

Abstract
Heterotrimeric G proteins, composed of α, β, and γ subunits, can transduce a variety of signals from seven-transmembrane-type receptors to intracellular effectors. By whole-exome sequencing and subsequent mutation screening, we identified de novo heterozygous mutations in GNAO1, which encodes a Gαo subunit of heterotrimeric G proteins, in four individuals with epileptic encephalopathy. Two of the affected individuals also showed involuntary movements. Somatic mosaicism (approximately 35% to 50% of cells, distributed across multiple cell types, harbored the mutation) was shown in one individual. By mapping the mutation onto three-dimensional models of the Gα subunit in three different complexed states, we found that the three mutants (c.521A>G [p.Asp174Gly], c.836T>A [p.Ile279Asn], and c.572_592del [p.Thr191_Phe197del]) are predicted to destabilize the Gα subunit fold. A fourth mutant (c.607G>A), in which the Gly203 residue located within the highly conserved switch II region is substituted to Arg, is predicted to impair GTP binding and/or activation of downstream effectors, although the p.Gly203Arg substitution might not interfere with Gα binding to G-protein-coupled receptors. Transient-expression experiments suggested that localization to the plasma membrane was variably impaired in the three putatively destabilized mutants. Electrophysiological analysis showed that Gαo-mediated inhibition of calcium currents by norepinephrine tended to be lower in three of the four Gαo mutants. These data suggest that aberrant Gαo signaling can cause multiple neurodevelopmental phenotypes, including epileptic encephalopathy and involuntary movements.
Introduction
Epileptic encephalopathy is a group of neurological disorders characterized by severe and progressive cognitive and behavioral impairments, which are most likely caused or made worse by epileptic activity.1 Ohtahara syndrome (OS [MIM 308350 and 612164]) is the most severe and the earliest form of epileptic encephalopathy and is characterized by tonic spasms mainly in the neonatal period, seizure intractability, and a suppression-burst pattern on electroencephalography (EEG).2 De novo mutations in three genes, ARX (MIM 300382), STXBP1 (MIM 602926), and KCNQ2 (MIM 602235), have been reported in individuals with OS.3–6
Heterotrimeric guanine-binding proteins (G proteins) are composed of α, β, and γ subunits. In its basal state, Gα is bound with guanosine diphosphate (GDP) and forms the Gαβγ complex. When a seven-transmembrane-type receptor binds its agonist, it activates G proteins by catalyzing the exchange of GDP for guanosine triphosphate (GTP) on the Gα subunit. Subsequently, GTP-bound Gα dissociates from Gβγ, and each of the two complexes activates distinct downstream effectors.7 In mammals, Gα subunits are divided into four classes: Gαi/o, Gαq/11, Gαs, and Gα12/13.7 Gαo, encoded by GNAO1 (MIM 139311), is extremely abundant in brain tissue, where it can constitute up to approximately 0.5% of membrane protein,8 suggesting important roles in brain function. In fact, mice lacking Gαo show multiple neurological abnormalities, including generalized tremor, occasional seizures, severe motor-control impairment, hyperalgesia, and behavioral abnormalities with early postnatal lethality.9,10
In this study, de novo GNAO1 mutations were identified in four epileptic-encephalopathy-affected individuals, three of whom were diagnosed with OS. In addition, two of the four individuals showed involuntary movements, suggesting that aberration of Gαo can cause multiple neurodevelopmental phenotypes.
Subjects and Methods
Subjects
Twelve individuals with OS were previously analyzed by whole-exome sequencing (WES).3,11 In addition, we analyzed parental samples from 5 of the 12 individuals by WES. Screening for GNAO1 mutations was performed in 367 individuals with epileptic encephalopathy (including 62 OS cases) by high-resolution-melting (HRM) analysis (339 cases) and/or WES (100 cases). The diagnosis was made on the basis of clinical features and characteristic patterns on EEG. Experimental protocols were approved by the institutional review board of Yokohama City University School of Medicine and Yamagata University Faculty of Medicine. Informed consent was obtained from the families of all individuals.
DNA Samples
Genomic DNA was obtained from peripheral-blood leukocytes by standard methods. For detection of a mosaic mutation in individual 2, genomic DNA from saliva and nails was isolated with an Oragene DNA kit (DNA Genotek) and an ISOHAIR kit (Nippon Gene), respectively.
WES
Genomic DNA was captured with the SureSelect Human All Exon v.4 Kit (Agilent Technologies) and sequenced with four samples per lane on an Illumina HiSeq 2000 (Illumina) with 101 bp paired-end reads. Image analysis and base calling were performed by Sequencing Control Software with Real-Time Analysis and CASAVA software v.1.8 (Illumina). Exome data processing, variant calling, and variant annotation were performed as previously described.12–14 Reads were aligned to GRCh37 with Novoalign (Novocraft Technologies). Duplicate reads were removed with Picard, and local realignments around indels and base-quality-score recalibration were performed with the Genome Analysis Toolkit (GATK).13 Single-nucleotide variants and small indels were identified with the GATK UnifiedGenotyper and were filtered according to the Broad Institute’s best-practice guidelines v.3. Not flagged as clinically associated, variants registered in dbSNP135 were filtered. Filter-passed variants were annotated with ANNOVAR.14 Pathogenic mutations detected by WES were confirmed by Sanger sequencing.
Mutation Screening
Genomic DNA was amplified with an illustra GenomiPhi V2 DNA Amplification Kit (GE Healthcare). Exons 1–8 covering the GNAO1 coding region of two transcript variants (transcript variant 1, RefSeq accession number NM_020988.2, encoding Gαo1; transcript variant 2, RefSeq accession number NM_138736.2, encoding Gαo2) were screened by HRM analysis. The last two exons differ between the transcript variants. HRM analysis was performed with a Light Cycler 480 (Roche Diagnostics). Samples showing an aberrant melting curve in the HRM analysis were sequenced. PCR primers and conditions are shown in Table S1, available online. All mutations not present in publically available databases were verified with original genomic DNA and were searched for in the variant database of our 408 in-house control exomes.
Deep Sequencing of a Mosaic Mutation
PCR products (length 178 bp) spanning the c.521A>G mutation were amplified with the use of blood, saliva, and nail DNA samples from individual 2 and blood DNA samples from her parents as a template. Adaptor ligation, nick repair, and amplification were performed with the Ion Xpress Plus Fragment Library Kit (Life Technologies) according to the manufacturer’s protocol (part no. 4471989 Rev. B). Indexing was carried out with the Xpress Barcode Adapters 1–16 Kit (Life Technologies). Emulsion PCR and enrichment steps were carried out with the Ion OneTouch 200 Template Kit v.2 (Life Technologies) according to the manufacturer’s protocol (part no. 4478371 Rev. A). Sequencing of the amplicon libraries was carried out on the Ion Torrent Personal Genome Machine (PGM) with the Ion 314 sequencing chip and the Ion PGM 200 Sequencing Kit (Life Technologies) according to the recommended protocol (part no. 4474246 Rev. B). Torrent Suite 2.2 was used for all analyses. The percentage of mosaicism was examined with the Integrative Genomics Viewer.15,16
Expression Vectors
A full-length human GNAO1 cDNA clone (transcript variant 1, encoding Gαo1) was purchased from Kazusa DNA Research Institute. Human GNAO1 cDNA was inserted into a pEF6/V5-His-C vector for the introduction of a C-terminal V5 epitope (Life Technologies). Site-directed mutagenesis using a KOD-Plus-Mutagenesis kit (Toyobo) was performed for generating GNAO1 mutants, including c.521A>G (p.Asp174Gly), c.572_592del (p.The191_Phe197del), c.836T>A (p.Ile279Asn), and c.607G>A (p.Gly203Arg). A c.607_609delinsACA (p.Gly203Thr) mutant, in which GTP binding was reversible in contrast to the WT,17 was also generated to serve as the known loss-of-function mutant.18 All variant cDNAs were confirmed by Sanger sequencing.
Immunofluorescence Microscopy
Mouse neuroblastoma 2A (N2A) cells were grown as previously described.4 N2A cells on glass coverslips were transfected with 200 ng of plasmid DNA with the use of X-tremeGENE 9 DNA Transfection Reagent (Roche Diagnostics). After 24 hr, cells were fixed in PBS containing 4% paraformaldehyde for 15 min and permeabilized in PBS containing 0.1% Triton X-100 for 5 min. Cells were then blocked with 10% normal goat serum for 30 min. V5-tagged Gαo1 was detected with a mouse V5 antibody (1:200 dilution; Life Technologies) and Alexa-488-conjugated goat anti-mouse IgG (1:1000 dilution; Life Technologies). Coverslips were mounted with Vectashield (Vector Laboratories) that contained DAPI and were visualized with an inverted FV1000-D confocal microscope (Olympus).
Structural Modeling and Free-Energy Calculations
We used FoldX software (version 3.0β5) to construct mutated molecular structures and calculate the free-energy changes caused by the mutations.19 We used crystal structures of the GDP-bound inactive Gαiβγ heterotrimer (Protein Data Bank [PDB] 1GG2),20 the nucleotide-free Gαsβγ in complex with agonist-occupied monomeric β2 adrenergic receptor (β2AR) (PDB 3SN6),21 and the transition-state analog of GTP (GDP+AlF4−)-bound Gαq in complex with its effector phospholipase C-β (PLCβ) (PDB 3OHM)22 as three-dimensional structure models of the Gαo subunit in different complexed states. Each of the mutations, corresponding to p.Asp174Gly, p.Ile279Asn, or p.Gly203Arg in the human Gαo subunit, was introduced into the Gα subunit of each complex, and the free-energy change upon the mutation was calculated with FoldX software. Note that ligands included in the complexes were ignored in the calculation because the FoldX energy function does not consider the contribution of ligands. The calculation was repeated three times, and the resultant data were presented as an average value with a SD.
Electrophysiology
For electrophysiological recording of calcium currents, we used NG108-15 cells transfected with individual GNAO1 mutants. Expression vectors were introduced by electroporation with the Lonza Nucleofector device and the Cell Line Nucleofector Kit V (Lonza) according to the manufacture’s protocol (program X-023). Two micrograms of plasmid DNA was used per transfection. The transfected cells were plated on poly-l-lysine-coated plastic coverslips (Cell Desk LF, MS-0113L; Sumitomo Bakelite) at a density of about 5 × 104 cells/cm2 and cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). One day after transfection, the cells were differentiated with DMEM supplemented with 10 μM prostaglandin E1, 50 μM IBMX, and 1% FBS for 3–7 days before recording. During the culture period, half of the medium was changed every other day.
The recording was made by the perforated whole-cell patch-clamp technique with amphotericin B. Cells on coverslips were perfused under an Olympus BX51W upright microscope (Olympus) with a bath solution containing 140 mM NaCl, 5 mM CaCl2, 4 mM KCl, 1 mM MgCl2, 10 mM HEPES, 10 mM TEACl, 8 mM glucose, and 0.0002 mM tetrodotoxin (pH 7.3 adjusted with NaOH). The patch pipette solution contained 100 mM CsCl, 10 mM EGTA, and 40 mM HEPES (pH 7.3 adjusted with CsOH). Amphotericin B was added to the pipette solution at 2 μl/ml just before the experiments. The pipettes were fabricated from borosilicate glass capillaries and had a resistance of 4–8 MΩ when backfilled with the amphotericin-B-containing pipette solution. The recording was started when the series resistance was reduced to <150 MΩ after gigaseal formation and clear cellular capacitive surges had appeared. Voltage-gated calcium currents were elicited by the application of 50 ms depolarizing pulses to +10 mV from the holding potential of −65 mV, recorded with a Multiclamp 700B (Molecular Devices) controlled via pCLAMP10 software (Molecular Devices), filtered at 2 kHz, and sampled at 10 kHz with 50% compensation for series resistance. Gαo-mediated current inhibition was elicited by the application of 10 μM norepinephrine via the bath solution. After 3–5 min, inhibition was assessed by measurement of the changes in current density just before the end of the depolarizing pulses. Recordings were made at room temperature.
Statistical multiple comparisons were made with ANOVA followed by Dunnett’s post hoc test, and the threshold p value for judging statistical significance was 0.05. The current inhibition induced by norepinephrine in individual mutant-expressing cells was assessed with a paired t test. The results are given as the mean ± SEM.
Results
GNAO1 Is Mutated in Individuals with Epileptic Encephalopathy
We previously performed WES of 12 individuals with OS.3,11 In this study, we analyzed parental samples from 5 of the 12 individuals by WES (mean RefSeq read depth of 109) to systematically screen de novo or recessive mutations. We found no recessive mutations in SLC25A22 (MIM 609302), PNPO (MIM 610090), PNKP (MIM 613402), PLCB1 (MIM 613722), or ST3GAL3 (MIM 615006), whose mutations were previously found in epileptic encephalopathy,23–27 but we did find one or two de novo mutations in each of the five trio exomes. Among them, a de novo missense mutation (c.836T>A [p.Ile279Asn]) in GNAO1 at 16q12.2 was identified in individual 1. In the exome data of the other seven original individuals, we also found in individual 2 a second missense mutation (c.521A>G [p.Asp174Gly]), which was confirmed as a de novo event by Sanger sequencing (Figure 1). Moreover, GNAO1 mutation screening in 367 individuals with epileptic encephalopathy by HRM analysis (339 individuals) and/or WES (100 individuals, mean read depth of 129) revealed two de novo mutations: c.572_592 del (p.Thr191_Phe197del) in individual 3 and c.607G>A (p.Gly203Arg) in individual 4 (Figure 1). One mutation (c.836T>A) specifically affects GNAO1 transcript variant 1, whereas the other three mutations affect both transcript variants 1 and 2. Web-based prediction tools suggested that these four mutations would be pathogenic (Table S2). None of the four mutations was found in the 6,500 exomes of the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project Exome Variant Server or among our 408 in-house control exomes. Interestingly, exome data and Sanger sequencing indicated that the c.521A>G mutation in individual 2 was somatic mosaic (Figure 1 and Table S3). We confirmed de novo somatic mosaicism of the c.521A>G mutation by deep sequencing of PCR products amplified with blood, nail, and saliva DNA from individual 2 and blood DNA from her parents, showing that approximately 35%–50% of cells harbored the mutation (Table S3).
Figure 1.
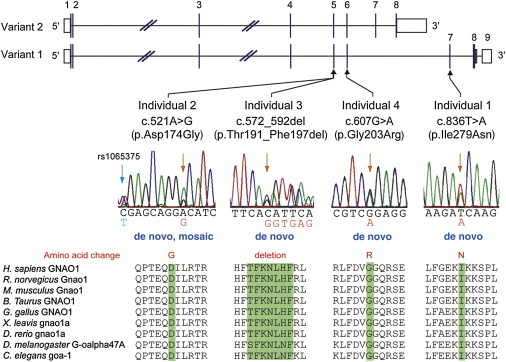
De Novo GNAO1 Mutations in Individuals with Epileptic Encephalopathy
Schematic representation of GNAO1, including two transcript variants: transcript variant 1 (RefSeq NM_020988.2) with nine exons and transcript variant 2 (RefSeq NM_138736.2) with eight exons. The UTRs and coding regions are shown in white and black rectangles, respectively. Three mutations occurred in common exons of two transcript variants, and one mutation occurred uniquely in transcript variant 1. Note that the electropherogram of individual 2 suggested mosaicism of the c.521A>G mutation, and a heterozygous C>T change (rs1065375) was clearly demonstrated. All mutations caused substitution or deletion of evolutionarily conserved amino acids. Homologous sequences were aligned with the use of the CLUSTALW web site.
Phenotypes Associated with GNAO1 Mutations
Neurological features of four female individuals with GNAO1 mutations are shown in Table 1. Three individuals (individuals 1–3) developed tonic seizures with suppression-burst pattern on EEG at the onset (range 4–29 days), leading to a diagnosis of OS. Individuals 2 and 3 transited to West syndrome, a common infantile epileptic syndrome, as revealed by hypsarrhythmia on EEG at 3–4 months of age (Figures 2A–2C). Individual 4 showed developmental delay and opisthotonic posture at 7 months of age, and complex partial seizures with epileptic discharge on EEG was observed at 5 years (Figure 2D). Of note, two individuals showed involuntary movements: individual 3 showed dystonia, and individual 4 displayed chorea and athetosis (Table 1 and Movie S1). Brain MRI showed delayed myelination in individuals 2 and 4, cerebral atrophy or reduced cerebral white matter in individuals 1 and 4, and thin corpus callosum in individuals 2 and 4 (Figures 2E–2I). Although seizures and EEG findings in two individuals with OS (individuals 2 and 3) were temporarily improved by adrenocorticotropic hormone therapy and valproic acid, all four individuals had intractable epileptic seizures in spite of combinatory therapy of antiepileptic drugs. All individuals had severe intellectual disability and motor developmental delay, and individual 3 died at 11 months because of respiratory-tract obstruction. These data suggest that GNAO1 mutations can cause multiple neurodevelopmental phenotypes, including epileptic encephalopathy and involuntary movements.
Table 1.
Clinical Features of Individuals with a GNAO1 Mutation
| Individual 1 | Individual 2 | Individual 3 | Individual 4 | |
|---|---|---|---|---|
| Gender | female | female | female | female |
| Age | 13 years | 4 years, 1 month | died at 11 months | 8 years |
| Mutation | c.836T>A (p.Ile279Asn) | c.521A>G (p.Asp174Gly) | c.572_592 del (p.Thr191_Phe197 del) | c.607G>A (p.Gly203Arg) |
| Inheritance | de novo | de novo, somatic mosaic | de novo | de novo |
| Diagnosis | Ohtahara syndrome | Ohtahara syndrome | Ohtahara syndrome | epileptic encephalopathy |
| Initial symptom | tonic seizure at 4 days | series of tonic seizures at 29 days (tonic upgaze, head nodding, extension of all extremities) | series of tonic seizures at 2 weeks (resemble spasms) | opisthotonic posture, developmental delay at 7 months |
| Initial EEG | suppression-burst pattern at 4 days | suppression-burst pattern at 29 days | suppression-burst pattern at 2 weeks | diffuse irregular spike-and-slow-wave complex at 5 years |
| Course of seizures | tonic seizure at 5 years | series of tonic seizures at 9 months | tonic seizure at 10 months | focal seizure (tonic upgaze), tonic seizure at 5 years |
| Course of EEG | multifocal sharp waves at 1 year, 4 months; suppression-burst pattern at 5 years, 6 months | hypsarrhythmia at 3 months; diffuse spike-and-slow-wave complex at 1 year, 7 months; sharp waves at frontal lobe at 3 years, 9 months | hypsarrhythmia at 4 months | not done |
| Involuntary movement | - | - | dystonia | severe chorea, athetosis |
| Seizure control | intractable (2–3 times per day) | intractable (0–2 times per day) | intractable | intractable (several times per day) |
| Development | ||||
| Head control | - | + | - | - |
| Sitting | - | - | - | - |
| Meaningful words | - | - | - | - |
| MRI | normal at 1 month; cerebral atrophy at 5 years, 6 months | delayed myelination and thin corpus callosum at 10 months | normal at 3 months | delayed myelination at 1 year, 3 months; reduced cerebral white matter, thin corpus callosum at 4 years, 8 months |
Figure 2.
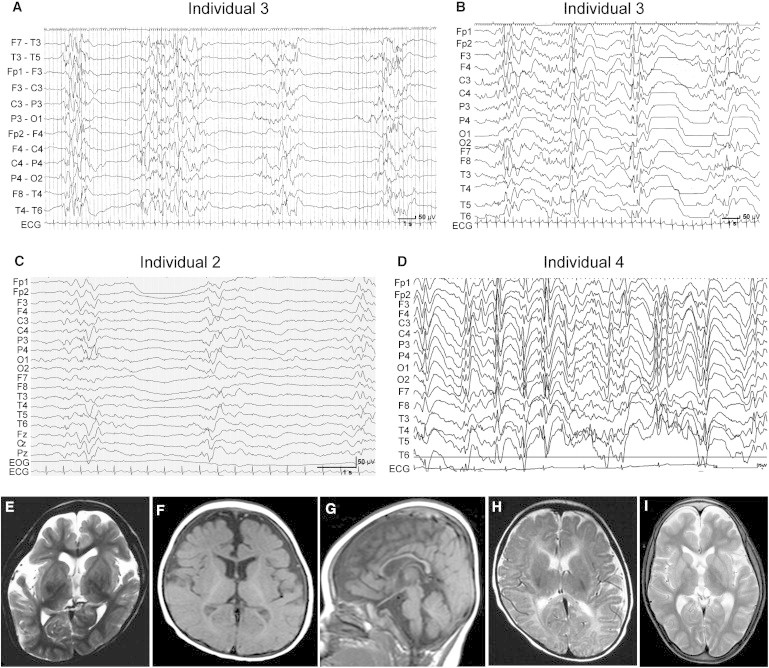
EEG and Brain MRI Features of Individuals with GNAO1 Mutations
(A and B) Interictal EEG of individual 3. A suppression-burst pattern was observed at 2 months of age (A), and transition to hypsarrhythmia was observed at 4 months (B).
(C) Interictal EEG of individual 2 shows a suppression-burst pattern at 2 months.
(D) Interictal EEG of individual 4 shows a diffuse spike- or sharp-and-slow-wave complex at 5 years.
(E–I) T2-weighted axial images through the basal ganglia (E, H, and I) and T1-weighted axial (F) and sagittal (G) images. Individual 1 showed cerebral atrophy at 5 years and 6 months (E). Individual 2 showed delayed myelination and thin corpus callosum at 10 months (F and G). Individual 3 showed normal appearance at 3 months (H). Individual 4 showed reduced white matter at 7 years (I).
Expression of Mutant Gαo1 in N2A Cells
To examine the mutational effect of four GNAO1 mutations, we performed transient expression experiments in N2A cells (Figure 3). C-terminally V5-epitope-tagged wild-type (WT) Gαo1, encoded by transcript variant 1, was clearly localized in the cell periphery, as previously reported.28 The p.Gly203Thr (with known loss of function)17 and p.Gly203Arg (in individual 4) altered proteins were also localized in the cell periphery. In contrast, the p.Thr191_Phe197del altered protein (in individual 3) accumulated in the cytosolic compartment. The p.Asp174Gly (individual 2) and p.Ile279Asn (individual 1) altered proteins were localized to the cell periphery and had weak signal in the cytosol, where more intense signal was observed in the p.Asp174Gly protein. Similar patterns of localization were observed for C-terminally AcGFP1-tagged Gαo1 (Figure S1). These localization patterns suggest that the function of the p.Thr191_Phe197del altered protein might be most severely affected.
Figure 3.
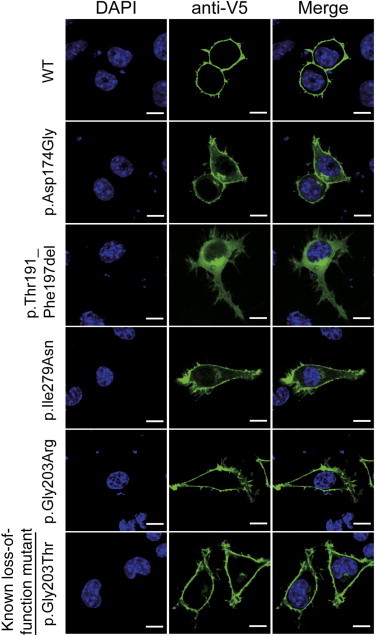
Localization of V5-Tagged Gαo1 Proteins in N2A Cells
Localization of WT and five altered Gαo1 proteins in N2A cells. The WT and p.Gly203Arg and p.Gly203Thr altered proteins were localized to the cell periphery. In contrast, the p.Thr191_Phe197del protein was localized to the cytosolic compartment. The other p.Asp174Gly and p.Ile279Asn proteins were localized to the cell periphery but were also observed in the cytosol. The scale bars represent 10 μm.
Structural Impacts of the Mutations on the Gα-Containing Complexes
To evaluate the impact of the GNAO1 mutations on specific functions at the atomic level, we mapped the substituted positions onto structures of the Gα subunit in complexed states representing the GDP-bound inactive state, the nucleotide-free Gαsβγ in complex with the receptor, and the GTP-bound active state. In the case of point mutations, we further estimated free-energy changes of the mutations by using FoldX software (version 3.0β5).19
The region corresponding to amino acid residues 191–197 of human Gαo1 is located in β strands and their connecting loop region and is involved in interactions with the G-protein-coupled receptor (GPCR) in the Gαβγ- β2AR complex (Figure 4A and Figure S2A). Thus, the deletion would affect secondary structure of the molecule and would not only impair the interaction with GPCR but also severely destabilize the Gα-subunit fold. The substituted residues corresponding to Asp174 and Ile279 of the human Gαo1 subunit are both buried inside the protein (Figure 4A) and are involved in hydrogen-bonding and hydrophobic interactions, respectively (Figure S2B). Therefore, the p.Asp174Gly and p.Ile279Asn substitutions would destabilize the Gα-subunit fold, as supported by FoldX calculations showing a more than 2 kcal/mol increase in free-energy changes for these substitutions (Figure 4B). It can be speculated that these altered proteins tend to be misfolded or denatured in N2A cells and thus have altered cellular localization (Figure 3).
Figure 4.
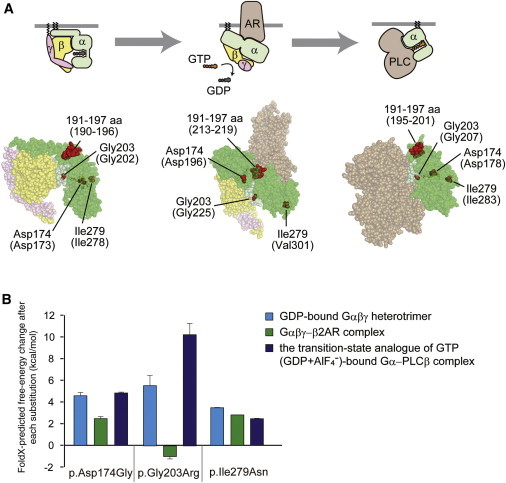
Structural Consideration of the Gα Amino Acid Substitutions in Some Complexed States
(A) Map of the amino acid substitution sites on the crystal structures of Gα-containing complexes: the GDP-bound inactive Gαiβγ heterotrimer (left), the nucleotide-free Gαsβγ in complex with an agonist-occupied monomeric β2AR (center), and the GDP+AlF4−-bound Gαq in complex with its effector PLCβ (right). Molecular structures are shown as space-filling representations (from PyMOL). Gα, Gβ, and Gγ subunits are colored green, yellow, and pink, respectively, and the switch I and switch II regions in the Gα subunit are in light green. The β2AR (center) and PLCβ (right) molecules are colored brown. The substituted sites are shown in red, and the indicated amino acid numbers correspond to human Gαo1 and, in parentheses, rat Gαi1 (UniProtKB/Swiss-Prot P10824) (left), bovine Gαs (UniProtKB/Swiss-Prot P04896) (center), and mouse Gαq (UniProtKB/Swiss-Prot P21279) (right). The illustrations above each model show the orientation of each subunit and the bound molecules.
(B) The free-energy change after each of the amino acid substitutions was estimated from calculations using FoldX software. Each error bar represents an average value with a SD.
The substituted residue corresponding to Gly203 of human Gαo1 is located within the highly conserved switch II region, responsible for activation of downstream effectors upon GTP binding (Figure 4A). Conformations of the switch regions differ depending on the complex state of the G protein. In the Gαβγ heterotrimer and the GDP+AlF4−-bound Gα-effector (PLCβ) complex, the glycine residues are closely surrounded by the switch I region and GTP (Figure 4A and Figure S2C). Thus, the p.Gly203Arg substitution would cause steric hindrance between the arginine side chain and the switch I region and/or GTP, destabilizing the complex, as supported by the FoldX calculations showing a remarkable increase in free-energy change upon the p.Gly203Arg substitution. By contrast, in the Gαβγ-receptor (β2AR) complex, no substantial steric hindrance was predicted from the structural modeling and FoldX calculations (Figure 4B and Figure S2C). These findings suggest that the p.Gly203Arg-substituted Gα subunit would impair GTP binding and/or activation of the downstream effectors, although it might still bind to GPCR. This prediction was supported by previous reports, in which GTP binding was weakened in the p.Gly203Thr altered Gα.17 This also appears to be consistent with the apparently normal cellular localization of the p.Gly203Arg altered protein in N2A cells (Figure 3).
Electrophysiological Evaluation of Gαo1 Mutants
It has been reported that N-type calcium channels are inhibited, at least in part, via Gαo-mediated signaling.7 Using NG108-15 cells, in which norepinephrine-induced calcium-current inhibition is mediated by Gαo (Figure 5A),29 we analyzed functional properties of altered Gαo1. Compared with cells expressing WT Gαo1 (the leftmost column in Figure 5B), NG108-15 cells expressing the p.Thr191_Phe197del substitution revealed a significant increase in calcium-current density before application of norepinephrine (p < 0.05 by Dunnett’s post hoc test; the second column from the right in Figure 5B), suggesting that localization of the altered Gαo1 might affect calcium-channel activity. In cells expressing the p.Asp174Gly substitution, a mild increase in the current density was also suggested, although the difference was not significant (the third column from the left in Figure 5B). The other two substitutions had no effects on the current (the second column from the left and the rightmost column in Figure 5B). Treatment with 10 μM norepinephrine reduced the calcium-current density by 19.0% ± 5.0% in cells expressing WT Gαo1 (p < 0.01 by paired t test; left panel in Figure 5A and the leftmost bar in Figure 5C). A similar reduction was observed in cells expressing the p.Ile279Asn alteration (18.5% ± 3.5%, p < 0.01 by paired t test; the rightmost bar in Figure 5C). In cells expressing the p.Thr191_Phe197del alteration, by contrast, the reduction was obscured (12.1% ± 5.0%, not significant by paired t test; right panel in Figure 5A and the second bar from the right in Figure 5C). In cells expressing the other two substitutions (p.Gly203Thr and p.Asp174Gly), weaker current inhibition by norepinephrine was suggested (9.9% ± 3.8% and 11.1% ± 3.5%, respectively; both were p < 0.05 by paired t test; the second and third bars from the left in Figure 5C), although compared with that in WT-expressing cells, the degrees of inhibition in Gly203Thr- and p.Asp174Gly-expressing cells did not reach statistical significance (not significant by ANOVA). These data suggest that GNAO1 mutations could hamper Gαo-mediated signaling.
Figure 5.
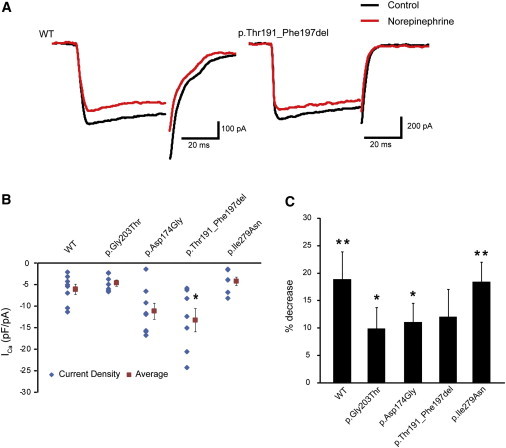
Evaluation of Gαo-Mediated Signaling in NG108-15 Cell Calcium-Current Generation
(A) Representative traces of voltage-gated calcium currents generated in NG108-15 cells expressing WT (left) or p.Thr191_Phe197del altered (right) Gαo1. Black and red traces represent the currents before and 3 min after application of 10 μM norepinephrine, respectively.
(B) Current densities of the calcium currents before norepinephrine treatment in cells expressing WT or altered Gαo1. Scatter plots represent the densities in individual cells. Red squares and bars represent the means and SEMs, respectively, of the densities in individual cell groups (WT, n = 8; p.Gly203Thr, n = 7; p.Asp174Gly, n = 8; p.Thr191_Phe197del, n = 7; p.Ile279Asn, n = 7). Compared with that in cells expressing WT Gαo1, the current density in cells expressing p.Thr191_Phe197del increased significantly (∗p < 0.05 by Dunnett’s post hoc test). The densities in the cells expressing other altered proteins did not vary significantly.
(C) Comparison of norepinephrine-induced inhibition of calcium currents in cells expressing altered Gαo1. Each error bar represents the mean and SEM of the percent decrease in current density induced by application of 10 μM norepinephrine. Paired t tests indicated that the inhibition induced by norepinephrine was significant in cells expressing WT (n = 8) and p.Gly203Thr (n = 7), p.Asp174Gly (n = 8), and p.Ile279Asn (n = 7) altered proteins (∗∗p < 0.01 and ∗p < 0.05), but not in cells expressing p.Thr191_Phe197del (n = 7). Although there was some tendency for decreased inhibition in cells expressing altered proteins, the tendency did not reach statistical significance compared with that in WT-expressing cells (p = 0.41 by ANOVA).
Discussion
We successfully identified four de novo heterozygous missense GNAO1 mutations in four individuals. All four individuals showed severe intellectual disability and motor developmental delay, demonstrating that aberration of Gαo affects intellectual and motor development. In addition, all four individuals showed epileptic encephalopathy, and two of them showed involuntary movements. Because Gαo-deficient mice show occasional seizures and generalized tremor,9,10 it is likely that epilepsy and involuntary movement are two of the characteristic features caused by GNAO1 mutations. Although Gαo-deficient mice also show hyperactivity and hyperalgesia,10 it is difficult to evaluate whether our individuals had these symptoms because of severe motor and cognitive impairment.
All four of these mutations, and especially two mutations leading to the p.Thr191_Phe197del and p.Gly203Arg substitutions, are predicted to affect Gαo function by structural evaluation. In fact, transient expression in N2A cells showed that localization of the p.Thr191_Phe197del altered protein was dramatically changed to the cytosolic compartment. Interestingly, two alterations (p.Ile279Asn and p.Asp174Gly) also showed weak signal in the cytosol, suggesting that localization to the plasma membrane was variably impaired in three altered proteins. Measurement of voltage-dependent calcium currents in NG108-15 cells also suggested impaired functions of altered Gαo1. The p.Thr191_Phe197del alteration significantly increased the basal calcium-current density, and compared with WT-expressing cells, cells expressing one of the three substitutions (p.Thr191_Phe197del, p.Asp174Gly, or p.Gly203Thr) showed a tendency towards weaker inhibition of calcium currents by norepinephrine. All these data suggest that the four GNAO1 mutations might cause loss of Gαo1 function.
Our experimental data suggest that Gαo function might be most severely affected in the p.Thr191_Phe197del altered protein. This appears to be correlated with the severity of clinical features because individual 3 showed both OS and involuntary movements and indeed died during the infantile period. Therefore, she might have had the most severe phenotype caused by a GNAO1 mutation. Another interesting finding is somatic mosaicism of the c.521A>G (p.Asp174Gly) mutation in individual 2, in whom approximately 35%–50% of cells harbored the mutation. Somatic mosaicism of responsive genes in infantile epilepsy, such as SCN1A (MIM 182389) and STXBP1, have been reported, explaining the presence of unaffected or mildly affected transmitting parents.30,31 However, individual 2 showed OS, delayed myelination, and thin corpus callosum. Although we did not determine the mosaic rate in brain tissues, the presence of 35%–50% of cells harboring the GNAO1 mutation in the brain might be sufficient to cause abnormal brain development.
It has been reported that activation of G-protein-coupled α2 adrenergic receptors by norepinephrine attenuates epileptiform activity in the hippocampal CA3 region.32 Gαo is known to be involved in this response,33 suggesting that alteration of pathways mediated by α2 adrenergic receptor and Gαo might contribute to the pathogenesis of epilepsy. Because calcium-current inhibition is a well-known consequence of Gαo-mediated signaling induced by norepinephrine, it is possible that epileptic seizures associated with GNAO1 mutations might be improved by calcium-channel modulators. For example, pregabalin and gabapentin act as selective calcium-channel blockers,34,35 and topiramate modulates high-voltage-activated calcium channels in dentate granule cells.36 Because our four individuals were not treated with these drugs, it is worth administrating these three drugs for examining putative protective effects.
In conclusion, de novo heterozygous GNAO1 mutations were identified in four individuals with epileptic encephalopathy. Furthering our understanding of abnormal Gαo-mediated heterotrimeric G protein signaling might provide new insights into the pathogenesis and treatment of epileptic encephalopathy.
Acknowledgments
We would like to thank the individuals and their families for their participation in this study. We also thank Aya Narita and Nobuko Watanabe for their technical assistance and Tohru Kozasa and Nobuchika Suzuki for their valuable comments. This work was supported by the Ministry of Health, Labour, and Welfare of Japan, the Japan Society for the Promotion of Science (Grants-in-Aid for Scientific Research (B) [25293085 and 25293235] and a Grant-in-Aid for Scientific Research (A) [13313587]), the Takeda Science Foundation, the Japan Science and Technology Agency, the Strategic Research Program for Brain Sciences (11105137), and a Grant-in-Aid for Scientific Research on Innovative Areas (Transcription Cycle) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (12024421).
Contributor Information
Naomichi Matsumoto, Email: naomat@yokohama-cu.ac.jp.
Hirotomo Saitsu, Email: hsaitsu@yokohama-cu.ac.jp.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
CLUSTALW, http://www.genome.jp/tools/clustalw/
GenBank, http://www.ncbi.nlm.nih.gov/Genbank/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
Picard, http://picard.sourceforge.net/
Protein Data Bank, http://www.rcsb.org/pdb/home/home.do
PyMOL, www.pymol.org
UniProtKB/Swiss-Prot, http://www.uniprot.org/
References
- 1.Dulac O. Epileptic encephalopathy. Epilepsia. 2001;42(Suppl 3):23–26. doi: 10.1046/j.1528-1157.2001.042suppl.3023.x. [DOI] [PubMed] [Google Scholar]
- 2.Ohtahara S., Yamatogi Y. Ohtahara syndrome: with special reference to its developmental aspects for differentiating from early myoclonic encephalopathy. Epilepsy Res. 2006;70(Suppl 1):S58–S67. doi: 10.1016/j.eplepsyres.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 3.Saitsu H., Kato M., Koide A., Goto T., Fujita T., Nishiyama K., Tsurusaki Y., Doi H., Miyake N., Hayasaka K., Matsumoto N. Whole exome sequencing identifies KCNQ2 mutations in Ohtahara syndrome. Ann. Neurol. 2012;72:298–300. doi: 10.1002/ana.23620. [DOI] [PubMed] [Google Scholar]
- 4.Saitsu H., Kato M., Mizuguchi T., Hamada K., Osaka H., Tohyama J., Uruno K., Kumada S., Nishiyama K., Nishimura A. De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat. Genet. 2008;40:782–788. doi: 10.1038/ng.150. [DOI] [PubMed] [Google Scholar]
- 5.Weckhuysen S., Mandelstam S., Suls A., Audenaert D., Deconinck T., Claes L.R., Deprez L., Smets K., Hristova D., Yordanova I. KCNQ2 encephalopathy: emerging phenotype of a neonatal epileptic encephalopathy. Ann. Neurol. 2012;71:15–25. doi: 10.1002/ana.22644. [DOI] [PubMed] [Google Scholar]
- 6.Kato M., Saitoh S., Kamei A., Shiraishi H., Ueda Y., Akasaka M., Tohyama J., Akasaka N., Hayasaka K. A longer polyalanine expansion mutation in the ARX gene causes early infantile epileptic encephalopathy with suppression-burst pattern (Ohtahara syndrome) Am. J. Hum. Genet. 2007;81:361–366. doi: 10.1086/518903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wettschureck N., Offermanns S. Mammalian G proteins and their cell type specific functions. Physiol. Rev. 2005;85:1159–1204. doi: 10.1152/physrev.00003.2005. [DOI] [PubMed] [Google Scholar]
- 8.Huff R.M., Axton J.M., Neer E.J. Physical and immunological characterization of a guanine nucleotide-binding protein purified from bovine cerebral cortex. J. Biol. Chem. 1985;260:10864–10871. [PubMed] [Google Scholar]
- 9.Valenzuela D., Han X., Mende U., Fankhauser C., Mashimo H., Huang P., Pfeffer J., Neer E.J., Fishman M.C. G alpha(o) is necessary for muscarinic regulation of Ca2+ channels in mouse heart. Proc. Natl. Acad. Sci. USA. 1997;94:1727–1732. doi: 10.1073/pnas.94.5.1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang M., Gold M.S., Boulay G., Spicher K., Peyton M., Brabet P., Srinivasan Y., Rudolph U., Ellison G., Birnbaumer L. Multiple neurological abnormalities in mice deficient in the G protein Go. Proc. Natl. Acad. Sci. USA. 1998;95:3269–3274. doi: 10.1073/pnas.95.6.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saitsu H., Kato M., Osaka H., Moriyama N., Horita H., Nishiyama K., Yoneda Y., Kondo Y., Tsurusaki Y., Doi H. CASK aberrations in male patients with Ohtahara syndrome and cerebellar hypoplasia. Epilepsia. 2012;53:1441–1449. doi: 10.1111/j.1528-1167.2012.03548.x. [DOI] [PubMed] [Google Scholar]
- 12.Saitsu H., Nishimura T., Muramatsu K., Kodera H., Kumada S., Sugai K., Kasai-Yoshida E., Sawaura N., Nishida H., Hoshino A. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat. Genet. 2013;45:445–449. doi: 10.1038/ng.2562. e1. [DOI] [PubMed] [Google Scholar]
- 13.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robinson J.T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thorvaldsdóttir H., Robinson J.T., Mesirov J.P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Slepak V.Z., Wilkie T.M., Simon M.I. Mutational analysis of G protein alpha subunit G(o) alpha expressed in Escherichia coli. J. Biol. Chem. 1993;268:1414–1423. [PubMed] [Google Scholar]
- 18.Williams D.J., Puhl H.L., Ikeda S.R. A Simple, Highly Efficient Method for Heterologous Expression in Mammalian Primary Neurons Using Cationic Lipid-mediated mRNA Transfection. Front Neurosci. 2010;4:181. doi: 10.3389/fnins.2010.00181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guerois R., Nielsen J.E., Serrano L. Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J. Mol. Biol. 2002;320:369–387. doi: 10.1016/S0022-2836(02)00442-4. [DOI] [PubMed] [Google Scholar]
- 20.Wall M.A., Coleman D.E., Lee E., Iñiguez-Lluhi J.A., Posner B.A., Gilman A.G., Sprang S.R. The structure of the G protein heterotrimer Gi alpha 1 beta 1 gamma 2. Cell. 1995;83:1047–1058. doi: 10.1016/0092-8674(95)90220-1. [DOI] [PubMed] [Google Scholar]
- 21.Rasmussen S.G., DeVree B.T., Zou Y., Kruse A.C., Chung K.Y., Kobilka T.S., Thian F.S., Chae P.S., Pardon E., Calinski D. Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Waldo G.L., Ricks T.K., Hicks S.N., Cheever M.L., Kawano T., Tsuboi K., Wang X., Montell C., Kozasa T., Sondek J., Harden T.K. Kinetic scaffolding mediated by a phospholipase C-beta and Gq signaling complex. Science. 2010;330:974–980. doi: 10.1126/science.1193438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Edvardson S., Baumann A.M., Mühlenhoff M., Stephan O., Kuss A.W., Shaag A., He L., Zenvirt S., Tanzi R., Gerardy-Schahn R., Elpeleg O. West syndrome caused by ST3Gal-III deficiency. Epilepsia. 2013;54:e24–e27. doi: 10.1111/epi.12050. [DOI] [PubMed] [Google Scholar]
- 24.Molinari F., Raas-Rothschild A., Rio M., Fiermonte G., Encha-Razavi F., Palmieri L., Palmieri F., Ben-Neriah Z., Kadhom N., Vekemans M. Impaired mitochondrial glutamate transport in autosomal recessive neonatal myoclonic epilepsy. Am. J. Hum. Genet. 2005;76:334–339. doi: 10.1086/427564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mills P.B., Surtees R.A., Champion M.P., Beesley C.E., Dalton N., Scambler P.J., Heales S.J., Briddon A., Scheimberg I., Hoffmann G.F. Neonatal epileptic encephalopathy caused by mutations in the PNPO gene encoding pyridox(am)ine 5′-phosphate oxidase. Hum. Mol. Genet. 2005;14:1077–1086. doi: 10.1093/hmg/ddi120. [DOI] [PubMed] [Google Scholar]
- 26.Shen J., Gilmore E.C., Marshall C.A., Haddadin M., Reynolds J.J., Eyaid W., Bodell A., Barry B., Gleason D., Allen K. Mutations in PNKP cause microcephaly, seizures and defects in DNA repair. Nat. Genet. 2010;42:245–249. doi: 10.1038/ng.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kurian M.A., Meyer E., Vassallo G., Morgan N.V., Prakash N., Pasha S., Hai N.A., Shuib S., Rahman F., Wassmer E. Phospholipase C beta 1 deficiency is associated with early-onset epileptic encephalopathy. Brain. 2010;133:2964–2970. doi: 10.1093/brain/awq238. [DOI] [PubMed] [Google Scholar]
- 28.Nakata H., Kozasa T. Functional characterization of Galphao signaling through G protein-regulated inducer of neurite outgrowth 1. Mol. Pharmacol. 2005;67:695–702. doi: 10.1124/mol.104.003913. [DOI] [PubMed] [Google Scholar]
- 29.McFadzean I., Mullaney I., Brown D.A., Milligan G. Antibodies to the GTP binding protein, Go, antagonize noradrenaline-induced calcium current inhibition in NG108-15 hybrid cells. Neuron. 1989;3:177–182. doi: 10.1016/0896-6273(89)90030-5. [DOI] [PubMed] [Google Scholar]
- 30.Saitsu H., Hoshino H., Kato M., Nishiyama K., Okada I., Yoneda Y., Tsurusaki Y., Doi H., Miyake N., Kubota M. Paternal mosaicism of an STXBP1 mutation in OS. Clin. Genet. 2011;80:484–488. doi: 10.1111/j.1399-0004.2010.01575.x. [DOI] [PubMed] [Google Scholar]
- 31.Marini C., Scheffer I.E., Nabbout R., Suls A., De Jonghe P., Zara F., Guerrini R. The genetics of Dravet syndrome. Epilepsia. 2011;52(Suppl 2):24–29. doi: 10.1111/j.1528-1167.2011.02997.x. [DOI] [PubMed] [Google Scholar]
- 32.Jurgens C.W., Hammad H.M., Lichter J.A., Boese S.J., Nelson B.W., Goldenstein B.L., Davis K.L., Xu K., Hillman K.L., Porter J.E., Doze V.A. Alpha2A adrenergic receptor activation inhibits epileptiform activity in the rat hippocampal CA3 region. Mol. Pharmacol. 2007;71:1572–1581. doi: 10.1124/mol.106.031773. [DOI] [PubMed] [Google Scholar]
- 33.Goldenstein B.L., Nelson B.W., Xu K., Luger E.J., Pribula J.A., Wald J.M., O’Shea L.A., Weinshenker D., Charbeneau R.A., Huang X. Regulator of G protein signaling protein suppression of Galphao protein-mediated alpha2A adrenergic receptor inhibition of mouse hippocampal CA3 epileptiform activity. Mol. Pharmacol. 2009;75:1222–1230. doi: 10.1124/mol.108.054296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sills G.J. The mechanisms of action of gabapentin and pregabalin. Curr. Opin. Pharmacol. 2006;6:108–113. doi: 10.1016/j.coph.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 35.Stefani A., Spadoni F., Giacomini P., Lavaroni F., Bernardi G. The effects of gabapentin on different ligand- and voltage-gated currents in isolated cortical neurons. Epilepsy Res. 2001;43:239–248. doi: 10.1016/s0920-1211(00)00201-1. [DOI] [PubMed] [Google Scholar]
- 36.Zhang X., Velumian A.A., Jones O.T., Carlen P.L. Modulation of high-voltage-activated calcium channels in dentate granule cells by topiramate. Epilepsia. 2000;41(Suppl 1):S52–S60. doi: 10.1111/j.1528-1157.2000.tb02173.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.