

Abstract
TGF-β can act as a tumor suppressor at early stages of cancer progression and as a tumor promoter at later stages. The E3 ubiquitin ligase Arkadia (RNF111) is a critical component of the TGF-β signaling pathway, being required for a subset of responses, those mediated by Smad3-Smad4 complexes. It acts by mediating ligand-induced degradation of Ski and SnoN (SKIL), which are two potent transcriptional repressors. Here we investigate the role of Arkadia in cancer, using model systems to address both potential tumor suppressive and tumor-promoting roles. Stable re-expression of Arkadia in lung carcinoma NCI-H460 cells, which we show contain a hemizygous nonsense mutation in the Arkadia/RNF111 gene efficiently restored TGF-β-induced Smad3-dependent transcription, and substantially decreased the ability of these cells to grow in soft agar in vitro. However, it had no effect on tumor growth in vivo in mouse models. Moreover, loss of Arkadia in cancer cell lines and human tumors is rare, arguing against a prominent tumor-suppressive role. In contrast, we have uncovered a potent tumor promoting function for Arkadia. Using three different cancer cell lines whose tumorigenic properties are driven by TGF-β signaling, we demonstrate that loss of Arkadia function, either by overexpression of dominant negative Arkadia or by siRNA-induced knockdown, substantially inhibited lung colonization in tail vein injection experiments in immunodeficient mice. Our findings indicate that Arkadia is not critical for regulating tumor growth per se, but is required for the early stages of cancer cell colonization at the sites of metastasis.
Keywords: Arkadia, TGF-β signaling, E3 ubiquitin ligase, cancer, tumor promoter
Introduction
The TGF-β signaling pathway has a dual role in tumorigenesis (1, 2). It can function as a tumor suppressor by inhibiting cell growth, inducing apoptosis, promoting differentiation, as well as acting on the stroma to suppress inflammation and the production of mitogens (2). Conversely, TGF-β can support tumor development by inhibiting immune responses and by regulating processes necessary for the colonization of distant tissues, such as angiogenesis, cancer cell migration and invasion (2, 3). At later stages of tumorigenesis the TGF-β signal is a major contributor to the transcriptional regulation of genes necessary for cancer cell migration and invasion, as well as microenvironment remodeling (3, 4).
TGF-β binds and activates complexes of serine/threonine kinase receptors comprising TβRII and TβRI (ALK5) at the cell surface. This leads to phosphorylation of receptor-regulated Smads, of which the best understood are Smad2 and Smad3 (5). These activated Smads complex with Smad4 and accumulate in the nucleus where they directly regulate the transcription of target genes (6). Ski and SnoN are potent transcriptional co-repressors that inhibit the transcription of a subset of TGF-β-responsive genes (7). In the absence of TGF-β, Ski and SnoN bind Smad Binding Elements (SBE) in the promoters/enhancers of target genes together with Smad4, forming a transcriptional repressor complex with histone deacetylases to silence basal transcription (8-15). The particular elements recognized by Ski and SnoN are those recognized by Smad3–Smad4 complexes, and also complexes of Smad4 with a splice form of Smad2, Smad2Δexon3 (12). In response to TGF-β, Ski and SnoN are rapidly degraded via the ubiquitin-proteasome pathway (8). This degradation allows the phosphorylated Smad3/Smad2Δexon3-containing complexes to bind SBEs in the promoters/enhancers of target genes (7, 8, 12).
While several ubiquitin ligases, namely Smurf2 and the anaphase-promoting complex were originally proposed to be responsible for regulating Ski and SnoN levels, (16-18), several years ago we and others established that the E3 ubiquitin ligase Arkadia (encoded by the RNF111 gene) was required for TGF-β-induced SnoN and Ski degradation (12, 19, 20). We showed that in response to TGF-β Arkadia interacts with SnoN and phosphorylated Smad2/Smad3, and this is necessary for SnoN degradation (12). As a result, Arkadia is essential for a subset of TGF-β-induced transcriptional responses, those mediated via Smad3/Smad2Δexon3.
Like the TGF-β pathway itself, SnoN also plays a dual role in cancer (21). Since Arkadia is a critical modulator of Ski and SnoN levels, deregulation of Arkadia function might be predicted to influence tumor development and/or dissemination. We previously described a lung carcinoma cell line, NCI-H460 (originally wrongly classified as the esophageal carcinoma cell line SEG-1 (22)) that lacked functional Arkadia, and hence did not exhibit TGF-β-induced SnoN degradation, and was deficient in Smad3-dependent transcriptional responses (12). We hypothesized that Arkadia might be a novel tumor suppressor, with specific loss of the Smad3/Smad2Δexon3-dependent arm of the TGF-β pathway through loss of Arkadia allowing cells to evade the tumor suppressive effects of TGF-β, whilst maintaining TGF-β’s tumor-promoting activities (12). Consistent with this, Arkadia+/− heterozygous mice are more susceptible to developing tumors in a colorectal tumor model after exposure to carcinogen, compared with wild-type mice (23). However, there was no evidence that the other allele of Arkadia was lost in these tumors, as might be expected for a classical tumor suppressor. Moreover, although a number of mutations in Arkadia were found in primary colorectal tumors from human patients, only one of them clearly resulted in a non-functional protein (23). An alternative possibility to the idea of the two arms of the TGF-β pathway having different functions in cancer, is that the pathway as a whole may have both tumor suppressive and tumor promoting functions, but which predominates depends on the context. If this were the case, then Arkadia, like SnoN and Smad4 (2, 21, 24, 25) might be expected to exhibit a dual role in cancer.
Here we dissect the role of Arkadia in tumorigenesis, using two model systems designed to examine both potential tumor suppressor and tumor promoting activities. Our data do not support a prominent tumor suppressive role. Instead we show that Arkadia is required for metastasis, probably at the level of extravasation.
Materials and Methods
Plasmids
The following plasmids were previously described: HA-SnoN, HA-Smad3, FLAG-Arkadia, CAGA12-Luciferase and TK-Renilla (12) and HA-Ski (9). To make the stable cell lines, wild-type Arkadia and Arkadia C937A (12) were subcloned into the 3 × Flag pBICEP-CMV2 vector (Sigma). FLAG-Arkadia 1–440 was generated by introducing a stop codon at amino acid 441 in the FLAG-Arkadia construct (12).
Cell lines and cell treatments
HaCaT, MDA-MB-231, 293T, B16, CACO-2 and HT29 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 2 mM glutamine and 10% fetal calf serum (FCS). NCI-H460 and COLO-205 cells were cultured in Roswell Park Memorial Institute (RPMI 1640) supplemented with 2 mM glutamine and 10% FCS. MTLN3E cells were cultured in α-MEM containing 2 mM glutamine 10% FCS. HT-55 cells were cultured in a 1:1 mix of DMEM and RPMI containing 2 mM glutamine and 10% FCS. Primary human umbilical vein endothelial cells (HUVEC; CC-2517, Lonza,) were grown in collagen precoated flasks (BD Biocoat) in EGM-2 Bullet-Kit media with supplements (CC-3162, Lonza) at 5% CO2. Stable cell lines were obtained by transfecting NCI-H460 or MDA-MB-231 cells with either pBICEP-CMV2-FLAG-Arkadia or pBICEP-CMV2-FLAG-Arkadia-C937A respectively and selecting clones with G418 (Gibco). MDA-MB-231 cells expressing GFP or mCherry were generated by transfecting pEGFP-N1 and pCherry-N1 plasmids (Clontech) into MDA-MB-231 cells, selected with 500 μg/ml G418 (Gibco) and by FACS sorting. The B16 cells were labeled with Cherry or EGFP in the same way. The MTLN3E cells were labelled with lentivirus containing either myr-GFP or myr-Cherry and FACS sorted. The MDA-MB-231 Arkadia C937A clones were labeled with a membrane associated GFP (GFP-CAAX) using the lentivirus system and were selected with blasticidin.
Cells were stimulated with 2 ng/ml of TGF-β (Peprotech EC Ltd) for the specified times. The ALK5 inhibitor SB-431542 (Tocris) was used at 10 μM. For proteasome inhibition, cells were treated with 50 μM of MG132 (Tocris) for 4 h.
Immunoprecipitations, Western blots, antibodies and luciferase assays
Whole cell extracts were prepared either using radioimmunoprecipitation assay buffer (RIPA) (12) or as described (26). Western blots were performed following standard procedures. For TMEPAI blots, extracts were treated with PNGase as described (27). Antibodies are listed in the Supplementary Methods. Immunoprecipitations and luciferase assays were as described (12). For luciferase assays TGF-β induction was for 8 h.
Xenografts and tail vein injection assays
For xenografts, cells were trypsinized and 5 × 106 cells were resuspended in 100 μl PBS and injected subcutaneously into the right and left flanks of ~ 6 week old female, Balb/c nu/nu mice (4 mice per condition). Tumor growth was measured with external calipers every two or three days for a maximum of six weeks.
For tail vein injections with unlabeled cells, the cells were trypsinized and 1 × 106 cells were injected into the tail vein of Balb/c nu/nu mice (4 or 5 mice per condition). Lungs were removed at 20 or 30 days post injection and fixed in neutral-buffered formalin. Three sections corresponding to different levels of the lungs were obtained, which were stained with hematoxylin and eosin. The number of tumors in each slide was determined by a pathologist. For the tail vein injections with fluorescent cells, 1 × 106 cells of a 1:1 mixture GFP- and mCherry-expressing cells (where either both were controls or one an experimental sample and the other a control) was injected into the tail vein of 6 week old female, ICRF nu/nu mice (MDA-MB-231 and B16) or Balb/c nu/nu (MTLN3E). Additional controls for the ratio of mCherry and GFP cells were performed by seeding 10 μl of the cell suspension into a glass-bottom dish coated with poly-lysine (Sigma); after 2 h, cells were fixed in 4% paraformaldehyde and imaged with a Zeiss LSM 780 confocal microscope using a Plan-Neofluar 10× 0.3 objective. 48 h post-injection the mice were culled, lungs extracted and representative images of the tumor distribution were analyzed by confocal microscopy. The area occupied by fluorescent tumor cells was calculated using Volocity software (PerkinElmer) and the GFP:mCherry ratio calculated based on the total area of the green and the red cells and normalized using the GFP:mCherry ratio observed in the control plates.
Cell line origin and authentication
The parental MDA-MB-231 cells were obtained from the ECACC/HPA culture collection and were banked at Cancer Research UK Cell Services. The MDA-MB-231 cells expressing Arkadia C937A were derived from these cells. These cell lines were authenticated using the STR Profiling and Isoenzyme Analysis. The NCI-H460 cells were obtained from the ATCC and were banked at Cancer Research UK Cell Services. The Arkadia-expressing clones were derived from these cells. The MTLN3E cells were obtained from John Condeelis. We have proven that they are syngeneic with Fisher 344 rats in agreement with their published history (28). The B16 cells were obtained from Cancer Research UK Cell Services. We have proven they are syngeneic with C57BL/6 mice and produce pigment consistent with their published history (29). All cell lines were frequently tested for mycoplasma and were negative.
For genomic sequencing, RNA-seq, qPCR, soft agar and cell cycle analyses, cell spreading and cell adhesion assays and lists of primers, antibodies and siRNAs, see Supplemental Methods.
Results
NCI-H460 cells express a truncated version of Arkadia that is catalytically inactive and are deficient in TGF-β-induced Smad3-dependent transcription
To investigate whether Arkadia might be a novel tumor suppressor gene, we utilized a lung cancer cell line, NCI-H460 that we previously demonstrated had lost expression of full-length Arkadia (12). Although NCI-H460 cells express Arkadia mRNA (Fig. 1A), they do not express full-length Arkadia protein. However, a faster migrating band was observed in extracts from these cells, that was absent in HaCaT extracts, suggesting that they might express a truncated version of Arkadia (Fig. 1B). This was confirmed using an siRNA SMARTpool against human Arkadia (Fig. S1A). Genomic sequencing of the Arkadia/RNF111 gene in NCI-H460 cells and HaCaT cells revealed a hemizygous single nucleotide deletion in the Arkadia/RNF111 gene in the NCI-H460 cells that creates a stop codon at amino acid 441 (Fig. S1B). Thus, NCI-H460 cells express an Arkadia protein lacking the C-terminal catalytic RING domain.
Figure 1. NCI-H460 cells express a truncated and catalytically inactive version of Arkadia.

A. Levels of Arkadia and GAPDH mRNA in HaCaT and NCI-H460 cells were assayed by RT-PCR. B. Whole cell extracts from HaCaT and NCI-H460 cells analyzed by Western Blot using an antibody against Arkadia. C. Whole cell extracts from 293T cells transfected with the plasmids indicated were immunoprecipitated (IP) using anti-FLAG beads and Western blotted (WB) using HA and FLAG antibodies. Inputs show expression levels of Arkadia, Smad3, Ski and SnoN. D. NCI-H460 and 293T cells were transfected with the CAGA12-Luciferease reporter and the TK-Renilla control along with plasmids expressing wild-type (WT), C937A, or truncated (1–440) Arkadia. Luciferase activity was normalized to Renilla activity. Means and standard deviations are shown of representative experiments performed in triplicate.
To investigate the biological relevance of this mutation, we tested the ability of this truncated Arkadia to interact with Ski, SnoN and Smad3 (12, 19, 20). In immunoprecipitations, Arkadia 1–440 did not interact with SnoN, and only very weakly interacted with Ski. However, it still retained its ability to interact with Smad3 (Fig. 1C). To assay the activity of Arkadia 1–440 we compared its ability to rescue CAGA12-luciferase activity in the NCI-H460 cells with that of wild-type Arkadia and a dominant negative Arkadia which has a point mutation in the RING domain (C937A) (12). Arkadia 1–440 was substantially less active than wild-type Arkadia, showing only residual activity comparable with that of Arkadia C937A (Fig. 1D). In 293T cells, which contain endogenous Arkadia, Arkadia 1–440 had very little activity, compared with wild-type Arkadia (Fig. 1D). However, unlike Arkadia C937A, it did not exhibit dominant negative activity, probably as a result of losing its ability to interact with Ski and SnoN.
Reintroduction of functional Arkadia in NCI-H460 cells restores specific TGF-β-responses and inhibits anchorage-independent growth, but has no effect on tumor growth in vivo
To determine whether loss of Arkadia in NCI-H460s might be responsible for their tumorigenic phenotype, we investigated the effect of restoring Arkadia function. We generated two cell lines (WT Ark 1 and WT Ark 11) that stably express FLAG-tagged Arkadia (Fig. 2A). Treatment of cells with the proteosome inhibitor MG132 increased protein levels of overexpressed Arkadia in these NCI-H460 cell lines and also endogenous Arkadia in control MDA-MB-231 cells, indicating that Arkadia is normally unstable, probably due to autoubiquitination (Fig. 2A; see Fig. 3A) (12). In both WT Ark 1 and WT Ark 11 cells, reintroduction of Arkadia restored TGF-β-induced SnoN and Ski degradation (Fig. 2B) and Smad3-dependent transcription (Fig. 2C). Induction of the TGF-β-dependent gene, transmembrane prostate androgen-induced RNA (TMEPAI) (30), was also recovered after reintroduction of Arkadia (Fig. 2D).
Figure 2. Reintroduction of Arkadia in NCI-H460 cells restores TGF-β-dependent Ski/SnoN degradation, transcriptional responses and inhibits anchorage independent growth, but has no effect on their tumorigenic properties in vivo.

A. Western blot showing Arkadia levels in two NCI-H460-derived cell lines expressing FLAG-tagged Arkadia. Cells were treated for 4 h ± MG132. Actin is a loading control. B. Whole cell extracts from cells treated ± TGF-β Western blotted using the antibodies indicated. C. Luciferase assays were performed as in Fig. 1. D. Whole cell extracts prepared from the cell lines indicated and Western blotted using antibodies shown. E. NCI-H460 parental cells or the clones expressing FLAG-tagged Arkadia were grown in soft agar for 10 days. The numbers of colonies were quantified and are shown in the bar graph (right). F. The cell lines shown were injected subcutaneously in both flanks of Balbc nu/nu mice. The data are the averages of the tumor volumes for four mice (8 tumors in total) per cell line.
Figure 3. Expression of Arkadia C937A abolishes Arkadia function in MDA-MB-231 cells and alters their adhesion and spreading properties.

A–D. NCI-H460 cells, parental MDA-MB-231 cells, and a stable MDA-MB-231 cell line expressing FLAG-tagged Arkadia C937A (clone 1) were treated ± MG132 for 4 h (A) or with TGF-β for the times indicated (B–D). Whole cell extracts were Western blotted using the antibodies indicated (A, B and D). In C, levels of mRNA for the genes shown were analyzed either by RNA-seq or by qPCR which was normalized to GAPDH. E. GFP-labeled parental MDA-MB-231 cells and Arkadia C937A cells (clone 1) were mixed with an equal number of parental cells expressing mCherry, seeded on a monolayer of HUVEC cells and incubated for 1 h, after which they were washed, fixed and counted. The mean ratio of GFP:mCherry cells and standard deviations from 6 wells is plotted. F. GFP-labeled parental MDA-MB-231 cells or Arkadia C937A cells (clone 1) were seeded on a layer of HUVEC cells, and movies were recorded. Cell spreading was measured by quantification of changes in the area of the cells over time using the Imaris software. The graphs show the means of the average cell area for 4 independent wells for each cell type with standard errors. Similar results were obtained in three independent experiments.
Tumor suppressive effects of the TGF-β pathway are thought to be at least partly mediated by effects on cell growth (2). However, we found no effect of Arkadia reintroduction into NCI-H460 cells on the rate of cell proliferation in vitro in the presence or absence of TGF-β (Fig. S2). We next determined whether restoring Arkadia altered the transformed phenotype of NCI-H460 cells in vitro by performing soft-agar assays, which measure anchorage-independent growth. NCI-H460 cells formed colonies in soft agar (Fig. 2E). This growth depends on autocrine TGF-β signaling, as it was abolished by SB-431542, a specific inhibitor of the TGF-β type I receptor (31) (data not shown). Exogenous TGF-β could not further promote colony formation, suggesting that the autocrine TGF-β activity in these cells is sufficient. Since the cells lack Arkadia activity, this growth is Arkadia independent. Strikingly, neither NCI-H460 clone in which Arkadia has been re-expressed were able to form a substantial number of colonies (Fig. 2E). Thus, anchorage-independent growth of NCI-H460 cells is inhibited by Arkadia.
We went on to test if restoring Arkadia activity in NCI-H460 cells had any effect on their tumorigenic properties in vivo. In xenograft assays in immunodeficient mice, primary tumor growth was not affected by Arkadia (Fig 2F). We also investigated whether the ability of cells to colonize and develop tumors in distant tissues was affected by Arkadia activity by performing tail vein injections in immunodeficient mice. However, no significant differences in the number of lung tumors derived from parental cells or from the clones expressing wild-type Arkadia were observed, either at 20 or 30 days post-injection (Table S1). We conclude therefore that although restoration of Arkadia activity in NCI-H460 cells at least partly reversed the transformed phenotype in vitro, it does not affect tumorigenicity in vivo. This might be explained by the gain of additional driving mutations after acquisition of the Arkadia mutation.
Mutations in Arkadia in human cancer are rare
To obtain a more comprehensive view of Arkadia mutation frequency in human cancer we analyzed Arkadia protein levels and TGF-β-induced SnoN degradation in a number of cancer cell lines of different tissue origin, focusing particularly on those identified from the Sanger Centre CGP LOH and Copy Number Analysis that displayed LOH at the 15q22.1 locus containing the Arkadia/RNF111 gene. We were unable to find another cancer cell line in which Arkadia was deleted or which contained a loss-of-function mutation in Arkadia (data not shown). Interestingly, we observed a direct correlation between loss of TGF-β-induced SnoN degradation and loss of Smad4 activity. Examples are the CACO-2 cell line, which contains a point mutation in Smad4 that renders it unable to form complexes with R-Smads (32), and the Colo-205 and HT-29 cell lines that are deleted for Smad4 (Fig. S3). Thus mutation or deletion of Smad4, which is common in certain tumors, has the same inhibitory effect on SnoN degradation as loss of Arkadia (see Discussion).
Inhibition of Arkadia activity in MDA-MB-231 cells alters their adherence and ability to spread on endothelial cells
The evidence presented above does not support the idea that Arkadia is primarily a tumor suppressor. Moreover, cancer cell lines that exhibit LOH at the Arkadia locus do not lose or acquire mutations in the other allele, suggesting the possibility that Arkadia might be important for mediating TGF-β’s tumor promoting functions (2). To address this we chose a well-characterized breast cancer cell line MDA-MB-231 that requires TGF-β signaling for metastasis (24, 33-35) and investigated how loss of Arkadia activity affected its tumorigenic properties.
Overexpression of Arkadia C937A acts dominant negatively to suppress the activity of an endogenous Arkadia (12) (Fig. 1C). We therefore used this construct to inactivate Arkadia in MDA-MB-231 cells (Fig. 3A). Arkadia C937A prevented TGF-β-induced Ski and SnoN degradation in three independent clones, (Fig. 3B; Fig. S4A). To determine the effect of dominant negative Arkadia on TGF-β-regulated target genes at the genomic scale we performed RNA-seq at 1 h and 24 h after TGF-β stimulation. Fig. S5 shows the filtered datasets presented as heatmaps and Fig. S6 shows qPCR validations for selected genes. The similarity between the qPCR data and the RNA-seq data gave us confidence in the RNA-seq dataset as a whole. We identified 36 genes that are significantly (more than 2-fold) up- or down-regulated by TGF-β at 1 h, 103 genes that are up-regulated by TGF-β at 24 h, and 70 genes down-regulated by TGF-β at 24 h (Fig. S5). Consistent with our previous data showing that Arkadia is required only for TGF-β responses that are dependent on Smad3/Smad2Δexon3 (12), we found that a subset of TGF-β responsive genes was strongly affected by dominant negative Arkadia, whilst other genes were only weakly affected, or not affected at all (Fig. S5). Examples of strongly affected genes are the two well-characterized TGF-β targets, PAI-1 and TMEPAI (Fig. 3C). This was corroborated at the protein level (Fig. 3D; Fig. S4B). We conclude that expression of Arkadia C937A efficiently inhibits endogenous Arkadia function.
MDA-MB-231 cells are resistant to TGF-β-induced growth arrest (21) and we noted an absence of genes involved in TGF-β-induced cell cycle arrest in the MDA-MB-231 cells in the RNA-seq analysis (Fig. S5). Inactivation of Arkadia is therefore unlikely to affect cell growth. Indeed, we found no difference in the growth rate of parental or Arkadia C937A-expressing cells in vitro on plastic, in soft agar or on the growth of these cells in xenograft assays in immunodeficient mice (data not shown), consistent with other studies showing that TGF-β signaling does not have a tumor suppressive effect in MDA-MB-231 cells (34).
To gain insight into the TGF-β-driven processes for which Arkadia is likely to be required we performed a MetaCore analysis of genes that significantly change in their TGF-β regulation between the parental and Arkadia C937A-expressing cells. This indicated an enrichment of genes involved in cell adhesion, cell matrix interactions, EMT and ECM remodeling (Table 1), processes involved in tumor cell dissemination from primary tumors to sites of metastasis (36). During metastasis, tumor cells enter the blood or lymphatic circulation (intravasation) and then extravasate at the site of metastasis (37). Since both of these processes involve invasion through a layer of endothelial cells, we attempted to mimic this in vitro by assessing cell adhesion and ability to spread on a confluent layer of endothelial cells (HUVEC). To visualize the cells we fluorescently labeled them with GFP and, in the case of the parental cells, also mCherry. Equal numbers of GFP- (parental or Arkadia C937A-expressing) and mCherry-labeled parental cells were plated onto a layer of HUVECs. We found that the Arkadia C937A-expressing cells adhered more strongly to the HUVEC cells than the parental MDA-MB-231 cells (Fig. 3E). When the GFP-labeled cells were plated onto confluent layers of HUVEC cells and filmed over a period of hours to assess cell spreading (38) (Supplemental Movies S1 and S2), we consistently observed an inhibition in the ability to spread of the Arkadia C937A-expressing cells compared with parental cells (Fig. 3F). Thus cells inhibited in Arkadia function are more adherent to endothelial cells, but have defects in spreading, possibly indicating a defect in remodeling of adhesions.
Table 1.
Gene enrichment analysis report. A MetaCore analysis of genes that significantly change in their TGF-β regulation (false discovery rate < 0.05 and fold change > 2) between the parental cells and Arkadia C937A-expressing cells. They are ordered with respect to p value
| Networks | Total1 | p value | No. genes in data |
Genes from active data2 |
|---|---|---|---|---|
|
Cell adhesion and
cell–matrix interaction |
213 | 7.096 × 10−4 | 8 | TGFB1; ADAM-TS14; Collagen V; MMP-8; COL5A1; COL1A2; LAMC2; SPOCK1 |
|
Proteolysis, ECM
remodeling |
85 | 1.07 × 10−3 | 5 | LRP1; ADAM-TS14; PAI1/Serpine1; MMP-8; SPOCK1 |
|
Proteolysis, connective tissue degradation |
119 | 4.69 × 10−3 | 5 | LRP1; ADAM-TS14; PAI1/Serpine1; MMP-8; SPOCK1 |
| Development, EMT | 232 | 5.31 × 10−3 | 7 | AP-1; KRT14; PAI1/Serpine1; Lysyl Oxidase; SNAIL1; PDGF-B; COL1A2 |
|
Cytoskeleton and
intermediate filaments |
81 | 6.49 × 10−3 | 4 | PPL (periplakin); KRT14; KRT17; KRT7 |
|
Cardiac
development, role of NADPH oxidase and ROS |
133 | 7.47 × 10−3 | 5 | MYH15; SNAIL1; NCF2; Gli-2; ID1 |
|
Reproduction,
gonadotrophin regulation |
199 | 9.82 × 10−3 | 6 | G-specific class A orphan/other GPCRs; AP-1; FosB; JunB; VIPR1 |
| Proliferation | 221 | 1.59x 10−2 | 6 | AP-1; G(i)-specific amine GPCRs; CSF1R; VIPR1; HTR1D; TACSTD2 |
|
Cardiac
development: TGF-β and BMP signaling |
117 | 2.26 × 10−2 | 4 | MYH15; SNAIL1; Gli-2; ID1 |
|
Development,
neurogenesis, synaptogenesis |
180 | 2.49 × 10−2 | 5 | LRP1; Syntrophin B; KIF17; Synaptotagmin II; Synaptotagmin |
Total refers to the number of MetaCore entries associated with a particular network.
In most cases these are individual genes. In some cases however multiple members of one family have been mapped to the same MetaCore database entry for example, Lysyl oxidase or AP-1.
Inhibition of Arkadia activity in MDA-MB-231 cells inhibits colonization of lungs of immunodeficient mice
The reduced ability of cells lacking Arkadia activity to spread on endothelial cells suggested that Arkadia could play a role in metastasis. We tested this directly, and observed a robust inhibition of lung colonization of the three individual clones of MDA-MB-231 cells expressing Arkadia C937A, compared with parental cells in tail vein injection assays performed over 20 days (Fig. 4A). To determine whether Arkadia activity is important for early stages of lung colonization, we performed these assays over a period of just 48 h, using the fluorescently-labeled cells described above. Mice were injected with a 1:1 ratio of GFP- and mCherry-labeled cells as described in the Materials and Methods, and after 48 h lung colonization was assessed. A dramatic decrease in the number of Arkadia C937A-expressing cells compared with the control mCherry-labeled parental cells was observed (Fig. 4B; Fig. 4C). Since the effects of dominant negative Arkadia were evident just 48 h after tail vein injection, we concluded that Arkadia is required for early stage colonization. Taken together with the in vitro cell spreading and adhesion data, it is likely that Arkadia is required for extravasation.
Figure 4. Inhibition of Arkadia activity in MDA-MB-231 by expression of Arkadia C937A inhibits lung colonization.

A. Parental MDA-MB-231 and three independent clones of Arkadia C937A-expressing cells were injected into the tail vein of immunodeficient mice. After 20 days, lungs were analyzed for number of tumors. B and C. GFP-labeled parental MDA-MB-231 cells and the three independent clones of Arkadia C937A-expressing cells were mixed with an equal number of parental cells expressing mCherry and injected into the tail vein of immunodeficient mice. After 2 days mice were culled and lungs directly analyzed by confocal microscopy. Representative pictures of tumor cell distribution for parental and one clone of Arkadia C937A-expressing cells were taken (B, left panels). As a control, aliquots of the mixed GFP/mCherry cells were plated prior to injection (B, right panels). The magnification scale bar corresponds to 100 μm. The ratio between the GFP/mCherry cells in the lungs from multiple mouse experiments is plotted (C). In A and C each point represents data from a single mouse, and means and standard deviation are plotted.
Arkadia C937A is catalytically inactive, but retains its ability to interact with partners such as SnoN and Smad2/3 (12). It was therefore important to exclude the possibility that the decrease in the efficiency of lung colonization by cells overexpressing Arkadia C937A was due to titration of some of Arkadia’s partners. We therefore downregulated Arkadia in parental MDA-MB-231 cells using two different siRNAs and investigated the effect on short-term lung colonization. Knockdown of Arkadia was efficient for both siRNAs, and TGF-β-induced SnoN degradation was inhibited (Fig. 5A), as was Smad3-dependent transcription (Fig. 5B). In lung colonization assays, we observed a significant decrease (20–30%) for the cells in which Arkadia was downregulated compared with control cells (Fig. 5C).
Figure 5. The ability of MDA-MB-231 cells to colonize lungs requires Arkadia function.
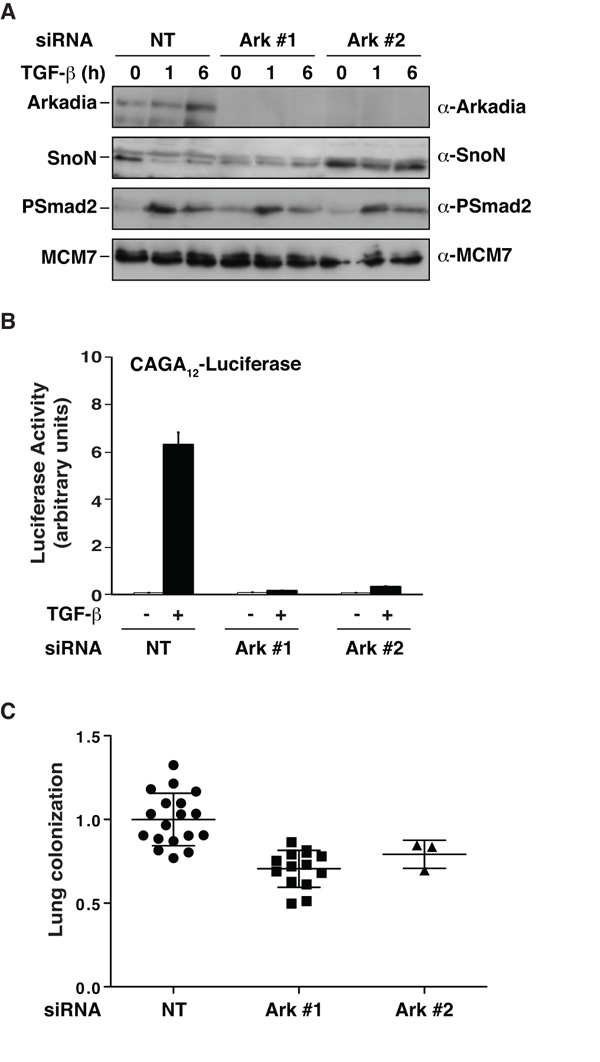
A–C. GFP and mCherry-labeled MDA-MB-231 cells were transfected with a control (NT), and GFP-labeled MDA-MB-231 cells were transfected with two different siRNAs against Arkadia (Ark #1 and #2). 24 h later cells were split for immunoblots, CAGA12-Luciferase reporter assays and mice experiments. A. Whole cell extracts were prepared from the GFP-labeled cells treated ± TGF-β and Western blotted using the antibodies indicated. B. The GFP-labeled cells were transfected with the CAGA12-Luciferase and TK-Renilla reporters then treated ± TGF-β. Coupled Luciferase/Renilla assays were as in Fig. 1. C. Equal numbers of NT mCherry cells were mixed with either NT GFP cells or Ark #1 and Ark #2 siRNA GFP cells and were injected into the tail vein of immunodeficient mice. Analysis and quantitation of the experiment was as in Fig. 4.
Finally, to confirm that Arkadia acts as a tumor promoter, we extended our analysis to two further cell lines for which metastasis is known to be driven by TGF-β: the rat mammary carcinoma cell line MTLN3E (39) and the mouse B16 melanoma cell line (29). In both cases, knockdown of Arkadia resulted in loss of TGF-β-induced Ski and SnoN degradation, reduction of Smad3-dependent transcription (Fig. S7), and most importantly, substantial inhibition (between 66% and 78%) in lung colonization (Fig. 6).
Figure 6. MTLN3E and B16 cells require Arkadia to colonize lungs.

A and B. GFP and mCherry-labeled MTLN3E cells or B16 cells were transfected with a control (NT), and GFP-labeled MTLN3E and B16 cells were transfected with siRNAs against Arkadia as indicated. 24 h later cells were split for immunoblots, qPCR for Arkadia/GAPDH and 48-h lung colonization mouse experiments, which were as described in Fig. 4.
Discussion
A role for Arkadia in tumorigenesis had been hypothesized since it was first described as the ubiquitin-ligase controlling the cellular levels of Ski and SnoN (12, 19, 20). Here we investigated a potential tumor suppressor function for Arkadia by restoring its activity in the NCI-H460 cell line that contains a hemizygous nonsense mutation. Although re-expression of Arkadia decreased the transformed phenotype of these cells in vitro, we found no effect on growth of tumors in xenograft assays, or in lung colonization assays. These results could indicate that the inactivating mutation we identified in Arkadia is not a cancer driver mutation (40). However, it is also possible that loss of Arkadia constitutes an early priming event for tumorigenesis, and that acquisition of subsequent mutations in this cell line prevent the re-expression of Arkadia reversing the tumorigenicity of these cells in vivo. In support of this, another recent study concluded that Arkadia has tumor suppressive activity in colorectal cancer (23). Interestingly, the Arkadia+/− mice used in that study were only susceptible to cancer when treated with a carcinogen (23), suggesting that loss of Arkadia is not sufficient for tumorigenesis, but may sensitize cells to other oncogenic signals. Moreover, unlike classical tumor suppressors there was no tendency for the tumor cells in the Arkadia+/− mice to lose the other allele. Consistent with this, complete loss of Arkadia appears to be very rare in both tumor samples and cancer cell lines. In this study we identified a cell line that exhibits a hemizigous nonsense mutation (S441*), but were unable to find other cell lines containing mutations in Arkadia, even in cell lines showing LOH at 15q22.1. Interestingly a small number of nonsense mutations: E389*, E561*, R598*, Q605*, Q722*, Q899* (of unknown zygosity), that would similarly delete the RING domain of Arkadia, have been found in tumors of the upper aerodigestive tract, large intestine and hematopoetic and lymphoid tissue sequenced by the cancer genome project at the Sanger Institute (reported in the COSMIC database) and in a colorectal tumor (23). In addition four missense mutations have also been reported in the COSMIC database, but how these mutations affect Arkadia function is unknown. These finding indicate that Arkadia mutations do occur in human cancer, but are rare.
It is well established that different components of the TGF-β pathway are mutated in cancer at different rates. Whereas inactivating mutations and deletions in Smad4 and TGFBR2 are common in certain cancers, mutations in ALK5, Smad2 and Smad3 are relatively rare (41-43). Arkadia appears to be in this latter class. In our search for cell lines containing mutations in Arkadia we made the unexpected discovery that deletion of Smad4, or acquisition of Smad4 mutations that abolish Smad–Smad interactions also abolished TGF-β-induced degradation of SnoN, i.e. it confers the same effect on Ski and SnoN levels as would loss of Arkadia. We therefore speculate that cancer cells may lose Smad4 in preference to Arkadia to achieve stabilization of Ski and SnoN.
The observation that cancer cells do not preferentially lose all Arkadia function led us to investigate whether it might play additional roles at later stages of tumorigenesis. To explicitly address this we have used three different tumor cell lines that metastasize in a TGF-β-dependent manner (29, 34, 39). Our results clearly demonstrate that Arkadia has a potent tumor-promoting activity. Arkadia inactivation has no effect on primary tumor growth, in agreement with previous work demonstrating that manipulation of the TGF-β pathway in these cells had no effect on mammary tumor growth (34). Instead, we show a dramatic effect of loss of Arkadia activity on lung colonization in tail vein injection experiments in immunodeficient mice. The fact that we can detect these effects within 48 h, coupled with our observation that MDA-MB-231 cells expressing dominant negative Arkadia adhere more strongly than parental cells to a confluent layer of HUVEC cells, which mimics the capillary wall, and do not spread as efficiently, leads us to conclude that Arkadia is likely important for extravasation, rather than growth and survival of the cells in the lungs.
Our RNA-seq analysis uncovered a large group of genes whose TGF-β regulation was perturbed by loss of Arkadia activity. Importantly, this list contained genes previously implicated in lung metastasis of MDA-MB-231 cells, such as ANGPTL4, Id1, LOX and SNAI1 (33, 34, 44, 45) (Figs S5; S6). We have used gene enrichment analysis to further define classes of genes that might be responsible for lung colonization and identified genes involved in cell adhesion, cell matrix interactions, EMT and ECM remodeling as particularly affected by loss of Arkadia activity. Specific combinations of these genes are likely to be responsible for driving metastasis in this tumor model.
Our data indicate that Arkadia regulates metastasis in mouse models of breast cancer and melanoma. TGF-β signaling has both tumor suppressive and tumor-promoting roles, and it is therefore difficult to target this pathway for cancer therapy (46). Since Arkadia is an enzyme that is required for only a subset of TGF-β responses, it might be amenable to inhibition by small molecules, and thus represent a possible therapeutic target for cancer.
Supplementary Material
Acknowledgments
We thank the Biological Resources Unit for help with the mouse experiments and Emma Nye, Bradley Spencer-Dene and Gordon Stamp from Experimental Histopathology for the analysis of the tumors. We thank Florence Tatin and Taija Makinen for the GFP-CAAX plasmid. We thank Nik Matthews and the advanced sequencing facility for the RNA-seq analysis and Richard Mitter for data analysis. We thank the Light Microscopy Facility, Equipment Park, Mike Howell and the High Throughput Screening Facility and FACS facility at the London Research Institute for the technical support. We are very grateful to the Hill lab, and Ilaria Malanchi for discussions and comments on the manuscript. We thank Becky Randall for help with the preparation of the manuscript. The work was funded by Cancer Research UK, a postdoctoral fellowship from CONACYT and a Marie Curie International Incoming Fellowship PIIF-GA-2009-235980 (to M.A.B-O), and a FEBS long term fellowship (to C.D.M). L.L. acknowledges funding from Marie Curie Actions (ERG FP7), la Ligue Nationale Contre le Cancer (LNCC) and l’Association pour la Recherche sur le Cancer (ARC). Y.E. was supported by the MRT.
Footnotes
There are no potential conflicts of interest.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Massague J. TGFβ in Cancer. Cell. 2008;134:215–30. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Padua D, Massague J. Roles of TGFβ in metastasis. Cell Res. 2009;19:89–102. doi: 10.1038/cr.2008.316. [DOI] [PubMed] [Google Scholar]
- 4.Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006;127:679–95. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 5.Feng XH, Derynck R. Specificity and versatility in TGF-β signaling through Smads. Annu Rev Cell Dev Biol. 2005;21:659–93. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- 6.Ross S, Hill CS. How the Smads regulate transcription. Int J Biochem Cell Biol. 2008;40:383–408. doi: 10.1016/j.biocel.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 7.Luo K. Ski and SnoN: negative regulators of TGF-β signaling. Curr Opin Genet Dev. 2004;14:65–70. doi: 10.1016/j.gde.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 8.Stroschein SL, Wang W, Zhou S, Zhou Q, Luo K. Negative feedback regulation of TGF-β signaling by the SnoN oncoprotein. Science. 1999;286:771–4. doi: 10.1126/science.286.5440.771. [DOI] [PubMed] [Google Scholar]
- 9.He J, Tegen SB, Krawitz AR, Martin GS, Luo K. The transforming activity of Ski and SnoN is dependent on their ability to repress the activity of Smad proteins. J Biol Chem. 2003;278:30540–7. doi: 10.1074/jbc.M304016200. [DOI] [PubMed] [Google Scholar]
- 10.Nomura T, Khan MM, Kaul SC, Dong HD, Wadhwa R, Colmenares C, et al. Ski is a component of the histone deacetylase complex required for transcriptional repression by Mad and thyroid hormone receptor. Genes Dev. 1999;13:412–23. doi: 10.1101/gad.13.4.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kokura K, Kaul SC, Wadhwa R, Nomura T, Khan MM, Shinagawa T, et al. The Ski protein family is required for MeCP2-mediated transcriptional repression. J Biol Chem. 2001;276:34115–21. doi: 10.1074/jbc.M105747200. [DOI] [PubMed] [Google Scholar]
- 12.Levy L, Howell M, Das D, Harkin S, Episkopou V, Hill CS. Arkadia activates Smad3/Smad4-dependent transcription by triggering signal-induced SnoN degradation. Mol Cell Biol. 2007;27:6068–83. doi: 10.1128/MCB.00664-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shinagawa T, Dong HD, Xu M, Maekawa T, Ishii S. The sno gene, which encodes a component of the histone deacetylase complex, acts as a tumor suppressor in mice. EMBO J. 2000;19:2280–91. doi: 10.1093/emboj/19.10.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Briones-Orta MA, Sosa-Garrocho M, Moreno-Alvarez P, Fonseca-Sanchez MA, Macias-Silva M. SnoN co-repressor binds and represses smad7 gene promoter. Biochem Biophys Res Commun. 2006;341:889–94. doi: 10.1016/j.bbrc.2006.01.041. [DOI] [PubMed] [Google Scholar]
- 15.Denissova NG, Liu F. Repression of endogenous Smad7 by Ski. J Biol Chem. 2004;279:28143–8. doi: 10.1074/jbc.M404961200. [DOI] [PubMed] [Google Scholar]
- 16.Bonni S, Wang HR, Causing CG, Kavsak P, Stroschein SL, Luo K, et al. TGF-β induces assembly of a Smad2-Smurf2 ubiquitin ligase complex that targets SnoN for degradation. Nat Cell Biol. 2001;3:587–95. doi: 10.1038/35078562. [DOI] [PubMed] [Google Scholar]
- 17.Wan Y, Liu X, Kirschner MW. The anaphase-promoting complex mediates TGF-β signaling by targeting SnoN for destruction. Mol Cell. 2001;8:1027–39. doi: 10.1016/s1097-2765(01)00382-3. [DOI] [PubMed] [Google Scholar]
- 18.Stroschein SL, Bonni S, Wrana JL, Luo K. Smad3 recruits the anaphase-promoting complex for ubiquitination and degradation of SnoN. Genes Dev. 2001;15:2822–36. doi: 10.1101/gad.912901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nagano Y, Mavrakis KJ, Lee KL, Fujii T, Koinuma D, Sase H, et al. Arkadia induces degradation of SnoN and c-Ski to enhance transforming growth factor-β signaling. J Biol Chem. 2007;282:20492–501. doi: 10.1074/jbc.M701294200. [DOI] [PubMed] [Google Scholar]
- 20.Le Scolan E, Zhu Q, Wang L, Bandyopadhyay A, Javelaud D, Mauviel A, et al. Transforming growth factor-β suppresses the ability of Ski to inhibit tumor metastasis by inducing its degradation. Cancer Res. 2008;68:3277–85. doi: 10.1158/0008-5472.CAN-07-6793. [DOI] [PubMed] [Google Scholar]
- 21.Zhu Q, Krakowski AR, Dunham EE, Wang L, Bandyopadhyay A, Berdeaux R, et al. Dual role of SnoN in mammalian tumorigenesis. Mol Cell Biol. 2007;27:324–39. doi: 10.1128/MCB.01394-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boonstra JJ, van Marion R, Beer DG, Lin L, Chaves P, Ribeiro C, et al. Verification and unmasking of widely used human esophageal adenocarcinoma cell lines. J Natl Cancer Inst. 2010;102:271–4. doi: 10.1093/jnci/djp499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sharma V, Antonacopoulou AG, Tanaka S, Panoutsopoulos AA, Bravou V, Kalofonos HP, et al. Enhancement of TGF-β signaling responses by the E3 ubiquitin ligase Arkadia provides tumor suppression in colorectal cancer. Cancer Res. 2011;71:6438–49. doi: 10.1158/0008-5472.CAN-11-1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deckers M, van Dinther M, Buijs J, Que I, Lowik C, van der Pluijm G, et al. The tumor suppressor Smad4 is required for transforming growth factor β-induced epithelial to mesenchymal transition and bone metastasis of breast cancer cells. Cancer Res. 2006;66:2202–9. doi: 10.1158/0008-5472.CAN-05-3560. [DOI] [PubMed] [Google Scholar]
- 25.Kang Y, He W, Tulley S, Gupta GP, Serganova I, Chen CR, et al. Breast cancer bone metastasis mediated by the Smad tumor suppressor pathway. Proc Natl Acad Sci U S A. 2005;102:13909–14. doi: 10.1073/pnas.0506517102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Randall RA, Howell M, Page CS, Daly A, Bates PA, Hill CS. Recognition of phosphorylated-Smad2-containing complexes by a novel Smad interaction motif. Mol Cell Biol. 2004;24:1106–21. doi: 10.1128/MCB.24.3.1106-1121.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dorey K, Hill CS. A novel Cripto-related protein reveals an essential role for EGF-CFCs in Nodal signalling in Xenopus embryos. Dev Biol. 2006;292:303–16. doi: 10.1016/j.ydbio.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 28.Chan AY, Raft S, Bailly M, Wyckoff JB, Segall JE, Condeelis JS. EGF stimulates an increase in actin nucleation and filament number at the leading edge of the lamellipod in mammary adenocarcinoma cells. J Cell Sci. 1998;111(Pt 2):199–211. doi: 10.1242/jcs.111.2.199. [DOI] [PubMed] [Google Scholar]
- 29.Zhang C, Zhang F, Tsan R, Fidler IJ. Transforming growth factor-β2 is a molecular determinant for site-specific melanoma metastasis in the brain. Cancer Res. 2009;69:828–35. doi: 10.1158/0008-5472.CAN-08-2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Watanabe Y, Itoh S, Goto T, Ohnishi E, Inamitsu M, Itoh F, et al. TMEPAI, a transmembrane TGF-β-inducible protein, sequesters Smad proteins from active participation in TGF-β signaling. Mol Cell. 2010;37:123–34. doi: 10.1016/j.molcel.2009.10.028. [DOI] [PubMed] [Google Scholar]
- 31.Inman GJ, Nicolas FJ, Callahan JF, Harling JD, Gaster LM, Reith AD, et al. SB-431542 is a potent and specific inhibitor of transforming growth factor-β superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol Pharmacol. 2002;62:65–74. doi: 10.1124/mol.62.1.65. [DOI] [PubMed] [Google Scholar]
- 32.De Bosscher K, Hill CS, Nicolas FJ. Molecular and functional consequences of Smad4 C-terminal missense mutations in colorectal tumour cells. Biochem J. 2004;379:209–16. doi: 10.1042/BJ20031886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gupta GP, Perk J, Acharyya S, de Candia P, Mittal V, Todorova-Manova K, et al. ID genes mediate tumor reinitiation during breast cancer lung metastasis. Proc Natl Acad Sci U S A. 2007;104:19506–11. doi: 10.1073/pnas.0709185104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Padua D, Zhang XH, Wang Q, Nadal C, Gerald WL, Gomis RR, et al. TGFβ primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell. 2008;133:66–77. doi: 10.1016/j.cell.2008.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Petersen M, Pardali E, van der Horst G, Cheung H, van den Hoogen C, van der Pluijm G, et al. Smad2 and Smad3 have opposing roles in breast cancer bone metastasis by differentially affecting tumor angiogenesis. Oncogene. 2010;29:1351–61. doi: 10.1038/onc.2009.426. [DOI] [PubMed] [Google Scholar]
- 36.Bravo-Cordero JJ, Hodgson L, Condeelis J. Directed cell invasion and migration during metastasis. Curr Opin Cell Biol. 2012;24:277–83. doi: 10.1016/j.ceb.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sahai E. Illuminating the metastatic process. Nature reviews Cancer. 2007;7:737–49. doi: 10.1038/nrc2229. [DOI] [PubMed] [Google Scholar]
- 38.Cain RJ, d’Agua BB, Ridley AJ. Quantification of transendothelial migration using three-dimensional confocal microscopy. Methods Mol Biol. 2011;769:167–90. doi: 10.1007/978-1-61779-207-6_12. [DOI] [PubMed] [Google Scholar]
- 39.Giampieri S, Manning C, Hooper S, Jones L, Hill CS, Sahai E. Localized and reversible TGFβ signalling switches breast cancer cells from cohesive to single cell motility. Nat Cell Biol. 2009;11:1287–96. doi: 10.1038/ncb1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stratton MR. Exploring the genomes of cancer cells: progress and promise. Science. 2011;331:1553–8. doi: 10.1126/science.1204040. [DOI] [PubMed] [Google Scholar]
- 41.Daly AC, Vizan P, Hill CS. Smad3 protein levels are modulated by Ras activity and during the cell cycle to dictate transforming growth factor-β responses. J Biol Chem. 2010;285:6489–97. doi: 10.1074/jbc.M109.043877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Levy L, Hill CS. Alterations in components of the TGF-β superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev. 2006;17:41–58. doi: 10.1016/j.cytogfr.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 43.Tian M, Neil JR, Schiemann WP. Transforming growth factor-β and the hallmarks of cancer. Cell Signal. 2011;23:951–62. doi: 10.1016/j.cellsig.2010.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Erler JT, Bennewith KL, Nicolau M, Dornhofer N, Kong C, Le QT, et al. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature. 2006;440:1222–6. doi: 10.1038/nature04695. [DOI] [PubMed] [Google Scholar]
- 45.Olmeda D, Moreno-Bueno G, Flores JM, Fabra A, Portillo F, Cano A. SNAI1 is required for tumor growth and lymph node metastasis of human breast carcinoma MDA-MB-231 cells. Cancer Res. 2007;67:11721–31. doi: 10.1158/0008-5472.CAN-07-2318. [DOI] [PubMed] [Google Scholar]
- 46.Hawinkels LJ, Ten Dijke P. Exploring anti-TGF-β therapies in cancer and fibrosis. Growth factors. 2011;29:140–52. doi: 10.3109/08977194.2011.595411. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.