

Abstract
There are many misconceptions surrounding the roles of protein phosphatases in the regulation of signal transduction, perhaps the most damaging of which is the erroneous view that these enzymes exert their effects merely as constitutively active housekeeping enzymes. On the contrary, the phosphatases are critical, specific regulators of signaling in their own right and serve an essential function, in a coordinated manner with the kinases, to determine the response to a physiological stimulus. This review is a personal perspective on the development of our understanding of the protein tyrosine phosphatase (PTP) family of enzymes. I have discussed various aspects of the structure, regulation and function of the PTP family, which I hope will illustrate the fundamental importance of these enzymes to the control of signal transduction.
I have studied protein phosphatases for my entire career in research. After completing my PhD with Phil Cohen in Dundee, working on Ser/Thr phosphatases, I was fortunate to have the opportunity to work as a postdoctoral fellow with Eddy Fischer at the University of Washington in Seattle, during which time we purified to homogeneity and characterized the first Protein Tyrosine Phosphatase (PTP) – PTP1B [1, 2]. Since then, I have continued to work on these enzymes in my own lab. This review is based upon an introductory lecture that I presented at the 2011 EMBO Europhosphatases conference, ”Protein Phosphatases: From Molecules to Networks”. As with the companion lecture and review on Protein Ser/Thr Phosphatases from David Brautigan [3], the goal was to introduce the PTPs to newcomers to the field. To try and accomplish this, I have presented a personal perspective on historical developments in the study of these enzymes, illustrated some general principles governing the regulation of PTP function and the role of these enzymes in the regulation of cell signaling, and described my view of some of the major challenges we face in the future.
Through the pioneering work of Eddy Fischer and Ed Krebs, who discovered protein phosphorylation in the 1950s [4, 5], the genesis of the signal tranduction field can be found in the study of glycogen metabolism. In the early 60s, Danforth, Helmreich and Cori were studying the regulation of glycogen phosphorylase and published that “kinetic analysis suggests that changes in the phosphorylase b kinase rather than phosphorylase phosphatase activity are responsible for the increase and decrease in phosphorylase a” [6]. This statement has been a thorn in the side of all who have worked on protein phosphatases ever since! It fostered the view that the sophistication in regulation of signaling was manifested at the level of the kinases, with the phosphatases serving a general housekeeping function associated with maintenance of the basal state. This promoted an emphasis on the study of kinases in the signal transduction community, and a somewhat dismissive attitude to phosphatases, which were regarded as being of lesser importance. In fact, nothing could be further from the truth! Over the years a substantial body of data has revealed exquisite specificity in substrate recognition and function of protein phosphatases, emphasizing that kinases and phosphatases play essential, competing roles that are coordinated to determine the outcome of a physiological stimulus. Nevertheless, in this post-genomic era, we are now witnessing the advent of “systems biology” approaches to try and understand how signal transduction pathways are integrated at the level of the whole organism. Although mathematical and computational modeling studies are revealing new complexities in the coordination and regulation of signaling circuits, such models often downplay the contribution of phosphatases. Recent analysis of the evolution of phosphotyrosine-based signals has described a three-part toolkit that involves a “writer” (kinase), “reader” (SH2 domain) and “eraser” (phosphatase) [7]. The choice of “eraser” to describe the PTPs is unfortunate and conjures up the old images of phosphatases as merely switching pathways off and cleaning up after kinases. Overall, I hope that the examples described in this review will illustrate that PTPs are critical regulators of signaling in their own right, playing an essential role under normal and pathophysiological conditions and providing the basis for novel approaches to therapeutic intervention in major human diseases.
In the beginning there was chaos!
When I joined Eddy Fischer’s lab in 1985, there were clear indications of the importance of tyrosine phosphorylation in triggering signaling pathways associated with cell proliferation. The SRC oncogene was known to encode a protein tyrosine kinase, and growth factor-receptor PTKs had been identified and characterized; nevertheless, the nature of the phosphatases that provided regulatory balance to these PTKs was unknown. That such enzymes existed was evident in studies using temperature-sensitive mutants of SRC. Transformation at the permissive temperature was associated with a robust increase in the levels of tyrosine phosphorylation; however, shift to the non-permissive temperature was accompanied by a rapid decline in phosphotyrosine, close to the levels in an uninfected cell, indicative of the action of powerful PTPs [8]. But what were these enzymes?
Initial attempts to identify the PTPs responsible for this activity focused on whether the known phosphatases were capable of dephosphorylating tyrosyl residues in proteins. Of the Ser/Thr phosphatases, PP1 was devoid of PTP activity when purified from tissue. Subsequently, improperly folded recombinant PP1 was shown to display a low level of PTP activity that was lost when the enzyme was taken through an inactivation/reactivation cycle with the chaperone inhibitor-2 [9]. Low levels of PTP activity were reported in PP2A and PP2C, but this was dependent on non-physiological levels of Mn2+ and alkaline pH [10]. PP2B/calcineurin was shown to dephosphorylate EGF receptor in vitro, displaying a similar specific activity as its Ser/Thr phosphatase activity [11, 12]. Attention was also paid to broad specificity acid and alkaline phosphatases. The standard way to assay phosphatase activity in vitro was to use low Mr phosphate esters such as p-nitrophenylphosphate (pNPP). Although these enzymes did dephosphorylate pNPP, the identity of their physiological substrates, and hence their physiological function, was unknown. Nevertheless, there were data to suggest that acid phosphatases had the ability to dephosphorylate pTyr residues in proteins. Prostatic acid phosphatase, in particular, has been linked to dephosphorylation of receptor PTKs [13].
In addition to the above, there were several preliminary observations that pointed to the potential for a new class of phosphatases that dephosphorylated tyrosyl residues in proteins. Multiple fractions containing PTP activity had been identified and partially purified from a variety of different sources [14–17], with some interesting, novel properties being revealed. Vanadate was recognized as a PTP inhibitor [18] and is still used as such to date in many studies. David Brautigan and colleagues also identified Zn2+ ions as specific inhibitors [19] and used this in an affinity purification step to enrich PTPs from extracts of rabbit kidney [20, 21]. In addition, there were indications that the PTPs were dependent upon reducing agents for activity. Nevertheless, the identity of the PTPs remained elusive.
Technical challenges associated with working with phosphatases in general, and PTPs in particular
The major problem facing those interested in the PTPs in the mid-80s was reminiscent of that encountered with the protein Ser/Thr phosphatases some twenty years earlier – the literature was replete with numerous reports of multiple PTPs, variously distributed between soluble and particulate fractions of extracts from a wide array of tissues and cells. How, or whether, such fractions were related was unclear, and it was likely that susceptibility to proteolysis added a further level of complication, as also encountered with the Ser/Thr phosphatases. Although some fractions had been significantly enriched [20, 22, 23], none were in homogeneous form.
In the study of kinases, use of γ32P-ATP permits the measurement of activity by following incorporation of radioactively labelled phosphate into a potential target substrate. In contrast, a major technical challenge facing those studying phosphatases is the requirement first to have a purified, suitably phosphorylated substrate with which to measure enzyme activity. At this time, the problem was even more acute in the study of PTPs because the physiological tyrosine phosphorylated protein substrates had yet to be fully characterized. The known substrates, such a receptor PTKs, had yet to be generated in recombinant form and so were present in limiting amounts that did not permit their use as substrates in routine assays. People turned to artificial substrates, including proteins that were chemically modified to expose tyrosyl residues for phosphorylation in vitro, such as BSA [20] and phosphorylase [14]; however, these faced the drawbacks of poor solubility, low stoichiometry of phosphorylation and labeling of multiple tyrosyl residues in the protein. Consequently, the range of concentrations at which such substrates could be used was restricted and the heterogeneous labeling could result in non-linear kinetics of dephosphorylation. The identification of a suitable substrate was the first major obstacle to be overcome.
Reduced, Carboxamidomethylated and Maleylated Lysozyme (RCML) – a significant tool in the characterization of PTP activity
We tested several potential substrates [1, 2], but focused primarily on chemically modified derivatives of lysozyme, which had been reported as substrates of the insulin receptor [24]. We encountered problems with poor solubility when using these published derivatives. To address this, we introduced a second chemical modification, maleylation of lysyl residues, which improved solubility and avoided the harsh treatments with acid and alkali that result in hydrolysis during the solubilization of the modified protein. In addition, we focused considerable attention on optimizing conditions for the phosphorylation of RCML, using insulin and EGF receptor preparations that were partially purified from human placenta membranes. The result was a substrate that could be readily prepared in large quantities, was phosphorylated on a single tyrosine to a high stoichiometry (>0.5 mol/mol), and which formed the basis for a robust, flexible assay over a wide range of substrate concentrations [1]. This was crucial for success in purifying a PTP to homogeneity.
We chose human placenta as the tissue source because it was known to be rich in protein tyrosine kinase activity [25] and we found high levels of PTP activity in tissue extracts [1]; also, there was the novelty of working on a human enzyme. We observed that the majority of the activity in a placental extract was retained on DEAE-cellulose, eluting before the known Ser/Thr phosphatases; the remainder could be resolved into a cationic pool, which bound to phosphocellulose, CM-Sepharose etc, and a neutral pool, which was not retained on ion exchange columns. Even these limited data pointed to diversity in the spectrum of cellular PTPs. We focused on the fraction retained on DEAE-cellulose and noted that two major peaks were eluted with a salt gradient. The first, eluting at 50–70 mM NaCl, was termed PTP1A (and was subsequently resolved into two fractions, 1A and 1A’, on poly-lysine Sepharose) and the second, quantitatively larger peak, eluting at 90–100 mM NaCl, was termed PTP1B.
We applied well-established chromatographic procedures to the purification of PTP1A and 1B, but even after seven steps the preparations were far from pure. What was needed was an effective affinity chromatography step, which is where RCML was again instrumental. During undergraduate [26] and PhD research [27], I had applied the ability of some kinases to use ATPγS, instead of ATP, to create thiophosphorylated proteins that are phosphatase-resistant. Thiophosphorylated “substrates” are recognized by phosphatases, but are resistant to de-thiophosphorylation. Using insulin and EGF receptor PTKs, which are able to utilize ATPγS as a phosphate donor, we produced thiophosphorylated RCML that was labeled to a relatively high stoichiometry. By immobilizing this derivative on a Sepharose support, we created a powerful substrate affinity column that was the critical final step in isolating PTP1B in a pure form, enriched some 23,000 fold from aqueous soluble placental extracts [1].
Both PTP1A and PTP1B were isolated as monomeric catalytic subunits of ~35kDa. Although the catalytic subunits of the Ser/Thr phosphatases are of similar size [3], they could be distinguished from these PTPs by their enzymatic properties and by peptide mapping. Unlike the Ser/Thr phosphatases, these PTPs were potently inhibited by Zn2+, vanadate and molybdate, and were unaffected by sodium fluoride and the thermostable inhibitors of PP1. They were also insensitive to classical inhibitors of acid and alkaline phosphatases, including tartrate and tetramisole, suggesting a new class of phosphatase. Interestingly, we purified these PTPs from both the soluble (extracted in aqueous buffer) and membrane/particulate (extracted in the same buffer containing Triton detergent) fractions of placenta, with approximately equal amounts in each; at each step, the behavior of the various PTP forms suggested that the soluble enzymes had similar counterparts in the particulate fraction. In fact, peptide mapping of PTP1B illustrated that the enzyme from each fraction was the same, raising interesting questions about the basis for this distribution.
We noted a number of striking features, including that these PTPs were unable to dephosphorylate Ser or Thr residues in proteins; however, although specific for tyrosyl residues in proteins, they displayed broad specificity for pTyr proteins in assays in vitro. In addition, we observed a high specific activity and high affinity (sub-micromolar Km) for substrate, suggesting that these enzymes had the potential to be a formidable barrier to PTK function in a cell. The enzymes were totally dependent on reducing agents for activity – simply diluting them into assay buffer in the absence of reducing agent resulted in a total, but reversible, inhibition. These data suggested that at least one reactive Cys residue was essential for catalysis, a point that ultimately led to the identification of a new level of control of tyrosine phosphorylation-dependent signaling.
The two fractions that we identified, PTP1A and PTP1B, displayed preferential recognition of substrates and differential responses to potential modulators of activity in vitro [2]. Particularly striking was the differential effects of highly charged compounds on activity. Polycationic compounds, such as spermine and spermidine, stimulated PTP1B to a greater extent than 1A. Furthermore, polyanionic compounds, such as random copolymers of glutamate and tyrosine, which were used as artificial substrates of PTKs, were potent non-competitive inhibitors of PTP1B (IC50 ~50 nM), whereas 1–2 orders of magnitude higher concentrations were required for inhibition of PTP1A. These data suggested that the two fractions represented distinct PTP enzymes. Unfortunately, due to the lower levels of the PTP1A fraction than PTP1B in placental extracts, we never succeeded in isolating it in sufficient quantities for detailed sequence analysis. All these years later, the identity of PTP1A remains unknown!
Out of the chaos came a new family of Cys-dependent protein phosphatases that play fundamental roles in the regulation of cell signaling
After many long hours in the cold room, we succeeded in purifying milligram quantities of PTP1B! In collaboration with Harry Charbonneau and Ken Walsh at the University of Washington, we determined its amino acid sequence – in fact, PTP1B was probably one the last proteins to have its complete amino acid sequence determined by Edman degradation [28]! Around this time the impact of molecular biology, and the more ready availability of sequence data, was just beginning to be felt in the signaling field. The primary sequence of the catalytic subunits of PP1, 2A and 2B/calcineurin were available and revealed the existence of a family of structurally related phosphatases. The sequences of several acid and alkaline phosphatases were now also known, and were distinct. Our work added a further level of complexity, demonstrating that PTP1B was a member of a new class of phosphatases. This illustrates a fundamental difference between the evolution of kinases and phosphatases – whereas the protein kinases have evolved from a common ancestor, the phosphatases have evolved in structurally and mechanistically distinct enzyme families (Figure 1). Although distinct from the Ser/Thr phosphatases, we noted that the sequence of PTP1B bore a striking similarity to a transmembrane, receptor-like molecule found on nucleated hematopoietic cells, termed CD45 [29]. This was an observation that would have a profound effect on the field.
Figure 1. Protein phosphatases in signal transduction.
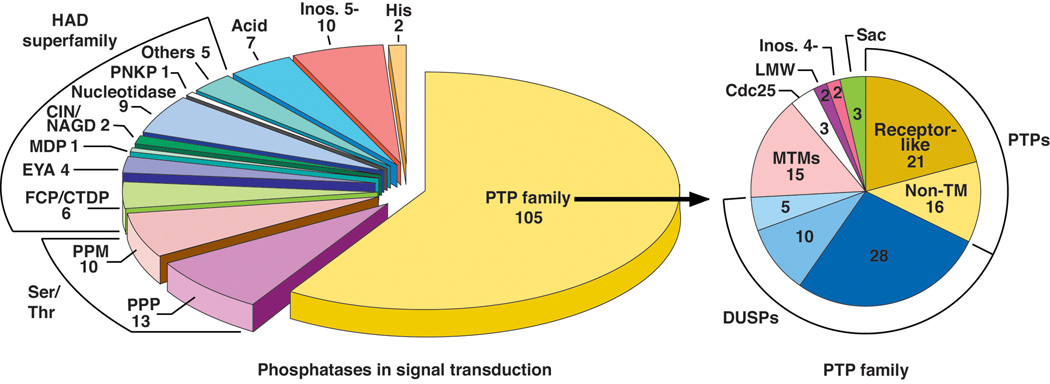
The phosphatases that are implicated in the regulation of signal transduction are highlighted on the left. They are represented by structurally and mechanistically distinct families. The major categories include the PPP and PPM Ser/Thr phosphatases [3, 47], the haloacid dehalogenase (HADS) [226] and the largest group, the Cys-dependent PTP family [44]. The breakdown of the PTPs into individual categories is shown on the right [44].
CD45, also known as T200 or the Leukocyte Common Antigen, was recognized as an abundant glycoprotein on the surface of leukocytes, which displayed cell type-specific heterogeneity in molecular weight and carbohydrate content. There was considerable interest in CD45 because antibodies that targeted the protein had been used to define functionally distinct populations of lymphocytes [30]. We were extremely fortunate that Matt Thomas, working both with Alan Williams and Ian Trowbridge, had defined the complete sequence of CD45 from cDNA clones and revealed its receptor-like structure [31, 32]. Variations in the highly glycosylated extracellular segment were explained by the alternative splicing of three exons, encoding sequences in the N-terminal portion of the protein [33], and suggested the possibility that different functions of CD45 could be elicited by interaction with distinct ligands, depending upon which CD45 isoform was expressed. Matt Thomas also noted that, as with the classical arrangement of growth receptor receptor PTKs, CD45 contained a single transmembrane domain and a conserved intracellular segment, the latter containing two tandemly repeated domains, each of ~300 residues [31, 32]. It was these repeated domains within the intracellular segment of CD45 that we noted as bearing a striking similarity to the sequence of PTP1B [28, 29]. Based on this observation, we went on to demonstrate that CD45 displayed intrinsic PTP activity [34]. This was important because it flew in the face of the notion of PTPs as housekeeping enzymes – instead, our data were consistent with the designation of CD45 as a receptor-linked PTP, suggesting that it may be a prototype for a family of proteins with the ability to regulate signal transduction directly through ligand-controlled dephosphorylation of tyrosyl residues in proteins [35, 36]. In further functional studies, Matt Thomas made another important contribution by generating cells that were deficient in expression of CD45 [37, 38]. These approaches not only revealed an important role for CD45 in the regulation of signaling through antigen receptors, but also demonstrated that CD45 could function positively, to promote signaling – further emphasizing an important direct role in switching on signaling pathways, rather than simply acting as a passive antagonist of PTK function (Figure 2). The mechanism for these effects lies in the regulation of SRC family PTKs [39, 40]. Dephosphorylation of an inhibitory, C-terminal site of phosphorylation leads to activation of SRC. Consequently, CD45 function illustrates the somewhat paradoxical situation that the activity of a PTP can result in enhanced tyrosine phosphorylation and cell signaling. These data illustrated that the response to a stimulus was governed by the coordinated and competing actions of both phosphatases and kinases, and emphasized the importance of characterizing tyrosine phosphorylation as a reversible process, with the PTPs serving as critical regulators of signaling in their own right.
Figure 2. Signaling function of Protein Tyrosine Phosphatases.
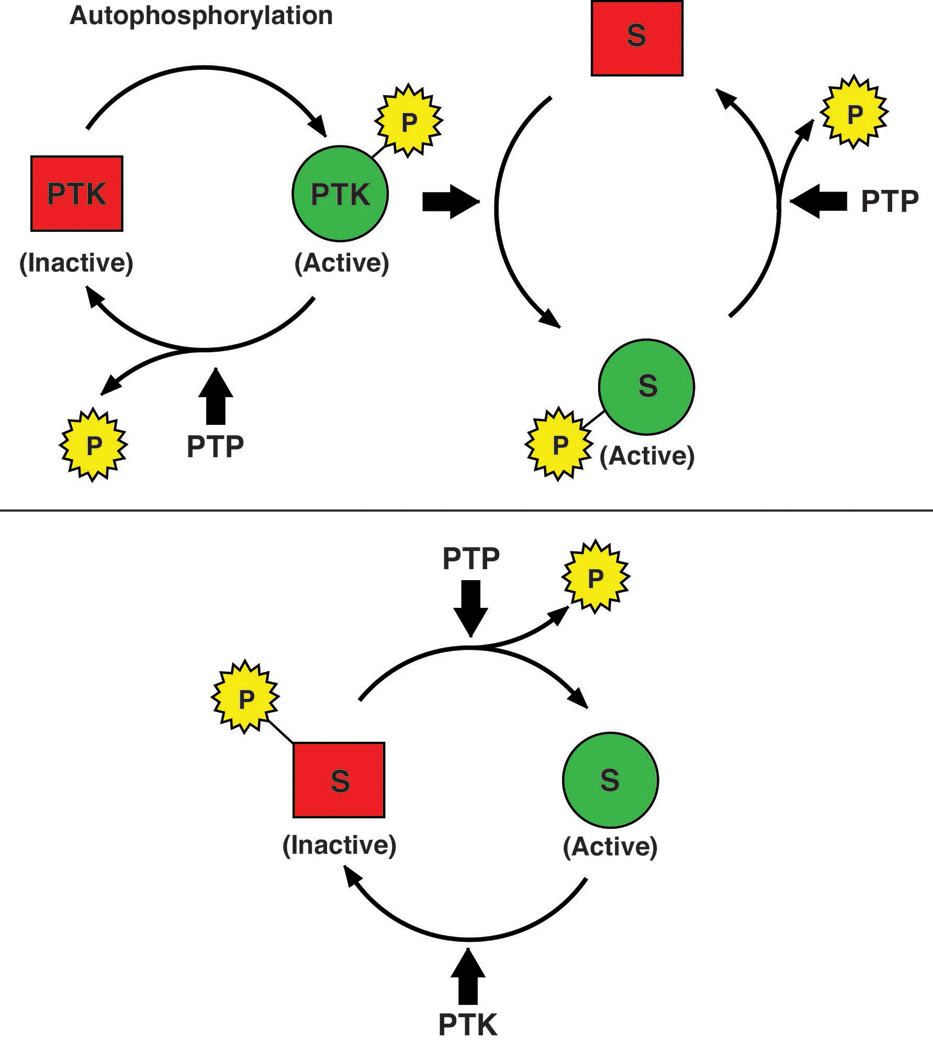
Members of the PTP family have the potential to act negatively in the regulation of signaling, by dephosphorylating autophosphorylation sites in PTKs themselves or phosphorylation sites in their downstream targets (upper panel). In addition, PTPs may play a positive role, for example by dephosphorylating an inhibitory site in a PTK, such as the C-terminal sites in SRC family PTKs, thereby activating the kinase and promoting phosphorylation and signaling (lower panel).
Receptor PTKs are widely distributed; consequently, if receptor PTPs were to be important regulators of signaling in general it would be expected that their expression would not be restricted to hematopoietic cells. The first non-hematopoietic cell RPTP to be identified was Leukocyte common Antigen-Related (LAR), which also displayed features of cell adhesion molecules [41]. This suggested that RPTPs may play an important role in regulating phenomena associated with cell-cell contact, such as contact inhibition of cell growth. What followed were contributions from many labs leading to the identification of multiple PTPs. The catalytic domain of the classical, pTyr specific PTPs spans ~280 residues and contains 10 conserved motifs [42–44] (Figure 3). All members of the PTP family are characterized by the presence of the signature motif, HC-(X5)-R (motif 9), which offered a structural explanation for the dependence of PTPs on reducing agents for activity [2]. Within this motif, the invariant Cys and Arg residues are essential for catalysis. This, together with two additional motifs, the WPD loop and the Q loop (Motifs 8 and 10) defines a minimal catalytic core. The pTyr-recognition loop, Motif 1, explains the selectivity for pTyr residues in substrates. The remaining motifs play a structural role in the catalytic domain. As for the protein kinases, the presence of these motifs allowed for the precise definition of a PTP domain based upon primary sequence. Ultimately, the PTP family was shown to comprise ~100 genes in humans [43–45]. Of these, 37 encode pTyr-specific enzymes, whereas the others are operationally defined as dual specificity phosphatases (DUSPs), which have the capacity to dephosphorylate both Tyr and Ser/Thr residues in proteins in vitro [46]. Nevertheless, as discussed below, these DUSPs may show preference for particular substrates in vivo. Overall, the PTPs are Cys-dependent enzymes that evolved independently three times to create three distinct groups [47]. The largest group comprises the classical pTyr-specific and DUSP enzymes. In addition, there is the Low Mr PTPs [48] and, finally, the rhodanese-derived PTP cdc25 [49]. The definition of the primary sequence of the PTPs also revealed distinct evolutionary routes to the current protein phosphatases. For the PTPs, an ancient phosphatase domain functionally evolved by fusing to additional domains that serve a regulatory function; in contrast, docking to novel subunits, giving rise to multi-subunit holoenzyme complexes, resulted in the Ser/Thr phosphatases [47].
Figure 3. The sequence motifs that define the conserved PTP catalytic domain.
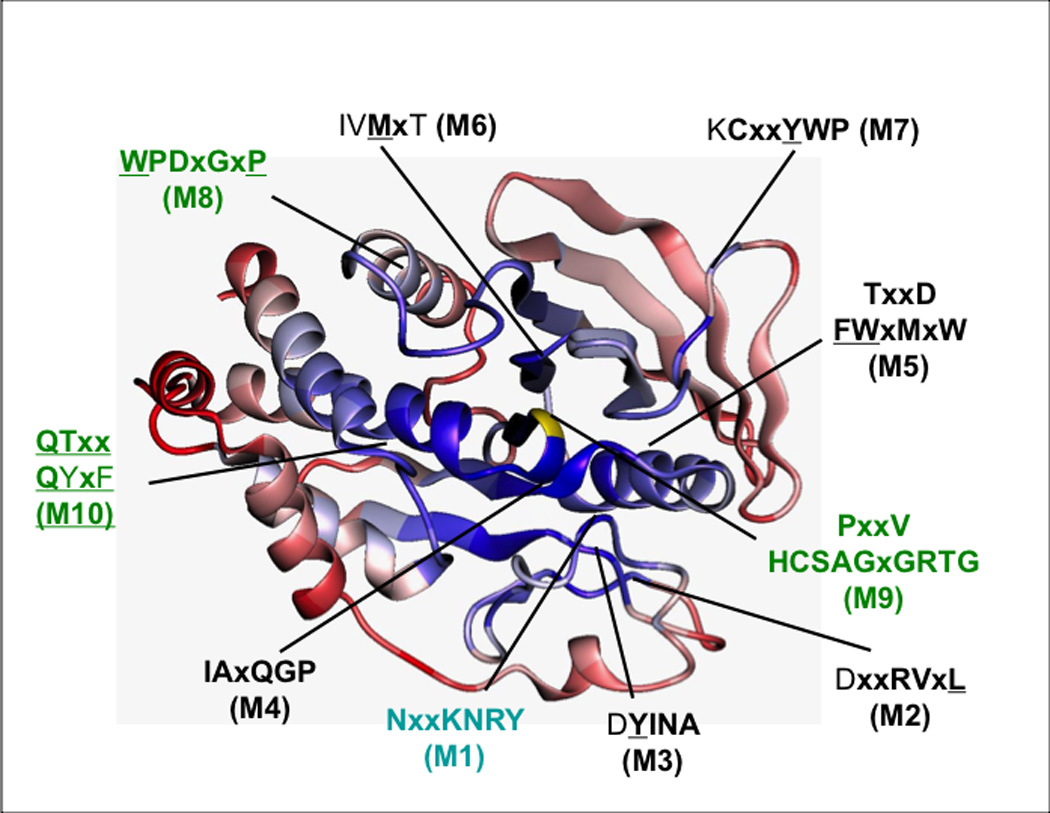
The figure illustrates a ribbon diagram of a PTP catalytic domain, highlighting the positions of the conserved motifs (M1-M10) that define the domain. Areas of conservation (blue = most conserved, red = least conserved) are illustrated using the catalytic domain of PTP1B as the reference. Reproduced from [42], copyright American Society for Microbiology.
Structure reveals mechanism
The first crystal structure of a protein phosphatase was actually that of a member of the PTP family, the 37kDa catalytic domain of PTP1B, the form of the enzyme we first isolated from human placenta [50]. This, coupled with extensive structural, enzymatic and kinetic analyses from several laboratories, has revealed the mechanism of PTP1B-mediated substrate recognition and catalysis [51]. In fact, through the structures of several mutant forms of PTP1B, which were all solved in collaboration with David Barford, we visualized each of the reaction steps in PTP-mediated catalysis [52, 53]. The signature motif, [I/V]HCXXGXXRS/T], which recognizes the dianionic phosphate moiety of the target substrate and contains the essential nucleophilic cysteinyl residue (Cys 215 in PTP1B) is located at the base of a pronounced cleft on the surface of the protein, the depth of which is determined by a tyrosine residue (Tyr 46 in PTP1B) in the pTyr loop. This contributes to the absolute specificity that PTP1B displays for pTyr-containing substrates, since the smaller phosphoserine and phosphothreonine residues would not reach down to the nucleophilic cysteine residue at the base of the cleft. In addition to the pTyr loop, the sides of the cleft are formed by the WPD loop and the Q loop, which contribute residues that are essential for catalysis [54].
PTP-mediated catalysis proceeds via a 2-step mechanism. In the first step, following substrate binding, the phosphate of the substrate undergoes nucleophilic attack by the sulphur atom of the thiolate side chain of the essential Cys residue. Substrate binding is accompanied by a large conformational change in the active site in which the WPD loop closes around the side chain of the pTyr residue of the substrate. In fact PTP1B represents an example of the concept of "induced fit", in which the conformational change induced by substrate binding creates the catalytically competent form of the enzyme. Closure of the WPD loop positions the invariant Asp residue (Asp181 in PTP1B) to function as a general acid in the first step of catalysis and protonate the tyrosyl leaving group of the substrate. The second step of catalysis involves hydrolysis of the cysteinyl-phosphate catalytic intermediate. The details were revealed in the structure of a PTP1B-orthovanadate complex, which is a mimic of the pentavalent phosphorus transition state, and the structure of a Gln 262 -> Ala mutant form of PTP1B, which allowed trapping and visualization of the catalytic intermediate in a crystal of the mutant protein because its hydrolysis is impaired [53]. Hydrolysis is mediated by Gln 262 from the Q loop, which coordinates a water molecule, and Asp 181, which now functions as a general base, culminating in release of phosphate. These structures also revealed that the WPD loop is closed over the entrance to the active site, thereby sequestering the cysteinyl-phosphate intermediate with water molecules at the catalytic centre, promoting its hydrolysis and preventing the transfer of phosphate to extraneous phosphoryl acceptors. This explains why the PTPs do not function as ‘kinases in reverse’, unlike isocitrate dehydrogenase kinase/phosphatase [55].
These general features of the catalytic mechanism are conserved throughout the PTP family, although the precise architecture of the active site may be fine-tuned to accommodate the requirements of individual enzymes. For example, although the pTyr specific PTPs possess a deep active site cleft, the active site of the DUSPs is shallower to allow accommodation of pSer and pThr residues. In PTEN, the cleft is broader to accommodate the sugar head group of the inositol phospholipid (Figure 4). In cdc25, protonation of the tyrosyl leaving group in the first step of catalysis is not mediated by a general acid on the phosphatase, but instead by the monoprotonated phosphate group on the substrate. The myotubularin-like enzymes (MTMs) also lack a conserved acidic residue on the loop that corresponds structurally to the WPD loop in classical PTPs. One of the conserved Asp residues in the signature motif of MTMs (VHCSDGWDRT) is required for phosphatase activity, and may function as the general acid in catalysis [56]. Unlike the other members of the PTP family, the signature motif of the low molecular weight LMW-PTP is at the N-terminus of the protein, with Cys 12 serving as the nucleophilic residue; furthermore, the Asp residue that serves as general acid/base in the LMW-PTPs is located C-terminal to the signature motif, unlike the classical PTPs in which it is located N-terminal to the signature. Nevertheless, the basic features of the catalytic mechanism are conserved throughout the family, as would be expected from their conserved 3-D structural folds.
Figure 4. Comparison of the active site of different members of the PTP family.
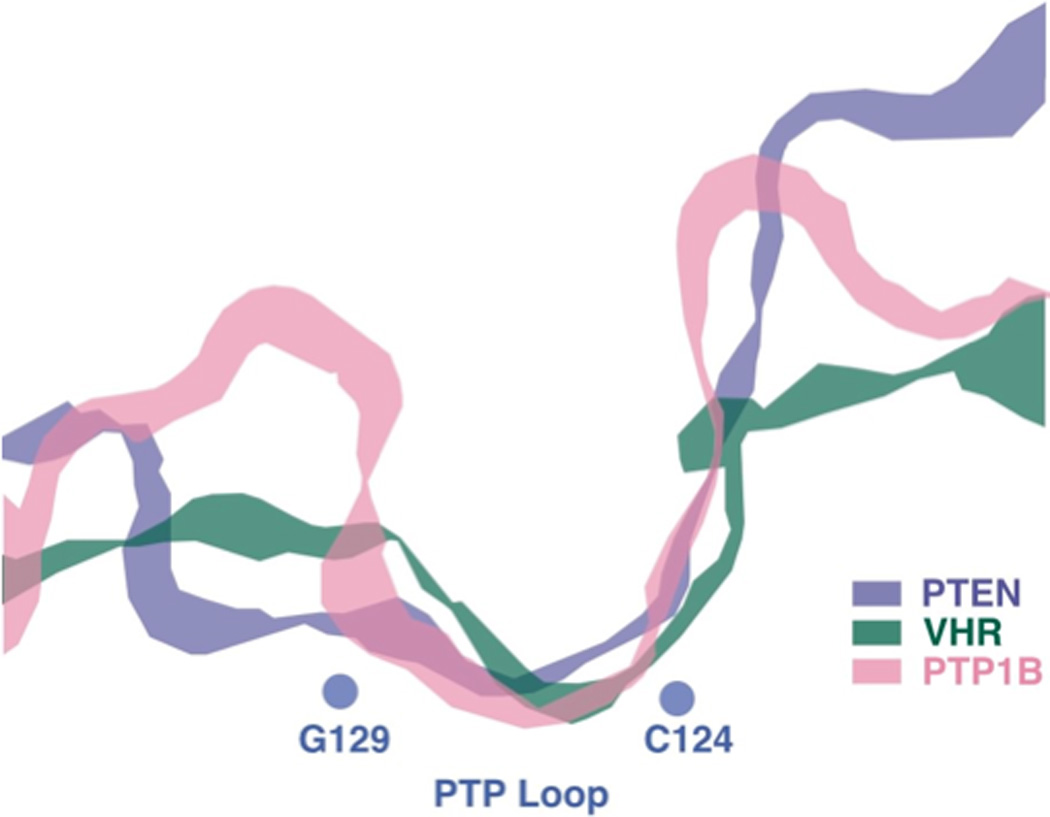
A section through the active site of PTP1B (classical, pTyr-specific enzyme), VHR (DUSP) and PTEN (specificity for inositol sugar head group of phosphatidylinositol phospholipid as substrate), to illustrate how the architecture of the active site of members of the PTP family is adapted for their substrate preference (re-drawn from [139]).
The classical, pTyr-specific PTPs
With the diversity of structure in the PTP family came diversity in function, again attesting to the fundamental importance of the PTPs in the regulation of cell signaling.
Receptor-like PTPs
Of the 37 classical PTP genes in humans, 21 encode transmembrane, receptor-like proteins (RPTPs). The diversity in the extracellular segments of the RPTPs presumably reflects a similar diversity in the nature of the ligands to which they respond; however, the identity and function of such ligands remains a largely unresolved issue in the field [57].
A characteristic feature of the extracellular segment of many RPTPs is the presence of immunoglobulin (Ig)-like and fibronectin type III (FNIII) domains. These are motifs that are commonly found in cell adhesion molecules, suggesting a role in regulating processes involving cell-cell contact. Early in the discovery of RPTPs we focused our attention on PTPμ, which comprises an extracellular segment containing a MAM domain, one Ig-like and four FNIII domains, a single transmembrane domain and an intracellular segment containing two PTP domains separated from the transmembrane domain by a juxtamembrane sequence with homology to the intracellular segment of the cadherin superfamily of cell adhesion molecules. In trying to identify the ligand for PTPμ, we demonstrated that when it was expressed in non-adhesive Sf9 cells, the formation of cell:cell aggregates was induced through a homophilic binding mechanism that involved only the extracellular segment of the PTP. Thus, the ligand for PTPμ on one cell is the extracellular segment of another PTPμ molecule on the surface of an adjacent cell [58]. In fact, it is Ig domain that is both necessary and sufficient for homophilic binding [59]. Yossi Schlessinger's group noted that when Sf9 cells expressing PTPμ and PTPκ (a close relative of PTPμ displaying ~75% overall identity) were mixed, they sorted independently [60]. In other words PTPμ binds to itself but not to PTPκ, suggesting that the homophilic binding interaction is highly specific. Nevertheless, aggregation, i.e. ligand binding to the extracellular segment of PTPμ, had no detectable direct effect on the activity of the intracellular PTP domains. It is likely that these interactions play a role in controlling activity by restricting its distribution in the membrane, perhaps targeting it to interactions with the cadherin-catenin cell adhesion complex [61, 62].
Another perspective on the issue of ligands comes from CD45, which is highly glycosylated and comprises up to 10% of the surface of a hematopoietic cell. In this case, although lectins, such as CD22, have been reported to bind to the extracellular segment, they do not appear to modulate PTP activity [63]. Dimerization of CD45 has been reported to vary according to the glycosylation of the extracellular segment, which is determined by the alternative splicing of 3 exons encoding sequences at the N-terminus. The larger, more highly glycosylated and sialylated forms, which expresses all 3 exons (RABC), are less efficient at forming dimers than the smallest form (RO). When a CD45-deficient T cell line was reconstituted with physiological levels of the RO and RABC isoforms, RO was found to dimerize more efficiently that RABC and was less effective than RABC at reconstituting signaling through the T cell receptor [64]. This suggests there may be an equilibrium between monomers and dimers of CD45 on the cell surface, with PTP activity determined by the differential dimerization of specific isoforms. Nevertheless, there have been reports of ligands that bind to the extracellular segment of RPTPs and alter directly the activity of the intracellular PTP domains. Perhaps the best characterized example came from Thomas Deuel’s lab who reported that binding of the soluble cytokine pleiotrophin (PTN) led to inhibition of RPTPζ activity, thereby promoting tyrosine phosphorylation [65]. It is thought that some of the effects of pleiotrophin on the cytoskeleton are mediated via RPTPζ-induced increases in tyrosine phosphorylation of β-catenin and β-adducin [65, 66]. It has also been reported that p190 Rho GAP is a PTN-modulated substrate of RPTPζ [67]. In an elegant study in Drosophila, David Van Vactor identified both soluble and surface-bound ligands that recognise the Ig domains LAR [68]. A high affinity interaction between LAR on neurons and syndecan (Sdc), a transmembrane heparin sulphate proteoglycan (HSPG) on muscle, serves to promote PTP function, whereas the interaction with the GPI-anchored protein Dallylike (Dlp) is inhibitory [68]. These ligands compete to regulate a pathway that integrates the effects of LAR with the ABL PTK via changes in the tyrosine phosphorylation of Enabled (Ena), which binds to the cytoplasmic segment of LAR and regulates the actin cytoskeleton and synaptic morphogenesis.
A characteristic feature of 12 of the RPTPs is a tandem arrangement of PTP domains in the intracellular segment [44]. The indication from phylogenetic analyses is that a PTP domain duplication occurred in an ancestral gene before the whole gene duplicated to give rise to other RPTPs [47]. Essentially all of the catalytic activity of these RPTPs resides in the membrane-proximal PTP domain (termed D1). Nevertheless, although the membrane-distal domains (termed D2) are inactive themselves, there are examples in which their structural integrity is required for the enzymatic activity of the PTP as a whole [69, 70]. Although the D2 domains lack intrinsic activity, there is considerable conservation of sequence, as well as secondary and tertiary structure, between domains D1 and D2; in fact, only two point mutations were required to convert LAR D2 into an active enzyme [71]. Nevertheless, there are also structural distinctions that suggest differences in function. For example, all the D2 domains are phylogenetically distinct from domain D1; i.e., D2 sequences do not cluster together with D1 sequences in the phylogenetic tree but define a separate subfamily of PTP domains [72]. Interestingly, within the LAR RPTP subtype, comprising LAR, PTPσ and PTPδ, the sequence similarity between domains D2 is even higher than between the corresponding domains D1 [42]. There have been suggestions that D2 domains serve a binding function [73]. More recently, based upon studies of RPTPα, it has been shown that the D2 domain displays greater sensitivity to oxidation than D1 and may serve as redox sensors [74]. Furthermore, oxidation induces a conformational change in D2 that can be transmitted to the extracellular segment of the receptor PTP [75, 76]. Nevertheless, elucidation of the functions of RPTP D2s remains an issue to be resolved in the PTP field.
The mechanism by which ligand binding to an RPTP may modulate the catalytic activity of the intracellular D1 domain remains a hot button issue in the field. The solution of the crystal structure of the membrane proximal PTP domain of RPTPα by Joe Noel’s group was an important development [77]. Within the crystal, the PTP D1 domains are organized in symmetrical dimers, in which an inhibitory helix-turn-helix “wedge” motif from one domain occludes the active site of the partner domain. This led to the proposal of a mechanism by which ligand binding may directly modulate PTP activity. If ligand binding was to induce dimerization, as it does for the PTKs, then the catalytic activity of RPTPs may be attenuated in a dimeric state by reciprocal occlusion of the active sites; it is notable that the effects on activity would be in contrast to RPTKs, which are stimulated by ligand-induced dimerization and trans-phosphorlyation [77]. This has proven to be a controversial model and the issue remains to be fully resolved. The first problem is that this structure describes only the membrane-proximal D1 domain in isolation. In a similar study of PTPμ, which shares 46% sequence identity with the D1 domain of PTPα, we found that the tertiary structures of the two were very similar (r.m.s.d. between equivalent Cα-atoms of 1.1Å); however, although the PTPμ D1 domain was also a dimer in the crystal, the dimer interface was distinct and the active site was present in an open, uninhibited conformation [78]. When both D1 and D2 domains were included in the construct to be crystallized, as reported for LAR [71] and CD45 [79], the wedge motif was present in the structure, but there was no evidence of dimerization in the crystal. In addition, the D1 and D2 domains were oriented in such a way that steric hindrance caused by the presence of D2 would prevent wedge-mediated dimerization of D1. These structures were seen in crystals in different space groups with distinct crystallographic contacts between neighbouring molecules. Also, in both LAR and CD45, the D1-D2 domain interfaces were stabilized by short linkers and extensive non-covalent interactions. Of course, within a cell these PTP domains are connected to transmembrane and extracellular segments, such that ligand binding may influence the relative orientations of D1 and D2. Nevertheless, the short linker, coupled with its limited flexibility, suggests that the relative orientations of D1 and D2 may be restricted [71, 79]. There are also conflicting data from cell-based studies. On the one hand, it has been suggested that in T cell lines CD45 is monomeric, as are other components of the T cell receptor signaling complex [80]. In contrast, studies in a “knock-in” mouse model highlight the importance of the inhibitory wedge motif in CD45 [81]. Glu 613, at the tip of the wedge, was mutated to Arg, in a mutation that would be expected to prevent wedge-mediated inhibition of CD45 in a dimer. It is interesting to note that the consequences of expressing CD45-E613R in the knock-in mice are essentially the opposite of those observed following knock-out of the CD45 gene, consistent with the wedge mutation having removed an inhibitory constraint upon CD45 function. Overall, the data clearly demonstrate the regulatory importance of the wedge, but whether or not the mechanism involves dimer-induced inhibition continues to fuel interesting debate.
Non-transmembrane/cytoplasmic PTPs
Although isolated originally as a 37kDa catalytic domain [1, 2], the cloning of cDNA encoding PTP1B by three separate groups, the labs of Ben Neel, Jack Dixon and Dave Hill, revealed a full-length form of the protein that also contains a regulatory segment of ~115 residues on the C-terminal side of the catalytic domain [82–84]. The C-terminal 35 residues are predominantly hydrophobic in nature and function in targeting the enzyme to the cytoplasmic face of membranes of the endoplasmic reticulum (ER) [85]. This was similar to the arrangement that had already been reported for the close relative of PTP1B, the 48 kDa form of TCPTP (T-Cell enriched PTP) [86]. In the latter case, alternative splicing generates two forms that differ in their extreme C-termini, but share a common catalytic domain in the N-terminal portion of the molecule. Whereas the 48kDa form (TC48) is targeted to the ER, a 45kDa form (TC45), which lacks the hydrophobic segment, is targeted to the nucleus [87] and is able to shuttle in and out of the nucleus in response to extracellular stimuli [88]. The C-terminal segment of PTP1B also contains sites of phosphorylation by Ser/Thr kinases [89] and a site of proteolytic cleavage by calpain, which generates a truncated, soluble form of the enzyme with enhanced activity [90]. This suggests a role for this segment not only in targeting, but also in the direct regulation of PTP1B activity. Such a direct role of the C-terminal segment in suppressing catalytic activity was defined for TCPTP [91].
Inspection of the sequences of the nontransmebrane PTPs reveals that the situation for PTP1B and TCPTP illustrates a general principle; sub-cellular targeting is now recognized as an important component of the regulation of PTP function. For example, the presence of SH2 domains In the N-terminal portion of the SHPs targets these PTPs to interact with sites of tyrosine phosphorylation in receptors and scaffolding adaptor proteins [92]. The presence of a FERM domain targets PTPs to interfaces between the plasma membrane and the cytoskeleton [93]. The SEC14 domain functions in lipid binding and membrane targeting [94]. The BRO1 domain has been implicated in targeting proteins to endosomes [95]. As Jack Dixon put it in his review in 1994 [96], these regulatory motifs function as zip-codes to direct the PTPs to the correct cellular address. Nevertheless, it is important to stress that the PTPs are not simply a collection of non-specific enzymes the activity of which is regulated indirectly by tethering. There is clear evidence of gene duplication in the nontransmembrane PTPs, giving rise to PTP1B and TCPTP, SHP1 and SHP2, as well as PTPD1 and PTPD2 [42]; these pairs have a high degree of sequence identity but distinct, non-redundant functions, consistent with specificity. There are examples of intrinsic specificity within the PTP catalytic domains themselves [97–99]. Furthermore, the regulatory sequences that flank the catalytic domain can also influence specificity, such as the KIM domain directing STEP and HePTP to dephosphorylate MAP kinases [100] and the poly-Pro sequences in PTP-PEST, which influence its interaction with p130cas [101].
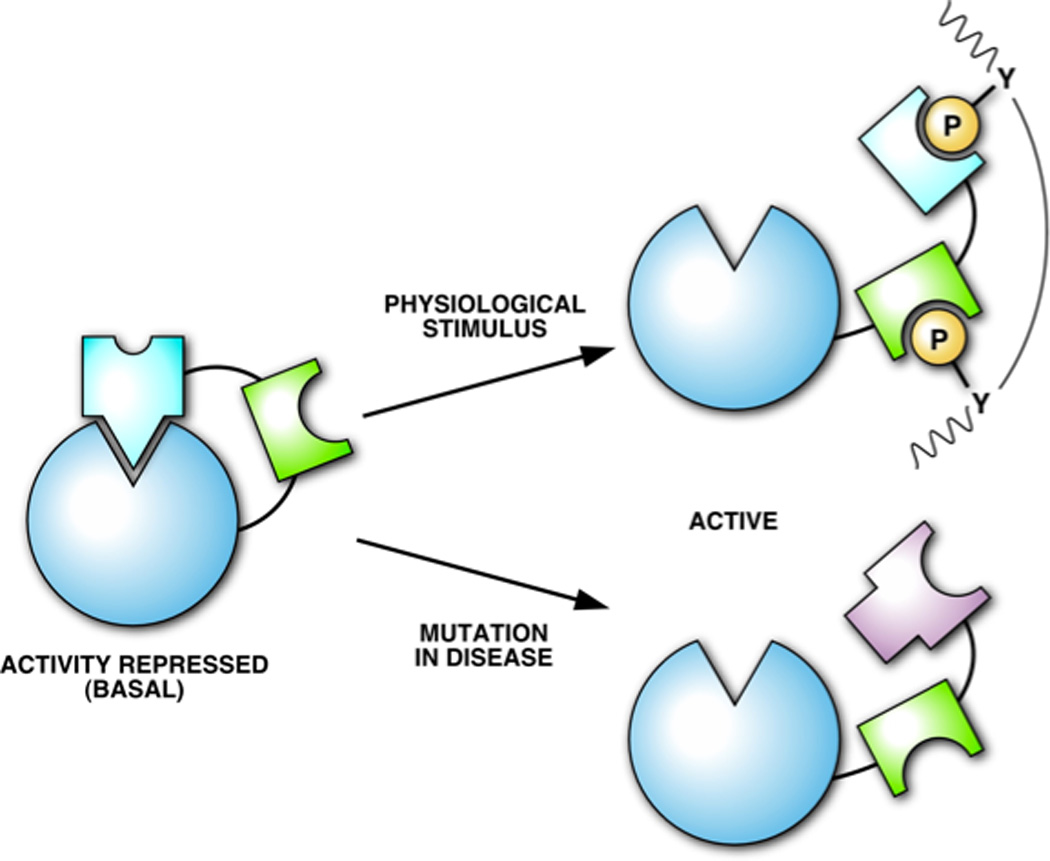
As illustrated for TCPTP [91], in addition to subcellular targeting, these non-catalytic segments of the PTPs may regulate activity directly. A clear example of this is the SH2 domain-containing PTP SHP2 [92]. Crystallographic analysis revealed that SHP2 exists in a low activity state under basal conditions because the active site is occluded by an intramolecular interaction with residues of the N-terminal SH2 domain, located on the opposite side to its pTyr binding site. Engagement of the SH2 domains by appropriate pTyr ligands induces a conformational change that releases the autoinhibitory interaction and creates a form of the phosphatase in which the active site is now open and can dephosphorylate substrates. Thus, SHP2 becomes activated once it has been recruited into the correct signaling complex. On the basis of this structure, Ben Neel designed mutants of SHP2 (D61A and E76A) that resulted in a constitutively active enzyme and found that they triggered FGF signaling in the absence of growth factor [102]. It is interesting to note that gain of function mutations in SHP2 have been identified in human disease, initially in Noonan syndrome. This includes mutations in residues in and around the N-SH2 domain, which may facilitate activation by pTyr ligands, and in key residues at the interface between the N-SH2 and catalytic domains, which would induce the active conformation in the absence of a stimulus. Particularly striking is the fact that those key residues that Ben Neel chose to mutate on the basis of the structure to create constitutively active forms of SHP2 are actually mutated in Noonan syndrome [92]. Mutations in SHP2 are now also associated with increased risk of certain childhood malignancies, such as juvenile myelomonocytic leukemia and acute myeloid leukemia. In fact, SHP2 was recognized as the first PTP oncogene, its positive role attesting further to the importance of PTPs as regulators of signaling in their own right.
The dual specificity phosphatases (DUSPs)
The prototypic dual specificity phosphatase was identified by Kunliang Guan and Jack Dixon as an open reading frame in the poxvirus vaccinia, termed VH1 [103]. Paula Traktman’s lab showed that this phosphatase plays an important role in controlling virion infectivity and the expression of viral genes [104], raising the possibility that inhibitors of the phosphatase may have anti-viral activity. VH1 has substrates in the virion itself, including the F18 DNA-binding protein [104] and the A17 pTyr protein [105], as well as the ability to antagonize interferon-γ signaling to STAT1 in the target cell [106].
In a collaboration with Lester Lau’s lab, we identified first mammalian DUSP, which was the product of the 3CH134 immediate early gene, and demonstrated that it functioned as a MAP kinase phosphatase, which we termed MKP1 [107, 108]. It dephosphorylated both the tyrosyl and threonyl residues in the TXY motif of ERK, thereby inactivating MAP kinase function [108]. Steve Keyse also characterized the human variant, termed CL100, as an MKP[109]. Now, VH1-like DUSPs have been shown to play important roles in many aspects of cellular function, including cytoskeleton reorganization, cell cycle control, apoptosis and RNA metabolism. Due to contributions from many labs, 43 DUSPs have been identified that display sequence homology to the Vaccinia virus H1 protein (VH1); however, a detailed classification of these enzymes was difficult because the domain that is conserved is relatively small and the primary sequences are far more diverse than those of the classical PTPs. Using bioinformatic tools, we formulated a classification system based on sequence homology and divided them into 3 classes, in which there were 28 Class I, 10 Class II and 5 Class III members [110].
Class I VH1-like DUSPs
MAP Kinase Phosphatases
A major functional theme in the Class I DUSPs is the regulation of signaling by Mitogen-Activated Protein Kinases (MAPKs). The stimulation of MAPKs is an important aspect of the cellular response to growth factors, hormones, cytokines and stresses. The MAPKs are the terminal component of “signaling modules” that comprise a series of three protein kinases (MAP3K, MAP2K and MAPK) that act sequentially. Triggering of these signaling modules by small GTP-binding proteins, such as those of the RAS superfamily, and STE20-like protein kinases, such as HPK and GCK, culminates in the activation of MAPKs by phosphorylation of both the tyrosine and the threonine residue of a conserved TxY motif in their activation loop. There are four major subgroups of MAPKs, ERK 1 and 2, ERK 5, p38 α, β, γ and δ and JNK 1, 2 and 3. These enzymes exert distinct effects in a physiological context through phosphorylation of a wide variety of substrates, including effector protein kinases, such as MAPK-activated protein kinases (MAPKAPs) and transcription factors, such as the AP-1 complex [111]. The physiological effects of a particular MAPK are regulated by the balance between the activity of the MAP2K that phosphorylates it and the protein phosphatases that dephosphorylate either or both of the phosphorylated residues in the TxY motif. Thus protein phosphatases are also critical regulators of MAPK-dependent signaling. Several classical PTPs have been implicated in dephosphorylation of the tyrosyl residue in the TxY activation loop motif of MAPKs, including PTP-SL, STEP and He-PTP [44]. A kinase interaction motif (KIM) in the non-catalytic segment of these PTPs, which interacts directly and specifically with the MAPK, serves as a determinant of the substrate specificity of the phosphatase. Several DUSPs are now known to function as MAP Kinase Phosphatases (MKPs) and are critical regulators of MAPK signaling, recognizing both the Thr and Tyr residues of the TxY activation loop motif. These MKPs display differences in expression (constitutive and inducible expression), tissue and subcellular distribution (nuclear vs cytosolic), and specificity for MAPK family members [46, 111]. It appears that the specificity of some of these enzymes for MAPKs is also determined, at least in part, by interactions between the non-catalytic segment of the DUSP (termed the cdc25 homology/CH2 motif, which contains a KIM motif) and the target MAPK. For example, the N-terminal segment of MKP3 was shown to bind specifically to Erk2, its physiological substrate, and to promote a conformational change leading to activation of the DUSP by Erk2 (Figure 5) [112].
Figure 5. Activation of SHP2.
In the basal state the active site of SHP2 is occluded by an intramolecular interaction with the N-terminal SH2 domain. The phosphatase may be activated either by engagement of the SH2 domains by pTyr sequence motifs in an RPTK or scaffolding molecule, or by mutations in either the N-SH2 or PTP domains that disrupt their interaction [92].
In addition to the CH2 domain-containing MKPs, this class of DUSPs contains small phosphatases with MKP activity that comprise isolated catalytic subunits. VHR is a classic example [46, 111]. Originally identified in Stuart Aaronson’s lab in an interesting expression cloning strategy that used decreased tyrosine phosphorylation of FGFR2 as a readout [113], VHR was recognized as a broadly expressed DUSP that preferentially dephosphorylated the phosphotyrosyl residue over the phosphothreonyl residue in the TxY motif of ERK [114, 115]. Therefore, although classified as a DUSP, it appeared to show preference of one particular phosphoamino acid. The structural basis for this was revealed in a co-crystal of VHR and a bisphosphorylated peptide substrate derived from the activation loop of the MAPK p38 [116]. The pTyr residue was bound at the active site, with a major determinant of this substrate selection being the narrow entrance to the active site that is created by two residues from the signature motif, Glu 126 and Tyr 128. When these residues were mutated to Ala and Ile, respectively, the catalytic efficiency was not altered but the ability to dephosphorylate pThr was increased 9 fold relative to wildtype VHR [114, 116]. In most Class I DSPs, Ala and Val/Ile residues, respectively, are found at these positions in the signature motif, suggesting a greater capacity for dephosphorylation of Ser and Thr residues in substrates than VHR. In addition, Arg 158 in VHR contributes to a positively charged pocket that coordinates the pThr residue in the bisphosphorylated MAPK peptide, thereby orienting the peptide substrate on the surface of the enzyme. In addition to ERK, VHR has been implicated in the dephosphorylation of JNK and regulation of apoptosis [117–120]. Overall, at least 14 VH1-like DUSPs are involved in the control of MAP kinase signaling. Therefore, these DUSPs represent a complex response network that implements the attenuation of specific MAPK-dependent signaling pathways in particular tissues and subcellular compartments following defined stimuli and plays a crucial role in determining the signaling outcome.
A different perspective on the regulation of MAPKs by DUSPs is provided by the JSP-1-like subclass, comprising JSP-1 (JNK Stimulatory Phosphatase-1, DUSP22) and VHY (DUSP15). We identified JSP-1 in humans as another broadly expressed, small (~20kDa) DUSP that lacked N-terminal CH2 domains and comprised primarily a catalytic domain [121]. The intriguing property of this phosphatase was that instead of directly inactivating MAP kinases, overexpression of JSP-1 led to a dose-dependent increase in the phoshorylation and activation of co-expressed JNK, whereas the activation of ERK and p38 were unaffected [121]. These effects, which required the phosphatase activity of JSP-1, were blocked by expression of a dominant-negative mutant form of MKK4, a MAP2K that phosphorylates and activates JNK, suggesting that the site of action of the phosphatase is upstream of MKK4 in the JNK signaling cascade. An enzyme that was originally identified as the murine orthologue of JSP-1, JKAP (JNK Pathway Associated Phosphatase) is actually a spliced variant that possesses an extension of 21 residues at the C-terminus. In this case, overexpression of an inactive C->S mutant form of JKAP blocked TNFα-induced activation of JNK in HEK293T cells [122]. Furthermore, ablation of JKAP in mouse embryonic stem cells abolished JNK activation in response to TNFα and TGFβ, but not UV [122]. Although additional publications that reported the isolation of human JSP-1, termed VHX [123], and murine JSP-1, termed LMW-DSP2 [124], have suggested that under certain conditions it may dephosphorylate ERK2 and p38, our data suggest the potential for a new aspect of the control of JNK. Although the mechanism remains to be defined, potential substrates include MAP3Ks, such as MLK3, ASK1 and the TAB1 regulatory subunit of TAK1, in which inhibitory sites of phosphorylation have been identified. Dephosphorylation of such sites would have the potential to augment signaling. Uniquely among the DUSPs, JSP-1 and VHY/DUSP15 contain a potential myristoylation site at the N-terminus [125]. Although active in the absence of myristoylation, the subcellular localization of JSP-1 and its ability to induce JNK activation and apoptosis were compromised [125], highlighting the functional importance of this modification.
Diversity in Class 1 VH1-like DUSP function
The effects of this class of VH1-like DUSPs are not restricted to the regulation of MAPK signaling. The Drosophila protein Slingshot was named after the split-ended hair/bristle phenotype in the mutant flies that lack this phosphatase. These structures are actin-based, and Slingshot is found to be a regulator of actin polymerization at such structures as growth cones and lamellipodia of migrating cells (Figure 7). There are three Slingshot-like DUSPs (SSH) in humans. Unphosphorylated cofilin binds to G actin and favors actin depolymerization, whereas phosphorylation of cofilin on Ser-3 by LIM kinase inhibits both its filament-severing and G actin-binding abilities. The SSH DUSPs specifically dephosphorylate this residue and reactivate the cofilin for microfilament reorganization (reviewed in [126]), providing another example of an enzyme that is operationally defined as a DUSP, but in practice favours one particular phosphoamino acid. This regulation of cofilin phosphorylation also provides a striking illustration of the integration of kinases and phosphatases to control a cell signaling pathway. The activity of LIM kinase is itself regulated by upstream kinases, becoming activated following phosphorylation by PAK or the RHO-dependent kinase ROCK. In turn, the activity of Slingshot-like DUSPs is also regulated by phosphorylation. In this case, phosphorylation of SSH by PAK4 triggers sequestration of the phosphatase in a complex with 14-3-3. Activation of SSH requires its dephosphorylation by another phosphatase, the Ca2+-dependent enzyme calcineurin, providing an example of a phosphatase-on-phosphatase cascade [127].
Figure 7. Regulation of the actin cytoskeleton by slingshot phosphatases.
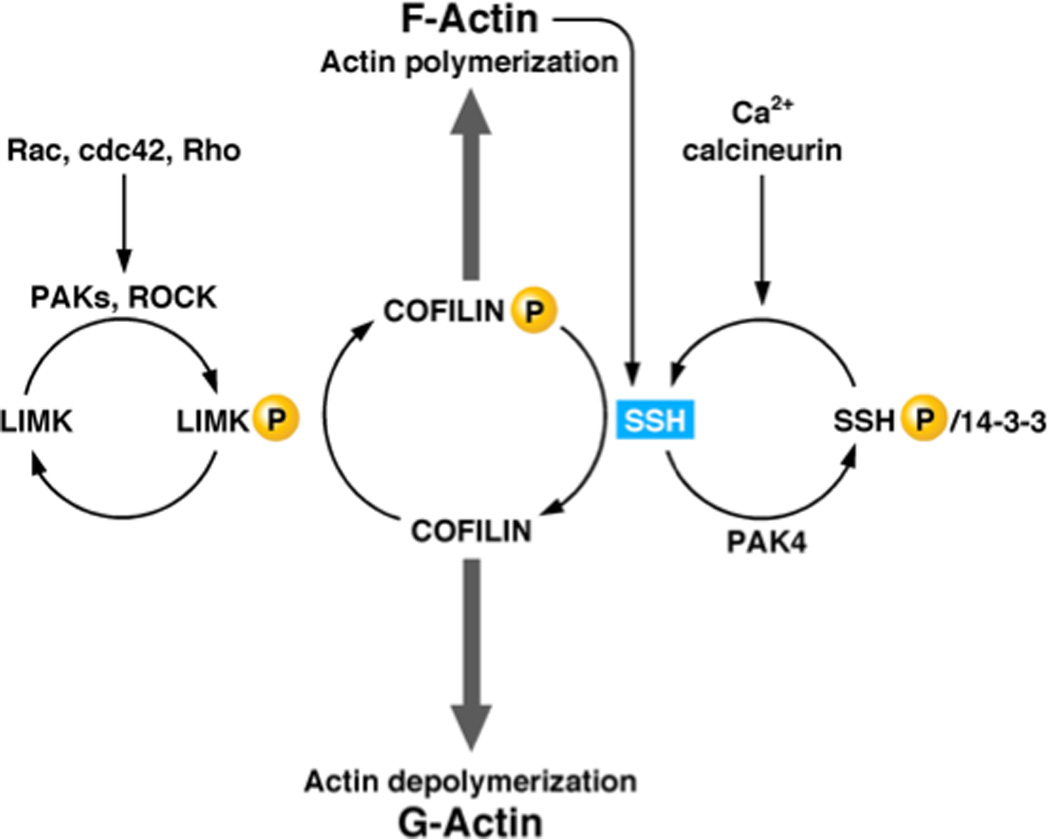
Slingshot regulates the actin cytoskeleton through dephosphorylation of cofilin as substrate. The regulation of slingshot illustrates cascade arrangements of both kinases and phosphatases.
An intriguing genomic arrangement is found at the DUSP13 locus on human chromosome 10q22.2, which utilizes alternative open reading frames to encode two of the DUSPs in the VHR-like subclass, MDSP (DUSP13a, muscle restricted DUSP) and TMDP (DUSP13b, testis and skeletal specific DUSP) [128]. This gene organization is conserved in mouse and, although reminiscent of the p16INK4/p19ARF locus that encodes two unrelated proteins [129], this was the first example of a gene from which two distinct proteins of the same family are expressed using alternative ORFs. Both MDSP and TMDP are restricted to specific tissues, skeletal muscle and testis, respectively, in the adult mouse. Expression of both proteins was markedly increased at approximately the third week after birth and continued to increase gradually into adulthood, implying that the physiological functions of both DUSPs are specific to the mature organs. Nevertheless, the function of these enzymes in vivo remains to be established. Although DUSP13b/TMDP has the capacity to inactivate JNK and p38 [130], DUSP13a/MDSP does not dephosphorylate MAPKs and its physiological substrates are unknown.
Finally, these class 1 VH1-like DUSPs also introduced us to the concept of “pseudophosphatases”, proteins in which the 3D fold of the phosphatase is maintained, but in which key residues that are critical for catalysis are missing. The prototype for these proteins was discovered by Jack Dixon’s lab and was named STYX [131]. STYX (phosphoSerine, Threonine or tYrosine interacting protein, is named after one of the five rivers separating the world of the living from Hades, the world of the dead in Greek mythology, to emphasize that these molecules are ‘dead’ phosphatases. In the signature motif of STYX, LVHGNAGISRS, Gly replaces the catalytic Cys that is conserved among PTP superfamily members. Mutation of this Gly to a Cys ‘restores’ phosphatase activity to this dead enzyme, suggesting that wildtype STYX may bind to phosphoproteins in the same manner that substrates bind to DUSPs, but without catalyzing dephosphorylation [131]. Therefore, it may function as naturally occurring “substrate trap”. Ablation of the STYX gene in mouse results in male infertility due to defects in spermatogenesis. Endogenous STYX interacts with Crhsp-24 (Calcium-Responsive Heat Stable Protein of 24 kDa) in vivo, a double stranded RNA binding phosphoprotein expressed at the same developmental stage as STYX in spermatogenesis; however, the physiological role of Crhsp-24 is not clear [132]. The second example, MK-STYX, contains an additional N-terminal domain similar to those found in the CH2 motif-containing MKPs; however, its signature motif is LIFSTQGISRS, in which the conserved His and Cys residues, both of which are required for catalysis, are replaced by Phe and Ser, respectively. As reported for STYX, we have observed that mutation of MK-STYX to a consensus active signature motif (LIHCTQGISRS) is sufficient to “restore” catalytic activity [133]. The RAS-GAP SH3 domain-binding protein-1 (G3BP1) has been identified as a binding partner of MK-STYX in the regulation of stress granule formation [133]. Nevertheless, the similarity between the N-terminal domain of MK-STYX and those of the 10 human CH2 motif-containing MKPs suggests the possibility that MK-STYX may also participate in protein-protein interactions with MAPKs in vivo, potentially adding a further level of complexity to the regulation of MAPK signaling.
Class 2 VH1-like DUSPs
There are 10 DUSPs in the human genome that we categorized as Class II, of which the PTEN-like and the capping enzyme-like DUSPs belong to one monophyletic group, whereas the PRLs and the CDC14s belong to a separate monophyletic group.
The PTEN tumor suppressor phosphatase
Perhaps the most famous member of this class of DUSPs is PTEN, also known originally as MMAC1 and TEP1, the gene for which is located at chromosome 10q23 and is one of the most frequently lost or mutated tumor suppressors in human cancer [134]. Germline mutations in PTEN are also responsible for a series of autosomal dominant cancer predisposition syndromes, called the PTEN Harmartoma Tumor Syndromes [135] which are associated with the formation of multiple benign tumors (harmartomas). PTEN is classified as being haploinsufficient in that inactivation of one allele, leaving only one functional copy, is insufficient to support the wild type condition. Overall, it is now clear that PTEN serves diverse roles in vivo and even minor disruption of its function can increase the risk of cancer and other diseases [136].
PTEN is a 403 residue protein comprising an N-terminal PTP domain, which forms an extensive interface with a C2 domain, which serves a phospholipid binding function and may facilitate the localization of the enzyme to cell membranes. In addition, the C-terminal segment of the protein, which is characterized by multiple phosphorylation sites and a -Thr-Lys-Val-COOH binding motif for PDZ domain-containing proteins, serves a regulatory function and is also important for its role as a tumor suppressor. The most striking feature of PTEN is its substrate specificity, which illustrates how a member of the PTP family can regulate signal transduction through dephosphorylation of non-protein substrates. We provided the first demonstration that PTEN possessed intrinsic phosphatase activity, displaying an unusual preference for highly acidic substrates [137]; however, in an important breakthrough for the field, Jack Dixon’s lab demonstrated that PTEN also has the ability to dephosphorylate the lipid second messenger phosphatidylinositol-3,4,5-trisphosphate (PIP3) [138]. In fact, the crystal structure of PTEN revealed an unusual architecture of the active site, which is sufficiently large to accommodate the sugar head-group of inositol phospholipids; this allows PTEN to dephosphorylate the 3 position in the inositol sugar ring specifically as a substrate and thereby to antagonize PI 3 kinase-dependent signaling [139]. In characterizing the activity of disease-derived mutations in PTEN, we demonstrated that the G129E mutant, identified in tumors and in patients with Cowden syndrome, is able to dephosphorylate protein substrates, but its ability to dephosphorylate inositol phospholipids is impaired [140]. This observation suggested that it was the lipid phosphatase activity of PTEN that was important for its tumor suppressor function.
There have also been reports of lipid phosphatase-independent functions of PTEN, including the observation that it can inhibit glioma cell migration through its C2 domain alone, in the absence of the catalytic domain [141]. In addition, recent studies have addressed the importance of its protein phosphatase activity. PTEN has been implicated in dephosphorylation of the PDGF receptor [142] and it was proposed to influence cell migration through dephosphorylation of the cytoskeletal-associated proteins FAK and p130CAS [143]. Intriguingly, a variety of studies have demonstrated that PTEN is itself a phosphoprotein with major sites of phosphorylation near the C-terminus, particularly an acidic stretch of residues (DHYRYSDTTDSDPENE) [134]. This phosphorylation may be of regulatory importance, including in controlling the stability of the protein [144] and its susceptibility to cleavage by caspases [145]. An intramolecular association was reported between the N-terminal PTP and C2 domains and the C-terminal portion of PTEN, resulting in a “closed” conformation that limits its association with the membrane. Dephosphorylation of the C-terminal tail serves to open up the conformation, promoting both membrane recruitment and activation, and regulating its ability to interact with binding proteins, via its C-terminal PDZ-binding domain, which may control its localization, stability and activity [146]. In an interesting twist, it appears that PTEN may autodephosphorylate these sites – thus, although PTEN may influence signaling through dephosphorylating inositol phospholipids, a major protein substrate for PTEN may be PTEN itself [147]. This suggests a striking parallel with PI 3 kinase, which phosphorylates exogenous lipid substrates and displays protein kinase activity through autophosphorylation [148], illustrating symmetry in the regulation of signaling via phosphorylation and dephosphorylation of phosphatidylinositol phospholipids.
CDC14 and regulation of the cell cycle
CDC14 was first identified in the classic screens for cell division cycle (CDC) regulators performed in budding yeast, S. cerevisiae, by Lee Hartwell. Entry into mitosis is accompanied by a burst of phosphorylation triggered by the cyclin-dependent kinase complex, cyclin B-CDK1; in order to get out of mitosis, the effects of the kinase must be reversed, involving dephosphorylation of multiple mitotic substrates. Cells lacking CDC14 arrest late in anaphase with high CDK activity whereas, conversely, cells in which the DUSP is overexpressed display inappropriate inactivation of CDKs, indicating that in budding yeast CDC14 is a critical phosphatase for the regulation of mitotic exit [149, 150]. The importance of localization in controlling function of a member of the PTP family is well illustrated in the example of CDC14. It is sequestered in the nucleolus until the onset of anaphase, when it is released throughout the nucleus and cytosol. This change in localization results in a change in activity because of the targeting function of the binding protein NET1, which serves as an inhibitor of CDC14. This association between NET1 and CDC14 is regulated by FEAR and MEN! FEAR (CDC Fourteen Early Anaphase Release) functions through inactivation of the PP2A holoenzyme that contains the CDC55 regulatory subunit, which promotes phosphorylation of NET1 and the release of CDC14. MEN (Mitotic Exit Network) is a RAS-like GTPase signaling module that is thought to culminate in the activation of a kinase that also phosphorylates NET1 to trigger CDC14 release. It appears that there may be different functions for CDC14 released by each pathway – FEAR, which regulates mitotic spindle and chromosome segregation, promotes activation of MEN, whereas MEN-activated CDC14 is associated with CDK1 inactivation and the exit from mitosis [151].
The evolutionary conservation of CDC14 structure and substrate specificity suggested that its biological role may also be conserved; however, advances in the recent years surprisingly revealed major differences in CDC14 function in different organisms. Although the function of CDC14 in mitotic exit in budding yeast is clear, its role in other systems is more controversial. The CDC14 orthologue in the fission yeast is FLP1/CLP1 (CDC Fourteen-Like-Phosphatase). In contrast to S. cerevisiae CDC14, CLP1/FLP1 is not essential and is not required for mitotic exit, but plays a more important role in regulating cytokinesis and G2-M transition by its down-regulation of CDK1 activity [152]. In humans, there are two CDC14s, termed A and B, which display ~50% sequence identity [151, 152]. There is also evidence for a third, CDC14C, which is closely related to CDC14B, having arisen through gene duplication. During the long-standing collaboration between my lab and David Barford, we determined the crystal structure of human CDC14B [153]. This revealed that its substrate specificity is partly determined by features of the active site. In the crystal structure of CDC14B there are two domains with DUSP-like folds; the C-terminal domain is the DUSP catalytic domain, whereas the N-terminal domain assumes a similar fold but contains no sequence homology to VH1-like DUSPs. The active site of CDC14B is located in a long groove at the interface between the two domains. The consensus phosphorylation site of CDKs, which is the in vivo substrate of CDC14, is characterized by a Pro at the +1 position and basic residues at +2 and +4 positions. Co-crystallization studies of Cdc14B with such phosphopeptides showed that a hydrophobic pocket binds to the Pro in its trans-conformation and the basic residues are then in the position to bind to an acidic groove leading to the active site. This structural analysis of CDC14B demonstrated yet another mode of substrate recognition for VH1-like DUSPs; furthermore, it also suggested that it may be possible to predict the features of the physiological substrate from the structure of the phosphatase. Recent studies have extended the function of CDC14 in mammals beyond the exit to mitosis to encompass DNA replication and repair, and the DNA damage checkpoint [151, 152], however, the details of the critical substrates for these effects remain to be established.
The PRLs
PRL-1 (Phosphatase in Regenerating Liver) was discovered as one of the immediate early genes induced upon liver regeneration following partial hepatectomy [154]. Three human PRL genes (PRL-1, 2 and 3, also known as PTP4A1, A2 and A3, and PTPCAAX1, 2 and 3) have been identified [155],[156]. PRL-1 and 2 are broadly expressed, whereas expression of PRL-3 is more restricted, primarily to heart and skeletal muscle. A unique feature of the PRLs is the presence of a poly-basic sequence preceding a CAAX motif at the C-terminus of the protein. The PRLs are the only farnesylated members of the PTP family, with prenylation being important for localization to the plasma membrane and early endosomes [155, 156]. It has been reported that the PRLs can oligomerize, particularly to form trimers, but the functional significance of this remains to be established. In addition, the PRLs are unusual in that the Ser/Thr residue following the invariant Arg in the signature motif (H-C-(X)5-R-[S/T]) is replaced by Ala. This hydroxyl residue functions in the second step of catalysis by facilitating hydrolysis of the cysteinyl-phosphate intermediate. It has been shown that mutation of this hydroxyl residue to Ala in active PTPs impairs catalysis by attenuating hydrolysis of the catalytic intermediate [157]. As expected, PRLs are characterized by low activity in vitro. Interestingly, although mutating the Ala to Ser did augment activity towards low Mr substrates in vitro, in PRL-3 this mutant was devoid of activity against PIP2, which has been proposed as a potential physiological substrate [158]. This raises questions about what contributes the important functionality of this Ser residue to catalysis in vivo. Structural studies have revealed an unusually wide and shallow active site cleft, with a signature motif characterized by hydrophobic residues; it is thought that this arrangement would allow the PRLs to accommodate a broad array of substrates, including pSer and pThr residues in proteins.
PRL-1 and PRL-2 have been shown to behave as oncogenes when overexpressed. Overexpression of PRL-1 in NIH3T3 cells induced anchorage independent growth, and D27 pancreatic cells overexpressing PRL-1 and PRL-2 formed tumors upon injection into nude mice [155, 156]. Both catalytic activity and farnesylation appear to be important for this oncogenic function. Most strikingly, PRL-3 has been identified as a potential cancer biomarker. Bert Vogelstein’s lab demonstrated that its expression is dramatically upregulated in metastatic colorectal cancer, but not in primary cancer [159], suggesting that it may be a therapeutic target for metastatic cancer. PRL-3 has now been implicated in progression and metastasis of other cancers, including gastric, ovarian and breast. The physiological substrates that underlie these effects of the PRLs are currently unclear. These DUSPs have been linked to regulation of PI3K/AKT signaling, but this may be an indirect effect due to down-regulation of the expression of PTEN [160, 161]. PRL-3 has also been implicated in the regulation of SRC function in integrin signaling and the actin cytoskeletal changes involved in cell invasion and motility. There are some data on a direct substrate, in particular the basic leucine zipper transcription factor ATF-7, which was found to interact with PRL-1 in a yeast 2-hybrid screen and could be dephosphorylated by PRL-1 in vitro; however, the significance of this interaction is unknown [162]. Overall, the identification of the physiological substrates remains one of the major challenges in the study of PRL function.
Class 3 VH1-like DUSPs
There are five Class III DUSPs present in both human and mouse genomes. These are intriguing enzymes because of similarities with the classical PTPs. For example, in the classical PTPs the Q-loop functions to position the water molecule for the second step of catalysis. Many of the Class III DUSPs retain either a partial, or an intact Q-loop sequence; however, this motif is missing from the Class I and II VH1-like DUSPs, in which, a conserved Ser residue preceding the invariant Arg in the signature motif ([I/V]-H-C-x-x-G-x-S-R-S) is believed to substitute for the missing Q loop. Although the Class I DSPs contain one invariant Gln residue in the sequence motif PNXXFXXQL, this Gln does not superimpose with Q262 in the Q-loop of PTP1B in either sequence or structural alignment and the function of this motif in Class I DSPs is unknown. In contrast, in the Class III DSP KAP1, the Q loop motif is conserved and superimposes perfectly in structural alignment with the classical PTP Q-loop [110]. In those Class III DSPs that have a Q-loop sequence, the hydroxyl residue preceding Arg in the signature sequence is always absent. However, Class III DSPs that contain this hydroxyl residue, such as PTPMT1 (human), lack the complete Q-loop sequence. This suggests that, when present, the Q-loops in the Class III DSPs may function similarly to those in the classical PTPs. Furthermore, these similarities suggest that motifs 8–10 in the classical PTPs, which represent the catalytic core, may have evolved from an ancient DUSP similar to the Class III enzymes.
KAP1/Cdi1
The progression through the cell cycle is governed by cyclin-dependent protein kinases. In addition to cyclin binding, and the dephosphorylation of Thr14 and Tyr 15 by cdc25, the full activity of CDKs requires the phosphorylation of Thr 160 in the kinase activation loop, which is catalyzed by CAK (Cdk-Associated Kinase). This DUSP, cyclin-dependent Kinase-Associated Phosphatase, which is product of the CDKN3 gene, is another example of a member of the PTP family that is of fundamental physiological importance, serving as a critical regulator of cell cycle progression. Although classified as a DUSP, it recognizes a pThr residue, specifically dephosphorylating CDK2 on Thr160 (Figure 8). The crystal structure of the KAP1-CDK2 complex revealed that substrate binding occurs between the C-terminal helix of KAP1 and the C-terminal lobe of CDK2, which positions the DUSP active site to dephosphorylate pThr 160. It is only the phosphate group of pThr 160 that interacts with the DUSP active site; hence, unlike the situation for protein kinases, there is no interaction with the residues flanking the phosphorylation site in the substrate [163]. KAP1 can bind to CDK2 in the presence or absence of cyclin A, but dephosphorylation requires removal of the cyclin, either by proteolysis or dissociation. A further element of control involves a four transmembrane domain protein family member, HTm4, which binds directly to the C-terminus of KAP1. In a KAP1-CDK2-cyclinA complex, HTm4 promotes the ability of KAP1 to dephosphorylate pThr 160 by facilitating access of the DUSP to the phosphorylated residue and excluding cyclin A from the KAP1-CDK2 complex [164]. Not unexpectedly, altered expression of KAP1 has been associated with cancer. Aberrant splicing has been shown to increase the levels of KAP1-related transcripts, but decrease KAP1 protein, in malignant astrocytomas [165].
Figure 8. KAP1 and regulation of the cell cycle.

KAP1 regulates the cell cycle through dephosphorylation of Thr 160 in the activation loop of the cyclin-dependent kinases.
PTPMT1
Originally noted for its similarity to PTEN, this DUSP, then termed PLIP (Phospho-Lipid Inositol Phosphatase), was first identified in the slime mold Dictyostelium as a putative transmembrane phosphatase required for aggregation at low cell density [166]. It is highly conserved in evolution, with orthologues identified in all phylogenetic kingdoms, suggesting that it serves a fundamental function. This enzyme was re-named PTPMT1 (PTP localized to the mitochondrion 1), reflecting the fact that, uniquely among the PTPs, it is targeted, via an N-terminal signal sequence, to the matrix face of the inner mitochondrial membrane together with the protein complexes responsible for electron transport and ATP production [167]. Initially, both human and Dictyostelium PTPMT1 were shown to be 5-position phosphoinositide phosphatases that specifically target the singly phosphorylated PI5P, expanding the repertoire of PTP family members that act through phospholipids [166]. Knockdown of PTPMT1 in pancreatic β cells enhanced ATP production and insulin secretion, coincident with changes in mitochondrial protein phosphorylation [167]. More recently, attention has focused back on phospholipid substrates with the demonstration by Jack Dixon’s lab that PTPMT1 plays an essential role in the regulation of cardiolipin biosynthesis [168]. Targeted deletion of the PTPMT1 gene in mice resulted in embryonic lethality, indicating an essential function in development. In cells lacking PTPMT1, complex I and II activity was inhibited, with disruption of mitochondrial respiration and morphology. The critical substrate for PTPMT1 appears to be phosphatidylglycerol phosphate (PGP); in the absence of the phosphatase, PGP accumulates and formation of phosphatidylglycerol (PG) is impaired, disrupting the pathway of cardiolipin biosynthesis. These studies highlight the critical role of cardiolipin, and in turn PTPMT1, in the regulation of mitochondrial morphology and metabolism.
Laforin
Lafora disease is an autosomal-recessive neurodegenerative disease that results in progressive neurological deterioration, myoclonus epilepsy and ultimately death. The disease is characterized by the accumulation of Lafora bodies in multiple tissues. These deposits comprise insoluble glucans made up of poorly branched glycogen, more similar to the plant starch amylopectin, with higher than normal levels of phosphate. Mutations in either of two genes, encoding EPM2A (Epilepsy of Progressive Myoclonus type 2) and EPM2B, results in Lafora disease. EPM2A encodes the DUSP laforin, whereas EPM2B encodes malin, an E3 ubiquitin ligase [169]. There are two aspects to the function of Laforin. It forms a complex with malin, in which it serves as an adaptor to target substrates to the ubiquitin ligase. In addition, and uniquely among the members of the PTP family, laforin contains an N-terminal carbohydrate binding domain and functions as a phosphatase to dephosphorylate glucans directly [170–172]. Peter Roach’s lab has recently reported that glycogen synthase, which normally transfers glucose to glycogen, also, in a rare side reaction, catalyzes the transfer of the β phosphate of UDP-glucose to glycogen [173]. In the absence Laforin, there is hyperphosphorylation of glycogen, the structure of which becomes compromised leading to its aggregation, with formation of Lafora bodies and disease. In the light of the fact that the Lafora disease-associated hyperphosphorylation of glycogen arises from a rare side reaction of glycogen synthase that may be regarded as a catalytic error, Peter Roach has made the intriguing suggestion that laforin may be viewed as a “repair enzyme”, functioning in a similar manner to proof-reading by DNA polymerases to ensure that the normal structure of the glycogen polymer is maintained [174]. Although Laforin itself would not be a therapeutic target, these data suggest that inhibition of brain glycogen synthase may offer an approach to treating this disease.
Other DUSPs
The Myotubularins (MTMs) are another category of DUSPs that exert their effects through dephosphorylation of non-protein substrates. In this case, the MTMs dephosphorylate the 3 position of the sugar ring in phosphatidylinositol (PtdIns) 3P and PtdIns(3.5)P2 [175–177]. The MTMs feature a variety of regulatory, non-catalytic domains that function in protein-protein and protein-lipid interactions and target the enzymes to specific cell membranes, including endosomes and the plasma membrane, which allows them to regulate particular pools of PtdIns3P and PtdIns3,5P2. A further striking characteristic is the prevalence of pseudophosphatases. Of the 15 MTMs, 9 are active enzymes, whereas the remaining 6 display no intrinsic phosphatase activity due to the absence of catalytically important residues in the primary sequence, in particular the active site cysteine. Heterodimerization of MTMs leads to formation of complexes between active and inactive MTMs. The structural basis for the recognition of specific phosphoinositide substrates by the active MTMs has been defined [178] and the residues that coordinate substrate binding are not conserved in the pseudophophatases. Consequently, it is unlikely that these pseudophosphatases function as substrate traps; instead, interaction between a positively charged face of the active MTM and the equivalent segment of the pseudophosphatase, which is negatively charged, most likely serves a scaffolding function that regulates the activity and subcellular location of the active phosphatase. The MTMs have been implicated in many fundamental physiological processes, including endocytosis and membrane trafficking, cell proliferation, differentiation, survival and autophagy. They are also intimately linked to human disease [177]. Defects in myotubularin (MTM1), the prototype of this family, cause X-linked myotubular myopathy. This is a rare genetic disease, characterized by the presence of immature myotubes, with resulting general muscle weakness and impaired motor skills. In many patients breathing is also impaired, resulting in death in early childhood. Mutations in another active member of the MTM family, MTMR2, have been found in type 4B Charcot-Marie-Tooth syndrome (CMT) [177]. CMT syndrome, which is a common hereditary disorder affecting one in every 2500 births, is a heterogeneous demyelinating disease characterized by myelin outfolding in peripheral nerves and progressive muscle atrophy. Type 4B CMT, which refers to the autosomal recessive disease, is caused by mutations in either MTMR2 or in the pseudophosphatase MTMR13 that heterodimerizes with MTMR2 [179]. Thus, mutation in either component of this phosphatase-pseudophosphatase complex gives rise to the disease, highlighting the functional importance of both constituents of the heterodimer.
The PTPs are thought to have evolved independently three times, to form three separate groups. Cdc25 represents the second such group and most likely evolved from a bacterial rhodanese-like enzyme. Rhodanese is a sulphur transferase that detoxifies cyanide to produce thiocyanate. A catalytic cysteine residue reacts with thiosulphate to generate a persulphide, which then transfers sulphur to cyanide. In cdc25, the catalytic cysteine is adapted for dephosphorylation. Cdc25 is part of the universal cell cycle engine conserved in eukaryotes from yeast to human. It is required for entry into mitosis because it removes phosphate from the inhibitory sites on cdc2 and thereby activates the cyclin-dependent kinase [49]. The three cdc25-like phosphatases in the human genome, designated A, B, and C, have all been shown to target the inhibitory phosphorylation sites on cyclin-dependent kinases, and are thus positive regulators of cell cycle progression and dual specificity phosphatases in vivo as well as in vitro. Many studies now point to increased expression of cdc25 in various cancers and consequently, cdc25-specific inhibitors are being explored as potential therapeutic reagents [180].
The third evolutionary event led to the phosphatase Ssu72, which targets the C-terminal domain of RNA polymerase II [181], and a pTyr-specific enzyme, the low molecular weight PTP (LMW-PTP), which is encoded by the ACP1 gene in humans and is highly conserved all the way to bacteria. The LMW-PTP is structurally related to bacterial arsenate reductase. Despite the low sequence identity, there is considerable overlap in the 3D structures of arsenate reductase and LMW-PTP, particularly in the area of the active site [182, 183]. In fact, arsenate reductase exhibits PTP activity in vitro [183]! The activity of arsenate reductase involves formation of an arsenocysteine covalent intermediate at the active site, equivalent to the phosphocysteine intermediate in PTP-mediated catalysis [183]. In contrast to the PTP, in which the cysteinyl phosphate intermediate undergoes hydrolysis, reactivation of arsenocysteine involves formation of a cascade of disulphide bonds through which the oxidative equivalents pass to the surface of the arsenate reductase for reaction with thioredoxin [184, 185]. Although implicated in the regulation of cell proliferation through dephosphorylation of several growth factor receptor PTKs, the function of the LMW-PTP remains unclear [48].
The issue of specificity: from the crystal structure of PTP1B to substrate trapping mutant PTPs
Although the classical PTPs are specific for pTyr residues in proteins, the Km for free pTyr is some 10,000-fold higher than that of optimal peptide substrates [186]. Interaction between residues flanking the pTyr in the primary sequence of the substrate and the residues surrounding the PTP active site contribute to the higher affinity for protein substrates. Furthermore, variability in the surface residues surrounding the active site between different PTP catalytic domains also has the potential to contribute to specificity in these interactions. In its recognition of the peptide DADEpYL from the EGF receptor as a substrate, there is an interaction between the acidic residues in the peptide and the side chain of Arg 47 in PTP1B [52]. In the example of recognition of the activation loop of the insulin receptor β-subunit (IRβ), there are interactions between PTP1B and residues on both N- and C-terminal sides of the substrate pTyr; in fact, we identified the sequence E/D-pY-pY-R/K as important for optimal recognition of the substrate [98]. In this case, the pTyr 1162 of the IRβ binds at the active site, whereas the adjacent pTyr 1163 binds within a shallow groove on the surface of PTP1B that is connected to the active site by a channel. In this groove, previously identified as “a second aryl-phosphate binding site”[187], the phosphate of pTyr 1163 forms salt bridges with the side chains of Arg 24 and Arg 254 of PTP1B, with specificity for pTyr binding to this site being determined by the length of the phosphorylated residue and the positioning of these arginines [97]. As a result of these interactions, PTP1B was observed to display 70 fold higher affinity for tandem pTyr-containing peptides compared to mono-pTyr derivatives [97]. On the basis of this E/D-pY-pY-R/K consensus motif, we predicted that the JAK subfamily of PTKs would be physiological substrates of PTP1B and demonstrated that PTP1B dephosphorylated JAK2 and TYK2 [98]. This suggested to us that PTP1B may function as a negative regulator of leptin signaling in the brain and that the resistance of PTP1B-knockout mice to high fat diet-induced obesity may result from aberrant effects on leptin signaling due to failure to down-regulate the JAK PTKs. In fact, experiments in the knockout mice did illustrate such a role for PTP1B [188, 189]. Although PTP specificity is not restricted to specific primary sequence motifs in substrates in the same way as for protein kinases, the importance of residues flanking sites of phosphorylation has been shown to contribute to specificity for other members of the PTP family [99, 190].
The critical step in the purification of PTP1B was affinity chromatography on an immobilized thiophosphorylated substrate. The underlying principle was that thiophosphorylated substrates retain the ability to bind tightly to the PTP, but are resistant to dephosphorylation. Therefore, the PTP binds the thiophosphorylated substrate in a stable complex from which it can be eluted with a salt gradient [1, 2]. The ability to define the substrate specificity of members of the PTP family is an essential aspect of gaining an understanding of the function of these enzymes. Using insights from the crystal structure, we undertook a mutational analysis to generate the converse of the affinity purification step – i.e. to produce a form of PTP1B that maintains its high affinity for substrate but does not catalyze dephosphorylation effectively, thereby to convert the active enzyme into a “substrate trap”. Mutation of the invariant catalytic acid (Asp181 in PTP1B) yielded such a substrate-trapping mutant [191]. The fact that the function of this Asp residue is conserved across nearly all members of the PTP family suggests that these mutants may offer an approach to identifying the physiological substrates of PTPs in general. The principle is that, following expression, the mutant PTP binds to its physiological substrates in the cell but, because it is unable to dephosphorylate the target efficiently, the mutant and substrate become locked in a stable, “dead-end” complex. These complexes can then be isolated, for example by immunoprecipitating the mutant PTP, and the substrates identified. This strategy has now been applied widely in the field [192]. Overall, the results illustrate that members of the PTP family display exquisite substrate specificity in a cellular context, consistent with a function as highly selective regulators of signal transduction.
Application of the PTP1B substrate trapping mutant (PTP1B-D181A) indicated a role for the enzyme in dephosphorylating the EGFR [191] insulin receptor [193] and JAK PTKs [98]. The localization of PTP1B to the ER has prompted questions regarding its importance as a regulator of PTK signaling. Using PTP1B−/− fibroblasts reconstituted with the substrate trapping mutant, the Neel and Bastiaens labs demonstrated that dephosphorylation of EGF and PDGF receptors occurred at specific sites in the ER following endocytosis of the PTK [194]. Their data illustrated that PTP1B has the potential to attenuate the activity of newly synthesized receptor PTKs, as well as functioning in a “dephosphorylation compartment” that is encountered by down-regulated PTKs before being directed either to the lysosome or recycled to the plasma membrane. This suggests that PTP1B plays an important role in terminating receptor-PTK signaling.
Regulation of PTP function – redox regulation reveals a new tier of control over pTyr-based signaling
As might be anticipated for a family of enzymes that play such fundamental physiological roles, their activity in vivo is tightly regulated. There is long term regulation at the level of expression, alternative promoters, alternative splicing and subcellular targeting, in addition to acute regulation of activity such as in response to ligand binding to RPTPs, or covalent modification, including phosphorylation and proteolysis. Over the last few years the importance of reversible oxidation of PTPs as a new tier of control of tyrosine phosphorylation-dependent signaling has become apparent [44, 195]. Due to the unique environment of the PTP active site, the invariant Cys residue in the signature motif displays an unusually low pKα, which enhances its nucleophilic properties but renders it susceptible to oxidation. Thus a stimulus (hormone, growth factor etc.) enhances tyrosine phosphorylation directly, by activation of a PTK, and/or indirectly, by production of Reactive Oxygen Species (ROS), which inactivate transiently the critical PTP(s) that provides the inhibitory constraint upon the system. Localized, stimulus-induced production of ROS leads to oxidation of the Cys residue at the PTP active site, converting it from a thiolate ion (the active form) to sulfinic acid, which can no longer function as a nucleophile, thus facilitating protein tyrosine phosphorylation and the initiation of the signalling response. Restoration of PTP activity following reduction back to the thiolate form of the active site Cys residue terminates the tyrosine phosphorylation-dependent signal. In collaboration with T.C. Meng and Kay-Hooi Khoo, we used mass spectrometry to characterize the oxidation status of each Cys residue in PTP1B that had been immunoprecipitated from HepG2 and A431 human cancer cells, which produce high levels of ROS constitutively. Our data revealed that the oxidation of PTP1B is specific to the active site Cys, with the other Cys residues in the protein remaining in a reduced state. This highlights the importance of the unique properties of this active site cysteine residue as the basis for this regulatory modification. In addition, we observed that up to 50% of PTP1B was reversibly oxidized and, due to the high constitutive levels of ROS, we observed that up to 40% of PTP1B was irreversibly oxidized in these cells [196]. These data suggest that the high level of intrinsic ROS may contribute to the transformed phenotype of HepG2 and A431 cells via constitutive inactivation of cellular PTPs, revealing another potential element of the etiology of cancer.
We developed approaches to identify those PTPs that are oxidized in response to a physiological stimulus, including modified “in-gel” phosphatase assays and the application of biotinylated small molecule probes [197–199]. We integrated these into an overall strategy involving RNA interference, combined with the use of substrate trapping mutants, to establish and define regulatory links between particular PTPs and specific signaling pathways. For example, we demonstrated that insulin induces the transient oxidation and inactivation of PTP1B, a PTP that normally attenuates insulin signaling[193]. More recently, Ben Neel’s lab developed a powerful mass spectrometry-based strategy for profiling both the total PTPome and the oxidized subset of PTPs, the oxPTPome, in cells and tissue samples [200, 201]. They have revealed that different cancer cells are characterized by distinct PTP oxidation profiles [200]. Further application of their approach is likely to generate fundamentally important insights into the importance of redox regulation of PTP function in various disease states.
In collaboration with David Barford, we investigated the oxidation of PTP1B from a structural perspective. We observed that oxidation of the nucleophilic Cys to sulfenic acid is accompanied by a rapid condensation reaction that generates a cyclic sulphenamide at the PTP active site [202]. This induces profound conformational changes that both protect the PTP from irreversible oxidation, and expose the oxidized Cys on the surface of the protein to facilitate reactivation by cellular reducing agents. The PTP loop, which contains the essential Cys residue, and the pTyr loop, in which Tyr46 defines the depth of the active site cleft, are normally buried in the structure. Upon formation of the cyclic sulphenamide, critical hydrogen bonds are broken triggering these residues to flip out of the active site and adopt solvent exposed positions. Thus, reversible oxidation generates a form of PTP1B in which the chemical properties of the active site of the native enzyme that are problematic for inhibitor development are circumvented, and unique new binding surfaces for small molecule inhibitors are presented. Consequently, if one could find a way to stabilize the oxidized, inactive form of PTP1B (PTP1B-OX), the result would be a novel mechanism through which to inhibit PTP function and promote the insulin signaling response – the property you would want in a treatment for insulin resistance.
Recently, we reported the use phage display technology to generate conformation-sensing antibodies that recognize oxidation-specific epitopes in PTP1B that would not be found in the active, reduced enzyme [203]. An important breakthrough was our discovery that a double point mutation in the PTP-loop of PTP1B (CASA mutant) induced a stable conformation that is indistinguishable in structure from the conformation that is induced by H2O2 and, therefore, could be used as an antigen. We generated a library of single chain fragment variable (scFv) antibodies, in which the variable light (VL) and variable heavy (VH) regions were amplified by PCR from spleen and bone marrow RNA of immunized chickens and joined with a neutral peptide linker. To isolate PTP1B-OX-specific antibodies from the scFv library we employed a subtractive panning strategy, in which N-terminally biotinylated PTP1B-CASA was mixed with the library under reducing conditions in the presence of up to 50-fold molar excess of wild type PTP1B. The PTP1B-CASA-scFv-phage complexes were isolated and amplified through four rounds of panning to enrich the library with pools of antibodies selective for the mutant form of the enzyme. We used an activity-based “in-solution” screen, in which we tested the ability of individual scFvs to stabilize the oxidized, inactive form of PTP1B in enzymatic assays in vitro; this allowed us to identify scFvs that bound PTP1B-OX and inhibited its reactivation by reducing agent, but did not exert any direct inhibitory effect on activity of the reduced, wild type enzyme. We observed that expression of the PTP1B-OX-specific scFvs in cells as “intrabodies” enhanced and extended the time course of insulin-induced phosphorylation the β-subunit of the insulin receptor and IRS-1, as well as insulin-induced downstream signaling (phosphorylation of PKB/AKT), in a manner that depended on production of H2O2; however, there was little or no impact on the basal level of tyrosyl phosphorylation and signaling. These data highlight the importance of redox regulation of PTP1B to insulin-induced signaling. Furthermore, they illustrate that stabilization of the inactive oxidized conformation of PTP1B, which is produced in response to insulin, potentiates the signaling response, suggesting a new approach for PTP-directed drug development.
PTPs and the etiology of disease
Perhaps not surprisingly considering their importance in normal physiology, it has become apparent over the years that disruption of PTP function has a major impact in human disease. In the early 90s, Kunliang Guan and Jack Dixon demonstrated that an essential virulence determinant of the bacterium Yersinia, the cause of the “Black Death” plague, was a highly active PTP encoded by the YOPH gene [204]. The bacterium uses a type III secretion system to inject effector proteins, including YopH, directly into the host cell, where is disrupts normal tyrosine phosphorylation-dependent signaling and inhibits the immune response [205]. The same authors also noted sequence similarity between the VH1 open reading frame of the poxvirus vaccinia and the PTP family, with VH1 representing the prototype DUSP [103]. These observations suggested that inhibitors of these PTPs may open up new approaches to counteracting infectious organisms.
Considering the prevalence of activated PTKs as oncoproteins, it was initially anticipated that many PTPs would be identified as tumour suppressors. It took a surprisingly long time to identify the first tumour suppressor PTP. Furthermore, the first example turned out to be a DUSP that regulated signaling through dephosphorylation of non-protein substrates - specifically PTEN, which regulates PI3 kinase-dependent signaling pathways associated with cell survival through dephosphorylation of the 3 position in the sugar head group of phosphatidylinositol phospholipids [138]. It has now been reported that 22 PTP genes map to chromosomal regions that are frequently deleted or amplified in human cancer [206]; however, many of these changes involve large segments of DNA and often the role of the PTPs remains to be established. More precise sequence analyses of individual PTPs have been conducted, including the identification of 83 somatic mutations in six PTP genes in colorectal cancer [207]. Ten somatic mutations were identified in the PTPRD gene alone in glioblastoma multiforme and malignant melanoma [208]; additional mutations have been identified in this gene in other cancers. A further level of control is exerted through epigenetic silencing, involving hypermethylation of CpG sites in the promoters of several members of the PTP family, as in the case of PTPRO and SHP1 [44, 206, 209]. Overall, several tumor suppressors have now been identified from among the classical PTPs [44, 206, 209]. In the case of some of the RPTPs, including DEP-1 (Density-Enhanced PTP)/PTPRJ and PTPRD, it is interesting to note clustering of mutations in the extracellular segments of these proteins, highlighting further the potential importance of ligand binding for the regulation of activity. As described above for SHP2, it is important also to recognise that PTPs may not only antagonize PTK function but also they have the capacity to function positively to promote signaling. Aberrant upregulation of PTPs in human disease has been detected in multiple cancers, such as overexpression of the cell cycle regulatory phosphatase cdc25, which often correlates with poor prognosis. Although there may be several mechanisms by which a PTP may act positively, one well established example involves the dephosphorylation of an inhibitory site at the C-terminus of SRC family PTKs, which promotes SRC activity and SRC-induced signaling. This mechanism is thought to underlie the potential tumorigenic effects of RPTPα [206, 209, 210]. Such positively acting PTPs may themselves prove to be important therapeutic targets for new ways to intervene in cancer.
Perhaps the major breakthrough that established PTPs as potential therapeutic targets was the definition of the role of PTP1B in down-regulating insulin and leptin signaling [44]. An important development came from the labs of Michel Tremblay and Ben Neel who demonstrated independently that targeted deletion of the PTP1B gene produced healthy mice that displayed increased insulin sensitivity and resistance to obesity induced by a high fat diet [211, 212]. This suggests that inhibitors of PTP1B may promote signaling in insulin resistant states and offer a novel approach to treating diabetes and obesity. Furthermore, PTP1B function is not restricted to metabolic regulation. The PTP1B gene is located at chromosome 20q13, which is a region that is frequently amplified in breast cancer and associated with poor prognosis. It has been reported that PTP1B is over-expressed in breast tumors, correlating with expression of HER2. Mice expressing activated alleles of HER2 in mammary glands develop multiple mammary tumors and frequent metastases to the lung; however, the Tremblay and Neel labs also demonstrated that when such mice were crossed with PTP1B-knockout mice, tumor development was delayed and the incidence of lung metastases was decreased [213, 214]. Conversely, MMTV-directed overexpression of PTP1B alone was sufficient to drive mammary tumorigenesis [213]. Although there remains some controversy regarding the underlying mechanism, the Tremblay group has proposed that one mechanism by which PTP1B exerts its effects is via dephosphorylation of p62DOK, and thereby regulation of the RAS/MAPK signaling pathway [213]. These observations illustrate than in addition to its role in down-regulating insulin signaling, PTP1B is an important positive regulator of HER2 signaling. Therefore, inhibition of PTP1B function with small molecule drugs would be expected to enhance signaling in conditions of insulin resistance and attenuate mammary tumorigenesis and malignancy in HER2-positive breast cancer.
The challenge of PTPs as therapeutic targets
Diabetes and obesity, together with cancer, represent huge healthcare challenges for the 21st century and the ability to modulate tyrosine phosphorylation-dependent signaling pathways selectively holds enormous therapeutic potential in these areas. The first drugs directed against protein tyrosine kinases (PTKs) have now entered the market and represent breakthroughs in cancer therapy. The classic example is Gleevec (STI-571), the small molecule inhibitor of the p210 BCR-ABL oncoprotein PTK for the treatment of chronic myelogenous leukemia [215]. In addition, there are antibody-based therapies including Herceptin (Trastuzumab), which targets the PTK HER2 that is overexpressed in ~25% of breast tumors [216]. Nevertheless, the overall success rate is limited and a major obstacle is that patients develop resistance to these therapies. Therefore, the development of novel approaches to target tyrosine phosphorylation-dependent signaling pathways will have a profound impact on drug development.
The emphasis in signal transduction-based drug discovery is currently placed on the kinases. Although PTPs have been garnering attention as potential therapeutic targets, they remain a largely untapped resource for drug development and are not without their challenges! The high degree of sequence similarity surrounding the active site of PTPs has been considered a problem. Nevertheless, it is interesting to consider that in the 1990s it was also thought that there was too much similarity between kinases to permit generation of specific inhibitors. Now, not only do most kinase inhibitors target the ATP-binding site, a feature they all share in common, but also some inhibitors, such as Gleevec, are known to act on more than one PTK. In fact, Lapatinib (Tykerb), which inhibits the EGFR and HER2 PTKs, shows efficacy in patients in which Herceptin has failed and was chosen because of its broad specificity!
The extensive biological data validating PTP1B as a therapeutic target for treating diabetes and obesity fueled considerable excitement in the pharmaceutical industry. Major programs were established in pharmaceutical companies, as well as biotechnology and academic groups, that were focused on developing small molecule inhibitors of PTP1B [217]. These programs followed standard procedures of looking for active site-directed inhibitors; however, such efforts have been frustrated by technical challenges arising from the chemical properties of the PTP active site. The susceptibility of PTPs to oxidation, coupled with the presence of oxidizing agents and peroxide generators in compound libraries, frequently confounded high throughput screens. Ultimately, it was possible to generate potent, specific and reversible inhibitors of PTP1B; in fact, nonhydrolyzable pTyr-mimetics, such as those pioneered by Zhong-Yin Zhang (Figure 9), were among the most potent [218]. Nevertheless, such molecules are highly charged, and thus of low oral bioavailability and limited drug development potential. Consequently, industry views PTPs as challenging, “undruggable” targets and new ideas are required to reinvigorate drug development efforts.
Figure 9. Specific inhibitors of PTP1B.
Potent, specific and reversible inhibitors of PTP1B have been generated, both in industry and in academia (illustrated here for the work of ZY Zhang [218]). Unfortunately, many, such as these, are analogues of a pTyr substrate and are thus highly charged, with limited bioavailability.
Back to the future
Despite the fact that “big pharma” are experts in drug development, they tend to be somewhat conservative in their approaches, focusing on active site-directed inhibitors of therapeutic targets. Therein lies the opportunity for those of us working on PTPs in academia. In the case of PTP1B, we need innovative approaches to generate inhibitors of this highly validated target that exhibit greater drug development potential. Antisense-based therapeutics directed against PTP1B have shown efficacy in clinical trials [219], providing further validation of this target in humans. In my lab, we are approaching this from two complementary perspectives. One strategy that would avoid targeting the catalytic center of PTP1B would be to look for allosteric inhibitors. These are inhibitors that bind at a site remote from the catalytic center, but induce conformational changes in the enzyme that result in inhibition. Precedence for such approaches comes from the work of Stig Hansen and his colleagues, who identified a novel allosteric site within the catalytic domain of PTP1B [220]. This is an approach that is also generating interesting results in the kinase field [221, 222]. We have identified novel allosteric sites in the regulatory C-terminal segment of PTP1B and are characterizing small molecules inhibitors that target this site. In a second, complementary approach, we are trying to harness the normal physiological regulatory mechanism through which the activity of PTP1B is attenuated by reversible oxidation. We have identified conformation-sensor antibodies that recognize and stabilize the reversibly oxidized form of PTP1B selectively, thereby inhibiting phosphatase activity and enhancing insulin-induced signal transduction [203]. Thus, stabilization of the oxidized, inactive form of PTP1B with appropriate therapeutic small molecules may offer a novel paradigm for phosphatase drug development. The search for drug-like small molecules that mimic the effects of the antibodies is underway.
There is more to PTP-based therapeutic development than just PTP1B! The ability to target the extracellular segment of RPTPs with therapeutic antibodies may be of benefit, as illustrated by the importance of PTPs in post-injury axon regrowth in the peripheral and central nervous system [223]. Great progress has been made in defining the physiological function of members of the PTP family, with the application of RNAi-based approaches [224] and animal models [225] continuing to reveal new functional insights. Nevertheless, many PTPs remain to be characterized extensively, resulting in new opportunities for biological insights. Overall, the days of phosphatases as housekeeping enzymes are over! I think there can be no doubt that PTPs display biochemical and biological specificity – but NOT at the level of one PTP for one PTK. Further characterization will help to reinforce that message. As further progress is made in defining the signalling function of PTPs and elucidating novel links to human disease, it is anticipated that new insights into therapeutic development will be revealed, either at the level of the PTPs themselves or from targets within the pathways they regulate.
Figure 6. Mutual regulation of MKP3 and ERK.
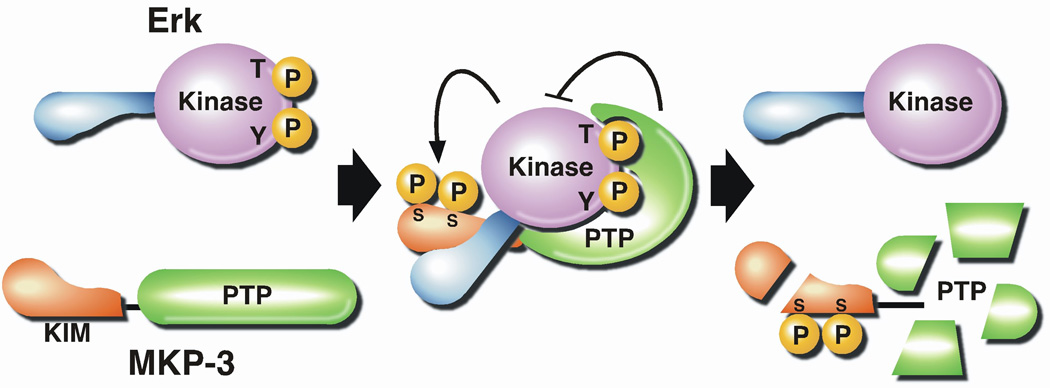
A Kinase Interaction Motif (KIM) in MKP3 interacts with ERK specifically, inducing a conformational change that promotes MKP function. Phosphorylation of MKP3 by the bound ERK promotes down regulation of MKP by proteolysis [227].
Acknowledgments
I am extremely grateful to all the excellent colleagues, past and present, for their important contributions to the research in my lab. I thank May Chen, in particular, for devising the classification of VH1-like DUSPs reported here as part of her PhD thesis research. Work in my laboratory is currently supported by NIH grants CA53840 and GM55989 and the CSHL Cancer Centre Support Grant CA45508. I am also grateful for support from the following foundations; The Gladowsky Breast Cancer Foundation, Hansen Foundation, Hartman Foundation, Irving Hansen Foundation, Islip Breast Cancer Coalition, Fannie Rippel Foundation, Robertson Research Fund and the Masthead Cove Yacht Club Carol Marcincuk Fund.
REFERENCES
- 1.Tonks NK, Diltz CD, Fischer EH. Purification of the major protein tyrosine phosphatases of human placenta. J Biol Chem. 1988;263:6722–6730. [PubMed] [Google Scholar]
- 2.Tonks NK, Diltz CD, Fischer EH. Characterization of the major protein tyrosine phosphatases of human placenta. J Biol Chem. 1988;263:6731–6737. [PubMed] [Google Scholar]
- 3.Brautigan DL. Protein Ser/ Thr phosphatases - the ugly ducklings of cell signalling. Febs J. 2012 doi: 10.1111/j.1742-4658.2012.08609.x. [DOI] [PubMed] [Google Scholar]
- 4.Krebs EG. Protein phosphorylation and cellular regulation I. Nobel Lecture. 1992:72–89. doi: 10.1007/BF01149958. [DOI] [PubMed] [Google Scholar]
- 5.Fischer EH. Protein phosphorylation and cellular regulation II. Nobel Lecture. 1992:95–113. [Google Scholar]
- 6.Danforth WH, Helmreich E, Cori CF. The effect of contraction and of epinephrine on the phosphorylase activity of frog Sartorius muscle. PNAS. 1962;48:1191–1199. doi: 10.1073/pnas.48.7.1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lim WA, Pawson T. Phosphotyrosine signaling: evolving a new cellular communication system. Cell. 2010;142:661–667. doi: 10.1016/j.cell.2010.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sefton BM, Hunter T, Beemon K, Eckhart W. Evidence that the phosphorylation of tyrosine is essential for cellular transformation by Rous sarcoma virus. Cell. 1980;20:807–816. doi: 10.1016/0092-8674(80)90327-x. [DOI] [PubMed] [Google Scholar]
- 9.MacKintosh C, Garton AJ, McDonnell A, Barford D, Cohen PTW, Tonks NK, Cohen P. Further evidence that inhibitor-2 acts like a chaperone to fold PP1 and its nature conformation. FEBS Lett. 1996;397:235–238. doi: 10.1016/s0014-5793(96)01175-1. [DOI] [PubMed] [Google Scholar]
- 10.Chernoff J, Li HC, Cheng YS, Chen LB. Characterization of a phosphotyrosyl protein phosphatase activity associated with a phosphoseryl protein phosphatase of Mr = 95,000 from bovine heart. J Biol Chem. 1983;258:7852–7857. [PubMed] [Google Scholar]
- 11.Pallen CJ, Valentine KA, Wang JH, Hollenberg MD. Calcineurin-mediated dephosphorylation of the human placental membrane receptor for epidermal growth factor urogastrone. Biochemistry. 1985;24:4727–4730. doi: 10.1021/bi00339a003. [DOI] [PubMed] [Google Scholar]
- 12.Chan CP, Gallis B, Blumenthal DK, Pallen CJ, Wang JH, Krebs EG. Characterization of the phosphotyrosyl protein phosphatase activity of calmodulin-dependent protein phosphatase. J Biol Chem. 1986;261:9890–9895. [PubMed] [Google Scholar]
- 13.Lin MF, Clinton GM. The epidermal growth factor receptor from prostate cells is dephosphorylated by a prostate-specific phosphotyrosyl phosphatase. Molecular and Cellular Biology. 1988;8:5477–5485. doi: 10.1128/mcb.8.12.5477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Horlein D, Gallis B, Brautigan DL, Bornstein P. Partial purification and characterization of phosphotyrosyl-protein phosphatase from Ehrlich ascites tumor cells. Biochemistry. 1982;21:5577–5584. doi: 10.1021/bi00265a030. [DOI] [PubMed] [Google Scholar]
- 15.Foulkes JG, Erikson E, Erikson RL. Separation of multiple phosphotyrosyl-and phosphoseryl-protein phosphatases from chicken brain. J Biol Chem. 1983;258:431–438. [PubMed] [Google Scholar]
- 16.Nelson RL, Branton PE. Identification, purification, and characterization of phosphotyrosine-specific protein phosphatases from cultured chicken embryo fibroblasts. Molecular and Cellular Biology. 1984;4:1003–1012. doi: 10.1128/mcb.4.6.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brunati AM, Pinna LA. Isolation and partial characterization of distinct species of phosphotyrosyl protein phosphatases from rat spleen. Biochem Biophys Res Commun. 1985;133:929–936. doi: 10.1016/0006-291x(85)91225-2. [DOI] [PubMed] [Google Scholar]
- 18.Swarup G, Cohen S, Garbers DL. Inhibition of membrane phosphotyrosyl-protein phosphatase activity by vanadate. Biochem Biophys Res Commun. 1982;107:1104–1109. doi: 10.1016/0006-291x(82)90635-0. [DOI] [PubMed] [Google Scholar]
- 19.Brautigan DL, Bornstein P, Gallis B. Phosphotyrosyl-protein phosphatase. Specific inhibition by Zn. J Biol Chem. 1981;256:6519–6522. [PubMed] [Google Scholar]
- 20.Shriner CL, Brautigan DL. Cytosolic protein phosphotyrosine phosphatases from rabbit kidney. Purification of two distinct enzymes that bind to Zn2+-iminodiacetate agarose. J Biol Chem. 1984;259:11383–11390. [PubMed] [Google Scholar]
- 21.Rotenberg SA, Brautigan DL. Membrane protein phosphotyrosine phosphatase in rabbit kidney. Proteolysis activates the enzyme and generates soluble catalytic fragments. Biochem J. 1987;243:747–754. doi: 10.1042/bj2430747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okada M, Owada K, Nakagawa H. [Phosphotyrosine]protein phosphatase in rat brain. A major [phosphotyrosine]protein phosphatase is a 23 kDa protein distinct from acid phosphatase. Biochem J. 1986;239:155–162. doi: 10.1042/bj2390155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tung HY, Reed LJ. Identification and purification of a cytosolic phosphotyrosyl protein phosphatase from bovine spleen. Anal Biochem. 1987;161:412–419. doi: 10.1016/0003-2697(87)90469-6. [DOI] [PubMed] [Google Scholar]
- 24.Kohanski RA, Lane MD. Kinetic evidence for activating and non-activating components of autophosphorylation of the insulin receptor protein kinase. Biochem Biophys Res Commun. 1986;134:1312–1318. doi: 10.1016/0006-291x(86)90393-1. [DOI] [PubMed] [Google Scholar]
- 25.Pike LJ, Eakes AT, Krebs EG. Characterization of affinity-purified insulin receptor/kinase. Effects of dithiothreitol on receptor/kinase function. J Biol Chem. 1986;261:3782–3789. [PubMed] [Google Scholar]
- 26.Tonks NK, Kearns A, Randle PJ. Pig heart [35S]thiophosphoryl pyruvate dehydrogenase complexes. Eur J Biochem. 1982;122:549–551. doi: 10.1111/j.1432-1033.1982.tb06472.x. [DOI] [PubMed] [Google Scholar]
- 27.Tonks NK, Cohen P. Calcineurin is a calcium ion-dependent, calmodulin-stimulated protein phosphatase. Biochim Biophys Acta. 1983;747:191–193. doi: 10.1016/0167-4838(83)90140-1. [DOI] [PubMed] [Google Scholar]
- 28.Charbonneau H, Tonks NK, Kumar S, Diltz CD, Harrylock M, Cool DE, Krebs EG, Fischer EH, Walsh KA. Human placenta protein-tyrosine-phosphatase: amino acid sequence and relationship to a family of receptor-like proteins. Proc Natl Acad Sci U S A. 1989;86:5252–5256. doi: 10.1073/pnas.86.14.5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Charbonneau H, Tonks NK, Walsh KA, Fischer EH. The leukocyte common antigen (CD45): a putative receptor-linked protein tyrosine phosphatase. Proc Natl Acad Sci U S A. 1988;85:7182–7186. doi: 10.1073/pnas.85.19.7182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trowbridge IS, Thomas ML. CD45: an emerging role as a protein tyrosine phosphatase required for lymphocyte activation and development. Annu Rev Immunol. 1994;12:85–116. doi: 10.1146/annurev.iy.12.040194.000505. [DOI] [PubMed] [Google Scholar]
- 31.Thomas ML, Barclay AN, Gagnon J, Williams AF. Evidence from cDNA clones that the rat leukocyte-common antigen (T200) spans the lipid bilayer and contains a cytoplasmic domain of 80,000 Mr. Cell. 1985;41:83–93. doi: 10.1016/0092-8674(85)90063-7. [DOI] [PubMed] [Google Scholar]
- 32.Ralph SJ, Thomas ML, Morton CC, Trowbridge IS. Structural variants of human T200 glycoprotein (leukocyte-common antigen) Embo J. 1987;6:1251–1257. doi: 10.1002/j.1460-2075.1987.tb02361.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thomas ML, Reynolds PJ, Chain A, Ben-Neriah Y, Trowbridge IS. B-cell variant of mouse T200 (Ly-5): evidence for alternative mRNA splicing. Proc Natl Acad Sci U S A. 1987;84:5360–5363. doi: 10.1073/pnas.84.15.5360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tonks NK, Charbonneau H, Diltz CD, Fischer EH, Walsh KA. Demonstration that the leukocyte common antigen CD45 is a protein tyrosine phosphatase. Biochemistry. 1988;27:8695–8701. doi: 10.1021/bi00424a001. [DOI] [PubMed] [Google Scholar]
- 35.Tonks NK, Charbonneau H. Protein tyrosine dephosphorylation and signal transduction. Trends Biochem Sci. 1989;14:497–500. doi: 10.1016/0968-0004(89)90184-9. [DOI] [PubMed] [Google Scholar]
- 36.Fischer EH, Charbonneau H, Tonks NK. Protein tyrosine phosphatases: a diverse family of intracellular and transmembrane enzymes. Science. 1991;253:401–406. doi: 10.1126/science.1650499. [DOI] [PubMed] [Google Scholar]
- 37.Pingel JT, Thomas ML. Evidence that the leukocyte-common antigen is required for antigen-induced T lymphocyte proliferation. Cell. 1989;58:1055–1065. doi: 10.1016/0092-8674(89)90504-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koretzky GA, Picus J, Thomas ML, Weiss A. Tyrosine phosphatase CD45 is essential for coupling T-cell antigen receptor to the phosphatidyl inositol pathway. Nature. 1990;346:66–68. doi: 10.1038/346066a0. [DOI] [PubMed] [Google Scholar]
- 39.Ostergaard HL, Shackelford DA, Hurley TR, Johnson P, Hyman R, Sefton BM, Trowbridge IS. Expression of CD45 alters phosphorylation of the lck-encoded tyrosine protein kinase in murine lymphoma T-cell lines. Proc Natl Acad Sci U S A. 1989;86:8959–8963. doi: 10.1073/pnas.86.22.8959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thomas ML, Brown EJ. Positive and negative regulation of Src-family membrane kinases by CD45. Immunol Today. 1999;20:406–411. doi: 10.1016/s0167-5699(99)01506-6. [DOI] [PubMed] [Google Scholar]
- 41.Streuli M, Krueger NX, Hall LR, Schlossman SF, Saito H. A new member of the immunoglobulin superfamily that has a cytoplasmic region homologous to the leukocyte common antigen. J Exp Med. 1988;168:1523–1530. doi: 10.1084/jem.168.5.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Andersen JN, Mortensen OH, Peters GH, Drake PG, Iversen LF, Olsen OH, Jansen PG, Andersen HS, Tonks NK, Moller NP. Structural and evolutionary relationships among protein tyrosine phosphatase domains. Molecular and Cellular Biology. 2001;21:7117–7136. doi: 10.1128/MCB.21.21.7117-7136.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Andersen JN, Jansen PG, Echwald SM, Mortensen OH, Fukada T, Del Vecchio R, Tonks NK, Moller NP. A genomic perspective on protein tyrosine phosphatases: gene structure, pseudogenes, and genetic disease linkage. Faseb J. 2004;18:8–30. doi: 10.1096/fj.02-1212rev. [DOI] [PubMed] [Google Scholar]
- 44.Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol. 2006;7:833–846. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- 45.Alonso A, Sasin J, Bottini N, Friedberg I, Osterman A, Godzik A, Hunter T, Dixon J, Mustelin T. Protein tyrosine phosphatases in the human genome. Cell. 2004;117:699–711. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 46.Patterson KI, Brummer T, O'Brien PM, Daly RJ. Dual-specificity phosphatases: critical regulators with diverse cellular targets. Biochem J. 2009;418:475–489. doi: 10.1042/bj20082234. [DOI] [PubMed] [Google Scholar]
- 47.Moorhead GB, De Wever V, Templeton G, Kerk D. Evolution of protein phosphatases in plants and animals. Biochem J. 2009;417:401–409. doi: 10.1042/BJ20081986. [DOI] [PubMed] [Google Scholar]
- 48.Raugei G, Ramponi G, Chiarugi P. Low molecular weight protein tyrosine phosphatases: small, but smart. Cell Mol Life Sci. 2002;59:941–949. doi: 10.1007/s00018-002-8481-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boutros R, Lobjois V, Ducommun B. CDC25 phosphatases in cancer cells: key players? Good targets? Nat Rev Cancer. 2007;7:495–507. doi: 10.1038/nrc2169. [DOI] [PubMed] [Google Scholar]
- 50.Barford D, Flint AJ, Tonks NK. Crystal structure of human protein tyrosine phosphatase 1B. Science. 1994;263:1397–1404. [PubMed] [Google Scholar]
- 51.Tonks NK. PTP1B: from the sidelines to the front lines! FEBS Lett. 2003;546:140–148. doi: 10.1016/s0014-5793(03)00603-3. [DOI] [PubMed] [Google Scholar]
- 52.Jia Z, Barford D, Flint AJ, Tonks NK. Structural basis for phosphotyrosine peptide recognition by protein tyrosine phosphatase 1B. Science. 1995;268:1754–1758. doi: 10.1126/science.7540771. [DOI] [PubMed] [Google Scholar]
- 53.Pannifer AD, Flint AJ, Tonks NK, Barford D. Visualization of the cysteinyl-phosphate intermediate of a protein-tyrosine phosphatase by x-ray crystallography. J Biol Chem. 1998;273:10454–10462. doi: 10.1074/jbc.273.17.10454. [DOI] [PubMed] [Google Scholar]
- 54.Barford D, Jia Z, Tonks NK. Protein tyrosine phosphatases take off. Nat Struct Biol. 1995;2:1043–1053. doi: 10.1038/nsb1295-1043. [DOI] [PubMed] [Google Scholar]
- 55.Zheng J, Yates SP, Jia Z. Structural and mechanistic insights into the bifunctional enzyme isocitrate dehydrogenase kinase/phosphatase AceK. Philos Trans R Soc Lond B Biol Sci. 2012;367:2656–2668. doi: 10.1098/rstb.2011.0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Begley MJ, Dixon JE. The structure and regulation of myotubularin phosphatases. Curr Opin Struct Biol. 2005;15:614–620. doi: 10.1016/j.sbi.2005.10.016. [DOI] [PubMed] [Google Scholar]
- 57.Mohebiany AN, Nikolaienko RM, Bouyain S, Harroch S. Receptor-type tyrosine phosphatase ligands: looking for the needle in the haystack. Febs J. 2012 doi: 10.1111/j.1742-4658.2012.08653.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brady-Kalnay SM, Flint AJ, Tonks NK. Homophilic binding of PTP mu, a receptor-type protein tyrosine phosphatase, can mediate cell-cell aggregation. J Cell Biol. 1993;122:961–972. doi: 10.1083/jcb.122.4.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brady-Kalnay SM, Tonks NK. Identification of the homophilic binding site of the receptor protein tyrosine phosphatase PTP mu. J Biol Chem. 1994;269:28472–28477. [PubMed] [Google Scholar]
- 60.Sap J, Jiang YP, Friedlander D, Grumet M, Schlessinger J. Receptor tyrosine phosphatase R-PTP-kappa mediates homophilic binding. Molecular and Cellular Biology. 1994;14:1–9. doi: 10.1128/mcb.14.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brady-Kalnay SM, Rimm DL, Tonks NK. Receptor protein tyrosine phosphatase PTPmu associates with cadherins and catenins in vivo. J Cell Biol. 1995;130:977–986. doi: 10.1083/jcb.130.4.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brady-Kalnay SM, Mourton T, Nixon JP, Pietz GE, Kinch M, Chen H, Brackenbury R, Rimm DL, Del Vecchio RL, Tonks NK. Dynamic interaction of PTPmu with multiple cadherins in vivo. J Cell Biol. 1998;141:287–296. doi: 10.1083/jcb.141.1.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hermiston ML, Xu Z, Weiss A. CD45: a critical regulator of signaling thresholds in immune cells. Annu Rev Immunol. 2003;21:107–137. doi: 10.1146/annurev.immunol.21.120601.140946. [DOI] [PubMed] [Google Scholar]
- 64.Xu Z, Weiss A. Negative regulation of CD45 by differential homodimerization of the alternatively spliced isoforms. Nat Immunol. 2002;3:764–771. doi: 10.1038/ni822. [DOI] [PubMed] [Google Scholar]
- 65.Meng K, Rodriguez-Pena A, Dimitrov T, Chen W, Yamin M, Noda M, Deuel TF. Pleiotrophin signals increased tyrosine phosphorylation of beta beta-catenin through inactivation of the intrinsic catalytic activity of the receptor-type protein tyrosine phosphatase beta/zeta. Proc Natl Acad Sci U S A. 2000;97:2603–2608. doi: 10.1073/pnas.020487997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pariser H, Perez-Pinera P, Ezquerra L, Herradon G, Deuel TF. Pleiotrophin stimulates tyrosine phosphorylation of beta-adducin through inactivation of the transmembrane receptor protein tyrosine phosphatase beta/zeta. Biochem Biophys Res Commun. 2005;335:232–239. doi: 10.1016/j.bbrc.2005.07.060. [DOI] [PubMed] [Google Scholar]
- 67.Tamura H, Fukada M, Fujikawa A, Noda M. Protein tyrosine phosphatase receptor type Z is involved in hippocampus-dependent memory formation through dephosphorylation at Y1105 on p190 RhoGAP. Neurosci Lett. 2006;399:33–38. doi: 10.1016/j.neulet.2006.01.045. [DOI] [PubMed] [Google Scholar]
- 68.Johnson KG, Tenney AP, Ghose A, Duckworth AM, Higashi ME, Parfitt K, Marcu O, Heslip TR, Marsh JL, Schwarz TL, et al. The HSPGs Syndecan and Dallylike bind the receptor phosphatase LAR and exert distinct effects on synaptic development. Neuron. 2006;49:517–531. doi: 10.1016/j.neuron.2006.01.026. [DOI] [PubMed] [Google Scholar]
- 69.Streuli M, Krueger NX, Thai T, Tang M, Saito H. Distinct functional roles of the two intracellular phosphatase like domains of the receptor-linked protein tyrosine phosphatases LCA and LAR. Embo J. 1990;9:2399–2407. doi: 10.1002/j.1460-2075.1990.tb07415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Felberg J, Johnson P. Characterization of recombinant CD45 cytoplasmic domain proteins. Evidence for intramolecular and intermolecular interactions. J Biol Chem. 1998;273:17839–17845. doi: 10.1074/jbc.273.28.17839. [DOI] [PubMed] [Google Scholar]
- 71.Nam HJ, Poy F, Krueger NX, Saito H, Frederick CA. Crystal structure of the tandem phosphatase domains of RPTP LAR. Cell. 1999;97:449–457. doi: 10.1016/s0092-8674(00)80755-2. [DOI] [PubMed] [Google Scholar]
- 72.Yang QT, Tonks NK. Adv Protein Phosphatases. Leuven University Press; 1993. Structural diversity within the protein tyrosine phosphatase family. In; pp. 359–372. [Google Scholar]
- 73.Blanchetot C, Tertoolen LG, Overvoorde J, den Hertog J. Intra- and intermolecular interactions between intracellular domains of receptor protein-tyrosine phosphatases. J Biol Chem. 2002;277:47263–47269. doi: 10.1074/jbc.M205810200. [DOI] [PubMed] [Google Scholar]
- 74.Persson C, Sjoblom T, Groen A, Kappert K, Engstrom U, Hellman U, Heldin CH, den Hertog J, Ostman A. Preferential oxidation of the second phosphatase domain of receptor-like PTP-alpha revealed by an antibody against oxidized protein tyrosine phosphatases. Proc Natl Acad Sci U S A. 2004;101:1886–1891. doi: 10.1073/pnas.0304403101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Blanchetot C, Tertoolen LG, den Hertog J. Regulation of receptor protein-tyrosine phosphatase alpha by oxidative stress. Embo J. 2002;21:493–503. doi: 10.1093/emboj/21.4.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.van der Wijk T, Overvoorde J, den Hertog J. H2O2-induced intermolecular disulfide bond formation between receptor protein-tyrosine phosphatases. J Biol Chem. 2004;279:44355–44361. doi: 10.1074/jbc.M407483200. [DOI] [PubMed] [Google Scholar]
- 77.Bilwes AM, den Hertog J, Hunter T, Noel JP. Structural basis for inhibition of receptor protein-tyrosine phosphatase-alpha by dimerization. Nature. 1996;382:555–559. doi: 10.1038/382555a0. [DOI] [PubMed] [Google Scholar]
- 78.Hoffmann KM, Tonks NK, Barford D. The crystal structure of domain 1 of receptor protein-tyrosine phosphatase mu. J Biol Chem. 1997;272:27505–27508. doi: 10.1074/jbc.272.44.27505. [DOI] [PubMed] [Google Scholar]
- 79.Nam HJ, Poy F, Saito H, Frederick CA. Structural basis for the function and regulation of the receptor protein tyrosine phosphatase CD45. J Exp Med. 2005;201:441–452. doi: 10.1084/jem.20041890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.James JR, McColl J, Oliveira MI, Dunne PD, Huang E, Jansson A, Nilsson P, Sleep DL, Goncalves CM, Morgan SH, et al. The T cell receptor triggering apparatus is composed of monovalent or monomeric proteins. J Biol Chem. 2011;286:31993–32001. doi: 10.1074/jbc.M111.219212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hermiston ML, Tan AL, Gupta VA, Majeti R, Weiss A. The juxtamembrane wedge negatively regulates CD45 function in B cells. Immunity. 2005;23:635–647. doi: 10.1016/j.immuni.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 82.Guan KL, Haun RS, Watson SJ, Geahlen RL, Dixon JE. Cloning and expression of a protein-tyrosine-phosphatase. Proc Natl Acad Sci U S A. 1990;87:1501–1505. doi: 10.1073/pnas.87.4.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chernoff J, Schievella AR, Jost CA, Erikson RL, Neel BG. Cloning of a cDNA for a major human protein-tyrosine-phosphatase. Proc Natl Acad Sci U S A. 1990;87:2735–2739. doi: 10.1073/pnas.87.7.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Brown-Shimer S, Johnson KA, Lawrence JB, Johnson C, Bruskin A, Green NR, Hill DE. Molecular cloning and chromosome mapping of the human gene encoding protein phosphotyrosyl phosphatase 1B. Proc Natl Acad Sci U S A. 1990;87:5148–5152. doi: 10.1073/pnas.87.13.5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Frangioni JV, Beahm PH, Shifrin V, Jost CA, Neel BG. The nontransmembrane tyrosine phosphatase PTP-1B localizes to the endoplasmic reticulum via its 35 amino acid C-terminal sequence. Cell. 1992;68:545–560. doi: 10.1016/0092-8674(92)90190-n. [DOI] [PubMed] [Google Scholar]
- 86.Cool DE, Tonks NK, Charbonneau H, Walsh KA, Fischer EH, Krebs EG. cDNA isolated from a human T-cell library encodes a member of the protein-tyrosine-phosphatase family. Proc Natl Acad Sci U S A. 1989;86:5257–5261. doi: 10.1073/pnas.86.14.5257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lorenzen JA, Dadabay CY, Fischer EH. COOH-terminal sequence motifs target the T cell protein tyrosine phosphatase to the ER and nucleus. J Cell Biol. 1995;131:631–643. doi: 10.1083/jcb.131.3.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lam MH, Michell BJ, Fodero-Tavoletti MT, Kemp BE, Tonks NK, Tiganis T. Cellular stress regulates the nucleocytoplasmic distribution of the protein-tyrosine phosphatase TCPTP. J Biol Chem. 2001;276:37700–37707. doi: 10.1074/jbc.M105128200. [DOI] [PubMed] [Google Scholar]
- 89.Flint AJ, Gebbink MF, Franza BR, Jr, Hill DE, Tonks NK. Multi-site phosphorylation of the protein tyrosine phosphatase, PTP1B: identification of cell cycle regulated and phorbol ester stimulated sites of phosphorylation. Embo J. 1993;12:1937–1946. doi: 10.1002/j.1460-2075.1993.tb05843.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Frangioni JV, Oda A, Smith M, Salzman EW, Neel BG. Calpain-catalyzed cleavage and subcellular relocation of protein phosphotyrosine phosphatase 1B (PTP-1B) in human platelets. Embo J. 1993;12:4843–4856. doi: 10.1002/j.1460-2075.1993.tb06174.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hao L, Tiganis T, Tonks NK, Charbonneau H. The noncatalytic C-terminal segment of the T cell protein tyrosine phosphatase regulates activity via an intramolecular mechanism. J Biol Chem. 1997;272:29322–29329. doi: 10.1074/jbc.272.46.29322. [DOI] [PubMed] [Google Scholar]
- 92.Neel BG, Gu H, Pao L. The 'Shp'ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci. 2003;28:284–293. doi: 10.1016/S0968-0004(03)00091-4. [DOI] [PubMed] [Google Scholar]
- 93.Chishti AH, Kim AC, Marfatia SM, Lutchman M, Hanspal M, Jindal H, Liu SC, Low PS, Rouleau GA, Mohandas N, et al. The FERM domain: a unique module involved in the linkage of cytoplasmic proteins to the membrane. Trends Biochem Sci. 1998;23:281–282. doi: 10.1016/s0968-0004(98)01237-7. [DOI] [PubMed] [Google Scholar]
- 94.Saito K, Tautz L, Mustelin T. The lipid-binding SEC14 domain. Biochim Biophys Acta. 2007;1771:719–726. doi: 10.1016/j.bbalip.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 95.Kim J, Sitaraman S, Hierro A, Beach BM, Odorizzi G, Hurley JH. Structural basis for endosomal targeting by the Bro1 domain. Dev Cell. 2005;8:937–947. doi: 10.1016/j.devcel.2005.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mauro LJ, Dixon JE. 'Zip codes' direct intracellular protein tyrosine phosphatases to the correct cellular 'address'. Trends Biochem Sci. 1994;19:151–155. doi: 10.1016/0968-0004(94)90274-7. [DOI] [PubMed] [Google Scholar]
- 97.Salmeen A, Andersen JN, Myers MP, Tonks NK, Barford D. Molecular basis for the dephosphorylation of the activation segment of the insulin receptor by protein tyrosine phosphatase 1B. Mol Cell. 2000;6:1401–1412. doi: 10.1016/s1097-2765(00)00137-4. [DOI] [PubMed] [Google Scholar]
- 98.Myers MP, Andersen JN, Cheng A, Tremblay ML, Horvath CM, Parisien J-P, Salmeen A, Barford D, Tonks NK. TYK2 and JAK2 are substrates of protein-tyrosine phosphatase 1B. J Biol Chem. 2001;276:47771–47774. doi: 10.1074/jbc.C100583200. [DOI] [PubMed] [Google Scholar]
- 99.Ren L, Chen X, Luechapanichkul R, Selner NG, Meyer TM, Wavreille AS, Chan R, Iorio C, Zhou X, Neel BG, Pei D. Substrate specificity of protein tyrosine phosphatases 1B, RPTPa, SHP-1, and SHP-2. Biochemistry. 2011;50:2339–2356. doi: 10.1021/bi1014453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pulido R, Zuniga A, Ullrich A. PTP-SL and STEP protein tyrosine phosphatases regulate the activation of the extracellular signal-regulated kinases ERK1 and ERK2 by association through a kinase interaction motif. Embo J. 1998;17:7337–7350. doi: 10.1093/emboj/17.24.7337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Garton AJ, Burnham MR, Bouton AH, Tonks NK. Association of PTP-PEST with the SH3 domain of p130cas; a novel mechanism of protein tyrosine phosphatase substrate recognition. Oncogene. 1997;15:877–885. doi: 10.1038/sj.onc.1201279. [DOI] [PubMed] [Google Scholar]
- 102.O'Reilly AM, Pluskey S, Shoelson SE, Neel BG. Activated mutants of SHP-2 preferentially induce elongation of Xenopus animal caps. Molecular and Cellular Biology. 2000;20:299–311. doi: 10.1128/mcb.20.1.299-311.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Guan KL, Broyles SS, Dixon JE. A Tyr/Ser protein phosphatase encoded by vaccinia virus. Nature. 1991;350:359–362. doi: 10.1038/350359a0. [DOI] [PubMed] [Google Scholar]
- 104.Liu K, Lemon B, Traktman P. The dual-specificity phosphatase encoded by vaccinia virus, VH1, is essential for viral transcription in vivo and in vitro. J Virol. 1995;69:7823–7834. doi: 10.1128/jvi.69.12.7823-7834.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Derrien M, Punjabi A, Khanna M, Grubisha O, Traktman P. Tyrosine phosphorylation of A17 during vaccinia virus infection: involvement of the H1 phosphatase and the F10 kinase. J Virol. 1999;73:7287–7296. doi: 10.1128/jvi.73.9.7287-7296.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Najarro P, Traktman P, Lewis JA. Vaccinia virus blocks gamma interferon signal transduction: viral VH1 phosphatase reverses Stat1 activation. J Virol. 2001;75:3185–3196. doi: 10.1128/JVI.75.7.3185-3196.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Charles CH, Sun H, Lau LF, Tonks NK. The growth factor-inducible immediate-early gene 3CH134 encodes a protein-tyrosine-phosphatase. Proc Natl Acad Sci U S A. 1993;90:5292–5296. doi: 10.1073/pnas.90.11.5292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sun H, Charles CH, Lau LF, Tonks NK. MKP-1 (3CH134), an immediate early gene product, is a dual specificity phosphatase that dephosphorylates MAP kinase in vivo. Cell. 1993;75:487–493. doi: 10.1016/0092-8674(93)90383-2. [DOI] [PubMed] [Google Scholar]
- 109.Alessi DR, Smythe C, Keyse SM. The human CL100 gene encodes a Tyr/Thr-protein phosphatase which potently and specifically inactivates MAP kinase and suppresses its activation by oncogenic ras in Xenopus oocyte extracts. Oncogene. 1993;8:2015–2020. [PubMed] [Google Scholar]
- 110.Chen H-H. Ph.D. Thesis. Stony Brook NY: Stony Brook University; 2004. Characterization of the dual specificity phosphatases. [Google Scholar]
- 111.Caunt CJ, Keyse SM. Dual-specificity MAP kinase phosphatases (MKPs): Shaping the outcome of MAP kinase signalling. Febs J. 2012 doi: 10.1111/j.1742-4658.2012.08716.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhou B, Zhang ZY. Mechanism of mitogen-activated protein kinase phosphatase-3 activation by ERK2. J Biol Chem. 1999;274:35526–35534. doi: 10.1074/jbc.274.50.35526. [DOI] [PubMed] [Google Scholar]
- 113.Ishibashi T, Bottaro DP, Chan A, Miki T, Aaronson SA. Expression cloning of a human dual-specificity phosphatase. Proc Natl Acad Sci U S A. 1992;89:12170–12174. doi: 10.1073/pnas.89.24.12170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Todd JL, Tanner KG, Denu JM. Extracellular regulated kinases (ERK) 1 and ERK2 are authentic substrates for the dual-specificity protein-tyrosine phosphatase VHR. A novel role in down-regulating the ERK pathway. J Biol Chem. 1999;274:13271–13280. doi: 10.1074/jbc.274.19.13271. [DOI] [PubMed] [Google Scholar]
- 115.Kang TH, Kim KT. Negative regulation of ERK activity by VRK3-mediated activation of VHR phosphatase. Nat Cell Biol. 2006;8:863–869. doi: 10.1038/ncb1447. [DOI] [PubMed] [Google Scholar]
- 116.Schumacher MA, Todd JL, Rice AE, Tanner KG, Denu JM. Structural basis for the recognition of a bisphosphorylated MAP kinase peptide by human VHR protein Phosphatase. Biochemistry. 2002;41:3009–3017. doi: 10.1021/bi015799l. [DOI] [PubMed] [Google Scholar]
- 117.Alonso A, Saxena M, Williams S, Mustelin T. Inhibitory role for dual specificity phosphatase VHR in T cell antigen receptor and CD28-induced Erk and Jnk activation. J Biol Chem. 2001;276:4766–4771. doi: 10.1074/jbc.M006497200. [DOI] [PubMed] [Google Scholar]
- 118.Todd JL, Rigas JD, Rafty LA, Denu JM. Dual-specificity protein tyrosine phosphatase VHR down-regulates c-Jun N-terminal kinase (JNK) Oncogene. 2002;21:2573–2583. doi: 10.1038/sj.onc.1205344. [DOI] [PubMed] [Google Scholar]
- 119.Rahmouni S, Cerignoli F, Alonso A, Tsutji T, Henkens R, Zhu C, Louis-dit-Sully C, Moutschen M, Jiang W, Mustelin T. Loss of the VHR dual-specific phosphatase causes cell-cycle arrest and senescence. Nat Cell Biol. 2006;8:524–531. doi: 10.1038/ncb1398. [DOI] [PubMed] [Google Scholar]
- 120.Arnoldussen YJ, Lorenzo PI, Pretorius ME, Waehre H, Risberg B, Maelandsmo GM, Danielsen HE, Saatcioglu F. The mitogen-activated protein kinase phosphatase vaccinia H1-related protein inhibits apoptosis in prostate cancer cells and is overexpressed in prostate cancer. Cancer Res. 2008;68:9255–9264. doi: 10.1158/0008-5472.CAN-08-1224. [DOI] [PubMed] [Google Scholar]
- 121.Shen Y, Luche R, Wei B, Gordon ML, Diltz CD, Tonks NK. Activation of the Jnk signaling pathway by a dual-specificity phosphatase, JSP-1. Proc Natl Acad Sci U S A. 2001;98:13613–13618. doi: 10.1073/pnas.231499098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chen AJ, Zhou G, Juan T, Colicos SM, Cannon JP, Cabriera-Hansen M, Meyer CF, Jurecic R, Copeland NG, Gilbert DJ, et al. The dual specificity JKAP specifically activates the c-Jun N-terminal kinase pathway. J Biol Chem. 2002;277:36592–36601. doi: 10.1074/jbc.M200453200. [DOI] [PubMed] [Google Scholar]
- 123.Alonso A, Merlo JJ, Na S, Kholod N, Jaroszewski L, Kharitonenkov A, Williams S, Godzik A, Posada JD, Mustelin T. Inhibition of T cell antigen receptor signaling by VHR-related MKPX (VHX), a new dual specificity phosphatase related to VH1 related (VHR) J Biol Chem. 2002;277:5524–5528. doi: 10.1074/jbc.M107653200. [DOI] [PubMed] [Google Scholar]
- 124.Aoyama K, Nagata M, Oshima K, Matsuda T, Aoki N. Molecular cloning and characterization of a novel dual specificity phosphatase, LMW-DSP2, that lacks the cdc25 homology domain. J Biol Chem. 2001;276:27575–27583. doi: 10.1074/jbc.M100408200. [DOI] [PubMed] [Google Scholar]
- 125.Schwertassek U, Buckley DA, Xu CF, Lindsay AJ, McCaffrey MW, Neubert TA, Tonks NK. Myristoylation of the dual-specificity phosphatase c-JUN N-terminal kinase (JNK) stimulatory phosphatase 1 is necessary for its activation of JNK signaling and apoptosis. Febs J. 2010;277:2463–2473. doi: 10.1111/j.1742-4658.2010.07661.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Huang TY, DerMardirossian C, Bokoch GM. Cofilin phosphatases and regulation of actin dynamics. Curr Opin Cell Biol. 2006;18:26–31. doi: 10.1016/j.ceb.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 127.Soosairajah J, Maiti S, Wiggan O, Sarmiere P, Moussi N, Sarcevic B, Sampath R, Bamburg JR, Bernard O. Interplay between components of a novel LIM kinase-slingshot phosphatase complex regulates cofilin. Embo J. 2005;24:473–486. doi: 10.1038/sj.emboj.7600543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chen HH, Luche R, Wei B, Tonks NK. Characterization of two distinct dual specificity phosphatases encoded in alternative open reading frames of a single gene located on human chromosome 10q22.2. J Biol Chem. 2004;279:41404–41413. doi: 10.1074/jbc.M405286200. [DOI] [PubMed] [Google Scholar]
- 129.Gil J, Peters G. Regulation of the INK4b-ARF-INK4a tumour suppressor locus: all for one or one for all. Nat Rev Mol Cell Biol. 2006;7:667–677. doi: 10.1038/nrm1987. [DOI] [PubMed] [Google Scholar]
- 130.Katagiri C, Masuda K, Nomura M, Tanoue K, Fujita S, Yamashita Y, Katakura R, Shiiba K, Nomura E, Sato M, et al. DUSP13B/TMDP inhibits stress-activated MAPKs and suppresses AP-1-dependent gene expression. Mol Cell Biochem. 2011;352:155–162. doi: 10.1007/s11010-011-0749-x. [DOI] [PubMed] [Google Scholar]
- 131.Wishart MJ, Denu JM, Williams JA, Dixon JE. A single mutation converts a novel phosphotyrosine binding domain into a dual-specificity phosphatase. J Biol Chem. 1995;270:26782–26785. doi: 10.1074/jbc.270.45.26782. [DOI] [PubMed] [Google Scholar]
- 132.Wishart MJ, Dixon JE. The archetype STYX/dead-phosphatase complexes with a spermatid mRNA-binding protein and is essential for normal sperm production. Proc Natl Acad Sci U S A. 2002;99:2112–2117. doi: 10.1073/pnas.251686198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hinton SD, Myers MP, Roggero VR, Allison LA, Tonks NK. The pseudophosphatase MK-STYX interacts with G3BP and decreases stress granule formation. Biochem J. 2010;427:349–357. doi: 10.1042/BJ20091383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN tumor suppression. Cell. 2008;133:403–414. doi: 10.1016/j.cell.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 135.Hollander MC, Blumenthal GM, Dennis PA. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat Rev Cancer. 2011;11:289–301. doi: 10.1038/nrc3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Leslie NR, Foti M. Non-genomic loss of PTEN function in cancer: not in my genes. Trends Pharmacol Sci. 2011;32:131–140. doi: 10.1016/j.tips.2010.12.005. [DOI] [PubMed] [Google Scholar]
- 137.Myers MP, Stolarov JP, Eng C, Li J, Wang SI, Wigler MH, Parsons R, Tonks NK. P-TEN, the tumor suppressor from human chromosome 10q23, is a dual-specificity phosphatase. Proc Natl Acad Sci U S A. 1997;94:9052–9057. doi: 10.1073/pnas.94.17.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 139.Lee JO, Yang H, Georgescu MM, Di Cristofano A, Maehama T, Shi Y, Dixon JE, Pandolfi P, Pavletich NP. Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell. 1999;99:323–334. doi: 10.1016/s0092-8674(00)81663-3. [DOI] [PubMed] [Google Scholar]
- 140.Myers MP, Pass I, Batty IH, Van der Kaay J, Stolarov JP, Hemmings BA, Wigler MH, Downes CP, Tonks NK. The lipid phosphatase activity of PTEN is critical for its tumor supressor function. Proc Natl Acad Sci U S A. 1998;95:13513–13518. doi: 10.1073/pnas.95.23.13513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Raftopoulou M, Etienne-Manneville S, Self A, Nicholls S, Hall A. Regulation of cell migration by the C2 domain of the tumor suppressor PTEN. Science. 2004;303:1179–1181. doi: 10.1126/science.1092089. [DOI] [PubMed] [Google Scholar]
- 142.Mahimainathan L, Choudhury GG. Inactivation of platelet-derived growth factor receptor by the tumor suppressor PTEN provides a novel mechanism of action of the phosphatase. J Biol Chem. 2004;279:15258–15268. doi: 10.1074/jbc.M314328200. [DOI] [PubMed] [Google Scholar]
- 143.Tamura M, Gu J, Takino T, Yamada KM. Tumor suppressor PTEN inhibition of cell invasion, migration, and growth: differential involvement of focal adhesion kinase and p130Cas. Cancer Res. 1999;59:442–449. [PubMed] [Google Scholar]
- 144.Vazquez F, Ramaswamy S, Nakamura N, Sellers WR. Phosphorylation of the PTEN tail regulates protein stability and function. Molecular and Cellular Biology. 2000;20:5010–5018. doi: 10.1128/mcb.20.14.5010-5018.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Torres J, Rodriguez J, Myers MP, Valiente M, Graves JD, Tonks NK, Pulido R. Phosphorylation-regulated cleavage of the tumor suppressor PTEN by caspase-3: implications for the control of protein stability and PTEN-protein interactions. J Biol Chem. 2003;278:30652–30660. doi: 10.1074/jbc.M212610200. [DOI] [PubMed] [Google Scholar]
- 146.Odriozola L, Singh G, Hoang T, Chan AM. Regulation of PTEN activity by its carboxyl-terminal autoinhibitory domain. J Biol Chem. 2007;282:23306–23315. doi: 10.1074/jbc.M611240200. [DOI] [PubMed] [Google Scholar]
- 147.Zhang XC, Piccini A, Myers MP, Van Aelst L, Tonks NK. Functional analysis of the protein phosphatase activity of PTEN. Biochem J. 2012;444:457–464. doi: 10.1042/BJ20120098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Vanhaesebroeck B, Higashi K, Raven C, Welham M, Anderson S, Brennan P, Ward SG, Waterfield MD. Autophosphorylation of p110delta phosphoinositide 3-kinase: a new paradigm for the regulation of lipid kinases in vitro and in vivo. Embo J. 1999;18:1292–1302. doi: 10.1093/emboj/18.5.1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Wurzenberger C, Gerlich DW. Phosphatases: providing safe passage through mitotic exit. Nat Rev Mol Cell Biol. 2011;12:469–482. doi: 10.1038/nrm3149. [DOI] [PubMed] [Google Scholar]
- 150.Queralt E, Uhlmann F. Cdk-counteracting phosphatases unlock mitotic exit. Curr Opin Cell Biol. 2008;20:661–668. doi: 10.1016/j.ceb.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Amon A. A decade of Cdc14--a personal perspective. Delivered on 9 July 2007 at the 32nd FEBS Congress in Vienna, Austria. Febs J. 2008;275:5774–5784. doi: 10.1111/j.1742-4658.2008.06693.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Mocciaro A, Schiebel E. Cdc14: a highly conserved family of phosphatases with non-conserved functions? J Cell Sci. 2010;123:2867–2876. doi: 10.1242/jcs.074815. [DOI] [PubMed] [Google Scholar]
- 153.Gray CH, Good VM, Tonks NK, Barford D. The structure of the cell cycle protein Cdc14 reveals a proline-directed protein phosphatase. Embo J. 2003;22:3524–3535. doi: 10.1093/emboj/cdg348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Diamond RH, Cressman DE, Laz TM, Abrams CS, Taub R. PRL-1, a unique nuclear protein tyrosine phosphatase, affects cell growth. Molecular and Cellular Biology. 1994;14:3752–3762. doi: 10.1128/mcb.14.6.3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Bessette DC, Qiu D, Pallen CJ. PRL PTPs: mediators and markers of cancer progression. Cancer Metastasis Rev. 2008;27:231–252. doi: 10.1007/s10555-008-9121-3. [DOI] [PubMed] [Google Scholar]
- 156.Rios P, Li X, Kohn M. Molecular mechanisms of the PRL phosphatases. Febs J. 2012 doi: 10.1111/j.1742-4658.2012.08565.x. [DOI] [PubMed] [Google Scholar]
- 157.Hengge AC, Denu JM, Dixon JE. Transition-state structures for the native dual-specific phosphatase VHR and D92N and S131A mutants. Contributions to the driving force for catalysis. Biochemistry. 1996;35:7084–7092. doi: 10.1021/bi960255i. [DOI] [PubMed] [Google Scholar]
- 158.McParland V, Varsano G, Li X, Thornton J, Baby J, Aravind A, Meyer C, Pavic K, Rios P, Kohn M. The metastasis-promoting phosphatase PRL-3 shows activity toward phosphoinositides. Biochemistry. 2011;50:7579–7590. doi: 10.1021/bi201095z. [DOI] [PubMed] [Google Scholar]
- 159.Saha S, Bardelli A, Buckhaults P, Velculescu VE, Rago C, St Croix B, Romans KE, Choti MA, Lengauer C, Kinzler KW, et al. A phosphatase associated with metastasis of colorectal cancer. Science. 2001;294:1343–1346. doi: 10.1126/science.1065817. [DOI] [PubMed] [Google Scholar]
- 160.Wang H, Quah SY, Dong JM, Manser E, Tang JP, Zeng Q. PRL-3 down-regulates PTEN expression and signals through PI3K to promote epithelial-mesenchymal transition. Cancer Res. 2007;67:2922–2926. doi: 10.1158/0008-5472.CAN-06-3598. [DOI] [PubMed] [Google Scholar]
- 161.Dong Y, Zhang L, Zhang S, Bai Y, Chen H, Sun X, Yong W, Li W, Colvin SC, Rhodes SJ, et al. Phosphatase of Regenerating Liver 2 (PRL2) Is Essential for Placental Development by Down-regulating PTEN (Phosphatase and Tensin Homologue Deleted on Chromosome 10) and Activating Akt Protein. J Biol Chem. 2012;287:32172–32179. doi: 10.1074/jbc.M112.393462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Peters CS, Liang X, Li S, Kannan S, Peng Y, Taub R, Diamond RH. ATF-7, a novel bZIP protein, interacts with the PRL-1 protein-tyrosine phosphatase. J Biol Chem. 2001;276:13718–13726. doi: 10.1074/jbc.M011562200. [DOI] [PubMed] [Google Scholar]
- 163.Song H, Hanlon N, Brown NR, Noble ME, Johnson LN, Barford D. Phosphoprotein-protein interactions revealed by the crystal structure of kinase-associated phosphatase in complex with phosphoCDK2. Mol Cell. 2001;7:615–626. doi: 10.1016/s1097-2765(01)00208-8. [DOI] [PubMed] [Google Scholar]
- 164.Chinami M, Yano Y, Yang X, Salahuddin S, Moriyama K, Shiroishi M, Turner H, Shirakawa T, Adra CN. Binding of HTm4 to cyclin-dependent kinase (Cdk)-associated phosphatase (KAP).Cdk2.cyclin A complex enhances the phosphatase activity of KAP, dissociates cyclin A, and facilitates KAP dephosphorylation of Cdk2. J Biol Chem. 2005;280:17235–17242. doi: 10.1074/jbc.M413437200. [DOI] [PubMed] [Google Scholar]
- 165.Yu Y, Jiang X, Schoch BS, Carroll RS, Black PM, Johnson MD. Aberrant splicing of cyclin-dependent kinase-associated protein phosphatase KAP increases proliferation and migration in glioblastoma. Cancer Res. 2007;67:130–138. doi: 10.1158/0008-5472.CAN-06-2478. [DOI] [PubMed] [Google Scholar]
- 166.Merlot S, Meili R, Pagliarini DJ, Maehama T, Dixon JE, Firtel RA. A PTEN-related 5-phosphatidylinositol phosphatase localized in the Golgi. J Biol Chem. 2003;278:39866–39873. doi: 10.1074/jbc.M306318200. [DOI] [PubMed] [Google Scholar]
- 167.Pagliarini DJ, Wiley SE, Kimple ME, Dixon JR, Kelly P, Worby CA, Casey PJ, Dixon JE. Involvement of a mitochondrial phosphatase in the regulation of ATP production and insulin secretion in pancreatic beta cells. Mol Cell. 2005;19:197–207. doi: 10.1016/j.molcel.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 168.Zhang J, Guan Z, Murphy AN, Wiley SE, Perkins GA, Worby CA, Engel JL, Heacock P, Nguyen OK, Wang JH, et al. Mitochondrial phosphatase PTPMT1 is essential for cardiolipin biosynthesis. Cell Metab. 2011;13:690–700. doi: 10.1016/j.cmet.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Tagliabracci VS, Girard JM, Segvich D, Meyer C, Turnbull J, Zhao X, Minassian BA, Depaoli-Roach AA, Roach PJ. Abnormal metabolism of glycogen phosphate as a cause for Lafora disease. J Biol Chem. 2008;283:33816–33825. doi: 10.1074/jbc.M807428200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Worby CA, Gentry MS, Dixon JE. Laforin, a dual specificity phosphatase that dephosphorylates complex carbohydrates. J Biol Chem. 2006;281:30412–30418. doi: 10.1074/jbc.M606117200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Tagliabracci VS, Turnbull J, Wang W, Girard JM, Zhao X, Skurat AV, Delgado-Escueta AV, Minassian BA, Depaoli-Roach AA, Roach PJ. Laforin is a glycogen phosphatase, deficiency of which leads to elevated phosphorylation of glycogen in vivo. Proc Natl Acad Sci U S A. 2007;104:19262–19266. doi: 10.1073/pnas.0707952104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Gentry MS, Roma-Mateo C, Sanz P. Laforin, a protein with many faces: glucan phosphatase, adapter protein, and others. Febs J. 2012 doi: 10.1111/j.1742-4658.2012.08549.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Tagliabracci VS, Heiss C, Karthik C, Contreras CJ, Glushka J, Ishihara M, Azadi P, Hurley TD, DePaoli-Roach AA, Roach PJ. Phosphate incorporation during glycogen synthesis and Lafora disease. Cell Metab. 2011;13:274–282. doi: 10.1016/j.cmet.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Roach PJ. Are there errors in glycogen biosynthesis and is laforin a repair enzyme? FEBS Lett. 2011;585:3216–3218. doi: 10.1016/j.febslet.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Vergne I, Deretic V. The role of PI3P phosphatases in the regulation of autophagy. FEBS Lett. 2010;584:1313–1318. doi: 10.1016/j.febslet.2010.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Mruk DD, Cheng CY. The myotubularin family of lipid phosphatases in disease and in spermatogenesis. Biochem J. 2011;433:253–262. doi: 10.1042/BJ20101267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Hnia K, Vaccari I, Bolino A, Laporte J. Myotubularin phosphoinositide phosphatases: cellular functions and disease pathophysiology. Trends Mol Med. 2012;18:317–327. doi: 10.1016/j.molmed.2012.04.004. [DOI] [PubMed] [Google Scholar]
- 178.Begley MJ, Taylor GS, Brock MA, Ghosh P, Woods VL, Dixon JE. Molecular basis for substrate recognition by MTMR2, a myotubularin family phosphoinositide phosphatase. Proc Natl Acad Sci U S A. 2006;103:927–932. doi: 10.1073/pnas.0510006103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Robinson FL, Dixon JE. The phosphoinositide-3-phosphatase MTMR2 associates with MTMR13, a membrane-associated pseudophosphatase also mutated in type 4B Charcot-Marie-Tooth disease. J Biol Chem. 2005;280:31699–31707. doi: 10.1074/jbc.M505159200. [DOI] [PubMed] [Google Scholar]
- 180.Lavecchia A, Giovanni CD, Novellino E. CDC25 phosphatase inhibitors: an update. Mini Rev Med Chem. 2012;12:62–73. doi: 10.2174/138955712798868940. [DOI] [PubMed] [Google Scholar]
- 181.Zhang Y, Zhang M. Crystal structure of Ssu72, an essential eukaryotic phosphatase specific for the C-terminal domain of RNA polymerase II, in complex with a transition state analogue. Biochem J. 2011;434:435–444. doi: 10.1042/BJ20101471. [DOI] [PubMed] [Google Scholar]
- 182.Bennett MS, Guan Z, Laurberg M, Su XD. Bacillus subtilis arsenate reductase is structurally and functionally similar to low molecular weight protein tyrosine phosphatases. Proc Natl Acad Sci U S A. 2001;98:13577–13582. doi: 10.1073/pnas.241397198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Zegers I, Martins JC, Willem R, Wyns L, Messens J. Arsenate reductase from S. aureus plasmid pI258 is a phosphatase drafted for redox duty. Nat Struct Biol. 2001;8:843–847. doi: 10.1038/nsb1001-843. [DOI] [PubMed] [Google Scholar]
- 184.Li R, Haile JD, Kennelly PJ. An arsenate reductase from Synechocystis sp. strain PCC 6803 exhibits a novel combination of catalytic characteristics. J Bacteriol. 2003;185:6780–6789. doi: 10.1128/JB.185.23.6780-6789.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Guo X, Li Y, Peng K, Hu Y, Li C, Xia B, Jin C. Solution structures and backbone dynamics of arsenate reductase from Bacillus subtilis: reversible conformational switch associated with arsenate reduction. J Biol Chem. 2005;280:39601–39608. doi: 10.1074/jbc.M508132200. [DOI] [PubMed] [Google Scholar]
- 186.Zhang ZY, Maclean D, McNamara DJ, Sawyer TK, Dixon JE. Protein tyrosine phosphatase substrate specificity: size and phosphotyrosine positioning requirements in peptide substrates. Biochemistry. 1994;33:2285–2290. doi: 10.1021/bi00174a040. [DOI] [PubMed] [Google Scholar]
- 187.Puius YA, Zhao Y, Sullivan M, Lawrence DS, Almo SC, Zhang ZY. Identification of a second aryl phosphate-binding site in protein-tyrosine phosphatase 1B: a paradigm for inhibitor design. Proc Natl Acad Sci U S A. 1997;94:13420–13425. doi: 10.1073/pnas.94.25.13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Zabolotny JM, Bence-Hanulec KK, Stricker-Krongrad A, Haj F, Wang Y, Minokoshi Y, Kim YB, Elmquist JK, Tartaglia LA, Kahn BB, et al. PTP1B regulates leptin signal transduction in vivo. Dev Cell. 2002;2:489–495. doi: 10.1016/s1534-5807(02)00148-x. [DOI] [PubMed] [Google Scholar]
- 189.Cheng A, Uetani N, Simoncic PD, Chaubey VP, Lee-Loy A, McGlade CJ, Kennedy BP, Tremblay ML. Attenuation of leptin action and regulation of obesity by protein tyrosine phosphatase 1B. Dev Cell. 2002;2:497–503. doi: 10.1016/s1534-5807(02)00149-1. [DOI] [PubMed] [Google Scholar]
- 190.Zhang ZY, Thieme-Sefler AM, Maclean D, McNamara DJ, Dobrusin EM, Sawyer TK, Dixon JE. Substrate specificity of the protein tyrosine phosphatases. Proc Natl Acad Sci U S A. 1993;90:4446–4450. doi: 10.1073/pnas.90.10.4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Flint AJ, Tiganis T, Barford D, Tonks NK. Development of "substrate-trapping" mutants to identify physiological substrates of protein tyrosine phosphatases. Proc Natl Acad Sci U S A. 1997;94:1680–1685. doi: 10.1073/pnas.94.5.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Blanchetot C, Chagnon M, Dube N, Halle M, Tremblay ML. Substrate-trapping techniques in the identification of cellular PTP targets. Methods. 2005;35:44–53. doi: 10.1016/j.ymeth.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 193.Meng TC, Buckley DA, Galic S, Tiganis T, Tonks NK. Regulation of insulin signaling through reversible oxidation of the protein-tyrosine phosphatases TC45 and PTP1B. J Biol Chem. 2004;279:37716–37725. doi: 10.1074/jbc.M404606200. [DOI] [PubMed] [Google Scholar]
- 194.Haj FG, Verveer PJ, Squire A, Neel BG, Bastiaens PI. Imaging sites of receptor dephosphorylation by PTP1B on the surface of the endoplasmic reticulum. Science. 2002;295:1708–1711. doi: 10.1126/science.1067566. [DOI] [PubMed] [Google Scholar]
- 195.Tonks NK. Redox redux: revisiting PTPs and the control of cell signaling. Cell. 2005;121:667–670. doi: 10.1016/j.cell.2005.05.016. [DOI] [PubMed] [Google Scholar]
- 196.Lou YW, Chen YY, Hsu SF, Chen RK, Lee CL, Khoo KH, Tonks NK, Meng TC. Redox regulation of the protein tyrosine phosphatase PTP1B in cancer cells. Febs J. 2008;275:69–88. doi: 10.1111/j.1742-4658.2007.06173.x. [DOI] [PubMed] [Google Scholar]
- 197.Meng TC, Fukada T, Tonks NK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell. 2002;9:387–399. doi: 10.1016/s1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- 198.Meng TC, Hsu SF, Tonks NK. Development of a modified in-gel assay to identify protein tyrosine phosphatases that are oxidized and inactivated in vivo. Methods. 2005;35:28–36. doi: 10.1016/j.ymeth.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 199.Boivin B, Zhang S, Arbiser JL, Zhang ZY, Tonks NK. A modified cysteinyl-labeling assay reveals reversible oxidation of protein tyrosine phosphatases in angiomyolipoma cells. Proc Natl Acad Sci U S A. 2008;105:9959–9964. doi: 10.1073/pnas.0804336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Karisch R, Fernandez M, Taylor P, Virtanen C, St-Germain JR, Jin LL, Harris IS, Mori J, Mak TW, Senis YA, et al. Global proteomic assessment of the classical protein-tyrosine phosphatome and "Redoxome". Cell. 2011;146:826–840. doi: 10.1016/j.cell.2011.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Karisch R, Neel BG. Methods to monitor classical protein-tyrosine phosphatase oxidation. Febs J. 2012 doi: 10.1111/j.1742-4658.2012.08626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Salmeen A, Andersen JN, Myers MP, Meng TC, Hinks JA, Tonks NK, Barford D. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature. 2003;423:769–773. doi: 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]
- 203.Haque A, Andersen JN, Salmeen A, Barford D, Tonks NK. Conformation-sensing antibodies stabilize the oxidized form of PTP1B and inhibit its phosphatase activity. Cell. 2011;147:185–198. doi: 10.1016/j.cell.2011.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Guan KL, Dixon JE. Protein tyrosine phosphatase activity of an essential virulence determinant in Yersinia. Science. 1990;249:553–556. doi: 10.1126/science.2166336. [DOI] [PubMed] [Google Scholar]
- 205.Bliska JB, Clemens JC, Dixon JE, Falkow S. The Yersinia tyrosine phosphatase: specificity of a bacterial virulence determinant for phosphoproteins in the J774A.1 macrophage. J Exp Med. 1992;176:1625–1630. doi: 10.1084/jem.176.6.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Julien SG, Dube N, Hardy S, Tremblay ML. Inside the human cancer tyrosine phosphatome. Nat Rev Cancer. 2011;11:35–49. doi: 10.1038/nrc2980. [DOI] [PubMed] [Google Scholar]
- 207.Wang Z, Shen D, Parsons DW, Bardelli A, Sager J, Szabo S, Ptak J, Silliman N, Peters BA, van der Heijden MS, et al. Mutational analysis of the tyrosine phosphatome in colorectal cancers. Science. 2004;304:1164–1166. doi: 10.1126/science.1096096. [DOI] [PubMed] [Google Scholar]
- 208.Solomon DA, Kim JS, Cronin JC, Sibenaller Z, Ryken T, Rosenberg SA, Ressom H, Jean W, Bigner D, Yan H, et al. Mutational inactivation of PTPRD in glioblastoma multiforme and malignant melanoma. Cancer Res. 2008;68:10300–10306. doi: 10.1158/0008-5472.CAN-08-3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Ostman A, Hellberg C, Bohmer FD. Protein-tyrosine phosphatases and cancer. Nat Rev Cancer. 2006;6:307–320. doi: 10.1038/nrc1837. [DOI] [PubMed] [Google Scholar]
- 210.Pallen CJ. Protein tyrosine phosphatase alpha (PTPalpha): a Src family kinase activator and mediator of multiple biological effects. Curr Top Med Chem. 2003;3:821–835. doi: 10.2174/1568026033452320. [DOI] [PubMed] [Google Scholar]
- 211.Elchebly M, Payette P, Michaliszyn E, Cromlish W, Collins S, Loy AL, Normandin D, Cheng A, Himms-Hagen J, Chan CC, et al. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science. 1999;283:1544–1548. doi: 10.1126/science.283.5407.1544. [DOI] [PubMed] [Google Scholar]
- 212.Klaman LD, Boss O, Peroni OD, Kim JK, Martino JL, Zabolotny JM, Moghal N, Lubkin M, Kim YB, Sharpe AH, et al. Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Molecular and Cellular Biology. 2000;20:5479–5489. doi: 10.1128/mcb.20.15.5479-5489.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Julien SG, Dube N, Read M, Penney J, Paquet M, Han Y, Kennedy BP, Muller WJ, Tremblay ML. Protein tyrosine phosphatase 1B deficiency or inhibition delays ErbB2-induced mammary tumorigenesis and protects from lung metastasis. Nat Genet. 2007;39:338–346. doi: 10.1038/ng1963. [DOI] [PubMed] [Google Scholar]
- 214.Bentires-Alj M, Neel BG. Protein-tyrosine phosphatase 1B is required for HER2/Neu-induced breast cancer. Cancer Res. 2007;67:2420–2424. doi: 10.1158/0008-5472.CAN-06-4610. [DOI] [PubMed] [Google Scholar]
- 215.Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, Zimmermann J, Lydon NB. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2:561–566. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- 216.Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341–354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- 217.He R, Zeng LF, He Y, Zhang S, Zhang ZY. Small molecule tools for functional interrogation of protein tyrosine phosphatases. Febs J. 2012 doi: 10.1111/j.1742-4658.2012.08718.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Shen K, Keng YF, Wu L, Guo XL, Lawrence DS, Zhang ZY. Acquisition of a specific and potent PTP1B inhibitor from a novel combinatorial library and screening procedure. J Biol Chem. 2001;276:47311–47319. doi: 10.1074/jbc.M106568200. [DOI] [PubMed] [Google Scholar]
- 219.Zinker BA, Rondinone CM, Trevillyan JM, Gum RJ, Clampit JE, Waring JF, Xie N, Wilcox D, Jacobson P, Frost L, et al. PTP1B antisense oligonucleotide lowers PTP1B protein, normalizes blood glucose, and improves insulin sensitivity in diabetic mice. Proc Natl Acad Sci U S A. 2002;99:11357–11362. doi: 10.1073/pnas.142298199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Wiesmann C, Barr KJ, Kung J, Zhu J, Erlanson DA, Shen W, Fahr BJ, Zhong M, Taylor L, Randal M, et al. Allosteric inhibition of protein tyrosine phosphatase 1B. Nat Struct Mol Biol. 2004;11:730–737. doi: 10.1038/nsmb803. [DOI] [PubMed] [Google Scholar]
- 221.Zhang J, Adrian FJ, Jahnke W, Cowan-Jacob SW, Li AG, Iacob RE, Sim T, Powers J, Dierks C, Sun F, et al. Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature. 2010;463:501–506. doi: 10.1038/nature08675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Iverson C, Larson G, Lai C, Yeh LT, Dadson C, Weingarten P, Appleby T, Vo T, Maderna A, Vernier JM, et al. RDEA119/BAY 869766: a potent, selective, allosteric inhibitor of MEK1/2 for the treatment of cancer. Cancer Res. 2009;69:6839–6847. doi: 10.1158/0008-5472.CAN-09-0679. [DOI] [PubMed] [Google Scholar]
- 223.Thompson KM, Uetani N, Manitt C, Elchebly M, Tremblay ML, Kennedy TE. Receptor protein tyrosine phosphatase sigma inhibits axonal regeneration and the rate of axon extension. Mol Cell Neurosci. 2003;23:681–692. doi: 10.1016/s1044-7431(03)00120-9. [DOI] [PubMed] [Google Scholar]
- 224.Lin G, Aranda V, Muthuswamy SK, Tonks NK. Identification of PTPN23 as a novel regulator of cell invasion in mammary epithelial cells from a loss-of-function screen of the 'PTP-ome'. Genes Dev. 2011;25:1412–1425. doi: 10.1101/gad.2018911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Hendriks WJ, Elson A, Harroch S, Pulido R, Stoker A, den Hertog J. Protein tyrosine phosphatases in health and disease. Febs J. 2012 doi: 10.1111/febs.12000. [DOI] [PubMed] [Google Scholar]
- 226.Seifried A, Schultz J, Gohla A. Human HAD phosphatases: structure, mechanism, and roles in health and disease. Febs J. 2012 doi: 10.1111/j.1742-4658.2012.08633.x. [DOI] [PubMed] [Google Scholar]
- 227.Pouyssegur J, Volmat V, Lenormand P. Fidelity and spatio-temporal control in MAP kinase (ERKs) signalling. Biochem Pharmacol. 2002;64:755–763. doi: 10.1016/s0006-2952(02)01135-8. [DOI] [PubMed] [Google Scholar]