

1. INTRODUCTION
The phospholipase C (PLC) isozymes catalyze conversion of phosphatidylinositol 4,5-bisphosphate (PtdIns(4,5)P2) into the Ca2+-mobilizing second messenger inositol 1,4,5-trisphosphate (inositol (1,4,5)P3) and the protein kinase-activating second messenger diacylglycerol (DAG).1 Many PLC-dependent cellular responses occur in addition to those mediated by these classical second messengers since the activities of a broad range of membrane, cytosolic, and cytoskeletal proteins are regulated by PtdIns(4,5)P2 binding.2–4 Mammals express 13 different PLCs (Figure 1), which differ markedly in their modes of upstream regulation and physiological functions.5 Many growth factors, antigens, and other extracellular stimuli signal through tyrosine phosphorylation of the PLC-γ isozymes, whereas an even larger group of hormones, neurotransmitters, chemoattractant and chemosensory molecules, and other extracellular stimuli promote physiological effects through heterotrimeric G protein-dependent activation of the PLC-β isozymes. These and other forms of receptor-promoted signaling pathways also communicate to Ras superfamily GTPases, which in turn directly activate certain PLC isoyzmes. Thus, PLC-dependent signal transduction provides one of the major fabrics for communication of cells, and delineation of its function at all levels from the intact animal to the atomic resolution of mechanism is fundamental to understanding mammalian biology.
Figure 1.
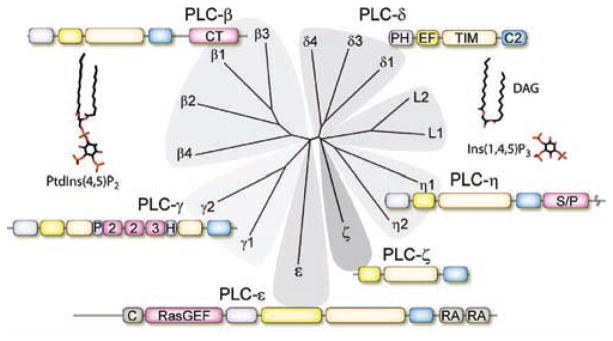
Conserved domain structure of the mammalian PLC isozymes. The 13 functional human PLC isozymes were aligned on the basis of the conservation of the protein sequence, and a dendrogram was constructed to cluster similar sequences into shared branches. The common core of the PLC isozymes includes a PH domain (purple), a series of four EF-hands (yellow), a catalytic TIM barrel (cadmium), and a C2 domain (blue). The four PLC-β isozymes contain a long C-terminal (CT) domain (pink). The two PLC-γ isozymes include conserved domains within the TIM barrel consisting of a split PH domain, two SH2 domains, and a single SH3 domain. PLC-ε contains a guanine nucleotide exchange domain (RasGEF) that activates Rap1 and possibly other GTPases and two C-terminal RA domains that bind activated Ras GTPases. A cysteine-rich (C) domain of unknown function occurs at the N-terminus. PLC-ζ is the only mammalian PLC that lacks a PH domain. PLC-η isozymes contain a serine/proline-rich (S/P) region in the C-terminus. Two PLC-like proteins, L1 and L2, are also depicted due to their sequence homology to active PLCs. These proteins have been reported to lack catalytic activity due to the absence of critical residues in the active site. The structures of the substrate of PLC, PtdIns(4,5)P2, and its second messenger products, Ins(1,4,5)P3 and DAG, are also depicted. PtdIns(4,5)P2 acts as a signaling molecule by binding to a broad range of membrane, cytoskeletal, and cytosolic proteins to change their activities, (Ins(1,4,5)P3) mobilizes intracellular Ca2+, and DAG activates protein kinase C isozymes.
Many excellent reviews on PLC-dependent signaling are available that focus, for example, on early aspects of the discovery and function of receptor-promoted formation of Ins(1,4,5)P3/diacylglycerol and mobilization of Ca2+,1,6,7 classification and regulation of the PLC isozymes,8–10 tyrosine phosphorylation-dependent activation,11,12 activation through Ras superfamily GTPases,5,13,14 physiology,15,16 and structure/function.17–19 We have limited this review to a general introduction of the domain and structural features of the PLC isozymes and then consider in detail recent advances made in understanding the mechanisms through which Gα-subunits of the Gq family bind to and activate PLC-β isozymes and how the PLC-β isozymes in turn promote inactivation of this signaling complex by stimulating GTP hydrolysis by the GTP-activated G protein.
2. CANONICAL STRUCTURE OF PLC ISOZYMES
The thirteen mammalian PLC isozymes (Figure 1) comprise six different subfamilies.5,9 Each of these isozymes contains a common catalytic core that includes an N-terminal pleckstrin homology (PH) domain (this is not present in PLC-ζ), an array of four EF-hands, a catalytic triose phosphate isomerase (TIM) barrel comprised of X and Y subdomains, and a C-terminal C2 domain. The three-dimensional structure of PLC-β3,20 shown in Figure 2 in an activated complex with Gαq, illustrates the conserved core structure found in all PLC isozymes.
Figure 2.
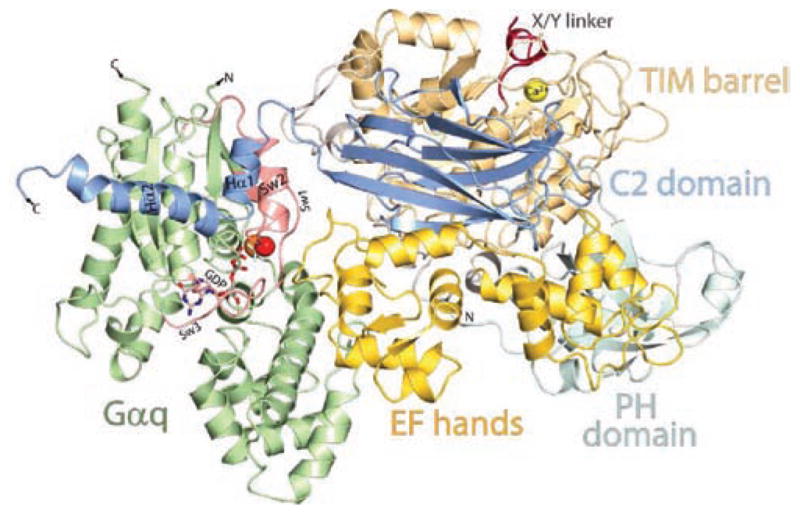
Three-dimensional structure of PLC-β3 in an activated complex with Gαq. A ribbon diagram is illustrated of the three dimensional structure (Protein Data Bank accession code 3OHM) of PLC-β3 in an activated complex with Gαq solved at 2.7 Å resolution by Waldo and co-workers.20 The PH domain (light purple), EF-hands (yellow), TIM barrel (cadmium), and C2 domain (blue) are colored as in Figure 1. The X/Y-linker region, which occludes the active site of the lipase, is depicted in red, and the Ca2+ cofactor (yellow sphere) of the active site is also shown. The approximate membrane-binding surface is across the top of the figure. Gαq is illustrated in green except for the switch regions (Sw1, Sw2, and Sw3), which are shown in red. GDP bound in the GTP-binding pocket is illustrated as a stick figure. AlF4, which mimics the γ-phosphate of GTP and promotes the transition state, is shown as a gray stick figure, and the Mg2+ cofactor (orange sphere) and catalytic H2O (red sphere) of the nucleotide-binding site are also shown. The helix—turn—helix region (in blue and labeled Hα1 and Hα2) at the end of the C2 domain interacts with Gαq in an effector-binding pocket approximately between α3 and switch 2 of the activated G protein, and a loop (yellow) between EF-hand 3 and EF-hand 4 of PLC-β3 interacts with the GTP-binding region of Gαq to promote GTP hydrolysis.
The PH domain of PLC-δ1 binds PtdIns(4,5)P2 with high affinity, and a three-dimensional crystal structure of this archetypal PH domain revealed a sandwich of seven β-strands capped by an α-helix at the C-terminus.21 Although PH domains are structurally conserved, they mediate diverse functions. The PH domain of PLC-δ1 is tethered to the core catalytic domain through a flexible linker and is likely to be mobile.21–23 PtdIns- (4,5)P2 binding to the PH domain anchors PLC-δ1 at the plasma membrane, and multiple PtdIns(4,5)P2 molecules are subsequently hydrolyzed by the catalytic site in a process sometimes referred to as membrane scooting. In contrast, the PH domains of PLC-β isozymes, which lack the basic residues necessary for PtdIns(4,5)P2 binding, are tightly associated with the other domains of the catalytic core.24 Although PH domain binding of PtdIns(4,5)P2 or other phosphoinositides is not important for regulation of PLC-βs, PLC-β2 is activated by high-affinity binding of Rac GTPases to this domain.24–26
EF-hands consist of two helices joined by a loop.27 Although EF-hands are known to be calcium-binding motifs, the three-dimensional structures of PLC isozymes illustrate a relatively high degree of flexibility in these domains, and little evidence for Ca2+ binding in these regions is available.22,24 However, a cassette between the third and fourth EF-hands of PLC-β isozymes plays an indispensible role in PLC-β-mediated stimulation of GTP hydrolysis by Gαq.20 Structure/function aspects of this signaling motif are discussed in detail below.
C2 domains are composed of an eight-β-stranded antiparallel sandwich, with three loops at one end of the sandwich typically forming calcium-binding sites.28 The C2 domain of PLC-δ1 binds Ca2+ and has been proposed to promote translocation to the plasma membrane.29 However, the residues that mediate Ca2+ binding are not generally conserved in other PLC isozymes, and little evidence of C2 domain-dependent regulation by Ca2+ exists for most PLC isozymes. The major binding determinants for activated Gαq are found just outside the N- and C-terminal ends of the C2 domain of PLC-β isozymes.20 Molecular aspects of this signaling surface are considered below.
The catalytic TIM barrel is the most highly conserved (60–70% identical) component of the 13 PLC isozymes, and the X and Y subdomains of this region fold to form the lipase active site.22,30 Residues that bind the 4′- and 5′-phosphorylated hydroxyl groups of the inositol ring of PtdIns(4,5)P2 are invariantly conserved in all PLC isozymes.22,31 Similarly conserved residues coordinate an essential Ca2+ cofactor. A conserved glutamine acts as a putative general base for deprotonation of the 2-hydroxyl group of the inositol group, and nucleophilic attack of the 2-hydroxyl group on the 1-phosphate, facilitated by the catalytic Ca2+, produces a cyclic intermediate.32,33 In the second step of the lipase reaction, a conserved histidine uses a proton from water to promote nucleophilic attack on the pentavalent cyclic intermediate, and diacylglycerol and Ins(1,4,5)P3 are formed.22,31,32,34 Not only are all residues participating in the catalytic reaction conserved in all PLCs, but they are also positioned similarly in X-ray crystal structures solved of PLC- δ1,22 PLC-β2,24 and PLC-β3.20
The core catalytic domain is essentially conserved in all PLC isozymes, but the six different subclasses of PLCs contain additional structural domains that mediate different forms of upstream and, at least in one case, downstream regulation (Figure 1). Most of the remainder of this review focuses on the structure of PLC-β isozymes and the function of these isozymes in signaling complexes with Gαq. The structure/function of the other subclasses of PLC isozymes is briefly summarized.
The two PLC-γ isozymes PLC-γ1 and PLC-γ2 provide major cell signaling responses downstream of receptor and nonreceptor tyrosine kinases.9,11 These isozymes uniquely contain conserved domains that “split” the X and Y subdomains of the TIM barrel. These include a second PH domain, two Src-homology 2 (SH2) domains, and an Src-homology 3 (SH3) domain. Activation of the two PLC-γ isozymes involves tyrosine phosphorylation-dependent binding of receptor and nonreceptor tyrosine kinases to the N-terminal SH2 domain of PLC-γ and subsequent tyrosine kinase-stimulated phosphorylation of a tyrosine residue- (s) in the region between the C-terminal SH2 domain and SH3 domain.9,11,35 This phosphotyrosine in turn interacts with the C-terminal SH2 domain to promote activation (see the discussion of activation below). Interestingly, PLC-γ2 also is activated by binding of the small GTPase Rac to the split PH domain found in the linker region between the X and Y subdomains.36,37
PLC-ε was discovered as a Ras-binding protein and contains two conserved Ras-associating domains at its C-terminus.38–41 H-Ras, K-Ras, Rap1A, Rap2A, and Rap2B have been reported to bind to the second Ras-association (RA) domain of this isozyme and to stimulate its lipase activity.39,42 A Cdc25 guanine nucleotide exchange domain (GEF) is present in the N-terminal region of PLC-ε. Although the selectivity and molecular details of this GEF function have not been completely defined, PLC-ε is an upstream activator of Ras- and Rap-dependent signaling43,44 and also apparently exhibits a feed-forward form of self-regulation, which may be important in, for example, localizing PLC-ε to different membrane compartments in the cell.
PLC-ζ is a sperm-specific signaling protein that plays an indispensible role in the acrosome reaction.45 This PLC lacks a PH domain and is uniquely sensitive to activation by Ca2+, although the molecular details of this process remain undefined. The two PLC-η isozymes PLC-η1 and PLC-η2 are recently discovered PLCs that have a long C-terminal domain.46–48 These isozymes are prominently expressed in neurons as well as in astrocytes, and PLC-η2 is directly activated by Gβγ-subunits of heterotrimeric G proteins.49
3. PLC-β-DEPENDENT REGULATION OF INOSITOL LIPID SIGNALING BY G PROTEIN-COUPLED RECEPTORS
Heterotrimeric G proteins, consisting of a guanine nucleotide- binding Gα-subunit and an obligate dimer of a Gβ- and a Gγ-subunit, were first discovered as components of hormone-receptor- regulated adenylyl cyclase and of the rhodopsin-promoted phototransduction cascade.50 Agonist-occupied G protein- coupled receptors (GPCRs) promote exchange of GTP for GDP on Gα-subunits, which bind and activate downstream effectors. Later work revealed that Gβγ-subunits released from activated heterotrimeric G proteins also bind to and activate downstream effectors.51,52
The obligatory involvement of PLC linking activation of many neurotransmitter and hormone receptors in mobilization of intracellular Ca2+ and activation of protein kinase C was elucidated in the work of Michell, Berridge, Nishizuka, and others in the 1970s and early 1980s.1,6,7 Indirect studies suggested involvement of a G protein in activation of PLC,53–56 and Litosch and Fain57 and Cockcroft and Gomperts58 first reported in 1985 the activation of PLC by GTP in cell-free preparations. This proved to be a general phenomenon illustrated with membranes from a variety of target tissues and with GTP-dependent activation observed with agonists for a broad range of GPCRs.59–62 Simon and his colleagues used degenerate primers to identify novel Gα-subunits,63,64 including the genes for Gα-subunits, i.e., Gαq, Gα11, Gα14, and Gα16, that comprise the Gq family of G proteins. Smrcka et al.,65 Taylor et al.,66 and Waldo et al.67 subsequently showed in 1991 that Gα-subunits of the Gq family directly activate PLC-β isozymes. Importantly, Gαq was shown to activate PLC-β without activating PLC-γ and PLC-δ isozymes.66 Several excellent reviews focus on early aspects of G protein-mediated regulation of PLC-β isozymes.68–70
GPCRs comprise up to 5% of mammalian genomes, and a large percentage of these signaling proteins, estimated to be up to 40% of those in the brain, signal primarily through the Gq family of Gα-subunits. The four PLC-β isozymes in turn receive their major form of upstream regulation through activation of Gq family heterotrimeric G proteins. These isozymes are widely, but differentially, expressed, and their broad involvement in many physiological responses is not surprising given the number and functional variety of the extracellular stimuli that activate Gq-coupled GPCRs.
A long (~350 residue), relatively poorly conserved, C-terminal domain has often been considered a key structural component of the PLC-β isozymes. This view was in part driven by observations that removal of this region led to loss of the capacity of Gαq to activate PLC-β after transient overexpression in cells71 or in assays with partially purified proteins.72 The capacity of PLC-β isozymes to promote GTP hydrolysis also was tentatively73,74 mapped to this region. The C-terminal domain also has long stretches of polybasic residues and has been proposed to be important for membrane association mediated by electrostatic interactions.75 An X-ray structure of a portion of this region was solved and was shown to take the form of a unique coiled-coil domain that was suggested to be important for dimerization, membrane association, and interaction with Gq familyGα-subunits.76 Although most research has suggested that the long C-terminal domain serves as the predominant Gαqinteracting region(s) of PLC-β isozymes, a recent crystal structure of an activated complex of Gαq with PLC-β3 surprisingly indicates that the Gαq-binding interface exists in a stretch of approximately 30 residues immediately C-terminal to the C2 domain.20 Thus, although it plays a role in activation, the function of the remainder of the C-terminal domain remains undefined. Does its propensity to interact with membranes simply provide a second component for membrane anchoring/active site orientation that is driven in a functional manner by binding of activated Gαq to other regions of PLC-β?
Giershick and his colleagues first suggested that Gβγ-subunits also activate PLC,77 and reconstitution assays with purified avian and mammalian PLC-β isozymes illustrated that Gβγ stimulates inositol lipid signaling by directly activating these signaling proteins.78–80 PLC-β2 and PLC-β3 are more sensitive to this mode of regulation than PLC-β1 and PLC-β4. Heterotrimeric G proteins of the Gi family often are expressed at relatively high levels, and Gβγ-dependent activation of effectors usually occurs through activation of Gi-coupled GPCRs. Thus, a broad range of Gi-coupled GPCRs have been reported to promote inositol lipid signaling responses that are sensitive to inactivation by pretreatment with pertussis toxin.52
The three-dimensional structure of a Gβγ/PLC-β signaling complex is not available. Therefore, the Gβγ-binding surface of PLC-β2 and PLC-β3 is not unambiguously known, although both the PH domain81 and a region of the Y domain82 in the TIM barrel have been implicated. The reader is referred to an excellent recent review on Gβγ-mediated signaling that details what is known about the interaction of Gβγ with PLC-β isozymes.52
4. PLC-β-PROMOTED GTP HYDROLYSIS BY Gαq
The capacity of Gα-subunits to hydrolyze GTP and therefore self-regulate their activation state was first realized almost three decades ago.83,84 Whether this process was regulated by other proteins was a matter of conjecture, but observations that the rate of hydrolysis of GTP by purified Gα-subunits was much lower than the turnoff rate of G protein-mediated signaling responses in intact cell systems were consistent with this idea.50,85 The discovery of GTPase-activating proteins (GAPs) for the small G protein Ras86 also added support to the concept that downstream regulators of the activity of heterotrimeric G proteins might exist.
Early observations on the properties of purified Gαq revealed a remarkably slow rate of basal guanine nucleotide exchange as well as a low rate of GTP hydrolysis observed in single turnover type assays.87 However, Bernstein and Ross and their colleagues88 reported ground-breaking studies in 1992 that provided an initial explanation for the differences between the rates of GTP hydrolysis by purified Gαq and the rates of turnoff of, for example, GPCR-promotedCa2+ signaling observed in vivo. A reconstitution system was developed in which purified M1 muscarinic cholinergic receptors were reconstituted in model phospholipid vesicles with purified heterotrimeric Gq. Steady-state hydrolysis of GTP was enhanced by addition of the agonist carbachol, which promoted an increase in guanine nucleotide exchange. However, the overall rate of GTP hydrolysis remained relatively slow due to the intrinsically low rate constant for GTP hydrolysis by Gαq. Addition of purified PLC-β1 caused a remarkable increase in GTP hydrolysis, and the concentration dependence for this effect was similar to the concentration dependence for Gαq-stimulated lipase activity. Whereas PLC-β1 promoted hydrolyis of GTP by Gαq, it had no effect on the rate of hydrolysis of GTP by Gαs or Gαo. The effect of PLC-β1 on steady-state GTP hydrolysis did not occur due to promotion of guanine nucleotide exchange since the rates of basal and agonist-stimulated [35S]GTPγS binding were essentially unchanged in the presence of PLC-β1. Moreover, the rate of hydrolysis of GTP observed with [γ32P]GTP-prelabeled Gαq was enhanced up to 50-fold by PLC-β1, illustrating that PLC-β directly stimulates GTP hydrolysis by, and therefore inactivation of, its upstream activator.
This early work identified the first GAP for a heterotrimeric G protein and provided the much needed explanation for differences in in vitro and in vivo turnoff reactions for activated heterotrimeric G proteins.88 An additional important new concept was conceived in this work. Thus, it was proposed that the GAP activity of PLC-β results in a rate of GTP hydrolysis that is sufficiently high to prevent activated receptor from dissociating from the Gq/PLC-β signaling complex during the lifetime of bound GTP (Figure 3). That is, PLC-β rapidly returns the activated Gα-subunit to the GDP-bound state, the activated receptor remains in the complex, where it promotes rapid dissociation of GDP, and the activation cycle is reinitiated. As a consequence, agonist binding to a single receptor results in multiple cycles of activation for the Gq/PLC-β complex. Conversely, lateral diffusion of proteins out of the complex and loss of signal acuity inherent in the process of reengaging another Gq/PLC-β pair are circumvented.
Figure 3.

Kinetic model of GPCR-promoted Gαq-dependent activation of PLC-β. Agonists (A) interact reversibly with GPCRs (R) to promote coupling to GDP-bound heterotrimeric Gq (G) and consequential dissociation of GDP and binding of GTP to Gαq. GTP—Gαq then binds with high affinity to PLC-β to activate the lipase and promote a high rate of conversion of PtdIns(4,5)P2 (PIP2) to Ins(1,4,5)P3 (IP3) and diacylglycerol. Bound PLC-β markedly enhances the rate of GTP hydrolysis by Gαq, resulting in a return of the G protein to the inactive GDP-bound state and dissociation of PLC-β. In the continued presence of agonist, activated R stays bound to Gq through multiple cycles of GTP binding and hydrolysis since the rate of dissociation of R from GTP-bound Gαq is much slower than that of the PLC-β- promoted return of the G protein to the GDP-bound state. This phenomenon whereby agonist-activated R is retained in a signaling complex with the G protein through multiple cycles of GTP binding and hydrolysis has been referred to by Ross,96, Neubig and co-workers, 122 and others as a “kinetic scaffold”.
The subsequent work of Ross and his colleagues elegantly expanded on their initial observation of GAP activity of PLC-β. Thus, precise regulation of signal initiation, termination, and amplitude occurs through coordinated effects of the activated GPCR and PLC-β on guanine nucleotide exchange and GTP hydrolysis.89 Quench flow techniques were applied to show that PLC-β-bound Gαq hydrolyzes GTP with a t1/2 of ~25 ms at 30 °C.90 This rate is 1000-fold greater than the basal rate of GTP hydrolysis by Gαq and clearly is consistent with a physiologically relevant inactivation rate for inositol lipid signaling in vivo.
A kinetic model for coordinate regulation of the GPCR/Gq/PLC-β signaling node has been described recently and tested with kinetic measurements in vitro.91 The results from these analyses provide details on how the GAP activity of the PLC-β effector can provide rapid shutoff of signaling response upon termination of agonist activation while simultaneously maintaining high signal response in the presence of an activating agonist. The model and related kinetic data also indicate that whereas the activated GPCR stays bound to Gαq throughout the GTPase cycle, PLC-β mainly binds during the GTP-bound state of Gαq.
5. STRUCTURAL BASIS FOR Gαq BINDING AND GAP ACTIVITY OF PLC-β
Receptor-promoted activation of Gq results in stimulation of PLC-β, and bound PLC-β inactivates its activating Gα-subunit by enhancing the rate of GTP hydrolysis by ~1000-fold. A snapshot of this process recently was revealed by Waldo et al.,20 who solved the three-dimensional structure of an activated complex of Gαq/PLC-β3 (Figure 2). This advance together with related biochemical analyses has delineated at the atomic level the mechanisms whereby PLC-β binds to activated Gαq and increases the rate of hydrolysis of bound GTP.
Activated Gαq interacts with three different regions of PLC-β 3. First, an approximately 30 amino acid cassette just C-terminal to the C2 domain forms a helix—turn—helix that binds in a shallow declivity between switch 2 and the third α-helix of activated Gαq (Figures 2 and 4). This region in Gα-subunits is known to be important for the binding of effectors. For example, cyclic GMP phosphodiesterase-γ binds activated Gαt (transducin) in a similar manner.92 The helix—turn—helix is highly conserved in all PLC-β isozymes, including the two found in Caenorhabditis elegans, and mutation of Gαq-interacting residues in this region of PLC-β isozymes causes loss of the capacity to bind Gαq. Binding to activated Gαq is supported by a second region in PLC isozymes that includes a number of charged residues just N-terminal to the C2 domain. These residues, which form salt bridges with complementary residues in Gαq, are largely conserved in the PLC-β isozymes, and their mutation reduces Gαq binding. Importantly, the helix—turn—helix of PLC-β3 or the helix—turn—helix plus the residues on the N-terminal side of the C2 domain were introduced into PLC-δ1, which does not bind Gαq. Addition of these regions from PLC-β into PLC-δ1 resulted in chimeric forms of the isozyme that bound Gαq in an AlF4-dependent manner. Moreover, whereas PLC-δ1 is insensitive to activation by Gαq, GTP- and agonist-dependent increases in lipase activity were observed with the chimeras using phospholipid vesicles reconstituted with purified heterotrimeric Gq and P2Y1 receptors.
Figure 4.

Key interfaces in Gαq-promoted activation of PLC-β and PLC-β-promoted hydrolysis of GTP by Gαq. Essentially the same orientation of the Gαq/PLC-β3 structure21 used in Figure 2 is shown as a surface map using the same colors as in Figure 2 to depict the various regions in PLC-β3 and Gαq. “Magnified” representations are utilized to show the key interactions responsible for Gαq binding (effector site), for autoinhibition of the lipase (PLC active site), and for PLC-β-mediated promotion of GTP hydrolysis by Gαq (Gαq active site). The effector site magnification highlights the interaction of the helix—turn—helix region of PLC-β3 with switch regions of activated Gαq. The PLC active site magnification highlights the occlusion of the active site of the lipase by the X/Y-linker region (red) of the PLC. The Gαq active site magnification highlights the interaction of the side chain nitrogen of Asn260 of the EF-hand 3/4 loop of PLC-β3 with the side chain carbonyl of Gln209 of the GTP-binding region of Gαq. The catalytic H2O (red sphere) orients the views of the effector-binding site and GTP-binding region of Gαq, and the Ca2+ cofactor (yellow sphere) orients the view of the PLC active site.
A number of studies had suggested as discussed above that the C-terminal region of PLC-β isozymes is important for Gαq binding as well as for the GAP activity of PLC-β against Gαq.71–74,76 The Gαq/PLC-β3 structure provided a surprise by revealing the prominent role of the helix—turn—helix region and sequence on the N-terminal side of the C2 domain in mediating Gαq binding.20 Indeed, wild-type PLC-β3 and a deletionmutant of PLC-β3 that lacked almost the entire C-terminal region bound to activated Gαq with essentially the same affinity. Equally surprisingly, a third region in PLC-β 600 residues away in the primary amino acid sequence from the C-terminal region interacts directly with GTP-binding residues in Gαq (Figures 2 and 4). This eight amino acid cassette is conserved in the loop between EF-hand 3 and EF-hand 4 in all PLC-β isozymes, including the two C. elegans proteins, but other PLC isozymes lack this cassette. This region was disordered in the structure of PLC-β2 alone93 as well as in the structure of PLC-β2 in the activated complex with Rac1.24 However, it becomes ordered in the process of PLC-β binding to activated Gαq, and examination of the interactions with Gαq mediated by residues in this region reveals the mechanism whereby PLC-β enhances the rate of hydrolysis of GTP (Figure 4). Thus, Asn260 is presented in a tight turn of the EF-hand 3/4 loop of PLC-β3, and the side chain nitrogen of this residue directly interacts with the side chain carbonyl of Gln209 of Gαq. This Gln functions similarly in Gαq and in all other Gα-subunits to position a catalytic water for nucleophilic attack on the γ-phosphate of GTP. Thus, Asn260 of PLC-β3 interacts with Gln209 in Gαq to fix its position, and consequently, the rate of GTP hydrolysis is dramatically increased. Other interactions in this region of PLC-β also appear to be important for promotion of GTP hydrolysis. For example, Asn260 interacts with Glu212 in Gαq, which also stabilizes switch 1 for GTP hydrolysis.
Results from biochemical analyses with purified proteins supported the model for the mechanism of PLC-β-dependent inactivation of its activating G protein derived from the Gαq/PLC-β3 structure.20 Phospholipid vesicles reconstituted with purified P2Y1 receptor and heterotrimeric Gq were utilized to quantify steady-state GTP hydrolysis. Mutation of Asn260 greatly decreased the maximal effect, but not apparent potency, of PLC-β3 for stimulation of GTP hydrolysis. Amutant of PLC-β3 that lacked the EF-hand 3/4 loop cassette almost completely lost GAP activity. Similarly, whereas agonist-stimulated phospholipase activity of wild-type PLC-β3 rapidly turned off after addition of a P2Y1 receptor antagonist, the EF-hand mutants of PLC-β3 remained active in the presence of antagonist. Thus, mutants of PLC-β3 deficient in capacity to stimulate GTP hydrolysis by their activating G protein remained active following termination of receptor-promoted exchange of guanine nucleotides on Gαq.
The role of the EF-hand region of PLC-β also was tested in vivo. Phototransduction in Drosophila melanogaster involves rhodopsin promoted activation of a Gq/PLC-β signaling complex.94 There-fore, transgenic flies were generated in which the endogenous PLC-β, NorpA, that mediates the light response was replaced with NorpA harboring a mutation of the relevant asparagine (Asn262) to alanine.20 Dark-adapted eyes were exposed to a 5 s flash of orange light, and retinograms were generated. Eyes with wild-type NorpA or with NorpA(N262A) exhibited similar amplitudes of the photoresponse to light. However, the rate of termination of the photoresponse after cessation of light was decreased by greater than 20-fold in NorpA(N262A) animals. Thus, the properties of reduced GAP activity in EF-hand 3/4 loop mutants observed with purified proteins are recapitulated in a physiological system.
Members of a large family of regulators of G protein signaling (RGS) proteins selectively interact with Gα-subunits to markedly stimulate their rates of GTP hydrolysis.85,95,96 The magnitude of the effect (>1000-fold stimulation of GTP hydrolysis) of RGS proteins is similar to that observed for the effect of PLC-β on Gαq.90 The RGS proteins not only function to promote rapid turnoff and/or inhibition of signaling mediated by most Gα-subunits, but also serve to both sharpen and spatially focus GPCR-promoted signal transduction.97 The mechanism where-by RGS proteins act as GAPs for Gα-subunits is identical to the mode of regulation that independently evolved in PLC isozymes. Thus, the interaction of Asn260 of PLC-β3 with Glu209 in Gαq is recapitulated by a functionally equivalent interaction of an Asn in the structures of, for example, RGS4/Gαi298 and RGS9/Gαt.92 Although the structure of an RGS protein in a complex with Gαq is not yet available, the RGS protein-interacting surface of other Gα-subunits overlaps with a portion of the PLC-β- interacting surface of Gαq.20
Phototransduction in mammals is mediated by Gαt-mediated activation of a cyclic GMP phosphodiesterase, and normal recovery of the photoresponse requires RGS9-mediated acceleration of GTP hydrolysis by Gαt.99,100 Although cyclic GMP phosphodiesterase is not aGAP, it allosterically promotes an increase in binding of RGS9 to Gαt and therein promotes its inactivation.92,101 The binding of phosphodiesterase in the effector pocket of Gαt occurs analogously to the binding of PLC-β within the effector pocket of Gαq. The capacity of PLC-β to be activated by Gαq and then function to turn off its activating Gprotein evolved as coordinating actions of two different surfaces of the PLC-β effector protein. In contrast, the high signal amplification of the photoresponse in mammals evolved in the activities of separate effectors and GAP proteins, which nonetheless interact allosterically to promote a high level of signal-to-noise ratio in phototransduction.
PLC-β is not the only direct effector of Gαq since members of the Trio family of guanine nucleotide exchange factors for Rho, i.e., Trio, Kalirin, and p63RhoGEF,102–104 and G protein receptor kinase 2 (GRK2) bind to activated Gαq with high affinity.105,106 Thus, biological effects downstream of the Gq-coupled GPCR also include Rho-mediated responses, e.g., smooth muscle contraction, cell migration, and cell proliferation, and desensitization of Gq-promoted signaling responses is regulated by direct interaction of the activated Gα-subunit with GRK2, which targets the GPCR for phosphorylation. Comparison of the structure of an activated complex of Gαq/p63RhoGEF104 with the structure of Gαq/PLC-β320 reveals that these diverse effector proteins independently evolved a Gαq-binding interface mediated by essentially identical residues in the two effector proteins. GRK2 also interacts with Gαq in a similar fashion.106
In summary, dynamic regulation of Gαq/PLC-β signaling complexes underpins the capacity of these signaling nodes to exhibit rapid activation and inactivation kinetics while maintaining high levels of response amplitude, signal-to-noise ratio, and spatial focusing. Importantly, precise interplay of two surfaces evolved in PLC-β isozymes to mediate this regulation through a PLC-β-mediated process that has been coined “catch and release”.20 This term followed from the concept of “fly-casting” in which conformationally flexible proteins sample a large space and then couple folding and binding in a mechanism that increases the rate of formation of a protein— protein complex.107,108 Thus, the helix—turn—helix region in PLC-β “catches” the effector region of GTP-bound Gαq (Figure 5A). The EF-hand 3/4 loop region then engages the switch regions of the activated G protein and stabilizes the transition state for GTP hydrolysis (Figure 4). “Release” then occurs with GTP hydrolysis, which changes the conformation of switch 2 of Gαq and therein markedly reduces its binding affinity for the helix—turn—helix region of PLC-β. The unbound lipase is then free to sample the activation state of the GPCR-regulated G protein once more. As discussed above, a helix—turn—helix in p63RhoGEF binds in the effector pocket of activated Gαq in an analogous manner. p63RhoGEF is not aGAP and does not bind to the surface of Gαq utilized by RGS proteins or the EF-hand 3/4 region of PLC-β. Thus, RGS proteins can bind to activated Gαq in a complex with p63RhoGEF, and negative allosteric regulation occurs between RGS2 binding and p63RhoGEF binding in this ternary complex of RGS2, Gαq, and p63RhoGEF.109
Figure 5.
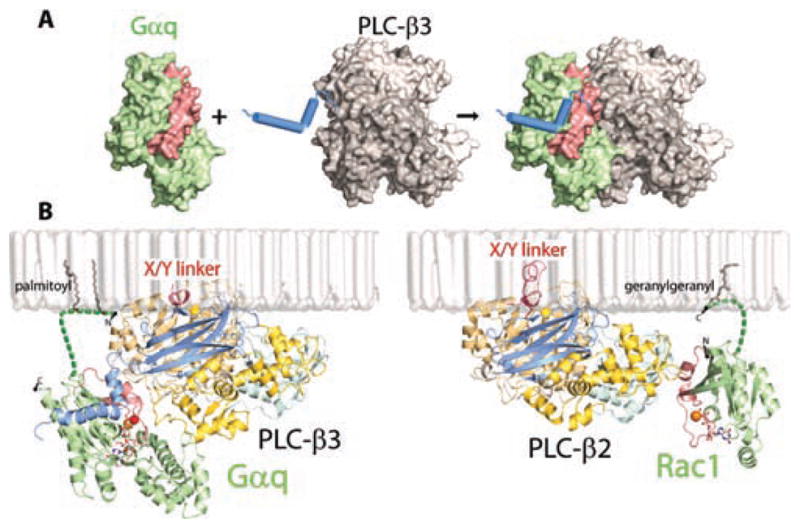
Binding of G proteins to PLC-β isozymes. (A) The helix—turn—helix region of PLC-β3 is shown engaging activated Gαq. (B) Membrane-anchored orientations are compared for PLC-β3 bound to activated Gαq (Protein Data Bank accession code 3OHM)21 and PLC-β2 bound to activated Rac1 (Protein Date Bank accession code 2FJU).25 The palmitoylated N-terminus of Gαq and geranylgeranylated C-terminus of Rac1 are modeled as sites of membrane attachment of the G proteins. The X/Y-linker, which occludes the active lipase site in both structures, is indicated in red, and the catalytic Ca2+ (yellow ball) present in the active site of both structures is also shown. The figure highlights the dramatic difference in PLC binding of the two activated G proteins: the switch regions (red) of activated Gαq (green) primarily engage PLC-β3 through the helix—turn—helix region at the C-terminal end of the C2 domain, whereas the switch regions (red) of activated Rac1 (green) engage PLC-β2 entirely through interactions with the PH domain. The structures also illustrate that activated Gproteins bind at great distance from the active site of the lipase. Activation in both cases involves an interfacial mechanism whereby the occluding X/Y-linker, which is highly negatively charged, is removed upon encountering the negative charge of the membrane surface.
The kinetics and mechanism of activation of PLC-β isozymes by Gβγ are much less well-defined than is the case for Gαq-mediated activation. Clearly, the “kinetic scaffolding” effect contributed by the Gαq-directed GAP activity of PLC-β is not operative during activation by Gβγ, and the same holds true during activation of PLC-β2 by Rac. Whereas the GAP activity of PLC-β against Gαq ensures that Gαq-activated PLC-β isozymes turn off on a millisecond time scale when the activating GPCR is no longer agonist-bound, the rate of turnoff of Gβγ- and Racactivated complexes must be much slower. Gβγ and Rac have been reported to activate PLC-β2 at least in an additive fashion,25 but the interplay of these two activators in PLC-β-mediated signaling has not been extensively studied. Interestingly, PLC-β2 and PLC-β3 can be simultaneously activated by Gαq and Gβγ, and studies with purified proteins illustrate that the combined presence of activated Gαq and Gβγ results in supra-additive stimulation of PLC-β3.110 This cooperative activation apparently accounts for synergistic activation of PLC-β often observed in cells during simultaneous activation of Gq- and Gi-activating GPCR.111
6. MECHANISM OF Gαq-MEDIATED ACTIVATION OF PLC-β ISOZYMES
PtdIns(4,5)P2 is membrane-bound, and the PLC isozymes are generally considered to be soluble proteins. Early models for receptor-promoted activation of PLC included the concept of recruitment to the plasma membrane. However, the mechanism- (s) of activation of these signaling proteins is clearly more complex than simple membrane association, and this is assuredly the case for PLC-β isozymes which are largely found at the plasma membrane under basal conditions. Simple presence at the membrane is not optimal for efficient enzyme activity since the binding pocket of the active site must be appropriately positioned to face PtdIns(4,5)P2 at the membrane surface. Thus, Gαq-dependent activation includes orientation of the lipase active site for substrate binding. Although bound Gαq also could propagate a conformational change to the active site, this does not appear to be the case on the basis of recent structures of PLC-β2 alone,93 PLC-β2 in an activated complex with Rac1,24 and PLC-β3 in an activated complex with Gαq.20 Indeed, the primary mechanism of activation of PLC-β by Gαq, and indeed the means whereby activation of most if not all PLC isozymes occurs, is through removal of autoinhibition mediated by the X/Y-linker region of these signaling proteins. We review the evidence for such a model below.
A broad range of studies over the past two decades illustrate that disruption of the X/Y-linker of PLC isozymes increases enzymatic activity. For example, limited proteolysis of PLC-δ,112 PLC-β,113 PLC-γ,114 and PLC-ζ115 isozymes resulted in higher basal activity. Functional lipase activity also was reassembled from independent expression of the X and Y domains of PLC-β 2116 or PLC-γ1,117 and the basal activities observed with these recombinant proteins lacking X/Y-linkers exceeded that observed with expression of the holoenzymes.
The X/Y-linker region in the first three-dimensional structure of a PLC isozyme, PLC-δ1, was completely disordered.22 The more recent structures of PLC-β2 alone93 and PLC-β2 in an activated complex with GTP-bound Rac124 revealed a slightly different situation. That is, no electron density was associated with most of the X/Y-linker in these basal and activated forms of PLC-β2, but importantly, a region (22 of the 70 residues) of the X/Y-linker was ordered and occluded the active site (Figure 5B). Fourteen of these residues form an α-helix that runs perpendicular to the TIM barrel, and the last eight residues are positioned on the surface of the catalytic cleft and form a small 310 helix that makes direct hydrogen-bond-mediated contacts with active site residues. These interactions within the active site occur in structures of both the basal and Rac-bound forms of PLC-β2.93 Importantly, the structure of PLC-β3 in complex with activated Gαq also displayed similar residues making direct contacts with the active site.20 Occurrence of X/Y-linker-mediated occlusion of the active site of PLC-β2 and PLC-β3 in structures of the G protein-bound enzymes indicates that binding of activators is insufficient to remove autoinhibition. Indeed, super-position of the three structures of PLC-β isozymes illustrates that no conformational changes occur within the catalytic TIM barrel.20,24,93
Observation of direct occlusion of the active site of PLC-β2 by residues in the X/Y-linker led Hicks and her co-workers93 to carry out a systematic deletion/mutation analysis of this region. Removal of the stretch of 22 occluding residues resulted in a PLC-β2 isozyme with remarkably higher basal activity when assessed by transient expression or as a purified protein. Removal of all of the disordered portion or all of the X/Y-linker resulted in PLC-β2 mutants with even higher basal activity. Disruption of the active-site-interacting small 310 helix by introduction of a proline in place of a glycine, i.e., PLC-β2(G530P), also resulted in a marked increase in enzyme activity. These data support the idea that basal activity of PLC-β isozymes is strongly controlled by an autoinhibitory mechanism. G protein activators do not directly interact with the X/Y-linker region (Figure 5B), and therefore, binding is not affected by its removal. Indeed, Gαq, Gβγ, and Rac all still activate PLC-β2 lacking the linker,93 apparently because G protein binding still optimally orients the active site of the X/Y-linker-deleted isozyme at the membrane surface.
Occurrence of X/Y-linker-mediated inhibition is not restricted to the PLC-β isozymes. Thus, systematic deletion of the X/Ylinker markedly enhances the lipase activity of PLC-δ1, -ε, -γ1, and -γ2 both in intact cells and with purified proteins.20,35,93 Removal of the X/Y-linker also robustly activates PLC-ε, and this linker-deleted isozyme retains the capacity to be activated by its upstream regulators RhoA and H-Ras.118
Although biochemical and structural studies highlight a common inhibitory role of the X/Y-linker in all PLC isozymes, how is this function mediated by the very divergent primary sequences of the various X/Y-linkers and how is this autoinhibition removed by Gαq binding or by other activators? The most parsimonious model for activation of these lipases involves an interfacial mechanism driven by electrostatic repulsions between the negatively charged X/Y-linker and negatively charged membrane surfaces. Thus, activators do not induce general conformational changes, but rather anchor and optimally orient PLC isoforms at the plasma membrane to facilitate electrostatic repulsion and removal of the X/Y-linker from the active site (Figure 5B). Consistent with this model, removal of monovalent acidic phospholipids from the monolayer decreased by 3-fold the initial rate of PtdIns(4,5)P2 hydrolysis by PLC-β1 and -δ1.119 Rac-mediated activation of PLC-β2 also was abolished in detergent- mixed micelles, but not in phospholipid vesicles, which is also consistent with the idea that recruitment to a membrane surface is critical for mediation of interfacial activation.93 Finally, PLC-δ does not bind Gαq and is not activated by guanine nucleotides.20 However, a chimeric recombinant form of PLC- δ1 that incorporates the Gαq-binding helix—turn—helix region from PLC-β3 is sufficient to engender GPCR- and Gαq-dependent lipase activation. Although additional mechanisms may support removal of X/Y-linker-mediated autoinhibition, results to date favor the idea that activation largely occurs through a membrane-surface-mediated mechanism.
The X/Y-linker region of PLC-γ isozymes differs dramatically from that of other PLC isozymes since it contains a split PH domain, two SH2 domains, and an SH3 domain.9,11 X/Y-linker-mediated autoinhibition of basal enzyme activity also occurs with these isozymes, and recent studies implicate the C-terminal SH2 domain (cSH2) in mediating this effect. Recruitment of PLC-γ isozymes to the plasma membrane occurs through engagement of the N-terminal SH2 domain by activated receptor or nonreceptor tyrosine kinases. These kinases in turn promote phosphorylation of a tyrosine in the intervening sequence between the cSH2 domain and SH3 domain which then interacts with the cSH2 domain to relieve autoinhibition.35
7. CONCLUSIONS
A panoply of extracellular stimuli activate several hundred different GPCRs that are known to activate Gq/PLC-β signaling complexes to promote inositol lipid signaling cascades. These signaling responses of course include the classical mobilization of Ca2+ and activation of protein kinase C that are cornerstones of PLC-dependent signaling, but the activities of a broad range of membrane, cytoskeletal, and cytosolic proteins also are directly regulated by PLC-mediated changes in membrane concentrations of PtdIns(4,5)P2. Thus, signaling responses exhibiting many different characteristics radiate from PLC-β isozymes. The forms that these signaling nodes take both spatially and functionally remain poorly understood, but application of increasingly sophisticated live cell-imaging capabilities offers great promise for new understanding. Strong insight into the physiological functions that depend on Gq/PLC-β signaling complexes is available, but much is yet to be learned about their roles in human physiology and pathophysiology. Understanding of these nodes at the mechanistic level has increased recently with the availability of three-dimensional structures of G protein-bound PLC-β isozymes. This information should in turn provide both reagents and new research directions that increase insight into inositol lipid signaling at the cellular and intact animal levels. The absence of pharmacological agents that selectively target PLC isozymes has definitely slowed research progress, and an out-standing contribution would be made by discovery of such molecules. PLC-β-selective inhibitors may also have clinical value. For example, a large preponderance of uveal melanomas have somatic mutations in Gαq or Gα11 that result in constitutive activation of these signaling proteins and their downstream signaling responses.120,121
Biographies

T. Kendall Harden is a Kenan Professor in the Department of Pharmacology at the University of North Carolina School of Medicine, where he has studied G protein-coupled receptors and their associated cell signaling mechanisms for three decades. His Ph.D. is from the University of Mississippi, and he trained as a postdoctoral fellow with Perry Molinoff at the University of Colorado School of Medicine.

Gary L. Waldo is a research analyst in the Department of Pharmacology at the University of North Carolina. He has focused on the biochemistry of G proteins and their downstream effectors for the past two decades. He was involved in early studies illustrating direct activation of phospholipase C isozymes by Gq family Gα-subunits and Gβγ-subunits. More recently, he solved an X-ray crystal structure and carried out biochemical analyses that defined the Gαq/PLC-β-binding interface and the mechanism whereby PLC-β acts as a GTPase-activating protein for Gαq.

Stephanie N. Hicks is a Research Assistant Professor in the Department of Pharmacology at the University of North Carolina. She received her Ph.D. in Biochemistry, Cellular, & Molecular Biology from the University of Tennessee in 2003. Her postdoctoral work with Drs. Sondek and Harden focused on the regulation of phospholipase C. She has applied biophysical and structural analyses to define the mechanism of autoinhibition of phospholipase C isozymes and to delineate the mechanism where by activators relieve this autoinhibition.

John Sondek received his Ph.D. in Biochemistry from Johns Hopkins University in 1992. Upon completing postdoctoral studies at Yale University in 1996 as a Damon-Runyon-Walter Winchell Fellow, he joined the University of North Carolina at Chapel Hill and was named a Pew Scholar in the Biomedical Sciences. He is now a Professor in the Departments of Pharmacology and of Biochemistry & Biophysics as well as a member of the Lineberger Comprehensive Cancer Center. His independent research program has been continuously funded since its inception from various agencies, including the National Institutes of Health, GlaxoSmithKline, and the Pew Charitable Trusts. His research is focused on understanding protein complexes involved in signal transduction cascades as potential targets for new therapeutics against cancer and neurological diseases. He has authored or coauthored over 80 original research articles and holds four patents related to the mutagenesis and screening of proteins.
References
- 1.Berridge MJ. Annu Rev Biochem. 1987;56:159. doi: 10.1146/annurev.bi.56.070187.001111. [DOI] [PubMed] [Google Scholar]
- 2.Yin HL, Janmey PA. Annu Rev Physiol. 2003;65:761. doi: 10.1146/annurev.physiol.65.092101.142517. [DOI] [PubMed] [Google Scholar]
- 3.Suh BC, Hille B. Annu Rev Biophys. 2008;37:175. doi: 10.1146/annurev.biophys.37.032807.125859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ling K, Schill NJ, Wagoner MP, Sun Y, Anderson RA. Trends Cell Biol. 2006;16:276. doi: 10.1016/j.tcb.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 5.Harden TK, Sondek J. Annu Rev Pharmacol Toxicol. 2006;46:355. doi: 10.1146/annurev.pharmtox.46.120604.141223. [DOI] [PubMed] [Google Scholar]
- 6.Michell RH. Biochim Biophys Acta. 1975;415:81. doi: 10.1016/0304-4157(75)90017-9. [DOI] [PubMed] [Google Scholar]
- 7.Nishizuka Y. Science. 1992;258:607. doi: 10.1126/science.1411571. [DOI] [PubMed] [Google Scholar]
- 8.Rebecchi MJ, Pentyala SN. Physiol Rev. 2000;80:1291. doi: 10.1152/physrev.2000.80.4.1291. [DOI] [PubMed] [Google Scholar]
- 9.Rhee SG. Annu Rev Biochem. 2001;70:281. doi: 10.1146/annurev.biochem.70.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rhee SG, Bae YS. J Biol Chem. 1997;272:15045. doi: 10.1074/jbc.272.24.15045. [DOI] [PubMed] [Google Scholar]
- 11.Horstman DA, Chattopadhyay A, Carpenter G. Arch Biochem Biophys. 1999;361:149. doi: 10.1006/abbi.1998.0978. [DOI] [PubMed] [Google Scholar]
- 12.Katan M, Rodriguez R, Matsuda M, Newbatt YM, Aherne GW. Adv Enzyme Regul. 2003;43:77. doi: 10.1016/s0065-2571(02)00027-4. [DOI] [PubMed] [Google Scholar]
- 13.Bunney TD, Katan M. Trends Cell Biol. 2006;16:640. doi: 10.1016/j.tcb.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 14.Harden TK, Hicks SN, Sondek J. J Lipid Res. 2009;50(Suppl):S243. doi: 10.1194/jlr.R800045-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakamura Y, Fukami K. Physiology (Bethesda) 2009;24:332. doi: 10.1152/physiol.00031.2009. [DOI] [PubMed] [Google Scholar]
- 16.Suh PG, Park JI, Manzoli L, Cocco L, Peak JC, Katan M, Fukami K, Kataoka T, Yun S, Ryu SH. BMB Rep. 2008;41:415. doi: 10.5483/bmbrep.2008.41.6.415. [DOI] [PubMed] [Google Scholar]
- 17.Bunney TD, Katan M. Trends Biochem Sci. 2011;36:88. doi: 10.1016/j.tibs.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 18.Drin G, Scarlata S. Cell Signalling. 2007;19:1383. doi: 10.1016/j.cellsig.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Katan M, Williams RL. Semin Cell Dev Biol. 1997;8:287. doi: 10.1006/scdb.1997.0150. [DOI] [PubMed] [Google Scholar]
- 20.Waldo GL, Ricks TK, Hicks SN, Cheever ML, Kawano T, Tsuboi K, Wang X, Montell C, Kozasa T, Sondek J, Harden TK. Science. 2010;330:974. doi: 10.1126/science.1193438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferguson KM, Lemmon MA, Schlessinger J, Sigler PB. Cell. 1995;83:1037. doi: 10.1016/0092-8674(95)90219-8. [DOI] [PubMed] [Google Scholar]
- 22.Essen LO, Perisic O, Cheung R, Katan M, Williams RL. Nature. 1996;380:595. doi: 10.1038/380595a0. [DOI] [PubMed] [Google Scholar]
- 23.Lomasney JW, Cheng HF, Wang LP, Kuan Y, Liu S, Fesik SW, King K. J Biol Chem. 1996;271:25316. doi: 10.1074/jbc.271.41.25316. [DOI] [PubMed] [Google Scholar]
- 24.Jezyk MR, Snyder JT, Gershberg S, Worthylake DK, Harden TK, Sondek J. Nat Struct Mol Biol. 2006;13:1135. doi: 10.1038/nsmb1175. [DOI] [PubMed] [Google Scholar]
- 25.Illenberger D, Walliser C, Nurnberg B, Diaz Lorente M, Gierschik P. J Biol Chem. 2003;278:3006. doi: 10.1074/jbc.M208282200. [DOI] [PubMed] [Google Scholar]
- 26.Snyder JT, Singer AU, Wing MR, Harden TK, Sondek J. J Biol Chem. 2003;278:21099. doi: 10.1074/jbc.M301418200. [DOI] [PubMed] [Google Scholar]
- 27.Kawasaki H, Kretsinger RH. Protein Profile. 1994;1:343. [PubMed] [Google Scholar]
- 28.Nalefski EA, Falke JJ. Protein Sci. 1996;5:2375. doi: 10.1002/pro.5560051201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Essen LO, Perisic O, Lynch DE, Katan M, Williams RL. Biochemistry. 1997;36:2753. doi: 10.1021/bi962466t. [DOI] [PubMed] [Google Scholar]
- 30.Wierenga RK. FEBS Lett. 2001;492:193. doi: 10.1016/s0014-5793(01)02236-0. [DOI] [PubMed] [Google Scholar]
- 31.Cheng HF, Jiang MJ, Chen CL, Liu SM, Wong LP, Lomasney JW, King K. J Biol Chem. 1995;270:5495. doi: 10.1074/jbc.270.10.5495. [DOI] [PubMed] [Google Scholar]
- 32.Ellis MVUS, Katan M. Biochem J. 1995;307:69. doi: 10.1042/bj3070069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ellis MV, James SR, Perisic O, Downes CP, Williams RL, Katan M. J Biol Chem. 1998;273:11650. doi: 10.1074/jbc.273.19.11650. [DOI] [PubMed] [Google Scholar]
- 34.Heinz DW, Essen LO, Williams RL. J Mol Biol. 1998;275:635. doi: 10.1006/jmbi.1997.1490. [DOI] [PubMed] [Google Scholar]
- 35.Gresset A, Hicks SN, Harden TK, Sondek J. J Biol Chem. 2010;285:35836. doi: 10.1074/jbc.M110.166512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Piechulek T, Rehlen T, Walliser C, Vatter P, Moepps B, Gierschik P. J Biol Chem. 2005;280:38923. doi: 10.1074/jbc.M509396200. [DOI] [PubMed] [Google Scholar]
- 37.Walliser C, Retlich M, Harris R, Everett KL, Josephs MB, Vatter P, Esposito D, Driscoll PC, Katan M, Gierschik P, Bunney TD. J Biol Chem. 2008;283:30351. doi: 10.1074/jbc.M803316200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shibatohge M, Kariya K, Liao Y, Hu CD, Watari Y, Goshima M, Shima F, Kataoka T. J Biol Chem. 1998;273:6218. doi: 10.1074/jbc.273.11.6218. [DOI] [PubMed] [Google Scholar]
- 39.Kelley GG, Reks SE, Ondrako JM, Smrcka AV. EMBO J. 2001;20:743. doi: 10.1093/emboj/20.4.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lopez I, Mak EC, Ding J, Hamm HE, Lomasney JW. J Biol Chem. 2001;276:2758. doi: 10.1074/jbc.M008119200. [DOI] [PubMed] [Google Scholar]
- 41.Song C, Hu CD, Masago M, Kariyai K, Yamawaki-Kataoka Y, Shibatohge M, Wu D, Satoh T, Kataoka T. J Biol Chem. 2001;276:2752. doi: 10.1074/jbc.M008324200. [DOI] [PubMed] [Google Scholar]
- 42.Kelley GG, Reks SE, Smrcka AV. Biochem J. 2004;378:129. doi: 10.1042/BJ20031370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jin TG, Satoh T, Liao Y, Song C, Gao X, Kariya K, Hu CD, Kataoka T. J Biol Chem. 2001;276:30301. doi: 10.1074/jbc.M103530200. [DOI] [PubMed] [Google Scholar]
- 44.Citro S, Malik S, Oestreich EA, Radeff-Huang J, Kelley GG, Smrcka AV, Brown JH. Proc Natl Acad Sci USA. 2007;104:15543. doi: 10.1073/pnas.0702943104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saunders CM, Larman MG, Parrington J, Cox LJ, Royse J, Blayney LM, Swann K, Lai FA. Development. 2002;129:3533. doi: 10.1242/dev.129.15.3533. [DOI] [PubMed] [Google Scholar]
- 46.Hwang JI, Oh YS, Shin KJ, Kim H, Ryu SH, Suh PG. Biochem J. 2005;389:181. doi: 10.1042/BJ20041677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nakahara M, Shimozawa M, Nakamura Y, Irino Y, Morita M, Kudo Y, Fukami K. J Biol Chem. 2005;280:29128. doi: 10.1074/jbc.M503817200. [DOI] [PubMed] [Google Scholar]
- 48.Zhou Y, Wing MR, Sondek J, Harden TK. Biochem J. 2005;391:667. doi: 10.1042/BJ20050839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou Y, Sondek J, Harden TK. Biochemistry. 2008;47:4410. doi: 10.1021/bi800044n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gilman AG. Annu Rev Biochem. 1987;56:615. doi: 10.1146/annurev.bi.56.070187.003151. [DOI] [PubMed] [Google Scholar]
- 51.Clapham DE, Neer EJ. Annu Rev Pharmacol Toxicol. 1997;37:167. doi: 10.1146/annurev.pharmtox.37.1.167. [DOI] [PubMed] [Google Scholar]
- 52.Smrcka AV. Cell Mol Life Sci. 2008;65:2191. doi: 10.1007/s00018-008-8006-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Goodhardt M, Ferry N, Geynet P, Hanoune J. J Biol Chem. 1982;257:11577. [PubMed] [Google Scholar]
- 54.Boyer JL, Garcia A, Posadas C, Garcia-Sainz JA. J Biol Chem. 1984;259:8076. [PubMed] [Google Scholar]
- 55.Evans T, Martin MW, Hughes AR, Harden TK. Mol Pharmacol. 1985;27:32. [PubMed] [Google Scholar]
- 56.Evans T, Hepler JR, Masters SB, Brown JH, Harden TK. Biochem J. 1985;232:751. doi: 10.1042/bj2320751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Litosch I, Wallis C, Fain JN. J Biol Chem. 1985;260:5464. [PubMed] [Google Scholar]
- 58.Cockcroft S, Gomperts BD. Nature. 1985;314:534. doi: 10.1038/314534a0. [DOI] [PubMed] [Google Scholar]
- 59.Uhing RJ, Jiang H, Prpic V, Exton JH. FEBS Lett. 1985;188:317. doi: 10.1016/0014-5793(85)80394-x. [DOI] [PubMed] [Google Scholar]
- 60.Straub RE, Gershengorn MC. J Biol Chem. 1986;261:2712. [PubMed] [Google Scholar]
- 61.Hepler JR, Harden TK. Biochem J. 1986;239:141. doi: 10.1042/bj2390141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Martin TF, Bajjalieh SM, Lucas DO, Kowalchyk JA. J Biol Chem. 1986;261:10141. [PubMed] [Google Scholar]
- 63.Strathmann M, Simon MI. Proc Natl Acad Sci USA. 1990;87:9113. doi: 10.1073/pnas.87.23.9113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wilkie TM, Scherle PA, Strathmann MP, Slepak VZ, Simon MI. Proc Natl Acad Sci USA. 1991;88:10049. doi: 10.1073/pnas.88.22.10049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Smrcka AV, Hepler JR, Brown KO, Sternweis PC. Science. 1991;251:804. doi: 10.1126/science.1846707. [DOI] [PubMed] [Google Scholar]
- 66.Taylor SJ, Chae HZ, Rhee SG, Exton JH. Nature. 1991;350:516. doi: 10.1038/350516a0. [DOI] [PubMed] [Google Scholar]
- 67.Waldo GL, Boyer JL, Morris AJ, Harden TK. J Biol [Google Scholar]
- 68.Sternweis PC, Smrcka AV. Ciba Found Symp. 1993;176:96. doi: 10.1002/9780470514450.ch7. [DOI] [PubMed] [Google Scholar]
- 69.Exton JH. Annu Rev Pharmacol Toxicol. 1996;36:481. doi: 10.1146/annurev.pa.36.040196.002405. [DOI] [PubMed] [Google Scholar]
- 70.Singer WD, Brown HA, Sternweis PC. Annu Rev Biochem. 1997;66:475. doi: 10.1146/annurev.biochem.66.1.475. [DOI] [PubMed] [Google Scholar]
- 71.Wu D, Jiang H, Katz A, Simon MI. J Biol Chem. 1993;268:3704. [PubMed] [Google Scholar]
- 72.Lee SB, Shin SH, Hepler JR, Gilman AG, Rhee SG. J Biol Chem. 1993;268:25952. [PubMed] [Google Scholar]
- 73.Paulssen RH, Woodson J, Liu Z, Ross EM. J Biol Chem. 1996;271:26622. doi: 10.1074/jbc.271.43.26622. [DOI] [PubMed] [Google Scholar]
- 74.Ilkaeva O, Kinch LN, Paulssen RH, Ross EM. J Biol Chem. 2002;277:4294. doi: 10.1074/jbc.M109612200. [DOI] [PubMed] [Google Scholar]
- 75.Kim CG, Park D, Rhee SG. J Biol Chem. 1996;271:21187. doi: 10.1074/jbc.271.35.21187. [DOI] [PubMed] [Google Scholar]
- 76.Singer AU, Waldo GL, Harden TK, Sondek J. Nat Struct Biol. 2002;9:32. doi: 10.1038/nsb731. [DOI] [PubMed] [Google Scholar]
- 77.Camps M, Hou C, Sidiropoulos D, Stock JB, Jakobs KH, Gierschik P. Eur J Biochem. 1992;206:821. doi: 10.1111/j.1432-1033.1992.tb16990.x. [DOI] [PubMed] [Google Scholar]
- 78.Camps M, Carozzi A, Schnabel P, Scheer A, Parker PJ, Gierschik P. Nature. 1992;360:684. doi: 10.1038/360684a0. [DOI] [PubMed] [Google Scholar]
- 79.Boyer JL, Waldo GL, Harden TK. J Biol Chem. 1992;267:25451. [PubMed] [Google Scholar]
- 80.Smrcka AV, Sternweis PC. J Biol Chem. 1993;268:9667. [PubMed] [Google Scholar]
- 81.Wang T, Dowal L, El-Maghrabi MR, Rebecchi M, Scarlata S. J Biol Chem. 2000;275:7466. doi: 10.1074/jbc.275.11.7466. [DOI] [PubMed] [Google Scholar]
- 82.Barr AJ, Ali H, Haribabu B, Snyderman R, Smrcka AV. Biochemistry. 2000;39:1800. doi: 10.1021/bi992021f. [DOI] [PubMed] [Google Scholar]
- 83.Cassel D, Selinger Z. Biochim Biophys Acta. 1976;452:538. doi: 10.1016/0005-2744(76)90206-0. [DOI] [PubMed] [Google Scholar]
- 84.Cassel D, Levkovitz H, Selinger Z. J Cyclic Nucleotide Res. 1977;3:393. [PubMed] [Google Scholar]
- 85.Ross EM, Wilkie TM. Annu Rev Biochem. 2000;69:795. doi: 10.1146/annurev.biochem.69.1.795. [DOI] [PubMed] [Google Scholar]
- 86.Bollag G, McCormick F. Annu Rev Cell Biol. 1991;7:601. doi: 10.1146/annurev.cb.07.110191.003125. [DOI] [PubMed] [Google Scholar]
- 87.Berstein G, Blank JL, Smrcka AV, Higashijima T, Sternweis PC, Exton JH, Ross EM. J Biol Chem. 1992;267:8081. [PubMed] [Google Scholar]
- 88.Berstein G, Blank JL, Jhon DY, Exton JH, Rhee SG, Ross EM. Cell. 1992;70:411. doi: 10.1016/0092-8674(92)90165-9. [DOI] [PubMed] [Google Scholar]
- 89.Biddlecome GH, Berstein G, Ross EM. J Biol Chem. 1996;271:7999. doi: 10.1074/jbc.271.14.7999. [DOI] [PubMed] [Google Scholar]
- 90.Mukhopadhyay S, Ross EM. Proc Natl Acad Sci USA. 1999;96:9539. doi: 10.1073/pnas.96.17.9539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Turcotte M, Tang W, Ross EM. PLoS Comput Biol. 2008;4:e1000148. doi: 10.1371/journal.pcbi.1000148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Slep KC, Kercher MA, He W, Cowan CW, Wensel TG, Sigler PB. Nature. 2001;409:1071. doi: 10.1038/35059138. [DOI] [PubMed] [Google Scholar]
- 93.Hicks SN, Jezyk MR, Gershburg S, Seifert JP, Harden TK, Sondek J. Mol Cell. 2008;31:383. doi: 10.1016/j.molcel.2008.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang T, Montell C. Pfluegers Arch. 2007;454:821. doi: 10.1007/s00424-007-0251-1. [DOI] [PubMed] [Google Scholar]
- 95.Dohlman HG, Thorner J. J Biol Chem. 1997;272:3871. doi: 10.1074/jbc.272.7.3871. [DOI] [PubMed] [Google Scholar]
- 96.Wang Q, Liu M, Mullah B, Siderovski DP, Neubig RR. J Biol Chem. 2002;277:24949. doi: 10.1074/jbc.M203802200. [DOI] [PubMed] [Google Scholar]
- 97.Ross EM. Curr Biol. 2008;18:R777. doi: 10.1016/j.cub.2008.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tesmer JJ, Berman DM, Gilman AG, Sprang SR. Cell. 1997;89:251. doi: 10.1016/s0092-8674(00)80204-4. [DOI] [PubMed] [Google Scholar]
- 99.Chen CK, Burns ME, He W, Wensel TG, Baylor DA, Simon MI. Nature. 2000;403:557. doi: 10.1038/35000601. [DOI] [PubMed] [Google Scholar]
- 100.Lishko PV, Martemyanov KA, Hopp JA, Arshavsky VY. J Biol Chem. 2002;277:24376. doi: 10.1074/jbc.M203237200. [DOI] [PubMed] [Google Scholar]
- 101.Arshavsky VY, Lamb TD, Pugh EN., Jr Annu Rev Physiol. 2002;64:153. doi: 10.1146/annurev.physiol.64.082701.102229. [DOI] [PubMed] [Google Scholar]
- 102.Lutz S, Freichel-Blomquist A, Yang Y, Rumenapp U, Jakobs KH, Schmidt M, Wieland T. J Biol Chem. 2005;280:11134. doi: 10.1074/jbc.M411322200. [DOI] [PubMed] [Google Scholar]
- 103.Rojas RJ, Yohe ME, Gershburg S, Kawano T, Kozasa T, Sondek J. J Biol Chem. 2007;282:29201. doi: 10.1074/jbc.M703458200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lutz S, Shankaranarayanan A, Coco C, Ridilla M, Nance MR, Vettel C, Baltus D, Evelyn CR, Neubig RR, Wieland T, Tesmer JJ. Science. 2007;318:1923. doi: 10.1126/science.1147554. [DOI] [PubMed] [Google Scholar]
- 105.Carman CV, Parent JL, Day PW, Pronin AN, Sternweis PM, Wedegaertner PB, Gilman AG, Benovic JL, Kozasa T. J Biol Chem. 1999;274:34483. doi: 10.1074/jbc.274.48.34483. [DOI] [PubMed] [Google Scholar]
- 106.Tesmer VM, Kawano T, Shankaranarayanan A, Kozasa T, Tesmer JJ. Science. 2005;310:1686. doi: 10.1126/science.1118890. [DOI] [PubMed] [Google Scholar]
- 107.Shoemaker BA, Portman JJ, Wolynes PG. Proc Natl Acad Sci USA. 2000;97:8868. doi: 10.1073/pnas.160259697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sugase K, Dyson HJ, Wright PE. Nature. 2007;447:1021. doi: 10.1038/nature05858. [DOI] [PubMed] [Google Scholar]
- 109.Shankaranarayanan A, Thal DM, Tesmer VM, Roman DL, Neubig RR, Kozasa T, Tesmer JJ. J Biol Chem. 2008;283:34923. doi: 10.1074/jbc.M805860200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Philip F, Kadamur G, Silos RG, Woodson J, Ross EM. Curr Biol. 2010;20:1327. doi: 10.1016/j.cub.2010.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rebres RA, Roach TI, Fraser ID, Philip F, Moon C, Lin KM, Liu J, Santat L, Cheadle L, Ross EM, Simon MI, Seaman WE. J Biol Chem. 2011;286:942. doi: 10.1074/jbc.M110.198200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ellis MV, Carne A, Katan M. Eur J Biochem. 1993;213:339. doi: 10.1111/j.1432-1033.1993.tb17767.x. [DOI] [PubMed] [Google Scholar]
- 113.Schnabel P, Camps M. Biochem J. 1998;330:461. doi: 10.1042/bj3300461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Fernald AW, Jones GA, Carpenter G. Biochem J. 1994;302(Part 2):503. doi: 10.1042/bj3020503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kurokawa M, Yoon SY, Alfandari D, Fukami K, Sato K, Fissore RA. Dev Biol. 2007;312:407. doi: 10.1016/j.ydbio.2007.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhang W, Neer EJ. J Biol Chem. 2001;276:2503. doi: 10.1074/jbc.M003562200. [DOI] [PubMed] [Google Scholar]
- 117.Horstman DA, DeStefano K, Carpenter G. Proc Natl Acad Sci USA. 1996;93:7518. doi: 10.1073/pnas.93.15.7518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Seifert JP, Zhou Y, Hicks SN, Sondek J, Harden TK. J Biol Chem. 2008;283:29690. doi: 10.1074/jbc.M805038200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Boguslavsky V, Rebecchi M, Morris AJ, Jhon DY, Rhee SG, McLaughlin S. Biochemistry. 1994;33:3032. doi: 10.1021/bi00176a036. [DOI] [PubMed] [Google Scholar]
- 120.Van Raamsdonk CD, Bezrookove V, Green G, Bauer J, Gaugler L, O’Brien JM, Simpson EM, Barsh GS, Bastian BC. Nature. 2009;457:599. doi: 10.1038/nature07586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Van Raamsdonk CD, Griewank KG, Crosby MB, Garrido MC, Vemula S, Wiesner T, Obenauf AC, Wackernagel W, Green G, Bouvier N, Sozen MM, Baimukanova G, Roy R, Heguy A, Dolgalev I, Khanin R, Busam K, Speicher MR, O’Brien J, Bastian BC. N Engl J Med. 2010;363:2191. doi: 10.1056/NEJMoa1000584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhong H, Wade SM, Woolf PJ, Linderman JJ, Traynor JR, Neubig RR. J Biol Chem. 2003;278:7278. doi: 10.1074/jbc.M208819200. [DOI] [PubMed] [Google Scholar]