

Abstract
G-protein-coupled receptors (GPCRs) activate the epidermal growth factor receptor (EGFR) and mediate EGFR-independent signaling pathways to promote the growth of a variety of cancers including head and neck squamous cell carcinoma (HNSCC). Identification of the common signaling mechanisms involved in GPCR-induced EGFR-dependent and independent processes will facilitate the development of more therapeutic strategies. In this study, we hypothesized that phosphoinositide-dependent kinase 1 (PDK1) contributes to GPCR-EGFR crosstalk and signaling in the absence of EGFR and suggests that inhibition of the PDK1 pathway may be effective in the treatment of HNSCC. The contribution of PDK1 to the EGFR-dependent and independent signaling in HNSCC was determined using RNAi, a kinase-dead mutant and pharmacological inhibition. In vivo xenografts studies were also performed to determine the efficacy of targeting PDK1 alone or in combination with the FDA-approved EGFR inhibitor cetuximab. PDK1 contributed to both GPCR-induced EGFR activation and cell growth. PDK1 also mediated activation of p70S6K in the absence of EGFR. Blockade of PDK1 with a small molecule inhibitor (AR-12) abrogated HNSCC growth, induced apoptosis and enhanced the anti-proliferative effects of EGFR tyrosine kinase inhibitors in vitro. HNSCC xenografts expressing kinase-dead PDK1 demonstrated increased sensitivity to cetuximab compared to vector-transfected controls. Administration of AR-12 substantially decreased HNSCC tumor growth in vivo. These cumulative results demonstrate that PDK1 is a common signaling intermediate in GPCR-EGFR crosstalk and EGFR-independent signaling, and where targeting the PDK1 pathway may represent a rational therapeutic strategy to enhance clinical responses to EGFR inhibitors in HNSCC.
Keywords: PDK1, Head and neck cancer, GPCR-EGFR crosstalk, AR-12
Introduction
G-protein-coupled receptors (GPCR) are seven-transmembrane receptors that mediate various signaling pathways that contribute to growth, survival and cellular motility. Several GPCR ligands including gastrin-releasing peptide (GRP), prostaglandin E2 (PGE2), bradykinin (BK) and lysophosphatidic acid (LPA) have all been shown to promote the growth of cancers including head and neck squamous cell carcinoma (HNSCC) (1–3). Combined inhibition of GPCRs and EGFR has been further reported to result in improved anti-tumor effects in HNSCC (2, 4). However, due to the heterogeneous expression of GPCRs in HNSCC, targeting of multiple GPCRs and EGFR is impractical in the clinical setting. Identification of a common signaling intermediate downstream of GPCR signaling that is amenable to inhibition, may represent a plausible therapeutic strategy.
We previously reported that GRP mediated the release of EGFR ligands in a phosphoinositide-dependent kinase 1 (PDK1)-dependent manner through phosphorylation of TNF-alpha converting enzyme (TACE) (5). Furthermore, downmodulation of PDK1 expression combined with erlotinib resulted in improved anti-proliferative and anti-invasive effects (5). We recently showed that GPCRs induce p70S6K activation in an EGFR-independent setting, where combined inhibition of p70S6K and EGFR resulted in decreased growth in vitro and in vivo (6). PDK1 is a serine/threonine kinase that has been demonstrated to activate multiple kinases from the AGC (Protein kinase A, protein kinase G, protein kinase C) family of kinases that also includes p70S6K, PKB/Akt and p21-activated kinase (PAK) (7). The pleiotropic capacity of PDK1 makes it a promising molecular and therapeutic target for HNSCC. In the present study, we investigated the contribution of PDK1 in pathways mediated by several GPCR agonists detected in HNSCC, including PGE2, BK and LPA. In addition, we assessed the contribution of PDK1 in activating EGFR-independent signaling. The contribution of PDK1 was tested using several approaches including siRNA, expression of a dominant-negative construct, and pharmacologic inhibition, alone and in combination with EGFR blockade. Our results validate PDK1 as a therapeutic target where strategies that target the PDK1 pathway may enhance EGFR blockade in HNSCC, where EGFR inhibition is an established therapeutic strategy.
Materials & Methods
Cell lines
All the HNSCC cell lines (PCI-37A, 1483, PCI-6B, UM-22A, UM-22B, UMSCC-1) were of human origin. 1483 cells were derived from an oropharyngeal tumor, UM-22B and PCI-6B cell lines were derived from metastatic lymph nodes and PCI-37A and UM-22A were from a primary tumor arising in the epiglottis (8). UMSCC-1 cells were derived from squamous cell carcinoma of the oral cavity. Cells were maintained in DMEM with 10% heat-inactivated FCS (Invitrogen, Carlsbad, CA) at 37°C with 5% CO2. All cell lines were validated by genotyping with the AmpFISTR Identifiler System (Applied Biosystems) within 6 months of their use for the studies described.
Reagents
Epidermal growth factor (EGF) and Prostaglandin E2 (PGE2) were obtained from Calbiochem (San Diego, CA). Bradykinin was obtained from Bachem (Torrance, CA). Lysophosphatidic acid (LPA) was obtained from Sigma-Aldrich Corporation (St. Louis, MO). C225 (cetuximab, ™Erbitux) was obtained from the University of Pittsburgh Cancer Institute pharmacy. The kinase-dead PDK1 (K110Q) cDNA plasmid was a kind gift from Dr. Alexandra Newton (University of California, San Diego). The kinase activity of this mutant was reported in the following publication (9). AR-12 (formally known as OSU-03012) was provided by Arno Therapeutics (Fairfield, NJ). The chemical structure of this compound has been previously published (10).
Establishment of PDK1 kinase-dead HNSCC cells
1483 cells were seeded in 6-well plates and transfected with 2 μg of pcDNA3.1-PDK1 (K110Q) or 2 μg of pcDNA3.1. Two days later, cells were selected with 1 mg/ml G418 until untransfected cells displayed 100% cell death. Individual clones were selected and grown before verification by immunoblotting for expression of the myc-tag.
Co-immunoprecipitation and western blotting
For immunoprecipitation, 300 μg of total protein were incubated overnight with 2 μg of EGFR antibody (BD Transduction, San Jose, CA) and incubated overnight at 4°C on a rotary shaker. Fourty μl of Protein G agarose beads (Upstate, Temecula, CA) were added to the lysates and allowed to incubate for 2 hours at 4°C on a rotary shaker. The beads were collected by centrifugation at 4°C, 14,000 rpm for 1 minute. The beads were resuspended and washed three times with lysis buffer. The beads were resuspended in 30 μL of lysis buffer and 8 μl of 4× loading dye and boiled for 10 minutes at 95°C, followed by Western blot analysis. The immunoprecipitated proteins were then resolved on an 8% SDS-PAGE gel. After being transferred onto a nitrocellulose membrane, the membrane was blocked in 5% milk and blotted with the antiphosphotyrosine antibody PY99 (Santa Cruz Biotechnology, Santa Cruz, CA) at 1:500 in 5% milk dissolved in TBST solution [0.9% NaCl, 0.5% Tween 20, and 50 mmol/L Tris (pH 7.4)]. After washing three times with TBST solution, the membrane was incubated with the secondary antibody (goat antirabbit/mouse IgG-horseradish peroxidase conjugate; Bio-Rad Laboratories) for 1 hour and washed three times for 10 minutes. The membrane was developed with Luminol Reagent (Santa Cruz Biotechnology) by autoradiography. Blots were stripped in Restore Western Blot Stripping buffer (Pierce, Rockford, IL) for 15 minutes at room temperature, blocked for 1 hour, and reprobed with EGFR antibody (Transduction Laboratories) at 1:500 dilution. Whole cell lysates were also resolved on 8% SDS-PAGE, transferred to nitrocellulose and probed for PDK1, (Cell Signaling, Danvers, MA), and β-tubulin (Abcam, Cambridge, MA).
siRNA transfection
PDK1 siRNA was designed to the target the following sequence: 5'-CUGGCAACCUCCAGAGAA-3' and obtained from Dharmacon (Lafayette, CO). 2 × 106 cells were seeded in 10 cm plates and allowed to incubate overnight at 37°C. Cells were transfected with siRNA using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions.
Cell viability assays
HNSCC cells were seeded and incubated overnight at 37°C. Cells were treated with inhibitors or transfected with siRNA for different time points. Cells were trypsinized and stained with trypan blue solution before being transferred to a hemocytometer and viable cells were counted. MTT assays were performed by adding the MTT solution (Sigma-Aldrich) for 30 minutes at 37°C. MTT solution was removed, followed by addition of dimethyl sulfoxide (DMSO) solution. The resulting formazan product was read at 595nm using the uQuant microplate spectrophotometer. Graphs were plotted using the GraphPad Prism Software.
Xenograft Studies
All animal studies were done according to the University of Pittsburgh Institutional Animal Care and Use Committee. Athymic nude mice were subcutaneously inoculated with UMSCC1, 1483VC or 1483 PDKM cells. Following observation of palpable tumors, xenografts were randomized into treatment groups. For AR-12 experiments, mice were randomized into 4 groups 1) Vehicle 2) AR-12 (100mg/kg daily p.o) 3) C225 (0.2mg i.p 3 times/week) 4) AR-12+C225. For the PDKM experiments, nude mice (n=5 per group) inoculated with 1483VC and 1483 PDKM were treated with either vehicle or C225 (0.8mg i.p 3 times/week). Tumors and mouse weights were measured three times per week. Tumor volume was calculated with the following formula: length/2 × width2.
Statistics
The differences between treatment groups in biochemical assays were determined by two-tailed Student's t-test. The differences between treatment groups in xenograft experiments were determined using the exact Wilcoxon Mann Whitney 1-sided test. A p-value less than 0.05 was considered to be statistically significant.
Results
PDK1 contributes to EGFR phosphorylation by several GPCR ligands
We previously reported that GRP stimulates the release of the EGFR ligand amphiregulin via phosphoinositide-dependent kinase 1(PDK1)-mediated TACE phosphorylation (5). To determine whether PDK1 is a common signaling intermediate in the activation of EGFR by other GPCR ligands, we examined the effect of PDK1 knockdown on GPCR-induced phosphorylation of EGFR. We used three HNSCC cell lines: PCI-37A, UM-22A, and PCI-6B. These HNSCC cell lines were treated with three different GPCR ligands, PGE2, BK or LPA followed by assessment of EGFR tyrosine phosphorylation. PGE2, BK and LPA have previously been shown to phosphorylate EGFR in several HNSCC cell lines in addition to the HNSCC models shown here (1–3). PGE2 and BK-mediated phosphorylation of EGFR was significantly abrogated in HNSCC cells transfected with PDK1 siRNA, (Figures 1A and 1B; p=0.048). LPA-mediated phosphorylation of EGFR was also reduced by PDK1 downmodulation (Figure 1C; p=0.0001). These findings suggest that PDK1 broadly contributes to GPCR-mediated EGFR transactivation in HNSCC.
Figure 1. PDK1 and PI3K contribute to GPCR-mediated EGFR phosphorylation and HNSCC growth.

(A) PCI-37A, (B) UM-22A and (C) PCI-6B cells were transfected with non-targeting control (NTC) or PDK1 siRNA followed by 72hr of serum starvation and stimulation with EGF (10 ng/ml), PGE2 (10 nM), BK (10 nM) or LPA (10 μM) for 5 minutes. Cell lysates were collected and immunoprecipitated with EGFR and immunoblotted with an anti-phosphotyrosine antibody (P-EGFR) or total EGFR antibody, respectively. PDK1 levels were determined by western blotting with an anti-PDK1 antibody. Densitometric analysis was performed to determine cumulative results. The experiments were repeated 3–4 times with similar findings (p= 0.048; p=0.0001). (D) UM-22A cells were transiently transfected with PDK1 or NTC siRNA, serum starved for 48 hrs, followed by stimulation with PGE2 (10 nM) or BK (10 nM) or vehicle for 24 hours. Percentage increase in proliferation was determined by trypan blue dye exclusion assay. Results were graphed using GraphPad Prism Software. The experiment was performed twice in triplicate with similar results (p=0.046; p=0.007).
PDK1 contributes to GPCR-induced proliferation
We and others have reported that GPCR ligands induce HNSCC proliferation (1, 2). To determine the role of PDK1 in GPCR-stimulated cell growth, HNSCC cells were transfected with control or PDK1 siRNA for 48 hours followed by 24-hour stimulation with PGE2 or BK. PGE2 or BK (Figure 1D) induced an approximate 20% increase in growth of control siRNA-transfected cells. However, in PDK1 siRNA-transfected cells, PGE2 or BK-mediated growth was abrogated to 8 and 5%, respectively (p=0.046, p=0.007). In UM-SCC1 cells, BK induced a 20% increase in growth. However, in the presence of PDK1 siRNA, BK failed to augment growth (Supplementary Figure 1; p = 0.003). These results indicate that PDK1 plays a role in GPCR-mediated proliferation of HNSCC cells.
PDK1 kinase activity contributes to GPCR signaling and HNSCC growth
Kinase inhibitors have emerged as potential cancer treatment strategies in tumors that demonstrate increased activation of oncogenic kinases. To determine the effect of PDK1 kinase inactivation, HNSCC cells (UM-22A) were transiently transfected with the PDK1 kinase-dead mutant (9) resulting in abrogation of PGE2-induced growth compared to vector-transfected control cells (Supplementary Figure 2; p=0.04). A HNSCC cell line (1483) was stably transfected with the kinase-dead PDK1 construct (PDKM) or control vector (VC). We validated expression of the kinase-dead mutant by assessing expression of the myc tag in two representative clones (Figure 2A). Next, we treated the control and kinase-dead PDK1 mutant-transfected cells with BK followed by assessment of EGFR phosphorylation. As shown in Figure 2B, EGFR phosphorylation was not induced by BK in the PDKM cells compared with a 2-fold increase in EGFR phosphorylation detected in the vector-transfected control cells. Next, we assessed the proliferation of the PDKM cells compared to the control cells under normal growth conditions. As shown in Figure 2C, the proliferation rate of 1483 PDKM cells was reduced compared to vector-transfected control cells after 4 days. These results indicate that PDK1 kinase activity contributes to HNSCC growth and GPCR-induced EGFR phosphorylation.
Figure 2. Kinase-dead PDK1 abrogates GPCR-mediated signaling and HNSCC growth.
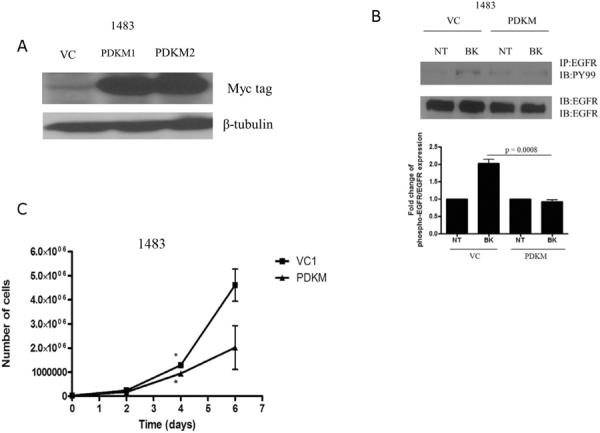
(A) 1483 cells were stably transfected with pcDNA3.1 (vector control; VC) or pcDNA3-PDK1M (K110Q-kinase dead) and selected with neomycin for 2 weeks. Lysates were resolved by SDS-PAGE and probed for myc-tag and β-tubulin. (B) 1483 vector-transfected control (VC) and 1483 kinase-dead PDK1 (PDKM) cells were seeded, serum-starved for 72 hours and stimulated with BK (10 nM) for 5 minutes. Lysates were collected and immunoprecipitated with anti-phosphotyrosine antibody (P-EGFR) and immunoblotted for EGFR. Lysates were separately immunoblotted for EGFR to determine equal EGFR expression levels. Experiment was performed twice with similar results (C) 1483 VC and PDKM cells were seeded and assessed for growth by trypan blue dye exclusion on days 1, 3 and 5. Cell counts were graphed using GraphPad Prism Software (Day 5, p=0.039).
PDK1 mediates EGFR-independent signaling in HNSCC
GPCRs have been reported to induce signaling via both EGFR-dependent and EGFR-independent mechanisms. We recently demonstrated that GPCR ligands induce EGFR-independent effects via the robust activation of p70S6K (6). PDK1 has been reported to activated p70S6K in AKT-dependent and independent manners (11). To determine whether PDK1 activation was exclusively downstream of EGFR in HNSCC, we examined the phosphorylation of p70S6K in the presence of siRNAs targeting EGFR and/or PDK1. EGFR siRNA induced p70S6K phosphorylation as previously reported (6), however in the presence of PDK1 siRNA, p70S6K phosphorylation was significantly abrogated (Figure 3A; p=0.02). Similar to the results observed with PDK1 siRNA, EGFR siRNA failed to induce p70S6K phosphorylation in PDKM cells (Figure 3B). These results suggest that PDK1 can mediate mitogenic signaling via EGFR-dependent and –independent routes.
Figure 3. PDK1 contributes to EGFR-independent p70S6K phosphorylation.

(A) HNSCC (1483) cells were transfected with NTC, EGFR, PDK1 or a combination of EGFR and PDK1 siRNA for 72 hours. Lysates collected were analyzed for phospho-p70S6K, p70S6K, PDK1 and β-tubulin. The results are representative of 3 independent experiments. (B) 1483 cells were stably transfected with pcDNA3 (VC) or pcDNA3-PDKM (kinase-dead). 1483 VC and PDKM cells were transfected with NTC or EGFR siRNA for 72 hours. Lysates were collected and resolved by SDS-PAGE. The results are representative of 2 experiments.
Pharmacological inhibition of the PDK1 pathway abrogates HNSCC growth, AKT phosphorylation and cyclin D1
Given the central role of PDK1 in both EGFR-dependent and EGFR-independent signaling, we next sought to evaluate the efficacy of pharmacological inhibition of the PDK1 pathway using the small molecule inhibitor AR-12 (Figure 4A). AR-12 is a celecoxib derivative with no activity against COX-2 (12). HNSCC cell lines displayed a range of IC50 values from 74 nM to 820 nM (Figure 4B and Supplementary Table 1). In contrast, inhibition of the normal mucosal epithelial cell line Het-1A required higher concentrations of AR-12 (IC50 = 6 μM). The effects of AR-12 was previously reported in oral cancer cells in vitro (13). To determine the in vivo efficacy of AR-12, HNSCC (UMSCC-1) cells were inoculated subcutaneously in athymic nude mice. After the formation of palpable tumor nodules 7 days later, mice were (9 mice per group) treated with vehicle or AR-12 (100mg/kg daily by oral gavage). The PDK1 pathway inhibitor AR-12 induced significant tumor regression as early as day 5 (Figure 4C; p < 0.001, Day 28). No toxicities were observed as measured by body weight analysis. To assess the biochemical effects of AR-12, we assessed cell lines for levels of Akt phosphorylation, which can serve as an indicator of PDK1 activity (14). AR-12 (1 μM) abrogated phospho-Akt (T308 and S473) expression levels in both HNSCC cell lines examined (Figure 4D). We next investigated the role of PDK1 as an AR-12 target by performing dose response studies in PDK1 siRNA and control siRNA-transfected HNSCC cells. PDK1 siRNA-transfected cells displayed greater IC50 values compared to control siRNA-transfected cells (Supplementary Figure 3). These observations indicate that PDK1 expression contributed to HNSCC sensitivity to AR-12. PDK1 has been reported to mediate cellular proliferation via cyclin D1 activity (15). Further, cyclin D1 expression has also been shown to confer resistance to the EGFR tyrosine kinase inhibitor gefitinib (15, 16). We compared the inhibitory effect of AR-12 on cyclin D1 expression in PCI-37A and UM-22B cells, which represented the most and least sensitive HNSCC cell lines to AR-12, respectively. As shown in Supplementary figure 4, AR-12 treatment was associated with downmodulation of cyclin D1 at 100 nM and 1μM in PCI-37A and UM-22B cells, respectively. These results suggest that HNSCC cells are sensitive to pharmacological inhibition of the PDK1 pathway. Furthermore, cyclin D1 may be a more sensitive readout for PDK1 inhibition in HNSCC models, than Akt phosphorylation.
Figure 4. AR-12 inhibits in vitro and in vivo HNSCC growth, AKT phosphorylation and cyclin D1.

(A) Chemical structure of AR-12 (B) HNSCC (PCI-37A, UM-22B, PCI-6B, 1483) and normal mucosal epithelial cells (Het-1A) were treated with increasing concentrations of AR-12. After 72 hours, MTT assay was performed and the IC50 values were determined using Prism (GraphPad) Software. The experiment was repeated 3 times with similar results. (C) UMSCC-1 cells (3 × 106) were inoculated into the flanks of athymic nude mice. After the formation of tumor nodules (7 days later), mice were divided into two groups (9 mice per group) Vehicle and AR-12 (100mg/kg daily by oral gavage. Tumors were measured 3 times weekly over 28 days and graphed using GraphPad Prism (p < 0.0001, Day 28). (D) PCI-37A and UM-22B cells were treated with increasing concentrations of AR-12 for 72 hours. Lysates were assessed by immunoblotting for phospho-Akt, total Akt and β-tubulin. Representative results from 3 independent experiments are shown.
AR-12 induces apoptotic signaling pathways in HNSCC cells
To determine whether the decreased growth rate induced by AR-12 was due to increased apoptosis we assessed PARP cleavage and expression of the anti-apoptotic molecule survivin in treated HNSCC cells. AR-12-induced PARP cleavage was observed at 300 nM and 1μM concentrations in PCI-37A and UM-22B cells, respectively (Figure 5A). In contrast, AR-12 failed to induce apoptosis in the Het-1A normal mucosal epithelial cell line, (Figure 5B). In addition to PARP, we examined the effect of AR-12 on survivin expression. Survivin is a member of the inhibitor of apoptosis (IAP) family of proteins, which is involved in mediating cell proliferation and inhibiting apoptosis (17,18). AR-12 decreased survivin expression at 100 nM and 1μM concentrations in PCI-37A and UM-22B cells, respectively (Figure 5C). TUNEL analysis of vehicle and AR-12 treated xenografts displayed no significant differences in apoptosis between treatment groups (data not shown). However, in the absence of serial determinations of apoptosis, it is possible that assessment of a dynamic process at a single four week time point may not be sensitive enough to assess the mechanism of reduced growth. These results indicate that the pharmacologic inhibition of PDK1 with AR-12 induces apoptosis by increasing PARP cleavage and decreasing expression of the anti-apoptotic marker survivin.
Figure 5. AR-12 induces pro-apoptotic signaling.

(A) PCI-37A (left panel), UM-22B (right panel) and (B) HET-1A cells were treated with increasing concentrations of AR-12 for 36 hours and analyzed by immunoblotting for cleaved PARP (116 and 89Kd bands). Representative results from three independent experiments are shown. (C) PCI-37A (left panel) and UM-22B (right panel) cells were treated with AR-12 and assessed for survivin expression by immunoblotting. The experiment was performed twice with similar results.
Targeting PDK1 enhances the anti-proliferative effects of EGFR inhibition
Since PDK1 mediates both EGFR-dependent and –independent signaling pathways, we determined the efficacy of PDK1 targeting in combination with EGFR inhibition strategies. To determine whether AR-12 treatment increased HNSCC sensitivity to EGFR inhibition, we first examined the effects of AR-12 in combination with the preclinical EGFR TKI, AG1478 or the clinical EGFR TKI, erlotinib. As shown in Figure 6A, combined treatment with sub-IC50 concentrations of each compound resulted in enhanced cell growth inhibition compared to either agent alone (p < 0.001). HNSCC cells have been shown to release GPCR ligands BK and PGE2 into the tumor microenvironment (19,20). Therefore, we next used an in vivo HNSCC model, where the autocrine and paracrine secretion of the GPCR ligands contributes to tumor growth. We used the EGFR monoclonal antibody cetuximab (C225), which was FDA-approved in 2006 for the treatment of HNSCC. As shown in Figure 6B, C225 treatment of HNSCC xenografts expressing kinase-dead PDK1 (PDKM1) resulted in decreased tumor growth compared to cetuximab treatment of vector control (VC) xenografts (p=0.048). Interestingly, vehicle-treated PDKM xenografts and C225-treated VC xenografts displayed increased tumor growth compared to vehicle-treated VC xenografts and C225-treated PDKM xenografts. In an attempt to determine the mechanism of enhanced tumor growth, we performed immunoblot analysis of vehicle and C225-treated xenografts. Decreased phospho-Akt was observed in PDKM xenografts, however, expression of phospho-MAPK was only abrogated in the C225-treated PDKM xenografts compared to vehicle-treated xenografts (Supplementary Figure 5). Additional investigation demonstrated that the antitumor effects of combined treatment with C225 and AR-12 was not significantly different compared with treatment using to either agent alone (Figure 6C). Since both AR-12 and C225 are highly potent in these HNSCC xenograft models, we were likely unable to observe a combined effect. These results suggest that inactivation of PDK1 kinase activity may enhance the efficacy of EGFR inhibitors in preclinical HNSCC models. Furthermore, inactivation of PDK1 kinase may not be sufficient to abrogate HNSCC tumor growth due to persistent MAPK phosphorylation, which can be abrogated by EGFR inhibition.
Figure 6. PDK1 targeting enhances EGFR inhibition in HNSCC.

(A) UM-22B cells were treated with AR-12 (200 nM), AG1478 (6 μM) or erlotinib (7 μM) or a combination of AR-12 plus AG1478 or erlotinib. MTT assay was performed after 72 hours and the data analysis was done using GraphPad Prism Software. Experiments were performed three times in 6 replicates (p=0.003). (B) Athymic nude mice were (n=5) were inoculated with 1.5 ×106 PDK1 kinase-dead transfected HNSCC cells (PDKM) or vector-transfected control cells (VC) in the left and right flanks, respectively. After the formation of tumor nodules (7 days later), mice were treated with vehicle (saline) or cetuximab three times a week (0.8 mg i.p.) in conjunction with tumor volume determinations. (C) Athymic nude mice were inoculated with UMSCC1 cells. After 7 days, mice were randomized into 4 treatment groups shown 1) vehicle 2) AR-12 3) C225 and 4) AR-12+C225. Tumors were measured every 3 days and tumor volume was calculated (length/2 × width2). Tumor volumes were graphed using Graph Pad prism Software.
Discussion
GPCR-EGFR crosstalk has been demonstrated to mediate tumorigenesis in a variety of cancer models via the extracellular release of EGFR ligands and EGFR-independent signaling (1, 21–28). Although the combined targeting of GPCRs and EGFR demonstrates additive or synergistic growth suppression effects, it is impractical to target multiple GPCRs and EGFR in the clinical setting. Identification of a common “druggable” signaling intermediate may represent a more rational co-targeting strategy to target GPCR and EGFR-mediated tumor progression. We previously reported that PDK1 mediated EGFR ligand release in response to GRP stimulation (5). PDK1 downmodulation also enhanced EGFR inhibition of proliferation and invasion. In the present study, we show that PDK1 represents a common signaling mediator of GPCR-mediated EGFR-dependent and –independent signaling in HNSCC. Furthermore, we demonstrate that targeting PDK1 expression and activity abrogated GPCR-mediated growth and enhanced EGFR inhibition in HNSCC pre-clinical models.
Previous studies demonstrated that GPCR ligands mediate release of EGFR ligands in a Src and MMP-dependent manner (1, 5, 29–31) in multiple HNSCC cell line models. We first reported the contribution of the PI3K-PDK1 signaling complex in the release of EGFR ligands in HNSCC mediated by the bombesin, GRP. Here, we observed that PDK1 contributed to PGE2, BK or LPA-mediated phosphorylation of EGFR. PGE2, BK and LPA-specifically mediate the EGFR phosphorylation via activation of EP, B2R and LPA1 receptors respectively (1, 19, 20). We have also reported that PGE2 and BK-mediated release of TGF-α was abrogated with TACE siRNA (2). Taken together, these cumulative results suggest that PDK1-mediated TACE phosphorylation appears to be critical for PGE2, BK and LPA–mediated EGFR activation and oncogenic signaling. In addition to EGFR ligands (32), TACE has been shown to mediate the release of heregulin leading to activation of HER3 (33). Targeting the release of these autocrine ligands via PDK1 inhibition may have therapeutic benefits over a broader range of tumor types.
PDK1 is a pleiotropic kinase that mediates proliferative, invasive and survival signaling pathways via activation of multiple substrates (7). We previously reported that PDK1 downmodulation decreased HNSCC growth (5), however, studies to date have not investigated the contribution of PDK1 in specific GPCR-mediated growth in any cancer. Using PDK1 siRNA, we found that GPCR-mediated growth was inhibited in HNSCC. Blockade of EGFR has been shown to significantly abrogate GPCR-mediated growth and invasion (2). The present study corroborates our earlier findings that GPCRs partially mediate HNSCC growth via EGFR ligand release and consequent EGFR activation (2). However, we observed that PDK1 mediated both EGFR-dependent and EGFR-independent signaling. GPCRs therefore most likely mediate HNSCC growth via both EGFR ligand release as well as EGFR-independent activation of p70S6K. We also observed that growth of PDKM cells was abrogated compared to control cells although PDK1 siRNA and PDKM displayed contrasting effects on phospho-Akt (Supplementary figure 6). Since these cells were cultured in serum-containing media, it would suggest that the partial growth inhibition was due to serum factors inducing EGFR-mediated growth. Interestingly, we did not observe the similar phenotype in vivo. PDKM xenografts displayed modestly enhanced tumorigenicity compared to vector control xenografts. We noted that vehicle-treated PDKM xenografts showed sustained levels of p-MAPK, which was diminished in C225-treated PDKM xenografts. Inhibition of MAPK activity by EGFR inhibitors was shown to improve the anti-tumor efficacy of breast cancer cells forced to express the PI3K inhibitor PTEN (34). Furthermore, monotherapeutic PDK1 inhibition was reported to increase MAPK phosphorylation levels and ineffectively prevent tumor inhibition in transgenic mouse models (35). The sustained activation of MAPK in PDKM cells may create an enriched tumor-promoting microenvironment in vivo compared to in vitro conditions.
Although we have shown that PDK1 mediates GPCR stimulated growth via EGFR activation, PDK1 may contribute to GPCR-mediated growth in the presence of EGFR blockade. We and others have reported that PGE2 and BK-mediate mitogenic signaling in the presence of EGFR inhibitors (2, 36). In NSCLC and an ovarian cancer cell line, PGE2 and BK mediated EGFR-independent signaling via PI3K and PKC (36–38). PI3K is upstream of PDK1 and PKC is also a PDK1 substrate (7, 39). Here, we show that PDK1 siRNA decreased p70S6K phosphorylation induced by EGFR downmodulation. Furthermore, we reported that EGFR downmodulation increased PKCδ activation, and PKCδ siRNA decreased p70S6K phosphorylation (6). Therefore, GPCR-mediated activation of PDK1 may also have phenotypic implications independent of EGFR ligand release.
Celecoxib has been demonstrated to inhibit Akt activity via blockade of PDK1 (40, 41). AR-12 is a celecoxib derivative that was reported to inhibit PDK1 activity in different cell models (10). However, in some cancer models, AR-12 also demonstrated cytotoxic effects via PDK1-independent pathways (12, 42–44). AR-12 was reported to activate CDKs and mediate oral cancer apoptosis in a p21-dependent manner (13). In the present study, we observed that AR-12 abrogated Akt phosphorylation. However, AR-12 also downmodulated cyclin D1 expression and induced PARP cleavage at much lower concentrations. These cumulative findings indicate that AR-12 induces cell death in HNSCC via multiple mechanisms. One report showed that AR-12 had a greater affinity for the PDK1 substrate, p21-activated kinase 1 (PAK1) than for PDK1 itself. Structurally, AR-12 was found to bind to the ATP binding pocket of PAK1 (45). Therefore, the enhanced sensitivity of HNSCC cells to AR-12 may be due to its ability to inhibit other AGC kinases with a greater affinity suggesting that this compound is a PDK1 pathway inhibitor. Alternatively, PDK1 may inhibit HNSCC cell proliferation and induce apoptosis through its effects on substrates other than Akt. To further delineate the efficacy of pharmacological PDK1 inhibition, use of more novel and specific inhibitors should be investigated (46).
Combined targeting of GPCRs and EGFR has demonstrated improved anti-tumor effects in many preclinical cancer models, including HNSCC (2, 4, 47, 48). Approximately 50% of new drugs developed target GPCRs, hence emphasizing their importance in treating human disease. The autocrine/paracrine release of TGF-α was reported to correlate with poor HNSCC patient prognosis (49). Therefore, inhibition of EGFR ligand release by both GPCR and EGFR-mediated activity represents a potential treatment strategy. In addition to abrogating PDK1-mediated activation of proliferative and motility effectors such as p70S6K and PAK1, inhibition of TACE-mediated release of TGF-α mediated by PGE2 and BK may enhance the inhibitory efficacy of EGFR blockade and its consequent autocrine release of EGFR ligands. In the present study, we found that the pharmacologic inhibitor AR-12 enhanced EGFR TKI inhibition, at least in part, through PDK1 targeting. AR-12 has also been reported to enhance the anti-tumor efficacy of erlotinib in NSCLC (44). Further, after observing that PDK1 can mediate mitogenic signaling in the absence of EGFR, the enhanced efficacy of PDK1 and EGFR inhibition in vitro may be due to combined inhibition of independent pathways. Upregulation of IGF1R which also signals via PDK1-PI3K pathway has been reported to contribute to EGFR TKI resistance in HNSCC (50). Therefore inhibition of a common downstream RTK mediator such as PDK1 will potently enhance EGFR inhibition strategies. However, it must be noted that we did not observe a combined effect with AR-12 and cetuximab in vivo, most likely due to the potent efficacy of AR-12 administered as a single agent. Due to its reported PDK1-independent effects, it is also possible that AR-12 potently suppresses both EGFR-dependent and –independent signaling cascades to induce tumor regression. While EGFR is a proven therapeutic target in HNSCC, only a subset of patients will respond to EGFR inhibitors. Targeting PDK1 by inhibition of its kinase activity or expression is a viable therapeutic option for HNSCC patients to enhance the effects of EGFR blockade.
Supplementary Material
ACKNOWLEDGEMENTS
Grant Support: NIH grants P50CA097190 and R01CA098372 and the American Cancer Society Clinical Research Professorship (to JRG)
Footnotes
CONFLICTS OF INTERESTS The authors have no conflict of interest to declare.
References
- 1.Gschwind A, Prenzel N, Ullrich A. Lysophosphatidic Acid-induced Squamous Cell Carcinoma Cell Proliferation and Motility Involves Epidermal Growth Factor Receptor Signal Transactivation. Cancer Res. 2002;62:6329–36. [PubMed] [Google Scholar]
- 2.Thomas SM, Bhola NE, Zhang Q, Contrucci SC, Wentzel AL, Freilino ML, et al. Cross-talk between G Protein-Coupled Receptor and Epidermal Growth Factor Receptor Signaling Pathways Contributes to Growth and Invasion of Head and Neck Squamous Cell Carcinoma. Cancer Res. 2006;66:11831–9. doi: 10.1158/0008-5472.CAN-06-2876. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Q, Thomas SM, Xi S, Smithgall TE, Siegfried JM, Kamens J, et al. Src Family Kinases Mediate Epidermal Growth Factor Receptor Ligand Cleavage, Proliferation, and Invasion of Head and Neck Cancer Cells. Cancer Res. 2004;64:6166–73. doi: 10.1158/0008-5472.CAN-04-0504. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Q, Bhola NE, Lui VWY, Siwak DR, Thomas SM, Gubish CT, et al. Antitumor mechanisms of combined gastrin-releasing peptide receptor and epidermal growth factor receptor targeting in head and neck cancer. Mol Cancer Ther. 2007;6:1414–24. doi: 10.1158/1535-7163.MCT-06-0678. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Q, Thomas S, Lui V, Xi S, Siegfried J, Fan H, et al. Phosphorylation of TNF-alpha converting enzyme by gastrin-releasing peptide induces amphiregulin and EGF receptor activation. Proc Natl Acad Sci. 2006;103:690–6906. doi: 10.1073/pnas.0509719103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhola NE, Thomas SM, Freilino M, Joyce S, Sahu A, Maxwell J, et al. Targeting GPCR-Mediated p70S6K Activity May Improve Head and Neck Cancer Response to Cetuximab. Clin Cancer Res. 2011;17:4996–5004. doi: 10.1158/1078-0432.CCR-10-3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mora A, Komander D, van Aalten DMF, Alessi DR. PDK1, the master regulator of AGC kinase signal transduction. Semin Cell Dev Biol. 2004;15:161–70. doi: 10.1016/j.semcdb.2003.12.022. [DOI] [PubMed] [Google Scholar]
- 8.Sacks PG, Parnes SM, Gallick GE, Mansouri Z, Lichtner R, Satya-Prakash KL, et al. Establishment and Characterization of Two New Squamous Cell Carcinoma Cell Lines Derived from Tumors of the Head and Neck. Cancer Res. 1988;48:2858–66. [PubMed] [Google Scholar]
- 9.Biondi RM, Cheung PCF, Casamayor A, Deak M, Currie RA, Alessi DR. Identification of a pocket in the PDK1 kinase domain that interacts with PIF and the C-terminal residues of PKA. Embo J. 2000;19:979–88. doi: 10.1093/emboj/19.5.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu J, Huang JW, Tseng PH, Yang YT, Fowble J, Shiau CW, et al. From the Cyclooxygenase-2 Inhibitor Celecoxib to a Novel Class of 3-Phosphoinositide-Dependent Protein Kinase-1 Inhibitors. Cancer Res. 2004;64:4309–18. doi: 10.1158/0008-5472.CAN-03-4063. [DOI] [PubMed] [Google Scholar]
- 11.Pullen N, Dennis PB, Andjelkovic M, Dufner A, Kozma SC, Hemmings BA, et al. Phosphorylation and Activation of p70s6k by PDK1. Science. 1998;279:707–10. doi: 10.1126/science.279.5351.707. [DOI] [PubMed] [Google Scholar]
- 12.Yacoub A, Park MA, Hanna D, Hong Y, Mitchell C, Pandya AP, et al. OSU-03012 Promotes Caspase-Independent but PERK-, Cathepsin B-, BID-, and AIF-Dependent Killing of Transformed Cells. Mol Pharmacol. 2006;70:589–603. doi: 10.1124/mol.106.025007. [DOI] [PubMed] [Google Scholar]
- 13.Haiming D, Chunhua H, Dongmei G, Dasheng W, Ching-Shih C, Steven MDA. OSU03012 activates Erk1/2 and Cdks leading to the accumulation of cells in the S-phase and apoptosis. Int J Cancer. 2008;123:2923–30. doi: 10.1002/ijc.23896. [DOI] [PubMed] [Google Scholar]
- 14.Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PRJ, Reese CB, et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase B[alpha] Curr Biol. 1997;7:261–9. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 15.Nakamura K, Sakaue H, Nishizawa A, Matsuki Y, Gomi H, Watanabe E, et al. PDK1 Regulates Cell Proliferation and Cell Cycle Progression through Control of Cyclin D1 and p27Kip1 Expression. J Biol Chem. 2008;283:17702–11. doi: 10.1074/jbc.M802589200. [DOI] [PubMed] [Google Scholar]
- 16.Kalish LH, Kwong RA, Cole IE, Gallagher RM, Sutherland RL, Musgrove EA. Deregulated Cyclin D1 Expression Is Associated with Decreased Efficacy of the Selective Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor Gefitinib in Head and Neck Squamous Cell Carcinoma Cell Lines. Clin Cancer Res. 2004;10:7764–74. doi: 10.1158/1078-0432.CCR-04-0012. [DOI] [PubMed] [Google Scholar]
- 17.Burkhard ML, Shirley KK, Verena F, Wolf M, Roland HS. Dynamic survivin in head and neck cancer: Molecular mechanism and therapeutic potential. Int J Cancer. 2007;121:1169–74. doi: 10.1002/ijc.22941. [DOI] [PubMed] [Google Scholar]
- 18.Marioni G, D'Alessandro E, Bertolin A, Staffieri A. Survivin multifaceted activity in head and neck carcinoma: Current evidence and future therapeutic challenges. Acta Otolaryngol. 2010;130:4–9. doi: 10.3109/00016480902856588. [DOI] [PubMed] [Google Scholar]
- 19.Abrahao AC, Castilho RM, Squarize CH, Molinolo AA, Santos-Pinto Dd, Jr, Gutkind JS. A role for COX2-derived PGE2 and PGE2-receptor subtypes in head and neck squamous carcinoma cell proliferation. Oral Oncol. 2010;46:880–7. doi: 10.1016/j.oraloncology.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang W, Bhola N, Kalyankrishna S, Gooding W, Hunt J, Seethala R, et al. Kinin B2 Receptor Mediates Induction of Cyclooxygenase-2 and Is Overexpressed in Head and Neck Squamous Cell Carcinomas. Mol Cancer Res. 2008;6:1946–56. doi: 10.1158/1541-7786.MCR-07-2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Amorino G, Deeble P, Parsons S. Neurotensin stimulates mitogenesis of prostate cancer cells through a novel c-Src/Stat5b pathway. Oncogene. 2007;26:745–56. doi: 10.1038/sj.onc.1209814. [DOI] [PubMed] [Google Scholar]
- 22.Barki-Harrington L, Daaka Y. Bradykinin induced mitogenesis of androgen independent prostate cancer cells. J Urol. 2001;165:2121–5. doi: 10.1097/00005392-200106000-00081. [DOI] [PubMed] [Google Scholar]
- 23.Bergmann S, Junker K, Henklein P, Hollenberg M, Settmacher U, Kaufmann R. PAR-type thrombin receptors in renal carcinoma cells: PAR1-mediated EGFR activation promotes cell migration. Oncol Rep. 2006;15:889–93. [PubMed] [Google Scholar]
- 24.Daub H, Ulrich Weiss F, Wallasch C, Ullrich A. Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature. 1996;379:557–60. doi: 10.1038/379557a0. [DOI] [PubMed] [Google Scholar]
- 25.Ding Y, Shi RH, Tong JD, Li X, Zhang GX, et al. PGE2 Up-regulates Vascular Endothelial Growth Factor Expression in MKN28 Gastric Cancer Cells via Epidermal Growth Factor Receptor Signaling System. Exp Oncol. 2005;27:108–13. [PubMed] [Google Scholar]
- 26.Fishman DA, Liu Y, Ellerbroek SM, Stack MS. Lysophosphatidic Acid Promotes Matrix Metalloproteinase (MMP) Activation and MMP-dependent Invasion in Ovarian Cancer Cells. Cancer Res. 2001;61:3194–9. [PubMed] [Google Scholar]
- 27.Luppi F, Longo AM, de Boer WI, Rabe KF, Hiemstra PS. Interleukin-8 stimulates cell proliferation in non-small cell lung cancer through epidermal growth factor receptor transactivation. Lung Cancer. 2006;56:25–33. doi: 10.1016/j.lungcan.2006.11.014. [DOI] [PubMed] [Google Scholar]
- 28.McCole DF, Keely SJ, Coffey RJ, Barrett KE. Transactivation of the Epidermal Growth Factor Receptor in Colonic Epithelial Cells by Carbachol Requires Extracellular Release of Transforming Growth Factor-alpha. J Biol Chem. 2002;277:42603–12. doi: 10.1074/jbc.M206487200. [DOI] [PubMed] [Google Scholar]
- 29.Gschwind A, Hart S, Fischer OM, Ullrich A. TACE cleavage of proamphiregulin regulates GPCR-induced proliferation and motility of cancer cells. Embo J. 2003;22:2411–21. doi: 10.1093/emboj/cdg231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luttrell LM, Hawes BE, van Biesen T, Luttrell DK, Lansing TJ, Lefkowitz RJ. Role of c-Src Tyrosine Kinase in G Protein-coupled Receptorand Gbeta gamma Subunit-mediated Activation of Mitogen-activated Protein Kinases. J Biol Chem. 1996;271:19443–50. doi: 10.1074/jbc.271.32.19443. [DOI] [PubMed] [Google Scholar]
- 31.Pages-Borrell M, Rojo F, Albanell J, Baselga J, Arribas J. TACE is required for activation of EGFR by TGF-alpha in tumors. Embo J. 2003;22:1114–24. doi: 10.1093/emboj/cdg111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kenny PA. Targeting TACE-dependent EGFR ligand shedding in breast cancer. J Clin Invest. 2007;117:337–45. doi: 10.1172/JCI29518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang SE, Xiang B, Guix M, Olivares MG, Parker J, Chung CH, et al. Transforming Growth Factor {beta} Engages TACE and ErbB3 To Activate Phosphatidylinositol-3 Kinase/Akt in ErbB2-Overexpressing Breast Cancer and Desensitizes Cells to Trastuzumab. Mol Cell Biol. 2008;28:5605–20. doi: 10.1128/MCB.00787-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.She QB, Solit DB, Ye Q, O'Reilly KE, Lobo J, Rosen N. The BAD protein integrates survival signaling by EGFR/MAPK and PI3K/Akt kinase pathways in PTEN-deficient tumor cells. Cancer Cell. 2005;8:287–97. doi: 10.1016/j.ccr.2005.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ellwood-Yen K, Keilhack H, Kunii K, Dolinski B, Connor Y, Hu K, et al. PDK1 Attenuation Fails to Prevent Tumor Formation in PTEN-Deficient Transgenic Mouse Models. Cancer Res. 2011;71:3052–65. doi: 10.1158/0008-5472.CAN-10-2282. [DOI] [PubMed] [Google Scholar]
- 36.Krysan K, Reckamp KL, Dalwadi H, Sharma S, Rozengurt E, Dohadwala M, et al. Prostaglandin E2 Activates Mitogen-Activated Protein Kinase/Erk Pathway Signaling and Cell Proliferation in Non-Small Cell Lung Cancer Cells in an Epidermal Growth Factor Receptor-Independent Manner. Cancer Res. 2005;65:6275–81. doi: 10.1158/0008-5472.CAN-05-0216. [DOI] [PubMed] [Google Scholar]
- 37.Adomeitt A, Graness A, Gros S, Seedorf K, Wetzker R, Liebmann C. Bradykinin B2 receptor-mediated mitogen-activated protein kinase activation in COS-7 cells requires dual signaling vis both protein kinase C pathway and epidermal growth factor receptor transactivation. Mol Cell Biol. 1999;19:5289–97. doi: 10.1128/mcb.19.8.5289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Graness A, Hanke S, Boehmer FD, Presek P, Liebmann C. Protein-tyrosinephosphatase-mediated epidermal growth factor(EGF) receptor transactivation and EGF receptor-independent stimulation of mitogen-activated protein kinase by bradykinin in A431 cells. Biochem J. 2000;347:441–7. doi: 10.1042/0264-6021:3470441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xia S, Chen Z, Forman LW, Faller DV. PKC[delta] survival signaling in cells containing an activated p21Ras protein requires PDK1. Cell Signal. 2009;21:502–8. doi: 10.1016/j.cellsig.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hsu AL, Ching TT, Wang DS, Song X, Rangnekar VM, Chen CS. The Cyclooxygenase-2 Inhibitor Celecoxib Induces Apoptosis by Blocking Akt Activation in Human Prostate Cancer Cells Independently of Bcl-2. J Biol Chem. 2000;275:11397–403. doi: 10.1074/jbc.275.15.11397. [DOI] [PubMed] [Google Scholar]
- 41.Arico S, Pattingre S, Bauvy C, Gane P, Barbat A, Codogno P, et al. Celecoxib Induces Apoptosis by Inhibiting 3-Phosphoinositide-dependent Protein Kinase-1 Activity in the Human Colon Cancer HT-29 Cell Line. J Biol Chem. 2002;277:27613–21. doi: 10.1074/jbc.M201119200. [DOI] [PubMed] [Google Scholar]
- 42.Park MA, Yacoub A, Rahmani M, Zhang G, Hart L, Hagan MP, et al. OSU-03012 Stimulates PKR-Like Endoplasmic Reticulum-Dependent Increases in 70-kDa Heat Shock Protein Expression, Attenuating Its Lethal Actions in Transformed Cells. Mol Pharmacol. 2008;73:1168–84. doi: 10.1124/mol.107.042697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ryan E, Bushnell T, Friedman A, Rahman I, Phipps R. Cyclooxygenase-2 independent effects of cyclooxygenase-2 inhibitors on oxidative stress and intracellular glutathione content in normal and malignant human B-cells. Cancer Immunol Immunother. 2008;57:347–58. doi: 10.1007/s00262-007-0374-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang YC, Kulp SK, Wang D, Yang CC, Sargeant AM, Hung JH, et al. Targeting Endoplasmic Reticulum Stress and Akt with OSU-03012 and Gefitinib or Erlotinib to Overcome Resistance to Epidermal Growth Factor Receptor Inhibitors. Cancer Res. 2008;68:2820–30. doi: 10.1158/0008-5472.CAN-07-1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Porchia LM, Guerra M, Wang YC, Zhang Y, Espinosa AV, Shinohara M, et al. 2-Amino-N-{4-[5-(2-phenanthrenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]-phenyl} Acetamide (OSU-03012), a Celecoxib Derivative, Directly Targets p21-Activated Kinase. Mol Pharmacol. 2007;72:1124–31. doi: 10.1124/mol.107.037556. [DOI] [PubMed] [Google Scholar]
- 46.Murphy ST, Bailey S, Burke BJ, Chappie TA, Ferre R, Greasley S, et al. Discovery of Novel, Potent and Selective Inhibitors of 3-Phosphoinositide-Dependent Kinase (PDK1) J Med Chem. 2011;54:8490–500. doi: 10.1021/jm201019k. [DOI] [PubMed] [Google Scholar]
- 47.Gadgeel S, Ruckdeschel J, Heath E, Heilbrun L, Venkatramanamoorthy R, Wozniak A. Phase II Study of Gefitinib, an Epidermal Growth Factor Receptor tyrosine kinase Inhibitor (EGFR-TKI), and Celecoxib, a Cyclooxygenase-2 (COX-2) Inhibitor, in Patients with Platinum Refractory Non-small Cell Lung Cancer (NSCLC) J Thoracic Oncol. 2007;2:299–305. doi: 10.1097/01.JTO.0000263712.61697.69. [DOI] [PubMed] [Google Scholar]
- 48.Reckamp KL, Krysan K, Morrow JD, Milne GL, Newman RA, Tucker C, et al. A Phase I Trial to Determine the Optimal Biological Dose of Celecoxib when Combined with Erlotinib in Advanced Non-Small Cell Lung Cancer. Clin Cancer Res. 2006;12:3381–8. doi: 10.1158/1078-0432.CCR-06-0112. [DOI] [PubMed] [Google Scholar]
- 49.Grandis JR, Melhem MF, Gooding WE, Day R, Holst VA, Wagener MM, et al. Levels of TGF-alpha and EGFR protein in head and neck squamous cell carcinoma and patient survival. J Natl Cancer Inst. 1998;90:824–32. doi: 10.1093/jnci/90.11.824. [DOI] [PubMed] [Google Scholar]
- 50.Jameson MJ, Beckler AD, Taniguchi LE, Allak A, VanWagner LB, Lee NG, et al. Activation of Epidermal Growth Factor Receptor Antagonism in Head and Neck Squamous Carcinoma Cells. Mol Cancer Ther. 2011;10:2124–34. doi: 10.1158/1535-7163.MCT-11-0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.