

Abstract
In this study we investigated the advantage of including network information in prioritizing disease genes of type 1 diabetes (T1D). First, a naïve Bayesian network (NBN) model was developed to integrate information from multiple data sources and to define a T1D-involvement probability score (PS) for each individual gene. The algorithm was validated using known functional candidate genes as a benchmark. Genes with higher PS were found to be more likely to appear in T1D-related publications. Next a new network activity metric was proposed to evaluate the T1D relevance of protein-protein interaction (PPI) subnetworks. The metric considered the contribution both from individual genes and from network topological characteristics. The predictions were confirmed by several independent datasets, including a genome wide association study (GWAS), and two large-scale human gene expression studies. We found that novel candidate genes in the T1D subnetworks showed more significant associations with T1D than genes predicted using PS alone. Interestingly, most novel candidates were not encoded within the human leukocyte antigen (HLA) region, and their expression levels showed correlation with disease only in cohorts with low-risk HLA genotypes. The results suggested the importance of mapping disease gene networks in dissecting the genetics of complex diseases, and offered a general approach to network-based disease gene prioritization from multiple data sources.
Introduction
Type 1 diabetes (T1D) is a polygenic disorder that results from the immune destruction of the insulin-releasing pancreatic islet beta cells. It is one of the most common chronic diseases in children. One of the major challenges in T1D research, as is true for many complex disorders, is to dissect the underlying genetic architecture. Identifying new disease genes can be useful in advancing our understanding of disease pathogenesis, and can be translated into new targets for therapeutic interventions. Traditional linkage mapping offers too many positional candidates, while the lack of an accurate understanding of the disease etiology makes it difficult to construct a comprehensive list of functional candidate genes for association analysis. Newer technologies such as the genome wide association study (GWAS) and the next-generation sequencing hold the promise of surveying the whole genome for sequence variants that are associated with disease risk. However, a lack of statistical power and multiple testing problems prevent them from mapping out the complete picture of the genetic predisposition of a complex trait.
In recent decades, advancements in high-throughput genomic technologies and bioinformatics have enabled the development of new approaches to the genetic study of complex disorders. Information from several data sources has been utilized to supplement the traditional mapping and association studies, including functional (ontological) annotation, gene expression analysis, sequence information, protein-protein interactions (PPI), phenotypic data, and text mining of the literature (Botstein and Risch, 2003; Eaves et al., 2002; Franke et al., 2006; Tiffin et al., 2006). Independently each type of data possesses high noise. For example, Jones and associates estimated that the error rate for some computationally-derived gene ontology (GO) annotations may reach 49% (Jones et al., 2007). In gene expression studies most of the differentially-expressed genes identified are likely not disease genes; therefore data integration is necessary. In an obesity study, English and colleagues found that combining 49 genome-wide experiments had much better predictive accuracy of obesity-associated genes than individual experiments, demonstrating the advantage of information integration (English and Butte, 2007).
A number of integrative genomics strategies have been developed to predict and prioritize candidate disease genes from multiple data sources (Oti and Brunner, 2007; Zhu and Zhao, 2007). These approaches usually adopt the idea that similar phenotypes are caused by genes with similar or related functions (Goh et al., 2007; Jimenez-Sanchez et al., 2001; Smith and Eyre-Walker, 2003). Prioritizer (Franke et al., 2006) and Endeavour (Aerts et al., 2006) are two representative examples. Using a Bayesian network to integrate data of gene expression, PPI, and functional annotation, Prioritizer evaluates functional similarities between positional candidate genes in different linkage regions and prioritizes them accordingly. Endeavour takes a set of user-provided training genes (presumably known disease genes), then builds models of their characteristics according to sequence similarity, expression data, PPI, functional and pathway annotation, and transcription binding information. It then utilizes the models to rank new candidate disease genes. The performance of these approaches is typically evaluated using the known disease genes as a benchmark, and normally 5- to 10-fold enrichment over random selection is observed. Nevertheless, it is still debatable whether the enrichment observed at a high-throughput level is meaningful to a specific disease (Oti and Brunner, 2007).
Most of the candidate gene prioritization approaches are based on individual genes. On the contrary, complex traits result from interactions of multiple genes. In fact, most cellular functions are carried out by groups of highly interconnected proteins that form modular networks. Phenotypic traits are properties that emerge from the interactions in these networks. A disease trait normally correlates with the inability of a particular functional network module to carry out its basic function, and the pathogenesis of a complex disease can involve perturbations to multiple modules. On the other hand, different combinations of gene variants may incapacitate a functional module in the same way, and lead to the same clinical phenotype. Gene expression studies show that disease genes do not always exhibit high differential expression; rather, they tend to interact with and regulate many genes that show significant changes (Nitsch et al., 2009). In T1D, in addition to the major genetic determinant the human leukocyte antigen (HLA) locus, recent studies have identified over 50 non-HLA regions that significantly affect disease risk (http://www.t1dbase.org; Pociot et al., 2010; Rich et al., 2009). The majority of novel variations are in intronic and/or regulatory gene regions, which highlights the importance of mapping disease gene networks (Pociot et al., 2010; Rich et al., 2009).
Recently several studies defined cancer phenotype-associated PPI subnetworks by their correlative changes in gene expression to phenotype variations (Chuang et al., 2007; Dao et al., 2010; Su et al., 2010). The PPI subnetworks provided insights into pathways involved in tumor progression and were more reproducible markers of cancer prognosis than individual genes selected without network information (Chuang et al., 2007; Dao et al., 2010; Su et al., 2010). Other studies have utilized disease association measurements to identify PPI subnetworks of interest (Luo et al., 2010; Wang and Xia, 2008; Wang et al., 2007). For instance, Wang and Xia (2008) used predicted gene-disease association confidence scores to identify type 2 diabetes–relevant PPI subnetworks. They found that the subnetworks might be better biomarkers than single proteins. In the analysis of genome wide association data, inclusion of interaction network information has facilitated the identification of likely disease genes from a large number of candidates with moderate p values and high false-positive rates (Baranzini et al., 2009; Zhong et al., 2010).
In initial studies to identify trait-relevant networks, whole network modules were scored through the sum or mean of the individual gene measurements (Ideker et al., 2002), without considering network structure. However, the interaction pattern is important in determining the outcome of interactions, and structure defines function (Gao and Wang, 2007; Massa et al., 2010; Thomas et al., 2009). Efforts have been made to incorporate network topology in the evaluation of a gene network (Carter et al., 2004; Liu et al., 2006; Massa et al., 2010; Thomas et al., 2009), such as ranking the contributions by individual genes by their topological positions in the network (Hung et al., 2010; Thomas et al., 2009). More recently, several groups proposed to dissect network measurements into contributions from nodes and edges, and developed metrics to score each separately (Ideker et al., 2002; Ma et al., 2011; Smoot et al., 2011; Wang and Xia, 2008). Previously, we showed that proteins encoded by known T1D genes interact with one another significantly more often than expected by chance, even after adjustment for their high network degrees (p<0.0001). We subsequently utilized this enhancement to identify novel candidate disease genes (Gao and Wang, 2009). In a separate effort, we proposed a new network metric, the Pathway Connectivity Index (PCI), to describe the collective state of genes in a pathway (Gao and Wang, 2007). It not only accounts for activity (such as expression) of individual genes, but also the topological properties of their interaction networks. These studies, by our group as well as others, have demonstrated the advantages of incorporating network structure in characterizing pathway/network states, and in linking them to phenotype variations.
In this study, we propose a new candidate disease gene prioritization approach and apply it to T1D. It first uses the naïve Bayesian network (NBN) to integrate functional, genetic, and sequence features of individual genes, and derives a T1D involvement probability score (PS) for each. Bayesian networks are efficient at integrating multiple data sources and have been widely used in systems biology, such as prediction of gene function, reconstruction of gene networks, and identification of disease genes (Aragues et al., 2008; Franke et al., 2006; Gao and Wang, 2011). Next, modules in the PPI network are scored according to the PS of module members and their interaction patterns using a new network metric, and the top T1D-relevant modules are identified. PPI is one of the strongest manifestations of a functional relationship between genes, and has been frequently utilized to assist in candidate disease gene prioritization (Gao and Wang, 2009; George et al., 2006; Kann, 2007; Xu and Li, 2006). Increasing evidence suggests that interacting proteins often share similar functions, participate in the same biological pathways and processes, and contribute to related phenotypes (Lage et al., 2007; Oti and Brunner, 2007). Lastly, we examine the potential role in T1D of novel candidate genes using independent data from one GWAS study and two large-scale gene expression studies. All algorithms are freely available at http://zen.dom.uab.edu:8080/t1dsource/index.jsp.
Materials and Methods
Known T1D candidates and their features
Prior to the GWAS era, numerous genetic studies of T1D have identified hundreds of functional candidate genes and 20 linkage regions. The complete list of 265 known functional candidates and 983 positional candidates that reside within linkage regions were obtained from T1Dbase (http://www.t1dbase.org). These are listed in Supplementary Tables S1 and S2, respectively (see online supplementary material at http://www.liebertonline.com). Sharing of GO terms by gene sets was evaluated using GOStat (http://gostat.wehi.edu.au), which employs the Fisher's exact test. A gene annotated with a GO term was also annotated with all the term's ancestors. The human PPI database was downloaded from the Human Protein Reference Database (HPRD; http://www.hprd.org/; Mishra et al., 2006). In all, 222 of the 265 functional candidates and 487 of the 983 positional candidate genes were annotated in the HPRD. Gene lengths were retrieved from Ensembl (http://uswest.ensembl.org/), and protein domain information was downloaded from InterPro (http://www.ebi.ac.uk/interpro). InterPro is an integrated documentation resource for protein families, domains, and sites, and has become an important protein function classification tool (Hunter et al., 2009). It integrates predictive models or “signatures,” representing protein domains, families, and functional sites from multiple, diverse source databases: Gene3D, PANTHER, Pfam, PIRSF, PRINTS, ProDom, PROSITE, SMART, SUPERFAMILY, and TIGRFAMs.
Genome-wide association study data of T1D
GWAS data were downloaded from the Wellcome Trust Case-Control Consortium (Wellcome Trust Case-Control Consortium, 2007; http://www.wtccc.org.uk), and consisted of 2000 T1D cases and 3000 healthy controls. SNP markers were mapped to genes with dbSNP build 129 (http://www.ncbi.nlm.nih.gov/projects/SNP/). For each gene, SNPs up to 5 kb upstream and 5 kb downstream were regarded its associated SNPs, since these regions are strongly enriched for gene regulatory elements (Veyrieras et al., 2008). The significance of each gene was assigned with the lowest p value of its associated SNPs.
Gene expression data of T1D
Two gene expression datasets were utilized in this study. The first came from our own study (Wang et al., 2008) of the global transcription profile induced in healthy unrelated peripheral blood mononuclear cells (PBMCs) by the serum of different T1D cohorts (http://www.ncbi.nlm.nih.gov/geo, GSE24147). The approach relies on the sensitivity of cells to respond to inflammatory factors in serum or plasma. For the responder cells, we chose to use cryopreserved PBMCs from Cellular Technologies Ltd. (Shaker Heights, OH), a provider of high-quality PBMCs. The cohort samples included 39 recent-onset (RO) T1Ds (mean age 9.97±2.89 years, collected 2–7 months post-diagnosis); 50 unrelated healthy controls (HC; mean age 14.98±4.13 years); 77 siblings of T1D patients (not part of the RO group) that had both high-risk (high-risk siblings [HRS]; DR3/4 haplotypes, 32) and low-risk (low-risk siblings [LRS]; non-DR3/4, 45) HLA genotypes. The RO cohort includes 27 with high-risk (RO-HR), and 12 with low-risk (RO-LR) HLA genotypes. The numbers for their HC cohorts were 2 (HC-HR) and 48 (HC-LR), respectively. Among the RO, HR, and LR cohorts, no two samples came from the same family. Gene expression was profiled with Affymetrix's human genome U133 plus 2.0 GeneChip array that interrogates 47,000 transcripts.
The second dataset was downloaded from Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo, GSE9006). In this study by Kaizer and associates, genome-wide gene expression levels in PBMCs were directly measured in 43 patients with newly-diagnosed T1D, between the ages of 2 and 18 years, and 24 healthy controls (Kaizer et al., 2007). Affymetrix's human U133A and U133B GeneChips were used, which analyze the expression level of 39,000 transcripts and variants.
Data were normalized with Robust Multichip Analysis (RMA; www.bioconductor.org/). The statistical significance of differential gene expression was derived through the Mann-Whitney test.
Naive Bayesian network to predict T1D involvement of individual genes from multiple data sources
Using all known T1D candidates as positive controls (training set), and the rest as negative controls, our NBN learns its parameters based on the following features:
GO molecular function and biological process. For each of the top 30 GO terms overrepresented in the training gene set, a feature is scored 1 if the gene is annotated with this term, and 0 otherwise, totaling 30 variables.
T1D linkage. A feature is scored 1 if the gene locates within a linkage locus, and 0 if not.
Gene length. Scored with the logarithm value of the nucleotide sequence length.
Protein sequence domains. For each of the top 10 InterPro terms overrepresented in the training gene set, a feature is scored 1 if the gene is annotated with the term, and 0 otherwise, totaling 10 variables.
Protein interaction. A feature is scored 1 if the gene interacts with known T1D candidates, and 0 otherwise.
Through back-inference the NBN then calculates a PS of T1D involvement for each gene given the above five features. The number of variables, such as 30 GO terms, and 10 InterPro terms, were chosen for points at which the improvement in performance plateaued.
New algorithm to extract T1D-relevant subnetworks
The T1D relevance of each network module is determined based on the PS of individual genes in the network and their interaction patterns. Briefly, given a network module A, the PS value of its members is first converted to a z-score with z=Φ−1 (1-PS), where Φ−1 is the inverse normal cumulative distribution function (CDF). Let (aij) be the adjacency matrix of A, where aij=1 if members i and j interact, and 0 otherwise, with all diagonal elements set to 1. We define the overall significance score 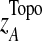 and the normalized score
and the normalized score 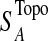 of A with:
of A with:
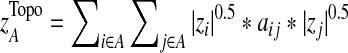 |
[Eq.1] |
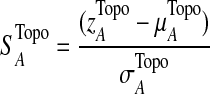 |
[Eq.2] |
where 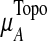 and
and 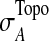 are the mean and standard deviation of
are the mean and standard deviation of 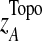 , from randomly-sampled PPI subnetworks of the same size.
, from randomly-sampled PPI subnetworks of the same size. 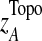 captures the topological properties of the network through the adjacency matrix, and hub genes contribute more to this metric. |zi|0.5 *aij*|zj|0.5 can be regarded as the confidence measure of T1D association of the interaction between genes i and j. Those interactions in which both genes have high z-scores contribute more to the network score (Gao and Wang, 2007). A heuristic search algorithm was used to find the maximally-scored subnetworks, since it is a NP-hard problem.
captures the topological properties of the network through the adjacency matrix, and hub genes contribute more to this metric. |zi|0.5 *aij*|zj|0.5 can be regarded as the confidence measure of T1D association of the interaction between genes i and j. Those interactions in which both genes have high z-scores contribute more to the network score (Gao and Wang, 2007). A heuristic search algorithm was used to find the maximally-scored subnetworks, since it is a NP-hard problem.
Results
Properties of the known functional candidates of T1D
We found that known T1D candidates share unique features in functional, structural, and sequence properties that separate them from other genes. This information in turn can be used to predict novel T1D candidate genes. Table 1 lists the top 30 shared GO terms. As expected, immune system process and immune response are the two top terms, consistent with the fact that T1D is an autoimmune disease. The third significant term is defense response, which is annotated as: “reactions, triggered in response to the presence of a foreign body or the occurrence of an injury, which result in restriction of damage to the organism attacked or prevention or recovery from the infection caused by the attack.” This is consistent with the hypothesis that T1D results from initial beta cell damage, and the failure of the immune system to respond appropriately (Mathis et al., 2001). Most of the remaining GO terms, such as regulation of cell proliferation, response to stress/external stimulus, leukocyte/lymphocyte activation, cytokine production/activity, and cell death, also fit well with the current understanding of T1D pathophysiology.
Table 1.
Top 30 Gene Ontology (GO) Terms Shared by the Known T1D Candidates
| GO ID | GO name | No. of related genes | No. of all genes | p value |
|---|---|---|---|---|
| GO:0002376 | Immune system process | 119 | 419 | 8.96623E-57 |
| GO:0006955 | Immune response | 107 | 324 | 1.11306E-56 |
| GO:0006952 | Defense response | 66 | 262 | 1.16757E-29 |
| GO:0048522 | Positive regulation of cellular process | 78 | 422 | 3.38945E-27 |
| GO:0048518 | Positive regulation of biological process | 87 | 527 | 3.5746E-27 |
| GO:0051707 | Response to other organism | 47 | 129 | 1.07228E-26 |
| GO:0045321 | Leukocyte activation | 42 | 96 | 2.88314E-26 |
| GO:0001775 | Cell activation | 43 | 107 | 1.01018E-25 |
| GO:0009607 | Response to biotic stimulus | 47 | 163 | 4.75999E-23 |
| GO:0046649 | Lymphocyte activation | 37 | 85 | 6.47356E-23 |
| GO:0005102 | Receptor binding | 63 | 351 | 2.22003E-21 |
| GO:0001816 | Cytokine production | 29 | 49 | 1.81449E-20 |
| GO:0009891 | Positive regulation of biosynthetic process | 24 | 30 | 7.94301E-19 |
| GO:0005126 | Hematopoietin cytokine receptor binding | 22 | 22 | 1.64226E-18 |
| GO:0005125 | Cytokine activity | 36 | 114 | 1.98231E-18 |
| GO:0051239 | Regulation of multicellular organismal process | 38 | 132 | 2.29147E-18 |
| GO:0005615 | Extracellular space | 47 | 237 | 3.02711E-17 |
| GO:0009893 | Positive regulation of metabolic process | 41 | 179 | 8.12675E-17 |
| GO:0031325 | Positive regulation of cellular metabolic process | 40 | 171 | 1.19356E-16 |
| GO:0042127 | Regulation of cell proliferation | 42 | 210 | 2.18148E-15 |
| GO:0009611 | Response to wounding | 40 | 193 | 4.7197E-15 |
| GO:0009605 | Response to external stimulus | 46 | 273 | 1.96122E-14 |
| GO:0008283 | Cell proliferation | 51 | 349 | 7.50038E-14 |
| GO:0044421 | Extracellular region part | 48 | 355 | 8.16255E-12 |
| GO:0005886 | Plasma membrane | 84 | 939 | 1.40211E-11 |
| GO:0044459 | Plasma membrane part | 73 | 778 | 6.85688E-11 |
| GO:0008219 | Cell death | 45 | 365 | 9.93575E-10 |
| GO:0016265 | Death | 45 | 365 | 9.93575E-10 |
| GO:0006950 | Response to stress | 51 | 458 | 1.07864E-09 |
| GO:0005515 | Protein binding | 163 | 2891 | 9.8504E-07 |
All human genes in the Human Protein Reference Database were used as reference.
The known T1D candidates are more likely to be within one of the linkage loci compared to random selections. Presently there are ∼20,152 known (Entrez GeneID unique) human genes, and 983 genes located within the 20 mapped T1D linkage loci. Out of the 265 known candidates, 66 are encoded within these linkage regions; this is a ∼5-fold enrichment (Fisher's exact test, p<1e-14). If we restrict to genes annotated in the HPRD, the numbers become 487 out of 9222, and 58 out of 222. This is again a ∼5-fold enrichment (Fisher's exact test, p<1e-12). This finding is somewhat expected, given the way disease gene mapping was conventionally carried out in the past. Often after linkage regions were identified, investigators then tried to make the most intelligent speculation on the etiological variants within the regions that might explain the linkage, followed by association tests. Many functional candidates were proposed this way.
We compared the protein structure and domain composition of the T1D candidates against that of all the other genes. Table 2 lists the top 10 protein domains significantly over-represented in the T1D candidates. Overall they are consistent with T1D being an autoimmune disease. Among the most significant domains are Ig-like, Immunoglobulin, and Ig_c1. These are structure domains possessed by proteins in the immunoglobulin superfamily, a large group of cell-surface and soluble proteins that are involved in the recognition, binding, or adhesion processes of cells. One end of the domain has a section called the complementarity-determining region, which is important for the specificity of antibodies for their ligands. Members of the family are commonly associated with roles in the immune system, and include cell surface antigen receptors, co-receptors, and co-stimulatory molecules of the immune system, and molecules involved in antigen presentation to lymphocytes. hLST1 is a well-known domain closely related with cell morphogenesis and immune response. MHC_I_alpha_A1A2, MHC_II_alpha_N, Ig_MHC, IL1_rcpt_1 (interleukin receptor 1), SEFIR (IL17 receptor) and TIR (Toll-Interleukin receptor), also agree with T1D being strongly associated with the major histocompatibility complex (MHC) molecules.
Table 2.
Top 10 Protein Domains Over-Represented in Known T1D Candidates
| InterPro ID | InterPro short description | InterPro description | p value |
|---|---|---|---|
| IPR003597 | Ig_c1 | Immunoglobulin C1 type | 8.93E-34 |
| IPR007110 | Ig-like | Immunoglobulin-like | 2.64E-23 |
| IPR003006 | Ig_MHC | Immunoglobulin/major histocompatibility complex | 2.89E-22 |
| IPR013151 | Immunoglobulin | Immunoglobulin | 7.90E-21 |
| IPR013568 | SEFIR | SEFIR | 3.37E-19 |
| IPR004075 | IL1_rcpt_1 | Interleukin-1 receptor, type I/Toll precursor | 4.49E-16 |
| IPR000157 | TIR | Toll-interleukin receptor | 1.17E-15 |
| IPR001039 | MHC_I_alpha_A1A2 | MHC class I, alpha chain, alpha1 and alpha2 | 1.17E-15 |
| IPR007775 | LST1 | LST-1 | 3.04E-15 |
| IPR001003 | MHC_II_alpha_N | MHC class II, alpha chain, N-terminal | 1.93E-14 |
It has been reported that disease genes tend to have longer nucleotide sequence length (Xu and Li, 2006). We found that this is also true for the T1D candidates. Their length distribution is significantly biased toward the long end (p<2e-11, KS-test).
Naïve Bayesian network algorithm: Optimization and cross-validation
Our NBN algorithm was trained using the following five features of the known T1D candidates: GO molecular function and biological process; T1D linkage; gene length; protein sequence domains; and protein interactions.
Through a standard cross-validation test we first investigated performance of each feature and the best feature combination for disease gene prediction. A fraction of the known disease genes (67%, or 2/3) were used as the positive learning set, and we examined how well the model is able to predict the remaining fraction (33%). The process was repeated 20 times. For features with continuous or high-dimension discrete values, we evaluated the performance using the receiver operating characteristic (ROC) curve. The ROC curve plots sensitivity (i.e., true-positive rate) of a classifier against 1 – specificity (i.e., false-positive rate). The area under the curve (AUC) of the ROC curve measures the performance, with AUC=0.5 for a random classification. Figure 1 presents the AUC for features GO and InterPro. The NBN predictions improve rapidly with the number of values, and plateaus around 30 for GO (Fig. 1A), and 10 for InterPro (Fig. 1B). Hence we kept the top 30 GO and top 10 InterPro terms for predicting novel candidate genes.
FIG. 1.

AUC of the cross-validation ROC curve shows that performance of the NBN depends on the number of feature values. (A) GO. (B) InterPro.
Figure 2 presents the ROC of the feature combination GO + linkage + PPI + InterPro. On average, we obtained 63.1% sensitivity/88.0% specificity with a PS cut-off at 0.5, and 66.6% sensitivity/86.4% specificity at 0.3. Table 3 lists the AUC of the top four feature combinations, and the individual features GO, InterPro, and Length, and GO was found to yield the best performance. Evidently the AUC of the feature combinations are higher than those of the individual ones. For the two binary-valued features, linkage and PPI, we determined their enrichment ratios, which were 3.7 and 3.4, respectively. In contrast, the enrichment ratios of feature combinations were significantly higher, with GO+linkage+PPI+InterPro reaching 7.2.
FIG. 2.

ROC curve of the feature combination GO+linkage+PPI+InterPro.
Table 3.
Performance in Disease Gene Prediction of the Top Four Feature Combinations and Individual Features
| Feature combination | AUC of ROC curve |
|---|---|
| GO+Linkage+PPI+InterPro | 0.824±0.034 |
| GO+Linkage+PPI+InterPro+Length | 0.823±0.020 |
| GO+Linkage+PPI | 0.823±0.032 |
| GO+Linkage | 0.816±0.035 |
| GO | 0.804±0.032 |
| InterPro | 0.594±0.022 |
| Length | 0.512±0.013 |
Overall we observed a significant improvement when the features were integrated. Among the top combinations, we decided to use GO+linkage+PPI+InterPro for novel disease gene prediction, as adding the remaining feature (length), which is not disease-specific, did not further improve the performance.
Individual candidate genes with high PS
Figure 3 presents the cumulative fraction plot of the predicted T1D involvement PS for all 9222 HPRD-annotated genes. The results are listed in Supplementary Table S3 (see online supplementary material at http://www.liebertonline.com), and are available at http://zen.dom.uab.edu:8080/t1dsource/index.jsp. We examined their citations in T1D-related publications. The 222 known T1D candidates were excluded from this analysis to avoid confounding contributions from them. In our previous study, we used the T1D publication citations annotated by T1Dbase to evaluate candidate disease gene prediction (Gao and Wang, 2009). The text mining algorithm of T1Dbase is not open, and we found that the annotation changes significantly over time. Therefore, we instead searched the PubMed database (abstract only) directly using the official gene name and the aliases. An article was termed T1D-related if its abstract contains the phrase “type 1 diabetes” or its aliases. We found a significant difference in the PS score distribution between genes cited or not cited by T1D-related publications (p=1.9e-57, KS test). Figure 4 illustrates the percent of genes in different PS intervals that are cited in T1D-related publications. A positive correlation is evident. We found a number of genes with high PS for which their potential role in T1D had been implied in the literature (highlighted in Supplementary Table S3; see online supplementary material at http://www.liebertonline.com).
FIG. 3.

The cumulative distribution plot of the predicted PS values for all human HPRD genes.
FIG. 4.

Genes with higher predicted probability scores are cited more often by T1D-related publications.
Predicted T1D subnetworks
Using our new network metric defined in [Eq. 2], six subnetworks with 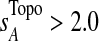 were predicted to be associated with T1D (Fig. 5; a complete list of genes is provided in Supplementary Table S4; see online supplementary material at http://www.liebertonline.com; additional details are available at http://zen.dom.uab.edu:8080/t1dsource/index.jsp). There are a total of 207 nodes in the 6 subnetworks, corresponding to 92 genes; 53 are novel candidates. Most of the network members appear in T1D publications, even after excluding the known T1D candidates (p<1e-9, Fisher's exact test).
were predicted to be associated with T1D (Fig. 5; a complete list of genes is provided in Supplementary Table S4; see online supplementary material at http://www.liebertonline.com; additional details are available at http://zen.dom.uab.edu:8080/t1dsource/index.jsp). There are a total of 207 nodes in the 6 subnetworks, corresponding to 92 genes; 53 are novel candidates. Most of the network members appear in T1D publications, even after excluding the known T1D candidates (p<1e-9, Fisher's exact test).
FIG. 5.

Subnetworks related to T1D. Round rectangles denote known T1D candidates collected in T1Dbase.
It is believed that hub genes play a central role in a network. Interestingly, 66.7% of those genes with network degree ≥5 appear in T1D-related abstracts, while only 31% of those with degree <5 do. Table 4 lists genes with degree ≥5 in the six subnetworks, excluding the known T1D candidates. Most of them participate in biological processes that are likely relevant to T1D pathophysiology. Though PIK3R1 did not appear in the T1D-related publications, it is part of the insulin receptor signaling KEGG pathway (Gustafson et al., 1995), plays an important role in the metabolic actions of insulin, and a mutation in this gene has been associated with insulin resistance. Furthermore, it participates in apoptotic processes, and the Mouse Genome Informatics (MGI) mutant studies have shown that its murine homologue affects the immune system. TRAF6 activates the IL-1 signaling pathway via MYD88 and IRAK kinases. Both TRAF6 and TRAF2 are involved in NF-κB activation during NF-κB-regulated apoptotic beta-cell death (Liuwantara et al., 2006). The mouse homologue of TNFR2 lies within the T1D susceptibility locus, and the diabetes-associated variant contains a mutation adjacent to the TRAF2 binding site. It is also part of the anti-apoptotic TNF/NF-kB/Bcl-2 pathway (Hill et al., 2007). SYK is required for signaling of several immunoreceptors of the antigen-presenting cells. Inhibition of SYK was recently found to interrupt the humoral contributions to T-cell-driven autoimmunity, and delayed spontaneous diabetes onset in the NOD mouse model of T1D (Colonna et al., 2010).
Table 4.
Genes with Degree ≥5 in the Predicted Subnetworks
| EntrezID | Gene name | No. of T1D publications | Degree | Subnetwork |
|---|---|---|---|---|
| 5295 | PIK3R1 | 0 | 14 | 4 |
| 7186 | TRAF2 | 3 | 13 | 5 |
| 7186 | TRAF2 | 3 | 12 | 2 |
| 7186 | TRAF2 | 3 | 11 | 4 |
| 7189 | TRAF6 | 1 | 9 | 5 |
| 6850 | SYK | 2 | 7 | 4 |
| 7189 | TRAF6 | 1 | 7 | 2 |
| 7189 | TRAF6 | 1 | 7 | 4 |
| 3556 | IL1RAP | 2 | 6 | 4 |
| 4615 | MYD88 | 4 | 6 | 5 |
| 6885 | MAP3K7 | 0 | 6 | 5 |
| 4615 | MYD88 | 4 | 5 | 4 |
| 7852 | CXCR4 | 4 | 5 | 1 |
| 7852 | CXCR4 | 4 | 5 | 2 |
| 7852 | CXCR4 | 4 | 5 | 3 |
| 8915 | BCL10 | 0 | 5 | 5 |
The administration of IL1RAP was found to improve beta-cell function in both type 1 and type 2 diabetes (Pfleger et al., 2008). Activation of MYD88 can lead to NF-κB activation, cytokine secretion, and inflammatory responses (Sharp et al., 2008). In the NOD mouse model of T1D, animals lacking MyD88 protein do not develop T1D. Also in this animal model, CXCR4 was found to contribute to the negative regulation of T-cell adhesion, which is important in the recruitment of diabetogenic T cells into islet cells (Sharp et al., 2008). It was found that signaling of CXCR4 together with SDF-1 protects against autoimmune diabetes, and inhibition of CXCR4 activity exacerbates the adoptive transfer of diabetes (Aboumrad et al., 2007). BCL10 participates in T- and B-cell-receptor signaling and the adaptive immune response, both of which are relevant to T1D pathogenesis (Ruefli-Brasse et al., 2004).
Among all human HPRD genes there were 430 (5.1%) with PS>0.95 (Fig. 2 and Supplementary Table S3; see online supplementary material at http://www.liebertonline.com). If we use this value as a cut-off, 349 novel candidates are predicted (after excluding the known T1D candidates). In each predicted subnetwork, there are genes that miss this cut-off. However, they interact with genes with the highest PS values. We found that their potential role in T1D is implicated by existing literature reports. IFNAR2 in subnetwork 6 has a moderate PS score of 0.8879, ranked #474 out of 9222, and #389 after excluding T1D candidates. Its neighbors IFNA5, IFNA8, IFNA2, and IFNB1, all have PS=1, and are all known T1D candidates. IFN-α is a group of pleiotropic cytokines in the type I family of IFNs. IFN-α exerts broad but distinct effects on the innate and adoptive immune responses by signaling through a heterodimeric receptor composed of IFN-α receptor 1 (IFNAR1) and IFNAR2. Many studies suggest that IFN-α is involved in the development of T1D (Li et al., 2008). SPG21 (PS=0.8826), ranked #477, interacts with two PS=1 genes, CD4 (a known T1D gene) and TRAF2. The noncatalytic alpha/beta hydrolase fold domain of this protein binds to the hydrophobic C-terminal amino acids of CD4, which are involved in repression of T-cell activation. It is thus proposed that this gene product modulates the stimulatory activity of CD4 (Zeitlmann et al., 2001). IGF1R (PS=0.9487), ranked #433, interacts with PIK3R1 (PS=0.99997). It is a transmembrane tyrosine kinase receptor that is activated by insulin-like growth factor-I (IGF-1), and by the related growth factor IGF-2. Decreases in IGF1R signaling causing insulin resistance is a major component in the development of type 2 diabetes.
In T1D, the HLA locus accounts for most of the genetic risk. Recent studies identified over 50 non-HLA regions that significantly affect disease risk (http://www.t1dbase.org; Pociot et al., 2010; Rich et al., 2009). Genes outside the HLA, though contributing less to the overall disease risk, are likely the major source for disease heterogeneity. The majority of novel variations recently discovered are in intronic and/or regulatory gene regions, indicating the importance of mapping disease gene networks (Pociot et al., 2010; Rich et al., 2009). Among the genes coding for the 9222 HPRD proteins, 115 (1.25%) are located in the HLA locus. The number of HLA genes among the known T1D genes is 22 out of 222 (10%), significantly higher than that expected by random chance (χ2=107.8, p<0.0001). This is consistent with the genetic importance of the HLA locus in T1D. Interestingly, the presence of the HLA genes is diminished among the novel candidate genes. At PS>0.95, 7 out of the 349 novel candidates (2.01%) are in the HLA locus, a frequency no different from that expected by chance (χ2=0.99, p ∼0.32), and is significantly less than that of the known T1D candidates (χ2=15.98, p<0.0001). In the six predicted network modules, only one, TNF-α, a known T1D gene, resides in the HLA region. Of the 53 novel candidates predicted by the six subnetworks, none reside within the HLA locus. Again, this is no different from the rate expected by chance (χ2=0.11, p ∼0.74), and significantly less than that of the known T1D candidates (χ2=6.22, p ∼0.013). In conventional genetic approaches, the presence of a dominating disease risk locus often makes it difficult to dissect the contributions from the minor loci. Our results indicate that mapping disease-associated networks may be an efficient approach to identify genes that contribute moderately to disease risk.
Predicted subnetworks were supported by GWAS and gene expression data
First, we examined evidence of T1D association using independent data from GWAS. A number of large-scale GWAS studies have been carried out for T1D, most notably the study conducted by the Wellcome Trust Case-Control Consortium (Wellcome Trust Case-Control Consortium, 2007), and those by the Type 1 Diabetes Genetics Consortium (T1DGC; Barrett et al., 2009; Morahan et al., 2011). To validate our algorithm prediction, we chose to use the WTCCC dataset over a meta-analysis of all studies, since the WTCCC cases were drawn from only two countries (U.K. and U.S.), whereas the T1DGC families were recruited from many different countries and diverse recruitment networks (Asia-Pacific, Europe, and North America). The markedly increased genetic and environmental heterogeneity in the latter could affect the penetrance of susceptibility alleles and confound the ability to detect genes that may affect risk in some populations but not others. Interestingly, although each individual gene did not meet the stringent p-value cut-off for disease association typically required for a GWA study (p>0.001 for all genes), as a group the 53 novel candidate genes from the six T1D subnetworks showed a shift toward to the low-p end compared to the rest (p=0.063, KS test). Figure 6A presents the distribution of their p values and those of all genes (excluding the known disease genes). We further examined the p values of genes with degree ≥2 (31 genes); the difference is more significant, with p=0.055 (Fig. 6A). For novel candidates with degree >2, the number of genes typed by the GWAS study was too few for a statistical test to be meaningful. For comparison we also evaluated the GWAS p values of the 222 known T1D candidates, and the 349 new candidates predicted with PS only at PS>0.95, and found that they were also shifted toward the low-p end, with p=0.083 and p=0.074, respectively. However, if we randomly picked 53 members from each gene list (to compare them at the same sample size), KS-test on average yielded p>0.2. These results suggest that the network-based candidate gene prioritization is likely more efficient at identifying true disease genes.
FIG. 6.

GWAS and gene expression studies indicate the potential disease involvement of the novel candidate genes in the predicted T1D network modules. (A) Cumulative distribution function of the T1D association p value from GWAS. Distribution of the novel candidates is shifted to the low-p-value end (p=0.063 and p=0.055 for those with network degree 2 or above). (B) Cumulative distribution function of the differential expression p value between the RO-LR and LRS cohorts. The distribution for the novel candidate genes shows significant (p=0.03) shift to the low end, indicating correlation with the T1D phenotype in cohorts with low-risk HLA genotypes.
Next we investigated expression of the novel candidate genes in two large microarray studies of T1D. The first dataset came from our own study, which profiled global transcription in healthy donor PBMCs stimulated by sera of different cohorts (Wang et al., 2008): RO T1D patients, unrelated HC with no family history of T1D, and healthy siblings of T1D patients that possess either high-risk (HRS) or low-risk (LRS) HLA genotypes. One-way ANOVA found that overall the new candidates exhibited significant variation in the four cohorts, with p=0.03 for the 53-gene list, and p=0.004 for the 349-gene list. Between the RO and HC samples, neither expression of the novel candidates, nor the known disease genes, showed significant correlation to disease phenotype (Table 5). However, when stratified by T1D risk defined by the DR3/4 HLA genotypes, the new candidates as a set showed significant correlations with disease in the low-risk cohorts (Table 5). In Figure 6B, the cumulative plot of the p values between the RO samples with low-risk HLA genotypes (RO-LR), and the LRS samples, is presented. The distribution of the novel candidates is shifted toward the low-p end (p ∼0.03). This finding of disease correlation in low-risk cohorts is likely due to the extremely low presence of the HLA genes in the novel candidates, and because the non-HLA genes contribute more to disease pathology in cohorts with low-risk HLA genotypes.
Table 5.
Gene Set Analysis of New and Known T1D Candidates
| Comparison | RO versus HC | RO-HR versus HC-HR | RO-HR versus HRS | RO-LR versus HC-LR | RO-LR versus LRS |
|---|---|---|---|---|---|
| 53 Novel candidates from the 6 T1D subnetworks | 0.76 | 0.43 | 0.19 | 0.26 | 0.03 |
| 349 Candidates with PS>0.95 | 0.86 | 0.93 | 0.91 | 0.06 | 0.07 |
| 222 Known T1D candidates | 0.23 | 0.20 | 0.12 | 0.52 | 0.85 |
The new candidates show signs of differential activity in cohorts with low-risk HLA genotypes.
The second microarray dataset came from a study by Kaizer and associates, in which genome-wide mRNA expression levels in PBMCs were directly measured in 43 RO and 24 HC samples (Kaizer et al., 2007). Again, few individual genes showed significant differential expression between RO and HC samples. As a group, 53 novel candidates exhibited marginal significance in disease correlation (p=0.11, KS test). The results from the known T1D disease genes are similar (p=0.09). The HLA genotypes are not available so we could not stratify the analysis into different HLA risk groups.
On-line resources of T1D genetics
The algorithms reported in this article are freely available at: http://zen.dom.uab.edu:8080/t1dsource/index.jsp. Interactive examination of the network context (such as neighboring genes), and properties of genes using several data sources, are enabled with an in-house developed Cytoscape plugin using the Java webstart technique, which is shown in Figure 7. Currently, data sources are also being collected that include: association studies, linkage analysis, gene expression, T1D-related literature, GO annotations, protein domains, and protein-protein interaction networks.
FIG. 7.

Snapshots from our T1D resources website. Top left is the homepage; top right is the web interface to examine information about the given genes from several data sources; bottom is the Cytoscape webstart interface to examine subnetworks of interest.
Discussion
In this study we demonstrated the importance of mapping disease gene networks in dissecting the genetics of a complex disease. First, an NBN model was developed to score human genes for their T1D relevance based on multiple features of known disease genes. The efficiency of the model was demonstrated through cross-validation, and by the significant appearance in T1D-related literature of the predicted novel candidate genes. Further, we proposed a new network activity metric, and used it to extract PPI subnetworks that are relevant to T1D. We showed that the novel candidates identified through the predicted subnetworks are more likely to contain true disease genes. Two major features distinguish our study from others. First, our network metric defined in [Eq. 1] and [Eq. 2] incorporates the consideration of both individual genes and topological characteristics of their interaction networks. Second, to evaluate the performance of our new algorithm, we went beyond the commonly used cross-validation approach, and utilized real, independent data to confirm the predictions. This includes the published peer-reviewed literature, GWAS, and functional genomics studies. Our algorithm offers a general approach to mapping disease-relevant gene networks and prioritizing candidate disease genes through integration of multiple data sources. It collects the existing data and knowledge relevant to a disease to train an NBN model, which in turn is used to identify new disease genes. The procedure does not depend on any specific characteristics of T1D, or its pathogenesis. For any disease for which there is existing genetic data and knowledge, even if only partial, a similar NBN model can be developed and trained to discriminate disease genes from the rest. Therefore our approach is generally applicable to other complex diseases.
In this study we used NBN to integrate multiple data sources that contain functional, genetic, and sequence information of genes. A number of data sources that may also offer valuable information on disease associations were not included, for instance, evolution conservation, protein stability, and post-translational modification of proteins. These will be investigated in our future studies. In training NBN, all known T1D candidates were used as positive controls and the rest as negative controls. Note that since the disease etiology is still not fully understood, the negative controls likely contain genes that are involved in T1D pathogenesis, although presently they are not known to be. Some of the false-positive predictions might represent true-positives. In fact, this is a common challenge in current integrative genomics approaches to complex diseases, in that we can be fairly confident of the positive controls, but the negative controls may still contain true disease genes. It would be of interest to investigate means to improve the procedure. One possibility is to carry out the novel disease gene prediction steps iteratively, and retrain the NBN by removing all positive predictions from the negative set, followed by recalculating the PS for all genes.
The NBN is theoretically optimal when the attributes are independent, but it performs well in many domains when there are moderate attribute dependences (Domingos and Pazzani, 1997; Friedman et al., 1997). The five features of the known T1D candidates being studied here show weak interdependence. In modeling some of the disease gene features, such as GO (Table 1) and InterPro (Table 2), we only retained the top annotation terms. Intuitively one may think that any contributing information should be considered, and more terms could be included to improve the performance. However, high dimensionality and sparse training data can result in over-fitting, especially when there is inter-dependency among data. In future studies we will examine how feature inter-dependence, and types of information (discrete versus continuous), affect the performance. We will also investigate the balance between amounts of prior information and the dimensionality issue. It is also worthwhile to investigate more sophisticated algorithms to address these issues in data integration. Examples include partial least square, Tree Augmented Naive Bayes (TAN), and neural-networks.
Validating computational algorithms for integrative genomics is a challenge, as directly testing the predictions (of novel candidate disease genes) is usually not immediately feasible. Presently the performance is typically evaluated using the known disease genes as a benchmark, and normally 5- to 10-fold enrichment over random selection is observed. It has been questioned whether the enrichment observed at a high-throughput level is meaningful to a specific disease (Oti and Brunner, 2007). To overcome this problem, we used the pre-GWAS genetic knowledge of T1D as input to our algorithm, and independent high-throughput association and gene-expression profiling studies to validate the network-based disease gene predictions. We demonstrated the value of incorporating network information in candidate gene prioritization.
Our algorithms can of course include the latest genetic results as input to further improve the efficiency and power of identifying true disease genes. Recent technological advancements, including the GWAS and the next-generation sequencing technologies, hold great promise for surveying the entire genome in one experiment for disease-causing variants. Some exciting results have already been published for T1D (Barrett et al., 2009; Morahan et al., 2011; Wellcome Trust Case-Control Consortium, 2007). However, because of the high number of variants and their combinations, high false-positive rates arising from multi-testing is a serious problem (McCarthy et al., 2008). Some have argued the advantages of focusing on shorter predefined gene lists consisting of the most probable candidates created by computational or pathogenomic approaches (Gaulton et al., 2008; Loza et al., 2007). This can significantly reduce the number of tests that need to be performed. One of the potential advantages of our study is to provide such a list (the 53 novel candidate genes from the six predicted T1D network modules, for instance) for T1D association studies. In addition, conventional single-marker-based analysis of sequence variant-disease association offers little insight into the disease mechanism. Network-based pathway analysis can provide a more comprehensive picture of the genetic architecture underlying a disease (Wang et al., 2007). Our network metric defined in [Eq. 1] and [Eq. 2] can be applied to ascertain the disease association of a network by replacing the z score of PS with that of the p value of association. Though network-based association analysis has been explored (Baranzini et al., 2009), our metric is unique in that it accounts for contributions from both the network topology and the individual genes.
Supplementary Material
Acknowledgments
This work was supported in part by National Institute of Diabetes and Digestive and Kidney Diseases grant R01DK080100 (X.W.), National Institute of Allergy and Infectious Diseases grant R01AI078713 (M.J.H.), and Juvenile Diabetes Research Foundation International grant 1-2008-1026 (M.J.H). We thank Ryan Kelley and Trey Ideker for kindly providing the source code of jActiveModules.
Author Disclosure Statement
The authors declare that no competing financial interests exist.
References
- Aboumrad E. Madec A.M. Thivolet C. The CXCR4/CXCL12 (SDF-1) signalling pathway protects non-obese diabetic mouse from autoimmune diabetes. Clin Exp Immunol. 2007;148:432–439. doi: 10.1111/j.1365-2249.2007.03370.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aerts S. Lambrechts D. Maity S., et al. Gene prioritization through genomic data fusion. Nat Biotechnol. 2006;24:537–544. doi: 10.1038/nbt1203. [DOI] [PubMed] [Google Scholar]
- Aragues R. Sander C. Oliva B. Predicting cancer involvement of genes from heterogeneous data. BMC Bioinformatics. 2008;9:172. doi: 10.1186/1471-2105-9-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranzini S.E. Galwey N.W. Wang J., et al. Pathway and network-based analysis of genome-wide association studies in multiple sclerosis. Hum Mol Genet. 2009;18:2078–2090. doi: 10.1093/hmg/ddp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett J.C. Clayton D.G. Concannon P., et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet. 2009;41:703–707. doi: 10.1038/ng.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botstein D. Risch N. Discovering genotypes underlying human phenotypes: past successes for mendelian disease, future approaches for complex disease. Nat Genet. 2003;33(Suppl):228–237. doi: 10.1038/ng1090. [DOI] [PubMed] [Google Scholar]
- Carter S.L. Brechbuhler C.M. Griffin M. Bond A.T. Gene co-expression network topology provides a framework for molecular characterization of cellular state. Bioinformatics (Oxf, England) 2004;20:2242–2250. doi: 10.1093/bioinformatics/bth234. [DOI] [PubMed] [Google Scholar]
- Chuang H.Y. Lee E. Liu Y.T. Lee D. Ideker T. Network-based classification of breast cancer metastasis. Mol Systems Biol. 2007;3:140. doi: 10.1038/msb4100180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colonna L. Catalano G. Chew C., et al. Therapeutic targeting of Syk in autoimmune diabetes. J Immunol. 2010;185:1532–1543. doi: 10.4049/jimmunol.1000983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dao P. Colak R. Salari R., et al. Inferring cancer subnetwork markers using density-constrained biclustering. Bioinformatics. 2010;26:i625–i631. doi: 10.1093/bioinformatics/btq393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingos P. Pazzani M. On the optimality of the simple Bayesian classifier under zero-one loss. Machine Learning. 1997;29:103–130. [Google Scholar]
- Eaves I.A. Wicker L.S. Ghandour G., et al. Combining mouse congenic strains and microarray gene expression analyses to study a complex trait: the NOD model of type 1 diabetes. Genome Res. 2002;12:232–243. [PubMed] [Google Scholar]
- English S.B. Butte A.J. Evaluation and integration of 49 genome-wide experiments and the prediction of previously unknown obesity-related genes. Bioinformatics (Oxf, England) 2007;23:2910–2917. doi: 10.1093/bioinformatics/btm483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke L. Bakel H. Fokkens L. De Jong E.D. Egmont-Petersen M. Wijmenga C. Reconstruction of a functional human gene network, with an application for prioritizing positional candidate genes. Am J Hum Genet. 2006;78:1011–1025. doi: 10.1086/504300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman N. Geiger D. Goldszmidt M. Bayesian network classifiers. Machine Learning. 1997;29:131–163. [Google Scholar]
- Gao S. Wang X. Predicting type 1 diabetes candidate genes using human protein-protein interaction networks. J Comput Sci Syst Biol. 2009;2:133–146. doi: 10.4172/jcsb.1000025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S. Wang X. Quantitative utilization of prior biological knowledge in the Bayesian network modeling of gene expression data. BMC Bioinformatics. 2011;12:359. doi: 10.1186/1471-2105-12-359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S. Wang X. TAPPA: topological analysis of pathway phenotype association. Bioinformatics. 2007;23:3100–3102. doi: 10.1093/bioinformatics/btm460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaulton K.J. Willer C.J. Li Y., et al. Comprehensive association study of type 2 diabetes and related quantitative traits with 222 candidate genes. Diabetes. 2008;57:3136–3144. doi: 10.2337/db07-1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George R.A. Liu J.Y. Feng L.L. Bryson-Richardson R.J. Fatkin D. Wouters M.A. Analysis of protein sequence and interaction data for candidate disease gene prediction. Nucleic Acids Res. 2006;34:e130. doi: 10.1093/nar/gkl707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh K.I. Cusick M.E. Valle D. Childs B. Vidal M. Barabasi A.L. The human disease network. Proc Natl Acad Sci USA. 2007;104:8685–8690. doi: 10.1073/pnas.0701361104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson T.A. He W. Craparo A. Schaub C.D. O'Neill T.J. Phosphotyrosine-dependent interaction of SHC and insulin receptor substrate 1 with the NPEY motif of the insulin receptor via a novel non-SH2 domain. Molec Cellular Biol. 1995;15:2500–2508. doi: 10.1128/mcb.15.5.2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill N.J. Stotland A. Solomon M. Secrest P. Getzoff E. Sarvetnick N. Resistance of the target islet tissue to autoimmune destruction contributes to genetic susceptibility in type 1 diabetes. Biol Direct. 2007;2:5. doi: 10.1186/1745-6150-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung J.H. Whitfield T.W. Yang T.H. Hu Z. Weng Z. Delisi C. Identification of functional modules that correlate with phenotypic difference: the influence of network topology. Genome Biol. 2010;11:R23. doi: 10.1186/gb-2010-11-2-r23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter S. Apweiler R. Attwood T.K., et al. InterPro: the integrative protein signature database. Nucleic Acids Res. 2009;37:D211–D215. doi: 10.1093/nar/gkn785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ideker T. Ozier O. Schwikowski B. Siegel A.F. Discovering regulatory and signalling circuits in molecular interaction networks. Bioinformatics (Oxf, England) 2002;18(Suppl 1):S233–S240. doi: 10.1093/bioinformatics/18.suppl_1.s233. [DOI] [PubMed] [Google Scholar]
- Jimenez-Sanchez G. Childs B. Valle D. Human disease genes. Nature. 2001;409:853–855. doi: 10.1038/35057050. [DOI] [PubMed] [Google Scholar]
- Jones C.E. Brown A.L. Baumann U. Estimating the annotation error rate of curated GO database sequence annotations. BMC Bioinformatics. 2007;8:170. doi: 10.1186/1471-2105-8-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaizer E.C. Glaser C.L. Chaussabel D. Banchereau J. Pascual V. White P.C. Gene expression in peripheral blood mononuclear cells from children with diabetes. J Clin Endocrinol Metab. 2007;92:3705–3711. doi: 10.1210/jc.2007-0979. [DOI] [PubMed] [Google Scholar]
- Kann M.G. Protein interactions and disease: computational approaches to uncover the etiology of diseases. Briefings Bioinformatics. 2007;8:333–346. doi: 10.1093/bib/bbm031. [DOI] [PubMed] [Google Scholar]
- Lage K. Karlberg E.O. Storling Z.M., et al. A human phenome-interactome network of protein complexes implicated in genetic disorders. Nat Biotech. 2007;25:309–316. doi: 10.1038/nbt1295. [DOI] [PubMed] [Google Scholar]
- Li Q. Xu B. Michie S.A. Rubins K.H. Schreriber R.D. McDevitt H.O. Interferon-α initiates type 1 diabetes in nonobese diabetic mice. Proc Natl Assn Sci USA. 2008;105:12439–12444. doi: 10.1073/pnas.0806439105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C.C. Chen W.S. Lin C.C., et al. Topology-based cancer classification and related pathway mining using microarray data. Nucleic Acids Res. 2006;34:4069–4080. doi: 10.1093/nar/gkl583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liuwantara D. Elliot M. Smith M.W., et al. Nuclear factor-kappaB regulates beta-cell death: a critical role for A20 in beta-cell protection. Diabetes. 2006;55:2491–2501. doi: 10.2337/db06-0142. [DOI] [PubMed] [Google Scholar]
- Loza M.J. McCall C.E. Li L. Isaacs W.B. Xu J. Chang B.L. Assembly of inflammation-related genes for pathway-focused genetic analysis. PloS One. 2007;2:e1035. doi: 10.1371/journal.pone.0001035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L. Peng G. Zhu Y. Dong H. Amos C.I. Xiong M. Genome-wide gene and pathway analysis. Eur J Hum Genet. 2010;18:1045–1053. doi: 10.1038/ejhg.2010.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H. Schadt E.E. Kaplan L.M. Zhao H. COSINE: COndition-SpecIfic sub-NEtwork identification using a global optimization method. Bioinformatics (Oxf, England) 2011;27:1290–1298. doi: 10.1093/bioinformatics/btr136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massa M.S. Chiogna M. Romualdi C. Gene set analysis exploiting the topology of a pathway. BMC Systems Biol. 2010;4:121. doi: 10.1186/1752-0509-4-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathis D. Vence L. Benoist C. Beta-cell death during progression to diabetes. Nature. 2001;414:792–798. doi: 10.1038/414792a. [DOI] [PubMed] [Google Scholar]
- McCarthy M.I. Abecasis G.R. Cardon L.R., et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet. 2008;9:356–369. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- Mishra G.R. Suresh M. Kumaran K., et al. Human protein reference database—2006 update. Nucleic Acids Res. 2006;34:D411–D414. doi: 10.1093/nar/gkj141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morahan G. Mehta M. James I., et al. Tests for genetic interactions in type 1 diabetes: linkage and stratification analyses of 4,422 affected sib-pairs. Diabetes. 2011;60:1030–1040. doi: 10.2337/db10-1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitsch D. Tranchevent L.C. Thienpont B., et al. Network analysis of differential expression for the identification of disease-causing genes. PloS One. 2009;4:e5526. doi: 10.1371/journal.pone.0005526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oti M. Brunner H.G. The modular nature of genetic diseases. Clin Genet. 2007;71:1–11. doi: 10.1111/j.1399-0004.2006.00708.x. [DOI] [PubMed] [Google Scholar]
- Pfleger C. Mortensen H.B. Hansen L., et al. Association of IL-1ra and adiponectin with C-peptide and remission in patients with type 1 diabetes. Diabetes. 2008;57:929–937. doi: 10.2337/db07-1697. [DOI] [PubMed] [Google Scholar]
- Pociot F. Akolkar B. Concannon P., et al. Genetics of type 1 diabetes: what's next? Diabetes. 2010;59:1561–1571. doi: 10.2337/db10-0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich S.S. Akolkar B. Concannon P., et al. Overview of the Type I Diabetes Genetics Consortium. Genes Immun. 2009;10(Suppl 1):S1–S4. doi: 10.1038/gene.2009.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruefli-Brasse A.A. Lee W.P. Hurst S. Dixit V.M. Rip2 participates in Bcl10 signaling and T-cell receptor-mediated NF-kappaB activation. J Biological Chem. 2004;279:1570–1574. doi: 10.1074/jbc.C300460200. [DOI] [PubMed] [Google Scholar]
- Sharp C.D. Huang M. Glawe J., et al. Stromal cell-derived factor-1/CXCL12 stimulates chemorepulsion of NOD/LtJ T-cell adhesion to islet microvascular endothelium. Diabetes. 2008;57:102–112. doi: 10.2337/db07-0494. [DOI] [PubMed] [Google Scholar]
- Smith N.G. Eyre-Walker A. Human disease genes: patterns and predictions. Gene. 2003;318:169–175. doi: 10.1016/s0378-1119(03)00772-8. [DOI] [PubMed] [Google Scholar]
- Smoot M. Ono K. Ideker T. Maere S. PiNGO: a Cytoscape plugin to find candidate genes in biological networks. Bioinformatics (Oxf, England) 2011;27:1030–1031. doi: 10.1093/bioinformatics/btr045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su J. Yoon B.J. Dougherty E.R. Identification of diagnostic subnetwork markers for cancer in human protein-protein interaction network. BMC Bioinformatics. 2010;11(Suppl 6):S8. doi: 10.1186/1471-2105-11-S6-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas R. Gohlke J. Stopper G. Parham F. Portier C. Choosing the right path: enhancement of biologically relevant sets of genes or proteins using pathway structure. Genome Biol. 2009;10:R44. doi: 10.1186/gb-2009-10-4-r44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiffin N. Adie E. Turner F., et al. Computational disease gene identification: a concert of methods prioritizes type 2 diabetes and obesity candidate genes. Nucleic Acids Res. 2006;34:3067–3081. doi: 10.1093/nar/gkl381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veyrieras J.-B. Kudaravalli S. Kim S.Y., et al. High-resolution mapping of expression-QTLs yields insight into human gene regulation. PLoS Genet. 2008;4:e1000214. doi: 10.1371/journal.pgen.1000214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K. Li M. Bucan M. Pathway-based approaches for analysis of genomewide association studies. Am J Hum Genet. 2007;81:1278–1283. doi: 10.1086/522374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. Jia S. Geoffrey R. Alemzadeh R. Ghosh S. Hessner M.J. Identification of a molecular signature in human type 1 diabetes mellitus using serum and functional genomics. J Immunol. 2008;180:1929–1937. doi: 10.4049/jimmunol.180.3.1929. [DOI] [PubMed] [Google Scholar]
- Wang Y. Xia Y. Condition specific subnetwork identification using an optimization model. The Second International Symposium on Optimization and Systems Biology 2008 [Google Scholar]
- Wellcome Trust Case-Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J. Li Y. Discovering disease-genes by topological features in human protein-protein interaction network. Bioinformatics (Oxf, England) 2006;22:2800–2805. doi: 10.1093/bioinformatics/btl467. [DOI] [PubMed] [Google Scholar]
- Zeitlmann L. Sirim P. Kremmer E. Kolanus W. Cloning of ACP33 as a novel intracellular ligand of CD4. J Biol Chem. 2001;276:9123–9132. doi: 10.1074/jbc.M009270200. [DOI] [PubMed] [Google Scholar]
- Zhong H. Beaulaurier J. Lum P.Y., et al. Liver and adipose expression associated SNPs are enriched for association to type 2 diabetes. PLoS Genet. 2010;6:e1000932. doi: 10.1371/journal.pgen.1000932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M. Zhao S. Candidate gene identification approach: progress and challenges. Int J Biol Sci. 2007;3:420–427. doi: 10.7150/ijbs.3.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.