

During the last decade, the classical view of a protein existing in equilibrium between discrete conformational states has undergone a radical transformation. Experimental observations, particularly those obtained from NMR-detected hydrogen/deuterium exchange, indicate that even under native conditions proteins need to be considered as complex statistical ensembles (see for example refs. 1–13). In the view pioneered by Englander and coworkers (1, 2, 4, 7, 8, 14) proteins undergo local unfolding reactions scattered throughout their entire structures. These unfolding reactions occur independently of each other, may involve only a few amino acids, and give rise to a large number of states in which each state is defined by the presence of one or several locally unfolded regions. This collection of states defines the native-state ensemble. The immediate, and perhaps most relevant, consequence of these observations is that the Gibbs energy of stabilization of a protein is not uniformly distributed throughout its three-dimensional structure. There are regions with high stability constants and regions with low stability constants (13, 15). Furthermore, under native conditions cooperativity appears to be local or regional rather than global. The implications of these findings have been mostly discussed in relation to the protein folding problem; however, their biological implications are tremendous because the native state is the functionally relevant state. If cooperativity is local or regional rather than global, how do distal sites communicate with each other? How do regulatory allosteric interactions take place? How are the functional properties of a protein related to the distribution of states within the native ensemble, and how does ligand binding affect this distribution and modulate function? Is the location of stable and unstable regions a random event or it is dictated by functional considerations? How are protein function and stability related?
The tools to begin addressing those issues in a systematic way became available with the observation that a structure-based algorithm (COREX) was able to model reasonably well the hydrogen exchange protection data obtained for many proteins (13, 15–19). Within the framework of the COREX algorithm the protein is considered as a statistical ensemble in which each state is characterized by having some region or regions in a nonfolded state. These regions can be as small as four or five amino acids or as large as the entire protein. Depending on the size of the protein the computation is performed either exhaustively or by using a sampling technique (19). The Gibbs energy of all of the resulting states and their respective probabilities are calculated in terms of an empirical structural parameterization of the Gibbs energy (summarized in ref. 20). What the COREX algorithm produces is a list of possible conformations and their respective probabilities, i.e., a probability distribution function. As such it can be used to examine the effects of ligands and other chemical or physical factors on that distribution. Previously (21) it was shown that the incorporation of the ligand linkage equations into the COREX algorithm (CORE_BIND) correctly predicted the propagation of binding effects through the structure of hen egg white lysozyme upon binding of a specific antibody. The paper by Pan et al. (22) in this issue of PNAS explores the linkage between binding sites in Escherichia coli dihydrofolate reductase and makes a convincing case that the coupling between these sites is also mediated by shifts in the probability distribution of the conformational ensemble.
The COREX algorithm produces a snapshot of the distribution of states existing under equilibrium conditions. For an ergodic system this distribution is identical to the one that would be obtained if a single protein molecule were observed over a period sufficiently long for thermodynamic averaging. Accordingly, ensemble properties can be mapped into individual molecules. This is illustrated in Fig. 1 for the structural stability of individual residues in a Src homology 3 molecule (18). In the ensemble view, the stability of an individual residue is proportional to the summed probabilities of all of the states in which that residue is in the native state. For an individual molecule, the structural stability of a residue is proportional to the fractional amount of time that the residue spends in the native-state conformation. A highly stable residue will spend most of the time in the native state, whereas a relatively unstable residue will make excursions into different regions of conformational space. These excursions may arise from different processes involving local as well as longer-range fluctuations. For an ergodic system, ensemble and time averages are equivalent.
Figure 1.

The COREX algorithm produces an ensemble representation of a protein. This representation can be mapped into a single molecule, because for an ergodic system ensemble and time averages are equivalent.
As pointed out by Pan et al. (22) classical linkage theories do not address the mechanism by which different binding sites communicate. The ensemble view created by the COREX algorithm provides an opportunity to examine the long-range effects of ligand binding and allosteric regulation. We can imagine that different conformational states might have different functional properties and that, consequently, a redistribution in the population of states may result in functional changes. A change in the distribution of states is triggered by changes in the Gibbs energy of the states that define the ensemble. States with lower Gibbs energies will be preferentially populated with respect to states with higher Gibbs energies. Therefore, the expression of a specific functional property can be triggered by a decrease in the Gibbs energy of those states that exhibit that property. In biological systems, this is usually accomplished by ligand molecules (activators or inhibitors) that selectively bind to those states that possess the selected property. In the presence of a ligand X, the Gibbs energy of any arbitrary state within the ensemble will be affected by an amount that depends on its binding affinity for the ligand:
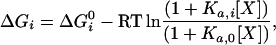 |
1 |
where ΔGi0 is the Gibbs energy of state i in the absence of the ligand, Ka,0 is the binding constant to the reference state, and Ka,i the binding constant to state i. Those states that are able to bind the ligand will be stabilized with respect to those states that are not able to bind the ligand, causing a change in the probability distribution of states. Fig. 2 illustrates this situation with the binding of a proline-rich helical peptide to a Src homology 3 domain. The protein ensemble can be considered as being composed of states in which the binding site is intact and therefore binding-competent and states in which the binding site is not formed and therefore unable to bind the ligand. The states that are binding-competent will be stabilized according to Eq. 1, resulting in a shift in the probability distribution. The residue stabilities calculated in the presence of the ligand can be mapped into an individual protein molecule as well as the differences in Gibbs energy per residue, as shown in Fig. 2. A notable result is that not all amino acids are affected equally, a consequence of the lack of global cooperativity in the native state. Under those conditions the transmission of binding signals to distal sites involves only a subset of residues. Their identification is important not only from the point of understanding a fundamental biological phenomena but also from the point of view of protein engineering and drug design.
Figure 2.

The binding of a proline-containing peptide (red) to the Src homology 3 domain (blue). Ligand binding stabilizes only those conformations that are binding-competent, inducing a redistribution in the conformational ensemble. The effects of binding can be mapped into a single molecule as shown on the right. The blue regions are the ones most affected by binding and the red regions the ones least affected. Notably, not all amino acids are predicted to be equally affected because of the lack of global cooperativity. A consequence of this result is that communication between distal sites involves only a subset of residues as elegantly shown by Pan et al. (22) for dihydrofolate reductase.
Communication between sites occurs when their behavior is coupled. For regulatory and active binding sites this coupling is expressed in a correlation between their binding competencies. For allosteric inhibition, binding to the regulatory site must trigger a destabilization of the active conformation of the catalytic site such that substrate either does not bind or binds in a nonproductive fashion. In this situation, the native-state ensemble can be considered as being primarily populated by two distinct subensembles: one subensemble of conformations that are catalytically active but are unable to bind the inhibitor, and one subensemble of molecules that lack biological activity but are able to bind the inhibitor. The presence of the inhibitor will shift the equilibrium toward the inactive conformations because those conformations are the ones that are able to bind the inhibitor. Again this redistribution of the ensemble can be mapped into a single molecule to identify those residues that are implicated in the communication between sites. The allosteric enzyme glycerol kinase provides a clear illustration of the above situation as illustrated in Fig. 3. An allosteric inhibitor of this enzyme is the protein IIAGlc. IIAGlc binds to glycerol kinase at a site located more than 30 Å away from the active site. The binding of IIAGlc stabilizes this region into a conformation capable of establishing specific contacts with adjacent regions that result into the stabilization of the inactive conformation of the catalytic site. According to the results of the COREX and CORE_BIND analysis only a small set of residues are involved in the allosteric coupling between regulatory and catalytic sites. The results obtained for dihydrofolate reductase by Pan et al. (22) emphasize those conclusions and provide new tools for the investigation of long-range communication in proteins.
Figure 3.
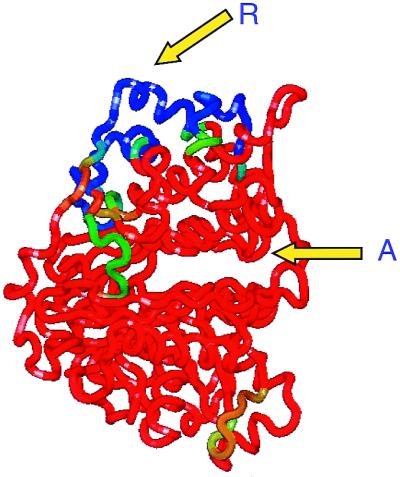
Results of COREX and CORE_BIND analysis of glycerol kinase and its allosteric inhibitor IIAGlc (19). The binding site for IIAGlc (R) to glycerol kinase contains regions of low structural stability. In the absence of IIAGlc, those regions do not interact strongly with the rest of the protein. Upon binding IIAGlc, those regions become structurally stable and initiate a sequence of interactions that shift the catalytic site into an inactive conformation. According to the computational results only a well-defined set of residues participates in the allosteric coupling of both sites.
Acknowledgments
This work was supported by grants from the National Institutes of Health (GM 51362 and GM 57144) and the National Science Foundation (MCB-9816661).
Footnotes
See companion paper on page 12020.
References
- 1.Jeng M-F, Englander S W, Elöve G A, Wand A J, Roder H. Biochemistry. 1990;29:10433–10437. doi: 10.1021/bi00498a001. [DOI] [PubMed] [Google Scholar]
- 2.Jeng M-F, Englander S W. J Mol Biol. 1991;221:1045–1061. doi: 10.1016/0022-2836(91)80191-v. [DOI] [PubMed] [Google Scholar]
- 3.Radford S E, Buck M, Topping K D, Dobson C M, Evans P A. Proteins. 1992;14:237–248. doi: 10.1002/prot.340140210. [DOI] [PubMed] [Google Scholar]
- 4.Bai Y, Milne J S, Mayne L, Englander S W. Proteins. 1993;17:75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim K-S, Woodward C. Biochemistry. 1993;32:9609–9613. doi: 10.1021/bi00088a013. [DOI] [PubMed] [Google Scholar]
- 6.Woodward C. Trends Biochem Sci. 1993;18:359–360. doi: 10.1016/0968-0004(93)90086-3. [DOI] [PubMed] [Google Scholar]
- 7.Bai Y, Sosnick T R, Mayne L, Englander S W. Science. 1995;269:192–197. doi: 10.1126/science.7618079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Milne J S, Mayne L, Roder H, Wand A J, Englander S W. Protein Sci. 1998;7:739–745. doi: 10.1002/pro.5560070323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schulman B A, Redfield C, Peng Z, Dobson C M, Kim P S. J Mol Biol. 1995;253:651–657. doi: 10.1006/jmbi.1995.0579. [DOI] [PubMed] [Google Scholar]
- 10.Chamberlain A K, Handel T M, Marqusee S. Nat Struct Biol. 1996;3:782–787. doi: 10.1038/nsb0996-782. [DOI] [PubMed] [Google Scholar]
- 11.Swint-Kruse L, Robertson A D. Biochemistry. 1996;35:171–180. doi: 10.1021/bi9517603. [DOI] [PubMed] [Google Scholar]
- 12.Dabora J M, Marqusee S. Protein Sci. 1994;3:1401–1408. doi: 10.1002/pro.5560030906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hilser V J, Oas T, Dowdy D, Freire E. Proc Natl Acad Sci USA. 1998;95:9903–9908. doi: 10.1073/pnas.95.17.9903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Milne J S, Xu Y, Mayne L C, Englander S W. J Mol Biol. 1999;290:811–822. doi: 10.1006/jmbi.1999.2924. [DOI] [PubMed] [Google Scholar]
- 15.Hilser V J, Freire E. J Mol Biol. 1996;262:756–772. doi: 10.1006/jmbi.1996.0550. [DOI] [PubMed] [Google Scholar]
- 16.Hilser V J, Townsend B D, Freire E. Biophys Chem. 1997;64:69–79. doi: 10.1016/s0301-4622(96)02220-x. [DOI] [PubMed] [Google Scholar]
- 17.Hilser V J, Freire E. Proteins. 1997;27:171–183. doi: 10.1002/(sici)1097-0134(199702)27:2<171::aid-prot3>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 18.Sadqi M, Casares S, Abril M A, Mayorga O L, Conejero-Lara F, Freire E. Biochemistry. 1999;38:8899–8906. doi: 10.1021/bi990413g. [DOI] [PubMed] [Google Scholar]
- 19.Luque I, Freire E. Proteins. 2000;4:63–71. doi: 10.1002/1097-0134(2000)41:4+<63::aid-prot60>3.3.co;2-y. [DOI] [PubMed] [Google Scholar]
- 20.Luque I, Freire E. Methods Enzymol. 1998;295:100–127. doi: 10.1016/s0076-6879(98)95037-6. [DOI] [PubMed] [Google Scholar]
- 21.Freire E. Proc Natl Acad Sci USA. 1999;96:10118–10122. doi: 10.1073/pnas.96.18.10118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pan H, Lee J C, Hilser V J. Proc Natl Acad Sci USA. 2000;97:12020–12025. doi: 10.1073/pnas.220240297. [DOI] [PMC free article] [PubMed] [Google Scholar]