

Abstract
The impact of loci that determine sexual identity upon the asexual, dominant stage of fungal life history has been well studied. To investigate their impact, expression differences between strains of different mating type during asexual development were assayed, with RNA sampled from otherwise largely isogenic mat A and mat a strains of Neurospora crassaat early, middle, and late clonal stages of development. We observed significant differences in overall gene expression between mating types across clonal development, especially at late development stages. The expression levels of mating-type genes and pheromone genes were assayed by reverse transcription and quantitative PCR, revealing expression of pheromone and receptor genes instrains of both mating types in all development stages, and revealing that mating type (mat) genes were increasingly expressed over the course of asexual development. Interestingly, among differentially expressed genes, the mat A genotype more frequently exhibited a higher expression level than mat a, and demonstrated greater transcriptional regulatory dynamism. Significant up-regulation of expression was observed for many late light-responsive genes at late asexual development stages. Further investigation of the impact of light and the roles of light response genes in asexual development of both mating types are warranted.
Keywords: mating type, pheromone, transcription, light, conidiation, microarray
1. Introduction
The genetics of sexual identity in most fungi are conferred by mating-type loci that exhibit diversity in size, number, and sequence among different fungal groups (Lee et al., 2010). Within the life cycle of heterothallic fungi, mating occurs when hyphae/conidiospores (conidia) from opposite mating-type strains meet at a pre-sexual stage, leading a diploid stage after fusion of nuclei from both mating-types (Esser, 1971). The function of mating-type genes has been intensively studied in fungal models during crossing and sexual development (Glass and Lee, 1992; Saupe et al., 1996; Ferreira et al., 1998; Heitman et al., 2007). However, vegetative stages of the life cycle, encompassing hyphal growth, branching, anastomosis, and asexual sporulation, are generally dominant in fungal life histories.
In general, fungal mating type is not considered to have a significant impact on the growth or phenotypic characteristics of individuals (Coppin et al., 1993; Brasier, 1999), a finding supported by research conducted on diverse fungal species (Dudzinski et al., 1993; Ahmed et al., 1996; Bardin et al., 1997). Nevertheless, an association between mating type and fungal pathogenicity has been demonstrated Kolmer and Ellingboe, 1988; Funnell et al., 2001; Lin et al., 2006). Furthermore, in Neurospora, a superiority in perithecial production of mating-type a (mat a) strains was observed in both intraspecific and interspecific crosses (Dettman et al., 2003). Indeed, genetic differences between mating types relating to the mating loci and their up/downstream regulated pathways have been investigated intensively in Neurospora, yeasts, and some other fungi (Heitman et al., 2007), whereas orthogonal studies of spatial differentiation in colony development without consideration of mating type have been performed in Aspergillus niger and N. crassa (Levin et al., 2007; Kasuga and Glass, 2008; Greenwald et al., 2010). Recently, regulation of mating types on their target genes were investigated using genome wide gene expression profiling for heterothallic fungus Podospora anserina, and mating-type transcription factors were found to have impact on genes not directly related to mating in P. anserina as well as in Gibberella moniliformis (Keszthelyi et al., 2007; Bidard et al., 2011). Although genome-wide transcriptional profiling in N. crassa has been applied to identify genes expressed in diverse stages of development (Bell-Pedersen et al., 1996; Nelson et al., 1997; Zhu et al., 2001; Kasuga and Glass, 2008; Greenwald et al., 2010), it has not been applied to ascertain life history differences between mating types. Further characterization of gene expression associated with mating type in N. crassa will be of great interests for future studies of the basic phenomena of life such as mating, asexual and sexual reproduction, and mitotic recombination in fungi.
In the heterothallic model fungus N. crassa, a bipolar mating system is conferred by the idiomorphic mat locus, which encodes mat a and mat A in opposite mating types. The mat a-1 gene, encoding a single HMG box protein called MAT a-1, has been identified as the major mating regulator in mat a strains (Chang and Staben, 1994), although the additional transcribed ORF mat a-2 is also identified in this region (Pöggeler and Kück, 2000). The genes of the mat A idiomorph encode three proteins: MAT A-1, A-2 and A-3 which are characterized by MAT - HMG (Martin et al., 2010), PPF (Kanematsu et al., 2007), and MATA-HMG domains (Ferreira et al., 1996, 1998). Differential gene regulation for the two mating types should be traceable to differential regulation by genes at the mat locus, which, in trans, is known to regulate the expression of presumably mating-type specific pheromone precursors and receptors during the process of mating (Pöggeler and Kück, 2001; Bobrowicz et al., 2002; Kim et al., 200; Kim and Borkovich, 2004, 2006; Pöggeler, 2011). Knockout strains of mat loci also show no morphological differences from wild-type in N. crassa during vegetative growth (Ferreira et al., 1998). Nevertheless, pheromone genes are maintained in the genomes of true homothallic fungi like Neurospora africana, Sordaria macrospora, and Anixiella sublineolata, and are expressed in the asexual life cycle of N. africana, Gibberella zeae, and S. macrospora, suggesting an alternate or pleiotropic function in vegetative development (Kim et al., 2002; Pöggeler et al., 2006; Lee et al., 2008). Although mat genes are undergoing genetic decay in some homothallic species, including N. africana, the ORF of mat A-1 was intact in all these species (Wik et al., 2008). Perhaps in part due to poorly understood pleiotropic functions, previous studies have revealed conflicting results with regard to the expression of pheromone precursors in strains of different mating types. Pheromone precursor gene mfa-1 has been attributed specific function only in mat a, yet it can be detected at a low expression level in mat A tissues (Kim et al., 2002).
The functions of N. crassa mating type proteins and pheromone precursors and receptors in the mating processes are well studied. However, their functions during pre-mating asexual development are not clear. In the recent studies of Podospora anserina and Sordaria macrospora, mating type specific expression was observed for genes with diverse function, includingmetabolism, information pathways, transport, and developmental processes (Bidard et al., 2011; Klix et al., 2010). However, these studies focused on crossing and sexual development, and no core genes active in asexual development, such as the cell division cycle genes (cdc), conidiation genes (con), and heat shock protein genes (hsp), were found differently expressed between mating types. During asexual development, these genes are of critical function, and regulation of many these genes, including clock-controlled genes (ccg) and con genes, has not been well understood. In most organisms, circadian oscillators regulate the rhythmic expression of ccgs, and the two best characterized ccgs in Neurospora areccg-1 andccg-2, known as morning-specific genes. Whilethe precise function of ccg-1, which is conserved among filamentous fungi and can be induced by heat shock, is elusive, the gene ccg-2 encodes a secreted hydrophobic protein belonging to the hydrophobins, which coat the outer cell wall of fungi and maintain the cell-surface hydrophobicity for air dispersal of mature conidiospores (Bell-Pedersen et al., 1992; Vitalini et al., 2006). The production and release of conidiospores in fungi is also subject to the circadian clock, and daily rhythms in spore development and spore discharge are common in fungi (Bell-Pedersen et al., 1996). At least four con genes, including con-6, con-8, con-10, and con-11, are known to be expressed during development of three types of spores in N. crassa (Sachs and Yanofsky, 1991; Springer, 1993). Nevertheless, disruption of these genes does not cause a discernible phenotype in spore morphology, abundance of spores, conidial germination efficiency, nor ability to function as either parent in sexual crosses (Springer and Yanofsky, 1992; Springer, 1993).
In this study, we investigated the global expression differences between largely isogenic strains of different mating type. Mating-type specific expression of genes was quantified using genomic microarrays. The minimal differences in genetic background between highly isogenic mating types provided a straightforward system for investigating transcriptomic shifts of metabolic and regulatory function during morphological development. To maintain a controlled environment for investigating asexual development, the light-induced internal oscillator of N. crassa was repressed by a long treatment of constant light, and to avoid temperature-retained clock, a constant temperature was maintained through the experiment. Even under such controlled constant-light conditions, nominally light-responsive genes continue to play a central role in fungal development, so we investigated the behavior of light responsive genes (Chen et al., 2009) for clonal development under a condition of constant light. Furthermore, we performed reverse transcription and quantitative polymerase chain reaction (RT-qPCR) to determine, for the first time, expression of mat genes, the pheromone precursor genes ccg-4 and mfa-1, and the receptor genes pre-1 and pre-2 at different stages of clonal development in N. crassa.
2. Experimental procedures
2.1. Strains and conditions
N. crassa strains FGSC 4200 (mat a) and FGSC 2489 (mat A) were obtained from the Fungal Genetics Stock Center (Kansas City, MO). FGSC 4200 was derived from a long series of recurrent backcrosses to strain FGSC 2489, and is generally regarded as highly isogenic to the latter (Mylyk et al., 1974; Newmeyer et al., 1987; Perkins, 2004; McCluskey et al., 2010). The strains were grown on Bird Medium (Metzenberg, 2004) covered by a cellophane membrane (Fisher Scientific) at 26°C under constant light. Light was provided by Ecolux bulbs (F17T8.SP41-ECO, General Electric Company) amounting to a net intensity of 14μMol/m2S at the media surface, measured at wavelengths between 400nm to 700nm. Mycelia were harvested from 90 mm Petri dishes at 36 h, 60 h, and 96 h, corresponding to vegetative growth (colony-size 30-50mm), start to middle of conidia production (50-70mm), and post-peak of conidia production (90 mm), respectively. From 96 h to 144 h in Bird Medium, no protoperithecia were formed. After inoculation, fungal tissues (mycelia) that covered the surface of cellophane membrane were collected with razor blade, snap frozen in liquid nitrogen, and stored at -80°C. We compared the overall gene expression patterns of the whole clones, which are composed of vegetative hyphae and asexual reproduction structures, among different time points for strains of both mating types. Our sampled gene expression pools were thus presumably more heterogeneous than in other sampling methods that address gene expression associated with development of specific hyphal morphology. Nevertheless, we demonstrated improved resolution in identifying differentially regulated genes.
2.2. Multi-targeted priming (MTP) design
We used multi-targeted primers (MTPs), degenerate oligonucleotides complementary to mRNAs but not non-coding RNAs, to facilitate selective reverse transcription of mRNA and elimination of contamination by rRNA and tRNA, leading to improved microarray assay sensitivity (Adomas et al., 2010). Adomas et al. (2010) identified an MTP (VWNVNNBDKGGC) that exactly targets 9826 ORFs in N. crassa (85%), that additionally showed strong binding (GC) in the 3′ end. It inexactly primes reverse transcription of all other known transcribed genes as well (Adomas et al., 2010 and unpublished data).
2.3. Sample preparation and hybridization microarrays
Total RNA was extracted from homogenized tissue using TRI REAGENT kits (Molecular Research Center) for three to ten biological replicates for the same time point, and was pooled together for next step. Messenger RNA was purified using Oligo(dT) Cellulose Columns (Molecular Research Center) as in Clark et al. (2008). For reverse transcription, we used 2 μg of purified mRNA and 0.25 μg oligo(dT) mixed with 0.25μg Neurospora-specific MTP. The resulting cDNA was labeled reciprocally with cyanine dyes (Townsend and Hartl, 2002) and used for hybridization. To rule out any accidental contamination with tissue or RNA from the opposite mating type, detection of expression of mating loci mat A and mat a was used, validating the purity of all RNA samples. Twenty-six hybridizations were performed, including dye-swaps originating from independent reverse transcription reactions and some technical replicates where three or more comparisons were made (Fig. 1). Microarrays were composed of 70mer oligonucleotides synthesized by Illumina (San Diego) for 9,826 ORFs identified by the Broad Institute (http://www.broad.mit.edu/annotation/genome/neurospora) as in Kasuga et al. (2005), robotically printed on CMT gamma-aminopolysilane-coated glass slides (Corning, Corning, NY) at the Yale University Center for Genomics and Proteomics.
Figure 1.
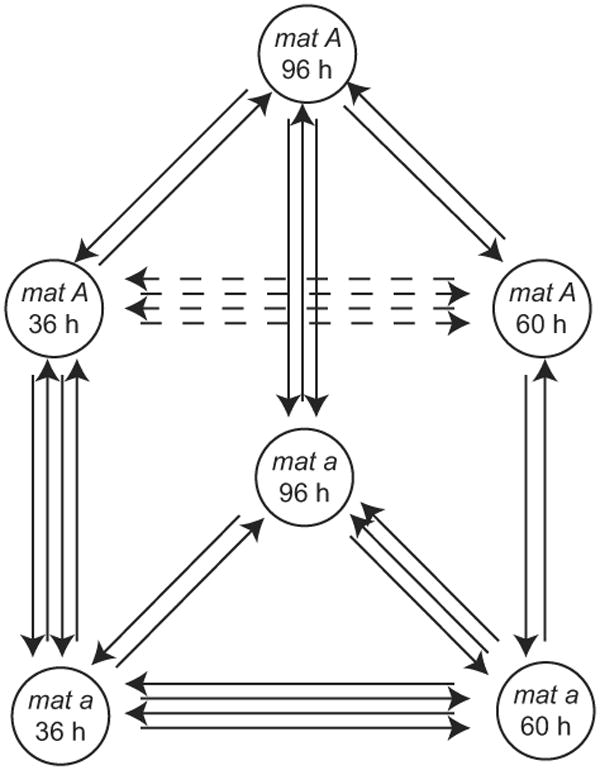
Experimental design for microarray comparisons of sex-specific gene expression during asexual development of Neurospora crassa. Each arrow represents an independent reverse transcription and microarray hybridization from numerous pooled biological replicates. Each arrowhead corresponds to the sample labeled with cyanine 5-dUTP, and each blunt end corresponds to the sample labeled with cyanine 3-dUTP. All samples were compared directly to neighboring time points, and the circuit design was closed by direct comparisons of 36 h to 96 h samples. Additionally, all time points featured direct comparisons between samples from strains of opposite mating type.
2.4. Microarray data acquisition and analysis
Hybridized microarray slides were scanned with a GenePix 4000B (Axon Instruments, Foster City, CA). Spots were located and expression was quantified by the GenePix 4000 software. All spots were verified by eye, and those with unusual morphology or with erratic signal intensity distribution were excluded from all analyses. Raw intensity was normalized as in Townsend (2004). Genes were considered expressed and deemed well-measured when the median spot foreground exceeded the median background plus two standard deviations of the background intensity. Normalized data were then statistically analyzed using Bayesian Analysis of Gene Expression Levels (BAGEL, Townsend and Hartl, 2002; Townsend, 2004). We additionally performed BAGEL analyses on subsets of the experiment. After normalization and BAGEL analyses, we obtained well-measured gene expression data for 4,491 genes out of 9,826 genes printed on the array (Table I, Supplemental Table S1). Well-measured genes were considered significantly differentially expressed when P ≤ 0.05.
Table I.
Numbers of genes well measured, and statistically significant, across the whole data set as well as subsets of nodes in the hybridization design.
| Feature | Whole set | 36-60 h | 60-96 h | A only | a only | |||
|---|---|---|---|---|---|---|---|---|
| Well-measured genes | 4491 | 4296 | 3403 | 3848 | 3553 | |||
| Gene detected only in the subset | NA* | 106 | 58 | 86(32)# | 11(6)# | |||
| Mating type | A | a | A | a | A | a | A | a |
| Genes significantly differentially expressed (p•0.05) | 2403 | 2012 | 1544 | 1411 | 1307 | 147 | 1601 | 1018 |
| Up-regulated genes for 36<60<96 (p•0.05) | 79 | 64 | NA | NA | NA | NA | 50 | 24 |
| Down-regulated genes for 36>60>96 (p•0.05) | 66 | 102 | NA | NA | NA | NA | 31 | 39 |
| Up-regulated genes for 36<60 (p•0.05) | 1190 | 1000 | 972 | 893 | NA | NA | 688 | 510 |
| Down-regulated genes for 36>60 (p•0.05) | 587 | 574 | 573 | 520 | NA | NA | 376 | 315 |
| Up-regulated genes for 60<96 (p•0.05) | 289 | 195 | NA | NA | 441 | 11 | 224 | 79 |
| Down-regulated genes for 60>96 (p•0.05) | 917 | 136 | NA | NA | 866 | 136 | 656 | 218 |
| GEL50 | 1.35 | 1.37 | 1.36 | 1.36 | 1.62 | 2.09 | 1.51 | 1.64 |
Not applicable.
Number of additional genes not detected as expressed at all time points; 36-60 h and 60-96 h results.
To quantify the power of the experiment, the gene expression level at which there was a 50% empirical probability of a significant call (GEL50) was estimated by a logistic regression of statistical significance against log2 fold-change (Townsend, 2004). To ensure that no subset of the data had greater experimental power due to technical differences in microarray quality, we examined the statistical powers for each subset. The GEL50 values were largely consistent among the subsets with one exception concerning the 60 to 96h subset (Table I). Thus, differences in numbers of detected genes were mostly biological in origin and not attributable to differences in quality of hybridization or statistical power applied to each subset.
2.5. Real-time quantitative PCR
Reporter oligonucleotides for genes mat a-1 and mat a-2 were not present on our microarray. Additionally, expression of mating type genes, pheromone precursor ccg-4, and receptor pre-2, was not detected with the microarray. Accordingly, we measured transcription of these genes and pheromone precursor mfa-1 and receptor pre-1 by reverse transcription and quantitative PCR. The function of mat a-2 has not been well characterized in Neurospora crassa, and we did not assess its expression level during asexual development in this study. RT-qPCR analyses were also used to verify microarray results on selected genes. For RT-qPCR analyses, 2μg of mRNA was reverse transcribed with Superscript II reverse transcriptase (Invitrogen, Carlsbad, CA) for each of two biological replicates, which were pooled mRNAs extracted from three to 10 plates under the same culture condition as for the microarray samples, and for three technical replicates with each of two concentrations (50nM and 300nM) of primers. Relative transcript abundance was quantified using the Applied Biosystems 7500 Fast Real-Time PCR System and SYBR Green PCR Master Mix (Applied Biosystems, Carlsbad, CA) according to the manufacturer's recommendations. The ATP citrate lyase and acyl-CoA dehydrogenase, whose expression was invariant in the two N. crassa strains in all time points for both mating types of the microarray experiment, were used as endogenous control references for qPCR. The comparative Ct (ΔΔCt) method (Livak and Schmittgen, 2001) was used to calculate transcript levels from triplicates. Real-time PCR normalization and standard error computation followed Applied Biosystems protocols (Table II) and statistical analyses were performed with BAGEL (Townsend, 2004) as well (Table I, Supplemental Table S1).
Table II.
Relative expression levels of pheromone precursors/receptors estimated with quantitative RT-PCR.
| Gene | NCU | mat a 36 h | mat a 60 h | mat a 96 h | mat A 36 h | mat A 60 h | mat A 96 h |
|---|---|---|---|---|---|---|---|
| ccg-4 | NCU02500 | 2.71±1.20 | 1.00 | 2760±1397 | 45.15±19.27 | 131.69±43.28 | 10078±3507 |
| mfa-1 | NCU01257 | 1.00 | 56.57±5.53 | 15.15±6.33 | 3.07±0.90 | 4.11±0.25 | 41.32±5.23 |
| pre-1 | NCU00138 | 1.00 | 2.30±0.36 | 14.96±7.06 | 2.01±0.72 | 8.09±1.45 | 5.57±1.02 |
| pre-2 | NCU05758 | 1.00 | 1.05±0.08 | 2.32±1.03 | 1.21±0.40 | 1.38±0.49 | 1.52±0.47 |
2.6. Functional and phylogenetic classification of genes with mating type dependent gene expression
The Functional Catalogue (FunCat: http://mips.helmholtz-muenchen.de/proj/funcatDB/) provided groupings of genes according to their cellular or molecular functions (Ruepp et al., 2004). We partitioned genes that showed monotonically increasing or monotonically decreasing patterns in expression for each mating type. We examined p-values for the comparisons of the neighboring stages (i.e., 36 h vs. 60 h and 60 h vs. 96 h) to identify genes showing increasing or decreasing patterns with credibility higher than 95% for each comparison (TableIII). Statistical significance of overrepresentation of gene groups in functional categories relative to the whole genome was calculated by applying the hypergeometric distribution to the MIPS gene groups (http://mips.gsf.de/proj/funcatDB/help_p-value.html). Further functional analysis of genes and pathways with significantly differential expression was performed using biochemical pathways and annotation from the Kyoto Encyclopedia of Genes and Genomes (KEGG, Kanehisa and Goto, 2000).
Table III.
Table III-A. Functional categories that were enriched for genes exhibiting increasing expression, for each mating type.
| Total number of genes in A = 78 out of 79, and in a =61 out of 64 annotated in FunCat. | No. of genes in A | No. of genes in a | P-value | |||
|---|---|---|---|---|---|---|
| Functional Category | Observed | Expected | Observed | Expected | mat A | mat a |
| 01.25.01 extracellular polysaccharide degradation | 3 | 0.1404 | 0 | NA | 0.0004 | NA |
| 01.04.04 regulation of phosphate metabolism | 3 | 0.1482 | 0 | NA | 0.0005 | NA |
| 01.05.03 polysaccharide metabolism | 6 | 1.3026 | 3 | 1.83 | 0.0019 | 0.0827 |
| 01.25 extracellular metabolism | 3 | 0.3042 | 0 | NA | 0.0036 | NA |
| 32.10.07 degradation / modification of foreign (exogenous) polysaccharides | 2 | 0.0936 | 0 | NA | 0.0044 | NA |
| 01.05 C-compound and carbohydrate metabolism | 13 | 5.8812 | 7 | 4.27 | 0.0052 | 0.1739 |
| 30.01.05.01.06 serine/threonine kinase | 2 | 0.1326 | 1 | 0.61 | 0.0084 | 0.1037 |
| 20.03 transport facilities | 7 | 2.3478 | 4 | 2.44 | 0.0089 | 0.1110 |
| 32.10 degradation / modification of foreign (exogenous) compounds | 2 | 0.1482 | 0 | NA | 0.0103 | NA |
| 01.06.10 regulation of lipid, fatty acid and isoprenoid metabolism | 2 | 0.1638 | 0 | NA | 0.0124 | NA |
| 34.01.01.01 homeostasis of metal ions (Na, K, Ca etc.) | 4 | 0.9048 | 0 | NA | 0.0128 | NA |
| 30.01.05.01.02 JNK cascade | 1 | 0.0078 | 0 | NA | 0.0154 | NA |
| 32.05.01.03 chemical agent resistance | 2 | 0.195 | 0 | NA | 0.0171 | NA |
| 10.03.02 meiosis | 3 | 0.5772 | 0 | NA | 0.0203 | NA |
| 34.11.03 chemoperception and response | 5 | 1.6146 | 3 | 1.83 | 0.0229 | 0.1330 |
| 01.04 phosphate metabolism | 6 | 2.223 | 4 | 2.44 | 0.0236 | 0.0957 |
| 20.01.01.07 anion transport | 2 | 0.234 | 0 | NA | 0.0238 | NA |
| 20.01.01 ion transport | 4 | 1.1544 | 1 | 0.61 | 0.0283 | 0.5985 |
| 16.17.03 potassium binding | 1 | 0.0234 | 0 | NA | 0.0306 | NA |
| 20.01.01.07.09 chloride transport | 1 | 0.0234 | 0 | NA | 0.0306 | NA |
| 30.05.01.18 transmembrane receptor protein serine/threonine kinase signaling pathways | 1 | 0.0234 | 0 | NA | 0.0306 | NA |
| 02.25 oxidation of fatty acids | 2 | 0.2886 | 0 | NA | 0.0348 | NA |
| 10.03.01.01.11 Mphase | 2 | 0.3354 | 0 | NA | 0.0455 | NA |
| 20.03.01 channel / pore class transport | 2 | 0.2028 | 3 | 1.83 | 0.0184 | 0.0006 |
| 32.07.07 oxygen and radical detoxification | 1 | 0.234 | 3 | 1.83 | 0.2146 | 0.0008 |
| 01.20.17.09 metabolism of alkaloids | 2 | 0.1092 | 2 | 1.22 | 0.0058 | 0.0036 |
| Table III-B. Functional categories that were enriched for genes exhibiting decreasing expression, for each mating type. | ||||||
|---|---|---|---|---|---|---|
| Total number of genes in A = 61 out of 66, and in a = 95 out of 102 annotated in FunCat. | No. of genes in A | No. of genes in a | P-value | |||
|
| ||||||
| Functional Category | Observed | Expected | Observed | Expected | mat A | mat a |
| 32.05.05 virulence, disease factors | 1 | 0.366 | 4 | 3.8 | 0.311 | 0.003 |
| 01.01.09.01.02 degradation of glycine | 0 | NA | 2 | 1.9 | NA | 0.004 |
| 01.01.09 metabolism of the cysteine - aromatic group | 1 | 0.6832 | 5 | 4.75 | 0.499 | 0.004 |
| 20.01.07 amino acid/amino acid derivatives transport | 1 | 0.2135 | 3 | 2.85 | 0.197 | 0.005 |
| 01.01.06.04 metabolism of threonine | 1 | 0.0671 | 2 | 1.9 | 0.070 | 0.005 |
| 20.03.02.02.02 sodium driven symporter | 0 | NA | 1 | 0.95 | NA | 0.009 |
| 01.01.06.06.01 biosynthesis of lysine | 1 | 0.0915 | 2 | 1.9 | 0.093 | 0.010 |
| 01.01.09.01 metabolism of glycine | 0 | NA | 2 | 1.9 | NA | 0.011 |
| 01.06.02.02 glycolipid metabolism | 0 | NA | 2 | 1.9 | NA | 0.011 |
| 01.01.09.02 metabolism of serine | 1 | 0.1098 | 2 | 1.9 | 0.109 | 0.014 |
| 01.01.06.06 metabolism of lysine | 1 | 0.1586 | 2 | 1.9 | 0.152 | 0.027 |
| 01.01 amino acid metabolism | 1 | 1.9276 | 7 | 6.65 | 0.861 | 0.031 |
| 14.13.01 cytoplasmic and nuclear protein degradation | 1 | 0.7686 | 4 | 3.8 | 0.540 | 0.032 |
| 01.01.06.05 metabolism of methionine | 1 | 0.183 | 2 | 1.9 | 0.172 | 0.034 |
| 42.10.07 nucleolus | 0 | NA | 1 | 0.95 | NA | 0.037 |
| 01.01.06.04.01 biosynthesis of threonine | 1 | 0.0244 | 1 | 0.95 | 0.030 | 0.046 |
| 34.01.03.03 homeostasis of phosphate | 0 | NA | 1 | 0.95 | NA | 0.046 |
| 20.09.04 mitochondrial transport | 4 | 0.549 | 2 | 0.855 | 0.002 | 0.212 |
| 20.03 transport facilities | 6 | 1.8361 | 4 | 2.8595 | 0.010 | 0.321 |
| 14.07.02.02 N-directed glycosylation, deglycosylation | 2 | 0.1525 | 0 | NA | 0.011 | NA |
| 01.05.03 polysaccharide metabolism | 4 | 1.0187 | 4 | 1.5865 | 0.019 | 0.076 |
| 02.07.01 pentose-phosphate pathway oxidative branch | 1 | 0.0244 | 0 | NA | 0.030 | NA |
| 32.07.07.05 peroxidase reaction | 1 | 0.0244 | 0 | NA | 0.030 | NA |
| 34.07.01 cell-cell adhesion | 1 | 0.0305 | 0 | NA | 0.036 | NA |
| 20.09 transport routes | 8 | 3.8491 | 7 | 5.9945 | 0.037 | 0.394 |
| 01.05 C-compound and carbohydrate metabolism | 9 | 4.5994 | 10 | 7.163 | 0.038 | 0.178 |
| 14.07.02 modification with sugar residues (e.g. glycosylation, deglycosylation) | 2 | 0.3355 | 0 | NA | 0.045 | NA |
| 20.01 transported compounds (substrates) | 8 | 4.0077 | 8 | 6.2415 | 0.045 | 0.286 |
| 20.03.02.03.01 proton driven antiporter | 1 | 0.0427 | 0 | NA | 0.047 | NA |
In this study, genes exhibiting different expression patterns between mating types during clonal development were mapped onto the classification of Kasuga et al. (2009) for their phylogenetic distribution (Table IV). Genes were classified into six mutually exclusive clade-specific groups: Eukaryote/Prokaryote-core, Dikarya-core, Ascomycota-core, Pezizomycotina-specific, N. crassa-orphans, and “others”. Three threshold values of length-adjusted protein identity were used by Kasuga et al. (2009) to group genes. We used the classification based on the lowest threshold values, which provided us the strictest list of Neurospora orphan genes from that study.
Table IV.
Expression patterns of genes with different levels of phylogenetic conservation.
| Phylogenetic classes of N. crassagenome (9127 genes)* | No difference between Mat-A and - a | Mat-A no change | Mat-a no change | Other differences | SUM** | |||
|---|---|---|---|---|---|---|---|---|
|
|
||||||||
| Mat-a up | Mat-a down | Mat-A up | Mat-A down | |||||
| NC orphans (24%) | 301 | 14 | 21 | 48 | 43 | 60 | 186 (11%) | 487 (12%) |
| Pezizo-specific (35%) | 701 | 39 | 79 | 127 | 84 | 151 | 480(27%) | 1181 (28%) |
| Ascomycota-core (2%) | 38 | 1 | 4 | 5 | 2 | 8 | 20 (1%) | 58 (1%) |
| Dikarya-core (11%) | 301 | 21 | 32 | 50 | 52 | 63 | 218 (11%) | 519 (12%) |
| Euk/Prok-core (26%) | 1000 | 64 | 164 | 194 | 123 | 257 | 802 (47%) | 1802 (43%) |
| others (2%) | 52 | 4 | 7 | 12 | 9 | 15 | 47 (3%) | 99 (2%) |
| 143 (8%) | 307 (18%) | 388 (24%) | 313 (18%) | 554 (32%) | 1705 | |||
| 2339 (59%) | 143 (3%) | 307 (8%) | 388 (9%) | 313 (8%) | 554 (13%) | 4146 analyzed | ||
Follows Kasuga et al. (2009).
Percentage of genes detected for each category was calculated based on total genes analyzed (4146 genes) and genes (highlighted) showing different expression patterns between mating types (1705 genes).
2.7. Supporting information and the Neurospora functional genomics microarray database
The complete expression data set is available in the supporting information (Table SI), on the Townsend Lab web site (http://www.yale.edu/townsend/), and in the filamentous fungal gene expression database (FFGED, Zhang and Townsend, 2010). NCU numbers correspond to the second NCU version of N. crassa genome annotation, unless otherwise specified. Specific primers designed for the RT-qPCR were provided as well (Supplemental Table S3). The data set is also available at the Gene Expression Omnibus of the National Center for Biotechnology Information (GEO http://www.ncbi.nlm.nih.gov/geo/) as accession GSE26209.
3. Results
3.1. Genome-wide quantified expression of genes associated with asexual development
A total of 4491 genes were well measured during asexual development (Table I), including genes playing key roles in asexual development. Among 3848 genes expressed in mat A during asexual development, 86 genes exhibited no detectable expression in any stage inmat a. Among 3553 genes expressed in mat a, 11 genes were not detected in any stage in mat A. Among 4296 genes detected for both mating types from 36 h to 60 h of development, expression of 106 genes was not detected at 96 h stage for at least one mating type. Among 3403 genes substantially expressed in both 60 h and 96 h stages, expression of 58 genes was not detected at 36 h stage for at least one of the mating types. We found 106 out of 4296, 58 out of 3402, 86 out of 3847, and 11 out of 3553 genes only detected in the subsets respectively (Table I). For 36 h, 60 h, and 96 h comparison between mating types, we observed 44, 61 and 469 genes expressed significantly higher in mat A strain while 233, 159, and 744 genes expressed significantly higher in mat a strain (Table I, Supplemental Table S1).
Genes of special interest in asexual development, including numerous hsp, cdc, con, ccg genes, and known transcription factors, showed significant changes of expression level across clonal development. Expression of ccg-1, ccg-2, and three con genes, con-6, con-8, and con-10, was well measured in this study (Table I, Supplemental Table S4). The expression of ccg-1 was generally similar between mating-types, but a significantly decreased expression during asexual development for ccg-2 was observed for only mat a in both microarray and RT-qPCR experiments. The expression pattern of con genes was also similar between mating-types, including down-regulated expression of con-6 and up-regulated expression of con-8 across asexual development. Genes of interest were further verified by RT-qPCR for all three time points (Table S2). Among the 17 selected genes, 9 genes, including hsp101, cdc3, cdc4a, cdc4b, C2H2 TFs (CRE-repressor, and fle), con-8, ccg-1, and ccg-2, exhibited corresponding expression patterns via RT-qPCR assay to those estimated from the microarray experiment, but with higher fold-changes. While the microarray result was not statistically significant, the genes encoding transcription factors NCU04179 and NCU03184 both exhibited up-regulation by RT-qPCR. Conidiation-related gene con-10 exhibited an increasing expression at later development stages for both mating types in the microarray experiment, and our RT-qPCR results agreed with this result except that highest expression for con-10 was measured at 36 hr for the mat a strain. Expression of some genes that were not detected by the microarray experiment in either mating type, like hsf2, or that were detected in just one of the mating types, such as cdc6, cdc12, tea1, and the ste18-like gene NCU00041, were detected by RT-qPCR. RT-qPCR provides substantial validation of microarray results, as it is a more sensitive assay than a microarray for detection of differences in expression for genes expressed at low levels (Wang et al., 2006), especially in this study, when more biological replicates were assayed performing real time PCR.
3.2. Real-time quantitative PCR for mating type genes, pheromone precursors and receptors
During asexual development, all mating type genes monotonically increased in expression during the course of asexual development (Fig. 2). In the mat A strain, the expression of all three mat A coding genes, mat A-1, mat A-2, and mat A-3, increased between 36 h and 96 h, and a 250 fold increase was measured for mat A-2 from 36 h to 96 h. The mat a-1 (GenBank P36981.2) gene exhibited a steady and linear increase in expression compared to the A mating type genes. Pheromone precursors ccg-4 and mfa-1 are regulated separately by mat A-1 and mat a-1 (Nelson et al., 1997; Bobrowicz et al., 2002; Kim et al., 2002; Kim and Borkovich 2006), and we observed previously reported expression patterns, but also found the pheromones and receptors to be regulated in the other mating-type (Table II). Consistent with previous studies, different levels of expression of pheromone precursors ccg-4 and mfa-1, and the receptors pre-1 and pre-2, were observed in all samples from both mating types (Table II). For the mat A strain, expression of ccg-4 is regulated by the mat A-1 gene products with a circadian rhythm (Bobrowicz et al., 2002). Our experiments demonstrated a strong correlation between the increasing expression patterns of ccg-4 andmat A-1, with a 3-fold increase of ccg-4 from 36 h to 60 h and a nearly 80-fold increase from 60 h to 96 h in mat A. A less than statistically significant increase (about 1.2 to 1.5 in fold change) of pre-2, the putative receptor of ccg-4, was observed. We detected about a ten-fold increase in expression of mfa-1, a pheromone precursor generally considered to be mat a specific, at the late development stage of the mat A strain. For the gene pre-1, the putative receptor of the MFA1 pheromone, a peak at 60 h samples of mat A was observed. For the mat a strain, expression of mfa-1 is probably regulated by the mat a-1 product and by the circadian clock (Bobrowicz et al., 2002). However, transcription of mfa-1 peaked at 60 h, then dropped significantly, while transcription of mat a-1 peaked at 96 h. Interestingly, transcription of both pre-1 and pre-2 increased during clonal development, peaking at 96 h in the mat a strain.
Figure 2.
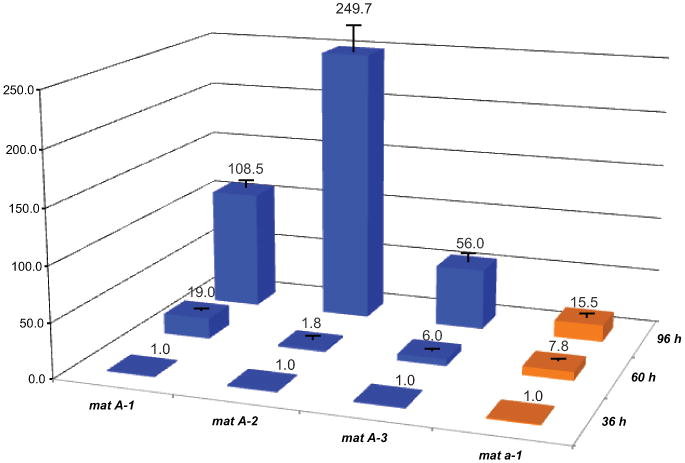
Real-time PCR estimates of the expression of mat genes in mating type A and a strains of Neurospora crassa at three time points during asexual development.
Transcription of mfa-1 and pre-1 was also measured with microarray analysis, and neither gene showed a significant change in transcription during the asexual development in either mating type, which was not consistent with our RT-qPCR results. Expression of pheromone precursors ccg-4 and mfa-1 was known to be low in vegetative growth and high in conidia (Bobrowicz et al., 2002; Kim et al., 2002; Kim and Borkovich 2006), but expression of these genes was not well-measured in reproduction of conidia using the same microarray (Kasuga and Glass, 2008). As our RT-qPCR results for all pheromone precursors/receptors and mating-type genes were based on more biological replicates, and showed corresponding expression patterns in that they were all significantly up-regulated during conidiation, we deemed the RT-qPCR measurements of transcription for these genes to be more accurate.
3.3. Up- and down-regulation of gene sets within functional categories of genes
Significant enrichment in different functional categories was observed for genes showing different expression patterns between mating types. The expression of 79 and 64 genes increased significantly, and 66 and 102 genes decreased significantly, across asexual development for mat A and mat a, respectively. Of those, we found 78, 61, 61, and 95 respectively, in the Functional Category database (Ruepp et al., 2004; Table III). Seventy-eight genes of increasing expression in mat A were classified into 25 functional categories as significantly enriched (P < 0.05, Table II Table III A), including genes involved in homeostasis of metal ions and meiosis. In contrast, for 61 genes of increasing expression in mat a, only three functional categories exhibited significant enrichment. In other words, the number of Funcat categories that showed significant enrichment was much higher for mat A, and most of these categories were not shared between mating types. Three categories exhibited significant enrichment in both: channel/pore class transport, oxygen and radical detoxification, and metabolism of alkaloids. For genes with decreasing patterns, 95 genes were classified into 17 functional categories as significantly enriched in mat a (Table III B). These included virulence and disease factors and metabolism of the cysteine – aromatic group. However, none of these categories was enriched with down-regulated genes in mat A, except for the biosynthesis of threonine. Down-regulated genes in mat A were enriched for 12 other functional categories, including mitochondrial and nuclear transport.
3.4. Expression of light response genes during the clonal development
Genes in N. crassa that are known to respond to induction by light exhibited a similar pattern of early and late induction, albeit over a much longer period of time, during asexual development under constant light. More than 300 early light responsive genes (ELRGs) and late light responsive genes (LLRGs) are known to change in expression after light induction (Chen et al., 2009). From 126 ELRGs and 157 LLRGs on the N. crassa array, expression of 59 ELRGs (Table S5) and 80 LLRGs (Table S6) were detected during asexual development for both mating types (Fig. 3). Among the 59 ELRGs detected, there were 13 genes in the mat a genotype and 25 genes in the mat A genotype exhibiting significant up-regulation from 36 h to 96 h (P < .05). The largest increase in expression was observed for a NonF related protein NCU06603 (2.7 in mat a and 3.1 in mat A) and for a probable cyanate lyase NCU01258 (1.8 in mat a and 2.7 in mat A). There were 11 genes in the mat a genotype and 12 genes in the mat A genotype showing significant down-regulation from 36 h to 96 h (P < .05). The largest decrease in expression was of a probable NADPH2 dehydrogenase NCU04452 (1.3 to 2.3-fold) in both mating types. Among the 80 LLRGs detected, there were 52 genes in the mat a genotype and 55 genes in mat A showing significant up-regulation from 36 h to 96 h (P < .05). The largest differences across time points in expression were of con-8 (16-fold in mat a and 19-fold in mat A), NCU05490 (7-fold in mat a and 16-fold in mat A), and NCU00322 (14-fold in mat a and 7.5-fold in mat A). Changing expression across asexual development in LLRGs was widespread. There were more than 20 LLRGs in both mating types that exhibited up-regulation over 3-fold from 36 h to 96 h, but we found no LLRGs showing significant down-regulation from 36 h to 96 h.
Figure 3.

Heat maps depicting comparative expression level (log2 ratio) of the early light responsive genes (ELRGs, Chen et al., 2009) and the late light responsive genes (LLRGs, Chen et al., 2009) in strains of mating type A and a of Neurospora across three time points of asexual development. Lighter color corresponds to a higher level of expression.
3.5. Phylogenetic depth of genes differentially expressed between mating types in asexual development
Because mating-type genes are fast evolving, it is helpful to the elucidation of their functional interactions with the rest of the genome to examine the phylogenetic depth of genes differentially expressed between mating types. Mating-type impacted expression of genes evolving at diverse rates. Divergent expression patterns for mating types during clonal development from this study were mapped onto the phylogenetic distribution classification of Kasuga et al. (2009, Table IV). Many differences between the two mating types in gene expression during asexual development did not correspond to fast-evolving, novel Neurospora- specific genes. Most expression differences between mat A and mat a were detected in genes with orthologues present in the core genomes of Eukaryotes and Prokaryotes (47%, 802 out of a total 1705 significant differences observed). A lower frequency of differential expression between mat A and mat a was observed for Pezizomyceta-specific genes (27%) and Neurospora orphan genes (11%).
4. Discussion
We have provided the first genomic characterization of gene expression differences during clonal development within both mating types of N. crassa. Gene expression levels for both mating types throughout vegetative development were characterized for mating type genes, pheromone precursors and receptors, for genes related to conidiation, for light responsive genes, and for genes showing different levels of phylogenetic affiliation (level of conservation) within eukaryotes. In general, fungal mating type has not been thought to affect individual fitness during clonal development, perhaps because morphological differences have not been observed between mating types of model filamentous fungi such as Neurospora crassa. However, genes close to the mating type locus have shown mating type-specific expression during sexual development in N. crassa (Randall and Metzenberg, 1998). Mating type genes' impacts on genes other than pheromone precursors and receptors have been reported in Fusarium, Sordaria and Podospora species (Pöggeler et al., 2006; Keszthelyi et al., 2007; Klix et al., 2010; Bidard et al., 2011). Our results demonstrate significant differences in gene expression between N. crassa strains of differing mating type during asexual development. Corresponding relations between mating types and fungal asexual development in diverse fungi have been previously reported (Kolmer and Ellingboe, 1988; Funnell et al., 2001), and, moreover, mating types are well known to exhibit differences in superiority of crossing and sexual development in Neurospora species (Dettman et al., 2003). In this study we made use of largely isogenic strains of N. crassa mating types. Sequence and hybridization data show that even the centromere-proximal flanks of the mat A and mat a idiomorphs are highly similar for N. crassa (Randall and Metzenberg, 1995), but full verification for the isogenic status requires genome sequencing of the mat a strain.
4.1. Regulatory interactions with mating type complicate conidial gene expression
Expression levels of the two best-characterized ccgs in N. crassa, ccg-1 and ccg-2, appear to be regulated by non-clock controlled factors under a constant light condition. Genes ccg-1 and ccg-2 are known as morning-specific genes, and were well measured in this experiment (Table S2,S4). The gene ccg-2 decreased significantly in expression in mat a during conidiation (Table S2, S4). Decreased expression of ccg-2 could be associated with lower conidial dispersal; in any case, the divergent expression between mating types is challenging to explain unless mating types have a proclivity toward playing different roles during life cycle in the natural ecosystem. For example, opposite mating types growing in the same environment may time their asexual and sexual reproduction differently to maximize mating success. In a daily rhythmic light environment, expression of ccg-1 and ccg-2 are regulated by frq-oscillator and some other factors (Arpaia et al., 1995; Vitalini et al., 2004). The observed divergence in expression between mating types suggests that there are likely regulators other than frq, wc-1, and wc-2, for expression of ccg-1 and ccg-2.
Typically, genes involved in conidia reproduction (con) were similarly expressed between the two mating types. Expression of three con genes, con-6, con-8, and con-10 was measured across asexual development for both mating types in this study (Table S4). The gene con-6 is expressed upon induction of conidiation, reaching high levels at the late stages of conidiation (White and Yanofsky, 1993) and in mature conidia, but is not detectably expressed in mycelium. The down-regulated expression pattern for con-6 found in this study was the same pattern as described by Greenwald et al. (2010), but differed from other studies that did not maintain a constant light environment (White and Yanofsky, 1993). The genes con-8 (up-regulated) and con-10 (no significant change) were also expressed in our study in a similar pattern to that described by Greenwald et al. (2010). For the gene con-8, there is no clarity about its function in conidiation and no evidence its expression is regulated by light or clock-controlled elements. Previous studies in different light environments also demonstrate that con-8 is expressed in older cultures at a high level (Roberts and Yanofsky, 1989; Sachs and Yanofsky 1991).
4.2. Mating type genes showed no mating-type specific regulation on expression of pheromone precursors and receptors
Transcription of pheromone genes could be more complex than just mating-type specific. Although mating-type specific expression of pheromone precursors was well established with mating-type knockouts (Bobrowicz et al., 2002), co-expression ofccg-4 and mfa-1 has been observed in wild type strains of mat A and mat a and mutant mat a-1m33 (Zhu et al., 2001; Bobrowicz et al., 2002; Kim et al., 2002). The co-expression of both ccg-4 and mfa-1 observed in this study implies transcription of these genes is not solely dependent on transcription of either mat A-1 or mat a-1 separately. Δccg-4 mat A and Δmfa-1 mat a strains completely lose male fertility, a trait attributable to the production of conidia that fail to induce trichogynes from female structures in opposite mating types (Kim et al., 2002; Kim and Borkovich, 2006). Δccg-4 mat a or Δmfa-1 mat A strains exhibit normal chemotropic attraction and male fertility and show no detectable phenotypic changes in their vegetative growth (Kim and Borkovich, 2006). Examination of female fertility of those mutants revealed that Δccg-4, Δmfa-1 and Δccg-4Δmfa-1 double mutants in mat A and mat a exhibit normal protoperithecial development (Kim and Borkovich, 2006). Based on observed differential transcription patterns of pheromone receptor genes, pre-2 and pre-1, in and between mating types in this study and previous studies (Pöggeler and Kück, 2001; Kim et al., 2002; Kim and Borkovich, 2004), regulation of transcription of pre-1 and pre-2 appears to be independent from the transcription of pheromone precursors. However, mRNA level of pre-2 was more consistent during asexual development than pre-1 between mating types. Unlike previous studies measuring pheromone precursors only in conidia and pre-conidiation mycelium in liquid cultures, we examined transcription of pheromone genes from the whole clone composed of a mixture of tissues undergoing vegetative growth and conidiation. Comparatively low transcription of ccg-4 in mat a (about 27% of that in mat A at the highest expression) and high expression of mfa-1 in mat A (about 72% of mfa-1 in mat a at the highest expression) during asexual development deserve further investigation to better characterize basal level of expression and the extent of stochastic variation in transcription of these genes. Identification of the location of the transcripts of these genes in surface hyphae and other mycelial tissues is thus called for. Interestingly, the transcription of mfa-1 in the mat a strain and its receptor pre-1 in mat A strain showed the same pattern across asexual development, with peak transcription at the 60 h stage, while transcription of ccg-4 and pre-2 also had matching patterns, but peaked at 96 h in mat A and mat a strains. This difference in peak transcription implies that there may be different timings of mating proclivity between mating types. If such differences operate in nature, it would challenge the paradigm of the independence of genetic mating type (mat A, mat a) and phenotypic gender (conidia or protoperithecia as in Neurospora crassa) in fungi. However, pre-pro-pheromones encoded by ccg-4 or mfa-1 in ascomycetes are translated and further processed to become mature pheromones excreted by appropriate cells (Pöggeler, 2011), and post-transcriptional repression of the expression of pheromone genes by opposite mating type was demonstrated in P. anserina (Coppin et al., 2005). Processing of pre-pro-pheromones in N. crassa is not yet well annotated, and transcription of many genes similar to those involved in S. cerevisiae pheromone processing, including endopeptidase (NCU03219), carboxypeptidase (NCU04316), and farnesyltransferase (NCU05999), was not detected in this study. However, transcription of genes similar to S. cerevisiae pheromone processing genes ram2 (NCU03632), ste13 (NCU02515), ste14 (NCU00034), ste24 (NCU03637), and AXL1 (NCU00481), was well measured, and transcription of NCU03637, NCU00034, and NCU00481 all showed significant increase during the asexual development in both mating-types (Table S1). Thus, further investigation of pheromone molecule concentration is required to test if transcription levels of pheromone precursors measured in this study directly reflect the protein level of mature pheromones in the system.
As our experiment was performed with samples exposed to constant light and temperature, the impact of the circadian clock on pheromone precursor genes is presumably muted. The overall transcription of mating locus and corresponding pheromone precursor genes and receptor genes was, however, clearly correlated with development stages. Intriguingly, these mating-type specific genes exhibit dramatic changes in transcription during “asexual” development. Accordingly, late asexual development may best be regarded as a time of pre-sexual development, where genetic preparation for mating and sexual reproduction starts. However, development of protoperithecia in N. crassa requires different growth conditions (Adomas et al., 2010), and we did not include any samples of protoperithecia in this study.
4.3. Functional category classification implies a need for further evaluation of the effects of mating type in studies of asexual development
Enrichments of genes in functional categories observed in this experiment imply distinct genetic regulatory mechanisms in mat A and mat a strains, mechanisms which up-regulate certain groups of genes during colony expansion in mat A genotypes, and mechanisms that down-regulate other groups of genes in mat a genotypes. Most genetic studies with Neurospora crassa have been carried out using the mat A mating type strains with little concern for the effect of this choice of genetic locus for sexual identity. However, our study shows that “asexual” development of N. crassa does have cryptic “sexual” components. These subtle differences associated with mating type will have implications for designing experiments for studying the genetic basis of asexual phenomena, such as conidiospore formation and protoperithecia development.
4.4. Expression of early and late light responsive genes during clonal development
Early and late light responsive genes are expressed differently during clonal development in both mating types. Light is suggested to play a critical role, via circadian systems in the mediation of sexual and asexual development in N. crassa, and the genetic basis of light responses in fungi has been studied for decades (Dunlap and Loros, 2004; Purschwitz et al., 2006, 2008; Chen et al., 2009). A constant light condition is generally used in the study of reproduction and associated morphological development in N. crassa and some other fungi (Hallen et al., 2007; Kasuga and Glass, 2008), yet the systemic effect of a constant light environment on N. crassa has not been examined. Underlying the complicated nature of biological reaction toward light, about 340 early light responsive genes (ELRGs) and late light responsive genes (LLRGs), over 92% of the total identified light-responsive genes, exhibit demonstrated changes in response to light (Chen et al., 2009). In experimental exposures to 4 hours of constant light following a long period of constant darkness, the ELRGs peaked between 15 and 45 min after the onset of light, and LLRGs peaked later: between 45 and 90 min (Chen et al., 2009). Although expression of wc-1, wc-2, and related frq were not measurably detected in this experiment, differences in expression patterns across asexual development between ELRGs and LLRGs were clear (Fig. 3, Table S5,S6).
We found that some LLRGs appear to be critical for late asexual development inNeurospora crassa. Expression of LLRGs is suggested to be regulated by light, probably via ELRGs (Chen et al., 2009). Via these mechanisms, the fungus is capable of sensing the light environment in a daily manner and is prepared to respond developmentally when appropriate levels of light and/or an appropriate stage of asexual development are reached. In Fusarium verticillioides, a fungal pathogen closely related to N. crassa, transcript levels of genes involved in photo-inducible carotenoid biosynthesis and of a green-light sensor similar gene carO were significantly reduced in a •FvMAT1-2-1 (mat a) knockout mutant (Keszthelyi et al., 2007; Adám et al., 2011). Our data showed a significant down-regulation for a carotenoid biosynthesis gene al-1 (NCU00552) in mat A, while a consistent expression level of this gene in mat a during the asexual development in N. crassa.
To summarize, mating loci were expressed at increasing levels during asexual development, and expression of pheromone precursors ccg-4 and mfa-1 and receptors pre-1 and pre-2 were detected in both mating types in all development stages. Moreover, expression of mating-type-specific pheromone-related genes occurred at different levels in the two mating types. This mating-type specific pheromone-related gene expression may relate to divergent roles of mating types in pre-sexual development and crossing. These divergent roles are also supported by the observation of significant differences in overall gene expression between the mating types across asexual development, especially at the late development stage prior to sexual differentiation. The mat A strain tended to exhibit heightened expression for many of the differentially expressed genes, and typically exhibited more dynamic regulation of gene activity. As we did not use completely isogenic strains of the two different mating-types, some of the differences observed may be attributed to other genetic variations between the two strains. Our results also call for further investigation of the impact of light and roles of light response genes in asexual development in this fungus, given significant up-regulation of expression was observed for many late light responsive genes at late asexual development stages in both mating types.
Supplementary Material
Supplemental Table 1 (Table S1): Gene expression for mat A and mat a of Neurospora crassa across 36hr, 60hr, and 96hr in asexual development.
Supplemental Table 2 (Table S2): Expression of selected genes measured with quantitative PCR. Statistical analysis was performed with BAGEL (Townsend, 2004).
Supplemental Table 3 (Table S3): Primers for genes investigated with quantitative RT- PCR.
Supplemental Table 4 (Table S4): Expression of genes of interest across asexual development in Neurospora crassa.
Supplemental Table 5 (Table S5): Expression of 59 Early Light Responsive Genes (ELRGs) in both mating types of Neurospora crassa.
Supplemental Table 6 (Table S6): Expression of 80 Late Light Responsive Genes (LLRGs) in both mating types of Neurospora crassa.
Highlights.
> Genome-wide, genes in mat A more frequently exhibited a higher expression level than genes in mat a. > Genome-wide, mat A demonstrated greater transcriptional regulatory dynamics. > Mating loci were increasingly expressed across asexual development for both mating types. > Pheromone and receptor genes were expressed within the whole clones for both mating types. > Early and late light responsive genes showed similar expression timing over asexual development.
Acknowledgments
The authors appreciate help on this project from Aleksandra Adomas, Travis Clark, and Zhang Zhang, as well as Frances Trail for extensive discussion of topics related to fungal sexual development. We also thank the Broad Institute and MIPS for making N. crassa gene and genomic data available for oligonucleotide prediction and MTP design. We are thankful to two reviewers and Associate Editor Mary Anne Nelson for detailed comments and suggestions with regard to biological interpretation and relevant citations. Special thanks go to one anonymous reviewer whose detailed and insightful comments were extraordinarily helpful to our accurate and comprehensive discussion of the broad impacts of mating type genes. This study was supported by NIH P01 grant GM068067 and NSF grant MCB 0923797 to JPT.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adám AL, García-Martínez J, Szücs EP, Avalos J, Hornok L. The MAT1-2-1 mating-type gene upregulates photo-inducible carotenoid biosynthesis in Fusarium verticillioides. FEMS Microbiol Lett. 2011;318:76–83. doi: 10.1111/j.1574-6968.2011.02241.x. [DOI] [PubMed] [Google Scholar]
- Adomas AB, Lopez-Giraldez F, Clark TA, Wang Z, Townsend JP. Multi-targeted priming for genome-wide gene expression assays. BMC Genomics. 2010;11:477. doi: 10.1186/1471-2164-11-477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed S, Morrall RAA, Kaiser WJ. Distribution of mating types of Ascochyta fabae f. sp. lentis. Can J Plant Pathol. 1996;18:347–353. [Google Scholar]
- Arpaia G, Loros JJ, Dunlap JC, Morelli G, Macino G. Light induction of the clock-controlled gene ccg-1 is not transduced through the circadian clock in Neurospora crassa. Mol Gen Genet. 1995;247:157–163. doi: 10.1007/BF00705645. [DOI] [PubMed] [Google Scholar]
- Bardin M, Nicot PC, Normand P, Lemaire JM. Virulence variation and DNA polymorphism in Sphaerotheca fuliginea, causal agent of powdery mildew of cucurbits. Eur J Plant Pathol. 1997;103:545–554. [Google Scholar]
- Bell-Pedersen D, Garceau N, Loros JJ. Circadian rhythms in fungi. J Genet. 1992;75:387–401. [Google Scholar]
- Bell-Pedersen D, Shinohara ML, Loros JJ, Dunlap JC. Circadian clock-controlled genes isolated from Neurospora crassa are late night to early morning specific. Proc Natl Acad Sci USA. 1996;93:13096–13101. doi: 10.1073/pnas.93.23.13096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidard F, Benkhali JA, Coppin E, Imbeaud S, Grognet P, Delacroix H, Debuchy R. Genome-wide gene expression profiling of fertilization competent mycelium in opposite mating types in the heterothallic fungus Podospora anserina. PLoS One. 2011;6:e21476. doi: 10.1371/journal.pone.0021476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobrowicz P, Pawlak R, Correa A, Bell-Pedersen D, Ebbole DJ. The Neurospora crassa pheromone precursor genes are regulated by the mating type locus and the circadian clock. Mol Microbiol. 2002;45:795–804. doi: 10.1046/j.1365-2958.2002.03052.x. [DOI] [PubMed] [Google Scholar]
- Brasier CM. Fitness continuous variation and selection in fungal populations an ecological perspective. In: Worrall JJ, editor. Structure and dynamics of fungal populations. Kluwer, Dordrecht; The Netherlands: 1999. pp. 307–339. [Google Scholar]
- Chang S, Staben C. Directed replacement of mt A by mt a-1 effects a mating type switch in Neurospora crassa. Genetics. 1994;138:75–81. doi: 10.1093/genetics/138.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Ringelberg CS, Gross RH, Dunlap JC, Loros JJ. Genome-wide analysis of light-inducible responses reveals hierarchical light signaling in Neurospora. EMBO J. 2009;28:1029–1042. doi: 10.1038/emboj.2009.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark TA, Guilmette JM, Renstrom D, Townsend JP. RNA extraction, probe preparation, and competitive hybridization for transcriptional profiling using Neurospora crassa long-oligomer DNA microarrays. Fungal Genet Rep. 2008;55:18–28. [Google Scholar]
- Coppin E, Arnaise S, Contamine V, Picard M. Deletion of the mating-type sequences in Podospora anserine abolishes mating without affecting vegetative functions and sexual differentiation. Mol Gen Genet. 1993;241:409–414. doi: 10.1007/BF00284694. [DOI] [PubMed] [Google Scholar]
- Coppin E, de Renty C, Debuchy R. The function of the coding sequences for the putative pheromone precursors in Podospora anserine is restricted to fertilization. Eukaryot Cell. 2005;4:407–420. doi: 10.1128/EC.4.2.407-420.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dettman JR, Jacobson DJ, Turner E, Pringle A, Taylor JW. Reproductive isolation and phylogenetic divergence in Neurospora, comparing methods of species recognition in a model eukaryote. Evolution. 2003;57:2721–2741. doi: 10.1111/j.0014-3820.2003.tb01515.x. [DOI] [PubMed] [Google Scholar]
- Dudzinski MJ, Old KM, Gibbs RJ. Pathogenic variability in Australian isolates of Phytophthora cinnamomi. Aust J Bot. 1993;41:721–732. [Google Scholar]
- Dunlap JC, Loros JJ. The Neurospora circadian system. J Biol Rhythms. 2004;19:414–424. doi: 10.1177/0748730404269116. [DOI] [PubMed] [Google Scholar]
- Esser K. Breeding systems in fungi and their significance for genetic recombination. Mol Gen Genet. 1971;110:86–100. doi: 10.1007/BF00276051. [DOI] [PubMed] [Google Scholar]
- Ferreira AV-B, An Z, Metzenberg RL, Glass NL. Characterization of mat A-2, mat A-3 and matA mating-type mutants of Neurospora crassa. Genetics. 1998;148:1069–1079. doi: 10.1093/genetics/148.3.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira AV-B, Saupe S, Glass NL. Transcriptional analysis of the mtA idiomorph of Neurospora crassa identifies two genes in addition to mtA-1. Mol Gen Genet. 1996;250:767–774. doi: 10.1007/BF02172989. [DOI] [PubMed] [Google Scholar]
- Funnell DL, Matthews PS, van Etten HD. Breeding for highly fertile isolates of Nectria haematococca MPVI that are highly virulent on pea and in planta selection for virulent recombinants. Phytopathology. 2001;91:92–101. doi: 10.1094/PHYTO.2001.91.1.92. [DOI] [PubMed] [Google Scholar]
- Glass NL, Lee L. Isolation of Neurospora crassa A mating type mutants by repeat induced point (RIP) mutation. Genetics. 1992;132:125–133. doi: 10.1093/genetics/132.1.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwald CJ, Kasuga T, Glass NL, Shaw BD, Ebbole DJ, Wilkinson HH. Temporal and spatial regulation of gene expression during asexual development of Neurospora crassa. Genetics. 2010;186:1217–1230. doi: 10.1534/genetics.110.121780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallen HE, Huebner M, Shiu SH, Guldener U, Trail F. Gene expression shifts during perithecium development in Gibberella zeae (anamorph Fusarium graminearum), with particular emphasis on ion transport proteins. Fungal Genet Biol. 2007;44:1146–1156. doi: 10.1016/j.fgb.2007.04.007. [DOI] [PubMed] [Google Scholar]
- Heitman J, Kronstad J, Taylor JW, Casselton LA. Sex in Fungi, Molecular Determination and Evolutionary Implications. Washington DC: ASM Press; 2007. [Google Scholar]
- Kanehisa M, Goto S. KEGG, Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanematsu S, Adachi Y, Ito T. Mating-type loci of heterothallic Diaporthe spp.: homologous genes are present in opposite mating-types. Curr Genet. 2007;52:11–22. doi: 10.1007/s00294-007-0132-3. [DOI] [PubMed] [Google Scholar]
- Kasuga T, Glass NL. Dissecting colony development of Neurospora crassa using mRNA profiling and comparative genomics approaches. Eukaryot, Cell. 2008;7:1549–1564. doi: 10.1128/EC.00195-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasuga T, Mannhaupt G, Glass NL. Relationship between phylogenetic distribution and genomic features in Neurospora crassa. PLoS One. 2009;4:e5286. doi: 10.1371/journal.pone.0005286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasuga T, Townsend JP, Tian C, Gilbert LB, Mannhaupt G, Taylor JW, Glass NL. Long-oligomer microarray profiling in Neurospora crassa reveals the transcriptional program underlying biochemical and physiological events of conidial germination. Nucleic Acids Res. 2005;33:6469–6485. doi: 10.1093/nar/gki953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keszthelyi A, Jeney A, Kerényi Z, Mendes O, Wallwijk C, Hornok L. Tagging target genes of the MAT1-2-1 transcription factor in Fusarium verticillioides (Gibberella fujikuroi MP-A) Antonie van Leeuwenhoek. 2007;91:373–391. doi: 10.1007/s10482-006-9123-5. [DOI] [PubMed] [Google Scholar]
- Kim H, Borkovich KA. A pheromone receptor gene, pre-1, is essential for mating type-specific directional growth and fusion of trichogynes and female fertility in Neurospora crassa. Mol Microbiol. 2004;52:1781–1789. doi: 10.1111/j.1365-2958.2004.04096.x. [DOI] [PubMed] [Google Scholar]
- Kim H, BorkovichK KA. Pheromones are essential for male fertility and sufficient to direct chemotropic polarized growth of trichogynes during mating in Neurospora crassa. Eukaryot Cell. 2006;5:544–554. doi: 10.1128/EC.5.3.544-554.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Metzenberg RL, Nelson MA. Multiple functions of mfa-1, a putative pheromone precursor gene of Neurospora crassa. Eukaryot Cell. 2002;1:987–999. doi: 10.1128/EC.1.6.987-999.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klix V, Nowrousian M, Ringelberg C, Loros JJ, Dunlap JC, Pöggeler S. Functional characterization of MAT1-1-specific mating-type genes in the homothallic ascomycete Sordaria macrospora provides new insights into essential and nonessential sexual regulators. Eukaryot Cell. 2010;9:894–905. doi: 10.1128/EC.00019-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolmer JA, Ellingboe AH. Genetic relationships between fertility and pathogenicity and virulence to rice in Magnaporthe grisea. Can J Bot. 1988;66:891–897. [Google Scholar]
- Lee J, Leslie JF, Bowden RL. Expression and function of sex pheromones and receptors in the homothallic ascomycete Gibberella zeae. Eukaryot Cell. 2008;7:1211–1221. doi: 10.1128/EC.00272-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SC, Ni M, Li W, Shertz C, Heitman J. The evolution of sex: a perspective from the fungal kingdom. Microbio Mol Biol Rev. 2010;74:298–340. doi: 10.1128/MMBR.00005-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin AM, de Vries RP, Conesa A, de Bekker C, Talon M, Menke HH, van Peij Noel NME, Wösten Han AB. Spatial differentiation in the vegetative mycelium of Aspergillus niger. Eukaryot Cell. 2007;6:2311–2322. doi: 10.1128/EC.00244-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Huang JC, Mitchell TG, Heitman J. Virulence attributes and hyphal growth of C neoformans are quantitative traits and the MATa allele enhances filamentation. PLoS Genet. 2006;2:e187. doi: 10.1371/journal.pgen.0020187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCt method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Martin T, Lu SW, van Tilbeurgh H, Ripoll DR, Dixelius C, Turgeon BG, Debuchy R. Tracing the origin of the fungal 1 domain places its ancestor in the HMG-box superfamily: implication for fungal mating-type evolution. PLoS One. 2010;5:e15199. doi: 10.1371/journal.pone.0015199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCluskey K, Wiest A, Plamann M. The Fungal Genetics Stock Center: a repository for 50 years of fungal genetics research. J Biosci. 2010;35:119–126. doi: 10.1007/s12038-010-0014-6. [DOI] [PubMed] [Google Scholar]
- Metzenberg RL. Bird medium: an alternative to Vogel medium. Fungal Genet Newslett. 2004;51:19–20. [Google Scholar]
- Mylyk OM, Barry EG, Galeazzi DR. New isogenic wild types in N. crassa. Neurospora Newslett. 1974;21:24. [Google Scholar]
- Nelson MA, Kang S, Braun EL, Crawford ME, Dolan PL, Leonard PM, Mitchell J, Armijo AM, Bean L, Blueyes E, Cushing T, Errett A, Fleharty M, Gorman M, Judson K, Miller R, Ortega J, Pavlova I, Perea J, Todisco S, Trujillo R, Valentine J, Wells A, Werner-Washburne M, Natvig DO. Expressed sequences from conidial, mycelial and sexual stages of Neurospora crassa. Fungal Genet Biol. 1997;21:348–363. doi: 10.1006/fgbi.1997.0986. [DOI] [PubMed] [Google Scholar]
- Newmeyer D, Perkins DD, Barry EG. An annotated pedigree of Neurospora crassa laboratory wild-types, showing the probable origin of the nucleolus satellite and showing that certain stocks are not authentic. Fungal Genet Newslett. 1987;34:46–51. [Google Scholar]
- Perkins DD. Wild type Neurospora crassa strains preferred for use as standards. Fungal Genet Newslett. 2004;51:7–8. [Google Scholar]
- Pöggeler S. Function and evolution of pheromones and pheromone receptors in filamentous ascomycetes. Pöggeler S, Wöstemeyer J, editors. Evolution of fungi and fungal-like organisms, The Mycota XIV. 2011:73–95. [Google Scholar]
- Pöggeler S, Kück U. Comparative analysis of the mating-type loci from Neurospora crassa and Sordaria macrospora: identification of novel transcribed ORFs. Mol Gen Genet. 2000;263:292–301. doi: 10.1007/s004380051171. [DOI] [PubMed] [Google Scholar]
- Pöggeler S, Kück U. Identification of transcriptionally expressed pheromone receptor genes in filamentous ascomycetes. Gene. 2001;280:9–17. doi: 10.1016/s0378-1119(01)00786-7. [DOI] [PubMed] [Google Scholar]
- Pöggeler S, Nowrousian M, Ringelberg C, Loros JJ, Dunlap JC, Kück U. Microarray and real-time PCR analyses reveal mating type-dependent gene expression in a homothallic fungus. Mol Genet Genomics. 2006;275:492–503. doi: 10.1007/s00438-006-0107-y. [DOI] [PubMed] [Google Scholar]
- Purschwitz J, Müller S, Kastner C, Fischer R. Seeing the rainbow: light sensing in fungi. Curr Opin Microbiol. 2006;9:566–571. doi: 10.1016/j.mib.2006.10.011. [DOI] [PubMed] [Google Scholar]
- Purschwitz J, Müller S, Kastner C, Schöser M, Haas H, Espeso EA, Atoui A, Calvo AM, Fischer R. Functional and physical interaction of blue- and red-light sensors in Aspergillus nidulans. Curr Biol. 2008;18:255–259. doi: 10.1016/j.cub.2008.01.061. [DOI] [PubMed] [Google Scholar]
- Randall TA, Metzenberg RL. Species-specific and mating type-specific DNA regions adjacent to mating type idiomorphs in the genus Neurospora. Genetics. 1995;141:119–136. doi: 10.1093/genetics/141.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall TA, Metzenberg RL. The mating type locus of Neurospora crassa: identification of an adjacent gene and characterization of transcripts surrounding the idiomorphs. Mol Gen Genet. 1998;259:615–621. doi: 10.1007/s004380050855. [DOI] [PubMed] [Google Scholar]
- Roberts AN, Yanofsky C. Genes expressed during conidiation in Neurospora crassa: characterization of con-8. Nucleic Acids Res. 1989;17:197–214. doi: 10.1093/nar/17.1.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruepp A, Zollner A, Maier D, Albermann K, Hani J, Mokrejs M, Tetko I, Güldener U, Mannhaupt G, Münsterkötter M, Mewes HW. The FunCat, a functional annotation scheme for systematic classification of proteins from whole genomes. Nucleic Acids Res. 2004;32:5539–5545. doi: 10.1093/nar/gkh894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs MS, Yanofsky C. Developmental expression of genes involved in conidiation and amino acid biosynthesis in Neurospora crassa. Dev Biol. 1991;148:117–128. doi: 10.1016/0012-1606(91)90322-t. [DOI] [PubMed] [Google Scholar]
- Saupe S, Stenberg L, Shiu KT, Griffiths AJF, Glass NL. The molecular nature of mutations in the mt A-1 gene of the Neurospora crassa A idiomorph and their relation to mating-type function. Mol Gen Genet. 1996;250:115–122. doi: 10.1007/BF02191831. [DOI] [PubMed] [Google Scholar]
- Springer ML. Genetic control of fungal differentiation: the three sporulation pathways of Neurospora crassa. BioEssays. 1993;15:365–374. doi: 10.1002/bies.950150602. [DOI] [PubMed] [Google Scholar]
- Springer ML, Yanofsky C. Expression of con genes along the three sporulation pathways of Neurospora crassa. Genes Dev. 1992;6:1052–1057. doi: 10.1101/gad.6.6.1052. [DOI] [PubMed] [Google Scholar]
- Townsend JP. Resolution of large and small differences in gene expression using models for the Bayesian analysis of gene expression levels and spotted DNA microarrays. BMC Bioinformatics. 2004;5:54. doi: 10.1186/1471-2105-5-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend JP, Hartl DL. Bayesian analysis of gene expression levels: statistical quantification of relative mRNA level across multiple strains or treatments. Genome Biol. 2002;3:research0071.1–0071.16. doi: 10.1186/gb-2002-3-12-research0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitalini MW, Morgan LW, March IJ, Bell-Pedersen D. A genetic selection for circadian output pathway mutations in Neurospora crassa. Genetics. 2004;167:119–129. doi: 10.1534/genetics.167.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitalini MW, de Paula RM, Park WD, Bell-Pedersen D. The rhythms of life: circadian output pathways in Neurospora. J Biol Rhythms. 2006;21:432–444. doi: 10.1177/0748730406294396. [DOI] [PubMed] [Google Scholar]
- Wang Y, Barbacioru C, Hyland F, Xiao W, Hunkapiller KL, Blake J, Chan F, Gonzalez C, Zhang L, Samaha RR. Large scale real-time PCR validation on gene expression measurements from two commercial long-oligonucleotide microarrays. BMC Genomics. 2006;7:59. doi: 10.1186/1471-2164-7-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White BT, Yanofsky C. Structural characterization and expression analysis of the Neurospora conidiation gene con-6. Dev Biol. 1993;160:254–264. doi: 10.1006/dbio.1993.1303. [DOI] [PubMed] [Google Scholar]
- Wik L, Karlsson M, Johannesson H. The evolutionary trajectory of the mating-type (mat) genes in Neurospora relates to reproductive behavior of taxa. BMC Evol Biol. 2008;8:109. doi: 10.1186/1471-2148-8-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Townsend JP. The filamentous fungal gene expression database (FFGED) Fungal Genet Biol. 2010;47:199–204. doi: 10.1016/j.fgb.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Nowrousian M, Kupfer D, Colot HV, Berrocal-Tito G, Lai H, Bell-Pedersen D, Roe BA, Loros JJ, Dunlap JC. Analysis of expressed sequence tags from two starvation, time-of-day-specific libraries of Neurospora crassa reveals novel clock-controlled genes. Genetics. 2001;157:1057–1065. doi: 10.1093/genetics/157.3.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1 (Table S1): Gene expression for mat A and mat a of Neurospora crassa across 36hr, 60hr, and 96hr in asexual development.
Supplemental Table 2 (Table S2): Expression of selected genes measured with quantitative PCR. Statistical analysis was performed with BAGEL (Townsend, 2004).
Supplemental Table 3 (Table S3): Primers for genes investigated with quantitative RT- PCR.
Supplemental Table 4 (Table S4): Expression of genes of interest across asexual development in Neurospora crassa.
Supplemental Table 5 (Table S5): Expression of 59 Early Light Responsive Genes (ELRGs) in both mating types of Neurospora crassa.
Supplemental Table 6 (Table S6): Expression of 80 Late Light Responsive Genes (LLRGs) in both mating types of Neurospora crassa.