

Abstract
Schizophrenia is a devastating neuropsychiatric disorder of unknown etiology. There is general agreement in the scientific community that schizophrenia is a disorder of neurodevelopmental origin in which both genes and environmental factors come together to produce a schizophrenia phenotype later in life. The challenging questions have been which genes and what environmental factors? Although there is evidence that different chromosome loci and several genes impart susceptibility for schizophrenia; and epidemiological studies point to broad aspects of the environment, only recently there has been an interest in studying gene × environment interactions. Recent evidence of a potential association between prenatal lead (Pb2+) exposure and schizophrenia precipitated the search for plausible neurobiological connections. The most promising connection is that in schizophrenia and in developmental Pb2+ exposure there is strong evidence for hypoactivity of the N-methyl-d-aspartate (NMDA) subtype of excitatory amino acid receptors as an underlying neurobiological mechanism in both conditions. A hypofunction of the NMDA receptor (NMDAR) complex during critical periods of development may alter neurobiological processes that are essential for brain growth and wiring, synaptic plasticity and cognitive and behavioral outcomes associated with schizophrenia. We also describe on-going proof of concept gene-environment interaction studies of early life Pb2+ exposure in mice expressing the human mutant form of the disrupted in schizophrenia 1 (DISC-1) gene, a gene that is strongly associated with schizophrenia and allied mental disorders.
Keywords: Schizophrenia – Lead – Pb2+, Early Life, NMDA Receptor, DISC1, gene, environment, interaction
Introduction
Schizophrenia is a severe neuropsychiatric disorder affecting approximately 1% of the world's population (Bromet and Fenning, 1999; McGrath et al., 2004). The symptoms typically begin in late adolescence or early adulthood, whereupon lifelong disability typically ensues. Its onset is defined by the emergence of psychosis in the setting of deteriorating function and other symptoms. Before psychosis onset, during a prodromal period of several weeks to years, nonspecific and variable subtle abnormalities worsen and coalesce into the classic disease features. These include alterations in the perception of reality, changes in the form and content of thoughts and speech, and social and emotional deficits including a disturbed sense of self, social dysfunction, apathy, and peculiar behavior. The symptoms of schizophrenia are often grouped into positive and negative subtypes, although there is substantial diversity in the pathophysiology of the symptoms within these groups. Positive symptoms include hallucinations and delusions, disorganized thinking or behavior, so denoted because these phenomena occur in addition to usual experiences. Negative symptoms are those that arise from the absence of normal behaviors or experiences, including affective flattening, alogia (impoverish thinking manifested by diminished speech output or content), apathy, avolition (lack of energy and drive), and social withdraw. In addition, schizophrenia patients express a pattern of cognitive dysfunction that represents a core feature of the disorder.
Genes and Environment: Etiological factors in Schizophrenia
The etiology of schizophrenia is not currently known, although there is strong evidence that genetic and environmental factors contribute to the expression of the disease (Tsuang, 2000; van Os et al., 2010; Brown, 2011). The identification of specific genes that cause schizophrenia has been less than satisfactory; although a number of genes associated with schizophrenia have been identified that together may contribute to the schizophrenia phenotype (Lewis and Lieberman 2000; Tsuang 2000; Horvath and Mirmics 2009; Nieratschker et al, 2010). Environmental risk factors have also been implicated to play a role in schizophrenia. These include birth in an urban environment, season of birth, viral infection during the prenatal or perinatal period, complications during pregnancy and delivery, nutritional and social factors (Bromet and Fenning, 1999; Tsuang, 2000; vanOs et al., 2010; Brown, 2011). It is generally thought that exposures to an environmental insult that damages or disrupts the development of the central nervous system may be associated with schizophrenia and allied mental disorders (Murray et al., 1992; Weinberger, 1996). To date, maternal exposures during the mid- to late-gestational period have been implicated. There are several reasons to suspect that chemical agents, in general, and particularly those associated with industrialization, may increase the risk of schizophrenia. Studies further suggest that some feature of urban environment may elevate the risk of schizophrenia (Pedersen et al., 2004; Pedersen and Mortensen, 2006). This may be due to chemicals or environmental agents that are more prevalent in urban settings, possibly ambient pollutants (Marcelis et al., 1998).
Are Specific Environmental Toxins Involved?
An environmental risk factor that has not been considered until recently is the possibility of early life exposure to a developmental neurotoxicant(s) pervasive in the global environment. This possibility has been brought to light by two recent studies by Opler and colleagues (2004; 2008). They showed a potential association between prenatal Pb2+ exposure and the increased likelihood of expressing a schizophrenic phenotype later in life. To our knowledge this is the first time that prenatal exposure to a specific environmental agent has been associated with schizophrenia. Lead (Pb2+) has been known as a developmental neurotoxin in the medical literature since the early part of the 20th century. It has been associated with psychosis following acute exposure in adults, and more recently, with deficits in intelligence, impaired attention and executive function, and juvenile delinquency following prenatal and perinatal exposure (Needleman et al., 1979; Dietrich et al., 2001; Kaneshiro-Olympio et al, 2009). While some studies have followed samples with perinatal exposure into adolescence (Ris et al., 2004), there is limited information on long-term effects of Pb2+ exposure, particularly on the subsequent risk of mental disorders in adulthood.
Relationships between early life exposure to Pb2+ and neuropsychological abnormalities have been observed from infancy to adolescence (Bellinger et al., 1991; Pocock et al., 1994; Kim et al., 1995). For example, the Yugoslavia Prospective Study reported that Pb2+ exposure during mid-pregnancy was associated with deficits in neuropsychiatric function at 24 months of age (Factor-Litvak et al., 1999). Further assessments of this cohort identified persistent decrements in measures of attention, cognition, and verbal comprehension at ages 4, 7, 10, and 12 (Wasserman et al., 2000).
Studies by Needleman and colleagues found associations between dentine Pb2+ levels measured in deciduous teeth (ages 6-8) and failure to graduate from high school (Needleman et al., 1990) and other studies have found poor end-of-grade performance in Pb2+-exposed children (Miranda et al, 2007). In a prospective study conducted in Cincinnati, prenatal childhood blood Pb2+ concentrations were reported to be associated with increased delinquent behavior later in life (Dietrich et al., 2001). Nevin (2007) has found a very strong association between preschool blood Pb2+ and subsequent crime rate trends over several decades in various countries. This suggests that prenatal Pb2+ exposure may be a risk factor for other adolescent and adult-onset outcomes, possibly psychiatric disorders. Schizophrenia is one plausible candidate, as some of its premorbid features such as reduced attention, neurocognitive impairment, and diminished educational attainment (Jones et al., 1993) strongly resembles the behavioral deficits associated with Pb2+ exposure.
Prenatal Lead Exposure and Schizophrenia
Opler and colleagues (2004; 2008) have conducted studies on prenatal cohorts to assess the risk of schizophrenia following Pb2+ exposure. The principle technique for assessing Pb2+ exposure is through direct measurement of Pb2+ in maternal blood. They used stored sera, not whole blood containing the Pb2+-sequestering erythrocytes required for direct analysis. A biological marker of Pb2+ exposure, δ-aminolevulinic acid (δ-ALA) can be detected in urine, plasma and serum. Feasibility studies were conducted to assess the utility of this marker in small volumes of stored maternal serum. It was determined that the second trimester serum was likely to be the best indicator of prenatal Pb2+ exposure, as both Pb2+ and corresponding δ-ALA levels are believed to be relatively stable at mid-pregnancy. A single aliquot of second trimester samples was made available for each subject and a cutoff value (15 μg/dL) was used to categorize the samples by Pb2+ exposure. Samples were coded and blinded with respect to case status. Using this approach, Pb2+ exposure as measured by elevated δ-ALA was associated with about a two-fold increase in risk of schizophrenia spectrum disorders in these studies (OR=2.3, 95% CI: 1.0-4.3; p=0.05). Because these studies used a biological marker of exposure, serum δ-ALA, rather than direct measure of Pb2+, an increase in risk in schizophrenia cannot be directly ascribed to Pb2+ exposure. Nevertheless, these findings illustrate how prenatal cohorts with archived specimens can take a leap forward in terms of defining and ascertaining exposure status and timing. At the very least, they allow researchers to say with more certainty than before that certain classes of exposure (e.g. infectious agents and/or environmental toxins) are risk factors that merit more detailed investigations. Although the studies by Opler and colleagues (2004; 2008) have certain limitations, it brings to light the possibility that prenatal Pb2+ exposure may be a putative risk factor for the expression of schizophrenia later in life. This potentially important connection between schizophrenia and exposure to a known developmental neurotoxicant should be re-examined using a larger cohort of subjects in which Pb2+ concentrations are directly measured in biological samples. Nevertheless, the potential link of expressing a schizophrenia phenotype as a result of prenatal exposure to a known developmental neurotoxicant that has been present in the global environment since antiquity deserves closer examination. Although impossible to do in a single document, the goal of this review is to examine the current understanding of the behavioral, anatomical, biochemical and neuropathological endpoints in schizophrenia and those resulting from developmental Pb2+ exposure. Despite the fact that research in these two arenas have occurred independently, there is a great deal of overlap in behavioral outcomes and neurochemical systems affected.
Behavioral manifestations in Schizophrenia and early life Pb2+ exposure
As previously noted, the clinical diagnosis of schizophrenia is based on behavioral observations and self-reported abnormal mental experiences (Pearlson, 2000). Symptoms of schizophrenia are divided into “positive” and “negative” types (Kay and Opler, 1987). Positive symptoms are usually defined as the presence of intrusive or abnormal neurobehavioral phenomenon, such as delusions (presence of false beliefs), hallucinations (including visual, auditory, olfactory, tactile, and gustatory modalities), formal thought disorder and unusual motoric and social behaviors. Negative symptoms are conceptualized as the absence of normal functions, including blunted affect, social withdrawal, apathy, poor initiative and motivation, difficulty in planning, impaired problem solving and abstract reasoning. Impairments in cognitive functions include those related to attention, executive functions and working memory. Keeping these symptoms in mind, there is evidence that some of these same behavioral and cognitive alterations have been described as a result of early life Pb2+ intoxication.
The effects of childhood Pb2+ intoxication on the central nervous system have been documented since the early part of the 20th century. In the classic paper by Byers and Lord (1943) it was recognized that Pb2+ intoxication in children produces mental retardation with cognitive impairments and maladaptive behaviors. In this classic paper, a series of 20 children exposed to Pb2+ from infancy (although it is highly likely that in utero exposure also occurred) were described as expressing “intellectual difficulties and sensorimotor deficits”. Behavioral difficulties were common in all children including “unreliable impulsive behavior”, “cruel impulsive behavior” and “short attention span”. It is interesting to quote some of the descriptions given for the Pb2+-intoxicated children: “It made several of the children friendless and difficult at home and in school. Three were excluded from school based on behavior, one for setting fires in the school, another for repeatedly getting up and dancing on the desks and other furniture and the third for sticking a fork into another's child face”. Another example provided was about a girl at 11 years of age that upon medical examination was described as “sullen, withdrawn and insecure”. A previous examination of the same girl at age of 5 did not find any signs of psychological difficulties. These observations suggest a developmental trajectory for the expression of the psychological symptoms. Based on this limited information it is difficult to classify any of these children as schizophrenics but clearly some of these behaviors can be categorized within the range of “positive” or “negative” symptoms of schizophrenia. Other cardinal features of schizophrenia were also described such as a child having “enlarged ventricles upon pneumoencephalographic examination at 12 years of age”.
The more recent human literature at Pb2+ exposures levels lower that those during the times of Byers and Lord (1943) confirms and extends the effects of childhood Pb2+ intoxication on behavioral and cognitive outcomes. Epidemilogical studies clearly show that besides the well-documented effects of Pb2+ on IQ (Bellinger et al 1991; Needleman et al 1979; Wasserman et al., 2000; Canfield et al 2003), children exposed to Pb2+ also perform poorly on tests of working memory, attentional flexibility, and planning and problem solving (Canfield et al., 2004). As these authors point out, the effects of early life Pb2+ exposure is not restricted to global indexes of general intellectual functioning, but executive processes may be at particular risk in Pb2+ intoxicated children (Canfield et al 2004). School children with elevated blood Pb2+ levels are also more likely to display antisocial behavior (Bellinger et al., 1994; Needleman et al., 1996; Roy et al., 2009) and perform poorly in end of grade examination (Miranda et al, 2007). These behavioral and cognitive problems are persistent even after cessation of Pb2+ exposure in early life (White et al., 2007). Prenatal and/or childhood Pb2+ exposure has also been associated with delinquent behavior at adolescense (Dietrich et al., 2001). They resemble endophenotypes described in schizophrenia in which poor performance on attentional, working memory, and executive functioning and increased incidence of violent behavior are observed (Pearlson, 2000; Ross et al., 2006).
Neuroimaging Studies of Brain Volume and Chemistry
Major advances in neuroimaging technologies such as Magnetic Resonance Imaging (MRI), Magnetic Resonance Spectroscopy (MRS) and Diffusion Tensor Imaging (DTI) during the last two decades have provided important clinical tools to probe the anatomical and biochemical bases of neurological and mental disease. Studies performed with these techniques in schizophrenia patients have provided valuable information that previously was only possible from post-mortem studies. The emerging evidence is that a common finding in schizophrenia patients is enlargement of the ventricles and decreased cerebral cortex volume especially in frontal and temporal lobes (Woods et al., 1996; Harrison, 1999; Pearlson, 2000; Andreasen et al., 2011; Horga et al., 2011). Reductions in the volume of the hippocampus, basal ganglia, thalamus and amygdala have also been documented (Harrison 1999; Horga et al., 2011). The loss of neurons or neuronal elements such as axons and dendrites has been demonstrated in vivo in the brain of schizophrenia patients by the use of MRS in which reductions in the levels of N-acetylaspartate (NAA)/creatine (Cr) ratios have been measured. The loss of the NAA signal has been associated with frank neuronal loss or reductions in brain neuropil in a number of neurodegenerative disorders (Ross et al., 1997; Block et al., 2002; Stork and Renshaw, 2005). At a different level of analysis, post-mortem studies have revealed abnormalities in neuronal architecture in many of these brain regions with alterations in neuronal density and reductions in the size of neurons (Arnold and Trajanowski, 1996; Cannon, 1996). Importantly, it appears that these changes in neuronal mass and size are present in the absence of gliosis suggesting that schizophrenia may not be due to an active neurodegenerative process but rather the result of an earlier developmental insult (Harrison, 1997; 1999; Schnieder and Dwork, 2011).
Relative to schizophrenia, there is a paucity of data on the neuroanatomical and brain chemistry changes in humans resulting from prenatal or perinatal Pb2+ exposure despite the fact that a high percentage of children and adults have been and continue to be exposed to this neurotoxicant in the United States and throughout the world. This is largely due to the fact that most human studies on the effects of Pb2+ exposure have been epidemiological in nature with little or no use of state-of-the-art neuroimaging techniques to examine brain structure and function. However, in the last decade, there are a growing number of research groups that are begining to utilize neuroimaging techniques in their studies and are providing valuable information on the effects of early life Pb2+ exposure and alterations in brain volume and chemistry.
The first literature reports were in small groups of Pb2+-exposed children (Trope et al., 1998; 2001) and one report on adult monozygotic twins with Pb2+ poisoning (Weisskopf et al., 2004). A more extensive series of studies in a larger cohort of Pb2+-exposed children has been reported by the Cincinnati group (Cecil et al., 2008; Brubaker et al., 2009; 2010). The two reports by Trope and colleagues in children used MRS to examine regional brain metabolite levels such as N-acetylaspartate (NAA), choline (Cho), myoInositol (mI) and creatine (Cr). In the first report, a 10 years old child with documented blood Pb2+ levels of 51 μg/dL at 38 months and 44 μg/dL at 41 months was compared to a 9 year-old cousin. Both children lived in the same household and shared the same socioeconomic background and home environment but one was exposed to Pb2+ during stays at his grandmother's house. The Pb2+-exposed child exhibited significant performance deficits in a number of neuropsychological and cognitive tests while his cousin's performance was in the normal range. The MRS results indicated a significantly lower NAA/Cr ratio in grey matter in the frontal cortex of the Pb2+-exposed child relative to his normal cousin. There were no apparent differences in the Cho/Cr ratio, but interestingly there was a significant decrease in the mI/Cr ratio in the Pb2+-exposed child. This latter observation is important because the mI peak consists of 70% mI, but 15% of the peak signal is also contributed by glycine an amino acid that is decreased in schizophrenia patients (Sumiyoshi et al., 2004). The MRI studies did not find any apparent structural abnormalities. In summary, this study indicates the possibility of neuronal loss or dysfunction in the absence of gliosis in the frontal cortex of a 10 year-old Pb2+-exposed child with decline in intellectual functioning.
In a second larger study of Pb2+-exposed children with blood Pb2+ levels in the 23-65 μg/dL range, the same group of investigators were able to confirm the Pb2+-induced decrease in NAA/Cr ratio in the grey matter relative to non-exposed controls (Trope et al., 2001). Thus, from these studies it appears that one consequence of early life Pb2+ exposure is the possible loss of neurons or neuronal elements such as axons and dendrites in the brain. The decrease in the NAA signal in the brain of Pb2+-exposed children is consistent with what is observed in the brain of schizophrenia subjects (Deicken et al., 1997; Bertolino et al., 1998). Although no apparent structural abnormalities were present in the brain of these Pb2+-exposed children, it may have been too early in the developmental process for measurable changes to occur in brain size and it is possible that at a latter age structural abnormalities may be apparent [See studies below].
Weisskopf and colleagues (2004) have presented a case report in which seventy-one year old monozygotic twins with Pb2+ poisoning were examined with MRI and MRS. Both brothers had elevated blood and bone Pb2+ concentrations relative to the general population of the same age and one brother had much higher levels than his sibling. Neurocognitive tests indicated that working memory/executive function were below expectations and the brother with the higher Pb2+ burden performed dramatically worse in tests of short-term memory indicative of frontal lobe dysfunction. Hippocampal dysfunction was also worse in the brother with higher Pb2+ levels. Importantly, the brother with the lower scores in the neurocognitive test battery and with a higher Pb2+ burden had a lower NAA/Cr ratio in the hippocampus, frontal cortex and midbrain. This finding is consistent with the previous two studies in children indicating that neuronal loss or dysfunction (based on decreased NAA/Cr ratio) is a consequence of Pb2+ exposure. Thus, one known aspect that is similar to schizophrenia is the decrease in NAA/Cr ratio in the hippocampus and cerebral cortex.
The most comprehensive series of studies on the effects of childhood Pb2+ exposure in brain chemistry changes in young adults comes from the Cincinnati group headed by Cecil and colleagues. In their first study (Cecil et al., 2008) using a total of 157 participants from the Cincinnati Lead Study ranging in age from 19 to 24 years of age with average blood Pb2+ levels of 13.3 μg/dL, they found significant decreases in grey matter volume in brain structures associated with executive function, mood regulation and decision-making. These included the anterior cingulate cortex, postcentral gyrus, inferior parietal lobe, medial frontal gyrus, and paracentral gyrus. Interestingly, they find that the loss in brain volume was greater in males than females independent of sex-related differences in blood Pb2+ concentrations and other demographic factors. This study provides neuroanatomical evidence that childhood Pb2+ exposure results in the loss of brain volume in regions affected in schizophrenia patients. It shows decreased brain volume in young adults with previous childhood Pb2+ exposure.
In a subsequent study from the same group using the same cohort of Pb2+-exposed subjects, they performed diffusion tensor imaging (DTI) to examine white matter effects (Brubaker et al., 2009). They found significant and persistent effects on white matter microstructure with evidence of axonal injury and myelin damage. Finally, in the most recent study, they find inverse associations between gray matter volume loss and yearly mean blood Pb2+ measurements that are most pronounced in the frontal lobes of males than females (Brubacker et al., 2011). These series of studies provide compelling evidence of the widespread impact of childhood Pb2+ exposure on brain volume loss in young adults and in particular the frontal cortex and hippocampus.
A series of reports from Schwartz and colleagues have found highly significant effects of previous Pb2+ exposure and longitudinal declines in cognitive function and loss of brain volume in aging. These studies made used of two population groups: 1) former organo-lead manufacturing workers, and 2) 50-70 year old Baltimore residents with environmental Pb2+ exposure. They find that previous exposure to Pb2+ results in longitudinal declines in cognitive function (Schwartz et al., 2000; Shih et al., 2006; Stewart and Schwartz, 2007; Bandeen-Roche et al., 2009) independent of socio-economic status and despite the fact that exposure to Pb2+ had stopped. They also show that previous cumulative Pb2+ dose was associated with persistent brain lesions, in particular, higher tibia Pb2+ levels was negatively associated with smaller total brain volume, frontal and total gray matter volume and parietal white matter volume (Stewart et al., 2006). Together, the MRI/MRS/DTI neuroimaging studies strongly suggest an association of cumulative Pb2+ exposure earlier in life and the loss of brain volume in young adults and in aging and these structural changes have been associated with cognitive decline.
Post-mortem studies
There is a lack of information on the effects of Pb2+ exposure on the human brain from post-mortem studies. The limited number of human brain samples from children that expressed Pb2+ encephalopathy were perform more than 25 years ago and they lacked the use of current advances in neuroanatomical methods such as unbiased stereological counting of cell number and size. Nevertheless, examination of the literature indicates that upon neuropathological examination, a common feature of the Pb2+-exposed brain in children is the presence of small brain infarcts (Winder et al., 1983). A common pathological abnormality present in the brain of patients with schizophrenia, are small infarcts that may be suggestive of vascular impairments (Harrison, 1999). Analysis of the same human brain samples from the Pb2+-exposed children did indicate gliosis, a condition that it does not appear to be an active event in the brain of schizophrenia patients (Harrison, 1999; Schnieder and Dwork, 2011).
Most of the knowledge base on the effects of Pb2+ exposure on brain pathology is provided by studies on experimental animals. Based on the Pb2+ dose given and time of exposure, studies have found some of the same features expressed in the brain of schizophrenia patients. These include: 1) reduction in brain weight, 2) reduced forebrain weight, 3) reduced cortical thickness, 4) reduced neuronal size, 5) increased neuronal packing density, 6) decreased synapse per neuron and decreased dendritic spine density (Krigman et al., 1974; Petit and LeBoutillier, 1979; Winder et al., 1983). The hippocampus is a brain structure with documented abnormalities as a result of early life Pb2+ exposure. For example, a number of studies have documented the loss of dendritic arborization and reductions in dendritic spines in the hippocampus of Pb2+-exposed rats (Alfano and Petit, 1982; Campbell et al., 1982; Kiraly and Jones, 1982; Petit et al., 1983). Similar changes have also been documented in the brain of schizophrenia patients (Harrison, 1999; Pearlson, 2000).
A more recent observation related to the hippocampus is that Pb2+-exposed rats exhibit reductions in mossy fiber innervation to the CA3 region of the hippocampus (Verina et al., 2007), a finding that has been documented in the brain of schizophrenia subjects (Kolomeets et al., 2005). The mossy fiber pathway is essential for sensory gating and memory and learning.
Behavioral findings in Animal Models of Schizophrenia and Developmental Pb2+ Exposure
One of the difficulties in developing animal models of schizophrenia is the complexity of the symptoms. Therefore, animal models of schizophrenia have for the most part attempted to model specific aspects of the disorder and there are many reviews on the topic (Marcotte et al., 2001; Boksa 2004; Lipska 2004; Ayhan et al., 2009; Inta et al., 2010). Schizophrenia patients have deficits in attention and sensory information processing such as prepulse inhibition (PPI). Prepulse inhibition is a model of sensorimotor gating mechanism in the brain that has been used in animals to study basic neuronal mechanisms in schizophrenia research (Weiss and Feldon, 2001; Van den Buuse et al., 2003). Prepulse inhibition of the acoustic response is the normal suppression of a startle response to a strong acoustic stimulus when it is preceded by a weak sound stimulus or prepulse. Alterations in PPI are commonly found in neurodevelopmental models of schizophrenia (Weiss and Feldon, 2001; Van den Buuse et al., 2003) and in rodents that have been exposed to Pb2+ during development (Commissaris et al., 2000).
Another common behavioral outcome in animal models of schizophrenia is increased locomotion or “hyperactivity” in response to a low dose of amphetamine (Snyder, 1972, Boksa, 2004) or to a novel environment (Kvajo et al., 2011). The response of increased locomotion to a low dose of amphetamine is used in rodents as a behavioral read out of activity of the dopaminergic system. Importantly, one of the well-documented effects of developmental Pb2+ exposure is hyperactivity (Winneke et al., 1977; Petit and Alfano, 1979; Burdette and Goldstein, 1986; Moreira et al., 2001), an effect that is believe to be mediated via a hyperactive mesolimbic dopaminergic system (Zuch et al., 1998).
Alterations in other behaviors that have been measured in animal models believed to be relevant to schizophrenia are deficits in social interaction, working memory, and spatial learning (Boksa 2004; Kvajo et al., 2011). Many of these same behaviors are also affected in developing animals exposed to Pb2+ (Brockel and Cory-Slechta, 1998, 1999; Nihei et al., 2000; Moreira et al., 2001). For example, several laboratories including our own have extensively documented the effects of developmental Pb2+ exposure on spatial learning (Nihei et al., 2000; Guilarte et al., 2003), a behavioral task that is mediated by the hippocampus.
The “dopaminergic” hypothesis of schizophrenia
The original hypothesis underlying the neurochemical abnormalities in schizophrenia was based on the use of antipsychotics as the first effective treatment (Carlsson and Lindqvist, 1963). Subsequent studies showed that the therapeutic value of antipsychotic drugs was mediated by blocking D2-dopamine receptors (Seeman and Lee, 1975; Creese et al., 1976; Snyder, 1981). Experimental evidence suggests that dysregulation of dopamine function plays an important role in the expression of positive symptoms in schizophrenia (Snyder, 1972, 1981; Lieberman et al 1987). PET studies in schizophrenia subjects and animal models of schizophrenia have provided evidence of hyperactivity of the dopaminergic system, with an apparent selectivity to the mesolimbic and mesocortical pathways (Laurelle et al., 1996; Abi-Dargham et al., 1998; Harrison, 1999; Lindstram et al., 1999; Pearlson, 2000; Thaker and Carpenter, 2001). In a similar fashion, the dopaminergic system is also affected by Pb2+ neurotoxicity. Seminal studies by Cory-Slechta and colleagues have shown that exposure to Pb2+ in early life results in hyperactivity of the dopaminergic system (Zuch et al., 1998; Pokora et al., 1996; Cory-Slechta et al., 2002; Bauter et al., 2003).
An important study by Cory-Slechta and colleagues (1997) is related to the modulation of dopaminergic drugs on the effects of Pb2+ exposure on NMDAR levels. They showed that Pb2+ exposure started at weaning resulted in significant increases (short term exposure) and decreases (long-term exposures) on NMDAR levels in the frontal cortex, nucleus accumbens and dorsal striatum. These Pb2+-induced changes in NMDAR levels as measured by [3H]-MK-801 binding could be abrogated by the administration of the D2-dopamine receptor agonist apomorphine, but not by the D1-receptor agonist SKF-82958. These findings clearly implicate interplay between the glutamatergic and dopaminergic systems in the Pb2+-exposed brain similar to what has been found in schizophrenia (Simpsom et al., 2010). However, despite the years of research on the involvement of the dopaminergic system in the pathophysiology of schizophrenia, it has become clear that dysregulation of the dopaminergic system alone does not explain the negative symptoms and cognitive dysfunction in schizophrenia and alterations of the glutamatergic system has emerged as a viable hypothesis.
The “glutamatergic” hypothesis of schizophrenia
The glutamatergic hypothesis of schizophrenia originated from the observations that administration of N-methyl-D-aspartate receptor (NMDAR) non-competitive antagonists such as phencyclidine (PCP) or ketamine exacerbates psychotic symptoms in schizophrenia patients and produces schizophrenia symptoms in normal subjects (Javitt and Zukin, 1991; Lahti et al., 1995; Thaker and Carpenter, 2001; Coyle et al., 2003). Compared to dopaminergic agents, NMDAR antagonists induce negative and cognitive symptoms of schizophrenia as well as positive symptoms. Thus, convergent evidence has accumulated to support a primary role of glutamatergic NMDAR hypofunction in schizophrenia (Marek et al., 2010; Javitt, 2010). Consistent with this notion, clinical trials with the NMDAR co-agonists glycine, d-serine and glycine transporter inhibitors that enhance endogenous glycine levels have provided encouraging reports in the treatment of schizophrenia (Javitt et al., 1994; Huresco-Levy et al., 1999; Lin et al., 2011).
The NMDAR is an excitatory amino acid receptor subtype that is known to play an essential role in neuronal development, synaptic plasticity and in learning and memory (Guilarte, 1998). The administration of PCP or other NMDA receptor antagonists to experimental animals, models certain aspects of the disease (Kilts, 2001). Further, genetic manipulation of the NMDAR to down regulate NR1 and NR2A subunit expression (Mohn et al., 1999; Miyamoto et al., 2001) or its selective deletion in the frontal cortex of mice (Belforte et al., 2010) results in behavioral manifestations consistent with those in schizophrenia providing further support to the important role of NMDAR hypoactivity in the pathophysiology of schizophrenia. In humans, a microsatellite repeat in the promoter of the NR2A subunit gene that represses transcriptional activity correlates with chronic outcomes in schizophrenia (Itokawa et al., 2003) indicating the importance of the NR2A subunit of the NMDAR in schizophrenia.
From an etiological perspective, NMDAR antagonists such as PCP, ketamine and MK-801 are drugs of abuse or anesthetic agents that are used to model schizophrenia symptoms, and humans are unlikely to be exposed at a global scale, although ketamine is still used as a pediatric anesthetic agent. This raises the question, is it possible that a chemical or classes of chemicals that are NMDAR antagonist and are ubiquitous in the global environment can result in widespread human exposures and be a risk factor for schizophrenia? The heavy metal lead (Pb2+) fits all of these characteristics. That is, Pb2+ is not only a well-recognized ubiquitous, global environmental and developmental neurotoxicant, but it is also a potent NMDAR antagonist. Since the early 1990s, there is experimental evidence that Pb2+ is a potent and selective non-competitive antagonist of the NMDAR (Alkondon et al., 1990; Guilarte and Miceli, 1992) and disrupts neuronal processes that are mediated via NMDAR activation.
A substantial body of evidence has shown that exposure to Pb2+ during development, in the same concentration range as implied in the work by Opler et al., (2004; 2008), alters gene and protein expression of NMDAR subunits in the developing and young adult rat hippocampus (Table 1). The hippocampus is part of the limbic system involved in learning and memory and it is a principal brain region affected in schizophrenia (Tsai and Coyle, 2002; Konradi and Heckers, 2003) and in Pb2+ neurotoxicity (Krigman et al 1974; Petit and LeBoutillier, 1979; Winder et al 1983; Guilarte and McGlothan, 1998; Nihei et al 2000).
Table 1.
NMDAR subunit gene and protein expression in the brain of schizophrenia patients or in the brain of rats exposed to lead during development. DG= dentate gyrus; CA= cornus ammonis; Hipp= Hippocampus; s.v= splice variants; PN= postnatal day.
| Schizophrenia | Developmental Pb2+ Exposure | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| NMDAR Subunit Effect |
Brain Region | Reference | NMDAR Subunit Effect |
Age | Brain Region | Reference |
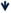 NR1 NR1 |
DG, Hipp | Gao et al., 2000 |
NR1 |
PN50 | CA, DG | Nihei et al., 2000 |
 NR2B NR2B |
CA2-CA3, Hipp |
NR2A |
PN50 | DG | ||
|
NR2B |
PN50 | CA3 | ||||
|
NR2B |
Thalamus | Clinton et al., 2006 |
NR1-4 s.v. |
PN50 | DG | |
|
NR1 |
superior temporal cortex – cognitive impairment with no cognitive impairment | Humphries et al., 1996 |
NR1 |
PN50 | CA, DG | Guilarte & McGlothan, 2003 |
 NR1 NR1 |
NR1-2 s.v. |
PN50 | CA, DG | |||
|
NR1-4 s.v. |
PN50 | CA, DG | ||||
|
NR1-b s.v. |
PN50 | CA, DG | ||||
|
NR1 |
dentate gyrus | Law & Deakin, 2001 |
NR1 |
PN50 | CA, DG | Guilarte et al., 2003 |
|
NR1 |
Superior frontal cortex | Sokolov, 1998 |
NR1 |
PN14 | CA1 | Guilarte & McGlothan, 1998 |
|
NR2A |
PN14 & 21 | CA1, DG | ||||
|
NR2B |
PN14 & 21 | -- | ||||
|
NR2A |
anterior cingulate cortex | Woo et al., 2004 | ||||
|
NR1 |
superior temporal gyrus | Le Corre et al., 2000 |
NR2A (protein) |
PN14, 21, 28 | Hipp | Nihei & Guilarte, 1999 |
|
NR1 |
dorsolateral prefrontal cortex/occipital cortex | Drachera et al., 2001 |
NR1 |
PN15, 20 | CA, DG | Zhang et al., 2002 |
|
NR2A |
PN15, 20 | CA, DG | ||||
|
NR2D |
PN20 | CA, DG | ||||
|
NR1-4 s.v. |
Hipp | Vrajova et al., 2010 (PCR) |
NR2B |
PN20 | CA, DG | |
|
NR1-2 s.v. |
||||||
|
NR2D |
prefrontal cortex | Akbarian et al., 1996 |
NR1-a s.v. |
PN14 | CA | Guilarte et al., 2000 |
|
NR1-a s.v. |
PN14, 21 | DG | ||||
|
NR1-b s.v. |
PN21 | CA | ||||
|
NR1-2 s.v. |
PN21 | CA4, DG | ||||
|
NR1-4 s.v. |
PN14 | CA3 | ||||
|
NR1-4 s.v. |
PN21 | CA3 | ||||
A consistent change in NMDAR subunits expression observed in Pb2+-exposed animals is alterations in the expression of the NR1 and NR2A subunits (See Tables 1 and 2). Guilarte and McGlothan (1998) have shown that developmental Pb2+ exposure increases the gene expression of the NR1 subunit but decreases NR2A subunit expression with no change in NR2B subunit in the hippocampus during early postnatal life. A subsequent study showed that Pb2+ exposure decreased both NR1 and NR2A subunit gene expression in several hippocampal regions in post adolescent rats (Nihei et al., 2000). Further, NR2B subunit expression was either not changed or slightly increased, but only in the CA3 region of the hippocampus (Nihei et al., 2000). Thus, there are age-dependent changes in the expression of the NR1, NR2A and NR2B subunit genes as a result of developmental Pb2+ exposure (Toscano and Guilarte, 2005). These findings resemble some of the changes in NMDAR subunit expression described in the brain of schizophrenia patients (Tsai and Coyle, 2002; Konradi and Heckers, 2003; Meador-Woodruff et al., 2003; Nudmamud-Thanoi and Reynolds, 2004; See Tables 1 and 2) and in the frontal cortex of rodents exposed to a sub-chronic administration of the NMDAR antagonist MK-801 (Xi et al., 2009). In the latter study they show that NMDAR subunit expression is dynamically regulated based on MK-801 dose. Together these studies suggest that based on the dose, timing of dose and chronicity of NMDAR antagonist dosing, NMDAR subunit expression are likely to be up or down regulated differentially.
Table 2.
Quantitative receptor autoradiography or radioligand receptor binding studies of NMDAR levels in the brain of schizophrenia patients or in the brain of rats exposed to lead during development. DG= dentate gyrus; CA= cornus ammonis; Hipp= Hippocampus; PN= postnatal day; EC= entorhinal cortex. (*) lead exposure started post-weaning.
| Schizophrenia | Developmental Pb2+ Exposure | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| Radioligand | Brain Region | Reference | Radioligand | Age | Brain Region | Reference |
|
3H-ifenprodil (NR1/NR2B) |
Superior temporal cortex | Grinwood et al., 1999 |
3H-MK801 No change if Pb started as adult |
PN14 | forebrain | Guilarte & Miceli, 1992 |
|
3H-MK801/3H-TCP |
cerebral cortex | Komhuber et al., 1989 |
3H-MK801 |
PN70-120 | cerebral cortex | Guilarte et al., 1993 |
|
3H-MK801/3H-TCP |
orbital frontal cortex | Simpson et al., 1992 |
3H-MK801 |
PN56-112 | occipital cortex entorhinal cortex temporal cortex | Ma et al., 1997 |
|
3H-TCP |
CA3-Hipp | Dean et al., 1999 | ||||
|
3H-MK801 |
Hipp | Beneyto et al., 2007 |
3H-Ifenprodil (NR1/NR2B) |
PN50 | cerebral cortex | Toscano et al., 2002 |
|
3H-CGP39653
|
adults | frontal cortex | Cory-Slechta et al., 1997a | |||
|
3H-TCP |
PN25 | CA3-Hipp | Brooks et al., 1993 | |||
|
3H-MK801 |
PN56-112 | DG CA1, CA2-Hipp | Ma et al., 1997 | |||
|
3H-MK801 |
PN14 | Hipp, EC | Jett & Guilarte, 1995 | |||
|
3H-ifenprodil |
PN50 | Hipp | Toscano et al., 2002 | |||
|
3H-MK801 |
adults | DG, CA1-Hipp | Cory-Slechta et al., 1997a | |||
Pb exposure started post-weaning (PN21)
It should be noted that a number of studies have shown that Pb2+ exposure decreases NR2A subunit gene and protein expression (Tables 1 and 2) and this effect results in a greater proportion of NR2B-NMDAR complexes. This is a relevant observation in the Pb2+ exposure/schizophrenia hypoglutamatergic hypothesis because NR2A-containing NMDAR complexes are responsible for the maintenance of paravalbumin (PV) and glutamic acid decarboxylase 67 (GAD67) positive interneurons (Kinney et al., 2006) and there is a selective decrease in these cortical GABAergic interneurons co-expressing the NR2A subunit (Woo et al., 2004) in the brain from schizophrenia patients. Further, Kocsis (2011) has demonstrated that the increase in aberrant gamma oscillations in the cerebral cortex that are associated with schizophrenia related behavioral and prepulse inhibition abnormalities was due to antagonist of NR2A-containing NMDAR complexes but not those containing the NR2B, NR2C, or NR2D subunits.
NR1 subunit splice variants mRNA expression in the brain of schizophrenia subjects showed a decrease in NR1 variants containing the C2 cassette (Clinton et al., 2003; Meador-Woodruff et al., 2003), a finding that is also observed in the brain of Pb2+-exposed rats (Guilarte et al., 2000; Guilarte and McGlothan, 2003). These Pb2+-induced changes in NMDAR subunit expression result in NMDAR complexes with different subunit composition (Toscano et al., 2002), synaptic expression (Guilarte and McGlothan, 2003) and alterations in calcium signaling downstream from the NMDA receptor (Toscano et al., 2002).
Examination of studies using tritiated-ligand binding to the NMDAR indicates similarities between the brain from schizophrenia subjects and brain tissue from Pb2+-exposed rats. For example, using the NR1/NR2B specific ligand [3H]-ifenprodil, there is increase levels of [3H]-ifenprodil binding in the cerebral cortex of both schizophrenia subjects and adult rats exposed to Pb2+ during development (See Table 2). Similarly, there is increased [3H]-MK-801 binding in the cerebral cortex of both schizophrenia subjects and adult rats exposed to Pb2+ during development (Table 2). These findings suggest that at least in the cerebral cortex binding studies provide evidence of an increase in the expression of NR1/NR2B-NMDAR receptors suggesting that out of the total pool of NMDAR, there is a greater proportion of receptors that express the NR2B subunit and a lower proportion of those containing the NR2A subunit.
Recent studies have also implicated NMDAR-dependent dysregulation of BDNF signaling and protein levels in the detrimental effects of Pb2+ on synaptic function (Neal et al., 2010; Neal and Guilarte, 2010). BDNF is a neurotrophin that is altered in the brain of schizophrenia patients (Weickert et al., 2003; Rizos et al., 2011; Buckley, 2011). A recent study has shown that BDNF exon IV mutant mice exhibit significant deficits in PV-positive GABAergic interneurons in the prefrontal cortex, an interneuron subtype that has been implicated in working memory/executive function impairment in schizophrenia (Sakata et al., 2009). These mice express impaired inhibitory transmission and abnormal appearance of spike-timing-dependent synaptic potentiation (Sakata et al., 2009). BDNF exon IV transcripts are decreased in the cerebral cortex of patients with schizophrenia with no history of anti-depressant treatment (Wong et al., 2010). This is particularly relevant to our work with Pb2+ exposure because we have recently shown a specific decrease of BDNF exon IV transcripts in neuronal cultures during synapse formation (Stansfield et al., 2011).
Consistent with the alterations in NMDAR function, several laboratories including our own have shown that developmental Pb2+ exposure produces deficits in NMDAR-dependent long-term potentiation in the hippocampus and impairments in spatial learning in young adult rats (Nihei et al., 2000; Nihei and Guilarte, 2001). Long-term potentiation is thought to represent a cellular correlate of learning and memory in the mammalian brain that is NMDAR dependent. Thus, young adult rats exposed to environmentally relevant levels of Pb2+ during development express some of the same neurobiological and behavioral deficits seen in animal models of schizophrenia (Tsai and Coyle, 2002; Konradi and Heckers, 2003). The common mechanism that links both conditions is dysregulation of the glutamatergic system and specifically hypoactivity of the NMDAR complex.
The glycine site of the NMDAR complex: a common molecular target?
Research in the molecular mechanisms associated with developmental Pb2+ exposure and schizophrenia have been performed independently, however, there is compelling evidence for a common molecular target, the glycine modulatory site of the NMDAR. The NMDAR is activated by the co-agonists glutamate and glycine in coincidence with depolarization of the neuronal membrane. This results in the removal of the magnesium block at the NMDAR channel and allows calcium entry into the cell. A proposed mechanism by which Pb2+ inhibits NMDAR function is by binding to a divalent cation site associated with the glycine site (Hashemzadeh-Gargari and Guilarte, 1999). The “antagonistic” effect of Pb2+ at the glycine site of the NMDA receptor is most effective at sub-saturating concentrations of glycine (Hashemzadeh-Gargari and Guilarte, 1999). This molecular interaction of Pb2+ at the glycine site is physiologically relevant since the glycine site of the NMDAR is not saturated under physiological conditions (Berger et al., 1998). Similar to the inhibitory effect of Pb2+ at the glycine site of the NMDA receptor, “negative” modulation of the glycine regulatory site of the NMDAR by putative endogenous antagonists has been proposed as a principal feature in the pathophysiology of schizophrenia (Coyle et al., 2003; Coyle and Tsai, 2003; 2004).
The significance of the antagonistic action of Pb2+ at the glycine site of the NMDAR to schizophrenia is that a potentially effective therapy that helps to ameliorate the negative symptoms and cognitive disability in schizophrenics is the activation of this site (Coyle and Tsai, 2003). The use of NMDAR glycine site agonists such as glycine, D-serine or D-cycloserine in clinical trials has demonstrated some degree of efficacy in ameliorating the negative symptoms and cognitive disabilities in schizophrenia subjects (Tsai and Coyle, 2002; Coyle and Tsai, 2004). Consistent with an important role of the glycine site of the NMDAR with the pathophysiology of schizophrenia, a recent animal model expressing an NMDAR complex with reduced glycine affinity produces some of the negative and cognitive symptoms of schizophrenia (Labrie et al., 2008). This and other evidence support the hypothesis that NMDAR hypofunction in schizophrenia is part of a complex pathophysiological network of neuronal systems that are affected in schizophrenia. For example, several schizophrenia risk genes such as DISC1, neuregulin and dysbindin interact with NMDAR to affect synaptic transmission, neuronal maturation and plasticity (Hayashi-Takagi et al., 2010; Geddes et al., 2011; Ramsey et al., 2011).
Another potential effect of Pb2+ exposure on the glycine regulatory site of the NMDAR is by modulating D-serine levels released from glial cells. Recent studies have shown that D-serine is synthesized in astrocytes and it is released to control NMDAR activity by being a co-agonist at the glycine site (Panatier et al., 2006; Oliet and Mothet, 2009). Since Pb2+ is actively taken up by astrocytes (Tiffany-Castiglion and Qian, 2001), it could potentially alter D-serine synthesis and release. Consistent with this hypothesis, Sun et al (2007) have shown that the addition of D-serine to hippocampal slices was able to reverse the Pb2+-induced impairment in CA1 long-term potentiation produced by in vivo Pb2+ exposure.
Testable hypothesis
Does exposure to Pb2+ alter D-serine synthetic and metabolic enzymes in astrocytes and/or D-serine release to modulate NMDAR activity. If dysregulation of D-serine metabolism or release are operational in Pb2+ exposed animals, then D-serine regulation of NMDAR complexes can be altered implicating another pathway for dysregulation of NMDAR activity by Pb2+.
NMDAR Antagonist-Induced Apoptosis in Early Life: A plausible mechanism for the loss of Brain Volume in Schizophrenia
It has been noted that schizophrenia is a neurodevelopmental disorder in which an early life event results in the loss of neurons without apparent glial cell activation suggestive of apoptotic cell death (Benes, 2004; Jarskog et al., 2004; 2005). It is possible that an early life event that causes apoptotic cell death could potentially explain the reductions in brain volume in schizophrenia in the absence of gliosis. Neuropathological examination of the brain from schizophrenia subjects has shown a relative lack of widespread neuron loss (Jarskog et al., 2004; 2005). However, other studies have shown selective reductions of neurons in discrete cortical layers and in other brain regions (Jarskog et al., 2004; 2005). Studies in rodents have shown that in the neonatal brain during a very specific window of vulnerability, the administration of NMDAR antagonists such as phencyclidine (PCP), ketamine, and MK-801 triggers apoptosis in multiple brain regions (Ikonomidou et al., 1999; Anastasio et al., 2009; Soriano et al., 2010) causing long-term behavioral deficits (Fredriksson and Archer, 2004; Yuede et al, 2010). Some of these same drugs have also been shown to produce similar apoptotic cell death in the developing primate brain (Slikker et al., 2007; Zou et al., 2009) and long-lasting cognitive deficits (Paule et al., 2011). Thus, they are likely to be relevant to the human condition. These drugs are the same types of NMDAR antagonists (PCP, ketamine, MK-801) that have been used to mimic schizophrenia symptoms in normal subjects and exacerbate symptoms in schizophrenia subjects that led to the glutamatergic hypothesis of schizophrenia. Now, a report by Dribben and colleagues (2011) shows that exposure of the neonatal mouse brain to Pb2+ during this same period of vulnerability reproduces the same pattern of apoptotic cell death as the other NMDAR antagonists noted above. This is an important finding because: 1) it provides additional support that in vivo, Pb2+ behaves as a potent NMDAR antagonist, and 2) it shows that Pb2+ exposure in early life enhances apoptotic cell death and this effect may help explain the loss in brain volume documented in Pb2+-exposed children later in life (Cecil et al., 2008). The brain regions affected by NMDAR antagonist (including Pb2+)-induced apoptosis in early life are some of the same brain regions in which neuronal cell loss has been documented in selected cortical layers in the schizophrenia brain (Jarskog et al., 2004). This includes layer II of the frontal cortex and layers II and IV of the cingulate cortex as well as striatum and hippocampal pyramidal cell layer (Ikonomidou et al., 1999; Jarskog et al., 2004; Dribben et al 2011). Consistent with the possibility of apoptotic cell death being an important part of the pathology of schizophrenia and as a result of early life Pb2+ exposure, the ratio of the pro-apoptotic Bax to the anti-apoptotic Bcl-2 protein has been shown to be increased in the brain of schizophrenia subjects (Jarskog et al., 2004; 2005), a phenomenon that has also been described in the Pb2+-exposed brain (Sharifi et al, 2002; 2010) and in PC12 cells exposed to Pb2+ (Xu et al., 2006). Other studies have shown that the ratio of Bax to Bcl-XL (a member of the anti-apoptotic Bcl-2 gene family) is increased in the neonatal rat cortex following NMDAR antagonist administration (Wang et al., 2001). These observations have important implications for the glutamatergic hypothesis of schizophrenia and suggest that enhancement of NMDAR antagonist-induced apoptotic cell death greater than it occurs naturally during brain development may be operational in schizophrenia. It could potentially explain the brain volume loss documented in schizophrenia and as a result of early life Pb2+ exposure. Alternatively, apoptosis has also been noted to occur in synaptic terminals in the absence of effects at the level of the soma (Mattson et al., 1998; Gliman and Mattson, 2002; Glantz et al., 2006). Such an event occurring in early life could result in the loss of brain neuropil and contribute to the loss of brain volume observed in schizophrenia and as a result of early life Pb2+ exposure.
Testable Hypothesis
An intriguing question is the possibility that mutations in schizophrenia susceptibility genes may impart an increased sensitivity of the neonatal brain to NMDAR-antagonist induced cellular or synaptic apoptosis. This is a testable hypothesis that could provide novel information about gene-environment interactions in the pathophysiology of schizophrenia.
The GABAergic hypothesis of Schizophrenia
Experimental and pathological evidence indicates that besides dysregulation of the dopaminergic and glutamatergic neuronal systems, other neurotransmitter systems are also affected in schizophrenia. Most prominently amongst these is the inhibitory neurotransmitter gamma-aminobutyric acid (GABA). Postmortem brain samples from schizophrenia patients suggest that dysfunction of cortical GABAergic interneurons, in particular those containing the calcium-binding protein parvalbumin (PV), may be a core feature of the working memory deficits in schizophrenia (Lewis et al., 2005; Uhlhaas and Singer, 2010). This is supported by evidence from rodent and non-human primate studies that administration of NMDAR antagonists decreases GAD67 and PV expression in cortical GABAergic interneurons (Cochran et al., 2003; Behrens et al., 2007; Morrow et al., 2007). More specifically the activity of NR2A subunit-containing NMDAR have been shown to have an important role in the maintenance of PV and GAD67 protein levels in cultured interneurons (Kinney et al., 2006). This is consistent with the selective loss of cortical GABAergic PV+ interneurons coexpressing the NR2A subunit in the schizophrenia brain (Woo et al., 2004). The GABAergic-PV+ interneurons in the cerebral cortex are also important for oscillation synchronization of pyramidal cells (Sohal et al., 2009).
Studies by Belforte and colleagues (2010) demonstrate that early postnatal ablation of the NR1 subunit of the NMDAR in corticolimbic interneurons produces the emergence of an schizophrenia phenotype and reductions in GAD67 and PV levels that was specific for cortical interneurons with deletion of the NR1 subunit. On the other hand, deletion of the NR1 subunit in corticolimbic neurons in postadolescence did not produce such abnormalities providing strong evidence for a neurodevelopmental basis of schizophrenia. These studies link GABAergic effects of NMDAR antagonists to pathological changes observed in schizophrenia.
Relevant to the potential schizophrenia-early life Pb2+ exposure hypothesis, our laboratory has previously shown that Pb2+ decreases NR2A-containing NMDAR (Tables 1 and 2). This suggests that PV and GAD67 levels should be affected in the brain of Pb2+-exposed animals since NR2A-containing NMDAR complex seems to regulate PV and GAD67 expression levels.
While an understanding of the effect of Pb2+ on GABAergic interneurons is much more limited, there is evidence that Pb2+ exposure decreases GAD protein levels and GABA release from rat brain (Lasley et al., 1999; Struzynska and Sulkowski, 2004). Electrophysiological studies by Albuquerque and colleagues have documented an inhibitory effect of Pb2+ on GABAergic neurotransmission in the hippocampus (Braga et al., 1999). This group of investigators also showed that Pb2+ increases spontaneous release of GABA from hippocampal neurons (Braga et al., 1999a). Decreased levels of brain GABA have been documented in Pb2+ intoxicated animals (Winder and Kitchen, 1984).
Testable hypothesis
If developmental Pb2+ exposure is an environmental risk factor for schizophrenia, then it should be able to reproduce the dysfunction of cortical GABergic interneurons expressing GAD67 and parvalbumin. This is a testable hypothesis that can be assessed by examining the developmental trajectory of parvalbumin and GAD67 expression in cortical GABAergic neurons in Pb2+ exposed animals.
Other neurotransmitter systems: Nicotinic Cholinergic Receptors
Emerging evidence suggests involvement of nicotinic cholinergic receptors in schizophrenia. Homomeric nicotinic cholinergic receptors composed of the α7 subunit and those with a α4β2 composition are the most abundantly expressed in the brain and play an important role in memory processes (Ripoll et al., 2004; Timofeeva and Levin, 2011). Recent studies have implicated a linkage between sensory gating defect and a locus of chromosome 15q14. This locus contains the gene encoding a α7 nicotinic cholinergic receptor subunit that is involved in sensory gating (Thaker and Carpenter, 2001). Studies have documented decreased levels of α7 nicotinic cholinergic receptors in thalamus, hippocampus and cerebral cortex in schizophrenic patients (Ripoll et al., 2004). Further, alterations in α4β2 nicotinic receptors have also been measured. In this instance, decreased levels have been documented in the hippocampus and striatum with increased levels in frontal and cingulate cortex (Ripoll et al., 2004). Therefore, it appears that alterations in nicotinic cholinergic receptors may play an important role in the pathophysiology of schizophrenia.
Studies have also shown that Pb2+ is a potent inhibitor of nicotinic cholinergic receptors in dissociated neurons from rat hippocampus (Ishihara et al., 1995). The inhibitory action of Pb2+ was dependent upon the subunit composition with α7 nicotinic receptors being the most sensitive (Ishihara et al 1995; Mike et al., 2000). Further, a study by Jett et al (2002) has shown that exposure to Pb2+ during brain development increases the levels of nicotinic cholinergic receptors that are labeled by [3H]-epibatidine. [3H]-epibatidine labels α4β2 nicotinic cholinergic receptors with some degree of overlap with other non-α7 receptors. In this study, α7 receptors were not measured, however, the increased levels of receptors labeled by [3H]-epibatidine in the cerebral cortex and thalamus of Pb2+-exposed rats suggests increased levels of α4β2 nicotinic cholinergic receptors in these brain structures. This is similar to the increased levels of α4β2 nicotinic cholinergic receptors in the cerebral cortex measured in schizophrenic patients (Ripoll et al., 2004).
Thus, consistent with the involvement of multiple neuronal systems in the pathophysiology of schizophrenia, Pb2+ exposure in early life also alters some of the same neuronal systems.
Disrupted in Schizophrenia 1 (DISC1) and early life Pb2+ exposure: A proof of concept gene-environment interaction study
There is evidence that understanding schizophrenia is likely to come from gene-environment interaction studies (Moffitt et al., 2005; Van Os et al., 2008; Brandon and Sawa, 2011). Schizophrenia is likely to be the result of complex interactions between many genes and multiple environmental factors that on their own they may make a small contribution but together contribute to an increased risk of schizophrenia. The main obstacle for mechanistic studies of gene-environment interplay has been the paucity of appropriate models to pinpoint the molecular mechanisms that mediate gene-environment interactions relevant to schizophrenia. Recent advances in psychiatric genetics and a plethora of experimental data from animal studies led us (TRG and MP) to suggest a new approach to gene-environment interactions in schizophrenia. We propose that in vivo and in vitro models based on genetic mutations with functional effects and measurable relevant environment factors will significantly advance studies of the molecular underpinnings of gene-environment interplay (Ayhan et al., 2009; Abazyan et al., 2010). Thus, we have initiated studies examining a potential interaction of early life Pb2+ exposure and mutant Disrupted in Schizophrenia 1 (DISC1).
Mutant DISC1 is the hypothetical protein product of a balanced chromosomal translocation [t(1;11)] in a Scottish pedigree with high load of major mental disorders (LOD=7.1 for all mental disorders and 3.6 for schizophrenia) [Millar et al., 2000a; Millar et al., 2001; Brandon and Sawa, 2011]. The breakpoint is in the middle of the open reading frame for the DISC1 gene, leading to truncation of the gene. Although expression of truncated proteins is theoretically possible, expression of truncated protein has not been demonstrated [Millar et al., 2000b]. The existence of a clear, identifiable mutation with the high LOD scores has put DISC1 in a unique position in schizophrenia research. In addition to the familial mutation of DISC1, multiple studies of associations of different DISC1 haplotypes or SNPs with mental disorders have stimulated studying the biology of DISC1 (Porteous et al., 2006; Ross et al., 2006; Mackie et al., 2007). Numerous investigations have implicated DISC1 and interacting proteins in neuronal differentiation, migration, synaptogenesis and adult neurogenesis in the hippocampus [Kamiya et al., 2005; Duan et al., 2007; Brandon, 2007; Faulkner et al., 2008; Brandon et al., 2009]. Recently generated DISC1 mouse models have advanced our understanding of the putative mechanisms whereby this protein and its interacting partners may be involved in abnormal neurodevelopment relevant to schizophrenia (Derosse et al., 2006; Koike et al., 2006; Li et al., 2007; Clapcote et al., 2007; Kvajo et al., 2008; Pletnikov et al., 2008; Shen et al., 2008; Brandon and Sawa, 2011).
We have generated a mouse model of inducible expression of mutant human DISC1 in forebrain neurons using the Tet-off system [Pletnikov et al., 2008]. In this model, expression of mutant DISC1 is regulated by the CAMK-II promoter and can be turned off by adding tetracycline or a related compound, doxycycline, to food or water. Similar to other DISC1 mouse models, expression of mutant DISC1 produced no gross developmental defects but significantly increased spontaneous locomotor activity in male but not female mice, decreased social interaction in male mice, enhanced their aggressive behavior and was associated with poorer spatial memory in Morris water maze task in female mice. The behavioral alterations have been accompanied by the enlargement of lateral ventricles in adult mice, reduced dendritic arborization in primary cortical neurons and decreased expression of a synaptic protein, SNAP-25, consistent with human post mortem studies that show decreased dendritic length and dendritic arborization in frontal cortical areas. The findings also suggest that binding of mutant human DISC1 to endogenous mouse DISC1 decreases expression of endogenous DISC1 and may disrupt its interactions with several partners (Pletnikov et al., 2008; Ayhan et al., 2011). These molecular disturbances might contribute to the observed neurobehavioral abnormalities.
An interesting feature of the model is that expression of mutant DISC1 is associated with relatively mild neurobehavioral abnormalities, consistent with the hypothesis that a genetic risk factor or a mutation is likely to interact with other genes and/or environmental factor(s) for a full-blown disease to develop (Moffitt et al., 2005; Van Os et al., 2008; Brandon and Sawa, 2011). In the context of the current review, another important aspect of the DISC1 model is that recent studies have shown that DISC1 interacts with several proteins of the NMDAR complex (e.g., PDS-95; Kalirin-7), indicating an intriguing converging target for molecular interactions between developmental Pb2+ exposure and mutant DISC1 (Hayashi-Takagi et al., 2010; Namba et al., 2011; Ramsey et al., 2011).
Thus, we have been using a DISC1 model to evaluate the effects of early life Pb2+ exposure on the neurobehavioral phenotype in mutant DISC1 mice. As our system is a double-transgenic (Tg) mouse model, we breed single mutant DISC1 mice and single transgenic tTA mice to produce double Tg animals that either express mutant DISC1 (mutants) or single Tg control mice that have the transgene in their genome but do not express mutant protein (controls). In order to mimic developmental exposure of humans to environmental Pb2+, we mated single tTA Tg female and single Tg mutant DISC1 male mice while they were given either control food or food containing low levels of Pb2+ (750 ppm lead acetate in the diet). Subsequently, pregnant dam and their male and female offspring were maintained on the same types of food throughout their life and while being tested in a series of behavioral tests relevant to aspects of a schizophrenia phenotype. We hypothesized that mutant DISC1 and developmental Pb2+ exposure will synergistically interact to produce an exaggerated or new phenotypic outcome in Tg mice.
Behavioral phenotyping included open field test to assess locomotor activity, elevated plus maze test to assess anxiety (Graeff et al., 1990; File, 1990), Y maze test to examine spatial working and recognition memory (Melnikova et al., 2006), pre-pulse inhibition (PPI) of the acoustic startle to evaluate sensorimotor gating (Swerdlow and Geyer, 1998), context- and cue-dependent fear conditioning to evaluate long-term hippocampus-dependent and independent learning and memory (Gerlai, 2001; Maren, 2008) as well as the NMDAR antagonist, MK-801 (0.3; mg/kg, ip),-induced locomotion to assess exacerbated responses to psychostimulants (Carlsson and Carlsson, 1990; Deutsch et al., 1997; Amann et al., 2010).
Although Pb2+ exposure produced increased anxiety in the elevated plus maze in control and mutant mice, significantly increased locomotor activity was found in Pb2+-exposed mutant mice only, with female mutant mice being affected more than male mice. A similar gender-related synergistic effect of Pb2+ and mutant DISC1 was observed in the forced swim test, mimicking aspects of affective and negative symptoms in schizophrenia. We also observed synergistic effects on impairment in pre-pulse inhibition of the acoustic startle and elevated locomotor response to MK-801, consistent with impaired sensorimotor gating and exacerbated responses to psychostimulants in schizophrenia patients. No synergistic effects were found on spontaneous alternation or spatial recognition tests. Our preliminary data suggest that developmental Pb2+ exposure and mutant DISC1 may synergistically interact in producing schizophrenia-like behavioral alterations in mice. Our on-going experiments are assessing the effect of interactions on cognitive and social behaviors, brain and lateral ventricle volume as well as regional brain expression of presynaptic markers and NMDAR expression.
Summary
Despite the fact that research efforts in schizophrenia and on the effects of developmental Pb2+ exposure on the brain have occurred independently, there is a remarkable number of similarities in outcomes between this neuropsychiatric disorder and exposure to Pb2+ in early life. The similarities at the behavioral, anatomical, biochemical and neuropathological level provides initial evidence that a neurobiological interaction may be plausible between a ubiquitous and pervasive global environmental pollutant and developmental neurotoxicant such as Pb2+ and the expression of schizophrenia later in life. Clearly, most of these putative neurobiological connections need further exploration and confirmation in animal models and in human studies. However, it is possible that individuals expressing mutations in certain genes (genetic component) may be more susceptible to the dysregulation of developmental processes produced by Pb2+ exposure (environmental trigger) alone or in combination with other environmental factors during a critical period of fetal or neonatal life. It is interesting to note that two environmental risk factors associated with schizophrenia may also be associated with an increased likelihood of being exposed to Pb2+ in the environment. These two factors are season of birth and living in an urban environment. It is widely recognized that there is a greater likelihood of a pregnant mother or a child being exposed to Pb2+ in urban environments. This is due to the high number of old housing units containing Pb2+-based paint as a continuing source of Pb2+ exposure in the United States and in many parts of the world (Jacobs et al., 2002). Second, environmental levels of Pb2+ in homes and in children's blood are highest during the summer months (July-August) (Yin, 2000). The latter observation makes another potentially important temporal connection between Pb2+ exposure and schizophrenia. The incidence of individuals with schizophrenia increases if born during the winter months (Torrey et al., 1997). For a child that is born during the winter months, the summer months in which increased exposure of the mother to Pb2+ is highly likely to occur in urban environments, places the exposure to the fetus during the early part of the second trimester of fetal life. This is the same gestational time point as the Pb2+ analysis in serum samples measured in the study by Opler et al., (2004; 2008) in which an association was noted with schizophrenia later in life, and the trimester in which the “insult” based on the neurodevelopmental hypothesis of schizophrenia is likely to occur (Roberts, 1991; Bloom, 1993).
Finally, while this review has concentrated on Pb2+ as a prototypical environmental toxicant and NMDAR antagonist that may be associated with schizophrenia, humans may be exposed to other heavy metals or environmental toxins that target the same neurobiological systems. For example, while Pb2+ is a potent NMDAR antagonist, another heavy metal manganese, with relevant exposures in children is also an NMDAR antagonist, albeit a weaker one (Guilarte and Chen, 2007). Nevertheless, several studies have recently shown that children exposed to manganese in drinking water express neuropsychological and cognitive abnormalities (Wasserman et al., 2006) and paranoid psychosis has been reported in humans occupationally exposed to high levels of manganese (Donaldson, 1987; Verhoeven et al., 2011).
Another class of environmental pollutant that has been shown to modulate NMDAR function and subunit expression is the polycyclic aromatic hydrocarbons (PAHs). Seminal studies by Hood and colleagues have shown that in utero exposure to Benzo(a)pyrene, a member of the PAH family results in downregulation of the NR1 subunit of the NMDAR and a subsequent impairment in hippocampal LTP (Wormley et al., 2004; Brown et al., 2007; McCallister et al., 2008). Therefore, there is significant evidence that a number of environmental anthropogenic pollutants are able to alter NMDAR function, a neuronal system whose hypofunction has been strongly implicated in the pathophysiology of schizophrenia.
While it is clear that genetics predispose humans to psychiatric disorders like schizophrenia, it is also highly likely that specific environmental chemicals or classes of chemicals may work in conjunction with genetic changes to facilitate full disease expression.
Acknowledgments
This work is supported by NIEHS grant ES06189 to TRG and a ViCTER supplement to ES06189 to TRG and MP to study DISC1- Pb2+ exposure gene-environment interaction. We also acknowledge the support of NIMH grant K01 MH080114-04 to MO. While we have made an effort to be comprehensive in our review, there is important and relevant work from many other scientists that we have not been able to cite or discuss. This is by no means an indication that their work is less important or valuable it is simply due to the large volume of published studies related to the various aspects of schizophrenia. In particular, we have left out studies related to adult neurogenesis in schizophrenia and in early life Pb2+ exposure where there are overlapping similarities in the outcome.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abazyan B, Nomura J, Kannan G, Ishizuka K, Tamashiro KL, Nucifora F, Pogorelov V, Ladenheim B, Yang C, Krasnova IN, Cadet JL, Pardo C, Mori S, Kamiya A, Vogel MW, Sawa A, Ross CA, Pletnikov MV. Prenatal interaction of mutant DISC1 and immune activation produces adult psychopathology. Biol Psychiatry. 2010;68:1172–1181. doi: 10.1016/j.biopsych.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abi-Dargham A, Gil R, Krystal J, Baldwin RM, Seibyl JP, Bowers M, van Dyck CH, Charney DS, Innis RB, Laurelle M. Increased striatal dopamine neurotransmission in schizophrenia; confirmation in a second cohort. Am J Psych. 1998;155:761–767. doi: 10.1176/ajp.155.6.761. [DOI] [PubMed] [Google Scholar]
- Akbarian S, Sucher NJ, Bradley D, Tafazzoli A, Trinh D, Hetrick WP, Potkin SG, Sandman CA, Bunney WE, Jones EG. Selective alterations in gene expression for NMDA receptor subunits in prefrontal cortex of schizophrenics. J Neurosci. 1996;16:19–30. doi: 10.1523/JNEUROSCI.16-01-00019.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfano DP, Petit TD. Neonatal lead exposure alters the dendritic development of hippocampal dentate granule cells. Exp Neurol. 1982;75:275–288. doi: 10.1016/0014-4886(82)90160-1. [DOI] [PubMed] [Google Scholar]
- Alkondon M, Costa ACS, Radhakrishnan V, Aronstam RS, Albuquerque ES. Selective block of NMDA-activated channel currents may be implicated in learning deficits caused by lead. Fed Eur Biochem Soc. 1990;261:124–130. doi: 10.1016/0014-5793(90)80652-y. [DOI] [PubMed] [Google Scholar]
- Amann LC, Gandal MJ, Halene TB, Ehrlichman RS, White SL, McCarren HS, Siegel SJ. Mouse behavioral endophenotypes for schizophrenia. Brain Res Bull. 2010;83:147–61. doi: 10.1016/j.brainresbull.2010.04.008. Epub 2010 Apr 28. Review. [DOI] [PubMed] [Google Scholar]
- Anastasio NC, Xia Y, O'Connor ZR, Johnson KM. Differential role of N-methyl-D-aspartate receptor subunits 2A and 2B in mediating phencyclidine-induced perinatal neuron apoptosis and behavioral deficits. Neurosci. 2009;163:1181–1191. doi: 10.1016/j.neuroscience.2009.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreasen NC, Nopoulos P, Magnotta V, Pierson R, Ziebell S, Ho BC. Progressive brain changes in schizophrenia: A prospective longitudinal study of first-episode schizophrenia. Biol Psych. 2011;70:672–679. doi: 10.1016/j.biopsych.2011.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold SE, Trojanowski JQ. Recent advances in defining the neuropathology of schizophrenia. Acta Neuropath. 1996;92:217–231. doi: 10.1007/s004010050512. [DOI] [PubMed] [Google Scholar]
- Ayhan Y, Abazyan B, Nomura J, Kim R, Ladenheim B, Krasnova IN, Sawa A, Margolis RL, Cadet JL, Mori S, Vogel MW, Ross CA, Pletnikov MV. Differentia effects of prenatal and postnatal expressions of mutant human DISC1 on neurobehavioral phenotypes in transgenic mice: evidence for neurodevelopmental origin of major psychiatric disorders. Mol Psychiatry. 2011;16:293–306. doi: 10.1038/mp.2009.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayhan Y, Sawa A, Ross CA, Pletnikov MV. Animal models of gene-environment interactions in schizophrenia. Beh Brain Res. 2009;204:274–281. doi: 10.1016/j.bbr.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandeen-Roche K, Glass TA, Bolla KI, Todd AC, Schwartz BS. Cumulative lead dose and cognitive function in older adults. Epidemiology. 2009;20:831–839. doi: 10.1097/EDE.0b013e3181b5f100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauter MR, Brockel BJ, Pankevich DE, Virgolini MB, Cory-Slechta DA. Glutamate and dopamine in the nucleus accumbens core and shell: sequence learning versus performance. NeuroToxicol. 2003;24:227–243. doi: 10.1016/S0161-813X(02)00167-5. [DOI] [PubMed] [Google Scholar]
- Behrens MM, Ali SS, Dao DN, Lucero J, Shekhtman G, Quick KL, Dugan LL. Ketamine-induced loss of phenotype of fast-spiking interneurons is mediated by NADPH-oxidase. Science. 2007;318:1645–1647. doi: 10.1126/science.1148045. [DOI] [PubMed] [Google Scholar]
- Belforte JE, Zsiros V, Sklar ER, Jiang Z, Yu G, Li Y, Quinlan EM, Nakazawa K. Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat Neurosci. 2010;13:76–83. doi: 10.1038/nn.2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellinger D, Sloman J, Leviton A, Rabinowitz M, Needleman HL, Waternaux C. Low-level lead exposure and children's cognitive function in the preschool years. Pediatrics. 1991;87:219–227. [PubMed] [Google Scholar]
- Bellinger D, Leviton A, Allred E, Rabinowitz M. Pre and postnatal lead exposure and behavior problems in school-age children. Env Res. 1994;66:12–30. doi: 10.1006/enrs.1994.1041. [DOI] [PubMed] [Google Scholar]
- Benes FM. The role of apoptosis in neuronal pathology in schizophrenia and bipolar disorder. Curr Opin Psych. 2004;17:189–190. [Google Scholar]
- Beneyto M, Kristiansen LV, Oni-Orisan A, McCullumsmith RE, Meador-Woodruff JH. Abnormal glutamate receptor expression in the medial temporal lobe in schizophrenia and mood disorders. Neuropsychopharmacol. 2007;32:1888–1902. doi: 10.1038/sj.npp.1301312. [DOI] [PubMed] [Google Scholar]
- Berger AJ, Dieudonne S, Aschner P. Glycine uptake governs glycine site occupancy at NMDA receptors of excitatory synapses. J Neurophysiol. 1998;80:3336–3340. doi: 10.1152/jn.1998.80.6.3336. [DOI] [PubMed] [Google Scholar]
- Bertolino A, Kumra S, Callicott JH, Mattay VS, Lestz RM, Jacobsen L, Barnett IS, Duyn JH, Frank JA, Rapaport JL, Weinberger DR. Common pattern of cortical pathology in childhood-onset and adult-onset schizophrenia as identified by proton magnetic resonance spectroscopic imaging. Am J Psychiatry. 1998;155:1376–1383. doi: 10.1176/ajp.155.10.1376. [DOI] [PubMed] [Google Scholar]
- Block W, Traber F, Flacke S, Jessen F, Pohl C, Schild H. In-vivo proton MR-spectroscopy of the human brain: assessment of N-acetylaspartate (NAA) reduction as a marker of neurodegeneration. Amino Acids. 2002;23:317–323. doi: 10.1007/s00726-001-0144-0. [DOI] [PubMed] [Google Scholar]
- Bloom FE. Advancing a neurodevelopmental origin for schizophrenia. Arch Gen Psych. 1993;50:224–227. doi: 10.1001/archpsyc.1993.01820150074008. [DOI] [PubMed] [Google Scholar]
- Boksa P. Animal models of obstetric complications in relation to schizophrenia. Brain Res Rev. 2004;45:1–17. doi: 10.1016/j.brainresrev.2004.01.001. [DOI] [PubMed] [Google Scholar]
- Braga MFM, Pereira EFR, Albuquerque EX. Nanomolar concentrations of lead inhibit glutamatergic and GABAergic transmission in hippocampal neurons. Brain Res. 1999;826:22–34. doi: 10.1016/s0006-8993(99)01194-4. [DOI] [PubMed] [Google Scholar]
- Braga MFM, Pereira EFR, Marchioro M, Albuquerque EX. Lead increases tetrodotoxin-insensitive spontaneous release of glutamate and GABA from hippocampal neurons. Brain Res. 1999a;826:10–21. doi: 10.1016/s0006-8993(99)01193-2. [DOI] [PubMed] [Google Scholar]
- Brandon NJ. Dissecting DISC1 function through protein-protein interactions. Biochem Soc Trans. 2007;35:1283–1286. doi: 10.1042/BST0351283. [DOI] [PubMed] [Google Scholar]
- Brandon NJ, Millar JK, Korth C, Sive H, Singh KK, Sawa A. Understanding the role of DISC1 in psychiatric disease and during normal development. J Neurosci. 2009;29(41):12768–75. doi: 10.1523/JNEUROSCI.3355-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandon NJ, Sawa A. Linking neurodevelopmental and synaptic theories of mental illness through DISC1. Nat Rev Neurosci. 2011;12:707–722. doi: 10.1038/nrn3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockel BJ, Cory-Slechta DA. Lead, attention, and impulsive behavior: changes in a fixed-ratio waiting for reward paradigm. Pharmacol Biochem Bev. 1998;60:545–552. doi: 10.1016/s0091-3057(98)00023-9. [DOI] [PubMed] [Google Scholar]
- Brockel BJ, Cory-Slechta DA. Lead-induced decrements in waiting behavior: involvement of D2-like dopamine receptors. Pharmacol Biochem Bev. 1999;63:423–434. doi: 10.1016/s0091-3057(99)00033-7. [DOI] [PubMed] [Google Scholar]
- Bromet EJ, Fennig S. Epidemiology and Natural History of Schizophrenia. Biol Psychiatry. 1999;46:871–881. doi: 10.1016/s0006-3223(99)00153-5. [DOI] [PubMed] [Google Scholar]
- Brooks WJ, Petit TL, Leboutillier JC, Nobrega JN, Jarvis MF. Differential effects of early chronic lead exposure on postnatal rat brain NMDA, PCP, and Adenosine A1 receptors: an autoradiography study. Drug Dev Res. 1993;29:40–47. [Google Scholar]
- Brown AS. The environment and susceptibility to schizophrenia. Prog Neurobiol. 2011;93:23–58. doi: 10.1016/j.pneurobio.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown LA, Khousbouei H, Goodwin JS, Irvin-Wilson CV, Ramesh A, Sheng L, McCallister MM, Jiang GC, Aschner M, Hood DB. Down-regulation of early ionotropic glutamate receptor subunit developmental expression as a mechanism for observed plasticity deficits following gestational exposure to benzo(a)pyrene. Neurotoxicol. 2007;28:965–978. doi: 10.1016/j.neuro.2007.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brubaker CJ, Schmithorst VJ, Haynes EN, Dietrich KN, Egelhoff JC, Lindquist DM, Lanphear BP, Cecil KM. Altered myelination and axonal integrity in adults with childhood lead exposure: a diffusion tensor imaging study. Neurotoxicol. 2009;30:867–975. doi: 10.1016/j.neuro.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brubaker CJ, Dietrich KN, Lanphear BP, Cecil KM. The influence of age and lead exposure on adult gray matter volume. Neurotoxicol. 2010;31:259–266. doi: 10.1016/j.neuro.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley PF, Pillai A, Howell KR. Brain-derived neurotrophic factor: findings in schizophrenia. Curr Opin Psych. 2011;24:122–127. doi: 10.1097/YCO.0b013e3283436eb7. [DOI] [PubMed] [Google Scholar]
- Burdette LJ, Goldstein R. Long-term exposure and electrophysiological changes associated with lead exposure at different stages of brain development in the rat. Dev Brain Res. 1986;29:101–110. doi: 10.1016/0165-3806(86)90086-6. [DOI] [PubMed] [Google Scholar]
- Byers RK, Lord EE. Late effects of lead poisoning on mental development. Am J Dis Children. 1943;66:471–494. [Google Scholar]
- Campbell JB, Woolley DE, Vijayan VK, Overmann SR. Morphometric effects of postnatal lead exposure on hippocampal development of the 15-day-old rat. Deve Brain Res. 1982;3:595–612. doi: 10.1016/0165-3806(82)90056-6. [DOI] [PubMed] [Google Scholar]
- Canfield RL, Henderson CR, Cory-Slechta DA, Cox C, Jusko TA, Lanphear BP. Intellectual impairment in children with blood lead concentrations below 10 μg per deciliter. New Engl J Med. 2003;348:1517–1526. doi: 10.1056/NEJMoa022848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canfield RL, Gendle MH, Cory-Slechta DA. Impaired neuropsychological functioning in lead-exposed children. Dev Neuropsych. 2004;26:513–540. doi: 10.1207/s15326942dn2601_8. [DOI] [PubMed] [Google Scholar]
- Cannon TD. Abnormalities of brain structure and function in schizophrenia: implications for aetiology and pathophysiology. Ann Med. 1996;28:533–539. doi: 10.3109/07853899608999117. [DOI] [PubMed] [Google Scholar]
- Carlsson M, Carlsson A. Interactions between glutamatergic and monoaminergic systems within the basal ganglia--implications for schizophrenia and Parkinson's disease. Trends Neurosci. 1990;7:272–276. doi: 10.1016/0166-2236(90)90108-m. [DOI] [PubMed] [Google Scholar]
- Carlsson A, Lundqvist M. Effect of chlorpromazine or haloperidol on formation of 3-methoxytyramine and norepinephrine in mouse brain. Acta Pharmacol Toxicol. 1963;20:140–144. doi: 10.1111/j.1600-0773.1963.tb01730.x. [DOI] [PubMed] [Google Scholar]
- Cecil KM, Brubaker CJ, Adler CM, Dietrich KN, Altaye M, Egelhoff JC, Wessel S, Elangovan I, Hornung R, Jarvis K, Lanphear BP. Decreased brain volume in aduts with childhood lead exposure. PLoS Med. 2008;5:e112. doi: 10.1371/journal.pmed.0050112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapcote SJ, Lipina TV, Millar JK, Mackie S, Christie S, Ogawa F, Lerch JP, Trimble K, Uchiyama M, Sakuraba Y, Kaneda H, Shiroishi T, Houslay MD, Henkelman RM, Sled JG, Gondo Y, Porteous DJ, Roder JC. Behavioral phenotypes of Disc1 missense mutations in mice. Neuron. 2007;54:387–402. doi: 10.1016/j.neuron.2007.04.015. [DOI] [PubMed] [Google Scholar]
- Clinton SM, Haroutunian V, Davis KL, Meador-Woodruff JH. Altered transcript expression of NMDA receptor-associated postsynaptic proteins in the thalamus of subjects with schizophrenia. Am J Psych. 2003;160:1100–1109. doi: 10.1176/appi.ajp.160.6.1100. [DOI] [PubMed] [Google Scholar]
- Clinton SM, Haroutunian V, Meador-Woodruff JH. Up-regulation of NMDA receptor subunit and post-synaptic density protein expression in the thalamus of elderly patients with schizophrenia. J Neurochem. 2006;98:1114–1125. doi: 10.1111/j.1471-4159.2006.03954.x. [DOI] [PubMed] [Google Scholar]
- Cochran SM, Kennedy M, McKerchar CE, Steward LJ, Pratt JA, Morris BJ. Induction of metabolic hypofunction and neurochemical deficits after chronic intermittent exposure to phencyclidine: differential modulation of antipsychotic drugs. Neuropsychopharmacol. 2003;28:265–275. doi: 10.1038/sj.npp.1300031. [DOI] [PubMed] [Google Scholar]
- Commissaris RL, Tavakoli-Nezhad M, Barron AJ, Pitts DK. Effects of chronic low-level lead exposure on prepulse inhibition of acoustic startle in the rat. Neurotoxicol Teratol. 2000;22:55–60. doi: 10.1016/s0892-0362(99)00042-2. [DOI] [PubMed] [Google Scholar]
- Cory-Slechta DA, McCoy L, Richfield EK. Time course and regional basis of Pb-induced changes in MK-801 binding: reversal by chronic treatment with the dopamine receptor agonist apomorphine but not the D1 agonist SKF-82958. J Neurochem. 1997;68:2012–2023. doi: 10.1046/j.1471-4159.1997.68052012.x. [DOI] [PubMed] [Google Scholar]
- Cory-Slechta DA, Brockel BJ, O'Mara DJ. Lead exposure and dorsomedial striatum mediation of fixed interval schedule-controlled behavior. NeuroToxicol. 2002;23:313–327. doi: 10.1016/s0161-813x(02)00059-1. [DOI] [PubMed] [Google Scholar]
- Cory-Slechta DA, Garcia-Osuna M, Greenamyre JT. Lead-induced changes in NMDA receptor complex binding: correlations with learning accuracy and with sensitivity to learning impairments caused by MK-801 and NMDA administration. Behav Brain Res. 1997;85:161–174. doi: 10.1016/s0166-4328(96)00174-x. [DOI] [PubMed] [Google Scholar]
- Coyle JT, Tsai G. The NMDA receptor glycine modulatory site: a therapeutic target for improving cognition and reducing negative symptoms in schizophrenia. Psychopharm. doi: 10.1007/s00213-003-1709-2. published on line. 2003; 10.1007/s00213-003-1709-2. [DOI] [PubMed] [Google Scholar]
- Coyle JT, Tsai G, Goff D. Converging evidence of NMDA receptor hypofunction in the pathophysiology of schizophrenia. Ann NY Acad Sci. 2003;1003:318–327. doi: 10.1196/annals.1300.020. [DOI] [PubMed] [Google Scholar]
- Coyle JT, Tsai G. NMDA receptor function, neuroplasticity, and the pathophysiology of schizophenia. Int Rev Neurobiol. 2004;59:491–515. doi: 10.1016/S0074-7742(04)59019-0. [DOI] [PubMed] [Google Scholar]
- Creese I, Burt DR, Snyder SH. Dopamine receptor binding predicts clinical and pharmacological potency of anti-schizophrenic drugs. Science. 1976;192:481–483. doi: 10.1126/science.3854. [DOI] [PubMed] [Google Scholar]
- Dean B, Scarr E, Bradbury R, Copolov D. Decreased hippocampal (CA3) NMDA receptors in Schizophrenia. Synapse. 1999;32:67–69. doi: 10.1002/(SICI)1098-2396(199904)32:1<67::AID-SYN9>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Deicken RF, Zhou L, Corwin F, Vinogradov S, Weiner MW. Decreased left frontal lobe N-acetylaspartate in schizophrenia. Am J Psychiatry. 1997;154:688–690. doi: 10.1176/ajp.154.5.688. [DOI] [PubMed] [Google Scholar]
- Derosse P, Hodgkinson CA, Lencz T, Burdick KE, Kane JM, Goldman D, Malhotra AK. Disrupted in Schizophrenia 1 Genotype and Positive Symptoms in Schizophrenia. Biol Psychiatry. 2006 doi: 10.1016/j.biopsych.2006.07.023. [DOI] [PubMed] [Google Scholar]
- Deutsch SI, Rosse RB, Mastropaolo J. Behavioral approaches to the functional assessment of NMDA-mediated neural transmission in intact mice. Clin Neuropharmacol. 1997;5:375–84. doi: 10.1097/00002826-199710000-00001. Review. [DOI] [PubMed] [Google Scholar]
- Dietrich KN, Ris MD, Succop PA, Berger OG, Bornschein RL. Early exposure to lead and juvenile delinquency. Neurotoxicol Teratol. 2001;23:511–518. doi: 10.1016/s0892-0362(01)00184-2. [DOI] [PubMed] [Google Scholar]
- Donaldson J. The physiopathologic significance of manganese in brain: its relation to schizophrenia and neurodegenerative disorders. Neurotoxicol. 1987;8:451–462. [PubMed] [Google Scholar]
- Dracheva S, Marras SAE, Elhakem SL, Kramer FR, Davis KL, Haroutunian V. N-methyl-D-aspartic acid receptor expression in the dorsolateral prefrontal cortex of elderly patients with schizophrenia. Am J Psych. 2001;158:1400–1410. doi: 10.1176/appi.ajp.158.9.1400. [DOI] [PubMed] [Google Scholar]
- Dribben WH, Creeley CE, Farber N. Low-level lead exposure triggers neuronal apoptosis in the developing mouse brain. Neurotoxicol Teratol. 2011;33:473–480. doi: 10.1016/j.ntt.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan X, Chang JH, Ge S, Faulkner RL, Kim JY, Kitabatake Y, Liu XB, Yang CH, Jordan JD, Ma DK, Liu CY, Ganesan S, Cheng HJ, Ming GL, Lu B, Song H. Disrupted-In-Schizophrenia 1 regulates integration of newly generated neurons in the adult brain. Cell. 2007;130:1146–1158. doi: 10.1016/j.cell.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Factor-Litvak P, Wasserman G, Kline JK, Graziano J. The Yugoslavia Prospective Study of environmental lead exposure. Environ Health Perspect. 1999 Jan;107(1):9–15. doi: 10.1289/ehp.991079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulkner RL, Jang MH, Liu XB, Duan X, Sailor KA, Kim JY, Ge S, Jones EG, Ming GL, Song H, Cheng HJ. Development of hippocampal mossy fiber synaptic outputs by new neurons in the adult brain. Proc Natl Acad Sci U S A. 2008;105:14157–11162. doi: 10.1073/pnas.0806658105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- File SE. New strategies in the search for anxiolytics. Drug Des Deliv. 1990;5(3):195–201. [PubMed] [Google Scholar]
- Fredriksson A, Archer T. Neurobehavioral deficits associated with apoptotic neurodegenration and vulnerability to ADHD. Neurotox Res. 2004;6:435–436. doi: 10.1007/BF03033280. [DOI] [PubMed] [Google Scholar]
- Gao X-M, Sakai K, Roberts RC, Conley RR, Dean B, Tamminga CA. Ionotropic glutamate receptors and expression of N-methyl-d-aspartate receptor subunits in subregions of human hippocampus: effects of schizophrenia. Am J Psych. 2000;157:1141–1149. doi: 10.1176/appi.ajp.157.7.1141. [DOI] [PubMed] [Google Scholar]
- Geddes AE, Huang X-F, Newell KA. Reciprocal signaling between NR2 subunits of the NMDA receptor and neuregulin 1 and their role in schizophrenia. Prog Neuro-Psychopharmacol Biol Psych. 2011;35:896–904. doi: 10.1016/j.pnpbp.2011.02.017. [DOI] [PubMed] [Google Scholar]
- Gerlai R. Behavioral tests of hippocampal function: simple paradigms complex problems. Behav Brain Res. 2001;125(1-2):269–77. doi: 10.1016/s0166-4328(01)00296-0. [DOI] [PubMed] [Google Scholar]
- Gilman CP, Mattson MP. Do apoptotic mechanisms regulate synaptic plasticity and growth-cone motility? NeuroMol Med. 2002;2:197–214. doi: 10.1385/NMM:2:2:197. [DOI] [PubMed] [Google Scholar]
- Glantz LA, Gilmore JH, Lieberman JA, Jarskog LF. Apopotic mechanisms and the synaptic pathology of schizophrenia. Schiz Res. 2006;81:47–63. doi: 10.1016/j.schres.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Graeff FG, Audi EA, Almeida SS, Graeff EO, Hunziker MH. Behavioral effects of 5-HT receptor ligands in the aversive brain stimulation, elevated plus-maze and learned helplessness tests. Neurosci Biobehav Rev. 1990;4:501–506. doi: 10.1016/s0149-7634(05)80074-0. [DOI] [PubMed] [Google Scholar]
- Grinwood S, Slater P, Deakin JFW, Hutson PH. NR2B-containing NMDA receptors are up-regulated in temporal cortex in schizophrenia. NeuroReport. 1999;10:461–465. doi: 10.1097/00001756-199902250-00004. [DOI] [PubMed] [Google Scholar]
- Guilarte TR. The N-methyl-D-aspartare receptor. Physiology and neurotoxicology in the developing brain. In: Slikker W Jr, Chang LW, editors. Handbook of Developmental Neurotoxicology. Academic Press; San Diego: 1998. pp. 285–304. [Google Scholar]
- Guilarte TR, Chen MK. Manganese inhibits NMDA receptor channel function: implications to psychiatric and cognitive effects. Neurotoxicol. 2007;28:1147–1152. doi: 10.1016/j.neuro.2007.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilarte TR, Miceli RC. Age-dependent effects of lead on [3H]-MK-801 binding to the NMDA receptor-gated ionophore: In vitro and in vivo studies. Neurosci Lett. 1992;148:27–30. doi: 10.1016/0304-3940(92)90796-a. [DOI] [PubMed] [Google Scholar]
- Guilarte TR, Miceli RC, Altmann L, Weinsberg F, Winneke G, Wiegand H. Chronic prenatal and postnatal Pb2+ exposure increases [3H]-MK-801 binding sites in the adult forebrain. Eur J Pharmacol. 1993;248:273–275. doi: 10.1016/0926-6917(93)90054-t. [DOI] [PubMed] [Google Scholar]
- Guilarte TR, McGlothan JL. Hippocampal NMDA receptor mRNA undergoes subunit specific changes during developmental lead exposure. Mol Brain Res. 1998;76:299–305. doi: 10.1016/s0006-8993(98)00054-7. [DOI] [PubMed] [Google Scholar]
- Guilarte TR, McGlothan JL, Nihei MK. Hippocampal expression of N-methyl-D-aspartare receptor (NMDAR1) subunit splice variant mRNA is altered by developmental exposure to Pb2+ Mol Brain Res. 2000;76:299–305. doi: 10.1016/s0169-328x(00)00010-3. [DOI] [PubMed] [Google Scholar]
- Guilarte TR, McGlothan JL. Selective decrease in NR1 subunit splice variant mRNA in the hippocampus of Pb2+-exposed rats: implications for synaptic targeting and cell surface expression of NMDAR complexes. Mol Brain Res. 2003;113:37–43. doi: 10.1016/s0169-328x(03)00083-4. [DOI] [PubMed] [Google Scholar]
- Guilarte TR, Toscano CD, McGlothan JL, Weaver SA. Environmental enrichment reverses cognitive and molecular deficits induced by developmental lead exposure. Ann Neurol. 2003;53:50–56. doi: 10.1002/ana.10399. [DOI] [PubMed] [Google Scholar]
- Harrison PJ. Schizophrenia: a disorder of neurodevelopment? Curr Opinion Neurobiol. 1997;7:285–289. doi: 10.1016/s0959-4388(97)80018-9. [DOI] [PubMed] [Google Scholar]
- Harrison PJ. The neuropathology of schizophrenia-a critical review of the data and their interpretation. Brain. 1999;122:593–624. doi: 10.1093/brain/122.4.593. [DOI] [PubMed] [Google Scholar]
- Hashemzadeh-Gargari H, Guilarte TR. Divalent cations modulate N-methyl-D-aspartare receptor function at the glycine site. J Pharm Exp Ther. 1999;290:1356–1362. [PubMed] [Google Scholar]
- Hayashi-Takagi A, Takaki M, Graziane N, Seshadri S, Murdoch H, Dunlop AJ, Makino Y, Seshadri AJ, Ishizuka K, Srivastava DP, Xie Z, Baraban JM, Houslay MD, Tomoda T, Brandon NJ, Kamiya A, Yan Z, Penzes P, Sawa A. Disrupted-in-Schizophrenia 1 (DISC1) regulates spines of the glutamate synapse via Rac1. Nat Neurosci. 2010;13:327–332. doi: 10.1038/nn.2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horga G, Bernacer J, Dusi N, Entis J, Chu K, Hazlett EA, Mehmet Haznedar M, Kemether E, Byne W, Buchsbaum MS. Correlations between ventricular enlargement and gray and white matter volumes of cortex, thalamus, striatum and internal capsule in schizophrenia. Eur Arch Psych Clin Neurosci. 2011 doi: 10.1007/s00406-011-0202-x. EPUb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S, Mirmics K. Breaking the gene barrier in schizophrenia. Nature Med. 2009;15:488–490. doi: 10.1038/nm0509-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphries C, Mortimer A, Hirsch S, de Belleroche J. NMDA receptor mRNA correlation with antemortem cognitive impairment in schizophrenia. NeuroReport. 1996;7:2051–2055. doi: 10.1097/00001756-199608120-00040. [DOI] [PubMed] [Google Scholar]
- Huresco-Levy U, Javitt DC, Ermilov M, Mordel C, Silipo G, Lichtenstein M. Efficacy of high-dose glycine in the treatment of enduring negative symptoms of schizophrenia. Arch Gen Psych. 1999;56:29–36. doi: 10.1001/archpsyc.56.1.29. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Busch F, Miksa M, Bittigan P, Vockler J, Dikranian K, Tenkova TI, Stefovska V, Turski L, Olney JW. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999;283:70–74. doi: 10.1126/science.283.5398.70. [DOI] [PubMed] [Google Scholar]
- Inta D, Monyer H, Sprengel R, Meyer-Lindenberg A, Gass P. Mice with genetically altered glutamate receptors as models of schizophrenia: a comprehensive review. Neurosci Beh Rev. 2010;34:285–294. doi: 10.1016/j.neubiorev.2009.07.010. [DOI] [PubMed] [Google Scholar]
- Ishihara K, Alkondon M, Montes JG, Albuquerque EX. Nicotinic responses in acutely dissociated rat hippocampal neurons and the selective blockade of fast-desensitizing nicotinic currents by Pb2+ J Pharm Exp Ther. 1995;273:1471–1482. [PubMed] [Google Scholar]
- Itokawa M, Yamada K, Yoshitsugu K, Toyota T, Suga T, Ohda H, Watanabe A, Hattori E, Shimizu H, Kamakura T, Ebihara M, Meerabux JMA, Toru M, Yoshikawa T. A microsatellite repeat in the promoter of the N-methyl-d-aspartate receptor 2A subunit (GRIN2A) gene suppresses transcriptional activity and correlates with chronic outcomes in schizophrenia. Phamacogenetics. 2003;13:271–278. doi: 10.1097/00008571-200305000-00006. [DOI] [PubMed] [Google Scholar]
- Jacobs DE, Clikner RP, Zhou JY, Viet SM, Marker DA, Rogers JW, Zeldin DC, Broene P, Friedman W. The prevalence of lead-based paint hazards in U.S. housing. Env Hlth Persp. 2002;110:A559–A606. doi: 10.1289/ehp.021100599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarskog LF, Glantz LA, Gilmore JH, Lieberman JA. Apoptotic mechanisms in the pathophysiology of schizophrenia. Prog Neuro-Psychopharmacol Biol Psych. 2005;29:846–858. doi: 10.1016/j.pnpbp.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Jarskog LF, Selinger ES, Liberman JA, Gilmore JH. Apoptotic proteins in the temporal cortex in schizophrenia: High Bax/Bcl-2 ratio without caspase-3 activation. Am J Psych. 2004;16:109–115. doi: 10.1176/appi.ajp.161.1.109. [DOI] [PubMed] [Google Scholar]
- Javitt DC. Glutamatergic theories of schizophrenia. Isr J Psych Relat Sci. 2010;47:4–16. [PubMed] [Google Scholar]
- Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psych. 1991;148:1301–1308. doi: 10.1176/ajp.148.10.1301. [DOI] [PubMed] [Google Scholar]
- Javitt DC, Zylberman I, Zukin SR, Heresco-Levy U, Lindermayer JP. Amelioration of negative symptoms in schizophrenia by glycine. Am J Psych. 1994;151:1234–1236. doi: 10.1176/ajp.151.8.1234. [DOI] [PubMed] [Google Scholar]
- Jett DA, Beckles RA, Navoa RV, McLemore GL. Increased high-affinity nicotinic receptor-binding in rats exposed to lead during development. Neurotoxicol Teratol. 2002;24:805–811. doi: 10.1016/s0892-0362(02)00314-8. [DOI] [PubMed] [Google Scholar]
- Jett DA, Guilarte TR. Developmental lead exposure alters N-methyl-D-aspartate and muscarinic cholinergic receptors in the rat hippocampus: an autoradiography study. Neurotoxicol. 1995;16:7–18. [PubMed] [Google Scholar]
- Jones PB, Bebbington P, Foerster A, Lewis SW, Murray RM, Russell A, Sham PC, Toone BK, Wilkins S. Premorbid social underachievement in schizophrenia. Results from the Camberwell Collaborative Psychosis Study. Br J Psychiatry. 1993;162:65–71. doi: 10.1192/bjp.162.1.65. [DOI] [PubMed] [Google Scholar]
- Kamiya A, Kubo K, Tomoda T, Takaki M, Youn R, Ozeki Y, Sawamura N, Park U, Kudo C, Okawa M, Ross CA, Hatten ME, Nakajima K, Sawa A. A schizophrenia-associated mutation of DISC1 perturbs cerebral cortex development. Nat Cell Biol. 2005;7:1167–1178. doi: 10.1038/ncb1328. [DOI] [PubMed] [Google Scholar]
- Kaneshiro-Olympio KP, Goncalves C, Risso Gunther WM, Henriques-Bechara EJ. Neurotoxicity and aggression triggered by low-level lead exposure in children: a review. Pam Amr J Public Hlth. 2009;26:266–275. doi: 10.1590/s1020-49892009000900011. [DOI] [PubMed] [Google Scholar]
- Koike H, Arguello PA, Kvajo M, Karayiorgou M, Gogos JA. Disc1 is mutated in the 129S6/SvEv strain and modulates working memory in mice. Proc Natl Acad Sci U S A. 2006;103:3693–3697. doi: 10.1073/pnas.0511189103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komhuber J, Mack-Burkhardt F, Riederer P, Hebenstreit GF, Reynolds GP, Andrews HB, Beckmann H. [3H]-MK-801 binding sites in postmortem brain regions of schizophrenia patients. J Neural Transm. 1989;77:231–236. doi: 10.1007/BF01248936. [DOI] [PubMed] [Google Scholar]
- Kinney JW, Davis CN, Tabarean I, conti B, Bartfai T, Behreus MM. A specific role of NR2A-containing NMDA receptors in the maintenance of Parvalbumin and GAD67 immunoreactivity in cultured inteneurons. J Neurosci. 2006;26:1604–1615. doi: 10.1523/JNEUROSCI.4722-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvajo M, McKellar H, Arguello PA, Drew LJ, Moore H, MacDermott AB, Karayiorgou M, Gogos JA. A mutation in mouse Disc1 that models a schizophrenia risk allele leads to specific alterations in neuronal architecture and cognition. Proc Natl Acad Sci U S A. 2008;105(19):7076–81. doi: 10.1073/pnas.0802615105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvajo M, McKellar H, Gogos JA. Avoiding mouse traps in schizophrenia genetics: Lessons and promises from current and emerging mouse models. Neurosci. 2011 doi: 10.1016/j.neuroscience.2011.07.051. EPub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay SR, Opler LA. The positive-negative dimension in schizophrenia: its validity and significance. Psychiatr Dev. 1987;5:79–103. [PubMed] [Google Scholar]
- Kilts CD. The changing roles and targets for animal models of schizophrenia. Biol Psychiatry. 2001;50:845–855. doi: 10.1016/s0006-3223(01)01286-0. [DOI] [PubMed] [Google Scholar]
- Kim R, Hu H, Rotnitzky A, Bellinger D, Needleman H. A longitudinal study of chronic lead exposure and physical growth in Boston children. Environ Health Perspect. 1995 Oct;103(10):952–7. doi: 10.1289/ehp.95103952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiraly E, Jones DG. Dendritic spine changes in rat hippocampal pyramidal cells after postnatal lead treatment: a golgi study. Exp Neuro. 1982;77:236–239. doi: 10.1016/0014-4886(82)90158-3. [DOI] [PubMed] [Google Scholar]
- Kocsis B. Differential role of NR2A and NR2B subunits in N-methyl-d-aspartate receptor antagonist-induced aberrant cortical gamma oscillations. Biol Psych. 2011 doi: 10.1016/j.biopsych.2011.10.002. EPub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolomeets NS, Orlovskaya DD, Rachmanova VI, Uranova NA. Ultrastructural alterations in hippocampal mossy fiber synapses in schizophrenia: a postmortem morphometric study. Synapse. 2005;57:47–55. doi: 10.1002/syn.20153. [DOI] [PubMed] [Google Scholar]
- Konradi C, Heckers S. Molecular aspects of glutamate dysregulation: implications for schizophrenia and its treatment. Pharmacol & Therap. 2003;97:153–179. doi: 10.1016/s0163-7258(02)00328-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krigman MR, Druse MJ, Traylor TD, Wilson MH, Newell LR, Hogan EL. Lead encephalopathy in the developing rat: effect on cortical ontogenesis. J Neuropath Exp Neurol. 1974;33:671–686. doi: 10.1097/00005072-197410000-00007. [DOI] [PubMed] [Google Scholar]
- Labrie V, Lipima T, Roder JL. Mice with reduced NMDA receptor glycine affinity model some of the negative and cognitive symptoms of schizophrenia. Neuropharmacol. 2008;200:217–2230. doi: 10.1007/s00213-008-1196-6. [DOI] [PubMed] [Google Scholar]
- Lahti AC, Koffel B, LaPorte D, Tamminga CA. Subanesthetic doses of ketamine stimulate psychosis in schizophrenia. Neuropsychopharmacol. 1995;13:9–19. doi: 10.1016/0893-133X(94)00131-I. [DOI] [PubMed] [Google Scholar]
- Lasley SM, Green MC, Gilbert ME. Influence of exposure period on in vivo hippocampal glutamate and GABA release in rats chronically exposed to lead. Neurotoxicology. 1999;20:619–629. [PubMed] [Google Scholar]
- Laurelle M, Abi-Dargham A, Van Dyck CH, Gil R, D'Souza CD, Erdos J, McCance E, Rosenblatt W, Fingado C, Zoghbi SS, Baldwin RM, Seibyl JP, Krystal JH, Charney DS, Innis RB. Single photon emission computed tomography imaging of amphetamine-induced dopamine release in drug-free schizophrenic subjects. Proc Nat Acad Sci. 1996;93:9225–9240. doi: 10.1073/pnas.93.17.9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law AJ, Deakin JFW. Asymmetrical reductions of hippocampal NMDAR1 glutamate receptor mRNA in the psychoses. NeuroReport. 2001;12:2971–2974. doi: 10.1097/00001756-200109170-00043. [DOI] [PubMed] [Google Scholar]
- Le Corre S, Harper CG, Lopez P, Ward P, Catts S. Increased levels of expression of an NMDAR1 splice variant in the superior temporal gyrus in schizophrenia. NeuroReport. 2000;11:983–986. doi: 10.1097/00001756-200004070-00017. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Lieberman JA. Catching up on Schizophrenia: Natural history and neurobiology. Neuron. 2000;28:325–334. doi: 10.1016/s0896-6273(00)00111-2. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci. 2005;6:312–324. doi: 10.1038/nrn1648. [DOI] [PubMed] [Google Scholar]
- Li W, Zhou Y, Jentsch JD, Brown RA, Tian X, Ehninger D, Hennah W, Peltonen L, Lönnqvist J, Huttunen MO, Kaprio J, Trachtenberg JT, Silva AJ, Cannon TD. Specific developmental disruption of disrupted-in-schizophrenia-1 function results in schizophrenia-related phenotypes in mice. Proc Natl Acad Sci U S A. 2007;104(46):18280–5. doi: 10.1073/pnas.0706900104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman JA, Kane JM, Alvir J. Provocative tests with psychostimulant drugs in schizophrenia. Psychopharmacol. 1987;91:415–433. doi: 10.1007/BF00216006. [DOI] [PubMed] [Google Scholar]
- Lin CH, Lane HY, Tsai GE. Glutamate signaling in the pathophysiology and therapy of schizophrenia. Pharm Biochem Beh. 2011 doi: 10.1016/jpbb.2011.03.023. EPub. [DOI] [PubMed] [Google Scholar]
- Lindstrom LH, Gefvert O, Hagberg G, Lundberg T, Hartvig P, Langstrom B. Increased dopamine synthesis rate in medial prefrontal cortex and striatum in schizophrenia indicated by L-(beta-11C) DOPA and PET. Biol Pscyh. 1999;46:681–688. doi: 10.1016/s0006-3223(99)00109-2. [DOI] [PubMed] [Google Scholar]
- Lipska BK. Using animal models to test a neurodevelopmental hypothesis of schizophrenia. Rev Psychiatry Neurosci. 2004;29:282–286. [PMC free article] [PubMed] [Google Scholar]
- Ma T, Chen HH, Chang HL, Hume AS, Ho IK. Effects of chronic lead exposure on [3H]-MK-801 binding in the rat brain. Toxicol Lett. 1997;92:59–66. doi: 10.1016/s0378-4274(97)00035-0. [DOI] [PubMed] [Google Scholar]
- Mackie S, Millar JK, Porteous DJ. Role of DISC1 in neural development and schizophrenia. Curr Opin Neurobiol. 2007;17(1):95–102. doi: 10.1016/j.conb.2007.01.007. [DOI] [PubMed] [Google Scholar]
- Marcelis M, Navarro-Mateu F, Murray R, Selten JP, Van Os J. Urbanization and psychosis: a study of 1942-1978 birth cohorts in The Netherlands. Psychol Med. 1998 Jul;28(4):871–9. doi: 10.1017/s0033291798006898. [DOI] [PubMed] [Google Scholar]
- Marcotte ER, Pearson DM, Srivastava LK. Animal models of schizophrenia: a critical review. J Psych Neurosci. 2001;26:395–410. [PMC free article] [PubMed] [Google Scholar]
- Marek GJ, Behl B, Bespalov AY, Gross G, Lee Y, Schomaker H. Glutamatergic (N-methyl-D-aspartate receptor) hypofontality in schizophrenia: too little juice or a miswired brain? Mol Pharm. 2010;77:317–326. doi: 10.1124/mol.109.059865. [DOI] [PubMed] [Google Scholar]
- Maren S. Pavlovian fear conditioning as a behavioral assay for hippocampus and amygdala function: cautions and caveats. Eur J Neurosci. 2008;28(8):1661–6. doi: 10.1111/j.1460-9568.2008.06485.x. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Keller JN, Begley JG. Evidence for synaptic apoptosis. Exp Neurol. 1998;153:35–48. doi: 10.1006/exnr.1998.6863. [DOI] [PubMed] [Google Scholar]
- McCallister MM, Maguire M, Ramesh A, Aimin Q, Liu S, Khoshbouei H, Achner M, Ebner FF, Hood DB. Prenatal exposure to benzo(a)pyrene impairs later-life cortical neuronal function. Neurotoxicol. 2008;29:846–854. doi: 10.1016/j.neuro.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Saha S, Welham J, El Saadi O, MacCauley C, Chant D. A systematic review of the incidence of schizophrenia: the distribution of rates and the influence of sex, urbanicity, migrant status and methodology. BMC Med. 2004;2:13. doi: 10.1186/1741-7015-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meador-Woodruff JH, Clinton SM, Beneyto M, McCullumsmith RE. Molecular abnormalities of the glutamate synapse in the thalamus in schizophrenia. Ann NY Acad Sci. 2003;1003:75–93. doi: 10.1196/annals.1300.005. [DOI] [PubMed] [Google Scholar]
- Melnikova T, Savonenko A, Wang Q, Liang X, Hand T, Wu L, Kaufmann WE, Vehmas A, Andreasson KI. Cycloxygenase-2 activity promotes cognitive deficits but not increased amyloid burden in a model of Alzheimer's disease in a sex-dimorphic pattern. Neuroscience. 2006;141(3):1149–62. doi: 10.1016/j.neuroscience.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Mike A, Pereira EFR, Albuquerque EX. Ca2+-sensitive inhibition by Pb2+ of α7-containing nicotinic acetylcholine receptors in hippocampal neurons. Brain Res. 2000;873:112–123. doi: 10.1016/s0006-8993(00)02533-6. [DOI] [PubMed] [Google Scholar]
- Millar JK, Wilson-Annan JC, Anderson S, Christie S, Taylor MS, Semple CA, Devon RS, Clair DM, Muir WJ, Blackwood DH, Porteous DJ. Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum Mol Genet. 2000a;9(9):1415–23. doi: 10.1093/hmg/9.9.1415. [DOI] [PubMed] [Google Scholar]
- Millar JK, Christie S, Semple CA, Porteous DJ. Chromosomal location and genomic structure of the human translin-associated factor X gene (TRAX; TSNAX) revealed by intergenic splicing to DISC1, a gene disrupted by a translocation segregating with schizophrenia. Genomics. 2000b;67(1):69–77. doi: 10.1006/geno.2000.6239. [DOI] [PubMed] [Google Scholar]
- Millar JK, Christie S, Anderson S, Lawson D, Hsiao-Wei Loh D, Devon RS, Arveiler B, Muir WJ, Blackwood DH, Porteous DJ. Genomic structure and localization within a linkage hotspot of Disrupted In Schizophrenia 1, a gene disrupted by a translocation segregating with schizophrenia. Mol Psychiatry. 2001;6(2):173–8. doi: 10.1038/sj.mp.4000784. [DOI] [PubMed] [Google Scholar]
- Miranda ML, Kim D, Galeano MA, Paul CJ, Hull AP, Morgan SP. The relationship between early childhood lead exposure and performance on end-of-grade tests. Env Hlth Persp. 2007;115:1242–1247. doi: 10.1289/ehp.9994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto Y, Yamada K, Noda Y, Mori H, Mishina M, Nabeshima T. Hyperfuntion of dopaminergic and serotonergic neuronal systems in mice lacking the NMDA receptor ε1 subunit. J Neurosci. 2001;21:750–757. doi: 10.1523/JNEUROSCI.21-02-00750.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffitt TE, Caspi A, Rutter M. Strategy for investigating interactions between measured genes and measured environments. Arch Gen Psychiatry. 2005;62:473–481. doi: 10.1001/archpsyc.62.5.473. [DOI] [PubMed] [Google Scholar]
- Mohn AR, Gainetdinov RR, Caron MG, Koller BH. Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell. 1999;98:427–436. doi: 10.1016/s0092-8674(00)81972-8. [DOI] [PubMed] [Google Scholar]
- Moreira EG, Vassilieff I, Vassilieff VS. Developmental lead exposure: behavioral alterations in the short and long term. Neurotoxicol Teratol. 2001;23:489–495. doi: 10.1016/s0892-0362(01)00159-3. [DOI] [PubMed] [Google Scholar]
- Morrow BA, Elsworth JD, Roth RH. Repeated phencyclidine in monkeys results in loss of parvalbumin-containing axo-axonic projections in the prefrontal cortex. Psychopharmacol. 2007;192:283–290. doi: 10.1007/s00213-007-0708-0. [DOI] [PubMed] [Google Scholar]
- Murray RM, Jones P, O'Callaghan E, Takei N, Sham P. Genes, viruses and neurodevelopmental schizophrenia. J Psychiatr Res. 1992 Oct;26(4):225–35. doi: 10.1016/0022-3956(92)90029-n. [DOI] [PubMed] [Google Scholar]
- Namba T, Ming GL, Song H, Waga C, Enomoto A, Kaibuchi K, Kohsaka S, Uchino S. NMDA receptor regulates migration of newly generated neurons in the adult hippocampus via Disrupted-In-Schizophrenia 1 (DISC1) J Neurochem. 2011;118(1):34–44. doi: 10.1111/j.1471-4159.2011.07282.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal AP, Guilarte TR. Molecular Neurobiology of Pb2+: effects on synaptic function. Mol Neurobiol. 2010;42:281–289. doi: 10.1007/s12035-010-8146-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal AP, Stansfield KH, Worley PF, Thompson RE, Guilarte TR. Lead exposure during synaptogenesis alters vesicular proteins and impairs vesicular release: potential role of NMDA receptor-dependent BDNF signaling. Tox Sci. 2010;116:249–263. doi: 10.1093/toxsci/kfq111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needleman HL, Gunnoe C, Leviton A, Reed R, Peresie H, Maher C, Barret P. Deficits in psychologic and classroom performance of children with elevated dentine lead levels. N Engl J Med. 1979;300:689–695. doi: 10.1056/NEJM197903293001301. [DOI] [PubMed] [Google Scholar]
- Needleman HL, Schell A, Bellinger D, Leviton A, Allred EN. The long-term effects of exposure to low doses of lead in childhood. An 11-year follow-up report. N Engl J Med. 1990 Jan 11;322(2):83–8. doi: 10.1056/NEJM199001113220203. [DOI] [PubMed] [Google Scholar]
- Needleman HL, Riess JA, Tobin MJ, Biesecker GE, Greenhouse JB. Bone lead levels and delinquent behavior. J Am Med Assoc. 1996;275:363–369. [PubMed] [Google Scholar]
- Nevin R. Understanding international crime rates: the legacy of preschool lead exposure. Env Res. 2007;104:315–336. doi: 10.1016/j.envres.2007.02.008. [DOI] [PubMed] [Google Scholar]
- Nieratschker V, Nothen MM, Rietschel M. New genetic findings in schizophrenia: is there still room for the dopamine hypothesis of schizophrenia? Frontiers Beh Neurosci. 2010;4:1–10. doi: 10.3389/fnbeh.2010.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nihei MK, Desmond NL, McGlothan JL, Kuhlmann AC, Guilarte TR. N-methyl-D-aspartare receptor subunit changes are associated with lead-induced deficits of long-term potentiation and spatial learning. Neurosci. 2000;99:233–242. doi: 10.1016/s0306-4522(00)00192-5. [DOI] [PubMed] [Google Scholar]
- Nihei MK, Guilarte TR. NMDAR-2A subunit protein expression is reduced in the hippocampus of rats exposed to Pb2+ during development. Brain Res Mol Brain Res. 1999;66:42–49. doi: 10.1016/s0169-328x(99)00005-4. [DOI] [PubMed] [Google Scholar]
- Nihei MK, Guilarte TR. Molecular changes in glutamatergic synapses induced by Pb2+: Association with deficits of LTP and spatial learning. NeuroToxicol. 2001;22:635–643. doi: 10.1016/s0161-813x(01)00035-3. [DOI] [PubMed] [Google Scholar]
- Nudmamud-Thanoi S, Reynolds GP. The NR1 subunit of the glutamate/NMDA receptor in the superior temporal cortex in schizophrenia and affective disorders. Neurosci Lett. 2004;372:173–177. doi: 10.1016/j.neulet.2004.09.035. [DOI] [PubMed] [Google Scholar]
- Oliet SHR, Mothet JP. Regulation of methyl-d-aspartate receptors by astrocytic D-serine. Neurosci. 2009:275–283. doi: 10.1016/j.neuroscience.2008.01.071. [DOI] [PubMed] [Google Scholar]
- Opler MGA, Brown AS, Graziano J, Desai M, Zheng W, Schaefer C, Factor-Litvak P, Susser ES. Prenatal lead exposure, δ-Aminolevulinic acid, and Schizophrenia. Env Hlth Persp. 2004;112:548–552. doi: 10.1289/ehp.6777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opler MG, Buka SL, Groeger J, McKeague I, Wei C, Factor-Litvak P, Bresnahan M, Graziano J, Goldstein JM, Seidman LJ, Brown AS, Susser ES. Prenatal exposure to lead, delta-aminolevulinic acid, and schizophrenia: further evidence. Environ Health Perspect. 2008;116:1586–90. doi: 10.1289/ehp.10464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panatier A, Theodosis DT, Mothet JP, Touquet B, Lollegioni L, Poulain DA, Oliet SHR. Glia-derived D-serine controls NMDA receptor activity and synaptic memory. Cell. 2006;125:775–784. doi: 10.1016/j.cell.2006.02.051. [DOI] [PubMed] [Google Scholar]
- Paule MG, Li M, Allen RR, Liu F, Zou X, Hotchkiss C, Hanig JP, Patterson TA, Slikker W, Jr, Wang C. Ketamine anesthesia during the first week of life can cause long-lasting cognitive deficits in rhesus monkeys. Neurotoxicol Teratol. 2011;33:220–230. doi: 10.1016/j.ntt.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearlson GD. Neurobiology of Schizophrenia. Ann Neurol. 2000;48:556–566. [PubMed] [Google Scholar]
- Pedersen CB, Mortensen PB. Urbanization and traffic related exposures as risk factors for schizophrenia. BMC Psychiatry. 2006 Jan 19;6:2. doi: 10.1186/1471-244X-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen CB, Raaschou-Nielsen O, Hertel O, Mortensen PB. Air pollution from traffic and schizophrenia risk. Schizophr Res. 2004 Jan 1;66(1):83–5. doi: 10.1016/s0920-9964(03)00062-8. [DOI] [PubMed] [Google Scholar]
- Pletnikov MV, Ayhan Y, Nikolskaia O, Xu Y, Ovanesov MV, Huang H, Mori S, Moran TH, Ross CA. Inducible expression of mutant human DISC1 in mice is associated with brain and behavioral abnormalities reminiscent of schizophrenia. Mol Psychiatry. 2008;13:173–186. 115. doi: 10.1038/sj.mp.4002079. [DOI] [PubMed] [Google Scholar]
- Petit TL, Alfano DP. Differential experience following developmental lead exposure: effects on brain and behavior. Pharmacol Biochem Bev. 1979;11:165–171. doi: 10.1016/0091-3057(79)90009-1. [DOI] [PubMed] [Google Scholar]
- Petit TL, Alfano DP, LeBoutillier JC. Early lead exposure and the hippocampus: a review and recent advances. NeuroToxicol. 1983;4:79–94. [PubMed] [Google Scholar]
- Petit TL, LeBoutillier JC. Effects of lead exposure during development on neocortical dendritic and synaptic structure. Exp Neurol. 1979;64:482–492. doi: 10.1016/0014-4886(79)90226-7. [DOI] [PubMed] [Google Scholar]
- Pocock SJ, Smith M, Baghurst P. Environmental lead and children's intelligence: a systematic review of the epidemiological evidence. BMJ. 1994;309:1189–97. doi: 10.1136/bmj.309.6963.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokora MJ, Richfield EK, Cory-Slechta DA. Preferantial vulnerability of nucleus accumbens dopamine binding sites to low-level lead exposure: time course of effects and interactions with chronic dopamine agonist treatment. J Neurochem. 1996;67:1540–1550. doi: 10.1046/j.1471-4159.1996.67041540.x. [DOI] [PubMed] [Google Scholar]
- Porteous DJ, Thomson P, Brandon NJ, Millar JK. The genetics and biology of DISC1--an emerging role in psychosis and cognition. Biol Psychiatry. 2006;60:123–131. doi: 10.1016/j.biopsych.2006.04.008. [DOI] [PubMed] [Google Scholar]
- Ramsey AJ, Milenkovic M, Oliveira AF, Escobedo-Lozoya Y, Seshadri S, Salahpour A, Sawa A, Yasuda R, Caron MG. Impaired NMDA receptor transmission alters striatal synapses and DISC1 protein in an age-dependent manner. Proc Natl Acad Sci U S A. 2011;108(14):5795–800. doi: 10.1073/pnas.1012621108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripoll N, Bronnec M, Bourin M. Nicotinic receptors and Schizophrenia. Curr Med Res Opinion. 2004;20:1057–1074. doi: 10.1185/030079904125004060. [DOI] [PubMed] [Google Scholar]
- Ris MD, Dietrich KN, Succop PA, Berger OG, Bornschein RL. Early exposure to lead and neuropsychological outcome in adolescence. J Int Neuropsychol Soc. 2004;10:261–70. doi: 10.1017/S1355617704102154. [DOI] [PubMed] [Google Scholar]
- Rizos EN, Papathanasian M, Michalopoulou PG, Mazioti A, Douzenis A, Kastania A, Nikolaidou P, Laskos E, Vasilopoulou K, Lykouras L. Association of serum BDNF levels with hippocampal volumes in first psychotic episode drug-naïve schizophrenia patients. Schiz Res. 2011 doi: 10.1016/j.schres.2011.03.011. EPub. [DOI] [PubMed] [Google Scholar]
- Roberts GW. Schizophrenia: a neuropathological perspective. Br J Psych. 1991;158:8–17. doi: 10.1192/bjp.158.1.8. [DOI] [PubMed] [Google Scholar]
- Ross BD, Blum S, Cowan R, Danielsen E, Farrow N, Gruetter R. In vivo magnetic resonance spectroscopy of human brain: the biophysical basis of dementia. Biophys Chem. 1997;68:161–172. doi: 10.1016/s0301-4622(97)00032-x. [DOI] [PubMed] [Google Scholar]
- Ross CA, Margolis RL, Reading SAJ, Pletnikov M, Coyle JT. Neurobiology of Schizophrenia. Neuron. 2006;52:139–153. doi: 10.1016/j.neuron.2006.09.015. [DOI] [PubMed] [Google Scholar]
- Roy A, Bellinger D, Hu H, Schwartz J, Ettinger AS, Wright RO, Bouchard M, Palaniappan K, Balakrishnan K. Lead exposure and behavior amoung young children in Chennai, India. Env Hlth Perp. 2009;107:1607–1611. doi: 10.1289/ehp.0900625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakata K, Woo NH, Martinowick K, Greene JS, Schloesser RJ, Shen L, Lu B. Critical role of promoter IV-driven BDNF transcription in GABAergic transmission and synaptic plasticity in prefrontal cortex. Proc Nat Acad Sci. 2009;106:5942–5947. doi: 10.1073/pnas.0811431106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnieder TP, Dwork AJ. Searching for Neuropathology: Gliosis in Schizophrenia. Biol Psych. 2011;69:134–139. doi: 10.1016/j.biopsych.2010.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz BS, Stewart WF, Simon PD, Bandeen-Roche K, Gordon PB, Links JM, Todd AC. Past adult lead exposure is associated with longitudinal decline in cognitive function. Neurology. 2000;55:1144–1150. doi: 10.1212/wnl.55.8.1144. [DOI] [PubMed] [Google Scholar]
- Seeman P, Lee T. Antipsychotic drugs: direct correlation between clinical potency and presynaptic action on dopamine neurons. Science. 1975;188:1217–1219. doi: 10.1126/science.1145194. [DOI] [PubMed] [Google Scholar]
- Sharifi AM, Baniasadi S, Jorjani M, Rahimi F, Bakhshayesh M. Investigation of acute lead poisoning on apoptosis in the rat hippocampus in vivo. Neurosci Lett. 2002;329:45–48. doi: 10.1016/s0304-3940(02)00576-1. [DOI] [PubMed] [Google Scholar]
- Sharifi AM, Hadi-Mousani S, Jorjani M. Effect of chronic lead exposure on pro-apoptotic Bax and anti-apoptotic Bcl-2 protein expression in rat hippocampus in vivo. Cell Mol Neurobiol. 2010;30:769–774. doi: 10.1007/s10571-010-9504-1. [DOI] [PubMed] [Google Scholar]
- Shen S, Lang B, Nakamoto C, Zhang F, Pu J, Kuan SL, Chatzi C, He S, Mackie I, Brandon NJ, Marquis KL, Day M, Hurko O, McCaig CD, Riedel G, St Clair D. Schizophrenia-related neural and behavioral phenotypes in transgenic mice expressing truncated Disc1. J Neurosci. 2008;28:10893–10904. doi: 10.1523/JNEUROSCI.3299-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih RA, Glass TA, Bandeen-Roche K, Carlson MC, Bolla KI, Todd AC, Schwartz BS. Environmental lead exposure and cognitive function in community-dwelling older adults. Neurology. 2006;67:1556–1562. doi: 10.1212/01.wnl.0000239836.26142.c5. [DOI] [PubMed] [Google Scholar]
- Simpsom EH, Kellendonk C, Kandel E. A possible role for the striatum in the pathogenesis of the cognitive symptoms of schizophrenia. Neuron. 2010;65:585–596. doi: 10.1016/j.neuron.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson MDC, Slater P, Royston MC, Deakin JFW. Alterations in phencyclidine and sigma binding sites in schizophrenic brains – Effects of disease process and neuroleptic medication. Schiz Res. 1992;6:41–48. doi: 10.1016/0920-9964(91)90019-n. [DOI] [PubMed] [Google Scholar]
- Slikker W, Jr, Zou H, Hotchkiss CE, Divine RL, Sadovova N, Twaddle NC, Doerge DR, Scallet AC, Patterson TA, Hanig JP, Paule MG, Wang C. Ketamine-induced neuronal cell death in the perinatal rhesus monkey. Tox Sci. 2007;98:145–158. doi: 10.1093/toxsci/kfm084. [DOI] [PubMed] [Google Scholar]
- Snyder SH. Catecholamines in the brain as mediators of amphetamine psychosis. Arch Gen Psychiatry. 1972;27:169–179. doi: 10.1001/archpsyc.1972.01750260021004. [DOI] [PubMed] [Google Scholar]
- Snyder SH. Dopamine receptors, neuroleptics, and schizophrenia. Am J Psych. 1981;138:460–464. doi: 10.1176/ajp.138.4.460. [DOI] [PubMed] [Google Scholar]
- Sohal VS, Zhang F, Yizhar O, Deisseroth K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature. 2009;459:698–702. doi: 10.1038/nature07991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolov BP. Expression of NMDAR1, GluR1, GluR7, and KA1 glutamate receptor mRNAs is decreased in frontal cortex of “neuroleptic-free” schizophrenics: evidence on reversible up-regulation by typical neuroleptics. J Neurochem. 1998;71:2454–2464. doi: 10.1046/j.1471-4159.1998.71062454.x. [DOI] [PubMed] [Google Scholar]
- Soriano SG, Lin Q, Li J, Lin JR, Han XH, Kanter JL, Bajic D, Ibla JC. Ketamine activates cell cycle signaling and apoptosis in the neonatal brain. Anesthesiol. 2010;112:1155–1163. doi: 10.1097/ALN.0b013e3181d3e0c2. [DOI] [PubMed] [Google Scholar]
- Stansfield KH, Pilsner JR, Lu Q, Wright RO, Guilarte TR. Dysregulation of BDNF-TrkB signaling in developing hippocampal neurons by Pb2+: implications for an environmental basis of neurodevelopmental disorders. 2011 doi: 10.1093/toxsci/kfs090. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart WF, Schwartz BS. Effects of lead on the adult brain: a 15-year exploration. Am J Ind Med. 2007;50:729–739. doi: 10.1002/ajim.20434. [DOI] [PubMed] [Google Scholar]
- Stewart WF, Schwartz BS, Davatzikos C, Shen D, Liu D, Wu X, Todd AC, Shi W, Bassett S, Youssem D. Past adult lead exposure is linked to neurodegeneration measured by brain MRI. Neurology. 2006;66:1476–1484. doi: 10.1212/01.wnl.0000216138.69777.15. [DOI] [PubMed] [Google Scholar]
- Stork C, Renshaw PF. Mitochondrial dysfunction in bipolar disorder: evidence from magnetic resonance spectroscopy research. Mol Psych. 2005;10:900–919. doi: 10.1038/sj.mp.4001711. [DOI] [PubMed] [Google Scholar]
- Struzynska L, Sulkowski G. Relationships between glutamine, glutamate ad GABA in nerve endings under Pb-toxicity conditions. J Inorg Biochem. 2004;98:951–958. doi: 10.1016/j.jinorgbio.2004.02.010. [DOI] [PubMed] [Google Scholar]
- Sumiyoshi T, Anil AE, Jin D, Jayathilake K, Lee M, Meltzer HY. Plasma glycine and serine levels in schizophrenia compared to normal controls and major depression: relation to negative symptoms. Int J neuropsychopharmacol. 2004;7:1–8. doi: 10.1017/S1461145703003900. [DOI] [PubMed] [Google Scholar]
- Sun H, Wang HL, Wang S. D-serine relieves lead exposure-impaired long-term potentiation in the CA1 region of the rat hippocampus in vitro. Neurosci Lett. 2007;417:118–122. doi: 10.1016/j.neulet.2007.01.085. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Geyer MA. Using an animal model of deficient sensorimotor gating to study the pathophysiology and new treatments of schizophrenia. Schizophr Bull. 1998;24(2):285–301. doi: 10.1093/oxfordjournals.schbul.a033326. [DOI] [PubMed] [Google Scholar]
- Thaker G, Carpenter WT., Jr Advances in schizophrenia. Nat Med. 2001;7:667–671. doi: 10.1038/89040. [DOI] [PubMed] [Google Scholar]
- Tiffany-Castiglion E, Qian Y. Astroglia as metal depots: molecular mechanisms for metal accumulation, storage and release. Neurotoxicol. 2001;22:577–592. doi: 10.1016/s0161-813x(01)00050-x. [DOI] [PubMed] [Google Scholar]
- Timofeeva OA, Levin ED. Glutamate and nicotinic receptor interactions in working memory: importance for cognitive impairment of schizophrenia. Neurosci. 2011 doi: 10.1016/j.neuroscience.2011.08.038. EPub. [DOI] [PubMed] [Google Scholar]
- Torrey EF, Miller J, Rawlings R, Yolken RH. Seasonality of births in schizophrenia and bipolar disorders: a review of the literature. Schizophr Res. 1997;28:1–38. doi: 10.1016/s0920-9964(97)00092-3. [DOI] [PubMed] [Google Scholar]
- Toscano CD, Guilarte TR. Lead neurotoxicity: from exposure to molecular effects. Brain Res Rev. 2005;49:529–554. doi: 10.1016/j.brainresrev.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Toscano CD, Hashemzadeh-Gargari H, McGlothan JL, Guilarte TR. Developmental Pb2+ exposure alters NMDAR subtypes and reduces CREB phosphorylation in the rat brain. Dev Brain Res. 2002;139:217–226. doi: 10.1016/s0165-3806(02)00569-2. [DOI] [PubMed] [Google Scholar]
- Trope I, Lopez-Villegas D, Lenkinski RE. Magnetic Resonance Imaging and Spectroscopy of regional brain structure in a 10-yeay-old boy with elevated lead levels. Pediatrics. 1998;101(6):e7. doi: 10.1542/peds.101.6.e7. [DOI] [PubMed] [Google Scholar]
- Trope I, Lopez-Villegas D, Cecil KM, Lenkinski RE. Exposure to lead appears to selectively alter metabolism of cortical gray matter. Pediatrics. 2001;107:1437–1442. doi: 10.1542/peds.107.6.1437. [DOI] [PubMed] [Google Scholar]
- Tsai G, Coyle JT. Glutamatergic mechanisms in Schizophrenia. Ann Rev Pharmacol Toxicol. 2002;42:165–179. doi: 10.1146/annurev.pharmtox.42.082701.160735. [DOI] [PubMed] [Google Scholar]
- Tsuang M. Schizophrenia: Genes and Environment. Biol Psychiatry. 2000;47:210–220. doi: 10.1016/s0006-3223(99)00289-9. [DOI] [PubMed] [Google Scholar]
- Uhlhaas PJ, Singer W. Abnormal neural oscillations and synchrony in schizophrenia. Nature Rev Neurosci. 2010;11:100–113. doi: 10.1038/nrn2774. [DOI] [PubMed] [Google Scholar]
- Van den Buuse M, Garner B, Koch M. Neurodevelopmental animal models of schizophrenia: effects on prepulse inhibition. Curr Mol Med. 2003;3:459–471. doi: 10.2174/1566524033479627. [DOI] [PubMed] [Google Scholar]
- Van Os J, Kenis G, Rutten PF. The environment and schizophrenia. Nature. 2010;468:203–212. doi: 10.1038/nature09563. [DOI] [PubMed] [Google Scholar]
- Van Os J, Rutten BP, Poulton R. Gene-environment interactions in schizophrenia: review of epidemiological findings and future directions. Schizophr Bull. 2008;34:1066–1082. doi: 10.1093/schbul/sbn117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoeven WM, Egger JI, Kuijpers HJ. Manganese and acute paranoid psychosis: a case report. J Med Case Rep. 2011;5:146–148. doi: 10.1186/1752-1947-5-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verina T, Rohde C, Guilarte TR. Environmental lead exposure in early life alters granule cell neurogenesis and morphology in the hippocampus of young adult rats. Neurosci. 2007;28:1147–1152. doi: 10.1016/j.neuroscience.2006.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrajova M, Stastny F, Horacek J, Lochman J, Sery O, Pekova J, Klaschka J, Hoschl C. Expression of the hippocampal NMDA receptor GluN1 subunit and its splicing isoforms in schizophrenia: postmortem study. Neurochem Res. 2010;35:994–1002. doi: 10.1007/s11064-010-0145-z. [DOI] [PubMed] [Google Scholar]
- Wang C, McInnis J, Ross-Sanchez M, Shinnich-Gallagher P, Wiley JL, Johnson KM. Long-term behavioral and neurodegenerative effects of perinatal phencyclidine administration: implications for schizophrenia. Neurosci. 2001;107:533–550. doi: 10.1016/s0306-4522(01)00384-0. [DOI] [PubMed] [Google Scholar]
- Wasserman GA, Liu X, Popovac D, Factor-Litvak P, Kline J, Waternaux C, LoIacono N, Graziano JH. The Yugoslavia Prospective Lead Study: contributions of prenatal and postnatal lead exposure to early intelligence. Neurotoxicol Teratol. 2000;22:811–8. doi: 10.1016/s0892-0362(00)00106-9. [DOI] [PubMed] [Google Scholar]
- Wasserman GA, Liu X, Parvez F, Ahsan H, Levy D, Factor-Litvak P, Kline J, van Geen A, Slavkovich V, Lolacono NJ, Cheng Z, Zheng Y, Graziano JH. Water manganese exposure and children's intellectual function in Araihazar, Bangladesh. Env Hlth Persp. 2006;114:124–129. doi: 10.1289/ehp.8030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weickert CS, Hyde TM, Lipska BK, Herman MM, Weinberger DR, Kleinman JE. Reduced brain-derived neurotrophic factor in prefrontal cortex of patients with schizophrenia. Mol Psych. 2003;8:592–610. doi: 10.1038/sj.mp.4001308. [DOI] [PubMed] [Google Scholar]
- Weinberger DR. On the plausibility of “the neurodevelopmental hypothesis” of schizophrenia. Neuropsychopharmacology. 1996 Mar;14(3 Suppl):1S–11S. doi: 10.1016/0893-133X(95)00199-N. [DOI] [PubMed] [Google Scholar]
- Weiss IC, Feldon J. Environmental animal models for sensorimotor gating deficiencies in schizophrenia: a review. Psychopharmacol. 2001;156:305–326. doi: 10.1007/s002130100800. [DOI] [PubMed] [Google Scholar]
- Weisskopf MG, Hu H, Mulken RV, White R, Aro A, Oliviera S, Wright RO. Cognitive deficits and Magnetic Resonance Spectroscopy in adult monozygotic twins with lead poisoning. Env Hlth Persp. 2004;112:620–625. doi: 10.1289/ehp.6687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White LD, Cory-Slechta DA, Gilbert ME, Tiffany-Castiglioni E, Zawia NH, Virgolini M, Rossi-George A, Lasley SM, Qian YC, Basha R. New and evolving concepts in the neurotoxicology of lead. Tox Appl Pharmacol. 2007;225:1–27. doi: 10.1016/j.taap.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Winder C, Garten LL, Lewis PD. The morphological effects of lead on the developing central nervous system. Neuropath Appl Neurobiol. 1983;9:87–108. doi: 10.1111/j.1365-2990.1983.tb00328.x. [DOI] [PubMed] [Google Scholar]
- Winder C, Kitchen I. Lead neurotoxicity: review of the biochemical and neurochemical and drug induced behavioural evidence. Prog Neurobiol. 1984;22:59–87. doi: 10.1016/0301-0082(84)90018-2. [DOI] [PubMed] [Google Scholar]
- Winneke G, Brockhaus A, Baltissen R. Neurobehavioral and systemic effects of long-term blood lead elevation in rats. Arch Toxicol. 1977;37:247–263. doi: 10.1007/BF00330817. [DOI] [PubMed] [Google Scholar]
- Wong J, Hyde TM, Cassano HL, Deep-Soboslay A, Kleinman JE, Shannon Weickert C. Promoter specific alterations of brain-derived neurotrophic factor mRNA in schizophrenia. Neurosci. 2010;169:1071–1084. doi: 10.1016/j.neuroscience.2010.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo TUW, Walsh JP, Benes FM. Density of glutamic acid decarboxylase 67 messenger RNA-containing neurons that express the N-methyl-d-aspartate receptor subunit NR2A in the anterior cingulate cortex in schizophrenia and bipolar disorder. Arch Gen Psych. 2004;61:649–657. doi: 10.1001/archpsyc.61.7.649. [DOI] [PubMed] [Google Scholar]
- Woods BT, Yurgelum-Todd D, Goldstein JM, Seidman LJ, Tsuang MT. MRI brain abnormalities in chronic schizophrenia: one process or more? Biol Psychiatry. 1996;40:585–596. doi: 10.1016/0006-3223(95)00478-5. [DOI] [PubMed] [Google Scholar]
- Wormley DD, Chirwa S, Nayvar T, Wu J, Johnson S, Brown LA, Harris E, Hood DB. Inhaled benzo(a)pyrene impairs long-term potentiation in the F1 generation rat dentate gyrus. Cell Mol Biol. 2004;50:715–721. [PubMed] [Google Scholar]
- Xi D, Zhang W, Wang HX, Stradman GG, III, Gao WJ. Dizocilpine (MK-801) induces distinct changes in N-methyl-d-aspartic acid receptor subunits in parvalbumin-containing interneurons in young adult rat prefrontal cortex. Int J Neuropsycopharm. 2009;12:1395–1408. doi: 10.1017/S146114570900042X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Ji LD, Xu LH. Lead-induced apoptosis in PC12 cells: involvement of p53, Bcl-2 family and caspase-3. Toxicol Lett. 2006;166:160–167. doi: 10.1016/j.toxlet.2006.06.643. [DOI] [PubMed] [Google Scholar]
- Yin LM, Rhoads GG, Lioy PJ. Seasonal influences on childhood lead exposure. Env Hlth Persp. 2000;108:177–182. doi: 10.1289/ehp.00108177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuede CM, Wozniak DF, Creeley CE, Taylor GT, Olney JW, Farber NB. Behavioral consequences of NMDA antagonist-induced neuroapoptosis in the infant mouse brain. PLoS One. 2010:e11374. doi: 10.1371/journal.pone.0011374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XY, Liu AP, Ruan DY, Liu J. Effect of developmental lead exposureon the expression of specific NMDA receptor subunit mRNAs in the hippocampus of neonatal rats by digoxigenin-labeled in situ hybridization histochemistry. Neurotoxicol Teratol. 2002;24:149–160. doi: 10.1016/s0892-0362(01)00210-0. [DOI] [PubMed] [Google Scholar]
- Zou X, Patterson TA, Divine RL, Sadovova N, Zhang X, Hanig JP, Paule MG, Slikker W, Jr, Wang C. Prolonged exposure to ketamine increases neurodegenration in the developing monkey brain. Int J Dev Neurosci. 2009;27:727–731. doi: 10.1016/j.ijdevneu.2009.06.010. [DOI] [PubMed] [Google Scholar]
- Zuch CL, O'Mara DJ, Cory-Slechta DA. Low-level lead exposure selectively enhances dopamine overflow in nucleus accumbens: an in vivo electrochemistry time course assessment. Toxicol Appl Pharmacol. 1998;150:174–185. doi: 10.1006/taap.1998.8396. [DOI] [PubMed] [Google Scholar]