

Abstract
3-nitrotyrosine (3NT) is an oxidative posttranslational modification associated with many diseases. Determining the specific sites of this modification remains a challenge due to the low stoichiometry of 3NT modifications in biological samples. Mass spectrometry-based proteomics is a powerful tool for identifying 3NT modifications, however several reports identifying 3NT sites were later demonstrated to be incorrect, highlighting that both the accuracy and efficiency of these workflows need improvement. To advance our understanding of the chromatographic and spectral properties of 3NT-containing peptides we have adapted a straightforward, reproducible procedure to generate a large set of 3NT peptides by chemical nitration of a defined, commercially available 48 protein mixture. Using two complementary LC-MS/MS platforms, a QTOF (QSTAR Elite) and dual pressure ion trap mass spectrometer (LTQ Velos), we detected over 200 validated 3NT-containing peptides with significant overlap in the peptides detected by both systems. We investigated the LC-MS/MS properties for each peptide manually using defined criteria and then assessed their utility to confirm that the peptide was 3NT modified. This broad set of validated 3NT-containing peptides can be utilized to optimize mass spectrometric instrumentation and data mining strategies or further develop 3NT peptide enrichment strategies for this biologically important, oxidative posttranslational modification.
Keywords: 3-nitrotyrosine, mass spectrometry, multiple reaction monitoring, posttranslational modification, spectral library
Introduction
The modification of tyrosine to 3-nitrotyrosine (3NT) under conditions of oxidative stress has become established as a significant event that occurs during the progression of many diseases [1] including, inflammation [2, 3], neurodegeneration [4-6], cardiovascular disease [7, 8] and aging [9, 10]. 3NT modification has been of significant interest due to the fact that a nitro group at the 3 position of the aromatic ring of tyrosine decreases the pKA of the neighboring hydroxyl moiety, and may alter protein structure, function, and interactions [11, 12]. Investigation into the formation of 3-nitrotyrosine has led to several proposed mechanisms [13-15] and uncovered numerous examples of specific proteins featuring a 3NT modification. Carboxypeptidase B1 [16], synuclein [17], synaptophysin [18], SERCA [10], muscle glycogen phosphorylase [19], manganese superoxide dismutase [9], and glutathione reductase [20] are some of the more important proteins that have been identified as being selectively nitrated and whose biological functions are affected by nitration.
While the availability of pan-3NT [21, 22] antibodies has led to the characterization of proteins that are 3NT modified, the low stoichiometry and extent of modifications in nitrated tyrosine residues (only 5 3NT residues per 10,000 tyrosines) [23] has hampered determination of the specific sites that are modified. Mass spectrometry based proteomics has become the predominant approach to identify sites of 3NT modification and dozens of proteomic studies have been undertaken with the aim of characterizing as much of the ‘nitroproteome’ as possible in various diseases and biological processes (reviewed in [24, 25]). These studies are limited by the fact that while numerous 3NT enrichment approaches using antibodies and chemical enrichment exist [26-31], none has been widely adopted or shown widespread success [32]. Approaches to improve 3NT peptide detection at the mass spectrometry level, such as use of precursor ion scanning, have also met with limited success to identify sites of 3NT modification [33]. Although matrix assisted laser desorption ionization time-of-flight (MALDI TOF) analysis of 3NT-containing peptides produces signature neutral loss ions due to laser-induced photochemical decomposition, its utility for complex samples is limited by the poor dynamic range of MALDI MS [34]. Nonetheless, proteomics approaches have been able to identify hundreds of 3NT sites in peroxynitrite treated cell lysates [29, 30], though considerably less success has been achieved in identifying these oxidative modifications in vivo such as those that accumulate during aging [35, 36], for example. It is generally assumed that the continued advances in the sensitivity, dynamic range and sampling efficiency of modern mass spectrometers and their proteomic workflows are poised to lead to the discovery of even more 3NT sites in the near future.
Large-scale mass spectrometry-based proteomics studies are often limited by the bioinformatic tools available to mine the hundreds of thousand or more MS/MS spectra collected during the course of a typical study. While identification of a protein in a complex sample is now fairly routine [37], typically requiring the identification of two or more proteotypic peptides, characterization of posttranslational modifications (PTMs) such as protein nitration is more difficult since it almost exclusively relies on the correct interpretation of only a single MS/MS spectrum. Several biologically interesting PTMs may overlap in mass with other modifications which requires careful consideration when interpreting spectra and evaluating potential ambiguity. For example, phosphorylation and sulfation can result in near isobaric peptides, as do trimethyl and acetylated lysines; although high mass accuracy can potentially differentiate among them. Teasing apart the PTM assignments is possible, however, through other experimental design features, such as specifically enriching for phosphopeptides with antibodies or chemical resins. In some cases, close inspection of the specific MS/MS fragmentation can differentiate between PTMs that may be close in mass. For example, the presence of an immonium ion at m/z 126.1 is only generated for acetylated lysine residues and can be used to differentiate these modified peptides from trimethyl-lysines [38, 39]. Acetylated and trimethylated peptides can also be chromatographically resolved by liquid chromatography [40].
The interpretation of MS/MS spectra is additionally complicated by the fact that mass spectrometry can detect unanticipated low abundant covalent peptide modifications. This underpins the important utility of mass spectrometry to discover authentic in vivo modifications but may also lead to spurious results or identification of artificially introduced modifications. Chemical artifacts introduced during SDS-PAGE are particularly problematic. A recent report by Nielsen et al. described that iodoacetamide commonly used for cysteine alkylation during sample preparation for proteomics experiments may mimic ubiquitination when non-specifically reacting with lysine residues [41]. Several 3NT proteomics studies in particular have been the subject of scrutiny in recent years [42-45] and have highlighted the need for careful evaluation of individual MS/MS spectra in order to eliminate false positive assignments. In these reported cases the nitrated peptides are at very low levels in vivo and a combination of several unexpected modifications on peptide sequences of interest led to MS/MS spectra that partially matched the theoretical spectra of a 3NT modified peptide. However, recognition of specific discrepancies in the spectra and attention to mass accuracy followed by peptide synthesis confirmed these misassignments by comparison of the resulting MS/MS spectra from the synthetic and endogenously generated spectra [44].
Manual interpretation of MS/MS spectra remains a commonly invoked method to better confirm PTM peptide assignments. However, a broadly applicable set of manual criteria for spectral evaluation are difficult to define and can vary dramatically depending on the mass spectrometric parameters such as mass accuracy, detectable mass range, etc. and the properties of gas phase ion chemistry of the modified amino acid residue in question (resulting neutral losses, effect on charge state, characteristic immonium ions, etc). 3NT peptides in particular have several critical LC-MS/MS characteristics that allow confirmation of their presence including a characteristic 3-nitrotyrosine immonium ion at m/z 181.1, increased chromatographic retention time of the more hydrophobic 3NT modified peptide relative to the unmodified peptide, and MS/MS fragmentation pattern similarity between the 3NT and unmodified peptide. Due to the typically low stoichiometry of the 3NT modification in biological samples there has been a lack of well-validated ‘positive controls’ of 3NT modified peptides to assess the LC-MS/MS characteristics and to determine their robustness and applicability as validating parameters. In order to more rigorously delineate a set of unbiased criteria for improved assignment of this modification, we generated a large number of 3NT modified peptides through chemical modification of a defined, equimolar mixture of 48 proteins, UPS1 (also known as Sigma-48) by treatment with tetranitromethane (TNM) and evaluation of this nitrated protein reference standard on two LC-MS/MS platforms, the QSTAR Elite (QTOF) and LTQ Velos (dual ion trap) as shown in Figure 1. We manually validated all MS/MS spectra of putative 3NT-modified peptides using an extensive set of defined LC-MS/MS criteria (See Table 1) and identified well over 200 validated 3NT modified peptides in the obtained spectral datasets. This approach also allowed us to compare and contrast the effectiveness of two widely utilized mass spectrometry platforms (QTOF and dual ion trap) for the detection of 3NT-containing peptides. The spectral library generated with Skyline, an open source program [46], is provided as a resource containing validated MS/MS spectra of all confidently identified 3NT peptides and their unmodified counterparts. Furthermore, this straightforward, reproducible nitration reaction of a commercially available protein standard makes it readily available to the proteomics field interested in studying or identifying nitrated proteins. This nitrated standard can also be used to optimize mass spectrometry data collection parameters and data mining to improve confident identification of 3NT-containing peptides and can also be spiked into biological matrices of interest (plasma, tissue, etc.) to optimize enrichment strategies for nitrated peptides.
Figure 1. Overview of experimental approach to generate, indentify, and validate 3NT peptides.

An equimolar mixture of 48 proteins, UPS1, was TNM or mock treated to generate a set of nitrated and control samples. Samples were trypsin digested and analyzed with by both QSTAR Elite (QTOF) and LTQ Velos (dual ion trap) LC-MS/MS systems. Data was searched with Mascot and the each resulting 3NT peptide was analyzed by manual inspection using the criteria defined in Table 1.
Table 1.
Criteria for 3-Nitrotyrosine (3NT) peptide identification by manual inspection of MS/MS spectra. Criteria were used to asses both QSTAR Elite (QTOF) and LTQ Velos (ion trap) spectra.
|
Corresponding unmodified peptides were identified on the QSTAR Elite for 70.4% of observed 3NT peptides and on the LTQ Velos for 78.7% of detected 3NT peptides.
Criteria applicable to QSTAR Elite data only since LTQ Velos data is searched using average mass of the precursor instead of the monoisotopic mass.
Materials and Methods
Bovine serum albumin (BSA, 98%), UPS1 (universal proteomics standard consisting of an equimolar mixture of 48 human proteins), tetranitromethane, sinapinic acid, dithiothreithol, and iodoacetamide were purchased from Sigma-Aldrich (St. Louis, MO). Recrystalized α-cyano-4-hydroxycinnamic acid (α-CHCA) and 2,5-dihydroxy benzoic acid (DHB) were purchased from LaserBio Labs (Valbonne, France). Trifluoroacetic acid (TFA), formic acid (FA) and phosphoric acid were from J. T. Baker, EMD and Aldrich, respectively. Sequencing grade trypsin was obtained from Promega (Madison, WI). C18 ZipTips and dialysis membrane filters (0.025 μm VSWP) were purchased from Millipore (Billerica, MA). Oasis HLB reversed phase extraction cartridges (1 cc, 30 mg) were purchased from Waters.
Protein nitration modification
BSA (50 μg, 753 pmol) was suspended in 50 μL (15.1 μM) of 10 mM ammonium bicarbonate (NH4HCO3, pH 8.0), and 5 μL of 8.2 mM tetranitromethane (TNM, in 95% ethanol) was added. The mixture was gently vortexed and incubated at room temperature for 1 h. The solution gained a pale yellow color during the nitration reaction. Glacial acetic acid (5 μL) was then added to quench the reaction. The nitrated protein mixture was desalted by drop dialysis against HPLC grade water with membrane filters. The desalted protein solution was concentrated to 20 μL (37.8 μM for total proteins) for downstream treatments. For nitration of UPS1, the protein mixture from one vial (240 pmol) was reconstituted in 25 μL of 20% acetonitrile (ACN)/80% 10 mM ammonium bicarbonate. To this solution, 20 μL of 8.2 mM TNM was introduced into the protein solution and allowed to react for 1 h at room temperature. The desalted nitrated UPS1 protein mixture was concentrated to 20 μL (12 µM) and digested with trypsin as described below. Mock treated samples were prepared as above without addition of TNM.
Trypsin proteolysis
Nitrated BSA (20 μL, 37.8 μM) and UPS1 (20 μL, 12.0 μM), respectively, were diluted to 10 μM with 10 mM NH4HCO3. Then, 132 μL or 12 μL of freshly prepared reducing agent 100 mM dithiothreitol (DTT), was added to the BSA and UPS1, respectively, followed by dilution with ACN to bring the organic solvent concentration to 40% (v/v). Proteins were gently vortexed and then incubated for 45 min at 56°C. To alkylate samples, freshly prepared 550 mM 2-iodoacetamide was added to each sample (49.3 μL to BSA or 4.5 μL to UPS1) and incubated in the dark for 45 min. Samples were diluted with 10 mM NH4HCO3 to a final concentration of 10% ACN (v/v) with a resulting pH of ~8. Finally, 150 μL or 10 μL of trypsin (20 μg resuspended in 1200 μL 25 mM NH4HCO3) were then added to the BSA or UPS1, respectively, to achieve a trypsin:protein ratio of 1:20 (wt/wt) and the mixture was incubated at 37°C overnight. In parallel, non-TNM treated BSA and UPS1 were prepared as controls in the same manner. Samples were acidified with TFA to pH <3.5 and desalted using C18 ZipTips according to the manufacturer’s instructions.
Protein mass profile and molecular weight measurement
The intact mass of proteins was determined using a Voyager-DE STR MALDI Plus (Applied Biosystems) instrument that allowed acquisition of spectra by MALDI-TOF mass spectrometry in linear mode. Desalted proteins (1 μL) and sinapinic acid (0.8 μL, saturated in 50% ACN 0.1% TFA) were briefly vortexed prior to spotting onto a MALDI target plate. The delay time was set as 750 nsec and the accelerating voltage was 25,000 V. Laser relative intensity was set at 2200, shots/spectrum were set to 100 for BSA and 1000 for the UPS1 mixture.
LC-MS/MS analysis of peptides using a QTOF (QSTAR Elite)
Digested, desalted peptides were analyzed by reverse-phase nano-HPLC-ESI-MS/MS using an Eksigent nano-LC 2D HPLC system (Eksigent, Dublin, CA) connected to a quadrupole time-of-flight (QTOF) QSTAR Elite mass spectrometer (MDS SCIEX, Concorde, Canada). Briefly, 3 replicates each with 10 μl of peptide mixtures (approximately 4-5 pmol total peptides in 0.1% formic acid) were loaded onto a guard column (C18 Acclaim PepMap100, 300 μm internal diameter (ID) × 5 mm long, 5 μm particle size, 100 Å pore size, Dionex, Sunnyvale, CA) and washed with the loading solvent (0.1 % formic acid, flow rate of 20 μL/min) for 10 min. Subsequently, samples were transferred onto the analytical C18-nanocapillary HPLC column (C18 Acclaim PepMap100, 75 μm ID × 15 cm long, 3 μm particle size, 100 Å pore size, Dionex, Sunnyvale, CA) and eluted at a flow rate of 300 nL/min using the following gradient: 0-2% solvent B (0.1% formic acid in ACN) in A (0.1% formic acid in H2O) (from 0-5 min), 2-40% solvent B in A (from 5-125 min), 40-90% solvent B in A (from 125-140 min) and at 90% solvent B in A (from 140-149 min), with a total runtime of 194 min (including mobile phase equilibration).
Mass spectra (ESI-MS) and tandem mass spectra (ESI-MS/MS) were recorded in positive-ion mode with a resolution of 12,000-15,000 full-width half-maximum. MS mass range was m/z 350-1600 and MS/MS mass range was m/z 100-1500. For collision induced dissociation tandem mass spectrometry (CID-MS/MS), the mass window for precursor ion selection of the quadrupole mass analyzer was set to ± 1 m/z. The precursor ions were fragmented in a collision cell using nitrogen as the collision gas. Advanced information dependent acquisition (IDA) was used for MS/MS collection, including QSTAR Elite (Analyst QS 2.0) specific features, such as “Smart Collision” and “Smart Exit” (fragment intensity multiplier set to 2.0 and maximum accumulation time at 1.5 sec), to obtain MS/MS spectra for the six most abundant parent ions following each survey scan. Dynamic exclusion features were based on value M not m/z and were set to exclusion mass width 50 mDa and exclusion duration of 60 sec. Under circumstance of multiple sample runs, in between a short blank gradient was run [mobile phase gradient: 0-30% B in A (from 0-15 min), 30-80% solvent B in A (from 15-17 min), 80% solvent B in A (from 17-20 min)].
LC-MS/MS analysis of peptides using a dual pressure linear ion trap (LTQ Velos)
Chromatographic separation of peptides was carried out using an Eksigent nanoLC-ultra 2D plus HPLC system directly connected to the LTQ Velos dual-pressure linear ion trap mass spectrometer (Thermo Scientific, San Jose, CA). Peptide mixtures were loaded onto a trap column (C18 Acclaim Pep-Map100; 300 μm inner diameter × 5 mm, 5-μm particle size, 100 Å pore size; Dionex) and washed with the loading solvent (0.1% formic acid, 98% H2O, and 2% acetonitrile; flow rate, 5 μL/min) for 20 min. Subsequently, samples were transferred onto the analytical C18-nanocapillary HPLC column (C18 Magic; 100 μM inner diameter × 15 cm, 3-μm particle size, 200-Å pore size; Michrom) and eluted at a flow rate of 500 nL/min using an identical gradient and solvents listed above for the QSTAR Elite. for the LTQ Velos mass spectra (ESI-MS) and tandem mass spectra (ESI-MS/MS) were recorded in positive-ion mode using an Nth Order Double Play experiment, where N=10 (1 MS survey scan followed by 10 MS/MS scans). The mass range (m/z) analyzed was 350-1600 for MS scans. Fragmentation parameters used were: activation type: CID, minimum signal required: 1000, precursor isolation width: 1.5, normalized collision energy: 30, activation time: 10 ms.
Multiple reaction monitoring (MRM) mass spectrometry assay for relative quantitation and targeted precursor ion analysis of peptides
MRM-MS assays of 3NT modified peptides were performed on a 4000 QTRAP hybrid triple quadrupole/linear ion trap mass spectrometer (Applied Biosystems/MDS Analytical Technologies). In a typical situation for an MRM assay, the instrument was operated with an ion spray voltage of 2450 V, curtain gas 12, nebulizer gas (GS1) 18, interface heater temperature (IHT) 150 °C. MRM precursor/fragment ion transitions were selected based on QSTAR Elite MS/MS fragment ion signals and charge states, and acquired at unit resolution in the first (Q1) and third (Q3) quadrupoles with the dwell times of 10 ms and cycle times of 4.5 s for all selected transitions. The collision energy (CE) was calculated by equations: [CE = m/z(Q1)×0.0625 − 3] for charge 2+ ions; [CE = m/z(Q1)×0.0625 − 8] for charge 3+ or higher precursor ions. Specifically to record transitions for the unmodified and 3NT modified tyrosine immonium ions (Q3 set to m/z 136.1 and m/z 181.1, respectively) the collision energy was set to 60 for all peptides. Peptides were loaded onto a trap column (C18 Acclaim Pep-Map100; 300 μm inner diameter × 5 mm, 5-μm particle size, 100-Å pore size; Dionex) at 20 μL/min for 5 minutes in 0.1% formic acid and eluted at 300 nL/min with a 75 μm inner diameter IntegraFrit analytical column (New Objective, Woburn, MA) packed in-house with 10-12 cm of ReproSil-Pur C18-AQ 3 μm reversed phase resin (Dr. Maisch GmbH, Germany) with a gradient of 2% ACN (0.1% formic acid) to 70% ACN over 32 min. MultiQuant software version 1.2 (Applied Biosystems) and Skyline [46] were used to quantify peak areas.
Protein identification and modification assignment through database searches
Both QSTAR Elite and LTQ Velos MS/MS data were searched using Mascot version 2.3.02 (Matrix Science) against Swissprot version 2010_10 specifying human taxonomy (20,259 sequence) using the following criteria: 2 missed cleavages (trypsin); fixed modification-carbamidomethyl (C); Variable modifications-Acetyl (Protein N-term), Oxidation (M), Deamidated (NQ), Nitro (Y). For the QSTAR Elite the precursor ion mass tolerance was 25 ppm and fragment ion mass tolerance 0.05 Da. For the LTQ Velos the precursor ion mass tolerance was 1.5 Da (using average mass value) and the fragment ion mass tolerance was 0.9 Da. The maximum expectation value (P value) for accepting spectra assigned to peptides by Mascot was 0.05. To assess false discovery rate (FDR) we did target-decoy database searches using a custom list of the 48 human proteins found in UPS1 standard mixture as the decoy database. All peptides presented in this manuscript were within a 5% FDR based on these searches.
Results
Optimization of chemical nitration reaction using BSA and UPS1
BSA was used to optimize conditions for partial protein nitration in vitro using tetranitromethane (TNM). The extent of nitration was determined by an increase in average mass of full length BSA as determined by MALDI-TOF mass spectrometry in linear mode. A time course of BSA nitration is shown in Supplementary Figure S1a. After 1, 2, and 3 h of TNM treatment the mass gains for the BSA protein were 392 Da, 500 Da, and 566 Da, respectively, which corresponded to the addition of 8-9, 11-12, and 12-13 nitro groups (NO2) to each BSA molecule. BSA that was nitrated with TNM for 1h was then trypsin digested and analyzed by LC-MS/MS on a QSTAR Elite LC-MS/MS to map the sites of 3NT modification. Nineteen 3NT sites in 24 BSA peptides were detected confirming that significant levels of modified tyrosine residues could be detected with a 1 h TNM treatment (Supplementary Table S1).
The UPS1 mixture contains 48 equal molar proteins of human origin that range in their molecular weights from 6.2 kDa (epidermal growth factor) to 83 kD (gelsolin). Eighteen proteins in the UPS1 mixture have a mass over 25 kDa while 30 proteins are smaller than 25 kDa. Nitration of the 48 proteins by incubation with TNM at room temperature for one hour resulted in a shift in the intact masses of all protonated molecular ion peaks detected by MALDI TOF MS in linear mode, confirming that significant levels of nitration had occurred (Supplementary Figure S1b).
Mass spectrometric characterization of 3NT modified UPS1
TNM treated and untreated UPS1 proteins were digested with trypsin and analyzed using two different LC-MS/MS platforms to maximize the number of 3NT peptides identified and to compare the instrument performance and efficiency for the detection of 3NT-containing peptides. Data was acquired using both a quadrupole time-of-flight (QTOF) mass spectrometer (QSTAR Elite) and a dual linear ion trap (LTQ Velos) which are common platforms in MS-based proteomics. A long LC gradient (120 min) was used to improve chromatographic separation of peptides and three mass spectrometric replicates were analyzed to maximize sampling of the mixture. The data was searched using an in-house Mascot MS/MS search algorithm as described in the methods. Initial examination of the search results for peptides with an expectation score below 0.05 determined that the QSTAR Elite and LTQ Velos detected 123 and 223 3NT-containing peptides in the treated, nitrated sample, respectively (Figure 2a). As expected, in the non-nitrated sample both the QSTAR Elite and LTQ Velos detected none or only one 3NT modified peptide, respectively. In our experiments TNM treatment generated a mixture of both unmodified and modified peptides with 77-81% of tyrosine-containing peptides in the TNM treated sample being 3NT modified. In many cases, peptides with multiple tyrosines were found to contain more than one nitration site. As expected, in the untreated sample only <1 % of peptides were 3NT modified (Figure 2b).
Figure 2. Peptides from UPS1 detected by LC-MS/MS after TNM or mock treatment.
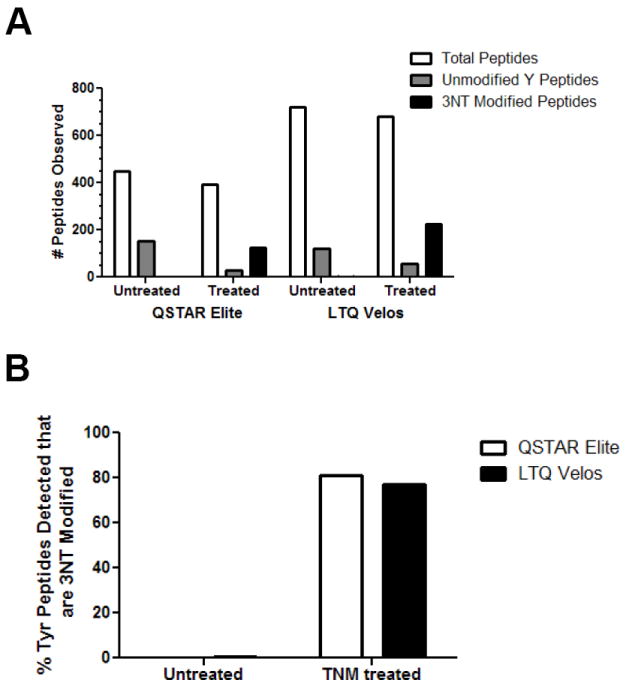
4.5 pmol of UPS1 either TNM (1 h) or mock treated was trypsin digested and analyzed using both a QTOF (QSTAR Elite) and dual ion trap (LTQ Velos) mass spectrometer. Three technical replicates were acquired. A) The total number of peptides indentified including ones not containing a Y, the number of peptides with no 3NT modified Ys, and the number of peptides with 3NT modified Ys are shown. B) The proportion of 3NT modified peptides shown as a percent of all Y containing peptides detected shown for both conditions. Distinct peptides were defined as differences in the site of nitration, missed cleavages, and methionine oxidation state
Criteria for 3NT peptide identification
An extensive set of criteria (Table 1) was used to manually inspect each MS/MS spectrum of the putative nitrated peptides as suggested from the database search. These criteria were created in order to (i) fully assess the analytical properties of 3NT-containing peptides relative to their unmodified counterparts, (ii) confirm that the search algorithm correctly identified the nitrated peptides, and (iii) prevent misassignment of the modified residue in the peptides detected. As nitration of tryptophan has been known to occur to a lesser degree during harsh chemical oxidation conditions and may lead to misinterpretation [47], this side reaction was closely considered in peptides that also contained a tryptophan residue. The criteria used to inspect mass spectral data for both MS instruments were the same except with regard to mass accuracy in which the data acquired by the instruments differ significantly.
Each 3NT-containing peptide underwent rigorous manual inspection of the corresponding MS/MS spectrum and the identification criteria were assessed for each. For example, the LC-MS/MS analysis of the doubly nitrated peptide 162Y(NO2)LY(NO2)EIAR168 from albumin and its unmodified counterpart from albumin are shown in Figure 3 to demonstrate the application of many of the criteria we used to identify 3NT modified peptides. The MS/MS spectra of the 3NT modified peptide (MH22+, 509.2 m/z) from both the QSTAR Elite and LTQ Velos closely match the MS/MS fragmentation pattern of the unmodified peptide (MH22+, 464.3 m/z). In addition, an appropriate mass increment corresponding to a 3NT residue (Δ208 Da) is observed between y4 and y5 flanking fragment ions in the MS/MS of the modified peptide compared to the mass increment of an unmodified tyrosine (Δ163 Da) in the unmodified peptide confirming the site of modification (Y164). There is also a mass difference of 45 m/z in the b2 ion due to the nitration of the N-terminal tyrosine. In both spectra of this 3NT modified peptide, the characteristic 3NT immonium ion at m/z 181.1 is relatively weak, but still observable above the noise, whereas the unmodified peptide has a tyrosine immonium ion at m/z 136.1. The extracted ion chromatograms (XIC) for the MS1 signal of the nitrated and unmodified peptide are shown in the insets and demonstrate the increased chromatographic retention time of the 3NT modified peptide due to the higher hydrophobicity of the nitrated peptide.
Figure 3. LC-MS/MS analysis the nitrated and unmodified peptide YLYEIAR and its unmodified counterpart by the QSTAR Elite and LTQ Velos.

A) MS/MS of the unmodified peptide YLYEIAR (MH22+, 464.3 m/z) from albumin detected by the QSTAR Elite. B) The MS/MS spectra of the 3NT modified peptide Y(NO2)LY(NO2)EIAR (MH22+, 509.2 m/z) from the QSTAR Elite and C) LTQ Velos. The Y and 3NT immonium ions are highlighted in bold as well as the y5 and b2 ions which are increased in mass due to the presence of the 3NT modification. Inset: Extracted ion chromatogram of the nitrated and unmodified peptide demonstrating the increased retention time that occurs after 3NT modification.
Manual inspection of the spectra obtained from QSTAR Elite and LTQ Velos were found to have identified 119 and 197 distinct 3NT peptides, respectively, from 40 of the 48 UPS1 proteins. 3% and 12% of the QSTAR Elite and LTQ Velos peptides, respectively, that initially had a Mascot search engine expectation value below 0.05 were removed. An example of an MS/MS spectrum with a significant expectation value that didn’t meet our criteria is shown in Supplementary Figure S2. There was an overlap of 95 peptides between the two MS platforms and a total of 221 3NT-containing peptides were detected (Figure 4a). Distinct peptides were defined as sequences with differences in the site of nitration and methionine oxidation state. There was no significant bias for detection of multiply nitrated peptides. We observed 23 multiply nitrated peptides with the QSTAR Elite and 36 multiply nitrated peptides with the LTQ Velos yielding an overlap of 17 peptides (Figure 4b). Four of the peptides identified have ambiguous sites of modification. All MS/MS spectra of the 3NT and corresponding unmodified peptides are included as a Skyline file in the Supplementary Data. Notably, the single 3NT-containing peptide initially suggested by the search engine from the LTQ Velos in the untreated sample did not meet our manual inspection criteria and was removed from the final list.
Figure 4. Overlap of validated 3NT peptides identified by the QSTAR Elite and LTQ Velos.
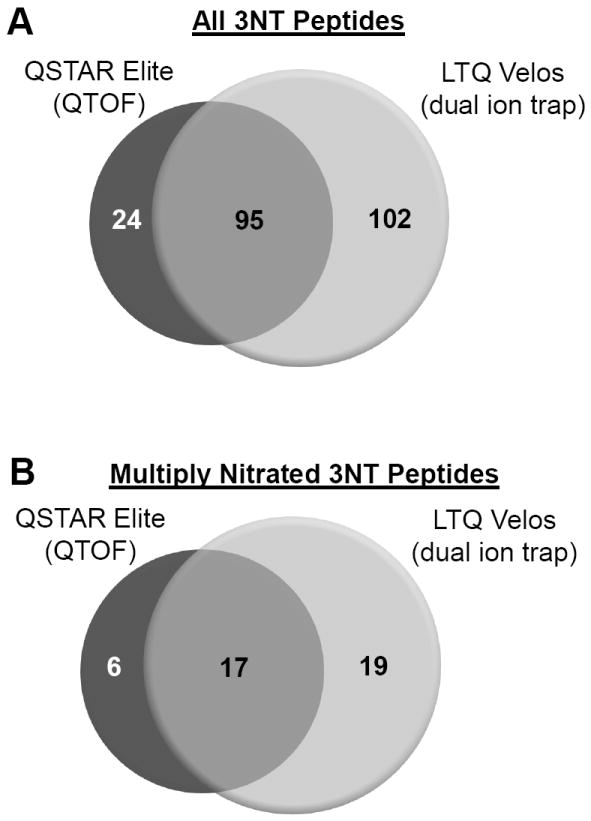
Venn diagram of the overlap of all validated 3NT (A) and multiply 3NT modified (B) peptides identified by the QSTAR Elite and LTQ Velos. Distinct peptides were defined as differences in the site of nitration, missed cleavages, and methionine oxidation state
LC-MS/MS characteristics of 3NT peptides
Next we determined the percentage of 3NT-containing peptides that met each of the specific criteria that we used to aid in the identification of a true 3NT modification (Figure 5, with a complete table including all criteria for every peptide listed in Supplementary Tables S2 and S3 for the QSTAR Elite and LTQ Velos, respectively). In addition, we are providing a Skyline spectral library file that contains all identified MS/MS spectra of nitrated and corresponding unmodified peptides. Criteria with the highest utility would be expected to be present in all 3NT peptides and be especially useful for assessing large proteomic datasets for 3NT modifications where false-positives present a strong possibility. Based on our dataset, increased retention time of the 3NT peptide and similarity of the dominant 3NT fragment ions to the unmodified peptide provided excellent validation criteria for putative 3NT peptides since these held true for all cases where we identified both a 3NT modified peptide and the unmodified counterpart (Figure 5a). The differences in chromatographic retention time for 3NT peptides relative to the unmodified peptides for both the QSTAR Elite and LTQ Velos are shown in Figure 5b. The wide range of retention time differences (ΔRT: 2.5-26 min) is likely due to the long (120 min) gradient used in this study. The retention difference between a pair of 3NT modified and intact peptides originates from introduction of the electron withdrawing group (-NO2) upon tyrosine residue, which makes the peptide of interest less polar and more hydrophobic as compared to the unmodified counterpart. Lastly, in instrument platforms with reasonably high mass accuracy such as the QSTAR Elite, in which 50 ppm precursor mass accuracy is routinely achievable, all peptides were within 25 parts per million (ppm) mass accuracy and most were within 10 ppm as shown in Figure 5c. It was also observed that the charge states of 3NT-modified peptides were generally not affected by the presence of the nitro group (data not shown).
Figure 5. LC-MS/MS properties of the 3NT peptides identified by the QSTAR Elite and LTQ Velos.

A) The percent of all the validated 3NT peptides which met each of the criteria assessed using Table 1 for both the QSTAR Elite and LTQ Velos mass spectrometers. B) The difference in chromatographic retention time (Δ RT) between 3NT modified peptides and their unmodified counterparts. C) Distribution of the precursor mass error (in ppm) for 3NT peptides indentified by the QSTAR Elite.
The presence of a 3NT immonium ion at m/z 181.1 in the MS/MS spectra typically provides a clear indication that a peptide is nitrated at tyrosine and was an important criterion for evaluation. Immonium ions are MS/MS fragment ions generated from a peptide by multiple internal cleavages and is a faithful signature for individual amino acids when observed in the MS/MS spectrum [48]. A schematic mechanism by which immonium ions for tyrosine and 3NT tyrosine are produced in MS/Ms spectra is shown in Supplementary Figure S3. The presence of 3NT immonium ions was not a conserved feature in our spectral datasets, with only 19.4% of QSTAR Elite spectra and 5.7% of LTQ Velos spectra showing a m/z 181,1 immonium ion with a signal to noise greater than 6 (Figure 5a). The percentage of LTQ Velos with the 3NT immonium ion would be expected to be lower than the QSTAR Elite due to its low mass cutoff which is a result of ion trap fragmentation. 72.8% of LTQ Velos MS/MS spectra collected data in the low mass region of interest.
Nitrated UPS1 is reproducibly generated and stable during storage
To determine the suitability of the nitrated UPS1 as a standard mixture of 3NT-containing peptides, we evaluated the reproducibility of nitration and the stability of the nitrated peptides using multiple reaction monitoring (MRM) mass spectrometry using a 4000 QTRAP. The MRM assay detected 47 pairs of 3NT modified and unmodified peptides representing 21 UPS1 proteins with Q1/Q3 transitions based on MS/MS data from the QSTAR Elite. The full transition list is detailed in Supplementary Table S4. Transitions included at least two y- or b-ions as well as the 3NT or tyrosine immonium ions for each peptide. The percent nitration was approximated by intensity of the 3NT peptide divided by the sum of the intensity of the 3NT and unmodified peptide. To determine reproducibility, the peak areas were quantified for two separate TNM reactions of UPS1 with a representative set of peptides shown in Figure 6a. The relative level of 3NT modification ranges from 0-100%, with most of the peptides nitrated over 70% (Figure 6a). The reproducibility for achieving the same nitration level per peptide comparing two independent TNM sample preparations deviates by less than 1%. To assess long-term stability, the level of nitration of the individual UPS1 peptides was evaluated after 4, 7, and 9 months of storage at -20°C with representative peptides shown in Figure 6b demonstrating that only very limited degradation of the 3NT peptides occurred.
Figure 6. UPS1 nitration is reproducible and stable.
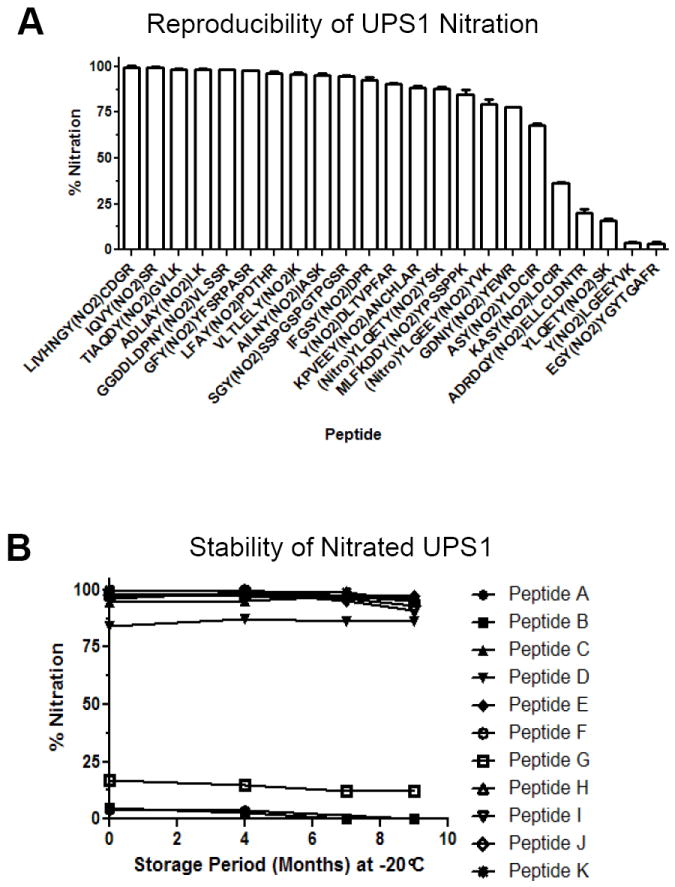
A) Two independent nitration reactions were performed on UPS1 and the levels of nitration was evaluated by quantitative MRM. The y-axis represents ion percentage of 3NT peptide relative to the unmodified peptide with representative peptides shown. Error bars represent the standard of deviation. B) Storage stability evaluation of 3NT modified UPS1 peptides by quantitative MRM assay of a set of ions on 4000 QTRAP MS platform. The y-axis represents the peak area percentage of the 3NT peptide divided by the total peak area of the unmodified and 3NT species of the peptide. The x-axis displays storage period in month at -20°C.
Discussion
There is growing interest in identifying tyrosine residues that are oxidatively modified by reactive nitrogen species in vivo. Tyrosine nitration among cellular proteins is likely to be an important hallmark of oxidative stress and contribute to cellular dysfunction and disease. Unbiased proteomics approaches are ideal for the detection of 3NT sites, though several recent reports have pointed out the importance of careful evaluation of MS/MS spectra to insure correct assignments [43, 44, 49]. With this in mind we generated a large LC-MS/MS dataset of 3NT modified peptides by chemical treatment of proteins with a commonly used nitration reagent, tetranitromethane, and validated the resulting protein nitration after trypsin digestion and LC-MS/MS analysis. This resulting peptide spectral dataset underwent rigorous manual interpretation and allowed us to investigate numerous chromatographic and fragmentation properties of 3NT peptides that could serve as criteria in their proper identification. Furthermore, we have developed a straightforward, reproducible means to prepare a complex nitrated protein standard that is well defined and validated. This standard can be easily generated by other laboratories to optimize their own mass spectrometry instrumentation or develop and evaluate new 3NT peptide enrichment strategies.
Choosing the appropriate type of mass spectrometer for a large-scale proteomic experiment is an important consideration in studies that seek to identify posttranslational modifications. There are many tradeoffs and each instrument has specific advantages and disadvantages. For example, mass accuracy and resolution are especially important parameters for the identification of PTMs that have very similar, but not identical masses. Analysis of complex biological samples requires MS/MS fragmentation and MS platforms which have shorter duty cycles comprising MS and data dependent MS/MS scans which are able to sample and identify more molecules in a fixed period of time. In this study we have utilized two different MS platforms; a hybrid quadrupole time of flight (QTOF) QSTAR Elite and a dual linear ion trap LTQ Velos. While the QTOF routinely acquires high resolution/high accuracy data, the LTQ Velos scans faster and is able to acquire more MS/MS spectra per unit time. We find that both systems perform quite well, though the LTQ Velos identified more peptides. Importantly, there is high overlap of peptides identified with ~80% of the QSTAR Elite peptides also detected by the LTQ Velos. While the mass accuracy in an ion trap is not as high as for a QTOF platform, peptides that were only identified in the LTQ Velos instrument had very convincing MS/MS spectral quality. Supplementary Figure S4 shows an annotated MS/MS spectrum of a peptide detected only by the LTQ Velos. An important conclusion from this is that while there is a large difference in the mass accuracy of the two instruments, using our stringent identification criteria results from both platforms are confident as also confirmed by the large overlap of independent peptide identifications between platforms.
Analysis of our dataset clearly demonstrates that an important aspect of validating 3NT peptides is the comparison to the unmodified form. All 3NT peptides we detected had increased chromatographic retention time relative to the unmodified peptides and also showed similar dominant fragment ions in their corresponding MS/MS spectra. Thus, 3NT peptides that do not conform to these two criteria relative to an unmodified form of the identified peptides in the dataset should be considered highly suspect assignments. This result also suggests that one drawback of enriching for 3NT-containing peptides is that comparison to the unmodified peptide is not possible unless one also runs a comparative analysis of the un-enriched sample and one is fortunate enough to detect the unmodified peptide as well. In contrast to relative retention time and spectral similarity, neither LC-MS/MS platform used in this study robustly detected the 3NT immonium ion at m/z 181.1 (Figure 2). However, other reports have indicated that the formation of immonium ions for 3NT is suppressed [33]. There is potential to enhance the formation of this low mass ion by increasing the collision energy used for MS/MS fragmentation [49]. However, there is a trade-off in this approach due to a decreased intensity of higher m/z fragment ions which are more informative of the peptide sequence and important for effective mining of the data with search algorithms. For some platforms, it may well be possible to alternate between high and low collision energies to obtain spectra where both features can be obtained consecutively. Indeed, such an approach has recently been reported for the acquisition of iTRAQ data sets where a similar trade-off in the low mass iTRAQ reporter ion region and overall fragmentation quality for proper spectral identification is present [50].
Proteomics often plays an important scientific role through the generation of candidate lists of proteins or PTMs with potential biological significance which warrant further investigation. There are many ways to generate these lists from proteomics experiments, and MS/MS search algorithms play an important role in this process. With regard to searching proteomic spectral data, we found that Mascot analysis of data from both LC-MS/MS systems was quite efficient and thorough in its assignments. However, manual validation of the spectra guided by the selection criteria that we assessed was a vital component to the proper assignment of these MS/MS spectra and resulted in removal of some peptides. Using our criteria no 3NT-containing peptides were detected in the negative control (the untreated UPS1 mixture) supporting the rigor of our validation approach (Figure 2). Nonetheless, additional validation beyond MS/MS spectra interpretation may be warranted for endogenous 3NT-containing peptides detected from in vivo sources considered for further characterization. The price of crude peptide synthesis on a 96 well scale is on par with the total investment in proteomics studies and synthesis of important 3NT target peptides can add additional confidence through retention time and MS/MS comparison with the endogenous analyte. While the in vitro nitration approach is not novel, the described study resulted in a well characterized standard protein mixture that can be generated reproducibly to yield a stable nitrotyrosine standard and this approach can be easily adapted by other laboratories to generate a standard mix of nitrated peptides to assess their own analytical workflows. We believe that with proper assessment of proteomic data and validation of workflows using defined standards, one can avoid many of the pitfalls that have plagued such studies in the past and allow for the development of truly efficient and comprehensive methodologies for identifying the nitroproteome.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Cancer Institute (CA0126477) and the National Institutes of Health, including the Geroscience Mass Spectrometry and Imaging Core (PL1 AG032118) and the NCRR shared instrumentation program (S10 RR024615 for the QSTAR Elite and S10 RR0021222 for the 4000 QTRAP).
Footnotes
Supplementary spectral library of validated 3NT peptides. The included Skyline file (Skyline software is freely available at proteome.gs.washington.edu/software/skyline/) includes the MS/MS data for all validated 3NT peptide and the corresponding non-modified peptides from both the QSTAR Elite and LTQ Velos. The *.sky file, the *.sky.view file, 4 *.blib bibliospec library files (Li_QSTAR_Untreated.blib, Li_QSTAR_Treated.blib, Li_Velos_Untreated.blib, and Li_Velos_Treated.blib, for either TNM treated or untreated samples run on either the QSTAR Elite or LTQ Velos) and the background proteome file (Sigma48.protdb) should all be saved in one folder and upon opening of the main *.sky file Skyline will find the corresponding related files. Extensive notes were used to clarify fragment ion assignments (particularly since Skyline currently does not include assignments for 3+ or higher charge state ions as displayed in the MS/MS viewer) or peptide-specific details.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Greenacre SA, Ischiropoulos H. Tyrosine nitration: localisation, quantification, consequences for protein function and signal transduction. Free Radic Res. 2001;34:541–81. doi: 10.1080/10715760100300471. [DOI] [PubMed] [Google Scholar]
- 2.Pittman KM, MacMillan-Crow LA, Peters BP, Allen JB. Nitration of manganese superoxide dismutase during ocular inflammation. Exp Eye Res. 2002;74:463–71. doi: 10.1006/exer.2002.1141. [DOI] [PubMed] [Google Scholar]
- 3.Wu W, Chen Y, Hazen SL. Eosinophil peroxidase nitrates protein tyrosyl residues. Implications for oxidative damage by nitrating intermediates in eosinophilic inflammatory disorders. J Biol Chem. 1999;274:25933–44. doi: 10.1074/jbc.274.36.25933. [DOI] [PubMed] [Google Scholar]
- 4.Reynolds MR, Berry RW, Binder LI. Nitration in neurodegeneration: deciphering the “Hows” “nYs”. Biochemistry. 2007;46:7325–36. doi: 10.1021/bi700430y. [DOI] [PubMed] [Google Scholar]
- 5.Sarchielli P, Galli F, Floridi A, Gallai V. Relevance of protein nitration in brain injury: a key pathophysiological mechanism in neurodegenerative, autoimmune, or inflammatory CNS diseases and stroke. Amino Acids. 2003;25:427–36. doi: 10.1007/s00726-003-0028-6. [DOI] [PubMed] [Google Scholar]
- 6.Ischiropoulos H, Beckman JS. Oxidative stress and nitration in neurodegeneration: cause, effect, or association? J Clin Invest. 2003;111:163–9. doi: 10.1172/JCI17638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turko IV, Murad F. Protein nitration in cardiovascular diseases. Pharmacol Rev. 2002;54:619–34. doi: 10.1124/pr.54.4.619. [DOI] [PubMed] [Google Scholar]
- 8.Peluffo G, Radi R. Biochemistry of protein tyrosine nitration in cardiovascular pathology. Cardiovasc Res. 2007;75:291–302. doi: 10.1016/j.cardiores.2007.04.024. [DOI] [PubMed] [Google Scholar]
- 9.Xu S, Ying J, Jiang B, Guo W, Adachi T, Sharov V, et al. Detection of sequence-specific tyrosine nitration of manganese SOD and SERCA in cardiovascular disease and aging. Am J Physiol Heart Circ Physiol. 2006;290:H2220–7. doi: 10.1152/ajpheart.01293.2005. [DOI] [PubMed] [Google Scholar]
- 10.Knyushko TV, Sharov VS, Williams TD, Schoneich C, Bigelow DJ. 3-Nitrotyrosine modification of SERCA2a in the aging heart: a distinct signature of the cellular redox environment. Biochemistry. 2005;44:13071–81. doi: 10.1021/bi051226n. [DOI] [PubMed] [Google Scholar]
- 11.Sokolovsky M, Riordan JF, Vallee BL. Conversion of 3-nitrotyrosine to 3-aminotyrosine in peptides and proteins. Biochem Biophys Res Commun. 1967;27:20–5. doi: 10.1016/s0006-291x(67)80033-0. [DOI] [PubMed] [Google Scholar]
- 12.Quint P, Reutzel R, Mikulski R, McKenna R, Silverman DN. Crystal structure of nitrated human manganese superoxide dismutase: mechanism of inactivation. Free Radic Biol Med. 2006;40:453–8. doi: 10.1016/j.freeradbiomed.2005.08.045. [DOI] [PubMed] [Google Scholar]
- 13.van der Vliet A, Eiserich JP, Halliwell B, Cross CE. Formation of reactive nitrogen species during peroxidase-catalyzed oxidation of nitrite. A potential additional mechanism of nitric oxide-dependent toxicity. J Biol Chem. 1997;272:7617–25. doi: 10.1074/jbc.272.12.7617. [DOI] [PubMed] [Google Scholar]
- 14.Smith CD, Carson M, van der Woerd M, Chen J, Ischiropoulos H, Beckman JS. Crystal structure of peroxynitrite-modified bovine Cu,Zn superoxide dismutase. Arch Biochem Biophys. 1992;299:350–5. doi: 10.1016/0003-9861(92)90286-6. [DOI] [PubMed] [Google Scholar]
- 15.Ischiropoulos H, Zhu L, Chen J, Tsai M, Martin JC, Smith CD, et al. Peroxynitrite-mediated tyrosine nitration catalyzed by superoxide dismutase. Arch Biochem Biophys. 1992;298:431–7. doi: 10.1016/0003-9861(92)90431-u. [DOI] [PubMed] [Google Scholar]
- 16.Chatterjee S, Lardinois O, Bonini MG, Bhattacharjee S, Stadler K, Corbett J, et al. Site-specific carboxypeptidase B1 tyrosine nitration and pathophysiological implications following its physical association with nitric oxide synthase-3 in experimental sepsis. J Immunol. 2009;183:4055–66. doi: 10.4049/jimmunol.0900593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao HM, Kotzbauer PT, Uryu K, Leight S, Trojanowski JQ, Lee VM. Neuroinflammation and oxidation/nitration of alpha-synuclein linked to dopaminergic neurodegeneration. J Neurosci. 2008;28:7687–98. doi: 10.1523/JNEUROSCI.0143-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mallozzi C, Ceccarini M, Camerini S, Macchia G, Crescenzi M, Petrucci TC, et al. Peroxynitrite induces tyrosine residue modifications in synaptophysin C-terminal domain, affecting its interaction with src. J Neurochem. 2009;111:859–69. doi: 10.1111/j.1471-4159.2009.06378.x. [DOI] [PubMed] [Google Scholar]
- 19.Dairou J, Pluvinage B, Noiran J, Petit E, Vinh J, Haddad I, et al. Nitration of a critical tyrosine residue in the allosteric inhibitor site of muscle glycogen phosphorylase impairs its catalytic activity. J Mol Biol. 2007;372:1009–21. doi: 10.1016/j.jmb.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 20.Savvides SN, Scheiwein M, Bohme CC, Arteel GE, Karplus PA, Becker K, et al. Crystal structure of the antioxidant enzyme glutathione reductase inactivated by peroxynitrite. J Biol Chem. 2002;277:2779–84. doi: 10.1074/jbc.M108190200. [DOI] [PubMed] [Google Scholar]
- 21.Beckmann JS, Ye YZ, Anderson PG, Chen J, Accavitti MA, Tarpey MM, et al. Extensive nitration of protein tyrosines in human atherosclerosis detected by immunohistochemistry. Biol Chem Hoppe Seyler. 1994;375:81–8. doi: 10.1515/bchm3.1994.375.2.81. [DOI] [PubMed] [Google Scholar]
- 22.Girault I, Karu AE, Schaper M, Barcellos-Hoff MH, Hagen T, Vogel DS, et al. Immunodetection of 3-nitrotyrosine in the liver of zymosan-treated rats with a new monoclonal antibody: comparison to analysis by HPLC. Free Radic Biol Med. 2001;31:1375–87. doi: 10.1016/s0891-5849(01)00712-2. [DOI] [PubMed] [Google Scholar]
- 23.Radi R. Nitric oxide, oxidants, and protein tyrosine nitration. Proc Natl Acad Sci U S A. 2004;101:4003–8. doi: 10.1073/pnas.0307446101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bigelow DJ, Qian WJ. Quantitative proteome mapping of nitrotyrosines. Methods Enzymol. 2008;440:191–205. doi: 10.1016/S0076-6879(07)00811-7. [DOI] [PubMed] [Google Scholar]
- 25.Kanski J, Schoneich C. Protein nitration in biological aging: proteomic and tandem mass spectrometric characterization of nitrated sites. Methods Enzymol. 2005;396:160–71. doi: 10.1016/S0076-6879(05)96016-3. [DOI] [PubMed] [Google Scholar]
- 26.Kim JK, Lee JR, Kang JW, Lee SJ, Shin GC, Yeo WS, et al. Selective enrichment and mass spectrometric identification of nitrated peptides using fluorinated carbon tags. Anal Chem. 83:157–63. doi: 10.1021/ac102080d. [DOI] [PubMed] [Google Scholar]
- 27.Lee JR, Lee SJ, Kim TW, Kim JK, Park HS, Kim DE, et al. Chemical approach for specific enrichment and mass analysis of nitrated peptides. Anal Chem. 2009;81:6620–6. doi: 10.1021/ac9005099. [DOI] [PubMed] [Google Scholar]
- 28.Abello N, Barroso B, Kerstjens HA, Postma DS, Bischoff R. Chemical labeling and enrichment of nitrotyrosine-containing peptides. Talanta. 80:1503–12. doi: 10.1016/j.talanta.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Q, Qian WJ, Knyushko TV, Clauss TR, Purvine SO, Moore RJ, et al. A method for selective enrichment and analysis of nitrotyrosine-containing peptides in complex proteome samples. J Proteome Res. 2007;6:2257–68. doi: 10.1021/pr0606934. [DOI] [PubMed] [Google Scholar]
- 30.Ghesquiere B, Colaert N, Helsens K, Dejager L, Vanhaute C, Verleysen K, et al. In vitro and in vivo protein-bound tyrosine nitration characterized by diagonal chromatography. Mol Cell Proteomics. 2009;8:2642–52. doi: 10.1074/mcp.M900259-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhan X, Desiderio DM. Nitroproteins from a human pituitary adenoma tissue discovered with a nitrotyrosine affinity column and tandem mass spectrometry. Anal Biochem. 2006;354:279–89. doi: 10.1016/j.ab.2006.05.024. [DOI] [PubMed] [Google Scholar]
- 32.Nuriel T, Deeb RS, Hajjar DP, Gross SS. Protein 3-nitrotyrosine in complex biological samples: quantification by high-pressure liquid chromatography/electrochemical detection and emergence of proteomic approaches for unbiased identification of modification sites. Methods Enzymol. 2008;441:1–17. doi: 10.1016/S0076-6879(08)01201-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Petersson AS, Steen H, Kalume DE, Caidahl K, Roepstorff P. Investigation of tyrosine nitration in proteins by mass spectrometry. J Mass Spectrom. 2001;36:616–25. doi: 10.1002/jms.161. [DOI] [PubMed] [Google Scholar]
- 34.Sarver A, Scheffler NK, Shetlar MD, Gibson BW. Analysis of peptides and proteins containing nitrotyrosine by matrix-assisted laser desorption/ionization mass spectrometry. J Am Soc Mass Spectrom. 2001;12:439–48. doi: 10.1016/S1044-0305(01)00213-6. [DOI] [PubMed] [Google Scholar]
- 35.Kanski J, Hong SJ, Schoneich C. Proteomic analysis of protein nitration in aging skeletal muscle and identification of nitrotyrosine-containing sequences in vivo by nanoelectrospray ionization tandem mass spectrometry. J Biol Chem. 2005;280:24261–6. doi: 10.1074/jbc.M501773200. [DOI] [PubMed] [Google Scholar]
- 36.Kanski J, Alterman MA, Schoneich C. Proteomic identification of age-dependent protein nitration in rat skeletal muscle. Free Radic Biol Med. 2003;35:1229–39. doi: 10.1016/s0891-5849(03)00500-8. [DOI] [PubMed] [Google Scholar]
- 37.Medzihradszky KF. Peptide sequence analysis. Methods Enzymol. 2005;402:209–44. doi: 10.1016/S0076-6879(05)02007-0. [DOI] [PubMed] [Google Scholar]
- 38.Zhang K, Yau PM, Chandrasekhar B, New R, Kondrat R, Imai BS, et al. Differentiation between peptides containing acetylated or tri-methylated lysines by mass spectrometry: an application for determining lysine 9 acetylation and methylation of histone H3. Proteomics. 2004;4:1–10. doi: 10.1002/pmic.200300503. [DOI] [PubMed] [Google Scholar]
- 39.Trelle MB, Jensen ON. Utility of immonium ions for assignment of epsilon-N-acetyllysine-containing peptides by tandem mass spectrometry. Anal Chem. 2008;80:3422–30. doi: 10.1021/ac800005n. [DOI] [PubMed] [Google Scholar]
- 40.Young NL, DiMaggio PA, Plazas-Mayorca MD, Baliban RC, Floudas CA, Garcia BA. High throughput characterization of combinatorial histone codes. Mol Cell Proteomics. 2009;8:2266–84. doi: 10.1074/mcp.M900238-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nielsen ML, Vermeulen M, Bonaldi T, Cox J, Moroder L, Mann M. Iodoacetamide-induced artifact mimics ubiquitination in mass spectrometry. Nat Methods. 2008;5:459–60. doi: 10.1038/nmeth0608-459. [DOI] [PubMed] [Google Scholar]
- 42.Gokulrangan G, Zaidi A, Michaelis ML, Schoneich C. Proteomic analysis of protein nitration in rat cerebellum: effect of biological aging. J Neurochem. 2007;100:1494–504. doi: 10.1111/j.1471-4159.2006.04334.x. [DOI] [PubMed] [Google Scholar]
- 43.Prokai L. Misidentification of nitrated peptides: comments on Hong SJ, Gokulrangan G, Schoneich C. Proteomic analysis of age-dependent nitration of rat cardiac proteins by solution isoelectric focusing coupled to nanoHPLC tandem mass spectrometry. Exp Gerontol. 2007;42:639–651. doi: 10.1016/j.exger.2007.03.005.Exp Gerontol. 2009;44:367–9. doi: 10.1016/j.exger.2009.02.014.
- 44.Stevens SM, Jr, Prokai-Tatrai K, Prokai L. Factors that contribute to the misidentification of tyrosine nitration by shotgun proteomics. Mol Cell Proteomics. 2008;7:2442–51. doi: 10.1074/mcp.M800065-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hong SJ, Gokulrangan G, Schoneich C. Proteomic analysis of age dependent nitration of rat cardiac proteins by solution isoelectric focusing coupled to nanoHPLC tandem mass spectrometry. Exp Gerontol. 2007;42:639–51. doi: 10.1016/j.exger.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26:966–8. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Herold S. Nitrotyrosine, dityrosine, and nitrotryptophan formation from metmyoglobin, hydrogen peroxide, and nitrite. Free Radic Biol Med. 2004;36:565–79. doi: 10.1016/j.freeradbiomed.2003.10.014. [DOI] [PubMed] [Google Scholar]
- 48.Falick AM, Hines WM, Medzihradszky KF, Baldwin MA, Gibson BW. Low-mass ions produced from peptides by high-energy collision-induced dissociation in tandem mass spectrometry. Journal of the American Society for Mass Spectrometry. 1993;4:882–93. doi: 10.1016/1044-0305(93)87006-X. [DOI] [PubMed] [Google Scholar]
- 49.Danielson SR, Held JM, Schilling B, Oo M, Gibson BW, Andersen JK. Preferentially increased nitration of alpha-synuclein at tyrosine-39 in a cellular oxidative model of Parkinson’s disease. Anal Chem. 2009;81:7823–8. doi: 10.1021/ac901176t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dayon L, Pasquarello C, Hoogland C, Sanchez JC, Scherl A. Combining low- and high-energy tandem mass spectra for optimized peptide quantification with isobaric tags. J Proteomics. 2010;73:769–77. doi: 10.1016/j.jprot.2009.10.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.