

Abstract
Iron (Fe0) corrosion in anoxic environments (e.g. inside pipelines), a process entailing considerable economic costs, is largely influenced by microorganisms, in particular sulfate-reducing bacteria (SRB). The process is characterized by formation of black crusts and metal pitting. The mechanism is usually explained by the corrosiveness of formed H2S, and scavenge of ‘cathodic’ H2 from chemical reaction of Fe0 with H2O. Here we studied peculiar marine SRB that grew lithotrophically with metallic iron as the only electron donor. They degraded up to 72% of iron coupons (10 mm × 10 mm × 1 mm) within five months, which is a technologically highly relevant corrosion rate (0.7 mm Fe0 year−1), while conventional H2-scavenging control strains were not corrosive. The black, hard mineral crust (FeS, FeCO3, Mg/CaCO3) deposited on the corroding metal exhibited electrical conductivity (50 S m−1). This was sufficient to explain the corrosion rate by electron flow from the metal (4Fe0 → 4Fe2+ + 8e−) through semiconductive sulfides to the crust-colonizing cells reducing sulfate (8e− + SO42− + 9H+ → HS− + 4H2O). Hence, anaerobic microbial iron corrosion obviously bypasses H2 rather than depends on it. SRB with such corrosive potential were revealed at naturally high numbers at a coastal marine sediment site. Iron coupons buried there were corroded and covered by the characteristic mineral crust. It is speculated that anaerobic biocorrosion is due to the promiscuous use of an ecophysiologically relevant catabolic trait for uptake of external electrons from abiotic or biotic sources in sediments.
Introduction
Iron, the fourth most abundant element in the earth's crust, is the principal redox-active metal in metabolic processes of essentially all living organisms. It is either involved in catalytic quantities as a component of a vast number of proteins, or in much higher, substrate quantities as the external electron donor or acceptor for specially adapted environmental microorganisms referred to as ‘iron bacteria’ (ferrotrophic bacteria, aerobic or anaerobic) or ‘iron-respiring bacteria’ respectively. In most biological functions, iron has the +II (ferrous) or +III (ferric) oxidation state. From a physiological point of view it appears astounding that also the native, metallic element (Fe0) can be involved in a biological process; this is anaerobic microbial corrosion. In technology, the process is often referred to as microbially influenced corrosion (MIC).
Iron is the technologically most widely employed metal, due to the abundance of its ores, straightforward melting and excellent mechanical properties. It is globally produced at a 25-fold higher extent (9.3 × 108 t year−1) than the second most widely employed metal, aluminum (US Geological Survey, 2011; data for 2009). Iron corrosion including MIC is thus of significant economic relevance. MIC affects industrial water-bearing systems such as oil and gas pipelines (Hamilton, 1985; Li et al., 2000; Schwermer et al., 2008). It therefore causes, besides economic losses, also failures that are of environmental concern or even hazardous (Duncan et al., 2009; Sherar et al., 2011). A critical feature of MIC is that it is not as visible as the commonly known rusting of iron under air, but usually occurs as a ‘hidden’ process in the interior of iron pipes or on iron constructions buried in aqueous underground. There is much agreement that sulfate-reducing bacteria (SRB; more generally also sulfate-reducing microorganisms, SRM) are the main culprits of MIC (Hamilton, 1985;Lee et al., 1995). Yet, the underlying mechanisms are apparently complex and insufficiently understood (Beech and Sunner, 2007). Their understanding is expected to contribute to the future development of effective mitigation strategies or causative counter measures.
The principal chemical feature in all models of MIC is that iron as a base metal easily gives off electrons, according to
| (1) |
(revised redox potential; Appendix S1). In rusting, which to our present knowledge is a purely chemical (abiotic) process, oxygen accepts electrons (4e− + O2 + 4H+ ⇄ 2H2O; E° = +1.229 V; E°′ = +0.815 V) and finally leads to the formation of brittle ferric oxides/hydroxides. Another ubiquitous electron acceptor is protons yielding hydrogen (2e− + 2H+ ⇄ H2; E° = ± 0.000 V; E°′ = −0.414 V). However, this is technologically only serious in rare instances of acidic surroundings. Proton reduction in circumneutral H2O and thus the net reaction
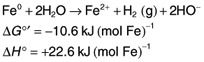 |
(2) |
are very slow (Fig. S1) so that iron in sterile anoxic water can, in principle, last for centuries. The corrosion risk for iron in the absence of acid or oxygen changes dramatically if constructions are exposed to non-sterile, ‘environmental’ aqueous surroundings where microorganisms such as SRB can grow and obviously accelerate iron oxidation enormously (Hamilton, 2003). Iron loss rates of 0.2–0.4 mm Fe0 year−1 are typically recorded in situ (Jack, 2002; Table S1). Two basically different modes by which SRB act upon iron have been envisaged (Dinh et al., 2004).
First, undissociated protons in H2S from respiratory reduction of natural sulfate (e.g. in seawater) with organic nutrients react more rapidly with iron-derived electrons than do protons from or in (circumneutral) H2O, according to
 |
(3) |
In such way, SRB act indirectly through an excreted chemical agent. We here refer to this process as ‘chemical microbially influenced corrosion’ (CMIC). The net reaction (Dinh et al., 2004) can be expressed, for instance with organic carbon of the oxidation state of carbohydrates (‘CH2O’, viz. the abundant building structure 〈H-C-OH〉) as
| (4) |
(details in Appendix S1).
Second, SRB can be involved more intimately in anaerobic iron corrosion by a mechanism that is fundamentally different from the above CMIC. This was first envisaged in a groundbreaking study of iron pipe corrosion in anoxic soil (von Wolzogen Kühr and van der Vlugt, 1934). SRB were suggested to use iron as the only source of reducing equivalents for sulfate reduction. The net stoichiometry of this purely lithotrophic process, here with the common carbonate (siderite) precipitation, is
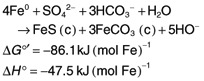 |
(5) |
Still, such bulk equation cannot provide hints as to the actual form of the reducing equivalents channelled from iron into sulfate reduction. It has been appealing to consider H2 (from H2O reduction; Eq. 2) as the intermediate (Booth and Tiller, 1960; von Wolzogen Kühr, 1961; Bryant and Laishley, 1990; Coetser and Cloete, 2005), indeed an excellent growth substrate of many SRB. Their high-affinity hydrogen scavenging (4H2 + SO42− + 2H+ → H2S + 4H2O) is thought to ‘pull’ the primary oxidation (von Wolzogen Kühr and van der Vlugt, 1934; von Wolzogen Kühr, 1961), an explanation also common in textbooks. On the other hand, accelerated anaerobic corrosion due to H2 utilization has been viewed critically (Costello, 1974; Hardy, 1983). In several kinetic studies, H2 scavenging did not accelerate iron oxidation (Spruit and Wanklyn, 1951; Dinh et al., 2004; Mori et al., 2010). Furthermore, novel marine deltaproteobacterial SRB enriched and isolated directly with metallic iron as the only electron donor reduced sulfate much faster than possible by mere scavenge of H2 and were more corrosive than conventional strains (Dinh et al., 2004). Moreover, they transiently formed much H2 rather than scavenged it, possibly due to an initial excess of iron-derived reducing power. Therefore, the ability to make use of Fe0 for sulfate respiration in a kinetically more efficient manner than via the slowly formed abiotic H2, viz. through a faster by-pass, was assumed, and direct electron uptake from iron has been suggested (Dinh et al., 2004). This theory is here referred to as ‘electrical microbially influenced corrosion’ (EMIC).
In this study, we investigated the extent of iron destruction by these strains of SRB as well as the postulated EMIC and its significance in more detail. First, we measured whether corrosion rates as high as observed in industrial settings can be also attained in vitro by appropriately adjusted cultivation conditions. Second, we examined whether and in which way the increasing coverage of the metallic substrate by the inorganic black corrosion crust (Dinh et al., 2004) is compatible with progressive corrosion and the hypothesized electron uptake from the metal. Third, we buried iron specimens in a field study in natural marine sediment to prove whether corrosion phenomena in situ were similar as observed in laboratory incubation experiments.
Results
To study the postulated EMIC by the previously isolated strains under experimentally defined conditions, metallic iron was provided in the form of coupons as the sole electron donor for sulfate reduction. The only added organic compounds were trace amounts of vitamins (totally 0.58 mg l−1, Table S2), and acetate (1 mM) provided as a biosynthetic building block to lithoheterotrophic strains IS5, HS3, and to Desulfopila inferna. Cultures incubated with 10 mM acetate without iron did not produce any sulfide, indicating that external acetate was not used as an electron donor. Measures of corrosion were the determination of iron mass loss at the end of incubation, a long-established routine method (Booth et al., 1967), and quantification of sulfate consumption, a more recently established method (Dinh et al., 2004) allowing highly resolved time-courses. Consumption of sulfate parallels production of sulfide that cannot be monitored directly due to precipitation as FeS (Eq. 5). An analytical control experiment verified that disappearance of sulfate was only due to reduction and not in addition to a certain co-precipitation in the forming corrosion crust (Fig. S2).
Iron corrosion rates in long-term incubation experiments
Metallic iron represents a very compact, dense form of an electron donor sufficient to reduce dissolved sulfate from a relatively large culture volume. In the initial study (Dinh et al., 2004), the culture volume (0.15 l) to metal (30 g) ratio was kept relatively small for clearly revealing the corrosive potential of novel marine SRB within 20 days. In such incubations, the sulfate reduction rate slowed down significantly after a while. Examination in more detail in the present study revealed that this drop in activity was mostly due to the pronounced alkalization and exhaust of counteracting CO2 (dissolved and gaseous). For the present biocorrosion experiments intended to examine iron destruction under conditions comparable to those in situ during much longer incubation, the ratio of the culture (and gas phase) volume to metal mass had to be increased. Because macroscopic corrosion phenomena were of central interest, the iron specimens (10 mm × 10 mm × 1 mm) could not be miniaturized to any extent, thus necessitating much bigger culture volumes. An appropriate medium volume was 1.4 l, which was still small enough for precise monitoring of sulfate consumption. Indeed, corrosion rates did not significantly decrease over months. Corrosive cultures reached values as high as 0.7 mm Fe0 year−1 and deposited steadily growing black crusts (Fig. 1A and B). After selective crust removal, severe metal loss was evident (Fig. 1B). In the present experiments, strain IS5 was more corrosive than strain IS4, whereas in the initial physiological characterization (Dinh et al., 2004), the latter was more corrosive. This may be due to the higher tolerance of strain IS4 to the significantly increasing pH in the previous incubations. ‘Conventional’ SRB (control strains), which were Desulfopila inferna (a phylogenetic relative of strain IS4; Gittel et al., 2010), and Desulfovibrio strain HS3 (an effective scavenger of H2 isolated in this study), showed essentially no signs of iron corrosion within the incubation period. Iron in these control cultures was not more affected than in sterile incubations (Fig. 1A and B; Fig. S3). The inability to make more efficient use of iron was not due to sensitivity towards Fe2+ ions. The control strains were able to scavenge H2 formed from iron and water (Eq. 2) below detection limit (40 ppmv) and grew readily in the presence of iron if H2 was supplied externally (Fig. S4).
Fig. 1.
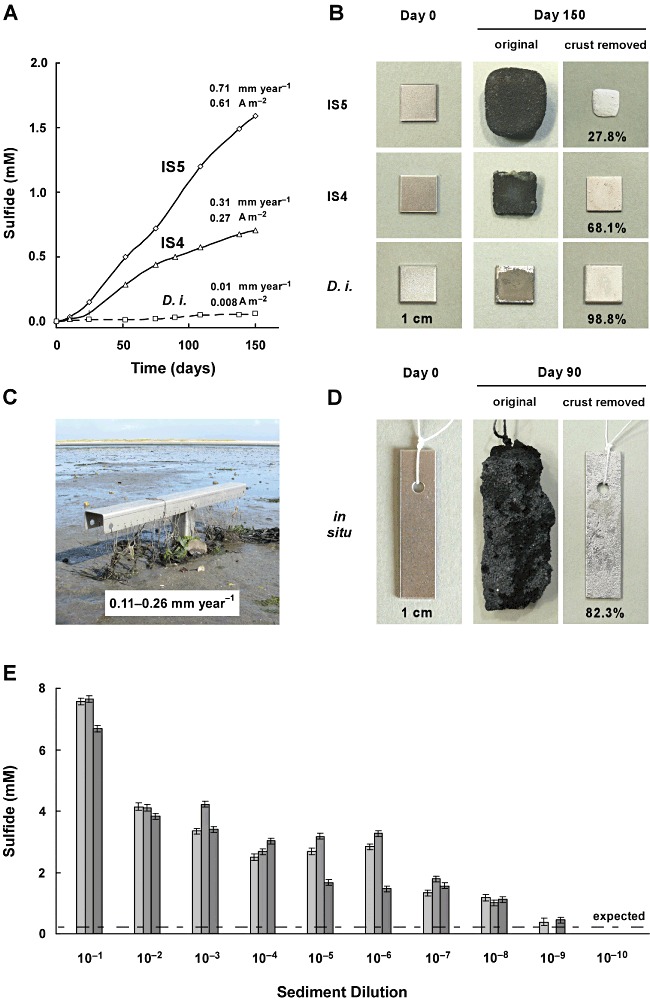
Corrosive sulfate-reducing bacteria in pure cultures and in situ. A. Long-term sulfide formation (measured as sulfate consumption) with iron coupons as the only electron donor in cultures of corrosive strains IS5 and IS4 (proposed: Desulfovibrio ferrophilus and Desulfopila corrodens respectively), and in a hydrogenotrophic control culture (Desulfopila inferna, D. i.). B. Thick corrosion crusts and metal loss in the same cultures. Residuary metal (% of initial) became obvious after crust removal by HCl-hexamine. C. Positioning device (stainless steel) for iron coupons in the Wadden Sea, island of Sylt (North Sea). Iron coupons were bound with threads to the device and buried for three months at ≥ 20 cm depth in anoxic sediment. D. Corrosion crust (with sand grains), and corroded metal (after crust dissolution) from (C). Here, the photographed fresh coupon (Day 0) is not the same as the incubated one. E. Sulfide formed (measured as sulfate consumed) after six months in serial dilutions (three in parallel) with native sediment (2 g, wet mass) from the same habitat. The line indicates sulfide expected solely by consumption of H2 formed from iron and seawater (based on independently measured H2-formation rates and experiments with merely H2-scavenging SRB).
Localization of corrosive cells, and determination of crust conductivity
If the pronounced corrosion is due to direct electron uptake by SRB, cells must be always electrically connected to their metallic substrate. This could be possible by direct attachment to the metal. However, such localization would implicate increasing coverage by the forming hard corrosion crust and cut-off from the medium which supplies sulfate and counteracts the strongly alkalizing effect of iron oxidation (Eq. 5). Progressive utilization of metallic iron despite coverage by crust would be possible if active cells would colonize the medium-exposed crust surface, and if the crust would be electrically conductive.
Indeed, virtually no planktonic (free-living) cells could be observed, and scanning electron microscopy revealed a densely colonized crust surface in the corrosive cultures of strains IS4 or IS5. Colonized areas of the structurally heterogeneous crust contained the element S in addition to Fe, C and O (details in Fig. 2), as revealed by energy-dispersive X-ray spectroscopy (EDX) of the uppermost (c. 5 µm) crust. Sulfur-free patches were never colonized.
Fig. 2.
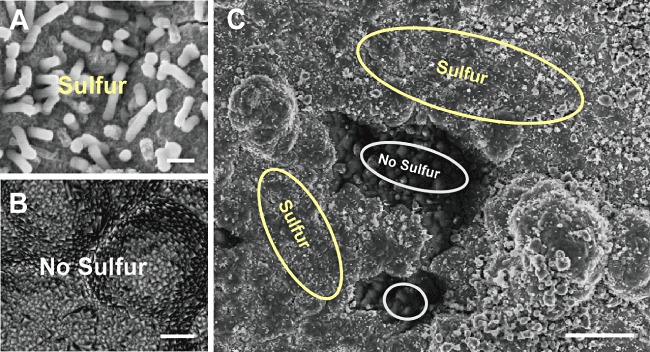
X-ray microanalysis (EDX) of crust surface in a culture of corrosive strain IS4. A. Site colonized by cells (Bar, 1 µm). B. Site without microbial colonization (Bar, 1 µm). C. Both sites in the same field of view (Bar, 20 µm). Surface-attached cells of strain IS4 colocalize with the element S. Cells were not detectable at sulfur-free sites. Both, sulfur-containing and sulfur-free sites contained the elements Fe, C and O. Sulfur-free sites contained in addition Mg and Ca. Thirty point spectra at 10 kV were collected for each site. Resolution (lateral and vertical), 3–5 µm.
Crust conductivity was evaluated as follows. Iron granules employed in previous cultivation (Dinh et al., 2004) tended to be cemented by the developing crust. This feature opened a simple way to measure conductivity of the crust in a non-invasive manner if the precipitate was allowed to cement two iron coupons fixed at defined distance and connected to monitoring wires protruding the stopper of the anoxic flask (Fig. 3, Fig. S5). The mounted coupons were only partly immersed so as to keep iron other than the slot-forming part outside of the medium. The conductivity of the biogenic crust measured at a voltage (< 0.2 V; DC) far below that for water electrolysis was around 50 S m−1 (Fig. 3, Table S3).
Fig. 3.
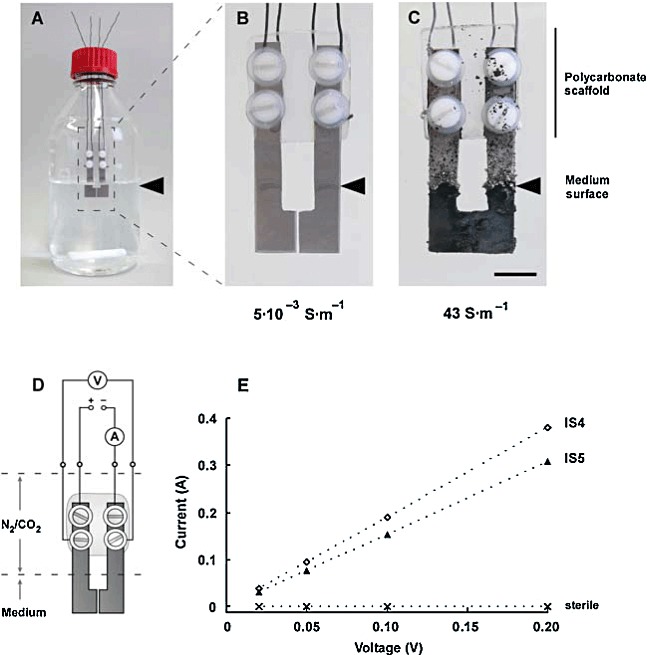
Determination of the conductivity of the corrosion crust. A. Anoxic bottle (600 ml medium) with two specially shaped fresh iron coupons. B. Coupons after three weeks of incubation in sterile medium. C. Coupons after three weeks of incubation with strain IS4. Bar, 1 cm. D. Scheme of the arrangement with voltage control and current measurement through separate circuits. E. Linear response of current to applied (non-electrolytic) voltage (0.02–0.2 V, DC).
Bulk composition and surface structures of biogenic corrosion crust
The bulk composition of the crust formed by strain IS4 was analysed quantitatively by combining EDX, X-ray diffraction (XRD), inductively coupled plasma optical emission spectroscopy (ICP-OES), and infrared spectroscopy. This revealed siderite (FeCO3) and amorphous ferrous sulfide at the expected ratio (Eq. 5; Table 1), and additional co-precipitated minerals such as calcite (CaCO3).
Table 1.
Analysis of corrosion products of iron coupons from cultures of strain IS4, and from burial in permanently anoxic sulfidic marine sediment (Sylt, North Sea)
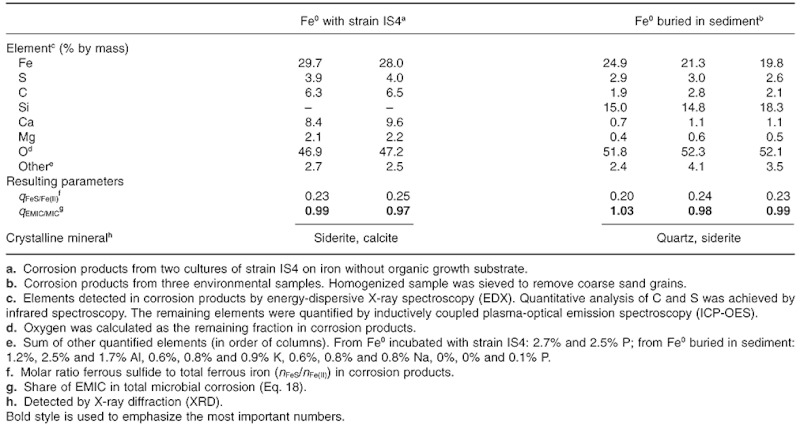 |
Figure 4 shows various images of the corrosion crust or coupon surface. On the crust covering coupons in cultures with strongly increased pH (as often observed in small culture volumes), ‘pustule’-like elevations appeared after several weeks of incubation (Fig. 4C, insert). The iron located underneath such ‘pustules’ exhibited a pronounced pitting area, as visualized upon crust removal (Fig. 4C). Strikingly shaped microscopic structures emerged on top of such ‘pustules’ at pH ≥ 9. In such cultures, the otherwise irregular crust exhibited round crater- or chimney-like structures (Fig. 4D–H, Fig. S6–8). Various growth stages of these structures were observed (Fig. 4).
Fig. 4.
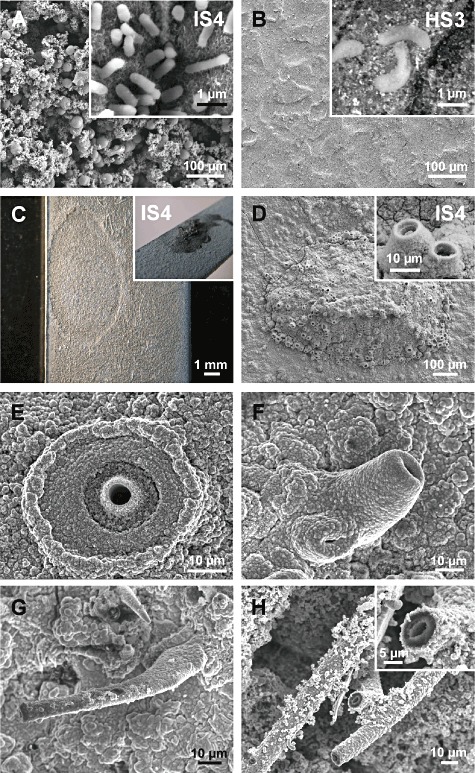
Scanning electron micrographs of crust surface, colonization and microchimneys. A. Cells of strain IS4 on corrosion crust after three months. B. Cells of non-corrosive hydrogenotrophic control strain HS3 on slightly corroded metal surface after three months. C. Anodic iron dissolution (crust removed) underneath a ‘pustule’ (insert) formed by strain IS4 at high pH (≍ 9; Fig. 5C). D. ‘Pustule’ with short microchimneys (magnified in insert) in a culture of strain IS4. E. Microchimney developing from a crater-like structure in a culture of strain IS4. F. Short microchimney in a culture of strain IS4. G. Long microchimney in a culture of strain IS4. H. Long microchimney (magnified in insert) in an alkalizing corrosive enrichment.
Field study of iron corrosion in marine sediment
To examine as to which extent the metallic iron used in laboratory experiments undergoes corrosion in a natural environment with sulfate reduction, iron coupons were buried in the dark (anoxic) part of a silty marine mud flat (Wadden Sea, island of Sylt, Germany) at c. 20 cm below the surface. Recovery was ensured by fixation via threads to a T-shaped positioning device (Fig. 1C). The coupons retrieved after three months were covered by thick black crusts (Fig. 1D). Their enormous thickness was largely due to sedimentary minerals (e.g. sand) cemented with the corrosion crust. Again, a characteristic crust composition of siderite and amorphous iron sulfide (Table 1) as well as surface pitting and mass or thickness loss ofthe metal were evident (up to 0.26 mm Fe0 year−1; Fig. 1D).
In addition, the natural abundance of SRB with corrosive potential was estimated via dilution series in anoxic tubes with iron coupons as the sole electron donor. The examined sediment sample was taken from the same site before the coupons were buried, viz. there was no artificial pre-enrichment of SRB with metallic iron. Development of sulfate reduction to a higher extent than would be possible by mere scavenging of chemically formed H2 (known from sterile control incubations) indicated corrosive SRB at numbers of more than 107 cells per gram wet sediment (Fig. 1E).
Discussion
In the present study, the ability of SRB to utilize metallic iron lithotrophically and thus cause corrosion was even more pronounced than previously expected (Fig. 1; Dinh et al., 2004). Apparently, only particular species of SRB can effectively exploit iron as an electron donor for fuelling their energy metabolism through sulfate reduction, so that distinction between corrosive and non-corrosive (‘conventional’) strains or species is justified. Corrosive SRB are not necessarily related on the basis of 16S rRNA-based phylogeny; they branch within distinct lineages of SRB (Dinh et al., 2004). Nevertheless, more extended comparative corrosion studies are needed to clarify whether corrosiveness is a genetically fixed trait, or whether also ‘conventional’ strains, for instance close relatives of strains IS4 and IS5, can gradually adapt to utilize and corrode iron if exposed to the metal over years. The ability to utilize iron directly as an electron donor for sulfate reduction primarily urges upon an understanding of the underlying mechanisms.
Towards an understanding of the corrosion mechanisms
Principal physico-chemical considerations as well as previous (Dinh et al., 2004) and present incubation experiments together with the measured electrical conductivity of the corrosion crust are strongly in favour of the EMIC hypothesis, that is direct electron gain for sulfate respiration from the metal via the crust.
The reduction of H+-ions (strictly, H3O+-ions) by Fe0-derived electrons on the metal surface is a par excellence example of a kinetically ‘impeded’, slow electrochemical reaction. Availability of H+ ions at the metal surface and combination of the primarily formed atomic hydrogen [e− + H+ → H(adsorbed)] to H2 are commonly understood as kinetic ‘bottle neck’ that also explains the high negative electrochemical overpotential (difference between potential during net reaction and equilibrium potential under the given conditions) of electrochemical H2-formation on iron (Bockris and Reddy, 1970; Hamann et al., 2007). Microbial scavenge of H2, viz. a product behind the ‘bottle neck’, is therefore not expected to accelerate the primary iron dissolution (Fig. S1). This is in accordance with experimental findings in evaluations of the ‘cathodic hydrogen’ theory of MIC (Costello, 1974; Hardy, 1983; Dinh et al., 2004). Some corrosive strains even formed significantly higher amounts of H2 than sterile incubations during the initial incubation phase with metallic iron. Deposition of some black FeS at the glass walls of the bottles indicated that a part of the SRB population grew distantly from the coupons with such biologically released H2. The assumption of a direct electron uptake by cells would not only explain the high corrosion rate of special SRB, but also the pronounced initial release of H2. This is possibly an ‘unavoidable’ side reaction via a hydrogenase because electron uptake from freshly supplied iron may be faster than electron consumption by sulfate reduction (Dinh et al., 2004).
Since electrons, unlike chemical compounds such as H2, cannot diffuse or flow through water, electron-conducting structures would be needed. On the side of the cell, these might be outer membrane and periplasmic membrane proteins investigated in various microorganisms in bioleaching of metals (Appia-Ayme et al., 1999; Auernik et al., 2008), extracellular iron(III) reduction or microbial fuel cells (Butler et al., 2010). Between cells and the corroding iron, which is being covered by a steadily growing sulfidic corrosion crust, the latter itself is envisaged as the electrical mediator. Metal sulfides, which tend to be non-stoichiometric, are long-known semiconductors (Braun, 1875; Pearce et al., 2006), and some earlier biocorrosion models based on H2 production and consumption hypothesized about a participation of semiconductive FeS (Booth et al., 1968; King and Miller, 1971) in abiotic H+ reduction.
The undisturbed corrosion crust in cultures of strains IS4 and IS5 indeed exhibited a conductivity of around 50 S m−1 (A V−1 m−1); this is even higher than that of many typical semiconductors (e.g. pure silicon, 1.6 × 10−3 S m−1; Table S4) or microbial biofilms with nanowires allowed to form between gold sheets mounted in cultures of Geobacter sulfurreducens (0.5 S m−1; Malvankar et al., 2011). Conductivity of the heterogeneous corrosion crust must be due to contained iron sulfides because FeCO3 and CaCO3 are essentially insulating minerals (10−10 and 10−14 S m−1 respectively; Table S4). This was confirmed in the present study by a conductivity test of siderite mineral (Fig. S9). Even though the measured biocorrosion rates of 0.71 mm Fe0 year−1 were high and technologically relevant, the corresponding current density of 0.61 A m−2 (Appendix S1) would need a voltage (potential difference) of only V = 1.2 × 10−4 V across a 1 cm crust. The calculated equilibrium potential at the corroding iron surface and the zone of sulfate reduction is around −0.60 and −0.25 V, respectively, viz. ΔE = 0.35 V (couples FeCO3/Fe0 and SO42−/FeS respectively; Appendix S1). Hence, there is significant leeway for the ‘self-adjusting’ potential difference driving the corrosion current through the crust. Crust conductivity is apparently not a rate-limiting factor. The model of corrosive SRB gaining electrons through semiconductive ferrous sulfide is further corroborated by the electron microscopic finding of cells attached mostly to the sulfide-rich islands within the predominantly carbonaceous structure (Fig. 2).
Electrons can only flow to cells if the crust also allows an equivalent ion flow via aqueous ‘bridges’ (maintenance of electroneutrality). These may be tiny interstices or fissures. An apparent, striking ion bridge, also strongly supporting the model of transcrustal electron flow, emerged at pH ≥ 9. In such cultures, the otherwise irregular crust exhibited round crater- or chimney-like structures (Fig. 4D–H, Fig. S6–8). Their formation is presently explained by a lowered, crust-preventing pH inside due to slightly acidic Fe2+-ions [Fe2+ + H2O ⇄ Fe(OH)+ + H+; pKa = 8.8] and precipitation of Fe2+ as soon as it enters the high-pH carbonate-containing medium (Fig. 5C, Fig. S10).
Fig. 5.
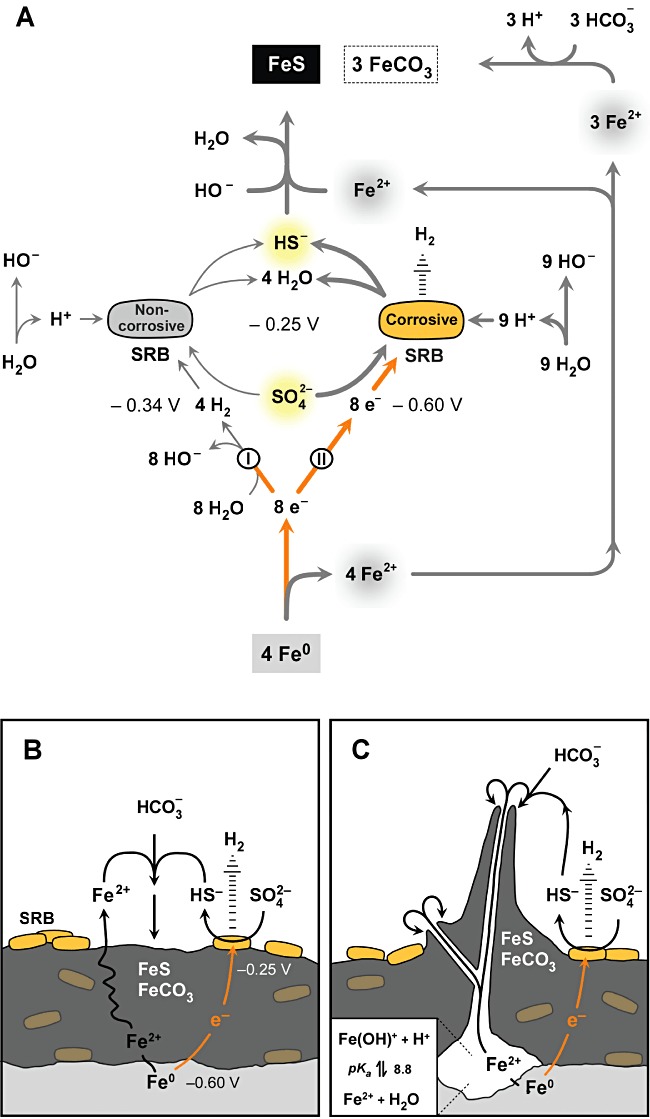
Scheme of the stoichiometry and topology of direct (lithotrophic) corrosion. A. Stoichiometry of iron dissolution and channelling of electrons via H2 (I; classical scheme) or directly (II; new model) into sulfate reduction. Bold lines indicate the much faster electric ‘bypass’. Equilibrium redox potentials are indicated for real conditions at pH = 8 (see Appendix S1). Direct electron utilization provides a higher metabolic driving force (voltage: ΔE = 0.35 V, compared with 0.09 V via H2 of assumed ≍ 40 ppmv). Fe2+ not precipitated as sulfide may enter solution or precipitate with naturally widespread inorganic carbon as FeCO3. B. Electron flow through the crust to attached cells at pH = 8 (simplified, non-stoichiometric). Crust may contain co-precipitated calcium and magnesium carbonate, and/or cemented sand. The equivalent ion flow may occur via aqueous interstices (not depicted). C. Build-up of chimney-like ion bridges at pH ≥ 9. Reactions are essentially as in (B); however, there are pronounced spots of anodic iron dissolution. A significant pH gradient (low inside, high outside) causes Fe(II) precipitation at the rim, leading to chimney growth. Schemes include the possibility of H2 release (that may foster remote bacterial cells) due to an imbalance between electron uptake and sulfate reduction. Participation of possibly buried (encrusted) cells in sulfate reduction and H2 release is unknown.
In conclusion, anaerobic corrosion caused by the direct, lithotrophic mode of iron utilization according to Eq. 5 can be only explained by direct electron uptake (Fig. 5), i.e. real occurrence of the electrochemical half-reaction
| (6) |
(E°′av = −0.218 V, average) coupled to iron dissolution (Eq. 1). Hence, the lithotrophic, direct corrosion is always EMIC.
Comment on direct corrosion by methanogenesis
There is first evidence that also special methanogenic archaea obtained through enrichment with metallic iron as the only source of reducing equivalents can bypass the slow abiotic H2 formation on iron in water by faster direct use of the electrons (Eq. 1) according to 8e− + HCO3− + 9H+ → CH4 + 3H2O (Dinh et al., 2004; Mori et al., 2010; Uchiyama et al., 2010). Here, the net reaction, 4Fe0 + 5HCO3− + 2H2O → 4FeCO3 (c) + CH4 (g) + 5HO−[ΔG°′ = −73.9 kJ (mol Fe)−1; ΔH° = −35.3 kJ (mol Fe)−1], does not lead to a conductive precipitate. One may speculate that in this case cell-metal contact must be sustained so that hindrance by crust coverage may become obvious during long-term incubations (which have not been carried out so far). Nevertheless, the process may play a role in MIC because methanogenic archaea may take advantage of electroconductive FeS precipitated by co-occurring sulfate reduction. In axenic laboratory cultures, there is some FeS precipitation by sulfide added as reductant.
Biosynthesis during direct iron corrosion by sulfate reduction
An understanding of the properties of the precipitate formed during corrosion also requires knowledge of the proportion of formed cell mass that may be embedded. Because there is presently no convenient method for determining cell mass in the solid corrosion crust, its organic content was estimated. According to the principle of bifurcate substrate flow in every chemotrophic organism, the amount (e.g. in mol or mmol) of total iron oxidized in cultures of SRB performing EMIC, nFeEMIC, is the sum of the amount oxidized catabolically by sulfate reduction, nFeCatab, and the amount oxidized due to the anabolic need of electrons, nFeAnab, i.e.
| (7) |
(basic scheme in Fig. S11). If EMIC is the only corrosion process, nFeEMIC is identical with the loss of metallic iron, nΔFe(0), and nFeCatab is fourfold higher than the amount of sulfate being reduced (Eq. 5), viz. nFeCatab = 4 nSR. Hence, anabolic iron oxidation can be expressed as the difference of two measurable parameters according to
| (8) |
With this, also a partition coefficient (quotient) or contribution of anabolic iron oxidation to total iron oxidation during EMIC can be formulated as
| (9) |
Iron loss is equivalent with ferrous iron formation, i.e. nΔFe(0) = nFe(II). The resulting nFeAnab (Eq. 8) can betranslated into formed biomass via assimilation equations if an elementary bulk composition and hence a formula mass (‘molecular’ mass) of bacterial dry mass is assumed. The assimilation equations express how much cell mass, mBio (e.g. in g or mg), is formed per amount of iron used for the anabolism, viz. they allow to formulate an anabolic yield coefficient,
| (10) |
Here we used the simplified bulk formula C4H8O2N (van Dijken and Harder, 1975; comments in Widdel and Musat, 2010) with ‘M’ = 102.1 g mol−1. Synthesis of cell carbon may occur with CO2 alone (lithoautotrophic growth; strain IS4), or require in addition an organic substrate such as acetate (lithoheterotrophic growth; strain IS5). The resulting assimilation equations are
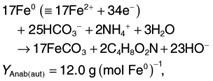 |
(11) |
and
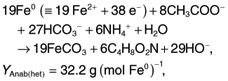 |
(12) |
for autotrophic and heterotrophic growth respectively. The latter equation is based on the observation that c. ⅔ of cell carbon in SRB is derived from acetate and c. ⅓ from bicarbonate (Sorokin, 1966; Badziong and Thauer, 1978). Through such theoretical assimilation equations and Eq. 8, the expected biomass can be calculated from nFeAnab as
| (13) |
The mass of the minerals (FeS, FeCO3, co-precipitated CaCO3 and possibly MgCO3) precipitated during lithotrophic corrosion, mMin, can be calculated from the same measurable parameters as nFeAnab (Eq. 8), viz. from nΔFe(0)[or nFe(II)] and nSR (Appendix S1). This further allows to express the biomass content as a partition coefficient (quotient) relating the biomass, mBio, to the total mass of precipitated crust, mMin + mBio. For this, the formulas
| (14) |
and
| (15) |
can be derived (Appendix S1) for autotrophic and heterotrophic growth respectively (again with nΔFe(0) = nFe(II)). They are applicable if EMIC is the only process of corrosion, as in our cultures, and if one assumes complete precipitation of ferrous iron (and variable precipitation of Ca/MgCO3).
Strains IS4 and IS5 corroded n▵Fe(0) = 4.31 and 9.86 mmol Fe0, respectively, during reduction of nSR = 0.95 (± 0.06) and 2.14 (± 0.08) mmol SO42− respectively. Hence, the amounts accounting for biosynthesis (Eq. 8) of strains IS4 and IS5 were nFeAnab = 0.51 and 1.3 mmol Fe0, respectively, which yields the following values for partition of anabolic in total iron oxidation (Eq. 9), and for the biomass content of the crust (Eqs 14 and 15):
| (16) |
| (17) |
These results indicate that the bulk of electrons is channelled into the catabolism (sulfate reduction), as common in the strictly anaerobic SRB, and that the corrosion crust is essentially inorganic, in agreement with its hard, mineral-like appearance. The corrosion crust thus profoundly differs from typical biofilms that are largely constituted of cells and an organic matrix.
Practical considerations
Protection of iron against corrosion by alloying or coating is technically and economically not possible at any extent. For instance, the most important alloying metal, chromium, is produced at a 48-fold lesser extent than iron (US Geological Survey, 2011; data for 2009). We therefore expect that advances in the analysis and control of MIC in situ will remain a relevant issue.
A long-term goal would be the design of specific counter measures based on detailed understanding of the environmental conditions and mechanisms leading to MIC. Such a ‘causative’ approach is confronted with a complexity of chemical conditions (e.g. availability of electron acceptors, or electron donors in addition to metallic iron) and chemical, electrochemical and microbial reactions in MIC. The most promising research strategy is, in the opinion of the authors, an experimental ‘dissection’ into individual and elementary processes for later synopsis. In the previous (Dinh et al., 2004) and present study, we focused on lithotrophic growth with metallic iron as the only electron donor under strictly anoxic conditions in the presence of sulfate as electron acceptor. Further, more comprehensive microbiological investigations would have to consider contributions to corrosion for instance by lithotrophic methanogenic archaea (Dinh et al., 2004; Mori et al., 2010; Uchiyama et al., 2010) if sulfate is limiting, or by CMIC due to organotrophic growth of SRB in organic-rich environments. Among the involved inorganic compounds, iron sulfides with their delicate, variable chemical and electrochemical properties are of central interest. They cannot only mediate electron flow. Under certain, high-sulfide conditions iron sulfides apparently protect the underlying metal against further rapid corrosion (Smith and Miller, 1975; Lee et al., 1995). Another, different role has been attributed to iron sulfides under intermittent anoxic-oxic conditions where corrosion can be particularly severe. This has been explained mainly by the reactivity of chemical oxidation products of ferrous sulfide towards iron (Jack, 2002; Hamilton, 2003; Beech and Sunner, 2007).
In view of the presently limited understanding of MIC in situ, recommendation of control measures is also limited. As long as MIC in a particular situation is largely CMIC, prevention would be possible if the organic nutrient of the anaerobic bacteria can be identified and eliminated. If MIC is essentially EMIC, avoidance of sulfate-containing water would be a causative solution. Extended in vitro and pilot studies may clarify as to which extent an addition of nitrate, which is applied to control biogenic H2S (souring) in petroleum reservoirs (Reinsel et al., 1996; Gieg et al., 2011), is also promising for controlling CMIC and EMIC (Hubert et al., 2005; Schwermer et al., 2008).
Despite the complexity of anaerobic corrosion processes in situ, the presently examined strains IS4 and IS5 are envisaged as representatives of the key players in MIC. These strains caused corrosion rates well matching those reported for the destruction of industrial iron structures in permanently anoxic aqueous surroundings (Table S1). Also, the rates of direct corrosion by strains IS4 and IS5 are similar as rates of chemical iron destruction by sulfide (Lee et al., 1995; Jack, 2002; Beech and Sunner, 2007; Table S1). This raises interest in an estimate of the contribution of EMIC in total MIC (EMIC plus CMIC by organotrophically formed sulfide) in cases of corrosion in situ. Because CMIC (Eq. 4) leads exclusively to sulfidic iron, whereas EMIC leads in addition to non-sulfidic (usually carbonaceous) iron, their measurable ratio in the crust of a corroded construction may be used for estimating the extent of EMIC. Such extent is quantitatively expressed as the quotient of the amounts (mol) of iron lost by EMIC, nFeEMIC, and iron lost totally by MIC, nFeMIC, and thus defined as qEMIC = nFeEMIC/nFeMIC. Assuming that all ferrous iron in the crust results from metal corrosion, nFeMIC = nFe(II), so that qEMIC = nFeEMIC/nFe(II). Further calculation so as to substitute the unknown nFeEMIC and introduce the measurable amount of precipitated sulfide, nFeS, leads to the formula (calculation in Appendix S1)
| (18) |
Hence, besides analysis of sulfidic and total ferrous iron in the crust, only the assumption of a qAnab value (see above) is needed. For approximate calculation, qAnab may be omitted (because qAnab << 3). Still, such formal treatment is only applicable for anoxic conditions and absence of processes other than sulfate reduction, for instance methanogenesis (Dinh et al., 2004; Mori et al., 2010; Uchiyama et al., 2010), and absence of secondary conversion of FeCO3 to FeS (further remarks in Appendix S1). Analysis of the crust on the coupons recovered from the field study revealed nFeS/nFe(II) = 0.20–0.24. By assuming, for convenience, qAnab ≍ 0.1, we obtain qEMIC = 0.98–1.03 (theoretically, according to Eq. 18, qEMIC always ≤ 1). This suggests that corrosion under the conditions prevailing at the studied marine sediment site was indeed only EMIC, viz. due to SRB capable of direct electron uptake. Such crust analysis (von Wolzogen Kühr and van der Vlugt, 1934; Spruit and Wanklyn, 1951), with awareness of its limits in view of additional processes, may be a more promising approach for understanding particular cases of corrosion than, for instance, the traditional analysis of aqueous phases (e.g. produced waters in oil fields). Because EDX as a semi-quantitative technique (Goldstein et al., 2003) is not applicable for determining mineral ratios in the crust, chemical analysis is the technique of choice.
The presence of SRB with the capability for EMIC such as strains IS5 and IS4 may be examined by a complementary cultivation-based approach. Such SRB may have been overlooked in microbiological monitoring studies of MIC which employ diagnostic methods based on fast growth with organic nutrients such as lactate and samples from water phases. The presently investigated corrosive strains grow relatively slowly and show pronounced surface attachment, so that they are easily out-competed in organic-rich diagnostic media by naturally widespread, rapidly growing planktonic SRB. Hence, development of convenient media with Fe0, inocula scraped off from surfaces, and longer than conventional incubation times should be envisaged.
If molecular, nucleic acid-based analyses of damages by microbial corrosion are of interest, again focus on the microorganisms in precipitated products rather than in aqueous phases is recommended for future studies (Skovhus et al., 2011). Still, molecular detection of directly corrosive SRB by long-established markers such as 16S rRNA or its genes is intrinsically limited. Already the first investigations into direct corrosion showed phylogenetic unrelatedness of corrosive SRB (Dinh et al., 2004). There is presently no target gene which among the more general marker genes of SRB (Stahl et al., 2007) could indicate the unique physiological capability for EMIC.
Physiological and ecological significance of the ability for iron corrosion
The specific ability to utilize metallic iron as an electron donor is a physiologically striking capability, the ecological significance of which is presently unknown. Apart from rare cases (meteorites, seldom rocks from deep subsurfaces; Deutsch et al., 1977; Haggerty and Toft, 1985), metallic iron has been introduced into the environment on a large scale only by industrialization, viz. very ‘recently’ from an evolutionary point of view. Yet, counting of corrosive SRB via dilution series with anoxic sediment and metallic iron revealed several 107 cells per gram wet mass (Fig. 1E), despite obvious absence of man-made iron constructions. One may speculate that corrosiveness represents the promiscuous use of a long-existing physiological trait for environmental electron uptake (‘electrotrophy’; Lovley, 2011) that is suited to also exploit the anthropogenically introduced metal as substrate. Normally, SRB with such trait may be involved in biogenic electron flow through sulfidic marine sediments and other ecosystems (Nakamura et al., 2009; Nielsen et al., 2010; Kato et al., 2011). Also, electron gain in direct contact with other bacteria with a surplus of catabolic electrons (Summers et al., 2010; Lovley, 2011) or from strongly reducing, reactive mineral surfaces, such as pyrite being formed from ferrous sulfide and free sulfide (FeS + H2S → FeS2 + H2 / FeS2 + 2H+ + 2e−; Wächtershäuser, 1992), can be envisaged as the genuine role of the electron uptake system underlying direct corrosion. Figure 6 summarizes a present hypothesis of the in situ function of SRB with the ability for electron uptake from external sources. Such SRB may represent a so far overlooked part of the anaerobic population. Still, more extended examinations (such as the above dilution series) of their abundance at various natural sites and physiological studies are needed to unravel their real significance in anaerobic mineralization.
Fig. 6.
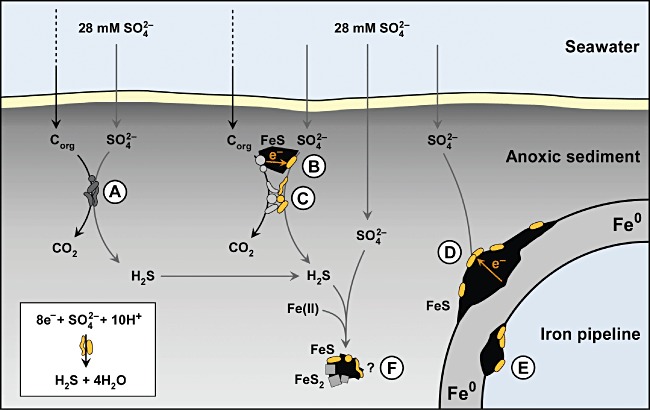
Present synoptic hypotheses of the role of sulfate-reducing bacteria (SRB) capable of electron uptake from external sources in anoxic marine sediment (not to scale). A. Conventional organotrophic SRB. B. SRB interacting via electroconductive ferrous sulfide with electron-donating organotrophic anaerobic microorganisms. C. SRB interacting in direct contact with electron-donating organotrophic anaerobic microorganisms. D. Special SRB exploit metallic iron as electron source at the outer surface of a pipeline. E. Special SRB exploit metallic iron as electron source inside of a pipeline. F. Speculative possibility of pyritization of FeS (FeS + H2S → FeS2 + 2H+ + 2e−) as a direct electron source for sulfate reduction.
From the viewpoint of mechanistic evolution, merely electron-donating and electron-accepting reactions can be regarded as simple or even primeval. Mere electron transfer does not require extra catalytic mechanisms like cleavage of C–H bonds (or of the H-H bond) and rearrangement of bound atoms. With many simple electron donors and acceptors, electron transfer takes place without specific catalysis simply according to redox potentials. This is a classical principle in biochemistry, for instance if electron accepting or donating dyes such as viologens or hexacyano-ferrates are used to react unspecifically with various redox proteins. In the cell, cofactors that can transfer electrons as such would be even critical in their free, dissolved form because of unspecific redox reactions. Their reducing or oxidizing power must be controlled for instance by embedding in a protein (haem, flavins) or by restriction to the lipophilic cytoplasmic membrane (quinones). Substances that merely accept or donate electrons are much less critical outside of the cell, and the ability to use them via transmembrane electron transport components is obviously a typical domain of environmental prokaryotes.
In conclusion, the study of microbial corrosion encompasses interesting mechanistic, ecological and evolutionary aspects, besides its obvious practical significance. It may become, besides microbial fuel cells (Lovley, 2006; Nealson and Finkel, 2011), microbial electrolysis cells for H2 production from H2O (Croese et al., 2011), and biogenic currents in environments (Nakamura et al., 2009; Nielsen et al., 2010; Kato et al., 2011), a fourth topic in the developing field of ‘electro-microbiology’ and in this way contribute to future synoptic views.
Experimental procedures
Organisms and cultivation
Strains IS4 and IS5, tentatively termed Desulfopila‘corrodens’ and Desulfovibrio ‘ferrophilus’, respectively, were re-activated from freeze-dried former cultures (Dinh et al., 2004). Desulfopila inferna was provided by Antje Gittel (University of Bergen, Norway; Gittel et al., 2010). SRB with high affinity for H2 were enriched from marine sediment with an H2-CO2 mixture (9/1, by volume) provided at growth-limiting rate through a silicon rubber membrane inside an anoxic cultivation device; some acetate (1 mM) was provided for cell synthesis. After two subcultures, the Desulfovibrio strain HS3 was isolated via agar dilution (Widdel and Bak, 1992).
Cultures were grown in CO2/bicarbonate-buffered artificial seawater medium (Widdel and Bak, 1992) in butyl rubber-stoppered bottles (routinely 150 ml) under an anoxic N2-CO2 (9/1, by volume) headspace at 28°C. SO42− was usuallyprovided at 28 mM; exceptions were cultures with 5 mM SO42− (not resulting in a noticeable decrease of the growth rate) for precise determination of its consumption. The reductant was usually Fe0 alone. However, some big cultures (1400 ml) received also 75 µM Na2S, which shortened the lag phase. All strains except strain IS4 were supplemented with 1 mM acetate for heterotrophic cell synthesis. H2-grown inocula were flushed with N2-CO2 to prevent transfer of H2 substrate and sulfide to Fe0 cultures. Iron specimens (mild steel EN 1.0330; > 99.37% Fe) were degreased with acetone (2 min), freed from oxide layers with 2 M HCl (2 min) and washed in sterile anoxic water (2 × 20 s). Coupons were then rapidly dried under N2, weighed (in sterile Petri dishes) and added to cultures. The ratio of medium volume to metal surface was between 15 ml cm−2 (for scanning electron microscopy; coupons: 30 mm × 10 mm × 1 mm) and 600 ml cm−2 (for corrosion rate determination).
Strain purity was routinely checked microscopically upon growth with H2 or lactate (+ yeast extract), and sequencing of 16S rRNA genes.
Cells of corrosive SRB in sediment were quantified via triplicate serial 1:10 dilutions with Fe0 as electron donor and 1 mM acetate as a carbon source. SO42− consumption was quantified after 6 months of incubation at 20°C. Sterile controls were included to calculate sulfate reduction solely based on the measured abiotic H2 formation. Marginal background sulfate reduction was measured in iron-free controls.
Processing of corroded iron coupons
Corrosion crust for analysis was (inside an N2 chamber) scraped off the water-rinsed and dried coupons, finely ground in a mortar, and kept under anoxic N2 until analysis to prevent secondary oxidation. Corrosion coupons for weight loss determination were freed from the crust in aqueous 2 M HCl and 0.7 M hexamethylenetetramine (hexamine). The freed iron was washed in anoxic water and dried under N2 before weighing.
Determination of corrosion crust conductivity
Shaped and polished mild steel coupons were fixed at exactly defined distance on a polycarbonate support (Fig. 2A and B). The four wires protruding the stopper of the anoxic flask (1 l, 0.6 l medium) for current and voltage control were also made of iron. Coupon-wire contacts were fixed with plastic screws. The device was sterilized with ethanol (2 h; dried afterwards). The lower, defined slot part was subjected to the pre-treatment of coupons described above and then immersed in artificial seawater medium (Fig. 2A and B). The medium was gently stirred during incubation. For conductivity measurement before and after crust formation, the current responding to 20, 50, 100 and 200 mV was measured. The instruments connected for this purpose were a TS3022S precision current (DC) supply (Thandar Instruments), an ammeter (± 0.2%, ± 10 µA), and a voltmeter (± 0.05%, ± 10 µV). Electrical conductivity was calculated as σ = I d/(V a); I, current; d, distance between coupons; V, voltage; a, split area (split height × coupon thickness). For photographic documentation, the mounted coupons were transferred to anoxic water.
The specific conductivity of siderite mineral (> 85% crystalline siderite with accompanying calcium and magnesium carbonates) was determined with specimens compressed (9 × 108 Pa) to smooth pills in a type 15.011 hydraulic press (Specac Ltd). Impedance was measured via a ‘piston electrode’ (Fig. S9) connected to a Model 1286 potentiostat with a 1255B frequency response analyser and a 1281 multiplexer (Solartron Analytical).
Field experiment
Mild steel coupons fixed by threads to a T-shaped scaffold (stainless steel; Fig. 1C) were buried during summer 2010 at 20 cm depth in silty black, anoxic sediment of the Wadden Sea (Tonnenlegerbucht, island of Sylt, Germany). Coupons recovered after three months were immediately transferred to anoxic artificial seawater and further processed as described for other coupons.
Scanning electron microscopy
Crust-covered iron specimens were fixed with 2% (v/v) glutaraldehyde in anoxic artificial seawater at 4°C (12 h), washed with anoxic seawater, dehydrated with anoxic ethanol at increasing concentration (25%, 50%, 75%, 90%, 100% and again 100%, v/v; each step 2 min), pre-dried chemically with hexamethyldisilazane (30 min; Araujo et al., 2003), and fully dried under N2. Scanning electron microscopy (SEM) at 5–15 kV was performed with a Leo 1550 FE instrument (Zeiss).
Mineral and solute analyses
For element identification, finely ground crust homogenate (see above) was compressed (9 × 108 Pa) to smooth pills in a type 15.011 hydraulic press (Specac Ltd) and surface-mapped (using triplicate samples) by energy-dispersive X-ray microanalysis (EDX) at 10 kV in combination with SEM. Data were analysed with the INCA software (Oxford Instruments). C and S were subsequently quantified after combustion (to CO2 and SO2 respectively) using a CS-444 infrared spectrometer (Leco). Na, Mg, Al, Si, P, K, Ca and Fe were quantified by inductively coupled plasma optical emission spectroscopy (ICP-OES) with an IRIS Intrepid HR Duo instrument (Thermo Fisher Scientific). Si was quantified after alkaline pulping. The O-content was calculated as the remaining fraction. H (not detectable with the used instruments) is assumed to constitute a marginal fraction of the dried mineral crust.
Crystalline mineral phases in finely ground corrosion crust were identified by means of X-ray diffraction (XRD) analysis in Bragg-Brentano geometry with CuKα radiation from 1 µm aperture in a D8 Advance diffractometer (Bruker). The 2θ angle was increased in 0.08° steps with 15 s counting time from 20 to 74.08°; the total scan time per diffractogram was 169 min. Data were analysed with the EVA software (Bruker).
H2 was quantified in samples withdrawn (with syringes through hypodermic needles) from the culture headspace via a GC-8A gas chromatograph (Shimadzu) equipped with a Porapak Q N80/100 (Machery-Nagel) column (temperature, 40°C; carrier gas, N2) and a thermal conductivity detector.
SO42− in filtered (0.45 µm pores) FeS-free samples was quantified via a 761 Compact Ion Chromatograph (Metrohm) by conductivity detection. Ions were separated via a Metrosep A Supp 5–100 column with an eluent of 3.2 mM Na2CO3 and 1 mM NaHCO3 at 0.7 ml min−1. Fe2+ dissolved in such filtrates was quantified by ICP-OES (see above).
Acknowledgments
We are indebted to Rebecca Ansorge for technical support. We thank Jens Harder and Thomas Holler for review of the manuscript. Andrea Mingers and Daniel Kurz conducted ICP-OES- and C/S-Analysis respectively. We thank Bernd Stickfort for support with the bibliography. The authors thank Justus van Beusekom, Ragnhild Asmus and their colleagues at the Alfred Wegener Institute for Polar and Marine Research for support of our field study. This work was supported by the Max Planck Society.
List of symbols and some abbreviations
| CMIC | Chemical microbially influenced corrosion (indirect corrosion) |
| ΔG° | Change of free energy at 298.15 K and standard activities (numerically ≍ 1 M, 1 atm) |
| ΔG°′ | As ΔG°, except {H+} = {HO−} = 10−7 (pH = 7) |
| ΔH° | Change of enthalpy at 298.15 K |
| EMIC | Electrical microbially influenced corrosion (direct corrosion) |
| E | Redox potential |
| E° | Redox potential at 298.15 K and standard activities (≍ 1 M, 1 atm) |
| E°′ | As E°, except {H+} = 10−7 (pH = 7) |
| i | Electrical current density |
| I | Electrical current |
| mBio | Biomass |
| mMin | Mass of precipitated minerals |
| MIC | Microbially influenced (usually anaerobic) corrosion (of iron) |
| nΔFe(0) | Loss of metallic iron |
| nFe(II) | Amount of (total) ferrous iron formed |
| nFeAnab | Amount of iron oxidized by the anabolism (cell synthesis) |
| nFeCatab | Amount of iron oxidized by the catabolism (sulfate reduction) |
| nFeS | Amount of ferrous sulfide (≡ nSR) |
| nSR | Amount of sulfate reduced (≡ nFeS) |
| qAnab | Partition of anabolic in total (anabolic + catabolic) electron consumption |
| qmBio | Proportion (by mass) of biomass in corrosion crust |
| qEMIC | Contribution (partition) of EMIC to total corrosion by SRB |
| v | Here: voltage (otherwise volume) |
| YAnab | Anabolic yield coefficient: cell mass per amount of anabolically oxidized iron |
Supporting information
Additional Supporting Information may be found in the online version of this article:
Fig. S1. Kinetic aspects of the abiotic reaction of iron in circumneutral water, and direct (lithotrophic) iron corrosion by SRB. Availability of H+ ions at the metal surface and combination of adsorbed H-atoms to adsorbed H2 are assumed to be rate-controlling steps (‘bottle necks’), thus also controlling liberation of H2 into water (Bockris and Reddy, 1970; Hamann et al., 2007). H2 consumption by SRB behind the bottle neck is therefore unlikely to promote iron dissolution. Direct consumption of electrons can oxidize the iron much faster. Thickness of arrows symbolizes speed. The net reaction is always 4Fe0 + SO42− +4H2O → FeS + 3Fe2+ +8HO−.
Fig. S2. Excluding disappearance of sulfate due to coprecipitation in the corrosion crust. Grown cultures were stepwise acidified with HCl until formed corrosion products were completely dissolved. Sulfate concentration of medium did not increase.
A. Culture of strain IS4.
B. Culture of strain IS5.
C. Control culture of strain IS4 which was not acidified.
Fig. S3. Abiotic anaerobic iron corrosion in sterile synthetic seawater medium.
A. Production of ‘cathodic’ hydrogen by reduction of H+-ions (Fig. S1), and sulfide that could be formed by subsequent H2 utilization by SRB (4H2 + SO42− + 2H+ → H2S + 4H2O).
B. Original iron specimen (day 0), specimen with precipitate after 5 months (original), and after removal of precipitate (using HCl-hexamine).
Fig. S4. Insensitivity of non-corrosive control strain HS3 towards Fe2+. Addition of H2 to the culture including iron granules leads to rapid sulfide production (measured as sulfate loss).
Fig. S5. Electro-technical scheme with approximate voltage drops of the split-coupon incubation device for conductance measurement of the biogenic corrosion crust formed on corroding iron. The device circumvents interference by the noticeable contact resistance between the iron wire and the iron coupon inside the incubated bottle (Fig. 3A). The plot in the lower part depicts the voltage drop along current flow. The outer voltage (Vo) is supplied and adjusted such that the voltage across the split (Vs) is kept at 0.20 V while the current (I) is being measured. The adjusted low voltage for measurement avoids electrolysis. Measurement of Vs is carried out with a high-resistance voltmeter. Vc1 and Vc4 are the voltage drops due to contact resistance between the iron wire and the iron coupon (around 1 Ω), and Vc2 and Vc3 the arbitrarily assumed voltage drops due to the contact resistance between iron and the sulfidic crust. Voltage drop along the iron wire and the iron coupons is negligible (resistance by two and four orders or magnitude lower, respectively, than resistance of wire-coupon contact and the crust).
Fig. S6. Pustule (elevated precipitate) with early stage of microchimney formation above an anodic site in a culture of strain IS4 after three months of incubation. Bar, 50μm.
Fig. S7. Early stage of microchimney formation above an anodic area in a culture of strain IS4 after three months of incubation. Bar, 10 μm.
Fig. S8. Late stage of microchimney formation in a culture of strain IS4 after six months of incubation. Bar, 200μm.
Fig. S9. Piston electrode set-up for measurement of conductivity of a compressed siderite mineral pill.
Fig. S10. Sulfide production (determined as sulfate consumption) and decrease of dissolved ferrous iron due to carbonate precipitation in long-term incubations of corrosive SRB. Strain IS4 (A) which was more alkali-tolerant than strain IS5 (B) grew up to higher pH (pH increase due to Eq. 5) thus promoting precipitation according to Fe2+ + HO− + HCO3− → FeCO3 + H2O. This favoured formation of microchimneys (Fig. 5C). Six cultures of each strain were incubated in parallel and sacrificed at different time points for SEM analysis (Fig. 4, Fig. S6–S8). Formation of crater- and chimney-like structures in cultures of strain IS4 coincided with the drop of [Fe2+(aq)] below detection limit (0.2 mg l−1). The initial pH was 7.3. Strain IS4 reached pH ≈ 9. Activity of strain IS5 ceased at pH ≈ 8.
Fig. S11. Electron flow from metallic iron into the catabolism and anabolism.
Table S1. Compilation of corrosion rates recorded for (anoxic) natural and engineered environments, and for laboratory cultures of sulfate-reducing bacteria.
Table S2. Vitamins in used media.
Table S3. Conductivity values measured in an incubation device with split coupon with corrosive cultures of strains IS4 and IS5, and with sterile artificial seawater. Iron is provided as the sole source of electrons.
Table S4. Electrical conductivity of selected substances.
Appendix S1. Data, calculations and combination of Fig. S1–11 and Tables S1–4.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Appia-Ayme C, Guiliani N, Ratouchniak J, Bonnefoy V. Characterization of an operon encoding two c-type cytochromes, an aa3-type cytochrome oxidase, and rusticyanin in Thiobacillus ferrooxidans ATCC 33020. Appl Environ Microbiol. 1999;65:4781–4787. doi: 10.1128/aem.65.11.4781-4787.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araujo JC, Teran FC, Oliveira RA, Nour EAA, Montenegro MAP, Campos JR, Vazoller RF. Comparison of hexamethyldisilazane and critical point drying treatments for SEM analysis of anaerobic biofilms and granular sludge. J Electron Microsc (Tokyo) 2003;52:429–433. doi: 10.1093/jmicro/52.4.429. [DOI] [PubMed] [Google Scholar]
- Auernik KS, Maezato Y, Blum PH, Kelly RM. The genome sequence of the metal-mobilizing, extremely thermoacidophilic archaeon Metallosphaera sedula provides insights into bioleaching-associated metabolism. Appl Environ Microbiol. 2008;74:682–692. doi: 10.1128/AEM.02019-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badziong W, Thauer RK. Growth yields and growth rates of Desulfovibrio vulgaris (Marburg) growing on hydrogen plus sulfate and hydrogen plus thiosulfate as sole energy sources. Arch Microbiol. 1978;117:209–214. doi: 10.1007/BF00402310. [DOI] [PubMed] [Google Scholar]
- Beech IB, Sunner IA. Sulphate-reducing bacteria and their role in corrosion of ferrous materials. In: Barton LL, Hamilton WA, editors. Sulphate-Reducing Bacteria: Environmental and Engineered Systems. Cambridge, UK: Cambridge University Press; 2007. pp. 459–482. [Google Scholar]
- Bockris JOM, Reddy AKN. Modern Electrochemistry. New York, USA: Plenum; 1970. [Google Scholar]
- Booth GH, Tiller AK. Polarization studies of mild steel in cultures of sulphate-reducing bacteria. Trans Faraday Soc. 1960;56:1689–1696. [Google Scholar]
- Booth GH, Cooper AW, Tiller AK. Criteria of soil aggressiveness towards buried metal. III. Verification of predicted behaviour of selected soils. Br Corros J. 1967;2:116–118. [Google Scholar]
- Booth GH, Elford L, Wakerley DS. Corrosion of mild steel by sulphate-reducing bacteria: an alternative mechanism. Br Corros J. 1968;3:242–245. [Google Scholar]
- Braun F. Ueber die Stromleitung durch Schwefelmetalle. Ann Phys. 1875;229:556–563. [Google Scholar]
- Bryant RD, Laishley EJ. The role of hydrogenase in anaerobic biocorrosion. Can J Microbiol. 1990;36:259–264. [Google Scholar]
- Butler JE, Young ND, Lovley DR. Evolution of electron transfer out of the cell: comparative genomics of six Geobacter genomes. BMC Genomics. 2010;11:1–12. doi: 10.1186/1471-2164-11-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coetser SE, Cloete TE. Biofouling and biocorrosion in industrial water systems. Crit Rev Microbiol. 2005;31:213–232. doi: 10.1080/10408410500304074. [DOI] [PubMed] [Google Scholar]
- Costello JA. Cathodic depolarization by sulphate-reducing bacteria. S Afr J Sci. 1974;70:202–204. [Google Scholar]
- Croese E, Pereira MA, Euverink G-JW, Stams AJM, Geelhoed JS. Analysis of the microbial community of the biocathode of a hydrogen-producing microbial electrolysis cell. Appl Microbiol Biotechnol. 2011;92:1083–1093. doi: 10.1007/s00253-011-3583-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch ER, Rao KV, Laurent R, Seguin MK. New evidence and possible origin of native iron in ophiolites of eastern Canada. Nature. 1977;269:684–685. [Google Scholar]
- van Dijken JP, Harder W. Growth yields of microorganisms on methanol and methane. A theoretical study. Biotechnol Bioeng. 1975;17:15–30. [Google Scholar]
- Dinh HT, Kuever J, Mußmann M, Hassel AW, Stratmann M, Widdel F. Iron corrosion by novel anaerobic microorganisms. Nature. 2004;427:829–832. doi: 10.1038/nature02321. [DOI] [PubMed] [Google Scholar]
- Duncan KE, Gieg LM, Parisi VA, Tanner RS, Tringe SG, Bristow J, Suflita JM. Biocorrosive thermophilic microbial communities in Alaskan North Slope oil facilities. Environ Sci Technol. 2009;43:7977–7984. doi: 10.1021/es9013932. [DOI] [PubMed] [Google Scholar]
- Gieg LM, Jack TR, Foght JM. Biological souring and mitigation in oil reservoirs. Appl Microbiol Biotechnol. 2011;92:263–282. doi: 10.1007/s00253-011-3542-6. [DOI] [PubMed] [Google Scholar]
- Gittel A, Seidel M, Kuever J, Galushko AS, Cypionka H, Konneke M. Desulfopila inferna sp. nov., a sulfate-reducing bacterium isolated from the subsurface of a tidal sand-flat. Int J Syst Evol Microbiol. 2010;60:1626–1630. doi: 10.1099/ijs.0.015644-0. [DOI] [PubMed] [Google Scholar]
- Goldstein J, Newbury D, Joy D, Lyman C, Echlin P, Lifshin E, et al. Scanning Electron Microscopy and X-Ray Microanalysis. 3rd. New York, USA: Springer; 2003. [Google Scholar]
- Haggerty SE, Toft PB. Native iron in the continental lower crust – petrological and geophysical implications. Science. 1985;229:647–649. doi: 10.1126/science.229.4714.647. [DOI] [PubMed] [Google Scholar]
- Hamann CH, Hamnett A, Vielstich W. Electrochemistry. 2nd. Weinheim, Germany: Wiley; 2007. [Google Scholar]
- Hamilton WA. Sulphate-reducing bacteria and anaerobic corrosion. Annu Rev Microbiol. 1985;39:195–217. doi: 10.1146/annurev.mi.39.100185.001211. [DOI] [PubMed] [Google Scholar]
- Hamilton WA. Microbially influenced corrosion as a model system for the study of metal microbe interactions: a unifying electron transfer hypothesis. Biofouling. 2003;19:65–76. doi: 10.1080/0892701021000041078. [DOI] [PubMed] [Google Scholar]
- Hardy JA. Utilization of cathodic hydrogen by sulphate-reducing bacteria. Br Corros J. 1983;18:190–193. [Google Scholar]
- Hubert C, Nemati M, Jenneman G, Voordouw G. Corrosion risk associated with microbial souring control using nitrate or nitrite. Appl Microbiol Biotechnol. 2005;68:272–282. doi: 10.1007/s00253-005-1897-2. [DOI] [PubMed] [Google Scholar]
- Jack TR. Biological corrosion failures. In: Shipley RJ, Becker WT, editors. ASM Handbook Volume 11: Failure Analysis and Prevention. Materials Park, OH, USA: ASM International; 2002. pp. 881–898. [Google Scholar]
- Kato S, Hashimoto K, Watanabe K. Methanogenesis facilitated by electric syntrophy via (semi)conductive iron-oxide minerals. Environ Microbiol. 2011 doi: 10.1111/j.1462-2920.2011.02611.x. doi: 10.1111/j.1462-2920.2011.02611.x. [DOI] [PubMed] [Google Scholar]
- King RA, Miller JDA. Corrosion by sulphate-reducing bacteria. Nature. 1971;233:491–492. doi: 10.1038/233491a0. [DOI] [PubMed] [Google Scholar]
- Lee W, Lewandowski Z, Nielsen PH, Hamilton WA. Role of sulfate-reducing bacteria in corrosion of mild-steel – a review. Biofouling. 1995;8:165–194. [Google Scholar]
- Li S, Kim Y, Jeon K, Kho Y. Microbiologically influenced corrosion of underground pipelines under the disbonded coatings. Met Mater. 2000;6:281–286. [Google Scholar]
- Lovley DR. Microbial fuel cells: novel microbial physiologies and engineering approaches. Curr Opin Biotechnol. 2006;17:327–332. doi: 10.1016/j.copbio.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Lovley DR. Reach out and touch someone: potential impact of DIET (direct interspecies energy transfer) on anaerobic biogeochemistry, bioremediation, and bioenergy. Rev Environ Sci Biotechnol. 2011;10:101–105. [Google Scholar]
- Malvankar NS, Vargas M, Nevin KP, Franks AE, Leang C, Kim BC, et al. Tunable metallic-like conductivity in microbial nanowire networks. Nat Nanotechnol. 2011;6:573–579. doi: 10.1038/nnano.2011.119. [DOI] [PubMed] [Google Scholar]
- Mori K, Tsurumaru H, Harayama S. Iron corrosion activity of anaerobic hydrogen-consuming microorganisms isolated from oil facilities. J Biosci Bioeng. 2010;110:426–430. doi: 10.1016/j.jbiosc.2010.04.012. [DOI] [PubMed] [Google Scholar]
- Nakamura R, Kai F, Okamoto A, Newton GJ, Hashimoto K. Self-constructed electrically conductive bacterial networks. Angew Chem Int Ed Engl. 2009;48:508–511. doi: 10.1002/anie.200804750. [DOI] [PubMed] [Google Scholar]
- Nealson KH, Finkel SE. Electron flow and biofilms. MRS Bull. 2011;36:380–384. [Google Scholar]
- Nielsen LP, Risgaard-Petersen N, Fossing H, Christensen PB, Sayama M. Electric currentscouple spatially separated biogeochemical processes in marine sediment. Nature. 2010;463:1071–1074. doi: 10.1038/nature08790. [DOI] [PubMed] [Google Scholar]
- Pearce CI, Pattrick RAD, Vaughan DJ. Electrical and magnetic properties of sulfides. Rev Mineral Geochem. 2006;61:127–180. [Google Scholar]
- Reinsel MA, Sears JT, Stewart PS, McInerney MJ. Control of microbial souring by nitrate, nitrite or glutaraldehyde injection in a sandstone column. J Ind Microbiol. 1996;17:128–136. [Google Scholar]
- Schwermer CU, Lavik G, Abed RMM, Dunsmore B, Ferdelman TG, Stoodley P, et al. Impact of nitrate on the structure and function of bacterial biofilm communities in pipelines used for injection of seawater into oil fields. Appl Environ Microbiol. 2008;74:2841–2851. doi: 10.1128/AEM.02027-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherar BWA, Power IM, Keech PG, Mitlin S, Southam G, Shoesmith DW. Characterizing the effect of carbon steel exposure in sulfide containing solutions to microbially induced corrosion. Corros Sci. 2011;53:955–960. [Google Scholar]
- Skovhus TL, Sørensen KB, Larsen J. Rapid diagnostics of microbiologically influenced corrosion (MIC) in oilfield systems with a DNA-based test kit. In: Whitby C, Skovhus TL, editors. Applied Microbiology and Molecular Biology in Oilfield Systems. Dordrecht, the Netherlands: Springer; 2011. pp. 133–140. [Google Scholar]
- Smith JS, Miller JDA. Nature of sulphides and their corrosive effect on ferrous metals: a review. Br Corros J. 1975;10:136–143. [Google Scholar]
- Sorokin YI. Role of carbon dioxide and acetate in biosynthesis by sulphate-reducing bacteria. Nature. 1966;210:551–552. doi: 10.1038/210551a0. [DOI] [PubMed] [Google Scholar]
- Spruit CJP, Wanklyn JN. Iron sulphide ratios in corrosion by sulphate-reducing bacteria. Nature. 1951;168:951–952. doi: 10.1038/168951a0. [DOI] [PubMed] [Google Scholar]
- Stahl DA, Loy A, Wagner M. Molecular strategies for studies of natural populations of sulphate-reducing microorganism. In: Barton LL, Hamilton WA, editors. Sulphate-Reducing Bacteria: Environmental and Engineered Systems. Cambridge, UK: Cambridge University Press; 2007. pp. 39–116. [Google Scholar]
- Summers ZM, Fogarty HE, Leang C, Franks AE, Malvankar NS, Lovley DR. Direct exchange of electrons within aggregates of an evolved syntrophic coculture of anaerobic bacteria. Science. 2010;330:1413–1415. doi: 10.1126/science.1196526. [DOI] [PubMed] [Google Scholar]
- Uchiyama T, Ito K, Mori K, Tsurumaru H, Harayama S. Iron-corroding methanogen isolated from a crude-oil storage tank. Appl Environ Microbiol. 2010;76:1783–1788. doi: 10.1128/AEM.00668-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- US Geological Survey. 2011. Mineral commodity summaries 2011. U.S. Geological Survey: 1–198 [WWW document]. URL http://minerals.usgs.gov/minerals/pubs/mcs/2011/mcs2011.pdf.
- Wächtershäuser G. Groundworks for an evolutionary biochemistry: the iron-sulphur world. Prog Biophys Mol Biol. 1992;58:85–201. doi: 10.1016/0079-6107(92)90022-x. [DOI] [PubMed] [Google Scholar]
- Widdel F, Bak F. Gram-negative mesophilic sulfate-reducing bacteria. In: Balows A, Trüper HG, Dworkin M, Harder W, Schleifer K-H, editors. The Prokaryotes. New York, USA: Springer; 1992. pp. 3352–3378. [Google Scholar]
- Widdel F, Musat F. Energetic and other quantitative aspects of microbial hydrocarbon utilization. In: Timmis KN, editor. Handbook of Hydrocarbon and Lipid Microbiology. Berlin, Germany: Springer; 2010. pp. 729–763. [Google Scholar]
- von Wolzogen Kühr CAH. Unity of anaerobic and aerobic iron corrosion process in the soil. Corrosion. 1961;17:119–125. [Google Scholar]
- von Wolzogen Kühr CAH, van der Vlugt LS. The graphitization of cast iron as an electrobiochemical process in anaerobic soil. Water. 1934;18:147–165. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.