

Abstract
The initial response of lymphoid malignancies to glucocorticoids (GCs) is a critical parameter predicting successful treatment. Although being known as a strong inducer of apoptosis in lymphoid cells for almost a century, the signaling pathways regulating the susceptibility of the cells to GCs are only partly revealed. There is still a need to develop clinical tests that can predict the outcome of GC therapy. In this paper, I discuss important parameters modulating the pro-apoptotic effects of GCs, with a specific emphasis on the microRNA world comprised of small players with big impacts. The journey through the multifaceted complexity of GC-induced apoptosis brings forth explanations for the differential treatment response and raises potential strategies for overcoming drug resistance.
1. Introduction
1.1. Glucocorticoids in the Treatment of Lymphoid Malignancies
Glucocorticoids (GCs) are among the most effective drugs used in the treatment of hematopoietic malignancies of the lymphoid lineage in virtue of their ability to induce apoptosis of these cancerous cells [1–3]. The main hematopoietic cancer types that respond well to GC therapy include T acute lymphoblastic leukemia (T-ALL), chronic B lymphocytic leukemia (CLL), multiple myeloma (MM), Hodgkin's lymphoma (HL), and non-Hodgkin's lymphoma (NHL). GCs appear, however, to have little value in the treatment of acute or chronic myeloid leukemia (AML/CML). A major drawback of GC therapy is the gradual development of resistance to GC during treatment that limits the clinical utility of this drug. Poor response to a 7-day monotherapy with the GC prednisone is one of the strongest predictors of adverse outcomes in the treatment of pediatric ALL [2, 4]. A great challenge today is to develop strategies that can overcome the drug resistant phenotype. For this purpose it is important to understand the underlying mechanisms of GC resistance and the signaling pathways regulating apoptosis induced by GCs.
Besides inducing apoptosis of lymphoid cells, GCs are used in palliative care. GC treatment produces rapid symptomatic improvements, including relief of fever, sweats, lethargy, weakness, and other nonspecific effects of cancer.GCs decrease the severity of chemotherapy-induced emesis. GCs are also used in the clinics for other medical conditions such as autoimmune diseases, asthma, ulcerative colitis, chronic obstructive pulmonary disease, kidney diseases, and rheumatologic disorders due to their strong anti-inflammatory and immunosuppressive properties. GC therapy is hampered by a variety of metabolic and medical complications, including insulin resistance, diabetes, hypertension, glaucoma, osteoporosis, and osteonecrosis with increased risk of bone fractures [5–10]. Diabetes may develop by direct GC-mediated induction of apoptosis in insulin-producing beta cells of the Langerhans islets [11–13], and osteoporosis may develop due to apoptosis of osteoblasts [14–16]. GCs also suppress cell growth and proliferation processes in the brain [17, 18].
Besides being used as monotherapy at high dosages, GCs are frequently combined with other chemotherapeutic drugs to achieve rapid and more efficient therapeutic effects. For the treatment of T-ALL, GCs such as prednisone, methylprednisolone, and dexamethasone are usually used in combination with other chemotherapeutic drugs such as vincristine, daunorubicine, L-asparaginase, cytosine arabinoside, doxorubicin, and cyclophosphamide. This multidrug regimen prolongs remission, minimizes the long-term use of prednisone, and thus reduces the steroid-mediated adverse effects.
Typical B-cell chronic lymphocytic leukemia (CLL) in the early stage of progression responds well to combination chemotherapy including an alkylating agent (such as chlorambucil) plus or minus prednisolone.Advanced stages of the disease often require the addition of an anthracycline and a vinca alkaloid for successful therapy. One commonly used combination is cyclophosphamide, doxorubicin, vincristine, and prednisolone, a drug combination termed CHOP. Rituximab, a chimeric monoclonal antibody directed against the B-cell specific antigen CD20, is often added to the therapy, which is here termed R-CHOP. Rituximab is also combined with fludarabine and cyclophosphamide in the treatment of CLL [19, 20]. Another antibody proved to be efficient against CLL in combination with methylprednisolone is alemtuzumab, which targets CD52. This combination is also effective in p53-defective CLLs [21]. However, alemtuzumab was not found to be superior to rituximab [22]. The immunomodulatory drug lenalidomide shows also good activity in relapse/refractory or treatment-naïve CLL [23, 24].
CHOP is also used for non-Hodgkin's lymphomas and anaplastic large cell lymphoma (ALCL). Sometimes interferon-α2b is added in the treatment of the former. GCs are also effective for the treatment of Hodgkin's lymphoma. Here, prednisone has been used in combination with carmustine, vincristine (Oncovin), procarbazine (MOPP), and rituximab. Recently, brentuximab vedotin (Adcetris), an antibody directed towards CD30 conjugated with the anti-tubulin chemotherapeutic agent monomethyl auristatin E [25], has been approved for the treatment of Hodgkin's lymphoma and systemic anaplastic large cell lymphoma. CD30 expression is restricted to only a relative small population of activated T and B cells, and therefore this treatment is expected to be more selective for CD30-positive tumor cells. Another monoclonal antibody entered the clinics is epratuzumab, which targets CD22 and is proved to be efficient in the treatment of adult non-Hodgkin's lymphoma as a single agent or in combination with chemotherapy. A phase II clinical trial showed that combining epratuzumab with rituximab and CHOP (ER-CHOP) may have a favorable response on diffusing large B-cell non-Hodgkin lymphoma (DLBCL) [26].
Multiple myeloma (MM) has frequently been treated with vincristine, doxorubicine (Adriamycin), and dexamethasone (VAD) or prednisone/melphalan. Bortezomib (Velcade), lenalidomide, and to a lesser extend thalidomide have proven efficient in the treatment of MM in combination with dexamethasone. This is in addition to autologous or allogeneic hematopoietic stem cell transplantation. Lenalidomide is a 4-amino-glutamyl analogue of thalidomide that lacks the neurological side effects of thalidomide and has emerged as a drug with activity against various hematological malignancies [27, 28]. Bortezomib is a selective inhibitor of the 26S proteasome that stabilizes many cell cycle-regulatory proteins. The antitumor effects of bortezomib in lymphoid tumors have been attributed to NFκB inhibition through stabilization of its inhibitor IκB. Other tumors that have been treated with combination chemotherapy involving a GC include medulloblastoma, primitive neuroectodermal tumors, and ependymomas.
1.2. Alternative Treatment Approaches for Overcoming GC Resistance
One major obstacle in the therapy of lymphoid malignancies is the appearance of GC resistant cells. Drug resistance may occur at the level of the glucocorticoid receptor (GR) or through alterations in downstream regulatory pathways. In most GC-resistant ALL primary biopsy specimens, GR was found to be functional [29], suggesting that pharmacological intervention may restore drug sensitivity. Several strategies have been developed that aim to overcome drug resistance through specifically targeting anti-apoptotic pathways. Below, three major strategies applicative for GC therapy are discussed.
1.2.1. Targeting Anti-Apoptotic Bcl-2 Members as a Therapeutic Approach for Overcoming GC Resistance
GC resistance may occur due to overexpression of anti-apoptotic proteins of the Bcl-2 superfamily [30, 31]. Among these, Bcl-2, Bcl-XL, and Mcl-1 are frequently overexpressed in lymphomas [32].
1.2.1.1. Targeting Bcl-2 with Small Molecular Inhibitors. Small molecules that target the anti-apoptotic proteins of the Bcl-2 family are attractive drugs that should be able to overcome GC resistance. One example is ABT-737, a BH3 mimetic that inhibits the pro-survival function of Bcl-2, Bcl-XL, and Bcl-w and induces apoptosis in a variety of cancer cell types including leukemias [33–35]. Treatment of the lymphoma-prone Eμ-Myc transgenic mice with ABT-737 prevented the development of Myc-driven lymphomagenesis [36], understating the need for these anti-apoptotic proteins. Combined use of ABT-737 and the dual specificity PI3K/mTOR inhibitor PI-103 led to loss of c-Myc expression and apoptosis of Burkitt's lymphoma cells, whose tumorigenicity is driven by overexpression of the c-Myc gene [37].
The pro-apoptotic effect of ABT-737 in CLL depends on sufficient amount of Bcl-2 that tonically sequesters the pro-apoptotic Bim protein [38]. Also, the sensitivity of lymphoma cell lines to Bcl-2 antagonism is directly related to the amount of Bcl-2 primed with Bim [35]. The sequestration of Bim may explain the marked chemosensitivity of CLL and follicular lymphoma (FL) that express abundant Bcl-2 [38]. This drug-responsive condition is termed “primed for death”.
ABT-737 potentiated the effect of vincristine, dexamethasone, and L-asparaginase (VXL) treatment on ALL cells [39] and could potentiate the effect of the VXL combination in chemoresistant human primary ALL xenografts [40]. This study also shows a synergistic effect between the three components of the VXL regimen. An additive effect was observed in primary MM cells when ABT-737 was combined with dexamethasone [41, 42].
ABT-263 (Navitoclax) is a second generation, orally bioavailable small molecule Bcl-2 family protein inhibitor that has entered clinical trials with promising efficacy on CLL [43–46]. ABT-263 has been shown to have synergistic effects with R-CHOP treatment on mantle cell lymphoma [45]. It also synergizes with rapamycin in killing lymphomas [47].
1.2.1.2. Overcoming ABT-737 Resistance by Targeting Mcl-1. Resistance to ABT-737 occurs in lymphoma cells with high expression of Mcl-1 and/or Bfl-1/A1 [48]. The pro-apoptotic Bim that is displaced from Bcl-2 by ABT-737, becomes captured by either Bfl-1 or Mcl-1. The resistance could be overcome by decreasing the Mcl-1 level with the cyclin-dependent kinase (Cdk) inhibitors flavopiridol and PHA767491 [48], or by inhibiting mTOR complex 1 (mTORC1) [49] or glycolysis [49, 50].
Another approach to overcome Mcl-1-dependent resistance is to use the small molecule obatoclax (GX15-070) that has entered clinical trials in the combined treatment of various hematopoietic neoplasms [51–53]. Obatoclax disrupts the interaction between Mcl-1 and its pro-apoptotic counterparts including Bak, Bax, and Noxa [54, 55]. Obatoclax and flavopiridol synergized in overcoming drug resistance in human myeloma cells through a mechanism involving Bim and Noxa [56]. The multikinase inhibitor sorafenib could synergize with Obatoclax in inducing apoptosis in acute myeloid leukemia (AML) through downregulating Mcl-1 [57]. Obatoclax could overcome GC resistance in ALL through induction of apoptosis and autophagy, an effect that depends on the pro-apoptotic Bak and to a certain extent also on Beclin-1 [58, 59], a mammalian orthologue of yeast Atg6 that plays a central role in autophagy [60]. Under certain conditions, cell death induced by Obatoclax and GC may be executed in the absence of both Bax and Bak [59]. Under these conditions, necroptosis ensues necroptosis ensues, a process mediated by RIP-1 (receptor-interacting protein-1) kinase and the cylindromatosis deubiquitinase CYLD [59]. RIP-1 kinase plays a dual role in determining the cell fate. It may promote either cell death or cell survival dependent on its ubiquitinated state, which is regulated by CYLD and A20, two NFκB target genes [61]. Altogether, there is a general consensus that Obatoclax might be a favorable drug that ought to be combined with dexamethasone/prednisone and/or rapamycin to overcome GC resistance in ALL cells and other hematological lymphoid malignancies.
1.2.1.3. Overcoming Bcl-2-Mediated Resistance with Small Molecular Inhibitors of XIAP (X-Linked Inhibitor of Apoptosis). Bcl-2-mediated resistance in CLL may also be overcome by small molecular inhibitors of the anti-apoptotic XIAP (X-linked inhibitor of apoptosis) when exposed to TRAIL [62, 63]. XIAP and the cellular cIAPs 1 and 2 are expressed at high levels in CLL cells [62, 63]. XIAP inhibitors enhanced Bcl-2 cleavage and induced a conformational change in Bax [62]. Similarly, XIAP inhibitors sensitized ALL for CD95-induced apoptosis [64]. In patients with T-ALL, poor prednisone response was associated with increased XIAP expression [65]. XIAP inhibition using the low-molecular-weight SMAC mimetic LBW242 resulted in increased prednisone-induced apoptosis in vitro [65].
1.2.2. Targeting Notch1 as a Therapeutic Approach for Overcoming GC Resistance
Another anti-apoptotic protein that negatively regulates GC-induced apoptosis is Notch1 [66–68]. Notch1 is indispensable for normal T-cell development [69–71] and is an attractive target in the treatment of hematopoietic malignancies of the T lineage [72]. Mice transplanted with bone marrow cells transduced with a constitutively active form of Notch1 develop T-cell neoplasms [73], while mice transgenic for constitutively active form of Notch3 develop thymic lymphomas [74]. Acute lymphoblastic T-cell leukemia is frequently associated with increased Notch signaling [75–79], which may be caused by the chromosomal translocation t(7; 9)(q34; q34.3) [80], gain-of-function mutations of Notch1 [81], and/or mutations in Fbw7 (F-box and WD repeat domain-containing 7), a negative regulator of Notch1 [82].
One approach to avoid Notch activation is to prevent its cleavage by the γ-secretase complex using γ-secretase inhibitors (GSI) [83]. GSIs can induce apoptosis of various lymphoma cell lines [84–87]. However, GSI as a monotherapeutic agent is often insufficient for inducing apoptosis. Rather, GSI can enhance the pro-apoptotic effect of GCs and other chemotherapeutic agents including the mTOR inhibitor rapamycin [84, 88]. GSI restored GR auto-upregulation and induced apoptosis through induction of Bim [88]. GSI does not overcome GC resistance in T-ALL deficient for PTEN [89, 90], supposedly due to elevated Akt activity. The constitutive Akt activation in the absence of PTEN leads to increased glucose metabolism and bypasses the requirement of Notch signaling to sustain cell growth [89]. In this context it should be noted that Notch1 by itself may upregulate the P13K/Akt pathway via its target gene Hes1 [89]. As PTEN is a target of several microRNAs that are often expressed abnormally in cancer (see Section 2.4.2.3), resistance to GSI may be far more prevalent. GSI is also not efficient in T-ALL carrying activating mutations in Notch1. Nevertheless, GSI compounds, such as PF-03084014, have entered clinical trials for refractory T-ALL [91]. Preclinical data do show a synergistic effect between GSI inhibition and GC in reducing xenografted T-ALL tumor burden [92]. Another concern associated with the clinical use of GSIs is severe toxicity to various organs at therapeutic doses, which may be explained by the broad action of Notch1 as well as γ-secretase on various biological systems. The simultaneous use of GCs may prevent the GSI-induced gastrointestinal toxicity via inhibition of goblet cell metaplasia [92]. A more specific inhibition of Notch1 can be achieved by the SAHM1 peptide that prevents Notch-mediated transcription by interfering with the Mastermind-Notch interaction essential for Notch-mediated transcription of target genes [93]. The effect of this peptide on GC sensitivity awaits examination as well as its toxicity. Since Notch signaling is intertwined with the PI3K/Akt/mTOR signaling axis [94–96], the inhibition of the latter has proven to be more efficient in overcoming GC resistance (see Section 1.2.3) and would be a better therapeutic choice.
1.2.3. Targeting Pro-Survival Protein Kinases
Accumulating data show that GC therapy can affect the activity of several protein kinases, and, vice versa, many protein kinases can affect GC-induced apoptosis [30, 31, 97–99]. The mTOR signaling pathway is frequently activated and found to be essential for cell growth and survival in lymphoid malignancies [100–106]. GC resistance frequently appears in malignant cells due to aberrant activation of various protein kinases that exert anti-apoptotic effects [30, 31, 67, 97, 107–109]. One strategy to overcome GC resistance would be to prevent the activities of the PI3K/Akt/mTOR, MEK1/ERK1/2, and other activated protein kinase pathways. The mTOR inhibitor rapamycin especially has proven efficient in sensitizing human GC-resistant T-ALL, B-ALL, MM, and NPM-ALK+ (nucleophosmin-anaplastic lymphoma kinase)-DLBCL to GC-induced apoptosis [110–117]. The combinatory therapy of rapamycin with dexamethasone was proven to be effective also in PTEN-negative cells [111]. A lower dose of dexamethasone was sufficient for reducing T-ALL burden in a xenograft model when used together with rapamycin [111]. One major drawback with rapamycin therapy is its immunosuppressive function, which adds to the immunosuppressive function of GCs.
The dual PI3K/mTOR inhibitor NVP-BEZ235 synergistically enhanced cytotoxicity of dexamethasone, doxorubicine, and cytosine arabinoside (AraC), even in GC-resistant ALL cells [118]. NVP-BEZ235 also overcomes bortezomib resistance in mantle cell lymphoma cells [119]. The broad-acting protein kinase staurosporine was especially effective in overcoming GC resistance in mouse lymphomas that overexpressed Notch-1, Bcl-2, and/or Bcl-XL [120]. This sensitization was achieved through prevention of Akt-mediated inhibition of GSK3 [67] and induction of the pro-apoptotic Nur77 [120]. However, staurosporine was less effective on human T-ALL cell lines (unpublished data), which could rather be sensitized to GC by rapamycin. In order to choose the right kinase inhibitor for combinatory therapy, it is important to determine the kinase responsible for GC resistance prior to therapy.
The cyclin-dependent kinase (Cdk) inhibitors flavopiridol (Alvocidib), BMS-387032 (SNS-032), sunitinib, and sorafenib are currently under clinical trials for relapsed/refractory CLL [121]. Multityrosine kinase inhibitors have also been developed for the treatment of lymphoid malignancies. These include Vandetanib (ZD6474), Bosutinib (SKI-606), TKI258 (CHIR-258), Pazopanib (GW786034), and Axitinib (AG013736). CHIR-258, a potent inhibitor of Flt3 (fms-like tyrosine kinase receptor-3), c-Kit tyrosine kinase, and fibroblast growth factor receptor 3 (FGFR3), prevented cell growth of FGFR3-positive human multiple myeloma cell lines and augmented their sensitivity to GC-induced apoptosis [122]. Importantly, neither interleukin-6 (IL-6) nor stromal cells conferred resistance to CHIR-258 [122].
Other protein kinase inhibitors with more cell-type specific effects have been developed, which are expected to have less adverse effects. The classical example for efficient use of a specific protein kinase inhibitor in the clinics is the Bcr-Abl kinase inhibitor STI-572 (Imatinib) used for the treatment of chronic myelogenic leukemia (CML) [123]. A similar strong response of a single agent was observed in ALK+-anaplastic large cell lymphoma (ALCL) patients treated with Crizotinib, an inhibitor of the ALK tyrosine kinase [124]. Two patients that relapsed after CHOP treatment received Crizotinib as a single agent. Both showed complete response [124].
Another promising target is the B-cell receptor (BCR) signaling, which is important during B-cell oncogenesis and is a key to the survival of malignant B cells, including CLL and DLBCL [125, 126]. The survival of DLBCL may depend on the nonligand-dependent (tonic) signals from the BCR. The BCR signaling can be targeted with small molecular inhibitors directed against Bruton's tyrosine kinase (Btk), spleen tyrosine kinase (Syk), or phosphoinositide 3′-kinase (PI3K) isoform p110δ (PI3Kδ), all being efficient in the treatment of CLL [125]. Targeting Btk with the inhibitor PCI-32765 leads to disruption of BCR signaling and was effective in a preclinical model of B cell non-Hodgin's lymphoma [127, 128]. PCI-32765 seems also to be promising for the treatment of CLL [128–131] and MM [132]. Importantly, PCI-32765 induced apoptosis in CLL cells even in the presence of various exogenous stimuli, including CD40L, BAFF, IL-6, and IL-4 and when cultivated together with stromal cells [131]. Two other Btk inhibitors, Ibrutinib and AVL-263, are also under investigation for CLL [121]. The Syk (spleen tyrosine kinase) inhibitor Fostamatinib had clinical activity in non-Hodgkin lymphoma and CLL [133]. Syk is a cytoplasmic tyrosine kinase that is important for immunoreceptor signaling in B cells. Syk has also been shown to be critical for the survival and maintenance of mature normal and malignant B cells [125, 134] and is frequently expressed at high levels in follicular lymphoma [135]. The PI3Kδ inhibitor GS-1101 (CAL-101) had preclinical and clinical activity against CLL, mantle cell lymphoma, and MM [121, 129, 136–138]. While the PI3Kα and β isoforms are ubiquitously expressed, PI3Kδ expression is largely restricted to hematopoietic cells, where it plays a role in B-cell homeostasis and function [139]. PI3Ks are constitutively activated in CLL cells [140–142]. The effect of the Btk, Syk, and PI3Kδ kinase inhibitors on the sensitivity to GCs warrants investigations.
Accordi et al. [143] found aberrant activation of protein kinases in poor prognosis pediatric B-cell precursor-ALL patients. The p56Lck (lymphocyte cell-specific tyrosine kinase) activity was enhanced in patients with poor clinical response to prednisone with respect to those with good response [143]. p56Lck is a nonreceptor tyrosine kinase of the Src oncogene family mostly expressed in T cells where it plays an essential role in activation and development, and in some B cells. Its activity is negatively regulated by the membrane-bound tyrosine kinase Csk (c-Src tyrosine kinase). The p56Lck inhibitor Dasatinib (BMS-354825) was shown to enhance apoptosis induction by dexamethasone in otherwise GC-resistant CLL cells [144]. This finding concurs with the observation by Sade et al. [68] showing that Notch-mediated resistance of a mouse lymphoma cell line could be overcome by inhibiting p56Lck. In MM, a synergistic effect was observed between the Aurora A kinase inhibitor MNL8237 (Alisertib) and dexamethasone [145].
AMPK (AMP activated protein kinase) activation has a dual effect on cell death and survival, which contextually depends on signaling alterations with related oncogenic pathways [146]. MLL-rearranged tumors showed Bcl-2 hyperphosphorylation through AMPK activation [143]. However, in ALL and CLL, activation of AMPK by AICAR (5-Aminoimidazole-4-carboxamide riboside or Acadesine), a cell-permeable nucleotide, induces growth inhibition and apoptosis [146–148]. However, AICAR prevented glucocorticoid-induced apoptosis [149] and thus cannot be combined with steroids in the treatment of lymphoid malignancies.
Of note, inhibition of either Bcl-2 family members, Notch1, or the Akt/mTOR survival pathways was independently sufficient for sensitizing resistant cells to GC, suggesting a tight crosstalk between these pathways, interruption of one of them being sufficient for abrogating the resistant phenotype. However, it is likely that using a combination of these three strategies together with GC should lead to a more efficient therapy, which may require lower dosages with reduced adverse effects.
2. Parameters Affecting the Susceptibility of Lymphoid Malignancies to GC-Induced Apoptosis
In order to develop strategies to overcome GC resistance, it is essential to understand the signaling network regulating GC-induced apoptosis. Main factors affecting the response to GC include the basal and inducible GR expression levels, the induction of and basal expression of genes involved in the intrinsic apoptotic pathway, the ability of GR to translocate to the mitochondria, the activity of GSK3 (glycogen synthase kinase 3), the general protein kinase activation profile of the cell prior to and following GC therapy, the expression profile of anti-apoptotic proteins, and the activities of pro-survival signaling pathways. The main traits will only be briefly described here as these have been extensively reviewed elsewhere [30, 31, 99, 150–153], and the scope of this paper is to provide updated data with a specific focus on the microRNA world that has emerged to comprise important regulators of most biological processes.
2.1. Sufficient Expression Levels of the Glucocorticoid Receptor (GR/NR3C1)
Numerous factors have been shown to affect GC responsiveness by regulating glucocorticoid receptor (GR) activity and expression level. These include GR co-activators and corepressors [154, 155], GR splice variants [156–159], GR isoforms [160, 161], and regulators of GC nucleocytoplasmic shuttle [162–164].
The transcription of human GR is regulated by at least 11 different promoters (1A1, 1A2, 1A3, 1B, 1C, 1D, 1E, 1F, 1H, 1I, and 1J) [155, 165], seven of them being embedded in a highly enriched CpG island region subjected to methylation and harbor single nucleotide polymorphisms (SNPs) that affect their activity [166]. Promoter 1A is involved in the upregulation of GR by GC in some kinds of T cells, while downregulated in other cell types [167–169]. GC resistance in primary pediatric T- and B-ALL could not be correlated with either basal or stimulated expression of the 1A-, 1B, or 1C transcripts [170].
The GR expression level prior and following GC therapy affects drug responsiveness. The cellular response to GCs depends on sufficient GR expression [30, 171–179], and resistance to GC therapy has been associated with downregulation and loss of GR expression in malignant plasma cells [180, 181]. However, most primary ALL cells showed upregulation of GR expression upon prednisolone treatment regardless of their phenotype or sensitivity to GC-induced apoptosis, suggesting that other factors are more dominant for conferring a GC-resistant phenotype in these cells [29, 170, 182–184]. Many glucocorticoid-regulated genes (e.g., FKBP5 and SOCS1) were upregulated by dexamethasone in all primary ALL xenografts tested, suggesting for a functional GR in these leukemic cells [29]. Also, Beesley et al. [185] observed that receptor mutation is not a common mechanism of GC resistant in primary ALL [185]. However, the minor C allele of rs10482605 (1C) has been associated with a higher complication rate in childhood ALL [186]. A BclI polymorphism in the NR3C1 gene was associated with increased lymphocyte response to methylprednisolone [187]. Also, initial good responder cells may develop resistance upon repeated GC dosages, a phenomenon that sometimes occurs due to downregulation of GR [156, 179, 188, 189]. Regulation of GR expression by microRNAs is discussed in Section 4.1.
Posttranslational modifications of GR are another way of regulating its target gene specificity and involve several cell-signaling cascades [30, 190, 191]. GR can be phosphorylated at Ser211 by CDKs and p38 MAP kinase, and at Ser226 by JNK. Phosphorylation of GR modulates its transcriptional activity, alters its protein stability and subcellular location [192–195]. GR phosphorylation appears to be cell-cycle dependent [196, 197] and may affect GC-sensitivity of T-ALL cells [98, 195].
2.2. The Ability to Upregulate the Pro-Apoptotic Gene Bim in Response to GC
2.2.1. GR as a Transcription Factor
GR is a well-known regulator of transcription. In the absence of ligand, GR is mostly located to the cytosol sequestered to heat-shock protein complexes [30, 162]. Following GC binding to GR, the receptor undergoes phosphorylation, dissociates from the heat-shock complexes, dimerizes, and translocates to the nucleus where it either promotes or represses a whole series of genes. Transcriptional activation is either directly mediated by binding of GR to glucocorticoid response elements (GREs), or through interaction with other transcription factors such as forkhead transcription factors, thereby increasing their transcriptional activity on target genes. GR may repress gene expression either through binding to negative GREs (nGREs) or through interaction with and inhibition of the transcription factors activating protein-1 (AP-1) and NFκB. The O-GlcNAc transferase (OGT) was found to be involved in GC-mediated transrepression [198]. Hundreds of genes are regulated by GCs [199–203], and some genes are differentially regulated in GC-sensitive versus GC-resistant cells [29, 199, 204].
2.2.2. Importance of Bim in GC-Induced Apoptosis
Of special importance is the induction of the pro-apoptotic Bim (BH3-only B-cell lymphoma 2 (Bcl-2) interacting mediator of cell death; or BCL2L11—Bcl-2-like apoptosis initiator-11) for achieving the propensity to undergo apoptosis in response to GC [29, 30, 67, 205–208]. The central role of Bim in GC-induced apoptosis is understated by the partial GC response of Bim−/− thymocytes [205], and GC resistance of lymphoma cells after knocking down Bim [67, 207]. Bim is often expressed at high basal levels in lymphoid cells [30, 120, 209, 210], and in these cells there is no further need for upregulating Bim in order to achieve an apoptotic response to GCs [30, 59]. However, in several T-ALL and B-ALL cells, an upregulation of Bim in response to GCs is an absolute must, especially when the basal level is low.
Bim was shown to be upregulated in GC-sensitive primary T-ALL samples, but not in resistant ones [29, 182]. Also, a comparison of established T-ALL cell lines, Bim was upregulated in the sensitive ones only [211]. When sufficient Bim expression cannot be achieved, GC resistance pursued. A significantly lower Bim expression was detected in high risk childhood ALL patients who exhibited slow early response to a standard 4-drug induction regimen compared with patients who responded rapidly [212].
Homozygous deletion of Bim has been seen in many mantle cell lymphomas [213] and silencing of Bim by promoter methylation and mutation is common in B-cell lymphomas [214]. However, in pediatric ALL, no correlation between Bim CpG methylation and GC resistance was found [29]. Rather, GC resistance in primary pediatric ALL samples correlated with decreased histone H3 acetylation [29]. The histone deacetylase inhibitor vorinostat relieved Bim repression and exerted synergistic antileukemic efficacy with dexamethasone both in vitro and in vivo using a xenograft model [29]. Bim has been shown to be a prognostic biomarker for early prednisolone response in pediatric ALL [4].
2.2.3. The Pro-Apoptotic Function of Bim and Other Proteins in GC-Induced Apoptosis
Bim is a potent pro-apoptotic protein belonging to the Bcl-2 protein family [215, 216]. Bim binds to the pro-survival proteins Bcl-2, Bcl-XL, and Mcl-1, thereby allowing Bax and Bak to promote apoptosis [217]. Bim may also directly bind to Bax and Bak, triggering a conformational change required for their subsequent oligomerization on the mitochondrial outer membrane [215]. Bim appears in various alternative splice variants, which exhibit different intrinsic toxicities and modes of regulation [218]. In GC-resistant primary CLL, Bim was upregulated by dexamethasone, but failed to activate Bax and Bak due to exclusive sequestration to Bcl-2 [219].
Bim may cooperate with the pro-apoptotic PUMA (p53 upregulated modulator of apoptosis) in mediating apoptosis induced by dexamethasone [220]. In B-lymphoid cells, Bmf (Bcl-2 modifying factor) is also important for GC-induced apoptosis [221]. Other pro-apoptotic members of the Bcl-2 family that is not directly upregulated by GCs, but may contribute to the cell death response, include Bid, Bad, and Noxa. Essential downstream mediators are Bak and Bax [222] that are activated by Bim. Also the thioredoxin-interacting protein Txnip (VDUP1/TBP-2) has been shown to be upregulated by GC and could contribute to GC-induced apoptosis in one mouse lymphoma cell line [223]. During GC monotherapy of childhood ALL, GC was found to repress the expression of the pro-apoptotic PMAIP/Noxa, which could be one mechanism leading to impaired GC sensitivity [224]. Conditional overexpression of Noxa restored GC sensitivity [224]. Another transcript of the Bim locus, termed “Bam,” is also induced by GCs in ALL cells, but its importance in GC-induced apoptosis is still not defined [225].
2.2.4. Regulation of Bim Expression by Transcription Factors
Bim expression is tightly regulated both at the transcription and posttranscriptional levels [215, 218] (Figure 1). No GRE element has been found in the Bim promoter. Rather, GC-induced Bim expression in lymphoid cells requires p38 activation and is mediated by the forkhead transcription factor FoxO3a/FKHR-L1 [226]. FoxO3a has also been shown to promote Bim transcription in various other cellular systems [227–229] and may cooperate with Runx1 (Runt-related transcription factor 1) [230]. Differential recruitment of FoxO3a to the Bim promoter was observed after dexamethasone treatment of GC-sensitive versus GC-resistant childhood ALL xenografts [29]. FoxO3 was found to be an immediate early GR target, whose transcription is further enhanced by stimuli that activate the AMP-activated protein kinase AMPK [231]. The activity of FoxO transcription factors is tightly regulated, inhibited by Akt and ERK signaling, while promoted by p38 signaling [232–236].
Figure 1.
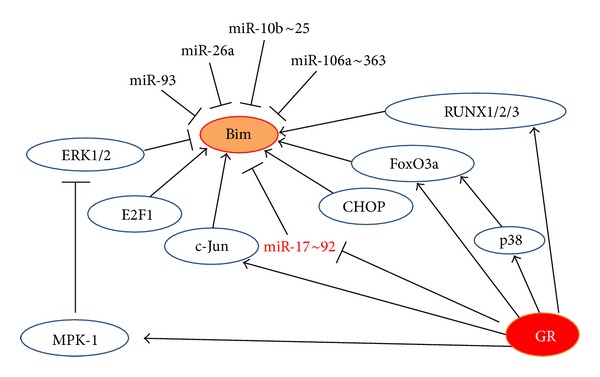
Regulation of Bim expression. Details are described in Sections 2.2.3–2.2.5.
Both ERK1/2 and Akt antagonize apoptosis by reducing the Bim expression level. ERK1/2 also directly phosphorylates Bim leading to its proteosomal-dependent degradation [237]. The ribosomal protein S6 kinase (RSK) activated downstream of ERK1/2, phosphorylates BimEL, providing a binding site for the F-box proteins beta-transducin repeat containing protein (βTrCP)1 and βTrCP2, which promote the polyubiquitination of BimEL [238]. ERK1/2 phosphorylates BimEL at Ser55, Ser69, and Ser73. The ERK1/2-mediated phosphorylation of BimEL at Ser69 facilitates optimal phosphorylation by RSK at Ser93, Ser94, and Ser98 and this motif serves as the binding sites for βTrCP1/2 [238]. While ERK1/2 lowers the affinity of Bim for Mcl-1 and Bcl-XL and targets Bim for degradation [239], phosphorylation of Bim by JNK increases the pro-apoptotic activity of Bim [240, 241]. GCs may repress ERK1/2 activity through upregulation of mitogen-activated protein kinase phosphatase 1 (MKP-1) [242]. Several drugs that inhibit the ERK1/2 and PKB/Akt pathways may facilitate upregulation of Bim expression. MEK inhibitor-induced Bim expression per se is usually insufficient to promote apoptosis. Additional signals are required, such as simultaneous inhibition of the PKB/Akt pathway or the downstream mammalian target of rapamycin (mTOR) kinase [218]. Apoptosis may be induced in a variety of ALL cells when cotreated with dexamethasone and a MEK/ERK inhibitor or an Akt inhibitor [67, 108, 243].
Early studies by the Thompson research group noticed that c-Jun played a role in GC-induced apoptosis [244]. An increase in c-Jun was observed in GC-sensitive, but not GC-resistant T-ALL cell lines, while c-Fos and JunD were unaffected by the steroid. Antisense to c-Jun conferred GC resistance [244]. Recently, the c-Jun issue was revisited. Chen et al. [204] reconfirmed that c-Jun was upregulated by GCs in GC-sensitive, but not GC-resistant ALL cells. They further showed that c-Jun is recruited to the AP-1 site of the Bim promoter upon GC treatment [204]. Another study showed that dexamethasone-induced Bim expression was decreased in cells harboring a dominant-negative c-Jun [245], suggesting a role for c-Jun in the upregulation of Bim. This research group also found a Runx2-dependent upregulation of Bim. A p38 inhibitor prevented dexamethasone-induced expression of Runx2, c-Jun, and Bim, suggesting that p38-MAPK activation acts upstream to the induction of these three molecules [245].
2.2.5. Regulation of Bim Expression by MicroRNAs
Another level of Bim regulation is through microRNAs. Bim transcription is repressed by the miR-17~92 microRNA cluster [246], which, in turn, is repressed by GCs [206]. Thus, one mechanism by which GCs upregulate Bim is through repression of miR-17~92. Of note, the miR-17~92 cluster is often overexpressed or amplified in human cancers [247–252], thereby preventing the upregulation of Bim required for an apoptotic response. Another microRNA that suppresses Bim expression is miR-26a, which is frequently upregulated in T-ALL patients [253]. In gastric cancer, miR-106a~363 targets Bim [254]. The miR-106a~363 cluster located at chromosome Xq26.2 is the paralogue of miR-17~92 and encodes for miR-363, miR-106a, and miR-20b [255]. In hepatocellular carcinoma, miR-25 of the miR-106b~25 cluster targets Bim [256]. Also, the miR-106b~25 cluster, which includes miR-106b, miR-93 and miR-25, is a paralogue of the miR-17~92 cluster and located on chromosome 7 within the thirteenth intron of the protein-coding gene Mcm7.
2.2.6. Regulation of FoxO Transcription Factors by MicroRNAs
Also, the FoxO transcription factors, important for Bim upregulation, are regulated by microRNAs [257] (Figure 2). FoxO1 and FoxO3 transcripts might be targeted by miR-182 [258–261], miR-1 [262], miR-27a [258], miR-96 [258], and miR-155 [263, 264]. miR-155 plays a role in the activation and function of B and T lymphocytes [265, 266] (see Section 3.1.6). miR-182 is upregulated in several human lymphoid cell lines [261]. miR-182 expression was higher in GC-resistant cells in comparison to GC sensitive ones [261]. Increased expression of miR-182 reduced total FoxO3a expression in T-ALL cells with consequent lower Bim expression. FoxO3a and Bim increased upon downregulation of miR-182, suggesting that miR-182 is involved in conferring GC resistance [261].
Figure 2.
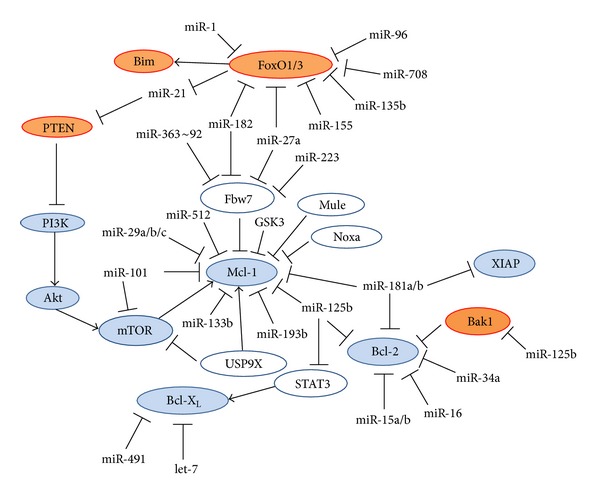
Interplay between microRNAs, pro- and anti-apoptotic proteins affecting GC-induced apoptosis. Details are described in Sections 2.2.6, 2.4.2, and 2.5.
The expression of the miR-182~96~183 cluster was induced in splenocytes from mouse with experimental systemic lupus erythematosus (SLE) [267], suggesting a role of these microRNAs in the breakdown of immunological tolerance and the manifestation of chronic autoimmune inflammation. This microRNA cluster was also upregulated upon T-cell activation by an IL-2-dependent manner. Prevention of the expression of the miR-182~96~183 cluster led to increased FoxO1 expression and limited population expansion of activated T-helper cells, due to increased cell death [260].
Vice versa, FoxO3a was found to negatively regulate the oncomiR miR-21, which may be one mechanism by which FoxO3a regulates apoptosis [268]. As miR-21 targets PTEN [269, 270], activation of FoxO3 by GCs [271] may be one mechanism responsible for the GC-induced reduction in Akt activity.
2.3. Mitochondrial Translocation of GR
Besides function as a transcription factor in the nucleus, GR was found to translocate to the mitochondria in GC-sensitive, but not GC-resistant, lymphoma cell lines [272]. GR was also found to translocate to the mitochondria in GC-sensitive thymocytes [272, 273]. Although there is one paper describing an interaction between GR and Bcl-2 in the mitochondria [274], GC-induced mitochondrial GR translocation in GC-sensitive thymocytes and lymphoma cells proceeded in the absence of Bcl-2 [272]. Exclusive overexpression of GR in the mitochondria was sufficient for inducing apoptosis [272], suggesting that mitochondrial GR may contribute to GC-induced apoptosis.
Glucocorticoids are known to exert multiple effects on the mitochondria. Glucocorticoid treatment inhibited Complex I and Complex III of the electron transport chain, and the mitochondria was found to be the primary source of H2O2 production required for GC-induced apoptosis of lymphoma cells [275, 276]. GCs may interact with the mitochondrial thioredoxin Trx2, a redox regulator [277], and directly modulate mitochondrial gene transcription [278]. Several mitochondrial metabolite and protein transporters and two subunits of the ATP synthase were downregulated in T-ALL and precursor B-ALL cells at the gene expression level by dexamethasone. These changes were observed in GC-sensitive, but not GC-resistant, cells [279]. Corticosterone and other steroids were found to directly act on mitochondria to inhibit mitochondrial ATP production by suppressing electron transfer from NADH to the electron transfer chain through complex I [280].
2.4. The Kinome
The cellular protein kinase network (kinome) has critical influence on the GC sensitivity of lymphoid cells [30, 31, 97, 281]. Above, I discussed the importance of p38 in Bim induction and activity. Below, I will provide data supporting an involvement of GSK3 (glycogen synthase kinase 3) in GC-induced apoptosis, and the antagonism of its activity by protein kinases such as Akt and mTOR, which leads to GC resistance.
2.4.1. GSK3 (Glycogen Synthase Kinase 3) Activity
The activity of GSK3 was found to be essential for GC-induced apoptosis [67, 282]. GSK3 inhibitors prevented GC-induced apoptosis, and GC resistance frequently occurs through inhibition of GSK activity. Reactivating GSK3 by using inhibitors of the PI3K-Akt or mTOR pathways sensitized GC-resistant cells to GC-induced apoptosis [67, 108, 115, 116, 243, 283]. GSK3α was found to interact with GR in the absence of ligand and released from GR following exposure to GC [67]. GC treatment led to interaction of both GSK3α and GSK3β with Bim [67]. GSK3β also regulates GR transcriptional activity of Bim, IAP1 (Inhibitor of Apoptosis 1), and GILZ (glucocorticoid-induced leucine zipper) [282, 284]. This effect of GSK3 on GR transactivation was independent of known GSK3β phosphorylation sites [284]. GSK3β was also shown to be involved in GC-induced bone lost [285].
2.4.2. Activity of the PI3K-PKB/Akt, mTOR, and ERK Pro-Survival Pathways
The PI3K/Akt and mTOR signaling pathways are frequently hyperactivated in GC-resistant T-ALL [104, 286, 287] and is associated with poor prognosis and chemotherapeutic resistance in pediatric B-precursor ALL [288]. mTOR is a crucial regulator of cell metabolism, growth, and proliferation and mTOR is positively regulated by PI3K/Akt and Notch1 [96, 289], while negatively regulated by the tuberous sclerosis tumor suppressor complex (TSC1/TSC2). mTORC2 activity was essential for Notch-driven T lymphomagenesis [290]. Activation of mTOR contributes to tumor cell survival in ALK (anaplastic lymphoma kinase)-positive ALCL (anaplastic large cell lymphoma) [102], mantle cell lymphoma [103], childhood B-precursor ALL [112], T-ALL [110], and AML [291]. Akt and mTOR confer drug resistance by phosphorylating a series of targets [292, 293]. Phosphorylation and inactivation of GSK3 is a major cause for GC resistance [67] that can be overcome by reactivating GSK3, for example, by Akt inhibitors or mTOR inhibitors. As mentioned in Section 1.2.3, the mTOR inhibitor Rapamycin is efficient in overcoming GC resistance in various lymphoid malignancies. GC resistance can also be overcome in Akt-active lymphoma cells by inhibiting Src members (e.g., by PP1), PI3K (e.g., Wortmannin), or an Akt inhibitor [67, 68].
Combination of GC with rapamycin or GC with Obatoclax led to reduced Akt phosphorylation at Ser473 [59], suggesting that mTOR may also act upstream to Akt [294]. mTORC1 directly phosphorylates Akt/PKB on Ser473 and facilitates Thr308 phosphorylation by PDK1 [295]. GCs could also independent of other cytotoxic agents reduce mTOR activity in lymphoid cells [296]. Low-dose arsenic trioxide could sensitize GC-resistant ALL to Dex through an Akt-dependent pathway [286]. Inhibition of mTOR with rapamycin, which binds to FKBP12, leads to increased Bim expression and overcomes Ras-dependent survival signals [297]. Synergy between mTOR inhibitors (e.g., rapamycin (Sirolimus) and CCI-779 (Temsirolimus)) and other chemotherapeutic agents has been observed in B- and T-lineage ALL cell lines and preclinical models [96, 298].
2.4.2.1. Negative Regulation of Akt by PTEN. The Akt activity is negatively regulated by PTEN (phosphatase and tensin homolog deleted on chromosome 10), a tumor suppressor gene that is suppressed, mutated, or deleted at high frequency in a large number of cancers [299]. PTEN mutations or deletions are frequent in T-ALL and PTEN deletions are associated with less favorable outcome in T-ALL [104, 300]. The PTEN status of the cell affects drug sensitivity. For instance, treatment of T-ALL with gamma secretase inhibitor (GSI) was only efficient if the cells expressed functional PTEN [90]. One mechanism by which Notch confers GC resistance is through PTEN inhibition leading to Akt activation. PTEN specifically catalyzes the dephosphorylation of 3′-phosphate of the inositol ring in phosphatidylinositol (3,4,5)-triphosphate (PIP3) resulting in the biphosphate product phosphatidyl (4,5)-biphosphate (PIP2). PIP3 is a second messenger generated by PI3K that binds to the pleckstrin homology (PH) domain of Akt, which allows its phosphorylation and activation by the 3-phosphoinositide-dependent protein kinase 1 (PDK1) [292].
2.4.2.2. Regulation of PTEN Stability by Phosphorylation and Ubiquitination. Taken into account the important role of PTEN in determining drug sensitivity, mechanisms regulating PTEN activity and stability have strong impact on the drug response. PTEN is regulated by several mechanisms [301]. Besides gene mutation and deletion, reduced PTEN expression has been attributed to epigenetic events such as promoter methylation [302, 303]. At the posttranslational level, phosphorylation and ubiquitination decrease PTEN protein levels, while oxidation and acetylation reduce PTEN activity [301]. Rak phosphorylation of PTEN at Tyr336 stabilizes the PTEN protein [304], while phosphorylation at Thr366, Ser370, Ser380, Thr382, and Ser385 by casein kinase 2 (CK2) and GSK3β reduces its stability [305, 306].
PTEN is regulated by the protooncogene ubiquitin ligase NEDD4-1 (neural precursor cell expressed, developmentally downregulated 4) that promotes PTEN for proteasomal degradation [307]. In multiple human cancer samples where the genetic background of PTEN was normal, but its protein level was low, NEDD4-1 was highly expressed [307]. Upon TCR/CD28 stimulation of T cells, PTEN undergoes inactivation by NEDD4-1 [308]. The association between PTEN and NEDD4 could be impeded by the E3 ubiquitin ligase Cbl-b (Casitas-B-lineage lymphoma protein-b) [308]. Cbl-b−/− T cells show elevated Akt activity, which was abrogated by simultaneous deficiency in NEDD4 [308]. PTEN is also negatively regulated by the anti-apoptotic XIAP (X-linked inhibitor of apoptosis) that promotes PTEN for polyubiquitination and proteosomal degradation [309]. Induction of apoptosis in B-CLL by arsenic trioxide was shown to lead to activation of c-Jun-NH2 terminal kinase (JNK), inactivation of AKT and NFκB, XIAP downregulation, and PTEN upregulation [310]. Two other E3 ligases downregulating PTEN include WWP2 (WW-domain containing protein-2 or AIP-2, atrophin-1-interacting protein 2) [311], and CHIP (chaperone-associated E3 ligase C terminus of Hsc70-interacting protein) [312]. Recently, PTEN was shown to be upregulated by dexamethasone [313].
2.4.2.3. Regulation of PTEN Stability by MicroRNAs. PTEN expression can also be repressed by a range of microRNAs including the miR-17~92 cluster [247, 248], miR-106b~25 [314], miR-21 [269], miR-26a [253, 315], miR-29b [316], miR-214 [317, 318], miR-216a and miR-217 [319], miR-212 [320], miR-221, and miR-222 [321] (Figure 3).
Figure 3.
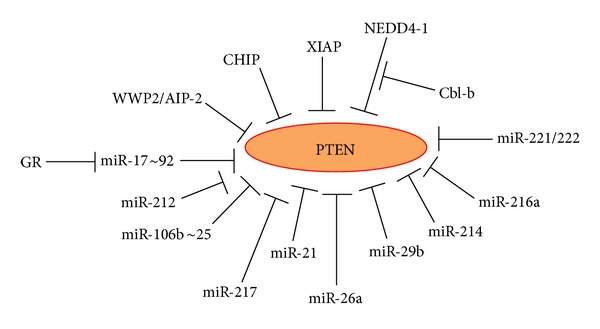
Regulation of PTEN expression. Details are described in Sections 2.4.2.1–2.4.2.3.
2.5. Expression Levels of Anti-Apoptotic Proteins of the Bcl-2 Superfamily
2.5.1. Bcl-2 and Bcl-X L
Bcl-2 and Bcl-XL are anti-apoptotic proteins residing in the mitochondrial outer membrane and in the endoplasmic reticulum. They prevent apoptosis of various chemotherapeutic drugs including GCs by capturing pro-apoptotic members of the Bcl-2 superfamily, including Bim, Bax, and Bak [215, 322, 323]. Bcl-2 may also regulate gene expression [324, 325], cell cycle [326–328], activate ERK1/2 [324, 329], and modulate the activities of transcription factors such as p53 [330], E2F [325], NFκB [331], and Notch [332, 333]. Bcl-2 promotes T-cell lymphoma in a p27Kip1-deficient background [334]. This may be explained by the ability of Bcl-2 to modulate p27Kip1 expression and promote G0 arrest [325, 327, 331, 335, 336].
Long-term exposure to GCs could overcome resistance caused by either Bcl-2 or Bcl-XL [30, 120, 337]. Overexpression of Bcl-2 is common in leukemias and lymphomas [338–341]. In follicular lymphoma (FL) and diffuse large B-cell lymphoma (DLBCL), Bcl-2 upregulation is commonly due to the t(14,18)(q32; q21) translocation, which places the Bcl-2 gene under the control of Ig heavy chain enhancers [342–344].
2.5.1.1. Targeting of Bcl-2 by MicroRNAs. Overexpression of Bcl-2 is common in CLL due to the loss or downregulation of the human chromosome 13q14 locus, which harbors the miR-15a and miR-16-1 cluster [345]. These microRNAs directly target the anti-apoptotic Bcl-2 protein [346]. Overexpression of either microRNA was sufficient to completely abrogate Bcl-2 expression in CLL cells. Overexpression of miR-15a and miR-16-1 in CLL cells led to cleavage of procaspase-9 and PARP (poly-ADP-ribose polymerase) and activation of the intrinsic apoptosis pathway. These two microRNAs could serve as natural antisense Bcl-2 actors that have potential use in the therapy of Bcl-2 overexpressing tumors [346].
The tumor-suppressor miR-34a, a pivotal member of the p53 network, also downregulates Bcl-2 [347, 348], which may be one mechanism by which p53 activation leads to downregulation of Bcl-2. Recent studies suggest that miR-125b also may contribute to Bcl-2 repression [349–351]. It also targets Mcl-1 and Bcl-w, and indirectly Bcl-XL by attenuating IL-6/STAT-3 (signal transducer and activator of transcription 3) signaling pathway [350, 352]. miR-125b may function both as tumor suppressor and as an oncogene [350] and has been widely considered as conferring drug resistance, among others by downregulating Bak1 (Bcl-2 antagonist killer 1) [353–355] and Bmf [356]. Over-expression of miR-125b could induce leukemia in a mouse model [357].
miR-181a/b that shows altered expression in CLL could also target Bcl-2, besides acting on Mcl-1 and XIAP [358–360]. Bcl-XL can be targeted by the tumor suppressor microRNA let-7 [361] and miR-491 [362]. A putative GR binding site was found within the promoter region of let7a2 [363].
2.5.2. Mcl-1
A predominant feature of the gene expression signature leading to GC resistance in ALL was found to be elevated expression of the anti-apoptotic Mcl-1 (myeloid cell leukemia sequence 1) [364, 365]. Mcl-1 expression is especially high in MLL-rearranged ALL, which represents an unfavorable type of leukemia that is often highly resistant to GCs [365]. Mcl-1 is also frequently overexpressed in B-cell and mantle-cell lymphomas, CML, CLL, and MM. Mcl-1 expression renders cancer cells resistant to the Bcl-2 antagonist ABT-737.
Mcl-1 is an anti-apoptotic protein that sequesters the pro-apoptotic proteins tBid, Bim, Puma, Noxa, and Bak [366]. Besides preventing GC-induced apoptosis [287], Mcl-1 confers resistance to TRAIL (tumor necrosis factor-related apoptosis inducing ligand)-induced cell death [367].
2.5.2.1. Regulation of Mcl-1 Stability. Mcl-1 differs from Bcl-2 and Bcl-XL in having a short protein turnover regulated by the 26S proteasome and its expression is tightly regulated [368]. Unlike Bcl-2, chromosomal translocations have not been implicated in dysregulated Mcl-1 levels. Rather, cellular signaling regulates Mcl-1 function and expression at the posttranslational level.
Rapamycin, a mTOR inhibitor that sensitizes resistant ALL cells to GC, reduces the expression level of Mcl-1 [113, 287]. Mcl-1 level could also be reduced by the protein kinase inhibitor Sorafenib. The degradation of Mcl-1 depends on GSK3-mediated phosphorylation of Mcl-1 at Ser159 [369, 370]. E3 ubiquitin ligases implicated in the regulation of Mcl-1 include Mule (Mcl-1-ubiquitinase ligase E3) [371], SCFβ-TrCP (Skp1/Cul1/F-box protein β-transducin repeat-containing protein) [369], and Fbw7 (F-box and WD repeat domain-containing 7) which is part of the Skp1-Cullin1-F-box (SCF) E3 ligase complex [372]. The deubiquitinase USP9X (ubiquitin specific peptidase 9 X-linked) is an important regulator of Mcl-1 stability [373]. Silencing of USP9X resulted in loss of Mcl-1. USP9X removes degradative Lys48-linked polyubiquitin chains on Mcl-1. High levels of Mcl-1 correlated with elevated USP9X expression in follicular lymphoma, diffuse large B-cell lymphoma, and some other cancer samples. Increased expression of USP9X mRNA was associated with poor prognosis of multiple myeloma [373]. USP9X also interacts with mTOR, negatively regulating its activity [374].
Interaction with BH3-only family members may also affect Mcl-1 stability. Whereas Noxa may destabilize Mcl-1, Bim increases its stabilization [375]. Noxa-induced degradation of Mcl-1 requires the E3 ligase Mule. Overexpression of Noxa triggered an increase in the Mule/Mcl-1 interaction in parallel to a decrease in Mule/USP9X complex formation [376].
In an Akt-driven, Eμ-Myc lymphoma mouse model, translational regulation of Mcl-1 by mTOR has been implicated in promoting lymphomagenesis [377]. As GC may activate GSK3 [67] and GSK3 inhibits mTOR through phosphorylation of TSC2 [378] and promotes Mcl-1 degradation [369, 370], Mcl-1 expressing lymphoid cells may ultimately undergo apoptosis if the exposure time to GC is sufficiently long. This may explain why many Mcl-1-positive ALL cells exhibit delayed response to GCs, and not complete resistance [67, 108]. Also, the anti-apoptotic function of Mcl-1 appears to require simultaneous expression of other anti-apoptotic Bcl-2 family members [379]. Similarly, overexpression of Mcl-1 in Bcl-2- and Bcl-XL-negative mouse double positive thymic lymphoma cells did not confer GC resistance upon these cells [120]. Usually, Mcl-1 is expressed together with other anti-apoptotic proteins in GC-resistant lymphoid malignancies.
2.5.2.2. Regulation of Mcl-1 by MicroRNAs. Mcl-1 is also regulated by microRNAs (Figure 2), including miR-29a [380], miR-29b [381–383], miR-101 [384], miR-125b [350], miR-181a/b [358, 385], miR-133b [386], miR-193b [387], and miR-512 [388]. ALK-positive anaplastic large cell lymphomas (ALCL) express low levels of miR-29a, whose downregulation requires an active NPM-ALK kinase, and may probably also be due to methylation repression [380]. Enforced miR-29a expression reduced Mcl-1 expression in ALCL cells and reduced tumor growth in a xenografted model [380]. miR-29b is downregulated in primary MM and AML samples and forced overexpression of miR-29b-induced apoptosis in MM and AML cells [381, 383]. miR-29b overexpression also downregulated the expression of the DNA methyltransferase isoforms DNMT1, DNMT3A, and 3B [383]. The global DNA hypomethylation induced by miR-29b led to reexpression of tumor suppressor genes such as the CDK inhibitor p15INK4b [383]. Altogether, these data propose that targeting Mcl-1 with microRNAs such as miR-29 represents a potential tool to constrict tumor growth of Mcl-1 positive lymphomas.
2.5.3. Effect of Bcl-2 Family Proteins on Intracellular Ca2+ Mobilization
GCs release Ca2+ from the endoplasmic reticulum into the cytosol, which in turn increases the amount of mitochondrial Ca2+. The increase in mitochondrial Ca2+ induces cytochrome C release and trigger apoptosis. Elevated expression of calcium-binding proteins S100A8 and S100A9 and of the anti-apoptotic Mcl-1 (myeloid cell leukemia-1) inhibits the free cytosolic Ca2+ and mitochondrial Ca2+ signals, respectively, thereby imposing GC resistance [287, 365, 389, 390]. Downregulation of S100A8 and S100A9 by the Src kinase inhibitor PP2 sensitized MLL-arranged ALL cells otherwise resistant to prednisolone-induced cell death [389]. Bcl-2 inhibits apoptosis in part by decreasing the size of Ca2+ stores in the endoplasmic reticulum resulting in reduced Ca2+ transfer to the mitochondria [391–393]. One mechanism is through interaction of Bcl-2 with IP3R (inositol 1,4,5-triphosphate (InsP3) receptor), which is the principle ER Ca2+ release channel in most cell types [394]. Also, Bcl-XL and Mcl-1 act in part by inhibiting IP3R [393, 395, 396]. Bcl-XL overexpression also leads to reduced expression of IP3R [397].
2.6. Presence of Reactive Oxygen Species (ROS) Scavengers
An increase in hydrogen peroxide (H2O2) is a necessary signal for GC-induced apoptosis [276]. The mitochondria is the source of this signal [275], GCs inhibit complex I and complex III of the electron transport chain [275]. Expression of anti-oxidant defense proteins such as manganese superoxide dismutase, thioredoxin, and catalase prevents GC-induced apoptosis [276, 398–400]. The anti-apoptotic Bcl-2 may regulate the mitochondrial redox state in cancer cells [323, 401].
2.7. Increased Notch Activity
Notch is frequently activated in T-ALL cells, which may be due to mutations in Notch1 (gain-of-function) and/or in the E3 ligase Fbw7 that targets Notch1 for degradation [76–78, 80, 81, 402–405]. Some other E3 ligases also regulate Notch signaling [406, 407]. For example LNX1 (ligand of Numb-protein X1) is a positive regulator of Notch signaling through degradation of Numb, a membrane-associated protein that inhibits the function of the Notch receptor [408]. Neuralized (neur) and Mind bomb (mib) promote the monoubiquitination and endocytosis of Delta [409, 410]. Itch binds to the N-terminal portion of the Notch intracellular domain via its WW domains and promotes ubiquitination of ICN-Notch1 through its HECT ubiquitin ligase domain [411]. Recent studies showed that Notch1 can be activated in leukemic cells through interaction with bone marrow stromal cells that express Notch receptors and ligands [412, 413]. Interaction with bone marrow stroma is also a mechanism for Notch activation in multiple myeloma [414]. The simultaneous expression of Bcl-2 may enforce Notch activity [332, 333]. Cyclin E, which is targeted for degradation by Fbw7 [415, 416], is expressed at higher levels in early relapsed pediatric B-cell precursor ALL patients, who usually show an unfavorable prognosis [143].
Notch1 prevents GC-induced apoptosis, among others, through activation of p56Lck, which activates the PI3K-Akt axis [68], and through the transactivation of its target genes Deltex and Hes1 [88]. Hes1 leads to downregulation of PTEN, thereby activating the PI3K/Akt pathway [88]. Deltex is a RING-domain ubiquitin ligase that may affect Notch activity [417], and its overexpression prevents GC-induced apoptosis [418]. Activation of the pro-survival PI3K/Akt/mTOR pathway by Notch has also been observed in other studies [95, 106, 419, 420] and may be responsible for Notch-mediated inhibition of the p53 tumor suppressor gene [95]. Another mechanism by which Notch1 protects T-ALL cells from GC-induced apoptosis, is through the anti-apoptotic GIMAP5/IAN5 (GTPase of the immunity-associated protein/immune-associated nucleotide-binding protein 5) [421, 422]. GIAMP5/IAN5 interacts with Bcl-2 and Bcl-XL and inhibits apoptosis during T-cell development [423] and is highly expressed in human B-cell lymphoid malignancies [424]. It is localized within the mitochondria and endoplasmic reticulum (ER) and regulates mitochondrial integrity [425]. GIMAP has been linked to immunological diseases such as T-cell lymphopenia and autoimmune diseases [426]. Notch also activates NFκB signaling [74, 427] and induces c-Myc expression [428–430], both contributing to apoptotic resistance. Long-term treatment with GCs can overcome Notch1 resistance [67]. This resistance can be overcome by the simultaneous exposure of the cells to Src inhibitors, PI3K/Akt inhibitors, or mTOR inhibitors [67, 68], understating the importance of the protein kinase network in regulating the effects of Notch1 on GC-induced apoptosis.
A recent report showed that GC sensitivity of T-ALL is associated with GR-mediated inhibition of Notch1 expression [431]. The serum- and glucocorticoid-inducible kinase 1 (SGK1) was also shown to control Notch1 signaling by downregulating its protein stability through Fbw7 ubiquitin ligase [432]. SGK1 phosphorylates Fbw7 at Ser227, an effect inducing ICN-Notch1 ubiquitination and degradation [432]. Despite GC resistance induced by Notch, Notch- and Fbw7-mutated T-ALL shows in general a favorable response to GC therapy and in some studies, but not all, also exhibits a better prognosis [405, 433–436]. This may be related to the fact that GCs may overcome Notch-dependent drug resistance, and in these T-ALL cases the cell survival depends on Notch signaling.
2.7.1. Regulation of Notch Activity by MicroRNAs
Notch activity may be affected by microRNAs [437]. Various microRNAs negatively regulate Fbw7 expression including miR-27a, miR-182, miR-363~92, and miR-223 [253, 438, 439] and may increase the expression of Fbw7-regulated target genes including Notch1, Mcl-1, c-Jun, c-Myc, and Cyclin E [438]. miR-451 and miR-709 suppressed oncogenesis in Notch1-induced mouse T-ALL [440]. miR-150, which is upregulated upon thymocyte maturation, targets Notch3 and thus regulates T-cell proliferation and survival [441]. miR-326 acts in a feedback loop with Notch signaling [442]. The p53-induced miR-34a also targets the Notch1 receptor as well as its ligand DLL1 (Delta like-1) [443, 444].
Prevention of Notch activation in cutaneous T-cell lymphoma (CTLC) by GSI (γ-secretase inhibitor) treatment led to alterations in the microRNA profile of the cell [445]. Among others, miR-27a, miR92b, miR-181a, miR-18a, miR-19b, miR-222, and miR-221 were downregulated, while miR-122 and miR-214 upregulated [445]. miR-27a targets Fbw7/hCDC4 [253, 438, 439], the substrate recognition component of the SCF (Skp1-Cullin-F-box) ubiquitin ligase complex that targets Notch1 for degradation [82]. The repressive effect of miR-27a on Fbw7 mRNA is especially pronounced at the G2/M and early G1 phases [438]. Thus, GSI may indirectly deregulate Notch1 through the miR-27a-Fbw7 pathway. Other targets of miR-27a includes ZBTB10 (zinc finger and BTB domain containing 10), which acts as a repressor of Sp (specificity proteins) transcription factors and induces G1 arrest, and the Myt-1 kinase, which inhibits the transition through G2-M by enhanced phosphorylation and inactivation of Cdc2 (Cdk1, cyclin-dependent kinase 1) [446]. miR-27a is frequently upregulated in pediatric B-ALL [438]. Upregulation of miR-122 by GSI seems to be mediated by p53 and has an antagonistic effect on apoptosis through activation of Akt [85].
2.8. c-Myc Overexpression
c-Myc is, among others, a target of Notch [428–430] and has broad effects on tumorigenesis [447] and modulates GC-induced apoptosis [99, 448]. Conditional overexpression of c-Myc in hematopoietic cells in mice culminated in the formation of malignant T-cell lymphomas and acute myeloid leukemias [449]. c-Myc may also be activated in T-ALL independently of Notch1 [450]. These authors demonstrated a role for the PI3K/Akt axis in c-Myc activation. Dysregulation of the c-Myc gene is a common trait of Burkitt's lymphoma due to chromosomal translocations, the most frequent one being t(8; 14)(q24:q32) involving c-Myc and IgH (Immunoglobulin heavy locus) [451–453]. Other hematopoietic malignancies characterized with c-Myc overexpression include diffuse large B-cell lymphoma (DLBCL), follicular lymphoma, CLL, B-cell lymphoma, and AML [454–459]. Earlier studies have shown that dexamethasone-induced apoptosis of a T-ALL cell line was associated with c-Myc suppression [460, 461]. The GC-mediated down-regulation of c-Myc expression was initially thought to be one mechanism that contributes to apoptosis. Not all studies have confirmed this finding [462], which may be explained by the many signaling pathways induced by GCs.
2.8.1. The c-Myc-E2F1-MicroRNA Network
c-Myc uses distinct mechanisms for activating and repressing gene expression. For transcriptional activation, c-Myc dimerizes with Max and binds to the promoters of its target genes [463–465]. Transcriptional repression is achieved through protein-protein interactions, where it antagonizes the activity of positive regulators of transcriptions [466]. c-Myc also regulates gene expression by regulating microRNA transcription [255]. The c-Myc-mediated upregulation of miR-17 and miR-20a (belonging to the miR-17~92 cluster) negatively regulates E2F1 translation by targeting the 3-UTR of E2F1 mRNA and may therefore fine tune the direct Myc-mediated transcriptional activation of E2F1, allowing a tightly regulated proliferative signal [255] (Figure 4). E2F1-3 also binds to the promoter of the miR-17~92 cluster and activates its transcription, thus generating an autoregulatory feedback loop [467]. Another target of the miR-17~92 cluster is cyclin D1, which also induces the expression of miR-17 and miR-20a by binding to the promoter regulatory region of the miR-17~92 cluster [468]. The miR-17~92 cluster prevents c-Myc-induced apoptosis [469]. The GC-induced down-regulation of miR-17~92 [206] should actually stimulate E2F1 expression, which under certain circumstances may exert pro-apoptotic effects [470]. E2F1 may promote apoptosis through transcriptional activation of the pro-apoptotic miR-15a~16 cluster [471] and by activating JNK [472]. In a B-cell lymphoma model, c-Myc down-regulated a series of microRNAs, an action that may contribute to tumorigenesis [473]. The c-Myc mediated repression of the miR-30 cluster [473] may affect autophagy, as Beclin-1 expression is regulated by miR-30a [474]. Some of the pro-autophagy activity of cancer therapy is mediated through down-regulation of miR-30a [475]. Also the down-regulation of miR-15a and miR-16 by c-Myc [473] is of interest as these microRNAs are deleted or downregulated in over two-thirds of individuals with CLL, and they target the anti-apoptotic Bcl-2 gene [345, 346]. A third miRNA downregulated by c-Myc is the tumor suppressor let-7 miRNA cluster [473], which targets, among others, the Ras oncogene [476], HMGA2 (high mobility group A2) [477, 478], Bcl-XL [361], Cdc25A, CDK6 (cyclin-dependent kinase 6), and cyclin D2 [479]. Other miRNAs repressed by Myc include miR-22, miR-23a/b, miR-26a/b, miR-29a/b/c, miR-34a, miR-146a, miR-150, and miR-195 [465, 473, 480].
Figure 4.
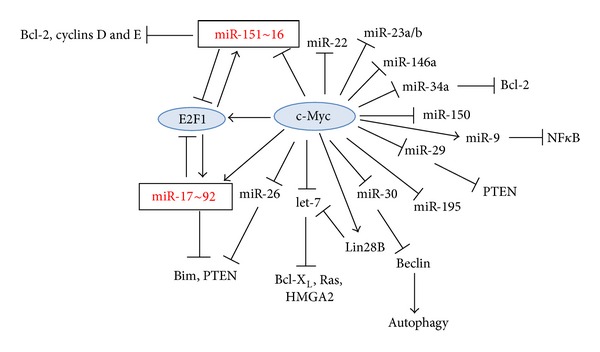
Interplay between microRNAs, c-Myc, and E2F1. Details are described in Section 2.8.1.
miR-26a levels were found to be reduced in various B-cell lymphomas, especially Burkitt lymphoma [465] as well as various solid tumors [481, 482]. B-CLL, which does not have a prominent pathological role of c-Myc, showed higher expression of miR-26a than Myc-dependent Burkitt lymphoma [465]. miR-26 restoration in Burkitt lymphoma or nasopharyngeal carcinomas reduced proliferation and colony formation through G1 arrest and repression of the histone-lysine N-methyltransferase EZH2, a global regulator of gene expression [465, 481, 483]. The tumor-suppression function was only seen in Myc-transformed cells, but not in v-Abl transformed cells [465, 483]. However, in T-ALL, miR-26a was one of five microRNAs that independently promoted tumorigenesis through inhibition of PTEN [253]. In the background of activating mutations in Notch1, miR-26a overexpression decreased the latency of T-ALL [253].
Forced overexpression of miR-34a, miR-150, and miR-15a/16-1 attenuated in vivo tumor growth of Myc-induced B-cell lymphoma [473]. miR-34a is a crucial component of the p53 tumor suppressor network with potential anti-proliferative and pro-apoptotic activity [484–486]. c-Myc transcriptionally induces Lin28B, which is an RNA-binding protein that suppresses the maturation of let-7 family microRNA precursors [487, 488]. This seems to be one mechanism used by c-Myc to repress let-7 [487]. Lin28 is involved in stem cell maintenance [489–491] and is a marker of cancer stem cells [492].
2.9. GC-Induced Autophagy
The effect of autophagy on the cellular response to chemotherapy is dual [493]. Under certain conditions, autophagy acts as a pro-survival mechanism to protect cancer cells from chemotherapy, whereas under other circumstances, autophagy mediates the therapeutic effects of the anticancer agents. Autophagy is regulated by Beclin-1 and autophagy-related genes (ATG) [60]. Another important regulator of autophagy is the activity of mTOR (mammalian target of rapamycin), which is a central element signaling cell growth and enhancing protein translation. When this kinase is inhibited, autophagy is promoted [60].
It should be noted that Beclin-1 may play a dual role in both regulating autophagy and apoptosis, thus being at the cross-road between these two physiological processes. Beclin-1 has recently been recognized as a BH3-only protein interacting with Bcl-2, Bcl-XL and Mcl-1 [59, 60, 494–496]. One report provides evidence that after initiating apoptosis, Beclin-1 is cleaved by caspases and the N-terminal fragment of Beclin can inhibit autophagy, while the C-terminal fragment can amplify mitochondrial-mediated apoptosis [497]. Perturbation of Beclin-1 cleavage by knockin mutation phenocopied the autophagy induction observed in apoptosis-defective cancer cells and rendered chemotherapy resistance both in vitro and in vivo [498]. A role for Beclin in regulating tumorigenesis has been demonstrated in mice with heterozygous disruption of Beclin-1 [499]. These mice have increased frequency of spontaneous malignancies. DLBCL expressing high Beclin-1 levels had a favorable clinical outcome with R-CHOP treatment than those with low Beclin-1 expression [500].
GCs have been shown to promote autophagy in lymphocyte cell lines and primary T-ALL cells [501, 502]. One mechanism for induction of autophagy is through upregulation of the mTOR-inhibitory stress protein Dig2 (dexamethasone-induced gene 2), also known as RTP801 and REDD1 (regulated in development and DNA damage responses 1) [503]. mTOR inhibition by dexamethasone was demonstrated by reduced phosphorylation of S6K (70kD ribosomal protein S6 kinase 1), a member of the RSK family of serine/threonine kinases [503]. Dig2 releases TSC2 from 14-3-3, thereby promoting the assembly of the TSC1/TSC2 complex, which inhibits mTOR [504]. Dig2 knockout thymocytes underwent more extensive dexamethasone-induced cell death, suggesting that autophagy promotes cell survival [503]. However, rapamycin, an inhibitor of mTOR and inducer of autophagy, strongly sensitizes resistant MM and T-ALL cells to GC-induced apoptosis [59, 111, 116, 117], suggesting that induction of autophagy does not always combat apoptosis. It could be that the higher degree of autophagy induced by rapamycin itself may be pro-apoptotic. Bonapace et al. [59] showed that rapamycin induces an autophagy-dependent necroptosis, which is required for childhood T-ALL to overcome GC resistance. Necroptosis is a form of programmed necrosis that occurs when apoptosis is abortive due to caspase inhibition [505]. The GC-mediated necroptosis was mediated by RIP-1 (receptor-interacting protein-1) and CYLD (cylindromatosis) [59]. miR-19, which is frequently overexpressed in T-ALL patients and cell lines, represses CYLD expression [506]. A miR-19 inhibitor induces CYLD expression with consequent decrease in NFκB expression [506]. Obatoclax, a putative antagonist of Bcl-2 family members, could also sensitize T-ALL cells to GC-induced apoptosis through induction of autophagy [59]. This effect was associated with dissociation of the autophagy inducer Beclin-1 from Mcl-1 and decreased mTOR activity [59]. The cell death process could proceed in the absence of Bax and Bak [59]. The apoptosis induced by GC in combination with Obatoclax or rapamycin could be prevented by the autophagy inhibitors 3-methyladenine and bafilomycin [59]. GCs may also induce autophagy by inhibiting Akt activity [501].
2.10. Additional Mechanisms Leading to GC Resistance
CDKN2/p16INK4a, which acts as a G0/G1 cycle inhibitor, is frequently lost in T-ALL [507, 508] and predicts relapse in children with ALL [508–510]. p16INK4a sensitizes T-ALL cell lines to GC-induced apoptosis through induction of BBC3/Puma and repression of Mcl-1 and Bcl-2 [511]. Noxa was repressed in p16INK4a transgenic cells, which could be a result of the simultaneous repression of E2F1 due to retinoblastoma protein and p130 activation [511]. The Bim level was unaffected by p16INK4a overexpression [511]. Diffuse large B-cell lymphoma with CDKN2A deletion had a poor prognosis under R-CHOP treatment [512]. Also, Myc gene arrangement in diffuse large B-cell lymphoma patients had a poor prognosis with R-CHOP chemotherapy [513].
3. MicroRNA in Normal and Malignant Lymphoid Cells
During the last decade, microRNAs have become the focus of having a central role in the pathogenesis of cancer including lymphoid malignancies, besides their role in regulating gene expression during cell division, development, and differentiation [514–523]. MicroRNAs are short noncoding RNAs that induce posttranscriptional gene silencing through base pairing with the 3′ untranslated region (UTR) of their target mRNAs, thereby inhibiting their translation, with subsequent reduced protein levels [524, 525]. Bases 2–7 or 2–8 of the microRNA are primary contributors to target specificity and are referred to as the microRNA seed region. The microRNAs are usually transcribed by RNA polymerase II, and sometimes by RNA polymerase III, into long primary precursor transcripts referred to as pri-miRNAs. miRNA are encoded by one arm of a stem loop structure embedded in introns or, less frequently, exons of protein-coding or noncoding transcripts. In the nucleus, the pri-miRNAs stem loop is cleaved by the nuclear RNase III enzyme Drosha together with its cofactor DGCR8 (DiGeorge syndrome critical region 8)/Pasha (the microprocessor complex) to generate ~70 nucleotides long precursors called pre-miRNAs. In some cases, an entire intron consists of such a stem loop structure, which is released by the splicing machinery in a Drosha-independent manner. Such miRNAs are referred to as mirtrons [526, 527]. Pre-miRNAs are exported by RanGTP/exportin-5 to the cytoplasm, where they are further processed by Dicer, another RNase III enzyme, to generate ~22 base pair microRNA duplexes that enter effector complexes called miRISC (miRNA-containing RNA-induced silencing complex). Here, they are converted into single-stranded mature miRNAs that target mRNAs and thereby affect their translation and stability [516, 528, 529].
Cancer cells frequently display reduced levels of microRNAs that act as tumor suppressors, while expressing elevated levels of oncogenic microRNAs, called “oncomiRs” that promote tumor development by negatively regulating tumor suppressor genes and/or genes that control cell differentiation and apoptosis. A network of oncomiRs expressed in lymphoid malignancies is depicted in Figure 5. Below I will describe briefly prominent microRNAs detected in normal and malignant lymphoid cells. There are variations in the microRNA expression pattern described between the various scientific reports, which can be explained by the use of different internal standards, different controls for comparison, and the use of sample materials of malignant cells at different developmental stage and at different ontogeny tumor grade.
Figure 5.
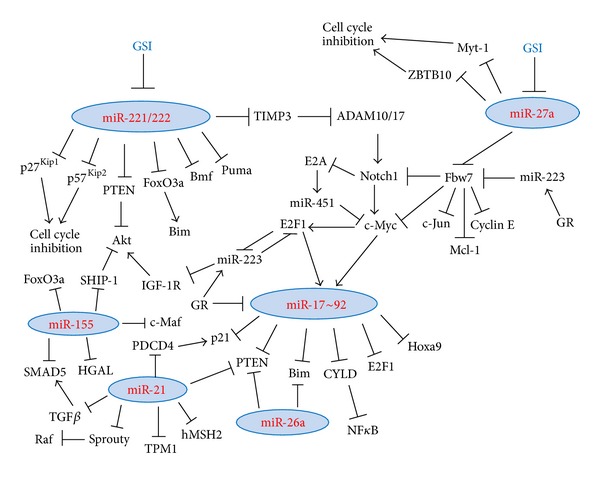
A network of oncomiRs expressed in lymphoid malignancies. A summary of details described in Section 3.
3.1. MicroRNAs in T- and B-Cell Development
Virtually every step in hematopoiesis seems to be finely tuned by specific microRNAs [514, 530–533]. Dicer has an essential role in the development of the adaptive immune system. Conditional deletion of Dicer expression in the T-cell compartment resulted in impaired T-cell development and diminished regulatory T-cell function [534–536], and ablation of Dicer in the B-cell compartment attenuates B-cell development and alters the antibody repertoire [537]. It should be noted that there exists an alternative microRNA processing pathway that is independent of Dicer, but dependent on Argonaute-2 [538].
3.1.1. MicroRNA during Thymocyte Development
MicroRNA expression is dynamically regulated during thymocyte development, with different enriched microRNAs expressed at each developmental stage [539] (Table 1). It should be emphasized that the CD4+CD8+ (double positive, DP) thymocytes are the most GC-sensitive thymocyte population [540–542]. Dicer-deficient DP thymocytes expressed higher levels of CD69 and TCR (T-cell receptor), but lower levels of Bcl-2 [539]. The Dicer-deficient thymocytes were more prone to apoptosis than control cells [539, 543], understating the role of microRNAs in regulating cell survival. Some microRNAs, such as miR-146a and miR-182, play a dominant role in the regulation of the innate and adaptive immune responses, respectively [544, 545].
Table 1.
Alterations in microRNA signature during T-cell development in the thymus. (according to Neilson et al. [539]).
| Thymocyte population | Relative high expression | Relative low expression |
|---|---|---|
| DN1 |
miR-21, miR-29b, miR-342, miR-221, miR-223 |
miR-16, miR-128b, miR-15b, miR-24 |
| DN3 | miR-191 | miR-142, miR-150 |
| DN4 | miR-142, miR-20a, miR-16, miR-128b | |
| DP | miR-92, miR-181a/b, miR-350, miR-15b, miR-16 | miR-21, miR-27a |
| CD4+ SP | miR-669c, miR-297, miR-142 | miR-142 |
| CD8+ SP | miR-15b, miR-150, miR-24, miR-27a, miR-142 | miR-142, miR-16, miR-128b, miR-92, miR-181b |
DN: double negative (CD4−8−)
DP: double positive (CD4+8+)
SP: single positive (CD4+8− or CD4−8+).
According to Neilson et al. [539], the pro-apoptotic miR-15b is almost not expressed at the immature DN1 (double negative 1) thymocyte stage but becomes gradually upregulated in DN3 and DN4, and further in DP cells. The pro-apoptotic miR-16 is also low in DP1 and reaches a maximum in DN4 cells, with a reduction upon transition to DP cells. The oncogenic miR-21 is expressed at the highest level in DN1 and becomes reduced upon transition to DN3 and is almost not expressed in DP cells. miR-181a/b is expressed at the highest level in DP thymocytes, together with miR-92 and miR-350. It should be noted that in this study the expression of each microRNA was determined relative to the general microRNA pool of each subpopulation. Since the amount of total microRNA becomes strongly reduced upon transition from DN4 to DP (a drop from 32000 to 5200 copies/cell), the absolute microRNA number in each cell population differs, which can be demonstrated by the miR-181a transcript. While miR-181a presents 15.6% of the microRNA in DP cells and 6.7% and 5% in DN3 and DN4, respectively, the numbers of miR-181a copies in these three populations were estimated to be 810 in DP, 1400 in DN3, and 1600 in DN4 [539]. Li et al. [546] showed that miR-181a is expressed at DN1 and becomes upregulated during DN2 and DN3 and then downregulated at DN4. miR-181 is still significantly expressed in DP cells, albeit at a slight lesser extent than in DN4 and becomes downregulated upon differentiation to the SP (single positive) stage [546]. miR-146 is upregulated in CD4+ T cells [547].
3.1.2. Differentiation Stage-Specific Expression of MicroRNAs in B Lymphocytes
Malumbres et al. [533] performed an extensive microRNA profiling to identify microRNAs specifically expressed in B-cell subsets during peripheral B-cell differentiation. Notably, miR-18a, miR-28, miR-15a~16-1, and miR-181 are expressed at higher levels in centroblasts (germinal center B lymphocytes) compared with memory B cells, whereas miR-101c, miR-150, miR-29a,b,c, and miR-23a~24 are enriched in memory B cells. miR-17~92, miR-363~106a, and miR25~106b are highly expressed in all B-cell subtypes. The high level of miR-15a~16-1 in germinal center B-cells corresponds with low Bcl-2 expression in these cells. miR-223 is highly expressed in naïve and memory B cells, but not in centroblasts. miR-125b is especially expressed in germinal center B lymphocytes [533].
3.1.3. miR-181a/b in T- and B-Cell Development
miR-181a represses the expression of Bcl-2, CD69, and the T-cell receptor (TCR) α-chain [539]. miR-181a augments the strength of TCR signaling and down-regulates several phosphatases including DUSP5, DUSP6, SHP-2, and PTPN22 that regulate the sensitivity of T cells to antigens [546]. The down-regulation of PTPN22 by miR-181a led to elevated phosphorylation of p56Lck at Y394 and the down-regulation of DUSP5/6 to increased ERK activation [546]. The normally high levels of miR-181a maintain T-cell tolerance to self-peptide/MHC molecules, with a reduction in this microRNA increasing the number of self-reactive T cells [548]. Also, dampening miR-181a expression using antagomiR-181a impaired positive selection with about a 70% reduction of mature CD4+ SP thymocytes [546]. Thus, miR-181a plays a role in regulating TCR response during T-cell development. Recently, miR-181a-1/b-1, but notmiR-181a-2b-2andmiR-181-c/d, was found to control the development of normal thymic T cells and leukemia cells [549]. EctopicmiR-181a-1expression in thymic progenitor cells potentiated DP cell development[549]. Conditional deletion of miR-181ab1 allele resulted in 50%–75% decrease in cellularity in the thymus and a significant reduction in the percentage of DP cells [549]. miR-181a expression decreased during the DN3a to DN3b transition during β-selection, and loss ofmir-181ab1resulted in a reduction in the percentage of DN3 and DN4 cells that expressed intracellular TCR-β, while preTα expression in DN3 thymocytes was normal [549].
miR-181a becomes downregulated when mouse T cells are costimulated with TCR and CD28 [317]. Other alterations occurring upon TCR/CD28 co-stimulation includes the upregulation of the miR-466 family, miR-574, miR-346, miR-214, miR-155, and miR-709, and the down-regulation of the miR-29 family, miR-15a, miR-15b, miR-16, miR-146b, miR-142, miR-27a, miR-150, and let-7 family [317].
Chen et al. [550] showed that miR-181 is expressed in the B-lymphoid cells of the mouse bone marrow, and its ectopic overexpression in hematopoietic stem/progenitor cells significantly increased B-cell production [550]. miR-181 also affects the development of NK cells through targeting the Nemo-like kinase (NLK), an inhibitor of Notch signaling [551]. miR-181 targets the RNA-binding protein Lin28, thereby disrupting the Lin28-let-7 reciprocal regulatory loop, with concomitant upregulation of let-7 and differentiation of megakaryocytes [552].
3.1.4. miR-150 in T- and B-Cell Development
miR-150 is highly expressed in mature and resting lymphocytes, but not in their progenitors [547, 553]. Overexpression of miR-150 led to a block in B-cell formation at the pro-B to pre-B-cell transition by downregulating c-Myb, among other targets [547], suggesting for a role for this microRNA in B-cell differentiation. Within the lymphoid lineage the choice between T and B cells is regulated by miR-150 [547, 553]. The T-cell population level was unaffected by overexpression of miR-150 in hematopoietic progenitor cells, while the mature B-cell levels were strongly reduced [553]. miR-150 drives megakaryocyte-erythrocyte progenitor (MEP) cells towards megakaryocytes at the expense of erythroid cells [554]. miR-150 also regulates the development of NK (natural killer) and iNKT (invariant NK) cells [555]. Mice with target deletion in miR-150 had a defect in their ability to generate mature NK cells, while overexpression of miR-150 resulted in a substantial reduction in iNKT in the thymus and in the peripheral lymphoid organs [555], supposedly through targeting of c-Myb by miR-150 [556].
3.1.5. miR-125b in T- and B-Cell Development
miR-125b affects T-cell differentiation through regulation of IFNγ (Interferon γ), IL-2Rβ, IL-10Rα, and PRDM1/Blimp1 (B lymphocyte-induced maturation protein-1) [592]. Ectopic expression of miR-125b in naïve lymphocytes inhibited differentiation to effector cells [592]. During normal B-cell development, miR-125b is enriched in germinal center B cells and keeps the transcription factor IRF4 and PRDM1/Blimp1 down, while miR-223 is enriched in memory B cells, where it targets the transcription factor LMO2, which is specifically expressed in germinal center B cells [533]. IRF4 and PRDM1/Blimp1 expression are repressed in centroblasts, but is necessary for differentiation into memory and plasma cells [593, 594]. Overexpression of miR-125b alone in mice causes an aggressive, transplantable myeloid leukemia [357]. Before leukemia, these mice did not display elevation of white blood cells in the spleen or bone marrow, rather the hematopoietic compartment showed lineage-skewing, with myeloid cell numbers dramatically increase and B-cell numbers severely diminished [357]. miR-125b targets Lin28A, an induced pluripotent stem cell gene [595]. Knockdown of Lin28A led to hematopoietic lineage-skewing similar to ectopic miR-125b overexpression, with increased myeloid and decreased B-cell number [595]. miR-125b is also a potent oncomiR in the development of megakaryoblastic leukemia [596].
3.1.6. miR-155 in T- and B-Cell Development
miR-155 is also important for lymphopoiesis and for preserving normal immune system responses [266, 597–599]. miR-155 is processed within the second exon of the nonprotein-encoding gene BIC (B-cell integration cluster). miR-155 is upregulated upon TCR/CD28 costimulation in mouse T cells [317], and in macrophages by several TLR (Toll-like receptor) pathways [600]. B cells require miR-155 for normal production of isotype-switched, high-affinity antibodies and for a memory response [599]. miR-155 knockout mice are immunocomprised owing to defects in B and T lymphocytes [597]. The transcription factor PU.1, which down-regulates IgG1 levels, is a target gene of miR-155 in B cells [599]. This may explain the reduced amount of circulating IgG1 in miR-155 knockout mice [599]. As with B cells, it seems that miR-155 is involved in T-cell differentiation [266, 597]. Naïve T cells derived from miR-155 knockout mice showed increased propensity to differentiate into Th2 rather than Th1 cells, with the concomitant production of Th2 cytokines such as IL-4, IL-5, and IL-10 [266, 597]. One explanation for this biased development of Th2 cells might be the miR-155 mediated targeting of c-Maf (musculoaponeurotic fibrosarcoma), a transcription factor that transactivates the IL-4 gene [597]. With regard to the acute immune response, the T cells had an impaired response and showed attenuated IL-2 and IFNγ release in response to antigens [266, 597]. miR-155 is upregulated by the transcription factor FoxP3 and critical for T regulatory cell function [601]. Mice overexpressing miR-155 in the B-cell lineage results in preleukemic pre-B-cell proliferation in the spleen and bone marrow, followed later in life by B-cell malignancy [602]. miR-155 represses genes encoding DNA damage response proteins [603].
3.1.7. miR-17~92 in T- and B-Cell Development
The miR-17~92 cluster located on chromosome 13 at locus q31.3 is essential for B-cell development [246]. The expression of miR-17~92 peaked in pre-B cells, where it inhibited cell death [246]. It is expressed at higher levels in normal germinal center B cells compared to naïve and memory B cells [533]. Knockout of miR-17~92 leads to increased Bim expression and inhibits B-cell development at the pro-B to pre-B transition [246], a step also blocked by miR-150 [553]. Mice overexpressing the miR-17~92 cluster in lymphocytes developed lymphoproliferative disease and autoimmunity and they died prematurely [247]. These animals were found to have increased numbers of activated B cells, and a higher ratio of activated CD4+ T cells versus CD8+ T cells. The enhanced proliferation and survival of B and T cells may result from the down-regulation of Bim and PTEN [247]. miR-17~92 expression is strongly induced after activation of CD8+ T cells, which is critical for the rapid clonal expansion of these cells [604]. However, following the clonal expansion, miR-17~92 is downregulated and further silenced during memory development [604].
3.2. MicroRNAs in Lymphoid Malignancies
Malignant lymphomas arise from normal B- or T-cell counterparts at different ontogeny stages and commonly continue to express gene signatures inherited from their nontransformed cellular progenitors. Extensive miRNA profiling studies have been performed on various lymphoid malignancies, including T-ALL [605], cutaneous T-cell lymphoma [606], CLL [557, 563], pre-B-ALL [557, 605, 607], diffuse large B-cell lymphoma (DLBCL) [564, 580–582, 608–610], anaplastic large cell lymphoma (ALCL) [587], multiple myeloma (MM) [574, 611, 612], mantle cell lymphoma (MCL) [583, 591, 613], Burkitt Lymphoma (BL) [564, 614], and follicular lymphoma (FL) [135, 582, 615]. A comprehensive study aimed to integrate the many miRNAs upregulated in T-ALL into a microRNA-transcription factor coregulatory network was performed by Ye et al. [506]. Various microRNAs have also been associated with poor prognosis [515]. A short description of some important microRNAs in malignant lymphoid diseases is described below and summarized in Tables 2 and 3.
Table 2.
MicroRNA signature in various lymphoid malignancies. The table shows microRNAs that have been detected at higher or lower levels in lymphoid malignancies according to data in the literature. More detailed description is found in Sections 3 and 4. It should be emphasized that the table presents microRNAs that are frequently dysregulated, and the microRNA expression pattern may vary during disease progression and depends on the ontogeny and tumor grade. Also, there are variabilities between the different studies which may be due to generalized classification or more specific classification of the given malignancy. Also, the reference gene and cell type used as control may affect the interpretation of microRNA profiling. MicroRNAs that can affect or are related to GC signaling and/or GC-induced apoptosis are highlighted in bold.
| Cancer type | Increased expression | Decreased expression | References |
|---|---|---|---|
| ALL | miR-17~92 cluster, miR-26a, miR-29a/b/c, miR-125b-1*, miR-128a, miR-128b, miR-146a, miR-204, miR-218, miR-331, miR-181a, miR-181b, miR-181c, miR-142-3P, miR-142, miR-150, miR-155, miR-193a, miR-196b, miR-30e-5p, miR-34b, miR-365, miR-582, miR-708, miR-223* | let-7b, miR-223*, miR-100, miR-125b*, miR-151, miR-99a, miR-124a | [253, 557–562] |
|
| |||
| CLL | miR-21, miR-23b, miR-24-1, miR-146a, miR-150*, miR-155, miR-106b, miR-195, miR-221*, miR-222*, miR-181a/b*, miR-19a, miR-20a, miR-106b, miR-142*, miR-29a/c*, miR-130, miR-26a, miR-197, miR-342, miR-483, miR-595 | miR-15a, miR-16-1, miR-29*, miR-34a, miR-143, miR-45, miR-30d, let-7a, miR-181a/b*, miR-223, miR-92, miR-150*, miR-126, miR-125b, miR-103, miR-572, miR-494, miR-923, miR-130a, miR-213, miR-17, miR-142*, miR-206, miR-220, miR-221*, miR-222*, miR-182, miR-199a, let7, miR-424, miR-10a, miR-7, miR-126, miR-218 | [345, 346, 358, 557, 558, 563–573] |
|
| |||
| MM | miR-21, miR-106b~25 cluster, miR-181a/b*, miR-20a, miR-19a, miR-19b, miR-93, miR-25, miR-92a, miR-19a, miR-19b, miR-32, miR-1, miR-133a, miR-193b~365, | let-7b, let-7-1, let-7c, miR-29a, and miR-29b, miR-328, miR-15a/16, miR-192~194~215, miR-181a/b* | [345, 574–579] |
|
| |||
| DLBCL | miR-155, miR-124a miR-125b*, miR-143, miR-451, miR-145, miR-10b, miR-34a, miR-100, miR-9, miR-21, miR-17~92, miR-128a, miR-106a/b, miR-425, miR-130b, miR-181b* | miR-27a/b, miR-29a/b/c; miR-142, miR-150, miR-125b*, miR-101, miR-28, miR-16, miR-189, miR-363, miR-223, miR-584, miR-361, miR-768, miR-625, miR-495, miR-181a*, miR-189, miR-363, miR-595, miR-663 | [533, 564, 580–585] |
|
| |||
| C-ALCL | miR-155, miR-27b, miR-30c, miR-29b | [586] | |
|
| |||
| ALK+-ALCL | miR-886-3p, miR-17, miR-18a, miR-20a, miR-363, miR-106a, miR-20a, miR-20b, miR-135b | miR-146a, miR-101, miR-29b, miR-26a, miR-29c, miR-29a, miR-22, miR-150, miR-125b | [587, 588] |
|
| |||
| ALK−-ALCL | miR-155 | miR-101 | [587] |
|
| |||
| cHL | miR-17~92 cluster, miR-16, miR-21, miR-24, miR27a, miR-124a, miR-134, miR-138, miR-155, miR-147, miR-182, miR-185, miR-198, miR-216, miR-220, miR-302a/b/c, miR-325 | miR-23b, miR-30b, miR-31, miR-96, miR-126, miR-128a/b, miR-135a, miR-183, miR-204, miR-205, miR-335, miR-150 | [584, 589, 590] |
|
| |||
| MCL | miR-124a, miR-155, miR-182, miR-183, miR-328, miR-326, miR-302c, miR-345, miR-373, miR-210, miR-617, miR-370, miR-654, miR-106b, miR-93, miR-25, miR-200c, miR-363, miR-181c, miR-654, miR-768 | miR-29a/b/c, miR-142, miR-150, miR-15a/b, miR-31, miR-148a, miR-27b, miR-126 | [564, 583, 591] |
|
| |||
| FL | miR-9, miR-20a/b, miR-301, miR-213, miR-330, miR-106a, miR-338, miR-155, miR-210, miR-138, miR-193a, miR-345, miR-513b, miR-574, miR-584, miR-663, miR-1287, miR-1295, miR-1471 | miR-30a, miR-33a, miR-106a, miR-141, miR-202, miR-205, miR-222, miR-301b, miR-320, miR-149, miR-139, miR-431, miR-570 | [135, 564, 582] |
Abbreviations: ALK: anaplastic lymphoma kinase; ALCL: anaplastic large cell lymphoma; C-ALCL: cutaneous large cell lymphoma; cHL: classical Hodgkin's lymphoma; DLBCL: diffuse large B-cell lymphoma; FL: follicular lymphoma; MCL: mantle cell lymphoma.
*Variation in expression, dependent on the tumor grade.
Table 3.
Target genes of prominent microRNAs in lymphoid malignancies and their role in regulating GC-mediated apoptosis. Relations to GC signaling and/or GC-induced apoptosis are highlighten in bold. More detailed description is found in Sections 3 and 4.
| miRNA | Important target genes |
Regulation/expression | Effect on GC-induced apoptosis | References |
|---|---|---|---|---|
| let-7 family |
K-Ras, Myc, HMGA2, PLCγ1, IMP-1, Dicer, IL-6, E2F2, CCND (Cyclin D2, Cdc25A, CDK6, Bcl- X L, PRDM1/Blimp1 |
↓ CLL ↓ MM ↓ T-ALL ↓ BL ↓Myc |
Anticipated to synergize with GC | [345, 361, 473, 476–479, 525, 564, 640] |
| miR-9 | PRDM1/Blimp1 NFκB |
↑ FL |
[564, 640–642] | |
| miR-15a~16 |
Bcl-2, CHEK1, CCND1 (Cyclin D1), CCND2 (Cyclin D2), CCND3 (Cyclin D3), CCNE (Cyclin E), CDK4, CDK6, Wnt3a, E2F, Cdc25A, Mcm5 |
↑↓ CLL ↓ MM ↑GC ↑E2F1-3 ↓c-Myc |
Promote GC-induced apoptosis | [255, 345, 346, 471, 473, 515, 525, 564, 566, 576, 643–646] |
| miR-17~92 cluster | Bim (Bcl2L11), PTEN, E2F1, Notch1, Hoxa9, CYLD, RUNX1, p21 | ↑ T-ALL ↑ CLL ↑ MM ↑ BCL ↑ ALK+-ALCL ↑ DLBCL ↑ BL ↓GC ↑ c-Myc ↑ E2F1 ↓ GSI |
Attenuates GC-induced apoptosis. Considered as an OncomiR. |
[135, 206, 246–248, 255, 445, 467, 469, 515, 525, 564, 574, 618, 647, 648] |
| miR-18 (member of the miR-17~92 cluster) |
GR | Reduced GR-mediated transactivation | [649] | |
| miR-21 |
PTEN, PDCD4, TPM-1, Tap63, SPRY2, Msh2, SHIP1, TRAIL-3 |
↑ CLL ↑ CML ↑ MM ↑ BCL ↑ DLBCL ↓FoxO3a |
Expected to prevent GC-induced apoptosis, due to increased Akt signaling. Considered as an OncomiR. |
[268, 269, 525, 566, 574, 579, 580, 650–654] |
| miR-23a/b | Notch1, PLK3, PAX, MTSS1 | ↑ CLL ↓ cHL ↓ Relapsed T-ALL |
[590, 655, 656] | |
| miR-26a |
PTEN, Bim, EZH2, c-Myc, CCND3 (Cyclin D3), CCNE2 (Cyclin E2) |
↑ T-ALL ↑ CLL ↓ BL ↓ Myc |
Expected to prevent GC-induced apoptosis. Considered as an OncomiR. |
[253, 315, 465, 473, 481, 565, 566, 629] |
| miR-27a | Fbw7, ZBTB10, Myt-1, MDR, BMI1, FoxO1/3 | ↑ B-ALL ↓ DLBCL ↓GSI ↓GC |
[253, 258, 438, 439, 445, 446, 657, 658] | |
| miR-29a/b |
Mcl-1, Tcl-1, CDK6, PTEN, DNMT1, DNMT3A, DNMT3B p85α, CDC42 |
↓ ALCL ↓ CLL ↓ MM ↓ MCL ↓ DLBCL ↓ BL ↓ Myc ↓ NFκB |
Expected to synergize with GC. | [316, 380–383, 473, 564–566, 569, 574, 583, 587, 643, 655, 659–662] |
| miR-34a/b/c |
Bcl-2, E2F1, c-Myb, B-Myb SIRT1, ZAP70, Notch1, Delta1, Jagged1 |
↑↓ CLL ↑p53 ↑PMA ↓Myc |
[347, 348, 443, 473, 570, 663–667] | |
| miR-101 | mTOR, Mcl-1, Cox2, Fos, EZH2 | ↓ ALCL | Expected to synergize with GC. | [384, 587] |
| miR-106a~363 and miR-106b~25 |
p21/CDKN1a Bim, PTEN |
↑ MCL ↑ MM ↑ DLBCL ↓GC |
Attenuates GC-induced apoptosis | [254–256, 314, 574, 591, 668, 669] |
| miR-124a | GR | ↑ MCL ↓ ALL |
Reduced GR-mediated transactivation | [560, 583, 649, 670] |
| CDK6 Hes-1 | ||||
| miR125a | PDPN, Bak1, KLF13, preproET1, ARID3B, HuR, ERBB2, ERBB3 | [576] | ||
| miR-125b | IRF4 PRDM1-Blimp1 Lin28, STAT3 Bak1, Bmf Mcl-1, Bcl-w, Bcl-2 |
↓ CLL | It has both pro- and anti-apoptotic effect. | [349–352, 356, 533, 571, 595, 671] |
| miR-128b | BMI1 | ↓ Relapsed T-ALL ↓ MLL-AF4-ALL ↓GC |
miR-128 sensitizes MLL-AF4 ALL to GC. | [646, 655, 672, 673] |
| miR-130b | GR | ↑ DLBCL ↑ Relapsed T-ALL |
Attenuates GC-induced apoptosis. | [655, 674–676] |
| RUNX3 p21 | ||||
| miR-135a/b | JAK2, |
↓ cHL | [590, 677] | |
| miR-142 | GR | ↓ MCL ↑ T-ALL ↓GC |
Confers GC resistance | [583, 657, 678–680] |
| AC9 | ||||
| miR-143 and miR145 | MLL-ALL ERK5 |
↓ CLL | [525, 573] | |
| miR-146a | TRAF6, IRAK1, Fas, Smad4, TBP, CCL8-MCP-2 | ↑ MM ↑ T-ALL ↑ CLL ↓ BL ↓Myc |
[473, 564, 576, 635, 637, 638, 681] | |
| miR-150 | c-Myb, DKC1 AKT2, Notch3 |
↑↓ CLL ↑ T-ALL ↓ MCL ↓ cHL ↓ DLBCL ↑GC ↓ Myc |
[441, 473, 547, 565, 566, 583, 589, 682–684] | |
| miR-155 | SOCS1, ETS1, c-MAF, HGAL, FoxO3a, SHIP1, SMAD5, PU.1, C/EBPβ, CSFR, KPC1, CEBPB, IL-13Rα1, CUTL1, CYR61, SMAD1, ETS1, SMAD2, |
↓↑ CLL ↑ DLBCL ↑ C-ALCL ↑ ALK−-ALCL ↑ MCL ↑ cHL ↑ NHL |
Expected to prevent GC-induced apoptosis. Considered as an OncomiR. |
[263, 264, 515, 525, 564, 566, 568, 576, 583, 584, 587, 589, 599, 602, 681, 685–691] |
| MEIS1, RUNX2, MYO10, PKIα, JARID2, AGTR1, PICALM, BACH1, ZIC3, | ↑↓ BL ↓GC (in MΦ) ↑ NFκB ↑ TLR4 ↑ c-Myb ↑ EBV |
|||
| miR-181a/b | Tcl1, Lin28, Bcl-2, Mcl-1, XIAP, CYLD, GR, CD69, TCR, Hoxa7, Hoxa9, Hoxa11, PBX3, NLK, TIMP3, Prox1, DUSP5, DUSP6, SHP-2, PTPN22, FoxP1, p27Kip1 |
↓↑ CLL ↓↑ DLBCL ↑ MM ↑ T-ALL ↓ GC ↓ GSI |
Dual role on GC-induced apoptosis: attenuation through repression of GR, but sensitization due to reduced expression of anti-apoptotic proteins. | [270, 358, 359, 385, 445, 539, 546, 549, 551, 552, 565, 568, 569, 574–576, 684, 692–694] |
| miR-182 |
FoxO1/3,
Fbw7 |
↑ T-ALL cell lines resistant to GC ↓ CLL ↑ MCL |
Confers GC resistance. |
[253, 258–261, 564] |
| miR-221 and miR-222 |
p27Kip1, p57Kip2, PTEN, TIPM3, FoxO3a, c-Kit, Puma, Dicer, APAF-1, WTAP, Ets1, Bmf, Mdm2 |
↑ CLL ↑ DLBCL ↑ MM ↓ MLL-AF4 ALL ↓GSI |
Dual effect on GC-induced apoptosis. Usually oncogenic with anti-apoptotic effect. In MLL-AF4 ALL, miR-221 sensitizes the cells to GC. Considered as an OncomiR. |
[321, 445, 525, 533, 563, 575, 576, 580, 672, 695–699] |
| miR-223 | LMO2, NFI-A, MYBL1, E2F1, Fbw7, Mef2c, IGFR |
↓↑ T-ALL ↓ CLL ↓ DLBCL ↓ Relapsed T-ALL ↑ GC ↑ C/EBPα ↓ NFI-A ↓ E2F1 |
May sensitize to GC-induced apoptosis by preventing Akt activation. | [515, 533, 565, 566, 646, 655, 700–706] |
| miR-708 | FoxO3 | ↑ Relapsed T-ALL |
May confer GC resistance. | [655] |
↑ upregulated, ↓ downregulated.
Abbreviations: AC9: adenylyl cyclase 9; BCL: B-cell lymphoma, Blimp1: B lymphocyte-induced maturity protein 1; cHL: classical Hodgkin's lymphoma; GSI- gamma secretase inhibitor; LMO2: LIM domain only 2; MDR: multidrug resistant gene; Msh2: DNA MutS homolog 2; MTSS1: Metastasis suppressor 1; NHL: non-Hodgkin's lymphoma; NLK: nemo-like kinase; PDCD4: programmed cell death 4; PMA: phorbol myristate acetate; SHIP1: SH2 (Src-homology 2) domain-containing inositol phosphatase 1; SOCS1: suppressor of cytokine signaling; SPRY2: Sprouty2; TPM-1: Tropomyosin 1; TRAF6: TNF receptor-associated factor 6; WTAP: Wilms' tumor-associated protein isoform 1; XIAP: X-linked inhibitor of apoptosis protein.
3.2.1. MicroRNAs in T-Acute Lymphoblastic Leukemia (T-ALL)
In general, T-ALL is characterized by upregulation of the miR-17~92 cluster, miR-26a, miR-128a/b, miR-146a, miR-181a/b, miR-150, and miR-155, while let-7b and miR-223 are downregulated [253, 557–561].
3.2.1.1. miR-17 ~92 in T-ALL. The miR-19, miR-20a, miR-92a, and miR-17 especially of the miR-17~92 cluster are upregulated in T-ALL [506]. All six miRNAs miR-17, miR-18a, miR-19a, miR-20a, miR-19b, and miR-92a, of the miR-17~92 cluster promoted leukemogenesis in Notch1-induced T-ALL in vivo [253, 616]. Among them, the miR-19 family has been considered the key oncogenic component [248, 506, 617]. The miR-17~92 cluster is located within a fragile site that is frequently amplified in a range of hematopoietic malignancies [618]. Paralogues to the miR-17~92 cluster include miR-106b~25 and miR-106a~363 [246, 619].
miR-19 represses Notch1, PTEN, Hoxa9, Cyld, Runx1, E2F1, and Bcl2L11 (Bim) [506, 616, 620]. Reduced expression of Bim attenuates GC-induced apoptosis. Posttranslational inactivation of PTEN by miR-19 promotes activation of the PI3K/Akt pathway, and incontrollable proliferation of T cells [104, 105]. Increased Akt signaling antagonizes GC-induced apoptosis by several mechanisms, including phosphorylation of FoxO3a, thus preventing its nuclear translocation and transcriptional activation of Bim, and through inactivation of GSK3, which is essential for GC-induced apoptosis [30, 67, 97].
Hoxa9 is a leukemogenic homeoprotein in T-ALL [621], and a target gene of the oncogenic MLL-AF4 fusion protein [622]. High expression of miR-196b was found in pediatric ALL with aberrant activation of Hoxa genes [623].
Notch1 plays a vital role in T-cell development and transformation, and about 50% of primary T-ALL samples show abnormal Notch1 expression [79]. Downstream transcriptional targets of Notch1 include Hes1 and c-Myc, the former affecting the PI3K/Akt and NFκB signaling pathways [624, 625]. c-Myc is a potent and direct transcriptional activator of miR-17~92, leading to modulation of E2F1 expression [255]. Deletion of miR-17~92 cluster repressed Myc-induced oncogenesis [246, 248]. GCs repress the expression of miR-17~92 [206], which may be one means to overcome the tumorigenicity of T-ALL cells and to elevate Bim expression [206].
In contrast to miR-19, miR-451, and miR-709 are potent suppressors of oncogenesis in Notch1-induced mouse T-ALL [440]. miR-451 represses c-Myc, while miR-709 represses Ras-GRF-1 that acts upstream to Ras and prevents Akt activation [625]. Both miR-451 and miR-709 are transcriptional targets of the bHLH E2A tumor suppressor, which is degraded upon Notch1 induction in mouse T-ALL cells [626, 627]. Repression of tumor suppressor miR-451 is essential for Notch1-induced oncogenesis in a murine model of T-ALL [440]. Human T-ALLs with activating Notch1 mutations have decreased miR-451 and increased c-Myc levels compared with T-ALLs with wild-type Notch1 [440]. One mechanism of the tumor suppressive action of miR-451 could be through down-regulation of the PI3K/Akt survival pathway [628].
3.2.1.2. miR-26a in T-ALL. Primary T-ALL cells also express elevated levels of miR-26a that suppresses PTEN and Bim [253]. miR-26a enhanced leukemogenesis in a mouse model of T-ALL [253]. miR-26a was found to be repressed by c-Myc in a mouse lymphoma model, leading to enhanced expression of the EZH2 oncogene, a component of the Polycomb repressive complex 2 [483]. c-Myc may also directly upregulate EZH2 [629]. In mantle cell lymphoma, miR-26a was found to affect NFκB nuclear translocation [591].
3.2.1.3. miR-146a in T-ALL. miR-146a, miR-181a/c, and miR-221 were associated with overall survival in ALL patients [559]. miR-146 seems to have opposing roles in tumorigenesis depending on the cellular context [517]. miR-146a and miR-146b are elevated in several types of solid tumor [630–633]. However, overexpression studies of miR-146a in transplanted bone marrow cells suggest a tumor-suppressive role for this microRNA [634]. miR-146 overexpression reduced the survival and engulfment of hematopoietic stem cells in recipient cells [634]. miR-146a knockout mice developed massive myeloproliferation followed by hematopoietic tumors, including myeloid sarcomas and lymphomas [635, 636]. The myeloproliferative phenotype correlated with enhanced NFκB signaling [636]. miR-146a suppresses the NFκB activators IRAK1 (interleukin 1 receptor-associated kinase 1) and TRAF6 (TNF receptor-associated factor 6) [635, 637, 638]. Thereby, overexpression of miR-146a leads to inhibition of NFκB activity. A negative feedback loop exists between NFκB and miR-146. Whereas miR-146 represses NFκB signaling, NFκB signaling upregulates miR-146 [637].
3.2.1.4. miR-181a in T-ALL. miR-181a family members are highly expressed in T-ALL leukemia cells and downregulated during remission[639]. Deletion ofmiR-181a-1/b-1expression inhibits the development ofNotch1 oncogene-induced T-ALL in a mouse model [549]. miR-181a/bcontrols the strength and threshold of Notch activity in tumorigenesis in part by dampening multiple negative feedback regulators downstream of Notch and pre-T-cell receptor (TCR) signaling pathways [549].
3.2.1.5. miR-124a in T-ALL. miR-124a has been shown to be downregulated in more than 50% of ALL cases and associated with higher relapse rate and mortality rate [560]. It targets CDK6 and reduces Rb (retinoblastoma) phosphorylation. Its down-regulation contributes to the abnormal proliferation of ALL. Inhibition of CDK6 by sodium butyrate or PD0332991 decreased ALL cell growth. Overexpression of miR-124a reduced tumorigenicity in a xenogeneic mouse model [560].
3.2.2. MicroRNAs in Chronic Lymphocytic Leukemia (CLL)
A comparison study of primary CLL samples with normal unstimulated or CpG-stimulated B cells showed high similarities between CLL and activated B cells, including upregulation of miR-34a, miR-155, and miR-342 and down-regulation of miR-103 and miR-181a/b [565]. Activation of normal B cells led to reduced miR-23a, miR-23b, miR-24, miR-27b, miR-181a/b, and miR-223 and increased miR-155 with all activation agents used. Differential effect on miR-29 family was observed with the different activation agents. One particular difference between activated B cells and CLL was seen with miR-150. miR-150 was reduced during B-cell activation, whereas it was upregulated in most CLL cases [565]. The latter confirms the study of Fulci et al. [566], but is opposed to Wang et al. [682] showing that miR-150 is downregulated in CLL. Ectopic miR-150 expression increased cell death in pro-B cells, while miR-150 deficiency led to B-cell expansion and an enhanced humoral immune response [547]. Some differences in miR-150 are observed between the mutated versus unmutated IgVH (immunoglobulin heavy chain variable-region genes) subgroups, where expression is higher at the average in the mutated IgVH subgroup [566].
CLL cases with unmutated IgVH or with high expression levels of ZAP-70 (70kD zeta-associated protein) show an unfavorable course with rapid progression in comparison to patients with a mutated IgVH [567]. Two research groups [565, 566] observed decreased levels of miR-29c and miR-223 in CLL with ZAP70+ and IgVH unmutated status. Calin et al. [643] observed that the unmutated IgVH CLL subgroup exhibited high levels of Tcl-1 due to low expression of miR-29 and miR-181 that negatively regulate this oncogene. miR-181 and miR-29 might therefore be considered to have tumor-suppressor functions. Tcl-1 functions as a coactivator of Akt [707], and B-cell forced expression of Tcl-1 in transgenic mice resulted in tumors that resembled CLL [567]. CLL with unmutated IgVH and high expression of ZAP-70 showed also relative high expression of miR-15a, miR-16-1, miR-16-2, miR-195, miR-23b, miR-155, miR24-1, and miR-146, while low expression of miR-223, miR-29a-2, miR-29b-2, and miR-29c [563]. In an aggressive subtype of CLL with abnormalities in the TP53 gene, the microRNAs miR-34a, miR-29c, and miR-17 were downregulated [567].
3.2.2.1. miR-15a~16 in CLL. CLL cases with good prognostic features are typically characterized by down-regulation of miR-15a and miR-16-1 [643, 708], located at the 13q14.3 locus. These miRNAs map to a region between exon 2 and 5 of the Leu2 gene. Deletion of 13q14.3 (del(13q)) is the most common cytogenetic abnormality in CLL occurring in more than 50% of the cases and implies for a favorable prognosis [709]. This deletion occurs also frequently in MM patients [575].Deletion in mice of the 13q14-minimal deleted region, which encompasses the miR-15a~16 cluster, caused the development of indolent B-cell-autonomous, clonal lymphoproliferative disorders, recapitulating the spectrum of CLL-associated phenotypes observed in humans [644]. Repression of miR-15a and miR-16-1, as well as miR-29b, in CLL may also be mediated by histone deacetylases (HDACs) [710]. HDAC inhibition triggered the accumulation of the transcriptionally activating chromatin modification H3K4me2 and restored the expression of miR-15a, miR-16-1, and miR-29b [710]. Deacetylase inhibition may therefore be an attractive therapeutic strategy.
Both miR-15a and miR-16-1 negatively regulate Bcl-2 [643], and miR-29 targets Mcl-1 [381, 382]. The expression of Bcl-2 in CLL cases is inversely correlated with the expression of miR-15a and miR-16-1 [563, 711]. Other targets of miR-15/16 include CHEK1 [615], CyclinD1, CyclinD2, and Cdc25A [525, 645]. Overexpression of miR-15a and miR-16-1 induced cell cycle arrest at G1/G0 in an Rb-dependent manner [712]. A germ-line mutation in the primary precursor of miR-15a/16-1 that impairs their processing was observed in familial CLL patients [563]. Targeting deletion of miR-15a~16 in mice led to the development of a spectrum of diseases resembling CLL-associated lymphoproliferation in humans, including CLL, CD5+ monoclonal B-cell lymphocytosis, and CD5− non-Hodgkin's lymphomas [644]. The New Zealand black (NZB) mouse that harbor a point mutation in the 3′-flanking region of miR-16 that leads to reduced miR-16 expression and develops symptoms similar to B-CLL in humans, further confirming the tumor suppressor function of this locus [713].
3.2.2.2. miR-181a/b in CLL. Underexpression of miR-181a/b was associated with shorter overall survival in CLL [358], while higher levels of miR-181a were associated with a shorter time from diagnosis to initial therapy [563]. During the course of CLL progression, the miR-181a/b levels were decreased, which inversely correlated with increased levels of its target genes Mcl-1 and Bcl-2 [385]. miR-181b was especially downregulated in treatment-refractory cases [714]. The study of Marton et al. [568] showed consistent underexpression of miR-181a, as well as let-7a and miR-30d in all CLL cases studied. However, increased expression of miR-181a/b was associated with favorable outcome in patients with cytogenetically normal acute myeloid leukemia (AML) [692].
Ectopic overexpression of miR-181a/b into primary CLL increased fludarabine-sensitivity in p53 wild-type cells, but not in CLL with attenuated p53 response [358]. The importance of the miR-181 target Mcl-1 in CLL survival was demonstrated by rapid apoptosis of CLL cells following siRNA-mediated down-regulation of Mcl-1 [715], and by the Mcl-1 transgenic mice, which developed B-cell lymphoma [715]. Thus, low miR-181 and miR-29 expression in CLL could confer drug resistance through upregulation of Mcl-1 expression.
3.2.2.3. miR-29 in CLL and Other B-Cell Malignancies. The miR-29 family consisting of miR-29a and miR-29b seems to play a dual role in tumorigenesis [517]. On the one hand, miR-29a and miR-29b are downregulated in mantle cell lymphoma [583], aggressive CLL samples (high ZAP-70 with unmutated IgVH) [659, 710, 716], ALK-positive anaplastic large cell lymphomas (ALCL) [380], MM [381], and AML [383]. On the other hand, miR-29a and b are expressed at higher degree in indolent CLL (low ZAP-70/mutated IgVH) than in normal CD19+ cells [563, 569, 716]. miR-29c together with miR-223 down-regulation is associated with higher tumor burden, disease aggressiveness, and poor prognosis in CLL [700].
Forced overexpression of miR-29b induced apoptosis in MM and AML cells [381, 383]. The tumor suppressor activity of miR-29 may be achieved through targeting cell cycle regulators and oncogenes such as Cdk6, DNA methyltransferase 3A (DNM3A) and 3B (DNMT3B), Mcl-1, and Tcl1A [382, 569, 583, 717]. Another tumor suppressor function of miR-29 is mediated through activation of p53, which is achieved by targeting p85α (the regulatory subunit of PI3K kinase) and CDC42 (a Rho family GTPase) [660].
However, in another setting miR-29 acts as an oncogene. miR29a overexpression in immature and mature B cells promoted CLL development [716], and transplantation of miR-29-transduced hematopoietic stem and progenitor cells into irradiated mice resulted in myeloproliferative disease and AML [661]. One mechanism for the oncogenic feature of miR-29 could be through repression of the tumor suppressor cell-adhesion molecule peroxidasin homologue (PXDN) [716]. Thus, depending on the cellular contexts,miR-29 can function as an oncogene or a tumor suppressor.
3.2.2.4. miR-221/222 in CLL. miR-221 and miR-222 are expressed at higher levels in CLL with unmutated IgVH and high expression of ZAP-70, the most aggressive CLL subtype with poor prognosis [563]. These microRNAs may contribute to oncogenesis by targeting the CDK inhibitor p27Kip1 [695, 696, 718, 719], FoxO3a [720, 721], Apaf-1 [721, 722], p57Kip2 [719], Bmf [723], PTEN [321], and TIMP3 (tissue inhibitor of metalloproteinase 3) [321]. In other CLL cases, the miR-222 was found to be lower than that of normal CD19+ cells [566]. miR-221 was expressed at reduced levels in CLL harboring the 13q14 deletion [711].
3.2.2.5. miR-34 in CLL. The p53 target miR-34a is decreased in CLL patients with 11q deletions, leading to increased ZAP-70 expression [663]. miR-34a also targets Bcl-2 [348, 484], and the E2F1 and B-Myb oncogenes in CLL [664]. Reduced miR-34a expression has been associated with resistance to DNA damage in CLL [570].
3.2.2.6. miR-17~92 in CLL. Members of the miR-17~92 polycistron are upregulated in B-cell lymphoma, as well as miR-155 [469, 568, 685]. Adoptive transfer of hematopoietic stem cells bearing a truncated portion of the miR-17~92 polycistron in c-Myc transgenic mice resulted in a more rapid onset of malignant B-cell lymphomas. These lymphomas exhibited resistance to apoptosis and increased proliferation [469]. Transgenic overexpression of the entire miR-17~92 in the murine hematopoietic compartment led to the development of lymphoproliferative disease and increased lethality [247]. The negative regulation of Bim by the miR-17~92 cluster seems to be a major mechanism by which B-cell lymphomas evade apoptosis [247]. Silencing of miR-17 and miR-20a in mantle cell lymphoma led to upregulation of the cyclin-dependent kinase (CDK) inhibitor p21, suggesting that p21 is an essential target of the miR-17~92 cluster during B-cell lymphomagenesis [647]. Overexpression of c-Myc mRNA together with miR-17-5p/miR-20a was associated with a more aggressive behavior in mantle cell lymphoma [724]. miR-17~92 confers chemoresistance in mantle cell lymphoma through activation of the PI3K/Akt pathway [725]. Knockdown of miR-17~92 inhibited tumor growth in a xenograft mantle cell lymphoma model [725].
3.2.2.7. miR-21 in CLL. miR-21 is commonly upregulated in CLL [650] as well as CML [726] and many other cancer cell types [525]. Forced overexpression of miR-21 under the control of the nestin promoter resulted in severe pre-B-cell lymphoma [727]. miR-21 overexpression potentiated lung tumorigenesis of a constitutively activated K-Ras proto-oncogene [728]. miR-21 deletion in mice reduced 7,12-dimethylbenz[a]anthracene (DMBA)/12-O-tetradecanoylphorbol-13-acetate(TPA) skin carcinogenesis [729]. miR-21-null mice exhibited an increase in cellular apoptosis and decrease in cell proliferation [729]. miR-21 is an oncomiR that promotes tumorigenesis by targeting a range of genes involved in regulating cell proliferation and/or survival, including PTEN [269], Sprouty (Spry2) [730], PDCD4 (programmed cell death 4) [731], TPM1 (tropomyosin 1) [651], and human DNA MutS homolog 2 (hMSH2) [732]. In glioblastoma cells, miR-21 also targets a network of p53 pathways, TGFβ, and mitochondrial tumor suppressor genes [733]. PDCD4 inhibits AP-1-mediated transactivation [734] and negatively regulates the pro-survival RAL guanine-nucleotide dissociation stimulator (RALGDS) signaling pathways [517, 729]. PDCD4 also induces the expression of the CDK inhibitor p21 [735]. Down-regulation of PDCD4 by miR-21 confers growth advantages to the cells. PDCD4 is a tumor suppressor that is upregulated during apoptosis [736] and downregulated in several cancer forms [737–739]. Spouty, which is downregulated by miR-21, negatively regulates the c-Raf pro-survival signaling pathway [729].
3.2.2.8. miR-125b in CLL. Both aggressive and indolent CLL patients showed reduced expression of miR-125b [571]. Overexpression of miR-125b in CLL-derived cell lines resulted in the repression of many transcripts encoding enzymes implicated in cell metabolism [571]. These authors proposed that miR-125b acts as a regulator for the adaptation of cell metabolism to a transformed state.
3.2.2.9. miR-150 in CLL. One microRNA consistently downregulated in most B-lymphomas is miR-150 [682], which is proposed to act as a tumor suppressor [523, 547, 589]. Mice lacking miR-150 have increased expression of its target transcription factor c-Myb, which plays an important role in lymphocyte development and maturation [547]. miR-150 is especially expressed in mature lymphocytes, but not in their progenitors [547]. Premature expression of miR-150 blocked the transition from pro-B to the pre-B stage [553]. Overexpression of miR-150 in NK/T lymphomas increased apoptosis and reduced cell proliferation, with concomitant reduction in DKC1 (Dyskeratosis congenita 1) and Akt2, reduced Akt phosphorylation, and elevated levels of Bim and p53 [683].
3.2.2.10. miR-155 in CLL. miR-155 is overexpressed in many B-cell lymphomas including CLL, primary mediastinal B-cell lymphoma (PMBL), aggressive activated B-cell like (ABC) subtype of DLBCL, Hodgkin's lymphoma, and pediatric Burkitt's lymphoma, but is almost absent in adult Burkitt's lymphoma [515, 566, 568, 576, 583, 584, 587, 602, 685, 686]. c-Myb (v-Myb myeloblastosis viral oncogene homolog), which is overexpressed in a subset of CLL patients, associates with the promoter of miR-155 host genes (miR155HG, also known as BIC, B-cell integration cluster) and stimulates its transcription [687]. Forced overexpression of miR-155 in B cells (Eμ-miR-155 transgenic mice) led to initial preleukemic pre-B-cell proliferation followed by frank B-cell malignancy [602]. The miR-155 orthologue miR-K12-11 in Kaposi sarcoma-associated herpes virus (KSHV) has been associated with B-cell tumors [740]. miR-155 is essential for immune function and is strongly induced in activated T and B cells [597]. miR-155 represses SH2-domain containing inositol-5-phosphatase-1 (SHIP-1), which is a critical phosphatase that negatively downmodulates Akt pathway and is involved in normal B cell development [688]. Thus, sustained overexpression of miR-155 in B cells unblocks Akt activity, inducing B-cell development. miR-155 targets c-Maf in lymphocytes [597], and HGAL and SMAD5 in diffuse large B-cell lymphoma (DLBCL) [741, 742]. HGAL, a germinal center (GC)-specific gene, inhibits lymphocyte and lymphoma cell motility by activating RhoA signaling cascade [743] and by interacting with actin and myosin proteins [744]. SMAD5 is a bone morphogenetic protein (BMP)-responsive transcription factor and is activated by various cytokines [745]. DLBCL expressing high levels of miR-155 concomitant with low HGAL expression showed high aggressiveness and cell dissemination [741]. siRNA-based SMAD5 knockdown recapitulated the effects of miR-155 overexpression in DLBCL [742]. Thus, down-regulation of SMAD5 in diffuse large B-cell lymphoma defines a unique mechanism used by the cancerous cells to escape TGFβ growth inhibitory effects [742]. In breast cancer, miR-155 targeted FoxO3a, thus modulating their response to chemotherapy [264]. As FoxO3a is a positive regulator of the pro-apoptotic Bim essential for GC-induced apoptosis [227–229], miR-155 overexpression may prevent Bim upregulation.
3.2.3. miRNAs in Multiple Myeloma (MM)
In one study, miR-93, miR-25, miR-92, miR-19a/b, miR-181a/b, and miR-32 were shown to be significantly overexpressed, while let7-b, let7-I, let7-c, miR-29a, and -29b significantly downregulated in MM [574]. Roccaro et al. [575] found decreased expression of miR-15a~16 and increased expression of miR-222, miR-221, miR-382, and miR-181a/b in their MM samples. Heterogeneous expression of miR-181a and -181b was observed in MM cells from many patients [574]. Also, the 13q14.3 locus containing the miR-15a and miR-16-1 is sometimes deleted in MM [345, 746–748]. The absence of miR-15a expression and overexpression of miR-181a/b correlated with worse prognosis of MM [575]. Antagonists especially to miR-19a/b and miR-181a/b (AntagomiRs) suppressed tumor growth of human myeloma cells implanted into nude mice [574]. This finding demonstrates the potential use of microRNAs in therapy.
Some differential miRNA expression was observed between malignant MM and MGUS (monoclonal gammopathy of undetermined significance) [574], which is the precancerous state preceding MM [749]. MGUS show already upregulation of miR-21, miR-106~25, miR-181a/b, miR-1, and miR-133a, while during the progression to malignant multiple myeloma miR-17~92, miR-32, miR-193b~365 are upregulated and miR-192~194~215 and miR-15a~16 are downregulated [574, 576, 577]. The upregulation of miR-17~92 could be related to the upregulation of c-Myc observed during MM progression [750, 751]. Upregulation of miR-1 and miR-133a correlated with t(14; 16) translocation in MM cases, suggesting that deregulation of microRNA expression could be associated with chromosomal abberations [578]. MGUS premalignant cases displayed higher levels of Dicer than MM cells [752]. Higher expression of Dicer was associated with improved progression-free survival in symptomatic MM cases [752].
The global increase in microRNA expression in high-risk MM patients with poor prognosis was associated with increased expression of Argonaute (AGO2/ElF2C2) [611], a master regulator of miRNA maturation and function [753, 754]. Silencing of AGO2 decreased viability in MM cell lines [611].
3.2.3.1. IL-6 and MM. Adhesion of multiple myeloma to bone marrow stroma triggers cytokine production and enhances cell proliferation and resistance to chemotherapy through IL-6-induced activation of NFκB, PI3K/Akt, and STAT3 pathways [755]. It should be noted that these pro-survival pathways antagonize GC-induced apoptosis in MM [756–760]. miR-19a and miR-19b that are part of the miR-17~92 cluster downregulate SOCS-1 (suppressor of cytokine signaling-1), a gene frequently silenced in MM that plays a critical role as inhibitor of IL-6 growth signaling [574], thus enforcing the IL-6-induced survival signals.
3.2.3.2. miR-21 in MM. The oncogenic miR-21 is upregulated in MM patient samples and cell lines [574, 579, 652]. In IL-6-dependent MM cell lines, miR-21 transcription is controlled by IL-6 through a STAT-3 mechanism. Ectopic miR-21 expression was sufficient to sustain growth of IL-6-dependent cell lines in the absence of IL-6 [761]. miR-21 is upregulated in a NFκB-dependent manner in MM cells upon cell adhesion to bone marrow stromal cells [762]. Combining miR-21 inhibition with dexamethasone inhibited MM cell survival more effectively than either treatment alone [762]. The p300-CBP-associated factor (PCAF) was found to be a target of the combined action of the miR106b~25 cluster and miR-32 [574]. PCAF is a positive regulator of p53 through ubiquitination activity on Hdm2 [763]. miR106b~25, miR-17, and miR-20a target the CDKN1A1/p21 cell cycle regulator, which prevents cell cycle progression in general and prevents the growth of MM cells [764, 765].
3.2.3.3. miR-15a~16 in MM. miR-15a~16 is a pro-apoptotic microRNA that targets Bcl-2, cyclin D1, cyclin D2, and Cdc25A [346, 748, 766–768]. Overexpression of miR-15a~16 in MM led to inhibition of Akt3, ribosomal protein S6, MAP kinases, and the NFκB-activator MAP3KIP3, ultimately resulting in an antiproliferative effect and apoptosis [575]. The anti-MM effect of miR-15a~16 was observed even in the context of the bone marrow microenvironment [575]. miR-15a~16 reduced VEGF secretion from MM cells, thereby reducing MM cell-induced pro-angiogenic activity on endothelial cells [575]. VEGF represents one of the major pro-angiogenic cytokines responsible for the induction of neoangiogenesis in MM patients [769, 770].
3.2.4. miRNAs in Anaplastic Large Cell Lymphoma (ALCL)
A distinct microRNA profile could distinguish between ALK+ and ALK− subtypes of ALCL, an aggressive form of non-Hodgkin's lymphoma (NHL) belonging to the T-cell lineage [587]. More than 80% of ALK+ ALCL harbor the t(2; 5)(p23; q35) translocation, resulting in the expression of the chimeric nucleophosmin (NPM)-ALK [771]. The constitutive ALK activity leads to the activation of many different growth-promoting and anti-apoptotic pathways including PI3K/Akt/mTOR, Jak/Stat, c-Jun, JunB, and c-Myc. The prognosis of ALK− ALCL is worse [772, 773]. ALK+ ALCL has a high cure rate with CHOP treatment, in contrast to ALK− cells that are relative resistant [774]. Five members of the miR-17~92 cluster were expressed higher in ALK+ ALCL, whereas miR-155 was expressed more than 10-fold higher in ALK− ALCL [587]. The upregulation of miR-17~92 cluster in ALK+ ALCL cells is in agreement with the observation that c-Myc is expressed in ALK+ ALCL and absent from ALK− samples [775]. miR-101 was downregulated in all ALCL tested [587]. miR-101 targets mTOR [776], Mcl-1 [777], and the histone methyltransferase EZH2 [778, 779]. Inhibition of mTOR, which is targeted by miR-101, led to reduced tumor growth in engrafted ALCL mouse models [587]. Overexpression of miR-101 reduced cell proliferation in ALK+, but not in ALK− [587]. The former was also more sensitive to mTOR inhibition by the rapamycin analogue CCI-779 [587]. miR-29a and miR29b down-regulation in ALK+ ALCL confer apoptotic resistance due to Mcl-1 upregulation [380, 587].
Another microRNA that has been implicated in NPM-ALK-driven oncogenicity is miR-135b [588]. miR-135b targets FoxO1 and promotes a IL-17-producing immunophenotype. miR-135b inhibition reduced tumor angiogenesis and growth in vivo, suggesting that targeting this microRNA has therapeutic potential [588].
3.2.5. miRNAs in Diffuse Large B-Cell Lymphoma (DLBCL)
A 9-miRNA signature (miR-146b-5p, miR-146a, miR-21, miR-155, miR-500, miR-222, miR-363, miR-574-3p, and miR-574-5p) could differentiate the diffuse large B-cell lymphoma (DLBCL), the most common subtype of non-Hodgkin's lymphoma, into ABC (activated B-cell) or GCB (germinal center B-cell) subtypes, with a general higher expression in the ABC subtype [533]. Another study [780] found that miR-331, miR-151, miR-28, and miR-454 were upregulated in the GCB type, whereas miR-222, miR-144, miR-451, and miR-221 upregulated in the ABC type. The microRNA expression of both GCB-like and ABC-like cells was more similar to germinal center lymphocytes than memory B-cells [533]. The region encoding the miR-17~92 cluster was more commonly amplified in GCB-like than ABC-like DLBCL [781]. Lawrie et al. [580] identified 3 miRNAs, miR-155, miR-21, and miR-221, more highly expressed in ABC type than GCB type cells. Expression of miR-21 was an independent prognostic indicator in DLBCL [580]. Expression of miR-155 and miR-21 was also higher in nonmalignant ABC than in GCB cells [580]. miR-150 was strongly downregulated in both ABC and GCB-like DLBCL cells [533]. Patients with GCB DLBCL have longer overall survival and event-free survival compared with patients with an ABC phenotype when treated with R-CHOP [782, 783]. Increased expression of miR-18a in DLBCL was associated with a shorter OS (overall survival) of patients receiving R-CHOP regimen [693]. Increased expression of miR-181a was associated with longer PFS (progression-free survival), while increased expression of miR-222 was associated with shorter PFS [693]. In DLBCL, miR-181a regulates FoxP1 (Forkhead Box protein P1) and MGMT (O6-methylguanine-DNA methyltransferase) expression in DLBCL cells [693]. FoxP1 is expressed in normal activated B cells, mantle zone B cells, and some germinal center B cells [784, 785]. FoxP1 is recurrently targeted by chromosomal translocations involving the immunoglobulin heavy chain locus in marginal zone lymphomas and DLBCL, suggesting a potential role for FoxP1 in lymphomagenesis [786, 787]. FoxP1 has in some studies been shown to be associated with poor prognosis and survival [788, 789]. MGMT encodes an enzyme that protects cells from the toxicity of alkylating agents. The ability of miR-181a to reduce MGMT protein expression may contribute to better cyclophosphamide chemosensitivity [693].
miR-222 is part of the miR-221/miR-222 cluster, which is highly expressed in ABC-like DLBCL cell lines [533] and ABC-like DLBCL tumors [580]. miR-222 regulates the expression of the stem cell factor c-Kit [697], and the cyclin-dependent kinase inhibitors p27Kip1 and p57Kip2 [695, 698]. High expression of miR-222 was associated with inferior overall survival and progression-free survival [533].
3.2.6. MicroRNA in Follicular Lymphoma (FL)
FL is characterized by high miR-9, miR-138, miR-20a/b, and miR-155 expression [135, 564, 582].
3.2.6.1. miR-9 in FL. miR-9, which is activated by c-Myc, regulates NFκB [641]. miR-9 targets also the transcription factor PRDM1/Blimp1 in lymphoma and may contribute to the phenotype maintenance and pathogenesis of lymphoma cells by interfering with normal B-cell terminal differentiation [582, 608]. BRDM1/Blimp1 has been considered to be a tumor suppressor [790, 791]. Besides miR-9, let7a and miR-125b regulate BRDM1/Blimp1 expression [533, 640]. BRDM1/Blimp1 and Bcl6 are critical regulators of germinal center B-cell differentiation [594, 792, 793]. BRDM1/Blimp1 and Bcl6 are expressed in a mutual exclusive pattern and evidence suggests that they repress each other in germinal center B cells [792, 794]. A marked decrease of BRDM1/Blimp1 and an increase of Bcl6 were observed in follicular lymphoma cells [135], which might be related to the increased miR-9 levels in these cells [564]. Mutations in BRDM1/Blimp1 are frequently found in activated B cell (ABC)-like DLBCL [790, 795].
3.2.7. miRNAs in Hodgkin's Lymphoma (HL)
The malignant Hodgkin's lymphoma cells are usually derived from B cells, but have lost the expression of typical B-cell genes. Multiple signaling pathways are deregulated, including NFκB, JAK (Janus kinase)/STAT (signal transducer and activator of transcription), PI3K/Akt, ERK, Notch1, and receptor tyrosine kinases [796]. Patients with low miR-135a expression had a higher probability of relapse and a shorter disease-free survival [677]. miR-135a targets JAK2, a cytoplasmic tyrosine kinase involved in a subset of cytokine receptor signaling pathways. Transfection of pre-miR-135a into classical HL (cHL) caused apoptosis and decreased cell growth [677]. The miR-135a-mediated JAK2 down-regulation led to decreased Bcl-XL expression [677], a downstream effector of JAK2 [797].
About 40%–60% of Hodgkin's lymphomas have EBV (Epstein-Barr virus) associated with the malignant cells. EBV could transactive miR-155 through NFκB activation [689]. Since miR-155 is overexpressed in Hodgkin's lymphoma [590] and promotes B-cell lymphoma formation [602, 798, 799], EBV may be important in the pathogenesis of cHL.
4. MicroRNA in Regulating GC-Induced Apoptosis of Lymphoid Malignancies
4.1. MicroRNAs in the Regulation of GR Expression
4.1.1. Downregulation of GR by miR-18 and miR-124a
MicroRNAs have been shown to modulate GR expression in neuronal tissue [649, 800, 801]. miR-18 and miR-124a especially reduced GR-mediated events in addition to decreasing GR protein levels [649]. miR-18 is part of the miR-17~92 cluster, which is repressed by GCs [206]. Upregulation of the miR-17~92 has causally been related to small cell lung cancer [802, 803], where reduced GR levels have been associated with GC resistance [804].
miR-124a was found to bind to the 3′ untranslated region of GR mRNA [649]. Activation of the GR-responsive glucocorticoid-induced leucine zipper (GILZ) was impaired by miR-124a and -18 overexpression, while miRs-22, -328, and -524 did not have any effect [649]. Of note, miR-124 regulates Hes1 expression in P19 teratocarcinoma cells [670], a transcription factor that negatively regulate GR expression [88]. GC resistance in sepsis patients was associated with miR-124-induced downregulation of GR [805].
4.1.2. Downregulation of GR by miR-130b
While miR-130b, -181a, and -636 have putative complimentary binding sites in the 3′-UTR of GRα, only miR-130b was found to down-regulate endogeneous GR protein expression in the multiple myeloma cell line MM.1 [674]. The miR-130b, -181a, and -636 were differentially expressed between GC-sensitive and GC-resistant MM.1 cell lines [674]. miR-130b was expressed at higher levels in the resistant MM cell line [674]. Overexpression of miR-130b in MM.1S cells resulted in decreased expression of endogeneous GR, decreased induction of the GR-target gene GILZ, and induction of GC resistance [674]. Expression of miR-130b was therefore suggested to be a potential biomarker for patients who could be refractory to GC therapy.
In gastric cancers, miR-130b regulated the tumor suppressor gene RUNX3 [675]. miR-130b may also down-regulate p21Waf1/Cip1, resulting in inhibition of cellular senescence [676, 806].
4.1.3. Downregulation of GR by miR-142 and miR-181a
Another study [678] showed that elevated miR-142 expression in human T-ALL cells confers GC resistance by reducing the GR expression level. Other mechanism for the oncogenic role of miR-142 might be explained by its targeting of adenylyl cyclase 9 mRNA [679] leading to reduced production of cyclic adenosine monophosphate (cAMP) production with concomitant inhibition of the protein kinase A (PKA) signaling pathway [678]. The reduction in cAMP levels and reduced PKA activity caused by miR-142 relieve the inhibitory effect of PKA on T-leukemic cell proliferation. T-ALL with poor prognosis expressed higher levels of miR-142 than those with good prognosis [678]. Also, miR-142 was expressed at higher levels in relapsed T-ALL than newly diagnosed samples [678]. Transfection of miR-142 inhibitor increased GRα expression levels and sensitized T-ALL cells to GC-induced apoptosis [678].
These findings are in accord with previous findings showing a synergistic effect of cAMP mimetics on GC-induced apoptosis [99, 460, 807]. cAMP signaling can also be negatively regulated by phosphodiesterase 4B (PDE4B) that is frequently overexpressed in diffuse large B-cell lymphoma (DLBCL) [808]. Pharmacological inhibition of PDE4 in a xenograft model of human lymphoma unleashed cAMP effects, inhibited Akt, and restored GC sensitivity [808]. PDE4 inhibitors may thus improve the clinical outcome of patients with B-cell malignancies.
Triptolide, a drug that overcomes dexamethasone-resistance in human multiple myeloma cells [809], was found to regulate GR expression in the MM1.S cell line by downregulating the expression of miR-142 and miR-181a [680]. miR-142 and miR-181a mimetics slightly attenuated, whereas miR-142 and miR-181a inhibitors enforced GC-induced apoptosis of MM1.S cells [680]. miR-181a/b can also increase GC-induced apoptosis in virtue of their ability to repress the expression of the anti-apoptotic Bcl-2, Mcl-1, and XIAP proteins [385, 539, 810].
4.2. MicroRNAs Affected by GCs in Lymphoid Cells
4.2.1. Repression of miR-17~92 by GCs
Smith et al. [648] showed that broad microRNA repression occurs during GC-induced apoptosis of rat thymocytes. This repression was associated with reduced expression of both nuclear (Drosha and DGCR8/Pasha) and cytoplasmic (Dicer) microRNA processing enzymes. Silencing of Dicer in two human leukemic cell lines (CEM-C7 and ectopic GRα-overexpressed Jurkat cells) led to enhanced sensitivity to GC-induced apoptosis [648]. Global downregulation of microRNA levels, especially the miR-17 family, by GCs was also observed in GC-sensitive ALL cell lines, with concomitant upregulation of Bim [657]. Later studies showed that GCs selectively upregulate and downmodulate specific miRNAs [646] that cannot be explained by altered Dicer expression.
One polycistron cluster repressed by GCs is miR-17~92 [648, 657], which regulates Bim expression [246, 247]. Downregulation of miR-17~92 contributes to the GC-mediated upregulation of Bim [206]. This microRNA cluster also represses PTEN [247], a negative regulator of the PI3K/Akt signaling pathway. The GC-mediated downregulation of miR-17~92 might be one mechanism responsible for the GC-induced dephosphorylation of Akt. Primary thymocytes derived from mice transgenic for the miR-17~92 polycistron members in the lymphocyte compartment exhibited diminished sensitivity to GC-induced apoptosis in lymphocytes, further supporting a role for GC-induced repression of miR-17~92 in promoting apoptosis [648]. Harada et al. [657] observed that GCs reduced miR-17 family expression in 50% of primary GC-sensitive ALL, but not in any of the GC-resistant ones. Overexpression of miR-17~92 attenuated GC-induced cell death, while inhibition of miR-17~92 increased the sensitivity to GC [657]. They also reported that in a pre-B ALL cell line, a 10-hour dexamethasone treatment led to a reduction in miR-142 and miR-27a, while miR-9 was induced. There is also some evidence that GCs can reduce miR-27a expression in mouse muscle cells [811].
4.2.2. Upregulation of miR-15~16 by GCs
Rainer et al. [646] reported an induction of the myeloid-specific miR-223 and the apoptosis and cell-cycle arrest inducing miR-15~16 clusters by GC in a subset of B- and T-ALL cells, together with downregulation of the miR-17~92 complex. A transient upregulation of miR-19b and miR-181a was also observed. Overexpression of miR-15b~16 mimics increased, whereas silencing by miR-15b~16 inhibitors decreased GC sensitivity [646]. The miRNAs of the miR-15~16 family are encoded in two clusters (15a~16-1 and 15b~16-2) embedded in the DLEU2 (deleted in leukemia 2) and SMC4 loci, respectively [766, 812]. They have been implicated in cell-cycle arrest [813] and in cell death/survival decisions, the latter supposedly by targeting Bcl-2 [346]. Other microRNAs affected by GCs in pediatric ALL include upregulation of miR-548d-1 and repression of miR-128b along with miR-106b~25~93, the paralogue of miR-17~92 [646].
4.2.3. Upregulation of miR-223 by GCs
It is still not known whether the GC-induced upregulation of miR-223 affects GC-induced apoptosis [646]. Increased expression of miR-223 is involved in the differentiation of myeloid precursors into granulocytes such as neutrophils [701, 814]. During granulopoiesis, miR-223 targets E2F1, which in turn represses miR-223 expression, creating an autoregulatory negative feedback loop [702]. A negative feedback loop also exists between miR-223 and the transcription factor NFI-A [814]. miR-223 is positively regulated by C/EBPα during differentiation to granulocytes [814] and negatively regulated by AML1/ETO in leukemia cells [703]. Moreover, miR-223 targets the myeloid ELF-1-like factor (Mef)-2c and IGFR (insulin-like growth factor receptor), which may account for some of its negative regulation of granulocyte proliferation [701]. Through suppression of IGF-1R, the downstream PI3K/Akt/mTOR/p70S6K pathway is suppressed, with consequent inhibition of cell proliferation [704]. miR-223 attenuates hematopoietic cell proliferation and positively regulates miR-142 through LMO2 isoforms and C/EBPβ [815]. Ectopic expression of miR-223 restores differentiation of AML leukemic cells [703]. miR-223 knockout mice showed increased numbers of granulocyte progenitors in the bone marrow and hypermature neutrophils in the circulation, suggesting that miR-223 is involved in the negative regulation of maturation rather than differentiation of granulocytes [701]. miR-223 may also target Fbw7 [705, 816], a negative regulator of the anti-apoptotic Mcl-1 [372]. Thus, it may indirectly increase apoptotic resistance by up-regulating Mcl-1.
4.2.4. Upregulation of miR-150 and miR-342 by GCs
Dexamethasone treatment of thymocytes led to upregulation of miR-150 and miR-342, while miR-181a and miR-181d were downregulated [684]. miR-181d represses CD69 and Prox-1 to a similar extent as miR-181a [684]. miR-181d, but not miR-181a, repressed Lif (leukemia inhibitory factor) [684]. Lif is a member of the IL-6 cytokine family expressed in thymic epithelial cells and T lymphocytes, which elevates GC levels following LPS exposure and is responsible for thymic atropy induced by stress [817–819]. Other effects of miR-181 are described in Sections 3.1.3 and 3.2.2.2. The effects of miR-150 are described in Sections 3.1.4 and 3.2.2.9.
4.2.5. Effect of GCs on MicroRNA Expression in Macrophages
A recent report showed that GCs could prevent lipopolysaccharide (LPS)-mediated inflammatory responses in macrophages by downregulating miR-155 [690]. LPS induces miR-155 expression in macrophages through TLR4-mediated activation of NFκB [690]. Overexpression of miR-155 reversed the suppressive action of GCs, while inhibition of miR-155 exhibited an effect similar to that of GCs on LPS-treated macrophages, suggesting that GC-induced repression of miR-155 is one mechanism for the immunosuppressive function of GC. This effect of GC on miR-155 was dependent on GR and NFκB [690]. miR-155 transgenic mice produced more proinflammatory cytokines in response to LPS [820]. miR-155 is transcribed from B-cell integration cluster (BIC) [584, 691] and targets among others SOCS1 (suppressor of cytokine signaling 1), which negatively regulates JAK/STAT signaling. GCs also prevented the LPS-mediated upregulation of miR-146, miR-147, miR-148, miR-32b, and miR-301 in macrophages [690].
4.2.6. Other MicroRNAs Affected by GCs
In the brain, GCs prevents BDNF (brain-derived neurotrophic factor)-regulated synaptic function through suppression of miR-132 expression [821]. miR-132 is increased by BDNF and is involved in promotion of neuronal outgrowth [822]. In some carcinoma cell lines, dexamethasone was shown to down-regulate miR-27b, miR-148a, and miR-451 [823].
4.3. MicroRNAs in the Regulation of Apoptotic GC-Sensitivity
From all we have learned above, any microRNA that modulates any of the many factors regulating GC-induced apoptosis may affect the apoptotic response to GCs (Figures 1–6). These include microRNAs that affect GR expression (e.g., miR-18, miR-124a, miR-130b, miR-142, and miR-181a), those affecting Bim expression (miR-26a, miR-93, miR-17~92, miR-106a~363, and miR-106b~25) or its transcription factor FoxO3 (e.g., miR-1, miR-21, miR-27a, miR-96, miR-135b, miR-155, and miR-182), those affecting PTEN expression (miR-17~92, miR-106b~25, miR-21, miR-26a, miR-29b, miR-214, miR-216a, miR-217, miR-221, and miR-222) or mTOR (e.g., miR-101), and those downregulating directly or indirectly the anti-apoptotic proteins Bcl-2, Bcl-XL, Mcl-1, XIAP, and CYLD (e.g., miR-15a~16, miR-181a/b, miR-34a, miR-125b, miR-29a/b/c, miR-101, miR-133b, miR-193b, miR-512, let-7, and miR-491). The effect of some of these microRNAs on GC-sensitivity has already been described above and will not be repeated here. Rather, I will present here some data from primary samples showing the influence of microRNAs on clinical outcome.
Figure 6.
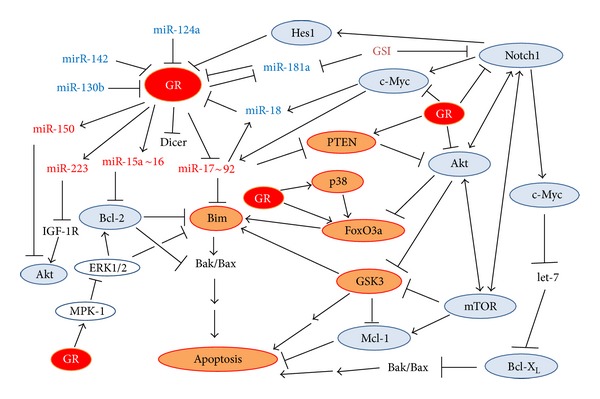
The complexity of GC-induced apoptosis. A summary of the main issue discussed in this paper.
A study searching for differential miRNAs expression in ALL relapse cells versus childhood ALL with complete remission showed significant associations for miR-708, miR-223, and miR-27a with individual relapse-free survival [655]. For samples at relapse versus diagnosis, the most differentially expressed microRNAs included miR-223, miR-23a, let-7g, miR-181, miR-708, and miR-130b, while comparison of complete response with diagnostic samples showed differential expression pattern of miR-27a, miR-223, miR-23a, miR-181, and miR-128b [655]. Among these microRNAs, miR-223, miR-128b, miR-23a, and let-7g were downregulated in the relapse samples compared with complete response samples, while miR-181 family members, miR-708, and miR-130b were upregulated in the relapse samples [655]. It should be remained here that miR-130b targets GR [674], RUNX3 [675], and p21 [676], and miR-223 is upregulated by GCs [646] and targets IGFR [701] and E2F1 [702]. E2F1 has a dual role in cell-cycle control, as it affects several cell processes. It can either act as a tumor-suppressor or oncogene depending on the cellular context [824]. Thus, the upregulation of miR-130b together with downregulation of miR-223 may contributes to GC resistance.
miR-708 was the most upregulated microRNA in the relapse samples, whereas miR-223 was significantly downregulated, suggesting that these two microRNAs may have important role in pediatric ALL relapse [655]. Moreover, upregulation of miR-708 was found to be associated with the in vivo GC therapy response and with disease risk stratification in childhood ALL [655]. Standard and middle risk stratification groups had a higher miR-708 expression at diagnosis than the high risk group. Interestingly, miR-708 was low in high relapse patients at diagnosis, while specimens of relapsed samples showed abundance of miR-708, suggesting for an upregulation of miR-708 during disease progression.
FoxO3, that is critical for hematopoietic stem cell self-renewal and mediates the initial apoptotic response [825–827], contains a conserved miR-708 response element in its 3′-UTR [655]. FoxO3 can act as either an oncogene or a tumor suppressor in leukemia [828, 829]. FoxO3 transcriptional activity was found to prevent B-CLL and CML proliferation [825, 828]. FoxO3a is also targeted by other microRNAs, including miR-27a (see Section 2.2.6).
Moreover, miR-27a directly regulates the drug-resistant factor P-glycoprotein, and overexpression of miR-27a increased sensitivity of leukemia cells to doxorubicin [658]. miR-27a is relevant to treatment outcome in vivo and may be involved in relapse of both lymphocytic leukemia and myeloid leukemia [658]. Low expression of miR-27a might promote ALL relapse [655, 658]. On the contrary, miR-27a exerts oncogenic effects by regulating ZBTB10 [446, 830] and Fbw7 [253, 438].
miR-128b, which was higher in relapse ALL and at diagnosis compared to complete response [655], has been reported to confer drug resistance in many cancers including ALL [672, 673]. Both miR-27a and miR-128b might target BMI1 [655], a transcription factor of the polycomb-group gene necessary for hematopoietic stem cell (HSC) and leukemia stem-cell self-renewal [831, 832]. Deletion of BMI1 inhibits self-renewal of tumor stem cells and prevents leukemia recurrence [833].
A role for miR-128 and miR-221 in regulating GC sensitivity in cells from MLL-AF4 ALL patients has been proposed [672]. miR-128b and miR-221 are downregulated in MLL-arranged ALL relative to other types of ALL [672]. The MLL gene is located at 11q23, a site frequently involved in chromosomal translocations in aggressive human lymphoid and myeloid leukemias. As a result of chromosomal translocations, a portion of MLL becomes fused to one among more than 40 different partner proteins. MLL-AF4 ALL, which results from the translocation between MLL and AF4, is associated with GC resistance and has a poor prognosis [834, 835]. Re-expression of mR-128 and miR-221 in cultured MLL-AF4 ALL cells sensitized them to GCs [672]. miR-128 targets MLL, AF4, and the MLL-AF4 pusion protein resulting in lower expression of HOXA9, whereas miR-221 downregulates CDKN1B (cyclin-dependent kinase inhibitor 1B, p27Kip1), another gene transcriptionally activated by MLL-AF4 as well as the wild-type MLL protein [672]. The targeting of different proteins may explain the cooperative effect of miR-128b and miR-221 on GC sensitization [672]. It should be noted that miR-221 in other settings, for example, CLL, has anti-apoptotic effects and functions as an oncogene.
4.4. Potential Use of miRNA Regulators in Therapy of Cancer Cells
In light of the multiple effects of various microRNAs on cell survival and apoptosis, modulating microRNA expression in tumor cells is an attractive approach for sensitizing the tumor cells to chemotherapeutic drugs. Inhibition of specific microRNAs is performed by using antisense sequences (termed antagomiRs) targeting the microRNA guide stand that blocks the interaction with the microRNA recognition elements within the 3′-UTR of the target mRNA genes [836]. To increase their binding affinity and stability in biological fluids, the antagomiRs are often modified with 2′-O-methyl-, phosphorothioate, or locked nucleic acid substitutions. To overexpress microRNAs, chemically synthesized microRNAs (called microRNA mimics) are used.
One potential use of microRNAs is to repress the expression of MLL-AF4 fusion protein in ALL that is responsible for GC resistance. This fusion protein can be repressed through overexpression of miR-143 [837], or miR-128 together with miR-221 [672]. The latter combination was shown to sensitize the MLL-AF4-carrying ALL cells to GCs [672].
Another promising approach is to target miR-155, an oncogenic microRNA often correlated with poor prognosis. A proof-of-principle was demonstrated by Babar et al. [838]. They showed that overexpression of miR-155 in lymphoid tissues resulted in disseminatedlymphomacharacterized by a clonal, transplantable pre-B-cell population of neoplastic lymphocytes. Withdrawal of miR-155 in mice with established disease resulted in rapid regression of lymphadenopathy. Systemic delivery of antisense peptide nucleic acids encapsulated in unique polymer nanoparticles inhibited miR-155 and slowed the growth of these pre-B-cell tumors in vivo [838].
5. Summary
Glucocorticoid-induced apoptosis appears to be a complex process involving several signaling pathways (Figure 6). These include (1) transactivation of pro-apoptotic genes (importantly Bim); (2) alterations in microRNA expression (upregulation of miR-15~16 that targets the pro-apoptotic Bcl-2; miR-223 that targets IGFR; miR-150 that targets Akt and Notch, while suppressing miR-17~92 that prevents Bim and PTEN translation); (3) direct action of GR on the mitochondria (including mitochondrial GR translocation and production of reactive oxygen species within the mitochondria); (4) activation of the protein kinases GSK3 and p38; (5) activation of the FoxO3a transcription factor that upregulates Bim; (6) inhibition of the Notch1, PI3/Akt/mTOR, and ERK1/2 survival pathways. Interruption of any of the pro-apoptotic processes may lead to drug resistance. Altered microRNA expression in malignant cells may modulate many of these processes thereby imposing apoptotic resistance (Figures 1–6).
GC-resistant lymphoid cells might be divided into two major subgroups according to the underlying mechanism of resistance. The first group consists of cancer cells whose drug resistance can be overcome by exposing the cells to GCs in combination with drugs that target protein kinases such as Akt, mTOR, Src, ALK, and/or BCR, or drugs antagonizing Bcl-2, Bcl-XL, Mcl-1, c-Myc, or Notch. These lymphoid malignancies show in general a more favorable response to combined GC therapy and in many cases may be explained by their growth dependency on these signaling molecules. The second group of GC-resistant cells exhibits an intrinsic defect in the GC-mediated apoptotic process and can thus not be turned sensitive to this drug. It is important to distinguish between these two subgroups prior to therapy initiation in order to choose the right drug combination. A diagnostic test needs to be developed that can distinguish between the different resistance backgrounds.
Recently, Burnsides et al. [839] have developed an ex vivo stimulation assay that determines the ability of leukocytes to upregulate anti-inflammatory genes such as GILZ and FKBP51 following exposure to dexamethasone. It is reasonable that a similar test may be developed to gene profiling lymphoid malignancies prior to and following GC treatment, where upregulation of the pro-apoptotic Bim gene would be a favorable predictor. Also, Bim induction may be measured after combining GC with a protein kinase inhibitor. Simultaneous expression profiling of microRNAs, Notch1, and Bcl-2 family proteins together with the activated protein kinase status in the malignant cell would provide valuable information for choosing the proper drug combination. A predictor for a good GC response would be to determine the ability of GCs to downregulate miR-17~92 and upregulate miR-15~16, miR-150, and miR-223.
A tentative therapeutic approach would be to modulate the microRNA status of the cell using microRNA mimics or antagomiRs as described in Section 4.4. What we have learned from the studies described in this paper is that it seems that in general it would be favorable to augment the expression of miR-29, miR-27, miR-15a~16, miR-34a, miR-150, and let-7, while suppressing miR-155, miR-181, miR-182, miR-21, and miR-221/222 as well as miR-17~92. Obviously, an initial microRNA profiling should be performed, and the cancer-type classification should be considered. Some microRNAs may have cell-type specific effects. While down-regulation of miR-181 may suppress the growth of T-ALL and MM, augmented miR-181 expression prevents the growth of unmutated IgVH CLL cases. Also, miR-26a has a dual effect. Its overexpression prevents growth of c-Myc-positive Burkitt lymphoma, while it must be downregulated in Notch-positive T-ALL to achieve growth inhibition. miR-451 and miR-709 could prevent growth of Notch-positive T-ALL. A reduction in miR-142, and maybe also of miR-708, which is highly expressed in relapsed childhood T-ALL, is anticipated to improve T-ALL therapy. For classical HL, miR-135a may cause apoptosis.
In conclusion, in certain types of lymphoid malignancies, GC resistance may be overcome by relieving the inhibitory effects of protein kinases and Bcl-2 family members. Both the activity of protein kinases and the expression of Bcl-2 members are affected by the microRNA network. Modulation of microRNA expression might increase GC drug responsiveness and thus improve the therapy of lymphoid malignancies.
Abbreviations
- ALCL:
Anaplastic large cell lymphoma
- BL:
Burkitt's lymphoma
- CLL:
Chronic B-lymphocytic leukemia
- DLBCL:
Diffuse large B-cell lymphoma
- FL:
Follicular lymphoma
- GC:
Glucocorticoid
- GR:
Glucocorticoid receptor
- GSI:
γ-secretase inhibitor
- HL:
Hodgkin's lymphoma
- MM:
Multiple myeloma
- NHL:
Non-Hodgkin's lymphoma
- NR3C1:
Nuclear receptor subfamily 3, group C, member 1
- NPM-ALK:
Nucleophosmin-anaplastic lymphoma kinase
- T-ALL:
T-cell acute lymphoblastic leukemia.
References
- 1.Pui CH, Gajjar AJ, Kane JR, Qaddoumi IA, Pappo AS. Challenging issues in pediatric oncology. Nature Reviews Clinical Oncology. 2011;8:540–549. doi: 10.1038/nrclinonc.2011.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Inaba H, Pui CH. Glucocorticoid use in acute lymphoblastic leukaemia. The Lancet Oncology. 2010;11(11):1096–1106. doi: 10.1016/S1470-2045(10)70114-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Armitage JO. The aggressive peripheral T-cell lymphomas: 2012 update on diagnosis, risk stratification, and management. American Journal of Hematology. 2012;87:511–519. doi: 10.1002/ajh.23144. [DOI] [PubMed] [Google Scholar]
- 4.Jiang N, Koh GS, Suang Lim JY, et al. BIM is a prognostic biomarker for early prednisolone response in pediatric acute lymphoblastic leukemia. Experimental Hematology. 2011;39(3):321.e3–329.e3. doi: 10.1016/j.exphem.2010.11.009. [DOI] [PubMed] [Google Scholar]
- 5.den Uyl D, Bultink IEM, Lems WF. Advances in glucocorticoid-induced osteoporosis. Current Rheumatology Reports. 2011;13(3):233–240. doi: 10.1007/s11926-011-0173-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dixon WG, Bansback N. Understanding the side effects of glucocorticoid therapy: shining a light on a drug everyone thinks they know. Annals of the Rheumatic Diseases. 2012;71(11):1761–1764. doi: 10.1136/annrheumdis-2012-202021. [DOI] [PubMed] [Google Scholar]
- 7.Beaudry JL, Riddell MC. Effects of glucocorticoids and exercise on pancreatic β-cell function and diabetes development. Diabetes/Metabolism Research and Reviews. 2012;28(7):560–573. doi: 10.1002/dmrr.2310. [DOI] [PubMed] [Google Scholar]
- 8.Jörns A, Sennholz C, Naujok O, Lenzen S. Beta cell mass regulation in the rat pancreas through glucocorticoids and thyroid hormones. Pancreas. 2010;39(8):1167–1172. doi: 10.1097/MPA.0b013e3181dfce4f. [DOI] [PubMed] [Google Scholar]
- 9.Weinstein RS. Glucocorticoid-induced osteoporosis and osteonecrosis. Endocrinology and Metabolism Clinics of North America. 2012;41(3):595–611. doi: 10.1016/j.ecl.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lansang MC, Hustak LK. Glucocorticoid-induced diabetes and adrenal suppression: how to detect and manage them. Cleveland Clinic Journal of Medicine. 2011;78(11):748–756. doi: 10.3949/ccjm.78a.10180. [DOI] [PubMed] [Google Scholar]
- 11.Reich E, Tamary A, Sionov RV, Melloul D. Involvement of thioredoxin-interacting protein (TXNIP) in glucocorticoid-mediated beta cell death. Diabetologia. 2012;55:1048–1057. doi: 10.1007/s00125-011-2422-z. [DOI] [PubMed] [Google Scholar]
- 12.Wang LX, Wang YP, Chen Z, et al. Exendin-4 protects murine pancreatic β-cells from dexamethasone-induced apoptosis through PKA and PI-3K signaling. Diabetes Research and Clinical Practice. 2010;90(3):297–304. doi: 10.1016/j.diabres.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 13.Ranta F, Avram D, Berchtold S, et al. Dexamethasone induces cell death in insulin-secreting cells, an effect reversed by exendin-4. Diabetes. 2006;55(5):1380–1390. doi: 10.2337/db05-1220. [DOI] [PubMed] [Google Scholar]
- 14.Moutsatsou P, Kassi E, Papavassiliou AG. Glucocorticoid receptor signaling in bone cells. Trends in Molecular Medicine. 2012;18:348–359. doi: 10.1016/j.molmed.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 15.Yun SI, Yoon HY, Jeong SY, Chung YS. Glucocorticoid induces apoptosis of osteoblast cells through the activation of glycogen synthase kinase 3β . Journal of Bone and Mineral Metabolism. 2009;27(2):140–148. doi: 10.1007/s00774-008-0019-5. [DOI] [PubMed] [Google Scholar]
- 16.Li H, Qian W, Weng X, et al. Glucocorticoid receptor and sequential P53 activation by dexamethasone mediates apoptosis and cell cycle arrest of osteoblastic MC3T3-E1 cells. PLoS ONE. 2012;7 doi: 10.1371/journal.pone.0037030.e37030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wong EYH, Herbert J. Raised circulating corticosterone inhibits neuronal differentiation of progenitor cells in the adult hippocampus. Neuroscience. 2006;137(1):83–92. doi: 10.1016/j.neuroscience.2005.08.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mayer JL, Klumpers L, Maslam S, de Kloet ER, Joëls M, Lucassen PJ. Brief treatment with the glucocorticoid receptor antagonist mifepristone normalises the corticosterone-induced reduction of adult hippocampal neurogenesis. Journal of Neuroendocrinology. 2006;18(8):629–631. doi: 10.1111/j.1365-2826.2006.01455.x. [DOI] [PubMed] [Google Scholar]
- 19.Hallek M, Fischer K, Fingerle-Rowson G, et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. The Lancet. 2010;376(9747):1164–1174. doi: 10.1016/S0140-6736(10)61381-5. [DOI] [PubMed] [Google Scholar]
- 20.Robak T, Dmoszynska A, Solal-Céligny P, et al. Rituximab plus fludarabine and cyclophosphamide prolongs progression-free survival compared with fludarabine and cyclophosphamide alone in previously treated chronic lymphocytic leukemia. Journal of Clinical Oncology. 2010;28(10):1756–1765. doi: 10.1200/JCO.2009.26.4556. [DOI] [PubMed] [Google Scholar]
- 21.Pettitt AR, Jackson R, Carruthers S, et al. Alemtuzumab in combination with methylprednisolone is a highly effective induction regimen for patients with chronic lymphocytic leukemia and deletion of TP53: final results of the national cancer research institute CLL206 trial. Journal of Clinical Oncology. 2012;30:1647–1655. doi: 10.1200/JCO.2011.35.9695. [DOI] [PubMed] [Google Scholar]
- 22.Skoetz N, Bauer K, Elter T, et al. Alemtuzumab for patients with chronic lymphocytic leukaemia. Cochrane Database of Systematic Reviews. 2012;2 doi: 10.1002/14651858.CD008078.pub2.CD008078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cortelezzi A, Sciume M, Reda G. Lenalidomide in the treatment of chronic lymphocytic leukemia. Advances in Hematology. 2012;2012 doi: 10.1155/2012/393864.393864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gentile M, Recchia AG, Vigna E, et al. Lenalidomide in the treatment of chronic lymphocytic leukemia. Expert Opinion on Investigational Drugs. 2011;20(2):273–286. doi: 10.1517/13543784.2011.546343. [DOI] [PubMed] [Google Scholar]
- 25.Younes A, Bartlett NL, Leonard JP, et al. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. The New England Journal of Medicine. 2010;363(19):1812–1821. doi: 10.1056/NEJMoa1002965. [DOI] [PubMed] [Google Scholar]
- 26.Micallef IN, Maurer MJ, Wiseman GA, et al. Epratuzumab with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone chemotherapy in patients with previously untreated diffuse large B-cell lymphoma. Blood. 2011;118:4053–4061. doi: 10.1182/blood-2011-02-336990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cives M, Milano A, Dammacco F, Silvestris F. Lenalidomide in multiple myeloma: current experimental and clinical data. European Journal of Haematology. 2012;88:279–291. doi: 10.1111/j.1600-0609.2011.01735.x. [DOI] [PubMed] [Google Scholar]
- 28.Scott LJ, Lyseng-Williamson KA. Spotlight on lenalidomide in relapsed or refractory multiple myeloma. BioDrugs. 2011;25:333–337. doi: 10.2165/11207120-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 29.Bachmann PS, Piazza RG, Janes ME, et al. Epigenetic silencing of BIM in glucocorticoid poor-responsive pediatric acute lymphoblastic leukemia, and its reversal by histone deacetylase inhibition. Blood. 2010;116(16):3013–3022. doi: 10.1182/blood-2010-05-284968. [DOI] [PubMed] [Google Scholar]
- 30.Sionov RV, Spokoini R, Kfir-Erenfeld S, Cohen O, Yefenof E. Chapter 6 mechanisms regulating the susceptibility of hematopoietic malignancies to glucocorticoid-induced apoptosis. Advances in Cancer Research. 2008;101:127–248. doi: 10.1016/S0065-230X(08)00406-5. [DOI] [PubMed] [Google Scholar]
- 31.Kfir-Erenfeld S, Sionov RV, Spokoini R, Cohen O, Yefenof E. Protein kinase networks regulating glucocorticoid-induced apoptosis of hematopoietic cancer cells: fundamental aspects and practical considerations. Leukemia and Lymphoma. 2010;51(11):1968–2005. doi: 10.3109/10428194.2010.506570. [DOI] [PubMed] [Google Scholar]
- 32.Reed JC. Bcl-2-family proteins and hematologic malignancies: history and future prospects. Blood. 2008;111(7):3322–3330. doi: 10.1182/blood-2007-09-078162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oltersdorf T, Elmore SW, Shoemaker AR, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435(7042):677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 34.Del Gaizo Moore V, Schlis KD, Sallan SE, Armstrong SA, Letai A. BCL-2 dependence and ABT-737 sensitivity in acute lymphoblastic leukemia. Blood. 2008;111(4):2300–2309. doi: 10.1182/blood-2007-06-098012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deng J, Carlson N, Takeyama K, Dal Cin P, Shipp M, Letai A. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell. 2007;12(2):171–185. doi: 10.1016/j.ccr.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 36.Kelly PN, Grabow S, Delbridge AR, Adams JM, Strasser A. Prophylactic treatment with the BH3 mimetic ABT-737 impedes Myc-driven lymphomagenesis in mice. Cell Death and Differentiation. 2013;20:57–63. doi: 10.1038/cdd.2012.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spender LC, Inman GJ. Phosphoinositide 3-kinase/AKT/mTORC1/2 signaling determines sensitivity of Burkitt's lymphoma cells to BH3 mimetics. Molecular Cancer Research. 2012;10:347–359. doi: 10.1158/1541-7786.MCR-11-0394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moore VDG, Brown JR, Certo M, Love TM, Novina CD, Letai A. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. Journal of Clinical Investigation. 2007;117(1):112–121. doi: 10.1172/JCI28281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kang MH, Kang HK, Szymanska B, et al. Activity of vincristine, L-ASP, and dexamethasone against acute lymphoblastic leukemia is enhanced by the BH3-mimetic ABT-737 in vitro and in vivo. Blood. 2007;110(6):2057–2066. doi: 10.1182/blood-2007-03-080325. [DOI] [PubMed] [Google Scholar]
- 40.Szymanska B, Wilczynska-Kalak U, Kang MH, et al. Pharmacokinetic modeling of an induction regimen for in vivo combined testing of novel drugs against pediatric acute lymphoblastic leukemia xenografts. PLoS ONE. 2012;7 doi: 10.1371/journal.pone.0033894.e33894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kline MP, Rajkumar SV, Timm MM, et al. ABT-737, an inhibitor of Bcl-2 family proteins, is a potent inducer of apoptosis in multiple myeloma cells. Leukemia. 2007;21(7):1549–1560. doi: 10.1038/sj.leu.2404719. [DOI] [PubMed] [Google Scholar]
- 42.Chauhan D, Velankar M, Brahmandam M, et al. A novel Bcl-2/Bcl-XL/Bcl-w inhibitor ABT-737 as therapy in multiple myeloma. Oncogene. 2007;26(16):2374–2380. doi: 10.1038/sj.onc.1210028. [DOI] [PubMed] [Google Scholar]
- 43.Gandhi L, Camidge DR, de Oliveira MR, et al. Phase I study of navitoclax (ABT-263), a novel bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. Journal of Clinical Oncology. 2011;29(7):909–916. doi: 10.1200/JCO.2010.31.6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roberts AW, Seymour JF, Brown J, et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. Journal of Clinical Oncology. 2012;30:488–496. doi: 10.1200/JCO.2011.34.7898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tse C, Shoemaker AR, Adickes J, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Research. 2008;68(9):3421–3428. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- 46.Wilson WH, O’Connor OA, Czuczman MS, et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. The Lancet Oncology. 2010;11(12):1149–1159. doi: 10.1016/S1470-2045(10)70261-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ackler S, Xiao Y, Mitten MJ, et al. ABT-263 and rapamycin act cooperatively to kill lymphoma cells in vitro and in vivo. Molecular Cancer Therapeutics. 2008;7(10):3265–3274. doi: 10.1158/1535-7163.MCT-08-0268. [DOI] [PubMed] [Google Scholar]
- 48.Yecies D, Carlson NE, Deng J, Letai A. Acquired resistance to ABT-737 in lymphoma cells that up-regulate MCL-1 and BFL-1. Blood. 2010;115(16):3304–3313. doi: 10.1182/blood-2009-07-233304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Coloff JL, Macintyre AN, Nichols AG, et al. Akt-dependent glucose metabolism promotes Mcl-1 synthesis to maintain cell survival and resistance to Bcl-2 inhibition. Cancer Research. 2011;71(15):5204–5213. doi: 10.1158/0008-5472.CAN-10-4531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Meynet O, Beneteau M, Jacquin MA, et al. Glycolysis inhibition targets Mcl-1 to restore sensitivity of lymphoma cells to ABT-737-induced apoptosis. Leukemia. 2012;26:1145–1147. doi: 10.1038/leu.2011.327. [DOI] [PubMed] [Google Scholar]
- 51.O’Brien SM, Claxton DF, Crump M, et al. Phase I study of obatoclax mesylate (GX15-070), a small molecule pan Bcl-2 family antagonist, in patients with advanced chronic lymphocytic leukemia. Blood. 2009;113(2):299–305. doi: 10.1182/blood-2008-02-137943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schimmer AD, O’Brien S, Kantarjian H, et al. A phase i study of the pan bcl-2FamilyInhibitor obatoclax mesylate in patients with advanced hematologic malignancies. Clinical Cancer Research. 2008;14(24):8295–8301. doi: 10.1158/1078-0432.CCR-08-0999. [DOI] [PubMed] [Google Scholar]
- 53.Joudeh J, Claxton D. Obatoclax mesylate: pharmacology and potential for therapy of hematological neoplasms. Expert Opinion on Investigational Drugs. 2012;21:363–373. doi: 10.1517/13543784.2012.652302. [DOI] [PubMed] [Google Scholar]
- 54.Nguyen M, Marcellus RC, Roulston A, et al. Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(49):19512–19517. doi: 10.1073/pnas.0709443104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pérez-Galán P, Roué G, Villamor N, Campo E, Colomer D. The BH3-mimetic GX15-070 synergizes with bortezomib in mantle cell lymphoma by enhancing Noxa-mediated activation of Bak. Blood. 2007;109(10):4441–4449. doi: 10.1182/blood-2006-07-034173. [DOI] [PubMed] [Google Scholar]
- 56.Chen S, Dai Y, Pei XY, et al. CDK inhibitors upregulate BH3-only proteins to sensitize human myeloma cells to BH3 mimetic therapies. Cancer Research. 2012;72:4225–4237. doi: 10.1158/0008-5472.CAN-12-1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rahmani M, Aust MM, Attkisson E, et al. Inhibition of Bcl-2 anti-apoptotic members by obatoclax potently enhances sorafenib-induced apoptosis in human myeloid leukemia cells through a Bim-dependent process. Blood. 2012;119:6089–6098. doi: 10.1182/blood-2011-09-378141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Heidari N, Hicks MA, Harada H. GX15-070 (obatoclax) overcomes glucocorticoid resistance in acute lymphoblastic leukemia through induction of apoptosis and autophagy. Cell Death and Disease. 2010;1(9, article e76) doi: 10.1038/cddis.2010.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bonapace L, Bornhauser BC, Schmitz M, et al. Induction of autophagy-dependent necroptosis is required for childhood acute lymphoblastic leukemia cells to overcome glucocorticoid resistance. Journal of Clinical Investigation. 2010;120(4):1310–1323. doi: 10.1172/JCI39987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death and Differentiation. 2011;18(4):571–580. doi: 10.1038/cdd.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Declercq W, Vanden Berghe T, Vandenabeele P. RIP kinases at the crossroads of cell death and survival. Cell. 2009;138(2):229–232. doi: 10.1016/j.cell.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 62.Fakler M, Loeder S, Vogler M, et al. Small molecule XIAP inhibitors cooperate with TRAIL to induce apoptosis in childhood acute leukemia cells and overcome Bcl-2-mediated resistance. Blood. 2009;113(8):1710–1722. doi: 10.1182/blood-2007-09-114314. [DOI] [PubMed] [Google Scholar]
- 63.Frenzel LP, Patz M, Pallasch CP, et al. Novel X-linked inhibitor of apoptosis inhibiting compound as sensitizer for TRAIL-mediated apoptosis in chronic lymphocytic leukaemia with poor prognosis. British Journal of Haematology. 2011;152(2):191–200. doi: 10.1111/j.1365-2141.2010.08426.x. [DOI] [PubMed] [Google Scholar]
- 64.Loeder S, Drensek A, Jeremias I, Debatin KM, Fulda S. Small molecule XIAP inhibitors sensitize childhood acute leukemia cells for CD95-induced apoptosis. International Journal of Cancer. 2010;126(9):2216–2228. doi: 10.1002/ijc.24816. [DOI] [PubMed] [Google Scholar]
- 65.Hundsdoerfer P, Dietrich I, Schmelz K, Eckert C, Henze G. XIAP expression is post-transcriptionally upregulated in childhood ALL and is associated with glucocorticoid response in T-cell ALL. Pediatric Blood and Cancer. 2010;55(2):260–266. doi: 10.1002/pbc.22541. [DOI] [PubMed] [Google Scholar]
- 66.Deftos ML, He YW, Ojala EW, Bevan MJ. Correlating notch signaling with thymocyte maturation. Immunity. 1998;9(6):777–786. doi: 10.1016/s1074-7613(00)80643-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Spokoini R, Kfir-Erenfeld S, Yefenof E, Sionov RV. Glycogen synthase kinase-3 plays a central role in mediating glucocorticoid-induced apoptosis. Molecular Endocrinology. 2010;24(6):1136–1150. doi: 10.1210/me.2009-0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sade H, Krishna S, Sarin A. The anti-apoptotic effect of Notch-1 requires p56lck-dependent, Akt/PKB-mediated signaling in T cells. The Journal of Biological Chemistry. 2004;279(4):2937–2944. doi: 10.1074/jbc.M309924200. [DOI] [PubMed] [Google Scholar]
- 69.Radtke F, Wilson A, Stark G, et al. Deficient T cell fate specification in mice with an induced inactivation of Notch1. Immunity. 1999;10(5):547–558. doi: 10.1016/s1074-7613(00)80054-0. [DOI] [PubMed] [Google Scholar]
- 70.Radtke F, Fasnacht N, MacDonald HR. Notch signaling in the immune system. Immunity. 2010;32(1):14–27. doi: 10.1016/j.immuni.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 71.Yin L, Velazquez OC, Liu ZJ. Notch signaling: emerging molecular targets for cancer therapy. Biochemical Pharmacology. 2010;80(5):690–701. doi: 10.1016/j.bcp.2010.03.026. [DOI] [PubMed] [Google Scholar]
- 72.Ersvaer E, Hatfield KJ, Reikvam H, Bruserud O. Future perspectives: therapeutic targeting of notch signalling may become a strategy in patients receiving stem cell transplantation for hematologic malignancies. Bone Marrow Research. 2011;2011:15 pages. doi: 10.1155/2011/570796.570796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pear WS, Aster JC, Scott ML, et al. Exclusive development of T cell neoplasms in mice transplanted with bone marrow expressing activated Notch alleles. Journal of Experimental Medicine. 1996;183(5):2283–2291. doi: 10.1084/jem.183.5.2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bellavia D, Campese AF, Alesse E, et al. Constitutive activation of NF-κB and T-cell leukemia/lymphoma in Notch3 transgenic mice. EMBO Journal. 2000;19(13):3337–3348. doi: 10.1093/emboj/19.13.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pear WS, Aster JC. T cell acute lymphoblastic leukemia/lymphoma: a human cancer commonly associated with aberrant NOTCH1 signaling. Current Opinion in Hematology. 2004;11(6):426–433. doi: 10.1097/01.moh.0000143965.90813.70. [DOI] [PubMed] [Google Scholar]
- 76.Ferrando AA. The role of NOTCH1 signaling in T-ALL. Hematology/the Education Program of the American Society of Hematology. 2009:353–361. doi: 10.1182/asheducation-2009.1.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tzoneva G, Ferrando AA. Recent advances on NOTCH signaling in T-ALL. Current Topics in Microbiology and Immunology. 2012;360:163–182. doi: 10.1007/82_2012_232. [DOI] [PubMed] [Google Scholar]
- 78.Pancewicz J, Nicot C. Current views on the role of Notch signaling and the pathogenesis of human leukemia. BMC Cancer. 2011;11, article 502 doi: 10.1186/1471-2407-11-502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Paganin M, Ferrando A. Molecular pathogenesis and targeted therapies for NOTCH1-induced T-cell acute lymphoblastic leukemia. Blood Reviews. 2011;25(2):83–90. doi: 10.1016/j.blre.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ellisen LW, Bird J, West DC, et al. TAN-1, the human homolog of the Drosophila Notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell. 1991;66(4):649–661. doi: 10.1016/0092-8674(91)90111-b. [DOI] [PubMed] [Google Scholar]
- 81.Weng AP, Ferrando AA, Lee W, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306(5694):269–271. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- 82.Wang Z, Inuzuka H, Zhong J, et al. Tumor suppressor functions of FBW7 in cancer development and progression. FEBS Letters. 2012;586:1409–1418. doi: 10.1016/j.febslet.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Palomero T, Ferrando A. Therapeutic targeting of NOTCH1 signaling in T-cell acute lymphoblastic leukemia. Clinical Lymphoma & Myeloma. 2009;9, supplement 3:S205–210. doi: 10.3816/CLM.2009.s.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cullion K, Draheim KM, Hermance N, et al. Targeting the Notch1 and mTOR pathways in a mouse T-ALL model. Blood. 2009;113(24):6172–6181. doi: 10.1182/blood-2008-02-136762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Manfè V, Biskup E, Rosbjerg A, et al. miR-122 regulates p53/Akt signalling and the chemotherapy-induced apoptosis in cutaneous T-cell lymphoma. PLoS ONE. 2012;7 doi: 10.1371/journal.pone.0029541.e29541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kamstrup MR, Ralfkiaer E, Skovgaard GL, Gniadecki R. Potential involvement of Notch1 signalling in the pathogenesis of primary cutaneous CD30-positive lymphoproliferative disorders. British Journal of Dermatology. 2008;158(4):747–753. doi: 10.1111/j.1365-2133.2007.08427.x. [DOI] [PubMed] [Google Scholar]
- 87.Kogoshi H, Sato T, Koyama T, Nara N, Tohda S. Gamma-secretase inhibitors suppress the growth of leukemia and lymphoma cells. Oncology Reports. 2007;18(1):77–80. [PubMed] [Google Scholar]
- 88.Real PJ, Tosello V, Palomero T, et al. γ-secretase inhibitors reverse glucocorticoid resistance in T cell acute lymphoblastic leukemia. Nature Medicine. 2009;15(1):50–58. doi: 10.1038/nm.1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Palomero T, Dominguez M, Ferrando AA. The role of the PTEN/AKT pathway in NOTCH1-induced leukemia. Cell Cycle. 2008;7(8):965–970. doi: 10.4161/cc.7.8.5753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Palomero T, Sulis ML, Cortina M, et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nature Medicine. 2007;13(10):1203–1210. doi: 10.1038/nm1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wei P, Walls M, Qiu M, et al. Evaluation of selective γ-secretase inhibitor PF-03084014 for its antitumor efficacy and gastrointestinal safety to guide optimal clinical trial design. Molecular Cancer Therapeutics. 2010;9(6):1618–1628. doi: 10.1158/1535-7163.MCT-10-0034. [DOI] [PubMed] [Google Scholar]
- 92.Samon JB, Castillo-Martin M, Hadler M, et al. Preclinical analysis of the gamma-secretase inhibitor PF-03084014 in combination with glucocorticoids in T-cell acute lymphoblastic leukemia. Molecular Cancer Therapeutics. 2012;11:1565–1575. doi: 10.1158/1535-7163.MCT-11-0938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Moellering RE, Cornejo M, Davis TN, et al. Direct inhibition of the NOTCH transcription factor complex. Nature. 2009;462:182–188. doi: 10.1038/nature08543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gutierrez A, Look AT. NOTCH and PI3K-AKT pathways intertwined. Cancer Cell. 2007;12(5):411–413. doi: 10.1016/j.ccr.2007.10.027. [DOI] [PubMed] [Google Scholar]
- 95.Mungamuri SK, Yang X, Thor AD, Somasundaram K. Survival signaling by Notch1: mammalian target of rapamycin (mTOR)-dependent inhibition of p53. Cancer Research. 2006;66(9):4715–4724. doi: 10.1158/0008-5472.CAN-05-3830. [DOI] [PubMed] [Google Scholar]
- 96.Chan SM, Weng AP, Tibshirani R, Aster JC, Utz PJ. Notch signals positively regulate activity of the mTOR pathway in T-cell acute lymphoblastic leukemia. Blood. 2007;110(1):278–286. doi: 10.1182/blood-2006-08-039883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sionov RV. The kinome and glucocorticoid-induced apoptosis. Chinese Journal of Cancer. 2008;27(11):1121–1129. [PubMed] [Google Scholar]
- 98.Garza AS, Miller AL, Johnson BH, Thompson EB. Converting cell lines representing hematological malignancies from glucocorticoid-resistant to glucocorticoid-sensitive: signaling pathway interactions. Leukemia Research. 2009;33(5):717–727. doi: 10.1016/j.leukres.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Thompson EB. Stepping stones in the path of glucocorticoid-driven apoptosis of lymphoid cells. Acta Biochimica et Biophysica Sinica. 2008;40(7):595–600. doi: 10.1111/j.1745-7270.2008.00433.x. [DOI] [PubMed] [Google Scholar]
- 100.Ruggero D, Montanaro L, Ma L, et al. The translation factor eIF-4E promotes tumor formation and cooperates with c-Myc in lymphomagenesis. Nature Medicine. 2004;10(5):484–486. doi: 10.1038/nm1042. [DOI] [PubMed] [Google Scholar]
- 101.Wendel HG, De Stanchina E, Fridman JS, et al. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature. 2004;428(6980):332–337. doi: 10.1038/nature02369. [DOI] [PubMed] [Google Scholar]
- 102.Vega F, Medeiros LJ, Leventaki V, et al. Activation of mammalian target of rapamycin signaling pathway contributes to tumor cell survival in anaplastic lymphoma kinase-positive anaplastic large cell lymphoma. Cancer Research. 2006;66(13):6589–6597. doi: 10.1158/0008-5472.CAN-05-3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Peponi E, Drakos E, Reyes G, Leventaki V, Rassidakis GZ, Medeiros LJ. Activation of mammalian target of rapamycin signaling promotes cell cycle progression and protects cells from apoptosis in mantle cell lymphoma. American Journal of Pathology. 2006;169(6):2171–2180. doi: 10.2353/ajpath.2006.051078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gutierrez A, Sanda T, Grebliunaite R, et al. High frequency of PTEN, PI3K, and AKT abnormalities in T-cell acute lymphoblastic leukemia. Blood. 2009;114(3):647–650. doi: 10.1182/blood-2009-02-206722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Silva A, Yunes JA, Cardoso BA, et al. PTEN posttranslational inactivation and hyperactivation of the PI3K/Akt pathway sustain primary T cell leukemia viability. Journal of Clinical Investigation. 2008;118(11):3762–3774. doi: 10.1172/JCI34616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Guo D, Teng Q, Ji C. NOTCH and phosphatidylinositide 3-kinase/phosphatase and tensin homolog deleted on chromosome ten/AKT/mammalian target of rapamycin (mTOR) signaling in T-cell development and T-cell acute lymphoblastic leukemia. Leukemia and Lymphoma. 2011;52(7):1200–1210. doi: 10.3109/10428194.2011.564696. [DOI] [PubMed] [Google Scholar]
- 107.Zhao WL. Targeted therapy in T-cell malignancies: dysregulation of the cellular signaling pathways. Leukemia. 2010;24(1):13–21. doi: 10.1038/leu.2009.223. [DOI] [PubMed] [Google Scholar]
- 108.Miller AL, Garza AS, Johnson BH, Thompson EB. Pathway interactions between MAPKs, mTOR, PKA, and the glucocorticoid receptor in lymphoid cells. Cancer Cell International. 2007;7, article 3 doi: 10.1186/1475-2867-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Beesley AH, Firth MJ, Ford J, et al. Glucocorticoid resistance in T-lineage acute lymphoblastic leukaemia is associated with a proliferative metabolism. British Journal of Cancer. 2009;100(12):1926–1936. doi: 10.1038/sj.bjc.6605072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Batista A, Barata JT, Raderschall E, et al. Targeting of active mTOR inhibits primary leukemia T cells and synergizes with cytotoxic drugs and signaling inhibitors. Experimental Hematology. 2011;39(4):457.e3–472.e3. doi: 10.1016/j.exphem.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 111.Zhang C, Ryu YK, Chen TZ, Hall CP, Webster DR, Kang MH. Synergistic activity of rapamycin and dexamethasone in vitro and in vivo in acute lymphoblastic leukemia via cell-cycle arrest and apoptosis. Leukemia Research. 2012;36:342–349. doi: 10.1016/j.leukres.2011.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Fuka G, Kantner HP, Grausenburger R, et al. Silencing of ETV6/RUNX1 abrogates PI3K/AKT/mTOR signaling and impairs reconstitution of leukemia in xenografts. Leukemia. 2012;26:927–933. doi: 10.1038/leu.2011.322. [DOI] [PubMed] [Google Scholar]
- 113.Gu L, Zhou C, Liu H, et al. Rapamycin sensitizes T-ALL cells to dexamethasone-induced apoptosis. Journal of Experimental and Clinical Cancer Research. 2010;29(1, article 150) doi: 10.1186/1756-9966-29-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gu L, Gao J, Li Q, et al. Rapamycin reverses NPM-ALK-induced glucocorticoid resistance in lymphoid tumor cells by inhibiting mTOR signaling pathway, enhancing G1 cell cycle arrest and apoptosis. Leukemia. 2008;22(11):2091–2096. doi: 10.1038/leu.2008.204. [DOI] [PubMed] [Google Scholar]
- 115.López-Royuela N, Balsas P, Galán-Malo P, Anel A, Marzo I, Naval J. Bim is the key mediator of glucocorticoid-induced apoptosis and of its potentiation by rapamycin in human myeloma cells. Biochimica et Biophysica Acta. 2010;1803(2):311–322. doi: 10.1016/j.bbamcr.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 116.Yan H, Frost P, Shi Y, et al. Mechanism by which mammalian target of rapamycin inhibitors sensitize multiple myeloma cells to dexamethasone-induced apoptosis. Cancer Research. 2006;66(4):2305–2313. doi: 10.1158/0008-5472.CAN-05-2447. [DOI] [PubMed] [Google Scholar]
- 117.Strömberg T, Dimberg A, Hammarberg A, et al. Rapamycin sensitizes multiple myeloma cells to apoptosis induced by dexamethasone. Blood. 2004;103(8):3138–3147. doi: 10.1182/blood-2003-05-1543. [DOI] [PubMed] [Google Scholar]
- 118.Schult C, Dahlhaus M, Glass A, et al. The dual kinase inhibitor NVP-BEZ235 in combination with cytotoxic drugs exerts anti-proliferative activity towards acute lymphoblastic leukemia cells. Anticancer Research. 2012;32:463–474. [PubMed] [Google Scholar]
- 119.Kim A, Park S, Lee JE, et al. The dual PI3K and mTOR inhibitor NVP-BEZ235 exhibits anti-proliferative activity and overcomes bortezomib resistance in mantle cell lymphoma cells. Leukemia Research. 2012;36:912–920. doi: 10.1016/j.leukres.2012.02.010. [DOI] [PubMed] [Google Scholar]
- 120.Kfir S, Sionov RV, Zafrir E, Zilberman Y, Yefenof E. Staurosporine sensitizes T lymphoma cells to glucocorticoid-induced apoptosis: role of Nur77 and Bcl-2. Cell Cycle. 2007;6(24):3086–3096. doi: 10.4161/cc.6.24.5023. [DOI] [PubMed] [Google Scholar]
- 121.Robak P, Robak T. A targeted therapy for protein and lipid kinases in chronic lymphocytic leukemia. Current Medicinal Chemistry. 2012;19(31):5294–5318. doi: 10.2174/092986712803833371. [DOI] [PubMed] [Google Scholar]
- 122.Trudel S, Li ZH, Wei E, et al. CHIR-258, a novel, multitargeted tyrosine kinase inhibitor for the potential treatment of t(4;14) multiple myeloma. Blood. 2005;105(7):2941–2948. doi: 10.1182/blood-2004-10-3913. [DOI] [PubMed] [Google Scholar]
- 123.Ohanian M, Cortes J, Kantarjian H, Jabbour E. Tyrosine kinase inhibitors in acute and chronic leukemias. Expert Opinion on Pharmacotherapy. 2012;13:927–938. doi: 10.1517/14656566.2012.672974. [DOI] [PubMed] [Google Scholar]
- 124.Carlo GP, Cristina M, Pogliani EM. Crizotinib in anaplastic large-cell lymphoma. The New England Journal of Medicine. 2011;364(8):775–776. doi: 10.1056/NEJMc1013224. [DOI] [PubMed] [Google Scholar]
- 125.Burger JA. Inhibiting B-cell receptor signaling pathways in chronic lymphocytic leukemia. Current Hematologic Malignancy Reports. 2012;7:26–33. doi: 10.1007/s11899-011-0104-z. [DOI] [PubMed] [Google Scholar]
- 126.Davis RE, Ngo VN, Lenz G, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010;463(7277):88–92. doi: 10.1038/nature08638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Honigberg LA, Smith AM, Sirisawad M, et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(29):13075–13080. doi: 10.1073/pnas.1004594107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Winer ES, Ingham RR, Castillo JJ. PCI-32765: a novel Bruton's tyrosine kinase inhibitor for the treatment of lymphoid malignancies. Expert Opinion on Investigational Drugs. 2012;21:355–361. doi: 10.1517/13543784.2012.656199. [DOI] [PubMed] [Google Scholar]
- 129.Woyach JA, Johnson AJ, Byrd JC. The B-cell receptor signaling pathway as a therapeutic target in CLL. Blood. 2012;120:1175–1184. doi: 10.1182/blood-2012-02-362624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ponader S, Chen SS, Buggy JJ, et al. The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood. 2012;119:1182–1189. doi: 10.1182/blood-2011-10-386417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Herman SEM, Gordon AL, Hertlein E, et al. Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood. 2011;117(23):6287–6296. doi: 10.1182/blood-2011-01-328484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Tai YT, Chang BY, Kong SY, et al. Bruton tyrosine kinase inhibition is a novel therapeutic strategy targeting tumor in the bone marrow microenvironment in multiple myeloma. Blood. 2012;120:1877–1887. doi: 10.1182/blood-2011-12-396853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Friedberg JW, Sharman J, Sweetenham J, et al. Inhibition of Syk with fostamatinib disodium has significant clinical activity in non-Hodgkin lymphoma and chronic lymphocytic leukemia. Blood. 2010;115(13):2578–2585. doi: 10.1182/blood-2009-08-236471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Young RM, Hardy IR, Clarke RL, et al. Mouse models of non-hodgkin lymphoma reveal Syk as an important therapeutic target. Blood. 2009;113(11):2508–2516. doi: 10.1182/blood-2008-05-158618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wang W, Corrigan-Cummins M, Hudson J, et al. MicroRNA profiling of follicular lymphoma identifies microRNAs related to cell proliferation and tumor response. Haematologica. 2012;97:586–594. doi: 10.3324/haematol.2011.048132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lannutti BJ, Meadows SA, Herman SEM, et al. CAL-101, a p110δ selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood. 2011;117(2):591–594. doi: 10.1182/blood-2010-03-275305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ikeda H, Hideshima T, Fulciniti M, et al. PI3K/p110δ is a novel therapeutic target in multiple myeloma. Blood. 2010;116(9):1460–1468. doi: 10.1182/blood-2009-06-222943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Castillo JJ, Furman M, Winer ES. CAL-101: a phosphatidylinositol-3-kinase p110-delta inhibitor for the treatment of lymphoid malignancies. Expert Opinion on Investigational Drugs. 2012;21:15–22. doi: 10.1517/13543784.2012.640318. [DOI] [PubMed] [Google Scholar]
- 139.Jou ST, Carpino N, Takahashi Y, et al. Essential, nonredundant role for the phosphoinositide 3-kinase p110δ in signaling by the B-cell receptor complex. Molecular and Cellular Biology. 2002;22(24):8580–8591. doi: 10.1128/MCB.22.24.8580-8591.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ringshausen I, Schneller F, Bogner C, et al. Constitutively activated phosphatidylinositol-3 kinase (PI-3K) is involved in the defect of apoptosis in B-CLL: association with protein kinase Cδ . Blood. 2002;100(10):3741–3748. doi: 10.1182/blood-2002-02-0539. [DOI] [PubMed] [Google Scholar]
- 141.Pauls SD, Lafarge ST, Landego I, Zhang T, Marshall AJ. The phosphoinositide 3-kinase signaling pathway in normal and malignant B cells: activation mechanisms, regulation and impact on cellular functions. Frontiers in Immunology. 2012;3, article 224 doi: 10.3389/fimmu.2012.00224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Baracho GV, Miletic AV, Omori SA, Cato MH, Rickert RC. Emergence of the PI3-kinase pathway as a central modulator of normal and aberrant B cell differentiation. Current Opinion in Immunology. 2011;23(2):178–183. doi: 10.1016/j.coi.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Accordi B, Espina V, Giordan M, et al. Functional protein network activation mapping reveals new potential molecular drug targets for poor prognosis pediatric BCP-ALL. PLoS ONE. 2010;5(10) doi: 10.1371/journal.pone.0013552.e13552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Harr MW, Caimi PF, McColl KS, et al. Inhibition of Lck enhances glucocorticoid sensitivity and apoptosis in lymphoid cell lines and in chronic lymphocytic leukemia. Cell Death and Differentiation. 2010;17(9):1381–1391. doi: 10.1038/cdd.2010.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Görgün G, Calabrese E, Hideshima T, et al. Anovel Aurora-A kinase inhibitor MLN8237 induces cytotoxicity and cell-cycle arrest in multiple myeloma. Blood. 2010;115(25):5202–5213. doi: 10.1182/blood-2009-12-259523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kuznetsov JN, Leclerc GJ, Leclerc GM, Barredo JC. AMPK and Akt determine apoptotic cell death following perturbations of one-carbon metabolism by regulating ER stress in acute lymphoblastic leukemia. Molecular Cancer Therapeutics. 2011;10(3):437–447. doi: 10.1158/1535-7163.MCT-10-0777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Santidrián AF, González-Gironès DM, Iglesias-Serret D, et al. AICAR induces apoptosis independently of AMPK and p53 through up-regulation of the BH3-only proteins BIM and NOXAin chronic lymphocytic leukemia cells. Blood. 2010;116(16):3023–3032. doi: 10.1182/blood-2010-05-283960. [DOI] [PubMed] [Google Scholar]
- 148.Campàs C, López JM, Santidrián AF, et al. Acadesine activates AMPK and induces apoptosis in B-cell chronic lymphocytic leukemia cells but not in T lymphocytes. Blood. 2003;101(9):3674–3680. doi: 10.1182/blood-2002-07-2339. [DOI] [PubMed] [Google Scholar]
- 149.Stefanelli C, Stanic’ I, Bonavita F, et al. Inhibition of glucocorticoid-induced apoptosis with 5-aminoimidazole-4-carboxamide ribonucleoside, a cell-permeable activator of AMP-Activated protein kinase. Biochemical and Biophysical Research Communications. 1998;243(3):821–826. doi: 10.1006/bbrc.1998.8154. [DOI] [PubMed] [Google Scholar]
- 150.Schlossmacher G, Stevens A, White A. Glucocorticoid receptor-mediated apoptosis: mechanisms of resistance in cancer cells. Journal of Endocrinology. 2011;211:17–25. doi: 10.1530/JOE-11-0135. [DOI] [PubMed] [Google Scholar]
- 151.Distelhorst CW. Recent insights into the mechanism of glucocorticosteroid-induced apoptosis. Cell Death and Differentiation. 2002;9(1):6–19. doi: 10.1038/sj.cdd.4400969. [DOI] [PubMed] [Google Scholar]
- 152.Smith LK, Cidlowski JA. Glucocorticoid-induced apoptosis of healthy and malignant lymphocytes. Progress in Brain Research. 2010;182:1–30. doi: 10.1016/S0079-6123(10)82001-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Schmidt S, Rainer J, Ploner C, Presul E, Riml S, Kofler R. Glucocorticoid-induced apoptosis and glucocorticoid resistance: molecular mechanisms and clinical relevance. Cell Death and Differentiation. 2004;11(1):S45–S55. doi: 10.1038/sj.cdd.4401456. [DOI] [PubMed] [Google Scholar]
- 154.Kumar R, Thompson EB. Transactivation functions of the N-terminal domains of nuclear hormone receptors: protein folding and coactivator interactions. Molecular Endocrinology. 2003;17(1):1–10. doi: 10.1210/me.2002-0258. [DOI] [PubMed] [Google Scholar]
- 155.Turner JD, Alt SR, Cao L, et al. Transcriptional control of the glucocorticoid receptor: CpG islands, epigenetics and more. Biochemical Pharmacology. 2010;80(12):1860–1868. doi: 10.1016/j.bcp.2010.06.037. [DOI] [PubMed] [Google Scholar]
- 156.Schaaf MJM, Cidlowski JA. Molecular mechanisms of glucocorticoid action and resistance. Journal of Steroid Biochemistry and Molecular Biology. 2002;83(1–5):37–48. doi: 10.1016/s0960-0760(02)00263-7. [DOI] [PubMed] [Google Scholar]
- 157.de Lange P, Segeren CM, Koper JW, et al. Expression in hematological malignancies of a glucocorticoid receptor splice variant that augments glucocorticoid receptor-mediated effects in transfected cells. Cancer Research. 2001;61(10):3937–3941. [PubMed] [Google Scholar]
- 158.Zhou J, Cidlowski JA. The human glucocorticoid receptor: one gene, multiple proteins and diverse responses. Steroids. 2005;70(5–7):407–417. doi: 10.1016/j.steroids.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 159.Lauten M, Fernandez-Munoz I, Gerdes K, et al. Kinetics of the in vivo expression of glucocorticoid receptor splice variants during prednisone treatment in childhood acute lymphoblastic leukaemia. Pediatric Blood and Cancer. 2009;52(4):459–463. doi: 10.1002/pbc.21867. [DOI] [PubMed] [Google Scholar]
- 160.Lu NZ, Cidlowski JA. Glucocorticoid receptor isoforms generate transcription specificity. Trends in Cell Biology. 2006;16(6):301–307. doi: 10.1016/j.tcb.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 161.Duma D, Jewell CM, Cidlowski JA. Multiple glucocorticoid receptor isoforms and mechanisms of post-translational modification. Journal of Steroid Biochemistry and Molecular Biology. 2006;102(1–5):11–21. doi: 10.1016/j.jsbmb.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 162.Vandevyver S, Dejager L, Libert C. On the trail of the glucocorticoid receptor: into the nucleus and back. Traffic. 2012;13:364–374. doi: 10.1111/j.1600-0854.2011.01288.x. [DOI] [PubMed] [Google Scholar]
- 163.Fitzsimons CP, Ahmed S, Wittevrongel CFW, et al. The microtubule-associated protein doublecortin-like regulates the transport of the glucocorticoid receptor in neuronal progenitor cells. Molecular Endocrinology. 2008;22(2):248–262. doi: 10.1210/me.2007-0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Daghestani HN, Zhu G, Johnston PA, Shinde SN, Brodsky JL, Day BW. Characterization of inhibitors of glucocorticoid receptor nuclear translocation: a model of cytoplasmic dynein-mediated cargo transport. ASSAY and Drug Development Technologies. 2012;10:46–60. doi: 10.1089/adt.2010.0367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Nicolaides NC, Galata Z, Kino T, Chrousos GP, Charmandari E. The human glucocorticoid receptor: molecular basis of biologic function. Steroids. 2010;75(1):1–12. doi: 10.1016/j.steroids.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Cao L, Leija SC, Kumsta R, et al. Transcriptional control of the human glucocorticoid receptor: identification and analysis of alternative promoter regions. Human Genetics. 2011;129(5):533–543. doi: 10.1007/s00439-011-0949-1. [DOI] [PubMed] [Google Scholar]
- 167.Pedersen KB, Vedeckis WV. Quantification and glucocorticoid regulation of glucocorticoid receptor transcripts in two human leukemic cell lines. Biochemistry. 2003;42(37):10978–10990. doi: 10.1021/bi034651u. [DOI] [PubMed] [Google Scholar]
- 168.Geng CD, Schwartz JR, Vedeckis WV. A conserved molecular mechanism is responsible for the auto-up-regulation of glucocorticoid receptor gene promoters. Molecular Endocrinology. 2008;22(12):2624–2642. doi: 10.1210/me.2008-0157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Geng CD, Vedeckis WV. A new, lineage specific, autoup-regulation mechanism for human glucocorticoid receptor gene expression in 697 pre-b-acute lymphoblastic leukemia cells. Molecular Endocrinology. 2011;25(1):44–57. doi: 10.1210/me.2010-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Tissing WJE, Meijerink JPP, Brinkhof B, et al. Glucocorticoid-induced glucocorticoid-receptor expression and promoter usage is not linked to glucocorticoid resistance in childhood ALL. Blood. 2006;108(3):1045–1049. doi: 10.1182/blood-2006-01-0261. [DOI] [PubMed] [Google Scholar]
- 171.Greenstein S, Ghias K, Krett NL, Rosen ST. Mechanisms of glucocorticoid-mediated apoptosis in hematological malignancies. Clinical Cancer Research. 2002;8(6):1681–1694. [PubMed] [Google Scholar]
- 172.Moalli PA, Rosen ST. Glucocorticoid receptors and resistance to glucocorticoids in hematologic malignancies. Leukemia and Lymphoma. 1994;15(5-6):363–374. doi: 10.3109/10428199409049738. [DOI] [PubMed] [Google Scholar]
- 173.Sánchez-Vega B, Gandhi V. Glucocorticoid resistance in a multiple myeloma cell line is regulated by a transcription elongation block in the glucocorticoid receptor gene (NR3C1) British Journal of Haematology. 2009;144(6):856–864. doi: 10.1111/j.1365-2141.2008.07549.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Sharma S, Liechtenstein A. Dexamethasone-induced apoptotic mechanisms in myeloma cells investigated by analysis of mutant glucocorticoid receptors. Blood. 2008;112(4):1338–1345. doi: 10.1182/blood-2007-11-124156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Geley S, Hartmann BL, Hala M, Strasser-Wozak EMC, Kapelari K, Kofler R. Resistance to glucocorticoid-induced apoptosis in human T-cell acute lymphoblastic leukemia CEM-C1 cells is due to insufficient glucocorticoid receptor expression. Cancer Research. 1996;56(21):5033–5038. [PubMed] [Google Scholar]
- 176.Ramdas J, Liu W, Harmon JM. Glucocorticoid-induced cell death requires autoinduction of glucocorticoid receptor expression in human leukemic T cells. Cancer Research. 1999;59(6):1378–1385. [PubMed] [Google Scholar]
- 177.Schwartz JR, Sarvaiya PJ, Vedeckis WV. Glucocorticoid receptor knock down reveals a similar apoptotic threshold but differing gene regulation patterns in T-cell and pre-B-cell acute lymphoblastic leukemia. Molecular and Cellular Endocrinology. 2010;320(1-2):76–86. doi: 10.1016/j.mce.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Gruber G, Carlet M, Türtscher E, et al. Levels of glucocorticoid receptor and its ligand determine sensitivity and kinetics of glucocorticoid-induced leukemia apoptosis. Leukemia. 2009;23(4):820–823. doi: 10.1038/leu.2008.360. [DOI] [PubMed] [Google Scholar]
- 179.Zilberman Y, Zafrir E, Ovadia H, Yefenof E, Guy R, Sionov RV. The glucocorticoid receptor mediates the thymic epithelial cell-induced apoptosis of CD4+8+ thymic lymphoma cells. Cellular Immunology. 2004;227(1):12–23. doi: 10.1016/j.cellimm.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 180.Moalli PA, Pillay S, Weiner D, Leikin R, Rosen ST. A mechanism of resistance to glucocorticoids in multiple myeloma: transient expression of a truncated glucocorticoid receptor mRNA. Blood. 1992;79(1):213–222. [PubMed] [Google Scholar]
- 181.Gomi M, Moriwaki K, Katagiri S, Kurata Y, Thompson EB. Glucocorticoid effects on myeloma cells in culture: correlation of growth inhibition with induction of glucocorticoid receptor messenger RNA. Cancer Research. 1990;50(6):1873–1878. [PubMed] [Google Scholar]
- 182.Bachmann PS, Gorman R, MacKenzie KL, Lutze-Mann L, Lock RB. Dexamethasone resistance in B-cell precursor childhood acute lymphoblastic leukemia occurs downstream of ligand-induced nuclear translocation of the glucocorticoid receptor. Blood. 2005;105(6):2519–2526. doi: 10.1182/blood-2004-05-2023. [DOI] [PubMed] [Google Scholar]
- 183.Bachmann PS, Gorman R, Papa RA, et al. Divergent mechanisms of glucocorticoid resistance in experimental models of pediatric acute lymphoblastic leukemia. Cancer Research. 2007;67(9):4482–4490. doi: 10.1158/0008-5472.CAN-06-4244. [DOI] [PubMed] [Google Scholar]
- 184.Haarman EG, Kaspers GJL, Pieters R, Rottier MMA, Veerman AJP. Glucocorticoid receptor alpha, beta and gamma expression vs in vitro glucocorticod resistance in childhood leukemia. Leukemia. 2004;18(3):530–537. doi: 10.1038/sj.leu.2403225. [DOI] [PubMed] [Google Scholar]
- 185.Beesley AH, Weller RE, Senanayake S, Welch M, Kees UR. Receptor mutation is not a common mechanism of naturally occurring glucocorticoid resistance in leukaemia cell lines. Leukemia Research. 2009;33(2):321–325. doi: 10.1016/j.leukres.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 186.Labuda M, Gahier A, Gagné V, Moghrabi A, Sinnett D, Krajinovic M. Polymorphisms in glucocorticoid receptor gene and the outcome of childhood acute lymphoblastic leukemia (ALL) Leukemia Research. 2010;34(4):492–497. doi: 10.1016/j.leukres.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 187.Cuzzoni E, De Iudicibus S, Bartoli F, Ventura A, Decorti G. Association between BclI polymorphism in the NR3C1 gene and in vitro individual variations in lymphocyte responses to methylprednisolone. British Journal of Clinical Pharmacology. 2012;73:651–655. doi: 10.1111/j.1365-2125.2011.04130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Burnstein KL, Jewell CM, Cidlowski JA. Human glucocorticoid receptor cDNA contains sequences sufficient for receptor down-regulation. The Journal of Biological Chemistry. 1990;265(13):7284–7291. [PubMed] [Google Scholar]
- 189.Burnstein KL, Jewell CM, Sar M, Cidlowski JA. Intragenic sequences of the human glucocorticoid receptor complementary DNA mediate hormone-inducible receptor messenger RNA down-regulation through multiple mechanisms. Molecular Endocrinology. 1994;8(12):1764–1773. doi: 10.1210/mend.8.12.7708063. [DOI] [PubMed] [Google Scholar]
- 190.Davies L, Karthikeyan N, Lynch JT, et al. Cross talk of signaling pathways in the regulation of the glucocorticoid receptor function. Molecular Endocrinology. 2008;22(6):1331–1344. doi: 10.1210/me.2007-0360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Faus H, Haendler B. Post-translational modifications ofsteroid receptors. Biomedicine and Pharmacotherapy. 2006;60(9):520–528. doi: 10.1016/j.biopha.2006.07.082. [DOI] [PubMed] [Google Scholar]
- 192.Wallace AD, Cidlowski JA. Proteasome-mediated glucocorticoid receptor degradation restricts transcriptional signaling by glucocorticoids. The Journal of Biological Chemistry. 2001;276(46):42714–42721. doi: 10.1074/jbc.M106033200. [DOI] [PubMed] [Google Scholar]
- 193.Itoh M, Adachi M, Yasui H, Takekawa M, Tanaka H, Imai K. Nuclear export of glucocorticoid receptor is enhanced by c-Jun N-terminal kinase-mediated phosphorylation. Molecular Endocrinology. 2002;16(10):2382–2392. doi: 10.1210/me.2002-0144. [DOI] [PubMed] [Google Scholar]
- 194.Chen W, Dang T, Blind RD, et al. Glucocorticoid receptor phosphorylation differentially affects target gene expression. Molecular Endocrinology. 2008;22(8):1754–1766. doi: 10.1210/me.2007-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Miller AL, Webb MS, Copik AJ, et al. p38 mitogen-activated protein kinase (MAPK) is a key mediator in glucocorticoid-induced apoptosis of lymphoid cells: correlation between p38 MAPK activation and site-specific phosphorylation of the human glucocorticoid receptor at serine 211. Molecular Endocrinology. 2005;19(6):1569–1583. doi: 10.1210/me.2004-0528. [DOI] [PubMed] [Google Scholar]
- 196.Hsu S, Qi M, DeFranco DB. Cell cycle regulation of glucocorticoid receptor function. EMBO Journal. 1992;11(9):3457–3468. doi: 10.1002/j.1460-2075.1992.tb05425.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Bodwell JE, Webster JC, Jewell CM, Cidlowski JA, Hu JM, Munck A. Glucocorticoid receptor phosphorylation: overview, function and cell cycle-dependence. Journal of Steroid Biochemistry and Molecular Biology. 1998;65(1–6):91–99. doi: 10.1016/s0960-0760(97)00185-4. [DOI] [PubMed] [Google Scholar]
- 198.Li MD, Ruan HB, Singh JP, et al. O-GlcNAc transferase is involved in glucocorticoid receptor-mediated transrepression. The Journal of Biological Chemistry. 2012;287:12904–12912. doi: 10.1074/jbc.M111.303792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Webb MS, Miller AL, Johnson BH, et al. Gene networks in glucocorticoid-evoked apoptosis of leukemic cells. Journal of Steroid Biochemistry and Molecular Biology. 2003;85(2–5):183–193. doi: 10.1016/s0960-0760(03)00194-8. [DOI] [PubMed] [Google Scholar]
- 200.Medh RD, Webb MS, Miller AL, et al. Gene expression profile of human lymphoid CEM cells sensitive and resistant to glucocorticoid-evoked apoptosis. Genomics. 2003;81(6):543–555. doi: 10.1016/s0888-7543(03)00045-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Schmidt S, Rainer J, Riml S, et al. Identification of glucocorticoid-response genes in children with acute lymphoblastic leukemia. Blood. 2006;107(5):2061–2069. doi: 10.1182/blood-2005-07-2853. [DOI] [PubMed] [Google Scholar]
- 202.Thompson EB, Johnson BH. Regulation of a distinctive set of genes in glucocorticoid-evoked apoptosis in CEM human lymphoid cells. Recent Progress in Hormone Research. 2003;58:175–197. doi: 10.1210/rp.58.1.175. [DOI] [PubMed] [Google Scholar]
- 203.Rainer J, Lelong J, Bindreither D, et al. Research resource: transcriptional response to glucocorticoids in childhood acute lymphoblastic leukemia. Molecular Endocrinology. 2012;26:178–193. doi: 10.1210/me.2011-1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Chen DW, Saha V, Liu JZ, Schwartz JM, Krstic-Demonacos M. Erg and AP-1 as determinants of glucocorticoid response in acute lymphoblastic leukemia. doi: 10.1038/onc.2012.321. Oncogene. In press. [DOI] [PubMed] [Google Scholar]
- 205.Bouillet P, Metcalf D, Huang DCS. Pro-apoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 1999;286(5445):1735–1738. doi: 10.1126/science.286.5445.1735. [DOI] [PubMed] [Google Scholar]
- 206.Molitoris JK, McColl KS, Distelhorst CW. Glucocorticoid-mediated repression of the oncogenic microRNA cluster miR-17∼92 contributes to the induction of Bim and initiation of apoptosis. Molecular Endocrinology. 2011;25:409–420. doi: 10.1210/me.2010-0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Abrams MT, Robertson NM, Yoon K, Wickstrom E. Inhibition of glucocorticoid-induced apoptosis by targeting the major splice variants of BIM mRNA with small interfering RNA and short hairpin RNA. The Journal of Biological Chemistry. 2004;279(53):55809–55817. doi: 10.1074/jbc.M411767200. [DOI] [PubMed] [Google Scholar]
- 208.Wang Z, Malone MH, He H, McColl KS, Distelhorst CW. Microarray analysis uncovers the induction of the pro-apoptotic BH3-only protein Bim in multiple models of glucocorticoid-induced apoptosis. The Journal of Biological Chemistry. 2003;278(26):23861–23867. doi: 10.1074/jbc.M301843200. [DOI] [PubMed] [Google Scholar]
- 209.Sionov RV, Kfir S, Zafrir E, Cohen O, Zilberman Y, Yefenof E. Glucocorticoid-induced apoptosis revisited: a novel role for glucocorticoid receptor translocation to the mitochondria. Cell Cycle. 2006;5(10):1017–1026. doi: 10.4161/cc.5.10.2738. [DOI] [PubMed] [Google Scholar]
- 210.O’Reilly LA, Cullen L, Visvader J, et al. The pro-apoptotic BH3-only protein Bim is expressed in hematopoietic, epithelial, neuronal, and germ cells. American Journal of Pathology. 2000;157(2):449–461. doi: 10.1016/S0002-9440(10)64557-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Zhao YN, Guo X, Ma ZG, Gu L, Ge J, Li Q. Pro-apoptotic protein BIM in apoptosis of glucocorticoid-sensitive and -resistant acute lymphoblastic leukemia CEM cells. Medical Oncology. 2011;28(4):1609–1617. doi: 10.1007/s12032-010-9641-x. [DOI] [PubMed] [Google Scholar]
- 212.Bhojwani D, Kang H, Menezes RX, et al. Gene expression signatures predictive of early response and outcome in high-risk childhood acute lymphoblastic leukemia: a Children’s Oncology Group Study on Behalf of the Dutch Childhood Oncology Group and the German Cooperative Study Group for Childhood Acute Lymphoblastic Leukemia. Journal of Clinical Oncology. 2008;26(27):4376–4384. doi: 10.1200/JCO.2007.14.4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Tagawa H, Karnan S, Suzuki R, et al. Genome-wide array-based CGH for mantle cell lymphoma: identification of homozygous deletions of the pro-apoptotic gene BIM. Oncogene. 2005;24(8):1348–1358. doi: 10.1038/sj.onc.1208300. [DOI] [PubMed] [Google Scholar]
- 214.Mestre-Escorihuela C, Rubio-Moscardo F, Richter JA, et al. Homozygous deletions localize novel tumor suppressor genes in B-cell lymphomas. Blood. 2007;109(1):271–280. doi: 10.1182/blood-2006-06-026500. [DOI] [PubMed] [Google Scholar]
- 215.Happo L, Strasser A, Cory S. BH3-only proteins in apoptosis at a glance. Journal of Cell Science. 2012;125:1081–1087. doi: 10.1242/jcs.090514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Molecular Cell. 2010;37(3):299–310. doi: 10.1016/j.molcel.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Willis SN, Fletcher JI, Kaufmann T, et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science. 2007;315(5813):856–859. doi: 10.1126/science.1133289. [DOI] [PubMed] [Google Scholar]
- 218.Gillings AS, Balmanno K, Wiggins CM, Johnson M, Cook SJ. Apoptosis and autophagy: BIM as a mediator of tumour cell death in response to oncogene-targeted therapeutics. FEBS Journal. 2009;276(21):6050–6062. doi: 10.1111/j.1742-4658.2009.07329.x. [DOI] [PubMed] [Google Scholar]
- 219.Melarangi T, Zhuang J, Lin K, et al. Glucocorticoid resistance in chronic lymphocytic leukaemia is associated with a failure of upregulated Bim/Bcl-2 complexes to activate Bax and Bak. Cell Death and Disease. 2012;3 doi: 10.1038/cddis.2012.102.e372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Erlacher M, Michalak EM, Kelly PN, et al. BH3-only proteins Puma and Bim are rate-limiting for γ-radiation- and glucocorticoid-induced apoptosis of lymphoid cells in vivo. Blood. 2005;106(13):4131–4138. doi: 10.1182/blood-2005-04-1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Labi V, Erlacher M, Kiessling S, et al. Loss of the BH3-only protein Bmf impairs B cell homeostasis and accelerates γ irradiation—induced thymic lymphoma development. Journal of Experimental Medicine. 2008;205(3):641–655. doi: 10.1084/jem.20071658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Rathmell JC, Lindsten T, Zong WX, Cinalli RM, Thompson CB. Deficiency in Bak and Bax perturbs thymic selection and lymphoid homeostasis. Nature Immunology. 2002;3(10):932–939. doi: 10.1038/ni834. [DOI] [PubMed] [Google Scholar]
- 223.Wang Z, Rong YP, Malone MH, Davis MC, Zhong F, Distelhorst CW. Thioredoxin-interacting protein (txnip) is a glucocorticoid-regulated primary response gene involved in mediating glucocorticoid-induced apoptosis. Oncogene. 2006;25(13):1903–1913. doi: 10.1038/sj.onc.1209218. [DOI] [PubMed] [Google Scholar]
- 224.Ploner C, Rainer J, Lobenwein S, Geley S, Kofler R. Repression of the BH3-only molecule PMAIP1/Noxa impairs glucocorticoid sensitivity of acute lymphoblastic leukemia cells. Apoptosis. 2009;14(6):821–828. doi: 10.1007/s10495-009-0355-5. [DOI] [PubMed] [Google Scholar]
- 225.Mansha M, Wasim M, Kofler A, Ploner C. Expression and glucocorticoid-regulation of, “Bam”, a novel BH3-only transcript in acute lymphoblastic leukemia. Molecular Biology Reports. 2012;39:6007–6013. doi: 10.1007/s11033-011-1414-x. [DOI] [PubMed] [Google Scholar]
- 226.Lu J, Quearry B, Harada H. p38-MAP kinase activation followed by BIM induction is essential for glucocorticoid-induced apoptosis in lymphoblastic leukemia cells. FEBS Letters. 2006;580(14):3539–3544. doi: 10.1016/j.febslet.2006.05.031. [DOI] [PubMed] [Google Scholar]
- 227.Dijkers PF, Medema RH, Lammers JWJ, Koenderman L, Coffer PJ. Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Current Biology. 2000;10(19):1201–1204. doi: 10.1016/s0960-9822(00)00728-4. [DOI] [PubMed] [Google Scholar]
- 228.Gilley J, Coffer PJ, Ham J. FOXO transcription factors directly activate bim gene expression and promote apoptosis in sympathetic neurons. Journal of Cell Biology. 2003;162(4):613–622. doi: 10.1083/jcb.200303026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Essafi A, Fernández de Mattos S, Hassen YAM, et al. Direct transcriptional regulation of Bim by FoxO3a mediates STI571-induced apoptosis in Bcr-Abl-expressing cells. Oncogene. 2005;24(14):2317–2329. doi: 10.1038/sj.onc.1208421. [DOI] [PubMed] [Google Scholar]
- 230.Wildey GM, Howe PH. Runx1 is a co-activator with FOXO3 to mediate transforming growth factor β(TGFβ)-induced Bim transcription in hepatic cells. The Journal of Biological Chemistry. 2009;284(30):20227–20239. doi: 10.1074/jbc.M109.027201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Lützner N, Kalbacher H, Krones-Herzig A, Rosl F. FOXO3 is a glucocorticoid receptor target and regulates lkb1 and its own expression based on cellular amp levels via a positive autoregulatory loop. PLoS ONE. 2012;7 doi: 10.1371/journal.pone.0042166.e42166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Tzivion G, Dobson M, Ramakrishnan G. FoxO transcription factors; Regulation by AKT and 14-3-3 proteins. Biochimica et Biophysica Acta. 2011;1813(11):1938–1945. doi: 10.1016/j.bbamcr.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 233.Zhang X, Tang N, Hadden TJ, Rishi AK. Akt, FoxO and regulation of apoptosis. Biochimica et Biophysica Acta. 2011;1813(11):1938–1945. doi: 10.1016/j.bbamcr.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 234.Ho KK, McGuire VA, Koo CY, et al. Phosphorylation of FOXO3a on Ser-7 by p38 promotes its nuclear localization in response to doxorubicin. The Journal of Biological Chemistry. 2012;287:1545–1555. doi: 10.1074/jbc.M111.284224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Cai B, Xia Z. p38 MAP kinase mediates arsenite-induced apoptosis through FOXO3a activation and induction of Bim transcription. Apoptosis. 2008;13(6):803–810. doi: 10.1007/s10495-008-0218-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Yang JY, Zong CS, Xia W, et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nature Cell Biology. 2008;10(2):138–148. doi: 10.1038/ncb1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Ley R, Balmanno K, Hadfield K, Weston C, Cook SJ. Activation of the ERK1/2 signaling pathway promotes phosphorylation and proteasome-dependent degradation of the BH3-only protein, Bim. The Journal of Biological Chemistry. 2003;278(21):18811–18816. doi: 10.1074/jbc.M301010200. [DOI] [PubMed] [Google Scholar]
- 238.Dehan E, Bassermann F, Guardavaccaro D, et al. βTrCP- and Rsk1/2-mediated degradation of BimEL inhibits apoptosis. Molecular Cell. 2009;33(1):109–116. doi: 10.1016/j.molcel.2008.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Ewings KE, Hadfield-Moorhouse K, Wiggins CM, et al. ERK1/2-dependent phosphorylation of BimEL promotes its rapid dissociation from Mcl-1 and Bcl-xL. EMBO Journal. 2007;26(12):2856–2867. doi: 10.1038/sj.emboj.7601723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Lei K, Davis RJ. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(5):2432–2437. doi: 10.1073/pnas.0438011100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Putcha GV, Le S, Frank S, et al. JNK-mediated BIM phosphorylation potentiates BAX-dependent apoptosis. Neuron. 2003;38(6):899–914. doi: 10.1016/s0896-6273(03)00355-6. [DOI] [PubMed] [Google Scholar]
- 242.Engelbrecht Y, De Wet H, Horsch K, Langeveldt CR, Hough FS, Hulley PA. Glucocorticoids induce rapid up-regulation of mitogen-activated protein kinase phosphatase-1 and dephosphorylation of extracellular signal-regulated kinase and impair proliferation in human and mouse osteoblast cell lines. Endocrinology. 2003;144(2):412–422. doi: 10.1210/en.2002-220769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Rambal AA, Panaguiton ZLG, Kramer L, Grant S, Harada H. MEK inhibitors potentiate dexamethasone lethality in acute lymphoblastic leukemia cells through the pro-apoptotic molecule BIM. Leukemia. 2009;23(10):1744–1754. doi: 10.1038/leu.2009.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Zhou F, Thompson EB. Role of c-jun induction in the glucocorticoid-evoked apoptotic pathway in human leukemic lymphoblasts. Molecular Endocrinology. 1996;10(3):306–316. doi: 10.1210/mend.10.3.8833659. [DOI] [PubMed] [Google Scholar]
- 245.Heidari N, Miller AV, Hicks MA, Marking CB, Harada H. Glucocorticoid-mediated BIM induction and apoptosis are regulated by Runx2 and c-Jun in leukemia cells. Cell Death and Disease. 2012;3 doi: 10.1038/cddis.2012.89.e349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Ventura A, Young AG, Winslow MM, et al. Targeted deletion reveals essential and overlapping functions of the miR-17∼92 family of miRNA clusters. Cell. 2008;132(5):875–886. doi: 10.1016/j.cell.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Xiao C, Srinivasan L, Calado DP, et al. Lymphoproliferative disease and autoimmunity in mice with increased miR-17∼92 expression in lymphocytes. Nature Immunology. 2008;9(4):405–414. doi: 10.1038/ni1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Mu P, Han YC, Betel D, et al. Genetic dissection of the miR-17∼92 cluster of microRNAs in Myc-induced B-cell lymphomas. Genes & Development. 2009;23:2806–2811. doi: 10.1101/gad.1872909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Olive V, Jiang I, He L. miR-17∼92, a cluster of miRNAs in the midst of the cancer network. International Journal of Biochemistry and Cell Biology. 2010;42(8):1348–1354. doi: 10.1016/j.biocel.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.van Haaften G, Agami R. Tumorigenicity of the miR-17∼92 cluster distilled. Genes and Development. 2010;24(1):1–4. doi: 10.1101/gad.1887110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Mi S, Li Z, Chen P, et al. Aberrant overexpression and function of the miR-17∼92 cluster in MLL-rearranged acute leukemia. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(8):3710–3715. doi: 10.1073/pnas.0914900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Lee SK, Calin GA. Non-coding RNAs and cancer: new paradigms in oncology. Discovery Medicine. 2011;11(58):245–254. [PubMed] [Google Scholar]
- 253.Mavrakis KJ, Van Der Meulen J, Wolfe AL, et al. A cooperative microRNA-tumor suppressor gene network in acute T-cell lymphoblastic leukemia. Nature Genetics. 2011;43(8):p. 815. doi: 10.1038/ng.858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Wu WKK, Lee CW, Cho CH, et al. MicroRNA dysregulation in gastric cancer: a new player enters the game. Oncogene. 2010;29(43):5761–5771. doi: 10.1038/onc.2010.352. [DOI] [PubMed] [Google Scholar]
- 255.O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435(7043):839–843. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- 256.Li Y, Tan W, Neo TWL, et al. Role of the miR-106b-25 microRNA cluster in hepatocellular carcinoma. Cancer Science. 2009;100(7):1234–1242. doi: 10.1111/j.1349-7006.2009.01164.x. [DOI] [PubMed] [Google Scholar]
- 257.Haftmann C, Stittrich AB, Sgouroudis E, et al. Lymphocyte signaling: regulation of FoxO transcription factors by microRNAs. Annals of the New York Academy of Sciences. 2012;1247:46–55. doi: 10.1111/j.1749-6632.2011.06264.x. [DOI] [PubMed] [Google Scholar]
- 258.Guttilla IK, White BA. Coordinate regulation of FOXO1 by miR-27a, miR-96, and miR-182 in breast cancer cells. The Journal of Biological Chemistry. 2009;284(35):23204–23216. doi: 10.1074/jbc.M109.031427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Segura MF, Hanniford D, Menendez S, et al. Aberrant miR-182 expression promotes melanoma metastasis by repressing FOXO3 and microphthalmia-associated transcription factor. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(6):1814–1819. doi: 10.1073/pnas.0808263106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Stittrich AB, Haftmann C, Sgouroudis E, et al. The microRNA miR-182 is induced by IL-2 and promotes clonal expansion of activated helper T lymphocytes. Nature Immunology. 2010;11(11):1057–1062. doi: 10.1038/ni.1945. [DOI] [PubMed] [Google Scholar]
- 261.Yang A, Ma J, Wu M, et al. Aberrant microRNA-182 expression is associated with glucocorticoid resistance in lymphoblastic malignancies. Leukemia & Lymphoma. 2012;53(12):2465–2473. doi: 10.3109/10428194.2012.693178. [DOI] [PubMed] [Google Scholar]
- 262.Elia L, Contu R, Quintavalle M, et al. Reciprocal regulation of microrna-1 and insulin-like growth factor-1 signal transduction cascade in cardiac and skeletal muscle in physiological and pathological conditions. Circulation. 2009;120(23):2377–2385. doi: 10.1161/CIRCULATIONAHA.109.879429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Yamamoto M, Kondo E, Takeuchi M, et al. miR-155, a modulator of FOXO3a protein expression, is underexpressed and cannot be upregulated by stimulation of HOZOT, a line of multifunctional Treg. PLoS ONE. 2011;6(2) doi: 10.1371/journal.pone.0016841.e16841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Kong W, He L, Coppola M, et al. MicroRNA-155 regulates cell survival, growth, and chemosensitivity by targeting FOXO3a in breast cancer. The Journal of Biological Chemistry. 2010;285(23):17869–17879. doi: 10.1074/jbc.M110.101055. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 265.O’Connell RM, Rao DS, Chaudhuri AA, Baltimore D. Physiological and pathological roles for microRNAs in the immune system. Nature Reviews Immunology. 2010;10(2):111–122. doi: 10.1038/nri2708. [DOI] [PubMed] [Google Scholar]
- 266.Thai TH, Calado DP, Casola S, et al. Regulation of the germinal center response by MicroRNA-155. Science. 2007;316(5824):604–608. doi: 10.1126/science.1141229. [DOI] [PubMed] [Google Scholar]
- 267.Dai R, Zhang Y, Khan D, et al. Identification of a common lupus disease-associated microRNA expression pattern in three different murine models of lupus. PLoS ONE. 2010;5(12):p. e14302. doi: 10.1371/journal.pone.0014302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Wang K, Li PF. Foxo3a regulates apoptosis by negatively targeting miR-21. The Journal of Biological Chemistry. 2010;285(22):16958–16966. doi: 10.1074/jbc.M109.093005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Meng F, Henson R, Wehbe-Janek H, Ghoshal K, Jacob ST, Patel T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology. 2007;133(2):647–658. doi: 10.1053/j.gastro.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Iliopoulos D, Jaeger SA, Hirsch HA, Bulyk ML, Struhl K. STAT3 activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Molecular Cell. 2010;39(4):493–506. doi: 10.1016/j.molcel.2010.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Ma J, Xie Y, Shi Y, Qin W, Zhao B, Jin Y. Glucocorticoid-induced apoptosis requires FOXO3A activity. Biochemical and Biophysical Research Communications. 2008;377(3):894–898. doi: 10.1016/j.bbrc.2008.10.097. [DOI] [PubMed] [Google Scholar]
- 272.Sionov RV, Cohen O, Kfir S, Zilberman Y, Yefenof E. Role of mitochondrial glucocorticoid receptor in glucocorticoid-induced apoptosis. Journal of Experimental Medicine. 2006;203(1):189–201. doi: 10.1084/jem.20050433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Talabér G, Boldizsár F, Bartis D, et al. Mitochondrial translocation of the glucocorticoid receptor in double-positive thymocytes correlates with their sensitivity to glucocorticoid-induced apoptosis. International Immunology. 2009;21(11):1269–1276. doi: 10.1093/intimm/dxp093. [DOI] [PubMed] [Google Scholar]
- 274.Du J, Wang Y, Hunter R, et al. Dynamic regulation of mitochondrial function by glucocorticoids. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(9):3543–3548. doi: 10.1073/pnas.0812671106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Tome ME, Lee K, Jaramillo MC, Briehl MM. Mitochondria are the primary source of the H2O2 signal for glucocorticoid-induced apoptosis of lymphoma cells. Experimental and Therapeutic Medicine. 2012;4:237–242. doi: 10.3892/etm.2012.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Tome ME, Jaramillo MC, Briehl MM. Hydrogen peroxide signaling is required for glucocorticoid-induced apoptosis in lymphoma cells. Free Radical Biology and Medicine. 2011;51:2048–2059. doi: 10.1016/j.freeradbiomed.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Psarra AMG, Hermann S, Panayotou G, Spyrou G. Interaction of mitochondrial thioredoxin with glucocorticoid receptor and NF-κB modulates glucocorticoid receptor and NF-κB signalling in HEK-293 cells. Biochemical Journal. 2009;422(3):521–531. doi: 10.1042/BJ20090107. [DOI] [PubMed] [Google Scholar]
- 278.Psarra AMG, Sekeris CE. Glucocorticoids induce mitochondrial gene transcription in HepG2 cells. Role of the mitochondrial glucocorticoid receptor. Biochimica et Biophysica Acta. 2011;1813(10):1814–1821. doi: 10.1016/j.bbamcr.2011.05.014. [DOI] [PubMed] [Google Scholar]
- 279.Eberhart K, Rainer J, Bindreither D, et al. Glucocorticoid-induced alterations in mitochondrial membrane properties and respiration in childhood acute lymphoblastic leukemia. Biochimica et Biophysica Acta. 2011;1807(6):719–725. doi: 10.1016/j.bbabio.2010.12.010. [DOI] [PubMed] [Google Scholar]
- 280.Fujita C, Ichikawa F, Teratani T, et al. Direct effects of corticosterone on ATP production by mitochondria from immortalized hypothalamic GT1-7 neurons. Journal of Steroid Biochemistry and Molecular Biology. 2009;117(1–3):50–55. doi: 10.1016/j.jsbmb.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 281.Beck IME, Berghe WV, Vermeulen L, Yamamoto KR, Haegeman G, De Bosscher K. Crosstalk in inflammation: the interplay of glucocorticoid receptor-based mechanisms and kinases and phosphatases. Endocrine Reviews. 2009;30(7):830–882. doi: 10.1210/er.2009-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Nuutinen U, Ropponen A, Suoranta S, et al. Dexamethasone-induced apoptosis and up-regulation of Bim is dependent on glycogen synthase kinase-3. Leukemia Research. 2009;33(12):1714–1717. doi: 10.1016/j.leukres.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 283.Nuutinen U, Postila V, Mättö M, et al. Inhibition of PI3-kinase-Akt pathway enhances dexamethasone-induced apoptosis in a human follicular lymphoma cell line. Experimental Cell Research. 2006;312(3):322–330. doi: 10.1016/j.yexcr.2005.10.023. [DOI] [PubMed] [Google Scholar]
- 284.Rubio-Patiño C, Palmeri CM, Perez-Perarnau A, et al. Glycogen synthase kinase-3beta is involved in ligand-dependent activation of transcription and cellular localization of the glucocorticoid receptor. Molecular Endocrinology. 2012;26:1508–1520. doi: 10.1210/me.2011-1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Wang FS, Ko JY, Weng LH, Yeh DW, Ke HJ, Wu SL. Inhibition of glycogen synthase kinase-3β attenuates glucocorticoid-induced bone loss. Life Sciences. 2009;85(19-20):685–692. doi: 10.1016/j.lfs.2009.09.009. [DOI] [PubMed] [Google Scholar]
- 286.Bornhauser BC, Bonapace L, Lindholm D, et al. Low-dose arsenic trioxide sensitizes glucocorticoid-resistant acute lymphoblastic leukemia cells to dexamethasone via an Akt-dependent pathway. Blood. 2007;110(6):2084–2091. doi: 10.1182/blood-2006-12-060970. [DOI] [PubMed] [Google Scholar]
- 287.Wei G, Twomey D, Lamb J, et al. Gene expression-based chemical genomics identifies rapamycin as a modulator of MCL1 and glucocorticoid resistance. Cancer Cell. 2006;10(4):331–342. doi: 10.1016/j.ccr.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 288.Morishita N, Tsukahara H, Chayama K, et al. Activation of Akt is associated with poor prognosis and chemotherapeutic resistance in pediatric B-precursor acute lymphoblastic leukemia. Pediatric Blood & Cancer. 2012;59:83–89. doi: 10.1002/pbc.24034. [DOI] [PubMed] [Google Scholar]
- 289.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes and Development. 2004;18(16):1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 290.Lee K, Nam KT, Cho SH, et al. Vital roles of mTOR complex 2 in Notch-driven thymocyte differentiation and leukemia. Journal of Experimental Medicine. 2012;209:713–728. doi: 10.1084/jem.20111470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Martelli AM, Evangelisti C, Chiarini F, McCubrey JA. The phosphatidylinositol 3-kinase/Akt/mTOR signaling network as a therapeutic target in acute myelogenous leukemia patients. Oncotarget. 2010;1(2):89–103. doi: 10.18632/oncotarget.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Carnero A. The PKB/AKT pathway in cancer. Current Pharmaceutical Design. 2010;16(1):34–44. doi: 10.2174/138161210789941865. [DOI] [PubMed] [Google Scholar]
- 293.Hafsi S, Pezzino FM, Candido S, et al. Gene alterations in the PI3K/PTEN/AKT pathway as a mechanism of drug-resistance. International Journal of Oncology. 2012;40:639–644. doi: 10.3892/ijo.2011.1312. [DOI] [PubMed] [Google Scholar]
- 294.Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12(1):9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 295.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307(5712):1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 296.Wang Z, Malone MH, Thomenius MJ, Zhong F, Xu F, Distelhorst CW. Dexamethasone-induced gene 2 (dig2) is a novel pro-survival stress gene induced rapidly by diverse apoptotic signals. The Journal of Biological Chemistry. 2003;278(29):27053–27058. doi: 10.1074/jbc.M303723200. [DOI] [PubMed] [Google Scholar]
- 297.Shinjyo T, Kuribara R, Inukai T, et al. Downregulation of Bim, a pro-apoptotic relative of Bcl-2, is a pivotal step in cytokine-initiated survival signaling in murine hematopoietic progenitors. Molecular and Cellular Biology. 2001;21(3):854–864. doi: 10.1128/MCB.21.3.854-864.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Argyriou P, Economopoulou P, Papageorgiou S. The role of mTOR inhibitors for the treatment of B-Cell lymphomas. Advances in Hematology. 2012;2012:13 pages. doi: 10.1155/2012/435342.435342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN tumor suppression. Cell. 2008;133(3):403–414. doi: 10.1016/j.cell.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 300.Jotta PY, Ganazza MA, Silva A, et al. Negative prognostic impact of PTEN mutation in pediatric T-cell acute lymphoblastic leukemia. Leukemia. 2010;24(1):239–242. doi: 10.1038/leu.2009.209. [DOI] [PubMed] [Google Scholar]
- 301.Fata JE, Debnath S, Jenkins EC, Jr, Fournier MV. Nongenomic mechanisms of PTEN regulation. International Journal of Cell Biology. 2012;2012:10 pages. doi: 10.1155/2012/379685.379685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.García JM, Silva J, Peña C, et al. Promoter methylation of the PTEN gene is a common molecular change in breast cancer. Genes Chromosomes and Cancer. 2004;41(2):117–124. doi: 10.1002/gcc.20062. [DOI] [PubMed] [Google Scholar]
- 303.Kang YH, Hye SL, Woo HK. Promoter methylation and silencing of PTEN in gastric carcinoma. Laboratory Investigation. 2002;82(3):285–291. doi: 10.1038/labinvest.3780422. [DOI] [PubMed] [Google Scholar]
- 304.Brauer PM, Tyner AL. RAKing in AKT: a tumor suppressor function for the intracellular tyrosine kinase FRK. Cell Cycle. 2009;8(17):2728–2732. doi: 10.4161/cc.8.17.9389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Torres J, Pulido R. The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus. Implications for PTEN stability to proteasome-mediated degradation. The Journal of Biological Chemistry. 2001;276(2):993–998. doi: 10.1074/jbc.M009134200. [DOI] [PubMed] [Google Scholar]
- 306.Maccario H, Perera NM, Davidson L, Downes CP, Leslie NR. PTEN is destabilized by phosphorylation on Thr366. Biochemical Journal. 2007;405(3):439–444. doi: 10.1042/BJ20061837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Wang X, Trotman LC, Koppie T, et al. NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell. 2007;128(1):129–139. doi: 10.1016/j.cell.2006.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Guo H, Qiao G, Ying H, et al. E3 ubiquitin ligase Cbl-b regulates Pten via Nedd4 in T cells independently of its ubiquitin ligase activity. Cell Reports. 2012;1:472–482. doi: 10.1016/j.celrep.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Van Themsche C, Leblanc V, Parent S, Asselin E. X-linked inhibitor of apoptosis protein (XIAP) regulates PTEN ubiquitination, content, and compartmentalization. The Journal of Biological Chemistry. 2009;284(31):20462–20466. doi: 10.1074/jbc.C109.009522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Redondo-Muñoz J, Escobar-Díaz E, Del Cerro MH, et al. Induction of B-chronic lymphocytic leukemia cell apoptosis by arsenic trioxide involves suppression of the phosphoinositide 3-kinase/Akt survival pathway via c-jun-NH2 terminal kinase activation and PTEN upregulation. Clinical Cancer Research. 2010;16(17):4382–4391. doi: 10.1158/1078-0432.CCR-10-0072. [DOI] [PubMed] [Google Scholar]
- 311.Maddika S, Kavela S, Rani N, et al. WWP2 is an E3 ubiquitin ligase for PTEN. Nature Cell Biology. 2011;13(6):728–733. doi: 10.1038/ncb2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Ahmed SF, Deb S, Paul I, et al. The chaperone-assisted E3 ligase C terminus of Hsc70-interacting protein (CHIP) targets PTEN for proteasomal degradation. The Journal of Biological Chemistry. 2012;287:15996–16006. doi: 10.1074/jbc.M111.321083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Ni Z, Tang J, Cai Z, et al. A new pathway of glucocorticoid action for asthma treatment through the regulation of PTEN expression. Respiratory Research. 2011;12:p. 47. doi: 10.1186/1465-9921-12-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Poliseno L, Salmena L, Riccardi L, et al. Identification of the miR-106b∼25 microRNA cluster as a proto-oncogenic PTEN-targeting intron that cooperates with its host gene MCM7 in transformation. Science Signaling. 2010;3 doi: 10.1126/scisignal.2000594.ra29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Huse JT, Brennan C, Hambardzumyan D, et al. The PTEN-regulating microRNA miR-26a is amplified in high-grade glioma and facilitates gliomagenesis in vivo. Genes and Development. 2009;23(11):1327–1337. doi: 10.1101/gad.1777409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Wang C, Bian Z, Wei D, Zhang JG. miR-29b regulates migration of human breast cancer cells. Molecular and Cellular Biochemistry. 2011;352(1-2):197–207. doi: 10.1007/s11010-011-0755-z. [DOI] [PubMed] [Google Scholar]
- 317.Jindra PT, Bagley J, Godwin JG, Iacomini J. Costimulation-dependent expression of microRNA-214 increases the ability of T cells to proliferate by targeting Pten. Journal of Immunology. 2010;185(2):990–997. doi: 10.4049/jimmunol.1000793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Yang H, Kong W, He L, et al. MicroRNA expression profiling in human ovarian cancer: miR-214 induces cell survival and cisplatin resistance by targeting PTEN. Cancer Research. 2008;68(5):p. 1609. doi: 10.1158/0008-5472.CAN-07-2488. [DOI] [PubMed] [Google Scholar]
- 319.Kato M, Putta S, Wang M, et al. TGF-β activates Akt kinase through a microRNA-dependent amplifying circuit targeting PTEN. Nature Cell Biology. 2009;11(7):881–889. doi: 10.1038/ncb1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Incoronato M, Garofalo M, Urso L, et al. miR-212 increases tumor necrosis factor-related apoptosis-inducing ligand sensitivity in non-small cell lung cancer by targeting the anti-apoptotic protein PED. Cancer Research. 2010;70(9):3638–3646. doi: 10.1158/0008-5472.CAN-09-3341. [DOI] [PubMed] [Google Scholar]
- 321.Garofalo M, Di Leva G, Romano G, et al. miR-221&222 Regulate TRAIL Resistance and Enhance Tumorigenicity through PTEN and TIMP3 Downregulation. Cancer Cell. 2009;16(6):498–509. doi: 10.1016/j.ccr.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 322.Brunelle JK, Letai A. Control of mitochondrial apoptosis by the Bcl-2 family. Journal of Cell Science. 2009;122(4):437–441. doi: 10.1242/jcs.031682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Krishna S, Low ICC, Pervaiz S. Regulation of mitochondrial metabolism: yet another facet in the biology of the oncoprotein Bcl-2. Biochemical Journal. 2011;435(3):545–551. doi: 10.1042/BJ20101996. [DOI] [PubMed] [Google Scholar]
- 324.Feng H, Xiang H, Mao YW, et al. Human Bcl-2 activates ERK signaling pathway to regulate activating protein-1, lens epithelium-derived growth factor and downstream genes. Oncogene. 2004;23(44):7310–7321. doi: 10.1038/sj.onc.1208041. [DOI] [PubMed] [Google Scholar]
- 325.Vairo G, Soos TJ, Upton TM, et al. Bcl-2 retards cell cycle entry through p27(Kip1), pRB relative p130, and altered E2F regulation. Molecular and Cellular Biology. 2000;20(13):4745–4753. doi: 10.1128/mcb.20.13.4745-4753.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Katayama N, Mahmud N, Nishi K, et al. Bcl-2 in cell-cycle regulation of hematopoietic cells by transforming growth factor-β1. Leukemia and Lymphoma. 2000;39(5-6):601–605. doi: 10.3109/10428190009113390. [DOI] [PubMed] [Google Scholar]
- 327.Janumyan Y, Cui Q, Yan L, Sansam CG, Valentin M, Yang E. G0 function of BCL2 and BCL-xL requires BAX, BAK, and p27 phosphorylation by mirk, revealing a novel role of BAX and BAK in quiescence regulation. The Journal of Biological Chemistry. 2008;283(49):34108–34120. doi: 10.1074/jbc.M806294200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Greider C, Chattopadhyay A, Parkhurst C, Yang E. BCL-xL and BCL2 delay Myc-induced cell cycle entry through elevation of p27 and inhibition of G1 cyclin-dependent kinases. Oncogene. 2002;21(51):7765–7775. doi: 10.1038/sj.onc.1205928. [DOI] [PubMed] [Google Scholar]
- 329.Wang HG, Rapp UR, Reed JC. Bcl-2 targets the protein kinase Raf-1 to mitochondria. Cell. 1996;87(4):629–638. doi: 10.1016/s0092-8674(00)81383-5. [DOI] [PubMed] [Google Scholar]
- 330.Reed JC. Double identity for proteins of the Bcl-2 family. Nature. 1997;387(6635):773–776. doi: 10.1038/42867. [DOI] [PubMed] [Google Scholar]
- 331.Batsi C, Markopoulou S, Kontargiris E, et al. Bcl-2 blocks 2-methoxyestradiol induced leukemia cell apoptosis by a p27Kip1-dependent G1/S cell cycle arrest in conjunction with NF-κB activation. Biochemical Pharmacology. 2009;78(1):33–44. doi: 10.1016/j.bcp.2009.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Sionov RV, Kfir-Erenfeld S, Spokoini R, Yefenof E. A role for bcl-2 in notch1-dependent transcription in thymic lymphoma cells. Advances in Hematology. 2012;2012:5 pages. doi: 10.1155/2012/435241.435241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Wang Z, Azmi AS, Ahmad A, et al. TW-37, a small-molecule inhibitor of Bcl-2, inhibits cell growth and induces apoptosis in pancreatic cancer: involvement of notch-1 signaling pathway. Cancer Research. 2009;69(7):2757–2765. doi: 10.1158/0008-5472.CAN-08-3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Cheng N, van de Wetering CI, Knudson CM. p27 deficiency cooperates with Bcl-2 but not Bax to promote T-cell lymphoma. PLoS ONE. 2008;3(4) doi: 10.1371/journal.pone.0001911.e1911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Tucker CA, Kapanen AI, Chikh G, et al. Silencing Bcl-2 in models of mantle cell lymphoma is associated with decreases in cyclin D1, nuclear factor-κB, p53, bax, and p27 levels. Molecular Cancer Therapeutics. 2008;7(4):749–758. doi: 10.1158/1535-7163.MCT-07-0302. [DOI] [PubMed] [Google Scholar]
- 336.Janumyan YM, Sansam CG, Chattopadhyay A, et al. Bcl-xL/Bcl-2 coordinately regulates apoptosis, cell cycle arrest and cell cycle entry. EMBO Journal. 2003;22(20):5459–5470. doi: 10.1093/emboj/cdg533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Hartmann BL, Geley S, Löffler M, et al. Bcl-2 interferes with the execution phase, but not upstream events, in glucocorticoid-induced leukemia apoptosis. Oncogene. 1999;18(3):713–719. doi: 10.1038/sj.onc.1202339. [DOI] [PubMed] [Google Scholar]
- 338.Rogalińska M, Kiliańska ZM. Targeting Bcl-2 in CLL. Current Medicinal Chemistry. 2012;19(30):5109–5115. doi: 10.2174/092986712803530566. [DOI] [PubMed] [Google Scholar]
- 339.Tzifi F, Economopoulou C, Gourgiotis D, Ardavanis A, Papageorgiou S, Scorilas A. The role of BCL2 family of apoptosis regulator proteins in acute and chronic leukemias. Advances in Hematology. 2012;2012:15 pages. doi: 10.1155/2012/524308.524308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Ploner C, Rainer J, Niederegger H, et al. The BCL2 rheostat in glucocorticoid-induced apoptosis of acute lymphoblastic leukemia. Leukemia. 2008;22(2):370–377. doi: 10.1038/sj.leu.2405039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Menendez P, Vargas A, Bueno C, et al. Quantitative analysis of bcl-2 expression in normal and leukemic human B-cell differentiation. Leukemia. 2004;18(3):491–498. doi: 10.1038/sj.leu.2403231. [DOI] [PubMed] [Google Scholar]
- 342.Tsujimoto Y, Finger LR, Yunis J. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science. 1984;226(4678):1097–1099. doi: 10.1126/science.6093263. [DOI] [PubMed] [Google Scholar]
- 343.Tsujimoto Y, Cossman J, Jaffe E, Croce CM. Involvement of the bcl-2 gene in human follicular lymphoma. Science. 1985;228(4706):1440–1443. doi: 10.1126/science.3874430. [DOI] [PubMed] [Google Scholar]
- 344.Nambiar M, Raghavan SC. Mechanism of fragility at BCL2 gene minor breakpoint cluster region during t(14,18) chromosomal translocation. Journal of Biological Chemistry. 2012;287:8688–8701. doi: 10.1074/jbc.M111.307363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Calin GA, Dumitru CD, Shimizu M, et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(24):15524–15529. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Cimmino A, Calin GA, Fabbri M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(39):13944–13949. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Hermeking H. p53 enters the microRNA world. Cancer Cell. 2007;12(5):414–418. doi: 10.1016/j.ccr.2007.10.028. [DOI] [PubMed] [Google Scholar]
- 348.Li L, Yuan L, Luo J, Gao J, Guo J, Xie X. MiR-34a inhibits proliferation and migration of breast cancer through down-regulation of Bcl-2 and SIRT1. doi: 10.1007/s10238-012-0186-5. Clinical and Experimental Medicine. In press. [DOI] [PubMed] [Google Scholar]
- 349.Willimott S, Wagner SD. miR-125b and miR-155 contribute to BCL2 repression and proliferation in response to CD40 ligand (CD154) in human leukemic B-cells. The Journal of Biological Chemistry. 2012;287:2608–2617. doi: 10.1074/jbc.M111.285718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Gong J, Zhang JP, Li B, et al. MicroRNA-125b promotes apoptosis by regulating the expression of Mcl-1, Bcl-w and IL-6R. doi: 10.1038/onc.2012.318. Oncogene. In press. [DOI] [PubMed] [Google Scholar]
- 351.Zhao A, Zeng Q, Xie X, et al. MicroRNA-125b induces cancer cell apoptosis through suppression of Bcl-2 expression. Journal of Genetics and Genomics. 2012;39:29–35. doi: 10.1016/j.jgg.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 352.Liu LH, Li H, Li JP, et al. miR-125b suppresses the proliferation and migration of osteosarcoma cells through down-regulation of STAT3. Biochemical and Biophysical Research Communications. 2011;416:31–38. doi: 10.1016/j.bbrc.2011.10.117. [DOI] [PubMed] [Google Scholar]
- 353.Zhou M, Liu Z, Zhao Y, et al. MicroRNA-125b confers the resistance of breast cancer cells to paclitaxel through suppression of pro-apoptotic Bcl-2 antagonist killer 1 (Bak1) expression. The Journal of Biological Chemistry. 2010;285(28):21496–21507. doi: 10.1074/jbc.M109.083337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Zhang H, Luo XQ, Feng DD, et al. Upregulation of microRNA-125b contributes to leukemogenesis and increases drug resistance in pediatric acute promyelocytic leukemia. Molecular Cancer. 2011;10, article 108 doi: 10.1186/1476-4598-10-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Surdziel E, Cabanski M, Dallmann I, et al. Enforced expression of miR-125b affects myelopoiesis by targeting multiple signaling pathways. Blood. 2011;117(16):4338–4348. doi: 10.1182/blood-2010-06-289058. [DOI] [PubMed] [Google Scholar]
- 356.Xia HF, He TZ, Liu CM, et al. MiR-125b expression affects the proliferation and apoptosis of human glioma cells by targeting Bmf. Cellular Physiology and Biochemistry. 2009;23(4–6):347–358. doi: 10.1159/000218181. [DOI] [PubMed] [Google Scholar]
- 357.Bousquet M, Harris MH, Zhou B, Lodish HF. MicroRNA miR-125b causes leukemia. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(50):21558–21563. doi: 10.1073/pnas.1016611107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Zhu DX, Zhu W, Fang C, et al. miR-181a/b significantly enhances drug sensitivity in chronic lymphocytic leukemia cells via targeting multiple anti-apoptosis genes. Carcinogenesis. 2012;33:1294–1301. doi: 10.1093/carcin/bgs179. [DOI] [PubMed] [Google Scholar]
- 359.Li H, Hui L, Xu W. miR-181a sensitizes a multidrug-resistant leukemia cell line K562/A02 to daunorubicin by targeting BCL-2. Acta Biochimica et Biophysica Sinica. 2012;44:269–277. doi: 10.1093/abbs/gmr128. [DOI] [PubMed] [Google Scholar]
- 360.Ouyang YB, Lu Y, Yue S, Giffard RG. miR-181 targets multiple Bcl-2 family members and influences apoptosis and mitochondrial function in astrocytes. Mitochondrion. 2012;12:213–219. doi: 10.1016/j.mito.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Shimizu S, Takehara T, Hikita H, et al. The let-7 family of microRNAs inhibits Bcl-xL expression and potentiates sorafenib-induced apoptosis in human hepatocellular carcinoma. Journal of Hepatology. 2010;52(5):698–704. doi: 10.1016/j.jhep.2009.12.024. [DOI] [PubMed] [Google Scholar]
- 362.Nakano H, Miyazawa T, Kinoshita K, Yamada Y, Yoshida T. Functional screening identifies a microRNA, miR-491 that induces apoptosis by targeting Bcl-XLin colorectal cancer cells. International Journal of Cancer. 2010;127(5):1072–1080. doi: 10.1002/ijc.25143. [DOI] [PubMed] [Google Scholar]
- 363.Guan H, Zhang P, Liu C, et al. Characterization and functional analysis of the human microRNA let-7a2 promoter in lung cancer A549 cell lines. Molecular Biology Reports. 2011;38(8):5327–5334. doi: 10.1007/s11033-011-0683-8. [DOI] [PubMed] [Google Scholar]
- 364.Holleman A, Cheok MH, Den Boer ML, et al. Gene-expression patterns in drug-resistant acute lymphoblastic leukemia cells and response to treatment. The New England Journal of Medicine. 2004;351(6):533–542. doi: 10.1056/NEJMoa033513. [DOI] [PubMed] [Google Scholar]
- 365.Stam RW, Den Boer ML, Schneider P, et al. Association of high-level MCL-1 expression with in vitro and in vivo prednisone resistance in MLL-rearranged infant acute lymphoblastic leukemia. Blood. 2010;115(5):1018–1025. doi: 10.1182/blood-2009-02-205963. [DOI] [PubMed] [Google Scholar]
- 366.Thomas LW, Lam C, Edwards SW. Mcl-1; the molecular regulation of protein function. FEBS Letters. 2010;584(14):2981–2989. doi: 10.1016/j.febslet.2010.05.061. [DOI] [PubMed] [Google Scholar]
- 367.Taniai M, Grambihler A, Higuchi H, et al. Mcl-1 mediates tumor necrosis factor-related apoptosis-inducing ligand resistance in human cholangiocarcinoma cells. Cancer Research. 2004;64(10):3517–3524. doi: 10.1158/0008-5472.CAN-03-2770. [DOI] [PubMed] [Google Scholar]
- 368.Opferman JT, Green DR. DUB-le trouble for cell survival. Cancer Cell. 2010;17(2):117–119. doi: 10.1016/j.ccr.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 369.Ding Q, He X, Hsu JM, et al. Degradation of Mcl-1 by β-TrCP mediates glycogen synthase kinase 3-induced tumor suppression and chemosensitization. Molecular and Cellular Biology. 2007;27(11):4006–4017. doi: 10.1128/MCB.00620-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Molecular Cell. 2006;21(6):749–760. doi: 10.1016/j.molcel.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 371.Zhong Q, Gao W, Du F, Wang X. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell. 2005;121(7):1085–1095. doi: 10.1016/j.cell.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 372.Inuzuka H, Fukushima H, Shaik S, Wei W. Novel insights into the molecular mechanisms governing Mdm2 ubiquitination and destruction. Oncotarget. 2010;1(7):685–690. doi: 10.18632/oncotarget.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Schwickart M, Huang X, Lill JR, et al. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature. 2010;463(7277):103–107. doi: 10.1038/nature08646. [DOI] [PubMed] [Google Scholar]
- 374.Agrawal P, Chen YT, Schilling B, Gibson BW, Hughes RE. Ubiquitin-specific peptidase 9, X-linked (USP9X) modulates activity of mammalian target of rapamycin (mTOR) Journal of Biological Chemistry. 2012;287:21164–21175. doi: 10.1074/jbc.M111.328021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Czabotar PE, Lee EF, Van Delft MF, et al. Structural insights into the degradation of Mcl-1 induced by BH3 domains. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(15):6217–6222. doi: 10.1073/pnas.0701297104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Gomez-Bougie P, Menoret E, Juin P, Dousset C, Pellat-Deceunynck C, Amiot M. Noxa controls Mule-dependent Mcl-1 ubiquitination through the regulation of the Mcl-1/USP9X interaction. Biochemical and Biophysical Research Communications. 2011;413:460–464. doi: 10.1016/j.bbrc.2011.08.118. [DOI] [PubMed] [Google Scholar]
- 377.Mills JR, Hippo Y, Robert F, et al. mTORC1 promotes survival through translational control of Mcl-1. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(31):10853–10858. doi: 10.1073/pnas.0804821105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Inoki K, Ouyang H, Zhu T, et al. TSC2 integrates wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 2006;126(5):955–968. doi: 10.1016/j.cell.2006.06.055. [DOI] [PubMed] [Google Scholar]
- 379.Willis SN, Chen L, Dewson G, et al. Pro-apoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes and Development. 2005;19(11):1294–1305. doi: 10.1101/gad.1304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Desjobert C, Renalier MH, Bergalet J, et al. MiR-29a down-regulation in ALK-positive anaplastic large cell lymphomas contributes to apoptosis blockade through MCL-1 overexpression. Blood. 2011;117(24):6627–6637. doi: 10.1182/blood-2010-09-301994. [DOI] [PubMed] [Google Scholar]
- 381.Zhang YK, Wang H, Leng Y, et al. Overexpression of microRNA-29b induces apoptosis of multiple myeloma cells through down regulating Mcl-1. Biochemical and Biophysical Research Communications. 2011;414:233–239. doi: 10.1016/j.bbrc.2011.09.063. [DOI] [PubMed] [Google Scholar]
- 382.Mott JL, Kobayashi S, Bronk SF, Gores GJ. mir-29 regulates Mcl-1 protein expression and apoptosis. Oncogene. 2007;26(42):6133–6140. doi: 10.1038/sj.onc.1210436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Garzon R, Heaphy CEA, Havelange V, et al. MicroRNA 29b functions in acute myeloid leukemia. Blood. 2009;114(26):5331–5341. doi: 10.1182/blood-2009-03-211938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Wang HJ, Ruan HJ, He XJ, et al. MicroRNA-101 is down-regulated in gastric cancer and involved in cell migration and invasion. European Journal of Cancer. 2010;46(12):2295–2303. doi: 10.1016/j.ejca.2010.05.012. [DOI] [PubMed] [Google Scholar]
- 385.Visone R, Rassenti LZ, Veronese A, et al. Karyotype-specific microRNA signature in chronic lymphocytic leukemia. Blood. 2009;114(18):3872–3879. doi: 10.1182/blood-2009-06-229211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Crawford M, Batte K, Yu L, et al. MicroRNA 133B targets pro-survival molecules MCL-1 and BCL2L2 in lung cancer. Biochemical and Biophysical Research Communications. 2009;388(3):483–489. doi: 10.1016/j.bbrc.2009.07.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Chen J, Zhang X, Lentz C, et al. miR-193b Regulates Mcl-1 in melanoma. American Journal of Pathology. 2011;179:2162–2168. doi: 10.1016/j.ajpath.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Saito Y, Suzuki H, Tsugawa H, et al. Chromatin remodeling at Alu repeats by epigenetic treatment activates silenced microRNA-512-5p with downregulation of Mcl-1 in human gastric cancer cells. Oncogene. 2009;28(30):2738–2744. doi: 10.1038/onc.2009.140. [DOI] [PubMed] [Google Scholar]
- 389.Spijkers-Hagelstein JA, Schneider P, Hulleman E, et al. Elevated S100A8/S100A9 expression causes glucocorticoid resistance in MLL-rearranged infant acute lymphoblastic leukemia. Leukemia. 2012;26:1255–1265. doi: 10.1038/leu.2011.388. [DOI] [PubMed] [Google Scholar]
- 390.Minagawa N, Kruglov EA, Dranoff JA, Robert ME, Gores GJ, Nathanson MH. The anti-apoptotic protein Mcl-1 inhibits mitochondrial Ca2+ signals. The Journal of Biological Chemistry. 2005;280(39):33637–33644. doi: 10.1074/jbc.M503210200. [DOI] [PubMed] [Google Scholar]
- 391.Bassik MC, Scorrano L, Oakes SA, Pozzan T, Korsmeyer SJ. Phosphorylation of BCL-2 regulates ER Ca2+ homeostasis and apoptosis. EMBO Journal. 2004;23(5):1207–1216. doi: 10.1038/sj.emboj.7600104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Thomenius MJ, Distelhorst CW. Bcl-2 on the endoplasmic reticulum: protecting the mitochondria from a distance. Journal of Cell Science. 2003;116(22):4493–4499. doi: 10.1242/jcs.00829. [DOI] [PubMed] [Google Scholar]
- 393.Giorgi C, Baldassari F, Bononi A, et al. Mitochondrial Ca2+ and apoptosis. Cell Calcium. 2012;52:36–43. doi: 10.1016/j.ceca.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Rong YP, Aromolaran AS, Bultynck G, et al. Targeting Bcl-2-IP3 receptor interaction to reverse Bcl-2’s inhibition of apoptotic calcium signals. Molecular Cell. 2008;31(2):255–265. doi: 10.1016/j.molcel.2008.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Li C, Wang X, Vais H, Thompson CB, Foskett JK, White C. Apoptosis regulation by Bcl-xL modulation of mammalian inositol 1,4,5-trisphosphate receptor channel isoform gating. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(30):12565–12570. doi: 10.1073/pnas.0702489104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Eckenrode EF, Yang J, Velmurugan GV, Kevin Foskett J, White C. Apoptosis protection by Mcl-1 and Bcl-2 modulation of inositol 1,4,5-trisphosphate receptor-dependent Ca2+ signaling. The Journal of Biological Chemistry. 2010;285(18):13678–13684. doi: 10.1074/jbc.M109.096040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Li C, Fox CJ, Master SR, Bindokas VP, Chodosh LA, Thompson CB. Bcl-XL affects Ca2+ homeostasis by altering expression of inositol 1,4,5-trisphosphate receptors. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(15):9830–9835. doi: 10.1073/pnas.152571899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Jaramillo MC, Frye JB, Crapo JD, Briehl MM, Tome ME. Increased manganese superoxide dismutase expression or treatment with manganese porphyrin potentiates dexamethasone-induced apoptosis in lymphoma cells. Cancer Research. 2009;69(13):5450–5457. doi: 10.1158/0008-5472.CAN-08-4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Tome ME, Baker AF, Powis G, Payne CM, Briehl MM. Catalase-overexpressing thymocytes are resistant to glucocorticoid-induced apoptosis and exhibit increased net tumor growth. Cancer Research. 2001;61(6):2766–2773. [PubMed] [Google Scholar]
- 400.Tome ME, Lutz NW, Briehl MM. Overexpression of catalase or Bcl-2 alters glucose and energy metabolism concomitant with dexamethasone resistance. Biochimica et Biophysica Acta. 2004;1693(1):57–72. doi: 10.1016/j.bbamcr.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 401.Low IC, Kang J, Pervaiz S. Bcl-2: a prime regulator of mitochondrial redox metabolism in cancer cells. Antioxidants & Redox Signaling. 2011;15:2975–2987. doi: 10.1089/ars.2010.3851. [DOI] [PubMed] [Google Scholar]
- 402.Aster JC, Blacklow SC, Pear WS. Notch signalling in T-cell lymphoblastic leukaemia/lymphoma and other haematological malignancies. Journal of Pathology. 2011;223(2):262–273. doi: 10.1002/path.2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Malyukova A, Dohda T, Von Der Lehr N, et al. The tumor suppressor gene hCDC4 is frequently mutated in human T-cell acute lymphoblastic leukemia with functional consequences for Notch signaling. Cancer Research. 2007;67:5611–5616. doi: 10.1158/0008-5472.CAN-06-4381. [DOI] [PubMed] [Google Scholar]
- 404.Öberg C, Li J, Pauley A, Wolf E, Gurney M, Lendahl U. The Notch intracellular domain is ubiquitinated and negatively regulated by the mammalian Sel-10 homolog. The Journal of Biological Chemistry. 2001;276(38):35847–35853. doi: 10.1074/jbc.M103992200. [DOI] [PubMed] [Google Scholar]
- 405.Ferrando A. NOTCH mutations as prognostic markers in T-ALL. Leukemia. 2010;24(12):2003–2004. doi: 10.1038/leu.2010.237. [DOI] [PubMed] [Google Scholar]
- 406.Lai EC. Protein degradation: four E3s for the Notch pathway. Current Biology. 2002;12(2):R74–R78. doi: 10.1016/s0960-9822(01)00679-0. [DOI] [PubMed] [Google Scholar]
- 407.Bray SJ. Notch signalling: a simple pathway becomes complex. Nature Reviews Molecular Cell Biology. 2006;7(9):678–689. doi: 10.1038/nrm2009. [DOI] [PubMed] [Google Scholar]
- 408.Nie J, McGill MA, Dermer M, Dho SE, Wolting CD, McGlade CJ. LNX functions as a RING type E3 ubiquitin ligase that targets the cell fate determinant Numb for ubiquitin-dependent degradation. EMBO Journal. 2002;21(1-2):93–102. doi: 10.1093/emboj/21.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Itoh M, Kim CH, Palardy G, et al. Mind bomb is a ubiquitin ligase that is essential for efficient activation of notch signaling by delta. Developmental Cell. 2003;4(1):67–82. doi: 10.1016/s1534-5807(02)00409-4. [DOI] [PubMed] [Google Scholar]
- 410.Pavlopoulos E, Pitsouli C, Klueg KM, Muskavitch MAT, Moschonas NK, Delidakis C. Neuralized encodes a peripheral membrane protein involved in delta signaling and endocytosis. Developmental Cell. 2001;1(6):807–816. doi: 10.1016/s1534-5807(01)00093-4. [DOI] [PubMed] [Google Scholar]
- 411.Qiu L, Joazeiro C, Fang N, et al. Recognition and ubiquitination of Notch by Itch, a Hect-type E3 ubiquitin ligase. The Journal of Biological Chemistry. 2000;275(46):35734–35737. doi: 10.1074/jbc.M007300200. [DOI] [PubMed] [Google Scholar]
- 412.Nwabo Kamdje AH, Krampera M. Notch signaling in acute lymphoblastic leukemia: any role for stromal microenvironment? Blood. 2011;118:6506–6514. doi: 10.1182/blood-2011-08-376061. [DOI] [PubMed] [Google Scholar]
- 413.Kamdje AHN, Mosna F, Bifari F, et al. Notch-3 and Notch-4 signaling rescue from apoptosis human B-ALL cells in contact with human bone marrow-derived mesenchymal stromal cells. Blood. 2011;118(2):380–389. doi: 10.1182/blood-2010-12-326694. [DOI] [PubMed] [Google Scholar]
- 414.Nefedova Y, Sullivan DM, Bolick SC, Dalton WS, Gabrilovich DI. Inhibition of notch signaling induces apoptosis of myeloma cells and enhances sensitivity to chemotherapy. Blood. 2008;111(4):2220–2229. doi: 10.1182/blood-2007-07-102632. [DOI] [PubMed] [Google Scholar]
- 415.Tetzlaff MT, Yu W, Li M, et al. Defective cardiovascular development and elevated cyclin E and Notch proteins in mice lacking the Fbw7 F-box protein. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(10):3338–3345. doi: 10.1073/pnas.0307875101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Koepp DM, Schaefer LK, Ye X, et al. Phosphorylation-dependent ubiquitination of cyctin E by the SCFFbw7 ubiquitin ligase. Science. 2001;294(5540):173–177. doi: 10.1126/science.1065203. [DOI] [PubMed] [Google Scholar]
- 417.Baron M. Endocytic routes to Notch activation. Seminars in Cell & Developmental Biology. 2012;23:437–442. doi: 10.1016/j.semcdb.2012.01.008. [DOI] [PubMed] [Google Scholar]
- 418.Jang J, Choi YI, Choi J, et al. Notch1 confers thymocytes a resistance to GC-induced apoptosis through Deltex1 by blocking the recruitment of p300 to the SRG3 promoter. Cell Death and Differentiation. 2006;13(9):1495–1505. doi: 10.1038/sj.cdd.4401827. [DOI] [PubMed] [Google Scholar]
- 419.Talora C, Campese AF, Bellavia D, et al. Notch signaling and diseases: n evolutionary journey from a simple beginning to complex outcomes. Biochimica et Biophysica Acta. 2008;1782(9):489–497. doi: 10.1016/j.bbadis.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 420.Palomero T, Ferrando A. Oncogenic NOTCH1 control of MYC and PI3K: challenges and opportunities for anti-NOTCH1 therapyinT-cell acute lymphoblastic leukemias and lymphomas. Clinical Cancer Research. 2008;14(17):5314–5317. doi: 10.1158/1078-0432.CCR-07-4864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421.Chadwick N, Zeef L, Portillo V, et al. Notch protection against apoptosis in T-ALL cells mediated by GIMAP5. Blood Cells, Molecules, and Diseases. 2010;45(3):201–209. doi: 10.1016/j.bcmd.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 422.Chadwick N, Zeef L, Portillo V, et al. Identification of novel Notch target genes in T cell leukaemia. Molecular Cancer. 2009;8, article 35 doi: 10.1186/1476-4598-8-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.Nitta T, Nasreen M, Seike T, et al. IAN family critically regulates survival and development of T lymphocytes. PLoS Biology. 2006;4(4):p. e103. doi: 10.1371/journal.pbio.0040103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Zenz T, Roessner A, Thomas A, et al. hlan5: the human ortholog to the rat lan4/lddm1/lyp is a new member of the Ian family that is overexpressed in B-cell lymphoid malignancies. Genes and Immunity. 2004;5(2):109–116. doi: 10.1038/sj.gene.6364044. [DOI] [PubMed] [Google Scholar]
- 425.Keita M, Leblanc C, Andrews D, Ramanathan S. GIMAP5 regulates mitochondrial integrity from a distinct subcellular compartment. Biochemical and Biophysical Research Communications. 2007;361(2):481–486. doi: 10.1016/j.bbrc.2007.07.048. [DOI] [PubMed] [Google Scholar]
- 426.Filén S, Lahesmaa R. GIMAP proteins in T-lymphocytes. Journal of Signal Transduction. 2010;2010:10 pages. doi: 10.1155/2010/268589.268589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Hyun MS, Minter LM, Ok HC, et al. Notch1 augments NF-κB activity by facilitating its nuclear retention. EMBO Journal. 2006;25(1):129–138. doi: 10.1038/sj.emboj.7600902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Palomero T, Wei KL, Odom DT, et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(48):18261–18266. doi: 10.1073/pnas.0606108103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Sharma VM, Calvo JA, Draheim KM, et al. Notch1 contributes to mouse T-cell leukemia by directly inducing the expression of c-myc. Molecular and Cellular Biology. 2006;26(21):8022–8031. doi: 10.1128/MCB.01091-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430.Weng AP, Millholland JM, Yashiro-Ohtani Y, et al. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes and Development. 2006;20(15):2096–2109. doi: 10.1101/gad.1450406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Cialfi S, Palermo R, Manca S, et al. Glucocorticoid sensitivity of T-cell lymphoblastic leukemia/lymphoma is associated with glucocorticoid receptor-mediated inhibition of Notch1 expression. doi: 10.1038/leu.2012.192. Leukemia. In press. [DOI] [PubMed] [Google Scholar]
- 432.Mo JS, Ann EJ, Yoon JH, et al. Serum- and glucocorticoid-inducible kinase 1 (SGK1) controls notch1 signaling by downregulation of protein stability through Fbw7 ubiquitin ligase. Journal of Cell Science. 2011;124(1):100–112. doi: 10.1242/jcs.073924. [DOI] [PubMed] [Google Scholar]
- 433.Kox C, Zimmermann M, Stanulla M, et al. The favorable effect of activating NOTCH1 receptor mutations on long-term outcome in T-ALL patients treated on the ALL-BFM 2000 protocol can be separated from FBXW7 loss of function. Leukemia. 2010;24(12):2005–2013. doi: 10.1038/leu.2010.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434.Zuurbier L, Homminga I, Calvert V, et al. NOTCH1 and/or FBXW7 mutations predict for initial good prednisone response but not for improved outcome in pediatric T-cell acute lymphoblastic leukemia patients treated on DCOG or COALL protocols. Leukemia. 2010;24(12):2014–2022. doi: 10.1038/leu.2010.204. [DOI] [PubMed] [Google Scholar]
- 435.Asnafi V, Buzyn A, Le Noir S, et al. NOTCH1/FBXW7 mutation identifies a large subgroup with favorable outcome in adult T-cell acute lymphoblastic leukemia (T-ALL): a Group for Research on Adult Acute Lymphoblastic Leukemia (GRAALL) study. Blood. 2009;113(17):3918–3924. doi: 10.1182/blood-2008-10-184069. [DOI] [PubMed] [Google Scholar]
- 436.Clappier E, Collette S, Grardel N, et al. NOTCH1 and FBXW7 mutations have a favorable impact on early response to treatment, but not on outcome, in children with T-cell acute lymphoblastic leukemia (T-ALL) treated on EORTC trials 58881 and 58951. Leukemia. 2010;24(12):2023–2031. doi: 10.1038/leu.2010.205. [DOI] [PubMed] [Google Scholar]
- 437.Tang Y, Wang Y, Chen L, Weintraub N, Pan Y. Cross talk between the notch signaling and noncoding RNA on the fate of stem cells. Progress in Molecular Biology and Translational Science. 2012;111:175–193. doi: 10.1016/B978-0-12-398459-3.00008-3. [DOI] [PubMed] [Google Scholar]
- 438.Lerner M, Lundgren J, Akhoondi S, et al. MiRNA-27a controls FBW7/hCDC4-dependent cyclin E degradation and cell cycle progression. Cell Cycle. 2011;10(13):2172–2183. doi: 10.4161/cc.10.13.16248. [DOI] [PubMed] [Google Scholar]
- 439.Wang Q, Li DC, Li ZF, et al. Upregulation of miR-27a contributes to the malignant transformation of human bronchial epithelial cells induced by SV40 small T antigen. Oncogene. 2011;30:3875–3886. doi: 10.1038/onc.2011.103. [DOI] [PubMed] [Google Scholar]
- 440.Li X, Sanda T, Thomas Look A, Novina CD, von Boehmer H. Repression of tumor suppressor miR-451 is essential for NOTCH1-induced oncogenesis in T-ALL. Journal of Experimental Medicine. 2011;208(4):663–675. doi: 10.1084/jem.20102384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Ghisi M, Corradin A, Basso K, et al. Modulation of microRNA expression in human T-cell development: targeting of NOTCH3 by miR-150. Blood. 2011;117(26):7053–7062. doi: 10.1182/blood-2010-12-326629. [DOI] [PubMed] [Google Scholar]
- 442.Kefas B, Comeau L, Floyd DH, et al. The neuronal microRNA miR-326 acts in a feedback loop with Notch and has therapeutic potential against brain tumors. Journal of Neuroscience. 2009;29(48):15161–15168. doi: 10.1523/JNEUROSCI.4966-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443.Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115(7):787–798. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 444.de Antonellis P, Medaglia C, Cusanelli E, et al. MiR-34a targeting of Notch ligand delta-like 1 impairs CD15+/CD133+ tumor-propagating cells and supports neural differentiation in medulloblastoma. PLoS ONE. 2011;6 doi: 10.1371/journal.pone.0024584.e24584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.Manfè V, Holst LM, Rosbjerg A, Kamstrup MR, Kaczkowski B, Gniadecki R. Changes in oncomiR expression in CTCL cell lines during apoptosis induced by Notch inhibition. Leukemia Research. 2010;34(9):e235–e236. doi: 10.1016/j.leukres.2010.03.035. [DOI] [PubMed] [Google Scholar]
- 446.Mertens-Talcott SU, Chintharlapalli S, Li X, Safe S. The oncogenic microRNA-27a targets genes that regulate specificity protein transcription factors and the G2-M checkpoint in MDA-MB-231 breast cancer cells. Cancer Research. 2007;67(22):11001–11011. doi: 10.1158/0008-5472.CAN-07-2416. [DOI] [PubMed] [Google Scholar]
- 447.Dang CV. MYC on the path to cancer. Cell. 2012;149:22–35. doi: 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448.Medh RD, Wang A, Zhou F, Thompson EB. Constitutive expression of ectopic c-Myc delays glucocorticoid-evoked apoptosis of human leukemic CEM-C7 cells. Oncogene. 2001;20(34):4629–4639. doi: 10.1038/sj.onc.1204680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449.Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Molecular Cell. 1999;4(2):199–207. doi: 10.1016/s1097-2765(00)80367-6. [DOI] [PubMed] [Google Scholar]
- 450.Bonnet M, Loosveld M, Montpellier B, et al. Posttranscriptional deregulation of MYC via PTEN constitutes a major alternative pathway of MYC activation in T-cell acute lymphoblastic leukemia. Blood. 2011;117(24):6650–6659. doi: 10.1182/blood-2011-02-336842. [DOI] [PubMed] [Google Scholar]
- 451.Battey J, Moulding C, Taub R, et al. The human c-myc oncogene: structural consequences of translocation into the IgH locus in Burkitt-lymphoma. Cell. 1983;34(3):779–787. doi: 10.1016/0092-8674(83)90534-2. [DOI] [PubMed] [Google Scholar]
- 452.Dominguez-Sola D, Dalla-Favera R. Burkitt lymphoma: much more than MYC. Cancer Cell. 2012;22:141–142. doi: 10.1016/j.ccr.2012.07.018. [DOI] [PubMed] [Google Scholar]
- 453.Molyneux EM, Rochford R, Griffin B, et al. Burkitt's lymphoma. The Lancet. 2012;379:1234–1244. doi: 10.1016/S0140-6736(11)61177-X. [DOI] [PubMed] [Google Scholar]
- 454.Yamamoto K, Matsuoka H, Yakushijin K, et al. A novel five-way translocation, t(3,9,13,8,14)(q27,p13,q32,q24,q32), with concurrent MYC and BCL6 rearrangements in a primary bone marrow B-cell lymphoma. Cancer Genetics. 2011;204:501–506. doi: 10.1016/j.cancergen.2011.08.017. [DOI] [PubMed] [Google Scholar]
- 455.Tomita N. BCL2 and MYC dual-hit lymphoma/leukemia. Journal of Clinical and Experimental Hematopathology. 2011;51:7–12. doi: 10.3960/jslrt.51.7. [DOI] [PubMed] [Google Scholar]
- 456.Snuderl M, Kolman OK, Chen YB, et al. B-cell lymphomas with concurrent IGH-BCL2 and MYC rearrangements are aggressive neoplasms with clinical and pathologic features distinct from Burkitt lymphoma and diffuse large B-cell lymphoma. American Journal of Surgical Pathology. 2010;34(3):327–340. doi: 10.1097/PAS.0b013e3181cd3aeb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457.Slack GW, Gascoyne RD. MYC and aggressive B-cell lymphomas. Advances in Anatomic Pathology. 2011;18(3):219–228. doi: 10.1097/PAP.0b013e3182169948. [DOI] [PubMed] [Google Scholar]
- 458.Shiratori S, Kondo T, Fujisawa S, et al. C-myc rearrangement in B-cell lymphoblastic lymphoma with the involvement of multiple extranodal lesions. Leukemia and Lymphoma. 2011;52(4):716–718. doi: 10.3109/10428194.2010.551158. [DOI] [PubMed] [Google Scholar]
- 459.Slovak ML, Ho JP, Pettenati MJ, et al. Localization of amplified MYC gene sequences to double minute chromosomes in acute myelogenous leukemia. Genes Chromosomes and Cancer. 1994;9(1):62–67. doi: 10.1002/gcc.2870090111. [DOI] [PubMed] [Google Scholar]
- 460.Medh RD, Saeed MF, Johnson BH, Thompson EB. Resistance of human leukemic CEM-C1 cells is overcome by synergism between glucocorticoid and protein kinase A pathways: correlation with c-Myc suppression. Cancer Research. 1998;58(16):3684–3693. [PubMed] [Google Scholar]
- 461.Thompson EB, Thulasi R, Saeed MF, Johnson BH. Glucocorticoid antagonist RU 486 reverses agonist-induced apoptosis and c-myc repression in human leukemic CEM-C7 cells. Annals of the New York Academy of Sciences. 1995;761:261–275. doi: 10.1111/j.1749-6632.1995.tb31383.x. [DOI] [PubMed] [Google Scholar]
- 462.Ausserlechner MJ, Obexer P, Böck G, Geley S, Kofler R. Cyclin D3 and c-MYC control glucocorticoid-induced cell cycle arrest but not apoptosis in lymphoblastic leukemia cells. Cell Death and Differentiation. 2004;11(2):165–174. doi: 10.1038/sj.cdd.4401328. [DOI] [PubMed] [Google Scholar]
- 463.Blackwood EM, Eisenman RN. Max: a helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science. 1991;251(4998):1211–1217. doi: 10.1126/science.2006410. [DOI] [PubMed] [Google Scholar]
- 464.Zeller KI, Zhao X, Lee CWH, et al. Global mapping of c-Myc binding sites and target gene networks in human B cells. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(47):17834–17839. doi: 10.1073/pnas.0604129103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465.Sander S, Bullinger L, Wirth T. Repressing the repressor—a new mode of MYC action in lymphomagenesis. Cell Cycle. 2009;8(4):556–559. doi: 10.4161/cc.8.4.7599. [DOI] [PubMed] [Google Scholar]
- 466.Kleine-Kohlbrecher D, Adhikary S, Eilers M. Mechanisms of transcriptional repression by Myc. Current Topics in Microbiology and Immunology. 2006;302:51–62. doi: 10.1007/3-540-32952-8_3. [DOI] [PubMed] [Google Scholar]
- 467.Sylvestre Y, De Guire V, Querido E, et al. An E2F/miR-20a autoregulatory feedback loop. The Journal of Biological Chemistry. 2007;282(4):2135–2143. doi: 10.1074/jbc.M608939200. [DOI] [PubMed] [Google Scholar]
- 468.Yu Z, Wang C, Wang M, et al. A cyclin D1/microRNA 17/20 regulatory feedback loop in control of breast cancer cell proliferation. Journal of Cell Biology. 2008;182(3):509–517. doi: 10.1083/jcb.200801079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.He L, Thomson JM, Hemann MT, et al. A microRNA polycistron as a potential human oncogene. Nature. 2005;435(7043):828–833. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Polager S, Ginsberg D. E2F—at the crossroads of life and death. Trends in Cell Biology. 2008;18(11):528–535. doi: 10.1016/j.tcb.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 471.Ofir M, Hacohen D, Ginsberg D. miR-15 and miR-16 are direct transcriptional targets of E2F1 that limit E2F-induced proliferation by targeting cyclin E. Molecular Cancer Research. 2011;9(4):440–447. doi: 10.1158/1541-7786.MCR-10-0344. [DOI] [PubMed] [Google Scholar]
- 472.Bashari D, Hacohen D, Ginsberg D. JNK activation is regulated by E2F and promotes E2F1-induced apoptosis. Cellular Signalling. 2011;23(1):65–70. doi: 10.1016/j.cellsig.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 473.Chang TC, Yu D, Lee YS, et al. Widespread microRNA repression by Myc contributes to tumorigenesis. Nature Genetics. 2008;40(1):43–50. doi: 10.1038/ng.2007.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474.Zhu H, Wu H, Liu X, et al. Regulation of autophagy by a beclin 1-targeted microRNA, miR-30a, in cancer cells. Autophagy. 2009;5(6):816–823. doi: 10.4161/auto.9064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Zou Z, Wu L, Ding H, et al. MicroRNA-30a sensitizes tumor cells to cis-platinum via suppressing beclin 1-mediated autophagy. The Journal of Biological Chemistry. 2012;287:4148–4156. doi: 10.1074/jbc.M111.307405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476.Johnson SM, Grosshans H, Shingara J, et al. RAS is regulated by the let-7 microRNA family. Cell. 2005;120(5):635–647. doi: 10.1016/j.cell.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 477.Yong SL, Dutta A. The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes and Development. 2007;21(9):1025–1030. doi: 10.1101/gad.1540407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478.Mayr C, Hemann MT, Bartel DP. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science. 2007;315(5818):1576–1579. doi: 10.1126/science.1137999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479.Johnson CD, Esquela-Kerscher A, Stefani G, et al. The let-7 microRNA represses cell proliferation pathways in human cells. Cancer Research. 2007;67(16):7713–7722. doi: 10.1158/0008-5472.CAN-07-1083. [DOI] [PubMed] [Google Scholar]
- 480.Gao P, Tchernyshyov I, Chang TC, et al. C-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458(7239):762–765. doi: 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481.Lu J, He ML, Wang L, et al. MiR-26a inhibits cell growth and tumorigenesis of nasopharyngeal carcinoma through repression of EZH2. Cancer Research. 2011;71(1):225–233. doi: 10.1158/0008-5472.CAN-10-1850. [DOI] [PubMed] [Google Scholar]
- 482.Ji J, Shi J, Budhu A, et al. MicroRNA expression, survival, and response to interferon in liver cancer. The New England Journal of Medicine. 2009;361(15):1437–1447. doi: 10.1056/NEJMoa0901282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483.Sander S, Bullinger L, Klapproth K, et al. MYC stimulates EZH2 expression by repression of its negative regulator miR-26a. Blood. 2008;112(10):4202–4212. doi: 10.1182/blood-2008-03-147645. [DOI] [PubMed] [Google Scholar]
- 484.Chang TC, Wentzel EA, Kent OA, et al. Transactivation of miR-34a by p53 broadlyinfluences gene expression andpromotesapoptosis. Molecular Cell. 2007;26(5):745–752. doi: 10.1016/j.molcel.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485.He L, He X, Lim LP, et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447(7148):1130–1134. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486.Raver-Shapira N, Marciano E, Meiri E, et al. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Molecular Cell. 2007;26(5):731–743. doi: 10.1016/j.molcel.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 487.Chang TC, Zeitels LR, Hwang HW, et al. Lin-28B transactivation is necessary for Myc-mediated let-7 repression and proliferation. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(9):3384–3389. doi: 10.1073/pnas.0808300106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488.Piskounova E, Polytarchou C, Thornton JE, et al. Lin28A and Lin28B inhibit let-7 microRNA biogenesis by distinct mechanisms. Cell. 2011;147:1066–1079. doi: 10.1016/j.cell.2011.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489.Viswanathan SR, Daley GQ, Gregory RI. Selective blockade of microRNA processing by Lin28. Science. 2008;320(5872):97–100. doi: 10.1126/science.1154040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490.Viswanathan SR, Powers JT, Einhorn W, et al. Lin28 promotes transformation and is associated with advanced human malignancies. Nature Genetics. 2009;41(7):843–848. doi: 10.1038/ng.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 491.Yu J, Vodyanik MA, Smuga-Otto K, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318(5858):1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 492.Dangi-Garimella S, Yun J, Eves EM, et al. Raf kinase inhibitory protein suppresses a metastasis signalling cascade involving LIN28 and let-7. EMBO Journal. 2009;28(4):347–358. doi: 10.1038/emboj.2008.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493.Wu WKK, Coffelt SB, Cho CH, et al. The autophagic paradox in cancer therapy. Oncogene. 2011;31:939–953. doi: 10.1038/onc.2011.295. [DOI] [PubMed] [Google Scholar]
- 494.Levine B, Sinha S, Kroemer G. Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy. 2008;4(5):600–606. doi: 10.4161/auto.6260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 495.Maiuri MC, Le Toumelin G, Criollo A, et al. Functional and physical interaction between Bcl-XL and a BH3-like domain in Beclin-1. EMBO Journal. 2007;26(10):2527–2539. doi: 10.1038/sj.emboj.7601689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 496.Pattingre S, Tassa A, Qu X, et al. Bcl-2 anti-apoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122(6):927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 497.Wirawan E, Vande Walle L, Kersse K, et al. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of pro-apoptotic factors from mitochondria. Cell Death and Disease. 2010;1(1, article e18) doi: 10.1038/cddis.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498.Wang J. Beclin 1 bridges autophagy, apoptosis and differentiation. Autophagy. 2008;4(7):947–948. doi: 10.4161/auto.6787. [DOI] [PubMed] [Google Scholar]
- 499.Qu X, Yu J, Bhagat G, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. Journal of Clinical Investigation. 2003;112(12):1809–1820. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 500.Huang JJ, Zhu YJ, Lin TY, Jiang WQ, Huang HQ, Li ZM. Beclin 1 expression predicts favorable clinical outcome in patients with diffuse large B-cell lymphoma treated with R-CHOP. Human Pathology. 2011;42(10):1459–1466. doi: 10.1016/j.humpath.2010.12.014. [DOI] [PubMed] [Google Scholar]
- 501.Laane E, Tamm KP, Buentke E, et al. Cell death induced by dexamethasone in lymphoid leukemia is mediated through initiation of autophagy. Cell Death and Differentiation. 2009;16(7):p. 1071. doi: 10.1038/cdd.2009.46. [DOI] [PubMed] [Google Scholar]
- 502.Swerdlow S, McColl K, Rong Y, Lam M, Gupta A, Distelhorst CW. Apoptosis inhibition by Bcl-2 gives way to autophagy in glucocorticoid-treated lymphocytes. Autophagy. 2008;4(5):612–620. doi: 10.4161/auto.5920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 503.Molitoris JK, McColl KS, Swerdlow S, et al. Glucocorticoid elevation of dexamethasone-induced gene 2 (Dig2/RTP801/REDD1) protein mediates autophagy in lymphocytes. The Journal of Biological Chemistry. 2011;286:30181–30189. doi: 10.1074/jbc.M111.245423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504.Deyoung MP, Horak P, Sofer A, Sgroi D, Ellisen LW. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes and Development. 2008;22(2):239–251. doi: 10.1101/gad.1617608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505.Galluzzi L, Kroemer G. Necroptosis: a specialized pathway of programmed Necrosis. Cell. 2008;135(7):1161–1163. doi: 10.1016/j.cell.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 506.Ye H, Liu X, Lv M, et al. MicroRNA and transcription factor co-regulatory network analysis reveals miR-19 inhibits CYLD in T-cell acute lymphoblastic leukemia. Nucleic Acids Research. 2012;40:5201–5214. doi: 10.1093/nar/gks175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 507.Otsuki T, Clark HM, Wellmann A, Jaffe ES, Raffeld M. Involvement of CDKN2 (p16(INK4A)/MTS1) and p15(INK4B)/MTS2 in human leukemias and lymphomas. Cancer Research. 1995;55(7):1436–1440. [PubMed] [Google Scholar]
- 508.Okuda T, Shurtleff SA, Valentine MB, et al. Frequent deletion of p16(INK4a)/MTS1 and p15(INK4b)/MTS2 in pediatric acute lymphoblastic leukemia. Blood. 1995;85(9):2321–2330. [PubMed] [Google Scholar]
- 509.Kees UR, Burton PR, Lü C, Baker DL. Homozygous deletion of the p16/MTS1 gene in pediatric acute lymphoblastic leukemia is associated with unfavorable clinical outcome. Blood. 1997;89(11):4161–4166. [PubMed] [Google Scholar]
- 510.Fizzotti M, Cimino G, Pisegna S, et al. Detection of homozygous deletions of the cyclin-dependent kinase 4 inhibitor (p16) gene in acute lymphoblastic leukemia and association with adverse prognostic features. Blood. 1995;85(10):2685–2690. [PubMed] [Google Scholar]
- 511.Obexer P, Hagenbuchner J, Rupp M, et al. p16INK4A sensitizes human leukemia cells to FAS- and glucocorticoid-induced apoptosis via induction of BBC3/Puma and repression of MCL1 and BCL2. The Journal of Biological Chemistry. 2009;284(45):30933–30940. doi: 10.1074/jbc.M109.051441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 512.Jardin F, Jais JP, Molina TJ, et al. Diffuse large B-cell lymphomas with CDKN2A deletion have a distinct gene expression signature and a poor prognosis under R-CHOP treatment: a GELA study. Blood. 2010;116(7):1092–1104. doi: 10.1182/blood-2009-10-247122. [DOI] [PubMed] [Google Scholar]
- 513.Savage KJ, Johnson NA, Ben-Neriah S, et al. MYC gene rearrangements are associated with a poor prognosis in diffuse large B-cell lymphoma patients treated with R-CHOP chemotherapy. Blood. 2009;114(17):3533–3537. doi: 10.1182/blood-2009-05-220095. [DOI] [PubMed] [Google Scholar]
- 514.Bissels U, Bosio A, Wagner W. MicroRNAs are shaping the hematopoietic landscape. Haematologica. 2012;97:160–167. doi: 10.3324/haematol.2011.051730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 515.Garzon R, Sandhu SK, Croce CM. Micro-RNA expression and function in lymphomas. Advances in Hematology. 2011;2011 doi: 10.1155/2011/347137.347137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516.Babashah S, Soleimani M. The oncogenic and tumour suppressive roles of microRNAs in cancer and apoptosis. European Journal of Cancer. 2011;47(8):1127–1137. doi: 10.1016/j.ejca.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 517.Kasinski AL, Slack FJ. Epigenetics and genetics. MicroRNAs en route to the clinic: progress in validating and targeting microRNAs for cancer therapy. Nature Reviews Cancer. 2011;11:849–864. doi: 10.1038/nrc3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 518.Ryan BM, Robles AI, Harris CC. Genetic variation in microRNA networks: the implications for cancer research. Nature Reviews Cancer. 2010;10(6):389–402. doi: 10.1038/nrc2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 519.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nature Reviews Cancer. 2006;6(11):857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 520.Schotte D, Chau JCK, Sylvester G, et al. Identification of new microRNA genes and aberrant microRNA profiles in childhood acute lymphoblastic leukemia. Leukemia. 2009;23(2):313–322. doi: 10.1038/leu.2008.286. [DOI] [PubMed] [Google Scholar]
- 521.Lu M, Zhang Q, Deng M, et al. An analysis of human microRNA and disease associations. PLoS ONE. 2008;3(10) doi: 10.1371/journal.pone.0003420.e3420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 522.Iorio MV, Croce CM. MicroRNAs in cancer: small molecules with a huge impact. Journal of Clinical Oncology. 2009;27(34):5848–5856. doi: 10.1200/JCO.2009.24.0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 523.Lu J, Getz G, Miska EA, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435(7043):834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 524.Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nature Reviews Genetics. 2008;9(2):102–114. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- 525.Di Leva G, Briskin D, Croce CM. MicroRNA in cancer: new hopes for antineoplastic chemotherapy. Upsala Journal of Medical Sciences. 2012;117:202–216. doi: 10.3109/03009734.2012.660551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526.Ruby JG, Jan CH, Bartel DP. Intronic microRNA precursors that bypass Drosha processing. Nature. 2007;448(7149):83–86. doi: 10.1038/nature05983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 527.Berezikov E, Chung WJ, Willis J, Cuppen E, Lai EC. Mammalian mirtron genes. Molecular Cell. 2007;28(2):328–336. doi: 10.1016/j.molcel.2007.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 528.Wu L, Belasco JG. Let me count the ways: mechanisms of gene regulation by miRNAs and siRNAs. Molecular Cell. 2008;29(1):1–7. doi: 10.1016/j.molcel.2007.12.010. [DOI] [PubMed] [Google Scholar]
- 529.Buchan JR, Parker R. The two faces of miRNA. Science. 2007;318(5858):1877–1878. doi: 10.1126/science.1152623. [DOI] [PubMed] [Google Scholar]
- 530.Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008;132(4):631–644. doi: 10.1016/j.cell.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531.Lodish HF, Zhou B, Liu G, Chen CZ. Micromanagement of the immune system by microRNAs. Nature Reviews Immunology. 2008;8(2):120–130. doi: 10.1038/nri2252. [DOI] [PubMed] [Google Scholar]
- 532.Lindsay MA. microRNAs and the immune response. Trends in Immunology. 2008;29(7):343–351. doi: 10.1016/j.it.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 533.Malumbres R, Sarosiek KA, Cubedo E, et al. Differentiation stage-specific expression of microRNAs in B lymphocytes and diffuse large B-cell lymphomas. Blood. 2009;113(16):3754–3764. doi: 10.1182/blood-2008-10-184077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 534.Muljo SA, Mark Ansel K, Kanellopoulou C, Livingston DM, Rao A, Rajewsky K. Aberrant T cell differentiation in the absence of Dicer. Journal of Experimental Medicine. 2005;202(2):261–269. doi: 10.1084/jem.20050678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535.Cobb BS, Hertweck A, Smith J, et al. A role for Dicer in immune regulation. Journal of Experimental Medicine. 2006;203(11):2519–2527. doi: 10.1084/jem.20061692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 536.Liston A, Lu LF, O’Carroll D, Tarakhovsky A, Rudensky AY. Dicer-dependent microRNA pathway safeguards regulatory T cell function. Journal of Experimental Medicine. 2008;205(9):1993–2004. doi: 10.1084/jem.20081062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 537.Koralov SB, Muljo SA, Galler GR, et al. Dicer ablation affects antibody diversity and cell survival in the B lymphocyte lineage. Cell. 2008;132(5):860–874. doi: 10.1016/j.cell.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 538.Cifuentes D, Xue H, Taylor DW, et al. A novel miRNA processing pathway independent of dicer requires argonaute2 catalytic activity. Science. 2010;328(5986):1694–1698. doi: 10.1126/science.1190809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 539.Neilson JR, Zheng GXY, Burge CB, Sharp PA. Dynamic regulation of miRNA expression in ordered stages of cellular development. Genes and Development. 2007;21(5):578–589. doi: 10.1101/gad.1522907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 540.Wiegers GJ, Knoflach M, Bock G, et al. CD4+CD8+TCRlow thymocytes express low levels of glucocorticoid receptors while being sensitive to glucocorticoid-induced apoptosis. European Journal of Immunology. 2001;31:2293–2301. doi: 10.1002/1521-4141(200108)31:8<2293::aid-immu2293>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 541.Rosenheimer-Goudsmid N, Haupt Y, Yefenof E, Zilberman Y, Guy R. p53 and thymic ‘death by neglect’: thymic epithelial cell-induced apoptosis of CD4+8+ thymocytes is p53-independent. Cell Death and Differentiation. 2000;7(3):241–249. doi: 10.1038/sj.cdd.4400657. [DOI] [PubMed] [Google Scholar]
- 542.Ashwell JD, Lu FWM, Vacchio MS. Glucocorticoids in T cell development and function. Annual Review of Immunology. 2000;18:309–345. doi: 10.1146/annurev.immunol.18.1.309. [DOI] [PubMed] [Google Scholar]
- 543.Cobb BS, Nesterova TB, Thompson E, et al. T cell lineage choice and differentiation in the absence of the RNase III enzyme Dicer. Journal of Experimental Medicine. 2005;201(9):1367–1373. doi: 10.1084/jem.20050572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 544.Ceribelli A, Satoh M, Chan EK. MicroRNAs and autoimmunity. Current Opinion in Immunology. 2012;24(6):686–691. doi: 10.1016/j.coi.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 545.Rusca N, Monticelli S. MiR-146a in immunity and disease. Molecular Biology International. 2011;2011:7 pages. doi: 10.4061/2011/437301.437301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 546.Li QJ, Chau J, Ebert PJR, et al. miR-181a is an intrinsic modulator of T cell sensitivity and selection. Cell. 2007;129(1):147–161. doi: 10.1016/j.cell.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 547.Xiao C, Calado DP, Galler G, et al. MiR-150 controls B cell differentiation by targeting the transcription factor c-Myb. Cell. 2007;131(1):146–159. doi: 10.1016/j.cell.2007.07.021. [DOI] [PubMed] [Google Scholar]
- 548.Ebert PJ, Jiang S, Xie J, Li QJ, Davis MM. An endogenous positively selecting peptide enhances mature T cell responses and becomes an autoantigen in the absence of microRNA miR-181a. Nature Immunology. 2009;10(11):1162–1169. doi: 10.1038/ni.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 549.Fragoso R, Mao T, Wang S, et al. Modulating the strength and threshold of NOTCH oncogenic signals by mir-181a-1/b-1 . PLOS Genetics. 2012;8 doi: 10.1371/journal.pgen.1002855.e1002855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 550.Chen CZ, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science. 2004;303(5654):83–86. doi: 10.1126/science.1091903. [DOI] [PubMed] [Google Scholar]
- 551.Cichocki F, Felices M, McCullar V, et al. Cutting edge: microRNA-181 promotes human NK cell development by regulating Notch signaling. Journal of Immunology. 2011;187:6171–6175. doi: 10.4049/jimmunol.1100835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 552.Li X, Zhang J, Gao L, et al. MiR-181 mediates cell differentiation by interrupting the Lin28 and let-7 feedback circuit. Cell Death & Differentiation. 2012;19:378–386. doi: 10.1038/cdd.2011.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 553.Zhou B, Wang S, Mayr C, Bartel DP, Lodish HF. miR-150, a microRNA expressed in mature B and T cells, blocks early B cell development when expressed prematurely. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(17):7080–7085. doi: 10.1073/pnas.0702409104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 554.Lu J, Guo S, Ebert BL, et al. MicroRNA-mediated control of cell fate in megakaryocyte-erythrocyte progenitors. Developmental Cell. 2008;14(6):843–853. doi: 10.1016/j.devcel.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 555.Bezman NA, Chakraborty T, Bender T, Lanier LL. miR-150 regulates the development of NK and iNKT cells. Journal of Experimental Medicine. 2011;208:2717–2731. doi: 10.1084/jem.20111386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 556.Zheng Q, Zhou L, Mi QS. MicroRNA miR-150 is involved in Valpha14 invariant NKT cell development and function. Journal of Immunology. 2012;188:2118–2126. doi: 10.4049/jimmunol.1103342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 557.Zanette DL, Rivadavia F, Molfetta GA, et al. miRNA expression profiles in chronic lymphocytic and acute lymphocytic leukemia. Brazilian Journal of Medical and Biological Research. 2007;40(11):1435–1440. doi: 10.1590/s0100-879x2007001100003. [DOI] [PubMed] [Google Scholar]
- 558.Zhao H, Wang D, Du W, Gu D, Yang R. MicroRNA and leukemia: tiny molecule, great function. Critical Reviews in Oncology/Hematology. 2010;74(3):149–155. doi: 10.1016/j.critrevonc.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 559.Wang Y, Li Z, He C, et al. MicroRNAs expression signatures are associated with lineage and survival in acute leukemias. Blood Cells, Molecules, and Diseases. 2010;44(3):191–197. doi: 10.1016/j.bcmd.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 560.Agirre X, Vilas-Zornoza A, Jiménez-Velasco A, et al. Epigenetic silencing of the tumor suppressor microRNA Hsa-miR-124a regulates CDK6 expression and confers a poor prognosis in acute lymphoblastic leukemia. Cancer Research. 2009;69(10):4443–4453. doi: 10.1158/0008-5472.CAN-08-4025. [DOI] [PubMed] [Google Scholar]
- 561.Mi S, Lu J, Sun M, et al. MicroRNA expression signatures accurately discriminate acute lymphoblastic leukemia from acute myeloid leukemia. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(50):19971–19976. doi: 10.1073/pnas.0709313104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 562.Babashah S, Sadeghizadeh M, Tavirani MR, Farivar S, Soleimani M. Aberrant microRNA expression and its implications in the pathogenesis of leukemias. Cellular Oncology. 2012;35(5):317–334. doi: 10.1007/s13402-012-0095-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 563.Calin GA, Ferracin M, Cimmino A, et al. A microRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. The New England Journal of Medicine. 2005;353(17):1793–1801. doi: 10.1056/NEJMoa050995. [DOI] [PubMed] [Google Scholar]
- 564.Di Lisio L, Sanchez-Beato M, Gomez-Lopez G, et al. MicroRNA signatures in B-cell lymphomas. Blood Cancer Journal. 2012;2 doi: 10.1038/bcj.2012.1.e57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 565.Li S, Moffett HF, Lu J, et al. Microrna expression profiling identifies activated B cell status in chronic lymphocytic leukemia cells. PLoS ONE. 2011;6(3) doi: 10.1371/journal.pone.0016956.e16956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 566.Fulci V, Chiaretti S, Goldoni M, et al. Quantitative technologies establish a novel microRNA profile of chronic lymphocytic leukemia. Blood. 2007;109(11):4944–4951. doi: 10.1182/blood-2006-12-062398. [DOI] [PubMed] [Google Scholar]
- 567.Mraz M, Pospisilova S, Malinova K, Slapak I, Mayer J. MicroRNAs in chronic lymphocytic leukemia pathogenesis and disease subtypes. Leukemia and Lymphoma. 2009;50(3):506–509. doi: 10.1080/10428190902763517. [DOI] [PubMed] [Google Scholar]
- 568.Marton S, Garcia MR, Robello C, et al. Small RNAs analysis in CLL reveals a deregulation of miRNA expression and novel miRNA candidates of putative relevance in CLL pathogenesis. Leukemia. 2008;22(2):330–338. doi: 10.1038/sj.leu.2405022. [DOI] [PubMed] [Google Scholar]
- 569.Pekarsky Y, Santanam U, Cimmino A, et al. Tcl1 expression in chronic lymphocytic leukemia is regulated by miR-29 and miR-181. Cancer Research. 2006;66(24):11590–11593. doi: 10.1158/0008-5472.CAN-06-3613. [DOI] [PubMed] [Google Scholar]
- 570.Zenz T, Mohr J, Eldering E, et al. miR-34a as part of the resistance network in chronic lymphocytic leukemia. Blood. 2009;113(16):3801–3808. doi: 10.1182/blood-2008-08-172254. [DOI] [PubMed] [Google Scholar]
- 571.Tili E, Michaille JJ, Luo Z, et al. The downregulation of miR-125b in chronic lymphocytic leukemias leads to metabolic adaptation of cells to a transformed state. Blood. 2012;120(13):2631–2638. doi: 10.1182/blood-2012-03-415737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 572.Barbarotto E, Schmittgen TD, Calin GA. MicroRNAs and cancer: profile, profile, profile. International Journal of Cancer. 2008;122(5):969–977. doi: 10.1002/ijc.23343. [DOI] [PubMed] [Google Scholar]
- 573.Akao Y, Nakagawa Y, Kitade Y, Kinoshita T, Naoe T. Downregulation of microRNAs-143 and -145 in B-cell malignancies. Cancer Science. 2007;98(12):1914–1920. doi: 10.1111/j.1349-7006.2007.00618.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 574.Pichiorri F, Suh SS, Ladetto M, et al. MicroRNAs regulate critical genes associated with multiple myeloma pathogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(35):12885–12890. doi: 10.1073/pnas.0806202105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 575.Roccaro AM, Sacco A, Thompson B, et al. MicroRNAs 15a and 16 regulate tumor proliferation in multiple myeloma. Blood. 2009;113(26):6669–6680. doi: 10.1182/blood-2009-01-198408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 576.Pichiorri F, De Luca L, Aqeilan RI. MicroRNAs: new players in multiple myeloma. Frontiers in Genetics. 2011;2, article 22 doi: 10.3389/fgene.2011.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 577.Unno K, Zhou Y, Zimmerman T, Platanias LC, Wickrema A. Identification of a novel microRNA cluster miR-193b-365 in multiple myeloma. Leukemia and Lymphoma. 2009;50(11):1865–1871. doi: 10.3109/10428190903221010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 578.Gutiérrez NC, Sarasquete ME, Misiewicz-Krzeminska I, et al. Deregulation of microRNA expression in the different genetic subtypes of multiple myeloma and correlation with gene expression profiling. Leukemia. 2010;24(3):629–637. doi: 10.1038/leu.2009.274. [DOI] [PubMed] [Google Scholar]
- 579.Lionetti M, Biasiolo M, Agnelli L, et al. Identification of microRNA expression patterns and definition of a microRNA/mRNA regulatory network in distinct molecular groups of multiple myeloma. Blood. 2009;114(25):e20–e26. doi: 10.1182/blood-2009-08-237495. [DOI] [PubMed] [Google Scholar]
- 580.Lawrie CH, Soneji S, Marafioti T, et al. MicroRNA expression distinguishes between germinal center B cell-like and activated B cell-like subtypes of diffuse large B cell lymphoma. International Journal of Cancer. 2007;121(5):1156–1161. doi: 10.1002/ijc.22800. [DOI] [PubMed] [Google Scholar]
- 581.Lawrie CH, Chi J, Taylor S, et al. Expression of microRNAs in diffuse large B cell lymphoma is associated with immunophenotype, survival and transformation from follicular lymphoma. Journal of Cellular and Molecular Medicine. 2009;13(7):1248–1260. doi: 10.1111/j.1582-4934.2008.00628.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 582.Roehle A, Hoefig KP, Repsilber D, et al. MicroRNA signatures characterize diffuse large B-cell lymphomas and follicular lymphomas. British Journal of Haematology. 2008;142(5):732–744. doi: 10.1111/j.1365-2141.2008.07237.x. [DOI] [PubMed] [Google Scholar]
- 583.Zhao JJ, Lin J, Lwin T, et al. MicroRNA expression profile and identification of miR-29 as a prognostic marker and pathogenetic factor by targeting CDK6 in mantle cell lymphoma. Blood. 2010;115(13):2630–2639. doi: 10.1182/blood-2009-09-243147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 584.Kluiver J, Poppema S, de Jong D, et al. BIC and miR-155 are highly expressed in Hodgkin, primary mediastinal and diffuse large B cell lymphomas. Journal of Pathology. 2005;207(2):243–249. doi: 10.1002/path.1825. [DOI] [PubMed] [Google Scholar]
- 585.Li C, Kim SW, Rai D, et al. Copy number abnormalities, MYC activity, and the genetic fingerprint of normal B cells mechanistically define the microRNA profile of diffuse large B-cell lymphoma. Blood. 2009;113(26):6681–6690. doi: 10.1182/blood-2009-01-202028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 586.Benner MF, Ballabio E, van Kester MS, et al. Primary cutaneous anaplastic large cell lymphoma shows a distinct miRNA expression profile and reveals differences from tumor-stage mycosis fungoides. Experimental Dermatology. 2012;21:632–634. doi: 10.1111/j.1600-0625.2012.01548.x. [DOI] [PubMed] [Google Scholar]
- 587.Merkel O, Hamacher F, Laimer D, et al. Identification of differential and functionally active miRNAs in both anaplastic lymphoma kinase (ALK)+ and ALK- anaplastic large-cell lymphoma. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(37):16228–16233. doi: 10.1073/pnas.1009719107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 588.Matsuyama H, Suzuki HI, Nishimori H, et al. miR-135b mediates NPM-ALK-driven oncogenicity and renders IL-17-producing immunophenotype to anaplastic large cell lymphoma. Blood. 2011;118:6881–6892. doi: 10.1182/blood-2011-05-354654. [DOI] [PubMed] [Google Scholar]
- 589.Gibcus JH, Tan LP, Harms G, et al. Hodgkin lymphoma cell lines are characterized by a specific miRNA expression profile. Neoplasia. 2009;11(2):167–176. doi: 10.1593/neo.08980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 590.Navarro A, Gaya A, Martinez A, et al. MicroRNA expression profiling in classic Hodgkin lymphoma. Blood. 2008;111(5):2825–2832. doi: 10.1182/blood-2007-06-096784. [DOI] [PubMed] [Google Scholar]
- 591.Di Lisio L, Gómez-López G, Sánchez-Beato M, et al. Mantle cell lymphoma: transcriptional regulation by microRNAs. Leukemia. 2010;24(7):1335–1342. doi: 10.1038/leu.2010.91. [DOI] [PubMed] [Google Scholar]
- 592.Rossi RL, Rossetti G, Wenandy L, et al. Distinct microRNA signatures in human lymphocyte subsets and enforcement of the naive state in CD4+ T cells by the microRNA miR-125b. Nature Immunology. 2011;12:796–803. doi: 10.1038/ni.2057. [DOI] [PubMed] [Google Scholar]
- 593.Klein U, Casola S, Cattoretti G, et al. Transcription factor IRF4 controls plasma cell differentiation and class-switch recombination. Nature Immunology. 2006;7(7):773–782. doi: 10.1038/ni1357. [DOI] [PubMed] [Google Scholar]
- 594.Angelin-Duclos C, Cattoretti G, Lin KI, Calame K. Commitment of B lymphocytes to a plasma cell fate is associated with Blimp-1 expression in vivo. Journal of Immunology. 2000;165(10):5462–5471. doi: 10.4049/jimmunol.165.10.5462. [DOI] [PubMed] [Google Scholar]
- 595.Chaudhuri AA, So AY, Mehta A, et al. Oncomir miR-125b regulates hematopoiesis by targeting the gene Lin28A. Proceedings of the National Academy of Sciences United States of America. 2012;109:4233–4238. doi: 10.1073/pnas.1200677109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 596.Klusmann JH, Li Z, Böhmer K, et al. miR-125b-2 is a potential oncomiR on human chromosome 21 in megakaryoblastic leukemia. Genes and Development. 2010;24(5):478–490. doi: 10.1101/gad.1856210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 597.Rodriguez A, Vigorito E, Clare S, et al. Requirement of bic/microRNA-155 for normal immune function. Science. 2007;316(5824):608–611. doi: 10.1126/science.1139253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 598.O’Connell RM, Rao DS, Chaudhuri AA, et al. Sustained expression of microRNA-155 in hematopoietic stem cells causes a myeloproliferative disorder. Journal of Experimental Medicine. 2008;205(3):585–594. doi: 10.1084/jem.20072108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 599.Vigorito E, Perks KL, Abreu-Goodger C, et al. microRNA-155 regulates the generation of immunoglobulin class-switched plasma cells. Immunity. 2007;27(6):847–859. doi: 10.1016/j.immuni.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 600.Ruggiero T, Trabucchi M, De Santa F, et al. LPS induces KH-type splicing regulatory protein-dependent processing of microRNA-155 precursors in macrophages. FASEB Journal. 2009;23(9):2898–2908. doi: 10.1096/fj.09-131342. [DOI] [PubMed] [Google Scholar]
- 601.Kohlhaas S, Garden OA, Scudamore C, Turner M, Okkenhaug K, Vigorito E. Cutting edge: the Foxp3 target miR-155 contributes to the development of regulatory T cells1. Journal of Immunology. 2009;182(5):2578–2582. doi: 10.4049/jimmunol.0803162. [DOI] [PubMed] [Google Scholar]
- 602.Costinean S, Zanesi N, Pekarsky Y, et al. Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in Eμ-miR155 transgenic mice. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(18):7024–7029. doi: 10.1073/pnas.0602266103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 603.Tili E, Michaille JJ, Wernicke D, et al. Mutator activity induced by microRNA-155 (miR-155) links inflammation and cancer. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(12):4908–4913. doi: 10.1073/pnas.1101795108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 604.Wu T, Wieland A, Araki K, et al. Temporal expression of microRNA cluster miR-17∼92 regulates effector and memory CD8+ T-cell differentiation. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:9965–9970. doi: 10.1073/pnas.1207327109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 605.Fulci V, Colombo T, Chiaretti S, et al. Characterization of B- and T-lineage acute lymphoblastic leukemia by integrated analysis of microRNA and mRNA expression profiles. Genes Chromosomes and Cancer. 2009;48(12):1069–1082. doi: 10.1002/gcc.20709. [DOI] [PubMed] [Google Scholar]
- 606.Ralfkiaer U, Hagedorn PH, Bangsgaard N, et al. Diagnostic microRNA profiling in cutaneous T-cell lymphoma (CTCL) Blood. 2011;118:5891–5900. doi: 10.1182/blood-2011-06-358382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 607.Cocco C, Airoldi I. Cytokines and microRNA in pediatric B-acute lymphoblastic leukemia. Cytokine and Growth Factor Reviews. 2011;22(3):149–156. doi: 10.1016/j.cytogfr.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 608.Zhang J, Jima DD, Jacobs C, et al. Patterns of microRNA expression characterize stages of human B-cell differentiation. Blood. 2009;113(19):4586–4594. doi: 10.1182/blood-2008-09-178186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 609.Jima DD, Zhang J, Jacobs C, et al. Deep sequencing of the small RNA transcriptome of normal and malignant human B cells identifies hundreds of novel microRNAs. Blood. 2010;116(23):e118–e127. doi: 10.1182/blood-2010-05-285403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 610.Culpin RE, Proctor SJ, Angus B, Crosier S, Anderson JJ, Mainou-Fowler T. A 9 series microRNA signature differentiates between germinal centre and activated B-cell-like diffuse large B-cell lymphoma cell lines. International Journal of Oncology. 2010;37(2):367–376. doi: 10.3892/ijo_00000685. [DOI] [PubMed] [Google Scholar]
- 611.Zhoua Y, Chena L, Barlogiea B, et al. High-risk myeloma is associated with global elevation of miRNAs and overexpression of EIF2C2/AGO2. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(17):7904–7909. doi: 10.1073/pnas.0908441107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 612.Corthals SL, Jongen-Lavrencic M, de Knegt Y, et al. Micro-RNA-15a and micro-RNA-16 expression and chromosome 13 deletions in multiple myeloma. Leukemia Research. 2010;34(5):677–681. doi: 10.1016/j.leukres.2009.10.026. [DOI] [PubMed] [Google Scholar]
- 613.Iqbal J, Shen Y, Liu Y, et al. Genome-wide miRNA profiling of mantle cell lymphoma reveals a distinct subgroup with poor prognosis. Blood. 2012;119:4939–4948. doi: 10.1182/blood-2011-07-370122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 614.Schiffman JD, Lorimer PD, Rodic V, et al. Genome wide copy number analysis of paediatric Burkitt lymphoma using formalin-fixed tissues reveals a subset with gain of chromosome 13q and corresponding miRNA over expression. British Journal of Haematology. 2011;155:477–486. doi: 10.1111/j.1365-2141.2011.08883.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 615.Leich E, Zamo A, Horn H, et al. MicroRNA profiles of t(14;18)-negative follicular lymphoma support a late germinal center B-cell phenotype. Blood. 2011;118:5550–5558. doi: 10.1182/blood-2011-06-361972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 616.Mavrakis KJ, Wolfe AL, Oricchio E, et al. Genome-wide RNA-mediated interference screen identifies miR-19 targets in Notch-induced T-cell acute lymphoblastic leukaemia. Nature Cell Biology. 2010;12(4):372–379. doi: 10.1038/ncb2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 617.Olive V, Bennett MJ, Walker JC, et al. miR-19 is a key oncogenic component of miR-17∼92. Genes and Development. 2009;23(24):2839–2849. doi: 10.1101/gad.1861409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 618.Ota A, Tagawa H, Karnan S, et al. Identification and characterization of a novel gene, C13orf25, as a target for 13q31-q32 amplification in malignant lymphoma. Cancer Research. 2004;64(9):3087–3095. doi: 10.1158/0008-5472.can-03-3773. [DOI] [PubMed] [Google Scholar]
- 619.Mourelatos Z, Dostie J, Paushkin S, et al. miRNPs: a novel class of ribonucleoproteins containing numerous microRNAs. Genes and Development. 2002;16(6):720–728. doi: 10.1101/gad.974702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 620.Nagel S, Venturini L, Przybylski GK, et al. Activation of miR-17∼92 by NK-like homeodomain proteins suppresses apoptosis via reduction of E2F1 in T-cell acute lymphoblastic leukemia. Leukemia and Lymphoma. 2009;50(1):101–108. doi: 10.1080/10428190802626632. [DOI] [PubMed] [Google Scholar]
- 621.Dorsam ST, Ferrell CM, Dorsam GP, et al. The transcriptome of the leukemogenic homeoprotein HOXA9 in human hematopoietic cells. Blood. 2004;103(5):1676–1684. doi: 10.1182/blood-2003-07-2202. [DOI] [PubMed] [Google Scholar]
- 622.Ferrando AA, Armstrong SA, Neuberg DS, et al. Gene expression signatures in MLL-rearranged T-lineage and B-precursor acute leukemias: dominance of HOX dysregulation. Blood. 2003;102(1):262–268. doi: 10.1182/blood-2002-10-3221. [DOI] [PubMed] [Google Scholar]
- 623.Schotte D, Lange-Turenhout EAM, Stumpel DJPM, et al. Expression of miR-196b is not exclusively MLL-driven but is especially linked to activation of HOXA genes in pediatric acute lymphoblastic leukemia. Haematologica. 2010;95(10):1675–1682. doi: 10.3324/haematol.2010.023481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 624.Wong GW, Knowles GC, Mak TW, Ferrando AA, Zuniga-Pflucker JC. HES1 opposes a PTEN-dependent check on survival, differentiation, and proliferation of TCRbeta-selected mouse thymocytes. Blood. 2012;120:1439–1448. doi: 10.1182/blood-2011-12-395319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 625.Koch U, Radtke F. Notch in T-ALL: new players in a complex disease. Trends in Immunology. 2011;32(9):434–442. doi: 10.1016/j.it.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 626.Li X, Gounari F, Protopopov A, Khazaie K, Von Boehmer H. Oncogenesis of T-ALL and nonmalignant consequences of overexpressing intracellular NOTCH1. Journal of Experimental Medicine. 2008;205(12):2851–2861. doi: 10.1084/jem.20081561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 627.Sharma VM, Draheim KM, Kelliher MA. The Notch1/c-Myc pathway in T cell leukemia. Cell Cycle. 2007;6(8):927–930. doi: 10.4161/cc.6.8.4134. [DOI] [PubMed] [Google Scholar]
- 628.Tian Y, Nan Y, Han L, et al. MicroRNA miR-451 downregulates the PI3K/AKT pathway through CAB39 in human glioma. International Journal of Oncology. 2012;40:1105–1112. doi: 10.3892/ijo.2011.1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 629.Salvatori B, Iosue I, Djodji Damas N, et al. Critical role of c-Myc in acute myeloid leukemia involving direct regulation of miR-26a and histone methyltransferase EZH2. Genes & Cancer. 2011;2:585–592. doi: 10.1177/1947601911416357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 630.Volinia S, Calin GA, Liu CG, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(7):2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 631.Dahiya N, Sherman-Baust CA, Wang TL, et al. MicroRNA expression and identification of putative miRNA targets in ovarian cancer. PLoS ONE. 2008;3(6) doi: 10.1371/journal.pone.0002436.e2436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 632.He H, Jazdzewski K, Li W, et al. The role of microRNA genes in papillary thyroid carcinoma. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(52):19075–19080. doi: 10.1073/pnas.0509603102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 633.Wang X, Tang S, Le SY, et al. Aberrant expression of oncogenic and tumor-suppressive microRNAs in cervical cancer is required for cancer cell growth. PLoS ONE. 2008;3(7) doi: 10.1371/journal.pone.0002557.e2557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 634.Starczynowski DT, Kuchenbauer F, Wegrzyn J, et al. MicroRNA-146a disrupts hematopoietic differentiation and survival. Experimental Hematology. 2011;39(2):167.e4–178.e4. doi: 10.1016/j.exphem.2010.09.011. [DOI] [PubMed] [Google Scholar]
- 635.Boldin MP, Taganov KD, Rao DS, et al. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. Journal of Experimental Medicine. 2011;208(6):1189–1201. doi: 10.1084/jem.20101823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 636.Zhao JL, Rao DS, Boldin MP, Taganov KD, O’Connell RM, Baltimore D. NF-κB dysregulation in microRNA-146a-deficient mice drives the development of myeloid malignancies. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(22):9184–9189. doi: 10.1073/pnas.1105398108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 637.Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-κB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(33):12481–12486. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 638.Paik JH, Jang JY, Jeon YK, et al. MicroRNA-146a downregulates NFκB activity via targeting TRAF6 and functions as a tumor suppressor having strong prognostic implications in NK/T cell lymphoma. Clinical Cancer Research. 2011;17(14):4761–4771. doi: 10.1158/1078-0432.CCR-11-0494. [DOI] [PubMed] [Google Scholar]
- 639.Landgraf P, Rusu M, Sheridan R, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129(7):1401–1414. doi: 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 640.Nie K, Gomez M, Landgraf P, et al. MicroRNA-mediated down-regulation of PRDM1/Blimp-1 in Hodgkin/Reed—sternberg cells: a potential pathogenetic lesion in Hodgkin lymphomas. American Journal of Pathology. 2008;173(1):242–252. doi: 10.2353/ajpath.2008.080009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 641.Guo LM, Pu Y, Han Z, et al. MicroRNA-9 inhibits ovarian cancer cell growth through regulation of NF-κB1. FEBS Journal. 2009;276(19):5537–5546. doi: 10.1111/j.1742-4658.2009.07237.x. [DOI] [PubMed] [Google Scholar]
- 642.Ruiz-Ballesteros E, Mollejo M, Mateo M, Algara P, Martínez P, Piris MA. MicroRNA losses in the frequently deleted region of 7q in SMZL. Leukemia. 2007;21(12):2547–2549. doi: 10.1038/sj.leu.2404853. [DOI] [PubMed] [Google Scholar]
- 643.Calin GA, Pekarsky Y, Croce CM. The role of microRNA and other non-coding RNA in the pathogenesis of chronic lymphocytic leukemia. Best Practice and Research in Clinical Haematology. 2007;20(3):425–437. doi: 10.1016/j.beha.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 644.Klein U, Lia M, Crespo M, et al. The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell. 2010;17(1):28–40. doi: 10.1016/j.ccr.2009.11.019. [DOI] [PubMed] [Google Scholar]
- 645.Salerno E, Scaglione BJ, Coffman FD, et al. Correcting miR-15a/16 genetic defect in New Zealand Black mouse model of CLL enhances drug sensitivity. Molecular Cancer Therapeutics. 2009;8(9):2684–2692. doi: 10.1158/1535-7163.MCT-09-0127. [DOI] [PubMed] [Google Scholar]
- 646.Rainer J, Ploner C, Jesacher S, et al. Glucocorticoid-regulated microRNAs and mirtrons in acute lymphoblastic leukemia. Leukemia. 2009;23(4):746–752. doi: 10.1038/leu.2008.370. [DOI] [PubMed] [Google Scholar]
- 647.Inomata M, Tagawa H, Guo YM, Kameoka Y, Takahashi N, Sawada K. MicroRNA-17-92 down-regulates expression of distinct targets in different B-cell lymphoma subtypes. Blood. 2009;113(2):396–402. doi: 10.1182/blood-2008-07-163907. [DOI] [PubMed] [Google Scholar]
- 648.Smith LK, Shah RR, Cidlowski JA. Glucocorticoids modulate microRNA expression and processing during lymphocyte apoptosis. The Journal of Biological Chemistry. 2010;285(47):36698–36708. doi: 10.1074/jbc.M110.162123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 649.Vreugdenhil E, Verissimo CSL, Mariman R, et al. MicroRNA 18 and 124a down-regulate the glucocorticoid receptor: implications for glucocorticoid responsiveness in the brain. Endocrinology. 2009;150(5):2220–2228. doi: 10.1210/en.2008-1335. [DOI] [PubMed] [Google Scholar]
- 650.Calin GA, Croce CM. Chronic lymphocytic leukemia: interplay between noncoding RNAs and protein-coding genes. Blood. 2009;114(23):4761–4770. doi: 10.1182/blood-2009-07-192740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 651.Zhu S, Si ML, Wu H, Mo YY. MicroRNA-21 targets the tumor suppressor gene tropomyosin 1 (TPM1) The Journal of Biological Chemistry. 2007;282(19):14328–14336. doi: 10.1074/jbc.M611393200. [DOI] [PubMed] [Google Scholar]
- 652.Lionetti M, Agnelli L, Mosca L, et al. Integrative high-resolution microarray analysis of human myeloma cell lines reveals deregulated miRNA expression associated with allelic imbalances and gene expression profiles. Genes Chromosomes and Cancer. 2009;48(6):521–531. doi: 10.1002/gcc.20660. [DOI] [PubMed] [Google Scholar]
- 653.Yamanaka Y, Tagawa H, Takahashi N, et al. Aberrant overexpression of microRNAs activate AKT signaling via down-regulation of tumor suppressors in natural killer-cell lymphoma/leukemia. Blood. 2009;114(15):3265–3275. doi: 10.1182/blood-2009-06-222794. [DOI] [PubMed] [Google Scholar]
- 654.Frankel LB, Christoffersen NR, Jacobsen A, Lindow M, Krogh A, Lund AH. Programmed cell death 4 (PDCD4) is an important functional target of the microRNA miR-21 in breast cancer cells. The Journal of Biological Chemistry. 2008;283(2):1026–1033. doi: 10.1074/jbc.M707224200. [DOI] [PubMed] [Google Scholar]
- 655.Han BW, Feng DD, Li ZG, et al. A set of miRNAs that involve in the pathways of drug resistance and leukemic stem-cell differentiation is associated with the risk of relapse and glucocorticoid response in childhood ALL. Human Molecular Genetics. 2011;20:4903–4915. doi: 10.1093/hmg/ddr428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 656.Wang Z, Wei W, Sarkar FH. miR-23a, a critical regulator of, “migR”ation and metastasis in colorectal cancer. Cancer Discovery. 2012;2:489–491. doi: 10.1158/2159-8290.CD-12-0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 657.Harada M, Pokrovskaja-Tamm K, Soderhall S, Heyman M, Grander D, Corcoran M. Involvement of miR17 pathway in glucocorticoid-induced cell death in pediatric acute lymphoblastic leukemia. Leukemia & Lymphoma. 2012;53:2041–2050. doi: 10.3109/10428194.2012.678004. [DOI] [PubMed] [Google Scholar]
- 658.Feng DD, Zhang H, Zhang P, et al. Down-regulated miR-331-5p and miR-27a are associated with chemotherapy resistance and relapse in leukaemia. Journal of Cellular and Molecular Medicine. 2011;15:2164–2175. doi: 10.1111/j.1582-4934.2010.01213.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 659.Pekarsky Y, Croce CM. Is miR-29 an oncogene or tumor suppressor in CLL? Oncotarget. 2010;1:224–227. doi: 10.18632/oncotarget.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 660.Park SY, Lee JH, Ha M, Nam JW, Kim VN. miR-29 miRNAs activate p53 by targeting p85α and CDC42. Nature Structural and Molecular Biology. 2009;16(1):23–29. doi: 10.1038/nsmb.1533. [DOI] [PubMed] [Google Scholar]
- 661.Han YC, Park CY, Bhagat G, et al. MicroRNA-29a induces aberrant self-renewal capacity in hematopoietic progenitors, biased myeloid development, and acute myeloid leukemia. Journal of Experimental Medicine. 2010;207(3):475–489. doi: 10.1084/jem.20090831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 662.Mott JL, Kurita S, Cazanave SC, Bronk SF, Werneburg NW, Fernandez-Zapico ME. Transcriptional suppression of mir-29b-1/mir-29a promoter by c-Myc, hedgehog, and NF-kappaB. Journal of Cellular Biochemistry. 2010;110(5):1155–1164. doi: 10.1002/jcb.22630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 663.Fabbri M, Bottoni A, Shimizu M, et al. Association of a microRNA/TP53 feedback circuitry with pathogenesis and outcome of b-cell chronic lymphocytic leukemia. Journal of the American Medical Association. 2011;305(1):59–67. doi: 10.1001/jama.2010.1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 664.Zauli G, Voltan R, Di Iasio MG, et al. miR-34a induces the downregulation of both E2F1 and B-Myb oncogenes in leukemic cells. Clinical Cancer Research. 2011;17(9):2712–2724. doi: 10.1158/1078-0432.CCR-10-3244. [DOI] [PubMed] [Google Scholar]
- 665.Hermeking H. The miR-34 family in cancer and apoptosis. Cell Death and Differentiation. 2010;17(2):193–199. doi: 10.1038/cdd.2009.56. [DOI] [PubMed] [Google Scholar]
- 666.Yamakuchi M, Ferlito M, Lowenstein CJ. miR-34a repression of SIRT1 regulates apoptosis. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(36):13421–13426. doi: 10.1073/pnas.0801613105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 667.Hashimi ST, Fulcher JA, Chang MH, Gov L, Wang S, Lee B. MicroRNA profiling identifies miR-34a and miR-21 and their target genes JAG1 and WNT1 in the coordinate regulation of dendritic cell differentiation. Blood. 2009;114(2):404–414. doi: 10.1182/blood-2008-09-179150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 668.Ivanovska I, Ball AS, Diaz RL, et al. MicroRNAs in the miR-106b family regulate p21/CDKN1A and promote cell cycle progression. Molecular and Cellular Biology. 2008;28(7):2167–2174. doi: 10.1128/MCB.01977-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 669.Petrocca F, Visone R, Onelli MR, et al. E2F1-regulated microRNAs impair TGFβ-dependent cell-cycle arrest and apoptosis in gastric cancer. Cancer Cell. 2008;13(3):272–286. doi: 10.1016/j.ccr.2008.02.013. [DOI] [PubMed] [Google Scholar]
- 670.Wang C, Yao N, Lu CL, Li D, Ma X. Mouse microRNA-124 regulates the expression of Hes1 in P19 cells. Frontiers in Bioscience. 2010;2:127–132. doi: 10.2741/e74. [DOI] [PubMed] [Google Scholar]
- 671.Bousquet M, Nguyen D, Chen C, Schields L, Lodish H. miR-125b transforms myeloid cell lines by repressing multiple mRNA targets. Haematologica. 2012;97(11):1713–1721. doi: 10.3324/haematol.2011.061515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 672.Kotani A, Ha D, Hsieh J, et al. miR-128b is a potent glucocorticoid sensitizer in MLL-AF4 acute lymphocytic leukemia cells and exerts cooperative effects with miR-221. Blood. 2009;114(19):4169–4178. doi: 10.1182/blood-2008-12-191619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 673.Kotani A, Ha D, Schotte D, Den Boer ML, Armstrong SA, Lodish HF. A novel mutation in the miR-128b gene reduces miRNA processing and leads to glucocorticoid resistance of MLL-AF4 acute lymphocytic leukemia cells. Cell Cycle. 2010;9(6):1037–1042. doi: 10.4161/cc.9.6.11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 674.Tessel MA, Benham AL, Krett NL, Rosen ST, Gunaratne PH. Role for microRNAs in regulating glucocorticoid response and resistance in multiple myeloma. Hormones and Cancer. 2011;2(3):182–189. doi: 10.1007/s12672-011-0072-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 675.Lai KW, Koh KX, Loh M, et al. MicroRNA-130b regulates the tumour suppressor RUNX3 in gastric cancer. European Journal of Cancer. 2010;46(8):1456–1463. doi: 10.1016/j.ejca.2010.01.036. [DOI] [PubMed] [Google Scholar]
- 676.Borgdorff V, Lleonart ME, Bishop CL, et al. Multiple microRNAs rescue from Ras-induced senescence by inhibiting p21 Waf1/Cip1. Oncogene. 2010;29(15):2262–2271. doi: 10.1038/onc.2009.497. [DOI] [PubMed] [Google Scholar]
- 677.Navarro A, Diaz T, Martinez A, et al. Regulation of JAK2 by miR-135a: prognostic impact in classic Hodgkin lymphoma. Blood. 2009;114(14):2945–2951. doi: 10.1182/blood-2009-02-204842. [DOI] [PubMed] [Google Scholar]
- 678.Lv M, Zhang X, Jia H, et al. An oncogenic role of miR-142-3p in human T-cell acute lymphoblastic leukemia (T-ALL) by targeting glucocorticoid receptor-alpha and cAMP/PKA pathways. Leukemia. 2012;26:769–777. doi: 10.1038/leu.2011.273. [DOI] [PubMed] [Google Scholar]
- 679.Huang B, Zhao J, Lei Z, et al. miR-142-3p restricts cAMP production in CD4+CD25− T cells and CD4+CD25+ TREG cells by targeting AC9 mRNA. EMBO Reports. 2009;10(2):180–185. doi: 10.1038/embor.2008.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 680.Huang X, Yang M, Jin J. Triptolide enhances the sensitivity of multiple myeloma cells to dexamethasone via microRNAs. Leukemia & Lymphoma. 2012;53:1188–1195. doi: 10.3109/10428194.2011.638069. [DOI] [PubMed] [Google Scholar]
- 681.Robertus JL, Kluiver J, Weggemans C, et al. MiRNA profiling in B non-Hodgkin lymphoma: a MYC-related miRNA profile characterizes Burkitt lymphoma. British Journal of Haematology. 2010;149(6):896–899. doi: 10.1111/j.1365-2141.2010.08111.x. [DOI] [PubMed] [Google Scholar]
- 682.Wang M, Tan LP, Dijkstra MK, et al. miRNA analysis in B-cell chronic lymphocytic leukaemia: proliferation centres characterized by low miR-150 and high BIC/miR-155 expression. Journal of Pathology. 2008;215(1):13–20. doi: 10.1002/path.2333. [DOI] [PubMed] [Google Scholar]
- 683.Watanabe A, Tagawa H, Yamashita J, et al. The role of microRNA-150 as a tumor suppressor in malignant lymphoma. Leukemia. 2011;25:1324–1334. doi: 10.1038/leu.2011.81. [DOI] [PubMed] [Google Scholar]
- 684.Belkaya S, Silge RL, Hoover AR, et al. Dynamic modulation of thymic microRNAs in response to stress. PLoS ONE. 2011;6 doi: 10.1371/journal.pone.0027580.e27580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 685.Metzler M, Wilda M, Busch K, Viehmann S, Borkhardt A. High expression of precursor MicroRNA-155/BIC RNA in children with Burkitt lymphoma. Genes Chromosomes and Cancer. 2004;39(2):167–169. doi: 10.1002/gcc.10316. [DOI] [PubMed] [Google Scholar]
- 686.Kluiver J, Haralambieva E, De Jong D, et al. Lack of BIC and microRNA miR-155 expression in primary cases of Burkitt lymphoma. Genes Chromosomes and Cancer. 2006;45(2):147–153. doi: 10.1002/gcc.20273. [DOI] [PubMed] [Google Scholar]
- 687.Vargova K, Curik N, Burda P, et al. MYB transcriptionally regulates the miR-155 host gene in chronic lymphocytic leukemia. Blood. 2011;117(14):3816–3825. doi: 10.1182/blood-2010-05-285064. [DOI] [PubMed] [Google Scholar]
- 688.Costinean S, Sandhu SK, Pedersen IM, et al. Src homology 2 domain-containing inositol-5-phosphatase and CCAAT enhancer-binding protein β are targeted by miR-155 in B cells of Eμ-MiR-155 transgenic mice. Blood. 2009;114(7):1374–1382. doi: 10.1182/blood-2009-05-220814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 689.Gatto G, Rossi A, Rossi D, Kroening S, Bonatti S, Mallardo M. Epstein-Barr virus latent membrane protein 1 trans-activates miR-155 transcription through the NF-κB pathway. Nucleic Acids Research. 2008;36(20):6608–6619. doi: 10.1093/nar/gkn666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 690.Zheng Y, Xiong S, Jiang P, et al. Glucocorticoids inhibit lipopolysaccharide-mediated inflammatory response by downregulating microRNA-155: a novel anti-inflammation mechanism. Free Radical Biology and Medicine. 2012;52:1307–1317. doi: 10.1016/j.freeradbiomed.2012.01.031. [DOI] [PubMed] [Google Scholar]
- 691.Eis PS, Tam W, Sun L, et al. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(10):3627–3632. doi: 10.1073/pnas.0500613102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 692.Li Z, Huang H, Li Y, et al. Up-regulation of a HOXA-PBX3 homeobox-gene signature following down-regulation of miR-181 is associated with adverse prognosis in patients with cytogenetically abnormal AML. Blood. 2012;119:2314–2324. doi: 10.1182/blood-2011-10-386235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 693.Alencar AJ, Malumbres R, Kozloski GA, et al. MicroRNAs are independent predictors of outcome in diffuse large B-cell lymphoma patients treated with R-CHOP. Clinical Cancer Research. 2011;17(12):4125–4135. doi: 10.1158/1078-0432.CCR-11-0224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 694.Wang X, Gocek E, Liu CG, Studzinski GP. MicroRNAs181 regulate the expression of p27Kip1 in human myeloid leukemia cells induced to differentiate by 1,25-dihydroxyvitamin D3. Cell Cycle. 2009;8(5):736–741. doi: 10.4161/cc.8.5.7870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 695.Le Sage C, Nagel R, Egan DA, et al. Regulation of the p27Kip1 tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. EMBO Journal. 2007;26(15):3699–3708. doi: 10.1038/sj.emboj.7601790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 696.Galardi S, Mercatelli N, Giorda E, et al. miR-221 and miR-222 expression affects the proliferation potential of human prostate carcinoma cell lines by targeting p27Kip1. The Journal of Biological Chemistry. 2007;282(32):23716–23724. doi: 10.1074/jbc.M701805200. [DOI] [PubMed] [Google Scholar]
- 697.Felli N, Fontana L, Pelosi E, et al. MicroRNAs 221 and 222 inhibit normal erythropoiesis and erythroleukemic cell growth via kit receptor down-modulation. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(50):18081–18086. doi: 10.1073/pnas.0506216102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 698.Medina R, Zaidi SK, Liu CG, et al. MicroRNAs 221 and 222 bypass quiescence and compromise cell survival. Cancer Research. 2008;68(8):2773–2780. doi: 10.1158/0008-5472.CAN-07-6754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 699.Miller TE, Ghoshal K, Ramaswamy B, et al. MicroRNA-221/222 confers tamoxifen resistance in breast cancer by targeting p27Kip1. The Journal of Biological Chemistry. 2008;283(44):29897–29903. doi: 10.1074/jbc.M804612200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 700.Stamatopoulos B, Meuleman N, Haibe-Kains B, et al. microRNA-29c and microRNA-223 down-regulation has in vivo significance in chronic lymphocytic leukemia and improves disease risk stratification. Blood. 2009;113(21):5237–5245. doi: 10.1182/blood-2008-11-189407. [DOI] [PubMed] [Google Scholar]
- 701.Johnnidis JB, Harris MH, Wheeler RT, et al. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature. 2008;451(7182):1125–1129. doi: 10.1038/nature06607. [DOI] [PubMed] [Google Scholar]
- 702.Pulikkan JA, Dengler V, Peramangalam PS, et al. Cell-cycle regulator E2F1 and microRNA-223 comprise an autoregulatory negative feedback loop in acute myeloid leukemia. Blood. 2010;115(9):1768–1778. doi: 10.1182/blood-2009-08-240101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 703.Fazi F, Racanicchi S, Zardo G, et al. Epigenetic silencing of the myelopoiesis regulator microRNA-223 by the AML1/ETO oncoprotein. Cancer Cell. 2007;12(5):457–466. doi: 10.1016/j.ccr.2007.09.020. [DOI] [PubMed] [Google Scholar]
- 704.Jia CY, Li HH, Zhu XC, et al. MiR-223 suppresses cell proliferation by targeting IGF-1R. PLoS ONE. 2011;6 doi: 10.1371/journal.pone.0027008.e27008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 705.Xu Y, Sengupta T, Kukreja L, Minella AC. MicroRNA-223 regulates cyclin E activity by modulating expression of F-box and WD-40 domain protein 7. The Journal of Biological Chemistry. 2010;285(45):34439–34446. doi: 10.1074/jbc.M110.152306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 706.Yuan JY, Wang F, Yu J, Yang GH, Liu XL, Zhang JW. MicroRNA-223 reversibly regulates erythroid and megakaryocytic differentiation of K562 cells. Journal of Cellular and Molecular Medicine. 2009;13(11-12):4551–4559. doi: 10.1111/j.1582-4934.2008.00585.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 707.Laine J, Künstle G, Obata T, Sha M, Noguchi M. The protooncogene TCL1 is an Akt kinase coactivator. Molecular Cell. 2000;6(2):395–407. doi: 10.1016/s1097-2765(00)00039-3. [DOI] [PubMed] [Google Scholar]
- 708.Munker R, Calin GA. MicroRNA profiling in cancer. Clinical Science. 2011;121(4):141–158. doi: 10.1042/CS20110005. [DOI] [PubMed] [Google Scholar]
- 709.Smonskey MT, Block AW, Deeb G, et al. Monoallelic and biallelic deletions of 13q14. 3 in chronic lymphocytic leukemia: FISH vs miRNA RT-qPCR detection. American Journal of Clinical Pathology. 2012;137:641–646. doi: 10.1309/AJCPP31FSSRQTTAQ. [DOI] [PubMed] [Google Scholar]
- 710.Sampath D, Liu C, Vasan K, et al. Histone deacetylases mediate the silencing of miR-15a, miR-16, and miR-29b in chronic lymphocytic leukemia. Blood. 2012;119:1162–1172. doi: 10.1182/blood-2011-05-351510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 711.Calin GA, Liu CG, Sevignani C, et al. MicroRNA profiling reveals distinct signatures in B cell chronic lymphocytic leukemias. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(32):11755–11760. doi: 10.1073/pnas.0404432101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 712.Bandi N, Zbinden S, Gugger M, et al. miR-15a and miR-16 are implicated in cell cycle regulation in a Rb-dependent manner and are frequently deleted or down-regulated in non-small cell lung cancer. Cancer Research. 2009;69(13):5553–5559. doi: 10.1158/0008-5472.CAN-08-4277. [DOI] [PubMed] [Google Scholar]
- 713.Raveche ES, Salerno E, Scaglione BJ, et al. Abnormal microRNA-16 locus with synteny to human 13q14 linked to CLL in NZB mice. Blood. 2007;109(12):5079–5086. doi: 10.1182/blood-2007-02-071225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 714.Rossi S, Shimizu M, Barbarotto E, et al. MicroRNA fingerprinting of CLL patients with chromosome 17p deletion identify a miR-21 score that stratifies early survival. Blood. 2010;116(6):945–952. doi: 10.1182/blood-2010-01-263889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 715.Longo PG, Laurenti L, Gobessi S, Sica S, Leone G, Efremov DG. The Akt/Mcl-1 pathway plays a prominent role in mediating anti-apoptotic signals downstream of the B-cell receptor in chronic lymphocytic leukemia B cells. Blood. 2008;111(2):846–855. doi: 10.1182/blood-2007-05-089037. [DOI] [PubMed] [Google Scholar]
- 716.Santanam U, Zanesi N, Efanov A, et al. Chronic lymphocytic leukemia modeled in mouse by targeted miR-29 expression. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(27):12210–12215. doi: 10.1073/pnas.1007186107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 717.Fabbri M, Garzon R, Cimmino A, et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(40):15805–15810. doi: 10.1073/pnas.0707628104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 718.Frenquelli M, Muzio M, Scielzo C, et al. MicroRNA and proliferation control in chronic lymphocytic leukemia: functional relationship between miR-221/222 cluster and p27. Blood. 2010;115(19):3949–3959. doi: 10.1182/blood-2009-11-254656. [DOI] [PubMed] [Google Scholar]
- 719.Fornari F, Gramantieri L, Ferracin M, et al. MiR-221 controls CDKN1C/p57 and CDKN1B/p27 expression in human hepatocellular carcinoma. Oncogene. 2008;27(43):5651–5661. doi: 10.1038/onc.2008.178. [DOI] [PubMed] [Google Scholar]
- 720.Di Leva G, Gasparini P, Piovan C, et al. MicroRNA cluster 221-222 and estrogen receptor α interactions in breast cancer. Journal of the National Cancer Institute. 2010;102(10):706–721. doi: 10.1093/jnci/djq102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 721.Hamada N, Fujita Y, Kojima T, et al. MicroRNA expression profiling of NGF-treated PC12 cells revealed a critical role for miR-221 in neuronal differentiation. Neurochemistry International. 2012;60:743–750. doi: 10.1016/j.neuint.2012.03.010. [DOI] [PubMed] [Google Scholar]
- 722.Garofalo M, Romano G, Di Leva G, et al. EGFR and MET receptor tyrosine kinase-altered microRNA expression induces tumorigenesis and gefitinib resistance in lung cancers. Nature Medicine. 2012;18:74–82. doi: 10.1038/nm.2577. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 723.Gramantieri L, Fornari F, Ferracin M, et al. MicroRNA-221 targets Bmf in hepatocellular carcinoma and correlates with tumor multifocality. Clinical Cancer Research. 2009;15(16):5073–5081. doi: 10.1158/1078-0432.CCR-09-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 724.Navarro A, Beà S, Fernández V, et al. MicroRNA expression, chromosomal alterations, and immunoglobulin variable heavy chain hypermutations in mantle cell lymphomas. Cancer Research. 2009;69(17):7071–7078. doi: 10.1158/0008-5472.CAN-09-1095. [DOI] [PubMed] [Google Scholar]
- 725.Rao E, Jiang C, Ji M, et al. The miRNA-17 approximately 92 cluster mediates chemoresistance and enhances tumor growth in mantle cell lymphoma via PI3K/AKT pathway activation. Leukemia. 2012;26:1064–1072. doi: 10.1038/leu.2011.305. [DOI] [PubMed] [Google Scholar]
- 726.Li Y, Zhu X, Gu J, et al. Anti-miR-21 oligonucleotide sensitizes leukemic K562 cells to arsenic trioxide by inducing apoptosis. Cancer Science. 2010;101(4):948–954. doi: 10.1111/j.1349-7006.2010.01489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 727.Medina PP, Nolde M, Slack FJ. OncomiR addiction in an in vivo model of microRNA-21-induced pre-B-cell lymphoma. Nature. 2010;467(7311):86–90. doi: 10.1038/nature09284. [DOI] [PubMed] [Google Scholar]
- 728.Hatley ME, Patrick DM, Garcia MR, et al. Modulation of K-Ras-dependent lung tumorigenesis by MicroRNA-21. Cancer Cell. 2010;18(3):282–293. doi: 10.1016/j.ccr.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 729.Ma X, Kumar M, Choudhury SN, et al. Loss of the miR-21 allele elevates the expression of its target genes and reduces tumorigenesis. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(25):10144–10149. doi: 10.1073/pnas.1103735108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 730.Sayed D, Rane S, Lypowy J, et al. MicroRNA-21 targets Sprouty2 and promotes cellular outgrowths. Molecular Biology of the Cell. 2008;19(8):3272–3282. doi: 10.1091/mbc.E08-02-0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 731.Asangani IA, Rasheed SAK, Nikolova DA, et al. MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene. 2008;27(15):2128–2136. doi: 10.1038/sj.onc.1210856. [DOI] [PubMed] [Google Scholar]
- 732.Valeri N, Gasparini P, Braconi C, et al. MicroRNA-21 induces resistance to 5-fluorouracil by down-regulating human DNA MutS homolog 2 (hMSH2) Proceedings of the National Academy of Sciences of the United States of America. 2010;107(49):21098–21103. doi: 10.1073/pnas.1015541107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 733.Papagiannakopoulos T, Shapiro A, Kosik KS. MicroRNA-21 targets a network of key tumor-suppressive pathways in glioblastoma cells. Cancer Research. 2008;68(19):8164–8172. doi: 10.1158/0008-5472.CAN-08-1305. [DOI] [PubMed] [Google Scholar]
- 734.Yang HS, Jansen AP, Nair R, et al. A novel transformation suppressor, Pdcd4, inhibits AP-1 transactivation but not NF-κB or ODC transactivation. Oncogene. 2001;20(6):669–676. doi: 10.1038/sj.onc.1204137. [DOI] [PubMed] [Google Scholar]
- 735.Göke R, Barth P, Schmidt A, Samans B, Lankat-Buttgereit B. Programmed cell death protein 4 suppresses CDK1/cdc2 via induction of p21Waf1/Cip1. American Journal of Physiology. 2004;287(6):C1541–C1546. doi: 10.1152/ajpcell.00025.2004. [DOI] [PubMed] [Google Scholar]
- 736.Yang HS, Knies JL, Stark C, Colburn NH. Pdcd4 suppresses tumor phenotype in JB6 cells by inhibiting AP-1 transactivation. Oncogene. 2003;22(24):3712–3720. doi: 10.1038/sj.onc.1206433. [DOI] [PubMed] [Google Scholar]
- 737.Chen Y, Knösel T, Kristiansen G, et al. Loss of PDCD4 expression in human lung cancer correlates with tumour progression and prognosis. Journal of Pathology. 2003;200(5):640–646. doi: 10.1002/path.1378. [DOI] [PubMed] [Google Scholar]
- 738.Gao F, Zhang P, Zhou C, et al. Frequent loss of PDCD4 expression in human glioma: possible role in the tumorigenesis of glioma. Oncology Reports. 2007;17(1):123–128. [PubMed] [Google Scholar]
- 739.Zhang H, Ozaki I, Mizuta T, et al. Involvement of programmed cell death 4 in transforming growth factor-β1-induced apoptosis in human hepatocellular carcinoma. Oncogene. 2006;25(45):6101–6112. doi: 10.1038/sj.onc.1209634. [DOI] [PubMed] [Google Scholar]
- 740.Gottwein E, Mukherjee N, Sachse C, et al. A viral microRNA functions as an orthologue of cellular miR-155. Nature. 2007;450(7172):1096–1099. doi: 10.1038/nature05992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 741.Dagan LN, Jiang X, Bhatt S, Cubedo E, Rajewsky K, Lossos IS. miR-155 regulates HGAL expression and increases lymphoma cell motility. Blood. 2012;119:513–520. doi: 10.1182/blood-2011-08-370536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 742.Rai D, Kim SW, McKeller MR, Dahia PLM, Aguiar RCT. Targeting of SMAD5 links microRNA-155 to the TGF-β pathway and lymphomagenesis. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(7):3111–3116. doi: 10.1073/pnas.0910667107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 743.Jiang X, Lu X, McNamara G, et al. HGAL, a germinal center specific protein, decreases lymphoma cell motility by modulation of the RhoA signaling pathway. Blood. 2010;116(24):5217–5227. doi: 10.1182/blood-2010-04-281568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 744.Lu X, Kazmierczak K, Jiang X, et al. Germinal center-specific protein human germinal center associated lymphoma directly interacts with both myosin and actin and increases the binding of myosin to actin. FEBS Journal. 2011;278(11):1922–1931. doi: 10.1111/j.1742-4658.2011.08109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 745.Massagué J, Seoane J, Wotton D. Smad transcription factors. Genes and Development. 2005;19(23):2783–2810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- 746.Fonseca R, Blood E, Rue M, et al. Clinical and biologic implications of recurrent genomic aberrations in myeloma. Blood. 2003;101(11):4569–4575. doi: 10.1182/blood-2002-10-3017. [DOI] [PubMed] [Google Scholar]
- 747.Kuehl WM, Bergsagel PL. Multiple myeloma: evolving genetic events and host interactions. Nature Reviews Cancer. 2002;2(3):175–187. doi: 10.1038/nrc746. [DOI] [PubMed] [Google Scholar]
- 748.Aqeilan RI, Calin GA, Croce CM. MiR-15a and miR-16-1 in cancer: discovery, function and future perspectives. Cell Death and Differentiation. 2010;17(2):215–220. doi: 10.1038/cdd.2009.69. [DOI] [PubMed] [Google Scholar]
- 749.Weiss BM, Abadie J, Verma P, Howard RS, Kuehl WM. A monoclonal gammopathy precedes multiple myeloma in most patients. Blood. 2009;113(22):5418–5422. doi: 10.1182/blood-2008-12-195008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 750.Chesi M, Robbiani DF, Sebag M, et al. AID-dependent activation of a MYC transgene induces multiple myeloma in a conditional mouse model of post-germinal center malignancies. Cancer Cell. 2008;13(2):167–180. doi: 10.1016/j.ccr.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 751.Chesi M, Bergsagel PL. Many multiple myelomas: making more of the molecular mayhem. Hematology/the Education Program of the American Society of Hematology. 2011;2011:344–353. doi: 10.1182/asheducation-2011.1.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 752.Sarasquete ME, Gutiérrez NC, Misiewicz-Krzeminska I, et al. Upregulation of dicer is more frequent in monoclonal gammopathies of undetermined significance than in multiple myeloma patients and is associated with longer survival in symptomatic myeloma patients. Haematologica. 2011;96(3):468–471. doi: 10.3324/haematol.2010.033845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 753.Liu J, Carmell MA, Rivas FV, et al. Argonaute2 is the catalytic engine of mammalian RNAi. Science. 2004;305(5689):1437–1441. doi: 10.1126/science.1102513. [DOI] [PubMed] [Google Scholar]
- 754.Diederichs S, Haber DA. Dual role for argonautes in microRNA processing and posttranscriptional regulation of microRNA expression. Cell. 2007;131(6):1097–1108. doi: 10.1016/j.cell.2007.10.032. [DOI] [PubMed] [Google Scholar]
- 755.Katz BZ. Adhesion molecules—the lifelines of multiple myeloma cells. Seminars in Cancer Biology. 2010;20(3):186–195. doi: 10.1016/j.semcancer.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 756.Frassanito MA, Cusmai A, Iodice G, Dammacco F. Autocrine interleukin-6 production and highly malignant multiple myeloma: relation with resistance to drug-induced apoptosis. Blood. 2001;97(2):483–489. doi: 10.1182/blood.v97.2.483. [DOI] [PubMed] [Google Scholar]
- 757.Hideshima T, Richardson P, Chauhan D, et al. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Research. 2001;61(7):3071–3076. [PubMed] [Google Scholar]
- 758.Bharti AC, Donato N, Aggarwal BB. Curcumin (diferuloylmethane) inhibits constitutive and IL-6-inducible STAT3 phosphorylation in human multiple myeloma cells. Journal of Immunology. 2003;171(7):3863–3871. doi: 10.4049/jimmunol.171.7.3863. [DOI] [PubMed] [Google Scholar]
- 759.Bharti AC, Donato N, Singh S, Aggarwal BB. Curcumin (diferuloylmethane) down-regulates the constitutive activation of nuclear factor-κB and IκBα kinase in human multiple myeloma cells, leading to suppression of proliferation and induction of apoptosis. Blood. 2003;101(3):1053–1062. doi: 10.1182/blood-2002-05-1320. [DOI] [PubMed] [Google Scholar]
- 760.Feinman R, Koury J, Thames M, Barlogie B, Epstein J, Siegel DS. Role of NF-κB in the rescue of multiple myeloma cells from glucocorticoid-induced apoptosis by bcl-2. Blood. 1999;93(9):3044–3052. [PubMed] [Google Scholar]
- 761.Löffler D, Brocke-Heidrich K, Pfeifer G, et al. Interleukin-6-dependent survival of multiple myeloma cells involves the Stat3-mediated induction of microRNA-21 through a highly conserved enhancer. Blood. 2007;110(4):1330–1333. doi: 10.1182/blood-2007-03-081133. [DOI] [PubMed] [Google Scholar]
- 762.Wang X, Li C, Ju S, Wang Y, Wang H, Zhong R. Myeloma cell adhesion to bone marrow stromal cells confers drug resistance by microRNA-21 up-regulation. Leukemia & Lymphoma. 2011;52:1991–1998. doi: 10.3109/10428194.2011.591004. [DOI] [PubMed] [Google Scholar]
- 763.Linares LK, Kiernan R, Triboulet R, et al. Intrinsic ubiquitination activity of PCAF controls the stability of the oncoprotein Hdm2. Nature Cell Biology. 2007;9(3):331–338. doi: 10.1038/ncb1545. [DOI] [PubMed] [Google Scholar]
- 764.Chen YH, Lavelle D, DeSimone J, Uddin S, Platanias LC, Hankewych M. Growth inhibition of a human myeloma cell line by all-trans retinoic acid is not mediated through downregulation of interleukin-6 receptors but through upregulation of p21(WAF1) Blood. 1999;94(1):251–259. [PubMed] [Google Scholar]
- 765.Lavelle D, Chen YH, Hankewych M, Desimone J. Histone deacetylase inhibitors increase p21WAF1 and induce apoptosis of human myeloma cell lines independent of decreased IL-6 receptor expression. American Journal of Hematology. 2001;68(3):170–178. doi: 10.1002/ajh.1174. [DOI] [PubMed] [Google Scholar]
- 766.Lerner M, Harada M, Lovén J, et al. DLEU2, frequently deleted in malignancy, functions as a critical host gene of the cell cycle inhibitory microRNAs miR-15a and miR-16-1. Experimental Cell Research. 2009;315(17):2941–2952. doi: 10.1016/j.yexcr.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 767.Xia L, Zhang D, Du R, et al. miR-15b and miR-16 modulate multidrug resistance by targeting BCL2 in human gastric cancer cells. International Journal of Cancer. 2008;123(2):372–379. doi: 10.1002/ijc.23501. [DOI] [PubMed] [Google Scholar]
- 768.Calin GA, Cimmino A, Fabbri M, et al. MiR-15a and miR-16-1 cluster functions in human leukemia. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(13):5166–5171. doi: 10.1073/pnas.0800121105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 769.Ria R, Vacca A, Russo F, et al. A VEGF-dependent autocrine loop mediates proliferation and capillarogenesis in bone marrow endothelial cells of patients with multiple myeloma. Thrombosis and Haemostasis. 2004;92(6):1438–1445. doi: 10.1160/TH04-06-0334. [DOI] [PubMed] [Google Scholar]
- 770.Vacca A, Ria R, Ribatti D, et al. A paracrine loop in the vascular endothelial growth factor pathway triggers tumor angiogenesis and growth in multiple myeloma. Haematologica. 2003;88(2):176–185. [PubMed] [Google Scholar]
- 771.Pearson JD, Lee JK, Bacani JT, Lai R, Ingham RJ. NPM-ALK: the prototypic member of a family of oncogenic fusion tyrosine kinases. Journal of Signal Transduction. 2012;2012:14 pages. doi: 10.1155/2012/123253.123253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 772.Campo E, Swerdlow SH, Harris NL, Pileri S, Stein H, Jaffe ES. The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Blood. 2011;117(19):5019–5032. doi: 10.1182/blood-2011-01-293050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 773.Carbone A, Gloghini A, Aiello A, Testi A, Cabras A. B-cell lymphomas with features intermediate between distinct pathologic entities. From pathogenesis to pathology. Human Pathology. 2010;41(5):621–631. doi: 10.1016/j.humpath.2009.10.027. [DOI] [PubMed] [Google Scholar]
- 774.Savage KJ. Therapies for peripheral T-cell lymphomas. Hematology American Society of Hematology Education Program. 2011;2011:515–524. doi: 10.1182/asheducation-2011.1.515. [DOI] [PubMed] [Google Scholar]
- 775.Raetz EA, Perkins SL, Carlson MA, Schooler KP, Carroll WL, Virshup DM. The nucleophosmin-anaplastic lymphoma kinase fusion protein induces c-Myc expression in pediatric anaplastic large cell lymphomas. American Journal of Pathology. 2002;161(3):875–883. doi: 10.1016/S0002-9440(10)64248-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 776.Wang FZ, Weber F, Croce C, Liu CG, Liao X, Pellett PE. Human cytomegalovirus infection alters the expression of cellular MicroRNA species that affect its replication. Journal of Virology. 2008;82(18):9065–9074. doi: 10.1128/JVI.00961-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 777.Su H, Yang JR, Xu T, et al. MicroRNA-101, down-regulated in hepatocellular carcinoma, promotes apoptosis and suppresses tumorigenicity. Cancer Research. 2009;69(3):1135–1142. doi: 10.1158/0008-5472.CAN-08-2886. [DOI] [PubMed] [Google Scholar]
- 778.Friedman JM, Liang G, Liu CC, et al. The putative tumor suppressor microRNA-101 modulates the cancer epigenome by repressing the polycomb group protein EZH2. Cancer Research. 2009;69(6):2623–2629. doi: 10.1158/0008-5472.CAN-08-3114. [DOI] [PubMed] [Google Scholar]
- 779.Varambally S, Cao Q, Mani RS, et al. Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science. 2008;322(5908):1695–1699. doi: 10.1126/science.1165395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 780.Montes-Moreno S, Martinez N, Sanchez-Espiridión B, et al. MiRNA expression in diffuse large B-cell lymphoma treated with chemoimmunotherapy. Blood. 2011;118(4):1034–1040. doi: 10.1182/blood-2010-11-321554. [DOI] [PubMed] [Google Scholar]
- 781.Lenz G, Wright GW, Emre NCT, et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(36):13520–13525. doi: 10.1073/pnas.0804295105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 782.Lenz G, Wright G, Dave SS, et al. Stromal gene signatures in large-B-cell lymphomas. The New England Journal of Medicine. 2008;359(22):2313–2323. doi: 10.1056/NEJMoa0802885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 783.Jais JP, Haioun C, Molina TJ, et al. The expression of 16 genes related to the cell of origin and immune response predicts survival in elderly patients with diffuse large B-cell lymphoma treated with CHOP and rituximab. Leukemia. 2008;22(10):1917–1924. doi: 10.1038/leu.2008.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 784.Banham AH, Beasley N, Campo E, et al. The FOXP1 winged helix transcription factor is a novel candidate tumor suppressor gene on chromosome 3p. Cancer Research. 2001;61(24):8820–8829. [PubMed] [Google Scholar]
- 785.Shaffer AL, Rosenwald A, Staudt LM. Lymphoid malignancies: the dark side of B-cell differentiation. Nature Reviews Immunology. 2002;2(12):920–932. doi: 10.1038/nri953. [DOI] [PubMed] [Google Scholar]
- 786.Wlodarska I, Veyt E, De Paepe P, et al. FOXP1, a gene highly expressed in a subset of diffuse large B-cell lymphoma, is recurrently targeted by genomic aberrations. Leukemia. 2005;19(8):1299–1305. doi: 10.1038/sj.leu.2403813. [DOI] [PubMed] [Google Scholar]
- 787.Streubel B, Vinatzer U, Lamprecht A, Raderer M, Chott A. T(3;14)(p14.1;q32) involving IGH and FOXP1 is a novel recurrent chromosomal aberration in MALT lymphoma. Leukemia. 2005;19(4):652–658. doi: 10.1038/sj.leu.2403644. [DOI] [PubMed] [Google Scholar]
- 788.Banham AH, Connors JM, Brown PJ, et al. Expression of the FOXP1 transcription factor is strongly associated with inferior survival in patients with diffuse large B-cell lymphoma. Clinical Cancer Research. 2005;11(3):1065–1072. [PubMed] [Google Scholar]
- 789.Barrans SL, Fenton JAL, Banham A, Owen RG, Jack AS. Strong expression of FOXP1 identifies a distinct subset of diffuse large B-cell lymphoma (DLBCL) patients with poor outcome. Blood. 2004;104(9):2933–2935. doi: 10.1182/blood-2004-03-1209. [DOI] [PubMed] [Google Scholar]
- 790.Mandelbaum J, Bhagat G, Tang H, et al. BLIMP1 is a tumor suppressor gene frequently disrupted in activated B cell-like diffuse large B cell lymphoma. Cancer Cell. 2010;18(6):568–579. doi: 10.1016/j.ccr.2010.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 791.Calado DP, Zhang B, Srinivasan L, et al. Constitutive canonical NF-κB activation cooperates with disruption of BLIMP1 in the pathogenesis of activated B cell-like diffuse large cell lymphoma. Cancer Cell. 2010;18(6):580–589. doi: 10.1016/j.ccr.2010.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 792.Martins G, Calame K. Regulation and functions of Blimp-1 in T and B lymphocytes. Annual Review of Immunology. 2008;26:133–169. doi: 10.1146/annurev.immunol.26.021607.090241. [DOI] [PubMed] [Google Scholar]
- 793.Kuo TC, Shaffer AL, Haddad J, Yong SC, Staudt LM, Calame K. Repression of BCL-6 is required for the formation of human memory B cells in vitro. Journal of Experimental Medicine. 2007;204(4):819–830. doi: 10.1084/jem.20062104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 794.Tunyaplin C, Shaffer AL, Angelin-Duclos CD, Yu X, Staudt LM, Calame KL. Direct repression of prdm1 by Bcl-6 inhibits plasmacytic differentiation. Journal of Immunology. 2004;173(2):1158–1165. doi: 10.4049/jimmunol.173.2.1158. [DOI] [PubMed] [Google Scholar]
- 795.Pasqualucci L, Compagno M, Houldsworth J, et al. Inactivation of the PRDM1/BLIMP1 gene in diffuse large B cell lymphoma. Journal of Experimental Medicine. 2006;203(2):311–317. doi: 10.1084/jem.20052204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 796.Küppers R. The biology of Hodgkin’s lymphoma. Nature Reviews Cancer. 2009;9(1):15–27. doi: 10.1038/nrc2542. [DOI] [PubMed] [Google Scholar]
- 797.Gozgit JM, Bebernitz G, Patil P, et al. Effects of the JAK2 inhibitor, AZ960, on Pim/BAD/BCL-xL survival signaling in the human JAK2 V617F cell line SET-2. The Journal of Biological Chemistry. 2008;283(47):32334–32343. doi: 10.1074/jbc.M803813200. [DOI] [PubMed] [Google Scholar]
- 798.Croce CM, Calin GA. miRNAs, cancer, and stem cell division. Cell. 2005;122(1):6–7. doi: 10.1016/j.cell.2005.06.036. [DOI] [PubMed] [Google Scholar]
- 799.Linnstaedt SD, Gottwein E, Skalsky RL, Luftig MA, Cullen BR. Virally induced cellular microRNA miR-155 plays a key role in B-cell immortalization by Epstein-Barr virus. Journal of Virology. 2010;84(22):11670–11678. doi: 10.1128/JVI.01248-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 800.de Kloet ER, Fitzsimons CP, Datson NA, Meijer OC, Vreugdenhil E. Glucocorticoid signaling and stress-related limbic susceptibility pathway: about receptors, transcription machinery and microRNA. Brain Research. 2009;1293:129–141. doi: 10.1016/j.brainres.2009.03.039. [DOI] [PubMed] [Google Scholar]
- 801.Uchida S, Nishida A, Hara K, et al. Characterization of the vulnerability to repeated stress in Fischer 344 rats: possible involvement of microRNA-mediated down-regulation of the glucocorticoid receptor. European Journal of Neuroscience. 2008;27(9):2250–2261. doi: 10.1111/j.1460-9568.2008.06218.x. [DOI] [PubMed] [Google Scholar]
- 802.Hayashita Y, Osada H, Tatematsu Y, et al. A polycistronic MicroRNA cluster, miR-17∼92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Research. 2005;65(21):9628–9632. doi: 10.1158/0008-5472.CAN-05-2352. [DOI] [PubMed] [Google Scholar]
- 803.Matsubara H, Takeuchi T, Nishikawa E, et al. Apoptosis induction by antisense oligonucleotides against miR-17-5p and miR-20a in lung cancers overexpressing miR-17∼92. Oncogene. 2007;26(41):6099–6105. doi: 10.1038/sj.onc.1210425. [DOI] [PubMed] [Google Scholar]
- 804.Sommer P, Le Rouzic P, Gillingham H, et al. Glucocorticoid receptor overexpression exerts an anti-survival effect on human small cell lung cancer cells. Oncogene. 2007;26(50):7111–7121. doi: 10.1038/sj.onc.1210524. [DOI] [PubMed] [Google Scholar]
- 805.Ledderose C, Mohnle P, Limbeck E, et al. Corticosteroid resistance in sepsis is influenced by microRNA-124-induced downregulation of glucocorticoid receptor-alpha. Critical Care Medicine. 2012;40(10):2745–2753. doi: 10.1097/CCM.0b013e31825b8ebc. [DOI] [PubMed] [Google Scholar]
- 806.Malzkorn B, Wolter M, Liesenberg F, et al. Identification and functional characterization of microRNAs involved in the malignant progression of gliomas. Brain Pathology. 2010;20(3):539–550. doi: 10.1111/j.1750-3639.2009.00328.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 807.Ji Z, Mei FC, Miller AL, Thompson EB, Cheng X. Protein kinase A (PKA) isoform RIIβ mediates the synergistic killing effect of cAMP and glucocorticoid in acute lymphoblastic leukemia cells. The Journal of Biological Chemistry. 2008;283(32):21920–21925. doi: 10.1074/jbc.M803193200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 808.Kim SW, Rai D, Aguiar RC. Gene set enrichment analysis unveils the mechanism for the phosphodiesterase 4B control of glucocorticoid response in B-cell lymphoma. Clinical Cancer Research. 2011;17:6723–6732. doi: 10.1158/1078-0432.CCR-11-0770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 809.Yang M, Huang J, Pan HZ, Jin J. Triptolide overcomes dexamethasone resistance and enhanced PS-341-induced apoptosis via PI3k/Akt/NF-κB pathways in human multiple myeloma cells. International Journal of Molecular Medicine. 2008;22(4):489–496. [PubMed] [Google Scholar]
- 810.Zhu W, Shan X, Wang T, Shu Y, Liu P. MiR-181b modulates multidrug resistance by targeting BCL2 in human cancer cell lines. International Journal of Cancer. 2010;127(11):2520–2529. doi: 10.1002/ijc.25260. [DOI] [PubMed] [Google Scholar]
- 811.Allen DL, Loh AS. Posttranscriptional mechanisms involving microRNA-27a and b contribute to fast-specific and glucocorticoid-mediated myostatin expression in skeletal muscle. American Journal of Physiology. 2011;300(1):C124–C137. doi: 10.1152/ajpcell.00142.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 812.Rodriguez A, Griffiths-Jones S, Ashurst JL, Bradley A. Identification of mammalian microRNA host genes and transcription units. Genome Research. 2004;14(10):1902–1910. doi: 10.1101/gr.2722704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 813.Linsley PS, Schelter J, Burchard J, et al. Transcripts targeted by the microRNA-16 family cooperatively regulate cell cycle progression. Molecular and Cellular Biology. 2007;27(6):2240–2252. doi: 10.1128/MCB.02005-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 814.Fazi F, Rosa A, Fatica A, et al. A minicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBPα regulates human granulopoiesis. Cell. 2005;123(5):819–831. doi: 10.1016/j.cell.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 815.Sun W, Shen W, Yang S, Hu F, Li H, Zhu TH. MiR-223 and miR-142 attenuate hematopoietic cell proliferation, and miR-223 positively regulates miR-142 through LMO2 isoforms and CEBP-β . Cell Research. 2010;20(10):1158–1169. doi: 10.1038/cr.2010.134. [DOI] [PubMed] [Google Scholar]
- 816.Li J, Guo Y, Liang X, et al. MicroRNA-223 functions as an oncogene in human gastric cancer by targeting FBXW7/hCdc4. Journal of Cancer Research and Clinical Oncology. 2012;138:763–774. doi: 10.1007/s00432-012-1154-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 817.Sempowski GD, Hale LP, Sundy JS, et al. Leukemia inhibitory factor, oncostatin M, IL-6, and stem cell factor mRNA expression in human thymus increases with age and is associated with thymic atrophy. Journal of Immunology. 2000;164(4):2180–2187. doi: 10.4049/jimmunol.164.4.2180. [DOI] [PubMed] [Google Scholar]
- 818.Sempowski GD, Rhein ME, Scearce RM, Haynes BF. Leukemia inhibitory factor is a mediator of Escherichia coli lipopolysaccharide-induced acute thymic atrophy. European Journal of Immunology. 2002;32:3066–3070. doi: 10.1002/1521-4141(200211)32:11<3066::AID-IMMU3066>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 819.Gearing DP, Gough NM, King JA, et al. Molecular cloning and expression of cDNA encoding a murine myeloid leukaemia inhibitory factor (LIF) EMBO Journal. 1987;6(13):3995–4002. doi: 10.1002/j.1460-2075.1987.tb02742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 820.Tili E, Michaille JJ, Cimino A, et al. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF- α stimulation and their possible roles in regulating the response to endotoxin shock. Journal of Immunology. 2007;179(8):5082–5089. doi: 10.4049/jimmunol.179.8.5082. [DOI] [PubMed] [Google Scholar]
- 821.Kawashima H, Numakawa T, Kumamaru E, et al. Glucocorticoid attenuates brain-derived neurotrophic factor-dependent upregulation of glutamate receptors via the suppression of microRNA-132 expression. Neuroscience. 2010;165(4):1301–1311. doi: 10.1016/j.neuroscience.2009.11.057. [DOI] [PubMed] [Google Scholar]
- 822.Vo N, Klein ME, Varlamova O, et al. A cAMP-response element binding protein-induced microRNA regulates neuronal morphogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(45):16426–16431. doi: 10.1073/pnas.0508448102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 823.Rodrigues AC, Li X, Radecki L, et al. MicroRNA expression is differentially altered by xenobiotic drugs in different human cell lines. Biopharmaceutics and Drug Disposition. 2011;32(6):355–367. doi: 10.1002/bdd.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 824.Biswas AK, Johnson DG. Transcriptional and nontranscriptional functions of E2F1 in response to DNA damage. Cancer Research. 2012;72:13–17. doi: 10.1158/0008-5472.CAN-11-2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 825.Hui RCY, Gomes AR, Constantinidou D, et al. The forkhead transcription factor FOXO3a increases phosphoinositide-3 kinase/Akt activity in drug-resistant leukemic cells through induction of PIK3CA expression. Molecular and Cellular Biology. 2008;28(19):5886–5898. doi: 10.1128/MCB.01265-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 826.Miyamoto K, Araki KY, Naka K, et al. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell. 2007;1(1):101–112. doi: 10.1016/j.stem.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 827.Tothova Z, Kollipara R, Huntly BJ, et al. FoxOs are critical mediators of hematopoietic stem cell resistance tophysiologic oxidative stress. Cell. 2007;128(2):325–339. doi: 10.1016/j.cell.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 828.Naka K, Hoshii T, Muraguchi T, et al. TGF-β-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature. 2010;463(7281):676–680. doi: 10.1038/nature08734. [DOI] [PubMed] [Google Scholar]
- 829.Myatt SS, Lam EWF. The emerging roles of forkhead box (Fox) proteins in cancer. Nature Reviews Cancer. 2007;7(11):847–859. doi: 10.1038/nrc2223. [DOI] [PubMed] [Google Scholar]
- 830.Chintharlapalli S, Papineni S, Abdelrahim M, et al. Oncogenic microRNA-27a is a target for anticancer agent methyl 2-cyano-3,11-dioxo-18β-olean-1,12-dien-30-oate in colon cancer cells. International Journal of Cancer. 2009;125(8):1965–1974. doi: 10.1002/ijc.24530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 831.Raaphorst FM. Self-renewal of hematopoietic and leukemic stem cells: a central role for the Polycomb-group gene Bmi-1. Trends in Immunology. 2003;24(10):522–524. doi: 10.1016/s1471-4906(03)00241-2. [DOI] [PubMed] [Google Scholar]
- 832.Park IK, Qian D, Kiel M, et al. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003;423(6937):302–305. doi: 10.1038/nature01587. [DOI] [PubMed] [Google Scholar]
- 833.Lessard J, Sauvageau G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature. 2003;423(6937):255–260. doi: 10.1038/nature01572. [DOI] [PubMed] [Google Scholar]
- 834.Pui CH, Chessells JM, Camitta B, et al. Clinical heterogeneity in childhood acute lymphoblastic leukemia with 11q23 rearrangements. Leukemia. 2003;17(4):700–706. doi: 10.1038/sj.leu.2402883. [DOI] [PubMed] [Google Scholar]
- 835.Pieters R, Schrappe M, De Lorenzo P, et al. A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (Interfant-99): an observational study and a multicentre randomised trial. The Lancet. 2007;370(9583):240–250. doi: 10.1016/S0140-6736(07)61126-X. [DOI] [PubMed] [Google Scholar]
- 836.Meister G, Landthaler M, Dorsett Y, Tuschl T. Sequence-specific inhibition of microRNA-and siRNA-induced RNA silencing. RNA. 2004;10(3):544–550. doi: 10.1261/rna.5235104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 837.Dou L, Zheng D, Li J, et al. Methylation-mediated repression of microRNA-143 enhances MLL-AF4 oncogene expression. Oncogene. 2011;31:507–517. doi: 10.1038/onc.2011.248. [DOI] [PubMed] [Google Scholar]
- 838.Babar IA, Cheng CJ, Booth CJ, et al. Nanoparticle-based therapy in an in vivo microRNA-155 (miR-155)-dependent mouse model of lymphoma. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E1695–E1704. doi: 10.1073/pnas.1201516109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 839.Burnsides C, Corry J, Alexander J, et al. Ex vivo stimulation of whole blood as a means to determine glucocorticoid sensitivity. Journal of Inflammation Research. 2012;5:89–97. doi: 10.2147/JIR.S33569. [DOI] [PMC free article] [PubMed] [Google Scholar]