

Abstract
Castellaniella defragrans is a Betaproteobacterium capable of coupling the oxidation of monoterpenes with denitrification. Geraniol dehydrogenase (GeDH) activity was induced during growth with limonene in comparison to growth with acetate. The N-terminal sequence of the purified enzyme directed the cloning of the corresponding open reading frame (ORF), the first bacterial gene for a GeDH (geoA, for geraniol oxidation pathway). The C. defragrans geraniol dehydrogenase is a homodimeric enzyme that affiliates with the zinc-containing benzyl alcohol dehydrogenases in the superfamily of medium-chain-length dehydrogenases/reductases (MDR). The purified enzyme most efficiently catalyzes the oxidation of perillyl alcohol (kcat/Km = 2.02 × 106 M−1 s−1), followed by geraniol (kcat/Km = 1.57 × 106 M−1 s−1). Apparent Km values of <10 μM are consistent with an in vivo toxicity of geraniol above 5 μM. In the genetic vicinity of geoA is a putative aldehyde dehydrogenase that was named geoB and identified as a highly abundant protein during growth with phellandrene. Extracts of Escherichia coli expressing geoB demonstrated in vitro a geranial dehydrogenase (GaDH) activity. GaDH activity was independent of coenzyme A. The irreversible formation of geranic acid allows for a metabolic flux from β-myrcene via linalool, geraniol, and geranial to geranic acid.
INTRODUCTION
A basic reaction in biochemistry is the oxidation of an alcohol to an aldehyde by an oxidoreductase. Alcohol dehydrogenases (ADHs; EC 1.1.1.x) are grouped into long-chain, medium-chain, and short-chain ADHs, according to their sequence length. Members of the medium-chain dehydrogenase/reductase superfamily (41, 45) are characterized by a Rossmann dinucleotide-binding domain (5) and two zinc ions as the structurally and catalytically acting transition metal in the active site (2). The medium-chain alcohol dehydrogenases evolved from a common ancestor into several families, and there is good evidence for different evolutionary rates within the families (45). The catalytic process is well understood (42). In bioconversion reactions, an ADH is usually followed by an aldehyde dehydrogenase (ALDH; EC 1.2.1.x) catalyzing the oxidation of the aldehyde to the corresponding carboxylic acid. The ALDH superfamily is ubiquitous in nature, oxidizing a wide range of aliphatic and aromatic aldehydes (34, 39, 56).
A number of oxidoreductases acting on alcohols with an adjacent carbon-carbon double bond, i.e., the allyl alcohol, have been isolated from different sources and characterized: allyl ADH (EC 1.1.1.54), retinol DH (EC 1.1.1.105), geraniol DH (GeDH; EC 1.1.1.183), coniferyl ADH (EC 1.1.1.194), cinnamyl ADH (EC 1.1.1.195), and farnesol DH (EC 1.1.1.216). The allyl alcohol motif is also present in benzyl alcohol, and therefore many benzyl ADHs, known as aryl ADHs (EC 1.1.1.90), can act on allyl alcohols.
In a Pseudomonas putida isolate, a 3-methyl-2-buten-1-ol dehydrogenase was found to be a benzyl ADH with broad specificity toward allyl and benzyl alcohols (36). Geraniol (3,7-dimethyl-trans-2,6-octadien-1-ol) is a C10 homologue of 3-methyl-2-buten-1-ol and is known to be an intermediate in the anaerobic degradation of β-myrcene by Castellaniella defragrans (7). This betaproteobacterium, originally named Alcaligenes defragrans (29), was isolated with various monoterpenes, natural unsaturated hydrocarbons (C10H16) that can be simply differentiated by their acyclic, monocyclic, or bicyclic structure (Fig. 1) (17). Plants synthesize monoterpenes for thermotolerance or other plant-environment interactions in amounts of over 100 Tg C year−1; thus, they represent an important component in the carbon cycle on earth (33, 51, 54). In insects, monoterpenes are synthesized as pheromones (4, 37, 50). Furthermore, these substances are widely used in the food, flavor, and fragrance industries due to their odorous properties (8). Geraniol exudes a sweet, rose-like scent and is commercially synthesized in amounts of 4,000 Mg year−1 (3).
Fig 1.

Monoterpene and monoterpenoid structures.
In C. defragrans, the linalool dehydratase-isomerase catalyzes the hydration of the acyclic β-myrcene to (S)-(+)-linalool as well as the isomerization to geraniol (7, 35). The formation of geranic acid was observed in vivo and in vitro (24), indicating the presence of dehydrogenases catalyzing the oxidation of the allyl alcohol geraniol to geranic acid, most probably via geranial. So far, the only geraniol dehydrogenases (GeDHs) characterized on the molecular level come from sweet basil, Ocimum basilicum (25), and the astigmatid mite Carpoglyphus lactis (37). An aldehyde dehydrogenase acting on geranial has never been reported. Therefore, we identified and characterized two relevant enzymes: an alcohol dehydrogenase with a remarkably high geraniol affinity and a geranial dehydrogenase, which specifically oxidizes geranial (Fig. 2). Both enzymes were induced in monoterpene-grown cells, and together with the linalool dehydratase-isomerase, they provide the molecular basis for our previous observation, the formation of geranic acid from β-myrcene in cell extracts (24).
Fig 2.
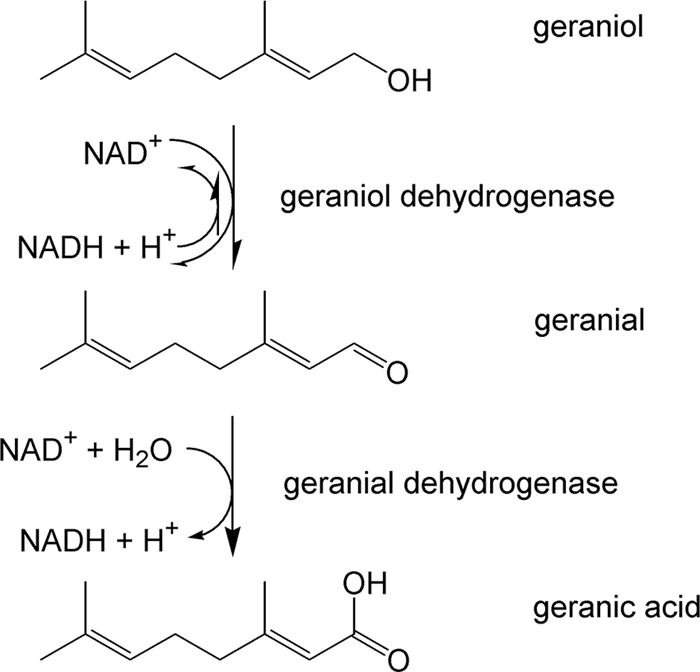
Geraniol oxidation pathway.
MATERIALS AND METHODS
Cultivation and biomass production.
Since its isolation in 1994, C. defragrans strain 65Phen has been maintained in the laboratory (17, 21) by four transfers per year, with 10% (vol/vol) inoculum. Anaerobic growth experiments were performed in 21-ml culture tubes with 15 ml aqueous medium, and the monoterpene was provided in 0.6 ml of the organic carrier phase 2,2,4,6,6,8,8-heptamethylnonane (HMN) (17). Biomass production yielded approximately 40 g (wet weight) cells grown on 15 mM limonene, α-phellandrene, or 100 mM acetate and 100 mM nitrate in a 10l fermentor (24). For large-scale, heterologous production of proteins, Escherichia coli BL21 Star(DE3) (Invitrogen, Darmstadt, Germany) carrying the overexpression plasmid was grown in terrific broth medium (7).
Enzyme assays and inhibition studies.
The NADH formation rate at 21°C in the standard assay for GeDH activity was photometrically measured at 340 nm. The assay mixture contained 100 mM glycine-NaOH, pH 9.4, 0.8 mM geraniol, and 1 mM NAD+ (final concentration). Rate constants were calculated based on a molar extinction coefficient for NADH of 6,220 M−1 cm−1. The catalytic properties (apparent Km and maximum rate of the enzyme system [Vmax]) were determined by the Hanes-Woolf algorithm (10). A dimer with a molecular mass of 76,544 Da (based on the deduced protein sequence) was used for the calculation of kcat. For inhibition studies with N-ethylmaleimide (NEM), diethylpyrocarbonate (DEPC), and 1-cyclohexyl-N-(2-morpholinoethyl)car-bodiimide (CMC), the purified recombinant GeDH was incubated together with the inhibitor for 1 h before the reaction was started by addition of geraniol and NAD+. Fifty and eighty percent inhibitory concentrations (I50 and I80, respectively) were graphically determined. The specificity of inhibition was proven by addition of a free amino acid (cysteine, histidine, or aspartate) at a concentration equimolar to the inhibitor at the I80.
Geranial is commercially available only as citral, a mixture of geranial and neral. Thus, GaDH activity was determined photometrically with 0.3 mM citral and 0.5 mM NAD+ in the aforementioned buffer at 30°C.
Chemical analyses.
Monoterpenes and monoterpenoids were analyzed in 1-μl volumes by gas chromatography (GC) with flame ionization detection (FID) (XL auto system; PerkinElmer, Überlingen, Germany). Separation was performed on an Optima-5 column (0.25-μm film thickness, 0.32-mm inside diameter [i.d.] by 50 m; Macherey-Nagel, Düren, Germany). The following temperature program was applied. The injection port temperature was 300°C, and the column start temperature was 85°C (held for 1 min). This was increased to 120°C at a rate of 15°C min−1 and held at 120°C for 0.1 min; the temperature was then increased to 320°C at a rate of 45°C min−1 and held at 320°C for 0.5 min. The detection temperature was 350°C. The split ratio was set to 1:10. For neral and geranial in citral, equal FID sensitivities were assumed and concentrations were calculated from the citral concentration and relative areas of geranial and neral. Other GC-FID and GC-mass spectrometry (MS) analyses were performed as previously reported (17, 21, 22).
Organic acids, i.e., geranic acid and neric acid, were analyzed on a reverse-phase high-performance liquid chromatography (HPLC) system using the method described by Heyen and Harder (24). For detection of geranic acid formation, a 10-fold-diluted sample of the assay mixture was injected on a Nucleodur C18 Isis column (Macherey-Nagel, Düren, Germany). Separation was performed with 1 mM H3PO4 at 1 ml min−1 in a water-acetonitrile gradient from 20 to 90% (vol/vol) acetonitrile at 25°C. UV detection was performed at 215 nm.
Protein purification and analyses.
For the wild-type GeDH, 40 g (wet weight) of frozen cells was thawed in 100 ml 100 mM potassium phosphate, pH 7.0, 2 mM dithiothreitol (DTT) (buffer A) and then homogenized and disintegrated in three passages through a French pressure cell press (Aminco, Rochester, NY) at 10.3 MPa. Ultracentrifugation for 90 min at 150,000 × g at 4°C yielded a soluble extract. The enzyme was purified at 4°C on a Pharmacia LC system (GE Healthcare, Freiburg, Germany). The extract was applied at a flow rate of 2.5 ml min−1 onto a DEAE fast-flow column (3-cm i.d., 200-ml column volume [CV]) and separated with a linear gradient of 0 to 1 M KCl in buffer A. Active fractions eluted early in the gradient and were directly applied to a phenyl-Sepharose 6 fast-flow column (2.6-cm i.d., 100-ml CV). Activity eluted at the end of a gradient of buffer A to 5 mM potassium phosphate, pH 7.0, 2 mM DTT, and 80% (vol/vol) ethylene glycol. After dialysis against 100 mM Na-HEPES, pH 7.0, 2 mM DTT (buffer B), the active fractions were purified on a DEAE fast-flow column (1.1-cm i.d., 10-ml CV) with a 0 to 1 M KCl gradient in buffer B. Molecular sieve chromatography was performed on a Superose 6 column (1.0-cm i.d., 47-ml CV) with buffer B containing 100 mM KCl.
The GeDH expressed in E. coli pET42a(+)geoA was purified at 4°C on an Äkta LC system (GE Healthcare, Freiburg, Germany) with filtered (0.2-μm pore size) and degassed buffers. Soluble extract was prepared as aforementioned and diluted to 10 mg protein ml−1. The extract contained 130 mg protein and was loaded at 4 ml min−1 on a butyl Sepharose 4 fast-flow column (2.6-cm i.d., 50-ml CV) that was equilibrated with 1.5 M (NH4)2(SO4) in 50 mM potassium phosphate buffer, pH 7.0, 2 mM DTT. Separation occurred in a gradient varying in both salt content and solvent polarity with 0 to 100% of 50% (vol/vol) ethylene glycol in 50 mM potassium phosphate buffer, pH 7.0, 2 mM DTT. Enzyme activity eluted at 42.5 vol% ethylene glycol. Active fractions were dialyzed against 10 mM potassium phosphate buffer, pH 7.0, 2 mM DTT (buffer C). Anion-exchange chromatography on a Source 15Q column (1.6-cm i.d., 20-ml CV) removed residual ethylene glycol. The enzyme activity eluted with 200 mM KCl in buffer C at 2 ml min−1. After concentration on a 10-Da membrane, gel filtration was performed on a Superdex 200 column (1.6-cm i.d., 120-ml CV) equilibrated in buffer C at 1 ml min−1. Standard proteins for the size determination were thyroglobulin (669 kDa), ferritin (440 kDa), catalase (232 kDa), aldolase (158 kDa), bovine serum albumin (67 kDa), ovalbumin (43 kDa), chymotrypsin (25 kDa), and RNase A (13.7 kDa).
Proteins were quantified according to the protocol of Bradford (6) with bovine serum albumin as a standard and visualized by SDS-PAGE according to the protocol of Laemmli (32). Native polyacrylamide gel electrophoresis was applied to determine independently the molecular mass of the GeDH. After separation in an 8% SDS-free polyacrylamide gel at 4°C, the gel was divided; one half was stained with Coomassie G250, and the other half was stained in 24.5 mM nitroblue tetrazolium chloride, 12 mM phenazine ethosulfate, 1 mM NAD+, and 0.4 mM geraniol in 100 mM glycine, pH 9.4, for 45 min in the dark. Afterwards, the gel was fixed with 7.5% acetic acid (modified from the protocol of Collins and Hegeman [9]).
Soluble extracts from cells grown on α-phellandrene or acetate were separated by anion-exchange chromatography on a Mono Q column (1-ml CV) with a NaCl gradient (1 to 400 mM, 40 fractions in a 10 mM range) in 50 mM Tris-HCl, pH 7.8, 2 mM DTT. Proteins in each fraction were separated by SDS-PAGE. The N termini of induced proteins as well as of native, purified GeDH were sequenced by Edman degradation after separation by SDS-PAGE and transfer by blotting onto a polyvinylidene difluoride (PVDF) membrane.
Soluble protein extracts for the detection of GaDH activity were prepared from E. coli pET42a(+)geoB as aforementioned with French pressure cell disintegration and ultracentrifugation, followed by removal of small molecules in a dialysis against 50 mM Na-HEPES, pH 7.0, 2 mM DTT (Visking dialysis tubing; Serva, Heidelberg, Germany).
Molecular biology and data deposition.
Standard techniques for molecular cloning and sequencing were applied (48) using the listed strains and plasmids (Table 1) and primers (Table 2). Fosmid libraries were prepared with the pCC1FOS vector using a CopyControl fosmid library production kit according to the manufacturer's instructions (Epicentre, Madison, WI) with the following modifications. Genomic DNA was embedded in low-melting-point agarose and equilibrated in 0.5× TE (48) and in end repair mix without enzyme. DNA strand ends were filled by incubation of 6 μl end repair enzyme mix together with the 40-μl agarose plug for 50 min at room temperature in 120 μl end repair mix containing a 0.5 mM concentration of the deoxynucleoside triphosphates (dNTPs). A transfer of the agarose plug in 500 μl 0.5 M EDTA stopped the reaction. DNA of about 25 to 48 kb was obtained on a preparative pulsed-field gel electrophoresis (PFGE) gel using 1% SeaPlaque GTG agarose (FMC BioProducts) and a Bio-Rad contour-clamped homogeneous electric field (CHEF) DRIII system (Bio-Rad) applying 0.5× TBE (48), a swtich time of 1 to 10 s, a reorientation angle of 120°, and 6 V/cm at 14°C for 16 h. The gel section of interest was excised, equilibrated in 1× TE, and digested with beta-agarase (New England BioLabs). The DNA was concentrated by drop dialysis on mixed cellulose ester membrane discs (Millipore; 0.025-μm pore size) against 30% polyethylene glycol 8000 in bidistilled water.
Table 1.
Strains and plasmids used in this study
| Strain or plasmid | Genotype, markers, and further characteristics | Source or reference |
|---|---|---|
| Strains | ||
| E. coli | ||
| One Shot TOP10 | F−mcrA Δ(mrr-hsdRMS-mcrBC) ϕ80lacZΔM15 ΔlacX74recA1araD139 Δ(ara leu)7697 galU galK rpsL (Strr) endA1 nupG | Invitrogen |
| BL21 Star(DE3) | F−ompT hsdSB (rB− mB−) gal dcm (DE3) | Invitrogen |
| DH5 α | supE44 ΔlacU169 ϕ80lacZΔM15 hsdR17 recA1 endA1 gyrA96 thi-1 relA1 | Invitrogen |
| EPI300-T1R | F−mcrA Δ(mrr-hsdRMS-mcrBC) (Strr) ϕ80d/acZΔM15 ΔlacX74recA1endA1araD139 Δ(ara-leu)7697 galUgalkλ−rpsLnupGtrfAtonAdhfr | Epicentre |
| C. defragrans 65Phen | Wild type | 17 |
| Plasmids | ||
| pCR2.1-TOPO | TOPO TA cloning vector; Amr KmrlacZα | Invitrogen |
| pCR4-TOPO | TOPO TA cloning vector; Amr KmrlacZα | Invitrogen |
| pBluescript SK(+/−) | Cloning vector; Amr KmrlacZα-ccdB | Stratagene |
| pCC1FOS | Cloning vector fosmid prepn, Chlr | Epicentre |
| pET42a(+) | Expression vector; Kmr | Novagen |
| pET42a(+)geoA | Expression vector; KmrgeoA | This study |
| pET42a(+)geoB | Expression vector; KmrgeoB | This study |
Table 2.
List of primers
| Name | Purpose | Sequencea |
|---|---|---|
| GDHFd1 | Nested PCR on N-terminal protein sequence | ATGAACTGTAC(GC)CA(AG)GA(CT)TT |
| GDHFd2 | AC(GC)CA(AG)GA(CT)TTCAT(CT)(AT)(GC)(GC)GC | |
| GDHRev | AC(GC)GG(CT)TC(GC)AC(GC)GC(GC)A(AG)(GC)GG | |
| GDH-F1 | PCR-mediated synthesis of DIG-labeled DNA probe | ACGCAGGATTTCATCAGG |
| GDH-R1 | TACGGGTTCGACGGCGAA | |
| geoA_BglII_R | Construction of expression vector pET42a(+)geoA | AGATCTTCAGAACACCAGCACCGGCTTG |
| geoA_NdeI_F | CATATGAACGACACCCAGGATTTCATTTCC | |
| geoB_NdeI_F | Construction of expression vector pET42a(+)geoB | CATATGACCATCGATCACCAGCACATCTTC |
| geoB_SalI_R | GTCGACCTAGCCAAGCAGGTACACTGAC |
Restriction sites are underlined.
Repetitive elements in the fosmid sequences required a semimanual assembly and contig closure by primer walking and gapping PCRs according to standard protocols. Sequence analyses and assembly were performed with Sequence Analysis 5.2 (Applied Biosystems, Foster City, CA), Sequencher 4.5 (Gene Codes, Ann Arbor, MI), and Lasergene (DNAStar, Madison, WI). The expression system used the vector pET42a(+) (Novagen, Darmstadt, Germany) in the host E. coli BL21 Star(DE3) (Invitrogen, Darmstadt, Germany).
Nucleotide sequence accession number.
The 50-kb genomic contig including geoA and geoB as well as the protein sequences were deposited within the EMBL nucleotide sequence database (accession number FR669447.2).
RESULTS
Geraniol dehydrogenase activity induced in monoterpene-grown cultures.
The identification of geranic acid as an intermediate in C. defragrans cells during the anaerobic mineralization of monoterpenes (24) suggested the presence of geraniol and geranial dehydrogenase activities (7). We had reported that geraniol did not support the growth of C. defragrans (17). In those experiments with HMN as an organic carrier phase, a calculated geraniol concentration of 4 mM corresponded to actual concentrations of 80 mM relative to that of the HMN phase and of 70 μM geraniol in the aqueous phase, according to the partial pressure of geraniol in the organic phase and a water solubility of geraniol of 2.62 mM (57). We then tested lower geraniol concentrations and observed geraniol utilization at aqueous concentrations of 5 μM geraniol. Higher concentrations of geraniol inhibited microbial growth, and nerol, geranial, and neral accumulated in the organic carrier phase with concomitant formation of traces of nitrite (see Tables S1 and S2 in the supplemental material).
We investigated geraniol biotransformations by soluble extracts of C. defragrans. Geranic acid was formed not only from β-myrcene, as previously reported (24), but also from geraniol and nerol. Nerol was also transformed to neric acid (see Table S3 in the supplemental material). Alcohol dehydrogenase activities were determined in soluble extracts by the reduction of NAD+. Geraniol dehydrogenase and benzyl alcohol dehydrogenase activities were 1.39 ± 0.10 and 24.6 ± 0.2 mU mg−1 protein (n = 2) in extracts from limonene-grown cells, in comparison to 0.14 ± 0.00 and 1.64 ± 0.09 mU mg−1 protein (n = 2) in extracts from acetate-grown cells. The 10- to 15-fold-higher rates of NADH formation indicate an inducible GeDH activity.
Identification of a GeDH and induced proteins.
GeDH activity was purified with the guidance of the purification protocol for a dehydrogenase from P. putida acting on 3-methyl-2-buten-1-ol, a homologue of geraniol (36). The 114-fold purification (Table 3) yielded a nearly homogenous protein with an apparent molecular mass of 39 kDa as determined by SDS-PAGE (Fig. 3A) and 52 kDa as determined by molecular sieve chromatography. The determination of the N-terminal protein sequence (MN-TQDFISAQA-VL-QVGGPLAVEPVI) by Edman degradation enabled the design of a degenerate primer and the amplification of a small DNA fragment 73 bp in length (Table 2). DNA sequencing of the cloned 73-bp fragment verified an open reading frame for the N-terminal protein sequence. Application of the 73-bp fragment as probe in Southern blot analysis revealed a location on a BamHI restriction fragment. A plasmid library with BamHI fragments of the C. defragrans genome was screened with the digoxigenin (DIG)-labeled 73-bp probe. A positive plasmid contained a genomic fragment 5,631 bp in length with the complete gene for the geraniol dehydrogenase at the 5′ end of the insert. We named the gene geoA, the first gene on the geraniol oxidation pathway.
Table 3.
Purification of GeDH from C. defragrans 65Phen
| Purification step | Total protein (mg) | Total activity (U) | Sp act (U/mg) | Yield (%) | Purification (fold) |
|---|---|---|---|---|---|
| Cell extract | 3,160 | 175 | 0.056 | 100 | 1 |
| 1st DEAE fast flow | 269 | 111 | 0.41 | 71 | 12 |
| Phenyl-Sepharose 6 fast flow | 46 | 101 | 2.19 | 58 | 69 |
| 2nd DEAE fast flow | 36 | 156 | 4.33 | 89 | 114 |
Fig 3.

(A) SDS-PAGE of the native GeDH purified from soluble extract of C. defragrans 65Phen (lane 1, soluble extract; lane 2, GeDH). (B) Induction of expression of GeDH (39 kDa) (lanes 3 and 4) in E. coli pET42a(+)geoA and GaDH (53 kDa) (lanes 5 and 6) in E. coli pET42a(+)geoB. The induced proteins are indicated by an arrow. Protein samples were taken just before (lanes 5 and 7) and after (lanes 6 and 8) 2 h of isopropyl-β-d-thiogalactopyranoside (IPTG) induction. (C) Native PAGE of GeDH. One microgram of purified recombinant GeDH was applied per lane; lane 7 is Coomassie G250 stained, and lane 8 is activity stained with geraniol as a substrate. Lane A, low-molecular-weight marker (Pharmacia); lane B, Page Ruler prestained protein ladder (Fermentas); lane C, native protein marker (Serva). Numbers at the left are molecular weights (in thousands).
For the analysis of the genomic neighborhood, we prepared a fosmid library and identified by PCR screening two fosmids that carried geoA. A continuous sequence of 50 kb was obtained by semimanual sequence assembly and used to interpret the results of a differential proteomic analysis. C. defragrans was grown with acetate or phellandrene. Proteins present in soluble extracts of these cells were compared by anion-exchange chromatography in combination with one-dimensional SDS-PAGE (see Fig. S1 in the supplemental material). The N termini of proteins specifically expressed in monoterpene-utilizing cells were sequenced by Edman degradation. Some amino acid sequences were coded by open reading frames annotated on the 50-kb contig. One of these proteins corresponded to a predicted aldehyde dehydrogenase (ALDH), initially named geoB. Other expressed genes located on the contig include oxidoreductases and acyl coenzyme A (acyl-CoA) dehydrogenases (Table 4), which may play a role in the further degradation of geranic acid.
Table 4.
N-terminal amino acid sequences and identified open reading frames of proteins found to be present during growth with phellandrene but not with acetate
| Position in contig | N-terminal amino acid sequence | N-terminal amino acid sequence of ORF in contig (GenBank accession no. FR669447) | Annotation |
|---|---|---|---|
| ORF4 | ND-TPPGQTWP(P)VV | MNDRTPPGQTWPPVD | Hypothetical protein |
| ORF11 | MANPKSEYDVIIVGGGLNGLA | MANPKSEYDVIIVGGGLNGLA | Phytoene dehydrogenase-like oxidoreductase |
| ORF12 | MSEVKQ-DVVVIGAG- | MSEVKQCDVVVIGAGH | Phytoene dehydrogenase-like oxidoreductase |
| ORF18 | (TA)I(D)(T)Q(H)IFVGGQWIAP(K) | MTIDHQHIFVGGQWIAPK | Geranial dehydrogenase geoB |
| ORF44 | MIE-LFGPE-(F)M(F)-(D)TV-K | MIERRLFGPEHEMFRDTVRK | Acyl-CoA dehydrogenase |
The genes geoA and geoB were introduced into the overexpression vector pET42a(+) and functionally expressed in E. coli BL21 Star(DE3) (Table 1). Induction of gene expression yielded proteins with the expected molecular masses (Fig. 3B). Soluble extracts of E. coli carrying geoA showed a NADH formation rate of 0.26 ± 0.03 mU mg−1 protein (n = 3) with geraniol. Soluble extracts of E. coli carrying geoB had an NADH formation rate of 3.7 ± 0.9 mU mg−1 protein (n = 3) with citral, the commercially available mixture of geranial and neral. Soluble extracts of E. coli pET42a(+) oxidizes neither geraniol nor citral, and therefore the inserted genes must be responsible for the monoterpenoid oxidation. The genes were named geraniol dehydrogenase (geoA/GeDH) and geranial dehydrogenase (geoB/GaDH).
Characterization of the geraniol dehydrogenase.
The identified open reading frame for the GeDH was 1,122 bp in length, had a GC content of 71.21% and a molecular mass of 38,272 Da, and coded for a protein of 373 amino acids (aa) (see Fig. S2 in the supplemental material). It displays specific motifs of medium-chain-length, Zn-containing, NAD+-dependent alcohol dehydrogenases (28) and affiliates within the mdr19 family, which comprises benzyl/aryl ADHs with a long quaternary structure-determining loop (QSDL) of more than 31 aa (30). The amino acid identity to eukaryotic geraniol dehydrogenases was low: 25.3% to the GeDH from sweet basil, Ocimum basilicum (25), and 27.1% to the GeDH from the astigmatid mite Carpoglyphus lactis (38). The C. defragrans GeDH was compared in a multiple-sequence alignment with ADHs of high similarity, namely, plant GeDHs and well-characterized ADHs, such as the horse liver ADH (HLADH), which is representative of Zn-containing, medium-chain ADHs of the MDR type (14, 27, 44) (Fig. S4). The coenzyme binding domain, which contains the Rossmann fold (46) with the glycine-rich phosphate binding loop (GXGXXG) as well as the catalytic zinc binding motif (GHEXXGXXXXXGXXV), was found to be conserved in C. defragrans GeDH (Gly200, Gly 202, Gly205, and Gly66-Val80). The structural zinc atom is coordinated by four conserved Cys residues (Cys96, Cys99, Cys102, Cys110).
The recombinant GeDH was purified to homogeneity. The native molecular mass was determined on different molecular sieve columns and varied between 52 and 91 kDa, likely indicating an equilibrium between the monomeric and dimeric state. Independently, native PAGE with subsequent GeDH activity staining revealed an apparent molecular mass of 85 ± 7 kDa, based on the size of marker proteins (Fig. 3C). These observations suggest that a dimer represents the native conformation of the active enzyme. Also the other members of the mdr19 family of the MDR ADH are active as dimers, with the exception of the benzyl ADH from Acinetobacter calcoaceticus (Protein Data Bank [PDB] accession number 1F8F) (30). Consequently, a dimer was used in the calculation of the catalytic efficiency.
According to gas chromatography analyses, the purified GeDH catalyzed the formation of geranial from geraniol. The cis isomer neral was formed in glycine buffer, pH 9.4, but not at pH 7.0 in 100 mM potassium phosphate, 2 mM DTT. Geraniol was not isomerized to nerol or linalool at pH 7.0. These results suggest the retention of the trans configuration of the alkene during biological oxidation and a chemical isomerization of geranial to neral under alkaline conditions. Citral, the commercially available mixture of geranial and neral, was not further oxidized.
Because GeDH is classified among the benzyl and aryl ADHs, the kinetic properties of the native and the recombinant purified GeDHs were determined for benzyl alcohol, cumic alcohol (p-isopropyl-benzyl alcohol), and (S)-(−)-perillyl alcohol in comparison with geraniol, nerol, and citronellol (Fig. 1). The purified GeDH exhibited typical Michaelis-Menten kinetics with all substrates (Table 5). The apparent Km values for geraniol and perillyl alcohol were around 5 μM, indicating a high affinity for these substrates. The affinities for nerol and citronellol were significantly lower. Benzyl alcohol had the highest Vmax value. The catalytic efficiency calculation identified perillyl alcohol as the best substrate for the enzyme, followed by geraniol and cumic alcohol.
Table 5.
Enzyme kinetics of native and recombinant GeDHa
| Substrate | Native GeDH |
Recombinant GeDH |
||||
|---|---|---|---|---|---|---|
| Km (μM) | Vmax (U mg−1) | kcat/Km(1 · 106 s−1 M−1) | Km (μM) | Vmax (U mg−1) | kcat/Km(1 · 106 s−1 M−1) | |
| Geraniol | 5 | 10 | 1.57 | 3.3 | 2.6 | 0.62 |
| Nerol | 45 | 18 | 0.31 | 23.2 | 9.1 | 0.31 |
| Citronellol | 86 | 11 | 0.10 | 57.5 | 3.7 | 0.05 |
| (S)-(−)-Perillyl alcohol | 7 | 18 | 2.02 | 4.4 | 19.9 | 3.55 |
| Cumic alcohol | 21 | 14 | 0.52 | 6.0 | 7.2 | 0.94 |
| Benzyl alcohol | 170 | 47 | 0.22 | 115.7 | 16.8 | 0.11 |
Values are apparent values.
The enzyme reduced one molecule of NAD+ to NADH per geraniol molecule provided in geraniol-limited assays. NADP+ was ineffective as a cosubstrate. The cofactor specificity for NAD+ was likely defined by the negative charge of Glu224, which is known to repel the additional phosphate of NADP+ (31, 44, 55). The pH optimum was, as expected for MDR ADHs, in the alkaline range at pH 9.4. Dichlorphenolindophenol (DCPIP), but not phenazine methosulfate (PMS), was accepted as an alternative electron acceptor in the enzyme reaction. GeDH is sensitive to chelating reagents, as expected for zinc-containing ADHs; 7 mM EDTA inhibited the enzyme by 96%. Inhibition by N-ethylmaleimide (I80 = <0.5 mM), diethylpyrocarbonate (I80 = 17 mM), and 1-cyclohexyl-N-(2-morpholinoethyl)carbodiimide (I80 = 34 mM) was partly suppressed by addition of equimolar amounts of cysteine, histidine, and aspartate, respectively. Inhibition was reduced from 80% to values between 43 and 58%. This indicates a participation of these amino acids in GeDH activity.
Characterization of the geranial dehydrogenase.
The geoB gene has 1,437 bp and codes for a protein of 478 aa (see Fig. S3 in the supplemental material) with a molecular mass of 50,637 Da, which coincides with the apparent molecular mass of 53 kDa from SDS-PAGE (Fig. 3B). ALDHs with related sequences originated from the Gram-positive order Actinomycetales, i.e., from Rhodococcus opacus (59% identity, GenBank accession number YP_002781874.1), Rhodococcus rhodochrous (58% identity, AAC15840.1), Rhodococcus jostii (59% identity, ABG99066.1), and “Streptomyces bingchenggensis” (57% identity, ADI11766.1). The C. defragrans GaDH is affiliated with the ALDH superfamily. A Clustal W alignment with ALDHs from different organisms revealed amino acid residues that are conserved in more than 95% of ALDHs (Fig. S5) (40). In C. defragrans GaDH, they are the following: Arg69, Gly146, Asn155, Pro157, Gly172, Lys178, Gly230, Gly254, Gly283, Cys286, Glu383, Phe385, Pro387, Gly433, Asn438, and Gly450. The Rossmann fold motif was shortened by one amino acid and only partially covered; Gly209 and Gly214 were present, but the second Gly residue was replaced by an Asp.
HPLC analyses of free medium-chain fatty acids revealed that only the trans-isomer geranic acid, and not the cis isomer neric acid, was the product of the oxidation catalyzed by soluble extracts of E. coli pET42a(+)geoB. Geranial disappeared faster than neral, suggesting that geranial is biologically oxidized and that neral is chemically isomerized to geranial and subsequently oxidized biologically (Table 6). The formation of geranic acid and NADH correlated in a 1:1 ratio (Fig. 4). Thus, the geranial dehydrogenase acted specifically on geranial.
Table 6.
Citral conversion by soluble extracts of E. coli pET42a(+) and pET42a(+)geoBa
| Plasmid | Time (h) | Geranial (mM) | Neral (mM) | Geraniol (mM) | Nerol (mM) | Geranic acid (mM) |
|---|---|---|---|---|---|---|
| pET42a(+)geoB | 0 | 11.60 ± 0.15 | 7.20 ± 0.20 | 0 | 0 | 0 |
| 2 | 2.95 ± 0.15 | 3.25 ± 0.05 | 2.3 ± 0.1 | 1.4 ± 0.1 | 7.65 ± 0.15 | |
| 24 | 0.32 ± 0.08 | 0.25 ± 0.05 | 0 | 0 | 8.90 ± 1.10 | |
| pET42a(+) | 0 | 11.50 ± 0.10 | 6.75 ± 0.05 | 0 | 0 | 0 |
| 2 | 11.32 ± 0.28 | 6.95 ± 0.35 | 0 | 0 | 0 | |
| 24 | 8.15 ± 0.55 | 5.20 ± 0.40 | 0 | 0 | 0 |
Soluble extracts were assayed in duplicate with 0.5 mM NAD+ in a two-phase system with 20 mM citral in HMN. The organic phase serves as reservoir for the substrate as well as for the products and was analyzed by gas chromatography.
Fig 4.

(A) Geranial dehydrogenase activity (NADH formation) at different citral concentrations, determined in triplicate measurements. (B) Geranic acid formation (●) was measured via RP-HPLC, and NADH formation (▵) was determined spectrophotometrically.
DISCUSSION
In this study, we identified a geraniol dehydrogenase and a geranial dehydrogenase of the β-myrcene degradation pathway present in denitrifying Castellaniella defragrans. The activities of both enzyme activities were specifically induced during growth with monoterpenes, and these enzymes were expressed in E. coli in their active forms. In contrast to the linalool dehydratase isomerase (7), the GeDH and the GaDH were not oxygen sensitive and were located in the cytoplasm. The genes did not code for a signal peptide for a periplasmic location. The involvement of alcohol and aldehyde dehydrogenase per se in degradation pathways is common, but the detailed characterization revealed the particular properties of GeDH and GaDH and emphasized their singularity.
The GeDH of C. defragrans has, among the GeDHs so far reported, the highest affinity for geraniol (Table 7). A number of other GeDHs from plant sources (25, 43, 49) and the insect pest Carpoglyphus lactis (37) are involved in geraniol synthesis and therefore may not have evolved a higher affinity for geraniol than enzymes in degradation pathways. The transformation of β-myrcene to geraniol by the linalool dehydratase isomerase is thermodynamically unfavorable and results in a steady state at low geraniol concentrations (7, 35), thus requiring a high-affinity GeDH for an efficient metabolic flux. Furthermore, geraniol was found to inhibit monoterpene metabolism at aqueous concentrations above 5 μM in the two-liquid-phase system with geraniol in the organic carrier phase. The molecular target for this specific inhibition remains unclear, but incorporation of the hydrophobic substances in membranes resulting in disruption of the proton motive force has often been reported (13, 53).
Table 7.
Apparent Km values, pH optima, and cofactor dependence for enzymes acting on geraniol
| Organism | Km (μM) | pH optimum | Cofactor | EC no. (enzyme) | Reference |
|---|---|---|---|---|---|
| C. defragrans | 3.3 | 10 | NAD+ | 1.1.1.183 | This study |
| Carpoglyphus lactis (prune mite) | 51 | 9 | NAD+ | 1.1.1.183 | 38 |
| Citrus sp. (orange) | 46.5 | 9 | NADP+ | 1.1.1.183 | 43 |
| Cymbopogon flexuosus (lemongrass) | 100 | NDa | NADP+ | 1.1.1.183 | 49 |
| Ocimum basilicum (basil) | 30 | 9.5 | NADP+ | 1.1.1.183 | 25 |
| Homo sapiens (human) | 25 | 9 | NADPH | 1.1.1.21 (aldehyde reductase) | 16 |
| Ipomoea batatas (sweet potato) | 729 | ND | NADP+ | 1.1.1.216 (farnesol dehydrogenase) | 26 |
| Arabidopsis thaliana | 800 | 10 | NAD+ | 1.1.1.284 [(S)-(hydroxymethyl) glutathione dehydrogenase] | 1 |
| Rosa hybrid | 2,783 | ND | ND | 2.3.1.84 (alcohol o-acetyltransferase) | 19 |
| Sorghum bicolor (sorghum) | 140 | ND | ND | 2.4.1.85 (cyanohydrin beta-glucosyltransferase) | 20 |
| Thea sinensis (tea) | 6,250 | ND | NAD | 1.1.1.1 (alcohol:NAD oxidoreductase) | 23 |
ND, not determined.
The enzyme possesses a higher affinity for the allyl alcohols geraniol and nerol than for the nonallylic citronellol. This may have a chemical explanation: the alkene bond donates overlapping π electron density to the carbon of the alcohol, thereby stabilizing the transient positive charge during hydride transfer of NAD+ and enhancing substrate binding as well as catalysis (12, 44). Alternatively, the binding pocket may be narrow and favor substrates with sp2-hybridized C-2 and C-3 atoms. Citronellol, with sp3-hybridized C-2 and C-3 atoms, is more space filling and may not fit perfectly into the binding pocket (15).
The preference for allyl alcohols over benzyl alcohols remains an open question. A multiple alignment with highly similar ADHs, i.e., plant GeDH and well-characterized ADHs, revealed some potentially important amino acids. His48, Arg229, and Lys368 are the positive charges interacting with the cofactor NAD+. Interestingly, mutation of Arg50 to His50 in benzyl ADH from A. calcoaceticus (18) decreased the catalytic activity of the perillyl-ADH activity 10-fold but that of the benzyl ADH activity 1,000-fold. This suggests a crucial role for His48 in the activity of C. defragrans GeDH. His51 of HLADH is replaced by Val52 in C. defragrans GeDH. The positive charge is not conserved among many benzyl ADHs and is not necessary for the reaction (18). The substrate specificity for MDR ADHs is defined by the hydrophobic cleft formed by residues of both domains and the cofactor itself. HLADH catalyzes the oxidation of many alcohols; however, an increase in the spatial volume of the alcohol correlates with a lower activity (44). The substrate binding site can be divided: the inner part is close to the catalytic zinc ion and formed by Ser48, Phe93, Phe140, and Leu141, the middle part by Leu57, Leu116, Val294, and Ile318, and the outer part by Phe110, Met306, and Leu309 (HLADH [14]). The C. defragrans GeDH may have a smaller, more polar, pocket with Thr49, Phe93, Phe141, and Phe142 in the interior and Phe57, Phe118, Leu293, and Ile317 in the middle part. Threonine replaces serine and reduces the available space for the alcohol, thus favoring smaller alcohols (11, 16, 58) or the less space-filling allyl and benzyl alcohols. The increase in phenylalanines enables interaction with π electrons of the substrate. The crystal structure of a Phe93Ala mutant revealed a loss of the ability to bind a benzyl alcohol in the perfect position for the catalysis (47). In fact, Thr49 and Phe93 are present in most of the bacterial benzyl ADHs (18, 52). Overall, the alignment together with our knowledge of ADHs suggests that in the C. defragrans GeDH, the amino acids His48, Thr49, and Phe93 play a crucial role in the substrate specificity of the enzyme.
In contrast to the often-observed operon organization of genes involved in metabolic pathways, the genes of the initial β-myrcene pathway, ldi, geoA, and geoB, are well separated on the genome but physically close in distance. The C. defragrans GaDH is not a citral dehydrogenase; it acts only on the trans-isomer geranial and affiliates with the ALDH superfamily. So far, the presence of a geranial dehydrogenase has not been reported. Future studies will deal with the characterization of this enzyme in more detail.
Thermodynamically, oxidation of the aldehyde to geranic acid yields sufficient free energy for ATP synthesis from inorganic phosphate and ADP. However, the hydration of β-myrcene, the isomerization to geraniol, and the coupling of geraniol oxidation to the reduction of NAD+ are three thermodynamically unfavorable reactions. Thus, geranial oxidation shifts the overall reaction from β-myrcene to geranic acid into a favorable reaction and allows the process to occur even at low myrcene concentrations.
Supplementary Material
ACKNOWLEDGMENTS
We thank Christina Probian for technical assistance. Jasmin Berg is acknowledged for critically reading the manuscript.
This study was financed by the Deutsche Forschungsgemeinschaft (grant Ha 1673/5-2) and the Max Planck Society.
Footnotes
Published ahead of print 27 January 2012
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1. Achkor H, et al. 2003. Enhanced formaldehyde detoxification by overexpression of glutathione-dependent formaldehyde dehydrogenase from Arabidopsis. Plant Phys. 132: 2248–2255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Auld DS, Bergman T. 2008. The role of zinc for alcohol dehydrogenase structure and function. Cell. Mol. Life Sci. 65: 3961–3970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Behr A, Johnen L. 2009. Myrcene as a natural base chemical in sustainable chemistry: a critical review. ChemSusChem 2: 1072–1095 [DOI] [PubMed] [Google Scholar]
- 4. Blomquist GJ, et al. 2010. Pheromone production in bark beetles. Insect Biochem. Mol. Biol. 40: 699–712 [DOI] [PubMed] [Google Scholar]
- 5. Bottoms CA, Smith PE, Tanner JJ. 2002. A structurally conserved water molecule in Rossmann dinucleotide-binding domains. Protein Sci. 11: 2125–2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bradford MM. 1976. A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72: 248–254 [DOI] [PubMed] [Google Scholar]
- 7. Brodkorb D, Gottschall M, Marmulla R, Lüddeke F, Harder J. 2010. Linalool dehydratase-isomerase, a bifunctional enzyme in the anaerobic degradation of monoterpenes. J. Biol. Chem. 285: 30406–30442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen W, Viljoen AM. 2010. Geraniol—a review of a commercially important fragrance material. S. Afr. J. Bot. 76: 643–651 [Google Scholar]
- 9. Collins J, Hegeman G. 1984. Benzyl alcohol metabolism by Pseudomonas putida: a paradox resolved. Arch. Microbiol. 138: 153–160 [Google Scholar]
- 10. Cornish-Bowden A. 1995. Fundamentals of enzyme kinetics. Portland Press, London, Great Britain [Google Scholar]
- 11. Creaser EH, Murali C, Britt KA. 1990. Protein engineering of alcohol dehydrogenases; effects of amino acid changes at positions 93 and 43 of yeast ADH1. Protein Eng. 3: 523–526 [DOI] [PubMed] [Google Scholar]
- 12. Curtis AJ, Shirk MC, Fall R. 1999. Allylic or benzylic stabilization is essential for catalysis by bacterial benzyl alcohol dehydrogenases. Biochem. Biophys. Res. Commun. 259: 220–223 [DOI] [PubMed] [Google Scholar]
- 13. di Pasqua R, et al. 2007. Membrane toxicity of antimicrobial compounds from essential oils. J. Agric. Food Chem. 55: 4863–4870 [DOI] [PubMed] [Google Scholar]
- 14. Eklund H, Ramaswamy S. 2008. Three-dimensional structures of MDR alcohol dehydrogenases. Cell. Mol. Life Sci. 65: 3907–3917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Eklund H, Horjales E, Vallee BL, Jörnvall H. 1987. Computer-graphics interpretations of residue exchanges between the α, β and γ subunits of human-liver alcohol dehydrogenase class I isozymes. Eur. J. Biochem. 167: 185–193 [DOI] [PubMed] [Google Scholar]
- 16. Endo ST, et al. 2009. Kinetic studies of AKR1B10, human aldose reductase-like protein: endogenous substrates and inhibition by steroids. Arch. Biochem. Biophys. 487: 1–9. [DOI] [PubMed] [Google Scholar]
- 17. Foss S, Heyen U, Harder J. 1998. Alcaligenes defragrans sp. nov., description of four strains isolated on alkenoic monoterpenes ((+)-menthene, α-pinene, 2-carene, and α-phellandrene) and nitrate. Syst. Appl. Microbiol. 21: 237–244 [DOI] [PubMed] [Google Scholar]
- 18. Gillooly DJ, Robertson AGS, Fewson CA. 1998. Molecular characterization of benzyl alcohol dehydrogenase and benzaldehyde dehydrogenase II of Acinetobacter calcoaceticus. Biochem. J. 330: 1375–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Guterman I, et al. 2006. Generation of phenylpropanoid pathway-derived volatiles in transgenic plants: rose alcohol acetyltransferase produces phenylethyl acetate and benzyl acetate in petunia flowers. Plant Mol. Biol. 60: 555–563 [DOI] [PubMed] [Google Scholar]
- 20. Hansen KS, et al. 2003. The in vitro substrate regiospecificity of recombinant UGT85B1, the cyanohydrin glucosyltransferase from Sorghum bicolour. Phytochemistry 64: 143–151 [DOI] [PubMed] [Google Scholar]
- 21. Harder J, Probian C. 1995. Microbial degradation of monoterpenes in the absence of molecular oxygen. Appl. Environ. Microbiol. 61: 3804–3808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Harder J, Heyen U, Probian C, Foß S. 2000. Anaerobic utilization of essential oils by denitrifying bacteria. Biodegradation 11: 55–63 [DOI] [PubMed] [Google Scholar]
- 23. Hatanaka A, Sekiya J, Kajiwara T. 1976. Subunit composition of alcohol dehydrogenase from Thea sinensis seeds and its substrate specificity for monoterpenes. Phytochemistry 15: 487–488 [Google Scholar]
- 24. Heyen U, Harder J. 2000. Geranic acid formation, an initial reaction of anaerobic monoterpene metabolism in denitrifying Alcaligenes defragrans. Appl. Environ. Microbiol. 66: 3004–3009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Iijima Y, Wang G, Fridman E, Pichersky E. 2006. Analysis of the enzymatic formation of citral in the glands of sweet basil. Arch. Biochem. Biophys. 448: 141–149 [DOI] [PubMed] [Google Scholar]
- 26. Inoue H, Tsuji H, Uritani I. 1984. Characterization and activity change of farnesol dehydrogenase in black rot fungus-infected sweet potato. Agric. Biol. Chem. 48: 733–738 [Google Scholar]
- 27. Jörnvall H. 1977. Differences between alcohol dehydrogenases-structural properties and evolutionary aspects. Eur. J. Biochem. 72: 443–452 [DOI] [PubMed] [Google Scholar]
- 28. Jörnvall H, Hempel J, Vallee B. 1987. Structures of human alcohol and aldehyde dehydrogenases. Enzyme 37: 5–18 [DOI] [PubMed] [Google Scholar]
- 29. Kämpfer P, et al. 2006. Castellaniella gen. nov., to accommodate the phylogenetic lineage of Alcaligenes defragrans, and proposal of Castellaniella defragrans gen. nov., comb. nov. and Castellaniella denitrificans sp. nov. Int. J. Syst. Evol. Microbiol. 56: 815–819 [DOI] [PubMed] [Google Scholar]
- 30. Knoll M, Pleiss J. 2008. The medium-chain dehydrogenase/reductase engineering database: a systematic analysis of a diverse protein family to understand sequence-structure-function relationship. Protein Sci. 17: 1689–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Korkhin Y, Kalb AJ, Bogin MO, Burstein Y, Frolow F. 1998. ADP-dependent bacterial alcohol dehydrogenases: crystal structure, cofactor-binding and cofactor specificity of the ADHs of Clostridium beijerinckii and Thermoanaerobacter brockii. J. Mol. Biol. 278: 967–981 [DOI] [PubMed] [Google Scholar]
- 32. Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680–685 [DOI] [PubMed] [Google Scholar]
- 33. Lathiere J, et al. 2006. Impact of climate variability and land use changes on global biogenic volatile organic compound emissions. Atmos. Chem. Phys. 6: 2129–2146 [Google Scholar]
- 34. Li X, et al. 2010. Characterization of a broad-range aldehyde dehydrogenase involved in alkane degradation in Geobacillus thermodenitrificans NG80-2. Microbiol. Res. 165: 706–7121 [DOI] [PubMed] [Google Scholar]
- 35. Lüddeke F, Harder J. 2011. Enantiospecific (S)-(+)-linalool formation from β-myrcene by linalool dehydratase-isomerase. Z. Naturforsch. C Biosci. 66c: 409–412 [DOI] [PubMed] [Google Scholar]
- 36. Malone VF, et al. 1999. Characterization of a Pseudomonas putida allylic alcohol dehydrogenase induced by growth on 2-methyl-3-buten-2-ol. Appl. Environ. Microbiol. 65: 2622–2630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Noge K, et al. 2005. Biosynthesis of neral by Carpoglyphus lactis (Acari: Carpoglyphidae) and detection of its key enzyme, geraniol dehydrogenase, by electrophoresis. J. Acarol. Soc. Jpn. 14: 75–81 [Google Scholar]
- 38. Noge K, et al. 2008. Geraniol dehydrogenase, the key enzyme in biosynthesis of the alarm pheromone, from the astigmatid mite Carpoglyphus lactis (Acari: Carpoglyphidae). FEBS J. 275: 2807–2817 [DOI] [PubMed] [Google Scholar]
- 39. Okibe N, et al. 1999. Gene cloning and characterization of aldehyde dehydrogenase from a petroleum-degrading bacterium, strain HD-1. J. Biosci. Bioeng. 88: 7–11 [DOI] [PubMed] [Google Scholar]
- 40. Perizoch J, Nicholas H, Wang BC, Lindahl R, Hempel J. 1999. Relationships within the aldehyde dehydrogenase extended family. Protein Sci. 8: 137–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Persson B, Zigler JS, Jr, Jörnvall H. 1994. A super-family of medium chain dehydrogenases/reductases (MDR). Sub-lines including zeta-crystallin, alcohol and polyol dehydrogenases, quinone oxidoreductase enoyl reductases, VAT-1 and other proteins. Eur. J. Biochem. 226: 15–22 [DOI] [PubMed] [Google Scholar]
- 42. Plapp BV. 2010. Conformational changes and catalysis by alcohol dehydrogenase. Arch. Biochem. Biophys. 493: 3–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Potty VH, Bruemmer H. 1970. Oxidation of geraniol by an enzyme system from orange. Phytochemistry 9: 1003–1007 [Google Scholar]
- 44. Reid MF, Fewson CA. 1994. Molecular characterization of microbial alcohol dehydrogenases. Crit. Rev. Microbiol. 20: 13–56 [DOI] [PubMed] [Google Scholar]
- 45. Riveros-Rosas H, Julian-Sanchez A, Villalobos-Molina R, Pardo JP, Pina E. 2003. Diversity, taxonomy and evolution of medium-chain dehydrogenase/reductase superfamily. Eur. J. Biochem. 270: 3309–3334 [DOI] [PubMed] [Google Scholar]
- 46. Rossmann MG, Moras D, Olsen KW. 1974. The chemical and biological evolution of a nucleotide-binding protein. Nature 250: 194–199 [DOI] [PubMed] [Google Scholar]
- 47. Rubach JK, Plapp BV. 2003. Amino acid residues in the nicotinamide binding site contribute to catalysis by horse liver alcohol dehydrogenase. Biochemistry 42: 2907–2915. [DOI] [PubMed] [Google Scholar]
- 48. Sambrook J, Russel DW. 2001: Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 49. Sangwan RS, Singh-Sangwan N, Luthra R. 1993. Metabolism of acyclic monoterpenes: partial purification and properties of geraniol dehydrogenase from lemongrass (Cymbopogon flexuosus Stapf.) leaves. J. Plant Physiol. 142: 129–134 [Google Scholar]
- 50. Seyboldt SJ, et al. 1995. De novo biosynthesis of the aggregation pheromone components ipsenol and ipsdienol by the pine bark beetles Ips paraconfusus Lanier and Ips pini (Say) (Coleoptera: Scolytidae). Proc. Natl. Acad. Sci. U. S. A. 92: 8393–8397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sharkey TD, Wiberly AE, Donohue AR. 2008. Isoprene emission from plants: why and how. Ann. Bot. 101: 5–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shaw JP, Harayama S. 1990. Purification and preliminary characterization of TOL plasmid-encoded benzyl alcohol dehydrogenase and benzaldehyde dehydrogenase of Pseudomonas putida. Eur. J. Biochem. 191: 705–714 [DOI] [PubMed] [Google Scholar]
- 53. Sikkema J, de Bont JAM, Poolman B. 1995. Mechanisms of membrane toxicity of hydrocarbons. Microbiol. Rev. 59: 201–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Smolander A, et al. 2006. Volatile monoterpenes in soil atmosphere under birch and conifers: effects on soil N transformations. Soil Biol. Biochem. 38: 3436–3442 [Google Scholar]
- 55. Sun HW, Plapp BV. 1992. Progressive sequence alignment and molecular evolution of the Zn-containing alcohol dehydrogenase family. J. Mol. Evol. 34: 522–535 [DOI] [PubMed] [Google Scholar]
- 56. Vasiliou V, Pappa A, Petersen DR. 2000. Role of aldehyde dehydrogenases in endogenous and xenobiotic metabolism. Chem. Biol. Interact. 129: 1–19 [DOI] [PubMed] [Google Scholar]
- 57. Weidenhamer JD, Macias FA, Fischer NH, Williamson GB. 1993. Just how insoluble are monoterpenes? J. Chem. Ecol. 19: 1799–1807 [DOI] [PubMed] [Google Scholar]
- 58. Xie P, Parsons SH, Speckhard DC, Bosron WF, Hurley TD. 1997. X-ray structure of human class IV σσ alcohol dehydrogenase-structural basis for substrate specificity. J. Bacteriol. Chem. 272: 18558–18563 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.