

TLR9 localizes to a novel intracellular compartment called the T granule to promote immune signaling by platelets.
Abstract
Human and murine platelets (PLTs) variably express toll-like receptors (TLRs), which link the innate and adaptive immune responses during infectious inflammation and atherosclerotic vascular disease. In this paper, we show that the TLR9 transcript is specifically up-regulated during pro-PLT production and is distributed to a novel electron-dense tubular system-related compartment we have named the T granule. TLR9 colocalizes with protein disulfide isomerase and is associated with either VAMP 7 or VAMP 8, which regulates its distribution in PLTs on contact activation (spreading). Preincubation of PLTs with type IV collagen specifically increased TLR9 and CD62P surface expression and augmented oligodeoxynucleotide (ODN) sequestration and PLT clumping upon addition of bacterial/viral ODNs. Collectively, this paper (a) tracks TLR9 to a new intracellular compartment in PLTs and (b) describes a novel mechanism of TLR9 organization and signaling in human PLTs.
Introduction
Human adults contain nearly one trillion blood PLTs in circulation. These small (2–3 µm), anuclear cells have a highly organized cytoskeleton, unique receptors, and specialized secretory granules. Among their primary functions, PLTs serve as the “Band-Aids” of the bloodstream and respond to blood vessel injury by changing shape, secreting their granule contents, and aggregating to form a PLT clot. PLTs also play secondary roles in lymphatic development and inflammation (Smyth et al., 2009) by expressing immunomodulatory molecules and cytokines with which they interact with various cells of the immune system (Semple et al., 2011). Among their most recognized and least understood immunomodulatory functions, PLTs express multiple members of a family of pattern recognition receptors called toll-like receptors (TLRs). TLRs have been best characterized in neutrophils, macrophages, and dendritic cells and promote immune activation in response to conserved molecular motifs expressed by pathogens (Janeway and Medzhitov, 2002). Although human and mouse PLTs express TLRs 1–9, our current understanding of TLR function in PLTs is limited to TLRs 1–6, which are typically expressed on the PLT surface and thought to trap bacteria for elimination by professional phagocytes (Andonegui et al., 2005; Aslam et al., 2006; Clark et al., 2007). Less understood is the role of TLR9, which is expressed in the endosomes of monocytes, macrophages, and plasmacytoid dendritic cells and is thought to act as a receptor for unmethylated CpG islands found in bacterial and viral DNA (Hennessy et al., 2010).
Although reported to localize internally in PLTs, neither the site nor function of subcellular TLR9 is currently known (Aslam et al., 2006). In macrophages and dendritic cells, TLR9 is recruited to lysosomes only after cells are stimulated with CpG DNA (Latz et al., 2004). The fact that all intracellular TLRs identified share specificity for nucleic acids suggests that this localization may be related to the recognition of this class of ligand (Barton et al., 2006). Synthetic oligodeoxynucleotides (ODNs) that contain unmethylated CpG motifs are typically used to activate TLR9. Type C CpG motifs contain a complete phosphorothioate backbone and a CpG-containing palindromic motif that is present at a 20-fold greater frequency in bacterial DNA compared with mammalian DNA (Bauer et al., 2001). Although type C CpG ODNs induce strong IFN-α production from both plasmacytoid dendritic cells and B cells, the effect of TLR9 signaling in PLTs remains to be established.
In this paper, we use confocal and electron microscopy to track TLR9 during pro-PLT production to a new electron-dense tubular system-related compartment in PLTs. Second, we use known PLT agonists to describe a novel mechanism of TLR9 signaling whereby PLTs are primed during activation to express TLR9 on their surface before secondary activation through pathogen-associated molecular patterns (PAMPs) on invading organisms. Finally, we reveal a unique role for human PLTs as mediators of innate immunity at sites of vascular damage.
Results
TLR9 is expressed in murine cell culture megakaryocytes (MKs) and is distributed to PLTs during pro-PLT production
Because TLR9 is a transmembrane receptor, we hypothesized that TLR9 should be translated by bone marrow MKs and transported along pro-PLT extensions to nascent PLTs. To test this hypothesis, mouse fetal liver cell cultures were generated, and round MKs, pro-PLT–producing MKs, released pro-PLTs, and individual PLTs were isolated on day 5 of culture. Next generation RNA sequencing of round MKs and pro-PLT–MKs confirmed the presence of the Tlr9 and Vamp8 transcripts in murine cell cultures (Fig. 1, A and B). Comparison of Tlr9 expression profiles between the two revealed that MKs experience a burst of Tlr9 mRNA expression during pro-PLT production (0.34 ± 0.21 vs. 2.44 ± 0.75 RPKM, n ≥ 3). Pro-PLT–producing MKs did not experience a similar burst of Vamp8 mRNA expression during pro-PLT production, which was used as an internal control (70.9 ± 14.5 vs. 60.4 ± 3.2 RPKM, n ≥ 3). These results confirm that TLR9 is generated by MKs during pro-PLT production. Intermediates in PLT production were probed for TLR9 and β1-tubulin to delineate the cell periphery, and samples were visualized by immunofluorescence microscopy (Fig. 1 C). TLR9 was present in mouse MKs, pro-PLT–MKs, released pro-PLTs, and individual PLTs and showed punctate/granular expression that partly colocalized with VAMP 8 at each stage of PLT production (Fig. 1 D). SNAREs, such as VAMP 8, associate with PLT granules and may mediate TLR9 localization to nascent PLT tips during thrombocytopoiesis.
Figure 1.

MKs up-regulate Tlr9 transcript expression during pro-PLT production and express TLR9 in distinct granules that partially colocalize with VAMP 8 at every stage in thrombocytopoiesis. (A and B) Shown is a screen shot from Integrated Genome Browser of reads distributing to the Tlr9 (A) and Vamp8 (B) locus. The black bars represent piled up sequencing reads aligning to the genomic coordinates encoding the respective RNAs. More reads will align to more highly expressed RNA regions, and the height of the black bars correlates with RNA expression level. Below the read distributions are the RefSeq annotations: thick horizontal lines represent exons, and thin horizontal lines represent introns. Quantification was based on three round MK and four pro-PLT–MK replicates and is expressed in reads per kilobase of exon model per million mapped reads. (C) Intermediates of PLT production from murine fetal liver cell cultures were spun down onto poly-l-lysine–coated glass cover slides, permeabilized with 0.5% Triton X-100 for 5 min, and probed for TLR9 and either β1-tubulin or VAMP 8. Slides were examined by fluorescence microscopy, and image fluorescence intensity is normalized to the round MK fraction. Round MKs, pro-PLT–MKs, released pro-PLTs, and individual PLTs revealed distinct, punctuate/granular localization of TLR9 similar to that observed in whole-blood PLTs. β1-Tubulin antibodies were used to delineate the cell periphery and denote the different intermediate stages in PLT production. Insets (from top to bottom) represent a PLT, pre-PLT, and barbell pro-PLT. (D) Background fluorescence was subtracted, and image brightness/contrast was adjusted linearly for each micrograph to resolve individual granules. TLR9 showed significant colocalization with VAMP 8 in round MKs, along the pro-PLT shafts of pro-PLT–MKs, and within released pro-PLTs and individual PLTs. A Manders’ coefficient of 0.64 was calculated for VAMP 8 colocalization with TLR9 throughout the entirety of MK cell culture. Insets represent the magnified region outlined by the yellow box for each image.
Human PLTs sequester TLR9 and protein disulfide isomerase (PDI) in previously undescribed electron-dense tubular system-related T granules
To confirm that the majority of TLR9 in resting PLTs is expressed intracellularly, washed PLTs were spun down onto poly-l-lysine–coated glass cover slides, permeabilized, and probed for either TLR9 (Fig. 2 A, green) alone or TLR9 (Fig. 2 B, green) and β1-tubulin (Fig. 2 B, red) together. Immunofluorescence microscopy revealed that both human (Fig. 2 A) and mouse (Fig. S1) whole-blood PLTs demonstrate punctuate/granular expression of TLR9 mostly (69 ± 5%) along the cell periphery, adjacent to the plasma membrane. TLR9 localization to the cell edge was confirmed in human PLTs by colabeling with β1-tubulin to demarcate the PLT border (Fig. 2 B). Unlike TLRs 1–6, which are always expressed on the cell surface, TLR9 is typically expressed in macrophage or mast cell endosomes. Interestingly, TLR9 was shown not to colocalize with known specific markers of α granules (fibrinogen, CD62P, PDGF-B, VEGF, CD42a, and CD42b), dense granule (serotonin), lysosome (LAMP-1), or endosome (M6P and Syntaxin-13) protein markers (Fig. 3 A). To validate this observation, TLR9 labeling of whole-blood PLTs from human normal controls was compared with Gray PLT and Hermansky–Pudlack syndrome patients, which are devoid of α-granule and dense-granule content, respectively (Fig. 3 B). Punctate/granular expression of TLR9 in these samples was undiminished and suggests that TLR9 is not contained in PLT α or dense granules.
Figure 2.
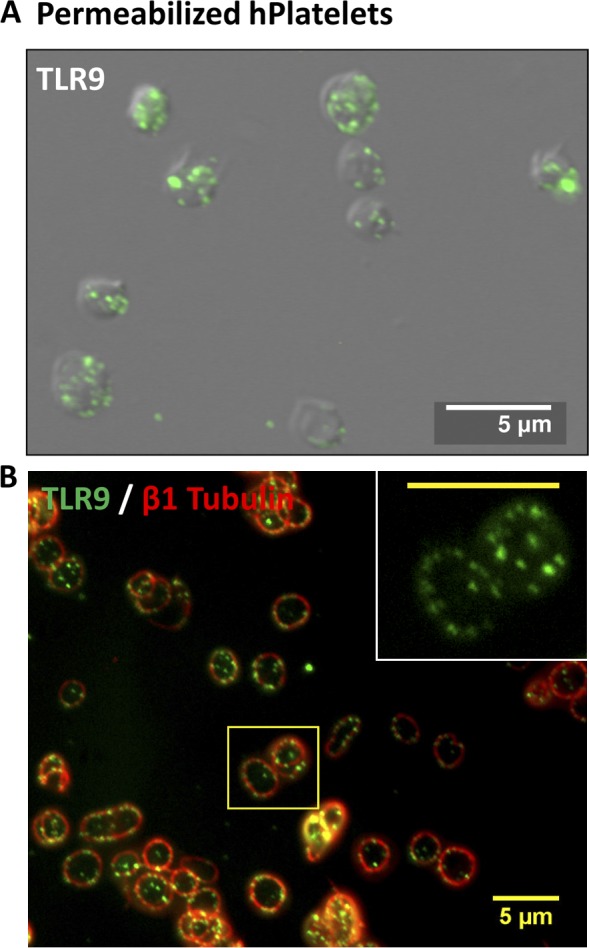
Human PLTs express TLR9 in distinct granules along the periphery of the cell, adjacent to the plasma membrane. (A and B) Washed human whole-blood PLTs were spun down onto poly-l-lysine–coated glass cover slides, permeabilized with 0.5% Triton X-100 for 5 min, and were probed for either TLR9 alone or TLR9 and β1-tubulin together. (A) Samples were examined by wide-field fluorescence microscopy and revealed peripheral labeling of TLR9 in distinct, punctate granules that localized mostly (69 ± 5%) along the cell periphery, adjacent to the plasma membrane. (B) TLR9 localization to the cell edge was confirmed in human PLTs by colabeling with β1-tubulin to demarcate the PLT border. Inset represents magnified region outlined by the yellow box. (inset) Bar, 5 µm.
Figure 3.

TLR9 does not colocalize with known PLT α granule, dense granule, lysosomal, or endosomal markers. Washed human whole-blood PLTs were spun down onto poly-l-lysine–coated glass cover slides, permeabilized with 0.5% Triton X-100 for 5 min, and probed for TLR9. (A) PLTs were colabeled for α granule (fibrinogen, CD62P, PDGF-B, VEGF, CD42a, and CD42b), dense granule (serotonin), lysosome (LAMP-1), or endosome (M6P and syntaxin-13) protein markers using two separate colors. Insets represent magnified regions outlined by the yellow boxes for each image. (B) TLR9 labeling of whole-blood PLTs from patients with Gray PLT and Hermansky–Pudlack syndromes was compared with human normal PLT controls. All samples were examined by wide-field fluorescence microscopy. (C) 2D intensity scatter plot analysis of image overlays reveal that, although TLR9 colocalizes well with PDI, it does not colocalize with either TLR7 or TLR8. These data suggest that TLR9 and PDI may be distributing to a unique intracellular body (T granule) underlying the plasma membrane in resting human PLTs. (insets) Bars, 2 µm.
In professional dendritic cells, TLR9 is distributed to intracellular endosomal compartments along with TLR7 and TLR8. PDI, by comparison, is highly expressed in the endoplasmic reticulum of endothelial cells and electron-dense tubular system-related compartments in PLTs (van Nispen Tot Pannerden et al., 2009). Interestingly, although PDI colocalized with TLR9 in resting human PLTs, TLR7 and TLR8 did not (Fig. 3 C and Videos 1 and 2). Representative maximum projection z series for TLR9 and PDI (Fig. 4 A) confirmed that TLR9 is distributing with PDI to a separate granular compartment.
Figure 4.

TLR9 colocalizes with PDI to electron-dense membrane-encapsulated regions adjacent the plasma membrane along the periphery of human PLTs. (A) Representative maximum projection z series for TLR9 and PDI by confocal immunofluorescence microscopy demonstrating colocalization. (B–E) Washed human whole-blood PLTs were fixed, frozen, and sectioned before mounting on Formvar carbon-coated copper grids. Ultrathin PLT sections were probed for TLR9, and bound antibody was labeled with immunogold. Samples were examined by electron microscopy and reveal the distribution of TLR9 (B), PDI (C), and colocalization of both (D and E) along the periphery of resting human PLTs. White arrows denote localization of TLR9 and PDI to limiting membrane of electron-dense regions adjacent the plasma membrane. (D and E) For colabeling, immunogold particles are 15 nm for PDI and 10 nm for TLR9.
To surpass the resolution limit of conventional fluorescence microscopy (200 nm) and because Gray PLT syndrome PLTs express some α-granule membrane proteins in normal amounts (Rosa et al., 1987; Maynard et al., 2010), TLR9 and PDI localization was examined by immunogold electron microscopy. PLT cryosections were probed for TLR9 (Fig. 4 B) and PDI (Fig. 4 C) separately and together (Fig. 4, D and E). TLR9 distributed to previously uncharacterized electron-dense granular compartments to which PDI was also shown to colocalize (Fig. 4, B–D). These were named T granules after TLR9 electron-dense tubular system-related granular compartments. TLR9/PDI labeling was associated with the limiting membrane, and T-granule identification was based on the colocalization of three or more immunogold molecules within a single electron-dense area (Fig. 4 B–E, denoted by white arrows). T granules had a mean cross-sectional area of 4,148 nm2, and there were a mean of eight T granules per PLT cross-sectional area (1,750,019 nm2) in 25 PLTs analyzed.
T-granule localization/plasma membrane fusion accompanies VAMP 7 and 8 reorganization on PLT activation
Because SNAREs mediate α-granule localization in PLTs, we hypothesized that these might also colocalize with T-granule reorganization during PLT activation and plasma membrane fusion. SNAREs are divided into t-SNAREs and v-SNAREs, which in PLTs include VAMPs 2, 3, 5, 7, and 8 (Graham et al., 2009). v-SNARE colocalization with TLR9 in resting and thrombin-activated human PLTs was determined by confocal immunofluorescence microscopy, and both risk ratio and Manders’ coefficients were calculated for each association. Representative figures are shown in Fig. 5, and results for colocalizations are summarized in Table 1. TLR9 did not associate with VAMPs 2 and 3 (not depicted) and shows poor association with VAMP 5, which was included as an internal threshold control (Fig. 5 A). Manders’ coefficients of 0.5 or greater were considered significant, and TLR9 showed partial colocalization with both VAMPs 7 and 8 in resting PLTs (Fig. 5, B and C). After PLT activation (spreading) on glass, VAMP 8 was redistributed to the PLT granulomere, whereas VAMPs 5 and 7 distributed to the PLT lamellipodium. TLR9 continued to show colocalization with VAMP 8 after spreading, which demonstrates that there are two major subpopulations of T granules in PLTs that can be differentially distributed on PLT activation. Localization of VAMPs to the PLT granulomere or lamellipodium on contact activation has been associated with granule membrane fusion with the plasma membrane or the open canalicular system, respectively (Flaumenhaft, 2003), and accounts for TLR9 surface expression.
Figure 5.

Human PLT TLR9 localization with VAMPs 5, 7, and 8 under resting conditions and when activated (spread) on glass surface. Samples were examined by confocal fluorescence microscopy. Image analysis was completed by using the JACoP (Just Another Colocalization Plugin) plugin for ImageJ as described in Table 1. Manders’ coefficients were used to compare the TLR9 relationship to the associated VAMP. (A) Micrographs demonstrating the relationship of VAMP 5 and TLR9. As demonstrated in the micrographs, in the resting PLT, ∼41% of TLR9 signal overlaps with that of VAMP 5. The portion of overlapping signal slightly increased in the adhered PLTs to 49%. (B) Additional micrographs demonstrating the relationship between VAMP 7 and TLR9. Image analysis confirms 82% of TLR9 overlapped with that of VAMP 7 in the resting PLT; however, upon spreading, the signal decreased to only 56%. (C) Micrographs of resting and spread PLTs probed with specific antiserum for VAMP 8 and TLR9. In the resting PLT, 77% of TLR9 signal overlapped with VAMP 8. Upon spreading, the signal decreased to 67% signal overlap.
Table 1.
Calculated risk ratio and Manders’ coefficients for each association of TLR9 with VAMPs 5, 7, and 8
| Resting | Spread (glass) | |||||
| Colocalization with TLR9 | Rr | M1 TLR9/VAMP | M2 VAMP/TLR9 | Rr | M1 TLR9/VAMP | M2 VAMP/TLR9 |
| VAMP 5 | 0.7 ± 0.1 | 0.4 ± 0.2 | 0.4 ± 0.2 | 0.5 ± 0.1 | 0.5 ± 0.2 | 0.14 ± 0.04 |
| VAMP 7 | 0.84 ± 0.02 | 0.82 ± 0.08 | 0.4 ± 0.1 | 0.69 ± 0.09 | 0.56 ± 0.08 | 0.32 ± 0.09 |
| VAMP 8 | 0.50 ± 0.03 | 0.77 ± 0.02 | 0.59 ± 0.02 | 0.4 ± 0.1 | 0.68 ± 0.04 | 0.4 ± 0.2 |
Calculated risk ratio (Rr) and Manders’ coefficients for each association of TLR9 with VAMPs 5, 7, and 8 in human whole-blood PLTs under resting conditions and when activated (spread) on glass surface. Manders’ coefficients of 0.5 or greater were considered significant and are highlighted in bold.
TLR9 is expressed on the PLT surface during activation
Unlike TLRs 1–6, TLR9 is exclusively expressed intracellularly in the professional dendritic cells (Barton et al., 2006). To determine whether PLTs express TLR9 on the plasma membrane under resting conditions and whether activation with a strong PLT agonist will cause TLR9 to relocate to the PLT surface, human PLTs were collected from whole blood and incubated with thrombin. Thrombin is a key regulator of the coagulation cascade and activates PLTs through the protease-activated receptors 1 and 4 in humans. Flow cytometry and immunofluorescence microscopy were used to measure the surface expression of TLR9 in human PLTs under resting conditions and after PLT activation (Fig. S3). Surprisingly, resting PLTs expressed basal levels of surface TLR9 that became significantly increased after thrombin activation, suggesting that although the majority of TLR9 is expressed intracellularly, some of it relocates to the plasma membrane on agonist exposure. Although it is unknown what local concentrations of bacterial DNA are achieved in vivo, increased TLR9 surface expression in thrombin-activated PLTs corresponded with increased surface binding of the synthetic TLR9 ligand, unmethylated CpG ODN, when ≥5 µM, and was therefore used at this concentration for all subsequent experiments (Fig. S3 A and Fig. S5). TLR9 surface expression was also regulated by PLT activation through other common PLT agonists that stimulate PLT activation and granule release through separate signaling pathways. These included ADP and type IV collagen, which were chosen because they represent well characterized, physiologically relevant agonists, PMA, which is a potent secretagogue used in both PLTs and leukocytes to evaluate granule exocytosis, and the GPVI agonist collagen-related peptide (CRP), given the importance of the GPVI receptor in PLT function. These data demonstrate that human PLTs may need to be primed to respond to bacterial/viral PAMPs in whole blood (Fig. 6). Moreover, although the direct addition of type IV collagen to resting PLTs resulted in much higher levels of TLR9 surface expression than did thrombin, these results were not matched with a significant increase in CD62P surface expression and revealed lower levels of CD61 surface expression relative to CRP controls, confirming that TLR9 and CD62P/CD61 localize to separate organelles. Conversely, CRP-mediated PLT activation resulted in significantly higher levels of both CD62P and CD61 surface expression than did type IV collagen but were met with relatively lower levels of TLR9 surface expression, as confirmed by quantitative immunofluorescence microscopy (Fig. S4).
Figure 6.

Human PLTs regulate surface expression of TLR9 on activation with select agonists. Human PLTs were collected from whole blood and examined under resting conditions or after activation with 1 mU/µl thrombin, 1 µM PMA, 3 µg/ml CRP, 20 µM ADP, or 50 µg/ml mouse type IV collagen for 5 min at 37°C. Samples were probed for TLR9 and either CD62P or CD61. PLT mean fluorescence intensity (relative surface expression of targeted receptor) was determined by flow cytometry for at least three different samples, and data were subject to one-way ANOVA and Tukey HSD analysis. Error bars represent one standard deviation about the mean for at least three independent samples. (A) Human PLT activation with all listed agonists results in increased surface expression of TLR9 relative to resting control. Strikingly, TLR9 surface expression on PLT activation did not correlate with CD62P/CD61 expression for the agonists tested and suggests that TLR9 and CD62P/CD61 may localize to separate granules. (B, left) Representative forward scatter versus side scatter dot plots highlight characteristic changes in PLT morphology on activation with the listed agonists relative to resting control. (right) Representative histograms demonstrate a shift in mean fluorescence intensity for TLR9 and CD62P/CD61 on PLT activation. Outlines represent PLT gate used for sample thresholding by forward and side scatter. Mean fluorescence intensity for resting PLTs is represented in gray. Mean fluorescence intensity for agonist-activated PLTs is represented in red.
TLR9 regulates DNA sequestration and CD62P surface expression in PLTs
Unmethylated ODNs, including a CpG motif, can mimic the effects of bacterial DNA, inducing B cell proliferation and activating cells of the myeloid lineage. We hypothesized that the addition of type C CpG to washed human PLTs may induce PLT activation and possibly result in increased surface expression of TLR9 and CD62P and ODN sequestration. Although incubation of resting human PLTs with synthetic unmethylated type C CpG did not cause PLT shape change (Fig. S2), it did result in a 37 ± 13% increase in TLR9 and 34 ± 15% increase in CD62P surface expression followed by a 31 ± 15% increase in FITC-conjugated type C CpG sequestration over 20 min (Fig. 7, A and C). Surprisingly, when TLR9 surface expression was specifically up-regulated by preincubating PLTs with type IV collagen, subsequent incubation with type C CpG resulted in 100% of PLTs demonstrating increased type C CpG sequestration, CD62P surface expression, and PLT clumping within 30 s of addition (Fig. 7, B and C; and Videos 3 and 4). This was inhibited by preincubating the PLT culture with 20 µM IRAK-1/4 (interleukin-1 receptor-associated kinase-1/4 inhibitor) for 1 h before collagen IV incubation and type C CpG addition (Videos 3 and 4). IRAK-1 and IRAK-4 are protein kinases that mediate signaling by TIR (Toll/IL1/Plant R) domain–containing receptors, including the IL-1, IL-8, and TLRs (Song et al., 2009). Pharmacological inhibition of both kinases blocked PLT clumping by type C CpG in the presence of type IV collagen and reveals downstream signaling effectors of ODN-mediated PLT activation.
Figure 7.

TLR9 signaling results in type C CpG sequestration, increased CD62P surface expression, and PLT clumping. (A and B) Flow cytometric analysis showing representative forward versus side scatter profiles of human washed PLTs under resting conditions and after incubation with synthetic unmethylated type C CpG ODNs (characteristic of bacterial/viral DNA) before (A) and after (B) type IV collagen preincubation. Outlines represent PLT gate used for sample thresholding by forward and side scatter. (C) Quantification of the percentage of PLTs expressing TLR9, CD62P, and type C CpG was normalized to resting levels to resolve the difference on agonist exposure over time. Incubation of resting washed human PLTs with type C CpG ODNs resulted in a 40% increase in TLR9 surface expression followed by a 30% increased type C CpG sequestration and CD62P surface expression over 20 min. (D) Type IV collagen preincubation resulted in considerably increased type C CpG sequestration, CD62P surface expression above resting PLT controls, and significant PLT clumping within 30 s of type C CpG addition. (E) Mouse PLTs show a more pronounced ODN sequestration and comparable CD62P expression after type C CpG incubation. Unlike in human PLTs, TLR9 surface expression is not changed in mice. Although TLR9 KO mice show reduced levels of ODN sequestration and CD62P surface expression 20 min after type C CpG addition, preincubation with type IV collagen still resulted in immediate PLT clumping when type C CpG was added (not depicted). Statistical significance for marked pairings was established using a one-tailed Student’s t test for paired samples (*, P < 0.05; **, P < 0.01). Error bars represent one standard deviation about the mean.
Unlike in human PLTs that contain ∼0.5 RPKM TLR9 RNA, TLR9 surface expression was not changed in mice after type C CpG incubation, and deep sequencing of mouse PLTs has shown that they do not contain detectable levels of TLR9 RNA (Rowley et al., 2011). Interestingly, although preincubation with type IV collagen still resulted in immediate PLT clumping when type C CpG was added (unpublished data), TLR9 knockout (KO) mice show reduced levels of ODN sequestration and CD62P surface expression relative to isotype-matched wild-type controls. To confirm that FITC-conjugated type C CpG behaves like nonconjugated type C CpG with regard to TLR9 binding (specificity control), resting/thrombin-activated human PLTs were incubated with 5 µM FITC-conjugated type C CpG and decreasing concentrations of unlabeled (cold) type C CpG and analyzed by flow cytometry (Fig. S5). Thrombin-activated PLTs showed fluorescence profiles that correlated exactly with the relative proportion of FITC-conjugated type C CpG in the culture. Resting human PLTs showed fluorescence profiles that were comparable with the 20-µM unlabeled type C CpG control at all the concentrations tested. To determine whether circulating ODNs also result in increased thrombus formation, washed human whole-blood PLTs and PLT-rich plasma (PRP) were pretreated with 5 µM control ODN, type C CpG ODN, or a vehicle control and perfused at a shear rate of 200 s−1 (flow rate of 18.7 µl/min) over a surface coated with type IV collagen or type I collagen for 10 min (Fig. 8, A–C; and Videos 5–11). Although addition of ODN to PLTs in the presence of type IV collagen resulted in PLT clumping and adhesion to the flow chamber surface (Fig. 8, A and B), these did not form thrombi as compared with type I collagen (Fig. 8 C). Addition of type C CpG did not result in increased thrombus formation relative to the ODN control in PRP on the type I collagen-coated surface.
Figure 8.
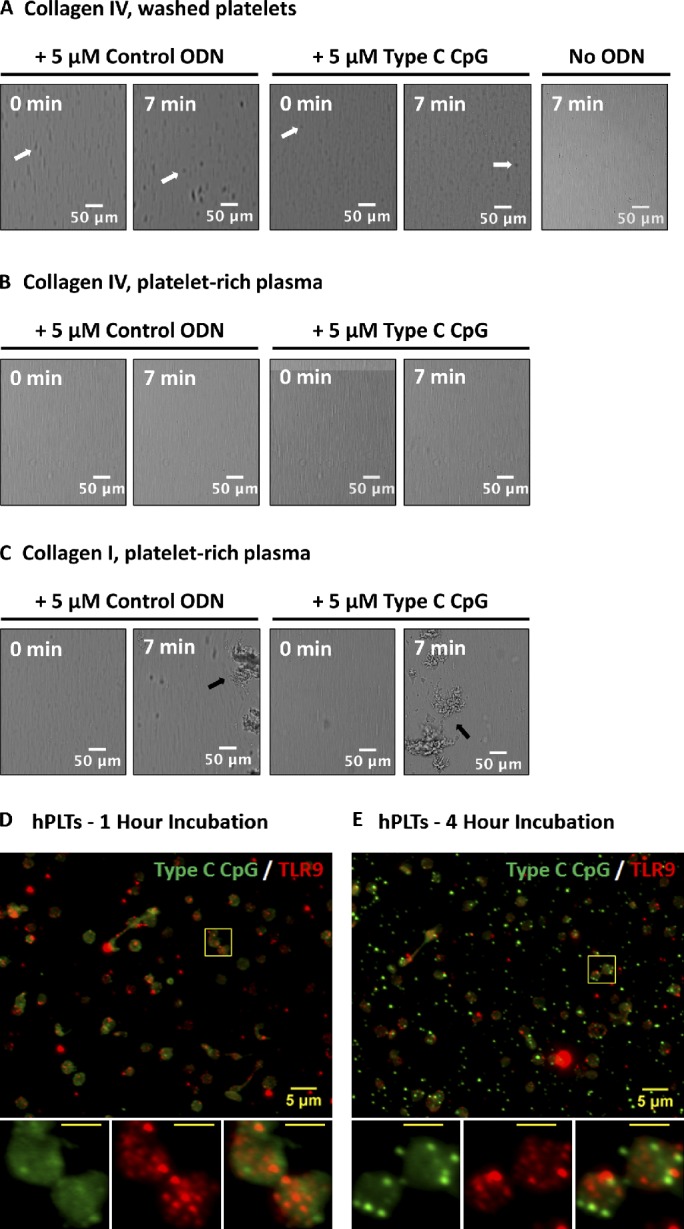
ODN-activated PLTs do not form thrombi on collagen type IV and endocytose type C CpG to distinct granules that do not colocalize with TLR9. (A–C) Washed human whole-blood PLTs (A) and PRP (B) were pretreated with 5 µM control ODN, type C CpG ODN, or a vehicle control and perfused at a shear rate of 200 s−1 (flow rate of 18.7 µl/min) over a surface coated with type IV collagen or type I collagen (C) for 10 min. Although addition of ODN to PLTs resulted in PLT clumping in the presence of type IV collagen (white arrows), these did not form thrombi as compared with type I collagen (positive control, black arrows). Addition of type C CpG did not result in increased thrombus formation relative to the ODN control in PRP on the type I collagen-coated surface. (D) PLTs were incubated with FITC-conjugated type C CpG at 37°C and 5% CO2 for a period of ≤4 h. PLTs were subsequently spun down onto poly-l-lysine–coated glass cover slides and probed for TLR9. After 1 h of incubation, the majority of FITC-conjugated type C CpG was associated with the PLT surface and did not colocalize with TLR9. (E) Samples were examined by wide-field fluorescence microscopy. After 4 h of incubation, FITC-conjugated type C CpG became endocytosed by resting human PLTs into distinct granules that showed minimal colocalization with TLR9. Insets represent (from left to right) type C CpG labeling, TLR9 labeling, and colabeling of magnified region outlined by the yellow boxes in the images. (insets) Bars, 2 µm.
In monocytes, macrophages, and plasmacytoid dendritic cells, TLR9 localizes to intracellular membranes, such as those of endosomes or phagosomes (Hennessy et al., 2010). This permits TLR9 to respond to nucleic acids from bacterium/virus that have been endocytosed/phagocytosed by the cell, while preventing recognition of self-DNA (Barton et al., 2006). To test whether this is the case in human PLTs, samples were incubated with FITC-conjugated type C CpG at 37°C and 5% CO2 for a period of ≤4 h. PLTs were subsequently spun down onto poly-l-lysine–coated glass cover slides and probed for TLR9 as described in the Immunofluorescence microscopy section of Materials and methods. Fig. 8 (D and E) shows that within the first hour of incubation the majority of type C CpG was sequestered to the PLT surface. Although prolonged exposure of human PLTs to FITC–type C CpG (4 h) induced endocytosis of the ODN, these showed minimal colocalization with intracellular PLT TLR9.
Discussion
Here, we show that TLR9 mRNA is expressed by mature MKs and becomes specifically up-regulated during pro-PLT production. Differential mRNA expression has previously been shown for TIMP-3 (Cecchetti et al., 2011) and suggests that TLR9 mRNA is selectively withheld or rapidly degraded in circulating PLTs. We speculate that increased TLR9 protein translation at this stage is necessary to package TLR9 protein into newly generated PLTs. VAMP 8 colocalizes with TLR9 protein throughout pro-PLT maturation and may be responsible for distributing T granules to PLT-sized swellings, where it is distributed with PDI to unique electron-dense membrane-delimited intracellular compartments we have named T granules. Localization of PDI to T granules suggests they may be derived from the endoplasmic reticulum of MKs and therefore related to the dense tubular system in PLTs (White and Gerrard, 1976). PDI facilitates the formation of disulfide bonds and proper folding of newly synthesized proteins. Secretion and targeting of PDI to the surface of activated PLTs is thought to contribute to isomerase activity, affect adhesion to CD41/61, α2-β1, and GPIb, and regulate the procoagulant activity of tissue factor (Chen et al., 1992, 1995; Essex et al., 1995; Burgess et al., 2000; Lahav et al., 2003; Ahamed et al., 2006). T granules can be organized in two major subpopulations that differentially associate with either VAMP 7 or VAMP 8. Although thought to reside exclusively in the endosomes of monocytes, macrophages, and plasmacytoid dendritic cells, TLR9 surface expression is significantly increased in PLTs after incubation with thrombin, ADP, PMA, CRP, and type IV collagen, demonstrating partial activation-mediated granule release. SNAREs represent the core of the fusion machinery, and the distribution and association of v-SNAREs, such as VAMP 8, with t-SNAREs provides the basis for α-granule localization and secretion (Blair and Flaumenhaft, 2009). Because TLR9 partly redistributes to the plasma membrane on contact activation with glass, T-granule colocalization with VAMP 8 and reposition to the PLT granulomere in spread PLTs suggest that increased TLR9 surface expression occurs through VAMP 8–mediated T-granule fusion with the open canalicular system (Flaumenhaft, 2003).
TLRs functionally modulate innate immunity by recognizing PAMPs on invading microorganisms (Hemmi et al., 2000). This is particularly true of bacterial/viral DNA, which depend on the presence of unmethylated CpG ODNs. By comparison, mammalian DNA has a lower frequency of CpG ODNs, is methylated, and does not have immune-stimulatory activity (Krieg, 1996; Lipford et al., 1998; Yamamoto et al., 2000). TLR9 has been identified as the receptor involved in the recognition of immunostimulatory CpG motifs (Hemmi et al., 2000; Bauer et al., 2001) and is uniquely expressed in peripheral granules in resting PLTs and on the human PLT surface after activation. Immunostimulatory CpG motifs have been implicated as a major contributor to the acute inflammatory response associated with nonviral vectors, most prominently seen after systemic delivery of cationic lipid–plasmid DNA complexes (Zhao et al., 2004). Unmethylated CpGs have been shown to stimulate B cell proliferation and macrophage activation and induce dendritic cell maturation (Krieg et al., 1995; Stacey et al., 1996; Sparwasser et al., 1998). Incubation of resting PLTs with synthetic unmethylated type C CpG ODNs (characteristic of bacterial/viral DNA) resulted in increased surface expression of TLR9 in humans and increased CD62P surface expression and type C CpG sequestration over 20 min in both humans and mice. Indeed, TLR9 KO mice sequester ∼50% less type C CpG and have significantly reduced levels of CD62P surface expression relative to isotype-matched wild-type controls.
Upon vascular damage, PLTs are recruited to the site of injury where they adhere to exposed basement membrane, become activated, and contribute to clot formation. The basement membrane is particularly rich in type IV collagen and PLT recruitment to a site of vascular injury may serve to sensitize the PLT to bacteria/virus DNA. Interestingly, when TLR9 surface expression was specifically increased by preincubating PLTs with type IV collagen, subsequent incubation with type C CpG resulted in considerably enhanced type C CpG sequestration, CD62P surface expression, and PLT clumping within 30 s of addition but did not result in thrombi formation. These results imply that PLTs must be primed to express TLR9 on their surface before signal transduction through TLR9 and provide a mechanism for PLT regulation of the immune response to infection in human whole blood by sequestering bacterial/viral DNA and marking themselves for clearance.
Interestingly, mouse PLTs did not show increased surface expression of TLR9 on addition of the ODN. Indeed, human PLTs express several surface receptors absent in mice (e.g., PAR1). With regard to TLR expression, human myeloid dendritic cells express functional TLR8, whereas mice do not. Conversely, although both myeloid and plasmacytoid dendritic cells in mice express intracellular TLR7 and TLR9, these are not present in human myeloid dendritic cells. Nevertheless, TLR9 KO mouse PLTs still aggregated when type C CpG was added after preincubation with type IV collagen, which substantiates the previously reported prevalence of non-TLR9–mediated effects of CpG. Indeed, although the absence of CpG recognition by TLR9 significantly reduces acute inflammatory responses to systematically delivered cationic lipid–plasmid DNA complexes, it does not eliminate them, and TLR9 KO mice still exhibit a pronounced loss of lymphocytes and PLTs (Zhao et al., 2004). Collectively, these findings suggest that signaling through TLR9 does not exclusively contribute to PLT activation when exposed to bacterial/viral DNA and predict the presence of at least one other yet-uncharacterized receptor of CpG on the PLT surface. Even so, increased surface expression of TLR9 in human PLTs, and type C CpG sequestration may help regulate circulating levels of bacterial DNA in whole blood and thus control the inflammatory response after bacterial cell lysis. The interaction of bacteria with human PLTs plays an important role in the pathogenesis of cardiovascular infection, and serious complications of bacteremia include life-threatening infective endocarditis, disseminated intravascular coagulation, immune thrombocytopenia purpura, and increased risk of myocardial infarction (Fitzgerald et al., 2006). A better understanding of the role TLRs play in regulating PLT activation will lead to targeted therapies for treating and preventing serious cardiovascular infections. Moreover, the development of small molecule inhibitors of PLT interactions with specific bacterial components have the advantage of bypassing present antibiotic resistance mechanisms and may be ideal for prophylaxis of infection in susceptible individuals. (Fitzgerald et al., 2006).
Materials and methods
Human/murine whole-blood PLTs
Human blood was obtained by venipuncture from healthy laboratory volunteers, a Gray PLT syndrome patient, and a Hermansky–Pudlack syndrome patient as previously described (Hartwig and DeSisto, 1991). Collections were performed in accordance with ethics regulation with International Review Board approval, and informed consent was provided according to the Declaration of Helsinki. Murine blood was obtained by retro orbital bleed into 0.1 vol Aster–Jandl anticoagulant from anesthetized mice as previously described (Hoffmeister et al., 2003). Gray PLT syndrome patient whole blood was provided by A. Michelson (Harvard Medical School, Boston, MA). Hermansky–Pudlack syndrome patient whole blood was provided by R. Flaumenhaft (Harvard Medical School, Boston, MA). TLR9 KO mice were provided by D. Golenbock and B. Caetano (University of Massachusetts Medical School, Worcester, MA).
MK suspension cultures
Mouse fetal liver cells were collected from wild-type CD1 mice (Charles River) on embryonic day 13.5 and cultured at 37°C and 5% CO2 in the presence of 0.1 µg/ml purified recombinant mouse c-Mpl ligand for 5 d. Round MKs, pro-PLT–MKs, and released pro-PLTs were isolated by BSA gradient sedimentation and cultured separately as previously described (Thon et al., 2010). In brief, fetal liver cell cultures were layered on a single-step gradient (1.5–3.0% BSA) on culture day 4, and MKs were allowed to sediment for 30 min. The MK pellet was then resuspended in fresh media and cultured for an additional 24 h during which pro-PLT production was readily observed and layered on a second single-step gradient (1.5–3.0% BSA) on culture day 5. MKs were allowed to sediment for 30 min, and MKs were isolated from the pelleted fraction, pro-PLT–producing MKs were isolated from the BSA fraction, and released pro-PLTs and PLTs were isolated from the upper (culture media) fraction. Preparation of polyadenylated RNA for sequencing on the Genome Analyzer IIx (Illumina) followed by alignment and analysis was performed as previously described (Rowley et al., 2011). In brief, cells were lysed in TRIZOL (Invitrogen) or in mirVana (Ambion) lysis buffer. RNA was isolated as described by each manufacturer, resuspended in RNase-free distilled H2O, and treated with reagent (TURBO DNase; Ambion). The DNase-treated RNA was precipitated with 3× vol ethanol and 1/10 vol sodium acetate followed by rigorous 70% ethanol washes. RNA integrity was evaluated on a bioanalyzer (2100 Bioanalyzer; Agilent Technologies), and samples with RNA integrity numbers >7.0 were used for sequencing. Poly(A)-tailed RNA was prepared by the University of Utah Core facility using the sample prep kit (mRNA-Seq Sample Prep; Illumina). All experiments complied with institutional guidelines approved by the Children’s Hospital animal care and use committee and the Institutional Animal Care and Use Committee.
Flow cytometry
Human/mouse PLTs were collected from whole blood and either (a) washed and examined under resting conditions or after activation (5 min at 37°C) with 1 mU/µl thrombin (Roche) or (b) isolated from PRP and examined under resting conditions or activation (5 min at 37°C) with 1 µM PMA (Sigma-Aldrich), 3 µg/ml CRP (Falet et al., 2010), 20 µM ADP (Bio/Data Corporation), or 50 µg/ml mouse type IV collagen (Gibco). Samples were probed with phycoerythrin (PE)-conjugated mouse anti-TLR9 antibodies (Imgenex) and one of the following: FITC-conjugated type C CpG (InvivoGen), FITC-conjugated mouse anti-CD62P antibodies, or FITC-conjugated mouse anti-CD61antibodies (BD) and run on a flow cytometer (FACSCalibur; BD). PLTs were gated by their characteristic forward and side scattering as they passed through the detector, and their total fluorescence intensity was calculated after subtraction of a PE- or FITC-conjugated IgG antibody specificity control (BD). Analyses of PLT mean fluorescence intensity were performed for at least three different samples. Where appropriate, data were subject to one-way analysis of variance (ANOVA) and Tukey honestly significant difference (HSD) analysis. Error bars represent one standard deviation about the mean for at least three independent samples.
For examination of PLT activation, TLR9 and CD62P surface expression, and type C CpG sequestration, washed human PLTs were incubated for 0, 5, 10, 20, 40, and 60 min at 37°C with either 5 µM FITC-conjugated type C CpG (InvivoGen) or a vector control. Samples were immediately fixed in 4% formaldehyde and probed with PE-conjugated mouse anti-TLR9 antibodies (Imgenex) or PE-conjugated mouse anti-CD61P antibodies (BD) before being run on a flow cytometer (FACSCalibur). PLTs were gated by their characteristic forward and side scattering as they passed through the detector, and the percentage of fluorescently conjugated PLT-sized events was calculated after subtraction of unlabeled type C CpG and FITC/PE-conjugated IgG antibody specificity controls. For type IV collagen activation experiments, PLTs were preincubated with 50 µg/ml type IV collagen for 5 min at 37°C before addition of type C CpG. Analyses of TLR9 and CD62P surface expression and type C CpG sequestration were performed for at least four different samples. Where appropriate, data were subject to one-way ANOVA and Tukey HSD analysis. Error bars represent one standard deviation about the mean for at least three independent samples.
Immunofluorescence microscopy
MKs, pro-PLT–MKs, released pro-PLTs, and whole-blood PLTs were isolated as described in MK suspension cultures. For type C CpG endocytosis experiments human whole blood–washed PLTs were incubated with FITC-conjugated type C CpG (InvivoGen) at 37°C and 5% CO2 for a period of ≤4 h. In brief, samples were fixed in 4% formaldehyde and centrifuged onto 1 µg/ml poly-l-lysine–coated coverslips, permeabilized with 0.5% Triton X-100, and blocked in immunofluorescence blocking buffer (0.5 g BSA, 0.25 ml of 10% sodium azide, and 5 ml FCS in 50 ml PBS) overnight before antibody labeling (Italiano et al., 2003). To demarcate permeabilized cells, samples were incubated with a rabbit polyclonal primary antibody for human or mouse β1-tubulin generated against the C-terminal peptide sequence CKAVLEEDEEVTEEAEMEPEDKGH and LEDSEEDAEEAEVEAEDKDH, respectively (Genemed Synthesis, Inc.). For granule localization, samples were incubated with primary antibodies against TLR9 (Imgenex), VEGF, fibrinogen, CD62P, CD42a, PDGF-B (Santa Cruz Biotechnology, Inc.), CD42b (Dako), M6P, LAMP-1 (Abcam), Syntaxin-13 (a gift from A. Peden, University of Cambridge, Cambridge, England, UK), serotonin (EMD Millipore), or VAMPs 5/7/8 (Novus Biologicals) alone or in combination. All samples were treated with a secondary goat anti–rabbit or mouse antibody conjugated to an Alexa Fluor 488 or 568 nm (Invitrogen; Molecular Probes). As background controls, slides were incubated with the appropriate secondary antibody alone, and all images were adjusted to account for nonspecific binding of antibodies. For granule localization experiments with known α or dense granule, lysosomal, and endosomal markers, samples were examined with a microscope (Axiovert 200; Carl Zeiss) equipped with a 63×, NA 1.4 Plan-Apochromat oil immersion objective, and images were obtained using a charged-coupled device (CCD) camera (ORCA-ER; Hamamatsu Photonics). Images were analyzed using the MetaMorph image analysis software (Molecular Devices) and ImageJ (National Institutes of Health).
For colocalization experiments with PDI, TLRs 7 and 8, and VAMPs 5, 7, and 8, 4.0 × 106 washed PLTs were spun down onto poly-l-lysine–coated cover slides in either 4% formaldehyde (resting PLTs) or in PLT resuspension buffer (10 mM Hepes, 140 mM NaCl, 3 mM KCl, 0.5 mM MgCl2, 5 mM NaHCO3, and 10 mM glucose, pH 7.4) and allowed to spread on a glass slide for 15 min at room temperature before fixation (spread PLTs). Samples were permeabilized with 0.5% Triton X-100 and blocked in immunofluorescence blocking buffer overnight before antibody labeling and then incubated with IgG matching the primary antibody’s species to block nonspecific binding. PLTs were subsequently probed with primary antibodies against TLR9 (Imgenex) and PDI (Abcam), TLR7 (Abcam), TLR8 (Abcam), VAMP 5 (Abcam), VAMP 7 (SYBL1; Abcam), or VAMP 8 (Synaptic Systems). Fluorescence images were visualized using a confocal microscope (BX62; Olympus) equipped with a 60×, NA 1.42 Plan-Apochromat oil immersion objective and captured with a cooled CCD camera (ORCA-ER). Image acquisitions were controlled by iVision-Mac (BioVision Technologies). Additional images were acquired by a confocal laser-scanning microscope with a TCS SP2 (for PDI, TLR7, and TLR8; Leica) or LSM 510 Meta system (for VAMPs 5, 7, and 8; Carl Zeiss). Fluorescence images of two colors were captured sequentially using laser lines at 488 nm (for Alexa Fluor 488) and 561 nm (for Alexa Fluor 568). Series of x-y images were collected along the z axis at 0.244-µm intervals using a Plan-Apochromat 63×, NA 1.4 oil immersion objective using an optical zoom of 2×. Image analysis and colocalization values were generated using ImageJ and Volocity (PerkinElmer) software. For PDI, TLR7, and TLR8 colocalization experiments, three individual images were averaged to generate a representative image for each sample plane. At least 11 averaged images were collected at 0.244-µm steps along the z axis of each sample for both fluorescence channels, and Pearson’s correlation coefficients above threshold were calculated using the Colocalization Threshold macro written by T. Collins and W. Rasband (National Institutes of Health, Bethesda, MD) for ImageJ.
Immunogold electron microscopy
Rapid-freeze immunogold electron microscopy of washed human PLTs was performed as previously described (Italiano et al., 1999). In brief, human PLTs were fixed with 1.25% paraformaldehyde, 0.03% picric acid, and 2.5% glutaraldehyde in 0.1 M cacodylate buffer, pH 7.4. Ultrathin sections were probed for TLR9 (Imgenex) and labeled with protein A–gold (British Biocell International) before staining/embedding. Grids were examined with an electron microscope (G2 Spirit BioTwin; Tecnai) at an accelerating voltage of 80 kV. Images were recorded with an Advanced Microscopy Techniques 2k CCD camera using Advanced Microscopy Techniques digital acquisition and analysis software.
Differential interference contrast live-cell imaging of PLT clumping
Washed human PLTs were transferred onto video chambers maintained at 37°C. Cells were viewed on an inverted microscope (TE-200; Nikon) equipped with a 63× oil immersion objective, and images were obtained using a CCD camera (ORCA-ER). Videos were prepared using the MetaMorph software package and ImageJ. Pictures of PLTs cultured with either 20 µM IRAK-1/4 for 1 h or a vehicle control and 50 µg/ml type IV collagen were captured at 30-s intervals over a course of 20 min. 5 µM type C CpG was then added to the PLT culture, and images were captured for an additional 20 min. Experiments were performed for at least three different samples.
Perfusion analysis of PLT thrombus formation
Washed human whole-blood PLTs or PRP were recalcified with 5 mM CaCl2 and pretreated with 5 µM of control ODN, type C CpG ODN, or a vehicle control at 37°C for 10 min. Samples were perfused at a rate of 18.7 µl/min over a 300 µg/ml type IV collagen-coated or type I collagen-coated (Takeda Pharmaceuticals International GmbH) perfusion chambers (0.1 µ-Slide IV; ibidi) for 10 min. Images were collected on an inverted microscope (Axiovert 200) with a monochrome camera (AxioCam MRm; Carl Zeiss) and processed using the AxioVision LE software package (Carl Zeiss).
Preparation of photomicrographs
The digital images produced in MetaMorph and AxioVision LE were assembled into composite images by using ImageJ and Photoshop CS3 (Adobe). Dividing lines explicitly separate different images or separate regions of the same image. No specific features within an image were enhanced, obscured, moved, removed, or introduced, and adjustments made to the brightness, contrast, and color balance were linearly applied to the whole image.
Online supplemental material
Fig. S1 shows surface expression of TLR9 and binding to type C CpG in resting and thrombin-activated human PLTs. Fig. S2 shows that increased surface expression of TLR9 in human whole-blood PLTs is not associated with increased surface expression of CD61. Fig. S3 shows that murine PLTs express TLR9 in distinct granules along the periphery of the cell, adjacent to the plasma membrane. Fig. S4 shows that incubation of resting human PLTs with type C CpG does not affect overall PLT morphology or TLR9 localization and expression. Fig. S5 shows that FITC-conjugated type C CpG behaves like nonconjugated type C CpG with regard to TLR9 binding. Video 1 shows TLR9 localization in resting human PLTs. Video 2 shows PDI localization in resting human PLTs. Video 3 shows that TLR9 PLT surface expression results in type C CpG-mediated PLT clumping. Video 4 shows that type C CpG-mediated PLT clumping is inhibited by IRAK-1/4. Video 5 shows that vehicle control-treated washed PLTs do not form thrombi on collagen type IV. Video 6 shows that type C CpG ODN-activated washed PLTs do not form thrombi on collagen type IV. Video 7 shows that control ODN-activated washed PLTs do not form thrombi on collagen type IV. Video 8 shows that type C CpG ODN-activated PRP PLTs do not form thrombi on collagen type IV. Video 9 shows that control ODN-activated PRP PLTs do not form thrombi on collagen type IV. Video 10 shows that type C CpG ODN-activated PRP PLTs form thrombi on collagen type I. Video 11 shows that control ODN-activated PRP PLTs form thrombi on collagen type I. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.201111136/DC1.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge Dr. Alan Michelson for access to PLTs from Gray PLT syndrome patients and Drs. Douglas Golenbock and Braulia Caetano at the University of Massachusetts Medical School for access to their TLR9 KO mice.
This work was supported in part by the National Institutes of Health grant Hl68130 (to J.E. Italiano Jr.). J.E. Italiano Jr. is an American Society of Hematology Junior Faculty Scholar. J.N. Thon is an American Society of Hematology Scholar. T.A. Fuchs is supported by the National Institutes of Health NHLBI R01 grant HL102101 (to D.D. Wagner).
Footnotes
Abbreviations used in this paper:
- ANOVA
- analysis of variance
- CCD
- charge-coupled device
- CRP
- collagen-related peptide
- HSD
- honestly significant difference
- KO
- knockout
- MK
- megakaryocyte
- ODN
- oligodeoxynucleotide
- PAMP
- pathogen-associated molecular pattern
- PDI
- protein disulfide isomerase
- PE
- phycoerythrin
- PLT
- platelet
- PRP
- PLT-rich plasma
- TLR
- toll-like receptor
References
- Ahamed J., Versteeg H.H., Kerver M., Chen V.M., Mueller B.M., Hogg P.J., Ruf W. 2006. Disulfide isomerization switches tissue factor from coagulation to cell signaling. Proc. Natl. Acad. Sci. USA. 103:13932–13937 10.1073/pnas.0606411103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andonegui G., Kerfoot S.M., McNagny K., Ebbert K.V., Patel K.D., Kubes P. 2005. Platelets express functional Toll-like receptor-4. Blood. 106:2417–2423 10.1182/blood-2005-03-0916 [DOI] [PubMed] [Google Scholar]
- Aslam R., Speck E.R., Kim M., Crow A.R., Bang K.W., Nestel F.P., Ni H., Lazarus A.H., Freedman J., Semple J.W. 2006. Platelet Toll-like receptor expression modulates lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-alpha production in vivo. Blood. 107:637–641 10.1182/blood-2005-06-2202 [DOI] [PubMed] [Google Scholar]
- Barton G.M., Kagan J.C., Medzhitov R. 2006. Intracellular localization of Toll-like receptor 9 prevents recognition of self DNA but facilitates access to viral DNA. Nat. Immunol. 7:49–56 10.1038/ni1280 [DOI] [PubMed] [Google Scholar]
- Bauer S., Kirschning C.J., Häcker H., Redecke V., Hausmann S., Akira S., Wagner H., Lipford G.B. 2001. Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc. Natl. Acad. Sci. USA. 98:9237–9242 10.1073/pnas.161293498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair P., Flaumenhaft R. 2009. Platelet alpha-granules: basic biology and clinical correlates. Blood Rev. 23:177–189 10.1016/j.blre.2009.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess J.K., Hotchkiss K.A., Suter C., Dudman N.P., Szöllösi J., Chesterman C.N., Chong B.H., Hogg P.J. 2000. Physical proximity and functional association of glycoprotein 1balpha and protein-disulfide isomerase on the platelet plasma membrane. J. Biol. Chem. 275:9758–9766 10.1074/jbc.275.13.9758 [DOI] [PubMed] [Google Scholar]
- Cecchetti L., Tolley N.D., Michetti N., Bury L., Weyrich A.S., Gresele P. 2011. Megakaryocytes differentially sort mRNAs for matrix metalloproteinases and their inhibitors into platelets: a mechanism for regulating synthetic events. Blood. 118:1903–1911 10.1182/blood-2010-12-324517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K., Lin Y., Detwiler T.C. 1992. Protein disulfide isomerase activity is released by activated platelets. Blood. 79:2226–2228 [PubMed] [Google Scholar]
- Chen K., Detwiler T.C., Essex D.W. 1995. Characterization of protein disulphide isomerase released from activated platelets. Br. J. Haematol. 90:425–431 10.1111/j.1365-2141.1995.tb05169.x [DOI] [PubMed] [Google Scholar]
- Clark S.R., Ma A.C., Tavener S.A., McDonald B., Goodarzi Z., Kelly M.M., Patel K.D., Chakrabarti S., McAvoy E., Sinclair G.D., et al. 2007. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 13:463–469 10.1038/nm1565 [DOI] [PubMed] [Google Scholar]
- Essex D.W., Chen K., Swiatkowska M. 1995. Localization of protein disulfide isomerase to the external surface of the platelet plasma membrane. Blood. 86:2168–2173 [PubMed] [Google Scholar]
- Falet H., Pollitt A.Y., Begonja A.J., Weber S.E., Duerschmied D., Wagner D.D., Watson S.P., Hartwig J.H. 2010. A novel interaction between FlnA and Syk regulates platelet ITAM-mediated receptor signaling and function. J. Exp. Med. 207:1967–1979 10.1084/jem.20100222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald J.R., Foster T.J., Cox D. 2006. The interaction of bacterial pathogens with platelets. Nat. Rev. Microbiol. 4:445–457 10.1038/nrmicro1425 [DOI] [PubMed] [Google Scholar]
- Flaumenhaft R. 2003. Molecular basis of platelet granule secretion. Arterioscler. Thromb. Vasc. Biol. 23:1152–1160 10.1161/01.ATV.0000075965.88456.48 [DOI] [PubMed] [Google Scholar]
- Graham G.J., Ren Q., Dilks J.R., Blair P., Whiteheart S.W., Flaumenhaft R. 2009. Endobrevin/VAMP-8-dependent dense granule release mediates thrombus formation in vivo. Blood. 114:1083–1090 10.1182/blood-2009-03-210211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwig J.H., DeSisto M. 1991. The cytoskeleton of the resting human blood platelet: structure of the membrane skeleton and its attachment to actin filaments. J. Cell Biol. 112:407–425 10.1083/jcb.112.3.407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmi H., Takeuchi O., Kawai T., Kaisho T., Sato S., Sanjo H., Matsumoto M., Hoshino K., Wagner H., Takeda K., Akira S. 2000. A Toll-like receptor recognizes bacterial DNA. Nature. 408:740–745 10.1038/35047123 [DOI] [PubMed] [Google Scholar]
- Hennessy E.J., Parker A.E., O’Neill L.A. 2010. Targeting Toll-like receptors: emerging therapeutics? Nat. Rev. Drug Discov. 9:293–307 10.1038/nrd3203 [DOI] [PubMed] [Google Scholar]
- Hoffmeister K.M., Felbinger T.W., Falet H., Denis C.V., Bergmeier W., Mayadas T.N., von Andrian U.H., Wagner D.D., Stossel T.P., Hartwig J.H. 2003. The clearance mechanism of chilled blood platelets. Cell. 112:87–97 10.1016/S0092-8674(02)01253-9 [DOI] [PubMed] [Google Scholar]
- Italiano J.E., Jr, Lecine P., Shivdasani R.A., Hartwig J.H. 1999. Blood platelets are assembled principally at the ends of proplatelet processes produced by differentiated megakaryocytes. J. Cell Biol. 147:1299–1312 10.1083/jcb.147.6.1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Italiano J.E., Jr, Bergmeier W., Tiwari S., Falet H., Hartwig J.H., Hoffmeister K.M., André P., Wagner D.D., Shivdasani R.A. 2003. Mechanisms and implications of platelet discoid shape. Blood. 101:4789–4796 10.1182/blood-2002-11-3491 [DOI] [PubMed] [Google Scholar]
- Janeway C.A., Jr, Medzhitov R. 2002. Innate immune recognition. Annu. Rev. Immunol. 20:197–216 10.1146/annurev.immunol.20.083001.084359 [DOI] [PubMed] [Google Scholar]
- Krieg A.M. 1996. Lymphocyte activation by CpG dinucleotide motifs in prokaryotic DNA. Trends Microbiol. 4:73–76 10.1016/0966-842X(96)81515-0 [DOI] [PubMed] [Google Scholar]
- Krieg A.M., Yi A.K., Matson S., Waldschmidt T.J., Bishop G.A., Teasdale R., Koretzky G.A., Klinman D.M. 1995. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature. 374:546–549 10.1038/374546a0 [DOI] [PubMed] [Google Scholar]
- Lahav J., Wijnen E.M., Hess O., Hamaia S.W., Griffiths D., Makris M., Knight C.G., Essex D.W., Farndale R.W. 2003. Enzymatically catalyzed disulfide exchange is required for platelet adhesion to collagen via integrin alpha2beta1. Blood. 102:2085–2092 10.1182/blood-2002-06-1646 [DOI] [PubMed] [Google Scholar]
- Latz E., Schoenemeyer A., Visintin A., Fitzgerald K.A., Monks B.G., Knetter C.F., Lien E., Nilsen N.J., Espevik T., Golenbock D.T. 2004. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat. Immunol. 5:190–198 10.1038/ni1028 [DOI] [PubMed] [Google Scholar]
- Lipford G.B., Heeg K., Wagner H. 1998. Bacterial DNA as immune cell activator. Trends Microbiol. 6:496–500 10.1016/S0966-842X(98)01408-5 [DOI] [PubMed] [Google Scholar]
- Maynard D.M., Heijnen H.F., Gahl W.A., Gunay-Aygun M. 2010. The α-granule proteome: novel proteins in normal and ghost granules in gray platelet syndrome. J. Thromb. Haemost. 8:1786–1796 10.1111/j.1538-7836.2010.03932.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa J.P., George J.N., Bainton D.F., Nurden A.T., Caen J.P., McEver R.P. 1987. Gray platelet syndrome. Demonstration of alpha granule membranes that can fuse with the cell surface. J. Clin. Invest. 80:1138–1146 10.1172/JCI113171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowley J.W., Oler A.J., Tolley N.D., Hunter B.N., Low E.N., Nix D.A., Yost C.C., Zimmerman G.A., Weyrich A.S. 2011. Genome-wide RNA-seq analysis of human and mouse platelet transcriptomes. Blood. 118:e101–e111 10.1182/blood-2011-03-339705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple J.W., Italiano J.E., Jr, Freedman J. 2011. Platelets and the immune continuum. Nat. Rev. Immunol. 11:264–274 10.1038/nri2956 [DOI] [PubMed] [Google Scholar]
- Smyth S.S., McEver R.P., Weyrich A.S., Morrell C.N., Hoffman M.R., Arepally G.M., French P.A., Dauerman H.L., Becker R.C.; 2009 Platelet Colloquium Participants 2009. Platelet functions beyond hemostasis. J. Thromb. Haemost. 7:1759–1766 10.1111/j.1538-7836.2009.03586.x [DOI] [PubMed] [Google Scholar]
- Song K.W., Talamas F.X., Suttmann R.T., Olson P.S., Barnett J.W., Lee S.W., Thompson K.D., Jin S., Hekmat-Nejad M., Cai T.Z., et al. 2009. The kinase activities of interleukin-1 receptor associated kinase (IRAK)-1 and 4 are redundant in the control of inflammatory cytokine expression in human cells. Mol. Immunol. 46:1458–1466 10.1016/j.molimm.2008.12.012 [DOI] [PubMed] [Google Scholar]
- Sparwasser T., Koch E.S., Vabulas R.M., Heeg K., Lipford G.B., Ellwart J.W., Wagner H. 1998. Bacterial DNA and immunostimulatory CpG oligonucleotides trigger maturation and activation of murine dendritic cells. Eur. J. Immunol. 28:2045–2054 [DOI] [PubMed] [Google Scholar]
- Stacey K.J., Sweet M.J., Hume D.A. 1996. Macrophages ingest and are activated by bacterial DNA. J. Immunol. 157:2116–2122 [PubMed] [Google Scholar]
- Thon J.N., Montalvo A., Patel-Hett S., Devine M.T., Richardson J.L., Ehrlicher A., Larson M.K., Hoffmeister K., Hartwig J.H., Italiano J.E., Jr 2010. Cytoskeletal mechanics of proplatelet maturation and platelet release. J. Cell Biol. 191:861–874 10.1083/jcb.201006102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Nispen Tot Pannerden H.E., van Dijk S.M., Du V., Heijnen H.F. 2009. Platelet protein disulfide isomerase is localized in the dense tubular system and does not become surface expressed after activation. Blood. 114:4738–4740 10.1182/blood-2009-03-210450 [DOI] [PubMed] [Google Scholar]
- White J.G., Gerrard J.M. 1976. Ultrastructural features of abnormal blood platelets. A review. Am. J. Pathol. 83:589–632 [PMC free article] [PubMed] [Google Scholar]
- Yamamoto S., Yamamoto T., Tokunaga T. 2000. The discovery of immunostimulatory DNA sequence. Springer Semin. Immunopathol. 22:11–19 10.1007/s002810000019 [DOI] [PubMed] [Google Scholar]
- Zhao H., Hemmi H., Akira S., Cheng S.H., Scheule R.K., Yew N.S. 2004. Contribution of Toll-like receptor 9 signaling to the acute inflammatory response to nonviral vectors. Mol. Ther. 9:241–248 10.1016/j.ymthe.2003.11.012 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.