

ABSTRACT
Microbes must assimilate carbon to grow and colonize their niches. Transcript profiling has suggested that Candida albicans, a major pathogen of humans, regulates its carbon assimilation in an analogous fashion to the model yeast Saccharomyces cerevisiae, repressing metabolic pathways required for the use of alterative nonpreferred carbon sources when sugars are available. However, we show that there is significant dislocation between the proteome and transcriptome in C. albicans. Glucose triggers the degradation of the ICL1 and PCK1 transcripts in C. albicans, yet isocitrate lyase (Icl1) and phosphoenolpyruvate carboxykinase (Pck1) are stable and are retained. Indeed, numerous enzymes required for the assimilation of carboxylic and fatty acids are not degraded in response to glucose. However, when expressed in C. albicans, S. cerevisiae Icl1 (ScIcl1) is subjected to glucose-accelerated degradation, indicating that like S. cerevisiae, this pathogen has the molecular apparatus required to execute ubiquitin-dependent catabolite inactivation. C. albicans Icl1 (CaIcl1) lacks analogous ubiquitination sites and is stable under these conditions, but the addition of a ubiquitination site programs glucose-accelerated degradation of CaIcl1. Also, catabolite inactivation is slowed in C. albicans ubi4 cells. Ubiquitination sites are present in gluconeogenic and glyoxylate cycle enzymes from S. cerevisiae but absent from their C. albicans homologues. We conclude that evolutionary rewiring of ubiquitination targets has meant that following glucose exposure, C. albicans retains key metabolic functions, allowing it to continue to assimilate alternative carbon sources. This metabolic flexibility may be critical during infection, facilitating the rapid colonization of dynamic host niches containing complex arrays of nutrients.
IMPORTANCE
Pathogenic microbes must assimilate a range of carbon sources to grow and colonize their hosts. Current views about carbon assimilation in the pathogenic yeast Candida albicans are strongly influenced by the Saccharomyces cerevisiae paradigm in which cells faced with choices of nutrients first use energetically favorable sugars, degrading enzymes required for the assimilation of less favorable alternative carbon sources. We show that this is not the case in C. albicans because there has been significant evolutionary rewiring of the molecular signals that promote enzyme degradation in response to glucose. As a result, this major pathogen of humans retains enzymes required for the utilization of physiologically relevant carbon sources such as lactic acid and fatty acids, allowing it to continue to use these host nutrients even when glucose is available. This phenomenon probably enhances efficient colonization of host niches where sugars are only transiently available.
Introduction
Carbon assimilation is fundamentally important for all organisms. When faced with choices of carbon source, microbes often assimilate preferred carbon sources to support the first phase of growth and then, having exhausted these carbon sources, turn to alternative energetically less favorable carbon sources to drive subsequent phases of diauxic growth. This selective carbon utilization is reflected in the differential regulation of genes and enzymes that support the uptake and catabolism of specific carbon sources. The Escherichia coli lac operon provides a classic example of this, mediating lactose utilization only after the preferred carbon source, glucose, is exhausted (1, 2). In Saccharomyces cerevisiae, glucose limits the assimilation of alternative carbon sources and represses respiration under aerobic conditions, promoting fermentative metabolism (the Crabtree effect [3, 4]).
S. cerevisiae is exquisitely sensitive to sugars: even glucose concentrations as low as 0.01% trigger the major redirection of cellular resources (5, 6). Glucose exerts its dramatic effects upon S. cerevisiae physiology via signaling pathways that include the glucose repression (Snf1 AMP kinase) pathway, cyclic AMP (cAMP)-protein kinase A signaling, and the sugar receptor repressor (Snf3-Rgt2) pathway (for reviews, see references 7 to 12). The cAMP-protein kinase A pathway activates ribosome biogenesis and downregulates stress responses in response to glucose (13–16). The sugar receptor repressor pathway modulates the expression of hexose transporters (9, 17, 18). Meanwhile, the glucose repression pathway represses the transcription of genes involved in the assimilation of alternative carbon sources, such as galactose, ethanol, and fatty acids (7, 8).
These sugar signaling mechanisms comprise an interlinked network rather than parallel signaling pathways (8, 10, 12, 18, 19). Furthermore, glucose regulation is imposed at multiple levels in S. cerevisiae. They include transcriptional (7–9), posttranscriptional (5, 6, 20, 21), translational (22), and posttranslational mechanisms (23–28). For example, glucose represses the transcription of genes encoding the gluconeogenic enzymes fructose 1,6-bisphosphatase (FBP1) and phosphoenolpyruvate carboxykinase (PCK1) (29, 30) and triggers the accelerated degradation of the FBP1 and PCK1 mRNAs (5, 20). Furthermore, glucose triggers the phosphorylation and inactivation of fructose 1,6-bisphosphatase (Fbp1) as well as its proteolytic degradation (31). The accelerated degradation of Fbp1 is mediated by vacuolar and ubiquitin-dependent mechanisms (28). Following glucose addition, Fbp1 is ubiquitinated and degraded via the proteasome (25, 28, 32). Like Fbp1, the glyoxylate cycle enzyme, isocitrate lyase (Icl1) is also subject to catabolite inactivation in S. cerevisiae (24). This tight control of central carbon metabolism is thought to reflect the evolution of this model yeast under conditions of “feast or famine” and to enhance the competitiveness of S. cerevisiae in sugar-rich niches containing complex microflora (9).
S. cerevisiae is often viewed as a paradigm for other yeasts (33). However, yeast species inhabit diverse niches and have evolved under contrasting selective pressures leading to differing strategies of carbon utilization (34). For example, the major systemic fungal pathogen of humans, Candida albicans, inhabits niches that contain complex mixtures of carbon sources. During commensalism and mucosal infection, C. albicans colonizes the oral cavity and the gastrointestinal and urogenital tracts, and during systemic infection, this pathogen can thrive in the bloodstream and most internal organs (35, 36). Few of these niches are rich in sugar. Blood glucose levels range from 4 to 7 mM (0.07 to 0.13%), whereas concentrations of about 111 mM (2%) are often used to impose glucose repression in in vitro experiments. Many niches are rich in alternative carbon sources, such as lactate, fatty acids, and amino acids. For example, lactic acid is found in ingested foods, is produced by host metabolic activity and by lactic acid bacteria in the gastrointestinal and urogenital tracts (37), and is essential for the proliferation of Candida glabrata in the intestinal tract (38). Also, glyoxylate cycle and fatty acid β-oxidation genes are expressed in the host and are required for the full virulence of C. albicans during systemic infections (39–42).
Although C. albicans occupies contrasting niches from S. cerevisiae, analogous sugar signaling pathways are thought to exist in these yeasts (33). Although there has been considerable rewiring of the regulatory circuitry that controls carbon metabolism (43–45), microarray experiments have revealed that C. albicans genes involved in the assimilation of alternative carbon sources are exquisitely sensitive to low concentrations of glucose (46, 47), like their orthologs in S. cerevisiae (6). However, unlike S. cerevisiae, C. albicans continues to respire in the presence of glucose, leading to its classification as a Crabtree-negative yeast (48).
These observations create an interesting conundrum relating to the carbon assimilation and pathogenicity of C. albicans: how can this yeast rapidly colonize niches that contain small amounts of glucose if many of the metabolic genes required for efficient growth in these niches are repressed by glucose? Could glucose regulation be relaxed at a posttranscriptional level in C. albicans, thereby facilitating simultaneous assimilation of sugars and alternative carbon sources in vivo? We have addressed these questions first by performing proteomic screens to identify proteins that are regulated in response to physiologically relevant carbon sources, revealing that gluconeogenic and glyoxylate cycle enzymes remain at high levels hours after glucose exposure. We then showed that C. albicans Pck1 and Icl1 are not destabilized by glucose, in contrast to S. cerevisiae Pck1 and Icl1, even though C. albicans has retained the molecular apparatus to program the accelerated, ubiquitin-mediated degradation of target proteins following glucose exposure. C. albicans Icl1 escapes degradation following glucose addition, because this enzyme lacks key ubiquitination sites required to target it for accelerated degradation. Our data show that there has been significant posttranscriptional rewiring during the evolution of this pathogen, thereby allowing C. albicans to continue to assimilate alternative carbon sources in the presence of glucose.
RESULTS
Carbon source has a major impact on the C. albicans proteome.
Microarray studies have shown that the C. albicans transcriptome is exquisitely sensitive to glucose and that genes involved in the assimilation of alternative carbon sources are subject to glucose repression (46, 47). Our first aim was to establish whether this transcriptional regulation was reflected in the C. albicans proteome. To test this, we grew prototrophic C. albicans NGY152 cells (see Table S1 in the supplemental material) for 20 h in minimal medium containing glucose, lactate, oleate, or amino acids as the sole carbon source, harvested them in mid-exponential phase, prepared protein extracts, and subjected them to two-dimensional (2D) gel electrophoresis (Materials and Methods). Principal component analysis confirmed the reproducibility of the 2D gels from the independent replicate experiments and showed that the carbon source had a significant impact upon the C. albicans proteome (see Fig. S1A in the supplemental material). Proteins that displayed statistically significant changes in level on the basis of three independent experiments (Fig. 1A) were identified by tryptic digestion and matrix-assisted laser desorption ionization–time of flight (MALDI-ToF) mass spectrometry. Positive identifications were obtained for 206 2D gel features, representing 152 different C. albicans proteins, some distinct features representing isoforms of the same protein. The list of C. albicans proteins identified is presented in Table S2 and submitted to the PRIDE (proteomics identifications database) proteomic data repository (http://www.ebi.ac.uk/pride/) (accession numbers 3186 to 3192).
FIG 1 .

Impact of carbon source on the C. albicans proteome. (A) Replicate 2D gels showing that Pox4 is more abundant during growth on oleic acid than on glucose and that Pox4 is retained after 2 h of exposure to glucose. (B) Network of C. albicans proteins regulated in response to carbon source. Nodes are connected by edges to the one or more conditions under which it was identified: upregulated ≥2-fold relative to growth on glucose (upward arrowhead); downregulated ≥2-fold (downward arrowhead). Functions that were differentially regulated on different carbon sources (rounded rectangle) have color-coded connecting lines: green for upregulated and red for downregulated. Functions regulated under all three conditions lie in three blocks in the center of the network interactions.
The network of proteins that displayed statistically significant changes in response to carbon source mainly comprised metabolic enzymes (Fig. 1B). Significant overlap was observed between the sets of proteins that were regulated in response to growth on lactate, oleate, or amino acids compared to the glucose condition. During growth on these organic acids, glycolytic enzymes were downregulated, and enzymes involved in gluconeogenesis, the glycoxylate cycle, the tricarboxylic acid (TCA) cycle and pathways involved in the assimilation of alternative carbon sources were upregulated (Fig. 1B and 2B; see Fig. S2 in the supplemental material). For example, gluconeogenic (Pck1 and Fbp1) and tricarboxylic acid cycle enzymes (Aco1, Kgd1, and Mdh11) displayed reduced levels, and the glycolysis-specific enzyme pyruvate kinase (Cdc19) was at elevated levels (see Fig. S2 and Table S2 in the supplemental material) during growth on glucose compared to lactate-, oleic acid-, and amino acid-grown cells. During growth on oleate, fatty acid β-oxidation (Faa21/23/24, Fox2, Pot11, Pox4, and Tes1), glyoxylate cycle (Icl1), and additional TCA cycle enzymes (Cit1 and Fum11) were present at elevated levels compared to the levels in glucose-grown cells (Fig. 2; Table S2). Enzymes on many amino acid biosynthetic pathways were downregulated during growth on amino acids (Arg5,6, Bat21, Cpa2, His1, Hom6, Ilv5, Lys12, and Met6/14/15), and this was also the case during growth on lactate (Arg5,6, Bat21, His1, and Met6) or oleic acid (Arg5,6, Bat21, His1, and Met6) (Fig. 1; Table S2). These data largely confirmed expectations based on our understanding of metabolism in other yeasts and the limited experimental data on C. albicans metabolic regulation (35, 49). The observed changes in Pck1 and Icl1 levels were validated by Western blot analysis of these proteins following Myc tagging in C. albicans (Fig. 2A). Note that Icl1 levels were below the limits of detection in the lactate proteome (Table S2).
FIG 2 .

Effect of carbon source on central carbon metabolic enzymes in C. albicans. (A) Western blots demonstrating the effects of overnight growth on different carbon sources on the levels of Myc-tagged Icl1 and Pck1 in C. albicans cells (CA1395 and CA1431 [see Table S1 in the supplemental material]). Control lanes contain extracts from the untagged parental strain (RM1000) control grown on lactate. (B) Effects of growth on oleic acid versus glucose on the levels of enzymes involved in central carbon metabolism. All enzymes shown were identified on the 2D gels. The effects of growth on oleic acid versus glucose on the levels of enzymes are indicated as follows: white, no significant change in the levels of glucose- and oleic acid-grown cells; red, protein level significantly elevated in oleic acid-grown cells; cyan, protein level significantly elevated in glucose-grown cells. For abbreviations, see the Candida Genome Database (http://www.candidagenome.org).
The levels of other types of protein were modulated in response to carbon source. These proteins included proteins involved in growth and cell polarity (Arp2, Cap2, Pfy1, Cof1, Crn1, and Rbp1), nuclear transport (Ntf2), DNA repair (Rad23), and protein folding (Pdi1 and Cyp51) (see Table S2 in the supplemental material). Interestingly, proteins involved in drug resistance (Pdr13 and Erg13) were also affected by carbon source, which is consistent with the recent finding that carbon source affects the antifungal drug resistance of C. albicans (50). Also, a number of stress functions were affected by carbon source (Table S2). The levels of Hsp70 family members (Hsp70/Ssa4, Ssa1, and Kar2) were differentially regulated in response to carbon source. The Trx1 thioredoxin was also dramatically upregulated during growth on lactate, oleic acid, or amino acids, while the glutaredoxin-like protein Grx3 was downregulated on lactate and oleic acid. The levels of catalase glutathione peroxidize (Gpx1) were also upregulated on lactate. These data were consistent with the observation that host carbon source affects stress resistance in C. albicans (50).
Transcript profiling data are available for the effects of glucose on lactate-grown C. albicans cells (47). We found the correlation between these transcript profiling data and our proteomic data for lactate- and glucose-grown cells to be modest at best (correlation coefficient = 0.25) (Fig. 3A). No doubt differential protein stabilities, alterations in posttranslational modifications, and the existence of multiple 2D gel features for some proteins contributed to this. Nevertheless, this was consistent with other comparisons of transcriptomic and proteomic data sets, which vary considerably in their degree of correlation (51–54).
FIG 3 .
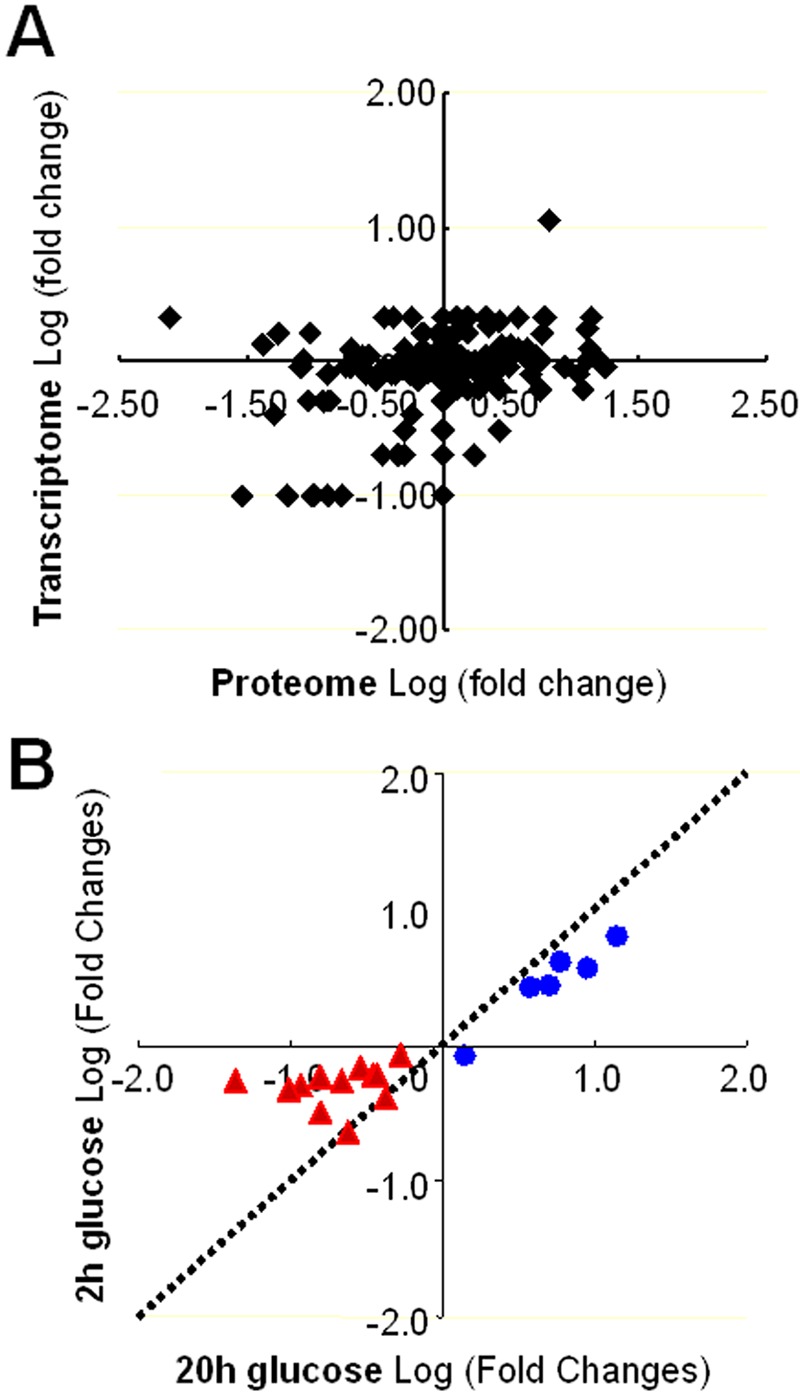
Comparisons of the C. albicans transcriptome and proteome. (A) The C. albicans transcriptome and proteome display limited correlation with respect to the observed log fold changes between cells grown on glucose or lactic acid. The transcriptomic data were taken from reference 47, and proteomic data were taken from Table S2 in the supplemental material. (B) Correlation between the short-term and long-term effects of glucose on central metabolic enzymes. Glycolytic enzymes are shown in blue. Gluconeogenic, glyoxylate cycle, and tricarboxylic acid (TCA) cycle enzymes are shown in red.
Differential effects of glucose on the C. albicans proteome in the short term and longer term.
Having tested the impact on the C. albicans proteome of growth on different carbon sources, we examined the effects of glucose addition to cells growing on alternative carbon sources. A similar experimental approach was taken except that C. albicans NGY152 cells were grown for 20 h to mid-exponential phase on lactate, oleate, or amino acids as the sole carbon source, and then 2% glucose was added 2 h before the cells were harvested for proteomic analyses. Once again, proteins that displayed statistically significant changes across three independent experiments (Fig. 1A) were identified by mass spectrometry (see Table S2 in the supplemental material; PRIDE accession numbers 3186 to 3192). Principal component analysis indicated that glucose addition exerted significant effects on the lactate, oleate, and amino acid proteomes and that these effects were distinct from long-term growth on glucose (Fig. 1B), although similar subsets of proteins were affected (Table S2).
We compared the short- and longer-term effects of glucose on the C. albicans proteome (Fig. 3B; see Fig. S1 in the supplemental material). Interestingly, the extent of correlation between the short- and long-term changes differed for different sets of functionally related proteins. Glycolytic enzymes showed a strong correlation, generally displaying glucose induction in both the short and longer terms (Fig. 3B). In contrast, gluconeogenic, glycoxylate cycle, TCA cycle, and fatty acid β-oxidation enzymes displayed a poor correlation. The levels of many of these enzymes were generally lower in glucose-grown cells, and their levels did not decline significantly after exposure to glucose for 2 h (Fig. S1).
Icl1 and Pck1 are not subject to catabolite inactivation in C. albicans.
The apparent stability of gluconeogenic and glyoxylate cycle enzymes following glucose exposure in C. albicans contrasted with observations in S. cerevisiae where glucose triggers catabolite inactivation of these enzymes (23–25, 31). Therefore, we examined the impact of glucose on the stability of C. albicans Icl1 and Pck1 by Western blotting of Myc-tagged versions of these proteins (Fig. 4). C. albicans CA1395 (ICL1-Myc3) and CA1431 (PCK1-Myc3) (see Table S1 in the supplemental material) were grown to mid-exponential phase in medium containing lactate as the sole carbon source, and then 2% glucose was added. Protein extracts were prepared at various times thereafter and subjected to Western blotting, revealing that Icl1 and Pck1 were stable following glucose addition (Fig. 4B). Similar observations were made in oleic acid-grown cells exposed to glucose (see Fig. S3 in the supplemental material). These data were consistent with our observation that Icl1 and Pck1 are retained at high levels in C. albicans cells 2 h after glucose addition (Table S2).
FIG 4 .
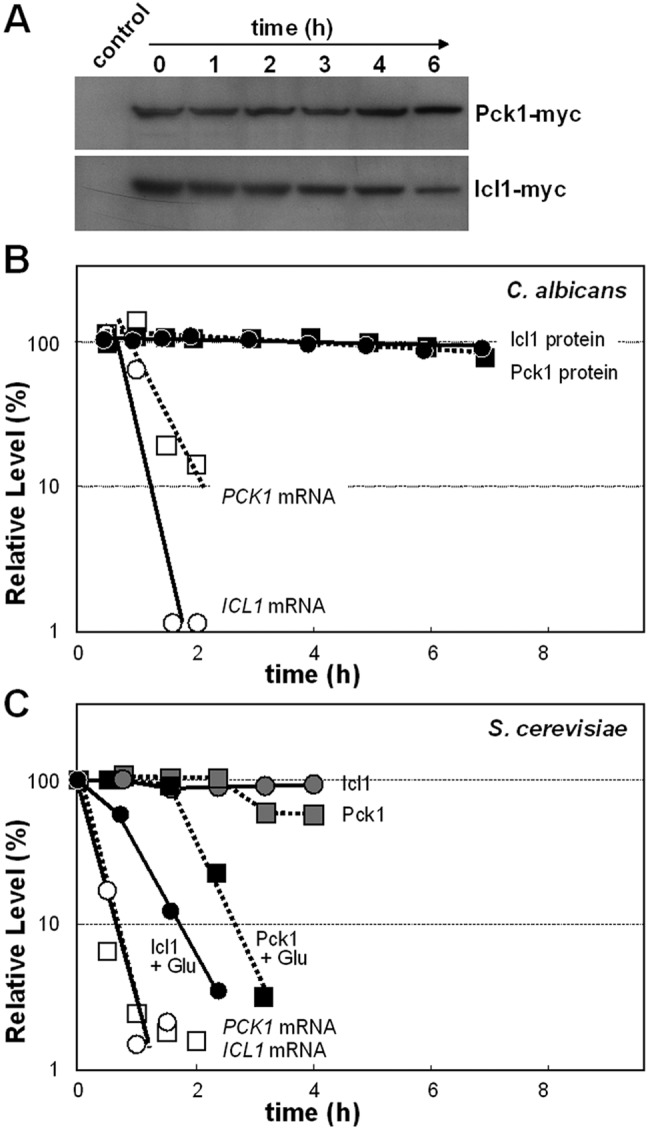
Icl1 and Pck1 protein stability and ICL1 and PCK1 mRNA turnover in C. albicans and S. cerevisiae following the addition of glucose. (A) CaIcl1-Myc and CaPck1-Myc protein levels were measured by Western blotting at various times after the addition of glucose to C. albicans cells grown on lactate. (B) CaIcl1-Myc and CaPck1-Myc were quantified by Western blotting relative to the Act1 internal control following glucose addition. Also CaICL1 and CaPCK1 mRNA levels were assayed in the same cultures by qRT-PCR and compared to the level in the ACT1 mRNA internal control after glucose addition. The relative levels of these mRNAs and proteins were expressed as a percentage of their abundance at time zero, which was set at 100%. (C) ScIcl1-Myc and ScPck1-HA protein levels were measured by Western blotting, and ScICL1 and ScPCK1 mRNA levels were assayed by qRT-PCR in S. cerevisiae cells grown on lactate and exposed to glucose at time zero. The relative levels of these mRNAs and proteins were expressed as a percentage of their abundance at time zero (which was set at 100%): circles, Icl1; squares, Pck1; black symbols, Icl1 and Pck1 proteins in lactate-grown cells after glucose addition; gray symbols, Icl1 and Pck1 proteins in control lactate-grown cells with no glucose addition; open symbols, ICL1 and PCK1 mRNAs in lactate-grown cells after glucose addition. Similar data were obtained from two independent replicate experiments.
We then measured the stability of the corresponding mRNAs under equivalent growth conditions by quantitative reverse transcription-PCR (qRT-PCR) relative to the internal ACT1 mRNA control. The ICL1 and PCK1 mRNAs were rapidly degraded following glucose addition to lactate-grown cells (Fig. 4B), and similar data were obtained for oleic acid-grown cells (see Fig. S3 in the supplemental material). These observations were entirely consistent with the results of previous microarray experiments, suggesting that these transcripts are strongly repressed by glucose in C. albicans (46, 47). Therefore, ICL1 and PCK1 transcription is strongly regulated by glucose, but this regulation is not reflected in the proteome.
These experiments were replicated in S. cerevisiae to exclude the possibility that the lack of catabolite inactivation in C. albicans was an experimental artifact. S. cerevisiae DS1-W10 (ICL1-Myc9) and DS1-W20 (PCK1-HA6) (see Table S1 in the supplemental material) were grown on lactate and exposed to 2% glucose, and at various times thereafter, Icl1 and Pck1 levels were examined by Western blotting (Fig. 4C). The Icl1 and Pck1 enzymes were stable in S. cerevisiae in the absence of glucose but were destabilized following glucose addition. Similar data were obtained for oleic acid-grown S. cerevisiae cells (see Fig. S3 in the supplemental material). These data recapitulate earlier observations (23, 24) and confirm that these gluconeogenic and glyoxylate cycle enzymes are subject to catabolite inactivation in S. cerevisiae. Furthermore, the ICL1 and PCK1 transcripts were rapidly degraded in S. cerevisiae following glucose addition (Fig. 4C), confirming earlier reports to this effect (5, 6, 20). Therefore, C. albicans differs significantly from S. cerevisiae in that Icl1 and Pck1 are not subject to catabolite inactivation in response to glucose in the pathogenic yeast.
C. albicans has retained the apparatus for catabolite inactivation.
In principle, the lack of glucose-accelerated degradation of Icl1 and Pck1 in C. albicans could have been due to the evolutionary loss of the catabolite inactivation apparatus in this pathogen. To test this, we asked whether S. cerevisiae Icl1 is subject to glucose-accelerated degradation in C. albicans.
C. albicans strains were constructed in which the S. cerevisiae ICL1 (ScICL1) open reading frame (ORF) was expressed from the C. albicans ICL1 (CaICL1) promoter at the native ICL1 locus. The functionality of three-Myc (Myc3)-tagged S. cerevisiae Icl1 (ScIcl1) in C. albicans was first confirmed in DSCO2 cells (icl1/ScICL1) (see Table S1 in the supplemental material), the ScICL1-Myc3 gene suppressing the growth defect of C. albicans icl1 cells on lactate, oleic acid, pyruvate, and acetate as the sole carbon source (Fig. 5A). The stability of the Myc3-tagged ScIcl1 was then examined in lactate-grown C. albicans DSCO1 cells (ICL1/ScICL1-Myc3). ScIcl1 was stable in C. albicans in the absence of glucose but was rapidly degraded following glucose addition (Fig. 5B). These observations were reproducible, and analogous observations were made in oleic acid-grown C. albicans DSCO1 cells (see Fig. S4 in the supplemental material). We conclude that ScIcl1 is subject to glucose-accelerated degradation in C. albicans, and hence that during evolution C. albicans has retained the apparatus for catabolite inactivation.
FIG 5 .

ScIcl1 is functional in C. albicans and rapidly degraded in response to glucose. (A) Expression of ScICL1 in C. albicans suppresses the carbon source conditional phenotypes of an icl1 mutant: ICL1, C. albicans CA510 (ICL1/icl1) (see Table S1 in the supplemental material); icl1, C. albicans CA517 (icl1/icl1); ScICL1, C. albicans DSCO1 (ScICL1/icl1). (B) Impact of glucose on the levels of the ScIcl1-Myc protein expressed in lactate-grown C. albicans DSCO1 cells relative to the abundance at time zero. (Top) Western blotting of ScIcl1-Myc levels. (Bottom) Quantification of ScIcl1-Myc levels expressed as a percentage of the abundance at time zero (100%): closed symbols, plus glucose; open symbols, control cells lacking glucose. Similar data were obtained from two independent replicate experiments.
In this case, has CaIcl1 lost the signal that triggers glucose-accelerated degradation? The stability of the CaIcl1-Myc3 protein was assayed in the S. cerevisiae strain DS4-Y40 in which the Myc3-tagged CaICL1 open reading frame was expressed from the ScICL1 promoter at the native ICL1/YDR477w locus (see Table S1 in the supplemental material). The CaIcl1-Myc3 protein was stable in lactate-grown DS4-Y40 cells and was not destabilized by glucose addition relative to internal loading controls (Fig. 6). Once again, these observations were reproducible, and analogous observations were made for oleic acid-grown S. cerevisiae DS4-Y40 cells (see Fig. S4 in the supplemental material). Therefore, CaIcl1 appears to have lost the signal that triggers glucose-accelerated degradation.
FIG 6 .
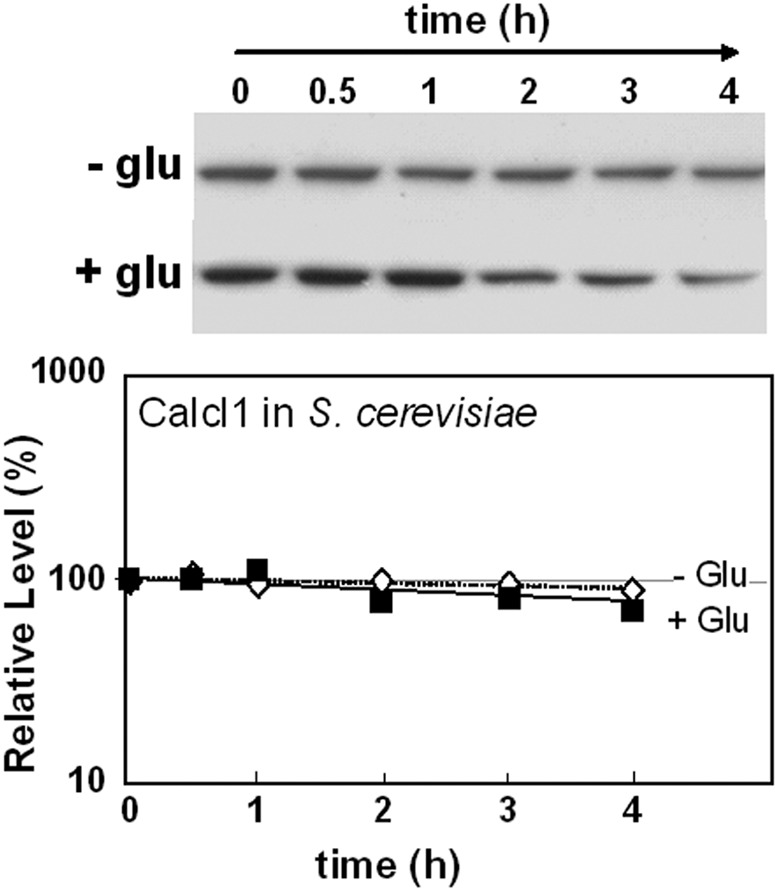
CaIcl1 is stable in S. cerevisiae following the addition of glucose. CaICL1-Myc3 was expressed in S. cerevisiae DS4-Y40 cells (see Table S1 in the supplemental material) grown on lactate, and CaIcl1-Myc protein levels were assayed by Western blotting after glucose addition. CaIcl1-Myc levels are expressed as a percentage of the abundance at time zero (which was set at 100%): closed symbols, plus glucose; open symbols, control cells lacking glucose. Similar data were obtained from two independent replicate experiments.
Ubiquitination contributes to glucose-accelerated protein degradation in C. albicans.
Ubiquitination contributes to glucose-mediated protein destabilization in S. cerevisiae (28, 32). Therefore, we tested whether ubiquitination contributes to glucose-accelerated degradation of ScIcl1 in C. albicans. The ScICL1-Myc3 cassette was integrated at the native ICL1 locus in a C. albicans ubi4 mutant that lacks polyubiquitin. Ubiquitination is significantly reduced in this ubi4 mutant, residual ubiquitin being expressed only from the UBI3 locus (55). The stability of the ScIcl1-Myc3 protein in these C. albicans DSCO3 cells (ubi4/ubi4 ICL1/ScICL1-Myc3) (see Table S1 in the supplemental material]) was compared to ScIcl1-Myc3 stability in DSCO1 cells (UBI4/UBI4 ICL1/ScICL1-Myc3) and the stability of the CaIcl1-Myc3 protein in CA1395 cells (UBI4/UBI4 ICL1/ICL1-Myc3). ScIcl1-Myc3 and CaIcl1-Myc3 were stable in these strains during growth on lactate in the absence of glucose (not shown). Once again, ScIcl1-Myc3 was destabilized following glucose addition to wild-type DSCO1 cells (Fig. 7). However, ScIcl1-Myc3 was partially stabilized in the polyubiquitin mutant, suggesting that glucose-accelerated degradation of ScIcl1 in C. albicans is at least partially dependent upon ubiquitination. This observation was reproducible in independent experiments, and furthermore, similar observations were made when the same strains were grown on oleic acid rather than lactate (see Fig. S5 in the supplemental material). The residual ScIcl1-Myc3 destabilization observed in lactate- or oleic acid-grown ubi4 cells could have been due either to the involvement of a second degradation pathway in this response (28) or to the residual ubiquitination that is observed in the C. albicans polyubiquitin mutant (55). We conclude that ubiquitination contributes to glucose-accelerated protein degradation in C. albicans.
FIG 7 .
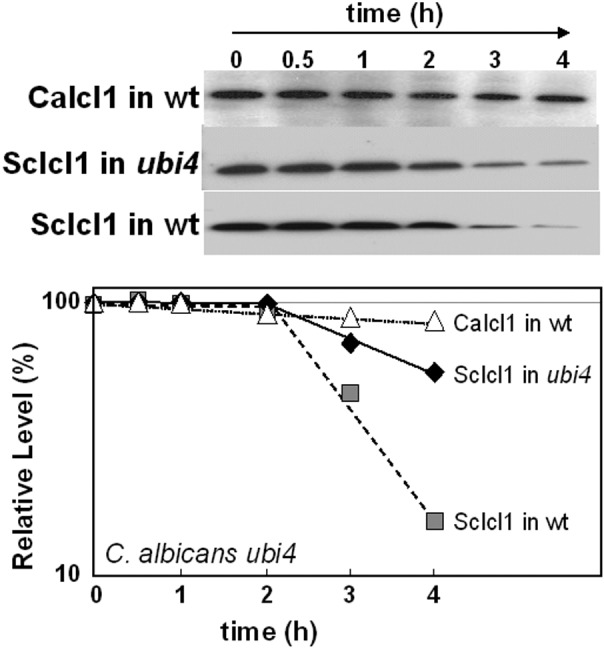
Inactivation of polyubiquitin inhibits glucose-accelerated ScIcl1 degradation in C. albicans. ScICL1-Myc3 was expressed in C. albicans DSCO3 (ubi4/ubi4) (see Table S1 in the supplemental material) and DSCO1 (wild type [wt]) (UBI4/UBI4) cells grown on lactate, and then ScIcl1-Myc protein levels were assayed after the addition of glucose by Western blotting. For a control, CaIcI1-Myc levels were assayed in C. albicans DSCO3 (ubi4/ubi4) cells grown on lactate and following glucose addition. ScIcl1-Myc and CaIcl1-Myc levels are expressed as a percentage of their abundance at time zero (which was set at 100%). Similar data were obtained from two independent replicate experiments.
C. albicans Icl1 lacks ubiquitination sites that trigger glucose-accelerated protein degradation.
Why does CaIcl1 escape glucose-accelerated ubiquitin-mediated protein degradation? We screened for consensus ubiquitination target sites in CaIcl1 and ScIcl1 using UbPred (predictor of protein ubiquitination sites) (http://www.ubpred.org/index.html) (56). The UbPred software, which was trained on 272 ubiquitination sites in S. cerevisiae, predicts ubiquitination sites based on numerous properties of the target amino acid sequence, including the net and total charge, aromatic content, charge/hydrophobicity ratio, sequence complexity, flexibility, amphipathic moment, and intrinsic disorder (56). The ScIcl1 and CaIcl1 proteins display strong amino acid sequence similarity (78% similarity and 67% identity). Yet while ScIcl1 contains two strong consensus ubiquitination sites at residues 158 and 551 according to UbPred, CaIcl1 carries no high-confidence ubiquitination targets (Fig. 8A). Therefore, we reasoned that the lack of such a ubiquitination site protected CaIcl1 from glucose-accelerated degradation.
FIG 8 .
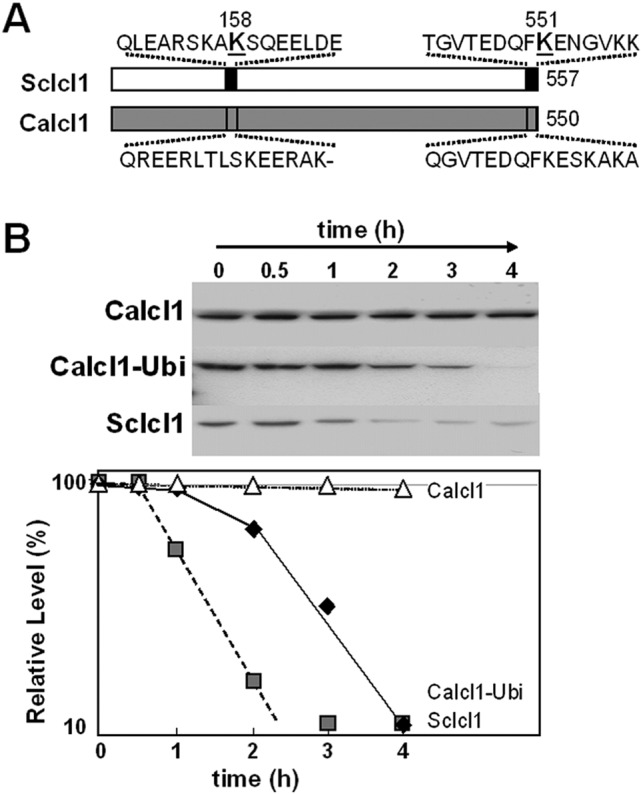
Addition of a consensus ubiquitin site stimulates glucose-accelerated degradation of CaICl1 in C. albicans. (A) Schematic representation illustrating the existence of high-confidence ubiquitination sites in ScIcl1 and the lack of such sites in CaIcl1 as predicted by UbPred (http://www.ubpred.org/index.html) (56). (B) The carboxy-terminal ubiquitination site from ScIcl1 was fused to CaIcl1 to create CaIcl1-Ubi-Myc in C. albicans DSCO4 (see Table S1 in the supplemental material). These cells were grown on lactate, and the levels of CaIcl1-Ubi-Myc were assayed by Western blotting after the addition of glucose. For controls, the stabilities of CaIcl1-Myc (CA1395) and ScIcl1-Myc (DSCO1) (gray squares) in C. albicans were compared under equivalent conditions. CaIcl1-Ubi-Myc, ScIcl1-Myc, and CaIcl1-Myc levels are expressed as a percentage of their abundance at time zero (which was set at 100%). Similar data were obtained from two independent replicate experiments.
To test this, we fused the carboxy-terminal ubiquitination site from ScIcl1 onto the carboxy terminus of CaIcl1 along with a Myc3 epitope tag and then compared the stability of this CaIcl1-Ubi-Myc3 protein with control CaIcl1-Myc3 and ScIcl1-Myc3 proteins in C. albicans. The stabilities of these proteins were first measured in mid-exponential C. albicans CA1395 (ICL1/ICL1-Myc3), DSCO1 (ICL1/ScICL1-Myc3), and DSCO4 (ICL1/ICL1-Ubi-Myc3) cells grown on lactate. All of the proteins were stable under these conditions (not shown). Once again, ScIcl1-Myc3 was destabilized following glucose addition, whereas CaIcl1-Myc3 remained stable (Fig. 8B). Interestingly, the CaIcl1-Ubi-Myc3 protein was reproducibly destabilized following glucose addition. Furthermore, analogous observations were made in C. albicans CA1395, DSCO1, and DSCO4 cells grown on oleic acid (see Fig. S6 in the supplemental material). Therefore, the addition of a ubiquitination site was sufficient to trigger glucose-accelerated degradation of the normally stable CaIcl1-Myc3 protein in C. albicans. These observations reinforced the view that C. albicans has retained the apparatus for glucose-accelerated protein degradation and suggested that the lack of an appropriate ubiquitination site in CaIcl1 prevents this protein from entering this degradation pathway.
C. albicans continues to assimilate alternative carbon sources following glucose exposure.
Is this evolutionary rewiring of ubiquitination targets between C. albicans and S. cerevisiae reflected in the metabolic activities of these yeasts? To test this, we compared [14C]lactate assimilation by mid-exponential C. albicans RM1000 and S. cerevisiae W303-1B cells that were grown on lactate in the presence and absence of glucose (Fig. 9). As expected, the presence of glucose inhibited the assimilation of [14C]lactate by S. cerevisiae. However, glucose did not inhibit [14C]lactate by C. albicans over the 4-h period examined, which was consistent with the retention by this pathogen of glyoxylate cycle and gluconeogenic enzymes after glucose addition. These reproducible observations were reinforced by analogous data on oleic acid assimilation by oleic acid-grown C. albicans and S. cerevisiae cells. Glucose inhibited [3H]oleic acid assimilation in S. cerevisiae W303-1B cells, but not in C. albicans RM1000 cells (see Fig. S7 in the supplemental material). These data indicate that, unlike S. cerevisiae, C. albicans continues to assimilate alternative carbon sources after exposure to glucose.
FIG 9 .
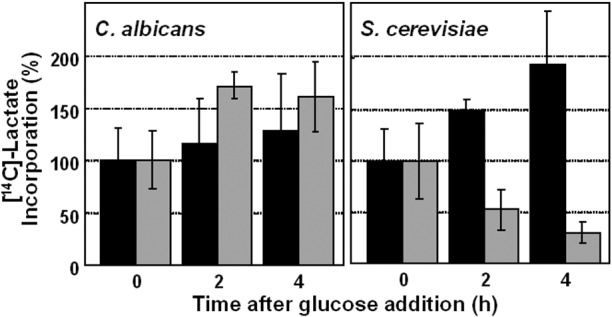
Glucose does not inhibit lactate assimilation by C. albicans. Exponential C. albicans RM1000 and S. cerevisiae W303-1B cells (see Table S1 in the supplemental material) grown on lactate were suspended in fresh medium containing [14C]lactate and 2% glucose (gray bars) or with no glucose (black bars). The assimilation of radiolabeled lactate by cells was assayed at various times thereafter (Materials and Methods). Similar data were obtained from two independent replicate experiments.
DISCUSSION
The prevailing view is that C. albicans fits the S. cerevisiae paradigm with regard to the impact of glucose upon central carbon metabolism. This influences current thinking about nutrient adaptation during infection and host-fungus interactions (46, 47, 57). However, this view does not resonate well with the evolution of these yeasts in contrasting niches that differ significantly with regard to carbon source availability (40) and with an early report that C. albicans is a Crabtree-negative yeast (48). We now show that, despite the similar effects that glucose exerts on the C. albicans and S. cerevisiae transcriptomes (46, 47), these yeasts display fundamental differences in the effects of glucose on central metabolic functions and carbon assimilation. The levels of many gluconeogenic, glyoxylate cycle, and fatty acid β-oxidation enzymes do decline in C. albicans after protracted growth on glucose, and the changes in the proteomic network (Fig. 1) reflect those observed previously during a comprehensive comparison of exponential- and stationary-phase C. albicans cells (58). However, these enzymes are stable during short-term exposure to glucose (Fig. 2 and 4; see Table S2 in the supplemental material), and this allows C. albicans cells to continue to assimilate alternative carbon sources even in the presence of glucose, unlike S. cerevisiae (Fig. 9).
Our analyses of Icl1 and Pck1 turnover have suggested that enzymes involved in the assimilation of alternative carbon sources are retained in C. albicans following glucose addition because these enzymes remain stable while their transcripts are degraded (Fig. 4). We show that this contrasts with S. cerevisiae cells that degrade these enzymes following glucose addition and their transcripts (Fig. 4). It is conceivable that carboxy-terminal tagging of CaIcl1 and ScIcl1 affected their localization and stability. Nevertheless, our data were consistent with the study of López-Boado et al. (24) who reported that Icl1 is subject to catabolite inactivation in S. cerevisiae. We then showed that, although CaIcl1 and CaPck1 are not subjected to glucose-accelerated protein degradation in C. albicans, this pathogen has retained the molecular apparatus to execute this function, as ScIcl1 is degraded following glucose addition when expressed as a functional enzyme in C. albicans (Fig. 5). Rather, CaIcl1 lacks the signal that triggers glucose-accelerated protein degradation in S. cerevisiae (Fig. 6).
Ubiquitin-dependent mechanisms contribute to the catabolite inactivation of Fbp1 in S. cerevisiae (28), and protein ubiquitination is known to be important for nutrient adaptation as well as growth, morphogenesis, and stress responses in C. albicans (55, 59–64). Therefore, we reasoned that ubiquitination might also contribute to glucose-accelerated protein degradation in C. albicans and that CaIcl1 might lack critical ubiquitination sites required to target this protein for catabolite inactivation. Three observations support this hypothesis. First, the inactivation of polyubiquitin (Ubi4) lowered the rate of ScIcl1 degradation in C. albicans (Fig. 7). ScIcl1 degradation was not completely blocked because the UBI3 gene would provide residual ubiquitin in C. albicans ubi4 cells (55, 65). Second, in silico analyses of the ScIcl1 and CaIcl1 sequences revealed that while ScIcl1 contains two high-confidence ubiquitination sites, CaIcl1 contains none (Fig. 8A) despite these proteins displaying a high degree of overall sequence identity (67%). Third, the addition of a carboxy-terminal ubiquitination site to the CaIcl1 protein was sufficient to program this protein for glucose-accelerated protein degradation in C. albicans (Fig. 8). Therefore, during the evolution of C. albicans, this pathogen has retained the molecular apparatus that mediates glucose-accelerated protein degradation, and CaIcl1 has evolved to escape this process.
Our proteomic analyses indicate that other C. albicans enzymes involved in the assimilation of alternative carbon sources lack ubiquitination sites are retained following glucose exposure (Fig. 3B; see Table S1 in the supplemental material). Furthermore, our in silico comparisons have suggested that enzymes specific for gluconeogenesis and the glyoxylate cycle lack the requisite ubiquitination sites required for glucose-accelerated protein degradation. S. cerevisiae malate synthase (ScMls1) contains three low-confidence ubiquitination sites according to UbPred (http://www.ubpred.org/index.html) (56), whereas CaMls1 contains no putative ubiquitination sites. UbPred also predicts that both ScFbp1 and ScPck1 contain high-confidence ubiquitination sites, whereas CaFbp1 and CaPck1 do not. This was consistent with our observation that, following glucose addition, ScPck1 is degraded in S. cerevisiae, but CaPck1 is not degraded in C. albicans (Fig. 4). Meanwhile, our previous work has indicated that a number of glycolytic enzymes are ubiquitinated in C. albicans (55). Therefore, significant evolutionary rewiring of ubiquitination targets appears to have occurred in this pathogen, allowing it to assimilate alternative carbon sources in the presence of glucose, unlike S. cerevisiae (Fig. 9).
These observations have several important implications for our understanding of C. albicans as a pathogen. First, our data indicate that under some conditions there is significant dislocation between the transcriptome and proteome. Microarray studies have contributed significantly to our understanding of C. albicans pathobiology (43, 44, 46, 66–68), and it is frequently presumed that changes in the transcriptome are reflected in corresponding changes in the C. albicans proteome and physiology. In some cases, there is a good correlation between the transcriptome and proteome, for example with regard to the induction of glycolytic enzymes in response to glucose (Fig. 3) and during amino acid starvation (54). However, this is not always the case (69), and this study demonstrates clearly that caution is required in making presumptions about the proteome based on the transcriptome. Indeed, this explains the conundrum as to why C. albicans is a Crabtree-negative yeast (48) even though its transcriptome is exquisitely sensitive to glucose (47). Thankfully, many studies provide experimental confirmation of working hypotheses on the basis of their microarray observations.
The second important implication relates to fungal nutrient assimilation during colonization and disease progression. C. albicans occupies dynamic host niches that contain complex mixtures of carbon sources that change over time through a combination of host and fungal metabolic activities. Many niches contain minimal sugar concentrations (e.g., the urogenital tract), while in others, C. albicans is transiently exposed to sugars (e.g., the oral cavity and gastrointestinal tract). This opportunistic pathogen probably evolved in niches such as these. Also, the evidence suggests that a proportion of C. albicans cells that infect internal organs actively assimilate alternative carbon sources rather than sugars (40, 70). Furthermore, lactate assimilation is essential for the proliferation of C. glabrata in the intestine (38). Our data now suggest that transient exposure to sugar does not prejudice the assimilation of alternative carbon sources and, as a result, the growth of the fungus in these niches. This explains why both gluconeogenic and glycolytic functions can be expressed in C. albicans cells infecting the kidney (40) and why mutations that block gluconeogenesis or the glyoxylate cycle partially attenuate the virulence of C. albicans (39–42). Clearly, C. albicans does not conform to the S. cerevisiae paradigm of glucose regulation, and this is important for the pathogenicity of this yeast.
MATERIALS AND METHODS
Strains and growth conditions.
C. albicans and S. cerevisiae strains (see Table S1 in the supplemental material) were grown at 30°C in minimal medium (0.67% yeast nitrogen base) containing glucose (2%) (SD), lactic acid (2%), oleic acid (0.2%), or mixed amino acids (2%) as the sole carbon source (71).
Strain construction.
C. albicans strains expressing Icl1-Myc3 (CA1395) or Pck1-Myc3 (CA1431) were made by PCR amplification of a Myc3-URA3 cassette and transforming these products into C. albicans RM1000 (see Table S1 in the supplemental material). Insertion at the correct locus was confirmed by diagnostic PCR, and expression of Icl1-Myc3 and Pck1-Myc3 was confirmed by Western blotting. Analogous controls were performed for all the following strain constructions.
To replace a CaICL1 allele in C. albicans with a Myc3-tagged ScICL1 open reading frame (ORF) from S. cerevisiae, the ScICL1 locus was first Myc3 tagged by PCR amplifying the Myc3-URA3 cassette with primers DS_3F (F stands for forward) and DS_3R (R stands for reverse) (see Table S3 in the supplemental material) and transforming this cassette into S. cerevisiae BY4743 to create strain DS3-Y30 (Table S1). The new ScICL1-Myc3-URA3 locus was then PCR amplified using primers DS_5F and DS_5R, and this CaICL1p-ScICL1-MYC3-URA3 cassette was then transformed into C. albicans RM1000 to create strain DSCO1 (CaICL1/CaICL1p-ScICL1-MYC3-URA3) (Table S1) and also transformed into C. albicans CA510 to create strain DSCO1 (Caicl1/CaICL1p-ScICL1-MYC3-URA3) (Table S1).
To introduce the CaICl1-ScICL1-Myc3-URA3 allele into the C. albicans ubi4/ubi4 background (55), the CaICl1-ScICL1-Myc3-URA3 cassette was PCR amplified using the DS_5F and DS_5R primers (see Table S3 in the supplemental material) and transformed into C. albicans DSCO to create strain DSCO3 (ubi4/ubi4 CaICL1/CaICl1-ScICL1-Myc3-URA3) (Table S1).
C. albicans DSCO4 expresses CaIcl1 with the carboxy-terminal ubiquitination site from ScIcl1 at its carboxy terminus (CaICL1/CaICl1-Ubi-Myc3-URA3) (see Table S1 in the supplemental material). This was achieved by PCR amplifying the 3′ end of the ScICL1 ORF along with the conjoined MYC3-URA3 sequences from S. cerevisiae DSCO3 cells using the DS_6F and DS_6Rprimers (Table S3) and transforming this CaICl1-Ubi-Myc3-URA3 cassette into C. albicans RM1000.
The ScICL1 and ScPCK1 loci were epitope tagged in S. cerevisiae. ScICL1 was tagged with Myc9 at its 3′ end by PCR amplification of the MYC9-NAT cassette in pYM21 (72) with primers DS_1F and DS_1R (see Table S3 in the supplemental material) and transformation of S. cerevisiae W303-1B to generate strain DS1-W10 (ScICL1-MYC9-NAT) (Table S1). ScPCK1 was tagged with hemagglutinin (a six-hemagglutinin tag [HA6]) by PCR amplification of the HA6-KlTRP1 (KlTRP1 stands for Kluyveromyces lactis TRP1) cassette in pYM3 (73) with primers DS_2F and DS_2R (Table S3) and transformation of S. cerevisiae W303-1B to create strain DS1-W20 (ScPCK1-HA6-KlTRP1) (Table S1).
The ScICl1 ORF in S. cerevisiae BY4743 was replaced with CaICL1-Myc3 to create the S. cerevisiae strain DS4-Y40 (ScICl1-CaICL1-Myc3-URA3) (see Table S1 in the supplemental material). The CaICL1-Myc3-URA3 ORF was PCR amplified from C. albicans CA1395 (Table S1) using the DS_4F and DS_4R primers (Table S3).
Proteomics.
Replicate cultures of C. albicans NGY152 (CAI4 containing CIp10 [74]) were grown in minimal medium containing glucose (2%), lactic acid (2%), oleic acid (0.2%), or mixed amino acids (2%) as the sole carbon source. Samples from the cultures were taken, then subcultured in the same medium, grown for a further 20 h, and harvested in exponential phase (optical density at 600 nm [OD600] of 0.8). Glucose (2%) was added to some cultures, and the cells were harvested after 2 h. Protein extracts were prepared, subjected to 2D gel electrophoresis, and stained with Coomassie blue as described previously (54). Independent triplicate experiments were done for each growth condition. Gel features were compared using Phoretix 2D Expression (Nonlinear Dynamics, Newcastle upon Tyne, United Kingdom) and GeneSpring (Silicon Genetics, San Carlos, CA). Principal component analysis scores were plotted in 3D (SIMCA-P version 11.0: Umetrics AB, Sweden). Proteins displaying statistically significant differences in mean spot volume (P ≤ 0.05) were identified by MALDI-ToF mass spectrometry of tryptic peptides (54) using MS-Fit and MASCOT (Matrix Science, Boston, MA). Protein annotations were from the Candida Genome Database (http://www.candidagenome.org/). The proteomic data set is available in the supplemental material (see Table S2 in the supplemental material) and at the PRIDE proteomic data repository (http://www.ebi.ac.uk/pride/) (accession numbers 3186 to 3192). The carbon source network was constructed using Cytoscape version (http://www.cytoscape.org/) (75).
Western blotting.
Protein extracts were subjected to Western blotting as described previously (76). Membranes were probed with a mouse anti-Myc antibody (diluted 1:10,000) (Sigma). The secondary antibody was peroxidase-conjugated rabbit anti-mouse IgG. Signals were detected with an enhanced chemiluminescence (ECL) Western blotting kit (Amersham, United Kingdom) and quantified using a Fuji FLA-3000 imager.
RNA analyses.
RNA was prepared (77, 78), yields were quantified using an RNA 6000 Nano assay, and RNA integrity was assessed using an Agilent 2100 bioanalyzer (79). ICL1 and PCK1 transcript levels were quantified to the internal ACT1 mRNA by qRT-PCR using primers (see Table S3 in the supplemental material). RNA samples (2 µg) were incubated in 20-µl reactions with DNase I (1.5 µl), RNase OUT (1.5 µl), and DNase I buffer (2 µl) (Invitrogen, United Kingdom) at room temperature for 15 min. cDNA was then made with SuperScript II reverse transcriptase (Invitrogen, United Kingdom) following the manufacturer’s protocols. Real-time RT-PCR SYBR green (Roche, United Kingdom) was performed using the manufacturer’s instructions with a LightCycler 480 real-time PCR system (Roche).
Lactate and oleic acid assimilation.
Lactate and oleic acid assimilation was assayed by measuring the incorporation by C. albicans and S. cerevisiae of [14C]lactate and [3H]oleic acid into trichloroacetic acid-precipitable material. Yeast cells were grown on YPLactate (2% Bacto peptone and 1% yeast extract containing 2% lactate) or YPOleic acid (2% Bacto peptone and 1% yeast extract containing 0.2% oleic acid) at 30°C to an OD600 of 1, harvested, and resuspended in 1 ml fresh prewarmed YPLactate or YPOleic acid. Glucose (2%) was added to half of the cells, and no glucose was added to control cells. Then, 1.85 MBq of [14C]lactic acid or 37 MBq of [3H]oleic acid was added at time zero, and samples were taken at various times thereafter. The samples were precipitated in 5% trichloroacetic acid at 0°C, washed at 0°C in a series of solutions [(i) fresh 5% trichloroacetic acid containing 0.1% SDS, (ii) 50% ethanol, and (iii) 100% ethanol], dried, and subjected to scintillation counting (Packard BioScience). The results are means and standard deviations from triplicate assays. Similar results were obtained in triplicate independent experiments.
SUPPLEMENTAL MATERIAL
(A) Principal component analysis confirms the significant impact of carbon source on the C. albicans proteome and the reproducibility of the replicate 2D gel profiles. (B) Correlation between the long-term and short-term effects of glucose on protein levels in C. albicans cells grown on oleic acid for all proteins for which changes in expression were detected. (C) Correlation for glycolytic enzymes. (D) Correlation for fatty acid β-oxidation enzymes. (E) Correlation for gluconeogenic, glyoxylate cycle, and TCA cycle enzymes. Download Figure S1, PDF file, 0.1 MB.
Impact of carbon sources on central carbon metabolism in C. albicans. (A) Glucose; (B) lactate; (C) amino acids. Download Figure S2, PDF file, 0.1 MB.
Icl1 and Pck1 protein stability and ICL1 and PCK1 mRNA turnover in oleic acid-grown C. albicans and S. cerevisiae following the addition of glucose. (A and B) CaIcl1-Myc and CaPck1-Myc protein levels (A) and CaICL1 and CaPCK1 mRNA levels (B) after the addition of glucose to C. albicans cells grown on oleic acid. (C and D) ScIcl1-Myc and ScPck1-HA protein levels (C) and ScICL1 and ScPCK1 mRNA levels (D) after glucose addition to S. cerevisiae cells grown on oleic acid. Similar data were obtained from two independent replicate experiments. Download Figure S3, PDF file, 0.1 MB.
ScIcl1 is degraded in C. albicans, but CaIcl1 is stable in S. cerevisiae. (A) ScIcl1 is rapidly degraded in oleic acid-grown C. albicans DSCO1 cells after glucose exposure. Similar data were obtained from two independent replicate experiments. (B) CaIcl1 is stable in oleic acid-grown S. cerevisiae DS4-Y40 cells after glucose exposure. Similar data were obtained from two independent replicate experiments Download Figure S4, PDF file, 0.1 MB.
Inactivation of polyubiquitin inhibits glucose-accelerated ScIcl1 degradation in oleic acid-grown C. albicans DSCO3 cells. Similar data were obtained from two independent replicate experiments. Download Figure S5, PDF file, 0.1 MB.
Addition of a consensus ubiquitin site stimulates glucose-accelerated degradation of the CaIcl1-Ubi-Myc protein in oleic acid-grown C. albicans. Similar data were obtained from two independent replicate experiments. Download Figure S6, PDF file, 0.1 MB.
Glucose does not inhibit [3H]oleic acid assimilation by C. albicans RM1000 but does block [3H]oleic acid assimilation in S. cerevisiae W303-1B. Similar data were obtained from two independent replicate experiments. Download Figure S7, PDF file, 0.1 MB.
C. albicans and S. cerevisiae strains
List of changes in the C. albicans proteome detected in response to alterations in carbon source
Primers used in this study
ACKNOWLEDGMENTS
We thank Esperanza Lopez-Franco for help with the construction of C. albicans Pck1-Myc and Icl1-Myc strains. We thank Phil Cash, Mike Lorenz, and Ben Distel for stimulating discussions and helpful advice. D.S. was supported by a scholarship from Universiti Sains, Malaysia. M.D.L. was the recipient of a Carnegie/Caledonian scholarship from the Carnegie Trust, and a Sir Henry Wellcome postdoctoral fellowship from the Wellcome Trust (096072). This work was also supported by the United Kingdom Biotechnology and Biological Research Council (BBS/B/06679 and BB/F00513X/1), the Wellcome Trust (080088 and 097377), and the European Commission (PITN-GA-2008-214004 and ERC-2009-AdG-249793).
Footnotes
Citation Sandai D, et al. 2012. The evolutionary rewiring of ubiquitination targets has reprogrammed the regulation of carbon assimilation in the pathogenic yeast Candida albicans. mBio 3(6):e00495-12. doi:10.1128/mBio.00495-12.
REFERENCES
- 1. Jacob F, Monod J. 1961. Genetic regulatory mechanisms in the synthesis of proteins. J. Mol. Biol. 3:318–356 [DOI] [PubMed] [Google Scholar]
- 2. Beckwith JR. 1967. Regulation of the lac operon. Recent studies on the regulation of lactose metabolism in Escherichia coli support the operon model. Science 156:597–604 [DOI] [PubMed] [Google Scholar]
- 3. Crabtree HG. 1928. The carbohydrate metabolism of certain pathological overgrowths. Biochem. J. 22:1289–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Barford JP, Hall RJ. 1978. An examination of the Crabtree effect in Saccharomyces cerevisiae: the role of respiratory adaptation. J. Gen. Microbiol. 114:267–275 [Google Scholar]
- 5. Yin Z, Smith RJ, Brown AJ. 1996. Multiple signalling pathways trigger the exquisite sensitivity of yeast gluconeogenic mRNAs to glucose. Mol. Microbiol. 20:751–764 [DOI] [PubMed] [Google Scholar]
- 6. Yin Z, et al. 2003. Glucose triggers different global responses in yeast, depending on the strength of the signal, and transiently stabilizes ribosomal protein mRNAs. Mol. Microbiol. 48:713–724 [DOI] [PubMed] [Google Scholar]
- 7. Gancedo JM. 1998. Yeast carbon catabolite repression. Microbiol. Mol. Biol. Rev. 62:334–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Carlson M. 1999. Glucose repression in yeast. Curr. Opin. Microbiol. 2:202–207 [DOI] [PubMed] [Google Scholar]
- 9. Johnston M. 1999. Feasting, fasting and fermenting. Glucose sensing in yeast and other cells. Trends Genet. 15:29–33 [DOI] [PubMed] [Google Scholar]
- 10. Thevelein JM, de Winde JH. 1999. Novel sensing mechanisms and targets for the cAMP-protein kinase A pathway in the yeast Saccharomyces cerevisiae. Mol. Microbiol. 33:904–918 [DOI] [PubMed] [Google Scholar]
- 11. Rolland F, Winderickx J, Thevelein JM. 2001. Glucose-sensing mechanisms in eukaryotic cells. Trends Biochem. Sci. 26:310–317 [DOI] [PubMed] [Google Scholar]
- 12. Gancedo JM. 2008. The early steps of glucose signalling in yeast. FEMS Microbiol. Rev. 32:673–704 [DOI] [PubMed] [Google Scholar]
- 13. Gounalaki N, Thireos G. 1994. Yap1p, a yeast transcriptional activator that mediates multidrug resistance, regulates the metabolic stress response. EMBO J. 13:4036–4041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Görner W, et al. 1998. Nuclear localization of the C2H2 zinc finger protein Msn2p is regulated by stress and protein kinase A activity. Genes Dev. 12:586–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stanhill A, Schick N, Engelberg D. 1999. The yeast ras/cyclic AMP pathway induces invasive growth by suppressing the cellular stress response. Mol. Cell. Biol. 19:7529–7538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Garreau H, et al. 2000. Hyperphosphorylation of Msn2p and Msn4p in response to heat shock and the diauxic shift is inhibited by cAMP in Saccharomyces cerevisiae. Microbiology 146(Part 9):2113–2120 [DOI] [PubMed] [Google Scholar]
- 17. Ozcan S, Johnston M. 1999. Function and regulation of yeast hexose transporters. Microbiol. Mol. Biol. Rev. 63:554–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kaniak A, Xue Z, Macool D, Kim JH, Johnston M. 2004. Regulatory network connecting two glucose signal transduction pathways in Saccharomyces cerevisiae. Eukaryot. Cell 3:221–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Usaite R, et al. 2009. Reconstruction of the yeast Snf1 kinase regulatory network reveals its role as a global energy regulator. Mol. Syst. Biol. 5:319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mercado JJ, Smith R, Sagliocco FA, Brown AJ, Gancedo JM. 1994. The levels of yeast gluconeogenic mRNAs respond to environmental factors. Eur. J. Biochem. 224:473–481 [DOI] [PubMed] [Google Scholar]
- 21. Scheffler IE, de la Cruz BJ, Prieto S. 1998. Control of mRNA turnover as a mechanism of glucose repression in Saccharomyces cerevisiae. Int. J. Biochem. Cell Biol. 30:1175–1193 [DOI] [PubMed] [Google Scholar]
- 22. Ashe MP, De Long SK, Sachs AB. 2000. Glucose depletion rapidly inhibits translation initiation in yeast. Mol. Biol. Cell 11:833–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Entian KD, Dröll L, Mecke D. 1983. Studies on rapid reversible and non-reversible inactivation of fructose-1,6-bisphosphatase and malate dehydrogenase in wild-type and glycolytic block mutants of Saccharomyces cerevisiae. Arch. Microbiol. 134:187–192 [DOI] [PubMed] [Google Scholar]
- 24. López-Boado YS, Herrero P, Gascón S, Moreno F. 1987. Catabolite inactivation of isocitrate lyase from Saccharomyces cerevisiae. Arch. Microbiol. 147:231–234 [DOI] [PubMed] [Google Scholar]
- 25. Hämmerle M, et al. 1998. Proteins of newly isolated mutants and the amino-terminal proline are essential for ubiquitin-proteasome-catalyzed catabolite degradation of fructose-1,6-bisphosphatase of Saccharomyces cerevisiae. J. Biol. Chem. 273:25000–25005 [DOI] [PubMed] [Google Scholar]
- 26. Jiang H, Tatchell K, Liu S, Michels CA. 2000. Protein phosphatase type-1 regulatory subunits Reg1p and Reg2p act as signal transducers in the glucose-induced inactivation of maltose permease in Saccharomyces cerevisiae. Mol. Gen. Genet. 263:411–422 [DOI] [PubMed] [Google Scholar]
- 27. Horak J, Regelmann J, Wolf DH. 2002. Two distinct proteolytic systems responsible for glucose-induced degradation of fructose-1,6-bisphosphatase and the Gal2p transporter in the yeast Saccharomyces cerevisiae share the same protein components of the glucose signaling pathway. J. Biol. Chem. 277:8248–8254 [DOI] [PubMed] [Google Scholar]
- 28. Regelmann J, et al. 2003. Catabolite degradation of fructose-1,6-bisphosphatase in the yeast Saccharomyces cerevisiae: a genome-wide screen identifies eight novel GID genes and indicates the existence of two degradation pathways. Mol. Biol. Cell 14:1652–1663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mercado JJ, Vincent O, Gancedo JM. 1991. Regions in the promoter of the yeast FBP1 gene implicated in catabolite repression may bind the product of the regulatory gene MIG1. FEBS Lett. 291:97–100 [DOI] [PubMed] [Google Scholar]
- 30. Mercado JJ, Gancedo JM. 1992. Regulatory regions in the yeast FBP1 and PCK1 genes. FEBS Lett. 311:110–114 [DOI] [PubMed] [Google Scholar]
- 31. Gancedo JM, Gancedo C. 1997. Gluconeogenesis and catabolite inactivation, p 359–377 In Zimmermann FK, Entian KD, Yeast sugar metabolism. Technomic Publishing, Basel, Switzerland [Google Scholar]
- 32. Schüle T, Rose M, Entian KD, Thumm M, Wolf DH. 2000. Ubc8p functions in catabolite degradation of fructose-1,6-bisphosphatase in yeast. EMBO J. 19:2161–2167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sabina J, Brown V. 2009. Glucose sensing network in Candida albicans: a sweet spot for fungal morphogenesis. Eukaryot. Cell 8:1314–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Merico A, Sulo P, Piskur J, Compagno C. 2007. Fermentative lifestyle in yeasts belonging to the Saccharomyces complex. FEBS J. 274:976–989 [DOI] [PubMed] [Google Scholar]
- 35. Odds FC. 1988. Candida and candidosis. Bailliere Tindall, London, United Kingdom [Google Scholar]
- 36. Calderone RA. 2002. Candida and candidiasis. ASM Press, Washington, DC [Google Scholar]
- 37. Buchalter SE, Crain MR, Kreisberg R. 1989. Regulation of lactate metabolism in vivo. Diabetes Metab. Rev. 5:379–391 [DOI] [PubMed] [Google Scholar]
- 38. Ueno K, et al. 2011. Intestinal resident yeast Candida glabrata requires Cyb2p-mediated lactate assimilation to adapt in mouse intestine. PLoS One 6:e24759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lorenz MC, Fink GR. 2001. The glyoxylate cycle is required for fungal virulence. Nature 412:83–86 [DOI] [PubMed] [Google Scholar]
- 40. Barelle CJ, et al. 2006. Niche-specific regulation of central metabolic pathways in a fungal pathogen. Cell. Microbiol. 8:961–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Piekarska K, et al. 2006. Peroxisomal fatty acid beta-oxidation is not essential for virulence of Candida albicans. Eukaryot. Cell 5:1847–1856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ramírez MA, Lorenz MC. 2007. Mutations in alternative carbon utilization pathways in Candida albicans attenuate virulence and confer pleiotropic phenotypes. Eukaryot. Cell 6:280–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ihmels J, et al. 2005. Rewiring of the yeast transcriptional network through the evolution of motif usage. Science 309:938–940 [DOI] [PubMed] [Google Scholar]
- 44. Martchenko M, Levitin A, Hogues H, Nantel A, Whiteway M. 2007. Transcriptional rewiring of fungal galactose-metabolism circuitry. Curr. Biol. 17:1007–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lavoie H, Hogues H, Whiteway M. 2009. Rearrangements of the transcriptional regulatory networks of metabolic pathways in fungi. Curr. Opin. Microbiol. 12:655–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lorenz MC, Bender JA, Fink GR. 2004. Transcriptional response of Candida albicans upon internalization by macrophages. Eukaryot. Cell 3:1076–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rodaki A, et al. 2009. Glucose promotes stress resistance in the fungal pathogen Candida albicans. Mol. Biol. Cell 20:4845–4855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Niimi M, Kamiyama A, Tokunaga M. 1988. Respiration of medically important Candida species and Saccharomyces cerevisiae in relation to glucose effect. J. Med. Vet. Mycol. 26:195–198 [PubMed] [Google Scholar]
- 49. Brown AJ. 2005. Integration of metabolism with virulence in Candida albicans, p 185–203 In Brown AJ, Fungal genomics. Mycota XIII. Springer-Verlag, Heidelberg, Germany [Google Scholar]
- 50. Ene IV, et al. 2012. Host carbon sources modulate cell wall architecture, drug resistance and virulence in a fungal pathogen. Cell. Microbiol. 14:1319–1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Futcher B, Latter GI, Monardo P, McLaughlin CS, Garrels JI. 1999. A sampling of the yeast proteome. Mol. Cell. Biol. 19:7357–7368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gygi SP, Rochon Y, Franza BR, Aebersold R. 1999. Correlation between protein and mRNA abundance in yeast. Mol. Cell. Biol. 19:1720–1730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. de Nobel H, et al. 2001. Parallel and comparative analysis of the proteome and transcriptome of sorbic acid-stressed Saccharomyces cerevisiae. Yeast 18:1413–1428 [DOI] [PubMed] [Google Scholar]
- 54. Yin Z, et al. 2004. Proteomic response to amino acid starvation in Candida albicans and Saccharomyces cerevisiae. Proteomics 4:2425–2436 [DOI] [PubMed] [Google Scholar]
- 55. Leach MD, Stead DA, Argo E, MacCallum DM, Brown AJ. 2011. Molecular and proteomic analyses highlight the importance of ubiquitination for the stress resistance, metabolic adaptation, morphogenetic regulation and virulence of Candida albicans. Mol. Microbiol. 79:1574–1593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Radivojac P, et al. 2010. Identification, analysis, and prediction of protein ubiquitination sites. Proteins 78:365–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fradin C, et al. 2005. Granulocytes govern the transcriptional response, morphology and proliferation of Candida albicans in human blood. Mol. Microbiol. 56:397–415 [DOI] [PubMed] [Google Scholar]
- 58. Kusch H, et al. 2008. A proteomic view of Candida albicans yeast cell metabolism in exponential and stationary growth phases. Int. J. Med. Microbiol. 298:291–318 [DOI] [PubMed] [Google Scholar]
- 59. Leng P, Sudbery PE, Brown AJ. 2000. Rad6p represses yeast-hypha morphogenesis in the human fungal pathogen Candida albicans. Mol. Microbiol. 35:1264–1275 [DOI] [PubMed] [Google Scholar]
- 60. Roig P, Gozalbo D. 2003. Depletion of polyubiquitin encoded by the UBI4 gene confers pleiotropic phenotype to Candida albicans cells. Fungal Genet. Biol. 39:70–81 [DOI] [PubMed] [Google Scholar]
- 61. Atir-Lande A, Gildor T, Kornitzer D. 2005. Role for the SCFCDC4 ubiquitin ligase in Candida albicans morphogenesis. Mol. Biol. Cell 16:2772–2785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Li WJ, et al. 2006. The F-box protein Grr1 regulates the stability of Ccn1, Cln3 and Hof1 and cell morphogenesis in Candida albicans. Mol. Microbiol. 62:212–226 [DOI] [PubMed] [Google Scholar]
- 63. Trunk K, et al. 2009. Depletion of the cullin Cdc53p induces morphogenetic changes in Candida albicans. Eukaryot. Cell 8:756–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Leach MD, Brown AJ. 2012. Posttranslational modifications of proteins in the pathobiology of medically relevant fungi. Eukaryot. Cell 11:98–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Roig P, Martínez JP, Gil ML, Gozalbo D. 2000. Molecular cloning and characterization of the Candida albicans UBI3 gene coding for a ubiquitin-hybrid protein. Yeast 16:1413–1419 [DOI] [PubMed] [Google Scholar]
- 66. Nantel A, et al. 2002. Transcription profiling of Candida albicans cells undergoing the yeast-to-hyphal transition. Mol. Biol. Cell 13:3452–3465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Enjalbert B, Nantel A, Whiteway M. 2003. Stress-induced gene expression in Candida albicans: absence of a general stress response. Mol. Biol. Cell 14:1460–1467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tsong AE, Miller MG, Raisner RM, Johnson AD. 2003. Evolution of a combinatorial transcriptional circuit: a case study in yeasts. Cell 115:389–399 [DOI] [PubMed] [Google Scholar]
- 69. Yin Z, et al. 2009. A proteomic analysis of the salt, cadmium and peroxide stress responses in Candida albicans and the role of the Hog1 stress-activated MAPK in regulating the stress-induced proteome. Proteomics 9:4686–4703 [DOI] [PubMed] [Google Scholar]
- 70. Thewes S, et al. 2007. In vivo and ex vivo comparative transcriptional profiling of invasive and non-invasive Candida albicans isolates identifies genes associated with tissue invasion. Mol. Microbiol. 63:1606–1628 [DOI] [PubMed] [Google Scholar]
- 71. Sherman F. 1991. Getting started with yeast. Methods Enzymol. 194:3–21 [DOI] [PubMed] [Google Scholar]
- 72. Janke C, et al. 2004. A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast 21:947–962 [DOI] [PubMed] [Google Scholar]
- 73. Knop M, et al. 1999. Epitope tagging of yeast genes using a PCR-based strategy: more tags and improved practical routines. Yeast 15:963–972 [DOI] [PubMed] [Google Scholar]
- 74. Walker LA, et al. 2009. Genome-wide analysis of Candida albicans gene expression patterns during infection of the mammalian kidney. Fungal Genet. Biol. 46:210–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Cline MS, et al. 2007. Integration of biological networks and gene expression data using cytoscape. Nat. Protoc. 2:2366–2382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Smith DA, Nicholls S, Morgan BA, Brown AJ, Quinn J. 2004. A conserved stress-activated protein kinase regulates a core stress response in the human pathogen Candida albicans. Mol. Biol. Cell 15:4179–4190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Enjalbert B, et al. 2006. Role of the Hog1 stress-activated protein kinase in the global transcriptional response to stress in the fungal pathogen Candida albicans. Mol. Biol. Cell 17:1018–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Enjalbert B, MacCallum DM, Odds FC, Brown AJ. 2007. Niche-specific activation of the oxidative stress response by the pathogenic fungus Candida albicans. Infect. Immun. 75:2143–2151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Imbeaud S, Auffray C. 2005. Functional annotation: extracting functional and regulatory order from microarrays. Mol. Syst. Biol. 1:2005, 0009 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Principal component analysis confirms the significant impact of carbon source on the C. albicans proteome and the reproducibility of the replicate 2D gel profiles. (B) Correlation between the long-term and short-term effects of glucose on protein levels in C. albicans cells grown on oleic acid for all proteins for which changes in expression were detected. (C) Correlation for glycolytic enzymes. (D) Correlation for fatty acid β-oxidation enzymes. (E) Correlation for gluconeogenic, glyoxylate cycle, and TCA cycle enzymes. Download Figure S1, PDF file, 0.1 MB.
Impact of carbon sources on central carbon metabolism in C. albicans. (A) Glucose; (B) lactate; (C) amino acids. Download Figure S2, PDF file, 0.1 MB.
Icl1 and Pck1 protein stability and ICL1 and PCK1 mRNA turnover in oleic acid-grown C. albicans and S. cerevisiae following the addition of glucose. (A and B) CaIcl1-Myc and CaPck1-Myc protein levels (A) and CaICL1 and CaPCK1 mRNA levels (B) after the addition of glucose to C. albicans cells grown on oleic acid. (C and D) ScIcl1-Myc and ScPck1-HA protein levels (C) and ScICL1 and ScPCK1 mRNA levels (D) after glucose addition to S. cerevisiae cells grown on oleic acid. Similar data were obtained from two independent replicate experiments. Download Figure S3, PDF file, 0.1 MB.
ScIcl1 is degraded in C. albicans, but CaIcl1 is stable in S. cerevisiae. (A) ScIcl1 is rapidly degraded in oleic acid-grown C. albicans DSCO1 cells after glucose exposure. Similar data were obtained from two independent replicate experiments. (B) CaIcl1 is stable in oleic acid-grown S. cerevisiae DS4-Y40 cells after glucose exposure. Similar data were obtained from two independent replicate experiments Download Figure S4, PDF file, 0.1 MB.
Inactivation of polyubiquitin inhibits glucose-accelerated ScIcl1 degradation in oleic acid-grown C. albicans DSCO3 cells. Similar data were obtained from two independent replicate experiments. Download Figure S5, PDF file, 0.1 MB.
Addition of a consensus ubiquitin site stimulates glucose-accelerated degradation of the CaIcl1-Ubi-Myc protein in oleic acid-grown C. albicans. Similar data were obtained from two independent replicate experiments. Download Figure S6, PDF file, 0.1 MB.
Glucose does not inhibit [3H]oleic acid assimilation by C. albicans RM1000 but does block [3H]oleic acid assimilation in S. cerevisiae W303-1B. Similar data were obtained from two independent replicate experiments. Download Figure S7, PDF file, 0.1 MB.
C. albicans and S. cerevisiae strains
List of changes in the C. albicans proteome detected in response to alterations in carbon source
Primers used in this study