

Abstract
The availability of the Arabidopsis thaliana genome sequence allows a comprehensive analysis of transcriptional regulation in plants using novel genomic approaches and methodologies. Such a genomic view of transcription first necessitates the compilation of lists of elements. Transcription factors are the most numerous of the different types of proteins involved in transcription in eukaryotes, and the Arabidopsis genome codes for more than 1,500 of them, or approximately 6% of its total number of genes. A genome-wide comparison of transcription factors across the three eukaryotic kingdoms reveals the evolutionary generation of diversity in the components of the regulatory machinery of transcription. However, as illustrated by Arabidopsis, transcription in plants follows similar basic principles and logic to those in animals and fungi. A global view and understanding of transcription at a cellular and organismal level requires the characterization of the Arabidopsis transcriptome and promoterome, as well as of the interactome, the localizome, and the phenome of the proteins involved in transcription.
Introduction.
Many of the biological processes in a plant are regulated at the level of transcription. Changes in gene expression have been shown to underlie the response to environmental cues and stresses (such as light, temperature, and nutrient availability), the defense response against pathogens, the regulation of metabolic pathways, the regulation of photosynthesis, or the establishment of symbiotic relationships, to name a few. In plants, as well as in animals, development is based on the cellular capacity for differential gene expression (reviewed in: Scott, 2000; Benfey and Weigel, 2001). Accordingly, many of the genes identified in screens for Arabidopsis mutants with altered, for example, flower or root development have been found to encode transcription factors. Alterations in gene expression are also emerging as a major source of the diversity and change that underlie the morphological evolution of eukaryotic organisms (Doebley and Lukens, 1998; Cubas et al., 1999b; Carroll, 2000; Tautz, 2000). In particular, morphological changes that occurred during plant domestication and crop improvement in agriculture have been associated with mutations in transcription factors (Peng et al., 1999), alterations in their expression (Doebley et al., 1997; Wang et al., 1999b), or changes in the expression of other types of regulatory proteins (Frary et al., 2000). Related transcription factors, such as the Arabidopsis MYB proteins WEREWOLF (WER) and GLABROUS1 (GL1), have been shown to be functionally equivalent, and owe their particular roles in plant development to differences in their expression patterns (Lee and Schiefelbein, 2001).
The availability of the Arabidopsis genome sequence (Lin et al., 1999; Mayer et al., 1999; Arabidopsis Genome Initiative, 2000; Salanoubat et al., 2000; Tabata et al., 2000; Theologis et al., 2000) allows a global, or genomic, analysis of transcriptional regulation in plants. Whereas the mechanisms of transcription are largely common across eukaryotes, their components vary among kingdoms. The complement of genes coding for transcriptional regulators in Arabidopsis has been described (Arabidopsis Genome Initiative, 2000; Riechmann et al., 2000). Their systematic functional characterization can be pursued with a variety of reverse genetic methods (Riechmann and Ratcliffe, 2000). In addition, gene expression profiling technologies, such as DNA microarrays, allow monitoring transcription factor activity at a genome-wide level. These studies should eventually lead to an understanding of the interplay of the transcription factors with the genome whose expression they control.
This chapter intends to provide a genomic perspective on transcriptional regulation in Arabidopsis. The first section briefly reviews the different types of proteins directly involved in transcription in eukaryotes, and our current understanding on how they function. The following sections consist of a description of the Arabidopsis complement of genes and proteins involved in transcriptional control, in particular sequence-specific DNA-binding transcription factors and chromatin-related proteins. Transcriptional regulators often act in a combinatorial fashion, and this mode of action is reviewed in the context of Arabidopsis promoters and cis-regulatory sequences, and of protein-protein interactions. Finally, genome-wide functional analyses of transcription factors, the characterization of the Arabidopsis promoterome, and of the transcriptome by gene expression profiling experiments, are considered. The availability of the genome sequence of different prokaryotic and eukaryotic organisms has provided for new ways of searching for unity and diversity among biological systems, and given birth to the field of comparative genomics. Although the subject of this book is Arabidopsis, reference is made in this chapter to other eukaryotic organisms, in order to situate the Arabidopsis genome information in a broader biological context.
2. Transcription machinery: concepts, components, and mechanisms
In eukaryotic organisms, regulation of gene expression proceeds through mechanisms that are fundamentally different from those in prokaryotes, which explains both the large number and diversity of proteins that are involved in the process, as well as how it can be tightly regulated to facilitate the diversification in expression patterns that is required for biological complexity (Struhl, 1999). In a prokaryote such as E. coli, the ground state for transcription is non-restrictive, that is, the RNA polymerase complex is not limited in its ability to gain access to the DNA and initiate RNA synthesis (Struhl, 1999). Negative regulation is rare, and exerted by sequence-specific repressors. Furthermore, it has been estimated that the global structure of the E. coli gene regulatory network possesses low complexity. On average, a transcription factor would regulate three genes, and an E. coli gene would be under the direct control of two transcription factors (Thieffry et al., 1998). There is a prominence of promoters controlled by a single regulator, and whereas many of the regulators regulate themselves (usually through auto-inhibitions), very rarely do they regulate other transcription factors (Thieffry et al., 1998).
In contrast, the ground state for transcription in eukaryotes is restrictive, as a result of the packing of the DNA into chromatin, which blocks the recognition of the core promoters by the basic transcription machinery (Kornberg, 1999; Struhl, 1999). The effects of chromatin structure on promoter accessibility makes chromatin modifying activities necessary for eukaryotic transcription, and has important implications for the way transcription factors act. In addition to the components of the basic transcription machinery and to scores of sequence-specific DNA-binding transcription factors, eukaryotic genomes contain a variety of genes that code for chromatin-related proteins. Furthermore, transcriptional regulators in eukaryotes operate following a combinatorial logic (an efficient way of increasing the number and diversity of gene regulatory activities), and the complexity of the regulatory networks can be great.
Prokaryotic sequence-specific DNA-binding transcription factors often recognize binding sites longer than 12 base-pairs (bp) (see RegulonDB, http://www.cifn.unam.mx/regulondb/ and DPInteract, http://arep.med.harvard.edu/dpinteract) (Robison et al., 1998; Salgado et al., 2001), whereas binding sites for eukaryotic transcription factors are usually shorter, 5 to 10 bp long. A combinatorial mechanism composed of factors that recognize short sequences is probably a more economical way (requires a reduced number of factors) of selectively regulating the expression of tens of thousands of genes, than a mechanism based upon factors that are each dedicated to control a small number of genes and operate through longer target sites. Thus, the DNA binding characteristics of eukaryotic transcription factors, and the mechanisms of transcription themselves, might be operationally and evolutionarily related to features of eukaryotic genomes such as the vast increase in genome size and in the number of genes to be regulated.
Briefly, the proteins involved in transcription in eukaryotes can be classified into four different functional groups: (1) the basic transcription apparatus and intrinsic associated factors (also known as general transcription factors, or GTFs); (2) large multi-subunit coactivators and other cofactors; (3) sequence-specific DNA-binding transcription factors; and (4) chromatin-related proteins. In contrast to the components of the basal transcription machinery, which in general are highly conserved, coregulators and transcription factors have diverged largely among eukaryotes. The roles that the proteins in these four classes play can be summarized as follows (after Lee and Young, 2000; Lemon and Tjian, 2000, and http://web.wi.mit.edu/young/pub/regulation.html) (for an extensive coverage of the mechanisms of eukaryotic transcription, see: Latchman, 1998; Elgin and Workman, 2000; White, 2001).
(1) The basic transcription apparatus, and intrinsic associated factors. In eukaryotic organisms, there are three different RNA polymerases, which are responsible for the synthesis of rRNA (Pol I), mRNA (Pol II), and tRNA, 5S rRNA, and other small RNA molecules (Pol III). The focus of this chapter is the transcription of protein-encoding genes, which is carried out by Pol II exclusively. Pol II is a multi-subunit enzyme (Cramer et al., 2001) that requires accessory factors to recognize promoter sequences and accurately initiate transcription. These general transcription factors (GTFs) include TFIIA, TFIIB, TFIID, TFIIE, TFIIF, and TFIIH. GTFs carry out a variety of different functions, from positioning the polymerase on the promoter (TFIIB) to unwinding its DNA (TFIIH). TFIID is a multi-subunit complex that is generally responsible for promoter recognition. It contains the TATA-box binding protein (TBP) and several TBP-associated factors (TAFs) (reviewed in Green, 2000). The TAF subunits of TFIID are critical for the responsiveness of the basic apparatus to transcriptional activators. However, individual TAFs are not essential for transcription of all genes in a genome. TAFs contribute to the specificity and variety of transcriptional responses: distinct TAFs can be targeted by different classes of activators, and individual TAFs can function as promoter selectivity factors. Furthermore, some TAFs can form part of other multi-subunit regulatory complexes, in addition to TFIID, such as the histone acetylation SAGA complex; and whereas most of the TAFs are ubiquitously expressed, some are expressed in a tissue or cell-type specific manner, which can lead to the formation of different TAF-containing complexes (for a review on gene-selective roles of GTFs and TAFs, see Veenstra and Wolffe, 2001).
(2) Large multi-subunit coactivators, and cofactors that bind sequence specific transcription factors. This heterogeneous class of regulatory proteins includes cofactors that interact with sequence-specific transcription factors and modulate their DNA binding or interaction with the core machinery, as well as large multi-subunit coactivators such as the Mediator complex, initially identified in yeast. Multi-subunit coactivators interact with Pol II and/or with multiple types of activators, serving as a modular adapter to regulate transcription initiation (Hampsey and Reinberg, 1999). The Mediator (or Mediator-like) complex is found in organisms from yeast to humans, but its number of subunits vary, and the complex from one organism might contain subunits that have no orthologs in another (Malik and Roeder, 2000; Rachez and Freedman, 2001).
(3) Sequence-specific DNA-binding transcription factors (activators and repressors). These are transcription factors of the classic type: usually defined as proteins that show sequence-specific DNA binding and are capable of activating and/or repressing transcription. They are responsible for the selectivity in gene regulation, and are often themselves expressed in a tissue, cell-type, temporal, or stimulus-dependent specific manner. Transcription factors are modular proteins, with distinct and functionally separable domains, such as DNA-binding and activation domains. Most known transcription factors can be grouped into families according to their DNA binding domain (Luscombe et al., 2000). Transcription factors can interact directly with different components of the general machinery and with coactivators, affecting complex formation. They can also interact with chromatin remodeling complexes.
(4) Chromatin-related proteins. This group includes factors that covalently modify histones (such as histone acetylases and deacetylases), and remodeling complexes that hydrolize ATP for reorganizing chromatin structure (such as the SWI/SNF and ISWI complexes). Histone acetylation is generally a characteristic of transcribed chromatin, whereas deacetylation is associated with repression. Accordingly, histone acetyltransferase activities are found in coactivators, and deacetylase activities in corepressors. Chromatin proteins usually form part of multi-subunit complexes.
Using the regulation of the HO endonuclease gene in yeast (Saccharomyces cerevisiae) as a paradigm, the steps leading to transcriptional activation can be summarized as follows (Cosma et al., 1999; Cosma et al., 2001). Upstream sequences are recognized by a transcription (enhancer-binding) factor, with accessibility to its targets sites despite the packing of the DNA into chromatin fibers. This transcription factor recruits the SWI/SNF complex, which then recruits SAGA, and results in the remodeling of chromatin and localized histone acetylation, which facilitates the access of additional transcription factors to cis-regulatory sequences. The secondary activators direct gene transcription through multiple interactions with cofactors and the core machinery, recruiting the RNA polymerase complex to the transcription initiation site. The specific order in which the different chromatin-modifying complexes are recruited can vary among promoters and organisms, but the dual role of activators, first enlisting chromatin modifying activities and then inducing localization of the basal transcription apparatus, appears to be widespread in eukaryotes, including plants (see below, and: Agalioti et al., 2000; Brown et al., 2001; Merika and Thanos, 2001).
In many instances, the correct functioning of a gene requires the termination of the activation of its transcription to be as rapid or precise as its initial triggering. Termination of activation can be accomplished by several mechanisms, among them the targeted destruction of transcription factors after their interaction with the basal transcription machinery. Phosphorylation of a transcription factor molecule by kinases that form part of the Pol II holoenzyme (such as Srb10 or TFIIH) would mark it for ubiquitin-mediated destruction, effectively preventing it from engaging into another Pol II initiation event, and freeing the promoter sequence to interact with another transcription factor molecule (reviewed in Tansey, 2001).
In addition to the mechanisms of transcriptional control that the classes of proteins described in this section mediate, there are at least two other possible levels of regulation of gene expression in eukaryotes: DNA methylation and nuclear organization. DNA methylation is associated with suppressed gene expression, and is reviewed in other chapters of this book (see also: Finnegan et al., 2000; Habu et al., 2001). Nuclear organization could provide for a higher level of regulation of gene expression, where different transcriptional functions might be segregated into distinct compartments (for models and reviews, see: Francastel et al., 2000; Lemon and Tjian, 2000; Cremer and Cremer, 2001; Misteli, 2001).
Of all the groups of proteins involved in transcription, the most numerous one is that of sequence-specific DNA-binding transcription factors. They are the principal factors upon which the mechanisms for selectivity of gene activation are built, and the basic (although not the only) protein components of the combinatorial logic of transcription.
3. Transcription factor gene content of the Arabidopsis genome
The analysis of the Arabidopsis genome sequence indicates that it codes for at least 1,572 transcription factors, which account for ∼6% of its estimated ∼26,000 genes (Arabidopsis Genome Initiative, 2000; Riechmann et al., 2000) (Table 1). This observation, however, represents an underestimate of the total number of transcription factors, given that, at present, approximately 40% of the proteins predicted from the genome sequence cannot be assigned to functional categories on the basis of sequence similarity to proteins of known biochemical function (Lin et al., 1999; Mayer et al., 1999; Arabidopsis Genome Initiative, 2000; Salanoubat et al., 2000; Tabata et al., 2000; Theologis et al., 2000). Some of those uncharacterized proteins are expected to be transcriptional regulators and, in fact, novel classes of transcription factors are still being discovered (for example: Boggon et al., 1999; Schauser et al., 1999; Kawaoka et al., 2000; Nagano et al., 2001; Windhövel et al., 2001). Therefore, the total number of transcription factor genes present in Arabidopsis (as well as, for the same reasons, in any other of the sequenced eukaryotic genomes) will be uncertain for some time.
Table 1.
Arabidopsis transcriptional regulators(a, b, c)
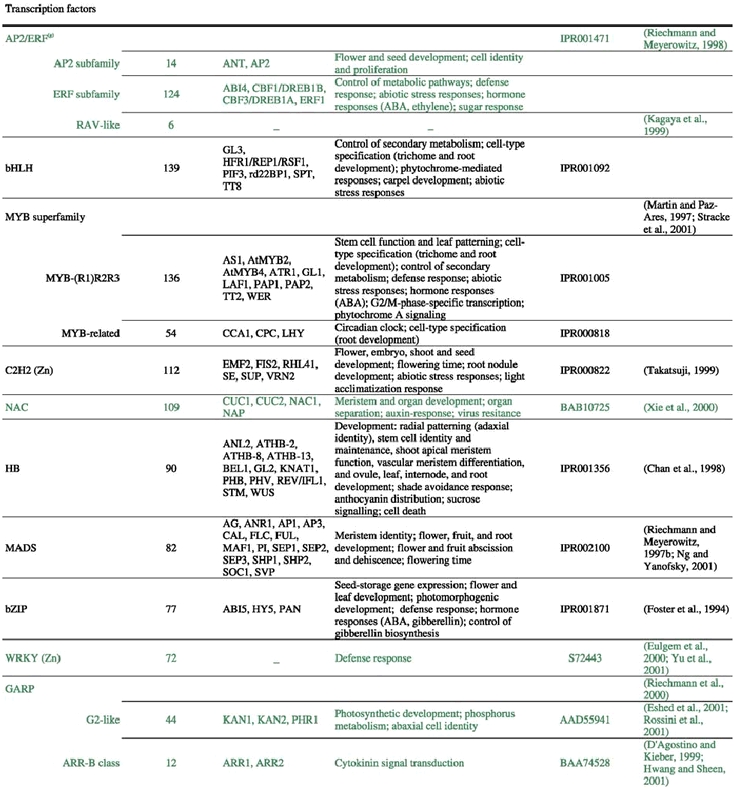
A question pertaining to genome-wide surveys is whether all the proteins identified by sequence similarity searches do indeed belong to the functional groups into which they are being catalogued. In the case of transcription factors, the answer depends on the particular gene family that is considered. If the conserved DNA-binding domain that defines a gene family is poor in sequence information (for example, some zinc-coordinating motifs), the ratio of false positives in the searches can be relatively high (although it can often be reduced by additional sequence comparison strategies that are beyond the scope of this chapter, see: Riechmann et al., 2000). On the other hand, many families are defined by long DNA-binding domains (50 to 70 amino acids) with multiple residues being highly conserved (that is, the domains are rich in sequence information). The three-dimensional structure of these domains might have been solved, and revealed the contacts between some of the conserved residues and the DNA. In cases like these, such as for example the homeobox and the AP2/ERF (APETALA2/ethylene response factor) families, it is reasonable to expect all the members of the gene family to be transcription factors (activators or repressors) (the AP2/ERF family was initially referred to as AP2/EREBP, for AP2/ethylene responsive element binding protein). However, there are cases in which a family of bona fide transcription factors might also contain members that have additional functions (for reviews on multifunctional transcription factors: Ladomery, 1997; Wilkinson and Shyu, 2001). For example, the Drosophila homeodomain protein Bicoid directs anterior embryo development both by regulating transcription and by interacting with Caudal mRNA and inhibiting its translation, thus restricting Caudal (which is another homeodomain protein) accumulation to the posterior part of the embryo through posttranscriptional control. Both DNA- and RNA-binding are specified by the Bicoid homeodomain, but by distinct subregions or residues in it (Niessing et al., 2000). The Arabidopsis MYB-related protein AtCDC5 is known to be homologous to the S. cerevisiae CEF1 and S. pombe Cdc5 proteins (Hirayama and Shinozaki, 1996; Ohi et al., 1998). Cdc5 proteins are essential for the G2/M progression, but their molecular functions are not completely understood, as they are required for pre-mRNA splicing and associate with core components of the splicing machinery, but also show sequence-specific binding to double stranded DNA and transactivation potential (Burns et al., 1999; Lei et al., 2000). It is thus possible that Cdc5 proteins are another example of factors with several distinct molecular functions. Two other members of the Arabidopsis MYB-related family, AtTRP1 and AtTBP1, have been identified as telomere-binding proteins (Chen et al., 2001a; Hwa,Ng et al., 2001), although a possible role in transcription cannot be ruled out because the 5′ regions of some Arabidopsis genes contain two or more non-contiguous telomeric repeats (Regad et al., 1994). These examples illustrate the limitations of using sequence similarity to assign potential roles to proteins that are otherwise uncharacterized, and also how the determination of their molecular functions can be elusive. Similar cases might occur within some of the zinc-coordinating protein families, since the same or related motifs can be involved in DNA- and RNA-binding, and may be present in proteins withfunctions involving nucleic acid binding but not necessarily transcriptional regulation. For example, vertebrate Y-box proteins contain a zinc-coordinating cold-shock domain, and are often dual DNA- and RNA-binding proteins that can regulate transcription and/or translation (reviewed in: Matsumoto and Wolffe, 1998; Sommerville, 1999).
With these caveats in mind, the Arabidopsis complement of transcription factors has been the subject of an extensive genome-wide descriptive analysis, which also included a comparison with those of Drosophila melanogaster, Caenorhabditis elegans, and Saccharomyces cerevisiae (Riechmann et al., 2000). The main conclusions of that study are summarized here.
The 1,572 transcriptional regulator genes identified in the Arabidopsis genome are classified into more than 45 different gene families (Table 1; Figure 1), all of which are scattered throughout the genome. In addition, there are a few single-copy or “orphan” genes, such as LEAFY (LFY). Transcriptional regulators represent approximately 4.6, 3.5, and 3.5% of the genes in Drosophila, C. elegans, and yeast, respectively (Riechmann et al., 2000). Thus, the Arabidopsis content of transcription factors is 1.3 times that of Drosophila, and 1.7 times that of C. elegans and yeast (Riechmann et al., 2000). The large number and diversity of transcription factors in Drosophila were proposed to be related to its substantial regulatory complexity (Adams et al., 2000). Applying the same logic to Arabidopsis suggests that the regulation of transcription in plants is as complex as that in Drosophila. Furthermore, if the estimated total number of genes in humans, 30,000–40,000, and of transcription factor genes, 1,850–2,000, are correct (International Human Genome Sequencing Consortium, 2001; Tupler et al., 2001; Venter et al., 2001), then the transcription factor gene content of Arabidopsis and of H. sapiens are similar (∼6% in Arabidopsis, versus 4.6–6.6% in humans). It should be noted, however, that there is a substantial degree of uncertainty about these estimates of gene numbers in humans (see, for example: Hogenesch et al., 2001; Wright et al., 2001).
Figure 1.
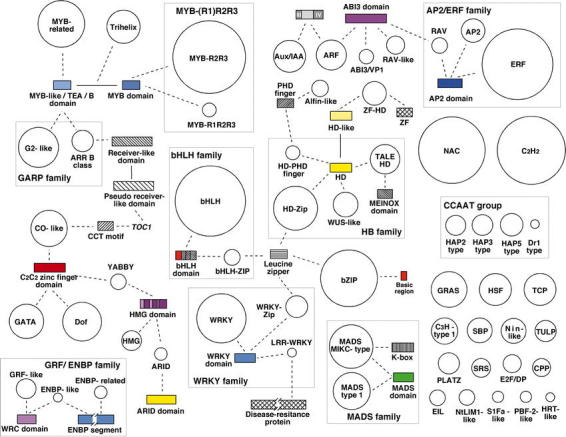
The Arabidopsis complement of transcription factors. Gene families are represented by circles, whose size is proportional to the number of members in the family. Domains that have been shuffled, and that therefore “connect” different groups of transcription factors are indicated with rectangles, whose size is proportional to the length of the domain. DNA binding domains are colored; other domains (usually protein-protein interaction domains) are shown with hatched patterns. Dashed lines indicate that a given domain is a characteristic of the family or subfamily to which it is connected. Gene names are written in italics. Whereas many of the indicated domain shuffling events are specific to plants, others likely predate the appearance of the three distinct eukaryotic kingdoms (for details, see Riechmann et al., 2000). This figure is an expanded and updated version of Figure 1 in Riechmann et al. (2000).
Transcription factors, the networks that they form, and the genes that they regulate, have been proposed as a possible objective measurement (connectivity of gene-regulation networks) of the biological complexity of an organism (Szathmáry et al., 2001). From that point of view, the large number of transcription factors in Arabidopsis was interpreted in the context of the complexity of secondary metabolism in plants (Szathmáry et al., 2001), but it might also be related to the complex interactions between plants and the environment (both biotic and abiotic) as well as to the degree of duplications in the genome (see below, and Arabidopsis Genome Initiative, 2000).
The extent to which the Arabidopsis complement of transcription factors represents that of other plants is still an open question. Since the evolutionary divergence between the monocot and dicot lineages is a relatively recent event, which perhaps occurred ∼200 million years ago (Savard et al., 1994), it could be expected that the overall composition of monocot and dicot transcription factor complements would be similar. In fact, the largest transcription factor families in Arabidopsis also appear to be the most prevalent ones in monocotyledonous plants. For example, the phylogenetic comparison of a subset of maize and Arabidopsis MYB-(R1)R2R3 sequences shows that the amplification of the gene family occurred prior to the separation of monocots and dicots (Rabinowicz et al., 1999). In addition, within phylogenetically well-studied families of transcription factors, such as the MADS-box family, many examples of orthology can be identified between Arabidopsis genes and those from rice or maize, and even from gymnosperms (reviewed in: Theissen et al., 2000; Ng and Yanofsky, 2001) (see also http://www.mpiz-koeln.mpg.de/mads). Putative orthologous MADS-box genes have regularly maintained conserved functions, even after substantial sequence divergence (Theissen et al., 2000). However, it is also apparent that diversity in transcriptional regulators will be found within the plant kingdom, and between monocots and dicots. Many MADS-box gene duplication and diversification events occurred after separation of the moss and fern lineages from the lineage that originated the flowering plants (Münster et al., 1997; Hasebe et al., 1998; Krogan and Ashton, 2000; Svensson et al., 2000), and at least two clades of MADS-box genes appear to have been amplified in the phylogenetic lineage that led to grasses with respect to Arabidopsis (Theissen et al., 2000). Similarly, whereas most of the amplification of the MYB-(R1)R2R3 gene family occurred prior to the separation between monocots and dicots, several subgroups in maize appear to have originated recently or undergone duplication (some of these duplications are likely to be associated with the allotetraploid origin of the maize genome, but others do not reflect it: Rabinowicz et al., 1999). These recent expansions could have allowed a functional diversification that might not be present in Arabidopsis.
An issue that impinges on the question of the similarity of the Arabidopsis complement of transcription factors with that of other plants is the degree of completeness of the current characterization (i. e., sequence determination and analysis) of the Arabidopsis genome, in particular if that question is to be addressed on a gene-by-gene basis. TRM1 is a maize C2H2 zinc finger transcription factor involved in the repression of rbcS gene expression in mesophyll cells that is related to the mammalian transcription activator-repressor YY1 (Xu et al., 2001). A BLAST search of the higher plant DNA sequences available in GenBank (July 2001) identifies homologous genes in other monocots (Triticum aestivum) as well as in dicotyledonous plants (Nicotiana tabacum, Solanum tuberosum), but not in Arabidopsis. It is possible that an Arabidopsis TRM1 homolog resides in one of the still unsequenced segments of the genome (see http://www.arabidopsis.org). Similarly, there are a few Arabidopsis transcription factor genes represented by ESTs or BAC-end sequences that still cannot be identified in the genome sequence. The limitations of the current sequencing technologies make it impractical or impossible to determine the sequence of eukaryotic genomes to absolute completeness. Thus, a failure to identify a particular gene in the genome sequence of an organism should not be taken as a definitive proof of the absence of that gene. In addition, gene sequences might diverge more than expected, which might result in the identification of homologous genes requiring more sophisticated sequence analysis than a standard BLAST search. For example, a homolog of the mammalian tumor suppressor gene p53 can be identified in the sequence of the C. elegans genome, despite initial reports that no p53-like gene was present in that organism (Derry et al., 2001; Schumacher et al., 2001).
The genome-wide comparison of transcription factors among eukaryotic organisms (Arabidopsis, Drosophila, C. elegans, and S. cerevisiae, encompassing the plant, animal, and fungal kingdoms) reveals the evolutionary generation of diversity in the regulation of transcription (Riechmann et al., 2000). Each of these eukaryotic kingdoms has its own set of particular transcription factor families and genes. Members of kingdom-specific families represent 45% of the Arabidopsis complement of transcriptional regulators, whereas those of families that are present in all four organisms account for 53% (Figure 2). In each organism, a minority (2–5%) of its transcription factors belong to families that are present in two of the three kingdoms: in animals and yeast (SOX/TCF, Fork head, and RFX1-like transcription factors) or in plants and animals (TULP, CPP, and E2F/DP families) (Figure 2) (Riechmann et al., 2000). This distribution of genes and gene families reflects the evolutionary history of eukaryotes. According to molecular phylogenetic analyses, plants, animals and fungi all diverged from a common ancestor during a short period of time, ∼1.5 billion years ago (Wang et al., 1999a; Philippe et al., 2000; Nei et al., 2001). Thus, most of the transcription factor families are either shared by the three kingdoms (those that were present in the common ancestor), or specific to each one (those families that arose independently following divergence).
Figure 2.

Distribution of transcriptional regulators in eukaryotic organisms (A. thaliana, D. melanogaster, C. elegans, and S. cerevisiae). Transcriptional regulators are kingdom-specific, common to plants, animals, and fungi, or present in only two of the three kingdoms. Members of kingdom-specific families represent only 14% of the total in Drosophila because of its extensive use of the C2H2 zinc finger proteins. The data represented in this figure are from Riechmann et al. (2000).
Many of the Arabidopsis transcription factor gene families are large (Table 1). However, none has been so disproportionately amplified as the nuclear hormone receptors in C. elegans (∼38% of its transcription factors), the C2H2 zinc finger proteins in Drosophila (∼46%), or the C6 and C2H2 families in yeast (∼25% each one) (Figure 3) (Riechmann et al., 2000). The three largest families of transcription factors in Arabidopsis, AP2/ERF, bHLH (basic-region helix-loop-helix), and MYB-(R1)R2R3, each represent only ∼9% of the total, and there are several other families with comparable numbers of genes (Figure 3) (Riechmann et al., 2000). The two transcription factor families that have been more substantially amplified in plants, as compared to animals and yeast, are the MYB and the MADS families. Another difference between the Arabidopsis complement of transcription factors and those of the other eukaryotes is that less than 25% of it consists of zinc coordinating proteins, whereas zinc coordinating transcription factors represent ∼51% of the total in Drosophila, ∼64% in C. elegans, and ∼56% in yeast (Riechmann et al., 2000).
Figure 3.
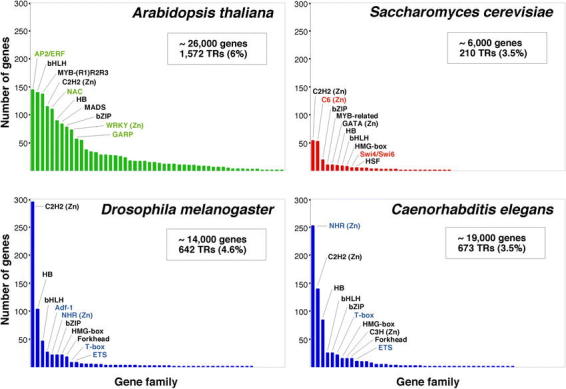
Content and distribution of transcriptional regulator genes in eukaryotic genomes. For each of the organisms considered (A. thaliana, D. melanogaster, C. elegans, and S. cerevisiae), the different families of transcription factors are ordered according to the number of members that they contain. The 10 largest families in each organism are identified. The names of those families that are specific to one kingdom are shown in color. The data represented in this figure are from Table 1 and from Riechmann et al. (2000). The number of genes in each of the genomes is given as an approximate number (Goffeau et al., 1997; The C. elegans Sequencing Consortium, 1998; Adams et al., 2000; Arabidopsis Genome Initiative, 2000). This is because the number of genes predicted at the time that a genome is sequenced is always an estimate that is refined over time. The number of genes that code for transcriptional regulators (TRs), and the percentage of the total number of genes that they represent, is indicated. (Zn) indicates a zinc coordinating DNA binding motif.
The Arabidopsis transcription factors that belong to families that are common to all eukaryotes do not share significant similarity with those from the other kingdoms, except in the conserved DNA binding domains that define the respective families (Riechmann et al., 2000). Furthermore, diversity in protein sequence and structure is increased by domain shuffling (Figure 1). Shuffling of some of the DNA-binding domains that are present in all eukaryotes has generated novel transcription factors with plant-specific combinations of modules, as for example in the homeodomain, MADS, and ARID protein families (Figures 1 and 4) (Riechmann et al., 2000).
Figure 4.
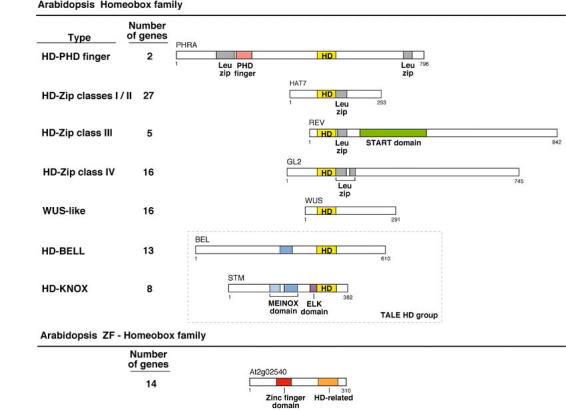
The Arabidopsis Homebox (HB) and ZF-HB gene families. The Arabidopsis homeobox gene family can be subdivided into different groups according to the combinations of domains that the corresponding proteins contain, and to the phylogenetic analysis of the homeodomain. The number of members in each Arabidopsis homeobox gene subfamily is indicated (except for three genes, whose classification is unclear and that are not represented in this figure). Most of the combinations of a homeodomain with a domain of a different type (leucine zipper, PHD finger, START domain) are the result of domain shuffling events specific to the plant kingdom: those combinations are not found in Drosophila, C. elegans, or yeast homeodomain proteins. The only Arabidopsis homeodomain proteins that have an additional motif also found in animal homeodomain proteins are those of the KNOX class, which contain a MEINOX domain (Bürglin, 1998). Conversely, homeodomains in animals are associated with a large variety of motifs, such as the paired and POU-specific domains (which are themselves specific to animals), the LIM motif, or C2H2 zinc fingers, in combinations that are not present in Arabidopsis (for a similar depiction of the animal homeodomain proteins, see Gehring et al., 1994). The START domain is a lipid-binding domain that could provide regulation of HD-Zip class III protein function by sterols. It is found in a variety of eukaryotic proteins, but has been found associated with transcription factor domains only in this class of plant homeobox genes (Ponting and Aravind, 1999). Proteins of the ZF-HB family contain a homeodomain-related sequence that is more divergent from all the different groups of homeodomain sequences of the HB family than these are among themselves (Windhövel et al., 2001). In addition, ZF-HB proteins contain a plant-specific zinc coordinating motif (Windhövel et al., 2001)
The Arabidopsis genome contains many tandem gene duplications and large-scale duplications on different chromosomes (Arabidopsis Genome Initiative, 2000; Blanc et al., 2000; Vision et al., 2000). Whereas some of these duplications have been followed by rearrangements and divergent evolution, up to 40 to 60% of the Arabidopsis genes might comprise pairs of highly related sequences (the percentage depending on the parameters used in the analyses) (Arabidopsis Genome Initiative, 2000; Blanc et al., 2000). Transcription factor genes follow these general observations. A comparison of the transcription factor complement to itself (all-against-all) revealed that, on average, closely related genes account for ∼45% of the total number in the major families (a pair of proteins was considered highly similar if they showed >60% amino acid sequence identity along at least two-thirds of the length of one of them) (Riechmann et al., 2000). The pairs or groups of closely related genes most often correspond to duplications in different chromosomes (∼65% on average), or to duplications in the same chromosome but at very large distances (∼22%), than to tandem repeats (∼13%) (Riechmann et al., 2000). In addition, clusters of three or more homologous transcription factor genes are very rare in the genome (Riechmann et al., 2000). This distribution indicates that it will be feasible to generate double or triple mutants for the majority of the pairs or groups of highly related genes that, because of their sequence similarity, might have overlapping or partially redundant functions (which might not be revealed by single mutant analyses; see below).
The analysis of ∼120,000 Arabidopsis expressed sequence tags (ESTs) (sequences available in GenBank in January 2001) suggests that, in terms of overall expression and considered as a whole, transcription factor genes are not substantially different from the rest of the genes in the genome. Approximately half of the ∼26,000 predicted genes are matched by an EST (Arabidopsis Genome Initiative, 2000; Theologis et al., 2000). Similarly, when the major Arabidopsis transcription families are considered, ∼47% of the genes are represented by an EST (Table 2). This observation is in contrast to the sometimes common assumption that, because of their regulatory nature, genes of this class are generally expressed at low levels.
Table 2.
EST representation of the major Arabidopsis transcription factor gene families(a)
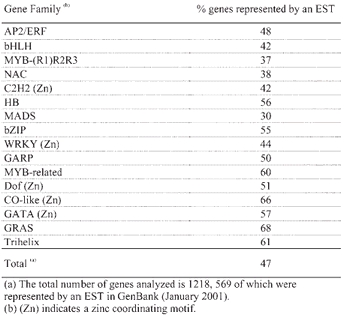
4. Chromatin remodeling proteins.
Chromatin structure is an important element of the mechanisms that determine gene expression patterns in eukaryotes, because nucleosome assembly eliminates the accessibility of promoter sequences for the basal transcription machinery. The unfolding of packed chromatin is necessary for gene expression and, conversely, repression requires the formation and maintenance of condensed chromatin structures. Gene silencing and epigenetic phenomena, in which chromatin structure and histone modifications play a role, are by themselves the subject of other chapters in this book.
As summarized above, one of the mechanisms of transcription factor action is the recruitment of chromatin remodeling complexes to target promoters. This mechanism has been deduced from research on transcription in yeast and mammalian cells, but studies on the regulation of the β-phaseolin (phas) gene in bean (Phaseolus vulgaris) suggest that it also operates in plants (reviewed in Li et al., 2001a). The phas gene is silenced in vegetative tissues as a consequence of the positioning of a nucleosome over the TATA boxes of the promoter, making them inaccessible to TBP, whereas nucleosome displacement allows the gene to be highly expressed during seed development (Li et al., 1998). Such modification in chromatin structure results from the presence of the seed-specific transcription factor PvALF, a member of the ABI3/VP1 family (Li et al., 1999). However, PvALF is not sufficient for phas transcriptional activation, which does not occur in the absence of abscisic acid (ABA) (Li et al., 1998). Thus, a plausible model is that PvALF mediates chromatin reconfiguration, then allowing the binding of ABA-responsive transcription factors and the recruitment and assembly of the basal transcription machinery on the phas promoter (Li et al., 2001a).
The remodeling or reconfiguration of chromatin involves different types of enzymes, such as members of the SWI2/SNF2 subfamily of the DEAD/H box superfamily of nucleic-acid stimulated ATPases, and proteins that covalently modify histones, such as acetyltranferases (HATs) and deacetylases (HDACs), kinases, and methyltransferases (for reviews: Kadonaga, 1998; Elgin and Workman, 2000; Fry and Peterson, 2001; Jenuwein, 2001; Urnov and Wolffe, 2001). All eukaryotes appear to contain several proteins belonging to each one of these types, and each type can be further divided into different structural subclasses. Such structural diversity allows different proteins with the same biochemical activity to be involved in specialized cellular functions. The chromatin proteins with enzymatic activity usually form part of multi-subunit complexes, which might be necessary for their specificity and functionality.
In general, histone acetylation is a characteristic of transcribed chromatin, whereas deacetylation is associated with repression. HATs acetylate the ε-amino groups of specific lysine residues in the amino-terminal tails of the histone proteins that form the octamer around which the DNA wraps in the nucleosomes. Histone deacetylase-containing complexes reverse this covalent modification (reviewed in Khochbin et al., 2001). The molecular mechanisms by which histone acetylation affects chromatin structure and influences transcription could involve the destabilization of interactions between the DNA and the histone octamer (by neutralizing positive charges), interference with the high-order packing of chromatin, or the modification of interactions between histones and other proteins (reviewed in: Marmorstein, 2001a; Marmorstein and Roth, 2001; Roth et al., 2001). Other types of post-translational modifications, such as phosphorylation and methylation, also occur on histones, and can regulate chromatin structure and transcriptional activation and repression (reviewed in Marmorstein, 2001a). Methylation of specific lysine residues in histone tails is a relatively stable modification, thus providing a stable epigenetic mark for transcriptional regulation and gene silencing via heterochromatin assembly (reviewed in: Jenuwein, 2001; Rice and Allis, 2001). Lysine methyltransferase activity has been demonstrated for several eukaryotic SET domain proteins (Table 3). In addition, histone tails can also be methylated at arginine residues by a different class of enzymes, that act as coactivators of transcription (Chen et al., 1999).
Table 3.
Structural domains frequently found in chromatin-related proteins(a)
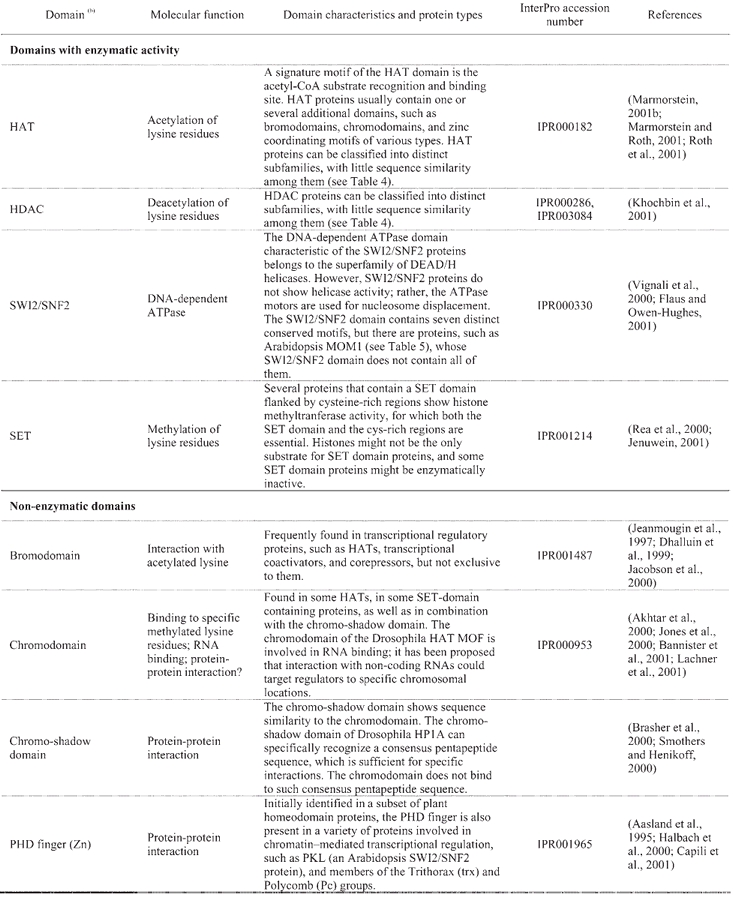
A “histone code” hypothesis has been proposed, suggesting that distinct covalent histone modifications might be used by the cell, sequentially or in combination, to generate a “code” that could be read by other proteins to produce different transcriptional outputs (Strahl and Allis, 2000). Reading the “histone code” would necessitate protein domains that recognize, in a receptor-ligand type of interaction, the different covalent modifications that can occur on histones (Strahl and Allis, 2000). Binding activities have been identified in several of the protein domains that are frequently found in chromatin-related proteins, such as the bromodomain and the chromodomain, which can recognize acetylated- and methylated-lysine residues of the histone tails, repectively (Table 3). A further level of complexity in regulatory mechanisms is inferred from the observation that the same covalent modifications that can be found on histones also occur on other proteins involved in transcriptional control. For example, histones are not the only targets for HATs, as HAT-catalyzed acetylation can also regulate the activity of transcription factors and co-factors (reviewed in: Sterner and Berger, 2000; Chen et al., 2001b). Lastly, another group of enzymes involved in chromatin remodeling is that of the DNA-dependent ATPases of the SWI2/SNF2 type. Yeast SWI2/SNF2 is the catalytic subunit of the multiprotein SWI/SNF remodeling complex, which can mediate the repositioning of nucleosomes by sliding histone octamers to other sites on the same DNA molecule, as well as by transferring them to other DNA molecules (reviewed in: Vignali et al., 2000; Flaus and Owen-Hughes, 2001).
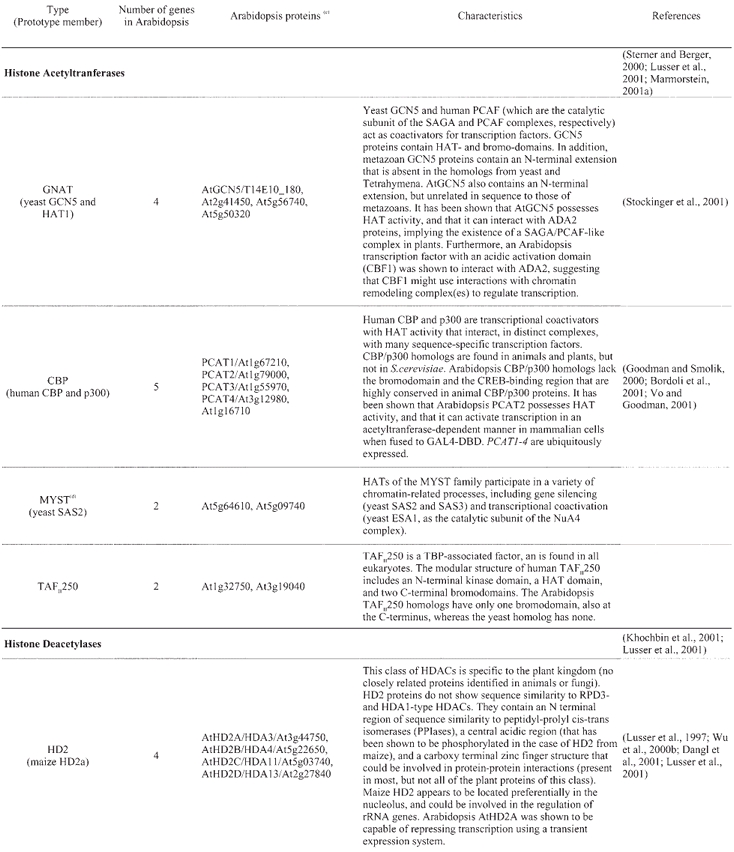
A catalogue of known and putative chromatin proteins in Arabidopsis and maize has been compiled in ChromDB, a database that aims to present information on the entire complement of chromatin proteins in plants (http://chromdb.biosci.arizona.edu/). ChromDB lists over 220 different Arabidopsis chromatin genes, including SWI2/SNF2 homologs (22 genes), HATs (12 genes; 10 are listed as HATs and 2 as TAFII250 homologs; Table 4), HDACs (17 genes; Table 4), and SET-domain-protein genes (29 genes), and also includes histones (50 genes) and homologs of subunits of global transcription factors.
Table 4.
Arabidopsis histone acetylases and deacetylases(a, b)
The definition and identification of the complement of chromatin proteins in Arabidopsis, or in any other eukaryotic organism, and in particular of the subset of those proteins that might be involved in transcriptional control, is complicated by several factors. First, chromatin remodeling is mediated by multiprotein complexes, some of which have already been purified and characterized from yeast and animal (mammalian, Drosophila) cells, but none from plants. Some of these complexes (for example, the yeast SAGA and human PCAF complexes) show a remarkable conservation in subunit composition, but there are also cases of proteins and complexes that are specific to one kingdom (Sterner and Berger, 2000, and see below). Thus, biochemical studies will be needed to obtain a complete description of the Arabidopsis complement of chromatin proteins. Another complication for the identification of bona fide chromatin proteins arises from their multi-domain architecture. Chromatin proteins frequently combine different domains or motifs of distinct molecular functions (Table 3). However, those domains are not necessarily unique to chromatin proteins, as they (or related sequences) can be present in other types of proteins. For example, an Arabidopsis protein that contains sequences related to the chromodomain is localized to the chloroplast and forms part of the chloroplast signal recognition particle pathway (Klimyuk et al., 1999). Lastly, the structure of chromatin influences not only transcription, but also other nuclear processes that are physically associated with the genome, such as replication, recombination, and DNA repair. Thus, that a protein is chromatin-related does not necessarily imply that it is involved in transcriptional control. For these different reasons, the identification and description by sequence similarity searches of the complement of chromatin proteins involved in transcriptional regulation, and of their biochemical and molecular functions, is more complicated than that of the sequence-specific DNA-binding transcription factors discussed above.
It is apparent from the content in known chromatin genes of the Arabidopsis genome that chromatin remodeling is important in plants for the control of gene expression. That some of the molecular mechanisms for chromatin reconfiguration and transcriptional control are conserved among plants and the other eukaryotic kingdoms can be deduced from the presence of orthologous genes. Furthermore, similarities or functional equivalence at the molecular or physiological level has been demonstrated in some cases, as illustrated with the following examples.
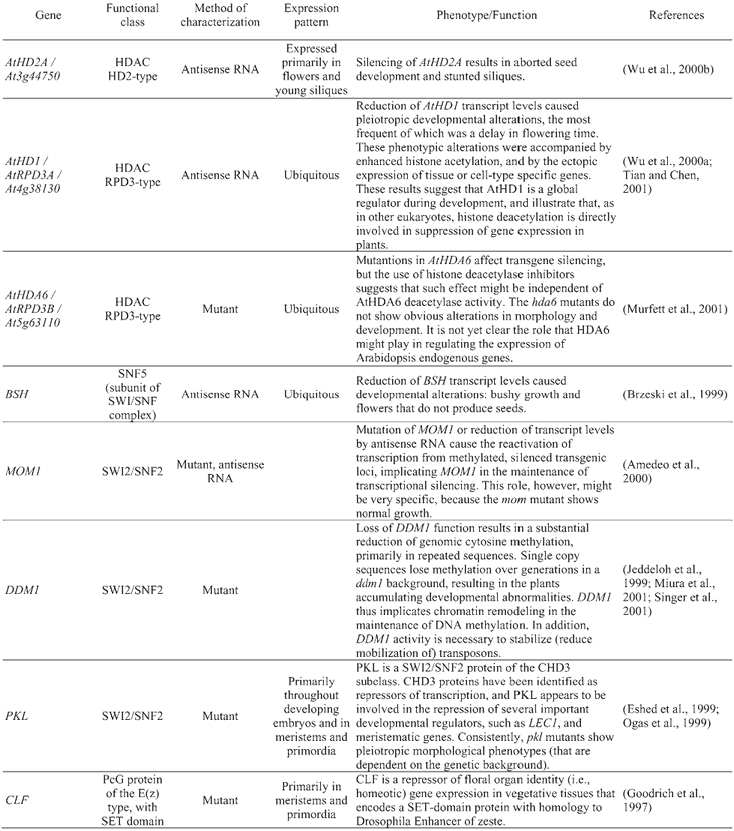
An RPD3-type maize histone deacetylase has been shown to complement a S. cerevisiae null mutant in the homologous RPD3 gene (Rossi et al., 1998).
The Arabidopsis gene BUSHY (BSH), which codes for a protein with high sequence similarity to S. cerevisiae SNF5 (a component of the SWI/SNF remodeling complex), can partially complement a snf5 mutation in yeast (Brzeski et al., 1999).
Arabidopsis homologs of human CBP/p300 proteins recapitulate the binding specificity of p300 for the adenoviral oncoprotein E1A, in addition to being capable of activating transcription in mammalian cells (Bordoli et al., 2001).
The Arabidopsis protein that is orthologous to yeast GCN5 possesses HAT activity, and can interact with Arabidopsis ADA2 proteins, suggesting that a complex analogous to yeast SAGA (of which GCN5 and ADA2 form part) and human PCAF also exists in plants (Stockinger et al., 2001).
PICKLE (PKL; also initially referred to as GYMNOS) is an Arabidopsis protein of the SWI2/SNF2-type that appears to be involved in the repression of several important developmental regulators, such as LEC1 and meristematic genes (Eshed et al., 1999; Ogas et al., 1999). PKL is homologous to human Mi-2, a component of the NuRD complex. By virtue of its different subunits, the NuRD complex combines both ATP-dependent chromatin remodeling and HDAC activity. The homology between PKL and Mi-2 suggests that a NuRD-like complex might exist in plants; thus, a plausible mechanism for gene repression by PKL is via histone deacetylation mediated by NuRD (reviewed in Ahringer, 2000).
The Polycomb group (PcG) and the trithorax group (trxG) of proteins in Drosophila and mammals control the cellular inheritance of mitotically stable states of gene expression, homeotic genes in particular. PcG and trxG proteins (repressors and activators, respectively) are thought to regulate transcription by modulating the structure of chromatin (reviewed in: Brock and van Lohuizen, 2001; Francis and Kingston, 2001; Mahmoudi and Verrijzer, 2001). The proteins within each group (Pc or trx) can be unrelated in sequence; rather, their relationship to each other comes from the fact that they operate together in the form of multi-subunit complexes of a genetically defined function (Gould, 1997). Homologous or related proteins for some PcG and trxG factors have been identified in Arabidopsis, and in some cases functionally characterized. Three Arabidopsis proteins show homology to the Drosophila SET-domain PcG protein Enhancer of zeste (E(z)), CURLY LEAF (CLF), CURLY LEAF LIKE (CLK), and MEDEA (MEA) (Goodrich et al., 1997; Grossniklaus et al., 1998) (Table 5). CLF is a repressor of floral organ identity (i.e., homeotic) gene expression in vegetative tissues (Goodrich et al., 1997), whereas MEA is involved in the maternal control of embryogenesis (Grossniklaus et al., 1998). Another Arabidopsis PcG protein involved in seed development is FERTILIZATION-INDEPENDENT ENDOSPERM (FIE), which shows homology to PcG proteins with WD repeats, such as Drosophila extra sex combs (esc) (Ohad et al., 1999). Animal E(z) and esc proteins have been shown to interact and to co-localize in unique complexes. Similarly, Arabidopsis FIE and MEA also interact, which provides a molecular explanation for the similarities between the fie and mea mutant phenotypes (Spillane et al., 2000; Yadegari et al., 2000). Other Arabidopsis proteins, such as EMBRYONIC FLOWER2 (EMF2), FERTILIZATION-INDEPENDENT SEED 2 (FIS2), and VERNALIZATION 2 (VRN2) are related to a different Drosophila PcG protein, Suppressor of zeste 12 (Su(z)12) (Luo et al., 1999; Birve et al., 2001; Gendall et al., 2001; Yoshida et al., 2001).
Table 5.
Arabidopsis chromatin genes functionally characterized
Despite these similarities, however, novel features in the chromatin-mediated regulation of gene expression have also evolved in plants. Plants contain what appears to be a kingdom-specific family of histone deacetylases, the HD2 class (Lusser et al., 1997; Aravind and Koonin, 1998; Dangl et al., 2001) (Table 4). Orthologs for some Arabidopsis chromatin proteins are not found in yeast or animals. This is the case, for example, of MOM1, a SWI2/SNF2-related protein that is involved in the maintenance of transcriptional gene silencing (Amedeo et al., 2000; Arabidopsis Genome Initiative, 2000). In addition, homologous chromatin proteins can show structural variation among the different eukaryotic kingdoms, and some of those variations appear to be specific to plants. In fact, eukaryotic chromatin proteins are a prominent example of evolutionary innovation by domain shuffling, deletion, and accretion (International Human Genome Sequencing Consortium, 2001). For example, Arabidopsis CBP/p300-like proteins lack the bromodomain and the CREB-binding region that are highly conserved in animal CBP/p300 proteins (Bordoli et al., 2001) (Table 4; CBP/p300 proteins are not found in yeast). Instead, one of these Arabidopsis proteins (PCAT1) contains a repeated motif of unknown function that does not show sequence similarity to any other known amino acid motif (Bordoli et al., 2001).
Other Arabidopsis chromatin genes that have already been genetically or functionally characterized further show the importance of chromatin-mediated regulation of gene expression in multiple aspects of the plant life cycle (Table 5). Reduction of AtHD1 (an HDAC-coding gene, also referred to as AtRPD3A) transcript levels by using antisense RNA caused pleiotropic developmental alterations, suggesting a global role for AtHD1 in regulating gene expression during development (Wu et al., 2000a; Tian and Chen, 2001). Similarly, reduction of AtHD2A activity (which codes for an HDAC of the plant-specific HD2 class) resulted in aborted seed development (Wu et al., 2000b). In another study, mutants in the HDAC gene AtHDA6 were isolated, which were morphologically wild-type but showed deregulated expression of transgenes, suggesting that AtHDA6 might be specifically involved in (transgene) silencing processes (Murfett et al., 2001). In addition to MOM1 and PKL, mentioned above, another Arabidopsis gene coding for a SWI2/SNF2-type protein that has been functionally characterized is DECREASE IN DNA METHYLATION1 (DDM1). DDM1 is required to maintain normal cytosine methylation patterns and to stabilize transposon behavior (Jeddeloh et al., 1999; Miura et al., 2001; Singer et al., 2001).
In summary, genetic studies on a variety of biological processes in Arabidopsis, the determination of its genome sequence, and biochemical studies performed in maize, have all started to illuminate the different physiological functions that chromatin remodeling might play in plants. However, our understanding of chromatin remodeling at the molecular level, and on how it influences plant nuclear processes, is extremely limited, and mostly derived from comparisons with the better-studied systems of yeast, Drosophila, and mammalian cells. If chromatin research in these model organisms is to be viewed as an example, it is clear that biochemical studies will be essential to understand chromatin in plants.
5. The combinatorial nature of transcriptional regulation: promoters, cis-elements and trans-acting factors.
Whereas plants and animals (or, to be more precise, Arabidopsis and Drosophila, C. elegans, and humans) might have comparable contents of transcription factors (3.5–6.6% of the total number of genes; see above), the organization of the regulatory sequences on which these transcription factors act can be different in the two kingdoms. In animals, the regulatory sequences that determine the correct temporal and spatial expression of a gene can extend over tens of kilobases (kbs) of DNA (for a review, see Bonifer, 2000). In contrast, regulatory sequences of plant genes usually span much shorter DNA intervals, often less than 1 or 2 kbs. This is reflected in the compact organization of the Arabidopsis genome, in which gene density is high. Out of the sequenced 115.4 megabases (Mb) of the 125 Mb genome, 51.2 Mb (or 44% of the sequenced regions) correspond to predicted exons and introns (Arabidopsis Genome Initiative, 2000). On average, there is one gene per 4.5 kb of DNA: the gene length (exons plus introns) is approximately 2 kb, and ∼2.5 kb correspond to intergenic regions. Considering the whole genome, transposons account for ∼20% of the intergenic DNA, resulting in an average of 2 kb of DNA for the 5′ and 3′ regions of a particular gene (Arabidopsis Genome Initiative, 2000). Other plants, maize for example, have genomes that are much larger than that of Arabidopsis, but with a similar organization of promoter sequences: in the maize genome, active genes are usually distributed in compact gene-rich islands, with much of the genomic DNA corresponding to repetitive sequences made up of retrotransposons (SanMiguel et al., 1996; Fu et al., 2001). As a result, regulatory sequences in Arabidopsis, and in plants in general, are easier to identify and delimit experimentally than in, for example, humans (for an introduction to the problem in mammals, see: Gumucio et al., 1993; Hardison et al., 1997; Bonifer, 2000; Fickett and Wasserman, 2000; Scherf et al., 2001). Compact Arabidopsis 5′ promoter sequences often recapitulate faithfully the expression of the native gene when assayed in transgenic plants by reporter gene fusions, that is, in a chromatin context. However, this is not always the case, because regulatory elements can also be localized downstream of the transcription start site: in introns, in the 5′ untranslated region, or in 3′ sequences (Larkin et al., 1993; Sieburth and Meyerowitz, 1997; Deyholos and Sieburth, 2000; Yu et al., 2001). For example, the large second intron of the MADS-box floral organ-identity gene AGAMOUS (AG) is essential for the correct expression of the gene, and contains binding sites for at least two AG regulators, LFY and WUSCHEL (WUS) (Sieburth and Meyerowitz, 1997; Bomblies et al., 1999; Busch et al., 1999; Deyholos and Sieburth, 2000; Lohmann et al., 2001).
In spite of the structural differences between animal and plant cis-regulatory and promoter regions, regulation of gene expression is often in both cases the result of multiple inputs, reflecting, or taking advantage of, the combinatorial nature of the mechanisms of eukaryotic transcription. Multiple stimuli can converge through different cis-acting elements on a promoter to coordinately regulate the expression of the corresponding gene (Arnone and Davidson, 1997; Yuh et al., 1998). The cis-acting elements are usually organized in a modular fashion: both in animals and plants, the regulatory region of a gene can be partitioned into discrete subelements, each one containing one or several binding sites for transcription factors and performing a certain regulatory function (Benfey and Chua, 1990; Arnone and Davidson, 1997). The modular nature of cis-regulatory systems is exemplified by the 2.3 kb promoter region of the sea urchin developmentally regulated Endo16 gene, one of the best characterized eukaryotic promoters (Yuh et al., 1998, 2001). It consists of six different regulatory modules, which provide different regulatory functions that are integrated through interrelations between the modules, and result in the spatial expression, and repression, of the gene, as well as on its variable rates of transcription (Yuh et al., 1998, 2001). This cis-regulatory system therefore acts like an information processing unit, and computational models for the modes of action of some of its modules have been established (Arnone and Davidson, 1997; Yuh et al., 1998, 2001). The view of cis-regulatory regions as information processing systems in which the output of developmental (or other) inputs is hardwired, is probably applicable to the eukaryotic genome as a whole (Arnone and Davidson, 1997; Davidson, 2001).
In plants, regulation of gene expression by systems of cis-acting modules, and the fact that these modules can interact synergistically (i. e., that combinations of modules direct gene expression in a manner not observed with the modules in isolation), was first described for the cauliflower mosaic virus (CaMV) 35S promoter (Benfey and Chua, 1990). The CaMV 35S promoter directs high levels of expression in most tissues and developmental stages when introduced as a transgene in plants, but can be dissected into subdomains that confer tissue-specific expression (Benfey and Chua, 1990; Benfey et al., 1990a, b).
The combinatorial interaction of cis-elements has also been demonstrated, for example, for Arabidopsis light-regulated promoters. Several consensus cis-sequences that are necessary for high activity in the light have been identified in the promoters of photosynthesis-associated nuclear genes (such as the rbcS and cab genes). These consensus sequences are referred to as ‘light responsive elements (LREs)’. Minimal promoters, sufficient to confer light-dependent expression, contain several LREs, but no single LRE is found in all light-regulated promoters (in fact, some LREs are also present in promoters that are not regulated by light) (Argüello-Astorga and Herrera-Estrella, 1998). LREs function combinatorially: whereas they cannot confer proper light responsiveness in isolation, paired combinations of them are able (1) to respond to a wide spectrum of light through the phytochrome signal transduction pathways, (2) to respond to the chloroplast developmental state, and (3) to confer a photosynthetic-cell specific expression pattern, therefore satisfying the strict definition of light-inducible (Puente et al., 1996; Chattopadhyay et al., 1998b). Thus, it is the combination of LREs in the promoter what serves as the integration system for the coordination of different light and developmental inputs to regulate the expression of the photosynthesis-related genes (Puente et al., 1996; Chattopadhyay et al., 1998b). Similarly, the promoter of the meristem identity gene LEAFY serves as the convergence point for different signals that control flowering time in Arabidopsis, including both environmental cues (daylength pathway) and endogenous signals (gibberellins) (Blázquez and Weigel, 2000), in accordance with the concept of promoters acting as information processing systems.
The combinatorial and synergistic function of cis-elements in eukaryotic promoters is logically accompanied by the combinatorial mode of action of the trans-acting factors that bind to those sites, and allows for the generation of regulatory diversity by a limited number of factors and binding sites. The requirement of several, often adjacent, cis-elements for the regulation of gene expression can be related to direct interactions between the proteins that bind to those elements. Direct interactions among transcription factors, however, is not the only molecular mechanism by which they can function combinatorially to regulate gene expression, since they can also interact with other components of the transcription machinery and with other classes of regulatory proteins. For example, LFY and WUS cooperatively participate in the regulation AG expression, yet they bind independently to AG cis-regulatory sequences and a direct interaction between the two proteins has not yet been detected (Lohmann et al., 2001).
Several examples of direct interactions between different Arabidopsis transcription factors have been reported, although the number is still small. In addition to increasing the regulatory repertoire, direct interactions between transcription factors are one of the mechanisms by which proteins with very similar DNA binding domains might achieve regulatory specificity (see, for example, Grotewold et al., 2000). Direct interactions can occur between members of the same protein family, to form dimeric complexes that bind to palindromic DNA sequences (such as in the case of the MADS domain proteins: Huang et al., 1996; Riechmann et al., 1996a; Riechmann et al., 1996b), or between transcription factors of different families. Examples of the latter include Arabidopsis, maize, and petunia proteins of the MYB and bHLH families (Table 6), interactions between bZIP and ABI3/VP1 proteins in rice and Arabidopsis (Hobo et al., 1999; Nakamura et al., 2001), between soybean C2H2 zinc finger and bZIP proteins (Kim et al., 2001), and between Dof and bZIP transcription factors in Arabidopsis and maize (Chen et al., 1996; Vicente-Carbajosa et al., 1997). The interaction between bZIP and ABI3/VP1 proteins (TRAB1 and VP1, respectively, in the case of rice, and ABI3 and ABI5 in Arabidopsis) provides a mechanism for VP1-mediated, ABA-inducible gene expression (Hobo et al., 1999; Nakamura et al., 2001). The interaction between bZIP and Dof proteins might mediate the endosperm-specific expression of seed-storage proteins (Vicente-Carbajosa et al., 1997).
Table 6.
Interactions between bHLH and MYB-R2R3 proteins(a)
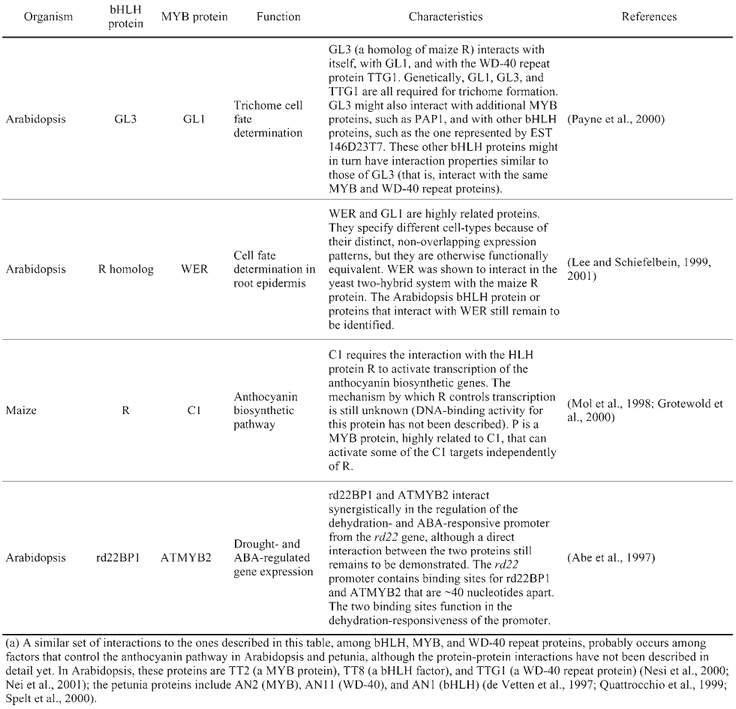
Within the MADS domain family, interactions are not limited to the formation of protein dimers, but also include the formation of ternary complexes. APETALA1 (AP1), APETALA3 (AP3), PISTILLATA (PI), and AGAMOUS (AG) are MADS domain proteins that, together with AP2, control the development of floral organs in Arabidopsis (Bowman et al., 1991; Coen and Meyerowitz, 1991; Goto et al., 2001; Jack, 2001; Theiben, 2001). AP3 and PI bind to DNA forming a heterodimer, whereas AP1 and AG can both bind to DNA as homodimers or as heterodimers with other MADS domain proteins (Huang et al., 1996; Riechmann et al., 1996a; Riechmann et al., 1996b). The activity of AP1, AP3, PI, and AG, however, requires of floral cofactors, also MADS domain proteins, that are encoded by the SEPALLATA genes, SEP1, SEP2, and SEP3 (Pelaz et al., 2000; Honma and Goto, 2001; Pelaz et al., 2001a; Pelaz et al., 2001b). In yeast two-hybrid experiments, AP3 and PI together, but neither one of the two proteins individually, can physically interact with AP1 and with SEP3 (Honma and Goto, 2001). The ectopic expression of AP3, PI, and AP1, or of AP3, PI, and SEP3 converts vegetative leaves into petaloid organs (Honma and Goto, 2001), whereas AP3 and PI alone are not sufficient for such organ conversion (Krizek and Meyerowitz, 1996). These results indicate that the formation of ternary complexes might be necessary for the function of AP3 and PI. The role that ternary complex formation might play in AP3 and PI function could be several fold: from providing an activation domain that AP3 and PI appear to lack, but that AP1 and SEP3 have (Honma and Goto, 2001), to increasing the DNA-binding specificity/affinity of the complex versus that of the protein dimers, given that the organ identity activity of AP1, AP3, PI, and AG is independent of their individual DNA-recognition properties (Riechmann and Meyerowitz, 1997a). These results with the Arabidopsis floral organ identity proteins parallel and expand previously obtained data for the Antirrhinum majus MADS-domain proteins SQUAMOSA, DEFICIENS, and GLOBOSA (SQUA, DEF, and GLO, which are AP1, AP3, and PI orthologs, respectively). DEF and GLO were also found to form ternary complexes with SQUA, and the three proteins together to bind DNA with increased affinity versus SQUA or DEF/GLO alone (Egea-Cortines et al., 1999).
Whereas these isolated examples illustrate the importance of interactions between transcription factors for the regulation of transcription, and how the combinatorial logic can operate, they do not convey the scope of the regulatory interactions in which transcription factors could be involved. For this, the whole complement of proteins should be considered (see below).
6. Genome-wide analyses of transcriptional regulation.
The future of biological sciences in the “post-genome era” has been anticipated as an endeavor to generate a collection of comprehensive “functional maps” (corresponding to the “transcriptome”, the “phenome”, the “interactome”, the “localizome”, and so on), that would be compiled into a “biological atlas” which would represent the modular nature of biological processes in a holistic manner and allow the formulation of new hypothesis (Greenbaum et al., 2001; Kim, 2001; Vidal, 2001). These maps could be visualized as two-dimensional matrices in which one axis represents all the genes or proteins that can be tested in an organism, and the other a comprehensive series of mutant backgrounds, conditions to which the organism can be exposed, etc. (Vidal, 2001). For instance, a yeast transcriptome map of this type is already being developed (Hughes et al., 2000b). The interactome would represent the map of physical interactions among all the proteins of a proteome (reviewed in Walhout and Vidal, 2001) (for attempts to construct the yeast interactome, see: Uetz et al., 2000; Ito et al., 2001). The localizome map would describe in what cells and cellular compartments, and when, all the proteins of an organism's proteome can be found; and to produce the phenome, a collection of mutants encompassing all the genes in a genome would be screened in a large series of phenotypic assays (for C. elegans and yeast, see: Ross-Macdonald et al., 1999; Fraser et al., 2000; Gönczy et al., 2000; Maeda et al., 2001).
Such view is also appropriate when considering Arabidopsis transcriptional regulation at a global level in a cellular and organismal context, for whose understanding several of those functional maps would be required: the genome-wide transcriptome map, the interactome and the phenome of the transcriptional regulators, as well as other “-ome” maps not previously considered, such as the promoterome. Intrinsic to this view is the realization that none of these different “-ome” maps would lead, in isolation from the others, to a comprehensive or even logical understanding of transcriptional regulation and of its role as a major determinant of cellular and organismal functions and phenotypes.
To generate these functional maps, the systematic investigation of transcription factor function and transcriptional regulation in Arabidopsis can be pursued with a variety of tools for functional genomic analyses, including reverse genetics methods, gene expression profiling experiments, and protein-protein interaction screens (reviewed in Riechmann and Ratcliffe, 2000). Whereas the availability of the Arabidopsis genome sequence allows us to compile lists of proteins that are involved in the regulation of transcription, and of putative promoter and cis-acting sequences, a global understanding of this process is still in its infancy. However, somewhere along the way of generating these functional maps, and once a sufficient amount of data has been collected, it should be possible to start decoding the “language” of transcriptional control, and to eventually be able, for instance, to build synthetic promoters directing gene expression in novel, designed spatial and temporal patterns (for an example of an initial attempt to design an artificial expression cassette in plants simply by statistical analysis of nucleotide sequences, see Sawant et al., 2001).
6.1 The transcription factor phenome map.
The number of Arabidopsis transcription factors that have been functionally characterized is still small, approximately 10% of the total (an incipient phenome; Table 1). Most of these genes were characterized through the traditional genetic approach, whereby genes are first defined by a mutant phenotype and then isolated. For the majority of these transcriptional regulators, functional characterization is limited to the description of phenotypic differences between mutant and wild-type plants, and determination of their expression pattern, but there is very little knowledge on their modes of action, that is, on the genes that they regulate (the transcriptome map) and on the mechanisms that they use to achieve that regulation (the interactome and the promoterome). As a result, the dynamic relationship between the genome, the transcriptional regulators, and the transcriptome, remains largely uncharacterized.
Different reverse genetics strategies are or can be used in plants to generate and isolate mutants in known genes: T-DNA or transposon insertional mutagenesis (Krysan et al., 1999; Maes et al., 1999; Parinov and Sundaresan, 2000; YouNg et al., 2001), fast neutron deletion mutagenesis (Li et al., 2001b), targeted screening for induced local lesions (TILLING) (Colbert et al., 2001), and DNA/RNA oligonucleotide-mediated site-directed mutagenesis (Oh and May, 2001). In addition, gene function can be inhibited by RNA interference (RNAi) or by virus–induced gene silencing (Baulcombe, 1999; Chuang and Meyerowitz, 2000; Levin et al., 2000; Hammond et al., 2001; Wesley et al., 2001). All these methods have been extensively reviewed and will not be discussed here. They are being used in several large-scale reverse genetics efforts to characterize the function of Arabidopsis transcription factors and chromatin-related proteins (for example, Meissner et al., 1999) (see also http://Ag.Arizona.Edu/chromatin/chromatin.html).
Probably the two main difficulties for generating a comprehensive phenome of Arabidopsis transcriptional regulators are the finite number of assays in which the mutants can usually be screened, and the existence of functional redundancy or overlap among different genes (Riechmann and Ratcliffe, 2000). Many of the Arabidopsis knockout mutants thus far isolated through reverse genetics approaches, in transcription factor genes as well as in genes of other classes, do not exhibit obvious morphological phenotypic alterations (Meissner et al., 1999; Bouche and Bouchez, 2001). This finding parallels what has been observed in other eukaryotic organisms, such as C. elegans, Drosophila, and yeast, in both forward and reverse genetics screens (for an overview, see Thatcher et al., 1998). For instance, the systematic analysis by RNAi in C. elegans of 4,590 genes (contained in chromosomes 1 and 3) only revealed mutant phenotypes in ∼14% of the cases (Fraser et al., 2000; Gönczy et al., 2000). However, it is likely that Arabidopsis mutants in “silent” or “nonessential” transcription factor genes (that is, they show no overt phenotype) might in fact reveal informative phenotypes when tested in comprehensive assays to characterize their physiology, metabolism, etc. (for an example of the use of metabolome data to reveal the phenotype of silent mutations in yeast, see Raamsdonk et al., 2001). For those genes that are involved in the plant's response to the environment, either biotic or abiotic, mutant phenotypes might not be revealed unless specific environmental conditions are used in the experiments. However, the assumption that if a gene is expressed or induced under a particular set of conditions then that gene is important for the organism's growth or survival in those conditions, should be taken with some caution: in yeast, there appears to be little correlation between the two when large sets of genes are considered (Winzeler et al., 1999). Lastly, detection of slightly deleterious effects caused by mutations in “silent” genes might require multigenerational competition studies in which fitness can be assessed, as shown in Arabidopsis (actin genes) and in yeast (Gilliland et al., 1998; Thatcher et al., 1998; Winzeler et al., 1999). The analysis by deletion of 2,026 genes in yeast indicated that ∼80% of them were nonessential for viability, but 40% of those silent deletants showed impaired growth in a simple competitive assay (Winzeler et al., 1999).
The extent of functional redundancy among related Arabidopsis transcription factors has been illustrated by several recent studies on factors from different groups, such as the MADS, GARP, YABBY, and GRAS gene families. MADS-box genes that act redundantly include: AP1, CAULIFLOWER (CAL), and FRUITFULL (FUL), in the control of floral meristem identity (Bowman et al., 1993; Kempin et al., 1995; Ferrándiz et al., 2000); the SHATTERPROOF genes (SHP1 and SHP2), which are required for proper development of the fruit-valve margin (Liljegren et al., 2000); and the SEPALLATA genes (SEP1, SEP2, and SEP3), which are cofactors or interactors for the floral organ identity genes AP1, AP3, PI, and AG (see above, and: Pelaz et al., 2000; Honma and Goto, 2001; Pelaz et al., 2001a; Pelaz et al., 2001b). The redundancy among AP1, CAL, and FUL in specifying floral meristem identity is partial. ap1 plants show a mutant phenotype (a partial conversion of flowers into inflorescences and a disruption of sepal and petal development), whereas a mutation in CAL results in a mutant phenotype only when combined with an ap1 allele (Bowman et al., 1993; Kempin et al., 1995). ap1 cal mutant plants show a complete conversion of the floral meristems into inflorescence meristems (Bowman et al., 1993). In other words, AP1 can completely compensate for the loss of CAL function, but CAL can only compensate for part of AP1 activity. A mutation in FUL does not alter floral meristem identity in the presence of a functional copy of AP1 or CAL (Ferrándiz et al., 2000). The SEP genes appear to have largely overlapping, although not identical, functions: the triple sep1 sep2 sep3 mutant shows a clear conversion of petals, stamens, and carpels, to sepals, whereas single or double sep mutants exhibit more subtle phenotypic alterations (Pelaz et al., 2000; Pelaz et al., 2001a) (for a review on the SEP genes and the ABC model of flower development: Jack, 2001). Similarly, only the shp1 shp2 double mutant, and not the single mutants, shows drastic phenotypic effects, in this case fruit that fails to dehisce (Liljegren et al., 2000).
Another example of related genes that act redundantly is provided by KANADI1 (KAN1) and KANADI2 (KAN2), which participate in the establishment of polarity in Arabidopsis lateral organs by determining abaxial cell fate (Eshed et al., 2001). KAN1 and KAN2 are members of the GARP family of plant-specific transcription factors, and they form part of a monophyletic group within the family (Eshed et al., 2001; Kerstetter et al., 2001). In fact, the genetic mechanism or network that controls lateral organ polarity in Arabidopsis appears to consist of multiple transcription factors from different gene families, with the corresponding genes within each group acting, at least in part, in a functionally overlapping manner (see below, and Eshed et al., 2001). Finally, GAI and RGA, which are highly related members of the GRAS gene family, have partially redundant functions as negative regulators of the gibberellin (GA) signaling pathway (Dill and Sun, 2001; King et al., 2001). In summary, situations of overlapping or partially redundant gene function among related genes are frequent within the different Arabidopsis transcription factor families (for general discussions on gene function after duplication, and on genetic redundancy and how it might be maintained by selection, see: Thomas, 1993; Cooke et al., 1997; Massingham et al., 2001).
Furthermore, in addition to redundancy resulting from the incomplete functional divergence between highly related (duplicated) members of a gene family, it can also arise from functional convergence of more distantly related genes (reviewed in: Pickett and Meeks-Wagner, 1995; Cooke et al., 1997). For instance, two divergent forkhead transcription factor genes from C. elegans, pes-1 and fkh-2, are partially redundant in embryonic development (Molin et al., 2000). Inactivation of pes-1 or fkh-2 alone caused no apparent phenotypic alteration during embryogenesis, whereas inactivation of both genes severely disrupted it. The functional association between pes-1 and fkh-2 was investigated because of the similarity in their expression patterns, but not because of sequence homology: pes-1 and fkh-2 belong to different clades within the forkhead gene family (Molin et al., 2000). The C. elegans genome contains 15 different forkhead genes (Riechmann et al., 2000), and the expression patterns had been determined for all of them. This example illustrates the limitations of sequence analysis as a tool to explore genetic redundancy. In addition, it suggests another reason why the complete functional characterization of the Arabidopsis complement of transcription factors will require the determination of the expression patterns of all of its members. Although the extent of this type of redundancy (genes that belong to the same family, but to different clades within it, and yet have the same or overlapping functions) in Arabidopsis is unknown, there is evidence that it exists. For example, AINTEGUMENTA (ANT) acts redundantly with APETALA2 (AP2) to repress AG expression in cells of the second whorl of developing Arabidopsis flowers (Krizek et al., 2000). Both ANT and AP2 are AP2/ERF proteins, but they belong to different clades within the AP2 subfamily.
Last, functional redundancy can also exist between genes of different classes or families (i.e., with distinct molecular functions), for instance if they form part of independent pathways controlling the same process. An example of this type is provided by members of the KANADI and YABBY gene families, involved in the determination of abaxial polarity in lateral organs (Eshed et al., 1999; Bowman et al., 2001; Eshed et al., 2001). For example, CRABS CLAW (CRC), the founding member of the YABBY family (Bowman and Smyth, 1999; Bowman, 2000), and KAN1 participate in the determination of abaxial polarity in the carpels (Eshed et al., 1999; Bowman et al., 2001). In contrast to redundancy between duplicated genes, it is not possible to predict functional redundancy between unrelated proteins by simply analyzing the genome sequence. Rather, these cases of functional overlap will be uncovered through classic mutagenesis screens for enhancers of a particular mutant phenotype (for example: Eshed et al., 1999; Bowman et al., 2001; Eshed et al., 2001), and as a result of genome-wide analyses of gene expression designed to identify the targets of different transcription factors (see below).
6.2 The transcriptome and promoterome maps.
Two of the different “-ome” maps are essential to understand transcriptional regulation at a molecular, genome-wide level and, ultimately, to explain the basis for the effects that differential gene expression has on the functions and phenotypes of cells and organisms: the transcriptome and the promoterome maps. The transcriptome can be viewed as the collection of transcripts that are expressed from the genome at any particular temporal and physiological instance, considering both transcript identity and abundance (i. e., a description both qualitative and quantitative). The promoterome, as defined in this chapter, would consist of all the promoters and cis-acting elements in a genome, and of their interactions with the complement of transcriptional regulators.
6.2.1 DNA microarrays.
A comprehensive characterization of the Arabidopsis transcriptome in its multiple forms can be achieved using DNA microarray technologies, which allow the parallel monitoring of the expression of thousands of genes and, eventually, of the complete Arabidopsis genome (for reviews on DNA microarrays and gene expression: Lockhart and Winzeler, 2000; Richmond and Somerville, 2000; Young, 2000; Altman and Raychaudhuri, 2001; Schulze and Downward, 2001) (for information on microarray resources, see: http://www.arabidopsis.org/links/microarrays.html). Currently, the expression of up to ∼8,500 different Arabidopsis genes, or approximately one third of the genome, has been monitored in DNA microarray experiments to generate catalogues of genes that are expressed in response to particular stresses or stimuli, or in certain tissues or developmental processes (Table 7). These early studies have included the response to different nutrient concentrations, to drought and cold stresses, to wounding and insect feeding, the disease response, and light-related processes, such as the circadian clock and phytochrome A signaling (Table 7). The most extensive dynamic reprogramming of the expression of the genome has been observed upon light stimulus or in light-related processes (Harmer et al., 2000; Schaffer et al., 2001; Tepperman et al., 2001) (Table 7). For instance, the analysis of circadian changes in the mRNA levels of more than 8,000 genes reveals how differential gene expression underlies many of the physiological changes that the plant undergoes in its daily life cycle. The expression of genes implicated in photosynthesis, in phenylpropanoid biosynthesis, in lipid modification, and in carbon, nitrogen, and sulfur pathways was found to be regulated by the circadian clock, and a physiological explanation can be reasoned for it: to prepare for light-harvesting, for protection against UV light, to increase chilling-resistance at night, and to coordinate the metabolism of the plant with its environment (Harmer et al., 2000).
Table 7.
Arabidopsis DNA microarray studies(a)
Catalogues of expressed genes provide insights into a variety of biological processes. However, elucidating the relationship between the transcriptome, the promoterome, and the complement of transcription factors, to eventually understand the logic of transcription, requires additional types of genome-wide analyses and experiments, such as the following.
6.2.2 Comparative analysis of promoter sequences of genes with similar expression profiles.
The results of multiple DNA microarray experiments can be combined and analyzed together using clustering techniques, by which those genes that show similar expression patterns across the set of experiments are identified and grouped (reviewed in: Sherlock, 2000; Quackenbush, 2001; Raychaudhuri et al., 2001). The set of experiments to be compared might consist of different cell types or tissue samples, different physiological conditions, or might characterize over a time course the transcriptional response to a given stimulus. One assumption underlying the clustering analysis of time course experiments is that genes with highly related expression profiles might be regulated by the same mechanism. Thus, once groups of co-regulated genes are established, their promoter sequences can be compared to identify common cis-acting elements. The success of the approach is determined, at least in part, by the structural organization (i.e., size and complexity) of the regulatory regions, and it has proven particularly fruitful in yeast (Cho et al., 1998; Roth et al., 1998; Spellman et al., 1998; Tavazoie et al., 1999; Wolfsberg et al., 1999; Gasch et al., 2000; Hughes et al., 2000a; Lyons et al., 2000; Jakt et al., 2001). In animals, in which complete regulatory regions are difficult to delimit from sequence information alone, and cis-acting elements might be distributed over very long distances, the analyses might have to be restricted to the proximal promoter sequences (for example, Livesey et al., 2000). In contrast, as discussed above, plant regulatory regions are more similar to those of yeast, in that they are often completely encompassed within a few hundred base pairs upstream from the transcription start site. The comparison of the promoter sequences of a group of Arabidopsis genes that are co-regulated by the circadian clock, and that in the experiment showed the highest level of expression near the end of the subjective day, identified a novel motif that is conserved among those promoters, and that was then experimentally shown to mediate their regulation (Harmer et al., 2000) (Table 7). Similarly, a group of Arabidopsis genes that co-regulate with PR-1 over a series of systemic acquired resistance (SAR) inducing or repressing conditions was identified, and their promoters searched for the presence of known cis-elements for transcription factors. W boxes, the binding site for WRKY proteins, were the only known cis-element that was present in all the promoters, suggesting that WRKY transcription factor(s) participate in the control of the PR-1 regulon (Maleck et al., 2000). This latter example also illustrates that the identification of common elements in the upstream regions of co-regulated genes will usually not be sufficient, in the absence of other information, to pinpoint the identity of the specific regulators, because the majority of the Arabidopsis transcription factors form part of multigene families (Table 1), in which different members have related or similar DNA-binding specificities. The same limitation applies to genome-wide searches for transcription factor binding sites that are carried out without reference to expression data (for an example in Arabidopsis, Du and Chen, 2000). Such searches can lead to the identification of potential downstream genes, especially if the target sites for the DNA-binding protein(s) or complex(es) of interest are well characterized, but additional experimentation is usually required to establish the association between the identified elements and the transcription factor(s) under study (for an example in yeast, Zhong et al., 1999).
6.2.3 Phylogenetic footprinting.
A valuable approach to identify unknown cis-regulatory regions and elements, and that can be applied at a genome-wide scale, is phylogenetic footprinting: sequence comparisons across phylogenetically related species that reveal conserved cis-elements in the non-coding regions of homologous genes. Phylogenetic footprinting is based on the observation that regulatory regions are more conserved throughout evolution than regions that do not have a function that is dependent on their sequence (for reviews: Gumucio et al., 1996; Duret and Bucher, 1997; Hardison et al., 1997; Fickett and Wasserman, 2000). A critical factor for the success of the phylogenetic footprinting method is the choice of species for the comparative analysis: at a genome-wide scale, they should be similar enough so that most sequences can be aligned with the corresponding ortholog(s), but distant enough so that non-functional sequences have diverged by accumulating mutations at neutral positions. Logically, the frequency of detectable conserved elements in the non-coding regions of orthologous genes decreases as species separated by increasing evolutionary distances are compared (Duret and Bucher, 1997). Identifying the optimal species (or group of species) for phylogenetic footprinting might require surveying several species within the corresponding genus or family (for an example of such survey in Saccharomyces, see Cliften et al., 2001). In fact, it appears that the most comprehensive and meaningful results will be obtained in comparisons that include several species of different evolutionary distances (Duret and Bucher, 1997; Cliften et al., 2001). The use of groups of species might be particularly important for genome-wide phylogenetic footprinting, because the optimal evolutionary distance for comparison might vary across genes. The phylogenetic footprinting method has been used to define regulatory elements by comparisons between human and mouse sequences and among mammals, and between C. elegans and C. briggsae, among other organisms (Gumucio et al., 1996; Thacker et al., 1999; Loots et al., 2000; Wasserman et al., 2000). Comparisons of the promoters of the CHALCONE SYNTHASE and AP3 genes across different cruciferous plant species has demonstrated the value of phylogenetic footprinting as a basis to functionally analyze Arabidopsis cis-regulatory regions (Koch et al., 2001), although the ideal species or group of species for that type of comparison with Arabidopsis at a genome-wide scale still needs to be identified.
Phylogenetic footprinting, however, will provide only a partial description of the promoterome, because only those elements that maintain similar functions across the species compared are likely to be conserved. Alterations in gene expression are an important mechanism of evolutionary change, and regulatory elements and functional features that arose after the divergence of the species used in the comparison might not be identified in the analysis (for variations of the phylogenetic footprinting method that, combined with experimental data, try to overcome these limitations, see Gumucio et al., 1996). Furthermore, there are instances in which a cis-region might maintain a given regulatory function despite considerable sequence variation.
6.2.4 Inducible activation of transcription factor activity.
The identification of the downstream genes of the many transcriptional regulators encoded by the genome is a necessary step to define the networks of gene activity that occur in a cell, tissue, or organism. The combination of DNA microarray technology with systems for the inducible activation of transcription factor function offers a way to dissect regulatory programs. The activity of transcription factors can be transcriptionally or posttranslationally regulated, using inducible gene expression systems or generating protein fusions to steroid-binding domains, such as the glucocorticoid receptor (GR) (Aoyama, 1999; Picard, 2000; Zuo and Chua, 2000). The advantage of using posttranslational regulation is that simple direct and indirect effects of transcription factor activity can be separated by using inhibitors of protein synthesis (for examples in Arabidopsis: Sablowski and Meyerowitz, 1998; Wagner et al., 1999; Samach et al., 2000; Sakai et al., 2001). Thus, an experiment to identify at a genome-wide scale the target genes of a particular transcription factor would consist of generating transgenic plants expressing a fusion of the factor to a steroid binding domain (ideally, in a mutant background in which the native transcription factor gene is inactivated), applying the hormone to the tissues under study (ideally, those in which the endogenous gene would be normally active), both in the presence and in the absence of an inhibitor of protein synthesis (cycloheximide), and following the effects on mRNA accumulation over time using DNA microarrays. Direct posttranslational regulation by fusion to GR has already been engineered for several plant transcription factors, including the maize bHLH R protein (Lloyd et al., 1994), the Arabidopsis homeodomain proteins ATHB-1, ATHB-2, and KNAT2 (Aoyama et al., 1995; Ohgishi et al., 2001; Pautot et al., 2001), CONSTANS, a zinc finger transcriptional regulator (Simon et al., 1996; Samach et al., 2000), the MADS domain protein AP3 (Sablowski and Meyerowitz, 1998), LFY (Wagner et al., 1999), and ARR1, a GARP transcription factor of the ARR-B subclass (Sakai et al., 2001). However, the technique might not be universally applicable, since some transcription factor-GR fusion proteins might be inactive, or constitutively active in the absence of the hormone.
6.2.5 Genome-wide maps of in vivo DNA binding by transcription factors.
An alternative approach to identify transcription factor downstream targets has been recently developed in yeast, combining chromatin immunoprecipitation and DNA microarrays. The underlying assumption is that transcription factors bind to the promoters or regulatory regions of the genes whose expression they control. In this method, proteins are crosslinked to genomic DNA in living cells using formaldehyde. The DNA that is specifically crosslinked to the protein of interest is then enriched by immunoprecipitation, amplified by PCR, and labeled for its use as a probe in dual-color microarray experiments (the corresponding control consisting of a sample DNA that was not enriched). The microarray used for the hybridization contains all the intergenic regions of the yeast genome, and might also contain the corresponding ORFs (Ren et al., 2000; Iyer et al., 2001; Lieb et al., 2001). This approach has been used to identify the binding sites for several transcriptional regulators in the yeast genome (Ren et al., 2000; Iyer et al., 2001; Lieb et al., 2001), and to study the targeted recruitment of the yeast histone acetylase Esa1 (Reid et al., 2000). In a more global study, the binding by the nine yeast transcription factors that are known to regulate the cell cycle (Mbp1, Swi4, Swi6, Mcm1, Fkh1, Fkh2, Ndd1, Swi5, and Ace2) was analyzed, showing that these factors form themselves a circular network of serial regulation (Simon et al., 2001). The results of all these experiments are also a testimony to the complexity of transcription, and to how much we still have to learn and to explain when considering the regulation of the expression of eukaryotic genomes as a whole. First, Gal4, SBF, MBF, and Rap1 were all found to bind preferentially to potential promoter regions, despite the fact that consensus binding site sequences for all of them are distributed all over the yeast genome (Ren et al., 2000; Iyer et al., 2001; Lieb et al., 2001). This biased recognition of binding sites suggests the existence of a superimposed level of regulation that might mark or distinguish regulatory regions from coding sequences, a component of which might be chromatin structure. The distribution of binding sites for a given transcription factor in the promoters of the genes that such factor regulates is not random either, suggesting the existence of, and constraints in, long-range interactions with other components of the transcription machinery. For example, Rap1 binding sequences were found to occur more often in tandem and at a certain upstream distance (250–450 bp), and to be located preferentially on the minus strand relative to the corresponding open reading frame (Lieb et al., 2001). In addition, not every promoter bound by, for example, SBF and MBF contains recognizable consensus sites (Iyer et al., 2001), indicating the existence of additional sources for specificity in transcription factor activity in vivo. Once again, the structural similarity between regulatory regions in yeast and plants suggests that the technique might in principle be applicable to Arabidopsis, provided that the corresponding experimental protocols (for crosslinking and immunoprecipiation) can be established, and with the caveat of the multicellular nature of plants (see below).
An alternative technique for the genome-wide identification of in vivo target loci has been developed in Drosophila, which makes use of E. coli DNA adenine methyltransferase (Dam). In this method, named DamID, a protein fusion between a chromatin protein of interest and Dam is expressed at low levels in Drosophila cells (in culture, or in the whole fly) (van Steensel and Henikoff, 2000; van Steensel et al., 2001). This leads to the methylation of the GATC sequences that might occur in the genome surrounding the binding sites of the target protein. Methylated regions are purified (by size fractionation of genomic DNA that has been cleaved with DpnI, which cuts at methylated GATC sites), and labeled for their use as a probe in dual-color microarray experiments (the corresponding control consisting of an equivalent sample from cells in which unfused Dam was expressed) (van Steensel and Henikoff, 2000; van Steensel et al., 2001). Chromatin profiling by DamID has not been developed for plants yet, and the method presents several potential technical difficulties. In particular, the fusion protein must be expressed at very low levels to allow distinguishing specific and non-specific methylation events, at least in Drosophila; and it is not yet known if the approach will work for proteins that bind as single molecules (or as dimers) to specific, short cis-elements (as many transcription factors do), since all the experiments reported so far involved potential cooperative binding that could ‘coat’ a region of DNA (for more information and discussion on DamID, see: http://blocks.fhrc.org/DamID). If these technical hurdles can be overcome, chromatin profiling by DamID could represent a valuable alternative to immunoprecipitation-based methods.
The genome-wide maps of in vivo protein-DNA association will contribute to clarify a longstanding unanswered question in transcriptional regulation: the correlation between the DNA binding properties of the transcription factors (i. e., affinities, which are measured in vitro), and their effects on transcription in vivo (for a discussion on this topic, see Biggin and Tjian, 2001). Ultimately, quantitative studies and information will be needed to understand the transcriptional code, and to be able to model transcriptional regulation, both in vivo and in silico.
The identification of the target genes for the many transcriptional regulators encoded by the Arabidopsis genome, the compilation of lists of genes that are differentially regulated (activated or repressed) in particular biological processes, and, most importantly, the integration and combined analysis of these large genome-wide data sets, will eventually define the networks by which transcription factors act, and the pathways downstream of them. Questions that are difficult to address in gene-by-gene studies can now be considered: to what extent different environmental responses, or distinct developmental pathways, share effector mechanisms (that is, the same target genes)?; how many different patterns of expression are triggered by a particular stimulus, and how are those differences achieved molecularly (that is, the complexity of response pathways at the level of the regulation of effector -or “realizator”- genes)? Combined with the characterization and analysis of the promoterome, such studies would lead to understand the transcriptional code, and eventually to rationally manipulate transcriptional regulation. These genome-wide studies would also assess the degree of connectivity among regulatory networks (and thus start defining the “networkome”). Furthermore, the identification of (direct) target genes that are shared by different transcription factors might also provide clues about what regulators act together, both molecularly (interacting proteins, or proteins binding to the same promoters but not interacting directly), and at the genetic level (either related genes that are (partially) redundant, or genes that form part of different pathways that control the same process). Such analysis would complement, and guide, other genetic studies to characterize overlapping functions among transcriptional regulators (for example, by identifying pairs or groups of genes to be analyzed in double, or multiple, mutant combinations; see above).
Last, it should be noted that the characterization of the Arabidopsis transcriptome, and its explanation in terms of transcription factor activity, faces one challenge not encountered in yeast, in which many of these types of studies have been pioneered: the multicellular nature of plants. The profiles of gene expression of different cell types and cells are logically different, and so can be their responses to the activity of particular transcription factors. Thus, in many instances the transcriptome that is characterized is in fact the average of those of the different cell types included in the study. It is still technically challenging, although not impossible, to achieve cellular resolution in genome-wide studies in multicellular organisms.
6.3 The transcription factor interactome map.
Genome-wide analyses of protein-protein interactions in eukaryotes have been pioneered for the proteomes of yeast and C. elegans using the yeast two-hybrid system. The general merits and problems of the approach in genome-wide screens have been discussed elsewhere (Riechmann and Ratcliffe, 2000; Hazbun and Fields, 2001; Legrain et al., 2001). For transcription factors, the two-hybrid system presents the added complication that their use as “baits” often requires the preparation of specialized constructs in which the sequences coding for activation domains have been removed (which is not a trivial hurdle if hundreds of proteins with presumed, but uncharacterized, activation domains have to be analyzed). This is because the read-out of the system consists on the transcriptional activation of reporter genes as a result of the interaction between the “bait” and “prey” fusion proteins. However, modifications of the two-hybrid system have already been devised (based on repression rather than on activation of transcription) that should be applicable for identifying interactions with transactivator proteins (for example: Hirst et al., 2001).
At present, there is very little data on the Arabidopsis transcription factor interactome, and only a small number of direct interactions between different plant transcription factors have been described (see above, and Singh, 1998). In addition to transcription factors directly interacting among themselves or with other components of the transcription machinery, they can also interact with other types of proteins, thus expanding the networks that would form the transcription factor interactome. Such interactions with proteins of other classes can be mechanistically important for the control of transcription, and they can also provide the link between transcription factor activity and signal transduction pathways, as for example in light- and disease-responses.
Arabidopsis perceives light using different types of light-absorbing photoreceptors, such as phytochromes (phyA through phyE), which absorb red and far-red light, and cryptochromes (cry1 and cry2), which absorb blue and UV-A light (for review, Nagy and Schäfer, 2000). Phytochrome- and cryptochrome-mediated light responses involve differential regulation of gene expression. PIF3 is a transcription factor of the bHLH family that is involved in phytochrome signal transduction, in particular signaling by phyB (Ni et al., 1998; Halliday et al., 1999). PIF3 binds to a cis-element present in several light-regulated promoters, and phyB (which is translocated to the nucleus in a light dependent manner: Kircher et al., 1999; Yamaguchi et al., 1999) reversibly binds to DNA-bound PIF3 upon the light-triggered conversion to its biologically active form (Ni et al., 1998; Martínez-García et al., 2000; Zhu et al., 2000). Thus, phytochromes might act as light-switchable components of transcription complexes, and their interaction with transcription factors might provide a short, direct pathway from light perception to photoresponsive nuclear gene expression (Martínez-García et al., 2000).
Another Arabidopsis transcription factor involved in light-mediated responses is HY5, which controls the photomorphogenic development that is undertaken by seedlings grown in the light. HY5 is a bZIP protein that binds to a cis-element present in several light-responsive promoters (Oyama et al., 1997; Chattopadhyay et al., 1998a). The regulation of HY5 activity by light involves its interaction with COP1, a RING-finger protein with WD-40 repeats whose subcellular localization is light-dependent (nuclear in the dark and cytoplasmic in the light), and that might target HY5 for proteasome-mediated degradation in the nucleus (Osterlund et al., 2000). In this case, it is the COP1 protein that interacts with light-activated photoreceptors (cryptochromes), which repress COP1 activity, thus permitting HY5 accumulation and induction of gene expression (WaNg et al., 2001).
The Arabidopsis ankyrin repeat-containing protein NPR1 has been shown to interact with some bZIP transcription factors of the TGA subfamily, which have been implicated in the activation of salicylic acid (SA)-responsive genes (Zhang et al., 1999). NPR1 is required for the induction of systemic acquired resistance (SAR) responses, such as the expression of pathogenesis-related (PR) genes, and it has been shown to act downstream of SAR-inducing agents (SA and avirulent pathogens) (Cao et al., 1997). NPR1 enhances the DNA binding activity of the interacting TGA bZIP proteins, and the in vivo relevance of the protein-protein interaction is demonstrated by the observation that point mutations in NPR1 that abolish its function also disrupt the interaction with the TGA factors (Després et al., 2000; Zhou et al., 2000). NPR1 is localized in both the cytoplasm and the nucleus of unstimulated cells, but concentrates in the nucleus in response to SA and, in fact, nuclear localization of NPR1 is required for PR gene expression (Kinkema et al., 2000). Furthermore, using an in vivo protein fragment complementation assay, based on association of reconstituted murine dihydrofolate reductase (mDHFR) with a fluorescent probe to detect protein-protein interactions, it has been shown that the interaction between NPR1 and the bZIP factor TGA2 is itself induced by SA and localized predominantly in the nucleus (Subramaniam et al., 2001). Thus, the interaction of transcription factor(s) with NPR1 provides a link between an SAR-inducing agent, SA, and the changes in gene expression that are associated with SAR. The mechanism by which the SA signal is transduced to NPR1 still remains to be determined, but additional two-hybrid screens have identified novel proteins of still uncharacterized function that interact with NPR1 and also accumulate in the nucleus (Weigel et al., 2001).
Another example of how an external trigger for a defense response might modulate gene expression through the interaction between a transcription factor and a protein of a different type is provided by the tomato AP2/ERF protein Pti4. Pti4 was first identified in a two-hybrid screen by virtue of its interaction with Pto, a protein kinase that confers resistance to Pseudomonas syringae carrying the corresponding avirulence gene, AvrPto (Zhou et al., 1997). Pti4 is phosphorylated by Pto, which enhances the binding of Pti4 to the GCC-box present in the promoter of PR genes (Gu et al., 2000).
In summary, it is clear from these different examples and from the published literature that a comprehensive description of the transcription factor “interactome” will encompass, in addition to the more than 1,500 transcriptional regulators encoded by the Arabidopsis genome, many other proteins from a variety of functional classes. It is also apparent that such comprehensive description will not be attained using only two-hybrid experiments, which will reveal only a subset of the interactions that occur in a cell, and that additional, alternative techniques are needed. The mDHFR-based protein fragment complementation assay mentioned above permits direct visualization (through spectroscopy, fluorescence-activated cell sorting, or fluorescence microscopy) of protein-protein interactions in living cells, and can be used to detect interactions in a quantitative manner and to follow the temporal pattern of the interaction (Subramaniam et al., 2001). Although the assay has been used in isolated protoplasts, and still needs to be developed to study interactions in whole plant tissues or organs, it represents a valuable alternative to the two-hybrid system for studying and dissecting signaling cascades, and could be developed into a high-throughput screening system for pathway and network mapping or for the identification of molecules that modify protein-protein interactions (Subramaniam et al., 2001). Additional techniques that will be useful to characterize the transcription factor interactome include fluorescence resonance energy transfer (FRET) by generating protein fusions to spectral variants of the jellyfish green fluorescent protein (GFP) (Gadella et al., 1999; Shah et al., 2001).
Conclusion
Genomics research, and functional genomics in particular, has often been hailed as the provider of a new paradigm in biology research; it has also been sometimes reviled by describing such type of research as consisting of little else than “fishing” experiments. Both opposing views are based on one common premise: the contrast between a frequently hypothesis-free genomics and the hypothesis-driven research that has been so much favored in molecular biology over the past decades. But genomics might not fit either one of these two disparate views. In many aspects, genomics bears many similarities with classic biology disciplines, such as genetics (in which the whole genome is blindly mutagenized at random to “fish” for interesting, novel phenotypes), and taxonomy and phylogeny (in which lists of elements are compiled and the relationships between them have to be established). It is in many instances the methodic collection of unanticipated data what allows the formulation of new hypothesis. However, if not radically different in concepts, genomics certainly changes the scales of biology research, and provides new dimensions to it. The relative simplicity of the Arabidopsis thaliana genome, together with the availability of many modern genetic and genomic research tools in that species, indicates that Arabidopsis is a premier organism to elucidate the complex logic of transcription at a genome-wide level in multicellular eukaryotes.
Acknowledgments
I wish to acknowledge my colleagues at Mendel Biotechnology for their input and work in our transcription factor genomics research program, as well as for their discussions, insight, and comments. I also wish to acknowledge the work of all those who participated in the Arabidopsis Genome Initiative and sequenced the Arabidopsis genome.
Footnotes
Citation: Riechmann J.L. (2002) Transcriptional Regulation: a Genomic Overview. The Arabidopsis Book 1:e0085. doi:10.1199/tab.0085
elocation-id: e0085
Published on: April 4, 2002
Table 1.
Continued
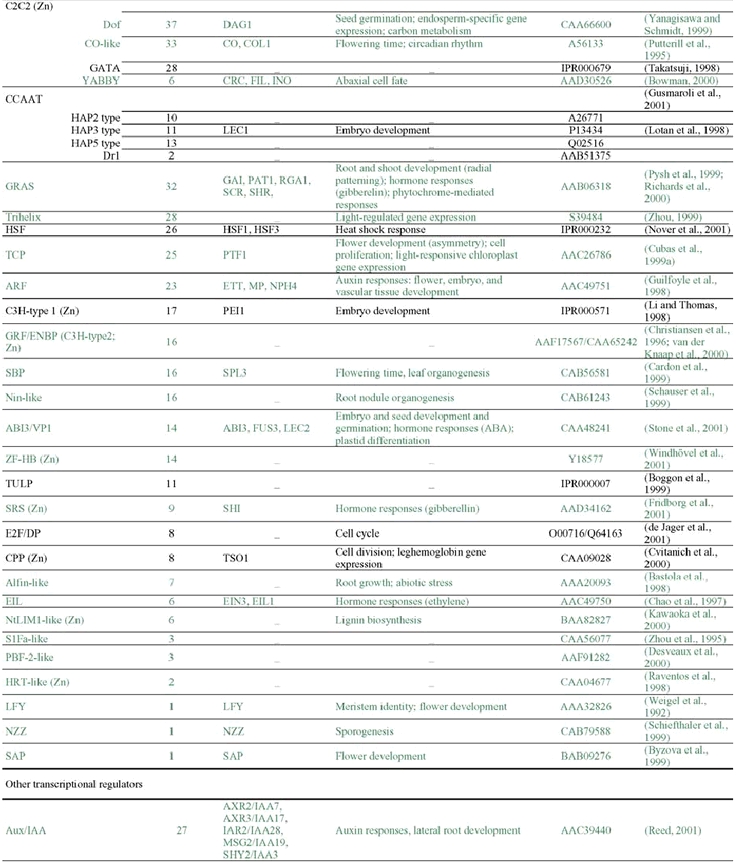
Table 1.
Continued
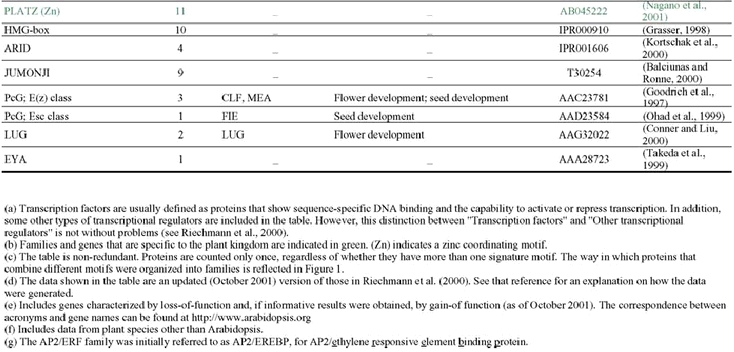
Table 3.
Continued
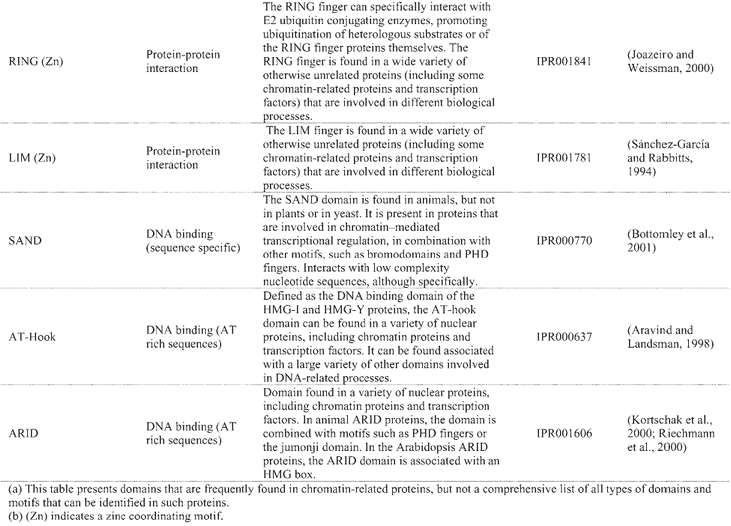
Table 4.
Continued

Table 5.
Continued

Table 7.
Continued
References
- Aasland R., Gibson T. J., Stewart A. F. The PHD finger: implications for chromatin-mediated transcriptional regulation. Trends Biochem. Sci. 1995;201(1):56–59. doi: 10.1016/s0968-0004(00)88957-4. [DOI] [PubMed] [Google Scholar]
- Abe H., Yamaguchi-Shinozaki K., Urao T., Iwasaki T., Hosokawa D., Shinozaki K. Role of Arabidopsis MYC and MYB homologs in drought- and abscisic acid- regulated gene expression. Plant Cell. 1997;91(1):1859–1868. doi: 10.1105/tpc.9.10.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams M. D., Celniker S. E., Holt R. A., Evans C. A., Gocayne J. D., Amanatides P. G., Scherer S. E., Li P. W., Hoskins R. A., Galle R. F. The genome sequence of Drosophila melanogaster. Science. 2000;2871(1):2185–2195. doi: 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- Agalioti T., Lomvardas S., Parekh B., Yie J., Maniatis T., Thanos D. Ordered recruitment of chromatin modifying and general transcription factors to the IFN-beta promoter. Cell. 2000;1031(1):667–678. doi: 10.1016/s0092-8674(00)00169-0. [DOI] [PubMed] [Google Scholar]
- Ahringer J. NuRD and SIN3 histone deacetylase complexes in development. Trends Genet. 2000;161(1):351–356. doi: 10.1016/s0168-9525(00)02066-7. [DOI] [PubMed] [Google Scholar]
- Akhtar A., Zink D., Becker P. B. Chromodomains are protein-RNA interaction modules. Nature. 2000;4071(1):405–409. doi: 10.1038/35030169. [DOI] [PubMed] [Google Scholar]
- Alabadí D., Oyama T., Yanovsky M. J., Harmon F. G., Más P., Kay S. A. Reciprocal regulation between TOC1 and LHY/CCA1 within the Arabidopsis circadian clock. Science. 2001;2931(1):880–883. doi: 10.1126/science.1061320. [DOI] [PubMed] [Google Scholar]
- Altman R. B., Raychaudhuri S. Whole-genome expression analysis: challenges beyond clustering. Curr. Opin. Struct. Biol. 2001;111(1):340–347. doi: 10.1016/s0959-440x(00)00212-8. [DOI] [PubMed] [Google Scholar]
- Amedeo P., Habu Y., Afsar K., Scheid O. M., Paszkowski J. Disruption of the plant gene MOM releases transcriptional silencing of methylated genes. Nature. 2000;4051(1):203–206. doi: 10.1038/35012108. [DOI] [PubMed] [Google Scholar]
- Aoyama T. Glucocorticoid-inducible gene expression in plants. 1999;1(1):43–59. In Inducible gene expression in plants, Reynolds, P. H. S. (Ed), (Wallingford, UK: CABI Publishing), pp. [Google Scholar]
- Aoyama T., Dong C-H., Wu Y., Carabelli M., Sessa G., Ruberti I., Morelli G., Chua N-H. Ectopic expression of the Arabidopsis transcription activator Athb-1 alters leaf cell fate in tobacco. Plant Cell. 1995;71(1):1773–1785. doi: 10.1105/tpc.7.11.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arabidopsis Genome Initiative Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;4081(1):796–815. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]
- Aravind L., Koonin E. V. Second family of histone deacetylases. Science. 1998;2801(1):1167a. [Google Scholar]
- Aravind L., Landsman D. AT-hook motifs identified in a wide variety of DNA-binding proteins. Nucleic Acids Res. 1998;261(1):4413–4421. doi: 10.1093/nar/26.19.4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argüello-Astorga G., Herrera-Estrella L. Evolution of light-regulated plant promoters. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1998;491(1):525–555. doi: 10.1146/annurev.arplant.49.1.525. [DOI] [PubMed] [Google Scholar]
- Arnone M. I., Davidson E. H. The hardwiring of development: organization and function of genomic regulatory systems. Development. 1997;1241(1):1851–1864. doi: 10.1242/dev.124.10.1851. [DOI] [PubMed] [Google Scholar]
- Balciunas D., Ronne H. Evidence of domain swapping within the jumonji family of transcription factors. Trends Biochem. Sci. 2000;251(1):274–276. doi: 10.1016/s0968-0004(00)01593-0. [DOI] [PubMed] [Google Scholar]
- Bannister A. J., Zegerman P., Partridge J. F., Miska E. A., Thomas J. O., Allshire R. C., Kouzarides T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;4101(1):120–124. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- Bastola D. R., Pethe V. V., Winicov I. Alfin1, a novel zinc-finger protein in alfalfa roots that binds to promoter elements in the salt-inducible MsPRP2 gene. Plant Mol. Biol. 1998;381(1):1123–1135. doi: 10.1023/a:1006081926699. [DOI] [PubMed] [Google Scholar]
- Baulcombe D. C. Fast forward genetics based on virus-induced gene silencing. Curr. Opin. Plant. Biol. 1999;21(1):109–113. doi: 10.1016/S1369-5266(99)80022-3. [DOI] [PubMed] [Google Scholar]
- Benfey P., Chua N. H. The Cauliflower Mosaic Virus 35S promoter: combinatorial regulation of transcription in plants. Science. 1990;2501(1):959–966. doi: 10.1126/science.250.4983.959. [DOI] [PubMed] [Google Scholar]
- Benfey P. N., Ren L., Chua N. H. Combinatorial and synergistic properties of CaMV 35S enhancer subdomains. EMBO J. 1990a;91(1):1685–1696. doi: 10.1002/j.1460-2075.1990.tb08292.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfey P. N., Ren L., Chua N. H. Tissue-specific expression from CaMV 35S enhancer subdomains in early stages of plant development. EMBO J. 1990b;91(1):1677–1684. doi: 10.1002/j.1460-2075.1990.tb08291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfey P. N., Weigel D. Transcriptional networks controlling plant development. Plant Physiol. 2001;1251(1):109–111. doi: 10.1104/pp.125.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggin M. D., Tjian R. Transcriptional regulation in Drosophila: the post genome challenge. Funct. Integr. Genomics. 2001;11(1):223–234. doi: 10.1007/s101420000021. [DOI] [PubMed] [Google Scholar]
- Birve A., Sengupta A. K., Beuchle D., Larsson J., Kennison J. A., Rasmuson-Lestander A., Muller J. Su(z)12, a novel Drosophila Polycomb group gene that is conserved in vertebrates and plants. Development. 2001;1281(1):3371–3379. doi: 10.1242/dev.128.17.3371. [DOI] [PubMed] [Google Scholar]
- Blanc G., Barakat A., Guyot R., Cooke R., Delseny M. Extensive duplication and reshuffling in the Arabidopsis genome. Plant Cell. 2000;121(1):1093–1101. doi: 10.1105/tpc.12.7.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blázquez M. A., Weigel D. Integration of floral inductive signals in Arabidopsis. Nature. 2000;4041(1):889–892. doi: 10.1038/35009125. [DOI] [PubMed] [Google Scholar]
- Boggon T. J., Shan W. S., Santagata S., Myers S. C., Shapiro L. Implication of tubby proteins as transcription factors by structure-based functional analysis. Science. 1999;2861(1):2119–2125. doi: 10.1126/science.286.5447.2119. [DOI] [PubMed] [Google Scholar]
- Bomblies K., Dagenais N., Weigel D. Redundant enhancers mediate transcriptional repression of AGAMOUS by APETALA2. Dev. Biol. 1999;2161(1):260–264. doi: 10.1006/dbio.1999.9504. [DOI] [PubMed] [Google Scholar]
- Bonifer C. Developmental regulation of eukaryotic gene loci: which cis-regulatory information is required? Trends Genet. 2000;161(1):310–315. doi: 10.1016/s0168-9525(00)02029-1. [DOI] [PubMed] [Google Scholar]
- Bordoli L., Netsch M., Luthi U., Lutz W., Eckner R. Plant orthologs of p300/CBP: conservation of a core domain in metazoan p300/CBP acetyltransferase-related proteins. Nucleic Acids Res. 2001;291(1):589–597. doi: 10.1093/nar/29.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottomley M. J., Collard M. W., Huggenvik J. I., Liu Z., Gibson T. J., Sattler M. The SAND domain structure defines a novel DNA-binding fold in transcriptional regulation. Nature Struct. Biol. 2001;81(1):626–633. doi: 10.1038/89675. [DOI] [PubMed] [Google Scholar]
- Bouche N., Bouchez D. Arabidopsis gene knockout: phenotypes wanted. Curr. Opin. Plant. Biol. 2001;41(1):111–117. doi: 10.1016/s1369-5266(00)00145-x. [DOI] [PubMed] [Google Scholar]
- Bowman J. L. The YABBY gene family and abaxial cell fate. Curr. Opin. Plant. Biol. 2000;31(1):17–22. doi: 10.1016/s1369-5266(99)00035-7. [DOI] [PubMed] [Google Scholar]
- Bowman J. L., Alvarez J., Weigel D., Meyerowitz E. M., Smyth D. Control of flower development in Arabidopsis thaliana by APETALA1 and interacting genes. Development. 1993;1191(1):721–743. [Google Scholar]
- Bowman J. L., Eshed Y., Baum S., Emery J. F., Floyd S. K., Alvarez J., Hawker N. P., Lee J-Y., Siegfried K. R., Khodosh R. The story of CRABS CLAW (or how we learned to love the mutagen). Flowering Newsletter. 2001;311(1):3–11. [Google Scholar]
- Bowman J. L., Smyth D. R. CRABS CLAW, a gene that regulates carpel and nectary development in Arabidopsis, encodes a novel protein with zinc finger and helix-loop-helix domains. Development. 1999;1261(1):2387–2396. doi: 10.1242/dev.126.11.2387. [DOI] [PubMed] [Google Scholar]
- Bowman J. L., Smyth D. R., Meyerowitz E. M. Genetic interactions among floral homeotic genes of Arabidopsis. Development. 1991;1121(1):1–20. doi: 10.1242/dev.112.1.1. [DOI] [PubMed] [Google Scholar]
- Brasher S. V., Smith B. O., Fogh R. H., Nietlispach D., Thiru A., Nielsen P. R., Broadhurst R. W., Ball L. J., Murzina N. V., Laue E. D. The structure of mouse HP1 suggests a unique mode of single peptide recognition by the shadow chromo domain dimer. EMBO J. 2000;191(1):1587–1597. doi: 10.1093/emboj/19.7.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brock H. W., van Lohuizen M. The Polycomb group-no longer an exclusive club? Curr. Opin. Genet. Dev. 2001;111(1):175–181. doi: 10.1016/s0959-437x(00)00176-3. [DOI] [PubMed] [Google Scholar]
- Brown C. E., Howe L., Sousa K., Alley S. C., Carrozza M. J., Tan S., Workman J. L. Recruitment of HAT complexes by direct activator interactions with the ATM-related Tra1 subunit. Science. 2001;2921(1):2333–2337. doi: 10.1126/science.1060214. [DOI] [PubMed] [Google Scholar]
- Brzeski J., Podstolski W., Olczak K., Jerzmanowski A. Identification and analysis of the Arabidopsis thaliana BSH gene, a member of the SNF5 gene family. Nucleic Acids Res. 1999;271(1):2393–2399. doi: 10.1093/nar/27.11.2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bürglin T. R. The PBC domain contains a MEINOX domain: coevolution of Hox and TALE homeobox genes? Dev Genes Evol. 1998;2081(1):113–116. doi: 10.1007/s004270050161. [DOI] [PubMed] [Google Scholar]
- Burns C. G., Ohi R., Krainer A. R., Gould K. L. Evidence that Myb-related CDC5 proteins are required for pre-mRNA splicing. Proc. Natl. Acad. Sci. U. S. A. 1999;961(1):13789–13794. doi: 10.1073/pnas.96.24.13789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch M. A., Bomblies K., Weigel D. Activation of a floral homeotic gene in Arabidopsis. Science. 1999;2851(1):585–587. doi: 10.1126/science.285.5427.585. [DOI] [PubMed] [Google Scholar]
- Byzova M. V., Franken J., Aarts M. G., de Almeida-Engler J., Engler G., Mariani C., Van Lookeren Campagne M. M., Angenent G. C. Arabidopsis STERILE APETALA, a multifunctional gene regulating inflorescence, flower, and ovule development. Genes Dev. 1999;131(1):1002–1014. doi: 10.1101/gad.13.8.1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao H., Glazebrook J., Clarke J. D., Volko S., Dong X. The Arabidopsis NPR1 gene that controls systemic acquired resistance encodes a novel protein containing ankyrin repeats. Cell. 1997;881(1):57–63. doi: 10.1016/s0092-8674(00)81858-9. [DOI] [PubMed] [Google Scholar]
- Capili A. D., Schultz D. C., Rauscher F. J., Borden K. L. B. Solution structure of the PHD domain from the KAP-1 corepressor: structural determinants for PHD, RING and LIM zinc-binding domains. EMBO J. 2001;201(1):165–177. doi: 10.1093/emboj/20.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardon G., Hohmann S., Klein J., Nettesheim K., Saedler H., Huijser P. Molecular characterisation of the Arabidopsis SBP-box genes. Gene. 1999;2371(1):91–104. doi: 10.1016/s0378-1119(99)00308-x. [DOI] [PubMed] [Google Scholar]
- Carroll S. B. Endless forms: the evolution of gene regulation and morphological diversity. Cell. 2000;1011(1):577–580. doi: 10.1016/s0092-8674(00)80868-5. [DOI] [PubMed] [Google Scholar]
- Chan R. L., Gago G. M., Palena C. M., Gonzalez D. H. Homeoboxes in plant development. Biochim. Biophys. Acta. 1998;14421(1):1–19. doi: 10.1016/s0167-4781(98)00119-5. [DOI] [PubMed] [Google Scholar]
- Chao Q., Rothenberg M., Solano R., Roman G., Terzaghi W., Ecker J. R. Activation of the ethylene gas response pathway in Arabidopsis by the nuclear protein ETHYLENE-INSENSITIVE3 and related proteins. Cell. 1997;891(1):1133–1144. doi: 10.1016/s0092-8674(00)80300-1. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay S., Ang L. H., Puente P., Deng X. W., Wei N. Arabidopsis bZIP protein HY5 directly interacts with light-responsive promoters in mediating light control of gene expression. Plant Cell. 1998a;101(1):673–683. doi: 10.1105/tpc.10.5.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay S., Puente P., Deng X. W., Wei N. Combinatorial interaction of light-responsive elements plays a critical role in determining the response characteristics of light-regulated promoters in Arabidopsis. Plant J. 1998b;151(1):69–77. doi: 10.1046/j.1365-313x.1998.00180.x. [DOI] [PubMed] [Google Scholar]
- Chen C. M., Wang C. T., Ho C. H. A plant gene encoding a myb-like protein that binds telomeric ggtttag repeats in vitro. J. Biol. Chem. 2001a;2761(1):16511–16519. doi: 10.1074/jbc.M009659200. [DOI] [PubMed] [Google Scholar]
- Chen D., Ma H., Hong H., Koh S. S., Huang S-M., Schurter B. T., Aswad D. W., Stallcup M. R. Regulation of transcription by a protein methyltransferase. Science. 1999;2841(1):2174–2177. doi: 10.1126/science.284.5423.2174. [DOI] [PubMed] [Google Scholar]
- Chen H., Tini M., Evans R. M. HATs on and beyond chromatin. Curr. Opin. Cell. Biol. 2001b;131(1):218–224. doi: 10.1016/s0955-0674(00)00200-3. [DOI] [PubMed] [Google Scholar]
- Chen W., Chao G., Singh K. B. The promoter of a H2O2-inducible, Arabidopsis glutathione S-transferase gene contains closely linked OBF- and OBP1-binding sites. Plant J. 1996;101(1):955–966. doi: 10.1046/j.1365-313x.1996.10060955.x. [DOI] [PubMed] [Google Scholar]
- Cho R. J., Campbell M. J., Winzeler E. A., Steinmetz L., Conway A., Wodicka L., Wolfsberg T. G., Gabrielian A. E., Landsman D., Lockhart D. J. A genome-wide transcriptional analysis of the mitotic cell cycle. Mol. Cell. 1998;21(1):65–73. doi: 10.1016/s1097-2765(00)80114-8. [DOI] [PubMed] [Google Scholar]
- Christiansen H., Hansen A. C., Vijn I., Pallisgaard N., Larsen K., Yang W. C., Bisseling T., Marcker K. A., Jensen E. O. A novel type of DNA-binding protein interacts with a conserved sequence in an early nodulin ENOD12 promoter. Plant Mol. Biol. 1996;321(1):809–821. doi: 10.1007/BF00020479. [DOI] [PubMed] [Google Scholar]
- Chuang C. F., Meyerowitz E. M. Specific and heritable genetic interference by double-stranded RNA in Arabidopsis thaliana. Proc. Natl. Acad. Sci. U. S. A. 2000;971(1):4985–4990. doi: 10.1073/pnas.060034297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliften P. F., Hillier L. W., Fulton L., Graves T., Miner T., Gish W. R., Waterston R. H., Johnston M. Surveying Saccharomyces genomes to identify functional elements by comparative DNA sequence analysis. Genome Res. 2001;111(1):1175–1186. doi: 10.1101/gr.182901. [DOI] [PubMed] [Google Scholar]
- Coen E. S., Meyerowitz E. M. The war of the whorls: genetic interactions controlling flower development. Nature. 1991;3531(1):31–37. doi: 10.1038/353031a0. [DOI] [PubMed] [Google Scholar]
- Colbert C., Till B. J., Tompa R., Reynolds S., Steine M. N., Yeung A. T., McCallum C. M., Comai L., Henikoff S. High-troughput screening for induced point mutations. Plant Physiol. 2001;1261(1):480–484. doi: 10.1104/pp.126.2.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conner J., Liu Z. LEUNIG, a putative transcriptional corepressor that regulates AGAMOUS expression during flower development. Proc. Natl. Acad. Sci. U. S. A. 2000;971(1):12902–12907. doi: 10.1073/pnas.230352397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke J., Nowak M. A., Boerlijst M., Maynard-Smith J. Evolutionary origins and maintenance of redundant gene expression during metazoan development. Trends Genet. 1997;131(1):360–364. doi: 10.1016/s0168-9525(97)01233-x. [DOI] [PubMed] [Google Scholar]
- Cosma M. P., Panizza S., Nasmyth K. Cdk1 triggers association of RNA polymerase to cell cycle promoters only after recruitment of the Mediator by SBF. Mol. Cell. 2001;71(1):1213–1220. doi: 10.1016/s1097-2765(01)00266-0. [DOI] [PubMed] [Google Scholar]
- Cosma M. P., Tanaka T., Nasmyth K. Ordered recruitment of transcription and chromatin remodeling factors to a cell cycle- and developmentally regulated promoter. Cell. 1999;971(1):299–311. doi: 10.1016/s0092-8674(00)80740-0. [DOI] [PubMed] [Google Scholar]
- Cramer P., Bushnell D. A., Kornberg R. D. Structural basis of transcription: RNA polymerase II at 2.8 A resolution. Science. 2001;191(1):19. doi: 10.1126/science.1059493. [DOI] [PubMed] [Google Scholar]
- Cremer T., Cremer C. Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat. Rev. Genet. 2001;21(1):292–301. doi: 10.1038/35066075. [DOI] [PubMed] [Google Scholar]
- Cubas P., Lauter N., Doebley J., Coen E. The TCP domain: a motif found in proteins regulating plant growth and development. Plant J. 1999a;181(1):215–222. doi: 10.1046/j.1365-313x.1999.00444.x. [DOI] [PubMed] [Google Scholar]
- Cubas P., Vincent C., Coen E. An epigenetic mutation responsible for natural variation in floral symmetry. Nature. 1999b;4011(1):157–161. doi: 10.1038/43657. [DOI] [PubMed] [Google Scholar]
- Cvitanich C., Pallisgaard N., Nielsen K. A., Hansen A. C., Larsen K., Pihakaski-Maunsbach K., Marcker K. A., Jensen E. O. CPP1, a DNA-binding protein involved in the expression of a soybean leghemoglobin c3 gene. Proc. Natl. Acad. Sci. U. S. A. 2000;971(1):8163–8168. doi: 10.1073/pnas.090468497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Agostino I. B., Kieber J. J. Phosphorelay signal transduction: the emerging family of plant response regulators. Trends Biochem. Sci. 1999;241(1):452–456. doi: 10.1016/s0968-0004(99)01465-6. [DOI] [PubMed] [Google Scholar]
- Dangl M., Brosch G., Haas H., Loidl P., Lusser A. Comparative analysis of HD2 type histone deacetylases in higher plants. Planta. 2001;2131(1):280–285. doi: 10.1007/s004250000506. [DOI] [PubMed] [Google Scholar]
- Davidson E. H. Genomic regulatory systems. Development and evolution. 2001. (San Diego: Academic Press) [DOI] [PubMed]
- de Jager S. M., Menges M., Bauer U-M., Murray J. A. H. Arabidopsis E2F1 binds a sequence present in the promoter of S-phase-regulated gene AtCDC6 and is a member of a multigene family with differential activities. Plant Mol. Biol. 2001;471(1):555–568. doi: 10.1023/a:1011848528377. [DOI] [PubMed] [Google Scholar]
- de Vetten N., Quattrocchio F., Mol J., Koes R. The an11 locus controlling flower pigmentation in petunia encodes a novel WD-repeat protein conserved in yeast, plants, and animals. Genes Dev. 1997;111(1):1422–1434. doi: 10.1101/gad.11.11.1422. [DOI] [PubMed] [Google Scholar]
- Derry W. B., Putzke A., Rothman J. H. Caenorhabditis elegans p53: role in apoptosis, meiosis, and stress resistance. Science. 2001;2941(1):591–595. doi: 10.1126/science.1065486. [DOI] [PubMed] [Google Scholar]
- Desikan R., A.-H.-Mackerness S., Hancock J. T., Neill S. J. Regulation of the Arabidopsis transcriptome by oxidative stress. Plant Physiol. 2001;1271(1):159–172. doi: 10.1104/pp.127.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Després C., DeLong C., Glaze S., Liu E., Fobert P. R. The Arabidopsis NPR1/NIM1 protein enhances the DNA binding activity of a subgroup of the TGA family of bZIP transcription factors. Plant Cell. 2000;121(1):279–290. [PMC free article] [PubMed] [Google Scholar]
- Desveaux D., Després C., Joyeux A., Subramaniam R., Brisson N. PBF-2 is a novel single-stranded DNA binding factor implicated in PR-10a gene activation in potato. Plant Cell. 2000;121(1):1477–1489. doi: 10.1105/tpc.12.8.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deyholos M. K., Sieburth L. E. Separable whorl-specific expression and negative regulation by enhancer elements within the AGAMOUS second intron. Plant Cell. 2000;121(1):1799–1810. doi: 10.1105/tpc.12.10.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhalluin C., Carlson J. E., Zeng L., He C., Aggarwal A. K., Zhou M. M. Structure and ligand of a histone acetyltransferase bromodomain. Nature. 1999;3991(1):491–496. doi: 10.1038/20974. [DOI] [PubMed] [Google Scholar]
- Dill A., Sun T-p. Synergistic derepression of gibberellin signaling by removing RGA and GAI function in Arabidopsis thaliana. Genetics. 2001;1591(1):777–785. doi: 10.1093/genetics/159.2.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doebley J., Lukens L. Transcriptional regulators and the evolution of plant form. Plant Cell. 1998;101(1):1075–1082. doi: 10.1105/tpc.10.7.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doebley J., Stec A., Hubbard L. The evolution of apical dominance in maize. Nature. 1997;3861(1):485–488. doi: 10.1038/386485a0. [DOI] [PubMed] [Google Scholar]
- Du L., Chen Z. Identification of genes encoding receptor-like protein kinases as possible targets of pathogen- and salicylic acid-induced WRKY DNA-binding proteins in Arabidopsis. Plant J. 2000;241(1):837–847. doi: 10.1046/j.1365-313x.2000.00923.x. [DOI] [PubMed] [Google Scholar]
- Duret L., Bucher P. Searching for regulatory elements in human noncoding sequences. Curr. Opin. Struct. Biol. 1997;71(1):399–406. doi: 10.1016/s0959-440x(97)80058-9. [DOI] [PubMed] [Google Scholar]
- Egea-Cortines M., Saedler H., Sommer H. Ternary complex formation between the MADS-box proteins SQUAMOSA, DEFICIENS and GLOBOSA is involved in the control of floral architecture in Antirrhinum majus. EMBO J. 1999;181(1):5370–5379. doi: 10.1093/emboj/18.19.5370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgin S. C. R., Workman J. L. Chromatin structure and gene expression. 2nd edn. 2000. (Eds) (Oxford, UK: Oxford University Press)
- Eshed Y., Baum S. F., Bowman J. L. Distinct mechanisms promote polarity establishment in carpels of Arabidopsis. Cell. 1999;991(1):199–209. doi: 10.1016/s0092-8674(00)81651-7. [DOI] [PubMed] [Google Scholar]
- Eshed Y., Baum S. F., Perea J. V., Bowman J. L. Establishment of polarity in lateral organs of plants. Curr. Biol. 2001;111(1):1251–1260. doi: 10.1016/s0960-9822(01)00392-x. [DOI] [PubMed] [Google Scholar]
- Eulgem T., Rushton P. J., Robatzek S., Somssich I. E. The WRKY superfamily of plant transcription factors. Trends Plant Sci. 2000;51(1):199–206. doi: 10.1016/s1360-1385(00)01600-9. [DOI] [PubMed] [Google Scholar]
- Ferrándiz C., Gu Q., Martienssen R., Yanofsky M. F. Redundant regulation of meristem identity and plant architecture by FRUITFULL, APETALA1 and CAULIFLOWER. Development. 2000;1271(1):725–734. doi: 10.1242/dev.127.4.725. [DOI] [PubMed] [Google Scholar]
- Fickett J. W., Wasserman W. W. Discovery and modeling of transcriptional regulatory regions. Curr. Opin. Biotechnol. 2000;111(1):19–24. doi: 10.1016/s0958-1669(99)00049-x. [DOI] [PubMed] [Google Scholar]
- Finnegan E. J., Peacock W. J., Dennis E. S. DNA methylation, a key regulator of plant development and other processes. Curr. Opin. Genet. Dev. 2000;101(1):217–223. doi: 10.1016/s0959-437x(00)00061-7. [DOI] [PubMed] [Google Scholar]
- Flaus A., Owen-Hughes T. Mechanisms for ATP-dependent chromatin remodelling. Curr. Opin. Genet. Dev. 2001;111(1):148–154. doi: 10.1016/s0959-437x(00)00172-6. [DOI] [PubMed] [Google Scholar]
- Foster R., Izawa T., Chua N. H. Plant bZIP proteins gather at ACGT elements. FASEB J. 1994;81(1):192–200. doi: 10.1096/fasebj.8.2.8119490. [DOI] [PubMed] [Google Scholar]
- Francastel C., Schubeler D., Martin D. I., Groudine M. Nuclear compartmentalization and gene activity. Nat. Rev. Mol. Cell Biol. 2000;11(1):137–143. doi: 10.1038/35040083. [DOI] [PubMed] [Google Scholar]
- Francis N. J., Kingston R. E. Mechanisms of transcriptional memory. Nat. Rev. Mol. Cell Biol. 2001;21(1):409–421. doi: 10.1038/35073039. [DOI] [PubMed] [Google Scholar]
- Frary A., Nesbitt T. C., Frary A., Grandillo S., van der Knaap E., Cong B., Liu J., Meller J., Elber R., Alpert K. B. fw2.2: a quantitative trait locus key to the evolution of tomato fruit size. Science. 2000;2891(1):85–88. doi: 10.1126/science.289.5476.85. [DOI] [PubMed] [Google Scholar]
- Fraser A. G., Kamath R. S., Zipperlen P., Martinez-Campos M., Sohrmann M., Ahringer J. Functional genomic analysis of C. elegans chromosome I by systematic RNA interference. Nature. 2000;4081(1):325–330. doi: 10.1038/35042517. [DOI] [PubMed] [Google Scholar]
- Fridborg I., Kuusk S., Robertson M., Sundberg E. The Arabidopsis protein SHI represses gibberellin responses in Arabidopsis and barley. Plant Physiol. 2001;1271(1):937–948. [PMC free article] [PubMed] [Google Scholar]
- Fry C. J., Peterson C. L. Chromatin remodeling enzymes: who's on first? Curr Biol. 2001;111(1):R185–R197. doi: 10.1016/s0960-9822(01)00090-2. [DOI] [PubMed] [Google Scholar]
- Fu H., Park W., Yan X., Zheng Z., Shen B., Dooner H. K. The highly recombinogenic bz locus lies in an unusually gene-rich region of the maize genome. Proc. Natl. Acad. Sci. U. S. A. 2001;981(1):8903–8908. doi: 10.1073/pnas.141221898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadella T. W. J., van der Krogt G. N. M., Bisseling T. GFP-based FRET microscopy in living plant cells. Trends Plant Sci. 1999;41(1):287–291. doi: 10.1016/s1360-1385(99)01426-0. [DOI] [PubMed] [Google Scholar]
- Gasch A. P., Spellman P. T., Kao C. M., Carmel-Harel O., Eisen M. B., Storz G., Botstein D., Brown P. O. Genomic expression programs in the response of yeast cells to environmental changes. Mol. Biol. Cell. 2000;111(1):4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring W. J., Affolter M., Bürglin T. Homeodomain proteins. Annu. Rev. Biochem. 1994;631(1):487–526. doi: 10.1146/annurev.bi.63.070194.002415. [DOI] [PubMed] [Google Scholar]
- Gendall A. R., Levy Y. Y., Wilson A., Dean C. The VERNALIZATION 2 gene mediates the epigenetic regulation of vernalization in Arabidopsis. Cell. 2001;1071(1):525–535. doi: 10.1016/s0092-8674(01)00573-6. [DOI] [PubMed] [Google Scholar]
- Gil P., Dewey E., Friml J., Zhao Y., Snowden K. C., Putterill J., Palme K., Estelle M., Chory J. BIG: a calossin-like protein required for polar auxin transport in Arabidopsis. Genes Dev. 2001;151(1):1985–1997. doi: 10.1101/gad.905201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilliland L. U., McKinney E. C., Asmussen M. A., Meagher R. B. Detection of deleterious genotypes in multigenerational studies. I. Disruptions in individual Arabidopsis actin genes. Genetics. 1998;1491(1):717–725. doi: 10.1093/genetics/149.2.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girke T., Todd J., Ruuska S., White J., Benning C., Ohlrogge J. Microarray analysis of developing Arabidopsis seeds. Plant Physiol. 2000;1241(1):1570–1581. doi: 10.1104/pp.124.4.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffeau A., Aert R., Agostini-Carbone M. L., Ahmed A., Aigle M., Alberghina L., Albermann K., Albers M., Aldea M., Alexandraki D. The yeast genome directory. Nature. 1997;3871(1):5. [Google Scholar]
- Gönczy P., Echeverri G., Oegema K., Coulson A., Jones S. J., Copley R. R., Duperon J., Oegema J., Brehm M., Cassin E. Functional genomic analysis of cell division in C. elegans using RNAi of genes on chromosome III. Nature. 2000;4081(1):331–336. doi: 10.1038/35042526. [DOI] [PubMed] [Google Scholar]
- Goodman R. H., Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000;141(1):1553–1577. [PubMed] [Google Scholar]
- Goodrich J., Puangsomlee P., Martin M., Long D., Meyerowitz E. M., Coupland G. A Polycomb-group gene regulates homeotic gene expression in Arabidopsis. Nature. 1997;3861(1):44–51. doi: 10.1038/386044a0. [DOI] [PubMed] [Google Scholar]
- Goto K., Kyozuka J., Bowman J. L. Turning floral organs into leaves, leaves into floral organs. Curr. Opin. Genet. Dev. 2001;111(1):449–456. doi: 10.1016/s0959-437x(00)00216-1. [DOI] [PubMed] [Google Scholar]
- Gould A. Functions of mammalian Polycomb group and trithorax group related genes. Curr. Opin. Genet. Dev. 1997;71(1):488–494. doi: 10.1016/s0959-437x(97)80075-5. [DOI] [PubMed] [Google Scholar]
- Grasser K. D. HMG1 and HU proteins: architectural elements in plant chromatin. Trends Plant Sci. 1998;31(1):260–265. [Google Scholar]
- Green M. R. TBP-associated factors (TAFIIs): multiple, selective transcriptional mediators in common complexes. Trends Biochem. Sci. 2000;251(1):59–63. doi: 10.1016/s0968-0004(99)01527-3. [DOI] [PubMed] [Google Scholar]
- Greenbaum D., Luscombe N. M., Jansen R., Qian J., Gerstein M. Interrelating different types of genomic data, from proteome to secretome: 'oming in on function. Genome Res. 2001;111(1):1463–1468. doi: 10.1101/gr.207401. [DOI] [PubMed] [Google Scholar]
- Grossniklaus U., Vielle-Calzada J. P., Hoeppner M. A., Gagliano W. B. Maternal control of embryogenesis by MEDEA, a polycomb group gene in Arabidopsis. Science. 1998;2801(1):446–450. doi: 10.1126/science.280.5362.446. [DOI] [PubMed] [Google Scholar]
- Grotewold E., Sainz M. B., Tagliani L., Hernandez J. M., Bowen B., Chandler V. L. Identification of the residues in the Myb domain of maize C1 that specify the interaction with the bHLH cofactor R. Proc. Natl. Acad. Sci. U. S. A. 2000;971(1):13579–13584. doi: 10.1073/pnas.250379897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y-Q., Yang C., Thara V. K., Zhou J., Martin G. B. Pti4 is induced by ethylene and salicylic acid, and its product is phosphorylated by the Pto kinase. Plant Cell. 2000;121(1):771–785. doi: 10.1105/tpc.12.5.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilfoyle T. J., Ulmasov T., Hagen G. The ARF family of transcription factors and their role in plant hormone-responsive transcription. Cell. Mol. Life Sci. 1998;541(1):619–627. doi: 10.1007/s000180050190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumucio D. L., Shelton D. A., Bailey W. J., Slightom J. L., Goodman M. Phylogenetic footprinting reveals unexpected complexity in trans factor binding upstream from the epsilon-globin gene. Proc. Natl. Acad. Sci. U. S. A. 1993;901(1):6018–6022. doi: 10.1073/pnas.90.13.6018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumucio D. L., Shelton D. A., Zhu W., Millinoff D., Gray T., Bock J. H., Slightom J. L., Goodman M. Evolutionary strategies for the elucidation of cis and trans factors that regulate the developmental switching programs of the beta-like globin genes. Mol. Phylogenet. Evol. 1996;51(1):18–32. doi: 10.1006/mpev.1996.0004. [DOI] [PubMed] [Google Scholar]
- Gusmaroli G., Tonelli C., Mantovani R. Regulation of the CCAAT-Binding NF-Y subunits in Arabidopsis thaliana. Gene. 2001;2641(1):173–185. doi: 10.1016/s0378-1119(01)00323-7. [DOI] [PubMed] [Google Scholar]
- Habu Y., Kakutani T., Paszkowski J. Epigenetic developmental mechanisms in plants: molecules and targets of plant epigenetic regulation. Curr. Opin. Genet. Dev. 2001;111(1):215–220. doi: 10.1016/s0959-437x(00)00182-9. [DOI] [PubMed] [Google Scholar]
- Halbach T., Scheer N., Werr W. Transcriptional activation by the PHD finger is inhibited through an adjacent leucine zipper that binds 14-3-3 proteins. Nucleic Acids Res. 2000;281(1):3542–3550. doi: 10.1093/nar/28.18.3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliday K. J., Hudson M., Ni M., Qin M., Quail P. H. poc1: an Arabidopsis mutant perturbed in phytochrome signaling because of a T DNA insertion in the promoter of PIF3, a gene encoding a phytochrome-interacting bHLH protein. Proc. Natl. Acad. Sci. U. S. A. 1999;961(1):5832–5837. doi: 10.1073/pnas.96.10.5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond S. M., Caudy A. A., Hannon G. J. Post-transcriptional gene silencing by double-stranded RNA. Nat. Rev. Genet. 2001;21(1):110–119. doi: 10.1038/35052556. [DOI] [PubMed] [Google Scholar]
- Hampsey M., Reinberg D. RNA polymerase II as a control panel for multiple coactivator complexes. Curr. Opin. Genet. Dev. 1999;91(1):132–139. doi: 10.1016/S0959-437X(99)80020-3. [DOI] [PubMed] [Google Scholar]
- Hardison R. C., Oeltjen J., Miller W. Long human-mouse sequence alignments reveal novel regulatory elements: a reason to sequence the mouse genome. Genome Res. 1997;71(1):959–966. doi: 10.1101/gr.7.10.959. [DOI] [PubMed] [Google Scholar]
- Harmer S. L., Hogenesch J. B., Straume M., Chang H. S., Han B., Zhu T., Wang X., Kreps J. A., Kay S. A. Orchestrated transcription of key pathways in Arabidopsis by the circadian clock. Science. 2000;2901(1):2110–2113. doi: 10.1126/science.290.5499.2110. [DOI] [PubMed] [Google Scholar]
- Hasebe M., Wen C. K., Kato M., Banks J. A. Characterization of MADS homeotic genes in the fern Ceratopteris richardii. Proc. Natl. Acad. Sci. U. S. A. 1998;951(1):6222–6227. doi: 10.1073/pnas.95.11.6222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazbun T. R., Fields S. Networking proteins in yeast. Proc. Natl. Acad. Sci. U. S. A. 2001;981(1):4277–4278. doi: 10.1073/pnas.091096398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirayama T., Shinozaki K. A cdc5+ homolog of a higher plant, Arabidopsis thaliana. Proc. Natl. Acad. Sci. U. S. A. 1996;931(1):13371–13376. doi: 10.1073/pnas.93.23.13371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst M., Ho C., Sabourin L., Rudnicki M., Penn L., Sadowski I. A two-hybrid system for transactivator bait proteins. Proc. Natl. Acad. Sci. U. S. A. 2001;981(1):8726–8731. doi: 10.1073/pnas.141413598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobo T., Kowyama Y., Hattori T. A bZIP factor, TRAB1, interacts with VP1 and mediates abscisic acid- induced transcription. Proc. Natl. Acad. Sci. U. S. A. 1999;961(1):15348–15353. doi: 10.1073/pnas.96.26.15348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogenesch J. B., Ching K. A., Batalov S., Su A. I., Walker J. R., Zhou Y., Kay S. A., Schultz P. G., Cooke M. P. A comparison of the Celera and Ensembl predicted gene sets reveals little overlap in novel genes. Cell. 2001;1061(1):413–415. doi: 10.1016/s0092-8674(01)00467-6. [DOI] [PubMed] [Google Scholar]
- Honma T., Goto K. Complexes of MADS-box proteins are sufficient to convert leaves into floral organs. Nature. 2001;4091(1):525–529. doi: 10.1038/35054083. [DOI] [PubMed] [Google Scholar]
- Huang H., Tudor M., Su T., Zhang Y., Hu Y., Ma H. DNA binding properties of two Arabidopsis MADS domain proteins: binding consensus and dimer formation. Plant Cell. 1996;81(1):81–94. doi: 10.1105/tpc.8.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes J. D., Estep P. W., Tavazoie S., Church G. M. Computational identification of cis-regulatory elements associated with groups of functionally related genes in Saccharomyces cerevisiae. J. Mol. Biol. 2000a;2961(1):1205–1214. doi: 10.1006/jmbi.2000.3519. [DOI] [PubMed] [Google Scholar]
- Hughes T. R., Marton M. J., Jones A. R., Roberts C. J., Stoughton R., Armour C. D., Bennett H. A., Coffey E., Dai H., He Y. D. Functional discovery via a compendium of expression profiles. Cell. 2000b;1021(1):109–126. doi: 10.1016/s0092-8674(00)00015-5. [DOI] [PubMed] [Google Scholar]
- Hugouvieux V., Kwak J. M., Schroeder J. I. An mRNA cap binding protein, ABH1, modulates early abscisic signal transduction in Arabidopsis. Cell. 2001;1061(1):477–487. doi: 10.1016/s0092-8674(01)00460-3. [DOI] [PubMed] [Google Scholar]
- Hwang I., Sheen J. Two-component circuitry in Arabidopsis cytokinin signal transduction. Nature. 2001;4131(1):383–389. doi: 10.1038/35096500. [DOI] [PubMed] [Google Scholar]
- Hwang M. G., Chung I. K., Kang B. G., Cho M. H. Sequence-specific binding property of Arabidopsis thaliana telomeric DNA binding protein 1 (AtTBP1). FEBS Lett. 2001;5031(1):35–40. doi: 10.1016/s0014-5793(01)02685-0. [DOI] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium Initial sequencing and analysis of the human genome. Nature. 2001;4091(1):860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Ito T., Chiba T., Ozawa R., Yoshida M., Hattori M., Sakaki Y. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc. Natl. Acad. Sci. U. S. A. 2001;981(1):4569–4574. doi: 10.1073/pnas.061034498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer V. R., Horak C. E., Scafe C. S., Botstein D., Snyder M., Brown P. O. Genomic binding sites of the yeast cell-cycle transcription factors SBF and MBF. Nature. 2001;4091(1):533–538. doi: 10.1038/35054095. [DOI] [PubMed] [Google Scholar]
- Jack T. Relearning our ABCs: new twists on an old model. Trends Plant Sci. 2001;61(1):310–316. doi: 10.1016/s1360-1385(01)01987-2. [DOI] [PubMed] [Google Scholar]
- Jacobson R. H., Ladurner A. G., King D. S., Tijan R. Structure and function of a human TAFII250 double bromodomain module. Science. 2000;2881(1):1422–1425. doi: 10.1126/science.288.5470.1422. [DOI] [PubMed] [Google Scholar]
- Jakt L. M., Cao L., Cheah K. S., Smith D. K. Assessing clusters and motifs from gene expression data. Genome Res. 2001;111(1):112–123. doi: 10.1101/gr.148301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeanmougin F., Wurtz J-M., Douarin B. L., Chambon P., Losson R. The bromodomain revisited. Trends Biochem. Sci. 1997;221(1):151–153. doi: 10.1016/s0968-0004(97)01042-6. [DOI] [PubMed] [Google Scholar]
- Jeddeloh J. A., Stokes T. L., Richards E. J. Maintenance of genomic methylation requires a SWI2/SNF2-like protein. Nature Genet. 1999;221(1):94–97. doi: 10.1038/8803. [DOI] [PubMed] [Google Scholar]
- Jenuwein T. Re-SET-ting heterochromatin by histone methyltransferases. Trends Cell Biol. 2001;111(1):266–273. doi: 10.1016/s0962-8924(01)02001-3. [DOI] [PubMed] [Google Scholar]
- Joazeiro C. A. P., Weissman A. M. RING finger proteins: mediators of ubiquitin ligase activity. Cell. 2000;1021(1):549–552. doi: 10.1016/s0092-8674(00)00077-5. [DOI] [PubMed] [Google Scholar]
- Jones D. O., Cowell I. G., Singh P. B. Mammalian chromodomain proteins: their role in genome organisation and expression. Bioessays. 2000;221(1):124–137. doi: 10.1002/(SICI)1521-1878(200002)22:2<124::AID-BIES4>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Kadonaga J. T. Eukaryotic transcription: an interlaced network of transcription factors and chromatin-modifying machines. Cell. 1998;921(1):307–313. doi: 10.1016/s0092-8674(00)80924-1. [DOI] [PubMed] [Google Scholar]
- Kagaya Y., Ohmiya K., Hattori T. RAV1, a novel DNA-binding protein, binds to bipartite recognition sequence through two distinct DNA-binding domains uniquely found in higher plants. Nucleic Acids Res. 1999;271(1):470–478. doi: 10.1093/nar/27.2.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaoka A., Kaothien P., Yoshida K., Endo S., Yamada K., Ebinuma H. Functional analysis of tobacco LIM protein Ntlim1 involved in lignin biosynthesis. Plant J. 2000;221(1):289–301. doi: 10.1046/j.1365-313x.2000.00737.x. [DOI] [PubMed] [Google Scholar]
- Kempin S. A., Savidge B., Yanofsky M. F. Molecular basis of the cauliflower phenotype in Arabidopsis. Science. 1995;2671(1):522–525. doi: 10.1126/science.7824951. [DOI] [PubMed] [Google Scholar]
- Kerstetter R. A., Bollman K., Taylor R. A., Bomblies K., Poethig R. S. KANADI regulates organ polarity in Arabidopsis. Nature. 2001;4111(1):706–709. doi: 10.1038/35079629. [DOI] [PubMed] [Google Scholar]
- Khochbin S., Verdel A., Lemercier C., Seigneurin-Berny D. Functional significance of histone deacetylase diversity. Curr. Opin. Genet. Dev. 2001;111(1):162–166. doi: 10.1016/s0959-437x(00)00174-x. [DOI] [PubMed] [Google Scholar]
- Kim J. C., Lee S. H., Cheong Y. H., Yoo C-M., Lee S. I., Chun H. J., Yun D-J., Hong J. C., Lee S. Y., Lim C. O. A novel cold-inducible zinc finger protein from soybean, SCOF-1, enhances cold tolerance in transgenic plants. Plant J. 2001;251(1):247–259. doi: 10.1046/j.1365-313x.2001.00947.x. [DOI] [PubMed] [Google Scholar]
- Kim S. K. Http://C. elegans: mining the functional genomic landscape. Nat. Rev. Genet. 2001;21(1):681–689. doi: 10.1038/35088523. [DOI] [PubMed] [Google Scholar]
- King K. E., Moritz T., Harberd N. P. Gibberellins are not required for normal stem growth in Arabidopsis thaliana in the absence of GAI and RGA. Genetics. 2001;1591(1):767–776. doi: 10.1093/genetics/159.2.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinkema M., Fan W., Dong X. Nuclear localization of NPR1 is required for activation of PR gene expression. Plant Cell. 2000;121(1):2339–2350. doi: 10.1105/tpc.12.12.2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita T., Yadegari R., Harada J. J., Goldberg R. B., Fischer R. L. Imprinting of the MEDEA polycomb gene in the Arabidopsis endosperm. Plant Cell. 1999;111(1):1945–1952. doi: 10.1105/tpc.11.10.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher S., Kozma-Bognar L., Kim L., Adam E., Harter K., Schäfer E., Nagy F. Light quality-dependent nuclear import of the plant photoreceptors phytochrome A and B. Plant Cell. 1999;111(1):1445–1456. doi: 10.1105/tpc.11.8.1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimyuk V. I., Persello-Cartieaux F., Havaux M., Contard-David P., Schuenemann D., Meiherhoff K., Gouet P., Jones J. D., Hoffman N. E., Nussaume L. A chromodomain protein encoded by the arabidopsis CAO gene is a plant-specific component of the chloroplast signal recognition particle pathway that is involved in LHCP targeting. Plant Cell. 1999;111(1):87–99. doi: 10.1105/tpc.11.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch M. A., Weisshaar B., Kroymann J., Haubold B., Mitchell-Olds T. Comparative genomics and regulatory evolution: conservation and function of the Chs and Apetala3 promoters. Mol. Biol. Evol. 2001;181(1):1882–1891. doi: 10.1093/oxfordjournals.molbev.a003729. [DOI] [PubMed] [Google Scholar]
- Kornberg R. D. Eukaryotic transcriptional control. Trends Cell Biol. 1999;91(1):M46–49. [PubMed] [Google Scholar]
- Kortschak R. D., Tucker P. W., Saint R. ARID proteins come in from the desert. Trends Biochem. Sci. 2000;251(1):294–299. doi: 10.1016/s0968-0004(00)01597-8. [DOI] [PubMed] [Google Scholar]
- Krizek B. A., Meyerowitz E. M. The Arabidopsis homeotic genes APETALA3 and PISTILLATA are sufficient to provide the B class organ identity function. Development. 1996;1221(1):11–22. doi: 10.1242/dev.122.1.11. [DOI] [PubMed] [Google Scholar]
- Krizek B. A., Prost V., Macias A. AINTEGUMENTA promotes petal identity and acts as a negative regulator of AGAMOUS. Plant Cell. 2000;121(1):1357–1366. doi: 10.1105/tpc.12.8.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogan N. T., Ashton N. W. Ancestry of plant MADS-box genes revealed by bryophite (Physcomitrella patens) homologues. New Phytol. 2000;1471(1):505–517. doi: 10.1046/j.1469-8137.2000.00728.x. [DOI] [PubMed] [Google Scholar]
- Krysan P. J., Young J. C., Sussman M. R. T-DNA as an insertional mutagen in Arabidopsis. Plant Cell. 1999;111(1):2283–2290. doi: 10.1105/tpc.11.12.2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachner M., O'Carroll D., Rea S., Mechtler K., Jenuwein T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 2001;4101(1):116–120. doi: 10.1038/35065132. [DOI] [PubMed] [Google Scholar]
- Ladomery M. Multifunctional proteins suggest connections between transcriptional and post-transcriptional processes. Bioessays. 1997;191(1):903–909. doi: 10.1002/bies.950191010. [DOI] [PubMed] [Google Scholar]
- Larkin J. C., Oppenheimer D. G., Pollock S., Marks M. D. Arabidopsis GLABROUS1 gene requires downstream sequences for function. Plant Cell. 1993;51(1):1739–1748. doi: 10.1105/tpc.5.12.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latchman D. S. Eukaryotic transcription factors. 3rd edn. 1998. (San Diego: Academic Press) [DOI] [PMC free article] [PubMed]
- Lee M. M., Schiefelbein J. WEREWOLF, a MYB-related protein in Arabidopsis, is a position-dependent regulator of epidermal cell patterning. Cell. 1999;991(1):473–483. doi: 10.1016/s0092-8674(00)81536-6. [DOI] [PubMed] [Google Scholar]
- Lee M. M., Schiefelbein J. Developmentally distinct MYB genes encode functionally equivalent proteins in Arabidopsis. Development. 2001;1281(1):1539–1546. doi: 10.1242/dev.128.9.1539. [DOI] [PubMed] [Google Scholar]
- Lee T. I., Young R. A. Transcription of eukaryotic protein-coding genes. Annu. Rev. Genet. 2000;341(1):77–137. doi: 10.1146/annurev.genet.34.1.77. [DOI] [PubMed] [Google Scholar]
- Legrain P., Wojcik J., Gauthier J. Protein-protein interaction maps: a lead towards cellular functions. Trends Genet. 2001;171(1):346–352. doi: 10.1016/s0168-9525(01)02323-x. [DOI] [PubMed] [Google Scholar]
- Lei X-H., Shen X., Xu X-Q., Bernstein H. S. Human Cdc5, a regulator of mitotic entry, can act as a site-specific DNA binding protein. J. Cell Sci. 2000;1131(1):4523–4531. doi: 10.1242/jcs.113.24.4523. [DOI] [PubMed] [Google Scholar]
- Lemon B., Tjian R. Orchestrated response: a symphony of transcription factors for gene control. Genes Dev. 2000;141(1):2551–2569. doi: 10.1101/gad.831000. [DOI] [PubMed] [Google Scholar]
- Levin J. Z., de Framond A. J., Tuttle A., Bauer M. W., Heifetz P. B. Methods of double-stranded RNA-mediated gene inactivation in Arabidopsis and their use to define an essential gene in methionine biosynthesis. Plant Mol. Biol. 2000;441(1):759–775. doi: 10.1023/a:1026584607941. [DOI] [PubMed] [Google Scholar]
- Li G., Bishop K. J., Chandrasekharan M. B., Hall T. C. β-Phaseolin gene activation is a two-step process: PvALF-facilitated chromatin modification followed by abscisic acid-mediated gene activation. Proc. Natl. Acad. Sci. U. S. A. 1999;961(1):7104–7109. doi: 10.1073/pnas.96.12.7104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G., Chandler S. P., Wolffe A. P., Hall T. C. Architectural specificity in chromatin structure at the TATA box in vivo: nucleosome displacement upon b-phaseolin gene activation. Proc. Natl. Acad. Sci. U. S. A. 1998;951(1):4772–4777. doi: 10.1073/pnas.95.8.4772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G., Chandrasekharan M. B., Wolffe A. P., Hall T. C. Chromatin structure and phaseolin gene regulation. Plant Mol. Biol. 2001a;461(1):121–129. doi: 10.1023/a:1010693703421. [DOI] [PubMed] [Google Scholar]
- Li X., Song Y., Century K., Straight S., Ronald P., Dong X., Lassner M., Zhang Y. A fast neutron deletion mutagenesis-based reverse genetics system for plants. Plant J. 2001b;271(1):235–242. doi: 10.1046/j.1365-313x.2001.01084.x. [DOI] [PubMed] [Google Scholar]
- Li Z., Thomas T. L. PEI1, an embryo-specific zinc finger protein gene required for heart-stage embryo formation in Arabidopsis. Plant Cell. 1998;101(1):383–398. doi: 10.1105/tpc.10.3.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieb J. D., Liu X., Botstein D., Brown P. O. Promoter-specific binding of Rap1 revealed by genome-wide maps of protein-DNA association. Nature Genet. 2001;281(1):327–334. doi: 10.1038/ng569. [DOI] [PubMed] [Google Scholar]
- Liljegren S. J., Ditta G. S., Eshed Y., Savidge B., Bowman J. L., Yanofsky M. F. SHATTERPROOF MADS-box genes control seed dispersal in Arabidopsis. Nature. 2000;4041(1):766–770. doi: 10.1038/35008089. [DOI] [PubMed] [Google Scholar]
- Lin X., Kaul S., Rounsley S., Shea T. P., Benito M. I., Town C. D., Fujii C. Y., Mason T., Bowman C. L., Barnstead M. Sequence and analysis of chromosome 2 of the plant Arabidopsis thaliana. Nature. 1999;4021(1):761–768. doi: 10.1038/45471. [DOI] [PubMed] [Google Scholar]
- Livesey F. J., Furukawa T., Steffen M. A., Church G. M., Cepko C. L. Microarray analysis of the transcriptional network controlled by the photoreceptor homeobox gene Crx. Curr. Biol. 2000;101(1):301–310. doi: 10.1016/s0960-9822(00)00379-1. [DOI] [PubMed] [Google Scholar]
- Lloyd A. M., Schena M., Walbot V., Davis R. W. Epidermal cell fate determination in Arabidopsis: patterns defined by a steroid-inducible regulator. Science. 1994;2661(1):436–439. doi: 10.1126/science.7939683. [DOI] [PubMed] [Google Scholar]
- Lockhart D. J., Winzeler E. A. Genomics, gene expression and DNA arrays. Nature. 2000;4051(1):827–836. doi: 10.1038/35015701. [DOI] [PubMed] [Google Scholar]
- Lohmann J. U., Hong R. L., Hobe M., Busch M. A., Parcy F., Simon R., Weigel D. A molecular link between stem cell regulation and floral patterning in Arabidopsis. Cell. 2001;1051(1):793–803. doi: 10.1016/s0092-8674(01)00384-1. [DOI] [PubMed] [Google Scholar]
- Loots G. G., Locksley R. M., Blankespoor C. M., Wang Z. E., Miller W., Rubin E. M., Frazer K. A. Identification of a coordinate regulator of interleukins 4, 13, and 5 by cross-species sequence comparisons. Science. 2000;2881(1):136–140. doi: 10.1126/science.288.5463.136. [DOI] [PubMed] [Google Scholar]
- Lotan T., Ohto M., Yee K. M., West M. A., Lo R., Kwong R. W., Yamagishi K., Fischer R. L., Goldberg R. B., Harada J. J. Arabidopsis LEAFY COTYLEDON1 is sufficient to induce embryo development in vegetative cells. Cell. 1998;931(1):1195–1205. doi: 10.1016/s0092-8674(00)81463-4. [DOI] [PubMed] [Google Scholar]
- Luo M., Bilodeau P., Dennis E. S., Peacock W. J., Chaudhury A. Expression and parent-of-origin effects for FIS2, MEA, and FIE in the endosperm and embryo of developing Arabidopsis seeds. Proc. Natl. Acad. Sci. U. S. A. 2000;971(1):10637–10642. doi: 10.1073/pnas.170292997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo M., Bilodeau P., Koltunow A., Dennis E. S., Peacock W. J., Chaudhury A. M. Genes controlling fertilization-independent seed development in Arabidopsis thaliana. Proc. Natl. Acad. Sci. U. S. A. 1999;961(1):296–301. doi: 10.1073/pnas.96.1.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscombe N. M., Austin S. E., Berman H. M., Thornton J. M. An overview of the structures of protein-DNA complexes. 2000;1 doi: 10.1186/gb-2000-1-1-reviews001. Genome Biol. reviews001.001-001.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusser A., Brosch G., Loidl A., Haas H., Loidl P. Identification of maize histone deacetylase HD2 as an acidic nucleolar phosphoprotein. Science. 1997;2771(1):88–91. doi: 10.1126/science.277.5322.88. [DOI] [PubMed] [Google Scholar]
- Lusser A., Kolle D., Loidl P. Histone acetylation: lessons from the plant kingdom. Trends Plant Sci. 2001;61(1):59–65. doi: 10.1016/s1360-1385(00)01839-2. [DOI] [PubMed] [Google Scholar]
- Lyons T. J., Gasch A. P., Gaither L. A., Botstein D., Brown P. O., Eide D. J. Genome-wide characterization of the Zap1p zinc-responsive regulon in yeast. Proc. Natl. Acad. Sci. U. S. A. 2000;971(1):7957–7962. doi: 10.1073/pnas.97.14.7957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda I., Kohara Y., Yamamoto M., Sugimoto A. Large-scale analysis of gene function in Caenorhabditis elegans by high-throughput RNAi. Curr. Biol. 2001;111(1):171–176. doi: 10.1016/s0960-9822(01)00052-5. [DOI] [PubMed] [Google Scholar]
- Maes T., De Keukeleire P., Gerats T. Plant tagnology. Trends Plant Sci. 1999;41(1):90–96. doi: 10.1016/s1360-1385(99)01375-8. [DOI] [PubMed] [Google Scholar]
- Mahmoudi T., Verrijzer C. P. Chromatin silencing and activation by Polycomb and trithorax group proteins. Oncogene. 2001;201(1):3055–3066. doi: 10.1038/sj.onc.1204330. [DOI] [PubMed] [Google Scholar]
- Maleck K., Levine A., Eulgem T., Morgan A., Schmid J., Lawton K. A., Dangl J. L., Dietrich R. A. The transcriptome of Arabidopsis thaliana during systemic acquired resistance. Nature Genet. 2000;261(1):403–410. doi: 10.1038/82521. [DOI] [PubMed] [Google Scholar]
- Malik S., Roeder R. G. Transcriptional regulation through Mediator-like coactivators in yeast and metazoan cells. Trends Biochem. Sci. 2000;251(1):277–283. doi: 10.1016/s0968-0004(00)01596-6. [DOI] [PubMed] [Google Scholar]
- Marmorstein R. Protein modules that manipulate histone tails for chromatin regulation. Nat. Rev. Mol. Cell Biol. 2001a;21(1):422–432. doi: 10.1038/35073047. [DOI] [PubMed] [Google Scholar]
- Marmorstein R. Structure of histone acetyltransferases. J. Mol. Biol. 2001b;3111(1):433–444. doi: 10.1006/jmbi.2001.4859. [DOI] [PubMed] [Google Scholar]
- Marmorstein R., Roth S. Y. Histone acetyltransferases: function, structure, and catalysis. Curr. Opin. Genet. Dev. 2001;111(1):155–161. doi: 10.1016/s0959-437x(00)00173-8. [DOI] [PubMed] [Google Scholar]
- Martin C., Paz-Ares J. MYB transcription factors in plants. Trends Genet. 1997;131(1):67–73. doi: 10.1016/s0168-9525(96)10049-4. [DOI] [PubMed] [Google Scholar]
- Martínez-García J. F., Huq E., Quail P. H. Direct targeting of light signals to a promoter element-bound transcription factor. Science. 2000;2881(1):859–863. doi: 10.1126/science.288.5467.859. [DOI] [PubMed] [Google Scholar]
- Massingham T., Davies L. J., Lio P. Analysing gene function after duplication. Bioessays. 2001;231(1):873–876. doi: 10.1002/bies.1128. [DOI] [PubMed] [Google Scholar]
- Matsumoto K., Wolffe A. P. Gene regulation by Y-box proteins: coupling control of transcription and translation. Trends Cell Biol. 1998;81(1):318–323. doi: 10.1016/s0962-8924(98)01300-2. [DOI] [PubMed] [Google Scholar]
- Mayer K., Schuller C., Wambutt R., Murphy G., Volckaert G., Pohl T., Dusterhoft A., Stiekema W., Entian K. D., Terryn N. Sequence and analysis of chromosome 4 of the plant Arabidopsis thaliana. Nature. 1999;4021(1):769–777. doi: 10.1038/47134. [DOI] [PubMed] [Google Scholar]
- Meissner R. C., Jin H., Cominelli E., Denekamp M., Fuertes A., Greco R., Kranz H. D., Penfield S., Petroni K., Urzainqui A. Function search in a large transcription factor gene family in Arabidopsis: assessing the potential of reverse genetics to identify insertional mutations in R2R3 MYB genes. Plant Cell. 1999;111(1):1827–1840. doi: 10.1105/tpc.11.10.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merika M., Thanos D. Enhanceosomes. Curr. Opin. Genet. Dev. 2001;111(1):205–208. doi: 10.1016/s0959-437x(00)00180-5. [DOI] [PubMed] [Google Scholar]
- Misteli T. Protein dynamics: implications for nuclear architecture and gene expression. Science. 2001;2911(1):843–847. doi: 10.1126/science.291.5505.843. [DOI] [PubMed] [Google Scholar]
- Miura A., Yonebayashi S., Watanabe K., Toyama T., Shimada H., Kakutani T. Mobilization of transposons by a mutation abolishing full DNA methylation in Arabidopsis. Nature. 2001;4111(1):212–214. doi: 10.1038/35075612. [DOI] [PubMed] [Google Scholar]
- Moazed D. Common themes in mechanisms of gene silencing. Mol. Cell. 2001;81(1):489–498. doi: 10.1016/s1097-2765(01)00340-9. [DOI] [PubMed] [Google Scholar]
- Mol J., Grotewold E., Koes R. How genes paint flowers and seeds. Trends Plant Sci. 1998;31(1):212–217. [Google Scholar]
- Molin L., Mounsey A., Aslam S., Bauer P., Young J., James M., Sharma-Oates A., Hope I. A. Evolutionary conservation of redundancy between a diverged pair of forkhead transcription factor homologues. Development. 2000;1271(1):4825–4835. doi: 10.1242/dev.127.22.4825. [DOI] [PubMed] [Google Scholar]
- Münster T., Pahnke J., Di Rosa A., Kim J. T., Martin W., Saedler H., Theiben G. Floral homeotic genes were recruited from homologous MADS-box genes preexisting in the common ancestor of ferns and seed plants. Proc. Natl. Acad. Sci. U. S. A. 1997;941(1):2415–2420. doi: 10.1073/pnas.94.6.2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murfett J., Wang X. J., Hagen G., Guilfoyle T. J. Identification of Arabidopsis histone deacetylase HDA6 mutants that affect transgene expression. Plant Cell. 2001;131(1):1047–1061. doi: 10.1105/tpc.13.5.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagano Y., Furuhashi H., Inaba T., Sasaki Y. A novel class of plant-specific zinc-dependent DNA-binding protein that binds to A/T-rich DNA sequences. Nucleic Acids Res. 2001;291(1):4097–4105. doi: 10.1093/nar/29.20.4097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy F., Schäfer E. Nuclear and cytosolic events of light-induced, phytochrome regulated signaling in higher plants. EMBO J. 2000;191(1):157–163. doi: 10.1093/emboj/19.2.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura S., Lynch T. J., Finkelstein R. R. Physical interactions between ABA response loci of Arabidopsis. Plant J. 2001;261(1):627–635. doi: 10.1046/j.1365-313x.2001.01069.x. [DOI] [PubMed] [Google Scholar]
- Nei M., Xu P., Glazko G. Estimation of divergence times from multiprotein sequences for a few mammalian species and several distantly related organisms. Proc. Natl. Acad. Sci. U. S. A. 2001;981(1):2497–2502. doi: 10.1073/pnas.051611498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesi N., Debeaujon I., Jond C., Pelletier G., Caboche M., Lepiniec L. The TT8 gene encodes a basic helix-loop-helix domain protein required for expression of DFR and BAN genes in Arabidopsis siliques. Plant Cell. 2000;121(1):1863–1878. doi: 10.1105/tpc.12.10.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng M., Yanofsky M. F. Function and evolution of the plant MADS-box gene family. Nat. Rev. Genet. 2001;21(1):186–195. doi: 10.1038/35056041. [DOI] [PubMed] [Google Scholar]
- Ni M., Tepperman J. M., Quail P. H. PIF3, a phytochrome-interacting factor necessary for normal photoinduced signal transduction, is a novel basic helix-loop-helix protein. Cell. 1998;951(1):657–667. doi: 10.1016/s0092-8674(00)81636-0. [DOI] [PubMed] [Google Scholar]
- Niessing D., Driever W., Sprenger F., Taubert H., Jackle H., Rivera-Pomar R. Homeodomain position 54 specifies transcriptional versus translational control by bicoid. Mol. Cell. 2000;51(1):395–401. doi: 10.1016/s1097-2765(00)80434-7. [DOI] [PubMed] [Google Scholar]
- Nover L., Bharti K., Döring P., Mishra S. K., Ganguli A., Scharf K. D. Arabidopsis and the heat stress transcription factor world: how many heat stress transcription factors do we need? Cell Stress Chaperones. 2001;61(1):177–189. doi: 10.1379/1466-1268(2001)006<0177:aathst>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogas J., Kaufmann S., Henderson J., Somerville C. PICKLE is a CHD3 chromatin-remodeling factor that regulates the transition from embryonic to vegetative development in Arabidopsis. Proc. Natl. Acad. Sci. U. S. A. 1999;961(1):13839–13844. doi: 10.1073/pnas.96.24.13839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh T. J., May G. D. Oligonucleotide-directed plant gene targeting. Curr. Opin. Biotechnol. 2001;121(1):169–172. doi: 10.1016/s0958-1669(00)00194-4. [DOI] [PubMed] [Google Scholar]
- Ohad N., Yadegari R., Margossian L., Hannon M., Michaeli D., Harada J. J., Goldberg R. B., Fischer R. L. Mutations in FIE, a WD polycomb group gene, allow endosperm development without fertilization. Plant Cell. 1999;111(1):407–416. doi: 10.1105/tpc.11.3.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohgishi M., Oka A., Morelli G., Ruberti I., Aoyama T. Negative autoregulation of the Arabidopsis homeobox gene ATHB-2. Plant J. 2001;251(1):389–398. doi: 10.1046/j.1365-313x.2001.00966.x. [DOI] [PubMed] [Google Scholar]
- Ohi R., Feoktistova A., McCann S., Valentine V., Look A. T., Lipsick J. S., Gould K. L. Myb-related Schizosaccharomyces pombe cdc5p is structurally and functionally conserved in eukaryotes. Mol. Cell. Biol. 1998;181(1):4097–4108. doi: 10.1128/mcb.18.7.4097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterlund M. T., Hardtke C. S., Wei N., Deng X. W. Targeted destabilization of HY5 during light-regulated development of Arabidopsis. Nature. 2000;4051(1):462–466. doi: 10.1038/35013076. [DOI] [PubMed] [Google Scholar]
- Oyama T., Shimura Y., Okada K. The Arabidopsis HY5 gene encodes a bZIP protein that regulates stimulus-induced development of root and hypocotyl. Genes Dev. 1997;111(1):2983–2995. doi: 10.1101/gad.11.22.2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parinov S., Sundaresan V. Functional genomics in Arabidopsis: large-scale insertional mutagenesis complements the genome sequencing project. Curr. Opin. Biotechnol. 2000;111(1):157–161. doi: 10.1016/s0958-1669(00)00075-6. [DOI] [PubMed] [Google Scholar]
- Pautot V., Dockx J., Hamant O., Kronenberger J., Grandjean O., Jublot D., Traas J. KNAT2: Evidence for a link between Knotted-like genes and carpel development. Plant Cell. 2001;131(1):1719–1734. doi: 10.1105/TPC.010184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne C. T., Zhang F., Lloyd A. M. GL3 encodes a bHLH protein that regulates trichome development in Arabidopsis through interaction with GL1 and TTG1. Genetics. 2000;1561(1):1349–1362. doi: 10.1093/genetics/156.3.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelaz S., Ditta G. S., Baumann E., Wisman E., Yanofsky M. F. B and C floral organ identity functions require SEPALLATA MADS-box genes. Nature. 2000;4051(1):200–203. doi: 10.1038/35012103. [DOI] [PubMed] [Google Scholar]
- Pelaz S., Gustafson-Brown C., Kohalmi S. E., Crosby W. L., Yanofsky M. F. APETALA1 and SEPALLATA3 interact to promote flower development. Plant J. 2001a;261(1):385–394. doi: 10.1046/j.1365-313x.2001.2641042.x. [DOI] [PubMed] [Google Scholar]
- Pelaz S., Tapia-Lopez R., Alvarez-Buylla E. R., Yanofsky M. F. Conversion of leaves into petals in Arabidopsis. Curr. Biol. 2001b;111(1):182–184. doi: 10.1016/s0960-9822(01)00024-0. [DOI] [PubMed] [Google Scholar]
- Peng J., Richards D. E., Hartley N. M., Murphy G. P., Devos K. M., Flintham J. E., Beales J., Fish L. J., Worland A. J., Pelica F. ‘Green revolution’ genes encode mutant gibberellin response modulators. Nature. 1999;4001(1):256–261. doi: 10.1038/22307. [DOI] [PubMed] [Google Scholar]
- Petersen M., Brodersen P., Naested H., Andreasson E., Lindhart U., Johansen B., Nielsen H. B., Lacy M., Austin M. J., Parker J. E. Arabidopsis MAP kinase 4 negatively regulates systemic acquired resistance. Cell. 2000;1031(1):1111–1120. doi: 10.1016/s0092-8674(00)00213-0. [DOI] [PubMed] [Google Scholar]
- Philippe H., Germot A., Moreira D. The new phylogeny of eukaryotes. Curr. Opin. Genet. Dev. 2000;101(1):596–601. doi: 10.1016/s0959-437x(00)00137-4. [DOI] [PubMed] [Google Scholar]
- Picard D. Posttranslational regulation of proteins by fusions to steroid-binding domains. Methods Enzymol. 2000;3271(1):385–401. doi: 10.1016/s0076-6879(00)27291-1. [DOI] [PubMed] [Google Scholar]
- Pickett F. B., Meeks-Wagner D. R. Seeing double: appreciating genetic redundancy. Plant Cell. 1995;71(1):1347–1356. doi: 10.1105/tpc.7.9.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponting C. P., Aravind L. START: a lipid-binding domain in StAR, HD-ZIP and signalling proteins. Trends Biochem. Sci. 1999;241(1):130–132. doi: 10.1016/s0968-0004(99)01362-6. [DOI] [PubMed] [Google Scholar]
- Puente P., Wei N., Deng X. W. Combinatorial interplay of promoter elements constitutes the minimal determinants for light and developmental control of gene expression in Arabidopsis. EMBO J. 1996;151(1):3732–3743. [PMC free article] [PubMed] [Google Scholar]
- Putterill J., Robson F., Lee K. J., Simon R., Coupland G. The CONSTANS gene of Arabidopsis promotes flowering and encodes a protein showing similarities to zinc finger transcription factors. Cell. 1995;801(1):847–857. doi: 10.1016/0092-8674(95)90288-0. [DOI] [PubMed] [Google Scholar]
- Pysh L. D., Wysocka-Diller J. W., Camilleri C., Bouchez D., Benfey P. N. The GRAS gene family in Arabidopsis: sequence characterization and basic expression analysis of the SCARECROW-LIKE genes. Plant J. 1999;181(1):111–119. doi: 10.1046/j.1365-313x.1999.00431.x. [DOI] [PubMed] [Google Scholar]
- Quackenbush J. Computational analysis of microarray data. Nat. Rev. Genet. 2001;21(1):418–427. doi: 10.1038/35076576. [DOI] [PubMed] [Google Scholar]
- Quattrocchio F., Wing J., van der Woude K., Souer E., de Vetten N., Mol J., Koes R. Molecular analysis of the anthocyanin2 gene of petunia and its role in the evolution of flower color. Plant Cell. 1999;111(1):1433–1444. doi: 10.1105/tpc.11.8.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raamsdonk L. M., Teusink B., Broadhurst D., Zhang N., Hayes A., Walsh M. C., Berden J. A., Brindle K. M., Kell D. B., Rowland J. J. A functional genomics strategy that uses metabolome data to reveal the phenotype of silent mutations. Nature Biotechnol. 2001;191(1):45–50. doi: 10.1038/83496. [DOI] [PubMed] [Google Scholar]
- Rabinowicz P. D., Braun E. L., Wolfe A. D., Bowen B., Grotewold E. Maize R2R3 Myb genes: Sequence analysis reveals amplification in the higher plants. Genetics. 1999;1531(1):427–444. doi: 10.1093/genetics/153.1.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachez C., Freedman L. P. Mediator complexes and transcription. Curr. Opin. Cell. Biol. 2001;131(1):274–280. doi: 10.1016/s0955-0674(00)00209-x. [DOI] [PubMed] [Google Scholar]
- Raventos D., Skriver K., Schlein M., Karnahl K., Rogers S. W., Rogers J. C., Mundy J. HRT, a novel zinc finger, transcriptional repressor from barley. J. Biol. Chem. 1998;2731(1):23313–23320. doi: 10.1074/jbc.273.36.23313. [DOI] [PubMed] [Google Scholar]
- Raychaudhuri S., Sutphin P. D., Chang J. T., Altman R. B. Basic microarray analysis: grouping and feature selection. Trends Biotech. 2001;191(1):189–193. doi: 10.1016/s0167-7799(01)01599-2. [DOI] [PubMed] [Google Scholar]
- Rea S., Eisenhaber F., O'Carroll D., Strahl B. D., Sun Z. W., Schmid M., Opravil S., Mechtler K., Ponting C. P., Allis C. D. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000;4061(1):593–599. doi: 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- Reed J. W. Roles and activities of Aux/IAA proteins in Arabidopsis. Trends Plant Sci. 2001;61(1):420–425. doi: 10.1016/s1360-1385(01)02042-8. [DOI] [PubMed] [Google Scholar]
- Regad F., Lebas M., Lescure B. Interstitial telomeric repeats within the Arabidopsis thaliana genome. J. Mol. Biol. 1994;2391(1):163–169. doi: 10.1006/jmbi.1994.1360. [DOI] [PubMed] [Google Scholar]
- Reid J. L., Iyer V. R., Brown P. O., Struhl K. Coordinate regulation of yeast ribosomal protein genes is associated with targeted recruitment of Esa1 histone acetylase. Mol. Cell. 2000;61(1):1297–1307. doi: 10.1016/s1097-2765(00)00128-3. [DOI] [PubMed] [Google Scholar]
- Ren B., Robert F., Wyrick J. J., Aparicio O., Jennings E. G., Simon I., Zeitlinger J., Schreiber J., Hannett N., Kanin E. Genome-wide location and function of DNA binding proteins. Science. 2000;2901(1):2306–2309. doi: 10.1126/science.290.5500.2306. [DOI] [PubMed] [Google Scholar]
- Reymond P., Weber H., Damond M., Farmer E. E. Differential gene expression in response to mechanical wounding and insect feeding in Arabidopsis. Plant Cell. 2000;121(1):707–720. doi: 10.1105/tpc.12.5.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice J. C., Allis C. D. Histone methylation versus histone acetylation: new insights into epigenetic regulation. Curr. Opin. Cell. Biol. 2001;131(1):263–273. doi: 10.1016/s0955-0674(00)00208-8. [DOI] [PubMed] [Google Scholar]
- Richards D. E., Peng J., Harberd N. P. Plant GRAS and metazoan STATs: one family? Bioessays. 2000;221(1):573–577. doi: 10.1002/(SICI)1521-1878(200006)22:6<573::AID-BIES10>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Richmond T., Somerville S. Chasing the dream: plant EST microarrays. Curr. Opin. Plant. Biol. 2000;31(1):108–116. doi: 10.1016/s1369-5266(99)00049-7. [DOI] [PubMed] [Google Scholar]
- Riechmann J. L., Heard J., Martin G., Reuber L., Jiang C., Keddie J., Adam L., Pineda O., Ratcliffe O. J., Samaha R. R. Arabidopsis transcription factors: genome-wide comparative analysis among eukaryotes. Science. 2000;2901(1):2105–2110. doi: 10.1126/science.290.5499.2105. [DOI] [PubMed] [Google Scholar]
- Riechmann J. L., Krizek B. A., Meyerowitz E. M. Dimerization specificity of Arabidopsis MADS domain homeotic proteins APETALA1, APETALA3, PISTILLATA, and AGAMOUS. Proc. Natl. Acad. Sci. U. S. A. 1996a;931(1):4793–4798. doi: 10.1073/pnas.93.10.4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riechmann J. L., Meyerowitz E. M. Determination of floral organ identity by Arabidopsis MADS domain homeotic proteins AP1, AP3, PI, and AG is independent of their DNA-binding specificity. Mol. Biol. Cell. 1997a;81(1):1243–1259. doi: 10.1091/mbc.8.7.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riechmann J. L., Meyerowitz E. M. MADS domain proteins in plant development. Biol. Chem. 1997b;3781(1):1079–1101. [PubMed] [Google Scholar]
- Riechmann J. L., Meyerowitz E. M. The AP2/EREBP family of plant transcription factors. Biol. Chem. 1998;3791(1):633–646. doi: 10.1515/bchm.1998.379.6.633. [DOI] [PubMed] [Google Scholar]
- Riechmann J. L., Ratcliffe O. J. A genomic perspective on plant transcription factors. Curr. Opin. Plant. Biol. 2000;31(1):423–434. doi: 10.1016/s1369-5266(00)00107-2. [DOI] [PubMed] [Google Scholar]
- Riechmann J. L., Wang M., Meyerowitz E. M. DNA-binding properties of Arabidopsis MADS domain homeotic proteins APETALA1, APETALA3, PISTILLATA and AGAMOUS. Nucleic Acids Res. 1996b;241(1):3134–3141. doi: 10.1093/nar/24.16.3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robison K., McGuire A. M., Church G. M. A comprehensive library of DNA-binding site matrices for 55 proteins applied to the complete Escherichia coli K-12 genome. J. Mol. Biol. 1998;2841(1):241–254. doi: 10.1006/jmbi.1998.2160. [DOI] [PubMed] [Google Scholar]
- Ross-Macdonald P., Coelho P. S., Roemer T., Agarwal S., Kumar A., Jansen R., Cheung K. H., Sheehan A., Symoniatis D., Umansky L. Large-scale analysis of the yeast genome by transposon tagging and gene disruption. Nature. 1999;4021(1):413–418. doi: 10.1038/46558. [DOI] [PubMed] [Google Scholar]
- Rossi V., Hartings H., Motto M. Identification and characterization of an RPD3 homologue from maize (Zea mays L. ) that is able to complement an rpd3 null mutant of Saccharomyces cerevisiae. Mol. Gen. Genet. 1998;2581(1):288–296. doi: 10.1007/s004380050733. [DOI] [PubMed] [Google Scholar]
- Rossini L., Cribb L., Martin D. J., Langdale J. A. The maize golden2 gene defines a novel class of transcriptional regulators in plants. Plant Cell. 2001;131(1):1231–1244. doi: 10.1105/tpc.13.5.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth F. P., Hughes J. D., Estep P. W., Church G. M. Finding DNA regulatory motifs within unaligned noncoding sequences clustered by whole-genome mRNA quantitation. Nature Biotechnol. 1998;161(1):939–945. doi: 10.1038/nbt1098-939. [DOI] [PubMed] [Google Scholar]
- Roth S. Y., Denu J. M., Allis C. D. Histone acetyltransferases. Annu. Rev. Biochem. 2001;701(1):81–120. doi: 10.1146/annurev.biochem.70.1.81. [DOI] [PubMed] [Google Scholar]
- Ruan Y., Gilmore J., Conner T. Towards Arabidopsis genome analysis: monitoring expression profiles of 1400 genes using cDNA microarrays. Plant J. 1998;151(1):821–833. doi: 10.1046/j.1365-313x.1998.00254.x. [DOI] [PubMed] [Google Scholar]
- Sablowski R. W., Meyerowitz E. M. A homolog of NO APICAL MERISTEM is an immediate target of the floral homeotic genes APETALA3/PISTILLATA. Cell. 1998;921(1):93–103. doi: 10.1016/s0092-8674(00)80902-2. [DOI] [PubMed] [Google Scholar]
- Sakai H., Honma T., Aoyama T., Sato S., Kato T., Tabata S., Oka A. ARR1, a transcription factor for genes immediately responsive to cytokinins. Science. 2001;2941(1):1519–1521. doi: 10.1126/science.1065201. [DOI] [PubMed] [Google Scholar]
- Salanoubat M., Lemcke K., Rieger M., Ansorge W., Unseld M., Fartmann B., Valle G., Blocker H., Perez-Alonso M., Obermaier B. Sequence and analysis of chromosome 3 of the plant Arabidopsis thaliana. Nature. 2000;4081(1):820–822. doi: 10.1038/35048706. [DOI] [PubMed] [Google Scholar]
- Salgado H., Santos-Zavaleta A., Gama-Castro S., Millan-Zarate D., Diaz-Peredo E., Sanchez-Solano F., Perez-Rueda E., Bonavides-Martinez C., Collado-Vides J. RegulonDB (version 3.2): transcriptional regulation and operon organization in Escherichia coli K-12. Nucleic Acids Res. 2001;291(1):72–74. doi: 10.1093/nar/29.1.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samach A., Onouchi H., Gold S. E., Ditta G. S., Schwarz-Sommer Z., Yanofsky M. F., Coupland G. Distinct roles of CONSTANS target genes in reproductive development of Arabidopsis. Science. 2000;2881(1):1613–1616. doi: 10.1126/science.288.5471.1613. [DOI] [PubMed] [Google Scholar]
- Sánchez-García I., Rabbitts T. H. The LIM domain: a new structural motif found in zinc-finger-like proteins. Trends Genet. 1994;101(1):315–320. doi: 10.1016/0168-9525(94)90034-5. [DOI] [PubMed] [Google Scholar]
- SanMiguel P., Tikhonov A., Jin Y. K., Motchoulskaia N., Zakharov D., Melake-Berhan A., Springer P. S., Edwards K. J., Lee M., Avramova Z. Nested retrotransposons in the intergenic regions of the maize genome. Science. 1996;2741(1):765–768. doi: 10.1126/science.274.5288.765. [DOI] [PubMed] [Google Scholar]
- Savard L., Li P., Strauss S. H., Chase M. W., Michaud M., Bousquet J. Chloroplast and nuclear gene sequences indicate late Pennsylvanian time for the last common ancestor of extant seed plants. Proc. Natl. Acad. Sci. U. S. A. 1994;911(1):5163–5167. doi: 10.1073/pnas.91.11.5163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawant S., Singh P. K., Madanala R., Tuli R. Designing an artificial expression cassette for the high-level expression of transgenes in plants. Theor. Appl. Genet. 2001;1021(1):635–644. [Google Scholar]
- Schaffer R., Landgraf J., Accerbi M., Simon V. V., Larson M., Wisman E. Microarray analysis of diurnal and circadian-regulated genes in Arabidopsis. Plant Cell. 2001;131(1):113–123. doi: 10.1105/tpc.13.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauser L., Roussis A., Stiller J., Stougaard J. A plant regulator controlling development of symbiotic root nodules. Nature. 1999;4021(1):191–195. doi: 10.1038/46058. [DOI] [PubMed] [Google Scholar]
- Schena M., Shalon D., Davis R. W., Brown P. O. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;2701(1):467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- Schenk P. M., Kazan K., Wilson I., Anderson J. P., Richmond T., Somerville S. C., Manners J. M. Coordinated plant defense responses in Arabidopsis revealed by microarray analysis. Proc. Natl. Acad. Sci. U. S. A. 2000;971(1):11655–11660. doi: 10.1073/pnas.97.21.11655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherf M., Klingenhoff A., Frech K., Quandt K., Schneider R., Grote K., Frisch M., Gailus-Durner V., Seidel A., Brack-Werner R. First pass annotation of promoters on human chromosome 22. Genome Res. 2001;111(1):333–340. doi: 10.1101/gr.154601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiefthaler U., Balasubramanian S., Sieber P., Chevalier D., Wisman E., Schneitz K. Molecular analysis of NOZZLE, a gene involved in pattern formation and early sporogenesis during sex organ development in Arabidopsis thaliana. Proc. Natl. Acad. Sci. U. S. A. 1999;961(1):11664–11669. doi: 10.1073/pnas.96.20.11664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze A., Downward J. Navigating gene expression using microarrays - a technology review. Nature Cell Biol. 2001;31(1):E190–E195. doi: 10.1038/35087138. [DOI] [PubMed] [Google Scholar]
- Schumacher B., Hofmann K., Boulton S., Gartner A. The C. elegans homolog of the p53 tumor supressor is required for DNA damage-induced apoptosis. Curr. Biol. 2001;111(1):1722–1727. doi: 10.1016/s0960-9822(01)00534-6. [DOI] [PubMed] [Google Scholar]
- Scott M. P. Development: the natural history of genes. Cell. 2000;1001(1):27–40. doi: 10.1016/s0092-8674(00)81681-5. [DOI] [PubMed] [Google Scholar]
- Seki M., Narusaka M., Abe H., Kasuga M., Yamaguchi-Shinozaki K., Carninci P., Hayashizaki Y., Shinozaki K. Monitoring the expression pattern of 1300 Arabidopsis genes under drought and cold stresses by using a full-length cDNA microarray. Plant Cell. 2001;131(1):61–72. doi: 10.1105/tpc.13.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah K., Gadella T. W. J., van Erp H., Hecht V., de Vries S. C. Subcellular localization and oligomerization of the Arabidopsis thaliana Somatic Embryogenesis Receptor Kinase 1 protein. J. Mol. Biol. 2001;3091(1):641–655. doi: 10.1006/jmbi.2001.4706. [DOI] [PubMed] [Google Scholar]
- Sherlock G. Analysis of large-scale gene expression data. Curr. Opin. Immunol. 2000;121(1):201–205. doi: 10.1016/s0952-7915(99)00074-6. [DOI] [PubMed] [Google Scholar]
- Sieburth L. E., Meyerowitz E. M. Molecular dissection of the AGAMOUS control region shows that cis elements for spatial regulation are located intragenically. Plant Cell. 1997;91(1):355–365. doi: 10.1105/tpc.9.3.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon I., Barnett J., Hannett N., Harbison C. T., Rinaldi N. J., Volkert T. L., Wyrick J. J., Zeitlinger J., Gifford D. K., Jaakkola T. S. Serial regulation of transcriptional regulators in the yeast cell cycle. Cell. 2001;1061(1):697–708. doi: 10.1016/s0092-8674(01)00494-9. [DOI] [PubMed] [Google Scholar]
- Simon R., Igeño M. I., Coupland G. Activation of floral meristem identity genes in Arabidopsis. Nature. 1996;3841(1):59–62. doi: 10.1038/384059a0. [DOI] [PubMed] [Google Scholar]
- Singer T., Yordan C., Martienssen R. A. Robertson's Mutator transposons in A. thaliana are regulated by the chromatin-remodeling gene Decrease in DNA Methylation (DDM1). Genes Dev. 2001;151(1):591–602. doi: 10.1101/gad.193701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh K. B. Transcriptional regulation in plants: the importance of combinatorial control. Plant Physiol. 1998;1181(1):1111–1120. doi: 10.1104/pp.118.4.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smothers J. F., Henikoff S. The HP1 chromo shadow domain binds a consensus sequence pentamer. Curr. Biol. 2000;101(1):27–30. doi: 10.1016/s0960-9822(99)00260-2. [DOI] [PubMed] [Google Scholar]
- Sommerville J. Activities of cold-shock domain proteins in translation control. Bioessays. 1999;211(1):319–325. doi: 10.1002/(SICI)1521-1878(199904)21:4<319::AID-BIES8>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Spellman P. T., Sherlock G., Zhang M. Q., Iyer V. R., Anders K., Eisen M. B., Brown P. O., Botstein D., Futcher B. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol. Biol. Cell. 1998;91(1):3273–3297. doi: 10.1091/mbc.9.12.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spelt C., Quattrocchio F., Mol J. N., Koes R. anthocyanin1 of petunia encodes a basic helix-loop-helix protein that directly activates transcription of structural anthocyanin genes. Plant Cell. 2000;121(1):1619–1632. doi: 10.1105/tpc.12.9.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillane C., MacDougall C., Stock C., Kohler C., Vielle-Calzada J. P., Nunes S. M., Grossniklaus U., Goodrich J. Interaction of the Arabidopsis polycomb group proteins FIE and MEA mediates their common phenotypes. Curr. Biol. 2000;101(1):1535–1538. doi: 10.1016/s0960-9822(00)00839-3. [DOI] [PubMed] [Google Scholar]
- Sterner D. E., Berger S. L. Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev. 2000;641(1):435–459. doi: 10.1128/mmbr.64.2.435-459.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockinger E. J., Mao Y., Regier M. K., Triezenberg S. J., Thomashow M. F. Transcriptional adaptor and histone acetyltransferase proteins in Arabidopsis and their interactions with CBF1, a transcriptional activator involved in cold-regulated gene expression. Nucleic Acids Res. 2001;291(1):1524–1533. doi: 10.1093/nar/29.7.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone S. L., Kwong L. W., Yee K. M., Pelletier J., Lepiniec L., Fischer R. L., Goldberg R. B., Harada J. J. LEAFY COTYLEDON2 encodes a B3 domain transcription factor that induces embryo development. Proc. Natl. Acad. Sci. U. S. A. 2001;981(1):11806–11811. doi: 10.1073/pnas.201413498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stracke R., Werber M., Weisshaar B. The R2R3-MYB gene family in Arabidopsis thaliana. Curr. Opin. Plant. Biol. 2001;41(1):447–456. doi: 10.1016/s1369-5266(00)00199-0. [DOI] [PubMed] [Google Scholar]
- Strahl B. D., Allis C. D. The language of covalent histone modifications. Nature. 2000;4031(1):41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- Struhl K. Fundamentally different logic of gene regulation in eukaryotes and prokaryotes. Cell. 1999;981(1):1–4. doi: 10.1016/S0092-8674(00)80599-1. [DOI] [PubMed] [Google Scholar]
- Subramaniam R., Desveaux D., Spickler C., Michnick S. W., Brisson N. Direct visualization of protein interactions in plant cells. Nature Biotechnol. 2001;191(1):769–772. doi: 10.1038/90831. [DOI] [PubMed] [Google Scholar]
- Svensson M. E., Johannesson H., Engstrom P. The LAMB1 gene from the clubmoss, Lycopodium annotinum, is a divergent MADS-box gene, expressed specifically in sporogenic structures. Gene. 2000;2531(1):31–43. doi: 10.1016/s0378-1119(00)00243-2. [DOI] [PubMed] [Google Scholar]
- Szathmáry E., Jordán F., Pál C. Can genes explain biological complexity? Science. 2001;2921(1):1315–1316. doi: 10.1126/science.1060852. [DOI] [PubMed] [Google Scholar]
- Tabata S., Kaneko T., Nakamura Y., Kotani H., Kato T., Asamizu E., Miyajima N., Sasamoto S., Kimura T., Hosouchi T. Sequence and analysis of chromosome 5 of the plant Arabidopsis thaliana. Nature. 2000;4081(1):823–826. doi: 10.1038/35048507. [DOI] [PubMed] [Google Scholar]
- Takatsuji H. Zinc-finger transcription factors in plants. Cell. Mol. Life Sci. 1998;541(1):582–596. doi: 10.1007/s000180050186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takatsuji H. Zinc-finger proteins: the classical zinc finger emerges in contemporary plant science. Plant Mol. Biol. 1999;391(1):1073–1078. doi: 10.1023/a:1006184519697. [DOI] [PubMed] [Google Scholar]
- Takeda Y., Hatano S., Sentoku N., Matsuoka M. Homologs of animal eyes absent (eya) genes are found in higher plants. Mol. Gen. Genet. 1999;2621(1):131–138. doi: 10.1007/s004380051067. [DOI] [PubMed] [Google Scholar]
- Tansey W. P. Transcriptional activation: risky business. Genes Dev. 2001;151(1):1045–1050. doi: 10.1101/gad.896501. [DOI] [PubMed] [Google Scholar]
- Tautz D. Evolution of transcriptional regulation. Curr. Opin. Genet. Dev. 2000;101(1):575–579. doi: 10.1016/s0959-437x(00)00130-1. [DOI] [PubMed] [Google Scholar]
- Tavares R., Aubourg S., Lecharny A., Kreis M. Organization and structural evolution of four multigene families in Arabidopsis thaliana: AtLCAD, AtLGT, AtMYST, and AtHD-GL2. Plant Mol. Biol. 2000;421(1):703–717. doi: 10.1023/a:1006368316413. [DOI] [PubMed] [Google Scholar]
- Tavazoie S., Hughes J. D., Campbell M. J., Cho R. J., Church G. M. Systematic determination of genetic network architecture. Nature Genet. 1999;221(1):281–285. doi: 10.1038/10343. [DOI] [PubMed] [Google Scholar]
- Tepperman J. M., Zhu T., Chang H. S., Wang X., Quail P. H. Multiple transcription-factor genes are early targets of phytochrome A signaling. Proc. Natl. Acad. Sci. U. S. A. 2001;981(1):9437–9442. doi: 10.1073/pnas.161300998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thacker C., Marra M. A., Jones A., Baillie D. L., Rose A. M. Functional genomics in Caenorhabditis elegans: an approach involving comparisons of sequences from related nematodes. Genome Res. 1999;91(1):348–359. [PMC free article] [PubMed] [Google Scholar]
- Thatcher J. W., Shaw J. M., Dickinson W. J. Marginal fitness contributions of nonessential genes in yeast. Proc. Natl. Acad. Sci. U. S. A. 1998;951(1):253–257. doi: 10.1073/pnas.95.1.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The C. elegans Sequencing Consortium Genome sequence of the nematode C. elegans: a platform for investigating biology. Science. 1998;2821(1):2012–2018. doi: 10.1126/science.282.5396.2012. [DOI] [PubMed] [Google Scholar]
- Theissen G. Development of floral organ identity: stories from the MADS house. Curr. Opin. Plant. Biol. 2001;41(1):75–85. doi: 10.1016/s1369-5266(00)00139-4. [DOI] [PubMed] [Google Scholar]
- Theiben G., Becker A., Di Rosa A., Kanno A., Kim J. T., Munster T., Winter K. U., Saedler H. A short history of MADS-box genes in plants. Plant Mol. Biol. 2000;421(1):115–149. [PubMed] [Google Scholar]
- Theologis A., Ecker J. R., Palm C. J., Federspiel N. A., Kaul S., White O., Alonso J., Altafi H., Araujo R., Bowman C. L. Sequence and analysis of chromosome 1 of the plant Arabidopsis thaliana. Nature. 2000;4081(1):816–820. doi: 10.1038/35048500. [DOI] [PubMed] [Google Scholar]
- Thieffry D., Huerta A. M., Pérez-Rueda E., Collado-Vides J. From specific gene regulation to genomic networks: a global analysis of transcriptional regulation in Escherichia coli. Bioessays. 1998;201(1):433–440. doi: 10.1002/(SICI)1521-1878(199805)20:5<433::AID-BIES10>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Thomas J. H. Thinking about genetic redundancy. Trends Genet. 1993;91(1):395–399. doi: 10.1016/0168-9525(93)90140-d. [DOI] [PubMed] [Google Scholar]
- Tian L., Chen Z. J. Blocking histone deacetylation in Arabidopsis induces pleiotropic effects on plant gene regulation and development. Proc. Natl. Acad. Sci. U. S. A. 2001;981(1):200–205. doi: 10.1073/pnas.011347998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tupler R., Perini G., Green M. R. Expressing the human genome. Nature. 2001;4091(1):832–833. doi: 10.1038/35057011. [DOI] [PubMed] [Google Scholar]
- Uetz P., Giot L., Cagney G., Mansfield T. A., Judson R. S., Knight J. R., Lockshon D., Narayan V., Srinivasan M., Pochart P. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature. 2000;4031(1):623–627. doi: 10.1038/35001009. [DOI] [PubMed] [Google Scholar]
- Urnov F. D., Wolffe A. P. Chromatin remodeling and transcriptional activation: the cast (in order of appearance). Oncogene. 2001;201(1):2991–3006. doi: 10.1038/sj.onc.1204323. [DOI] [PubMed] [Google Scholar]
- van der Knaap E., Kim J. H., Kende H. A novel gibberellin-induced gene from rice and its potential regulatory role in stem growth. Plant Physiol. 2000;1221(1):695–705. doi: 10.1104/pp.122.3.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel B., Delrow J., Henikoff S. Chromatin profiling using targeted DNA adenine methyltransferase. Nature Genet. 2001;271(1):304–308. doi: 10.1038/85871. [DOI] [PubMed] [Google Scholar]
- van Steensel B., Henikoff S. Identification of in vivo DNA targets of chromatin proteins using tethered Dam methyltransferase. Nature Biotechnol. 2000;181(1):424–428. doi: 10.1038/74487. [DOI] [PubMed] [Google Scholar]
- Veenstra G. J. C., Wolffe A. P. Gene-selective developmental roles of general trasncription factors. Trends Biochem. Sci. 2001;261(1):665–671. doi: 10.1016/s0968-0004(01)01970-3. [DOI] [PubMed] [Google Scholar]
- Venter J. C., Adams M. D., Myers E. W., Li P. W., Mural R. J., Sutton G. G., Smith H. O., Yandell M., Evans C. A., Holt R. A. The sequence of the human genome. Science. 2001;2911(1):1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- Vicente-Carbajosa J., Moose S. P., Parsons R. L., Schmidt R. J. A maize zinc-finger protein binds the prolamin box in zein gene promoters and interacts with the basic leucine zipper transcriptional activator Opaque2. Proc. Natl. Acad. Sci. U. S. A. 1997;941(1):7685–7690. doi: 10.1073/pnas.94.14.7685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal M. A biological atlas of functional maps. Cell. 2001;1041(1):333–339. doi: 10.1016/s0092-8674(01)00221-5. [DOI] [PubMed] [Google Scholar]
- Vignali M., Hassan A. H., Neely K. E., Workman J. L. ATP-dependent chromatin remodeling complexes. Mol. Cell. Biol. 2000;201(1):1899–1910. doi: 10.1128/mcb.20.6.1899-1910.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vision T. J., Brown D. G., Tanksley S. D. The origins of genomic duplications in Arabidopsis. Science. 2000;2901(1):2114–2117. doi: 10.1126/science.290.5499.2114. [DOI] [PubMed] [Google Scholar]
- Vo N., Goodman R. H. CREB-binding protein and p300 in transcriptional regulation. J. Biol. Chem. 2001;2761(1):13505–13508. doi: 10.1074/jbc.R000025200. [DOI] [PubMed] [Google Scholar]
- Wagner D., Sablowski R. W., Meyerowitz E. M. Transcriptional activation of APETALA1 by LEAFY. Science. 1999;2851(1):582–584. doi: 10.1126/science.285.5427.582. [DOI] [PubMed] [Google Scholar]
- Walhout A. J., Vidal M. Protein interaction maps for model organisms. Nat. Rev. Mol. Cell Biol. 2001;21(1):55–62. doi: 10.1038/35048107. [DOI] [PubMed] [Google Scholar]
- Wang D. Y., Kumar S., Hedges S. B. Divergence time estimates for the early history of animal phyla and the origin of plants, animals and fungi. Proc. R. Soc. Lond. B Biol. Sci. 1999a;2661(1):163–171. doi: 10.1098/rspb.1999.0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Ma L-G., Li J-M., Zhao H-Y., Deng X. W. Direct interaction of Arabidopsis cryptochromes with COP1 in mediation of photomorphogenic development. Science. 2001;2941(1):154–158. doi: 10.1126/science.1063630. [DOI] [PubMed] [Google Scholar]
- Wang R., Guegler K., LaBrie S. T., Crawford N. M. Genomic analysis of a nutrient response in Arabidopsis reveals diverse expression patterns and novel metabolic and potential regulatory genes induced by nitrate. Plant Cell. 2000;121(1):1491–1509. doi: 10.1105/tpc.12.8.1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R. L., Stec A., Hey J., Lukens L., Doebley J. The limits of selection during maize domestication. Nature. 1999b;3981(1):236–239. doi: 10.1038/18435. [DOI] [PubMed] [Google Scholar]
- Wasserman W. W., Palumbo M., Thompson W., Fickett J. W., Lawrence C. E. Human-mouse genome comparisons to locate regulatory sites. Nature Genet. 2000;261(1):225–228. doi: 10.1038/79965. [DOI] [PubMed] [Google Scholar]
- Weigel D., Alvarez J., Smyth D. R., Yanofsky M. F., Meyerowitz E. M. LEAFY controls floral meristem identity in Arabidopsis. Cell. 1992;691(1):843–859. doi: 10.1016/0092-8674(92)90295-n. [DOI] [PubMed] [Google Scholar]
- Weigel R. R., Bäuscher C., Pfitzner A. J. P., Pfitzner U. M. NIMIN-1, NIMIN-2 and NIMIN-3, members of a novel family of proteins from Arabidopsis that interact with NPR1/NIM1, a key regulator of systemic acquired resistance in plants. Plant Mol. Biol. 2001;461(1):143–160. doi: 10.1023/a:1010652620115. [DOI] [PubMed] [Google Scholar]
- Wesley S. V., Helliwell C. A., Smith N. A., Wang M., Rouse D. T., Liu Q., Gooding P. S., Singh S. P., Abbott D., Stoutjesdijk P. A. Construct design for efficient, effective and high-throughput gene silencing in plants. Plant J. 2001;271(1):581–590. doi: 10.1046/j.1365-313x.2001.01105.x. [DOI] [PubMed] [Google Scholar]
- White R. J. Gene transcription. Mechanisms and control. 2001. (Oxford: Blackwell Science)
- Wilkinson M. F., Shyu A-B. Multifunctional regulatory proteins that control gene expression in both the nucleus and the cytoplasm. Bioessays. 2001;231(1):775–787. doi: 10.1002/bies.1113. [DOI] [PubMed] [Google Scholar]
- Windhövel A., Hein I., Dabrowa R., Stockhaus J. Characterization of a novel class of plant homeodomain proteins that bind to the C4 phosphoenolpyruvate carboxylase gene of Flaveria trinervia. Plant Mol. Biol. 2001;451(1):201–214. doi: 10.1023/a:1006450005648. [DOI] [PubMed] [Google Scholar]
- Winzeler E. A., Shoemaker D. D., Astromoff A., Liang H., Anderson K., Andre B., Bangham R., Benito R., Boeke J. D., Bussey H. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 1999;2851(1):901–906. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- Wolfsberg T. G., Gabrielian A. E., Campbell M. J., Cho R. J., Spouge J. L., Landsman D. Candidate regulatory sequence elements for cell cycle-dependent transcription in Saccharomyces cerevisiae. Genome Res. 1999;91(1):775–792. [PMC free article] [PubMed] [Google Scholar]
- Wright F. A., Lemon W., Zhao W. D., Sears R., Zhuo D., Wang J-P., Yang H-Y., Baer T., Stredney D., Spitzner J. A draft annotation and overview of the human genome. 2001;2 doi: 10.1186/gb-2001-2-7-research0025. Genome Biol. research0025.0021-0025.0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K., Malik K., Tian L., Brown D., Miki B. Functional analysis of a RPD3 histone deacetylase homologue in Arabidopsis thaliana. Plant Mol. Biol. 2000a;441(1):167–176. doi: 10.1023/a:1006498413543. [DOI] [PubMed] [Google Scholar]
- Wu K., Tian L., Malik K., Brown D., Miki B. Functional analysis of HD2 histone deacetylase homologues in Arabidopsis thaliana. Plant J. 2000b;221(1):19–27. doi: 10.1046/j.1365-313x.2000.00711.x. [DOI] [PubMed] [Google Scholar]
- Xie Q., Frugis G., Colgan D., Chua N. H. Arabidopsis NAC1 transduces auxin signal downstream of TIR1 to promote lateral root development. Genes Dev. 2000;141(1):3024–3036. doi: 10.1101/gad.852200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T., Purcell M., Zucchi P., Helentjaris T., Bogorad L. TRM1, a YY1-like suppressor of rbcS-m3 expression in maize mesophyll cells. Proc. Natl. Acad. Sci. U. S. A. 2001;981(1):2295–2300. doi: 10.1073/pnas.041610098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadegari R., Kinoshita T., Lotan O., Cohen G., Katz A., Choi Y., Nakashima K., Harada J. J., Goldberg R. B., Fischer R. L. Mutations in the FIE and MEA genes that encode interacting polycomb proteins cause parent-of-origin effects on seed development by distinct mechanisms. Plant Cell. 2000;121(1):2367–2382. doi: 10.1105/tpc.12.12.2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi R., Nakamura M., Mochizuki N., Kay S. A., Nagatani A. Light-dependent translocation of a phytochrome B-GFP fusion protein to the nucleus in transgenic Arabidopsis. J. Cell Biol. 1999;1451(1):437–445. doi: 10.1083/jcb.145.3.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagisawa S., Schmidt R. J. Diversity and similarity among recognition sequences of Dof transcription factors. Plant J. 1999;171(1):209–214. doi: 10.1046/j.1365-313x.1999.00363.x. [DOI] [PubMed] [Google Scholar]
- Yoshida N., Yanai Y., Chen L., Kato Y., Hiratsuka J., Miwa T., Sung Z. R., Takahashi S. EMBRYONIC FLOWER2, a novel polycomb group protein homolog, mediates shoot development and flowering in Arabidopsis. Plant Cell. 2001;131(1):2471–2481. doi: 10.1105/tpc.010227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young J. C., Krysan P. J., Sussman M. R. Efficient screening of Arabidopsis T-DNA insertion lines using degenerate primers. Plant Physiol. 2001;1251(1):513–518. doi: 10.1104/pp.125.2.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young R. A. Biomedical discovery with DNA arrays. Cell. 2000;1021(1):9–15. doi: 10.1016/s0092-8674(00)00005-2. [DOI] [PubMed] [Google Scholar]
- Yu D., Chen C., Chen Z. Evidence for an important role of WRKY DNA binding proteins in the regulation of NPR1 gene expression. Plant Cell. 2001;131(1):1527–1539. doi: 10.1105/TPC.010115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuh C. H., Bolouri H., Davidson E. H. Genomic cis-regulatory logic: experimental and computational analysis of a sea urchin gene. Science. 1998;2791(1):1896–1902. doi: 10.1126/science.279.5358.1896. [DOI] [PubMed] [Google Scholar]
- Yuh C. H., Bolouri H., Davidson E. H. Cis-regulatory logic in the endo16 gene: switching from a specification to a differentiation mode of control. Development. 2001;1281(1):617–629. doi: 10.1242/dev.128.5.617. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Fan W., Kinkema M., Li X., Dong X. Interaction of NPR1 with basic leucine zipper protein transcription factors that bind sequences required for salicylic acid induction of the PR-1 gene. Proc. Natl. Acad. Sci. U. S. A. 1999;961(1):6523–6528. doi: 10.1073/pnas.96.11.6523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong H., McCord R., Vershon A. K. Identification of target sites of the a2-Mcm1 repressor complex in the yeast genome. Genome Res. 1999;91(1):1040–1047. doi: 10.1101/gr.9.11.1040. [DOI] [PubMed] [Google Scholar]
- Zhou D. X. Regulatory mechanism of plant gene transcription by GT-elements and GT-factors. Trends Plant Sci. 1999;41(1):210–214. doi: 10.1016/s1360-1385(99)01418-1. [DOI] [PubMed] [Google Scholar]
- Zhou D. X., Bisanz-Seyer C., Mache R. Molecular cloning of a small DNA binding protein with specificity for a tissue-specific negative element within the rps1 promoter. Nucleic Acids Res. 1995;231(1):1165–1169. doi: 10.1093/nar/23.7.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J., Tang X., Martin G. B. The Pto kinase conferring resistance to tomato bacterial speck disease interacts with proteins that bind a cis-element of pathogenesis-related genes. EMBO J. 1997;161(1):3207–3218. doi: 10.1093/emboj/16.11.3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J-M., Trifa Y., Silva H., Pontier D., Lam E., Shah J., Klessig D. F. NPR1 differentially interacts with members of the TGA/OBF family of transcription factors that bind an element of the PR-1 gene required for induction by salicylic acid. Mol. Plant Microbe Interact. 2000;131(1):191–202. doi: 10.1094/MPMI.2000.13.2.191. [DOI] [PubMed] [Google Scholar]
- Zhu T., Budworth P., Han B., Brown D., Chang H-S., Zou G., Wang X. Toward elucidating the global gene expression patterns of developing Arabidopsis: parallel analysis of 8300 genes by a high-density oligonucleotide probe array. Plant Physiol. Biochem. 2001;391(1):221–242. [Google Scholar]
- Zhu Y., Tepperman J. M., Fairchild C. D., Quail P. H. Phytochrome B binds with greater apparent affinity than phytochrome A to the basic helix-loop-helix factor PIF3 in a reaction requiring the PAS domain of PIF3. Proc. Natl. Acad. Sci. U. S. A. 2000;971(1):13419–13424. doi: 10.1073/pnas.230433797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo J., Chua N-H. Chemical-inducible systems for regulated gene expression of plant genes. Curr. Opin. Biotechnol. 2000;111(1):146–152. doi: 10.1016/s0958-1669(00)00073-2. [DOI] [PubMed] [Google Scholar]