

Abstract
Altered function of ion channels in the heart can increase the risk of sudden arrhythmic death. Hundreds of genetic variants exist in these cardiac ion channel genes. The challenge is how to interpret the effects of multiple conductance perturbations on the complex multi-variable cardiac electrical system? In theory, sensitivity analysis can address this question. However, to date this approach has been restricted by computational overheads to analysis of isolated cells, which has limited extrapolation to physiologically relevant scales. The goal of this study was to extend existing sensitivity analyses to electrocardiogram (ECG) signals derived from multicellular systems and quantify the contribution of ionic conductances to emergent properties of the ECG. To achieve this, we have developed a highly parallelised simulation environment using unconventional high performance computing architectures to analyse the emergent electrical properties of a multicellular system. This has permitted the first systematic analysis of the molecular basis of the T wave amplitude, revealing important but distinct roles for delayed rectifier and inward rectifier K+ currents. In addition to quantifying how interactions between multiple ion channels influence ECG parameters we show that these sensitivities are dynamic functions of heart rate. This study provides a significant advance in our understanding both of how individual ion conductances define ECG signals and of epistatic modification of cardiac electrical phenotypes. The parallelised simulation environment we have developed removes the computational roadblock that has limited this approach and so provides the framework for future analysis of more complex tissue and whole organ systems.
Key points
We used a novel high performance computing approach to conduct a sensitivity analysis of emergent properties of simulated ECGs from a transmural cable of cells.
The rapid delayed rectifier and inward rectifying potassium currents are the primary determinants of the height of the T wave in this system.
Theight is correlated with the temporal dispersion of repolarisation in the transmural cable while Tpeak– Tend is correlated with the interval from the time of maximum total rate of repolarisation to the end of repolarisation in the cable of cells.
This study advances our understanding of the molecular basis of T wave morphology and the role of epistatis in the modification of cardiac electrical phenotypes.
Introduction
Sudden cardiac death caused by abnormal electrical signalling in the heart accounts for approximately 10–15% of deaths in the developed world (Knollmann & Roden, 2008). Despite intensive investigation at both the tissue level (Hondeghem et al. 2001) and the molecular/cellular level (Keating & Sanguinetti, 2001) we still cannot predict accurately who is at greatest risk and whom we should target for use of implantable cardiac defibrillators. In recent years there has been considerable interest in identifying the genetic factors that may influence the risk of sudden cardiac death (Grace & Roden, 2012). What is not well understood is how this genetic variation translates to altered electrical phenotype of cardiac tissue.
One of the most commonly used techniques for measurement of electrical activity of the heart is the surface electrocardiogram (ECG). Many studies have shown that parameters measured from the ECG show significant variability across the population. For example, the QT interval, a measure of the repolarisation time of the heart, shows a distribution of 385 ± 24 ms in the normal male population (Gallagher et al. 2006). It is likely that some of this variability is acquired, e.g. related to myocardial injury or remodelling. Twin studies suggest that there is also a significant genetic component (Carter et al. 2000; Haarmark et al. 2011). However, twin studies do not identify the basis of this genetic contribution. At the other extreme, the advent of whole genome sequencing has highlighted the fact that genetic variation is extraordinarily extensive, and polymorphisms occur on average once every 17 bp in coding regions (Nelson et al. 2012). Whilst the vast majority of these are benign (it is estimated that ∼2% of variants in coding regions are functional, Tennessen et al. 2012), the likelihood is that an average-sized gene will have multiple functional variants from person to person. It has been suggested that combinations of these multiple small perturbations in function can account for the variability in a phenotype (Mann et al. 2012). In this study, we set out to test this hypothesis with respect to how individual ion currents affect cardiac repolarisation parameters determined from the ECG.
The electrical properties of the heart are a complex function of the spatial distribution of approximately 5–10 billion cells (Adler & Costabel, 1975), which themselves contain multiple ion channels and pumps. Predicting the effects of changes to even one component in such a complex multivariable system is very difficult, and realistically only tractable through use of computer modelling (Bailey, 1999). At the single cell level, sensitivity analysis of computational models is one approach that has been used to quantify the contribution of ionic conductance levels to cellular electrical outputs such as action potential duration (APD). Specifically, partial least squares (PLS) regression is a statistical technique that has been used to highlight the differences between cellular models (Sobie, 2009) and demonstrate how epistatic variability can produce a phenotype similar to that observed with single gene mutations of large effect (Mann et al. 2012). These studies, at the single cell level, have demonstrated that it is possible to investigate how simultaneous changes in multiple inputs can influence the output of a complex system.
The emergent electrical properties of the heart are more than the sum of individual myocytes. At the simplest level, we know that there are multiple distinct subtypes of myocytes including epicardial, mid-myocardial and endocardial myocytes and that electrotonic interactions between them modulate the properties of the individual cells (Glukhov et al. 2010). As a result, analysis of single cell systems provides only limited insight into whole heart physiology. Unfortunately, to date, sensitivity analysis of more complex multicellular systems has been prevented by the immense computational load of simulating hundreds of thousands of heart beats.
In this study we have overcome this computational obstacle by utilising the large scale parallelisation afforded by general purpose computing on graphics processing units (GPGPU) (Owens et al. 2007; Kirk & Hwu, 2010). Specifically, we have extended the application of partial least squares sensitivity analysis to the more complex and more clinically relevant scenario of a pseudo ECG generated from a transmural cable of ventricular myocytes that reflects the heterogeneity of endocardial, mid-myocardial and epicardial cells. Use of this novel computational framework has allowed us, for the first time, to analyse the molecular determinants of the amplitude of the T wave, a signal that cannot be inferred from single cell studies. We were also able to quantify the effects of individual ion channel conductance changes on ECG parameters at different pacing rates and assess the effect of multiple ‘genetic hits’ on ECG waveforms. This work also provides proof-of-principle that it is possible to analyse the molecular basis of emergent properties of multicellular preparations.
Methods
Ventricular cell model
Cellular simulations were carried out using the O’Hara ventricular model (O’Hara et al. 2011). This cellular model was chosen since it is based entirely on data from healthy human tissue. The sodium channel in the O’Hara model was replaced with the Markov description from Clancy and Kass (Clancy et al. 2002) to enable future incorporation of long QT syndrome (LQTS) type 3 mutants genotypes. Likewise the slow delayed rectifying current (IKs) in the model was replaced with the human IKs from Terrenoire et al. to allow future incorporation of adrenergic effects (Terrenoire et al. 2005). Both currents were scaled such that peak current amplitudes during the action potential (AP) were unaltered. Recent results suggest that Nav1.8 is responsible for the late sodium current in cardiomyocytes (Yang et al. 2012). We therefore left the existing persistent sodium current of the model unaltered. The eight most significant ionic conductances in the O’Hara cell model analysed as part of this study were: GNa (cardiac sodium current), GNaL (persistent sodium current), GCaL (L-type calcium current), GKr (rapid delayed rectifying potassium current), GKs (slow delayed rectifying potassium current), GK1 (inward rectifier), GKb (background potassium current), GJup (calcium uptake into sarcoplasmic reticulum via sarco/endoplasmic reticulum Ca2+-ATPase (SERCA)), Gncx (maximum current of sodium–calcium exchanger).
Cable simulations/ECG calculations
Cable simulations were performed as previously described (Shaw & Rudy, 1997; Gima & Rudy, 2002). Cables consisted of 165 O’Hara model cells, with cells 1–60 in endocardial configuration, cells 61–105 in mid-myocardial configuration, and cells 106–165 in epicardial configuration. Cables were stimulated from the endocardial terminal of the cable with a current of –80 m μA μF−1 for 0.5 ms and propagation calculated according to the partial differential equation (PDE):
| (1) |
Where Cm is the membrane capacitance, V is the transmembrane voltage, a is the radius of the fibre (11 μm), Rcg is the ratio between capacitive and geometric areas (Rcg= 2), σ is effective conductivity (composed of myoplasmic conductivity and gap junction conductivity) and Iion and Istim represent ionic and stimulus currents at position x, respectively.
To calculate V along the cable, eqn (1) was made discrete in space and time using the finite difference method (assuming Δt= 0.01 ms, Cm= 1 μF and Δx=L= 0.01 cm, where L is cell length). Therefore, at time t we can obtain 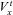 from the equation:
from the equation:
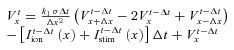 |
(1a) |
The value of 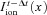 is calculated from the cell model (at position x) by applying the forward Euler integration time step (i.e. explicit Euler).
is calculated from the cell model (at position x) by applying the forward Euler integration time step (i.e. explicit Euler).
We calculated pseudo ECGs by determining a spatially weighted sum of the voltage gradient at a point x′= 2 cm away from the endocardial end of the cable along the fibre axis, according to the equation (Plonsey & Barr, 2007):
| (2) |
The first and last 15 cells were excluded from the pseudo ECG calculations due to edge effects.
Partial least squares analysis
To assess the sensitivity of action potential and ECG parameters to changes in individual ionic currents in the cardiac myocyte we employed partial least squares (PLS) analysis (Sobie, 2009). This technique correlates the effects of variation of input parameters of a model (ionic conductances through channels and pumps) with output parameters (action potential duration, QT interval, Theight, Tpeak– Tend). For each instance of the PLS analysis, the system (either individual cell or cable) was solved for 100 individual runs. In each run, all ionic conductances in the model were individually scaled by a random number drawn from a log–normal distribution centred around a mean value of 1, with standard deviation of 5%. A single scaling factor was applied to each conductance across the entire cable, regardless of whether the particular conductance varied depending on cell type (i.e. epi- mid-myo- and endocardial cells). For every run, the system was equilibrated for 500 beats after parameter randomisation to account for long-term changes in ionic concentrations (see Supplemental Fig. 1; Supplemental material is available online only). After equilibration, ECG or APD parameters were measured from the 500th beat and saved with the corresponding set of eight scaling factors. Matrices containing APD/ECG parameters and scaling factors were log-transformed, centred on their mean values, and normalised to their means. These values were used as inputs for the partial least squares function PLSREGRESS in the Matlab Statistics Toolbox (Mathworks, MA, USA). The output of this function is an array of PLS regression coefficients (ρ) that describes the model's sensitivity for each ionic conductance. To get an estimate of error in PLS regressions, datasets of 100 simulations were randomly split into three and PLS analysis carried out on each sub-dataset. Means and standard deviations of PLS coefficients were then derived from the three outputs.
‘Wheel of fortune’ plots
To generate ‘wheel of fortune’ plots we examined five ionic conductances in each case. Each of these conductances were varied by ±10% which, together with the baseline level, gives a total of 35 (three levels of five different conductances), or 243 different combinations for each plot. For each of these 243 combinations we simulated ECGs (each equilibrated for 500 beats at 1 Hz). We then used ECG parameters measured from these simulations to generate the ‘genetic wheel of fortune’ plots. In each plot, each of the 243 circle segments represents one of the possible combinations of conductance level variation in our five selected ion channels. Segments are coloured according to conductance level such that the baseline level of each conductance is shown in grey, an increase of 10% in white, and a decrease of 10% in black. Combinations are then sorted according to the magnitude of the measured parameter (QT interval, Tpeak– Tend and Theight), from highest (red, at the three o’clock position) to the lowest (blue, at the four o’clock position).
Graphics processing unit (GPU) simulation environment
To overcome the computational challenge of large-scale simulation, we exploited two levels of parallelism that naturally exist in the problem. First, in each simulation set, all of the 100 randomised simulation scenarios were executed concurrently. Second, at every time step of each simulation scenario, each cell model was solved independently from the rest of the cells in the cable (before solution of the PDE). It should be noted that even in a conventional sequential approach, each individual cell in the cable is solved independently. The difference in this approach is that the individual cells are solved simultaneously, rather than one after the other before solution of the PDE.
We exploited the massively parallel computing capability of the GPU architecture (Owens et al. 2007; Kirk & Hwu, 2010) to develop a custom parallel simulation system. The implementation consisted of two main subsystems: a simulation engine implemented in CUDA C 4.1 (NVIDIA, CA, USA) and a pre/post-processing subsystem implemented in Matlab. Communication between the two subsystems was achieved by exchanging text files in comma-separated format. Simulations were performed on a GPU-based supercomputer (Bragg, CSIRO, Australia), where individual cluster nodes, each comprising a Dual Xeon 8-Core E5-2650 CPU and three NVIDIA Tesla M2050 GPU cards, were assigned to each set of simulations (i.e. for the 100 scenarios at each pacing rate and the 243 scenarios for each wheel of fortune).
Results
Electrotonic coupling modulates sensitivity to conductance variability
A comparison of AP waveforms extracted from a cable in the middle of the endocardial, mid-myocardial and epicardial regions with AP waveforms from isolated single cell simulations of the same cell type is shown in Fig. 1. Electrotonic coupling resulted in significant shortening of the mid-myocardial action potential as well as more subtle effects on plateau and notch morphologies of all cell types.
Figure 1. Effect of electrotonic coupling.
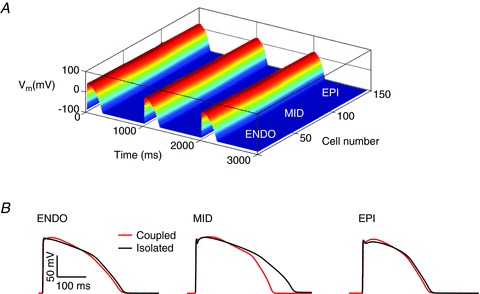
A, simulation of action potential propagation in a cable of ventricular cells. Three beats of the simulation paced at 1 Hz at the endocardial terminal of the cable are shown. B, comparison of action potential waveforms for endocardial (ENDO), mid-myocardial (MID) and epicardial (EPI) cells either simulated in isolation, or in the presence of electrotonic coupling (extracted from the cable in A).
To investigate how electrotonic coupling affects the sensitivity of AP waveforms to changes in individual ionic conductances we undertook a partial least squares (PLS) regression analysis of action potential duration at 90% repolarisation (APD90) in isolated versus coupled cells (see Fig. 2). Specifically, each of the ionic conductances in the cell models was scaled according to a randomised factor taken from a log–normal distribution, meaning the probability of the conductance being doubled was the same as that of it being halved (Fig. 2Ab). We focused our investigation on variations in conductance, rather than channel kinetics, since this is representative of most disease-causing mutations and variants. For example, the most thorough analysis of the mechanism of disease in mutations that cause long QT syndrome type 2 showed that the vast majority (80%) of mutants exerted their effect via reduced trafficking of proteins, resulting in altered membrane conductance, not via effects on channel kinetics (Anderson et al. 2006). This process was repeated 100 times in order to simulate families of APs that varied according to the combination of inputs (Fig. 2Ac). An example of how the output from these simulations can be analysed is illustrated in Fig. 2Ad, which shows a scatter plot of the simulated APD90 versus the conductance for GKr in each simulation. Each of the data points represents a different combination of the remaining (non-GKr) input parameters. This plot provides a crude estimate of the effect of changes in GKr on APD90. Similar plots can be done where each of the other ion conductances is made the independent variable. PLS regression takes the analysis one step further to present these data in terms of a sensitivity coefficient, ρ, which allows direct quantitative comparison between different input parameters.
Figure 2. PLS regression.
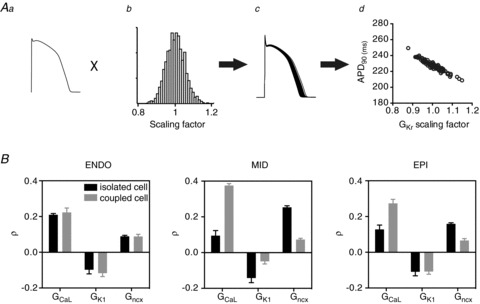
A, simulation pipeline for PLS analysis. Ionic conductances in the baseline model defining the action potential (a) are modified according to a log–normal distribution of randomised scaling factors (b). This process is repeated 100 times to yield a family of action potentials (c). The relationship between action potential parameters such as APD90 and variability in individual ionic conductances can be represented on a scatter plot (d). B, comparison of PLS sensitivities (ρ) for APD90 of action potentials simulated in isolation or as part of a cable for endo-, mid-myo- and epicardial cells.
PLS outputs describing the sensitivity of APD90 to changes in GCaL, GK1 and Gncx, for isolated versus coupled epi-, mid-myo- and endocardial cells are presented in Fig. 2B. The magnitude of the bars in Fig. 2B indicates how sensitive the output is to changes in the corresponding input. For example, the PLS regression coefficient of +0.4 for GCaL in coupled cells, tells us that for a 1 standard deviation (SD) increase in the conductance of GCaL (5% in our analysis), the APD90 increases by 0.4 SD. Conversely, the negative regression coefficients for GK1 indicate that an increase in GK1 causes a decrease in APD90. Furthermore, since in each case we know the distribution of measured output parameters, the PLS regression coefficient can be converted to an absolute value. For example, if the simulated APD90 values had a SD of 10 ms and ρGCaL was 0.4, the absolute change associated with a 1 SD increase in GCaL would be 4 ms.
The plots in Fig. 2B clearly show that the PLS regression coefficients differ for isolated cells compared to the more physiologically relevant multicellular arrangement. For example, in mid-myocardial cells from a cable, the APD90 is approximately fourfold more sensitive to changes in GCaL than in isolated cells but less sensitive to changes in GK1 and Gncx. While the exact sensitivities will vary depending on the cellular model used, as has been shown at the single cell level (Sobie, 2009), the results in Fig. 2 demonstrate that it is not possible to simply extrapolate sensitivity parameters from single cell studies to multicellular systems.
Partial least squares analysis of ECG parameters
A linear cable of cardiac myocytes such as that illustrated in Fig. 1A is the simplest system that can be used to generate a first approximation of an ECG, often referred to as a pseudo ECG (see Fig. 3Aa). These pseudo ECGs reproduce the important features of a full surface ECG: a tall, narrow QRS complex and a T wave, corresponding to depolarisation and repolarisation within the cable. By identifying three points, 1: the start of the Q-wave, 2: the peak of the T wave and 3: the end of the T wave, from each ECG trace (see Fig. 3Aa) we can measure the QT interval (time at 3 – time at 1), the Theight (amplitude at 2), and Tpeak– Tend (time at 3 – time at 2). We can then use PLS to analyse the sensitivities of each of these emergent properties of the whole, coupled heterogeneous system, to changes in individual ion conductances, thereby overcoming the limitations imposed by analysis of isolated cells.
Figure 3. PLS analysis of ECG waveforms.
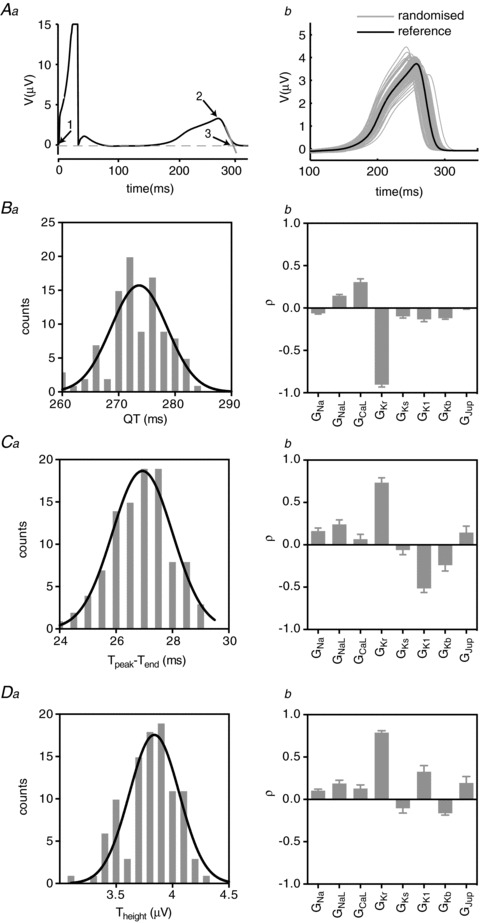
Aa, pseudo ECG generated from simulation of electrical propagation in a cable of cells as shown in Fig. 1A. Numbered arrows indicate points required in measuring ECG parameters: 1, onset of depolarisation; 2, peak of T wave; 3, end of T wave, defined as extrapolation of a tangent from the steepest part of the T wave slope to the isoelectric line. Ab, family of T waves from ECGs generated according to the randomisation pipeline in Fig. 2A from a transmural cable paced at 1 Hz. B–D, distributions (a) and PLS coefficients (b) for QT interval, Tpeak– Tend and Theight, respectively, for the family of ECGs shown in Ab.
It is important to note that it was necessary to equilibrate each instance of the cable for 500 beats to take into account long-term changes in ion concentrations, especially [Na+]i and [Ca2+]i (see Supplemental Fig. 1). The T waves of the 500th beat at 1 Hz pacing for each of the 100 scenarios generated using the same randomisation pipeline illustrated in Fig. 2A are illustrated in Fig. 3Ab. These data clearly illustrate that small changes in underlying ionic conductances can cause significant modification of the T wave of the ECG. The distributions of QT interval, Theight and Tpeak– Tend derived from the family of ECGs are shown in Fig. 3Ba–Da. The means ± SD for each of the parameters were 273.6 ± 5.0 ms, 3.8 ± 0.2 V and 26.9 ± 1.1 ms for QT interval, Theight and Tpeak– Tend, respectively.
The PLS regression coefficients for the eight most sensitive parameters derived from these distributions are shown in Fig. 3Bb–Db. Given this model is representative of baseline activity without adrenergic stimulation (O’Hara et al. 2011), it is unsurprising that the most important determinant for QT interval duration is GKr with a PLS regression coefficient, ρGKr,QT, of −0.91. This reflects the well-known relationship between decreased IKr and increased QT interval (Keating & Sanguinetti, 2001). Many of the other significant contributors to variation in ECG parameters, however, are less intuitive. For example, GKr was also an important determinant of Tpeak– Tend, although the sensitivity was in the opposite direction to that for QT interval, i.e. ρGKr,T(peak)-T(end) was +0.73 compared to −0.91 for the QT interval. Given that there is no single cell equivalent parameter to Theight, our data provide the first insights into the relative contributions of different conductances to this parameter. The major determinants of variation in Theight are GKr (ρGKr,T(height)=+0.78) and GK1 (ρGK1,T(height)=+0.33). Perhaps more surprising is that increased activity of SERCA (GJup) is also positively correlated with changes in Theight (ρGJup,T(height) of +0.194).
Relationship between cellular parameters and ECG waveforms
The datasets generated in the sensitivity analysis above (i.e. the sets of 100 cable simulations and corresponding ECGs) also provide a powerful means of correlating ECG parameters with the characteristics of the cable to yield some insight into how cellular action potential characteristics define the ECG waveform. The relationship between the three ECG parameters, QT interval, Theight and Tpeak– Tend and their correlated cable parameters at 1 Hz pacing rate are shown in Fig. 4. The QT interval on the ECG is well known to correspond to the period from the onset of depolarisation to the end of repolarisation. As would be expected therefore, a strong correlation (r2= 0.9941) exists between the QT interval and the time from the stimulus (tstim) to the APD90 of the last cell in the cable to repolarise (tlast) (Fig. 4Ba). Tpeak– Tend, on the other hand, is correlated with the period from the time of maximum total repolarisation rate (Repolmax) to tlast (r2= 0.5738, Fig. 4Bb). To calculate Repolmax we summed the first derivatives of all action potential waveforms in the cable (Σ(dV/dt)) at each time point. Repolmax was identified as the time at which the minimum of Σ(dV/dt) was reached (see Supplemental Fig. 2). The lower goodness of fit for this correlation is due to the error in measurement of Tend (through extrapolation of the tangent of the steepest part of the T wave, see Fig. 3Aa) relative to the absolute value of Tpeak– Tend. However, since we can unambiguously assign Repolmax to Tpeak by other means (there is a strong correlation between Q – Tpeak measured from the ECG and tstim– Repolmax in the cable, see Supplemental Fig. 2), we are confident that the time from Repolmax to tlast in the cable defines Tpeak– Tend. Lastly Theight is strongly correlated (r2= 0.9590) with the dispersion of repolarisation through the cable (i.e. the time from the APD90 of the earliest repolarising cell in the cable (tfirst) to tlast) (Fig. 4Bc).
Figure 4. Correlation of ECG waveform morphology and cellular AP characteristics.
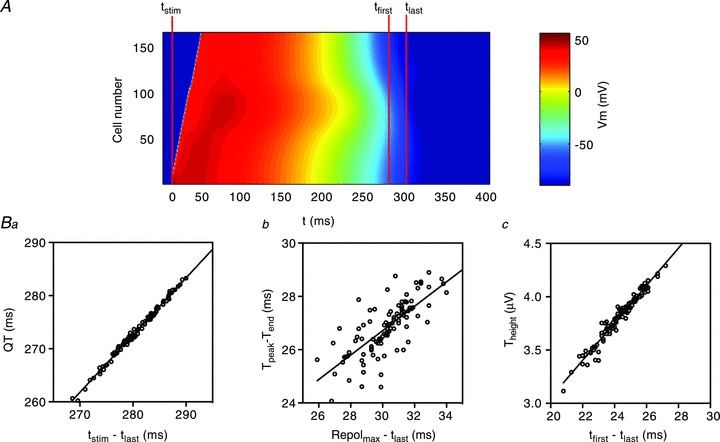
A, heatmap showing propagation of AP waveforms in the cable. Data are coloured according to voltage. Time points used to correlate cable/cellular characteristics with ECG waveforms are marked. Specifically, tstim is the time the stimulus is applied to the endocardial terminus of the cable, tfirst is the time at which the first cell in the cable repolarises to its APD90 and tlast is the time at which the last cell in the cable repolarises to its APD90. Ba, correlation between QT interval and the time from tstim to tlast. Bb, correlation between Tpeak– Tend and the time from Repolmax (see Supplemental Fig. 2) and tlast. Bc, correlation between Theight and the time from tfirst to tlast.
Quantification of additive epistatic modulation of ECG parameters
So far we have quantified how small variations (±5% SD) in individual ionic conductances can contribute to ECG parameter variability (Fig. 3). The absolute changes associated with this level of variation are small. For example, for the strongest determinant of QT interval, GKr, a 5% decrease (i.e. 1 SD change) results in a ∼5 ms, or 1.8%, increase in QT interval (i.e. 0.9 standard deviations of the distribution of measured QT intervals, see Fig. 3Ba). It is well known, however, that ECG signal parameters vary considerably more than this in the normal population, e.g. the standard deviation of the QT interval in the normal population is ∼±7% (Gallagher et al. 2006).
To quantifiably test the hypothesis that additive effects of changes in multiple conductances could explain the observed population variation in ECG parameters we took the five ionic conductances that had the greatest contribution to QT interval, Tpeak– Tend and Theight (from Fig. 3) and varied them ±10%. With the inclusion of the baseline level of each conductance, this gives a total of 35, or 243 different combinations for each parameter, for which we then simulated ECGs (each equilibrated for 500 beats at 1 Hz). We measured ECG parameters from each of these simulations and used them to generate the ‘genetic wheel of fortune’ plots shown in Fig. 5. In these plots each of the 243 circle segments represents one of the possible combinations of conductance level variation in our five selected ion channels. In each plot, the combinations are sorted according to the magnitude of the measured parameter, from highest (red, at the three o’clock position) to the lowest (blue, at the four o’clock position) and the ECG waveforms corresponding to the two extremes are shown below. Overall, the relative ranges of the variabilities for each of the three parameters observed as a result of additive modification of phenotype were mean ± 7% for QT interval, ±15% for Tpeak– Tend and ± 23% for Theight.
Figure 5. Genetic wheel of fortune plots.
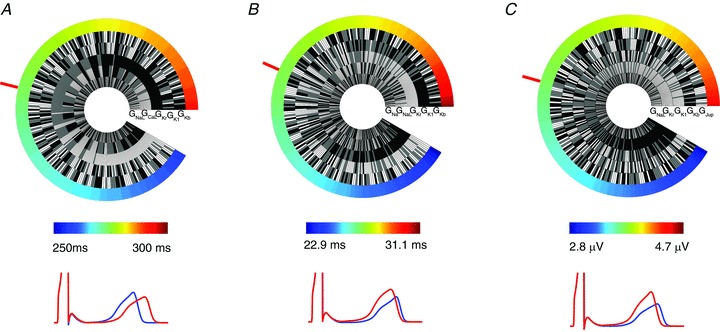
Data are shown for QT interval (A), Tpeak– Tend (B) and Theight (C). In each case the mean value is identified with a red bar. Pseudo ECGs simulated using the ionic conductance combinations from the extreme minimum (blue) and maximum (red) of the wheel are shown in each case. Grey, black and white segments represent conductance levels of baseline, 10% decreased and 10% increased, respectively.
The pattern of current segments in the QT interval wheel of fortune plot (Fig. 5A) is consistent with the data in Fig. 3, i.e. low GKr (black segments) dominate the long QT end of the circular plot. GCaL is also important, but less so, and reversed relative to GKr. The remaining three currents are more randomly distributed. The important output from this analysis is that a QT interval range from 257.8 ms to 293.5 ms is obtained using just ±10% changes in five currents. This very effectively demonstrates how multiple small perturbations can add up to a significant effect. Similar data for Tpeak– Tend is presented in Fig. 5B. The total range of the variability observed in our simulations for this parameter was 22.8–31 ms, with GKr and GK1 being the most important input parameters. Finally, the Theight observed in our simulations ranged from 2.8 to 4.52 V with a relatively small change in the QT interval between these extremes (Fig. 5C). Increased levels of GKr and GK1 are also the primary determinants of Theight. However, in this case, both GKr and GK1 correlate positively with Theight, the opposite to the relationship for QT duration.
Effect of pacing rate/heart rate on ECG parameter sensitivity
The rhythm of the heartbeat is highly dynamic with the typical heart rate ranging between ∼40 and ∼150 beats min-1. To examine how changes in heart rate influence ECG parameter sensitivity we repeated our PLS analysis on pseudo ECGs paced at 0.25, 0.5, 1, 1.5 and 2 Hz. The signals simulated at different pacing rates show obvious differences (Fig. 6A). For example, the QT interval and Theight both decrease with increasing heart rate, as has been previously reported physiologically (Bazett, 1920; Couderc et al. 2007), and also demonstrated for this model (O’Hara et al. 2011). However, there is also an apparent change in the magnitude of variability at different pacing rates. For example, there is greater variability in Theight at slower compared to higher heart rates (Fig. 6B).
Figure 6. Effect of heart rate on ECG sensitivities.
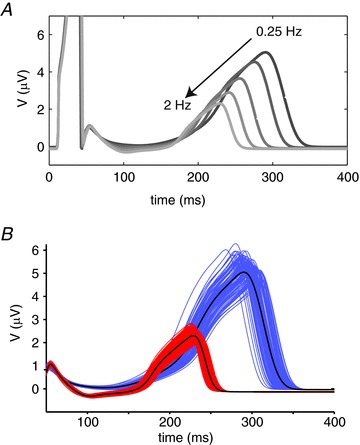
A, pseudo ECGs simulated at pacing frequencies between 0.25 and 2 Hz. B, T waves from families of 100 ECGs simulated according to the randomisation pipeline in Fig. 2A at 0.25 Hz (blue) and 2 Hz (red) after equilibration for 500 beats.
A selection of relationships derived from the PLS analysis of ECG parameters at all pacing rates are presented in Figs 7 and 8, while the full datasets are included in the Supplemental material (Supplemental Figs 3–5). The effect of heart rate on the distributions of the QT interval is illustrated in Fig. 7Aa. The QT interval both prolongs and shows greater variability at slower heart rates (239 ± 4 ms at 2 Hz compared to 318 ± 8.4 ms at 0.25 Hz). GKr and GCaL are the most important parameters influencing QT interval at all rates. Although the PLS regression coefficients for GKr and GCaL change only slightly with rate, the absolute change in the QT interval measured in response to a 1 SD change in each of these parameters decreases at higher pacing rates, consistent with the narrower distributions of QT interval at the higher rates. For example, a 1 SD (5%) increase in GKr causes a 6.7 ± 0.1 ms shortening of the QT interval at a pacing rate of 0.25 Hz compared to 2.98 ± 0.01 ms at a pacing rate of 2 Hz.
Figure 7. Rate dependence of QT and Tpeak– Tend sensitivities.
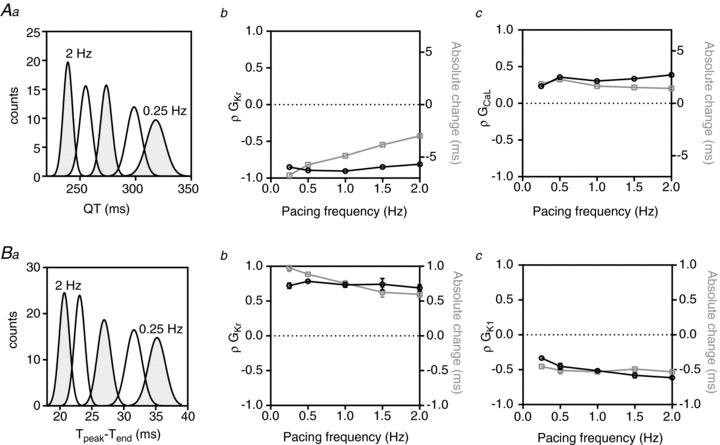
Aa, QT interval distributions at pacing rates between 0.25 and 2 Hz. Ab and c, rate dependence of PLS regression coefficients (ρ, black line) and absolute changes (grey line) in QT interval associated with GKr (b) and GCaL (c). Ba, Tpeak– Tend distributions at pacing rates between 0.25 and 2 Hz. Bb and c, rate dependence of PLS regression coefficients (ρ, black line) and absolute changes in Tpeak– Tend (grey line) associated with GKr (b) and GK1 (c).
Figure 8. Rate dependence of Theight sensitivities.
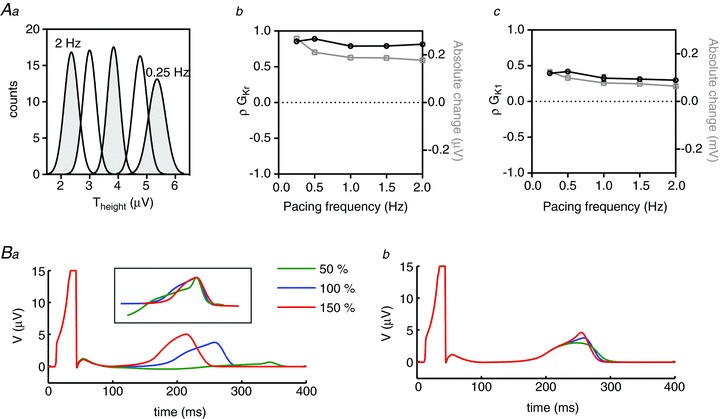
Aa, Theight distributions at pacing rates between 0.25 and 2 Hz. Ab and c, rate dependence of PLS regression coefficients (ρ, black line) and absolute changes (grey line) in Theight associated with GKr (b) and GK1 (c). B, pseudo ECGs simulated with baseline and ±50%GKr (a) and GK1 (b). The inset in Ba shows T waves normalised to their peaks illustrating that GKr variability primarily affects the early part of the T wave.
Tpeak– Tend is a measure of the temporal spread of the terminal phase of the T wave. Increasing pacing rate resulted in an increase in magnitude and a narrower distribution (i.e. reduced variability) for Tpeak– Tend (Fig. 7Ba). The two major contributors to variability in Tpeak– Tend at all pacing rates are GK1 and GKr. ρGKr is relatively rate independent (ρGKr,T(peak)-T(end) 0.72 ± 0.04 and 0.69 ± 0.04 at 0.25 and 2 Hz, respectively) whereas ρGK1,T(peak)-T(end) increases with increasing rate (ρGK1,T(peak)-T(end)–0.33 ± 0.01 and −0.63 ± 0.02 at 0.25 Hz and 2 Hz, respectively). As a result of these trends the ratio of the magnitude of sensitivities to GKr versus GK1 changes from approximately 2:1 at 0.25 Hz to 1:1 at 2 Hz.
The rate-dependent distributions of Theight are shown in Fig. 8Aa. There is a steady increase in the amplitude of the T wave in response to slowing heart rate, while the distribution of Theight only starts to widen at the slowest rate (2.36 ± 0.24 V and 5.26 ± 0.3 V at 2 Hz and 0.25 Hz, respectively). As is the case for Tpeak– Tend, GKr and GK1 are the main determinants of Theight; however, the GK1 sensitivity factors are reversed (compare Figs 7Bc and 8Ac). ρGKr does not vary significantly with heart rate, meaning the trend in absolute change in Theight directly reflects the rate-dependent change in distributions in Fig. 8Aa, e.g. a 1 SD change in conductance (5%) of GKr causes a 0.26 ± 0.007 μV change in Theight at 0.25 Hz but only a 0.18 ± 0.002 μV change at 2 Hz. ρGK1 shows a modest (25%) rate-dependent decrease from 0.4 ± 0.017 at 0.25 Hz to 0.3 ± 0.005 at 2 Hz resulting in a 50% decrease in the absolute effect at faster rates (0.12 ± 0.005 V at 0.25 Hz compared to 0.06 ± 0.001 V at 2 Hz).
Although both GKr and GK1 correlate positively with Theight, the mechanism by which they alter the T wave amplitude, as well as the overall ECG signal is very different. To highlight these differences, we performed simulations with larger perturbations to a conductance of ±50% for the two individual conductances (Fig. 8B). Changes in GKr primarily affect the early part of the T wave whereas changes in GK1 result in a distinct peaking of the terminal phase. These marked differences in the T wave responses can be more readily appreciated when the T waves for the GKr simulations are plotted with the peaks shifted to coincide and the magnitudes normalised as shown in the inset in Fig. 8Ba.
Discussion
Over the last 10–20 years significant progress has been made in elucidating the molecular and cellular basis of cardiac electrical activity (Keating & Sanguinetti, 2001). The challenge now is to utilise this knowledge to better understand the integrated activity of the whole heart both in health and disease. Computational simulation provides an ideal framework to address this challenge. Indeed, computational simulation of the heart is unique in that it is the only example in physiology where a clinically observable output of a whole organ, i.e. the ECG, can be modelled from the constituent molecular and cellular mechanisms (Noble, 2002). At the single cell level, models of cardiac APs are numerous and sensitivity analysis of these models is an approach that has been employed to examine how the network of interactions between the component ionic currents influences AP parameters. This has enabled some level of clinical insight particularly with regard to the role of epistasis in modulating genotype–phenotype presentation (Sobie, 2009; Mann et al. 2012). However, to date, this type of analysis has been limited to individual cells as a consequence of the immense computational burden of simulating multicellular systems. This in turn has limited our ability to extrapolate findings in single cells to intact cardiac tissue.
Here, we have provided the first sensitivity analysis, specifically using PLS regression, of ECG signals derived from a multicellular arrangements of cells. This was made possible by taking advantage of a new computational approach – general purpose computing on graphics processing units (GPGPU) (Owens et al. 2007). Through application of this approach we can now start to investigate the molecular basis of emergent phenomena such as QT interval, Tpeak– Tend and Theight, phenomena that are all complex functions of the arrangements of different cell types within the heart and are not properties that can be easily predicted from knowledge of how individual cells behave.
The computational approach
Simulating propagation in a multicellular environment comes with the added computational load of increased cell numbers, as well as solving the partial differential describing electrical propagation between cells. While multicellular simulations are often employed to answer specific questions in this research field (Gima & Rudy, 2002), there is a vast difference between simulating individual scenarios to answer specific questions and simulating the hundreds of thousands of beats necessary for a full sensitivity analysis. The key to our approach is taking advantage of recent developments in parallel computing hardware. Parallel computing revolves around solving multiple parts of a problem concurrently, traditionally on an array of separate connected computers, to accelerate the solution of the overall problem. Graphics processing units (GPUs) offer another alternative for parallelisation. GPUs have large scale, inbuilt parallel properties, since each card typically has hundreds of processing cores. While this hardware has historically been used for rendering graphics, recent developments now allow researchers to exploit this parallelisation for general purpose computing (Owens et al. 2007; Kirk & Hwu, 2010). Furthermore, this parallelisation can be further amplified by using connected arrays of these devices in GPU-based supercomputers (Top500: http://www.top500.org (2010)).
In relation to cardiac simulations, this parallelism can be exploited by two means: (1) by increasing the scale of parallelism, whereby a massive problem is subdivided and distributed to separate processors that each compute individual parts of one overall problem, and (2) increasing the order of parallelism, whereby many instances of the same problem can be solved concurrently, each on separate parallel processors. The former is an approach that is being actively pursued for whole organ simulation on GPU-based machines (Nimmagadda et al. 2012), but technical factors still restrict the scale of the parallelisation, meaning that practical simulation of whole organs for any extended period is still limited. In this study we have taken the second approach and increased the order of parallelisation. We have developed a parallel implementation of a multicellular simulation, deployed on a GPU-based supercomputer, to allow us to simulate the hundreds of thousands of beats necessary to perform a sensitivity analysis of how changes in individual ionic conductances affect multicellular ECG signals. In doing so, this work also lays out the computational framework for achieving the ultimate goal of this approach, large-scale analysis of whole organ cardiac systems, which will no doubt be achievable as this technology develops in the future.
Ionic conductance contributions to ECG waveforms
The data presented here provide the first quantification of the sensitivity of ECG parameters to variability in the individual ionic conductances of a computational model. Many of the conductance sensitivities and rate-dependent trends we have identified in ECG parameters are consistent with existing clinical and experimental data. Furthermore, the ECG parameter sensitivities we have identified are consistent with the basic single cell sensitivity analysis carried out in the original publication of the cell model (O’Hara et al. 2011). This acts as validation for both the original cell model and our analysis. More importantly, many of the sensitivities we identified, e.g. the sensitivities for Theight, were only made possible by the approach we have used.
The QT interval is effectively set by the action potential durations across the myocardium. It is not surprising that many of the trends in sensitivities for QT interval could be inferred from previous single cell simulations. Specifically, changes in GKr are the most important determinant of QT interval duration in this system, which correlates with the well-described effect of hERG mutations in long QT syndrome type 2 (Sanguinetti & Tristani-Firouzi, 2006). GCaL plays a lesser but nevertheless significant role in determining QT interval duration. This is also consistent with previously reported clinical observations: loss of function mutations in calcium channel subunits and diltiazem (a specific L-type calcium channel antagonist) both result in reduced QT duration (Kageyama et al. 1995; Antzelevitch et al. 2007), whereas gain of function mutations in calcium channel subunits lead to increased QT duration (Splawski et al. 2004). Finally, the negative correlation between GK1 and QT length is consistent with the QT prolongation seen clinically in patients with Andersen syndrome (Tristani-Firouzi et al. 2002).
In contrast to the ionic basis of the QT interval duration, much less is known about the origin of variability in Theight and Tpeak– Tend. Our analysis of these parameters therefore illustrates the power of the computational approach to provide novel biological insights. The major contributors to variation in Tpeak– Tend are changes in GKr and GK1, with these two parameters show differing rate dependences. At low rates, GKr is the primary determinant, while at fast rates, both conductances contribute equally to Tpeak– Tend.
Lastly, the rate-dependent sensitivities of Theight show that the two major contributors to variability in Theight are also GKr and GK1. The positive sensitivities for both GKr and GK1 are consistent with the clinically reported observations of low amplitude T waves in many patients with long QT syndrome type 2 (Zhang et al. 2000) as well as peaked, upright T waves seen in patients with short QT syndrome due to gain of function mutations in the genes coding for IKr (Brugada et al. 2004) and IK1 (Priori et al. 2005). It is clear from Fig. 8B that although both conductances contribute to variability in Theight, the associated ECG waveforms, specifically with regard to repolarisation, are very different. In the case of GK1, the T wave is primarily altered in its terminal phase, while GKr primarily affects the early part of the T wave, consistent with observations that LQTS2, as well as GKr block, alter the left slope of the T wave (Couderc et al. 2011).
Additive effects of conductance variability
In vitro characterisations of cardiac ion channel gene variants have shown that the functional effects on ionic conductances are often very small (Mann et al. 2012). Based on our quantification of the sensitivity of ECG parameters to conductance variations, this would suggest that the effects on measured ECG parameters would be correspondingly small. Indeed this is often what is observed, and extremely large population studies are required to have sufficient power to conclusively measure phenotypic changes associated with such variants (Lunetta, 2008). However, based on recent analysis of the frequency of single nucleotide polymorphisms (SNPs) in the population (Nelson et al. 2012), it is likely that we all have multiple variants in the genes that code for proteins, which contribute to cardiac electricity, i.e. the so-called ‘rhythmonome’. This leads to the question of whether multiple small variations in ion conductances can combine to give more significant modification of the ECG waveform.
Clinically, a quantitative analysis of the interaction between functional variants in the rhythmonome is important. First, in developing our understanding of the spectrum of ECG phenotypes observed in ‘normal’ and disease populations, and second for prediction of how ion channel conductance variability can alter the importance of other ion channels in the context of a ‘second hit’, whether that be from multiple gene defects or acquired defects such as drug block (as has previously been discussed in the literature, Sarkar & Sobie, 2011). The ‘genetic wheel of fortune’ plots shown in Fig. 5 illustrate how small variability in just a few ion channels can add up to significant changes in ECG parameters consistent with the degree of variability observed in the population (Gallagher et al. 2006; Viskin, 2009). This observation quantitatively demonstrates that combinations of small alterations in channel expression levels can give apparently pathogenic indicators on the ECG (a prolonged QT interval for example). Our data also illustrate how other combinations can be protective; for example, a reduced ICaL conductance can offset the QT prolongation caused by a reduction in IKr. Such combinations could easily explain how SNPs could modify the phenotype associated with disease-causing mutations even within the same family (Zhao et al. 2009) and why some mutants show apparently low penetrance (Priori et al. 1999).
Cellular electrical basis of ECG waveforms
In addition to quantifying the ionic basis of ECG parameter variability, the datasets generated during our analysis also allow us to correlate ECG parameters with characteristics of the action potential waveforms in the cable. Previous studies based on recordings from wedges of canine ventricular tissue have suggested that Tpeak– Tend is a measure of dispersion of repolarisation, from the epicardium to the mid-myocardium, across the wedge (Yan & Antzelevitch, 1998). Our analysis suggests that rather than being a measure of the total dispersion, Tpeak– Tend is determined by the interval from the time of maximum repolarisation, i.e. when the sum of the rates of repolarisation of all the cells in the cable is greatest, to the end of mid-myocardial repolarisation. In relation to Theight, much less is known about the cellular origins of this ECG waveform. Our analysis suggested that Theight is directly related to the total dispersion of repolarisation in the cable. That is, the shorter the time between the first repolarising cell and the last repolarising cell, the higher the T wave. This is in agreement with observations made in previous modelling and experimental studies where decreased dispersion of repolarisation (as a result of hyperkalaemia and the subsequent increased conductance of IKr) correlated with an increased amplitude of the T wave (Yan & Antzelevitch, 1998; Gima & Rudy, 2002).
Limitations
In this study we focused on a single cellular model of the ventricular action potential (O’Hara et al. 2011). Clearly, different models may give different sensitivities, based on the relative conductance levels of different ion channels in the models. The O’Hara–Rudy model is the only model based entirely on data recorded from healthy human hearts, with the heterogeneous expression of ion channel proteins between cell types (e.g. epi-, endo- and mid-myocardial cells) based on expression data from human tissue. With this in mind we considered it an appropriate model for extrapolation to human ECG signals. Similarly, in the construction of our cables we have included mid-myocardial cells whose functional role in the myocardium of the intact heart is still controversial (Janse et al. 2011; Wilson et al. 2011). Much like the choice of the specific cellular model, the construction of the cable in relation to cell type and number may affect the outcomes. Our primary goal, however, was not to provide a comparison of different cellular models (indeed this has already been carried out using PLS analysis at the single cell level, Sobie, 2009) or cable assemblies, but rather demonstrate the potential of the computational framework we have developed when applied to emergent properties of the cardiac electrical system.
A further point for consideration is that the expression levels of ionic currents involved in repolarisation (specifically IKs) in the O’Hara–Rudy model are representative of low adrenergic tone. Previous studies have demonstrated that IKs has little contribution to the cardiac AP under these conditions. As a result, the AP and ECG parameters examined in our study are relatively insensitive to variability in GKs (see Supplemental Figs 3–5). We would anticipate, however, that addition of adrenergic signalling to the O’Hara–Rudy model would result in GKs being a significant determinant of ECG parameters. Previous studies that have compared different cardiac AP models have indeed confirmed that GKs makes an important contribution when its level of activity is increased (Sarkar & Sobie, 2011).
Finally, calculating pseudo ECGs from a transmural cable is clearly a simplification relative to a full ECG recorded from a real heart. The consequences of this are twofold. First, only information related to transmural propagation can be inferred. As a result, while we are able to analyse contributions of transmural cellular heterogeneity to T wave morphology, as discussed above, we cannot examine other scenarios, such as how spatial heterogeneity from apex to base of the intact heart contributes to T wave parameters such as Tpeak– Tend. Second, pseudo ECGs are calculated as the spatially weighted sum of the voltage gradient across the cable measured from a single point. As a result of this, factors that contribute to the shape of the T wave in real ECGs such as T loop morphology (Malik & Batchvarov, 2000) cannot be examined. Either of these limitations can be addressed in future studies by extending our technique to whole organs, as technology progresses.
Summary
The concept of multiple gene variants in the rhythmonome contributing to population variability in ECG parameters is one that has been qualitatively discussed for some time (Grace & Roden, 2012). Furthermore, sensitivity analysis of isolated cardiac cell models over the past few decades has given us a semi-quantitative understanding of how variability in the ionic conductances in the cell can alter the electrical output at the level of the action potential. In this study we have developed a novel, highly parallelised simulation environment based on GPGPU, which has enabled the application of PLS to quantify the effects of individual ion channel conductance changes on the electrical signals manifest in the ECG. In addition to confirming many well-known clinical observations, we have been able to perform the first quantitative analysis of the molecular contributions to T wave amplitude in the ECG. Our results demonstrate how multiple small effect variants can combine to account for the significant ECG parameter variability observed in the population. In doing so this study provides the proof-of-principle that it is possible to analyse the molecular basis of emergent properties of multicellular preparations and provides the computational framework for future studies, that will no doubt be possible with further hardware development, to achieve the ultimate goal of this approach, sensitivity analysis of whole organ systems.
Translational perspective
The power of computational modelling in developing our understanding of cardiac function lies in the ability to evaluate relationships between parameters that are not experimentally tractable. Sensitivity analysis of cellular models of electrical activity is one approach that has been employed in understanding population variability in electrical phenotypes as well as epistatic modification of disease. However, until this study, this type of analysis has been restricted to single cells as a result of computational limitations and meaningful extrapolation to the whole heart has been limited. Using an unconventional parallel computing approach, ours is the first study to complete a sensitivity analysis of the emergent properties of an ECG signal.
From a translational perspective, this type of quantitative analysis of the molecular and cellular basis of the T wave paves the way for better diagnostic reading of the ECG. For example, an in-depth understanding of how individual ionic conductances in the cardiomyocyte influence T wave morphology will allow faster and more accurate identification of the mechanism of pathogenesis in patients. Furthermore, our analysis of the additive contribution of multiple gene variants to ECG parameters provides a quantitative understanding of the role of epistatis in modification of cardiac electrical phenotypes and acts as a framework for risk stratification in the context of a ‘second hit’, either from multiple gene defects or acquired defects such as drug block.
Acknowledgments
We thank Stefano Charissis for assistance with computational simulations and Emily Hodkinson for valuable discussions. This work was supported by grants from the National Health and Medical Research Council of Australia (no. 1006016). J.I.V. is supported by an NHMRC Senior Research Fellowship (no. 1019693) and A.P.H. is supported by an Australian Research Council Future Fellowship (FT110100075).
Glossary
- AP
action potential
- APD
action potential duration
- APD90
action potential duration at 90% repolarisation
- ECG
electrocardiogram
- GCaL
conductance of L-type calcium channel current
- GJup,
maximum current of SERCA component
- GKr
conductance of rapid delayed rectifier current
- GKs
conductance of slow delayed rectifier current
- GK1
conductance of inward rectifier current
- GNa
conductance of voltage-gated sodium channel current
- GNaL
conductance of persistent sodium current
- Gncx
maximum current of sodium–calcium exchanger
- GPU
graphics processing unit
- GPGPU
general purpose computing on graphics processing unit
- LQTS
long QT syndrome
- PDE
partial differential equation
- PLS
partial least squares regression
- SERCA
sarco/endoplasmic reticulum Ca2+-ATPase
Additional information
Author contributions
A.S. developed the GPU simulation environment used in most aspects of this project and analysed data. S.A.M. implemented single cell simulations, analysed data and prepared all figures. R.N.S. contributed clinical interpretation of data. L.D. and J.A.T. administered GPU cluster implementation of simulations. J.I.V. analysed data and drafted the manuscript. S.A.M., A.S. and A.P.H. designed the project and took primary responsibility for writing the manuscript. All authors contributed to drafting relevant sections of the manuscript and approving the final version of the manuscript. All pseudo ECG simulations were carried out on the CSIRO 'Bragg' GPU cluster. Single cell simulations, data analysis and experimental design were carried out at the Victor Chang Cardiac Research Institue.
Supplementary material
Supplemental Fig. 1
Supplemental Fig. 2
Supplemental Fig. 3–5
References
- Adler CP, Costabel U. Cell number in human heart in atrophy, hypertrophy, and under the influence of cytostatics. Recent Adv Stud Cardiac Struct Metab. 1975;6:343–355. [PubMed] [Google Scholar]
- Anderson CL, Delisle BP, Anson BD, Kilby JA, Will ML, Tester DJ, Gong Q, Zhou Z, Ackerman MJ, January CT. Most LQT2 mutations reduce Kv11.1 (hERG) current by a class 2 (trafficking-deficient) mechanism. Circulation. 2006;113:365–373. doi: 10.1161/CIRCULATIONAHA.105.570200. [DOI] [PubMed] [Google Scholar]
- Antzelevitch C, Pollevick GD, Cordeiro JM, Casis O, Sanguinetti MC, Aizawa Y, Guerchicoff A, Pfeiffer R, Oliva A, Wollnik B, Gelber P, Bonaros EP, Burashnikov E, Wu YS, Sargent JD, Schickel S, Oberheiden R, Bhatia A, Hsu LF, Haissaguerre M, Schimpf R, Borggrefe M, Wolpert C. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation. 2007;115:442–449. doi: 10.1161/CIRCULATIONAHA.106.668392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey JE. Lessons from metabolic engineering for functional genomics and drug discovery. Nat Biotechnol. 1999;17:616–618. doi: 10.1038/10794. [DOI] [PubMed] [Google Scholar]
- Bazett HC. An analysis of the time-relations of electrocardiograms. Heart. 1920;7:353–373. [Google Scholar]
- Brugada R, Hong K, Dumaine R, Cordeiro J, Gaita F, Borggrefe M, Menendez TM, Brugada J, Pollevick GD, Wolpert C, Burashnikov E, Matsuo K, Wu YS, Guerchicoff A, Bianchi F, Giustetto C, Schimpf R, Brugada P, Antzelevitch C. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation. 2004;109:30–35. doi: 10.1161/01.CIR.0000109482.92774.3A. [DOI] [PubMed] [Google Scholar]
- Carter N, Snieder H, Jeffery S, Saumarez R, Varma C, Antoniades L, Spector TD. QT interval in twins. J Hum Hypertens. 2000;14:389–390. doi: 10.1038/sj.jhh.1001026. [DOI] [PubMed] [Google Scholar]
- Clancy CE, Tateyama M, Kass RS. Insights into the molecular mechanisms of bradycardia-triggered arrhythmias in long QT-3 syndrome. J Clin Invest. 2002;110:1251–1262. doi: 10.1172/JCI15928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couderc JP, Vaglio M, Xia X, McNitt S, Wicker P, Sarapa N, Moss AJ, Zareba W. Impaired T-amplitude adaptation to heart rate characterizes IKr inhibition in the congenital and acquired forms of the long QT syndrome. J Cardiovasc Electrophysiol. 2007;18:1299–1305. doi: 10.1111/j.1540-8167.2007.00960.x. [DOI] [PubMed] [Google Scholar]
- Couderc JP, Xia X, Peterson DR, McNitt S, Zhao H, Polonsky S, Moss AJ, Zareba W. T-wave morphology abnormalities in benign, potent, and arrhythmogenic Ikr inhibition. Heart Rhythm. 2011;8:1036–1043. doi: 10.1016/j.hrthm.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher MM, Magliano G, Yap YG, Padula M, Morgia V, Postorino C, Di Liberato F, Leo R, Borzi M, Romeo F. Distribution and prognostic significance of QT intervals in the lowest half centile in 12,012 apparently healthy persons. Am J Cardiol. 2006;98:933–935. doi: 10.1016/j.amjcard.2006.04.035. [DOI] [PubMed] [Google Scholar]
- Gima K, Rudy Y. Ionic current basis of electrocardiographic waveforms: a model study. Circ Res. 2002;90:889–896. doi: 10.1161/01.res.0000016960.61087.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glukhov AV, Fedorov VV, Lou Q, Ravikumar VK, Kalish PW, Schuessler RB, Moazami N, Efimov IR. Transmural dispersion of repolarization in failing and nonfailing human ventricle. Circ Res. 2010;106:981–991. doi: 10.1161/CIRCRESAHA.109.204891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace AA, Roden DM. Systems biology and cardiac arrhythmias. Lancet. 2012;380:1498–1508. doi: 10.1016/S0140-6736(12)61462-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haarmark C, Kyvik KO, Vedel-Larsen E, Budtz-Jørgensen E, Kanters JK. Heritability of Tpeak-Tend interval and T-wave amplitude: a twin study. Circ Cardiovasc Genet. 2011;4:516–522. doi: 10.1161/CIRCGENETICS.111.959551. [DOI] [PubMed] [Google Scholar]
- Hondeghem LM, Carlsson L, Duker G. Instability and triangulation of the action potential predict serious proarrhythmia, but action potential duration prolongation is antiarrhythmic. Circulation. 2001;103:2004–2013. doi: 10.1161/01.cir.103.15.2004. [DOI] [PubMed] [Google Scholar]
- Janse MJ, Coronel R, Opthof T. Counterpoint: M cells do not have a functional role in the ventricular myocardium of the intact heart. Heart Rhythm. 2011;8:934–937. doi: 10.1016/j.hrthm.2010.10.048. [DOI] [PubMed] [Google Scholar]
- Kageyama M, Yanagisawa T, Taira N. Effects of semotiadil fumarate, a novel Ca2+ antagonist, on cytosolic Ca2+ level and force of contraction in porcine coronary arteries. Br J Pharmacol. 1995;114:1289–1295. doi: 10.1111/j.1476-5381.1995.tb13345.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating MT, Sanguinetti MC. Molecular and cellular mechanisms of cardiac arrhythmias. Cell. 2001;104:569–580. doi: 10.1016/s0092-8674(01)00243-4. [DOI] [PubMed] [Google Scholar]
- Kirk D, Hwu W. Programming Massively Parallel Processors: A Hands-on Approach. NVIDIA; 2010. [Google Scholar]
- Knollmann BC, Roden DM. A genetic framework for improving arrhythmia therapy. Nature. 2008;451:929–936. doi: 10.1038/nature06799. [DOI] [PubMed] [Google Scholar]
- Lunetta KL. Genetic association studies. Circulation. 2008;118:96–101. doi: 10.1161/CIRCULATIONAHA.107.700401. [DOI] [PubMed] [Google Scholar]
- Malik M, Batchvarov VN. Measurement, interpretation and clinical potential of QT dispersion. J Am Coll Cardiol. 2000;36:1749–1766. doi: 10.1016/s0735-1097(00)00962-1. [DOI] [PubMed] [Google Scholar]
- Mann SA, Otway R, Guo G, Soka M, Karlsdotter L, Trivedi G, Ohanian M, Zodgekar P, Smith RA, Wouters MA, Subbiah R, Walker B, Kuchar D, Sanders P, Griffiths L, Vandenberg JI, Fatkin D. Epistatic effects of potassium channel variation on cardiac repolarization and atrial fibrillation risk. J Am Coll Cardiol. 2012;59:1017–1025. doi: 10.1016/j.jacc.2011.11.039. [DOI] [PubMed] [Google Scholar]
- Nelson MR, Wegmann D, Ehm MG, Kessner D, St Jean P, Verzilli C, Shen J, Tang Z, Bacanu SA, Fraser D, Warren L, Aponte J, Zawistowski M, Liu X, Zhang H, Zhang Y, Li J, Li Y, Li L, Woollard P, Topp S, Hall MD, Nangle K, Wang J, Abecasis G, Cardon LR, Zollner S, Whittaker JC, Chissoe SL, Novembre J, Mooser V. An abundance of rare functional variants in 202 drug target genes sequenced in 14,002 people. Science. 2012;337:100–104. doi: 10.1126/science.1217876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmagadda VK, Akoglu A, Hariri S, Moukabary T. Cardiac simulation on multi-GPU platform. J Supercomput. 2012;59:1360–1378. [Google Scholar]
- Noble D. Modeling the heart–from genes to cells to the whole organ. Science. 2002;295:1678–1682. doi: 10.1126/science.1069881. [DOI] [PubMed] [Google Scholar]
- O’Hara T, Virag L, Varro A, Rudy Y. Simulation of the undiseased human cardiac ventricular action potential: model formulation and experimental validation. PLoS Comput Biol. 2011;7:e1002061. doi: 10.1371/journal.pcbi.1002061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens JD, Luebke D, Govindaraju N, Harris M, Krüger J, Lefohn AE, Purcell T. A survey of general-purpose computation on graphics hardware. Comput Graph Forum. 2007;26:80–113. [Google Scholar]
- Plonsey R, Barr RC. Bioelectricity. A Quantitative Approach. Springer; 2007. [Google Scholar]
- Priori SG, Napolitano C, Schwartz PJ. Low penetrance in the long-QT syndrome: clinical impact. Circulation. 1999;99:529–533. doi: 10.1161/01.cir.99.4.529. [DOI] [PubMed] [Google Scholar]
- Priori SG, Pandit SV, Rivolta I, Berenfeld O, Ronchetti E, Dhamoon A, Napolitano C, Anumonwo J, di Barletta MR, Gudapakkam S, Bosi G, Stramba-Badiale M, Jalife J. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ Res. 2005;96:800–807. doi: 10.1161/01.RES.0000162101.76263.8c. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature. 2006;440:463–469. doi: 10.1038/nature04710. [DOI] [PubMed] [Google Scholar]
- Sarkar AX, Sobie EA. Quantification of repolarization reserve to understand interpatient variability in the response to proarrhythmic drugs: a computational analysis. Heart Rhythm. 2011;8:1749–1755. doi: 10.1016/j.hrthm.2011.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RM, Rudy Y. Ionic mechanisms of propagation in cardiac tissue. Roles of the sodium and L-type calcium currents during reduced excitability and decreased gap junction coupling. Circ Res. 1997;81:727–741. doi: 10.1161/01.res.81.5.727. [DOI] [PubMed] [Google Scholar]
- Sobie EA. Parameter sensitivity analysis in electrophysiological models using multivariable regression. Biophys J. 2009;96:1264–1274. doi: 10.1016/j.bpj.2008.10.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K, Tager-Flusberg H, Priori SG, Sanguinetti MC, Keating MT. CaV1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119:19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Tennessen JA, Bigham AW, O’Connor TD, Fu W, Kenny EE, Gravel S, McGee S, Do R, Liu X, Jun G, Kang HM, Jordan D, Leal SM, Gabriel S, Rieder MJ, Abecasis G, Altshuler D, Nickerson DA, Boerwinkle E, Sunyaev S, Bustamante CD, Bamshad MJ, Akey JM, Broad GO, Seattle GO, Project NES. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrenoire C, Clancy CE, Cormier JW, Sampson KJ, Kass RS. Autonomic control of cardiac action potentials: role of potassium channel kinetics in response to sympathetic stimulation. Circ Res. 2005;96:e25–34. doi: 10.1161/01.RES.0000160555.58046.9a. [DOI] [PubMed] [Google Scholar]
- Tristani-Firouzi M, Jensen JL, Donaldson MR, Sansone V, Meola G, Hahn A, Bendahhou S, Kwiecinski H, Fidzianska A, Plaster N, Fu YH, Ptacek LJ, Tawil R. Functional and clinical characterization of KCNJ2 mutations associated with LQT7 (Andersen syndrome) J Clin Invest. 2002;110:381–388. doi: 10.1172/JCI15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viskin S. The QT interval: too long, too short or just right. Heart Rhythm. 2009;6:711–715. doi: 10.1016/j.hrthm.2009.02.044. [DOI] [PubMed] [Google Scholar]
- Wilson LD, Jennings MM, Rosenbaum DS. Point: M cells are present in the ventricular myocardium. Heart Rhythm. 2011;8:930–933. doi: 10.1016/j.hrthm.2011.01.026. [DOI] [PubMed] [Google Scholar]
- Yan GX, Antzelevitch C. Cellular basis for the normal T-wave and the electrocardiographic manifestations of the long-QT syndrome. Circulation. 1998;98:1928–1936. doi: 10.1161/01.cir.98.18.1928. [DOI] [PubMed] [Google Scholar]
- Yang T, Atack TC, Stroud DM, Zhang W, Hall L, Roden DM. Blocking Scn10a channels in heart reduces late sodium current and is antiarrhythmic. Circ Res. 2012;111:322–332. doi: 10.1161/CIRCRESAHA.112.265173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Timothy KW, Vincent GM, Lehmann MH, Fox J, Giuli LC, Shen J, Splawski I, Priori SG, Compton SJ, Yanowitz F, Benhorin J, Moss AJ, Schwartz PJ, Robinson JL, Wang Q, Zareba W, Keating MT, Towbin JA, Napolitano C, Medina A. Spectrum of ST-T-wave patterns and repolarization parameters in congenital long-QT syndrome: ECG findings identify genotypes. Circulation. 2000;102:2849–2855. doi: 10.1161/01.cir.102.23.2849. [DOI] [PubMed] [Google Scholar]
- Zhao JT, Hill AP, Varghese A, Cooper AA, Swan H, Laitinen-Forsblom PJ, Rees MI, Skinner JR, Campbell TJ, Vandenberg JI. Not All hERG pore domain mutations have a severe phenotype: G584S has an inactivation gating defect with mild phenotype compared to G572S, which has a dominant negative trafficking defect and a severe phenotype. J Cardiovasc Electrophysiol. 2009;20:923–930. doi: 10.1111/j.1540-8167.2009.01468.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.