoire, occurrence of somatic hypermutation, and finally manifestation of diabetes were maintained. Thus, the authors concluded that the significance of organized TLOs in modulating autoimmune responses remains questionable. In contrast, other studies suggested that formation of TLOs in affected organs contributes essentially to the pathogenesis of human autoimmune diseases by enhancing the efficiency of autoantigen presentation at the site of inflammation. In this process, autoreactive T lymphocytes and autoantibody-producing B cells could be generated.7 Thus, TLOs may play a role in maintaining an autoimmune response against persistent self-antigens and selection of autoreactive B and T cells.4 However, the contribution of autoantibodies to disease progression is still controversially discussed. There is evidence that lymphoid neogenesis is associated with local antibody production but not with autoimmune activity or severity.7
AIG represents an organ-specific autoimmune disease in which lymphocytes massively infiltrate the gastric mucosal (GM) tissue, which eventually leads to gastric lymphoid neogenesis. Loss of gastric parietal and zymogenic cells and the prevalence of circulating autoantibodies to gastric parietal antigen are specific characteristics.10 The morphology of mouse gastritis shares many pathohistologic and immunologic characteristics with the human disease and can be induced in a variety of experimental approaches, including lymphopenic and nonlymphopenic AIG models, or in T-cell receptor α-subunit and T-cell receptor β-subunit transgenic mice.11 Models of spontaneous AIG disease pathogenesis are rare, and disease incidence is often very low.11,12
We report the development of an early, spontaneous AIG with almost complete penetrance in mice lacking the homeostatic chemokine receptor CCR7. CCR7 is a major lymph node–homing receptor for lymphocytes and dendritic cells (DCs), and mice that lack CCR7 display not only a delayed induction of adaptive immune responses13,14 but also impaired lymphocyte recirculation through nonlymphoid organs.15,16 Lack of CCR7 is associated with a moderate multiorgan autoimmunity17 and formation of TLOs at mucosal sites.16,18 Our results show that AIG in CCR7−/− mice occurred as early as 4 weeks of age characterized by lymphocytic mucosal infiltrates, profound loss of parietal cells, increase in the population of mucous cells, and the occurrence of circulating autoantibodies to gastric antigen. AIG manifestation in these mice was associated with the development of gastric TLOs. We focused on the requirement of TLOs and lymph nodes (LNs) for the pathogenesis of spontaneous AIG. We show that local accumulation of autoreactive T lymphocytes promoted the development of spontaneous AIG in CCR7-deficient mice, a process that was independent from the occurrence of TLOs or classic peripheral LNs.
Materials and Methods
Mice
C57BL/6 Ly5.1 (CD45.1) congenic mice, lymphotoxin (Lt) α−/− mice,19 and Rag2−/− mice were obtained from Jackson Laboratories (Bar Harbor, ME). CCR7−/− were generated as previously described13 and backcrossed onto a C57BL/6 background for 12 generations. CCR7−/− mice were crossed with Ltα−/− mice to generate double knockout mice. CD11c.DTR transgenic mice were obtained from Natolio Garbi.20 Animals were housed in a specific pathogen-free facility at the Max-Delbrück-Center for Molecular Medicine, Berlin, Germany. All animal studies were performed according to institutional and Berlin State guidelines (registered under LaGeSo 0184/04 and 0371/05).
Determination of Gastric pH
Mice were fasted overnight and sacrificed, 50 μL of gastric contents was collected, and the pH was determined using pH paper (Whatman; GE Healthcare, Munic, Germany).
Antibodies
The following primary antibodies were used: fluorescein isothiocyanate (FITC)–conjugated Armenian hamster-anti-mouse CD3ε; phycoerythrin (PE)-labeled Armenian hamster-anti-mouse CCR5 (CD195); biotinylated Armenian hamster anti-mouse CXCR3 (CD183); FITC-labeled Armenian hamster anti-mouse CD69; PE-conjugated rat anti-mouse CD44; allophycocyanin (APC)-labeled rat anti-mouse CD4, CD8, and CD19; FITC-labeled rat anti-mouse CD86; PE-labeled rat anti-mouse CD23; PE-labeled rat-anti-mouse CD8α; peridinin chlorophyll protein complex (PerCP)-labeled rat anti-mouse CD8α; PerCP-labeled Armenian hamster anti-mouse CD3ε; PE-coupled Armenian hamster anti-mouse CD103 (all from BioLegend, Kithorn, the Netherlands); FITC-labeled rat anti-mouse CD21/CD35, IgD; FITC-labeled mouse anti-mouse I-Ab; PE-labeled rat anti-mouse IgM (all from BD Biosciences, Heidelberg, Germany); FITC-conjugated rat anti-mouse interferon (IFN)-γ and APC-conjugated rat anti-mouse interleukin 17A (IL-17A) (eBioscience, Frankfurt, Germany); and biotinylated Armenian hamster anti-mouse CD11c (Caltag, Hamburg, Germany). Biotinylated primary antibodies were detected with streptavidin conjugates (BioLegend).
For immunohistology, the following primary antibodies were used: mouse-anti-proton pump/H,K-adenosine triphosphatase (ATPase) α-subunit (MBL International Corporation, Woburn, MA); purified rat-anti-mouse CD138; FITC-labeled mouse-anti-mouse CD45.1; Pacific blue–labeled mouse-anti-mouse CD45.2 (all from Biolegend); Alexa 488–labeled rat-anti-mouse CD45R/B220; Pacific blue–labeled rat-anti-mouse CD3; biotinylated rat-anti-mouse CD45R/B220; and biotinylated Armenian hamster-anti-mouse CD11c (all from BioLegend). Biotinylated Ulex europaeus agglutinin I (UEAI; Vector Laboratories, Loerrach, Germany) and FITC-labeled Griffonia simplicifolia lectin (Invitrogen, Darmstadt, Germany) were used to identify pit and mucous neck cells, respectively.
Flow Cytometry Analysis
Data were acquired on a FACSCanto II flow cytometer (BD Biosciences) and analyzed with Flowjo software versions 8.8.2 and 9.2 (TreeStar, Ashland, OR).
Immunohistology
For Alcian blue staining, paraffin sections were deparaffinized, incubated with Alcian blue solution (pH 2.5) for 30 minutes at room temperature, rinsed in water, dehydrated, and mounted in Entellan (Merck, Darmstadt, Germany). Images were acquired with a Zeiss Axiophot fluorescence microscope with an Axiocam HRc camera and further processed using AxioVision 4.5 software (all from Carl Zeiss, Göttingen, Germany).
Mononuclear Cell Preparation from Mucosal Tissue and SLOs
Minced tissue of murine stomachs was incubated in RPMI 1640 medium and supplemented with 10% fetal calf serum and 6 mg/mL of collagenase NB4 (Serva, Heidelberg, Germany) for 30 minutes at 37°C. Spleen and mesenteric LNs (mLNs) were injected with 2 mg/mL of collagenase NB4, cut into small pieces, and incubated at 37°C for 30 minutes. Single-cell suspensions were applied on a Ficoll density gradient.
Detection of IFN-γ– or IL-17A–Producing Gastric T Cells
Mononuclear cells were isolated from the stomach and incubated for 4 hours at 37°C in RPMI 1640 medium containing 10% fetal calf serum in the presence or absence of a cocktail of ionomycin (1.5 μg/mL), phorbol myristate acetate (40 ng/mL), and brefeldin A (20 μg/mL) (Sigma-Aldrich, Munich, Germany). Surface expression of CD3, CD4, and CD8 and intracellular expression of IFN-γ and IL-17A (Fix & Perm cell permeabilization kit; Caltag) were analyzed by flow cytometry.
Determination of Secreted Inflammatory Cytokines
Stomach tissue of 2-month-old mice was cut into pieces and incubated in 1.25 mL of serum-free RPMI 1640 medium containing penicillin and streptomycin for 31 hours at 37°C and 5% CO2. Supernatants were harvested and assayed for secreted IL-1β, tumor necrosis factor (TNF)-α, IFN-γ, IL-6, IL-10, CCL2 (monocyte chemotactic protein 1), and IL-12p70 using the Cytometric Bead Array Mouse Inflammation Kit and FCAP Array Software (all from BD Biosciences) according to the manufacturer's instructions. Cytokine concentration was indicated by the fluorescent intensity and finally normalized to stomach weight.
Laser Capture Microdissection and Quantitative PCR
Gastric epithelial tissue from C57BL/6 wild-type (Wt) and CCR7−/− mice and TLOs from stomachs of CCR7−/− mice were laser microdissected using the PixCell IIe LCM System (Arcturus, Mörfelden-Walldorf, Germany). Cryosections of gastric tissue were mounted on diethyl pyrocarbonate–treated slides, stained, and dehydrated using the HistoGene LCM frozen section staining kit (Arcturus). RNA was isolated using the PicoPure RNA Isolation Kit, followed by two-round amplification of RNA using the RiboAmp Plus Kit (both from Arcturus). For gene expression analysis, antisense RNA was reverse transcribed to cDNA and quantitative PCR was performed in an array format (SABiosciences, Qiagen GmbH, Hilden, Germany).
Immunoblotting
Stomach tissue lysates were prepared in lysis buffer (100 mmol/L Tris, pH 7.5, 150 mmol/L NaCl, 1 mmol/L EDTA-Na2, 1 mmol/L sodium orthovanadate, 1 mmol/L NaF, 1% Triton X-100, and protease inhibitors), and 30 μg of protein was separated by SDS-PAGE. Blots were probed with mouse-anti-proton pump/H,K-ATPase α-subunit antibody (MBL International Corporation) and polyclonal rabbit-anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody (Abcam), followed by horseradish peroxidase (HRP)–coupled goat anti-mouse or donkey anti-rabbit secondary antibody. Proteins were visualized by chemiluminescence (ECL kit; Thermo Scientific, Bonn, Germany). Densitometric quantification of blot bands was performed using the ImageJ software version 1.42q (NIH).
Mixed Bone Marrow Chimeras
Bone marrow (BM) from 6- to 12-week-old Wt and CCR7−/− mice was transplanted into 8- to 12-week-old, sex-matched Rag2−/− mice. Recipients were sublethally irradiated (7 grays) and reconstituted with 2 to 3 × 107 mixed BM cells (1:1) from Wt C57BL/6/CD45.1 and CCR7−/−C57BL/6/CD45.2 donor mice.
Detection of Autoantibodies and Cytokines by Enzyme-Linked Immunosorbent Assay
Microtiter plates were coated with 10 μg/mL of whole Wt stomach tissue lysate and incubated with sera at a dilution of 1:50. Bound IgG antibodies were detected using HRP-conjugated rabbit anti-mouse antiserum (1:4000, Southern Biotechnology, Aachen, Germany) in conjunction with 3,3′,5,5′-tetramethylbenzidine (TMB) substrate (BD).
To quantitate serum IL-17 levels, microtiter plates were coated with rat anti-mouse IL-17A (BioLegend). Serum samples were diluted 1:30 and recombinant mouse IL-17A served as a standard. Bound cytokines were detected with biotinylated rat-anti-mouse IL-17A (BioLegend), followed by HRP-conjugated streptavidin and TMB substrate.
Depletion of CD4+ Lymphocytes and CD11c+ DCs
CCR7−/− mice were injected weekly with 50 to 100 μg of CD4+ cell-depleting antibody GK1.5 antibody (BioLegend) or isotype control antibody (rat IgG2b, κ) starting at 2 weeks of age. CD11c.DTR mice, in which the diphtheria toxin receptor (DTR) is under the control of the CD11c promoter, were used to deplete DCs.20 Eight DT (8 ng/g of body weight) i.p. injections in CCR7−/− CD11c.DTR transgenic mice every second day induced the depletion of CD11c+ DCs.
Statistical Analysis
Results are expressed as arithmetic mean ± SD or SEM, as indicated. P < 0.05 was considered statistically significant and determined by either the Mann-Whitney U-test (one-tailed or two-tailed) or an unpaired, two-tailed Student's t-test, as indicated.
Results
Spontaneous Development of AIG in CCR7-Deficient Mice with Profound Parietal Cell Loss and Mucosal Lymphoid Neogenesis
We previously reported that the gastrointestinal mucosal tissue of CCR7−/− mice is highly permissive for the formation of TLOs. In aged mice, histologic analysis revealed enlarged stomachs with profound cystic hyperplasia and an increased rate of mucosal proliferation.16 Here, immunohistologic analysis of 1- to 12-month-old CCR7−/− mice showed that all animals developed distinct histologic and immunohistologic features characteristic of an AIG. Staining for H+/K+-ATPase α, a marker for parietal cells, revealed a selective loss of parietal cells in CCR7−/− mice (Figure 1A). Atrophy of parietal cells was accompanied by profound lymphoid neogenesis (Figure 1B) exhibiting GC formation in gastric TLOs of aged mice (see Supplemental Figure S1A at http://ajp.amjpathol.org). The incidence of AIG manifestation was more than 95% in all age groups of CCR7−/− mice (1 month, n = 5; 2 months, n = 13; 10 to 16 months, n = 6), whereas Wt mice did not develop any signs of AIG up to 12 months of age. Parietal cell loss was quantitated by immunoblot analysis showing significantly less protein expression for parietal cell antigen H+/K+-ATPase α in whole stomach tissue lysates derived from CCR7−/− compared with Wt mice (Figure 2, A and B).
Figure 1.
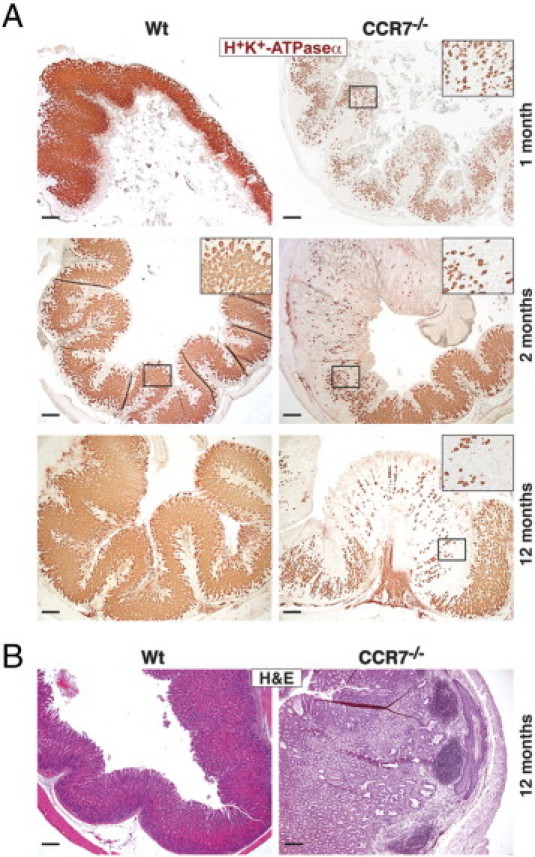
Lack of CCR7 causes early development of AIG. A: Paraffin-embedded stomach sections of 1-, 2-, and 12-month-old Wt (n = 3 to 5 per age group) or CCR7−/− mice (n = 3 to 5 per age group) were immunohistochemically stained for H+/K+-ATPase α as a marker for parietal cells. Representative stains are shown. Magnification of representative areas in the insets. Scale bar = 200 μm. B: Paraffin-embedded stomach sections of age-matched (12-month-old) Wt (n = 3) and CCR7−/− mice (n = 5) were stained with H&E (one representative animal of each group is shown).
Figure 2.
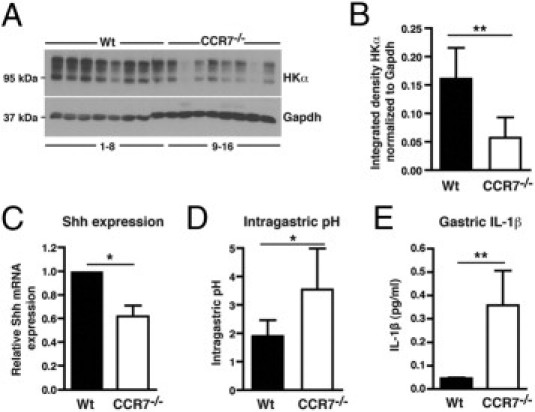
Parietal cell loss in CCR7−/− mice is associated with decreased gastric Shh mRNA levels, hypochlorhydria, and increased levels of IL-1β. A: Immunoblotting for parietal H+/K+-ATPase α (HKα) and GAPDH in stomach tissue lysates of 11- to 23-month-old Wt (n = 8; lane 1–8) and CCR7−/− (n = 8; lane 9–16) mice. B: Quantitation of H+/K+-ATPase α protein expression normalized to GAPDH is shown. C: Quantitative PCR for Shh mRNA transcript in stomachs of 2-month-old Wt (n = 6) and CCR7−/− (n = 6) mice. Expression of Shh mRNA was normalized to GAPDH and is depicted as relative gastric Shh mRNA expression in CCR7−/− compared with Wt (set arbitrarily to 1) mice. D: Measurement of intragastric pH in 11- to 23-month-old Wt (n = 7) and CCR7−/− (n = 6) mice using pH indicator paper. E: Determination of IL-1β secretion from stomachs of 2-month-old Wt (n = 5) and CCR7−/− (n = 5) mice. Stomach-derived cells were cultured for 31 hours in RPMI 1640 medium without serum at 37°C. IL-1β release was measured in supernatants by cytokine bead array. Bars represent means ± SD (B and D) or SEM (C and E). *P < 0.05, **P < 0.01; Mann-Whitney U-test.
Sonic hedgehog (Shh) is abundantly expressed in the gastric fundic glands, and signaling by Shh protein plays an important role in the maintenance of gastric fund gland homeostasis.21,22 Because it was described that parietal cell loss is correlated with reduced expression levels of Shh,23,24 we quantitated Shh expression in CCR7−/− mice. CCR7−/− mice exhibited decreased gastric Shh gene expression levels (Figure 2C) and hypochlorhydria (Figure 2D) compared with Wt controls. The inflammatory cytokine IL-1β can inhibit parietal cell acid secretion by promoting gastric atrophy through suppression of Shh.24 Thus, we analyzed whether local gastric IL-1β levels might be altered in CCR7−/− mice. CCR7−/− mice revealed a significant increase in gastric IL-1β production compared with Wt mice (Figure 2E), suggesting that enhanced IL-1β expression induces down-regulation of Shh. Gastric secretion of additional proinflammatory cytokines, ie, IL-6, IL-10, TNF-α, CCL2 (monocyte chemotactic protein 1), and IL-12p70, was not enhanced in CCR7−/− mice (data not shown).
Replacement of Parietal Cells by Aberrant Mucous-Producing Cells
Loss of gastric glandular cells frequently leads to a severe alteration of the normal gastric pit architecture, most likely through replacement by intestinal or fibrous tissues.25 Here, stomach sections of 2-month-old Wt or knockout mice were stained for CD138 expression, a marker for gastric pit (foveolar epithelial) cells. The CD138+ cell population was markedly increased in CCR7−/− compared with Wt mice (Figure 3A). In addition, single CD138+ plasma cells were frequently detected within the lamina propria of CCR7−/− mice (Figure 3A, right panel). To further differentiate between pit cells and mucous neck cells, lectin staining was performed. Pit cells (UEAI+) lined the gastric surface and were present along pit walls, whereas mucous neck cells (GSII+) marked the neck region of the gastric gland in Wt mice (Figure 3B, left panel). In sharp contrast, surface pit cells in CCR7−/− mice were found in the vicinity of mucous neck cells and both cell lineages were markedly expanded (Figure 3B, middle and right panels) compared with Wt mice.
Figure 3.
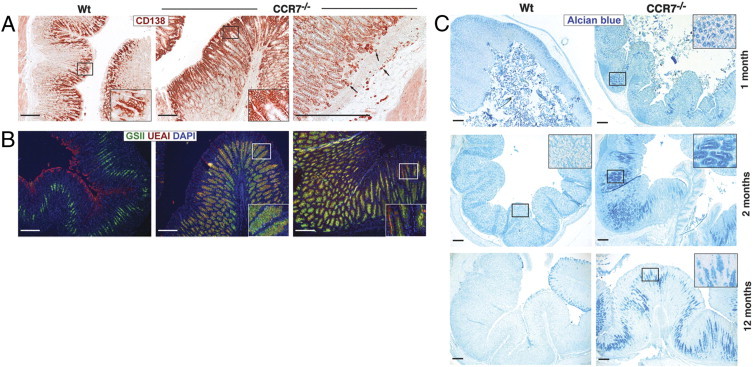
Lack of CCR7 leads to gastric intestinal metaplasia. Stomach sections of 2-month-old Wt or CCR7−/− mice (n = 5 per group) were stained for CD138 expression (pit and plasma cells) (A) or triple-stained with lectins UEAI (red, pit cells), GSII (green, mucous neck cells), and DAPI (blue) (B). C: Paraffin-embedded stomach sections of 1-, 2-, and 12-month-old Wt or CCR7−/− mice (n = 3 to 5 per group) were stained for acidic mucopolysaccharides (Alcian blue; pH 2.5). Magnification of representative areas in the insets. Scale bar = 200 μm.
Next, we stained stomach sections of CCR7−/− mice and Wt controls (1 to 12 months old) for acidic mucopolysaccharides (Figure 3C). Extended Alcian blue staining in young and elder CCR7−/− mice was indicative of an expansion of acidic mucous-secreting cells, demonstrating metaplasia in CCR7−/− mice. In conclusion, CCR7−/− mice exhibited a disturbed gastric fundic gland homeostasis with severely impaired differentiation of mucous-producing cells.
Activated Immune Cell Subsets in the Gastric Mucosa of CCR7−/− Mice Are Associated with an Inflammatory Milieu
Lymphocytes that infiltrate the GM can contribute to epithelial cell destruction and to gastric atrophy during chronic gastritis.26 To determine the transcriptional profile of the gastric ectopic lymphoid aggregates in CCR7−/− mice versus Wt GM tissue, we microdissected the gastric TLOs and GM tissue of 4- and 12-month-old CCR7−/− and Wt mice, respectively. Quantitative reverse transcription–PCR array analysis focused on genes that were informative on immunoregulatory and proliferative processes. Within gastric TLOs, we detected genes that are commonly associated with an inflammatory milieu, including ILs and IL receptors, ie, IFN-γ, inflammatory chemokines (CCL3, CCL4, CCL5, CCL6, CCL8, CCL9, CXCL9, CXCL10), chemokine receptors (CCR1, CCR4, CCR5, CXCR3), integrins, and complement factor C3 (see Supplemental Table S1 at http://ajp.amjpathol.org). Genes that were up-regulated in the GM of CCR7−/− mice involved growth factors, adhesion molecules, IFN-γ, TNF-α, proapoptotic genes (Fas, Caspase 1), angiogenesis-associated genes (CXCL5), and the inflammatory cytokine IL-1f6 (see Supplemental Table S2 at http://ajp.amjpathol.org). Taken together, these results are indicative of a prevalent inflammatory milieu in the GM tissue of CCR7−/− mice.
Using flow cytometry, we analyzed the activation status of CCR7−/− GM-infiltrating and mLN-derived lymphocytes compared with Wt lymphocytes. Because naive Wt mice do not show any intragastric formation of TLOs, the total number of gastric B and T lymphocytes was small and significantly lower compared with CCR7−/− mice (Figure 4A). Most gastric CCR7−/− CD19+ B cells expressed the common B-cell markers I-Ab (major histocompatibility complex [MHC] class II), CD21, and CD23 and were IgM+/IgD+ (Figure 4B). A low expression level was found for the co-stimulatory molecule CD86, and no significant staining was detectable for the early activation antigen CD69 (Figure 4B). Hence, CCR7-deficient GM-infiltrating B lymphocytes represent a mature but predominantly nonactivated phenotype comparable to those of Wt mice. The only differences between CCR7−/− and Wt B lymphocytes were a higher percentage of CD23+ and IgD+ B lymphocytes in CCR7−/− mice, which is because the TLOs in CCR7−/− mice predominantly contain classic B-2 B lymphocytes (CD21intCD23hi/IgM/IgDhi) (Figure 4B). In both strains, CCR7−/− and Wt animals, most GM-derived CD4+ and CD8+ T lymphocytes expressed the early activation antigen CD69 (Figure 4C, left panels). Importantly, the overall total number of activated CD69+CD44highCD4+ or CD69+CD44highCD8+ T cells was significantly increased in CCR7−/− compared with Wt mice (Figure 4C, right panel). CD69 expression was also up-regulated in 40% to 60% of mLN-derived CD4+ and in 20% to 30% of mLN-derived CD8+ T cells of CCR7−/− mice compared with mLN-derived CD4+ (20%-40%) and CD8+ (10%-15%) T lymphocytes of Wt mice (Figure 4D, left panels). The percentage of CD69+ CD4+ or CD8+ T cells that co-expressed CD44 was additionally increased in mLNs of CCR7−/− compared with Wt mice [6.75% ± 0.64% (Wt) and 12.8% ± 0.65% (CCR7−/−) for CD4+ T cells and 2.26% ± 0.35% (Wt) and 4.36% ± 0.38% (CCR7−/−) for CD8+ T cells] (Figure 4D, right panel).
Figure 4.
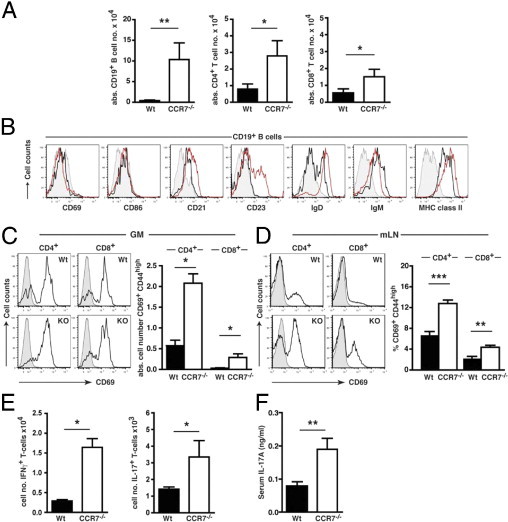
Immune cell subsets of CCR7−/− mice exhibit an activated and inflammatory phenotype within the GM and mLNs. A: Arithmetic means of absolute gastric CD19+ B cells and CD4+ and CD8+ T cells in the GM tissue of Wt (black bars; n = 12 to 13) and CCR7−/− (open bars; n = 5) mice. Data were calculated from flow cytometric analysis of CD3+CD4+ and CD3+CD8+ expression for T-cell subtypes and CD19+ expression for B cells of size-gated gastric lymphocytes. B: Flow cytometric analysis of activation and maturation markers on CD19+-gated B cells isolated from the GM of Wt (black curve; n = 6 to 9) or CCR7−/− mice (n = 7 to 10; red curve: isotype control). GM-derived (C) or mLN-derived (D) T-cell subsets (gated on CD4+ or CD8+) of 6- to 10-month-old CCR7−/− (GM, n = 12; mLN, n = 12 to 14) and Wt mice (GM, n = 9; mLN, n = 6 to 8) were stained for CD69 expression and analyzed by flow cytometry (shaded curve: isotype control). Total numbers of activated CD69+ CD44high T-cell subsets in GM (C; right panel) and percentages of activated CD69+ CD44high T-cell subsets in mLN (D; right panel) of Wt and CCR7−/− mice are shown. E: Flow cytometric analysis of IFN-γ or IL-17A production in GM-derived CD4+ T cells from Wt (n = 9) and CCR7−/− mice (n = 3) after in vitro culture with (+) or without (-) brefeldin, ionomycin, and PMA. Total numbers of gastric CD4+ IFN-γ+ or CD4+ IL-17A+ cells are shown. F: Serum samples from 4- to 6-week-old Wt (black bar; n = 9) and CCR7−/− mice (open bar; n = 9) were analyzed for the TH17 cytokine IL-17A by enzyme-linked immunosorbent. Bars in A–E represent mean ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001; Mann-Whitney U-test.
The total number of IFN-γ– or IL-17A–producing gastric CD4+ T cells was also significantly elevated in CCR7−/− mice (Figure 4E). In addition, increased IL-17A levels were detected in the serum of young (4 to 6 weeks of age) (Figure 4F) and old (12 months of age; data not shown) CCR7−/− mice compared with Wt mice. The inflammatory phenotype of CCR7−/− T lymphocytes was further supported by chemokine receptor expression. Approximately 10% of GM-derived T lymphocytes expressed the TH1-associated chemokine receptors CCR5 and/or CXCR3 (see Supplemental Figure S2 at http://ajp.amjpathol.org). The percentage of mLN-derived CD4+ T cells expressing CCR5 and/or CXCR3 was also enhanced in CCR7−/− compared with Wt mice (see Supplemental Figure S2 at http://ajp.amjpathol.org).
CD4+ T Cells Are Crucial for the Development of AIG and Ectopic Follicular Aggregates in CCR7−/− Mice
GM-infiltrating T cells, which exhibited an activated phenotype, were most likely involved in the production of inflammatory mediators. To assess the role of CD4+ T lymphocytes, we depleted CD4+ T cells with an anti-CD4 T-cell antibody starting at the age of 2 weeks until 6 or 8 weeks of age (Figure 5A). Strikingly, the formation of ectopic lymphoid follicles in the GM of CCR7−/− mice was completely abolished, whereas CCR7−/− mice treated with an isotype control antibody revealed normal development of TLOs as observed before (Figure 5A). Furthermore, CD4+ T-cell–depleted mice did not show any sign of parietal cell loss (Figure 5B, upper panel), and also no development of GM metaplasia (lower panel) could be observed compared with isotype-treated mice. Hence, activated CD4+ T cells act as crucial effectors and mediators in the development of AIG, including gastric TLO formation in CCR7−/− mice.
Figure 5.
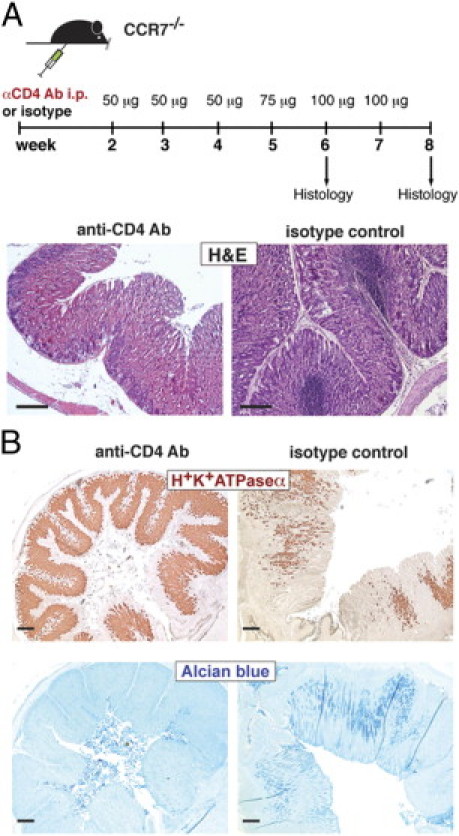
CD4+ T cells are crucial for the development of AIG and ectopic follicular aggregates. A: Schematic representation of CD4+ T-cell depletion in CCR7−/− mice. CCR7−/− mice were treated with 50 to 100 μg (as indicated) of anti-CD4 antibody or isotype control antibody i.p. starting at 2 weeks of age up to 6 or 8 weeks of age (once per week). B: Paraffin-embedded stomach sections of anti-CD4 Ab- (n = 5) or isotype control antibody-treated mice (n = 7) were stained with H&E for parietal cells (brown; H+/K+-ATPase α; upper panel) or acidic mucopolysaccharides (Alcian blue; lower panel). Scale bar = 200 μm.
Gastrointestinal accumulation of regulatory T cells (Tregs) is thought to reinforce the delicate balance between immune activation and tolerance at mucosal sites.27 To determine whether Tregs were present in the GM tissue of CCR7−/− mice, we stained stomach sections for the presence of Foxp3+ Tregs. Foxp3+ Tregs were frequently found within and adjacent to the gastric TLOs in CCR7−/− mice. In comparison, only a few Foxp3+ Tregs were detected in the GM of Wt mice (see Supplemental Figure S1B at http://ajp.amjpathol.org).
To analyze the contribution of DCs to gastric TLO formation, we backcrossed CCR7−/− mice with a CD11c DTR transgenic mouse strain.20 In nondepleted CCR7−/− CD11c.DTR mice, most of the CD11c+ DCs resided within or in close proximity to gastric TLOs (Figure 6A, left panel). Mice at 3 months of age were depleted of DCs over a time period of 14 to 16 days (Figures 6A, right panel, and 6B). Whereas CD11c+ DC depletion in the spleen was efficient (Figure 6B, upper panel), CD11c+ DC depletion in mucosal TLOs of CCR7−/− mice was incomplete [Figure 6B (lower panel) and Figure 6C (lower panel)]. Partial depletion of DCs was associated with smaller gastric TLOs in DT-treated CCR7−/−CD11c.DTR mice, but complete resolution of TLOs could not be observed (Figure 6A, right panel). Next, we characterized DC subsets in the gastrointestinal tissue of CCR7−/− mice compared with DC subsets in mLNs. A unique population of gastric CD11cint CD103+ MHC class IIvariable CD3− DC-like cells was found in CCR7−/− mice, which was absent from mLNs and from GM tissue of Wt control animals (Figure 6, D and E; see also Supplemental Figure S3 at http://ajp.amjpathol.org).
Figure 6.
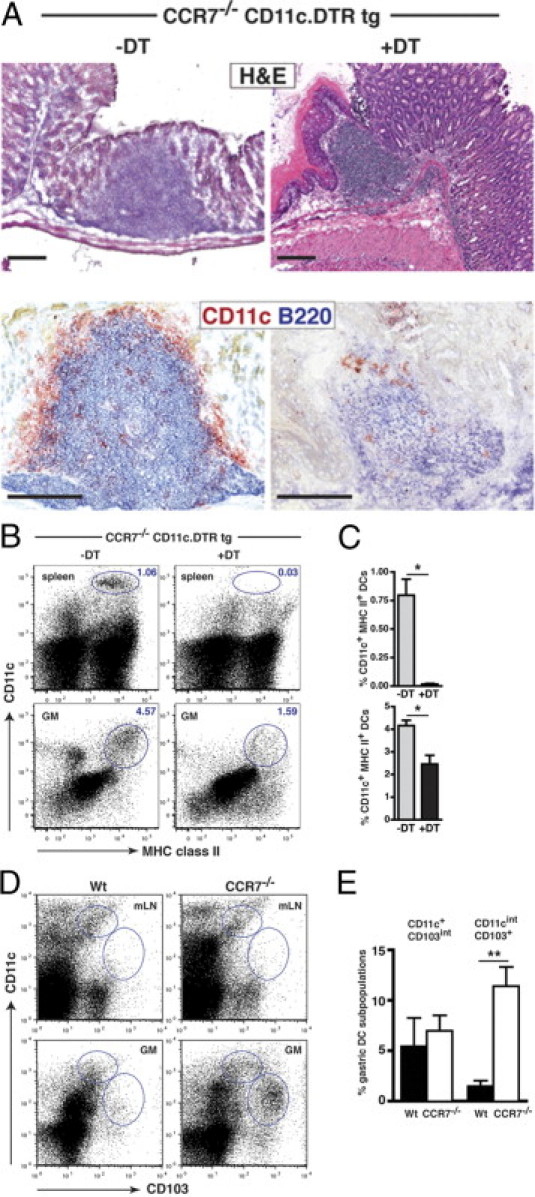
Gastric TLOs of CCR7−/− mice contain an intestinal type of CD11cint CD103+ DCs. A: Stomach sections of 3-month-old CCR7−/−CD11c.DTR transgenic mice, treated with PBS (-DT) or treated with DT (+DT) were stained with H&E or for CD11c+ DCs (red) and B220+ B cells (blue). Scale bar = 200 μm. B: CD11c+ DC depletion was analyzed in the spleen and in the GM of 3-month-old CCR7−/−CD11c.DTR transgenic mice, treated with PBS (-DT) or DT (+DT), by flow cytometric analysis. Representative dot plots for CD11c and MHC class II expression are shown. C: Percentage of CD11c+ MHC class II+ DCs in the spleen (upper panel) and GM (lower panel) of PBS-treated CCR7−/−CD11c.DTR transgenic (gray bars; n = 4) and DT-treated CCR7−/−CD11c.DTR transgenic mice (black bars; n = 4) mice. D: Dendritic cells isolated from GM and mLNs of Wt (GM, n = 3; mLN, n = 4) and CCR7−/− (GM, n = 8; mLN, n = 4) mice (2 to 3 months old) were analyzed by flow cytometry analysis. Representative data for CD103 and CD11c expression are shown. E: Quantification of CD11c+CD103int and CD11cintCD103+ DCs in the GM of Wt (black bars) and CCR7−/− (open bars) mice. Bars represent mean ± SEM. **P < 0.01; Mann-Whitney U-test.
CCR7 Deficiency Is a Prerequisite for Lymphocyte Retention within Gastric TLOs
To study whether gastric TLOs would also be permissive for nonautoimmune-reactive Wt lymphocytes, we generated mixed BM chimeras by transplanting BM from CCR7−/− (CD45.2) together with BM from congenic B6 (CD45.1) mice in a 1:1 ratio into sublethally irradiated Rag2−/− mice (Figure 7A). As a control, sublethally irradiated Rag2−/− mice were injected with Wt BM only. Three to 8 months after transfer, the frequency of gastric Wt and CCR7−/− lymphocytes and the formation of gastric TLOs were analyzed. Rag2−/− recipients of mixed BM cells exhibited the formation of large TLOs in the submucosa and lamina propria of the stomach accompanied by severe parietal cell loss and gastric metaplasia (Figure 7, B and C). Transfer of Wt BM alone did not cause lymphoid neogenesis (data not shown) or altered mucosal tissue architecture (Figure 7C). The proportion of Wt BM-derived and CCR7−/− BM-derived lymphocytes within mucosal TLOs revealed a striking difference. Most lymphocytes contributing to the follicular aggregates were of CCR7−/− origin, whereas only single Wt lymphocytes were found scattered throughout the lymphoid follicles and within the gastric lamina propria (Figure 7B). In contrast, the ratio of Wt to CCR7−/− lymphocytes in the mLNs was increased, thus underlining the role of CCR7 as a major LN-homing receptor (data not shown). These results support the notion that only autoreactive CD4+CCR7−/− T cells have the ability to lodge within the GM tissue and subsequently give rise to TLOs.
Figure 7.
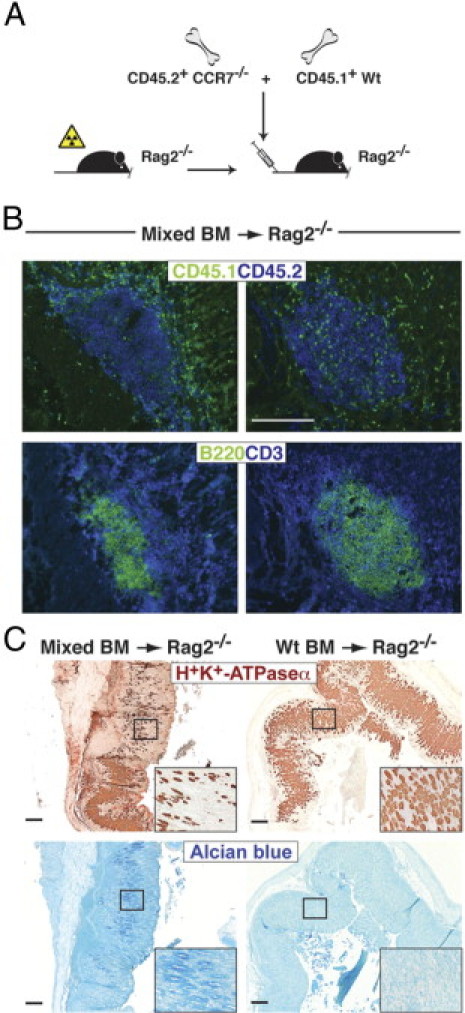
Autoreactive CCR7-deficient lymphocytes induce AIG and gastric TLO formation in Rag2−/− mice. A: Schematic representation for generation of BM chimeric mice by reconstitution of sublethally irradiated Rag2−/− mice with equal numbers of CD45.2+ C57BL/6 CCR7−/− and congenic CD45.1+ C57BL/6 Wt BM cells. B: Stomach sections were stained for CD45.1+ (green) and CD45.2+ (blue) cells (upper panel) or B220+ B (green) and CD3+ T (blue) cells (lower panel). Shown are representative stains of ectopic follicular aggregates in two of seven mice analyzed. C: Paraffin-embedded stomach sections were stained for H+/K+-ATPase α (brown) or acidic mucopolysaccharides (Alcian blue). Scale bar = 200 μm.
Manifestation of AIG Is Independent from Organized TLOs and from Peripheral LNs as Inductive Sites
The involvement of TLOs in the initiation and maintenance of chronic inflammatory autoimmune diseases is still unclear.7 To determine whether local TLOs and peripheral LNs are a prerequisite for the development of AIG in CCR7−/− mice, we crossed CCR7−/− mice into the Ltα−/− mouse strain to generate CCR7/Ltα double-deficient (CCR7−/−Ltα−/−) mice. Ltα, which is a member of the TNF family of cytokines, facilitates the development of organized secondary and tertiary lymphoid tissues. Subsequently, Ltα−/− mice entirely lack LNs and Peyer's patches (PPs) and have a disrupted splenic architecture.19 As expected, CCR7−/−Ltα−/− mice also lacked all peripheral LNs and failed to develop organized gastric TLOs. However, they displayed unorganized lymphoid infiltrates consisting of B and T lymphocytes (Figure 8A) and macrophages (data not shown) within the GM and submucosa. The frequency of naïve to activated T cells was not significantly altered in peripheral blood and splenic T lymphocytes of CCR7−/−Ltα−/− mice compared with Wt mice (data not shown). This was in contrast to the enhanced activation status that we found in peripheral and LN-derived T cells of CCR7 single-deficient mice and suggests that peripheral T cells are predominantly activated in SLOs. Nevertheless, despite the lack of organized gastric TLOs in CCR7−/−Ltα−/− mice, we observed substantial loss of parietal cells (Figure 8B, upper panel) accompanied by the development of GM metaplasia (Figure 8B, lower panel). We also determined serum autoantibody titers against gastric antigen. Gastric antigen-specific IgG was significantly increased in CCR7−/− compared with Wt mice, whereas CCR7−/−Ltα−/− (double knockout) mice only showed a minor increase (Figure 8C). We suggest that in our AIG model, the induction of humoral immune responses is more dependent on organized SLO or TLO structures compared with the induction of T-cell–mediated autoimmune responses.
Figure 8.
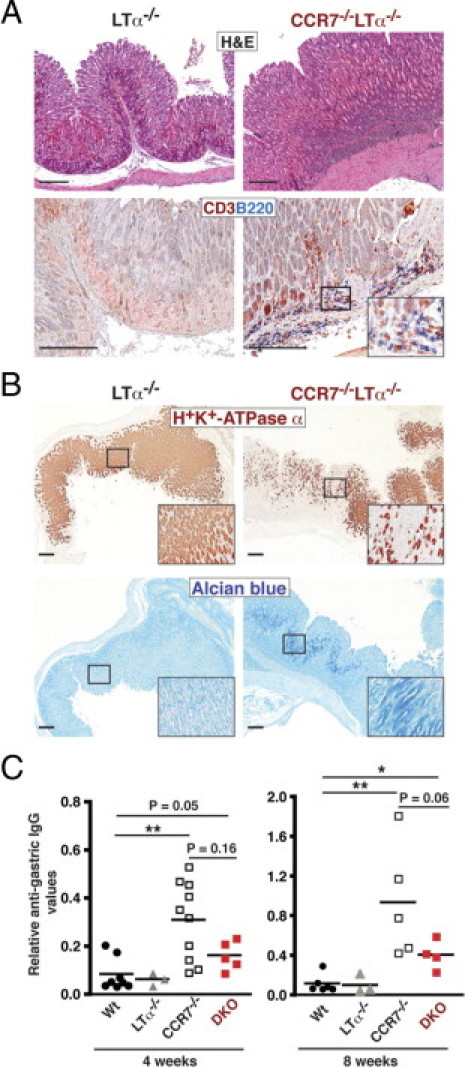
Development of AIG in CCR7−/−Ltα−/− mice is independent of organized lymph nodes and gastric TLOs. A: Paraffin-embedded stomach sections from 1- to 2-month-old Ltα−/− and CCR7−/−Ltα−/− double-deficient mice were stained with H&E or for B220+ B (blue) and CD3+ T (red) cells. B: Sections from 1- to 2-month-old Ltα−/− and CCR7−/−LTα−/− mice were stained for H+/K+-ATPase α (brown) and acidic mucopolysaccharides (Alcian blue). Scale bar = 200 μm. C: Relative serum titers of IgG autoantibodies directed against gastric-derived antigen of 1-month-old (left panel) and 2-month-old (right panel) Wt, Ltα−/−, CCR7−/−, and CCR7−/−Ltα−/− (double knockout; DKO) mice. Serum samples were used at a dilution of 1:50. Absorbance values at 450 nm represent the relative antigastric IgG. Mean values are shown; *P < 0.05, **P < 0.01; Mann-Whitney U-test.
Discussion
In the present study, we demonstrate that CCR7−/− mice develop an early spontaneous AIG with near-complete penetrance. This spontaneous model allowed us to address whether the development of gastric TLOs might contribute to the induction and maintenance of AIG.
The local inflammatory microenvironment, which promotes the formation of TLOs, is mediated by interactions of immune effector cells and tissue-resident cells. Hence, we analyzed the cellular requirements for TLO formation in CCR7−/− mice, a process that is believed to mimic several aspects of SLO development. The function of homeostatic chemokines in SLO development and organization is mutually dependent on signaling pathways activated by the TNF/Lt family of cytokines.28,29 Ltα1β2 expressing lymphoid tissue inducer (LTi) cells are the first cells to colonize the LN anlagen and are absolutely required for neonatal SLO development. However, it is less clear whether LTi cells are required for the formation of TLOs.7,30 Transgenic mice overexpressing IL-7 but lacking either RORγt, a transcription factor needed for LTi cell development, or Ltα1β2 did not support the generation of LNs, PPs, or TLOs.31 However, in CCR7−/−/RORγt−/− double knockout mice, we still observed the development of ectopic follicular aggregates within the GM tissue (U.E. Höpken et al, unpublished data). Thus, in our spontaneous AIG model, the development of gastric TLOs was independent of LTi cells. This is in line with a recent publication by Lochner and coworkers, which showed that in RORγt−/− mice microbiota-induced TLOs could be observed in the absence of LTis.32
In transgenic mice that express CCL21 in the thyroid gland, it was found that TLO formation involved the interaction of mature CD4+ T cells with clusters of DCs, a process mediated by homeostatic chemokines.33 In support of a decisive role of CD4+ T cells during TLO formation, depletion of all CD4+ T cells led to a complete abrogation of TLO formation in the GM tissue of CCR7−/− mice. Notably, on transplantation of CCR7−/−/Wt mixed BM into Rag−/− recipients, gastric TLOs frequently developed but were populated predominantly by CCR7−/−-derived lymphocytes. This suggests that only activated and putatively autoreactive CCR7−/− lymphocytes contribute to ectopic gastric lymphoid cell clusters. In addition, we and others recently showed that CCR7 deficiency led to an impaired lymphocytic recirculation and subsequently caused accumulation of lymphocytes in nonlymphoid tissues.15,34 This might cause an extended residence time of putatively autoreactive lymphocytes at mucosal sites and thereby support the development of AIG and the formation of gastric TLOs.
A recent publication showed that CD11c+ DCs are essential for the maintenance of TLOs in the lungs of influenza virus–infected CD11c.DTR transgenic mice.35 To further address the cellular requirements for TLO formation, we depleted DCs via administration of DT in CCR7−/−CD11c.DTR transgenic mice. We obtained an ineffective depletion of CD11c+ DCs in gastric TLOs compared with a near-complete depletion of DCs in the spleen. Incomplete depletion of a CD11cint/lo DC population has already been shown by Garbi et al,20 an effect that was explained by lower expression of the DTR in these cells. In CCR7−/− mice, we showed a unique population of gastric CD11cint CD103+ DCs, which was clearly absent from mLNs and GM tissue in Wt mice. Thus, in our model we envisage that reduced CD11c expression of this gastric TLO-associated subpopulation of DCs is responsible for their ineffective depletion. A functional subset of intestinal CD11c+CD103+ MHC class II+ DCs with classic DC function was previously described.36,37 The distinct subpopulation of gastric CD11cint CD103+ DCs in the CCR7−/− mice differs from those intestinal CD11c+CD103+ MHC class II+ DCs with regard to their lower CD11c and variable MHC class II expression levels. Hence, this gastric DC cell subpopulation most likely represents an immature DC subtype and might not serve classic DC function. CD11c intermediate expressing DCs have also been described in lungs infected with Mycobacterium tuberculosis38 and recently in murine Listeriosis.39 The authors demonstrated that these CD11cint DCs are critical for the recruitment of both T cell and myeloid cell migration to the lung38and that they possessed T-cell–priming abilities.39 We hypothesize that gastric CD11cint CD103+ MHC class IIvariable DCs support the recruitment of autoreactive T cells toward and within the TLOs of CCR7−/− mice and might also support induction and maintenance of gastric TLO formation.
However, because of ineffective DC depletion the question of whether interaction of mature and potentially autoreactive T cells with DCs is crucial for the initial development and maintenance of gastric TLOs cannot be definitely answered. Taken together, we suggest that during autoimmune inflammation autoreactive CD4+ T cells mediate TLO development in GM tissue, most likely in cooperation with local DCs, macrophages, and B cells.
Strikingly, manifestation of AIG clearly preceded the development of TLO formation in CCR7−/− mice. Thus, the central question arises of whether autoreactive lymphocytes must be structurally organized to initiate disease pathogenesis.
In all other animal models of AIG, the generation of gastric antigen-specific CD4+ T cells is pivotal for the pathology. Furthermore, mononuclear infiltrates within the GM, loss of parietal and zymogenic cells, and the production of parietal cell autoantibodies are indicative of gastric autoimmunity.11 We showed that activated and putatively autoreactive CD4+ T cells act as crucial effectors and mediators in the development of AIG in CCR7−/− mice. Accordingly, we suggest that the autoreactive CD4+ T cells mediate the formation of an inflammatory gastric milieu characterized by the production of proinflammatory cytokines and adhesion molecules. Notably, we found enhanced gastric expression of the inflammatory cytokine IL-1β in CCR7−/− mice. This result provides a mechanistic link to the development of gastric atrophy, including parietal cell loss and metaplasia in CCR7−/− mice. In a recent study, an intimate relationship between acid secretion and expression of the gastric morphogenic factor Shh was shown, and it was suggested that IL-1β promotes Helicobacter pylori–induced gastric atrophy through suppression of Shh.24 Reduced expression of Shh by parietal cells together with increased expression of transcriptional activators of intestinal and pancreatic differentiation (ie, CDX2 and PDX1) could also lead to gastric atrophy and metaplasia.40 Here, we show that the mechanism by which IL-1β induces gastric atrophy during H. pylori–induced gastritis also applies to AIG. We envisage that loss of parietal cells in CCR7−/− mice is linked to the down-regulation of Shh through an IL-1β–mediated and acid-dependent mechanism.
Impaired function or removal of CD4+CD25+ Tregs can also initiate AIG. In a lymphopenic model of AIG induced by neonatal thymectomy, AIG is mainly caused by extirpation of Tregs through prevention of their export from the thymus.11 In CCR7−/− mice, naturally occurring Tregs develop normally in the thymus, and their in vitro suppressive activity is unhampered, whereas their suppressive function in vivo is severely impaired.18 This functional impairment is mainly due to the inability of CCR7-deficient Tregs to home to the T-cell zone of LNs, where Tregs proliferate and expand on contact with their cognate antigen. In accordance, we detected numerous Tregs within loose gastric lymphoid aggregates and in structured TLOs of CCR7−/− mice. However, their suppressive activity might be impaired due to the inability of CCR7−/− Tregs to recirculate properly through tissue-associated LNs, which could explain why AIG evolves in the presence of numerous Tregs.
To investigate whether activation of autoreactive T cells can occur in the GM without structured TLOs and in the absence of classic LNs, we generated CCR7−/−LTα−/− double knockout mice. Despite the absence of all LNs, PPs, and structured gastric TLOs, these mice spontaneously developed severe AIG, comparable to CCR7 single knockout mice. In contrast, former studies in aly/aly mice, which completely lack LNs and PPs, suggested that cell-mediated immunity, including autoimmune responses, cannot be initiated in the absence of LNs.41 This view was challenged by a recent study showing that aly/aly mice, which had been reconstituted with Wt hematopoietic cells and were additionally splenectomized, developed pathogenic T-cell responses.42 In agreement, initiation of cellular T-cell responses to neoantigens and autoantigens was seen in LtβR−/− and Ltα−/− mice strains that lack all peripheral LNs.41,43 Although we have not yet investigated the spleen as an effector site for AIG development in the CCR7−/−LTα−/− mice, previous data of splenectomized Ltα−/− and LtβR−/− mice suggested that the spleen, PPs, and LNs are not absolutely required for the development of gastrointestinal immune responses. Instead, other tissues, including parenchymal organs, can develop into alternative sites of antigen processing and presentation.41
Taken together, we define the observed pathomorphologic phenotype in CCR7−/− mice as an autoimmune disease that results in atrophic gastritis. We show that immunopathogenetic steps that lead to spontaneous AIG can proceed at sites that are separate from classic LNs and organized TLOs.
Acknowledgments
We thank Kerstin Krüger, Katrin Räbel, Kerstin Gerlach, and Heike Schwede for excellent technical assistance and Christoph Loddenkemper for pathohistologic evaluation and helpful discussions.
Footnotes
Supported by the Priority Program SFB 633 of the German Research Foundation.
Supplemental material for this manuscript can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.04.012.
Supplementary data
GC formation and Foxp3+ Tregs in gastric TLOs of CCR7−/− mice. Stomach sections were stained for GC with peanut agglutinin (PNA) (in red) (A) and for Foxp3+ Tregs (in brown) and B220+ B cells (in blue) (B). Representative photographs are shown. The arrows show the GC or Foxp3+ Tregs. Scale bar = 200 μm.
Enhanced expression of the TH1-associated chemokine receptors CCR5 and/or CXCR3 on GM- and mLN-derived T lymphocytes of CCR7−/− mice. CD3+ CD4+-gated T cells from GM of CCR7−/− or from mLN of Wt and CCR7−/− mice (n = 3 per group) were analyzed for expression of CCR5 and CXCR3 (left panel). Percentages of CCR5+ or CXCR3+ CD4+ T cells are indicated and differences between age-matched Wt and CCR7−/− mLNs are shown (right panel). Bars represent mean ± SEM.
The CD11cintCD103+ DC-like cell subpopulation in the GM of CCR7−/− mice was largely CD3− and expressed variable levels of MHC class II. GM-derived leukocytes were stained for CD11c, CD103, CD3, and MHC class II expression. Cells were gated for CD11cint CD103+ (left panel) and further analyzed for CD3 and MHC class II expression (right panel). One representative dot plot is shown (n = 4).
References
- 1.Kim H.J., Berek C. B cells in rheumatoid arthritis. Arthritis Res. 2000;2:126–131. doi: 10.1186/ar77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barone F., Bombardieri M., Rosado M.M., Morgan P.R., Challacombe S.J., De Vita S., Carsetti R., Spencer J., Valesini G., Pitzalis C. CXCL13. CCL21, and CXCL12 expression in salivary glands of patients with Sjögren's syndrome and MALT lymphoma: association with reactive and malignant areas of lymphoid organization. J Immunol. 2008;180:5130–5140. doi: 10.4049/jimmunol.180.7.5130. [DOI] [PubMed] [Google Scholar]
- 3.Wu Q., Salomon B., Chen M., Wang Y., Hoffman L.M., Bluestone J.A., Fu Y.X. Reversal of spontaneous autoimmune insulitis in nonobese diabetic mice by soluble lymphotoxin receptor. J Exp Med. 2001;193:1327–1332. doi: 10.1084/jem.193.11.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kendall P.L., Yu G., Woodward E.J., Thomas J.W. Tertiary lymphoid structures in the pancreas promote selection of B lymphocytes in autoimmune diabetes. J Immunol. 2007;178:5643–5651. doi: 10.4049/jimmunol.178.9.5643. [DOI] [PubMed] [Google Scholar]
- 5.Katakai T., Hara T., Sugai M., Gonda H., Shimizu A. Th1-biased tertiary lymphoid tissue supported by CXC chemokine ligand 13-producing stromal network in chronic lesions of autoimmune gastritis. J Immunol. 2003;171:4359–4368. doi: 10.4049/jimmunol.171.8.4359. [DOI] [PubMed] [Google Scholar]
- 6.Drayton D.L., Liao S., Mounzer R.H., Ruddle N.H. Lymphoid organ development: from ontogeny to neogenesis. Nat Immunol. 2006;7:344–353. doi: 10.1038/ni1330. [DOI] [PubMed] [Google Scholar]
- 7.Carragher D.M., Rangel-Moreno J., Randall T.D. Ectopic lymphoid tissues and local immunity. Semin Immunol. 2008;20:26–42. doi: 10.1016/j.smim.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Katakai T., Shimizu A. Undesired meeting of lymphocytes: organ-specific infiltration and the organization of ectopic lymphoid tissue in a murine experimental autoimmune gastritis. Immunol Lett. 2008;118:103–109. doi: 10.1016/j.imlet.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 9.Henry RA, Kendall PL: CXCL13 blockade disrupts B lymphocyte organization in tertiary lymphoid structures without altering B cell receptor bias or preventing diabetes in nonobese diabetic mice. J Immunol 185:1460–1465 [DOI] [PMC free article] [PubMed]
- 10.van Driel I.R., Baxter A.G., Laurie K.L., Zwar T.D., La Gruta N.L., Judd L.M., Scarff K.L., Silveira P.A., Gleeson P.A. Immunopathogenesis, loss of T cell tolerance and genetics of autoimmune gastritis. Autoimmun Rev. 2002;1:290–297. doi: 10.1016/s1568-9972(02)00066-6. [DOI] [PubMed] [Google Scholar]
- 11.Field J., Biondo M.A., Murphy K., Alderuccio F., Toh B.H. Experimental autoimmune gastritis: mouse models of human organ-specific autoimmune disease. Int Rev Immunol. 2005;24:93–110. doi: 10.1080/08830180590884585. [DOI] [PubMed] [Google Scholar]
- 12.Alderuccio F., Toh B.H. Spontaneous autoimmune gastritis in C3H/He mice: a new mouse model for gastric autoimmunity. Am J Pathol. 1998;153:1311–1318. doi: 10.1016/S0002-9440(10)65676-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Forster R., Schubel A., Breitfeld D., Kremmer E., Renner-Muller I., Wolf E., Lipp M. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell. 1999;99:23–33. doi: 10.1016/s0092-8674(00)80059-8. [DOI] [PubMed] [Google Scholar]
- 14.Hopken U.E., Droese J., Li J.P., Joergensen J., Breitfeld D., Zerwes H.G., Lipp M. The chemokine receptor CCR7 controls lymph node-dependent cytotoxic T cell priming in alloimmune responses. Eur J Immunol. 2004;34:461–470. doi: 10.1002/eji.200324690. [DOI] [PubMed] [Google Scholar]
- 15.Debes G.F., Arnold C.N., Young A.J., Krautwald S., Lipp M., Hay J.B., Butcher E.C. Chemokine receptor CCR7 required for T lymphocyte exit from peripheral tissues. Nat Immunol. 2005;6:889–894. doi: 10.1038/ni1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hopken U.E., Wengner A.M., Loddenkemper C., Stein H., Heimesaat M.M., Rehm A., Lipp M. CCR7 deficiency causes ectopic lymphoid neogenesis and disturbed mucosal tissue integrity. Blood. 2007;109:886–895. doi: 10.1182/blood-2006-03-013532. [DOI] [PubMed] [Google Scholar]
- 17.Davalos-Misslitz A.C., Rieckenberg J., Willenzon S., Worbs T., Kremmer E., Bernhardt G., Forster R. Generalized multi-organ autoimmunity in CCR7-deficient mice. Eur J Immunol. 2007;37:613–622. doi: 10.1002/eji.200636656. [DOI] [PubMed] [Google Scholar]
- 18.Forster R., Davalos-Misslitz A.C., Rot A. CCR7 and its ligands: balancing immunity and tolerance. Nat Rev Immunol. 2008;8:362–371. doi: 10.1038/nri2297. [DOI] [PubMed] [Google Scholar]
- 19.De Togni P., Goellner J., Ruddle N.H., Streeter P.R., Fick A., Mariathasan S., Smith S.C., Carlson R., Shornick L.P., Strauss-Schoenberger Abnormal development of peripheral lymphoid organs in mice deficient in lymphotoxin. Science. 1994;264:703–707. doi: 10.1126/science.8171322. [DOI] [PubMed] [Google Scholar]
- 20.Hochweller K., Striegler J., Hammerling G.J., Garbi N. A novel CD11c. DTR transgenic mouse for depletion of dendritic cells reveals their requirement for homeostatic proliferation of natural killer cells. Eur J Immunol. 2008;38:2776–2783. doi: 10.1002/eji.200838659. [DOI] [PubMed] [Google Scholar]
- 21.van den Brink G.R., Hardwick J.C., Nielsen C., Xu C., ten Kate F.J., Glickman J., van Deventer S.J., Roberts D.J., Peppelenbosch M.P. Sonic hedgehog expression correlates with fundic gland differentiation in the adult gastrointestinal tract. Gut. 2002;51:628–633. doi: 10.1136/gut.51.5.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van den Brink G.R., Hardwick J.C., Tytgat G.N., Brink M.A., Ten Kate F.J., Van Deventer S.J., Peppelenbosch M.P. Sonic hedgehog regulates gastric gland morphogenesis in man and mouse. Gastroenterology. 2001;121:317–328. doi: 10.1053/gast.2001.26261. [DOI] [PubMed] [Google Scholar]
- 23.Xiao C., Ogle S.A., Schumacher M.A., Orr-Asman M.A., Miller M.L., Lertkowit N., Varro A., Hollande F., Zavros Y. Loss of parietal cell expression of Sonic hedgehog induces hypergastrinemia and hyperproliferation of surface mucous cells. Gastroenterology. 2010;138:550–561. doi: 10.1053/j.gastro.2009.11.002. 561.e551-558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Waghray M., Zavros Y., Saqui-Salces M., El-Zaatari M., Alamelumangapuram C.B., Todisco A., Eaton K.A., Merchant J.L. Interleukin-1beta promotes gastric atrophy through suppression of Sonic Hedgehog. Gastroenterology. 2010;138:562–572. doi: 10.1053/j.gastro.2009.10.043. 572.e561-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marshall A.C., Alderuccio F., Murphy K., Toh B.H. Mechanisms of gastric mucosal cell loss in autoimmune gastritis. Int Rev Immunol. 2005;24:123–134. doi: 10.1080/08830180590884567. [DOI] [PubMed] [Google Scholar]
- 26.van Driel I.R., Read S., Zwar T.D., Gleeson P.A. Shaping the T cell repertoire to a bona fide autoantigen: lessons from autoimmune gastritis. Curr Opin Immunol. 2005;17:570–576. doi: 10.1016/j.coi.2005.09.016. [DOI] [PubMed] [Google Scholar]
- 27.Izcue A., Coombes J.L., Powrie F. Regulatory lymphocytes and intestinal inflammation. Annu Rev Immunol. 2009;27:313–338. doi: 10.1146/annurev.immunol.021908.132657. [DOI] [PubMed] [Google Scholar]
- 28.Muller G., Lipp M. Concerted action of the chemokine and lymphotoxin system in secondary lymphoid-organ development. Curr Opin Immunol. 2003;15:217–224. doi: 10.1016/s0952-7915(03)00014-1. [DOI] [PubMed] [Google Scholar]
- 29.Ware C.F. Network communications: lymphotoxins. LIGHT, and TNF. Annu Rev Immunol. 2005;23:787–819. doi: 10.1146/annurev.immunol.23.021704.115719. [DOI] [PubMed] [Google Scholar]
- 30.Kim M.Y., Kim K.S., McConnell F., Lane P. Lymphoid tissue inducer cells: architects of CD4 immune responses in mice and men. Clin Exp Immunol. 2009;157:20–26. doi: 10.1111/j.1365-2249.2009.03932.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meier D., Bornmann C., Chappaz S., Schmutz S., Otten L.A., Ceredig R., Acha-Orbea H., Finke D. Ectopic lymphoid-organ development occurs through interleukin 7-mediated enhanced survival of lymphoid-tissue-inducer cells. Immunity. 2007;26:643–654. doi: 10.1016/j.immuni.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 32.Lochner M, Ohnmacht C, Presley L, Bruhns P, Si-Tahar M, Sawa S, Eberl G: Microbiota-induced tertiary lymphoid tissues aggravate inflammatory disease in the absence of RORgamma t and LTi cells. J Exp Med 208:125–134 [DOI] [PMC free article] [PubMed]
- 33.Marinkovic T., Garin A., Yokota Y., Fu Y.X., Ruddle N.H., Furtado G.C., Lira S.A. Interaction of mature CD3+CD4+ T cells with dendritic cells triggers the development of tertiary lymphoid structures in the thyroid. J Clin Invest. 2006;116:2622–2632. doi: 10.1172/JCI28993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hopken UE, Winter S, Achtman AH, Kruger K, Lipp M: CCR7 regulates lymphocyte egress and recirculation through body cavities. J Leukoc Biol 87:671–682 [DOI] [PubMed]
- 35.GeurtsvanKessel C.H., Willart M.A., Bergen I.M., van Rijt L.S., Muskens F., Elewaut D., Osterhaus A.D., Hendriks R., Rimmelzwaan G.F., Lambrecht B.N. Dendritic cells are crucial for maintenance of tertiary lymphoid structures in the lung of influenza virus-infected mice. J Exp Med. 2009;206:2339–2349. doi: 10.1084/jem.20090410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schulz O., Jaensson E., Persson E.K., Liu X., Worbs T., Agace W.W., Pabst O. Intestinal CD103+, but not CX3CR1+, antigen sampling cells migrate in lymph and serve classical dendritic cell functions. J Exp Med. 2009;206:3101–3114. doi: 10.1084/jem.20091925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jaensson E., Uronen-Hansson H., Pabst O., Eksteen B., Tian J., Coombes J.L., Berg P.L., Davidsson T., Powrie F., Johansson-Lindbom B., Agace W.W. Small intestinal CD103+ dendritic cells display unique functional properties that are conserved between mice and humans. J Exp Med. 2008;205:2139–2149. doi: 10.1084/jem.20080414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peters W., Cyster J.G., Mack M., Schlondorff D., Wolf A.J., Ernst J.D., Charo I.F. CCR2-dependent trafficking of F4/80dim macrophages and CD11cdim/intermediate dendritic cells is crucial for T cell recruitment to lungs infected with Mycobacterium tuberculosis. J Immunol. 2004;172:7647–7653. doi: 10.4049/jimmunol.172.12.7647. [DOI] [PubMed] [Google Scholar]
- 39.Dresing P, Borkens S, Kocur M, Kropp S, Scheu S: A fluorescence reporter model defines “Tip-DCs” as the cellular source of interferon beta in murine listeriosis. PLoS One 5:e15567 [DOI] [PMC free article] [PubMed]
- 40.Faller G., Kirchner T. Immunological and morphogenic basis of gastric mucosa atrophy and metaplasia. Virchows Arch. 2005;446:1–9. doi: 10.1007/s00428-004-1157-3. [DOI] [PubMed] [Google Scholar]
- 41.Hofmann J, Greter M, Du Pasquier L, Becher B: B-cells need a proper house, whereas T-cells are happy in a cave: the dependence of lymphocytes on secondary lymphoid tissues during evolution. Trends Immunol 31:144–153 [DOI] [PubMed]
- 42.Greter M., Hofmann J., Becher B. Neo-lymphoid aggregates in the adult liver can initiate potent cell-mediated immunity. PLoS Biol. 2009;7:e1000109. doi: 10.1371/journal.pbio.1000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lund F.E., Partida-Sanchez S., Lee B.O., Kusser K.L., Hartson L., Hogan R.J., Woodland D.L., Randall T.D. Lymphotoxin-alpha-deficient mice make delayed, but effective: T and B cell responses to influenza. J Immunol. 2002;169:5236–5243. doi: 10.4049/jimmunol.169.9.5236. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
GC formation and Foxp3+ Tregs in gastric TLOs of CCR7−/− mice. Stomach sections were stained for GC with peanut agglutinin (PNA) (in red) (A) and for Foxp3+ Tregs (in brown) and B220+ B cells (in blue) (B). Representative photographs are shown. The arrows show the GC or Foxp3+ Tregs. Scale bar = 200 μm.
Enhanced expression of the TH1-associated chemokine receptors CCR5 and/or CXCR3 on GM- and mLN-derived T lymphocytes of CCR7−/− mice. CD3+ CD4+-gated T cells from GM of CCR7−/− or from mLN of Wt and CCR7−/− mice (n = 3 per group) were analyzed for expression of CCR5 and CXCR3 (left panel). Percentages of CCR5+ or CXCR3+ CD4+ T cells are indicated and differences between age-matched Wt and CCR7−/− mLNs are shown (right panel). Bars represent mean ± SEM.
The CD11cintCD103+ DC-like cell subpopulation in the GM of CCR7−/− mice was largely CD3− and expressed variable levels of MHC class II. GM-derived leukocytes were stained for CD11c, CD103, CD3, and MHC class II expression. Cells were gated for CD11cint CD103+ (left panel) and further analyzed for CD3 and MHC class II expression (right panel). One representative dot plot is shown (n = 4).