

1. INTRODUCTION
The focus of this review is on the biophysical studies of the channel-forming bacterial toxins that shed light upon the mechanisms of their toxicity and propose new approaches to block their virulent action. Extensive data on the structural features of these toxins, the mechanisms of their secretion, proteolytic activation, extracellular receptors, enzymatic intracellular action, and cellular responses will be mostly omitted here. We refer our reader to several excellent specialized books and reviews that cover this material in the finest detail1–15.
1.1. Channel-blocking and channel-forming toxins
In the course of evolution, Nature created numerous toxins, which selectively target ion channels of excitable cells16–18. Regardless of which version of Cleopatra's self-poisoning is true, the toxin-triggered modification of channel function was certainly involved. She either suffered from intoxication following blockage of her ligand-gated channels by the Egyptian cobra venom or from poisonous action of the chloride channel inhibitor extracted from cicuta. Numerous studies of the neurotoxins' biological functions allowed not only for deeper understanding of the structural and functional features of several channels of excitable membranes, but also for developing the pharmaceutical approaches to use these toxins for therapeutic purposes. Conversely, among the great variety of virulence factors secreted by different organisms, there is a significant group of toxins (mostly bacterial) that, instead of blocking channels, are able to form ion-conductive pores in membranes of the targeted cells. For most of them, there are no antidotes or antitoxins developed and approved for human use. At the same time, one of the possible ideas to target the bacterial exotoxins is quite simple. Following Nature, which created the channel-inhibiting toxins, it should be possible to design the potent antitoxins specifically blocking the conductive pathways of the channel-forming toxins with an ultimate goal to defend from the cytotoxic action of these agents. This idea is neither preposterous nor new. Indeed, amantadine, the small-molecule blocker of the tetrameric proton-selective M2 channel from the viral envelope of influenza A virus19–21 had been approved for human use back in 1965. Interestingly, even though amantadine is no longer recommended by the CDC for use to treat seasonable influenza due to developed antidrug resistance, the exact mechanism of the amantadine interaction with the M2 channel is still a matter of debates22–29. Even so, the M2 channel is the main target for virtual screening for powerful channel blockers30–34. Therefore, the idea to design and develop the effective blockers of the channels formed by bacterial toxins in target membranes enjoys a well-appreciated attention. Not being able to use the evolutionary path, we can employ the modern single-molecule biophysical approaches to study the pore-forming toxins and their potential inhibitors.
1.2. Classification of channel-forming toxins
All bacterial toxins can be divided into two functionally different groups: endotoxins and exotoxins13. The endotoxins are components of the outer membrane of Gram-negative bacteria (i.e. lipopolysaccharide). The exotoxins are proteins secreted by a number of Gram-positive and Gram-negative bacteria, which act on eukaryotic cells far off from the host bacterium. A crucial property of many exotoxins is their ability to exist in two states: a stable water-soluble conformation and an integral membrane pore1. There are several ways to classify the channel-forming proteins. The classifications are usually based on the nature of the toxin secreting organisms (e.g. clostridial toxins14), on the mechanism of their cytotoxicity (e.g. membrane-damaging toxins35; ADP-ribosylating toxins36), or on their structural types (e.g. α-helical and β-barrel pore-forming toxins7, 35). For the specific task of this review, we will use a combined structural/functional classification to discriminate between the following three groups of bacterial toxins (Figure 1).
Figure 1.

Typical membrane-perforating, AB-type, and binary bacterial toxins α-hemolysin of Staphylococcus aureus, diphtheria toxins of Corynebacterium diphtheriae, and the channel-forming PA63 component of anthrax toxin of Bacillus anthracis. A: Ribbon representation of 1.9 Å crystal structure of heptameric α-hemolysin 167. PDB ID: 7AHL. B: 2.3 Å nucleotide-free crystal structure of monomeric single-chain diphtheria toxin292. PDB ID: 1SGK. C, left: Molecular model of the heptameric PA63 pore. Reprinted with permission from ref385. Taylor & Francis Copyright 2004. C, right: Three-dimensional reconstruction of the PA63 pore based on the electron microscopy structure. Reprinted with permission from ref376. Copyright 2008. Nature Publishing Group.
Membrane-perforating bacterial toxins (Figure 1A)
Membrane-perforating bacterial toxins represent more than one third of the protein toxins identified so far7, 35. These toxins insert into target membranes and form aqueous ion-permeable pores. Subsequently, these pores can compromise the membrane barrier function and cause ion imbalance by allowing the flow of ions down their electrochemical gradient. In accordance with the mechanism of their action, they are often called the pore-forming toxins (PFT).
AB-type bacterial toxins (Figure 1B)
In contrast to the PFTs that damage the target cells membranes by forming ion-permeable pores, certain exotoxins act in the cytosol of mammalian cells enzymatically modifying specific intracellular substrates. Many of them are secreted in a so-called AB-type form, which contains at least two functionally distinct domains. The binding B-domain docks to a receptor on the surface of the target cells and mediates intracellular transport of the toxin, whereas the active/enzymatic A-domain modifies certain components in the cytosol3.
Binary bacterial toxins (Figure 1C)
Several pathogenic species of Bacillus and Clostridium secrete binary exotoxins which consist of two (or three) individual non-linked proteins, an active/enzymatic A component and a binding/translocation B component3, 13, 37, 38. The B component of these toxins binds to a receptor on the surface of the target cells, self-assembles to form a ring-shaped oligomeric prepore able to bind the A components, and, after receptor-mediated endocytosis, is converted into an ion-conductive pore, which mediates A component translocation from acidified endosomal vesicles into the cytosol. Note that even though formally the binary toxins can be placed among the AB-type toxins, for the special purpose of this review we will describe these two groups separately.
This review aims to discuss the well-studied and/or intriguing channel-forming bacterial toxins, for which electrophysiological measurements coupled with X-ray crystallography provided important details of their structure and function. We do not intend to give comprehensive description of all membrane-perforating, AB-type, and binary toxins but prefer to focus on those examples of the channel-forming toxins that could serve as attractive candidates for the targeted design of effective channel-blocking agents.
1.3. Planar lipid bilayer technique
The planar lipid bilayer technique is a unique approach allowing incorporating purified bacterial protein toxins into artificial membranes formed across an aperture in a film (or wall) separating two compartments (cis and trans) of a bilayer chamber (Figure 2). The alternative terms are “black lipid membrane” or “bilayer lipid membrane” (BLM) techniques. Two versions of the technique are routinely used by different groups. First, the bilayer membranes can be formed by brushing (or “painting”) a solution of lipid dissolved in an organic solvent (often in decane) across the aperture39. In time, a lipid bilayer membrane spontaneously forms across the aperture (for details see40). The second approach is often referred to as the “solvent-free” monolayer opposition technique41. In that case, lipid solution in an organic solvent (often in pentane) is applied on top of the bathing electrolyte solution in cis and trans compartments of the bilayer chamber. A pair of syringes is used to keep electrolyte solution level below the aperture. After pentane evaporates, lipid monolayers are formed on top of the cis and trans electrolyte solutions. The aperture is pretreated at this point with a small amount of a solution containing a less volatile component (often with 1% hexadecane in pentane) to insure better contact of the bilayer with the edges of the aperture42. Solution levels in the both compartments of the chamber are then raised above the aperture, and a bilayer lipid membrane forms across the aperture with both monolayers of lipid contributing. Each of the existing lipid bilayer techniques has its advantages and weaknesses. Thus, due to presence of the islets of organic solvent and larger sizes of the aperture (often 0.2–1 mm), channel incorporations is achieved easier with the brushed/painted bilayer technique. This is especially important for multichannel measurements, which often involve 100–1000 channels. On the other hand, the monolayer opposition technique allows one to minimize the membrane area by reducing the aperture size, so that a higher time resolution (e.g., 15 μs) of single-channel recordings can be often achieved. The measurements are regularly performed using specially prepared Teflon films with a diameter of the aperture not exceeding 50 μm. In many cases, these unique low-noise high-resolution conditions allow for recording of the short individual events of a single molecule metabolite and macromolecular transport across a channel reconstituted into the planar membrane. Importantly, this technique also allows formation of asymmetrical membranes in which cis and trans monolayers are formed from lipid mixtures of different compositions.
Figure 2.

Schematic illustration of planar lipid bilayer membrane formation (1, 2) and high-resolution single-channel recordings (3, 4).
2. CHANNEL-FORMING BACTERIAL TOXINS
A unique ability of a number of bacterial toxins to exist both in a stable water-soluble conformation and in a membrane imbedded conformation allowed to perform numerous in vitro studies on the purified toxins. It turned out that the channel-forming toxins or their components are able to form stable ion-conductive structures in the bilayer lipid membranes lacking any extracellular receptors, which are normally required for their binding in cell assays or in vivo. The protein recombinant technology allowed for a large number of different mutants to be synthesized, in which the various amino acids were mutated in order to understand their possible role in the channel function. These studies not only provided the essential channels' properties, such as single channel conductance, ion selectivity, and current noise characteristics, but also allowed to reveal some interesting molecular details defining their complex behavior. One of the most fascinating examples is the binary (or tripartite) anthrax toxin where the meticulous biophysical studies yielded fine kinetics and structural details of the A component translocation through the channel formed by the B component43–50. Structure-based drug design approaches focused on single-channel single-molecule interactions had also allowed for designing a number of very efficient blockers, which act against several channel-forming toxins51–62. Moreover, a number of additional applications, which are not directly related to the toxicity of the channel-forming proteins, were developed. Perhaps one of the most notable applications involves the use of Staphylococcus aureus α-hemolysin channel as a sensor for different analytes63–90.
The largest group of bacterial toxins, PFTs act at the cell surface level. They are secreted as water-soluble single proteins, which target eukaryotic cells by embedding into their cell membrane and forming large water-filled ion-permeable pores. These pores can significantly modulate one of the most important functions of the plasma membrane – its ability to maintain the membrane potential. The latter is determined by movement of ions across the cellular membrane, which is mostly regulated by a number of small highly ion-selective channels. The formation of the large and, to a certain extent, nonspecific pores results in a significant disturbance of the cell membrane integrity, which leads to a membrane depolarization due to fast inflow of Na+ and efflux of Cl− across cell walls91. Larger extraneous pores would also allow for the leakage of the intracellular macromolecules, essential for the cellular metabolic integrity, which eventually leads to cell death91. Structurally, the PFTs can be separated into two families: α-PFTs and β-PFTs1, 10, 92. α-PFTs cross the membrane as α-helices and β-PFTs as β-sheets, which form β-barrels11. Note that channel-forming domains or components of the bacterial AB-type or binary toxins can be also ascribed to one of these groups.
2.1. Colicins as α-helix membrane-perforating toxins
Colicins represent a class of antibiotics produced by many strains of Escherichia coli in times of stress. Colicins are lethal for other E.coli strains or closely related bacteria93–95. The first colicin was discovered in 192596 and later named “colicine” because its activity against E.coli. The host bacteria also produce small polypeptides, called immunity proteins, to defend themselves from their own toxins95, 97. After secretion, the water-soluble colicin molecules bind to specific outer membrane protein receptors of a target bacteria and are subsequently translocated across the outer membrane98 into the intermembrane space using one of the two distinct bacterial transport systems, Tol or TonB99. Once in the periplasmic compartment, colicins cause bacterial cell death via variety of mechanisms100–104. Some colicins, such as colicins A, N, Ia, and E1, form pores in the cytoplasmic membranes105, while others translocate into the cytoplasm. They degrade peptidoglycan, inhibit protein synthesis, or function as DNAse or RNAse (recently reviewed95). Regardless of the cytotoxic mechanism, all colicins need to insert or pass through the inner membrane of a bacterium they target1. Interestingly, several of them were shown to be able to mediate their own transport across the inner membrane12, 106, 107. Therefore, it is not surprising that different aspects of colicins' structure and function have been intensively investigated in the last decades. A comprehensive 70-page review covering all the details of this research published in 2007 contains more than 700 references95, and the numerous papers published since then are reviewed in ref98. In particular, colicins were used as model systems for understanding the basic principles of protein incorporation into the lipid membranes as well as protein transmembrane transport1, 93, 108, 109. X-ray crystallography studies101, 110–118 revealed a three-domain structure of the colicins with several characteristic features of the domain organization; each domain is involved in a certain step in its toxic activity such as receptor binding, translocation, and cell death1 (Figure 3A). In pore-forming colicins, the cell death domain corresponds to a channel-forming part that is highly conserved among all pore-forming colicins. At that, no detectable sequence similarities were observed among the receptor binding and translocation domains of these molecules. This difference explains the narrow target range of each individual colicin that is usually able to bind only to specific receptors at the surface of a particular E.coli strain95. The striking similarities in amino acid sequence as well as the known crystal structures of colicin pore-forming domains suggest that all of the pore-forming colicins employ similar mechanisms of inserting into the inner membranes95. Interestingly, the pore-forming domains of the colicins101, 110, 114, 118, 119 are able to embed into the bilayer membranes even in the absence of a transmembrane potential when isolated from the rest of the protein1, 120, 121. However, despite the fact that the structures of the pore-forming domains of colicins A, Ia, E1, N, and B were solved by crystallography and extensively studied in bilayer lipid membranes, the mechanism of the transformation of a compact, water-soluble form of colicin into an ion channel in the inner membrane is far from being clear95. It is known that colicin membrane insertion is facilitated by an acidic pH, which may initiate a partial destabilization of the soluble colicin molecules resulting in rearrangement of the protein tertiary structure into a membrane-embedding configuration1, 121, 122. A single pore-forming colicin molecule is able to kill a bacterial cell by single-hit kinetics1; therefore, one molecule has to be sufficient to form a pore in the inner membrane or artificial bilayer. Formation of oligomeric transmembrane colicin pores has been also reported123. When inserted in the bilayer, membrane-perforating colicins form large stable voltage-gated ion pores122 permeable to both cations and anions124–126 and allowing for the passage of organic molecules of up to 9 Å diameter1, 124, 127, 128. Anomalous selectivity to protons over other cations (and anions) was reported for the pore-forming colicin A129, 130.
Figure 3.

Pore-forming colicin Ia. A: Colicin Ia crystal structure at the 3 Å resolution114. PDB ID: 1CII. B: Schematic diagram of the open state of the whole colicin Ia molecule inserted into a planar bilayer. Reprinted with permission from ref151. Copyright 2000. The Rockefeller University Press.
Voltage-gated opening and closing of the colicin-formed pores is believed to be principally different from that of both the “classical” small ion-selective channels94 and ion channels formed by the β-barrels131, 132. Gating of the classical ion channels, including many channel of excitable cells, is determined by a miniature conformational changes in the membrane spanning fragments of these channels133. The picture is entirely different for the voltage-gated colicin channels, where gating involves import of a large part of the protein molecule from the membrane surface. This is considered to be an essential part of their translocation across the membrane134–136. Structural organization and activity of pore-forming colicins (primarily of colicin Ia) were qualitatively described by Finkelstein and coworkers in a series of articles published in the last decades129, 135–154. Briefly, like many colicins studied so far, colicin Ia has three distinct domains: the middle receptor binding region, the N-terminal region which together with proteins on the target bacterial cells transports colicin inside the cell, and C-terminal region, which is the channel-forming domain made of 10 α-helices. All channel-forming domains carry a distinctive short hydrophobic segment near the C-terminus with 31–49 consecutive uncharged residues identified as a hydrophobic hairpin (helix 8 – helix 9, or H8–H9)153. The rest of the toxin is highly charged. Measurement with colicin Ia in planar lipid membranes led to an intriguing finding135, 148. It was demonstrated that a stretch of at least 31 amino acid residues (residues 474–541) of colicin Ia is translocated back and forth across the bilayer, which is accompanied by channel opening and closing. At the same time, residues 544–572 are moved in and out of the bilayer but not entirely across it135, 148. Measurements were performed in a series of experiments in planar lipid bilayers using cis and trans streptavidin trapping of biotin-labeled single-cysteine mutants of the C-terminal channel domain of colicin Ia135. Both the whole colicin Ia and its truncated C-domain are able to form voltage-gated ion channels with four membrane-spanning segments, all contributed by a single protein molecule136. At that, channel formation by colicin Ia in planar membranes was suggested to occur in several steps153. Once colicin is added to the cis side of the membrane, the hydrophobic hairpin H8–H9 inserts into the membrane in a voltage-dependent manner forming two transmembrane segments. Triggered by the positive voltage at the cis side, an additional part of the C-domain inserts, contributing two transmembrane segments (helices H1 and H6–H7). The helices H2–H5 are concurrently translocated across the membrane to the trans side (Figure 3B)153. This voltage-dependent insertion is recorded in the planar bilayer lipid membranes as a step-like opening of a conductive colicin Ia channel136, 152, 153. Interestingly, channel formation by the carboxyl-terminal colicin Ia fragment containing 345 residues is also similar to channel formation by the whole colicin or by the isolated C-domain151, 152. However, the portion of helix 1 is also translocated across the membrane and the channel is formed by three transmembrane segments only. The ability of the large transmembrane channel to be formed by only four (or even three) transmembrane segments created a fundamental problem in understanding the nature of the colicin Ia pore structure (see related discussions in refs.1, 93, 94, 153). It is still hard to find an explanation for a paradox of forming a channel permeable for large, folded proteins (up to 26 Å in diameter) by a small protein152. Different models attempting to resolve this paradox either suggest that the channel wall can be partially lined by membrane lipids155, 156 or advocate a possibility of oligomer formation94. Note that formation of multimeric colicin Ia channels was directly visualized recently with two-dimensional crystals of colicin Ia inserted into bilayer lipid membranes by electron crystallography123. However, the authors emphasize that despite their data indicate that colicin Ia channels exist as multimers, it does not imply that formation of an oligomer is required for the channel to function.
2.2. β-barrel membrane-perforating toxins
β-PFTs are secreted as water-soluble proteins that, in order to form a pore, need to oligomerize into multimeric complexes on the mammalian plasma membranes2. Each monomer of this oligomeric complex contributes one or two amphipathic β-hairpins to the pore thus forming a β-barrel. A hydrophobic outer surface of the β-hairpins favors insertion of these oligomeric pores into the lipid membrane2, 92, 157. The number of subunits composing the β-barrel can vary significantly ranging from 7 for α-hemolysin of Staphylococcus aureus to 50 for a family of cholesterol-dependent cytolysins. The variety in the subunit numbers results in a significant range of pore sizes2, from 2 to 50 nm.
2.2.1. Staphylococcal toxins
Along with Pseudomonas aeruginosa and Escherichia coli, Staphylococcus aureus is the most frequently isolated bacteria in routine clinical laboratory hospital testing1, 158. In recent years, significant attention has been attracted to the toxins produced by S. aureus due to the wide spread of the multi-drug resistant type of the bacterium, the so-called methicillin resistant Staphylococcus aureus, or MRSA159, 160, which is often associated with high mortality161. Among numerous virulence factors, S. aureus produces a number of pore-forming toxins (PFTs) that include α-hemolysin (sometimes referred as α-toxin or α-haemolysin), γ-hemolysin, and leukocidins. The cytolytic effect of these β-PFTs has been first described more than 100 years ago when the ability of Panton-Valentine leukocidin (PVL) of S. aureus to lyse leukocytes was demonstrated162, 163. The importance of each individual toxin secreted by S. aureus varies dramatically between different strains of the bacterium. For instance, a role of the pore-forming PVL in virulence of community associated MRSA (CA-MRSA) infection has been a subject of significant debates in recent years159, 160, 164.
Staphylococcal α-hemolysin (α-HL) is released from bacteria as a water-soluble 293-amino acid monomeric polypeptide with molecular mass of 33 kDa. Upon binding to a cell membrane, it oligomerizes and forms heptameric complexes on the surface of a target cell165–170. Formation of hexameric α-hemolysin complexes has also been reported171–173. α-hemolysin oligomers demonstrate the ability to insert into lipid bilayers forming large water-filled pores that are slightly anion selective. Pioneering electrophysiological recordings on α-hemolysin pores in bilayer lipid membranes were reported by Krasilnikov and co-authors some thirty years ago171, 174 followed by further intensive studies of the properties of the channel175–180. When inserted into a planar lipid bilayer, α-HL forms a stable channel of about 1 nS conductance in 1 M KCl at room temperature. The crystal structure of detergent-solubilized heptameric α-hemolysin has been solved to 1.9 Å resolution167 showing a hollow 100 Å×100 Å heptamer. It was demonstrated that the heptamer has a mushroom-like shape consisting of the stem, cap, and rim domains (Fig. 1A). The stem part of the α-HL is a 14-stranded β-barrel made from 7 β-hairpins each contributed by an individual monomer. The cap, which together with the rim forms the core of the protein, is composed of a β-sandwich and has a diameter of ~ 100 Å1. The internal diameter of the α-HL pore ranges from ~ 6 to ~ 50 Å. Two apparent constrictions with radii of 0.9 nm and 0.6 – 0.7 nm were reported to be present in the channel lumen, the larger one being closer to the cis side. Measurements were performed using an asymmetrical (one-sided) application of water-soluble polymers, polyethylene glycols (PEGs)178 as first described in ref.128 This approach explores the ability of polymers to partition into the channel lumen and reduce its conductance in a molecular weight-dependent way69, 181–190, while reducing solution conductivity based only on their monomer concentration191. Channel dimensions and robustness determine the wide usage of this PFT in a variety of applications that are not directly related to the toxicity of this protein (see section 4).
The most interesting property of a family of Staphylococcal pore-forming cytolysins: γ-hemolysin (Hlg), leukocidin (Luk), and PVL is their bi-component structural organization. These toxins are formed as a result of interaction of two distinct polypeptides, so called class F component and class S component192. First crystal structure of a β-barrel transmembrane protein γ-hemolysin composed of two proteins was reported recently193 (Figure 4), showing an octameric pore structure at 2.5 Å resolution. The measurements with planar lipid bilayers performed on another bi-component pore-forming octameric leukocidin, Luk showed a conductance of 2.5 nS in 1 M KCl, which is more than two-fold larger compared with that of α-hemolysin72, 194, 195. At the same time, the pore diameter was estimated as 28 Å indicating that geometrically an additional subunit contributes only slightly to the pore size and conductance increase194.
Figure 4.

Ribbon representation of the 2.5Å crystal structure of bi-component octameric γ-hemolysin193. PDB ID: 3B07. Side (A) and top (B) views are shown.
2.2.2. Epsilon toxin of Clostridium perfringens
Epsilon toxin (ETX) is the major virulence factor secreted by Gram-positive, spore-forming anaerobic bacteria Clostridium perfringens types B and D196. In 2011, two excellent reviews discussing every known aspect of ETX's toxicity were published197, 198. ETX is responsible for a rapidly fatal enterotoxaemia in herbivores when their gastrointestinal tracts are colonized by this bacterium leading to in situ toxin production14, 199, 200. ETX is secreted in a poorly active form called prototoxin201 and is activated into a highly potent toxin by proteolytic removal of 11 or 13 N-terminal and 29 C-terminal amino acid residues202. The activated ETX is one of the most potent bacterial toxins after botulinum and tetanus neurotoxins203; an estimated lethal human dose is 7 μg via the intravenous route199. Due to ETX's high potency and lethality, it has been classified as a CDC category B agent. The structure of the monomeric ETX204 has a similarity to aerolysin, a 100-fold less potent pore-forming protein produced by the Gram-negative pathogen Aeromonas hydrophila (see section 2.2.3 below). ETX consists of three structural domains: N-terminal domain, which may participate in receptor binding, domain II, which is thought to contain a transmembrane stem involved in pore formation and, probably, also takes part in oligomer formation, and C-terminal domain, which likely helps to mediate ETX membrane insertion. ETX activation triggers its oligomerization in the synaptosomal membrane within the detergent-insoluble microdomains (lipid rafts) of MDCK cells205, 206. ETX was reported to form aerolysin-like204 β-barrel heptameric205–207 transmembrane pores that increase cell permeability to small molecules and ions14, 208–210. Surprisingly, in vitro, only a few cell lines such as MDCK, mouse kidney cells, and human renal leiomyoblastoma G-402 cells were found to retain susceptibility to ETX207, 209 due to the presence of specific ETX-binding receptors. Moreover, several studies have reported that ETX was not cytotoxic for sensitive cell lines at 4°C207, 208, 211–213. This finding was recently extended to provide an evidence of a prepore stage in the channel formation by the ETX213. According to the suggested model, the toxin, when bound to an uncharacterized receptor, is first assembled into a heptameric prepore on the surface of the membrane. At 4°C, the process stops at this stage; however at 37°C, the heptameric prepore significantly changes its conformation and inserts into the membrane, forming an active pore that rapidly depolarizes the membrane213. The sequence of the assembly and membrane insertion steps represent one of the most significant problems in toxicology. For instance, clear evidence of prepore formation was provided for the β-barrel channel-forming component of the anthrax toxin, PA63 and prepore's crystal structure was resolved214. However, no prepore step was identified for membrane insertion α-hemolysin, where the heptameric channel is believed to be assembled directly from the monomers inserted into the membrane.
No receptors are needed for ETX incorporation into artificial lipid bilayers215, 216 or liposomes217. In bilayer lipid membranes, ETX forms wide, slightly anion-selective general diffusion pores with a single-channel conductance in the range of 440 – 640 pS in 1 M KCl215, 216. Based on the structural and functional similarities with oligomeric aerolysin of Aeromonas hydrophila and α-hemolysin of Staphylococcus aureus, ETX was supposed to be permeable to solutes up to a molecular mass of at least 1 kDa215. Recently a polymer partitioning study to access the ETX's pore functional shape and size has been conducted58. It was shown that PEG partitioning was highly asymmetric, as revealed by the dependence of ion current through the pore on the mode of asymmetric addition of polyethylene glycols to the membrane-bathing solutions (Figure 5). The trans opening of the ETX pore allowed for penetration of much larger polymer molecules than its cis opening (Figure 6A). Therefore, the partitioning data are suggestive of an asymmetrical, e.g., conical shape of the pore with the tentative radii of the openings of 0.4 nm and 1.0 nm on the cis and trans sides, respectively. In addition, the ionic selectivity of the ETX pore was explored by measuring reversal potentials in the oppositely directed gradients of potassium chloride aqueous solutions58 (Figure 6B). As it was shown previously218, such measurements allow one to judge upon the charge distribution along the channel pore. Interestingly, the asymmetry of the reversal potential in the salt gradient was found to be opposite to what is reported for the conical nanopores with a uniformly spread surface charge219, 220. In the case of the ETX pore, the selectivity is salted-out more easily from the wide trans opening of the channel. This suggests that the residues carrying the positive charge responsible for the anionic selectivity of the ETX pore215, 216 are not localized at its cis opening but are shifted toward the trans side.
Figure 5.

A: The effect of symmetrical addition of polyethylene glycol (PEG) of different molecular weights in 15% w/w concentration on the ion current through a single ETX channel. It is seen that PEGs not only change the average conductance but, depending on molecular weight, induce significant fluctuations. B and C: Experiments performed under asymmetrical PEG additions demonstrate that these fluctuations are mostly caused by permeant PEGs added to the trans side of the membrane. Time resolution was 0.1 ms, transmembrane voltage −100 mV. Reprinted with permission from ref58. Copyright 2010. Biophysical Society.
Figure 6.

A: The relative change in ETX channel conductance as a function of PEG molecular weight. The trans and cis side applications of polymers of varying molecular weights, solid triangles and open squares, respectively, have different effects on the channel conductance. The impermeant PEG 8000 was on the opposite side of the membrane. The effect of symmetrical addition of PEG is shown by open circles. Dotted line at 0.6 corresponds to the ratio of bulk solution conductivities with and without polymers. B: Channel reversal potential as a function of the concentration ratio for two series of measurements with the oppositely directed gradients. Erev was obtained in the series of experiments where ccis was kept constant at 0.1 M KCl and ctrans was varied from 0.01 M KCl to 3 M KCl (solid circles). –Erev was measured in the reversed gradient where ctrans = 0.1 M KCl and ccis was changed from 0.01 M KCl to 3 M KCl (open squares). The channel is asymmetric: the absolute value of the reversal potential is smaller when the more concentrated solution is on the trans side of the membrane. Open triangles show the difference. Reprinted with permission from ref58. Copyright 2010. Biophysical Society.
2.2.3. Aerolysin of Aeromonas hydrophila
Aeromonads are gram-negative bacteria frequently found in aqueous environments and mainly associated with gastrointestinal diseases35. Among the variety of virulent factors secreted by these bacteria, the aerolysin is one of the best-characterized pore-forming toxins. Aerolysin (for review see221) is produced by Aeromonas hydrophila as a water-soluble inactive precursor named proaerolysin, which can exist as a dimer or a monomer92, 222. It is known, that many toxins are synthesized by pathogenic organisms in an inactive form most likely to protect the host bacterial cells from self-destruction or to improve the efficiency of their delivery to the target cells223. Proaerolysin specifically binds to glycosylphosphatidylinositol-anchored receptors on the surface of target cells224, 225. Activation of the inactive aerolysin precursors involves proteolytic removal of a C-terminal peptide226, 227. The activated aerolysin then oligomerizes and incorporates into target cell membranes forming heptameric β-barrel channels. The structure of the proaerolysin was solved by X-ray crystallography at a 2.8-Å resolution228 showing a structural similarly to the ETX monomer described above. When inserted into bilayer membranes, aerolysin makes stable, voltage-sensitive, slightly anion-selective channels229–231. Even though the pore structure of aerolysin heptamers is not yet well established, a statement about an α-HL-like organization of the aerolysin heptamer had been recently formulated221. However, the structure and effective charge of aerolysin and α-HL are significantly different (Figure 7). The electron microscopy studies of aerolysin channels demonstrated that they lack the vestibule domain228, 232, resulting in a rivet-like model of the channel92. Despite anionic selectivity possessed both by aerolysin and α-HL channels, α-HL has a slightly positive global net charge (Z = +7e) whereas aerolysin is essentially negative (Z = −52e). The aerolysin pore diameter was also reported to be smaller compared with the α-HL, while their height is about the same231, 233. Due to the distinctive properties of the aerolysin, this PFT had recently been suggested232 as an alternative to α-HL, which is traditionally used as a biological nanopore sensor to study peptide translocation, peptide-pore interaction, and protein unfolding 75, 82, 234−236. The electrical properties of the aerolysin channel in the presence of two different proteins, a wild-type maltose-binding protein (MalEwt) and its destabilized variant (MalE219), were probed in denaturing conditions in the presence of guanidium chloride232 (Figure 8). While MalE219 is completely unfolded at 0.7 M Gdm-HCl, MalEwt required 1.5 M Gdm-HCl; at that, the aerolysin pore was proved to stay stable. After the addition of unfolded proteins, the authors detected two types of ionic current blockages with different ion current amplitudes and blockage duration. One was attributed to a situation when a protein chain diffuses close to the pore (bumping or straddling event) and another one to the protein chain transportation through the channel. The unfolded proteins were transported more slowly through the aerolysin channel compared with α-HL channel, thus making aerolysin a promising biological sensor for polymer analysis.
Figure 7.
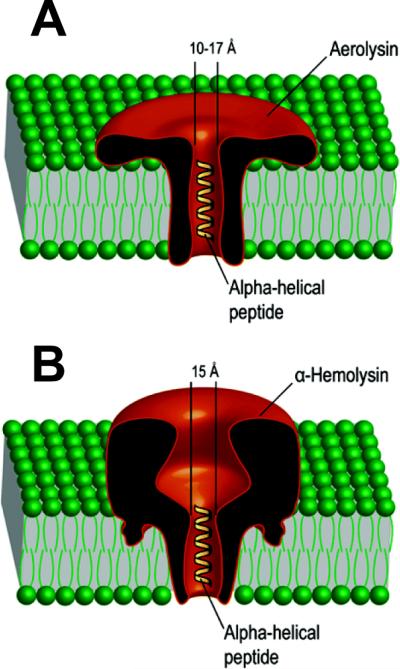
Schematic representation of the single oligomeric aerolysin (left) and α-hemolysin (right) pores incorporated into planar lipid bilayers with linear α-helical peptides traversing the channels. Adapted with permission from ref234. Copyright 2006. American Chemical Society.
Figure 8.

Single-channel current recordings showing unfolded protein transport across the aerolysin pore as a function of applied voltage: V = 80 mV (middle), V = 110 mV (top), and V = 130 mV (bottom), for the wild-type protein MalEwt (black), and mutant protein, MalE219 (blue). The stem domain of the aerolysin pore was on the cis side of the lipid bilayer, and the proteins entered by the stem side. Analysis of the current tracks (top) shows the difference between bumping or translocation events. An increase in the applied voltage results in the increase of the frequency of single channel ionic current blockades (middle and bottom). Reprinted with permission from ref.232 Copyright 2011. American Chemical Society.
2.2.4. Vibrio cholerae cytolysin
Recently resolved 2.9-Å crystal structure of another member of PFTs family, heptameric237Vibrio cholerae cytolysin (VCC) boosted interest to this toxin (Figure 9)238. High degree of structural similarity with α-HL167 was observed; at the same time, these two toxins display a rather weak sequence similarity (~15%). The oligomeric VCC was purified and crystallized in the presence of detergent, which allowed for determining the structure of the membrane-embedded oligomeric configuration of this toxin238 – one of the main challenges in toxin's crystallography. Previously the same group resolved a 2.3-Å structure of the VCC water-soluble monomer239. As a result, VCC provides one of just a few examples of β-PFT for which both soluble and membrane-assembled structures are resolved238, 239, which provides an excellent base for investigation of the intermediate steps of VCC oligomerization and membrane assembly238. X-ray crystallography revealed one interesting structural detail of the VCC pore – a narrow constriction region formed by an unexpected aromatic tryptophan W318 ring of residues within the pore that is rich in charged amino acid residues238. Authors compare this region with the famous phenylalanine clamp (ϕ-clamp) of the β-barrel PA63 component of anthrax toxin240 (see section 2.4.1). However, in the case of binary toxins, such as anthrax and clostridial C2, the ϕ-clamp was shown to be essential in channel-mediated translocation of the enzymatic components of these toxins. So far, there is no evidence indicating that VCC serves as a transmembrane protein translocase238. The ability of VCC to form channels was probed with the planar lipid bilayers241–246. The most interesting feature observed was a superlinear dependence of the rate of VCC channel formation on the fraction of cholesterol in the both monolayers of the membrane244, 246. Remarkably, methyl-β-cyclodextrin (MβCD), which removes cholesterol from membranes, rapidly inhibited formation of the VCC pores, even when MβCD was added to the side opposite to VCC addition244. This cholesterol-dependence, however, was not observed in an earlier study247, which may or may not be explained by the difference in the planar lipid membrane techniques used, namely, the monolayer opposition versus painted membrane techniques (see section 1.3).
Figure 9.

Ribbon representation of the 2.88 Å crystal structure of heptameric Vibrio cholerae cytolysin238. Side (A) and top (B) views are shown. PDB ID: 3044.
Note that VCC is distinct from the main virulent factor of the human pathogen Vibrio cholerae, which is cholera toxin. The involvement of VCC in the pandemic of this devastating disease remains unclear238. However, keeping in mind that Vibrio cholerae is widespread in many parts of the globe and responsible for thousands of deaths every year238, 248, it is important to focus on the secondary factors of virulence, such as the membrane-perforating toxin, VCC. It is not unusual when evolution brings bacterial toxins of secondary importance into the forefront. The binary CDT toxin secreted by hypervirulent Clostridium difficile pathogen is an excellent present-day example249–253.
2.2.5. Cholesterol-dependent cytolysins
The cholesterol-dependent cytolysins (CDCs) (for review see refs.9, 254–256) belong to a large family of PFTs that have been identified in five different genera of Gram-positive bacteria including Clostridium, Bacillus, Streptococcus, Listeria, and Arcanobacterium1, 256, 257. Up to now, 20 members of the CDCs family have been discovered that include perfringolysin O (PFO) from C. perfringens, streptolysin O (SLO) from Streptococcus pyogenes, pneumolysin from S. pneumonia, and listeriolysin O (LLO) from Listeria monocytogenes. The so far identified CDCs share a high level of amino acid sequence homology (40–80%), which suggests a certain degree of similarity in their structural and functional properties12, 256. The pore-forming mechanism of the CDCs exhibits two unique features: an absolute requirement of the presence of cholesterol in a membrane and formation of very large multimeric transmembrane pores. Note that even though the rate of channel formation by Vibrio cholerae cytolysin (section 2.2.4) and by several other toxins in planar lipid bilayers was shown to be cholesterol-dependent, they do not belong to the CDC family of toxins. The CDC pores are currently the largest known toxin pores. CDCs associate with the cholesterol-enriched membrane domains118,119. It was initially suggested that cholesterol acted as a receptor for a CDC binding, however, eventually it was shown that the exact step at which cholesterol is required (cell surface binding, oligomerization or membrane insertion) can vary between CDCs12. Thus, perfringolysin O can indeed bind to cholesterol directly258, however cholesterol is not a receptor for listeriolysin O and intermedilysin CDCs. Cholesterol is still required for pore formation by these two CDCs. The second hallmark CDC feature mentioned above is the ability to form extraordinary large pores. The CDC pores are composed of up to 50 monomers, though the number is somewhat variable259 and more often ranges between 30 and 40 monomers254, and can achieve about 480 Å in diameter1, 12. It is fascinating that the CDCs not only form oligomers with significantly larger number of identical subunits compared with the other β-PFTs, but also that each monomer contributes two β-hairpins to the transmembrane β-barrel channel259, 260. This structural arrangement leads to the unique β-barrels composed of up to 200 β-strands261.
X-ray crystal structures of several monomeric CDCs in a water-soluble form are currently available262–267. As was predicted from the sequence similarity, the CDCs share a similar global structure12. CDCs are elongated molecules composed mostly of β-sheets and divided into 4 distinct domains where domain 3 provides the segments that form the two transmembrane β-hairpins. The numerous electron microscopy studies of CDC oligomers revealed pores of 240 to 480 Å in diameter (Figure 10)268, that are big enough to allow the passage of large macromolecules. The pore-forming mechanism of the CDCs has been an object of intense studies and debates for the past two decades (recently reviewed in ref.254). The initial interaction of the CDC molecules with the membrane surface is mediated by hydrophobic loops on the tip of domain 4269–274. CDC membrane binding initiates changes in the monomeric CDC, leading to the formation of intermolecular contacts between different membrane-bound monomers275. The oligomeric complex continues to expand by incorporating multiple additional monomers up to the point when it is locked to a ring-shaped structure. This structure is usually referred to as a CDC “prepore complex” that has not yet embedded into the bilayer membrane as a β-barrel channel. Prepore to pore transition of CDC requires significant structural changes but proceeds in a cooperative and rapid manner as was visualized by electron microscopy276. In vitro electrophysiological measurements with CDCs are quite limited but those performed provide an interesting insight into the pore's physical properties277–280. In particular, perfringolysin O (PFO), one of the most studied members of the cholesterol-dependent cytolysin family, has been shown to form channels in planar bilayers277. PFO was found to increase the ion current through a lipid membrane by a number of discrete stepwise changes in current. These current steps were associated with the consecutive insertion of the large preassembled pore complexes into the bilayer. No small conductance patterns were ever recorded. At low PFO concentrations, when only a small number of channels were present, the conductivity values did not show the insertion of small channels growing into larger channels35. This study allowed the authors to support one of the two existing models of the cytolysins pore formation (Figure 11), namely, oligomeric prepore to pore transition263versus a continuous growth model281. Electrophysiological properties of the pores formed by another member of the CDC family, pneumococcal toxin pneumolysin in the membranes of nucleated cells were evaluated using a patch clamp technique278. Both the wild type pneumolysin and the lytic-deficient pneumolysin mutant, W433F, were studied to investigate if the lytic deficiency correlates with the absence of pore-forming capability. In contrast to the PFO study discussed above, the authors reported that a spectrum of differently sized channels was observed both with the WT and W433F pneumolysin.
Figure 10.

Perforin pore structure. A: Negative stain and cryo-electron microscopy images of perforin-containing liposomes. Surface (B) and cut-away (C) views of a cryo-electron microscopy reconstruction of the perforin pore with 20-fold symmetry. The map resolution was 28.5 Å. Reprinted and modified with permission from ref268. Copyright 2002. Nature Publishing Group.
Figure 11.

Two models of membrane insertion for the cholesterol-dependent cytolysins. A: the prepore model263 for the assembly and insertion of CDC. B: continuous growth model281. The number of monomers comprising the multimer is designated as (n). Reprinted with permission from ref277. Copyright 2000. American Chemical Society.
2.3. AB-type bacterial toxins
In contrast to the PFTs, a fundamental property of intracellularly active bacterial toxins, such as AB-type toxins, is that the enzymatic A domain or component has to be specifically delivered across the cell membrane into the cytosol of target cells7. AB toxins are secreted by a variety of bacterial pathogens in two forms. First, single-chain AB toxins can be comprised of two connected parts: part A, or an active enzymatic domain responsible for targeting the specific substrates in the cytosol, and part B, or a binding domain, which docks to certain cell surface receptors. The single-chain AB-type proteins will be reviewed in this section. The second form of AB-type toxins is represented by the binary toxins (reviewed in section 2.4) where the active and binding components are secreted as non-linked individual proteins. It is noteworthy that the B components of the binary toxins not only bind to the cell surface but also serve as receptors for the enzymatic A components. Moreover, following receptor-mediated endocytosis, B components form oligomeric transmembrane channels that facilitate translocation of the A components into cytosol of a target cell. The role of the B domain in transport of the single-chain AB-type toxins is not so obvious, namely it is not always clear if intracellular trafficking of these toxins involves formation of ion channels in target cell membranes. However, there are a number of single-chain AB-type toxins, such as botulinum neurotoxin, BoNT of Clostridium botulinum and diphtheria toxin, DT of Corynebacterium diphtheriae, for which channel formation was documented. Therefore, in this section of the review, we will focus on these two single-chain AB toxins. Investigation of the AB toxin intracellular transport is related to one of the most important problems in cell biology – understanding the mechanisms of protein transport across bilayer membranes. Insights into this process that constitutes a crucial intoxication step could provide the lacking knowledge needed for antidote/antitoxin discoveries282.
2.3.1. Diphtheria toxins of Corynebacterium diphtheriae
Diphtheria toxin (DT) is a highly efficient toxin secreted by toxigenic strains of Corynebacterium diphtheriae bacterium as a single-chain protein1, 283. It is the major virulent factor of diphtheria. It was estimated that a single DT molecule is enough to kill a cell, which makes the diphtheria toxin one of the most toxic proteins identified284. DT destroys human and animal cells by inactivating elongation factor 2, EF-2, which is an essential protein of the translocation machinery1. As many other bacterial toxins, DT is secreted in an inactive form that needs to be activated by proteolysis to be able to cross the cellular membrane285–287. X-ray crystallography studies on DT demonstrated that this single-chain protein consists of three distinct domains, each domain responsible for a specific biological function (Fig. 1B)288–294. The N-terminal catalytic, or C-domain catalyzes the NAD+-dependent ADP-ribosylation of EF-2, which completely shuts down protein synthesis and kills the cell295. The C-domain consists of both α-helices and β-sheets. The part of the protein corresponding to a fragment B of this AB-type toxin carries both the T- and R-domains289, 296–298. The A- and B-fragments of the DT toxin are connected by a disulfide bonds, and their reduction is important for the C-domain transport across the membrane. The central translocational, or T-domain (entirely α-helical) mediates protein translocation across the cell membrane. The C-terminal receptor-binding, or R-domain is rich of the β-sheets, which allows it to adopt a β-barrel-like conformation. The R-domain acts as the receptor-binding domain to the toxin molecule interacting with a 20 kDa heparin-binding epidermal growth factor-like precursor hb-EGF299. As with many other intracellularly acting toxins, this binding triggers receptor-mediated endocytosis, the mechanism of which is not completely understood as of yet300. It is generally accepted, that the acidification of the early endosomes triggers the unfolding of the transmembrane translocation T-domain283, 301, 302 followed by its incorporation into the endosomal membrane.
The precise mechanism for the catalytic domain translocation across the early endosomal membrane is still debated. The discussions mainly swing between two possible scenarios of C-domain transport. The first hypothesis suggests that the C-domain of DT is threaded through the channel by a process, which is mediated by the Cytosolic Translocation Factor complex303, 304. The second one assumes that the internal chaperone-like activity of the partially unfolded channel-forming T-domain facilities the transmembrane delivery of the C-domain303, 305. Somehow or other, it is widely accepted that the formation of the transmembrane channels by the T-domain of DT is a critical step mediating C-domain trafficking283. Furthermore, the transport of the catalytic domain is believed to be followed by the disulfide bond reduction (the famous “weak link” in biology) between fragments A (C-domain) and B (T- and R-domains), which leads to the release of the C-domain into the cytoplasm283.
The first planar lipid bilayer measurements on DT channels suggested that a transmembrane pH gradient was required to facilitate C-domain transport306–308. The diameter of the pore was estimated to range between 18 and 22 Å. Studies with asolectin vesicles showed that no additional proteins or factors were needed for the C-domain transmembrane trafficking: DT was able to deliver its catalytic domain across the bilayer in a pH-dependent manner309. It is remarkable that not only the full-length toxin, but also the T-domain alone and a mutant lacking the receptor-binding R-domain were shown to form channels in planar membranes under conditions of low pH (below 6) at the side of protein addition306, 307. Moreover, the transmembrane channel formed by a T-domain in planar lipid bilayers was shown to be fully functional, mediating translocation of the entire catalytic domain along with about 70 residues of the N-terminus of the T-domain across the membrane (Figure 12)305. The study was performed using DT labeled with an N-terminal His (H6) tag in the presence of Ni2+ (which binds to polyhistidine) in the trans compartment (opposite to DT addition). Alternatively, the authors used trans streptavidin addition when a residue near the 6 histidines was biotinylated. Ni2+ or streptavidin addition inhibited the rapid closure of the DT channels. These results indicated that the H6 tag had been translocated from the cis to trans side of the membrane. Since no additional cellular components or even the R-domain of the toxin were used, this fascinating study clearly demonstrated that the T-domain contains all of the required translocation machinery. However, the autonomous versus the facilitated mechanisms of DT catalytic domain delivery to the cytosol are still under discussion (recently reviewed283). The main argument against the autonomous mechanism involves the notion regarding limitations of the planar bilayer technique, where influence of the numerous membrane-associated proteins, known to serve as mediators of endocytosis and vesicular trafficking, cannot be directly examined283. Still, we believe that the planar lipid membrane approach is a powerful technique, which enables direct evaluations of the intermolecular forces involved in translocation processes and interactions of proteins with small molecules and other proteins.
Figure 12.

Schematic representation of the diphtheria toxin transmembrane domain incorporated into the endosomal vesicle membrane, which results in the formation of a transmembrane pore. Reprinted with permission from ref283. Copyright 2011. MDPI AG.
Indeed, several consecutive studies exploring the DT channel in planar bilayers contributed significantly to the current understanding of the catalytic domain intracellular transport310--327. One example includes an elegant study where a number of subunits of the T-domain of diphtheria toxin composing the channel were determined328. The paradigm addressed in that work dealt with the fact that the T-domain contributes only three transmembrane segments; however, the channel is permeable to ions as large as glucosamine+ and NAD−. To determine if the T-domain can form oligomeric channels in planar membranes, mixtures of two T-domain constructs with distinct voltage-gating characteristics were tried (Figure 13). One of the constructs contained an N-terminal H6 tag that blocked the channel at positive voltages. The other one had an H6 tag at the C-terminal end. If the channels could be assembled from multiple T-domain subunits, the authors expected to see a population of single channels that are blocked both at positive and negative voltages. The possibility of oligomer formation was completely ruled out since the observed single channels were blocked at either negative or positive voltages but never at both.
Figure 13.

Gating characteristics of the different T-domain constructs studied on a single-channel level 328. A: A single channel formed by wild-type T-domain (lacking H6 tag) remains open at both +60 and −60 mV, with irresolvably brief flickerings to a zero-conductance closed state. B: A single channel formed by T-domain with an N-terminal H6 tag remains open at +65 mV but rapidly closes to zero conductance at −65 mV. C: A single channel formed by T-domain with a C-terminal H6 tag spends a good deal of time in the zero-conductance closed state at +65 mV and remains open at −65 mV like the wild-type channel. D: A single channel formed by T-domain with both N- and C-terminal H6 tags is blocked at both +65 and −65 mV. At positive voltages, the channel fluctuates rapidly between the open and closed states, spending about half of its time in each. At negative voltage pulses, the channel remains open briefly, before fully closing for the duration of the pulse. Reprinted with permission from ref328. Copyright 2001. The Rockefeller University Press.
2.3.2. Botulinum neurotoxin of Clostridium botulinum
Clostridial bacteria C. botulinum and C. tetani produce two very potent neurotoxins, BoNT and TeNT that cause serious neurological disorders, botulism and tetanus2, 14. Both toxins were reported to form similar ion channel in the planar lipid bilayer membranes. Here we focus on the better studied BoNT channels (recently reviewed282, 329). The BoNT is the most toxic protein identified so far, which is characterized as a category A agent by the CDC. Moreover, scientific interest to this toxin is explained by its wide use in cosmetic industry for facial esthetics330, 331 and by the growing number of its applications in medicine332, 333. Just as the DT, BoNT is an AB-type single-chain protein with little proteolytic activity. It is activated to a dichain protein that is linked by a disulfide bond with AB structure-function properties2, 334. The N-terminal catalytic A-domain (light chain) is a ~ 50 kDa zinc metalloprotease335; the ~ 100 kDa C-terminal B domain (heavy chain) is made of two functional domains that are required for the receptor recognition and A-domain translocation across the endosomal membrane2, 334. As mentioned above, receptor-mediated endocytosis is an important step in intracellular trafficking of the AB toxins. While in cytosol, the BoNT proteases target their cytosolic SNARE (soluble NSF attachment protein receptor) substrates336–338 that form a coil-coil, which underlies the assembly of the synaptic fusion core complex important for synaptic vesicle assembly282. Cleavage of the SNARE components by BoNT disrupts membrane fusion and neurotransmitter release282. The heavy chains of the toxins were shown to form tetramers339 and to insert into the lipid membranes, forming cation-selective channels340 permeable to small molecules (< 700 Da)337. The mechanism of BoNT translocation is still not completely understood14. However, the essential molecular details of the mechanism underlying BoNT translocation across endosomal membranes were obtained from single-molecule studies in planar lipid bilayers340–348. The nicked BoNT molecule is believed to act as a nanomachine349–353 where the B-domain formed by the heavy-chain fragment acts as a specific protein-translocation chaperone for the light chain protease282, 342–345, 351, 354, 355. Translocation of the BoNT light-chain catalytic domain by the heavy chain was observed in real time using excised patches of BoNT-sensitive Neuro 2A343, 345 neuroblastoma cells. It manifested itself by a progressive increase in membrane conductance: the channel formed by the B component of BoNT was transiently blocked by the catalytic domain and then unblocked after completion of translocation and release of the light chain fragment282. Maintaining the right condition, namely mimicking the pH gradient across the endosome (acidic inside and neutral in the cytosol) and inside-positive transmembrane potential was the central requirement triggering the translocation.
The suggested consequence of events underlying BoNT transport across the membrane is shown in Figure 14282, 345. Step 1 demonstrates free BoNT represented by its crystal structure351. The catalytic light chain and translocation- and receptor-binding fragments of the heavy chain are shown in purple, orange, and red, respectively. Step 2 illustrates a schematic representation of BoNT inserted into the membrane during an event of entry where the enzymatic fragment (purple) is trapped within the channel formed by the heavy chain. A series of further steps of the light chain trafficking (3–4) and release (5) are shown. Remarkably, each step could be associated with the high-resolution real-time consecutive single channel recordings. Similarly to the DT transport, the disulfide bridge (shown in green) connecting the light and heavy chains of the toxin is stable at the low pH oxidizing environment of the cis compartment of the bilayer chamber that mimic the acidic endosomal pH. However, the disulfide bond is reduced by a reductant pH in the trans compartment (mimicking the pH of the cytosol), which promotes the release of the light chain. The authors observed the progressive stepwise increase in originally quite low single channel conductance (Fig. 14, current tracks from left to right). After the enzymatic fragment translocation is complete, the channel is free and shows a high (~66 pS) conductance value (step 5) typical for the heavy chain BoNT channel formed by the B-domain only342, 343. The half-time for the completion of one individual event of light chain translocation was found to be ~10 s. Numerous additional details of BoNT intracellular transport studied on a single channel level by Montal's group were recently reviewed and we address a zealous reader to these publications282, 329.
Figure 14.

Sequence of events showing BoNT light chain translocation through the heavy chain channel. Step1. BoNT/A holotoxin prior to insertion in the membrane (grey bar with magenta boundaries); BoNT/A is represented by the crystal structure rendered on YASARA using PDB accession code 3BTA351. Step 2. Schematic representation of the membrane inserted BoNT/A during an entry event. Steps 3 and 4. A series of transfer steps. Step 5. An exit event. Segments of typical single channel recordings are displayed under the corresponding interpretation for each step. Reprinted with permission from ref282. Copyright 2009. Elsevier.
It is clear that the planar lipid bilayer studies were able to provide an important insight into the AB toxins translocation mechanism contributing significantly to our understanding of the molecular events underlying protein-mediated protein transport. Several attempts to explore these processes using single-chained AB toxins were performed. One study reported measurements on a Pseudomonas exotoxin A secreted by Pseudomonas aeruginosa, where the burst-like single channel events were seen356. Other studies include planar lipid bilayer experiments with two major toxins of Clostridium difficile: Toxin A357, and Toxin B358. Membrane insertion of the both toxins was shown to be facilitated by a low solution pH and in the case of Toxin A turned out to be cholesterol-dependent. Despite of the evident interaction with the lipid membrane manifested by the increased ion-current noise and membrane conductance, no well-defined conductance steps representing consequent insertion of the individual channels were recorded.
2.4. Binary toxins
Certain pathogenic species of Bacillus and Clostridium families employ a unique and refined way for targeting mammalian cells – they produce binary exotoxins, which are composed of two separate non-linked proteins, an active/enzymatic A component and a binding/translocation B component (for detailed reviews see refs.3, 13, 37, 38). In contrast to single-chain AB toxins, A and B components of the binary toxins are secreted as individual unbound proteins. Each component itself does not exhibit toxic effect; however, together the A and B components are cytotoxic. Note that as an exception from the rule, the binding/translocation B component of clostridial Iota toxins was shown to produce cytotoxic activity through necrosis with certain cell lines359. To gain access for their A components to the cytosol, all binary toxins rely on a similar cellular uptake mechanism (Figure 15). The B component of these toxins binds to a receptor on the surface of the target cells, self-assembles to form a ring-shaped oligomeric prepore (usually heptameric) able to bind the A components, and, after receptor-mediated endocytosis, is converted into an ion-conductive pore, which mediates A component translocation from acidified endosomal vesicles into the cytosol. Remarkably, the binding/translocation B components are structurally conserved between the Bacillus and Clostridium families. They share a high level of amino acid homology and numerous functional similarities 13, whereas the enzymatic A components of these toxins are quite distinct and target different cell functions.
Figure 15.

Basic mechanisms of cell intoxication by Clostridium and Bacillus binary toxins. Cell-binding B-precursors are first activated by proteolytic cleavage in solution or on the cell surface (B. anthracis PA83 only). Subsequently, activated B components interact with specific cell surface receptors as either preformed ring-shaped heptamers or monomers that form heptamers on the cell surface. The enzymatic A components bind to the cell-associated B heptamer, and the receptor-toxin complex then undertakes receptor-mediated endocytosis. An acidic endosomal environment is essential for translocating the A components into the cytosol. Reprinted with permission from ref3. Copyright 2004. ASM Press.
2.4.1. Anthrax toxin of Bacillus anthracis
Recent progress made in understanding of anthrax toxin intracellular translocation is beyond any comparison. To the large extent, this was due to the well thought-out and elegant experiments performed with the planar lipid membranes, where protein translocation can be directly electrically monitored and managed, and the pH and transmembrane voltage can be precisely controlled (recently reviewed in refs.37,50, see also the subsequent publications46–49, 360–365). Anthrax toxin, as a member of the bacterial AB exotoxin family, consists of three proteins that self-assemble at the surface of the cell. It is comprised of two enzymatic A components: Lethal Factor (LF), a Zn-metalloprotease that cleaves MAP kinase kinases and induces the cell death of macrophages, and Edema Factor (EF) (sometimes named Oedema Factor, or OF), which is a Ca2+- and calmodulin-activated adenylyl cyclase366–369, and one translocation/binding B-component (83 kDa Protective Antigen, or PA). Note that the name of “protective antigen” originates from its use as an active component of anthrax vaccine and does not refer to any protective properties of this protein in the course of anthrax toxin intoxication. Because instead of one A and one B component, Bacillus anthracis secretes three individual factors (two enzymatic and one binding), anthrax toxin, being a member of binary toxin family, is often referred to as a tripartite toxin. X-ray crystallography of the channel-forming B component of anthrax toxin shows that PA contains four distinct domains involved in cellular receptor binding, oligomerization, pore formation, and A component binding214, 370. Following proteolytic activation by a furin-like protease of the host cell371, 372, the truncated B component, PA63, forms ring-shaped oligomers, so called prepores, on the surface of eukaryotic cells or in solution (Figure 16). The population of oligomeric PA63 prepores for a long time was believed to be exclusively 7-fold symmetrical, or heptameric214; however, formation of 8-fold symmetrical, or octameric forms both in solution and on cell surfaces was recently discovered373 and investigated44, 364, 365. Once assembled, the oligomeric (PA63)7 and (PA63)8 prepores can bind several copies of LF and EF45, 374 and undergo endocytosis being transferred into an acidic compartment of the intracellular endosome. As discussed above, the last step is critical for the toxicity of anthrax toxin375 as well as for several other intracellularly acting toxins. Under the acidic endosomal environment, the PA63 prepore endures substantial structural changes that allow it to embed into the endosomal membrane, forming an elongated mushroom-like cation-selective channel. The protein wall of the channel forms a single tunnel, a water-filled pore that connects solutions on both sides of the membrane. The mushroom-like (125 Å diameter and 70 Å long cap, and 100 Å long stem) membrane-spanning (PA63)7 pores were seen by the negative-stain electron microscopy376 (Fig. 1C, right). Instead of being a passive tunnel, PA then acts as an effective translocase, which, using the proton gradient across the endosomal membrane (pHendosome < pHcytosol), unfolds and translocates LF and EF into the cytosol of the target cell (Figure 17).
Figure 16.

Ribbon representation of 3.6 Å crystal structure of heptameric prepore of channel-forming component of anthrax toxin, PA63.607 Side (A) and top (B) views are presented. PDB ID: 1TZN.
Figure 17.
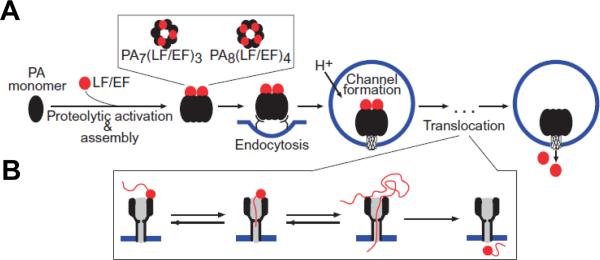
A: Schematic representation for anthrax toxin receptor-mediated cell assembly and entry into a target cell50. First, proteolytically activated PA monomers (PA63) oligomerize into the ring-shaped heptameric, (PA63)7, or octameric, (PA63)8, complexes, which are able to dock 3 or 4 enzymatic A components, LF and/or EF, respectively. These oligomeric prepores are then endocytosed and, under the acidic endosomal pH, converted to the transmembrane channels, which transport LF and EF into the cytosol. B: A possible protein unfolding and translocation pathway for anthrax toxin depicted in three successive steps: docking, protein unfolding, and translocation of the unfolded chain. Adapted with permission from ref.50 Copyright 2011. Wiley.
The peculiar molecular details of the PA63 channel acting as the translocase have emerged as result of its intense studies in bilayer membranes, where anthrax toxin intracellular transport was characterized as ΔpH-driven Brownian-ratchet mechanism48, 49, 377, 378. Briefly, LF transport across the PA channel was directly observed by monitoring the resumption of ion current, originally reduced by channel occlusion by LF, after LF was translocated43, 44, 240, 377, 379, 380. Moreover, the translocation of LF was proven to be initiated by entry of its N-terminus into the PA63 channel380 and driven through by either transmembrane potential379 or by proton gradient377 across the membrane. Most importantly, the transport of LF through the PA63 channel, and, as a result, the anthrax toxin's toxicity was shown to be significantly suppressed by mutating phenylalanine residues at position 427240. In general, the seven F427 residues are believed to form a narrow constriction region inside the channel lumen (ϕ-clamp) that acts as a translocase active site crucial for the A components transport.
To address the fundamental question of the translocation driving force, a charge state-dependent Brownian-ratchet model was suggested377. The PA63 channel is known to be preferentially selective to cations. Therefore, acidic residues in the translocating polypeptide are expected to protonate when entering the channel to avoid being repulsed by negatively charged groups in the lumen of the channel cap381. Right after the protonated acidic groups reach the cytosolic part of the membrane (or trans side of the bilayer membrane, where higher pH value is intentionally maintained), they deprotonate becoming negatively charged again. As a result, the negatively charged LF chain, emerging from the channel into the cytosol, should be electrostatically repulsed from the cation-selective channel, which carries negatively charged residues in the lumen. The authors of the model suggest that this electrostatic repulsion in the presence of Brownian motion drives the translocation per se and enforces its directionality50, 377. Remarkably, recent experiments with planar lipid bilayers have imparted support to this model using semi-synthetic variant of LFN(12–263), in which selected acidic residues were replaced with the unnatural amino acid, cysteic acid. This acid has a negatively charged side chain protonated only at pH values below the physiological range362. Depending on the number of acidic residues mutated, transport of these variants was either significantly suppressed or completely inhibited, whereas their binding and channel-blocking properties were comparable with those of WT LFN. In another study, when an essentially non-titratable negatively charged SO3− group was introduced at most positions in LFN, the voltage-driven LFN translocation was drastically reduced382.
To find out if the secondary structure of LF is preserved during the PA mediated transport, a method of trapping the polypeptide chain of a translocating protein within the channel was developed47. By attaching biotin to the N terminus of LFN and using molecular stoppers at different positions, the authors determined the minimum number of residues that could traverse the channel. Streptavidin added to the trans side of the bilayer chamber was used as a probe. If the N terminus – stopper distance was long enough for LFN to emerge from the channel, streptavidin was able to bind to the biotin. If the distance was not long enough, no biotin binding was recorded (Figure 18). The conclusion that the polypeptide chain can adopt a fully extended conformation as it translocates through the channel's stem was an instructive result of this elegant study. A kinetic analysis of protein transport via the PA63 channels, performed both in macroscopic and single-channel experiments, shows that the kinetics of channel-mediated translocation of a single LFN protein molecule are S-shaped49. A simple drift-diffusion model of LFN transport was also reported49. In this model, LFN is considered as a charged rod that translocates through the channel being governed by the combined influence of random thermal motion and an applied transmembrane electrical field.
Figure 18.

Representative example of a truncated H6-LFN construct (LFN 1–83), whose N terminus reached the trans solution47. The N-terminal H6 tag is depicted in blue. The recording starts at the moment when an appropriate level of PA63-induced current is reached and the cis compartment is perfused to remove the unbound (PA63)7 from the solution. The voltage protocol of + 80 mV for 5 s and −40 mV for 15 s was then applied. At the first arrow, the LFN (1–83), with the YFP stopper (depicted as a β barrel) attached to the C-terminus and biotin (orange) attached at residue 1, is added to the cis solution, and a constant level of unblocking is obtained. At the second arrow, streptavidin (four green balls) is added to the trans compartment. A dramatic decrease of unblocking over time is recorded, thus demonstrating that the biotin at residue 1 has reached the trans solution and has been grabbed by streptavidin, thereby preventing those channels from becoming unblocked at −40 mV. Reprinted with permission from ref47. Copyright 2011. The Rockefeller University Press.
Two types of PA63 channels insertions with slightly different conductances were reported in several recent publications57, 373. The observation was explained by formation of both the heptameric and octameric channels in planar lipid membranes44, 45, 364, 365, 373, 383. However, the channels of lower conductance, when studied on a single-channel level, were noticed to exhibit the spontaneous reversible stepwise transitions to a substate of higher conductance (also observed in ref.384) that exactly corresponded to the conductance of the higher conductive channels57. Kinetic characteristics of blockage of these two types of channels by seven-fold positively charged cyclodextrins were indistinguishable. It is unclear if these apparent discrepancies were caused by the different PA samples used in the above studies. In addition to the two insertion types, two types of complex non-Markovian channel gating were also reported. We will discuss these features below together with the similar observations for clostridial binary toxins (see section 2.4.2). Nonelectrolyte polymers of poly-(ethylene glycol) successfully used before to determine the diameter of several channel-forming proteins incorporated into planar lipid membranes were also used to size the PA63189 giving the PEG molecular mass cutoff of ~1400 Da, which approximately corresponds to the limiting diameter of the PA63 channel being less than 20 Å (Figure 19). This study is in a very close agreement with an all-atom model of the PA63 channel385 (Fig. 1C, left) and with planar lipid membrane measurements where channel diameter was estimated with the tetraalkylammonium ions of different size384, 386. Interestingly, PEG molecules were shown to strongly interact with the channel giving a dissociation constant of 9 mM.
Figure 19.
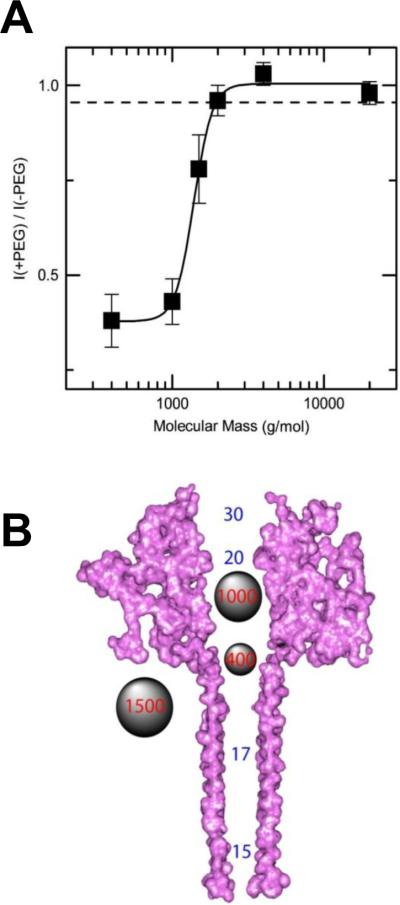
A: The molecular mass cutoff of the single heptameric PA63 channel as estimated from the effect of differently sized PEG molecules in planar bilayers189. It is seen that the PEGs with MM <2000 Da are able to enter into the channel, which results in conductance decrease. The PEG concentration was 1.2% (w/w), and the applied potential was V = +70 mV. B: A cross-sectional view of the heptameric PA63 channel model385 and spherical representations of PEG 400, 1000, and 1500 molecules. Reprinted with permission from ref189. Copyright 2008. Biophysical Society.
2.4.2. Clostridial binary toxins' B components are close analogs of the PA channel
Binary toxins secreted by a family of clostridial bacteria are closely related to anthrax toxin. They include C2 toxin of Clostridium botulinum, Iota toxin of Clostridium perfringens, CDT toxin of Clostridium difficile and CST toxin of Clostridium spiroforme, with C2 and Iota toxin showing well-defined stable ion channels when incorporated into planar bilayers. Therefore, here we will focus on these two binary toxins. In contrast to the anthrax toxin, which is a tripartite toxin formed by the two enzymatic and one binding/translocation components, clostridial C2 and Iota toxins are made of the two components only. Moreover, their A components (C2I and Ia) act through mono-ADP-ribosylation of G-actins, which causes a complete destruction of the actin cytoskeleton and caspase-dependent cell death13, 387–395. Just as with PA, X-ray crystallography of the B component of C2 toxin (C2II) has revealed four distinct domains involved in cellular receptor binding, oligomerization, pore formation, and A component binding214, 370. No crystal structure of the Iota toxin B component (Ib) is available, but there is a good reason to believe that the important structural details of PA and C2II, such as 4-domain organization, are also relevant for the Ib component. It has been shown that PA, C2II, and Ib share from 27% to 38% of amino acid homology. Remarkably, there is relatively little homology of domains 1 (A component binding) and 4 (cellular receptor binding) between PA, C2, and Ib, which is not surprising taking into account that these proteins are evolved to dock significantly different enzymatic A components and to bind to different cellular receptors. Thus, the amino acid sequence similarity of these three components is primarily localized within two central domains: domain 2, which participates in pore formation and A component translocation, and domain 3, which is important for the oligomerization of the monomers. Following proteolytic activation, the truncated B components of these toxins (C2IIa, and Ib) form ring-shaped heptamers – so called prepores on the surface of eukaryotic cells or in solution396. In contrast to the PA oligomer, formation of the 8-fold symmetrical C2IIa and Ib oligomers was not reported. A model structure of the (C2IIa)7 prepore was constructed based on the corresponding structural assembly of the (PA63)7 prepore214, 370. The cellular uptake mechanism of these two toxins is also similar to that of anthrax toxin (Fig. 15) with several distinctions reviewed elsewhere3, 13. Following the A component binding, the cell-bound C2I/C2IIa and Ia/Ib complexes are internalized by receptor-mediated endocytosis397–400 and reach endosomal vesicles where C2I and Ia translocate across the endosomal membranes into the cytosol using the C2IIa and Ib pores as translocation corridors396, 400–405. Interestingly, in vitro planar bilayer measurements showed that PA63 is able to bind C2I, whereas C2II binds both EF and LF406. The authors also reported that PA, but not C2II, has the ability to transport the non-native enzymatic component into target cells, where it causes actin modification and cell rounding.
In mildly acidic conditions, the B components of C2 and Iota toxins form ion-permeable, cation-selective oligomeric channels in planar lipid membranes401, 402, 407, 408. It is unclear to what extend the C2IIa and Ib channels serve as the active translocase of the A components, similarly to the PA function discussed above. However, the preserved phenylalanine clamp (ϕ-clamp) in position 428 was found to catalyze the unfolding and translocation of the C2I component moieties across the membrane409, 410. The Phe residue at the corresponding position is also conserved in Ib410, but the importance of the ϕ-clamp for the Iota toxin transport is not clear as of yet.
It is interesting to compare the salt concentration dependence of the PA, C2IIa, and Ib channel conductance because this dependence, together with ionic selectivity of the channel, may report on the sign and effective number of fixed charges inside the channel pore. Note that almost ideal cationic selectivity determined by the negative charges inside the PA63 channel lumen and, therefore, the requirements for the acidic residues of LF being protonated in order to pass through the channel, were persuasively used in the Brownian ratchet model of the PA-mediated LF transport (see discussion above)37, 47, 48, 363, 377, 378, 382. As shown in Figure 20A, conductances of the PA63 and C2IIa channels, in accordance with earlier studies384, 401, 411, demonstrate non-linear dependence on salt concentration in the membrane-bathing solution, which is characteristic of charged pores, whereas the Ib channel exhibits close to linear behavior. The data on the reversal potential measurements for the three channels in changing KCl concentration gradients are shown in Figure 20B. Two features are clearly seen: (i) the channel cationic selectivity drops in the sequence PA63 < C2IIa < Ib, and (ii) all three channels exhibit similar asymmetry. The channel selectivity is “salted out”218 more efficiently from the trans side of the channel, which corresponds to the neutral-pH cytosolic side. The channel selectivity is systematically smaller when the salt concentration is higher at the trans compartment of the reconstitution chamber. Interestingly, the ability of small molecules carrying one or two positive charges to block ion current through the PA63, C2IIa, and Ib channels was shown earlier240, 384, 386, 402, 407, 410, 411 (see section 3.2.1 below) with the equilibrium binding constants decreasing in the order PA63 > C2IIa >> Ib. The difference in the affinity of cationic blockers towards these channels was earlier attributed to the decreasing number of negatively charged amino acids in the lumens of these pores407, 410. The conductance salt dependence and selectivity measurements summarized in Fig. 20 strongly support this observation. The question that remains open is if the ΔpH-driven Brownian ratchet model is still relevant for the less cation-selective C2 and Ib toxins. Remarkably, the host cell chaperone Hsp90 and the peptidyl-prolyl cis/trans isomerase cyclophilin A were shown to be crucial for membrane translocation of the enzymatic components of clostridial C2, Iota, and CDT toxins but not for LF of anthrax toxin412–415. However, cyclophilin A and Hsp90 did facilitate translocation of the fusion protein LFNDTA (where DTA is a catalytic domain of diphtheria toxins)414. These interesting observations introduce some new details in the long-lasting discussion about autonomous versus facilitated toxin intracellular delivery mechanisms.
Figure 20.

A: Conductances of the PA63, C2IIa, and Ib channels as functions of salt concentrations demonstrate different behaviors at small salts. The dependences are close to linear for Ib but show significant deviations from linearity for the PA63 and C2IIa channels. B: Single channel reversal potentials as functions of the ratio of salt concentrations on the different sides of the membrane for two series of measurements with oppositely directed gradients. Open circles: Reversal potential (Erev) obtained in the series of experiments where KCl concentration in the cis compartment exceeds that in the trans compartment; at that ctrans = 0.1 M KCl. Solid circles: The values of the reversal potential (-Erev) obtained for the reversed gradient ctrans > ccis = 0.1 M KCl. The sign of the reversal potential is inverted in the latter case to facilitate comparison; the selectivity stays cationic in both cases. The channels are asymmetric because the absolute value of the reversal potential is greater when the more concentrated solution is on the cis side of the membrane. Membranes were formed from the DPhPC at pH 6.
The salt dependences of conductance and selectivity of PA63, C2IIa, and Ib are quite different, however current noise characteristics are alike. Interestingly, the voltage-independent flickering of the channels between open and completely closed states was observed in high time-resolution recordings57, 384, 401, 407, 416. Typical ion currents through the three single channels, reconstituted into planar lipid membranes are presented in Figure 21. For all three channels, the flickering was described by the complex non-Markovian kinetics which are manifested by a 1/f-type shape of the current power spectra57. To the best of our knowledge, the 1/f fast flickering between the open and completely closed states is unique for the family of channel-forming B components of binary toxins. For instance, α-hemolysin (see section 2.2.1 of this review), which similarly to PA63, C2IIa, and Ib forms heptameric channels, does not show this type of gating417, 418 and neither do many other β-barrel pore-forming proteins.
Figure 21.

A: In the absence of blockers, ion currents through the PA63, C2IIa, and Ib single channels reconstituted into planar lipid membranes demonstrate fast flickering between the open and closed states. The currents are given at 1 ms time resolution. B: Power spectral densities of the currents shown in panel A display 1/f behavior. Measurements were taken in 1 M KCl solutions at pH 6 buffered by 5 mM MES. The applied voltage was 50 mV.
In addition, PA63, C2IIa, and Ib exhibited strong voltage-dependent gating that is observed with many β-barrel channels including artificial ones131, 132, 419. This type of gating is highly asymmetrical (more pronounced at cis-negative voltages)420 and decreases in the order PA63 > C2IIa >> Ib. After the closure at negative voltages, the channels, especially PA63, tended to stay in a low-conductive state for minutes, and in a number of cases reopening was not observed on a timescale of several hours even after the applied voltage was reduced to zero. However, second-long pulses of voltages of −250 mV often made channel reopening possible. Interestingly, the voltage-dependent gating of cysteine-substituted PA63 channels was reported to be abolished by a reaction with a methanethiosulfonate reagent that catalyzes the formation of intersubunit disulfide bonds421. This perturbation was observed with cysteines substituted at sites all along the 100 Å length of the β-barrel stem region, which allowed the authors to draw a conclusion that the channel's entire β-barrel participates in the gating process. Moreover, the PA63 voltage gating was found to be lipid-dependent57. The channels were much more sensitive to the applied voltage if reconstituted in negatively charged PS membranes compared with zwitterionic PC membranes. The rate of PA63 channel insertions was also dramatically increased in the PS membranes.
3. INHIBITION OF CHANNEL-FORMING BACTERIAL TOXINS
Discovery and development of small-molecule antitoxins represent a high-priority task in modern drug design and medicinal chemistry422, 423. A number of bacterial toxins, such as BoNT of Clostridium botulinum can be aerosolized and directly used as biological weapons. With several bacterial diseases, such as anthrax, flu-like symptoms appear only after bacteria have multiplied inside the infected organism and started to produce the toxin that eventually causes death424. Even though aggressive antibiotic therapy inhibits Bacillus anthracis bacterium growth, patients can still die because of intoxication425. Moreover, engineered strains of Bacillus anthracis resistant to the existing antibacterial agents had been already developed422, 426. Besides the formation of the native toxic complexes, several binary toxins are able to cross-react interchanging their A and B components, which creates a danger of their potential misuse406, 427.
Most importantly, a number of existing and emergent multi-drug resistant bacteria, so-called “superbugs”, such as Pseudomonas aeruginosa, Clostridium difficile, Staphylococcus aureus, and Escherichia coli secrete a variety of highly potent exotoxins with no antitoxins currently available on the market. Vaccination, clearly being one of the most significant medical achievements of 20th century, is not always available, sometimes reactogenic428, or economically unpractical. Therefore, to combat the emergent infectious diseases and to counter a terrorist's deployment of bacteria whose pathogenicity relies upon secreted toxins, stable broad-spectrum antitoxins must be stockpiled. Small molecules are especially attractive as antitoxins, since their room temperature shelf-life far exceeds that of the current solution, antisera to toxins. We will focus here on the existing efforts to design the antitoxins counteracting the channel-forming bacterial toxins.
3.1. Role of interactions in channel-facilitated transport
Theoretical modeling of ion channels has long been recognized as an important tool in the studies of channel-facilitated transport133. Progress in this direction, quite substantial already, is being currently accelerated by the newly revealed three-dimensional structures of many channels. The availability of the high-resolution structures allows researchers to address and understand the structure-function relationships in channel functioning at the atomic level429, 430. One of the most successful approaches is all-atom molecular dynamics simulations, in which not only the molecules comprising the channel structure but also ions and water are explicitly taken into account431–437. Though this methodology has brought a number of important insights in ion channel functioning, in the present review, for the sake of clarity, we will restrict ourselves to a simple model of channel-facilitated transport, namely, the continuum diffusion model438–443. This model allows for analytical consideration of the problem, leading, in a number of cases, to simple algebraic expressions for the main transport characteristics. It does not involve any sofisticated state-of-the-art molecular dynamics simulations or complex numerical solutions of electrodiffusion equations and, for this reason, is very transparent in what regards the assumptions and approximations used in its formulation.
Interactions between particular solute molecules and the channel that define its major transport characteristics can be separated into different types not only by the nature of the underlying physical forces but also by their functional role. Perhaps the simplest one is related to steric limitations. The straightforward sieving principle is that solute molecules, which are smaller than the narrowest aperture of the channel pore, are able to partition into the channel and use it as a pathway from one side of the membrane to the other. The molecules that are too big are excluded from either any part of the pore or its constriction region. However, in reality, even this simple picture is complicated by at least two factors: (i) the dynamic structure of some pores whose aperture dimensions fluctuate and are able to adjust to the solute size and (ii) the entropic cost of molecule partitioning into the pore. For a hypothetical case of a regular cylinder of radius Rch and a spherical particle of radius r the entropic cost can be described by the following potential of mean force
| (1) |
where kB and T have their usual meaning of the Boltzmann constant and absolute temperature. To enter the channel, the particle has to climb up this potential barrier, which could be quite substantial. For example, for a molecule whose radius is 10% smaller that the radius of the channel, Eq. (1) gives a barrier height of 4.6 kBT or 2.7 kcal/mol. Such a potential will deplete molecule concentration in the channel compared with its concentration in the bulk by two orders of magnitude, rendering the channel inefficient.
To overcome this limitation, channels exhibit a significant affinity to the molecules they evolved to transport. A straightforward strategy is to create some kind of attractive interactions that would extend to the aqueous solutions at the channel entrances to increase the effective concentration of the molecules. This kind of attraction was recently demonstrated with the major β-barrel channel of mitochondria – voltage dependent anion channel (VDAC) of the outer mitochondrial membrane – in the light of its interaction with dimeric tubulin444. It was shown that phosphorylation of VDAC's cytosolic loops increases the on-rate of VDAC blockage by tubulin by orders of magnitude. It is clear, however, that the strength of such transport-facilitating interactions has an optimal value. When the attraction between the molecule and the channel is too strong, the molecule stays in the channel for too long, thus blocking translocation of other molecules.
Analytical considerations of the problem in the case when the attractive interaction is limited to the channel interior was given in a series of publications based on the continuum diffusion model of particle interaction with the channel438,439–443. The model describes particle motion in the channel as one-dimensional diffusion with the position-dependent diffusion coefficient D(x), where x is the particle coordinate measured along the channel axis, and the position-dependent interaction potential U(x) of mean force. The nature of the physical forces that could be responsible for the interaction potential is briefly discussed in Section 3.2.2. Motion of the particle in the channel is characterized by the Green's function G(x,t; x0)which is the probability density of finding the particle at point x at time t on condition that it was at x0 at t = 0 and it has not escaped from the channel during time t. The Green's function satisfies the Smoluchowski equation
| (2) |
with the initial condition G(x,0;x0) = δ(x−x0) and radiation boundary conditions at the cylindrical channel ends, x = 0 and,x=L that take care of the three-dimensional nature of the problem
| (3) |
where Db is the particle diffusion coefficient in the bulk. Introduction of the effective pore radius, Reff=Rch−r, accounts for the entropic contribution discussed above. Analysis can be easily extended to non-cylindrical, e.g., conical channels by including the position-dependent entropic potential into U(x)445, 446.
This model has been used to derive general expressions for the particle translocation probability439,Ptr, and the mean time of particle residence in the channel, τ. For the case of zero external field,U(0)=U(L) (Figure 22A), we have
| (4) |
The general expression for the mean time of particle residence in the channel can be found in refs.440, 447.˙ In the case of a deep rectangular potential well of depth ΔU occupying the entire length of a cylindrical channel (Figure 22B) and a position-independent diffusion coefficient in the channel,D(x)=Dch, the expression simplifies to the following
| (5) |
Then, assuming that the channel occupied by one molecule is blocked for other molecules, for the unbiased diffusion we can write down the flux of molecules in a simple algebraic form441, 442
| (6) |
The flux in Eq. (6) is driven by the difference in molecule concentrations on the cis and trans sides of the membrane, ccis–ctrans. It is seen that the flux depends not only on the geometric parameters of the channel,Reff and L, but also on the strength of the molecule-channel attraction, ΔU, and on the molecule diffusion coefficients both in the bulk and in the channel, Db and Dch. It is important that the flux is a non-monotonic function of the interaction strength. The depth of the potential well that maximizes the flux at Ctrans= 0 is
| (7) |
This value of ΔU provides a compromise between sufficiently high translocation probability and not too long blockage of the channel, Eqs. (4) and (5).
Figure 22.

Schematic view of the particle's potential of mean force in a membrane channel. A: An arbitrary potential demonstrating different level of interaction with different parts of the channel along its axis (x coordinate). B: A hypothetical square-well potential.
The non-monotonic behavior of the flux is shown in Figure 23441, 442. The parameters are: L = 5 nm, to put it close to the thickness of a lipid bilayer, Reff= 0.2 nm, to account for the fact that many solutes exhibit a tight fit to the channel pore, and Db=2Dch=3×10−10 m2/s, to follow the idea that a molecule in the channel moves slower than in bulk.
Figure 23.
Non-monotonic behavior of the flux given by Eq. (6) as a function of potential well depth at three different concentrations of translocating molecules and = 0. Reprinted with c2 permission from ref441. Copyright 2005. Biophysical Society.
Though the analysis reviewed above pertains to translocating molecules, the results can be easily extended to the channel blockage by non-penetrating molecules. The dynamics of the molecules, whose trajectories in the channel are limited to lengthB, can be described by assuming the reflecting boundary condition at x=LB, so that the mean residence time of the molecule in the channel is twice the time given by Eq. (5) with the integration range limited to LB. Assuming a square-well potential of depth ΔU occupying the entire blocker-accessible length of the channel, we arrive at
| (8) |
The characteristic on-rate time τon, that is, the time between successive blockages, is given by
| (9) |
where c is the blocker concentration in the bulk. It is reasonable to assume that the channel is closed for translocation of any molecules of interest while it is occupied by a blocker. Then for the efficiency of blockage, defined as an inverse probability to find the channel open, PO=τon/(τon+τr), minus one, one can write
| (10) |
Defined this way, the efficiency of blockage changes from zero to infinity, grows linearly with the bulk blocker concentration, and is exponential in the well depth.
Blockage by non-translocating molecules can be considered as an equilibrium process in which distribution of the blocker molecules between the bulk and the channel is characterized by equilibrium constants and thermodynamic potentials, which generally are the functions of the transmembrane voltage, temperature, and salt concentration. The equilibrium dissociation constant of the blockage is
| (11) |
which, using Eqs. (8) and (9), leads to
| (12) |
In the hypothetical case of the temperature-independent depth of the potential well, the first term of the right-hand side of Eq. (12) allows evaluation of the enthalpy change ΔH, while the second term – of the entropy change ΔS of the reaction, according to the main equation of thermodynamic analysis of binding reactions used in the van't Hoff plots448
| (13) |
where R is the molar gas constant.
3.2. Targeting bacterial binary toxins
With the binary anthrax toxin being an exception, no extensive studies searching for effective therapies against the binary toxins have been reported. The similarities between the channel-forming B components (see section 2.4 of this review) suggest that these channels can be a specific universal target in the search for new broad-spectrum antitoxins against the Bacillus and Clostridium pathogenic species.
3.2.1. Small cationic blockers
Nearly any tested compound, which is positively charged at mildly acidic pH, was shown to interact with PA63 channels incorporated into planar bilayers. Moreover, cells were shown to be protected from lethal and edema toxin action by weak bases such as ammonium chloride or antimalarial drug chloroquine375, 420, 449. The PA63 channel is permeable for and interacting with symmetrical tetraalkylammonium ions (Table 1, compounds 1–6)384, 386, 450. The ability of tetraalkylammonium ions to reversibly block the K+ current was used to determine the physical size of the PA63 channel lumen – the value that was later confirmed with several independent approaches189, 376, 385 and used for the rational design of anthrax toxin blockers51. It was shown that the tetraalkylammonium binding site was accessible from either cis or trans compartments of a bilayer chamber, showing the ability of these ions to permeate through the channel. The tetraalkylammonium residence time as a function of transmembrane voltage had a pronounced maximum, which is typical for the permeable versus impermeable blocking compounds451–454. It was also determined that an ion as large as tetraheptylammonium, which has a diameter of ~12 Å, could translocate through the channel; there was an impetuous fall in the entry rate from tetrahexylammonium to tetraheptylammonium ions420, 450. The interaction of tetraalkylammonium ions was described as a diffusion-controlled binding reaction450.
Table 1.
PA63 conductance block by small cationic compounds
| # | Cationic Compounds | Structure | IC50 |
|---|---|---|---|
| 1 | Tetramethylammonium |
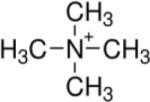
|
1.6 mM |
| 2 | Tetraethylammonium |
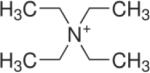
|
224 μM |
| 3 | Tetrapropylammonium |
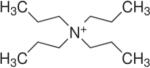
|
350 ± 10 nM |
| 4 | Tetrabutylammonium |
|
7.3 ± 0.2 μM |
| 5 | Tetrapentylammonium |
|
2 μM |
| 6 | Tetrahexylammonium |
|
3.8 ± 0.3 μM |
| 7 | (2-acetylamino-2,2-bis-ethoxycarbonyl-ethyl)-trimethyl-ammonium |
|
1 ± 0.1 mM |
| 8 | Tetraphenylphosphonium |
|
46 ± 2 nM |
| 9 | Chloroquine |
|
510 ± 30 nM |
| 10 | Quinacrine |
|
60 ± 5 nM |
| 11 | Benzyltriphenylphosphonium |
|
110 ± 30 nM |
| 12 | Butyltriphenylphosphonium |
|
88 ± 5 nM |
| 13 | Isoamyltriphenylphosphonium |
|
35 ± 6 nM |
| 14 | Methyltriphenylphosphonium |
|
370 ± 60 nM |
The ability of small molecules carrying one or two positive charges to block ion conductance through the channel-forming B components of the anthrax, C2, and Iota toxins was reported in several publications240, 402, 407, 410, 411, 416. Moreover, antimalarial drug chloroquine not only inhibited PA63 (KDPA = 0.51 μM in 0.1 M KCl) and C2IIa (KDC2IIa = 10 μM in 0.15 M KCl) channels in vitro but also prevented translocation of enzymatic component of C2 toxin, C2I across the cell membrane when studied with intact cultured cells. However, only a weak binding was reported for chloroquine with Iota toxin's channel forming component, Ib in planar lipid bilayers (KDIb = 0.22 μM in 0.1 M KCl) and, correspondingly, chloroquine did not efficiently protect cells from Iota toxin intoxication407. Parameters of the binding kinetics of chloroquine and related compounds (Figure 24) to the PA63 and C2II channels were obtained from the ligand-induced current noise on a multi-channel level using the planar bilayer technique410, 411, 416. The measurements showed that the spectral density of the open PA63 and C2IIa channels in the ligand-free solutions could be described by 1/f noise at low frequencies not exceeding about 100 Hz. As described above, this finding was later confirmed for the single PA63, C2IIa, and Ib channels (see Fig. 21), thus demonstrating that the origin of 1/f noise in the present systems is not related to channel-channel interactions. Rather, it is an inherent property of individual channels455–457. Analysis of the single-channel currents clearly shows that 1/f current noise of PA63, C2IIa, and Ib channels is caused by voltage-independent flickering between their open and completely closed states, which was never observed with β-barrel proteins of bacterial outer membranes. It would be interesting to see if high-resolution single-channel recording of the ϕ-clamp mutants of PA63, C2IIa, and Ib channels possess this type of fast flickering. The addition of interacting ligands led to the current noise increase with the spectral density of the Lorentzian type, characteristic of a single binding-site model411, 416. Strong dependence of the binding reaction on-rate, characterizing the frequency of blockage events, on the ionic strength was attributed to the involvement of ion-ion electrostatic interactions responsible for blocker binding. At that, the reaction off-rate, characterizing the residence time of the blocker in the channel, was dependent on the structural properties of the blocker. Interestingly, binding of chloroquine both to the PA63 and C2IIa channels was highly asymmetrical when the ligand was added either to cis or trans side of the bilayer chamber. Addition of chloroquine to the trans side of the membranes resulted in a significantly diminished binding affinity. However, fluphenazine binding was quite symmetrical. Note that the insertion of the channel into the planar lipid membranes is usually highly unidirectional, so it is expected that the “cap” region of the channels (assuming a mushroom-like shape for both PA63 and C2IIa) faces the cis side solution, the side of protein addition, and the membrane-inserted β-barrel stem emerges on the trans side. Generally, binding affinity of the cationic compounds decreases in the order PA63 > C2IIa >> Ib, which was explained by a decreasing number of the binding sites formed by negatively charged amino acid residues in the cis entry region of these channels.
Figure 24.

A: Structures of chloroquine and related compounds, which are able to block channel-forming B components of the binary toxins in the planar bilayers. B: Titration of membrane conductance induced by C2II with fluphenazine in 1 M KCl at the applied voltage of 20 mV. Reprinted with permission from ref416. Copyright 2003. Elsevier.
An alternative explanation of the binding of positively charged compounds to the PA63 pore was introduced in a study where the importance of the ϕ-clamp in PA63 translocase activity was first described240. A library of 35 cationic quaternary ammonium and phosphonium ion compounds was examined to compare their blocking activity towards the PA63 channel. Several interesting observations were made. First, mutations in the ϕ-clamp were shown to significantly affect binding affinity of hydrophobic cations, such as tetrabutylammonium (TBA). For instance, TBA's affinity to F427A channels was greatly reduced from that of the wild type (about 4000 times). Considering the hydrophobicity of the ϕ-clamp, authors hypothesized that this site may also be a binding site for the hydrophobic cations such as TBA. At that, the TBA blocking mechanism includes cation-π interactions between aromatic residues interacting with cations through their delocalized negative π-electron cloud. Second, it was shown that among the 35 compounds studied, the more hydrophobic ones possessed higher binding affinity towards the PA63 channel. For instance, introduction of the hydrophilic amide and ester groups to a TBA analog of a similar size, led to a 140-fold reduction in the binding affinity of this molecule (Table 1, compound 7). At the same time, the wild type PA63 channels preferred tetraphenylphosphonium to TBA by 160-fold (Table 1, compound 8). Across the studied library of compounds, the ϕ-clamp preferred aromatic moieties by 0.7 kcal/mol per aromatic ring. Table 1 (compounds 8–13) illustrates examples of the most effective (KD < 1 μM) small cationic blockers selected from the library of tested 35 compounds (for the full data see Table S1, supplemental material in ref.240). Several compounds with multiple aromatic rings (3 or 4) were the ones that showed the nM-range binding affinity towards the channel. Again, this activity was reduced drastically with the F427A mutants. The authors made an interesting suggestion that the ϕ-clamp can be exploited in the development of channel-blocking drugs as a binding site responsible for nonspecific hydrophobic interactions, although its negative π-clouds could also contribute through aromatic-aromatic π-π and cation-π interactions.
Amino acid residues involved in binding of cationic chloroquine to the C2IIa channel were also investigated410. It turned out that mutation of the negatively charged amino acids leads to a dramatic decrease in their affinity for binding chloroquine and its analogs, as well as the ϕ-clamp mutations (F428A, F428D, F428Y, and F428W) do. Note that the ϕ-clamp is preserved within the C2II as F428. The authors showed that residues Glu399, Asp426 (probably localized in the vestibule near the channel entrance), and Phe428, but not Glu272, Glu280, Asp341, or Glu346 are important for biding of chloroquine and 4-aminoquinolones, which act as C2IIa channel blockers. However the F428A mutation effect was the strongest, increasing KD values by a factor of almost 400, which emphasized once again the important role of the ϕ-clamp in binding of cationic compounds.
3.2.2. Cationic cyclodextrin derivatives
A substantial progress in disabling anthrax lethal toxin by the high-affinity blockage of the PA63 pore with cationic molecules was achieved using a rational drug design approach51. The idea was to block the oligomeric pore by a low-molecular non-peptide compound of the same symmetry as the target pore. In particular, it was shown that tailor-made 7-fold symmetrical 7-positively charged derivatives of β-cyclodextrin (7+ βCD) blocked the pore of anthrax's PA63 in planar bilayers (KD = 0.13 ± 0.1 nM) and protected cultured macrophage-like cells from intoxication with anthrax lethal toxin (PA + LF) (IC50 = 0.5 ± 0.2 μM)52, 62. As far as we are aware, these blockage and inhibition parameters are the best among the published data for the small-molecule blockers. Moreover, the most effective 7+βCD, per-6-S-(3-aminomethyl)benzylthio-β-cyclodextrin (AMBnTβCD) completely protected Fisher F344 rats from intoxication with lethal toxin and, in combination with the antibiotic ciprofloxacin, significantly increased the survival of mice in an infection model of anthrax56. These findings demonstrate a value of the 7+βCD as a potential scaffold for designing drugs against anthrax toxins, especially when an appropriate lead optimization and pharmacokinetic studies are conducted. Interestingly, about 2/3 of over 100 custom-synthesized 7+βCD compounds were protective against the lethal toxins with cell assays and nearly all tested so far showed strong or moderate channel blocking in planar lipid membranes. This finding once again showed an advantage of the rational drug design approach over the traditional time- and money-consuming high-throughput screening of libraries of thousands of compounds, which often produces a hit rate lower than 1%.
The idea to use 7+βCD cationic derivatives as inhibitors of anthrax was based on the wealth of earlier research51. First, as described in the Introduction, the high-affinity blockage of transmembrane channels formed by infectious agents with the anti-influenza M2 channel-blocking drug amantadine is the most prominent example19–21, 28. Second, in the particular case of heptameric pores, it was shown that the pore of β-barrel PFT α-hemolysin of S. aureus (see section 2.2.1) can be partially blocked by β-cyclodextrins65. Third, the positively charged tetraalkylammonium ions were reported to block PA63 channels384, 386, 450 interacting with the negative charges on the channel walls. Finally, the PA63 prepore internal diameter was estimated to be between 20 – 35 Å214 with the channel's constriction region not exceeding 12 Å384, 386, 450. These findings guided the choice of the ~15 Å βCD molecule carrying seven positive charges covalently linked to a cyclodextrin's core by a hydrocarbon links of the different length and nature (Figure 25A). From the point of view of medicinal chemistry, it is important to emphasize that cyclodextrins, a cyclic oligomers of glucose that can form water-soluble inclusion complexes with small molecules and portions of large molecules, have a long history of usage in pharmaceutical, agrochemical, environmental, cosmetic, and food industries458. The cyclodextrins do not elicit immune response and have low toxicity in animals and humans. There are thousands of variations of the CDs with different ring sizes and random or specific chemical modifications; as a result, reliable methods for cyclodextrin syntheses and selective modifications have been developed459.
Figure 25.

A, top: Blocking anthrax on a single-channel level. A heptameric mushroom-like channel of PA63 produced by Bacillus anthracis believed to be a translocation pathway for lethal and edema factor, LF and EF, inside the cell under attack. The idea is to design complementary heptameric low-molecular weight compounds – cationic cyclodextrins (A, bottom) that enter the pore and block it as molecular plugs. Note that the cartoon is a simplified illustration of the LF and EF penetration into the mammalian cell. In reality, the process is much more complex (Figs. 15 and 17). Adapted with permission from ref57. Copyright 2010. Ciophysical Society. B: Two 7-fold symmetrical synthetic molecules, per-6-S-(3-aminomethyl)thio-β-cyclodextrin (AMBnTβCD), left panel, and per-6-S-(3-amino)propylthio-β-cyclodextrin (AmPrβCD), right panel, were used as blockers of the PA63, C2IIa, and Ib pores. C: Planar lipid bilayer membrane containing about 60 PA63 channels in 0.1 M KCl. The downward arrow indicates the addition of AmPrβCD to the cis side of the membrane (side of PA addition). The dashed line shows zero current level. Adopted with permission from ref51. Copyright 2005. National Academy of Sciences.
Several 7-positively charged βCD derivatives as candidate antitoxins were custom-synthesized53 and tested51 (see Figure 25B for two examples). As Figure 25C shows, the addition of per-6-(3-aminopropylthio)- β-cyclodextrin (AmPrβCD, Fig. 25B, left) to the cis side of a membrane containing about 60 PA63 channels in 0.1 M KCl inhibits the ion channel activity in a step-like manner with a step amplitude of 87 ± 13 pS. This amplitude coincides with the PA63 single-channel conductance in 0.1 M KCl, implying that AmPrβCD acts on individual channels51. To identify more potent inhibitors of anthrax toxin compared with the reported AmPrβCD, the effect of the positively charged pendant groups and the length and nature of alkyl spacers on the activity of βCDs was estimated52, 60, 62 (Table 2). β-cyclodextrin is a naturally occurring cyclooligosaccharide containing seven α-(1,4)-D-glucopyranose subunits linked through α-(1,4) glucosidic bonds460. The primary (C-6) and secondary (C-2 and C-3) hydroxyl groups may be used as points of functionalization. The hydroxyl groups at positions 2 and 3 form hydrogen bonds and are required to keep the molecule rigid, making the 6-OH group a favorable site for modifications. First, a group of hepta-6-thioaminoalkyl derivatives with alkyl spacers of various lengths was tested for their ability to inhibit the cytotoxicity of lethal toxin and to block ion conductance through PA63 channels in planar lipid membranes (Table 2, compounds 1–11). From a combination of measurements with planar lipid membranes and cell assays, it was shown that there is some optimal length of the alkyl spacers (3–8 CH2-linkers) connecting an amino-group with the cyclodextrin core. Shorter spacers were less effective in inhibition of the channels and longer ones exhibited higher toxicity to the RAW cells, which was probably related to derivative-induced instability of the membranes observed in the bilayer measurements52. In order to check if the nature of the positively charged groups carried by the βCDs is an important factor in compound's efficiency, a group of hepta-6-guanidine β-cyclodextrin derivatives, in which positive charges were distributed between the nitrogens of the guanidine moiety was tested (Table 2, compounds 11, 12). The activity of these compounds was slightly decreased compared to their aminoalkyl analogs. Remarkably, hepta-6-arylamine βCD derivatives, which in addition to the pendant positively charged amino-groups contained one phenyl group carried by each thio-hydrocarbon linker, possessed significantly enhanced binding affinity both in vitro and with cell assays. One of the derivatives, per-6-S-(3-aminomethyl)benzylthio-β-cyclodextrin (AMBnTβCD) was chosen for the further development. In most of the experiments performed on planar membranes either AmPrβCD (Fig. 25B, left and Table 2, compound 3) or AMBnTβCD (Fig. 25B, right and Table 2, compound 13) or both were used as model molecules to study physico-chemical parameters of the blockage.
Table 2.
PA63 conductance block and cytotoxicity inhibition by cationic cyclodextrins
| |||||
|---|---|---|---|---|---|
| # | n | R1 | R2, R3 | Inhibition of conductance IC50, nM | Inhibition of cytotoxicity IC50, μM |
| I. Hepta-6-aminoalkyl β-cyclodextrin derivatives 52 | |||||
| 1 | 7 | -NH2 | -H | 140 ± 90 | 20 ± 9 |
| 2 | 7 | -S(CH2)2NH2 | -H | 3.5 ± 0.9 | 7.8 ± 2.4 |
| 3 | 7 | -S(CH2)3NH2 | -H | 0.57 ± 0.39 | 2.9 ± 1.0 |
| 4 | 7 | -S(CH2)4NH2 | -H | 1.1 ± 0.5 | 5.1 ± .2.4 |
| 5 | 7 | -S(CH2)5NH2 | -H | 3.8 ± 1.0 | 7.5 ± 2.4 |
| 7 | 7 | -S(CH2)6NH2 | -H | 0.97 ± 0.38 | 0.6 ± 0.3 |
| 8 | 7 | -S(CH2)7NH2 | -H | 4.6 ± 3.2 | 1.9 ± 1.1 |
| 9 | 7 | -S(CH2)8NH2 | -H | 2.4 ± 0.95 | 0.3 ± 0.1 |
| 10 | 7 | -S(CH2)10NH2 | -H | 27.0 ± 17.0 | 2.6 ± 0.1 |
| II. Hepta-6-guanidinealkyl β-cyclodextrin derivatives 52 | |||||
| 11 | 7 |
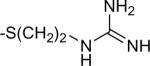
|
-H | 5.3±3.2 | 8.9±6.0 |
| 12 | 7 |
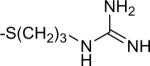
|
-H | 12.6±9.0 | 12.2±2.9 |
| III. Hepta-6-arylamine β-cyclodextrin derivative 52,62 | |||||
| 13 | 7 |
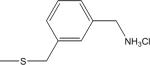
|
-H | 0.13±0.10 | 0.8±0.5 |
| IV. Cationic α- and γ cyclodextrin derivatives 62 | |||||
| 14 | 6 | -NH2 | -H | 1200 ± 300 | >100 |
| 15 | 8 | -NH2 | -H | 170 ± 50 | 12 ± 3 |
| 16 | 6 |
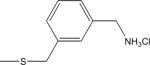
|
-H | 29±5 | 45±13 |
| 17 | 8 |
|
-H | 2.8±1.3 | 5.4±0.8 |
| V. Novel β-cydodextrin derivatives 60 | |||||
| 18 | 7 |
|
-H | n/a | >100 |
| 19 | 7 |
|
-H | n/a | >100 |
| 20 | 7 |
|
-H | n/a | >100 |
| 21 | 7 |
|
-H | n/a | >100 |
| 22 | 7 |
|
-H | n/a | >100 |
| 23 | 7 |
|
-H | n/a | >100 |
| 24 | 7 |
|
-H | n/a | 26 ± 21 |
| 25 | 7 |
|
-H | n/a | 3.2 ± 1.9 |
| 26 | 7 |
|
-H | n/a | 20 ± 14 |
| 27 | 7 |
|
-H | n/a | >100 |
| 28 | 7 | -H |
|
n/a | 4.1 ± 0.4 |
| 29 | 7 | -H |
|
n/a | 2.1 ± 0.2 |
| VI. α-, β-, and γ-cyclodextrin derivatives tested against α-hemolysin 62 | |||||
| 30 | 6 |
|
-H | >5000 | >100 |
| 31 | 7 |
|
-H | ~50 | 3.3±2.3 |
| 32 | 8 |
|
-H | >5000 | >100 |
It was shown that in addition to inhibition of anthrax toxin, cationic β-cyclodextrin derivatives were also effective against clostridial binary toxins. In particular, AMBnTβCD efficiently protected cultured epithelial cells from intoxication with two clostridial binary toxins (C2 and Iota) and blocked the ion current through heptameric channels formed by C2IIa and Ib in planar lipid membranes in vitro61. The compound acted by inhibiting the membrane translocation of C2 and Iota toxin A components (C2I and Ia) into the cytosol of intact cultured cells showing that it might serve as a universal pharmacological inhibitor against binary pore-forming toxins produced by pathogenic bacteria. To examine the nature of the physical forces involved in the blocker interactions, the voltage and salt dependence of the rate constants of binding and dissociation reactions for the two structurally different β-cyclodextrins (AmPrβCD and AMBnTβCD) (Fig. 25B) and the PA63, C2IIa, and Ib channels were recently investigated (submitted to Biophysical Journal). It turned out that with all three channels, AMBnTβCD, carrying extra hydrophobic aromatic groups on the thio-alkyl linkers of positively charged amino groups, showed significantly stronger binding compared with AmPrβCD. This finding is in a good agreement with the data reported earlier on a group of positively charged blockers. The blocking efficiency directly correlated with the number of aromatic groups carried by the molecules240 (section 3.2.1). The effect of increased affinity of the AMBnTβCD blocker to the channel is manifested by an increase in the residence time of the blocker in the channels, whereas the time between blockages (Figure 26A), which characterizes the binding reaction on-rate, stays practically unchanged. This result indicated that the capture rate of the blocker molecules by the channels did not significantly depend on the chemical structure of the blocker and probably was only determined by the relative size of the blocker and the channel. However, the time the blocker spends in the channel is significantly influenced by the structural changes in the blocker molecule.
Figure 26.

A: Ion currents through single PA63, C2IIa, and Ib channels in the absence (upper row of tracks) and in the presence of 0.135 μM AmPrβCD (middle row) and AMBnTβCD (bottom row) blockers in the cis side of the chamber. Measurements were taken in 1 M KCl solutions at pH 6 and 50 mV applied voltage. Recordings are shown at 10 ms time resolution. AMBnTβCD displays a significantly longer binding lifetime with all channels compared with AmPrβCD, whereas the time between the blockage events, characterizing the on-rate of the binding reaction, seems to be practically unchanged. B: Residence times of blocker binding to the channels plotted as functions of the transmembrane voltage reveal exponential voltage dependence. C: Residence times of blocker binding to the channels as functions of bulk salt concentration show different degrees of salt dependence for the three channels and two blockers. The salt dependence is most pronounced for AmPrβCD interacting with the PA63 pore. For the more efficient AMBnTβCD, the dependence is weaker.
Interestingly, the voltage sensitivity turned out to be practically the same for all six cases studied. The logarithm of the blocker residence time plotted as a function of transmembrane voltage, Figure 26B, displays practically identical slopes for both blockers and all three channels. Note that in contrast to tetraalkylammonium studies384, 386, 450, the residence times as functions of voltage lack any maxima, the circumstance that was used as a proof of the “blockage without translocation” mechanism of ion channel inhibiting57. It was also shown that the more effective AMBnTβCD blocker demonstrates weaker salt dependence of the binding and dissociation rate constants compared with AmPrβCD. Moreover, PA63 channel behavior was explored under conditions in which the blocker was added only to the trans side of the membrane57. Since the insertion of PA63 is always directional, the cis side application of the blocker is the physiologically relevant one. The trans addition was shown to irreversibly bring the channel to a low-conductance sub-state. This may indicate that the blocker is able to enter the channel from either side, but binds to different sets of amino acid residues without translocating through.
At moderate and low salts, the dependence of blockers' residence times on KCl concentration (Figure 26C) revealed the contribution of long-range Coulomb interactions. Particularly, at physiological salts, these interactions increase the binding efficiency by orders of magnitude. At salt concentrations lower than 0.5 M, the AmPrβCD residence time increases in the order Ib < C2IIa < PA63 (Fig. 26C), and so does the cationic selectivity of the channels (Fig. 20B). This pattern is Ib ≈ C2IIa < PA63 for the AMBnTβCD binding. The difference in the affinity of positively charged blockers towards the PA63, C2IIa, and Ib channels was earlier attributed to the different number of negatively charged amino acids on the lumen of these pores407, 410, 411. The preservation of the affinity pattern for the 7+βCD binding may indicate that the positive charges of the βCD blockers interact with the negatively charged amino acids in the channel lumen. At high salt concentrations, interpolation of the residence time to zero voltage allowed for an estimation of the salt-concentration-independent short-range interactions which predominate in all six cases studied.
In the case of the more effective AMBnTβCD blocker, its binding to the PA63, C2IIa, and Ib pores is further enhanced by the presence of the aromatic groups presumably interacting with a certain conserved group of residues (for instance with the ϕ-clamp240) in the lumen of these channels. Alternatively, the presence of aromatic groups could change the conformation of the linkers creating additional stabilizing interactions. The blocker's pharmacophoric pattern (three-dimensional arrangements of the several functional groups) is most probably involved in a number of Coulomb and salt-concentration-independent short-range interactions acting simultaneously within a single binding pocket of the channels. Clearly, the strong short-range interactions, added to the relatively weak Coulomb ones, determine enhanced affinity of the AMBnTβCD compound towards the Ib channel as well as the inhibitory properties of this compound against Iota toxin in cell assays61.
To summarize, analysis of voltage- and salt-dependence of the blockage demonstrated that even though both long-range Coulomb forces and interactions of the blocker charge with the transmembrane field are able to significantly increase the residence time of the blocker in the channel, the leading interactions are determined by salt-concentration-independent short-range forces for both AmPrβCD and AMBnTβCD blockers. As for the particular origin of these forces, the answer to this question will require further experimental and theoretical effort. Many factors complicate quantitative interpretation of blocker-channel interactions. Among them are the hydration state of the blocker molecule461, a sophisticated interplay between the hydrophobic effects and different electrostatic components, as, for example, in the case of polyamines binding to kainate subtype glutamate receptor channels462, and the uncertainties in the interaction-induced changes in the blocker and channel structures. At this moment, guided by the structural features of the efficient cyclodextrin-based blockers of Table 2, we can only speculate that in addition to hydrophobic interactions many others such as aromatic-aromatic π-π and cation-π interactions, hydrogen bonding, van der Waals interactions, etc. might be involved. We hope that careful Monte-Carlo and molecular dynamics simulations and multi-scale modeling combined with the results of existing and future experiments will shed light on the relative importance of different contributions.
The analysis given above accounts only for the reversible blockage obeying a two-state Markov process. It was also reported that the presence of blockers enhanced voltage gating (described in section 2.4.2) of these channels by making their closed state more favorable57. The closed state of the channel possessed the characteristic properties of a typical voltage-induced closed state of a β-barrel channel. For instance, it was hard to reopen the channel by keeping it at 0 mV, but application of −250 mV pulses often allowed channel reopening. Among the studied channels, the blocker-enhanced gating was minimal for Ib and maximal for PA63. Moreover, with all three channels studied, the voltage gating increase was more obvious with the more effective AMBnTβCD blocker. Thus with measurements on multi-channel membranes, which did not discriminate between the two modes of action, for AMBnTβCD this resulted in IC50 = (0.13 ± 0.1) nM for PA63, IC50 = (1.5 ± 0.5) nM for C2IIa, and IC50 = (23 ± 10) nM for Ib channels in “physiological” 0.1 M KCl at +20 mV of applied voltage. For the reason that the very nature of the voltage gating of β-barrel channels remains vague, it is unclear to what extent this second type of 7+βCD action could influence the blocker activity in vivo.
Finally, it was demonstrated that even though the 7-fold symmetry of the blocker molecules complementing heptameric structure of binary toxins' translocation components was important, it was not an absolute requirement for the effective blockage. Both 6-fold symmetrical αCD carrying 6 positive charges and 8-fold symmetrical γCD carrying 8 positive charges were able to block the PA63 channel in planar lipid membranes62 (Table 2, compounds 14–17). Note that 6- and 8-fold symmetrical compounds 14 and 15 are analogs of the 7-fold symmetrical compound 1, whereas compounds 16 and 17 are αCD and γCD analogs of the AMBnTβCD (compound 13). The activity of the 8+γCDs tested with cell assays was equal or even higher compared with their 7+βCD analogs, whereas binding of 6+αCD was not strong enough to make these compounds protective against anthrax toxin. Supposedly, the pronounced activity of the 8-fold symmetrical γCDs could be related to the recently reported observation of PA63 octameric pores373: however, both positively charged βCDs and γCDs were able to block any single PA63 channel incorporated into the planar lipid bilayers. The cyclodextrin molecules serve as ring-shaped platforms carrying multiple positive charges and aromatic groups that are able to interact with hydrophobic and negatively charged groups provided by the seven subunits of the PA63 channel. The less active derivative of αCD contributes only six substituents to the blocker-channel binding reaction. Moreover, its 13.7 Å external diameter compared to the 15.3 Å βCD diameter may not be large enough to allow for the strong interactions with the residues located in the channel lumen. The situation is different for the 8+γCDs, the compounds showing similar activity as their 7+βCDs analogs. The external diameter of γCD is 16.9 Å, which may limit the flexibility of the blocker inside the channel. However, the presence of an additional linker carrying the functional groups may increase the resultant strength of the channel-blocker interactions. Recently, several new groups of potential βCD-based inhibitors differing in the number, arrangement, and face location of the cationic elements were reported and tested using cell assays (Table 2, compounds 18–29)60. The results indicated that the development of new blocking agents couldn't rely exclusively on the symmetry and the putative electrostatic interactions. First of all, the cationic compounds synthesized based upon the symmetry complementarity concept alone did not provide an increased affinity towards the PA63 pore60. Second, introduction of additional positive charges (14+βCD) did not lead to more potent compounds (IC50 > 100 μM), however significantly increased their cytotoxicity (compounds # 19, 21, 23, 27)60. Interestingly, several derivatives, where positively charged groups were introduced in the (C-2) and (C-3) positions of the each α-(1,4)-d-glucopyranose subunit, turned out to be highly efficient as antitoxin agents (Table 2, compounds 28, 29).
In contrast to the cyclodextrin-based neutral derivatives interacting with the α-HL pores in planar lipid bilayers65, reversible blockages of the binary toxins' translocation components by cationic cyclodextrins were always complete, that is, they showed fluctuations between the current of the open state and zero. In the case of α-HL, the residual conductance that was observed in the reconstitution experiments was explained by the ability of ions to pass through the cyclodextrin cavity. This is obviously not the case for the blockage of the PA63, C2IIa, and Ib pores by the cationic CD derivatives. It is not clear what the reason for such a difference is. One tentative explanation would be that, in contrast to the α-HL – cyclodextrin complex, the cationic cyclodextrins are oriented in the channel in a way that prevents the ionic flow through their internal cavity. There is however no real evidence that would support this explanation.
3.2.3. Other agents against anthrax toxin
The current situation, when a vaccine is not available to the general public and only a limited number of antibiotics are approved by the FDA for the treatment of inhalational anthrax, had stimulated intense search for potent and selective antitoxins (see resent reviews)463, 464. Most of the efforts are focused on neutralizing and inhibiting the different basic steps in a multi-stage cell journey of the lethal and edema anthrax toxins (Fig. 15). In addition to the small molecule blockers directly obstructing the translocation channel-forming component of the toxin reviewed above, the therapies under development include employing toxin-neutralizing antibodies465–470, receptor decoy-based antitoxins471, 472, blockers of PA cleavage and oligomerization473, and inhibitors of LF and EF association with the PA63 prepore422, 474. Several interesting approaches utilize targeting of the channel-forming component of the anthrax toxin by means other than occluding it. PA as a potential target for the antitoxin therapy can be inhibited in a number of ways; several promising anti-PA strategies had been reviewed recently463, 464, 475, 476. For instance, a polyvalent peptide inhibitor that binds to the PA prepore preventing its interaction with LF has been reported477. The screening of a phage display library of mutant peptides able to interact with the heptameric component of anthrax toxin revealed a novel peptide that can block toxin assembly478. A series of mutant peptides were attached to polymer backbones to estimate their in vitro inhibitory activity. This approach allowed for identification of a minimal peptide sequence, TYWWLD, that can be used in developing of polyvalent anthrax toxin inhibitors. PA oligomer formation was shown to be inhibited by the one of the most effective chemotherapeutic anticancer agent, cisplatin that modifies PA in a reversible manner479. Dominant-negative mutants of protective antigen that co-assemble with the wild-type PA63 protein and block its ability to translocate the LF and EF components have also been developed474, 480–482. Interestingly, a single dominant-negative subunit with the key amino acid residues mutated was sufficient to block the translocase function of the PA63 heptamers. Moreover, dominant negative mutants of anthrax toxin potentially can be used both as an anthrax vaccine and in therapy463. In another study, the mouse monoclonal anti-PA antibody 1G3 was demonstrated to severely perturb the receptor-bound heptameric PA63 complex creating a PA supercomplex, which was directly visualized by electron microscopy483. De novo computer-aided drug design of small molecule inhibitors of the PA heptamerization had been recently reported484. Two molecular scaffolds were first identified using the CAVEAT molecular design package, and then seven candidate inhibitors were synthesized based on the discovered scaffolds and tested for their ability to inhibit anthrax toxin. Three of the designed agents demonstrated modest inhibition of the anthrax toxin in murine J774A.1 macrophage cells.
3.2.4. Polyvalent inhibitors of anthrax toxin
The idea of attaching multiple interacting ligands onto a suitable scaffold, similarly to the one realized with cyclodextrin-based molecules carrying 7 functional group clusters51, 52 or with the peptide-based inhibitors477, 478, 485, seems to be very helpful for the drug design objectives. These multi-ligand structures often possess an affinity toward multiple binding sites or receptors that is significantly higher than that of a single functional group interacting with a single receptor (recently reviewed in refs.486, 487). Different strategies exploring this approach had been suggested488–497. The first strategy is based on neutralization of PA63 heptameric complexes by peptide-functionalized liposomes of ~50 nm in size (Figure 27)489. The liposomes were allowed to interact with a cysteine-derivatized version of a heptameric PA63 binding peptide477, which acts by inhibiting lethal factor binding. Remarkably, peptide-functionalized liposomes inhibited the intoxication of murine macrophage RAW264.7 cells by the lethal toxins at extremely low concentrations (IC50 = 20 nM on a per-peptide basis), whereas the monovalent peptide did not inhibit lethal toxin activity at concentrations as high as 250 µM. Moreover, the peptide-functionalized liposomes were active in vivo protecting rats from the lethal toxin intoxication. Because several liposome-based drug formulations had already received an approval as vaccines adjuvants498 and the nanoparticles for drug delivery499–501, liposome-based scaffolds represent a promising application in search for polyvalent inhibitors including antitoxins. Furthermore, the liposome-based inhibitor study489 also indicated the several important aspects of the biophysics of polyvalent recognition, such as the influence of the density of the ligand and the membrane fluidity and heterogeneity on polyvalent inhibition (reviewed in ref.486).
Figure 27.
Neutralization of the PA63 heptameric complexes by peptide-functionalized liposomes of about 50 nm in size489. Left: The structure of the enzyme-binding face of heptameric PA63. Residues 197, 200, 207, 210 and 214, which form part of the LF-binding site, are highlighted in red. Approximate distances between residues 200 (30 Å) on adjacent monomers and residues 210 (40 Å) on adjacent monomers are indicated. Right: A schematic representation of a liposome-based anthrax toxin inhibitor. Reprinted with permission from ref489. Copyright 2006. Nature Publishing Group.
Another strategy used guided synthesis of polyvalent inhibitors of the controlled molecular weight and ligand density and placement along an inert polymeric scaffold491, 495. Such control was achieved by using reversible addition-fragmentation chain transfer polymerization (so called RAFT technique)493–495. The advantage of designing polyvalent inhibitors with the control over molecular weight and ligand spacing is illustrated in Figure 28. It is seen that the spacing between peptides on the inert linear framework is either too short (left panel) or is sufficient (right panel) to allow for polyvalent inhibitors to bind at the peptide-binding sites of the PA63 heptamer495.
Figure 28.
Design of polyvalent inhibitors with control over the molecular weight and ligand spacing491. The linear polyvalent inhibitors displaying peptides (black ovals) are shown bound to the PA63 heptamer at the peptide-binding sites (circles). The spacing between peptides on the linear scaffold is either too short (left panel) or is sufficient (right panel) to allow a polyvalent interaction. Reprinted with permission from ref491. Copyright 2006. American Chemical Society.
In rational drug design of polyvalent inhibitors, once a biospecific ligand is identified, the next important step is a search for a suitable and inert scaffold to attach the ligands486. Recently, the idea about advantage of matching the symmetry of the target with the symmetry of the interacting polyvalent molecule51 was developed further with β-cyclodextrin chosen as the core for the inhibitor scaffold497 (Figure 29). However, in contrast to the previous βCD-based studies, where the substituents consisted of positively charged groups linked to the core via hydrocarbon and aromatic linkers, seven copies of inhibitory peptide were linked to the cyclodextrin scaffold via polyethylene glycol linkers. A linker length was manipulated to insure an optimal fit of the polyvalent inhibitor to the heptameric PA63. The important objective of the study was to design the effective polyvalent ligands by matching the pattern of binding sites on the heptameric PA63. To achieve this goal, experimental mutagenic studies were accompanied by computational docking experiments searching for a suitable binding site for the inhibitory peptide on the heptameric component. The root-mean square distance from the center of cyclodextrin core to the end of the PEG11 linker was estimated as 30 Å, which matched the distance from the center of the PA63 oligomer to the identified peptide-binding residues. Note that unlike in the previous studies on cyclodextrin-based blockers, in this application the heptavalent inhibitors were designed to target lethal factor binding sites on the PA63 complex rather than to block the channel translocation pathway497. Again, the idea of a 7-fold symmetrical functional group placement allowed reaching the IC50 values as low as 10 nM on a per-peptide basis in vitro by incubating RAW264.7 cells with lethal toxin. Most importantly, the heptameric inhibitor provided a more than 100,000-fold increase in the activity compared with the monomeric peptide. At the same time, it did not show any major decline in activity over a three-day period and was also effective in vivo protecting rats from intoxication by the lethal toxin.
Figure 29.
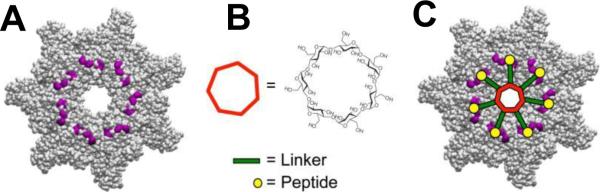
Structure-based design of the heptavalent anthrax toxin inhibitors497. A: The structure of the LF-binding face of heptameric PA63. Residues 184, 187, 197, and 200, which form part of the peptide-binding site are shown in purple. B: The structure of 7-fold symmetrical β-cyclodextrin, which was used as a scaffold for the heptameric inhibitor. C: A scheme illustrating the binding of a heptavalent inhibitor, synthesized by the attachment of seven inhibitory peptides to the β-cyclodextrin via an appropriate PEG linker, to heptameric PA63. Reprinted with permission from ref497. Copyright 2011. American Chemical Society.
To summarize, several studies to design PA-based anthrax toxin inhibitors showed the value of polyvalent interactions. These cooperative interactions can be significantly stronger than the corresponding monovalent interactions of the same ligands486. The examples include potent biospecific peptide-based or small-molecule ligands (or functional groups) attached to a variety of scaffolds formed by the liposomes489, polymers491 or cyclodextrins51, 52, 497. Note that the idea of using the polyvalent interactions was also explored in the design of potent inhibitors for other bacterial toxins502–506.
Meanwhile a thermodynamic analysis of multivalent interactions aiming to clarify the theoretical basis for the large enhancement in avidity as a result of multivalency had been recently published507. In particular, the author investigates the influence of a linker – the structural element that connects the binding fragments with the inert scaffold – on the activity of a resulting multivalent compound to address the existing controversy about the extent to which the loss of conformational entropy of the linker may influence the increase in binding. The simple thermodynamic analysis of a heterodivalent ligand-receptor interaction demonstrated that even if the loss in conformational entropy of the linker on binding is extremely unfavorable, the dependence of the free energy of multivalent binding on linker length can still be weak507. Most importantly, the predicted weak dependence of the free energy is consistent with other studies showing that flexible linkers of different length can be successfully used to design effective multivalent inhibitors.
3.3. Targeting membrane-perforating bacterial toxins
Surprisingly, the number of molecules under development that specifically target the pore-forming toxins is relatively low. At the same time, experimental studies of these toxins, particularly in the planar lipid bilayers, are significantly simplified by the fact that PFT's biological action is based on permeabilization of the target cell membranes. As a result, the complex multi-stage transport of enzymatic domains and components described above does not apply to these toxins. For this reason, their disabling could simply imply a physical obstruction of the “virulent” ion conductive pores formed by these toxins as opposed to the more elaborate approaches to either single-chain AB (diphtheria toxin and botulinum neurotoxins) or binary (anthrax) toxins.
3.3.1. Small molecule blockers of α–hemolysin and epsilon toxins
Currently used approaches to prevent the toxic effects of α–hemolysin and epsilon toxin include polyclonal and monoclonal antibodies508–517. Several compounds were shown to inhibit α–HL production518, 519. Dominant-negative inhibitors of ETX were also reported520. However, the studies searching for small-molecule channel-blocking inhibitors are very limited.
As described above (section 2.2.1), α–HL is a heptameric pore-forming cytotoxin secreted by gram-positive S. aureus bacterium, which is essential in the pathogenesis of pneumonia521. The high-resolution crystal structure of the membrane form of α–HL (Fig. 1A) revealed a mushroom-like heptameric channel with a radius changing from 7 Å to 23 Å in the cap diameter and about 100 Å in the total height167. Recently, the idea of enhancing the blocking ability of compounds by having the same symmetry as the target pore was extended to the design of βCD-based inhibitors of α–HL55. Interestingly, several unsubstituted cyclodextrins were earlier used as molecular adapters docking into the β-barrel of the α–HL65, 522. The approach allowed designing a new hepta-6-substituted β-cyclodextrin derivative, named IB201 (compound 31, Table 2), which was able to protect rabbit red blood cells from α–HL-induced hemolysis in the low micromolar range55, prevented α–HL-mediated alveolar epithelial cell lysis and mortality associated with S. aureus pneumonia in a murine model of infection59. IB201 was assayed for its ability to block ion conductance through the pores formed in artificial membranes by α–HL55. In contrast to 7+βCD binding to the PA63, the α–HL interaction with IB201 was irreversible within the limit of the time period of the experiment. Addition of this compound caused the channel to switch to a weakly conductive sub-state (Figure 30). The residual conductance of the closed state was between 1% and 15% of the open channel conductance and displayed a current noise typical for the voltage-gated closed state seen commonly with α–HL at high voltages. Thus, introduction of positive charges to the cyclodextrin molecule led to its ability to block this anion-selective channel from the physiologically relevant cis side, not from the trans side as in the case of molecular adapters65, 522. Since only a very limited number of compounds from the available library of 7+βCDs acted similarly to IB201, it is unclear what particular interactions are involved in the α–HL blocking. Remarkably, in contrast to PA63, α–HL was selectively blocked only by β-cyclodextrin derivatives, whereas αCDs and γCDs carrying the same substituents groups were not effective with cells assays and showed no interaction with the channel in planar membranes62 (Table 2, compounds 30–32).
Figure 30.
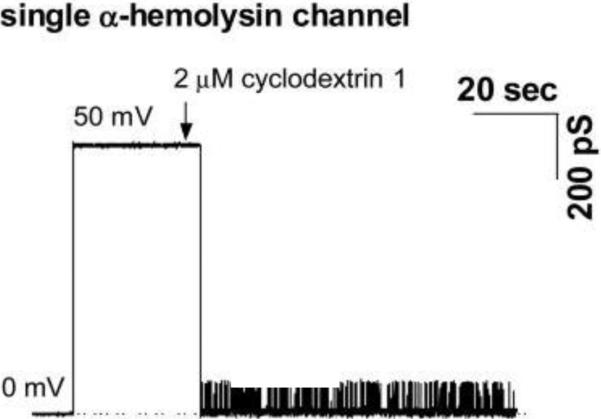
Modulation of the ion current through a single α-HL channel by 7-fold symmetrical βCD IB201 (Table 2, compound 31). In the absence of βCD, the current through the single α-HL channel is rather stable (no significant current fluctuations at 10 ms time resolution are seen). IB201 addition leads the channel to switch to a weakly conductive blocked sub-state. Reprinted with permission from ref55. Copyright 2007. Elsevier.
A different approach was used to search for the small-molecule inhibitors of epsilon toxin. A cell-based high-throughput screening of a 151,616-compound library led to identification of three compounds inhibiting ETX523. Those were N-cycloalkylbenzamide, furo[2,3-b]quinoline, and 6H- anthra[1,9-cd]isoxazol. It appeared that none of these three compounds inhibited ETX cell binding or oligomerization, making the authors to suggest that they might act by blocking the ETX pores.
3.4. Blockage of ion-selective channels of neurophysiology: a comparison
It is interesting to compare the affinity of the blockers of the channel-forming toxins discussed above with a wealth of data on the blockage of ion-selective channels of neurophysiology. The well-being of a multi-cellular organism involves significant coordination of the functions of many organs, which, in its own turn, relies on efficient transmission of information between the various cells in different parts of the body. In vertebrates, transduction of electrical signals and the corresponding calcium transients are the major mechanisms of processing information in the brain and environment sensing. They are also responsible for delivering signals from the brain to peripheral organs including controlling muscle contraction and release of hormones. Unsurprisingly, the ion-selective channels of neurophysiology, which are vital for the information transfer function, represent the most extensively studied membrane proteins, in both functional and structural aspect133.
Impressive advances in the structure-inspired design of new drugs that modulate properties of the ion-selective channels are reviewed in a number of recent instructive publications524–535, which discuss a variety of blockers and activators of different channel species. Most of the modulators discovered up to date are used as versatile research tools, but quite a remarkable number have found their way into pharmacology for treating a broad spectrum of diseases such as epilepsy, chronic pain, cardiac arrhythmia, ischemia, memory disorders, hypertension, type 2 diabetes, and many others. For example, in the case of potassium channels, the effect of the blocker is to increase cell excitability, which may facilitate signal transduction. On the contrary, channel activation leads to a decreased excitability, which could help in certain disorders. In the light of the present review, we are mostly interested in the channel blockers. Moreover, among the modulators of different nature we will restrict ourselves to the non-peptide “small-molecule” blockers only.
The comprehensive list of available IC50 values for blockers of potassium, sodium, and calcium channels is quite lengthy, even if restricted to non-peptide compounds. Because of this, in Table 3 we give only representative examples of the blockers, which, however, are intended to include the most efficient ones. It is seen that the IC50 values for the small-molecule blockers of ion-selective channels of neurophysiology are higher than that for compound #13 interaction with the anthrax PA63 channel (Table 2), characterized by IC50 = 0.13 ± 0.10 nM. The hepta-6-arylamine β-cyclodextrin derivative has nearly an order-of-magnitude stronger affinity to the PA63 channel than the most potent small-molecule blockers in Table 3 to their corresponding channels with only one exception of UCL1684 interaction with the calcium-activated potassium channel KCa2.2, which is characterized by about the same affinity.
TABLE 3.
Some of the most potent non-peptide blockers of classical ion-selective channels
| Channel | Compound | IC50 | Comments |
|---|---|---|---|
| Potassium, KV4.3 | Nicotine | 34 nM | ref.597* |
| Potassium, KV11.1 | Astemizole | 48 nM | ref.598* |
| Potassium, KV11.3 | Sertindole | 43 nM | ref.599* |
| Sodium, NaV1.1 | Tetrodotoxin | 6 nM | ref.527,600 |
| Sodium, NaV1.6 | Tetrodotoxin | 1 nM | ref. 527,601 |
| Calcium, CaV1.2 | Devapamil | 50 nM | ref.528,602 |
| Inwardly Rectifying Potassium, Kir2.1 | Spermine | 0.9 nM | ref.534,603 |
| Calcium-Activated Potassium, KCa1.1 | Paxilline | 1.9 nM | ref.532,604 |
| Calcium-Activated Potassium, KCa2.1 | UCL1684 | 0.8 nM | ref.532,605 |
| Calcium-Activated Potassium, KCa2.2 | UCL1684 | 0.28 nM | ref.532,606 |
Additional information can be found in a recent review533; however, it has to be used with caution as is contains a number of confusing misprints. The original papers cited there should be consulted to avoid misleading conclusions.
4. DOUBLE LIFE OF CHANNEL-FORMING BACTERIAL TOXINS
Unique intrinsic properties inherent to the channel-forming bacterial toxins, such as their ability to respond to electrical, chemical, or mechanical stimuli536, were utilized in several nanobiotechnological applications (recently reviewed in ref.537). The potential applications range from molecular sensing and detection, DNA sequencing, monitoring chemical and biochemical reactions, and development of biocompatible nanotransistors to drug delivery and targeted killing of cancer cells537. It is remarkable that most applications are not necessarily based on toxic pathways these pores are involved in, but rather benefit from their unique electrical properties. These applications truly bring about “second life” for these biological toxins, where genetic engineering and covalent attachments or non-covalent adapters are often used to generate desired novel features.
4.1. Bacterial toxins as biosensors
Without any doubt, of all the PFTs, α-hemolysin of S. aureus, suggested to serve as a “nanoscopic cuvette” to study reaction dynamics some 20 years ago417, 418, is an absolute leader in this field. The crystal structure of this heptameric pore resolved more than 15 years ago,167 as well as its ability to form large stable ion channels in bilayers174, 538 contributed to the wide usage of this toxin in the various biotechnological applications under development. Wild type or genetically engineered α-HL serves as a molecular sensor for the detection of ionic species and organic molecules including divalent metal ion64, 539, phosphate anions73, trinitrotoluene74, styryl dyes87, proteins78, 81, 90 and nitrogen mustards, which are chemical warfare agents80.
In another remarkable application, β-cyclodextrin-based molecular adapters were shown to reversibly interact with the β-barrel of the α-HL channel65, 522, 540. As a result, modification of the channel by βCD created a binding site for a number of small organic molecules. Transient formation of the host-guest complexes of small organic compounds with the cyclodextrin (Figure 31) by embedding into its internal cavity could be registered through conductance fluctuations. In particular, the α-HL channel – βCD complex allowed for stochastic sensing of adamantane derivatives and other small therapeutic molecules65, 541. This approach also allowed studying the changes in preferential solvation during association reactions at the singe-molecule level542, 543. Cyclodextrin adapters within the modified α-HL pores were used to discriminate between the S- and R-enantiomers of the ibuprofen and thalidomide drug molecules77. The enantiomers of these small chiral molecules generated distinctly different current signals upon their reversible binding to the α-HL-βCD complex. Keeping in mind that thalidomide, which is responsible for one of the most tragic medical failures of the 20th century, is now brought back to market for the treatment of leprosy and certain types of cancer544, the ability to rapidly distinguish between the chiral forms of the drug molecules is very important. Note that teratogenic and antitumor properties of this compound are sometimes attributed to the thalidomide S-enantiomer545. Novel α-HL-βCD based adapter systems with the asymmetrical cavity have been suggested recently88. Besides, α-HL was engineered to accommodate two different cyclodextrin molecules (βCD and hepta-6-sulfato βCD) simultaneously at the different binding sites541. As a result, the space between these adapters formed a nanocavity able to trap organic molecules, which shuttled back and forth between the adapters for hundreds of milliseconds.
Figure 31.

Bilayer recordings showing the interaction of a single α-HL pore with βCD and the model analytes 2-adamantanamine (A1) and 1-adamantanecarboxylic acid (A2) at −40 mV applied voltage65. α-HL was added to the cis compartment of the chamber and βCD and the adamantine derivatives were in the trans compartment. A: Control, single α-HL pore is unblocked (level 1). B: 20 mM trans-addition of βCD generates transient partial blockages of the channel (level 2). C: 80 mM trans-addition of 2-adamantanamine does not affect the fully open channel (level 1), but produces an additional block of α-HL- βCD complex (level 3). D: 20 mM trans-addition of 1-adamantanecarboxylic acid produces additional blockades (level 4), of the longer duration than those produced by 2-adamantanamine (level 3). Reprinted with permission from ref65. Copyright 1999. Nature Publishing Group.
Figure 32 illustrates the use of the cyclodextrin-based molecular adapters to study the dynamic aspects of the Hofmeister effect542. The thermodynamic and kinetic parameters of the adamantane binding to a cyclodextrin molecule residing in the β-barrel of the α-HL channel were explored as functions of addition of different salts to the initial membrane-bathing solution of 1.0 M KCl (or NaCl). The linearity of the changes in the free energy of the adamantane-cyclodextrin complexation with solution osmolarity (Fig. 32B) allowed the authors to characterize the reaction by a constant preferential hydration coefficient Δnwater, shown in the uppermost Table on the right. Similar formal procedure was applied to describe the sensitivity of the kinetic parameters to the added salts (Figs. 32C, D). It was found that not only the off-rate, characterized by the residence time of adamantane in cyclodextrin (τr), but also the on-rate, characterized by the times between consecutive complexations when channel is open (τo), are changed by salt addition. The change in the on-rate induced by KBr is small and might be well within the measurement error, but the effect of KCl and, especially, of Na2SO4 is clear. Thus, the change in the free energy of the complexation is due to both the change in the complex stability, as quantified by the off-rate, and the change in the accessibility of the complexation site, as quantified by the on-rate.
Figure 32.

The single-molecule nanopore approach illustrated in Fig. 31 was used to study the dynamic side of the Hofmeister effect542. A: Cartoon of the complexation process between the cyclodextrin hosted by the α-HL pore and adamantane carboxylate. B: Changes in the free energy of cyclodextrin– adamantane complexation, δΔG, versus changes in salt osmolarity. C: Effect of increasing osmolarity on the average residence time of adamantane in the channel-bound cyclodextrin, τr. D: Effect on the average time between the successive complexation events, τo. Both times are normalized by their values measured prior to extra salt addition. Tables on the right show the effective numbers of excluding water molecules. It is seen that not only the complex stability, as measured by the average adamantane residence time, but also the on-rates, represented by inverse τo, are influenced by salt addition and thus contribute to the changes in the complexation free energy. Reprinted with permission from ref542. Copyright 2009. Wiley.
Sensing with biological nanopores was also exploited to detect the DNA and RNA biopolymers with an ultimate goal of ultrafast sequencing (reviewed in refs.536, 537, 546–550). Since first electrical recording of polynucleotides passing through the α-HL pore were pioneered more than 15 years ago63, this application had been significantly advanced66–68, 70, 71, 76, 79, 83–85, 89, 551–555 mostly focusing on overcoming serious challenges related to the ability of α-HL (and other biological and engineered nanopores) to detect current signals generated by individual nucleotides. While α-HL undoubtedly bears the palm, pore-forming bacterial toxin aerolysin of Aeromonas hydrophila (see section 2.2.3 of this review) and heptameric channel-forming component of anthrax toxin, PA63 had been suggested for use as molecular biosensors232, 234, 556. The use of biological nanopores as molecular biosensors has been challenged by the emergent technology of synthetic solid-state nanopores, including the first demonstration of DNA sensing557 (recently reviewed in ref.536, 549). At the same time, biological nanopores, including bacterial toxins, bring several important advantages to the biosensing field. Among those is an atomic level of similarity between individual molecules of the same protein that often can be investigated with X-ray crystallography. Besides, modern genetic tools, such as site-directed mutagenesis, allow for specific modification of physico-chemical properties of the channel-forming proteins. On the other hand, the lipid bilayers are mechanically less stable compared to the solid-state nanopores. An interesting new approach that combines the advantages of two techniques (so called “hybrid biological-solid state nanopores) suggested incorporating a genetically engineered α-HL channel into a small nanopore fabricated within a thin SiN membrane 85.
4.2. Bacterial toxins for cancer treatment
Many existing cancer therapies have significant limitations because of the side effects on the fast-growing healthy cells. Several therapeutic agents, which specifically target cancer cells, are under development558. One of the novel emergent approaches involves the application of channel-forming bacterial toxins, rationally modified to selectively destroy cancer cells (reviewed in refs.537, 559). The later can be achieved by either genetic engineering or chemical modification of the bacterial toxins to provide these proteins with the cell-selective ligands or domains allowing them to effectively recognize certain membrane receptors or antigens that are typically overexpressed by malignant cells560. For this reason, many bacterial toxins for cancer cell targeting were designed as the fusion proteins composed of several parts with different functions. In a way, AB-type bacterial toxins are naturally made “fusion” proteins, where membrane binding domain translocates enzymatic domain into the cytosol. The same idea is exploited with the engineered “tailor-made” fusion therapeutic proteins13, where the cell specificity of one fragment can be successfully combined with the toxicity of another. Naturally, the mechanism of self-mediated delivery of the foreign proteins into the cytosol of mammalian cells is sometimes compared with the famous Trojan Horse5, 13. There are several strategies to generate fusion proteins, also termed “chimeras” (recently reviewed in refs.13, 561–565). One approach presumes integration of the enzymatic and binding components of different bacterial toxins, which allows for generation of the novel fusion proteins with a desired property being enhanced. The very first fusion protein that effectively destroyed cancer cells was generated from diphtheria toxins of Corynebacterium diphtheriae and exotoxin A of Pseudomonas aeruginosa566. Alternative (and often less toxic to the healthy cells) approach involves substitution of toxin's binding and/or enzymatic domains with eukaryotic proteins. Thus, the B-domain is intended to be replaced by a specific cell-binding protein that selectively targets the malignant cells only. Alternatively, specific antibodies are sometimes fused into the A-domains creating the chimeras referred as “immunotoxins”562, 563, 565. In several studies, epidermal growth factors or interleukin 2 as binding domains were conjugated with diphtheria toxin; a number of these chimeras are very effective for the treatment of hematopoietic malignancies567–570.
A series of interesting anthrax fusion toxins for targeting cancer or intracellular delivery of macromolecules were generated571–574, 574–577. Clostridial binary ADP-ribosylating toxins, which share many structural and functional similarities with the anthrax toxin (mostly at the stages of their cell binding and intracellular trafficking), represent another ideal system for transporting foreign molecules into the cell (recently reviewed in ref.13). In particular, the chimera toxin, based on the clostridial C2 toxin delivery system and enzymatic activity of C3 transferase from C. limosum, C2I1–255-C3lim + C2IIa, effectively delivers the C3 transferase into all targeted vertebrate cells, making this chimeric toxin 300-fold more active compared to C3lim transferase alone578, 579. Moreover, the binary Iota toxin of C. perfringens was also used to transport the C. botulinum C3 enzyme580. Besides AB and binary toxins, PFTs are also considered as functional components of the chimeric toxins. For instance, certain mutations of the pore-forming α-HL made this protein sensitive to activation by the tumor-specific protease, cathepsin B, which triggered the pore formation selectively in the cancer cells581. Recently, aerolysin of Aeromonas hydrophila was coupled as an inactive precursor of this protein to a peptide that could be cleaved by a prostate cancer cells protease582.
Detailed description of various chimeric toxins made of the enzymatic, binding, or channel-forming domains or components of bacterial toxins is beyond the scope of this review. The additional applications not mentioned above include targeted drug delivery, for instance to generate protective antiviral immunity583–585. Besides biomedical applications, chimeric proteins are of significant scientific interest, for instance the clostridial C2 toxin fused with the virulence factor SpvB of Salmonella enterica, C2I1–255 – C/SpvB is used to study the consequences of actin-ADP-ribosylation by SpvB in mammalian cells36, 564, 586, 587.
5. CONCLUDING REMARKS
For the last several decades, the structure-inspired drug design has been successfully used to develop small-molecule modulators of classical ion-selective channels of neurophysiology524–535. The emerging structural details of potassium, sodium, chloride, and various ligand-gated channels are crucially important in the virtual screening of molecular libraries and creating new compounds. Relatively recently51, the structure-inspired drug design was extended to include the pores of “virulence” – those of the channel-forming bacterial toxins. The newly synthesized compounds, which mimic the symmetry of the pores and are complementary to their structures in charge, size, and hydrophobicity, are quite effective in channel-blocking activity; by the potency (Table 2) several of these compounds compare well with the blockers of classical channels of electrophysiology (Table 3).
At the same time, the general approach to the development of efficient blockers of toxin pores is considerably different. Indeed, while one of the main goals of designing blockers of ion-selective channels of excitable cells is to achieve high specificity, with only one type of the channels targeted, the toxin pore blockers are expected to be universal. The first successful steps in the search for such wide-spectrum blockers have been reported recently in a study where cationic βCD-based compounds were shown to be protective against the cytotoxicity induced by the anthrax, C2, and Iota toxins61. The universality requirement is easy to appreciate, because a potent wide-spectrum antidote would be expected to be active against a number of different toxic agents. In the search for such a compound, researchers rely on the knowledge of the structure and function of bacterial toxins, focusing on the mechanisms of those shared intoxication steps that could be directionally targeted. For instance, formation of the β-barrel transmembrane pores by the membrane-perforating (section 2.2 and 3.3) and binary toxins (section 2.4 and 3.2) can be considered as a targetable universal property. On the other hand, such an ideal antidote is expected to be harmless against mammalian channels, including the β-barrel voltage dependent anion channel (VDAC)588–590 of the outer mitochondrial membrane, based on the fundamental differences in their structural organization. According to numerous studies, VDAC is a wide monomeric channel591, which does not possess the symmetry features characteristic of the oligomeric toxin channels discussed above. VDAC's functional properties can be modulated by a number of factors, including the membrane lipid composition and cytosolic proteins444, 592–596, but the effect of the cyclodextrin blockers on VDAC is negligible (Philip Gurnev, private communication). This leaves us with a hope that the inhibitive action of the advanced, wide-spectrum antidotes of the future will be mostly limited to toxin channels, thus paving the way for the eventual use of these drugs in clinical settings.
Acknowledgements
EMN research is supported by startup funds from The Catholic University of America. SMB research is supported by the Intramural Research Program of the NIH, Eunice Kennedy Shriver National Institute of Child Health and Human Development.
Biographies
 Nestorovich's Biosketch: Ekaterina M. Nestorovich earned her Ph.D. in electrochemistry from St. Petersburg State University, Russia under supervision of Prof. Valery Malev. She performed a postdoctoral research in biophysics with Dr. Sergey Bezrukov at the National Institutes of Health. While at the NIH, she mastered the art of ion channel reconstitution into planar lipid bilayers (the models of biological membranes) and modern methods of statistical analysis of ionic currents – powerful tools which allowed her to study kinetic and transport properties of channel-forming proteins at the single-molecule level. In January 2011, she joined the faculty in the Department of Biology at The Catholic University of America as Assistant Professor of the new Biotechnology program. Her teaching interests include Biotechnology Project Managements and Rational Drug Design. She also holds a professional certificate in Advanced Project Management from Stanford University. The direction of her research is best described as medical biotechnology and biophysics. From the biomedical science perspective, she searches for novel effective approaches to make good use of ion-conducting nanostructures in a variety of medical, chemical, and biotechnological applications. From the biophysical perspective, she pursues a new level of understanding of biological structures through the physical forces that animate them. By learning the physics and chemistry of biological structures' functioning, Dr. Nestorovich strives to determine how to design new agents that effectively correct the deviant interactions associated with diseases.
Nestorovich's Biosketch: Ekaterina M. Nestorovich earned her Ph.D. in electrochemistry from St. Petersburg State University, Russia under supervision of Prof. Valery Malev. She performed a postdoctoral research in biophysics with Dr. Sergey Bezrukov at the National Institutes of Health. While at the NIH, she mastered the art of ion channel reconstitution into planar lipid bilayers (the models of biological membranes) and modern methods of statistical analysis of ionic currents – powerful tools which allowed her to study kinetic and transport properties of channel-forming proteins at the single-molecule level. In January 2011, she joined the faculty in the Department of Biology at The Catholic University of America as Assistant Professor of the new Biotechnology program. Her teaching interests include Biotechnology Project Managements and Rational Drug Design. She also holds a professional certificate in Advanced Project Management from Stanford University. The direction of her research is best described as medical biotechnology and biophysics. From the biomedical science perspective, she searches for novel effective approaches to make good use of ion-conducting nanostructures in a variety of medical, chemical, and biotechnological applications. From the biophysical perspective, she pursues a new level of understanding of biological structures through the physical forces that animate them. By learning the physics and chemistry of biological structures' functioning, Dr. Nestorovich strives to determine how to design new agents that effectively correct the deviant interactions associated with diseases.
 Bezrukov's Biosketch: Sergey M. Bezrukov is a Senior Investigator and chief of the Section on Molecular Transport, Program in Physical Biology, NICHD/NIH, a position he has held since October 2002. In order to address fundamental questions of membrane transport, Dr. Bezrukov's section combines physical theory with experiments on bacterial, mitochondrial, and toxin-induced membrane channels, reconstituted in planar lipid bilayers. This line of research serves as the basis for the development of new approaches to treatment of many diseases where regulation of transport through ion channels plays the key role. Dr. Bezrukov received his M.S. in Physics from St. Petersburg Polytechnic University, Russia, Ph.D. in Biophysics from Moscow State University, and D.Sci. in Condensed Matter Physics from the St. Petersburg Nuclear Physics Institute of the Russian Academy of Sciences. He started his career as a researcher in Russia and moved to United States in 1990, first as a visiting research professor at the University of Maryland. In 1992, he joined the National Institutes of Health as a visiting scientist. He was awarded NIH tenure in 2002. Dr. Bezrukov has authored numerous scientific papers, five of them published in Nature (London). He has organized and chaired many international meetings and workshops. Dr. Bezrukov's recent honors include election to Fellowship in the American Physical Society (2009), the NIH Director's Award in Science and Medicine (2010), and appointment to the Senior Biomedical Researcher Service (2011).
Bezrukov's Biosketch: Sergey M. Bezrukov is a Senior Investigator and chief of the Section on Molecular Transport, Program in Physical Biology, NICHD/NIH, a position he has held since October 2002. In order to address fundamental questions of membrane transport, Dr. Bezrukov's section combines physical theory with experiments on bacterial, mitochondrial, and toxin-induced membrane channels, reconstituted in planar lipid bilayers. This line of research serves as the basis for the development of new approaches to treatment of many diseases where regulation of transport through ion channels plays the key role. Dr. Bezrukov received his M.S. in Physics from St. Petersburg Polytechnic University, Russia, Ph.D. in Biophysics from Moscow State University, and D.Sci. in Condensed Matter Physics from the St. Petersburg Nuclear Physics Institute of the Russian Academy of Sciences. He started his career as a researcher in Russia and moved to United States in 1990, first as a visiting research professor at the University of Maryland. In 1992, he joined the National Institutes of Health as a visiting scientist. He was awarded NIH tenure in 2002. Dr. Bezrukov has authored numerous scientific papers, five of them published in Nature (London). He has organized and chaired many international meetings and workshops. Dr. Bezrukov's recent honors include election to Fellowship in the American Physical Society (2009), the NIH Director's Award in Science and Medicine (2010), and appointment to the Senior Biomedical Researcher Service (2011).
References
- (1).Parker MW, Feil SC. Prog. Biophys. Mol. Biol. 2005;88:91. doi: 10.1016/j.pbiomolbio.2004.01.009. [DOI] [PubMed] [Google Scholar]
- (2).Henkel JS, Baldwin MR, Barbieri JT. EXS. 2010;100:1. doi: 10.1007/978-3-7643-8338-1_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Barth H, Aktories K, Popoff MR, Stiles BG. Microbiol. Mol. Biol. Rev. 2004;68:373. doi: 10.1128/MMBR.68.3.373-402.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Menetrey J, Gillet D, Menez A. Toxicon. 2005;45:129. doi: 10.1016/j.toxicon.2004.09.004. [DOI] [PubMed] [Google Scholar]
- (5).Trujillo C, Ratts R, Tamayo A, Harrison R, Murphy JR. Neurotox Res. 2006;9:63. doi: 10.1007/BF03033924. [DOI] [PubMed] [Google Scholar]
- (6).Rádis-BaptistaI G, Kerkis A, Prieto-Silva A, Hayashi M, Kerkis I, Yamane T. J. Braz. Chem. Soc. 2008;19:221. [Google Scholar]
- (7).Geny B, Popoff MR. Biol. Cell. 2006;98:667. doi: 10.1042/BC20050082. [DOI] [PubMed] [Google Scholar]
- (8).Gilbert RJ. Cell Mol. Life Sci. 2002;59:832. doi: 10.1007/s00018-002-8471-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Gilbert RJ. Adv. Exp. Med. Biol. 2010;677:56. doi: 10.1007/978-1-4419-6327-7_5. [DOI] [PubMed] [Google Scholar]
- (10).Gouaux E. Curr. Opin. Struct. Biol. 1997;7:566. doi: 10.1016/s0959-440x(97)80123-6. [DOI] [PubMed] [Google Scholar]
- (11).Gonzalez MR, Bischofberger M, Pernot L, van der Goot FG, Freche B. Cell Mol. Life Sci. 2008;65:493. doi: 10.1007/s00018-007-7434-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Iacovache I, van der Goot FG, Pernot L. Biochim. Biophys. Acta. 2008;1778:1611. doi: 10.1016/j.bbamem.2008.01.026. [DOI] [PubMed] [Google Scholar]
- (13).Barth H, Stiles BG. Curr. Med. Chem. 2008;15:459. doi: 10.2174/092986708783503195. [DOI] [PubMed] [Google Scholar]
- (14).Popoff MR, Bouvet P. Future Microbiol. 2009;4:1021. doi: 10.2217/fmb.09.72. [DOI] [PubMed] [Google Scholar]
- (15).Alouf JE, Popoff MR, editors. Bacterial Protein Toxins. Third Edition Academic Press; San-Diego, California: 2006. [Google Scholar]
- (16).Karalliedde L. Br. J. Anaesth. 1995;74:319. doi: 10.1093/bja/74.3.319. [DOI] [PubMed] [Google Scholar]
- (17).Mouhat S, Jouirou B, Mosbah A, De Waard M, Sabatier JM. Biochem. J. 2004;378:717. doi: 10.1042/BJ20031860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Dutertre S, Lewis RJ. J. Biol. Chem. 2010;285:13315. doi: 10.1074/jbc.R109.076596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Sugrue RJ, Hay AJ. Virology. 1991;180:617. doi: 10.1016/0042-6822(91)90075-M. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Pinto LH, Holsinger LJ, Lamb RA. Cell. 1992;69:517. doi: 10.1016/0092-8674(92)90452-i. [DOI] [PubMed] [Google Scholar]
- (21).Wang C, Takeuchi K, Pinto LH, Lamb RA. J. Virol. 1993;67:5585. doi: 10.1128/jvi.67.9.5585-5594.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Leonov H, Astrahan P, Krugliak M, Arkin IT. J. Am. Chem. Soc. 2011;133:9903. doi: 10.1021/ja202288m. [DOI] [PubMed] [Google Scholar]
- (23).Pielak RM, Chou JJ. Protein Cell. 2010;1:246. doi: 10.1007/s13238-010-0025-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Rosenberg MR, Casarotto MG. Proc. Natl. Acad. Sci. U. S. A. 2010;107:13866. doi: 10.1073/pnas.1002051107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Boltz DA, Aldridge JRJ, Webster RG, Govorkova EA. Drugs. 2010;70:1349. doi: 10.2165/11537960-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Kozakov D, Chuang GY, Beglov D, Vajda S. Trends Biochem. Sci. 2010;35:471. doi: 10.1016/j.tibs.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Cady SD, Schmidt-Rohr K, Wang J, Soto CS, DeGrado WF, Hong M. Nature. 2010;463:689. doi: 10.1038/nature08722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Chuang GY, Kozakov D, Brenke R, Beglov D, Guarnieri F, Vajda S. Biophys. J. 2009;97:2846. doi: 10.1016/j.bpj.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Balannik V, Carnevale V, Fiorin G, Levine BG, Lamb RA, Klein ML, DeGrado WF, Pinto LH. Biochemistry. 2010;49:696. doi: 10.1021/bi901799k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Tran L, Choi SB, Al-Najjar BO, Yusuf M, Wahab HA, Le L. Molecules. 2011;16:10227. doi: 10.3390/molecules161210227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Wang J, Ma C, Fiorin G, Carnevale V, Wang T, Hu F, Lamb RA, Pinto LH, Hong M, Klein ML, DeGrado WF. J. Am. Chem. Soc. 2011;133:12834. doi: 10.1021/ja204969m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Du QS, Huang RB, Wang SQ, Chou KC. PLoS One. 2010;5:e9388. doi: 10.1371/journal.pone.0009388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Duque MD, Ma C, Torres E, Wang J, Naesens L, Juarez-Jimenez J, Camps P, Luque FJ, DeGrado WF, Lamb RA, Pinto LH, Vazquez S. J. Med. Chem. 2011;54:2646. doi: 10.1021/jm101334y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Balannik V, Wang J, Ohigashi Y, Jing X, Magavern E, Lamb RA, DeGrado WF, Pinto LH. Biochemistry. 2009;48:11872. doi: 10.1021/bi9014488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Van der Goot G, editor. Pore-forming toxins. Berlin Heidelberg New York. Springer - Verlag; 2001. p. 168. [Google Scholar]
- (36).Aktories K, Lang AE, Schwan C, Mannherz HG. FEBS J. 2011;278:4526. doi: 10.1111/j.1742-4658.2011.08113.x. [DOI] [PubMed] [Google Scholar]
- (37).Collier RJ. Mol. Aspects Med. 2009;30:413. doi: 10.1016/j.mam.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Sakurai J, Nagahama M, Oda M, Tsuge H, Kobayashi K. Toxins (Basel) 2009;1:208. doi: 10.3390/toxins1020208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Mueller P, Rudin DO, Tien HT, Wescott WC. Nature. 1962;194:979. doi: 10.1038/194979a0. [DOI] [PubMed] [Google Scholar]
- (40).White SH. Biophys. J. 1972;12:432. doi: 10.1016/S0006-3495(72)86095-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Montal M, Mueller P. Proc. Natl. Acad. Sci. U. S. A. 1972;69:3561. doi: 10.1073/pnas.69.12.3561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).White SH, Petersen DC, Simon S, Yafuso M. Biophys. J. 1976;16:481. doi: 10.1016/S0006-3495(76)85703-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Thoren KL, Worden EJ, Yassif JM, Krantz BA. Proc. Natl. Acad. Sci. U. S. A. 2009;106:21555. doi: 10.1073/pnas.0905880106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Feld GK, Thoren KL, Kintzer AF, Sterling HJ, Tang II, Greenberg SG, Williams ER, Krantz BA. Nat. Struct. Mol. Biol. 2010;17:1383. doi: 10.1038/nsmb.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Kintzer AF, Sterling HJ, Tang II, Williams ER, Krantz BA. PLoS One. 2010;5:e13888. doi: 10.1371/journal.pone.0013888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Blaustein RO. J. Gen. Physiol. 2011;137:337. doi: 10.1085/jgp.201110622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Basilio D, Jennings-Antipov LD, Jakes KS, Finkelstein A. J. Gen. Physiol. 2011;137:343. doi: 10.1085/jgp.201010578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Brown MJ, Thoren KL, Krantz BA. J. Biol. Chem. 2011;286:23189. doi: 10.1074/jbc.M111.231167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Basilio D, Kienker PK, Briggs SW, Finkelstein A. J. Gen. Physiol. 2011;137:521. doi: 10.1085/jgp.201110627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Thoren KL, Krantz BA. Mol. Microbiol. 2011;80:588. doi: 10.1111/j.1365-2958.2011.07614.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Karginov VA, Nestorovich EM, Moayeri M, Leppla SH, Bezrukov SM. Proc. Natl. Acad. Sci. U. S. A. 2005;102:15075. doi: 10.1073/pnas.0507488102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Karginov VA, Nestorovich EM, Yohannes A, Robinson TM, Fahmi NE, Schmidtmann F, Hecht SM, Bezrukov SM. Antimicrob. Agents Chemother. 2006;50:3740. doi: 10.1128/AAC.00693-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Karginov VA, Yohannes A, Robinson TM, Fahmi NE, Alibek K, Hecht SM. Bioorg. Med. Chem. 2006;14:33. doi: 10.1016/j.bmc.2005.07.054. [DOI] [PubMed] [Google Scholar]
- (54).Backer MV, Patel V, Jehning BT, Claffey KP, Karginov VA, Backer JM. Antimicrob. Agents Chemother. 2007;51:245. doi: 10.1128/AAC.00983-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Karginov VA, Nestorovich EM, Schmidtmann F, Robinson TM, Yohannes A, Fahmi NE, Bezrukov SM, Hecht SM. Bioorg. Med. Chem. 2007;15:5424. doi: 10.1016/j.bmc.2007.05.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Moayeri M, Robinson TM, Leppla SH, Karginov VA. Antimicrob. Agents Chemother. 2008;52:2239. doi: 10.1128/AAC.00009-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Nestorovich EM, Karginov VA, Berezhkovskii AM, Bezrukov SM. Biophys. J. 2010;99:134. doi: 10.1016/j.bpj.2010.03.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Nestorovich EM, Karginov VA, Bezrukov SM. Biophys. J. 2010;99:782. doi: 10.1016/j.bpj.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Ragle BE, Karginov VA, Bubeck Wardenburg J. Antimicrob. Agents Chemother. 2010;54:298. doi: 10.1128/AAC.00973-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Diaz-Moscoso A, Mendez-Ardoy A, Ortega-Caballero F, Benito JM, Ortiz Mellet C, Defaye J, Robinson TM, Yohannes A, Karginov VA, Garcia Fernandez JM. ChemMedChem. 2011;6:181. doi: 10.1002/cmdc.201000419. [DOI] [PubMed] [Google Scholar]
- (61).Nestorovich EM, Karginov VA, Popoff MR, Bezrukov SM, Barth H. PLoS One. 2011;6:e23927. doi: 10.1371/journal.pone.0023927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Yannakopoulou K, Jicsinszky L, Aggelidou C, Mourtzis N, Robinson TM, Yohannes A, Nestorovich EM, Bezrukov SM, Karginov VA. Antimicrob. Agents Chemother. 2011;55:3594. doi: 10.1128/AAC.01764-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Kasianowicz JJ, Brandin E, Branton D, Deamer DW. Proc. Natl. Acad. Sci. U. S. A. 1996;93:13770. doi: 10.1073/pnas.93.24.13770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Braha O, Walker B, Cheley S, Kasianowicz JJ, Song L, Gouaux JE, Bayley H. Chem. Biol. 1997;4:497. doi: 10.1016/s1074-5521(97)90321-5. [DOI] [PubMed] [Google Scholar]
- (65).Gu LQ, Braha O, Conlan S, Cheley S, Bayley H. Nature. 1999;398:686. doi: 10.1038/19491. [DOI] [PubMed] [Google Scholar]
- (66).Akeson M, Branton D, Kasianowicz JJ, Brandin E, Deamer DW. Biophys. J. 1999;77:3227. doi: 10.1016/S0006-3495(99)77153-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Henrickson SE, Misakian M, Robertson B, Kasianowicz JJ. Phys. Rev. Lett. 2000;85:3057. doi: 10.1103/PhysRevLett.85.3057. [DOI] [PubMed] [Google Scholar]
- (68).Meller A, Nivon L, Brandin E, Golovchenko J, Branton D. Proc. Natl. Acad. Sci. U. S. A. 2000;97:1079. doi: 10.1073/pnas.97.3.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Movileanu L, Bayley H. Proc. Natl. Acad. Sci. U. S. A. 2001;98:10137. doi: 10.1073/pnas.181089798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Kasianowicz JJ, Henrickson SE, Weetall HH, Robertson B. Anal. Chem. 2001;73:2268. doi: 10.1021/ac000958c. [DOI] [PubMed] [Google Scholar]
- (71).Meller A, Nivon L, Branton D. Phys. Rev. Lett. 2001;86:3435. doi: 10.1103/PhysRevLett.86.3435. [DOI] [PubMed] [Google Scholar]
- (72).Miles G, Cheley S, Braha O, Bayley H. Biochemistry. 2001;40:8514. doi: 10.1021/bi010454o. [DOI] [PubMed] [Google Scholar]
- (73).Cheley S, Gu LQ, Bayley H. Chem. Biol. 2002;9:829. doi: 10.1016/s1074-5521(02)00172-2. [DOI] [PubMed] [Google Scholar]
- (74).Guan X, Gu LQ, Cheley S, Braha O, Bayley H. Chembiochem. 2005;6:1875. doi: 10.1002/cbic.200500064. [DOI] [PubMed] [Google Scholar]
- (75).Movileanu L, Schmittschmitt JP, Scholtz JM, Bayley H. Biophys. J. 2005;89:1030. doi: 10.1529/biophysj.104.057406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Mathe J, Aksimentiev A, Nelson DR, Schulten K, Meller A. Proc. Natl. Acad. Sci. U. S. A. 2005;102:12377. doi: 10.1073/pnas.0502947102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Kang XF, Cheley S, Guan X, Bayley H. J. Am. Chem. Soc. 2006;128:10684. doi: 10.1021/ja063485l. [DOI] [PubMed] [Google Scholar]
- (78).Wolfe AJ, Mohammad MM, Cheley S, Bayley H, Movileanu L. J. Am. Chem. Soc. 2007;129:14034. doi: 10.1021/ja0749340. [DOI] [PubMed] [Google Scholar]
- (79).Hornblower B, Coombs A, Whitaker RD, Kolomeisky A, Picone SJ, Meller A, Akeson M. Nat. Methods. 2007;4:315. doi: 10.1038/nmeth1021. [DOI] [PubMed] [Google Scholar]
- (80).Wu HC, Bayley H. J. Am. Chem. Soc. 2008;130:6813. doi: 10.1021/ja8004607. [DOI] [PubMed] [Google Scholar]
- (81).Mohammad MM, Prakash S, Matouschek A, Movileanu L. J. Am. Chem. Soc. 2008;130:4081. doi: 10.1021/ja710787a. [DOI] [PubMed] [Google Scholar]
- (82).Mohammad MM, Movileanu L. Eur. Biophys. J. 2008;37:913. doi: 10.1007/s00249-008-0309-9. [DOI] [PubMed] [Google Scholar]
- (83).Stoddart D, Heron AJ, Mikhailova E, Maglia G, Bayley H. Proc. Natl. Acad. Sci. U. S. A. 2009;106:7702. doi: 10.1073/pnas.0901054106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Stoddart D, Heron AJ, Klingelhoefer J, Mikhailova E, Maglia G, Bayley H. Nano Lett. 2010;10:3633. doi: 10.1021/nl101955a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Hall AR, Scott A, Rotem D, Mehta KK, Bayley H, Dekker C. Nat. Nanotechnol. 2010;5:874. doi: 10.1038/nnano.2010.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Banerjee A, Mikhailova E, Cheley S, Gu LQ, Montoya M, Nagaoka Y, Gouaux E, Bayley H. Proc. Natl. Acad. Sci. U. S. A. 2010;107:8165. doi: 10.1073/pnas.0914229107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Wu Y, Ma L, Cheley S, Bayley H, Cui Q, Chapman ER. Biochemistry. 2011;50:7493. doi: 10.1021/bi2006288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Li WW, Claridge TD, Li Q, Wormald MR, Davis BG, Bayley H. J. Am. Chem. Soc. 2011 doi: 10.1021/ja1100867. [DOI] [PubMed] [Google Scholar]
- (89).Hammerstein AF, Jayasinghe L, Bayley H. J. Biol. Chem. 2011;286:14324. doi: 10.1074/jbc.M111.218164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Rotem D, Jayasinghe L, Salichou M, Bayley H. J. Am. Chem. Soc. 2012;134:2781. doi: 10.1021/ja2105653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Bashford C. Cell. Biol. Mol. Lett. 2001;6:328. [Google Scholar]
- (92).Iacovache I, Paumard P, Scheib H, Lesieur C, Sakai N, Matile S, Parker MW, van der Goot FG. EMBO J. 2006;25:457. doi: 10.1038/sj.emboj.7600959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Lakey JH, Slatin SL. Curr. Top. Microbiol. Immunol. 2001;257:131. doi: 10.1007/978-3-642-56508-3_7. [DOI] [PubMed] [Google Scholar]
- (94).Zakharov SD, Cramer WA. Biochim. Biophys. Acta. 2002;1565:333. doi: 10.1016/s0005-2736(02)00579-5. [DOI] [PubMed] [Google Scholar]
- (95).Cascales E, Buchanan SK, Duche D, Kleanthous C, Lloubes R, Postle K, Riley M, Slatin S, Cavard D. Microbiol. Mol. Biol. Rev. 2007;71:158. doi: 10.1128/MMBR.00036-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Gratia A, Fredericq P. C. R. Soc. Biol. (Paris) 1946;140:1032. [Google Scholar]
- (97).Lloubes R, Baty D, Lazdunski C. Nucleic Acids Res. 1986;14:2621. doi: 10.1093/nar/14.6.2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Kleanthous C. Nat. Rev. Microbiol. 2010;8:843. doi: 10.1038/nrmicro2454. [DOI] [PubMed] [Google Scholar]
- (99).Bouveret E, Journet L, Walburger A, Cascales E, Benedetti H, Lloubes R. Biochimie. 2002;84:413. doi: 10.1016/s0300-9084(02)01423-2. [DOI] [PubMed] [Google Scholar]
- (100).Spangler R, Zhang SP, Krueger J, Zubay G. J. Bacteriol. 1985;163:167. doi: 10.1128/jb.163.1.167-173.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Vetter IR, Parker MW, Tucker AD, Lakey JH, Pattus F, Tsernoglou D. Structure. 1998;6:863. doi: 10.1016/s0969-2126(98)00088-4. [DOI] [PubMed] [Google Scholar]
- (102).Jakes KS. Mol. Cell. 2001;8:4. doi: 10.1016/s1097-2765(01)00290-8. [DOI] [PubMed] [Google Scholar]
- (103).James R, Penfold CN, Moore GR, Kleanthous C. Biochimie. 2002;84:381. doi: 10.1016/s0300-9084(02)01450-5. [DOI] [PubMed] [Google Scholar]
- (104).Housden NG, Loftus SR, Moore GR, James R, Kleanthous C. Proc. Natl. Acad. Sci. U. S. A. 2005;102:13849. doi: 10.1073/pnas.0503567102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Ridley H, Johnson CL, Lakey JH. Adv. Exp. Med. Biol. 2010;677:81. doi: 10.1007/978-1-4419-6327-7_7. [DOI] [PubMed] [Google Scholar]
- (106).Mosbahi K, Lemaitre C, Keeble AH, Mobasheri H, Morel B, James R, Moore GR, Lea EJ, Kleanthous C. Nat. Struct. Biol. 2002;9:476. doi: 10.1038/nsb797. [DOI] [PubMed] [Google Scholar]
- (107).Walker D, Mosbahi K, Vankemmelbeke M, James R, Kleanthous C. J. Biol. Chem. 2007;282:31389. doi: 10.1074/jbc.M705883200. [DOI] [PubMed] [Google Scholar]
- (108).Parker MW, Tucker AD, Tsernoglou D, Pattus F. Trends Biochem. Sci. 1990;15:126. doi: 10.1016/0968-0004(90)90205-p. [DOI] [PubMed] [Google Scholar]
- (109).Vetter IR, Parker MW, Pattus F, Tsernoglou D. Insights into membrane insertion based on studies of colicins. Protein Toxin Structure; RG Landes Company; Austin, Texas and Springer: Heidelberg: 1996. pp. 5–23. [Google Scholar]
- (110).Parker MW, Pattus F, Tucker AD, Tsernoglou D. Nature. 1989;337:93. doi: 10.1038/337093a0. [DOI] [PubMed] [Google Scholar]
- (111).Wormald MR, Merrill AR, Cramer WA, Williams RJ. Eur. J. Biochem. 1990;191:155. doi: 10.1111/j.1432-1033.1990.tb19105.x. [DOI] [PubMed] [Google Scholar]
- (112).Parker MW, Postma JP, Pattus F, Tucker AD, Tsernoglou D. J. Mol. Biol. 1992;224:639. doi: 10.1016/0022-2836(92)90550-4. [DOI] [PubMed] [Google Scholar]
- (113).Ghosh P, Mel SF, Stroud RM. Nat. Struct. Biol. 1994;1:597. doi: 10.1038/nsb0994-597. [DOI] [PubMed] [Google Scholar]
- (114).Wiener M, Freymann D, Ghosh P, Stroud RM. Nature. 1997;385:461. doi: 10.1038/385461a0. [DOI] [PubMed] [Google Scholar]
- (115).Kleanthous C, Kuhlmann UC, Pommer AJ, Ferguson N, Radford SE, Moore GR, James R, Hemmings AM. Nat. Struct. Biol. 1999;6:243. doi: 10.1038/6683. [DOI] [PubMed] [Google Scholar]
- (116).Ko TP, Liao CC, Ku WY, Chak KF, Yuan HS. Structure. 1999;7:91. doi: 10.1016/s0969-2126(99)80012-4. [DOI] [PubMed] [Google Scholar]
- (117).Soelaiman S, Jakes K, Wu N, Li C, Shoham M. Mol. Cell. 2001;8:1053. doi: 10.1016/s1097-2765(01)00396-3. [DOI] [PubMed] [Google Scholar]
- (118).Hilsenbeck JL, Park H, Chen G, Youn B, Postle K, Kang C. Mol. Microbiol. 2004;51:711. doi: 10.1111/j.1365-2958.2003.03884.x. [DOI] [PubMed] [Google Scholar]
- (119).Elkins P, Bunker A, Cramer WA, Stauffacher CV. Structure. 1997;5:443. doi: 10.1016/s0969-2126(97)00200-1. [DOI] [PubMed] [Google Scholar]
- (120).Mel SF, Falick AM, Burlingame AL, Stroud RM. Biochemistry. 1993;32:9473. doi: 10.1021/bi00087a027. [DOI] [PubMed] [Google Scholar]
- (121).Muga A, Gonzalez-Manas JM, Lakey JH, Pattus F, Surewicz WK. J. Biol. Chem. 1993;268:1553. [PubMed] [Google Scholar]
- (122).Wilmsen HU, Pugsley AP, Pattus F. Eur. Biophys. J. 1990;18:149. doi: 10.1007/BF02427374. [DOI] [PubMed] [Google Scholar]
- (123).Greig SL, Radjainia M, Mitra AK. J. Biol. Chem. 2009;284:16126. doi: 10.1074/jbc.M900292200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Bullock JO, Kolen ER, Shear JL. J. Membr. Biol. 1992;128:1. doi: 10.1007/BF00231866. [DOI] [PubMed] [Google Scholar]
- (125).Bullock JO. J. Membr. Biol. 1992;125:255. doi: 10.1007/BF00236438. [DOI] [PubMed] [Google Scholar]
- (126).Bullock JO, Kolen ER. J. Membr. Biol. 1995;144:131. doi: 10.1007/BF00232799. [DOI] [PubMed] [Google Scholar]
- (127).Krasilnikov OV, Yuldasheva LN, Nogueira RA, Rodrigues CG. Braz. J. Med. Biol. Res. 1995;28:693. [PubMed] [Google Scholar]
- (128).Krasilnikov OV, Da Cruz JB, Yuldasheva LN, Varanda WA, Nogueira RA. J. Membr. Biol. 1998;161:83. doi: 10.1007/s002329900316. [DOI] [PubMed] [Google Scholar]
- (129).Slatin SL, Finkelstein A, Kienker PK. Biochemistry. 2008;47:1778. doi: 10.1021/bi701900x. [DOI] [PubMed] [Google Scholar]
- (130).Slatin SL, Duche D, Baty D. Biochemistry. 2010;49:4786. doi: 10.1021/bi100122g. [DOI] [PubMed] [Google Scholar]
- (131).Bainbridge G, Gokce I, Lakey JH. FEBS Lett. 1998;431:305. doi: 10.1016/s0014-5793(98)00761-3. [DOI] [PubMed] [Google Scholar]
- (132).Delcour AH. J. Mol. Microbiol. Biotechnol. 2002;4:1. [PubMed] [Google Scholar]
- (133).Hille B. Ion channels of excitable membranes. Siunderland; Siunderland, MA: 2001. [Google Scholar]
- (134).Merrill AR, Cohen FS, Cramer WA. Biochemistry. 1990;29:5829. doi: 10.1021/bi00476a026. [DOI] [PubMed] [Google Scholar]
- (135).Qiu XQ, Jakes KS, Kienker PK, Finkelstein A, Slatin SL. J. Gen. Physiol. 1996;107:313. doi: 10.1085/jgp.107.3.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (136).Jakes KS, Kienker PK, Finkelstein A. Q. Rev. Biophys. 1999;32:189. doi: 10.1017/s0033583599003492. [DOI] [PubMed] [Google Scholar]
- (137).Schein SJ, Kagan BL, Finkelstein A. Nature. 1978;276:159. doi: 10.1038/276159a0. [DOI] [PubMed] [Google Scholar]
- (138).Weaver CA, Kagan BL, Finkelstein A, Konisky J. Biochim. Biophys. Acta. 1981;645:137. doi: 10.1016/0005-2736(81)90521-6. [DOI] [PubMed] [Google Scholar]
- (139).Cleveland MV, Slatin S, Finkelstein A, Levinthal C. Proc. Natl. Acad. Sci. U. S. A. 1983;80:3706. doi: 10.1073/pnas.80.12.3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (140).Finkelstein A. Ann. N. Y. Acad. Sci. 1985;456:26. doi: 10.1111/j.1749-6632.1985.tb14840.x. [DOI] [PubMed] [Google Scholar]
- (141).Raymond L, Slatin SL, Finkelstein A. J. Membr. Biol. 1985;84:173. doi: 10.1007/BF01872215. [DOI] [PubMed] [Google Scholar]
- (142).Liu QR, Crozel V, Levinthal F, Slatin S, Finkelstein A, Levinthal C. Proteins. 1986;1:218. doi: 10.1002/prot.340010304. [DOI] [PubMed] [Google Scholar]
- (143).Raymond L, Slatin SL, Finkelstein A, Liu QR, Levinthal C. J. Membr. Biol. 1986;92:255. doi: 10.1007/BF01869394. [DOI] [PubMed] [Google Scholar]
- (144).Slatin SL, Raymond L, Finkelstein A. J. Membr. Biol. 1986;92:247. doi: 10.1007/BF01869393. [DOI] [PubMed] [Google Scholar]
- (145).Jakes KS, Abrams CK, Finkelstein A, Slatin SL. J. Biol. Chem. 1990;265:6984. [PubMed] [Google Scholar]
- (146).Abrams CK, Jakes KS, Finkelstein A, Slatin SL. J. Gen. Physiol. 1991;98:77. doi: 10.1085/jgp.98.1.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (147).Qiu XQ, Jakes KS, Finkelstein A, Slatin SL. J. Biol. Chem. 1994;269:7483. [PubMed] [Google Scholar]
- (148).Slatin SL, Qiu XQ, Jakes KS, Finkelstein A. Nature. 1994;371:158. doi: 10.1038/371158a0. [DOI] [PubMed] [Google Scholar]
- (149).Kienker PK, Qiu X, Slatin SL, Finkelstein A, Jakes KS. J. Membr. Biol. 1997;157:27. doi: 10.1007/s002329900213. [DOI] [PubMed] [Google Scholar]
- (150).Jakes KS, Kienker PK, Slatin SL, Finkelstein A. Proc. Natl. Acad. Sci. U. S. A. 1998;95:4321. doi: 10.1073/pnas.95.8.4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (151).Kienker PK, Jakes KS, Finkelstein A. J. Gen. Physiol. 2000;116:587. doi: 10.1085/jgp.116.4.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (152).Kienker PK, Jakes KS, Blaustein RO, Miller C, Finkelstein A. J. Gen. Physiol. 2003;122:161. doi: 10.1085/jgp.200308852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (153).Kienker PK, Jakes KS, Finkelstein A. J. Gen. Physiol. 2008;132:693. doi: 10.1085/jgp.200810042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (154).Jakes KS, Finkelstein A. Mol. Microbiol. 2010;75:567. doi: 10.1111/j.1365-2958.2009.06966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (155).Slatin SL. Int. J. Biochem. 1988;20:737. doi: 10.1016/0020-711x(88)90058-4. [DOI] [PubMed] [Google Scholar]
- (156).Sobko AA, Kotova EA, Antonenko YN, Zakharov SD, Cramer WA. J. Biol. Chem. 2006;281:14408. doi: 10.1074/jbc.M513634200. [DOI] [PubMed] [Google Scholar]
- (157).Nassi S, Collier RJ, Finkelstein A. Biochemistry. 2002;41:1445. doi: 10.1021/bi0119518. [DOI] [PubMed] [Google Scholar]
- (158).Neary M. Nurs. Stand. 2011;26:57. doi: 10.7748/ns2011.12.26.15.57.c8868. [DOI] [PubMed] [Google Scholar]
- (159).Deleo FR, Otto M, Kreiswirth BN, Chambers HF. Lancet. 2010;375:1557. doi: 10.1016/S0140-6736(09)61999-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (160).Otto M. Annu. Rev. Microbiol. 2010;64:143. doi: 10.1146/annurev.micro.112408.134309. [DOI] [PubMed] [Google Scholar]
- (161).Otto M. Future Microbiol. 2012;7:189. doi: 10.2217/fmb.11.156. [DOI] [PubMed] [Google Scholar]
- (162).Van de Velde H. La Cellule. 1894;10:403. [Google Scholar]
- (163).Denys J, Van de Velde H. La Cellule. 1895;11:359. [Google Scholar]
- (164).Otto M. Nat. Med. 2011;17:169. doi: 10.1038/nm0211-169. [DOI] [PubMed] [Google Scholar]
- (165).Gouaux JE, Braha O, Hobaugh MR, Song L, Cheley S, Shustak C, Bayley H. Proc. Natl. Acad. Sci. U. S. A. 1994;91:12828. doi: 10.1073/pnas.91.26.12828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (166).Valeva A, Weisser A, Walker B, Kehoe M, Bayley H, Bhakdi S, Palmer M. EMBO J. 1996;15:1857. [PMC free article] [PubMed] [Google Scholar]
- (167).Song L, Hobaugh MR, Shustak C, Cheley S, Bayley H, Gouaux JE. Science. 1996;274:1859. doi: 10.1126/science.274.5294.1859. [DOI] [PubMed] [Google Scholar]
- (168).Valeva A, Pongs J, Bhakdi S, Palmer M. Biochim. Biophys. Acta. 1997;1325:281. doi: 10.1016/s0005-2736(96)00266-0. [DOI] [PubMed] [Google Scholar]
- (169).Valeva A, Palmer M, Bhakdi S. Biochemistry. 1997;36:13298. doi: 10.1021/bi971075r. [DOI] [PubMed] [Google Scholar]
- (170).Fang Y, Cheley S, Bayley H, Yang J. Biochemistry. 1997;36:9518. doi: 10.1021/bi970600j. [DOI] [PubMed] [Google Scholar]
- (171).Bhakdi S, Fussle R, Tranum-Jensen J. Proc. Natl. Acad. Sci. U. S. A. 1981;78:5475. doi: 10.1073/pnas.78.9.5475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (172).Czajkowsky DM, Sheng S, Shao Z. J. Mol. Biol. 1998;276:325. doi: 10.1006/jmbi.1997.1535. [DOI] [PubMed] [Google Scholar]
- (173).Thelestam M, Olofsson A, Blomqvist L, Hebert H. Biochim. Biophys. Acta. 1991;1062:245. doi: 10.1016/0005-2736(91)90399-s. [DOI] [PubMed] [Google Scholar]
- (174).Krasilnikov O, Ternovsky V, Musaev Y. Dokl AN UzSSR. 1980;N7:66. [Google Scholar]
- (175).Krasilnikov OV, Merzliak PG, Sabirov RZ, Tashmuk-Hamedov BA. Gen. Physiol. Biophys. 1990;9:569. [PubMed] [Google Scholar]
- (176).Krasilnikov OV, Ternovsky VI, Merzliak PG, Zachidova LT, Hungerer KD. Biochim. Biophys. Acta. 1993;1182:94. doi: 10.1016/0925-4439(93)90158-w. [DOI] [PubMed] [Google Scholar]
- (177).Krasilnikov OV, Merzlyak PG, Yuldasheva LN, Azimova RK, Nogueira RA. Med. Microbiol. Immunol. 1997;186:53. doi: 10.1007/s004300050046. [DOI] [PubMed] [Google Scholar]
- (178).Merzlyak PG, Yuldasheva LN, Rodrigues CG, Carneiro CM, Krasilnikov OV, Bezrukov SM. Biophys. J. 1999;77:3023. doi: 10.1016/S0006-3495(99)77133-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (179).Krasilnikov OV, Merzlyak PG, Yuldasheva LN, Rodrigues CG, Nogueira RA. Biochim. Biophys. Acta. 1999;1417:167. doi: 10.1016/s0005-2736(98)00244-2. [DOI] [PubMed] [Google Scholar]
- (180).Krasilnikov OV, Merzlyak PG, Yuldasheva LN, Rodrigues CG, Bhakdi S, Valeva A. Mol. Microbiol. 2000;37:1372. doi: 10.1046/j.1365-2958.2000.02080.x. [DOI] [PubMed] [Google Scholar]
- (181).Krasilnikov OV, Sabirov RZ, Ternovsky VI, Merzliak PG, Tashmukhamedov BA. Gen. Physiol. Biophys. 1988;7:467. [PubMed] [Google Scholar]
- (182).Bezrukov SM, Vodyanoy I. Biophys. J. 1993;64:16. doi: 10.1016/S0006-3495(93)81336-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (183).Korchev YE, Bashford CL, Alder GM, Kasianowicz JJ, Pasternak CA. J. Membr. Biol. 1995;147:233. doi: 10.1007/BF00234521. [DOI] [PubMed] [Google Scholar]
- (184).Bezrukov S, Vodyanoy I, Kasianowicz J. Macromolecules. 1996;29:8517. [Google Scholar]
- (185).Berestovsky GN, Ternovsky VI, Kataev AA. J. Exp. Bot. 2001;52:1173. [PubMed] [Google Scholar]
- (186).Rostovtseva TK, Nestorovich EM, Bezrukov SM. Biophys. J. 2002;82:160. doi: 10.1016/S0006-3495(02)75383-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (187).Ternovsky VI, Okada Y, Sabirov RZ. FEBS Lett. 2004;576:433. doi: 10.1016/j.febslet.2004.09.051. [DOI] [PubMed] [Google Scholar]
- (188).Nestorovich EM, Sugawara E, Nikaido H, Bezrukov SM. J. Biol. Chem. 2006;281:16230. doi: 10.1074/jbc.M600650200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (189).Nablo BJ, Halverson KM, Robertson JW, Nguyen TL, Panchal RG, Gussio R, Bavari S, Krasilnikov OV, Kasianowicz JJ. Biophys. J. 2008;95:1157. doi: 10.1529/biophysj.107.121715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (190).Duret G, Delcour AH. Biophys. J. 2010;98:1820. doi: 10.1016/j.bpj.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (191).Stojilkovic K, Berezhkovskii A, Bezrukov S. J. Chem. Phys. 2003;119:6973. [Google Scholar]
- (192).Kaneko J, Kamio Y. Biosci. Biotechnol. Biochem. 2004;68:981. doi: 10.1271/bbb.68.981. [DOI] [PubMed] [Google Scholar]
- (193).Yamashita K, Kawai Y, Tanaka Y, Hirano N, Kaneko J, Tomita N, Ohta M, Kamio Y, Yao M, Tanaka I. Proc. Natl. Acad. Sci. U. S. A. 2011;108:17314. doi: 10.1073/pnas.1110402108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (194).Miles G, Movileanu L, Bayley H. Protein Sci. 2002;11:894. doi: 10.1110/ps.4360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (195).Miles G, Jayasinghe L, Bayley H. J. Biol. Chem. 2006;281:2205. doi: 10.1074/jbc.M510842200. [DOI] [PubMed] [Google Scholar]
- (196).Sayeed S, Fernandez-Miyakawa ME, Fisher DJ, Adams V, Poon R, Rood JI, Uzal FA, McClane BA. Infect. Immun. 2005;73:7413. doi: 10.1128/IAI.73.11.7413-7421.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (197).Popoff MR. FEBS J. 2011;278:4602. doi: 10.1111/j.1742-4658.2011.08145.x. [DOI] [PubMed] [Google Scholar]
- (198).Bokori-Brown M, Savva CG, Fernandes da Costa SP, Naylor CE, Basak AK, Titball RW. FEBS J. 2011;278:4589. doi: 10.1111/j.1742-4658.2011.08140.x. [DOI] [PubMed] [Google Scholar]
- (199).Marks JD. Anesthesiol. Clin. North America. 2004;22:509. doi: 10.1016/j.atc.2004.05.010. [DOI] [PubMed] [Google Scholar]
- (200).Songer JG. Vet. Microbiol. 2010;140:399. doi: 10.1016/j.vetmic.2009.07.003. [DOI] [PubMed] [Google Scholar]
- (201).Hunter SE, Clarke IN, Kelly DC, Titball RW. Infect. Immun. 1992;60:102. doi: 10.1128/iai.60.1.102-110.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (202).Minami J, Katayama S, Matsushita O, Matsushita C, Okabe A. Microbiol. Immunol. 1997;41:527. doi: 10.1111/j.1348-0421.1997.tb01888.x. [DOI] [PubMed] [Google Scholar]
- (203).Gill DM. Microbiol. Rev. 1982;46:86. doi: 10.1128/mr.46.1.86-94.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (204).Cole AR, Gibert M, Popoff M, Moss DS, Titball RW, Basak AK. Nat. Struct. Mol. Biol. 2004;11:797. doi: 10.1038/nsmb804. [DOI] [PubMed] [Google Scholar]
- (205).Miyata S, Matsushita O, Minami J, Katayama S, Shimamoto S, Okabe A. J. Biol. Chem. 2001;276:13778. doi: 10.1074/jbc.M011527200. [DOI] [PubMed] [Google Scholar]
- (206).Miyata S, Minami J, Tamai E, Matsushita O, Shimamoto S, Okabe A. J. Biol. Chem. 2002;277:39463. doi: 10.1074/jbc.M206731200. [DOI] [PubMed] [Google Scholar]
- (207).Petit L, Gibert M, Gillet D, Laurent-Winter C, Boquet P, Popoff MR. J. Bacteriol. 1997;179:6480. doi: 10.1128/jb.179.20.6480-6487.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (208).Petit L, Gibert M, Gourch A, Bens M, Vandewalle A, Popoff MR. Cell. Microbiol. 2003;5:155. doi: 10.1046/j.1462-5822.2003.00262.x. [DOI] [PubMed] [Google Scholar]
- (209).Chassin C, Bens M, de Barry J, Courjaret R, Bossu JL, Cluzeaud F, Ben Mkaddem S, Gibert M, Poulain B, Popoff MR, Vandewalle A. Am. J. Physiol. Renal Physiol. 2007;293:F927. doi: 10.1152/ajprenal.00199.2007. [DOI] [PubMed] [Google Scholar]
- (210).Borrmann E, Gunther H, Kohler H. FEMS Immunol. Med. Microbiol. 2001;31:85. doi: 10.1111/j.1574-695X.2001.tb00503.x. [DOI] [PubMed] [Google Scholar]
- (211).Soler-Jover A, Blasi J, Gomez de Aranda I, Navarro P, Gibert M, Popoff MR, Martin-Satue M. J. Histochem. Cytochem. 2004;52:931. doi: 10.1369/jhc.4A6254.2004. [DOI] [PubMed] [Google Scholar]
- (212).Lindsay CD. Hum. Exp. Toxicol. 1996;15:904. doi: 10.1177/096032719601501107. [DOI] [PubMed] [Google Scholar]
- (213).Robertson SL, Li J, Uzal FA, McClane BA. PLoS One. 2011;6:e22053. doi: 10.1371/journal.pone.0022053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (214).Petosa C, Collier RJ, Klimpel KR, Leppla SH, Liddington RC. Nature. 1997;385:833. doi: 10.1038/385833a0. [DOI] [PubMed] [Google Scholar]
- (215).Petit L, Maier E, Gibert M, Popoff MR, Benz R. J. Biol. Chem. 2001;276:15736. doi: 10.1074/jbc.M010412200. [DOI] [PubMed] [Google Scholar]
- (216).Knapp O, Maier E, Benz R, Geny B, Popoff MR. Biochim. Biophys. Acta. 2009;1788:2584. doi: 10.1016/j.bbamem.2009.09.020. [DOI] [PubMed] [Google Scholar]
- (217).Nagahama M, Hara H, Fernandez-Miyakawa M, Itohayashi Y, Sakurai J. Biochemistry. 2006;45:296. doi: 10.1021/bi051805s. [DOI] [PubMed] [Google Scholar]
- (218).Alcaraz A, Nestorovich EM, Aguilella-Arzo M, Aguilella VM, Bezrukov SM. Biophys. J. 2004;87:943. doi: 10.1529/biophysj.104/043414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (219).Siwy Z, Kosinska ID, Fulinski A, Martin CR. Phys. Rev. Lett. 2005;94:048102. doi: 10.1103/PhysRevLett.94.048102. [DOI] [PubMed] [Google Scholar]
- (220).Cervera J, Alcaraz A, Ramirez P. J. Phys. Chem. C. 2007;111:12265. [Google Scholar]
- (221).Knapp O, Popoff M. The Open Toxinology Journal. 2010;3:53. [Google Scholar]
- (222).Abrami L, Fivaz M, van der Goot FG. Trends Microbiol. 2000;8:168. doi: 10.1016/s0966-842x(00)01722-4. [DOI] [PubMed] [Google Scholar]
- (223).Abrami L, Fivaz M, Decroly E, Seidah NG, Jean F, Thomas G, Leppla SH, Buckley JT, van der Goot FG. J. Biol. Chem. 1998;273:32656. doi: 10.1074/jbc.273.49.32656. [DOI] [PubMed] [Google Scholar]
- (224).Abrami L, Fivaz M, Glauser PE, Parton RG, van der Goot FG. J. Cell Biol. 1998;140:525. doi: 10.1083/jcb.140.3.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (225).Lacy DB, Stevens RC. Curr. Opin. Struct. Biol. 1998;8:778. doi: 10.1016/s0959-440x(98)80098-5. [DOI] [PubMed] [Google Scholar]
- (226).Howard SP, Buckley JT. J. Bacteriol. 1985;163:336. doi: 10.1128/jb.163.1.336-340.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (227).van der Goot FG, Lakey J, Pattus F, Kay CM, Sorokine O, Van Dorsselaer A, Buckley JT. Biochemistry. 1992;31:8566. doi: 10.1021/bi00151a026. [DOI] [PubMed] [Google Scholar]
- (228).Parker MW, Buckley JT, Postma JP, Tucker AD, Leonard K, Pattus F, Tsernoglou D. Nature. 1994;367:292. doi: 10.1038/367292a0. [DOI] [PubMed] [Google Scholar]
- (229).Wilmsen HU, Pattus F, Buckley JT. J. Membr. Biol. 1990;115:71. doi: 10.1007/BF01869107. [DOI] [PubMed] [Google Scholar]
- (230).Wilmsen HU, Buckley JT, Pattus F. Mol. Microbiol. 1991;5:2745. doi: 10.1111/j.1365-2958.1991.tb01983.x. [DOI] [PubMed] [Google Scholar]
- (231).Wilmsen HU, Leonard KR, Tichelaar W, Buckley JT, Pattus F. EMBO J. 1992;11:2457. doi: 10.1002/j.1460-2075.1992.tb05310.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (232).Pastoriza-Gallego M, Rabah L, Gibrat G, Thiebot B, van der Goot FG, Auvray L, Betton JM, Pelta J. J. Am. Chem. Soc. 2011;133:2923. doi: 10.1021/ja1073245. [DOI] [PubMed] [Google Scholar]
- (233).Tsitrin Y, Morton CJ, el-Bez C, Paumard P, Velluz MC, Adrian M, Dubochet J, Parker MW, Lanzavecchia S, van der Goot FG. Nat. Struct. Biol. 2002;9:729. doi: 10.1038/nsb839. [DOI] [PubMed] [Google Scholar]
- (234).Stefureac R, Long YT, Kraatz HB, Howard P, Lee JS. Biochemistry. 2006;45:9172. doi: 10.1021/bi0604835. [DOI] [PubMed] [Google Scholar]
- (235).Stefureac R, Waldner L, Howard P, Lee JS. Small. 2008;4:59. doi: 10.1002/smll.200700402. [DOI] [PubMed] [Google Scholar]
- (236).Oukhaled G, Mathe J, Biance AL, Bacri L, Betton JM, Lairez D, Pelta J, Auvray L. Phys. Rev. Lett. 2007;98:158101. doi: 10.1103/PhysRevLett.98.158101. [DOI] [PubMed] [Google Scholar]
- (237).He Y, Olson R. J. Struct. Biol. 2010;169:6. doi: 10.1016/j.jsb.2009.07.015. [DOI] [PubMed] [Google Scholar]
- (238).De S, Olson R. Proc. Natl. Acad. Sci. U. S. A. 2011;108:7385. doi: 10.1073/pnas.1017442108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (239).Olson R, Gouaux E. J. Mol. Biol. 2005;350:997. doi: 10.1016/j.jmb.2005.05.045. [DOI] [PubMed] [Google Scholar]
- (240).Krantz BA, Melnyk RA, Zhang S, Juris SJ, Lacy DB, Wu Z, Finkelstein A, Collier RJ. Science. 2005;309:777. doi: 10.1126/science.1113380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (241).Krasilnikov OV, Sabirov RZ, Ternovsky VI, Merzliak PG, Muratkhodjaev JN. FEMS Microbiol. Immunol. 1992;5:93. doi: 10.1111/j.1574-6968.1992.tb05891.x. [DOI] [PubMed] [Google Scholar]
- (242).Krasilnikov OV, Muratkhodjaev JN, Zitzer AO. Biochim. Biophys. Acta. 1992;1111:7. doi: 10.1016/0005-2736(92)90268-q. [DOI] [PubMed] [Google Scholar]
- (243).Yuldasheva LN, Merzlyak PG, Zitzer AO, Rodrigues CG, Bhakdi S, Krasilnikov OV. Biochim. Biophys. Acta. 2001;1512:53. doi: 10.1016/s0005-2736(01)00302-9. [DOI] [PubMed] [Google Scholar]
- (244).Krasilnikov OV, Merzlyak PG, Lima VL, Zitzer AO, Valeva A, Yuldasheva LN. Biochimie. 2007;89:271. doi: 10.1016/j.biochi.2006.12.003. [DOI] [PubMed] [Google Scholar]
- (245).Krasilnikov OV, Muratkhodjaev JN, Voronov SE, Yezepchuk YV. Biochim. Biophys. Acta. 1991;1067:166. doi: 10.1016/0005-2736(91)90039-b. [DOI] [PubMed] [Google Scholar]
- (246).Krasilnikov OV, Yuldasheva LN. Biochimie. 2009;91:620. doi: 10.1016/j.biochi.2009.03.005. [DOI] [PubMed] [Google Scholar]
- (247).Menzl K, Maier E, Chakraborty T, Benz R. Eur. J. Biochem. 1996;240:646. doi: 10.1111/j.1432-1033.1996.0646h.x. [DOI] [PubMed] [Google Scholar]
- (248).Mandal S, Mandal MD, Pal NK. Asian Pac. J. Trop. Med. 2011;4:573. doi: 10.1016/S1995-7645(11)60149-1. [DOI] [PubMed] [Google Scholar]
- (249).Schwan C, Stecher B, Tzivelekidis T, van Ham M, Rohde M, Hardt WD, Wehland J, Aktories K. PLoS Pathog. 2009;5:e1000626. doi: 10.1371/journal.ppat.1000626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (250).Cartman ST, Heap JT, Kuehne SA, Cockayne A, Minton NP. Int. J. Med. Microbiol. 2010;300:387. doi: 10.1016/j.ijmm.2010.04.008. [DOI] [PubMed] [Google Scholar]
- (251).Sundriyal A, Roberts AK, Ling R, McGlashan J, Shone CC, Acharya KR. Protein Expr. Purif. 2010;74:42. doi: 10.1016/j.pep.2010.04.014. [DOI] [PubMed] [Google Scholar]
- (252).Carman RJ, Stevens AL, Lyerly MW, Hiltonsmith MF, Stiles BG, Wilkins TD. Anaerobe. 2011;17:161. doi: 10.1016/j.anaerobe.2011.02.005. [DOI] [PubMed] [Google Scholar]
- (253).Shen A. J. Innate Immun. 2012;4:149. doi: 10.1159/000332946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (254).Hotze EM, Tweten RK. Biochim. Biophys. Acta. 2012;1818:1028. doi: 10.1016/j.bbamem.2011.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (255).Heuck AP, Moe PC, Johnson BB. Subcell. Biochem. 2010;51:551. doi: 10.1007/978-90-481-8622-8_20. [DOI] [PubMed] [Google Scholar]
- (256).Tweten RK. Infect. Immun. 2005;73:6199. doi: 10.1128/IAI.73.10.6199-6209.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (257).Tweten RK, Parker MW, Johnson AE. Curr. Top. Microbiol. Immunol. 2001;257:15. doi: 10.1007/978-3-642-56508-3_2. [DOI] [PubMed] [Google Scholar]
- (258).Heuck AP, Savva CG, Holzenburg A, Johnson AE. J. Biol. Chem. 2007;282:22629. doi: 10.1074/jbc.M703207200. [DOI] [PubMed] [Google Scholar]
- (259).Shatursky O, Heuck AP, Shepard LA, Rossjohn J, Parker MW, Johnson AE, Tweten RK. Cell. 1999;99:293. doi: 10.1016/s0092-8674(00)81660-8. [DOI] [PubMed] [Google Scholar]
- (260).Shepard LA, Heuck AP, Hamman BD, Rossjohn J, Parker MW, Ryan KR, Johnson AE, Tweten RK. Biochemistry. 1998;37:14563. doi: 10.1021/bi981452f. [DOI] [PubMed] [Google Scholar]
- (261).Tilley SJ, Orlova EV, Gilbert RJ, Andrew PW, Saibil HR. Cell. 2005;121:247. doi: 10.1016/j.cell.2005.02.033. [DOI] [PubMed] [Google Scholar]
- (262).Feil SC, Rossjohn J, Rohde K, Tweten RK, Parker MW. FEBS Lett. 1996;397:290. doi: 10.1016/s0014-5793(96)01200-8. [DOI] [PubMed] [Google Scholar]
- (263).Rossjohn J, Feil SC, McKinstry WJ, Tweten RK, Parker MW. Cell. 1997;89:685. doi: 10.1016/s0092-8674(00)80251-2. [DOI] [PubMed] [Google Scholar]
- (264).Polekhina G, Giddings KS, Tweten RK, Parker MW. Proc. Natl. Acad. Sci. U. S. A. 2005;102:600. doi: 10.1073/pnas.0403229101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (265).Rossjohn J, Polekhina G, Feil SC, Morton CJ, Tweten RK, Parker MW. J. Mol. Biol. 2007;367:1227. doi: 10.1016/j.jmb.2007.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (266).Xu L, Huang B, Du H, Zhang XC, Xu J, Li X, Rao Z. Protein Cell. 2010;1:96. doi: 10.1007/s13238-010-0012-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (267).Feil SC, Lawrence S, Mulhern TD, Holien JK, Hotze EM, Farrand S, Tweten RK, Parker MW. Structure. 2012;20:248. doi: 10.1016/j.str.2011.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (268).Law RH, Lukoyanova N, Voskoboinik I, Caradoc-Davies TT, Baran K, Dunstone MA, D'Angelo ME, Orlova EV, Coulibaly F, Verschoor S, Browne KA, Ciccone A, Kuiper MJ, Bird PI, Trapani JA, Saibil HR, Whisstock JC. Nature. 2010;468:447. doi: 10.1038/nature09518. [DOI] [PubMed] [Google Scholar]
- (269).Ramachandran R, Heuck AP, Tweten RK, Johnson AE. Nat. Struct. Biol. 2002;9:823. doi: 10.1038/nsb855. [DOI] [PubMed] [Google Scholar]
- (270).Heuck AP, Tweten RK, Johnson AE. J. Biol. Chem. 2003;278:31218. doi: 10.1074/jbc.M303151200. [DOI] [PubMed] [Google Scholar]
- (271).Ramachandran R, Tweten RK, Johnson AE. Proc. Natl. Acad. Sci. U. S. A. 2005;102:7139. doi: 10.1073/pnas.0500556102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (272).Czajkowsky DM, Hotze EM, Shao Z, Tweten RK. EMBO J. 2004;23:3206. doi: 10.1038/sj.emboj.7600350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (273).Sekino-Suzuki N, Nakamura M, Mitsui KI, Ohno-Iwashita Y. Eur. J. Biochem. 1996;241:941. doi: 10.1111/j.1432-1033.1996.00941.x. [DOI] [PubMed] [Google Scholar]
- (274).Nakamura M, Sekino-Suzuki N, Mitsui K, Ohno-Iwashita Y. J. Biochem. 1998;123:1145. doi: 10.1093/oxfordjournals.jbchem.a022054. [DOI] [PubMed] [Google Scholar]
- (275).Ramachandran R, Tweten RK, Johnson AE. Nat. Struct. Mol. Biol. 2004;11:697. doi: 10.1038/nsmb793. [DOI] [PubMed] [Google Scholar]
- (276).Dang TX, Hotze EM, Rouiller I, Tweten RK, Wilson-Kubalek EM. J. Struct. Biol. 2005;150:100. doi: 10.1016/j.jsb.2005.02.003. [DOI] [PubMed] [Google Scholar]
- (277).Shepard LA, Shatursky O, Johnson AE, Tweten RK. Biochemistry. 2000;39:10284. doi: 10.1021/bi000436r. [DOI] [PubMed] [Google Scholar]
- (278).El-Rachkidy RG, Davies NW, Andrew PW. Biochem. Biophys. Res. Commun. 2008;368:786. doi: 10.1016/j.bbrc.2008.01.151. [DOI] [PubMed] [Google Scholar]
- (279).Menestrina G, Bashford CL, Pasternak CA. Toxicon. 1990;28:477. doi: 10.1016/0041-0101(90)90292-f. [DOI] [PubMed] [Google Scholar]
- (280).Korchev YE, Bashford CL, Pederzolli C, Pasternak CA, Morgan PJ, Andrew PW, Mitchell TJ. Biochem. J. 1998;329:571. doi: 10.1042/bj3290571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (281).Palmer M, Harris R, Freytag C, Kehoe M, Tranum-Jensen J, Bhakdi S. EMBO J. 1998;17:1598. doi: 10.1093/emboj/17.6.1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (282).Montal M. Toxicon. 2009;54:565. doi: 10.1016/j.toxicon.2008.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (283).Murphy JR. Toxins (Basel) 2011;3:294. doi: 10.3390/toxins3030294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (284).Yamaizumi M, Mekada E, Uchida T, Okada Y. Cell. 1978;15:245. doi: 10.1016/0092-8674(78)90099-5. [DOI] [PubMed] [Google Scholar]
- (285).Smith WP, Tai PC, Murphy JR, Davis BD. J. Bacteriol. 1980;141:184. doi: 10.1128/jb.141.1.184-189.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (286).Kaczorek M, Delpeyroux F, Chenciner N, Streeck RE, Murphy JR, Boquet P, Tiollais P. Science. 1983;221:855. doi: 10.1126/science.6348945. [DOI] [PubMed] [Google Scholar]
- (287).Greenfield L, Bjorn MJ, Horn G, Fong D, Buck GA, Collier RJ, Kaplan DA. Proc. Natl. Acad. Sci. U. S. A. 1983;80:6853. doi: 10.1073/pnas.80.22.6853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (288).Kantardjieff K, Collier RJ, Eisenberg D. J. Biol. Chem. 1989;264:10402. [PubMed] [Google Scholar]
- (289).Choe S, Bennett MJ, Fujii G, Curmi PM, Kantardjieff KA, Collier RJ, Eisenberg D. Nature. 1992;357:216. doi: 10.1038/357216a0. [DOI] [PubMed] [Google Scholar]
- (290).Bennett MJ, Eisenberg D. Protein Sci. 1994;3:1464. doi: 10.1002/pro.5560030912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (291).Bell CE, Eisenberg D. Biochemistry. 1996;35:1137. doi: 10.1021/bi9520848. [DOI] [PubMed] [Google Scholar]
- (292).Bell CE, Eisenberg D. Adv. Exp. Med. Biol. 1997;419:35. doi: 10.1007/978-1-4419-8632-0_4. [DOI] [PubMed] [Google Scholar]
- (293).Bell CE, Eisenberg D. Biochemistry. 1997;36:481. doi: 10.1021/bi962214s. [DOI] [PubMed] [Google Scholar]
- (294).Pohl E, Qui X, Must LM, Holmes RK, Hol WG. Protein Sci. 1997;6:1114. doi: 10.1002/pro.5560060519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (295).Kochi SK, Collier RJ. Exp. Cell Res. 1993;208:296. doi: 10.1006/excr.1993.1249. [DOI] [PubMed] [Google Scholar]
- (296).Collier RJ, Kandel J. J. Biol. Chem. 1971;246:1496. [PubMed] [Google Scholar]
- (297).Gill DM, Pappenheimer AMJ. J. Biol. Chem. 1971;246:1492. [PubMed] [Google Scholar]
- (298).Uchida T, Gill DM, Pappenheimer AMJ. Nat. New Biol. 1971;233:8. doi: 10.1038/newbio233008a0. [DOI] [PubMed] [Google Scholar]
- (299).Shen WH, Choe S, Eisenberg D, Collier RJ. J. Biol. Chem. 1994;269:29077. [PubMed] [Google Scholar]
- (300).Naglich JG, Metherall JE, Russell DW, Eidels L. Cell. 1992;69:1051. doi: 10.1016/0092-8674(92)90623-k. [DOI] [PubMed] [Google Scholar]
- (301).Boquet P, Silverman MS, Pappenheimer AMJ, Vernon WB. Proc. Natl. Acad. Sci. U. S. A. 1976;73:4449. doi: 10.1073/pnas.73.12.4449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (302).Sandvig K, Olsnes S. J. Cell Biol. 1980;87:828. doi: 10.1083/jcb.87.3.828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (303).Ratts R, Zeng H, Berg EA, Blue C, McComb ME, Costello CE, vanderSpek JC, Murphy JR. J. Cell Biol. 2003;160:1139. doi: 10.1083/jcb.200210028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (304).Ratts R, Trujillo C, Bharti A, vanderSpek J, Harrison R, Murphy JR. Proc. Natl. Acad. Sci. U. S. A. 2005;102:15635. doi: 10.1073/pnas.0504937102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (305).Oh KJ, Senzel L, Collier RJ, Finkelstein A. Proc. Natl. Acad. Sci. U. S. A. 1999;96:8467. doi: 10.1073/pnas.96.15.8467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (306).Donovan JJ, Simon MI, Draper RK, Montal M. Proc. Natl. Acad. Sci. U. S. A. 1981;78:172. doi: 10.1073/pnas.78.1.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (307).Kagan BL, Finkelstein A, Colombini M. Proc. Natl. Acad. Sci. U. S. A. 1981;78:4950. doi: 10.1073/pnas.78.8.4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (308).Deleers M, Beugnier N, Falmagne P, Cabiaux V, Ruysschaert JM. FEBS Lett. 1983;160:82. doi: 10.1016/0014-5793(83)80941-7. [DOI] [PubMed] [Google Scholar]
- (309).Shiver JW, Donovan JJ. Biochim. Biophys. Acta. 1987;903:48. doi: 10.1016/0005-2736(87)90154-4. [DOI] [PubMed] [Google Scholar]
- (310).Misler S. Proc. Natl. Acad. Sci. U. S. A. 1983;80:4320. doi: 10.1073/pnas.80.14.4320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (311).Kagan BL, Reich KA, Collier RJ. Biophys. J. 1984;45:102. doi: 10.1016/S0006-3495(84)84126-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (312).Falnes PO, Madshus IH, Sandvig K, Olsnes S. J. Biol. Chem. 1992;267:12284. [PubMed] [Google Scholar]
- (313).Mindell JA, Silverman JA, Collier RJ, Finkelstein A. Biophys. J. 1992;62:41. doi: 10.1016/S0006-3495(92)81772-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (314).Cabiaux V, Mindell J, Collier RJ. Infect. Immun. 1993;61:2200. doi: 10.1128/iai.61.5.2200-2202.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (315).Mindell JA, Silverman JA, Collier RJ, Finkelstein A. J. Membr. Biol. 1994;137:29. doi: 10.1007/BF00234996. [DOI] [PubMed] [Google Scholar]
- (316).Mindell JA, Zhan H, Huynh PD, Collier RJ, Finkelstein A. Proc. Natl. Acad. Sci. U. S. A. 1994;91:5272. doi: 10.1073/pnas.91.12.5272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (317).Silverman JA, Mindell JA, Zhan H, Finkelstein A, Collier RJ. J. Membr. Biol. 1994;137:17. doi: 10.1007/BF00234995. [DOI] [PubMed] [Google Scholar]
- (318).Kaul P, Silverman J, Shen WH, Blanke SR, Huynh PD, Finkelstein A, Collier RJ. Protein Sci. 1996;5:687. doi: 10.1002/pro.5560050413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (319).Oh KJ, Zhan H, Cui C, Hideg K, Collier RJ, Hubbell WL. Science. 1996;273:810. doi: 10.1126/science.273.5276.810. [DOI] [PubMed] [Google Scholar]
- (320).Huynh PD, Cui C, Zhan H, Oh KJ, Collier RJ, Finkelstein A. J. Gen. Physiol. 1997;110:229. doi: 10.1085/jgp.110.3.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (321).Senzel L, Huynh PD, Jakes KS, Collier RJ, Finkelstein A. J. Gen. Physiol. 1998;112:317. doi: 10.1085/jgp.112.3.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (322).Oh KJ, Zhan H, Cui C, Altenbach C, Hubbell WL, Collier RJ. Biochemistry. 1999;38:10336. doi: 10.1021/bi990520a. [DOI] [PubMed] [Google Scholar]
- (323).Finkelstein A, Oh KJ, Senzel L, Gordon M, Blaustein RO, Collier RJ. Int. J. Med. Microbiol. 2000;290:435. doi: 10.1016/S1438-4221(00)80059-4. [DOI] [PubMed] [Google Scholar]
- (324).Wu Z, Jakes KS, Samelson-Jones BS, Lai B, Zhao G, London E, Finkelstein A. Biophys. J. 2006;91:3249. doi: 10.1529/biophysj.106.085753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (325).Senzel L, Gordon M, Blaustein RO, Oh KJ, Collier RJ, Finkelstein A. J. Gen. Physiol. 2000;115:421. doi: 10.1085/jgp.115.4.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (326).Rodnin MV, Kyrychenko A, Kienker P, Sharma O, Posokhov YO, Collier RJ, Finkelstein A, Ladokhin AS. J. Mol. Biol. 2010;402:1. doi: 10.1016/j.jmb.2010.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (327).Rodnin MV, Kyrychenko A, Kienker P, Sharma O, Vargas-Uribe M, Collier RJ, Finkelstein A, Ladokhin AS. Biophys. J. 2011;101:L41. doi: 10.1016/j.bpj.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (328).Gordon M, Finkelstein A. J. Gen. Physiol. 2001;118:471. doi: 10.1085/jgp.118.5.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (329).Montal M. Annu. Rev. Biochem. 2010;79:591. doi: 10.1146/annurev.biochem.051908.125345. [DOI] [PubMed] [Google Scholar]
- (330).Flynn TC. J. Cosmet. Dermatol. 2012;11:42. doi: 10.1111/j.1473-2165.2011.00593.x. [DOI] [PubMed] [Google Scholar]
- (331).Nguyen AT, Ahmad J, Fagien S, Rohrich RJ. Plast. Reconstr. Surg. 2012;129:142e. doi: 10.1097/PRS.0b013e3182362c63. [DOI] [PubMed] [Google Scholar]
- (332).Pickett A, Perrow K. Toxins (Basel) 2011;3:63. doi: 10.3390/toxins3010063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (333).Dhaked RK, Singh MK, Singh P, Gupta P. Indian J. Med. Res. 2010;132:489. [PMC free article] [PubMed] [Google Scholar]
- (334).Bandyopadhyay S, Clark AW, DasGupta BR, Sathyamoorthy V. J. Biol. Chem. 1987;262:2660. [PubMed] [Google Scholar]
- (335).Schiavo G, Rossetto O, Santucci A, DasGupta BR, Montecucco C. J. Biol. Chem. 1992;267:23479. [PubMed] [Google Scholar]
- (336).Rossetto O, Montecucco C. Handb. Exp. Pharmacol. 2008;184:129. doi: 10.1007/978-3-540-74805-2_6. [DOI] [PubMed] [Google Scholar]
- (337).Schiavo G, Matteoli M, Montecucco C. Physiol. Rev. 2000;80:717. doi: 10.1152/physrev.2000.80.2.717. [DOI] [PubMed] [Google Scholar]
- (338).Simpson LL. Annu. Rev. Pharmacol. Toxicol. 2004;44:167. doi: 10.1146/annurev.pharmtox.44.101802.121554. [DOI] [PubMed] [Google Scholar]
- (339).Schmid MF, Robinson JP, DasGupta BR. Nature. 1993;364:827. doi: 10.1038/364827a0. [DOI] [PubMed] [Google Scholar]
- (340).Oblatt-Montal M, Yamazaki M, Nelson R, Montal M. Protein Sci. 1995;4:1490. doi: 10.1002/pro.5560040806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (341).Montal MS, Blewitt R, Tomich JM, Montal M. FEBS Lett. 1992;313:12. doi: 10.1016/0014-5793(92)81173-j. [DOI] [PubMed] [Google Scholar]
- (342).Koriazova LK, Montal M. Nat. Struct. Biol. 2003;10:13. doi: 10.1038/nsb879. [DOI] [PubMed] [Google Scholar]
- (343).Fisher A, Montal M. Neurotox Res. 2006;9:93. doi: 10.1007/BF03033926. [DOI] [PubMed] [Google Scholar]
- (344).Fischer A, Montal M. J. Biol. Chem. 2007;282:29604. doi: 10.1074/jbc.M703619200. [DOI] [PubMed] [Google Scholar]
- (345).Fischer A, Montal M. Proc. Natl. Acad. Sci. U. S. A. 2007;104:10447. doi: 10.1073/pnas.0700046104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (346).Fischer A, Mushrush DJ, Lacy DB, Montal M. PLoS Pathog. 2008;4:e1000245. doi: 10.1371/journal.ppat.1000245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (347).Fischer A, Nakai Y, Eubanks LM, Clancy CM, Tepp WH, Pellett S, Dickerson TJ, Johnson EA, Janda KD, Montal M. Proc. Natl. Acad. Sci. U. S. A. 2009;106:1330. doi: 10.1073/pnas.0812839106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (348).Fischer A, Sambashivan S, Brunger AT, Montal M. J. Biol. Chem. 2012;287:1657. doi: 10.1074/jbc.C111.319400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (349).Chai Q, Arndt JW, Dong M, Tepp WH, Johnson EA, Chapman ER, Stevens RC. Nature. 2006;444:1096. doi: 10.1038/nature05411. [DOI] [PubMed] [Google Scholar]
- (350).Jin R, Rummel A, Binz T, Brunger AT. Nature. 2006;444:1092. doi: 10.1038/nature05387. [DOI] [PubMed] [Google Scholar]
- (351).Lacy DB, Tepp W, Cohen AC, DasGupta BR, Stevens RC. Nat. Struct. Biol. 1998;5:898. doi: 10.1038/2338. [DOI] [PubMed] [Google Scholar]
- (352).Swaminathan S, Eswaramoorthy S. Acta Crystallogr. D Biol. Crystallogr. 2000;56:1024. doi: 10.1107/s0907444900006764. [DOI] [PubMed] [Google Scholar]
- (353).Swaminathan S, Eswaramoorthy S. Nat. Struct. Biol. 2000;7:693. doi: 10.1038/78005. [DOI] [PubMed] [Google Scholar]
- (354).Fischer A, Garcia-Rodriguez C, Geren I, Lou J, Marks JD, Nakagawa T, Montal M. J. Biol. Chem. 2008;283:3997. doi: 10.1074/jbc.M707917200. [DOI] [PubMed] [Google Scholar]
- (355).Hoch DH, Romero-Mira M, Ehrlich BE, Finkelstein A, DasGupta BR, Simpson LL. Proc. Natl. Acad. Sci. U. S. A. 1985;82:1692. doi: 10.1073/pnas.82.6.1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (356).Gambale F, Rauch G, Belmonte G, Menestrina G. FEBS Lett. 1992;306:41. doi: 10.1016/0014-5793(92)80833-3. [DOI] [PubMed] [Google Scholar]
- (357).Giesemann T, Jank T, Gerhard R, Maier E, Just I, Benz R, Aktories K. J. Biol. Chem. 2006;281:10808. doi: 10.1074/jbc.M512720200. [DOI] [PubMed] [Google Scholar]
- (358).Genisyuerek S, Papatheodorou P, Guttenberg G, Schubert R, Benz R, Aktories K. Mol. Microbiol. 2011;79:1643. doi: 10.1111/j.1365-2958.2011.07549.x. [DOI] [PubMed] [Google Scholar]
- (359).Nagahama M, Umezaki M, Oda M, Kobayashi K, Tone S, Suda T, Ishidoh K, Sakurai J. Infect. Immun. 2011;79:4353. doi: 10.1128/IAI.05677-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (360).Fischer A, Holden MA, Pentelute BL, Collier RJ. Proc. Natl. Acad. Sci. U. S. A. 2011;108:16577. doi: 10.1073/pnas.1113074108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (361).Janowiak BE, Jennings-Antipov LD, Collier RJ. Biochemistry. 2011;50:3512. doi: 10.1021/bi1017446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (362).Pentelute BL, Sharma O, Collier RJ. Angew. Chem. Int. Ed Engl. 2011;50:2294. doi: 10.1002/anie.201006460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (363).Feld GK, Brown MJ, Krantz BA. Protein Sci. 2012;21:606. doi: 10.1002/pro.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (364).Feld GK, Kintzer AF, Tang II, Thoren KL, Krantz BA. J. Mol. Biol. 2012;415:159. doi: 10.1016/j.jmb.2011.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (365).Sterling HJ, Kintzer AF, Feld GK, Cassou CA, Krantz BA, Williams ER. J. Am. Soc. Mass Spectrom. 2012;23:191. doi: 10.1007/s13361-011-0301-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (366).Duesbery NS, Webb CP, Leppla SH, Gordon VM, Klimpel KR, Copeland TD, Ahn NG, Oskarsson MK, Fukasawa K, Paull KD, Vande Woude GF. Science. 1998;280:734. doi: 10.1126/science.280.5364.734. [DOI] [PubMed] [Google Scholar]
- (367).Vitale G, Pellizzari R, Recchi C, Napolitani G, Mock M, Montecucco C. Biochem. Biophys. Res. Commun. 1998;248:706. doi: 10.1006/bbrc.1998.9040. [DOI] [PubMed] [Google Scholar]
- (368).Leppla SH. Adv. Cyclic Nucleotide Protein Phosphorylation Res. 1984;17:189. [PubMed] [Google Scholar]
- (369).Leppla SH. Proc. Natl. Acad. Sci. U. S. A. 1982;79:3162. doi: 10.1073/pnas.79.10.3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (370).Schleberger C, Hochmann H, Barth H, Aktories K, Schulz GE. J. Mol. Biol. 2006;364:705. doi: 10.1016/j.jmb.2006.09.002. [DOI] [PubMed] [Google Scholar]
- (371).Singh Y, Chaudhary VK, Leppla SH. J. Biol. Chem. 1989;264:19103. [PubMed] [Google Scholar]
- (372).Klimpel KR, Molloy SS, Thomas G, Leppla SH. Proc. Natl. Acad. Sci. U. S. A. 1992;89:10277. doi: 10.1073/pnas.89.21.10277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (373).Kintzer AF, Thoren KL, Sterling HJ, Dong KC, Feld GK, Tang II, Zhang TT, Williams ER, Berger JM, Krantz BA. J. Mol. Biol. 2009;392:614. doi: 10.1016/j.jmb.2009.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (374).Pimental RA, Christensen KA, Krantz BA, Collier RJ. Biochem. Biophys. Res. Commun. 2004;322:258. doi: 10.1016/j.bbrc.2004.07.105. [DOI] [PubMed] [Google Scholar]
- (375).Friedlander AM. J. Biol. Chem. 1986;261:7123. [PubMed] [Google Scholar]
- (376).Katayama H, Janowiak BE, Brzozowski M, Juryck J, Falke S, Gogol EP, Collier RJ, Fisher MT. Nat. Struct. Mol. Biol. 2008;15:754. doi: 10.1038/nsmb.1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (377).Krantz BA, Finkelstein A, Collier RJ. J. Mol. Biol. 2006;355:968. doi: 10.1016/j.jmb.2005.11.030. [DOI] [PubMed] [Google Scholar]
- (378).Janowiak BE, Fischer A, Collier RJ. J. Biol. Chem. 2010;285:8130. doi: 10.1074/jbc.M109.093195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (379).Zhang S, Udho E, Wu Z, Collier RJ, Finkelstein A. Biophys. J. 2004;87:3842. doi: 10.1529/biophysj.104.050864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (380).Zhang S, Finkelstein A, Collier RJ. Proc. Natl. Acad. Sci. U. S. A. 2004;101:16756. doi: 10.1073/pnas.0405754101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (381).Benson EL, Huynh PD, Finkelstein A, Collier RJ. Biochemistry. 1998;37:3941. doi: 10.1021/bi972657b. [DOI] [PubMed] [Google Scholar]
- (382).Basilio D, Juris SJ, Collier RJ, Finkelstein A. J. Gen. Physiol. 2009;133:307. doi: 10.1085/jgp.200810170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (383).Kintzer AF, Sterling HJ, Tang II, Abdul-Gader A, Miles AJ, Wallace BA, Williams ER, Krantz BA. J. Mol. Biol. 2010;399:741. doi: 10.1016/j.jmb.2010.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (384).Blaustein RO, Lea EJ, Finkelstein A. J. Gen. Physiol. 1990;96:921. doi: 10.1085/jgp.96.5.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (385).Nguyen TL. J. Biomol. Struct. Dyn. 2004;22:253. doi: 10.1080/07391102.2004.10531226. [DOI] [PubMed] [Google Scholar]
- (386).Blaustein RO, Finkelstein A. J. Gen. Physiol. 1990;96:905. doi: 10.1085/jgp.96.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (387).Ohishi I, Odagiri Y. Infect. Immun. 1984;43:54. doi: 10.1128/iai.43.1.54-58.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (388).Aktories K, Barmann M, Ohishi I, Tsuyama S, Jakobs KH, Habermann E. Nature. 1986;322:390. doi: 10.1038/322390a0. [DOI] [PubMed] [Google Scholar]
- (389).Vandekerckhove J, Schering B, Barmann M, Aktories K. FEBS Lett. 1987;225:48. doi: 10.1016/0014-5793(87)81129-8. [DOI] [PubMed] [Google Scholar]
- (390).Aktories K, Wegner A. J. Cell Biol. 1989;109:1385. doi: 10.1083/jcb.109.4.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (391).Popoff MR, Milward FW, Bancillon B, Boquet P. Infect. Immun. 1989;57:2462. doi: 10.1128/iai.57.8.2462-2469.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (392).Gulke I, Pfeifer G, Liese J, Fritz M, Hofmann F, Aktories K, Barth H. Infect. Immun. 2001;69:6004. doi: 10.1128/IAI.69.10.6004-6011.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (393).Sundriyal A, Roberts AK, Shone CC, Acharya KR. J. Biol. Chem. 2009;284:28713. doi: 10.1074/jbc.M109.043018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (394).Chowdhury HH, Popoff MR, Zorec R. J. Physiol. 1999;521(Pt 2):389. doi: 10.1111/j.1469-7793.1999.00389.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (395).Papatheodorou P, Wilczek C, Nolke T, Guttenberg G, Hornuss D, Schwan C, Aktories K. Infect. Immun. 2012 doi: 10.1128/IAI.06378-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (396).Barth H, Blocker D, Behlke J, Bergsma-Schutter W, Brisson A, Benz R, Aktories K. J. Biol. Chem. 2000;275:18704. doi: 10.1074/jbc.M000596200. [DOI] [PubMed] [Google Scholar]
- (397).Nagahama M, Hagiyama T, Kojima T, Aoyanagi K, Takahashi C, Oda M, Sakaguchi Y, Oguma K, Sakurai J. Infect. Immun. 2009;77:5139. doi: 10.1128/IAI.00638-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (398).Pust S, Barth H, Sandvig K. Cell. Microbiol. 2010;12:1809. doi: 10.1111/j.1462-5822.2010.01512.x. [DOI] [PubMed] [Google Scholar]
- (399).Blocker D, Behlke J, Aktories K, Barth H. Infect. Immun. 2001;69:2980. doi: 10.1128/IAI.69.5.2980-2987.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (400).Stiles BG, Hale ML, Marvaud JC, Popoff MR. Biochem. J. 2002;367:801. doi: 10.1042/BJ20020566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (401).Schmid A, Benz R, Just I, Aktories K. J. Biol. Chem. 1994;269:16706. [PubMed] [Google Scholar]
- (402).Bachmeyer C, Benz R, Barth H, Aktories K, Gilbert M, Popoff MR. FASEB J. 2001;15:1658. doi: 10.1096/fj.00-0671fje. [DOI] [PubMed] [Google Scholar]
- (403).Blocker D, Pohlmann K, Haug G, Bachmeyer C, Benz R, Aktories K, Barth H. J. Biol. Chem. 2003;278:37360. doi: 10.1074/jbc.M305849200. [DOI] [PubMed] [Google Scholar]
- (404).Blocker D, Bachmeyer C, Benz R, Aktories K, Barth H. Biochemistry. 2003;42:5368. doi: 10.1021/bi034199e. [DOI] [PubMed] [Google Scholar]
- (405).Gibert M, Marvaud JC, Pereira Y, Hale ML, Stiles BG, Boquet P, Lamaze C, Popoff MR. FEBS Lett. 2007;581:1287. doi: 10.1016/j.febslet.2007.02.041. [DOI] [PubMed] [Google Scholar]
- (406).Kronhardt A, Rolando M, Beitzinger C, Stefani C, Leuber M, Flatau G, Popoff MR, Benz R, Lemichez E. PLoS One. 2011;6:e23133. doi: 10.1371/journal.pone.0023133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (407).Knapp O, Benz R, Gibert M, Marvaud JC, Popoff MR. J. Biol. Chem. 2002;277:6143. doi: 10.1074/jbc.M103939200. [DOI] [PubMed] [Google Scholar]
- (408).Blaustein RO, Koehler TM, Collier RJ, Finkelstein A. Proc. Natl. Acad. Sci. U. S. A. 1989;86:2209. doi: 10.1073/pnas.86.7.2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (409).Lang AE, Neumeyer T, Sun J, Collier RJ, Benz R, Aktories K. Biochemistry. 2008;47:8406. doi: 10.1021/bi800615g. [DOI] [PubMed] [Google Scholar]
- (410).Neumeyer T, Schiffler B, Maier E, Lang AE, Aktories K, Benz R. J. Biol. Chem. 2008;283:3904. doi: 10.1074/jbc.M709807200. [DOI] [PubMed] [Google Scholar]
- (411).Orlik F, Schiffler B, Benz R. Biophys. J. 2005;88:1715. doi: 10.1529/biophysj.104.050336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (412).Kaiser E, Pust S, Kroll C, Barth H. Cell. Microbiol. 2009;11:780. doi: 10.1111/j.1462-5822.2009.01291.x. [DOI] [PubMed] [Google Scholar]
- (413).Kaiser E, Kroll C, Ernst K, Schwan C, Popoff M, Fischer G, Buchner J, Aktories K, Barth H. Infect. Immun. 2011;79:3913. doi: 10.1128/IAI.05372-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (414).Dmochewitz L, Lillich M, Kaiser E, Jennings LD, Lang AE, Buchner J, Fischer G, Aktories K, Collier RJ, Barth H. Cell. Microbiol. 2011;13:359. doi: 10.1111/j.1462-5822.2010.01539.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (415).Barth H. Naunyn Schmiedebergs Arch. Pharmacol. 2011;383:237. doi: 10.1007/s00210-010-0581-y. [DOI] [PubMed] [Google Scholar]
- (416).Bachmeyer C, Orlik F, Barth H, Aktories K, Benz R. J. Mol. Biol. 2003;333:527. doi: 10.1016/j.jmb.2003.08.044. [DOI] [PubMed] [Google Scholar]
- (417).Bezrukov SM, Kasianowicz JJ. Phys. Rev. Lett. 1993;70:2352. doi: 10.1103/PhysRevLett.70.2352. [DOI] [PubMed] [Google Scholar]
- (418).Kasianowicz JJ, Bezrukov SM. Biophys. J. 1995;69:94. doi: 10.1016/S0006-3495(95)79879-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (419).Sakai N, Mareda J, Matile S. Acc. Chem. Res. 2008;41:1354. doi: 10.1021/ar700229r. [DOI] [PubMed] [Google Scholar]
- (420).Finkelstein A. Toxicology. 1994;87:29. doi: 10.1016/0300-483x(94)90153-8. [DOI] [PubMed] [Google Scholar]
- (421).Anderson DS, Blaustein RO. J. Gen. Physiol. 2008;132:351. doi: 10.1085/jgp.200809984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (422).Rainey GJ, Young JA. Nat. Rev. Microbiol. 2004;2:721. doi: 10.1038/nrmicro977. [DOI] [PubMed] [Google Scholar]
- (423).Ivarsson ME, Leroux JC, Castagner B. Angew. Chem. Int. Ed Engl. 2012;51:4024. doi: 10.1002/anie.201104384. [DOI] [PubMed] [Google Scholar]
- (424).Bartlett JG, Inglesby TVJ, Borio L. Clin. Infect. Dis. 2002;35:851. doi: 10.1086/341902. [DOI] [PubMed] [Google Scholar]
- (425).Mock M, Fouet A. Annu. Rev. Microbiol. 2001;55:647. doi: 10.1146/annurev.micro.55.1.647. [DOI] [PubMed] [Google Scholar]
- (426).Gilligan PH. Curr. Opin. Microbiol. 2002;5:489. doi: 10.1016/s1369-5274(02)00359-4. [DOI] [PubMed] [Google Scholar]
- (427).Popoff MR, Boquet P. Biochem. Biophys. Res. Commun. 1988;152:1361. doi: 10.1016/s0006-291x(88)80435-2. [DOI] [PubMed] [Google Scholar]
- (428).Turnbull PC. Curr. Opin. Infect. Dis. 2000;13:113. doi: 10.1097/00001432-200004000-00004. [DOI] [PubMed] [Google Scholar]
- (429).Roux B, Allen T, Berneche S, Im W. Q. Rev. Biophys. 2004;37:15. doi: 10.1017/s0033583504003968. [DOI] [PubMed] [Google Scholar]
- (430).Eisenberg B. J. Phys. Chem. C. Nanomater Interfaces. 2010;114:20719. doi: 10.1021/jp106760t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (431).Roux B. Biophys. J. 1996;71:3177. doi: 10.1016/S0006-3495(96)79511-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (432).Edwards S, Corry B, Kuyucak S, Chung SH. Biophys. J. 2002;83:1348. doi: 10.1016/S0006-3495(02)73905-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (433).Joseph S, Mashl RJ, Jakobsson E, Aluru NR. Nano Letters. 2003;3:1399. [Google Scholar]
- (434).Majumder M, Zhan X, Andrews R, Hinds BJ. Langmuir. 2007;23:8624. doi: 10.1021/la700686k. [DOI] [PubMed] [Google Scholar]
- (435).Beu TA. J. Chem. Phys. 2010;132:164513. doi: 10.1063/1.3387972. [DOI] [PubMed] [Google Scholar]
- (436).Hilder TA, Gordon D, Chung SH. J. Chem. Phys. 2011;134:045103. doi: 10.1063/1.3524310. [DOI] [PubMed] [Google Scholar]
- (437).De Biase PM, Solano CJ, Markosyan S, Czapla L, Noskov SY. J. Chem. Theory Comput. 2012;8:2540. doi: 10.1021/ct3004244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (438).Bezrukov SM, Berezhkovskii AM, Pustovoit MA, Szabo A. Journal of Chemical Physics. 2000;113:8206. [Google Scholar]
- (439).Berezhkovskii A, Pustovoit M, Bezrukov S. J. Chem. Phys. 2002;116:9952. [Google Scholar]
- (440).Berezhkovskii AM, Pustovoit MA, Bezrukov SM. J. Chem. Phys. 2003;119:3943. [Google Scholar]
- (441).Berezhkovskii AM, Bezrukov SM. Biophys. J. 2005;88:L17. doi: 10.1529/biophysj.104.057588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (442).Berezhkovskii A, Bezrukov S. Chemical Physics. 2005;319:342. [Google Scholar]
- (443).Bezrukov SM, Berezhkovskii AM, Szabo A. J. Chem. Phys. 2007;127:115101. doi: 10.1063/1.2766720. [DOI] [PubMed] [Google Scholar]
- (444).Sheldon KL, Maldonado EN, Lemasters JJ, Rostovtseva TK, Bezrukov SM. PLoS One. 2011;6:e25539. doi: 10.1371/journal.pone.0025539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (445).Berezhkovskii AM, Pustovoit MA, Bezrukov SM. Phys. Rev. E. Stat. Nonlin Soft Matter Phys. 2009;80:020904. doi: 10.1103/PhysRevE.80.020904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (446).Berezhkovskii AM, Pustovoit MA, Bezrukov SM. Chem. Phys. 2010;375:523. doi: 10.1016/j.chemphys.2010.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (447).Zitserman VY, Berezhkovskii AM, Pustovoit MA, Bezrukov SM. J. Chem. Phys. 2008;129:095101. doi: 10.1063/1.2972981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (448).Voet D, Voet JG. Biochemistry. John Wiley; New York: 1995. [Google Scholar]
- (449).Gordon VM, Leppla SH, Hewlett EL. Infect. Immun. 1988;56:1066. doi: 10.1128/iai.56.5.1066-1069.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (450).Blaustein RO, Finkelstein A. J. Gen. Physiol. 1990;96:943. doi: 10.1085/jgp.96.5.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (451).Woodhull AM. J. Gen. Physiol. 1973;61:687. doi: 10.1085/jgp.61.6.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (452).Tikhonov DB, Magazanik LG. J. Membr. Biol. 1998;161:1. doi: 10.1007/s002329900309. [DOI] [PubMed] [Google Scholar]
- (453).Nestorovich EM, Danelon C, Winterhalter M, Bezrukov SM. Proc. Natl. Acad. Sci. U. S. A. 2002;99:9789. doi: 10.1073/pnas.152206799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (454).Bezrukov SM, Krasilnikov OV, Yuldasheva LN, Berezhkovskii AM, Rodrigues CG. Biophys. J. 2004;87:3162. doi: 10.1529/biophysj.104.044453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (455).Bezrukov SM, Winterhalter M. Phys. Rev. Lett. 2000;85:202. doi: 10.1103/PhysRevLett.85.202. [DOI] [PubMed] [Google Scholar]
- (456).Nekolla S, Andersen C, Benz R. Biophys. J. 1994;66:1388. doi: 10.1016/S0006-3495(94)80929-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (457).Wohnsland F, Benz R. J. Membr. Biol. 1997;158:77. doi: 10.1007/s002329900245. [DOI] [PubMed] [Google Scholar]
- (458).Davis ME, Brewster ME. Nat. Rev. Drug Discov. 2004;3:1023. doi: 10.1038/nrd1576. [DOI] [PubMed] [Google Scholar]
- (459).Uekama K. Chem. Pharm. Bull. (Tokyo) 2004;52:900. doi: 10.1248/cpb.52.900. [DOI] [PubMed] [Google Scholar]
- (460).Szejtli J. Chem. Rev. 1998;98:1743. doi: 10.1021/cr970022c. [DOI] [PubMed] [Google Scholar]
- (461).Crouzy S, Berneche S, Roux B. J. Gen. Physiol. 2001;118:207. doi: 10.1085/jgp.118.2.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (462).Cu C, Bahring R, Mayer ML. Neuropharmacology. 1998;37:1381. doi: 10.1016/s0028-3908(98)00112-9. [DOI] [PubMed] [Google Scholar]
- (463).Bouzianas DG. J. Med. Chem. 2010;53:4305. doi: 10.1021/jm901024b. [DOI] [PubMed] [Google Scholar]
- (464).Beierlein JM, Anderson AC. Curr. Med. Chem. 2011;18:5083. doi: 10.2174/092986711797636036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (465).Sawada-Hirai R, Jiang I, Wang F, Sun SM, Nedellec R, Ruther P, Alvarez A, Millis D, Morrow PR, Kang AS. J. Immune Based. Ther. Vaccines. 2004;2:5. doi: 10.1186/1476-8518-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (466).Zhao P, Liang X, Kalbfleisch J, Koo HM, Cao B. Hum. Antibodies. 2003;12:129. [PubMed] [Google Scholar]
- (467).Maynard JA, Maassen CB, Leppla SH, Brasky K, Patterson JL, Iverson BL, Georgiou G. Nat. Biotechnol. 2002;20:597. doi: 10.1038/nbt0602-597. [DOI] [PubMed] [Google Scholar]
- (468).Wild MA, Xin H, Maruyama T, Nolan MJ, Calveley PM, Malone JD, Wallace MR, Bowdish KS. Nat. Biotechnol. 2003;21:1305. doi: 10.1038/nbt891. [DOI] [PubMed] [Google Scholar]
- (469).Chen Z, Moayeri M, Purcell R. Toxins (Basel) 2011;3:1004. doi: 10.3390/toxins3081004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (470).Froude JW, 2nd, Thullier P, Pelat T. Toxins (Basel) 2011;3:1433. doi: 10.3390/toxins3111433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (471).Bradley KA, Mogridge J, Mourez M, Collier RJ, Young JA. Nature. 2001;414:225. doi: 10.1038/n35101999. [DOI] [PubMed] [Google Scholar]
- (472).Scobie HM, Rainey GJ, Bradley KA, Young JA. Proc. Natl. Acad. Sci. U. S. A. 2003;100:5170. doi: 10.1073/pnas.0431098100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (473).Sarac MS, Peinado JR, Leppla SH, Lindberg I. Infect. Immun. 2004;72:602. doi: 10.1128/IAI.72.1.602-605.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (474).Sellman BR, Mourez M, Collier RJ. Science. 2001;292:695. doi: 10.1126/science.109563. [DOI] [PubMed] [Google Scholar]
- (475).Bouzianas DG. Expert Rev. Anti Infect. Ther. 2007;5:665. doi: 10.1586/14787210.5.4.665. [DOI] [PubMed] [Google Scholar]
- (476).Bouzianas DG. Trends Microbiol. 2009;17:522. doi: 10.1016/j.tim.2009.08.006. [DOI] [PubMed] [Google Scholar]
- (477).Mourez M, Kane RS, Mogridge J, Metallo S, Deschatelets P, Sellman BR, Whitesides GM, Collier RJ. Nat. Biotechnol. 2001;19:958. doi: 10.1038/nbt1001-958. [DOI] [PubMed] [Google Scholar]
- (478).Gujraty K, Sadacharan S, Frost M, Poon V, Kane RS, Mogridge J. Mol. Pharm. 2005;2:367. doi: 10.1021/mp050040f. [DOI] [PubMed] [Google Scholar]
- (479).Moayeri M, Wiggins JF, Lindeman RE, Leppla SH. Antimicrob. Agents Chemother. 2006;50:2658. doi: 10.1128/AAC.01412-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (480).Singh Y, Khanna H, Chopra AP, Mehra V. J. Biol. Chem. 2001;276:22090. doi: 10.1074/jbc.M010222200. [DOI] [PubMed] [Google Scholar]
- (481).Yan M, Collier RJ. Mol. Med. 2003;9:46. [PMC free article] [PubMed] [Google Scholar]
- (482).Cao S, Guo A, Liu Z, Tan Y, Wu G, Zhang C, Zhao Y, Chen H. Infect. Immun. 2009;77:4679. doi: 10.1128/IAI.00264-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (483).Radjainia M, Hyun JK, Leysath CE, Leppla SH, Mitra AK. Proc. Natl. Acad. Sci. U. S. A. 2010;107:14070. doi: 10.1073/pnas.1006473107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (484).Rubert Perez C, Lopez-Perez D, Chmielewski J, Lipton M. Chem. Biol. Drug Des. 2012;79:260. doi: 10.1111/j.1747-0285.2011.01285.x. [DOI] [PubMed] [Google Scholar]
- (485).Mourez M, Collier RJ. Methods Mol. Biol. 2004;261:213. doi: 10.1385/1-59259-762-9:213. [DOI] [PubMed] [Google Scholar]
- (486).Vance D, Shah M, Joshi A, Kane RS. Biotechnol. Bioeng. 2008;101:429. doi: 10.1002/bit.22056. [DOI] [PubMed] [Google Scholar]
- (487).Vance D, Martin J, Patke S, Kane RS. Adv. Drug Deliv. Rev. 2009;61:931. doi: 10.1016/j.addr.2009.06.002. [DOI] [PubMed] [Google Scholar]
- (488).Yanjarappa MJ, Gujraty KV, Joshi A, Saraph A, Kane RS. Biomacromolecules. 2006;7:1665. doi: 10.1021/bm060098v. [DOI] [PubMed] [Google Scholar]
- (489).Rai P, Padala C, Poon V, Saraph A, Basha S, Kate S, Tao K, Mogridge J, Kane RS. Nat. Biotechnol. 2006;24:582. doi: 10.1038/nbt1204. [DOI] [PubMed] [Google Scholar]
- (490).Joshi A, Saraph A, Poon V, Mogridge J, Kane RS. Bioconjug. Chem. 2006;17:1265. doi: 10.1021/bc060042y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (491).Gujraty KV, Joshi A, Saraph A, Poon V, Mogridge J, Kane RS. Biomacromolecules. 2006;7:2082. doi: 10.1021/bm060210p. [DOI] [PubMed] [Google Scholar]
- (492).Basha S, Rai P, Poon V, Saraph A, Gujraty K, Go MY, Sadacharan S, Frost M, Mogridge J, Kane RS. Proc. Natl. Acad. Sci. U. S. A. 2006;103:13509. doi: 10.1073/pnas.0509870103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (493).Rai PR, Saraph A, Ashton R, Poon V, Mogridge J, Kane RS. Angew. Chem. Int. Ed Engl. 2007;46:2207. doi: 10.1002/anie.200604317. [DOI] [PubMed] [Google Scholar]
- (494).Rai P, Vance D, Poon V, Mogridge J, Kane RS. Chemistry. 2008;14:7748. doi: 10.1002/chem.200801097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (495).Gujraty KV, Yanjarappa MJ, Saraph A, Joshi A, Mogridge J, Kane RS. J. Polym. Sci. A. Polym. Chem. 2008;46:7246. doi: 10.1002/pola.23031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (496).Joshi A, Vance D, Rai P, Thiyagarajan A, Kane RS. Chemistry. 2008;14:7738. doi: 10.1002/chem.200800278. [DOI] [PubMed] [Google Scholar]
- (497).Joshi A, Kate S, Poon V, Mondal D, Boggara MB, Saraph A, Martin JT, McAlpine R, Day R, Garcia AE, Mogridge J, Kane RS. Biomacromolecules. 2011;12:791. doi: 10.1021/bm101396u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (498).Christensen D, Korsholm KS, Andersen P, Agger EM. Expert Rev. Vaccines. 2011;10:513. doi: 10.1586/erv.11.17. [DOI] [PubMed] [Google Scholar]
- (499).Gonzalez-Rodriguez ML, Rabasco AM. Expert Opin. Drug Deliv. 2011;8:857. doi: 10.1517/17425247.2011.574610. [DOI] [PubMed] [Google Scholar]
- (500).Henriksen-Lacey M, Korsholm KS, Andersen P, Perrie Y, Christensen D. Expert Opin. Drug Deliv. 2011;8:505. doi: 10.1517/17425247.2011.558081. [DOI] [PubMed] [Google Scholar]
- (501).Muller RH, Shegokar R, Keck CM. Curr. Drug Discov. Technol. 2011;8:207. doi: 10.2174/157016311796799062. [DOI] [PubMed] [Google Scholar]
- (502).Merritt EA, Zhang Z, Pickens JC, Ahn M, Hol WG, Fan E. J. Am. Chem. Soc. 2002;124:8818. doi: 10.1021/ja0202560. [DOI] [PubMed] [Google Scholar]
- (503).Polizzotti BD, Maheshwari R, Vinkenborg J, Kiick KL. Macromolecules. 2007;40:7103. doi: 10.1021/ma070725o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (504).Gargano JM, Ngo T, Kim JY, Acheson DW, Lees WJ. J. Am. Chem. Soc. 2001;123:12909. doi: 10.1021/ja016305a. [DOI] [PubMed] [Google Scholar]
- (505).Kitov PI, Sadowska JM, Mulvey G, Armstrong GD, Ling H, Pannu NS, Read RJ, Bundle DR. Nature. 2000;403:669. doi: 10.1038/35001095. [DOI] [PubMed] [Google Scholar]
- (506).Polyzos A, Alderton MR, Dawson RM, Hartley PG. Bioconjug. Chem. 2007;18:1442. doi: 10.1021/bc0700640. [DOI] [PubMed] [Google Scholar]
- (507).Kane RS. Langmuir. 2010;26:8636. doi: 10.1021/la9047193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (508).Clarkson MJ, Faull WB, Kerry JB. Vet. Rec. 1985;116:467. doi: 10.1136/vr.116.17.467. [DOI] [PubMed] [Google Scholar]
- (509).Harshman S, Alouf JE, Siffert O, Baleux F. Infect. Immun. 1989;57:3856. doi: 10.1128/iai.57.12.3856-3862.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (510).Odendaal MW, Visser JJ, Bergh N, Botha WJ. Onderstepoort J. Vet. Res. 1989;56:251. [PubMed] [Google Scholar]
- (511).Percival DA, Shuttleworth AD, Williamson ED, Kelly DC. Infect. Immun. 1990;58:2487. doi: 10.1128/iai.58.8.2487-2492.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (512).Heveker N, Kiessig ST, Glaser R, Hungerer KD, Von Baehr R. Hybridoma. 1994;13:263. doi: 10.1089/hyb.1994.13.263. [DOI] [PubMed] [Google Scholar]
- (513).Heveker N, Hansen A, Hungerer KD, von Baehr R, Glaser RW. Hum. Antibodies Hybridomas. 1994;5:18. [PubMed] [Google Scholar]
- (514).Cifrian E, Guidry AJ, O'Brien CN, Marquardi WW. Am. J. Vet. Res. 1996;57:1308. [PubMed] [Google Scholar]
- (515).El-Enbaawy MI, Abdalla YA, Hussein AZ, Osman RM, Selim SA. Egypt. J. Immunol. 2003;10:77. [PubMed] [Google Scholar]
- (516).McClain MS, Cover TL. Infect. Immun. 2007;75:1785. doi: 10.1128/IAI.01643-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (517).Ragle BE, Bubeck Wardenburg J. Infect. Immun. 2009;77:2712. doi: 10.1128/IAI.00115-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (518).Wang J, Qiu J, Dong J, Li H, Luo M, Dai X, Zhang Y, Leng B, Niu X, Zhao S, Deng X. J. Appl. Microbiol. 2011;111:1551. doi: 10.1111/j.1365-2672.2011.05170.x. [DOI] [PubMed] [Google Scholar]
- (519).Hanada Y, Sekimizu K, Kaito C. J. Biol. Chem. 2011;286:39360. doi: 10.1074/jbc.M111.278416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (520).Pelish TM, McClain MS. J. Biol. Chem. 2009;284:29446. doi: 10.1074/jbc.M109.021782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (521).Kollef MH, Shorr A, Tabak YP, Gupta V, Liu LZ, Johannes RS. Chest. 2005;128:3854. doi: 10.1378/chest.128.6.3854. [DOI] [PubMed] [Google Scholar]
- (522).Gu LQ, Bayley H. Biophys. J. 2000;79:1967. doi: 10.1016/S0006-3495(00)76445-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (523).Lewis M, Weaver CD, McClain MS. Toxins (Basel) 2010;2:1825. doi: 10.3390/toxins2071825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (524).Wulff H, Zhorov BS. Chem. Rev. 2008;108:1744. doi: 10.1021/cr078234p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (525).Zhorov BS, Tikhonov DB. J. Neurochem. 2004;88:782. doi: 10.1111/j.1471-4159.2004.02261.x. [DOI] [PubMed] [Google Scholar]
- (526).Yu FH, Yarov-Yarovoy V, Gutman GA, Catterall WA. Pharmacol. Rev. 2005;57:387. doi: 10.1124/pr.57.4.13. [DOI] [PubMed] [Google Scholar]
- (527).Catterall WA, Goldin AL, Waxman SG. Pharmacol. Rev. 2005;57:397. doi: 10.1124/pr.57.4.4. [DOI] [PubMed] [Google Scholar]
- (528).Catterall WA, Perez-Reyes E, Snutch TP, Striessnig J. Pharmacol. Rev. 2005;57:411. doi: 10.1124/pr.57.4.5. [DOI] [PubMed] [Google Scholar]
- (529).Clapham DE, Julius D, Montell C, Schultz G. Pharmacol. Rev. 2005;57:427. doi: 10.1124/pr.57.4.6. [DOI] [PubMed] [Google Scholar]
- (530).Clapham DE, Garbers DL. Pharmacol. Rev. 2005;57:451. doi: 10.1124/pr.57.4.7. [DOI] [PubMed] [Google Scholar]
- (531).Hofmann F, Biel M, Kaupp UB. Pharmacol. Rev. 2005;57:455. doi: 10.1124/pr.57.4.8. [DOI] [PubMed] [Google Scholar]
- (532).Wei AD, Gutman GA, Aldrich R, Chandy KG, Grissmer S, Wulff H. Pharmacol. Rev. 2005;57:463. doi: 10.1124/pr.57.4.9. [DOI] [PubMed] [Google Scholar]
- (533).Gutman GA, Chandy KG, Grissmer S, Lazdunski M, McKinnon D, Pardo LA, Robertson GA, Rudy B, Sanguinetti MC, Stuhmer W, Wang X. Pharmacol. Rev. 2005;57:473. doi: 10.1124/pr.57.4.10. [DOI] [PubMed] [Google Scholar]
- (534).Kubo Y, Adelman JP, Clapham DE, Jan LY, Karschin A, Kurachi Y, Lazdunski M, Nichols CG, Seino S, Vandenberg CA. Pharmacol. Rev. 2005;57:509. doi: 10.1124/pr.57.4.11. [DOI] [PubMed] [Google Scholar]
- (535).Goldstein SA, Bayliss DA, Kim D, Lesage F, Plant LD, Rajan S. Pharmacol. Rev. 2005;57:527. doi: 10.1124/pr.57.4.12. [DOI] [PubMed] [Google Scholar]
- (536).Gu LQ, Shim JW. Analyst. 2010;135:441. doi: 10.1039/b907735a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (537).Majd S, Yusko EC, Billeh YN, Macrae MX, Yang J, Mayer M. Curr. Opin. Biotechnol. 2010;21:439. doi: 10.1016/j.copbio.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (538).Krasilnikov O, Ternovsky V, Tashmukhamedov B. Biofisica. 1981;26:271. [Google Scholar]
- (539).Braha O, Gu LQ, Zhou L, Lu X, Cheley S, Bayley H. Nat. Biotechnol. 2000;18:1005. doi: 10.1038/79275. [DOI] [PubMed] [Google Scholar]
- (540).Gu LQ, Dalla Serra M, Vincent JB, Vigh G, Cheley S, Braha O, Bayley H. Proc. Natl. Acad. Sci. U. S. A. 2000;97:3959. doi: 10.1073/pnas.97.8.3959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (541).Gu LQ, Cheley S, Bayley H. Science. 2001;291:636. doi: 10.1126/science.291.5504.636. [DOI] [PubMed] [Google Scholar]
- (542).Gurnev PA, Harries D, Parsegian VA, Bezrukov SM. ChemPhysChem. 2009;10:1445. doi: 10.1002/cphc.200900312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (543).Gurnev PA, Harries D, Parsegian VA, Bezrukov SM. J. Phys. Condens Matter. 2010;22:454110. doi: 10.1088/0953-8984/22/45/454110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (544).Kim JH, Scialli AR. Toxicol. Sci. 2011;122:1. doi: 10.1093/toxsci/kfr088. [DOI] [PubMed] [Google Scholar]
- (545).Wnendt S, Finkam M, Winter W, Ossig J, Raabe G, Zwingenberger K. Chirality. 1996;8:390. doi: 10.1002/(SICI)1520-636X(1996)8:5<390::AID-CHIR6>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- (546).Efcavitch JW, Thompson JF. Annu. Rev. Anal. Chem. (Palo Alto Calif) 2010;3:109. doi: 10.1146/annurev.anchem.111808.073558. [DOI] [PubMed] [Google Scholar]
- (547).Wang HY, Ying YL, Li Y, Long YT. Chem. Asian J. 2010;5:1952. doi: 10.1002/asia.201000279. [DOI] [PubMed] [Google Scholar]
- (548).Shendure JA, Porreca GJ, Church GM, Gardner AF, Hendrickson CL, Kieleczawa J, Slatko BE. Curr. Protoc. Mol. Biol. 2011;Chapter 7(Unit7.1) doi: 10.1002/0471142727.mb0701s96. [DOI] [PubMed] [Google Scholar]
- (549).Venkatesan BM, Bashir R. Nat. Nanotechnol. 2011;6:615. doi: 10.1038/nnano.2011.129. [DOI] [PubMed] [Google Scholar]
- (550).Luan B, Stolovitzky G, Martyna G. Nanoscale. 2012;4:1068. doi: 10.1039/c1nr11201e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (551).Deamer DW, Branton D. Acc. Chem. Res. 2002;35:817. doi: 10.1021/ar000138m. [DOI] [PubMed] [Google Scholar]
- (552).Purnell RF, Schmidt JJ. ACS Nano. 2009;3:2533. doi: 10.1021/nn900441x. [DOI] [PubMed] [Google Scholar]
- (553).Astier Y, Uzun O, Stellacci F. Small. 2009;5:1273. doi: 10.1002/smll.200801779. [DOI] [PubMed] [Google Scholar]
- (554).de Zoysa RS, Jayawardhana DA, Zhao Q, Wang D, Armstrong DW, Guan X. J Phys Chem B. 2009;113:13332. doi: 10.1021/jp9040293. [DOI] [PubMed] [Google Scholar]
- (555).Manrao EA, Derrington IM, Laszlo AH, Langford KW, Hopper MK, Gillgren N, Pavlenok M, Niederweis M, Gundlach JH. Nat. Biotechnol. 2012;30:349. doi: 10.1038/nbt.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (556).Halverson KM, Panchal RG, Nguyen TL, Gussio R, Little SF, Misakian M, Bavari S, Kasianowicz JJ. J. Biol. Chem. 2005;280:34056. doi: 10.1074/jbc.M507928200. [DOI] [PubMed] [Google Scholar]
- (557).Li J, Stein D, McMullan C, Branton D, Aziz MJ, Golovchenko JA. Nature. 2001;412:166. doi: 10.1038/35084037. [DOI] [PubMed] [Google Scholar]
- (558).Fuchs H, Bachran C. Curr. Drug Targets. 2009;10:89. doi: 10.2174/138945009787354557. [DOI] [PubMed] [Google Scholar]
- (559).Panchal RG, Smart ML, Bowser DN, Williams DA, Petrou S. Curr. Pharm. Biotechnol. 2002;3:99. doi: 10.2174/1389201023378418. [DOI] [PubMed] [Google Scholar]
- (560).Panchal RG. Biochem. Pharmacol. 1998;55:247. doi: 10.1016/s0006-2952(97)00240-2. [DOI] [PubMed] [Google Scholar]
- (561).Potala S, Sahoo SK, Verma RS. Drug Discov. Today. 2008;13:807. doi: 10.1016/j.drudis.2008.06.017. [DOI] [PubMed] [Google Scholar]
- (562).Mathew M, Verma RS. Cancer. Sci. 2009;100:1359. doi: 10.1111/j.1349-7006.2009.01192.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (563).Kreitman RJ. BioDrugs. 2009;23:1. doi: 10.2165/00063030-200923010-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (564).Barth H, Aktories K. Eur. J. Cell Biol. 2011;90:944. doi: 10.1016/j.ejcb.2010.11.007. [DOI] [PubMed] [Google Scholar]
- (565).Adkins I, Holubova J, Kosova M, Sadilkova L. Curr. Pharm. Biotechnol. 2012;13:1446. doi: 10.2174/138920112800784835. [DOI] [PubMed] [Google Scholar]
- (566).Fitzgerald D, Pastan I. Ann. N. Y. Acad. Sci. 1993;685:740. doi: 10.1111/j.1749-6632.1993.tb35935.x. [DOI] [PubMed] [Google Scholar]
- (567).Murphy JR, Lakkis FG, vanderSpek JC, Anderson P, Strom TB. Targeted Diagn. Ther. 1992;7:365. [PubMed] [Google Scholar]
- (568).Murphy JR, vanderSpek JC. Semin. Cancer Biol. 1995;6:259. doi: 10.1006/scbi.1995.0034. [DOI] [PubMed] [Google Scholar]
- (569).Foss FM, Saleh MN, Krueger JG, Nichols JC, Murphy JR. Curr. Top. Microbiol. Immunol. 1998;234:63. doi: 10.1007/978-3-642-72153-3_5. [DOI] [PubMed] [Google Scholar]
- (570).vanderSpek JC, Murphy JR. Methods Enzymol. 2000;327:239. doi: 10.1016/s0076-6879(00)27280-7. [DOI] [PubMed] [Google Scholar]
- (571).Arora N, Klimpel KR, Singh Y, Leppla SH. J. Biol. Chem. 1992;267:15542. [PubMed] [Google Scholar]
- (572).Arora N, Williamson LC, Leppla SH, Halpern JL. J. Biol. Chem. 1994;269:26165. [PubMed] [Google Scholar]
- (573).Arora N, Leppla SH. Infect. Immun. 1994;62:4955. doi: 10.1128/iai.62.11.4955-4961.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (574).Leppla SH, Arora N, Varughese M. J. Appl. Microbiol. 1999;87:284. doi: 10.1046/j.1365-2672.1999.00890.x. [DOI] [PubMed] [Google Scholar]
- (575).Liu S, Bugge TH, Leppla SH. J. Biol. Chem. 2001;276:17976. doi: 10.1074/jbc.M011085200. [DOI] [PubMed] [Google Scholar]
- (576).Frankel AE, Powell BL, Duesbery NS, Vande Woude GF, Leppla SH. Curr. Protein Pept. Sci. 2002;3:399. doi: 10.2174/1389203023380567. [DOI] [PubMed] [Google Scholar]
- (577).Liu S, Schubert RL, Bugge TH, Leppla SH. Expert Opin. Biol. Ther. 2003;3:843. doi: 10.1517/14712598.3.5.843. [DOI] [PubMed] [Google Scholar]
- (578).Barth H, Hofmann F, Olenik C, Just I, Aktories K. Infect. Immun. 1998;66:1364. doi: 10.1128/iai.66.4.1364-1369.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (579).Barth H, Roebling R, Fritz M, Aktories K. J. Biol. Chem. 2002;277:5074. doi: 10.1074/jbc.M109167200. [DOI] [PubMed] [Google Scholar]
- (580).Marvaud JC, Stiles BG, Chenal A, Gillet D, Gibert M, Smith LA, Popoff MR. J. Biol. Chem. 2002;277:43659. doi: 10.1074/jbc.M207828200. [DOI] [PubMed] [Google Scholar]
- (581).Panchal RG, Cusack E, Cheley S, Bayley H. Nat. Biotechnol. 1996;14:852. doi: 10.1038/nbt0796-852. [DOI] [PubMed] [Google Scholar]
- (582).Williams SA, Merchant RF, Garrett-Mayer E, Isaacs JT, Buckley JT, Denmeade SR. J. Natl. Cancer Inst. 2007;99:376. doi: 10.1093/jnci/djk065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (583).Goletz TJ, Klimpel KR, Arora N, Leppla SH, Keith JM, Berzofsky JA. Proc. Natl. Acad. Sci. U. S. A. 1997;94:12059. doi: 10.1073/pnas.94.22.12059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (584).Goletz TJ, Klimpel KR, Leppla SH, Keith JM, Berzofsky JA. Hum. Immunol. 1997;54:129. doi: 10.1016/s0198-8859(97)00081-5. [DOI] [PubMed] [Google Scholar]
- (585).Varughese M, Chi A, Teixeira AV, Nicholls PJ, Keith JM, Leppla SH. Mol. Med. 1998;4:87. [PMC free article] [PubMed] [Google Scholar]
- (586).Hochmann H, Pust S, von Figura G, Aktories K, Barth H. Biochemistry. 2006;45:1271. doi: 10.1021/bi051810w. [DOI] [PubMed] [Google Scholar]
- (587).Pust S, Hochmann H, Kaiser E, von Figura G, Heine K, Aktories K, Barth H. J. Biol. Chem. 2007;282:10272. doi: 10.1074/jbc.M610254200. [DOI] [PubMed] [Google Scholar]
- (588).Schein SJ, Colombini M, Finkelstein A. J. Membr. Biol. 1976;30:99. doi: 10.1007/BF01869662. [DOI] [PubMed] [Google Scholar]
- (589).Colombini M, Mannella CA. Biochim. Biophys. Acta. 2011;1818:1438. doi: 10.1016/j.bbamem.2011.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (590).Colombini M. Biochim. Biophys. Acta. 2012 [Google Scholar]
- (591).Ujwal R, Cascio D, Colletier JP, Faham S, Zhang J, Toro L, Ping P, Abramson J. Proc. Natl. Acad. Sci. U. S. A. 2008;105:17742. doi: 10.1073/pnas.0809634105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (592).Rostovtseva TK, Kazemi N, Weinrich M, Bezrukov SM. J. Biol. Chem. 2006;281:37496. doi: 10.1074/jbc.M602548200. [DOI] [PubMed] [Google Scholar]
- (593).Rostovtseva TK, Sheldon KL, Hassanzadeh E, Monge C, Saks V, Bezrukov SM, Sackett DL. Proc. Natl. Acad. Sci. U. S. A. 2008;105:18746. doi: 10.1073/pnas.0806303105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (594).Rostovtseva TK, Bezrukov SM. J. Bioenerg. Biomembr. 2008;40:163. doi: 10.1007/s10863-008-9145-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (595).Rostovtseva TK, Bezrukov SM. Biochim. Biophys. Acta. 2012;1818:1526. doi: 10.1016/j.bbamem.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (596).Gurnev PA, Rostovtseva TK, Bezrukov SM. FEBS Lett. 2011;585:2363. doi: 10.1016/j.febslet.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (597).Wang H, Shi H, Wang Z. Life Sci. 1999;65:PL143. doi: 10.1016/s0024-3205(99)00370-7. [DOI] [PubMed] [Google Scholar]
- (598).Suessbrich H, Waldegger S, Lang F, Busch AE. FEBS Lett. 1996;385:77. doi: 10.1016/0014-5793(96)00355-9. [DOI] [PubMed] [Google Scholar]
- (599).Kang J, Chen XL, Rampe D. Biochem. Biophys. Res. Commun. 2001;286:499. doi: 10.1006/bbrc.2001.5434. [DOI] [PubMed] [Google Scholar]
- (600).Clare JJ, Tate SN, Nobbs M, Romanos MA. Drug Discov. Today. 2000;5:506. doi: 10.1016/s1359-6446(00)01570-1. [DOI] [PubMed] [Google Scholar]
- (601).Dietrich PS, McGivern JG, Delgado SG, Koch BD, Eglen RM, Hunter JC, Sangameswaran L. J. Neurochem. 1998;70:2262. doi: 10.1046/j.1471-4159.1998.70062262.x. [DOI] [PubMed] [Google Scholar]
- (602).Hockerman GH, Johnson BD, Abbott MR, Scheuer T, Catterall WA. J. Biol. Chem. 1997;272:18759. doi: 10.1074/jbc.272.30.18759. [DOI] [PubMed] [Google Scholar]
- (603).Yang J, Jan YN, Jan LY. Neuron. 1995;14:1047. doi: 10.1016/0896-6273(95)90343-7. [DOI] [PubMed] [Google Scholar]
- (604).Sanchez M, McManus OB. Neuropharmacology. 1996;35:963. doi: 10.1016/0028-3908(96)00137-2. [DOI] [PubMed] [Google Scholar]
- (605).Strobaek D, Jorgensen TD, Christophersen P, Ahring PK, Olesen SP. Br. J. Pharmacol. 2000;129:991. doi: 10.1038/sj.bjp.0703120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (606).Fanger CM, Rauer H, Neben AL, Miller MJ, Rauer H, Wulff H, Rosa JC, Ganellin CR, Chandy KG, Cahalan MD. J. Biol. Chem. 2001;276:12249. doi: 10.1074/jbc.M011342200. [DOI] [PubMed] [Google Scholar]
- (607).Lacy DB, Wigelsworth DJ, Melnyk RA, Harrison SC, Collier RJ. Proc. Natl. Acad. Sci. U. S. A. 2004;101:13147. doi: 10.1073/pnas.0405405101. [DOI] [PMC free article] [PubMed] [Google Scholar]