

Abstract
Tumor infiltration with effector CD8+ T cells (Teff) predicts longer recurrence-free survival in mmany types of human cancer, illustrating the broad significance of Teff for effective immunosurveillance. Colorectal tumors with reduced accumulation of Teff express low levels of Teff-attracting chemokines such as CXCL10/IP10 and CCL5/RANTES. In this study, we investigated the feasibility of enhancing tumor production of Teff-attracting chemokines as a cancer therapeutic strategy, using a tissue explant culture system to analyze chemokine induction in intact tumor tissues. In different tumor explants, we observed highly heterogeneous responses to IFNα or poly-I:C (a TLR3 ligand) when they were applied individually. In contrast, a combination of IFNα and poly-I:C uniformly enhanced the production of CXCL10 and CCL5 in all tumor lesions. Moreover, these effects could be optimized by the further addition of COX inhibitors. Applying this triple combination also uniformly suppressed the production of CCL22/MDC, a chemokine associated with infiltration of T regulatory cells (Treg). The Teff-enhancing effects of this treatment occurred selectively in tumor tissues, as compared to tissues derived from tumor margins. These effects relied on the increased propensity of tumor-associated cells (mostly fibroblasts and infiltrating inflammatory cells) to hyper-activate NF-κB and produce Teff-attracting chemokines in response to treatment, resulting in an enhanced ability of the treated tumors to attract Teff cells and reduced ability to attract Tregs. Together, our findings suggest the feasibility of exploiting NF-κB hyper-activation in the tumor microenvironment to selectively enhance Teff entry into colon tumors.
Keywords: tumor microenvironment, immunomodulation, chemokines, effector T cells, regulatory T cells, colorectal cancer, indomethacin, celecoxib, PGE2, COX2, interferons
Introduction
The ability of CD8+ T cells to infiltrate cancer lesions is essential for anti-tumor immunity, as evidenced by studies highlighting the prognostic value of effector T (Teff) cells in multiple cancer types, including colorectal cancer (CRC) (1–4). In contrast, tumor infiltration with regulatory T cells (Tregs) predicts poor outcomes (5–8). Chemokines and their respective receptors are critical for T cell migration and homing (9–14). High levels of CCL5/RANTES (CCR5 ligand) and CXCL9/MIG and CXCL10/IP10 (ligands for CXCR3) in tumor tissues are associated with enhanced infiltration of CD8+ T cells in CRC (15), melanoma and gastric cancer (16, 17). In contrast to the benefits of intra-tumoral expression of CCL5 and CXCL9-11 (18), high levels of CCL22/MDC, the CCR4 ligand preferentially attracting Tregs, can be associated with reduced survival, as shown in ovarian cancer patients (19).
Several studies have indicated the propensity of colorectal tumors to over-express COX2 and its product PGE2 (20, 21), the factor shown to promote the induction of CCL22 in dendritic cell (DC) cultures (22). Prompted by these reports, and by our observations of the reciprocal impact of IFNα versus PGE2 on the production of Teff- and Treg-attracting chemokines in isolated DCs (22), we tested the feasibility of using these factors to manipulate tumor microenvironment to enhance the production of Teff-attracting chemokines in intact human tumor tissues. We used an ex vivo tumor/tissue explant culture system previously applied to study migration of DCs (23), to avoid spontaneous activation of the chemokine-producing cells in the process of tumor dissociation.
Guided by reports showing common hyper-activation of NF-κB in cancer tissues (24–27), and the requirement for this factor in the induction of both Treg- and Teff-attracting classes of chemokines (28–30), we tested whether the selected PGE2- and IFNα-targeting strategies can be used to selectively enhance the production of Teff-attracting chemokines in tumor tissues, rather than marginal tissues, in order to selectively direct Teff cells to tumors.
Materials and Methods
Patients
72 colorectal patients were involved in the study. Tumors and marginal tissues were harvested during routine surgery. The patient profile is presented in Table 1. All patients signed a consent approved by the Institutional Review Board of the University of Pittsburgh for collection of tumor samples (UPCI 02-077).
Table 1.
Demographic profile and clinical status of the 72 colorectal cancer patients involved in the study.
| N=72 | ||
|---|---|---|
| Sex | Number | % |
| Male | 38 | 52.78 |
| Female | 34 | 47.22 |
| Median age at surgery | 58.5 | |
| Stage | Number | % |
| I | 4 | 5.56 |
| IIA | 2 | 2.78 |
| IIB | 4 | 5.56 |
| IIIA | 1 | 1.39 |
| IIIB | 16 | 22.22 |
| IIIC | 6 | 8.33 |
| IV | 37 | 51.39 |
| Metastasis sites | Number | % |
| Liver | 30 | 41.67 |
| Ovary | 6 | 8.33 |
| Peritoneal Carcinomatosis | 14 | 19.44 |
| Primary | 4 | 5.56 |
| Rectal recurrence | 4 | 5.56 |
| Other | 14 | 19.44 |
Culture of macrophages, fibroblasts, HUVEC cells and colon cancer cell lines
For preparation of macrophages, monocytes were cultured in AIM-V with GM-CSF for 6 days. Fibroblasts (Cascade Biologicals, Portland, Oregon) and colon cancer cell lines CACO-2, HCT116, HT29, SW480 and SW620 (ATCC, Manassas, VA) were grown in IMDM+10% FBS, while HUVECs (AllCells, Emeryville, CA) were cultured in HUVEC complete media (Basal media supplemented with HUVEC stimulatory supplement; AllCells). All were washed, reseeded at 20,000 cells in 300μL in 96 well plates and treated with IFNα, poly-I:C, and/or indomethacin as indicated for 48hrs, and supernatants were analyzed for chemokine production by ELISA.
Ex vivo cultures of tumor- and marginal tissue explants
Using a 4mm biopsy puncher, the cubes of tumor or marginal tissue were prepared and placed in antibiotic-containing IMDM/10% FBS (typically 3 cubes/well in 24 well plates) for 24–48 hours, as indicated. When indicated, the tissues were treated with 10,000 units of IFNα, 20μg/mL of poly-I:C, 50μM of indomethacin or 10μM of celecoxib. Biopsies were harvested at 0 and 24hrs for mRNA analysis and confocal microscopy analysis. Culture supernatants were harvested at 24–48 hours, as indicated (all groups in a single type of experiment were harvested at the same time point) for ELISA and chemotaxis assays. The detailed work flow is depicted in Supplementary Fig. S1A. The system used, based on our previously-developed ex vivo whole tissue culture system (23), allowed us to avoid spontaneous induction of chemokine production by the process of tumor dissociation (Supplementary Fig S1B and data not shown)
Taqman analysis of mRNA expression in tumors and marginal tissues
4mm biopsies were placed in lysing Matrix E tubes (MP Biologicals, Solon, OH) containing RLT buffer (RNeasy kit, Qiagen, Valencia, CA), and agitated using a FP120 homogenizer (MP Biologicals). Debris-free supernatant from the lysis matrix tubes were transferred into new tubes and the total RNA was extracted using the RNeasy kit. 1μg of RNA extracted by the above described method was used for cDNA synthesis, and 25–50 ng of subsequent cDNA was used to perform mRNA expression analysis by Taqman analysis on the Step One Plus system (Applied Biosystems). All the primers used for the analysis were standard, purchased from Applied Biosystems.
ELISA analysis of chemokines in tumor ex vivo culture supernatants
Culture supernatants from tumor ex vivo cultures were analyzed by ELISAs for the presence of chemokine proteins CCL5, CCL22 and CXCL10, using primary and secondary antibodies from Peprotech, Rocky Hill, NJ. Detection was done using Streptavidin-HRP conjugate and TMB substrate from Pierce Biotechnology Inc, Rockford, IL.
Isolation of tumor infiltrating CD8+ T cells
Tumor infiltrating lymphocytes were isolated as described by Dudley et al (31), with the following modifications: Tumor was cut into 4mm cubes using a biopsy punch, and each 4mm tumor piece cultured in 1mL of IMDM + 5% human AB serum with 1000U/mL IL-2 for 2 weeks. Medium was changed twice a week, until lymphocytes were extruding from tumor and formed proliferating clusters.
Chemotaxis
Chemotaxis assays were performed in 24 transwell plates with 5μm pore size polycarbonate filters (Corning Inc, Corning, NY). The lower chambers were filled with 600μL of tumor supernatants. As indicated, 2x105 of either isolated tumor-infiltrating lymphocytes or αDC1-activated CD8+ Teff cells (32), in 200μL of IMDM 10% FCS, were added to the upper chambers and incubated for 3hrs at 37°C. Migrated cells were harvested from the lower chambers and stained for CD8. Cell counts were performed by a 60 second limited run on a BD Beckman Coulter XL cytometer. For analysis of Treg cell migration, bulk CD4+ T cells were isolated by negative selection using EasySep CD4 enrichment kits (StemCell), and 1x106 of the isolated cells in 200μL were allowed to migrate towards 600μL of tumor supernatants in the bottom chambers. The migrated cells in the bottom chambers were harvested and FOXP3/GITR frequencies were determined by Taqman analysis or flow cytometry.
In situ hybridization
Tissue specimens were fixed in 4% para-formaldehyde, processed and pre-treated as described (33), except that tissues were sectioned on a cryostat at 5μm. Gene-specific riboprobes were synthesized by in vitro transcription using a Maxiscript SP6/T7 kit (Ambion) and unincorporated nucleotides were removed using RNA Mini Quick Spin Columns (Roche). In situ hybridization with 35S-labeled riboprobes was performed as described (33, 34), with 0.1M dithiothreitol included in the hybridization mix. Hybridizations were performed at 50°C overnight. Tissue sections were coated with NTB emulsion (Kodak) and exposed at 10°C for 7–14d. Simultaneous in situ hybridization and immunohistochemistry were performed as described (33, 34), except that the dithiothreitol concentrations were 0.01M in the hybridization mix and 1mM in the washes. An antibody against HLA-DR (Dako) was used at a dilution of 1:25.
Confocal microscopy analysis of tumor and marginal tissues
4mm tumor punches, either untreated or treated, were embedded in OCT medium-containing cryomolds and immediately frozen in 2-methyl-butane. 6μm frozen sections of the tissues were made using the cryostat and layered on superfrost® plus slides (Thermo Scientific, Rockford, IL). The slides were incubated in 4% para-formaldehyde for 15 minutes, washed and blocked for 60 minutes at RT. The slides were then stained for 3hrs at RT with antibodies for P65 (ab16502) or for CD8 (ab4055), CXCL10 (ab8098) and CCL5 (ab10590; both AbcamCambridge, MA). The slides were washed 3 times with 1x PBS and incubated with secondary antibodies anti-rabbit (Alexa 647), anti-mouse (Alexa 488; both Cell Signal, Danvers, MA) and anti-goat (Alexa 488; Invitrogen, Carlsbad, CA) for 30 minutes at RT. The slides were washed 3 times with 1x PBS and once with high salt PBS. Cover slips were mounted on the sections using prolong gold anti-fade solution (Invitrogen). Confocal analyses of stained slides were performed using a LEICA TCS SL DMRE Microsystems. To quantify the numbers of cells showing nuclear NF-κB translocation, images were taken of 10 different fields (63x magnification) of the tumor and marginal tissue sections (untreated or treated). To identify the cells showing P65 translocation and chemokine production in response to treatment, tumor tissues were stained with CD45 (H130, BioLegend, San Deigo, CA), CD326 (9C4, BioLegend), fibroblast marker (TE-7, EMD Millipore, Billerica, MA) and CCL5 (ab9679, Abcam) and CXCL10 (ab9807, Abcam), and cells were enumerated as described above.
Statistical analysis
Pearson rank correlations between the chemokine genes and T cell markers were calculated on logarithmically transformed data. In situations where significant between-batch variation was observed, the correlation was adjusted for the batch effect by performing a multivariate analysis of variance (MANOVA) of each pair of variables on batch, and deriving the correlation from the MANOVA residual matrix. Comparisons of continuous variables between groups were performed by two-tailed paired t tests. P <0.05 was considered significant. Analyses were performed using SAS v9.2 (SAS Institute, Cary, NC) or GraphPad Prism 5 software.
Results
The expression of effector T cell (Teff)-recruiting chemokines in colorectal tumor samples correlates with effector CD8+ T cell markers
Using resected tumor material from 72 patients with advanced colorectal cancer (metastatic in 68 patients), we observed that local expression of two Teff cell markers (CD8 and Granzyme B; GZMB) is strongly correlated with the expression of two Teff-attracting chemokines, CCL5 and CXCL10 (Fig. 1A). In contrast, the Treg markers FOXP3 and GITR were correlated with CCL22 (Fig. 1B), a known Treg attractant (19, 22). Additional correlations were observed between CXCL9 (alternative CXCR3 ligand) and Teff markers and between CCL22 and the CCL22− inducing factor (22) COX2 (Supplementary Fig. S2A–B). Confocal microscopy analysis of the tumor sections revealed that all CXCL10-producing cells (Supplementary Fig. S2D; right panel) and a significant proportion of CCL5-producing cells (left panel) were CD8-negative, arguing against the possibility that the above correlations result from the production of these chemokines by CD8+ T cells themselves, instead suggesting their causative role in mediating CD8+ T cell infiltration.
Figure 1. Presence of Teff- and Treg markers in tumors correlates with intra-tumoral expression of, respectively, Teff- or Treg-attracting chemokines.

Tumor biopsies from colon cancer patients were lysed, RNA extracted and Taqman analysis of various markers was performed. (A) Correlation between Teff markers (CD8 and Granzyme B; GZMB) and Teff-attracting chemokines (CCL5 and CXCL10) in tumor lesions. (B) Correlation between Treg markers (FOXP3 and GITR) and the chemokine CCL22 in tumor lesions.
The phenotypic analysis of CD8+ tumor-infiltrating lymphocytes (TIL) obtained from colon cancer patients (see M&M) revealed that the majority of CD8+ TILs are CCR5+ CXCR3+ (Supplementary Fig. S2E), and Granzyme B+ (Supplementary Fig. S2F), which further indicates that the intra-tumoral expression of the CCR5− and CXCR3-ligands was responsible for recruiting the effector T cells into the tumor.
Combination of IFNα, indomethacin and poly-I:C selectively enhances the production of Teff-recruiting chemokines in tumor tissues and suppresses Treg-recruiting chemokines
In order to test the possibility of correcting the chemokine environment in the tumors with low ratios of Teff-to Treg-attracting chemokines, we tested in pilot studies the feasibility of modulating their production using different combinations of IFNα, indomethacin (COX1/2 inhibitor) and poly-I:C in individual populations of tumor-relevant cells, such as colon cancer cells, macrophages, fibroblasts and HUVECs. We observed strong synergy between IFNα and poly-I:C in the induction of CCL5 and CXCL10, and a strong suppressive effect of IFNα on the production of CCL22 in macrophages and fibroblasts (Supplementary Fig. S3A–B). These desirable effects were further potentiated in the presence of indomethacin (Supplementary Fig. S3B). In contrast, none of the long-term-cultured colon cancer cell lines tested (CACO-2, HCT116, HT29, SW480 and SW620) or HUVECs produced any detectable CCL5, CXCL10 or CCL22 (data not shown).
In order to test the feasibility of using these factors to manipulate the complex microenvironment of whole tumor tissues, involving all the above cell types and their interactions, we used an ex vivo tumor/tissue explant culture system previously developed to study migration of DCs (23). This system allowed us to avoid nonspecific activation of the chemokine-producing cells during tumor dissociation (see Supplementary Figure 1B and data not shown).
As shown in Figure 2, different tumor tissues treated with IFNα or poly-I:C alone showed variable chemokine expression, falling into three different patterns: minimal induction of CCL5 and CXCL10; minimal induction of CCL5 but significant induction of CXCL10; or significant induction of both CCL5 and CXCL10 (Fig. 2A). This heterogeneity was observed between tumors from different patients, and even between different lesions within a single patient (Fig. 2A and Supplementary Fig. S3C). However, combining IFNα and poly-I:C resulted in uniformly high expression of both CCL5 and CXCL10 in all tumors tested (Fig. 2A and Supplementary Fig. S3C).
Figure 2. Heterogenous response pattern of different tumor tissues to individual chemokine modulators and their uniform response to the combination of IFNα, poly-I:C and indomethacin.

(A) Fresh tumor samples from 11 patients with metastatic colorectal cancer were untreated or treated with IFNα and poly-I:C either individually or in combination for 48 hours. The release of CCL5 and CXCL10 into culture media was analyzed by ELISA. Numbers indicate the prevalence of tumors with each chemokine pattern (respective patterns A, B or C). (B) ELISA analysis of CCL5, CCL22 and CXCL10 in tumors untreated or treated with IFNα+ pI:C, with or without indomethacin. *denotes P<0.05 (the presence or absence of indomethacin).
Additional exposure to indomethacin (which blocks COX1 and COX2) further enhanced the production of CCL5 and CXCL10 induced by the combination IFNα and poly-I:C and reduced CCL22 in whole tumor tissues (Fig. 2B), with similar results obtained using a selective COX2 blocker, celecoxib (Supplementary Fig. S3D).
Based on these data, we selected the triple combination of IFNα, poly-I:C and indomethacin as the preferred treatment for all subsequent experiments. This combination consistently enhanced CXCL10 and CCL5 production and suppressed the production of CCL22 in all tumor samples, as shown by individual chemokine gene expression at the single cell level using in-situ hybridization (ISH; Fig. 3A) and at the level of chemokine secretion, using ELISA (Fig. 3B). Similar observations were also made in case of CXCL9 (data not shown).
Figure 3. Combination of IFNα, poly-I:C and indomethacin, consistently up regulates Teff-attracting chemokines and suppresses Treg-attracting chemokines in tumor tissues.

(A) In-situ hybridization for respective chemokine mRNA (black grains) in tumor biopsies which were either left untreated or treated with the combination of indomethacin, IFNα and poly-I:C (IAP). (B) ELISA analysis of the chemokine contents in the supernatants of 48 hour-cultured tumor tissues (untreated or treated) from 10 different patients.
The dual staining for HLA-DR (immunohistochemistry) and chemokine mRNA (ISH) demonstrated that CCL22 was expressed predominantly by HLA-DR+ APCs, while CXCL10 and CCL5 were expressed by both HLA-DR+ and HLA-DR− cells (Supplementary Fig. S4), indicating the contribution of multiple tumor-associated cell types to the production of Teff-recruiting chemokines within the tumor microenvironment.
Enhanced activation of tumor-associated NF-κB by the chemokine-modulatory regimen results in preferential induction of CXCL10 in tumors, rather than marginal healthy tissues
Using matched tissue samples from 10 patients with metastatic colon cancer, we compared the responsiveness to the chemokine-modulating regimen between liver-metastatic tumor tissues and marginal tissues. As shown in Fig. 4A (and Supplementary Fig. S5), while the baseline differences in chemokine production between the untreated liver-metastatic tumors and marginal liver tissues did not reach significance (P=0.12), tumor treatment with the combination of IFNα, poly-I:C and indomethacin induced much more pronounced secretion of CXCL10 by tumor tissues compared to the marginal tissues (P<0.01). Similar observations at the protein and chemokine gene expression level were made in the case of CCL5 (Supplementary Fig. S5A&B). This increased responsiveness of tumors compared to marginal tissues was not due to decreased survival of the marginal tissues, as determined by undisturbed expression levels of glycogen phosphorylase (Supplementary Fig. S5C). Similarly, the differences in the responsiveness to the chemokine-modulatory regimen between tumors and marginal tissues could not be explained by potential differences between their expression of the IFNα receptor, TLR3, IRF1, IRF3, or the differential infiltration with APCs or NK cells, which were all similar between tumors and marginal tissues (data not shown).
Figure 4. NF-κB-dependent selective enhancement of CXCL10 production in tumor tissues following exposure to the combination of IFNα, poly-I:C and indomethacin.

(A) ELISA for CXCL10 expression in matched normal liver and liver metastatic tissues from 10 different patients either untreated (left panel) or treated (right panel). (B) Average number of cells counted per field (confocal microscopy; in a total of 10 fields) showing nuclear translocation of NF-κB in normal liver or liver metastatic tissues either untreated or treated (right panel). Representative images of each condition are shown in the left panel. (C) ELISA analysis of CXCL10 production by the matched normal liver and liver-metastatic colorectal cancer tissues, either untreated or treated (IFNα, poly-I:C and indomethacin), in the absence or presence of 20μM CAY10470 (NF-κB inhibitor). D) Colorectal cancer tissues from three colorectal cancer patients were treated with indomethacin + IFNα + Poly-I:C for 30 minutes (p65 translocation) and 24 hours (chemokine production) and analyzed by confocal microscopy for the translocation of p65 and production of CCL5 and CXCL10 by infiltrating inflammatory cells (CD45+), tumor-associated fibroblasts (TE-7-binding cells) and cancer cells (CD326). Representative data from one of three experiments. Left panel: representative sections of the activated tumor tissue. Right panel: Numbers per vision field of the individual cells types showing p65 translocation and chemokine production. Data from 10 vision fields is expressed as average +/− SEM.
Driven by the previously reported key role of NF-κB in the induction of CXCL10 and other chemokines (28–30), and the ubiquitous enhancement of the NF-κB signaling in cancer lesions critically needed for tumor survival and growth (24–27), we tested whether potential differences in NF-κB activation could be responsible for the differential ability of the tumors versus marginal tissues to respond to the chemokine modulatory regimen.
In accordance with this possibility, we observed that the colorectal cancer tissues showed not only elevated baseline levels of NF-κB activation (measured by the rate of its nuclear translocation; see Fig. 4B, left), but an even more pronounced ability to further activate NF-κB after the IFNα/poly-I:C/indomethacin treatment (Fig. 4B, right). The key role of NF-κB in CXCL10 production by tumor tissues was validated by using an NF-κB inhibitor, CAY10470, which completely abrogated CXCL10 induction (Fig. 4C).
CCL5 regulation showed a similar pattern (treatment-induced up-regulation in tumors, rather than in marginal tissues) and was also blocked by CAY10470 (Supplementary Fig. S5B), showing the general role of the tumor-associated NF-κB deregulation in the selective induction of Teff-attracting chemokines by the chemokine-modulating regimen. CAY10470, used in these experiments (at 20μM), was non-toxic, as shown by similar expression of glycogen phosphorylase mRNA in untreated and treated tissues (Supplementary Fig. S5C).
Interestingly, our confocal microscopy analysis revealed that most of the cells that showed nuclear translocation of NF-κB and produced CCL5 and CXCL10 represented CD45+ infiltrating inflammatory cells and (TE-7-binding) tumor-associated fibroblasts, with only some of the CD326/EpCAM+ cancer cells producing CCL5 (Fig. 4D; also see Supplementary Fig. S5D for example of single-color analyses).
IFNα/poly-I:C/indomethacin-treated colorectal tumors preferentially attract effector CD8+ T cells
In order to demonstrate that the modulation of chemokine achieved by the combination of IFNα, poly-I:C and indomethacin is indeed sufficient to affect the ability of tumors to attract different subsets of T cells, we used an ex-vivo chemotaxis assay involving the supernatants from differentially-treated tumors and either expanded tumor-infiltrating CD8+ T cells (TILs; see Supplementary Fig. S2F) or polyclonal ex-vivo-induced effector CD8+ T cells induced by superantigen-loadedαDC1s (32). As shown in Fig. 5A, each type of effector CD8+ T cell showed strongly enhanced migratory responsiveness uniformly to all the IFNα/poly-I:C/indomethacin-treated tumors. In contrast, CD4+FOXP3+ T cells preferentially migrated to untreated tumors, as determined by Taqman analysis of the migrated blood-isolated CD4+ T cells (Fig. 5B), or flow cytometry (Supplementary Fig. S6A). As expected, Taqman analysis of another Treg marker, GITR, yielded similar results (Supplementary Fig. S6B).
Figure 5. IFNα, poly-I:C and indomethacin-treated tumors show enhanced ability to attract Teff, but strongly-reduced ability to attract Tregs.
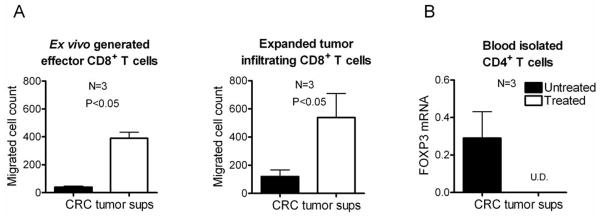
(A) Ex vivo generated Teff (left) or isolated CD8+ tumor-infiltrating lymphocytes (right) (see Materials and Methods) were allowed to migrate towards supernatants from either untreated or treated tumors from 3 different patients in transwell chemotaxis assays. (B) Negatively-isolated total CD4+ T cells were allowed to migrate towards the treated- or untreated tumor supernatants. Migrating cells were lysed and analyzed for FOXP3 expression by Taqman. U.D.: undetectable.
Discussion
Our data demonstrate the feasibility of tumor-selective modulation of the chemokine environment, using clinically applicable combinations of pharmacologic and biologic factors to correct the balance between tumor-infiltrating Teff- and Treg- cells, the types of immune cells known to differentially affect the clinical course of cancer (1–8). Importantly for the clinical application of this strategy, we observed that while the responses of the individual tumor lesions (even in the same patient) to the individual chemokine-modulators were highly variable (consistent with the limited clinical effectiveness of such factors applied individually), the combination of IFNα, poly-I:C and cyclooxygenase inhibitors allowed for highly consistent and selective enhancement of Teff-attracting chemokines (CCL5 and CXCL9-10) within tumor lesions tested, with the concomitant uniform suppression of local CCL22, the Treg-attracting chemokine.
The IFNα/poly-I:C/indomethacin-induced production of Teff-attracting chemokines was highly tumor-selective, suggesting that even systemic administration of these chemokine-modulating factors can preferentially direct effector cells to tumors. While the attraction of different subsets of T cells to different tumor types is known to be regulated by a complex network of additional chemokines not included in our current analysis (35, 36) and can be subject to regulation at the level of chemokine receptor expression, for example by CCR5 polymorphism (37), our current functional data (Fig 5) indicate that the proposed regimen can uniformly promote the influx of effector CD8+ T cells (both spontaneously-arising TILs and αDC1 vaccine-induced CTLs). The known role of CXCR3 and CCR5 in the attraction of Th1 cells and NK cells (12, 13, 38, 39) suggests that the proposed regimen may also be able to promote the entry of these additional types of desirable cells into tumors.
We observed that the tumor-selectivity of the proposed regimen depends on the propensity of tumor-associated fibroblasts and infiltrating inflammatory cells (with lesser involvement of tumor cells themselves) to not only spontaneously hyper-activate NF-κB, but also respond to treatment with further-enhanced levels of NF-κB activation. Since NF-κB activation, critically involved in tumor survival and growth, represents an intrinsic feature of many tumor types (24–27), the current data suggest that the currently-described NF-κB-targeting modulation of the tumor microenvironment may be applicable to multiple types of cancer. The currently-developed chemokine-modulating regimen consists of the combination of IFNα (type 1 interferon), poly-I:C (TLR3 ligand), and either indomethacin (COX1 and COX2 inhibitor) or a selective COX2 inhibitor, celecoxib. While our data demonstrate that interferons and prostanoids differentially regulate the NF-κB-driven production of Teff-, and Treg-attracting chemokines, the specific mechanisms and the molecular level of interplay between these factors remain subjects of our current research. Our analyses performed so far did not reveal any differences between the expression of the IFNα receptor, TLR3, IRF1, or IRF3 between tumors and marginal tissues (data not shown), but our current work focuses on the differential regulation of each of the pathways (poly-I:C, IFNα and PGE2 responsiveness) in whole tumor tissues and different types of tumor-associated cells. Similarly, we are also evaluating the mechanisms underlying the increased sensitivity of tumor-related cells to activate NF-κB and the relative heterogeneity of different tumors with regard to the requirement for poly-I:C activation, which may help us to identify new strategies of chemokine regulation and of targeting NF-κB in tumor therapy.
The combination of IFNα, poly-I:C and COX inhibition will be evaluated in clinical trials in patients with metastatic colorectal cancer, as a standalone treatment or in combination withαDC1 vaccines (32, 40), to enhance the numbers of circulating effector-type tumor reactive CD8+ T cells that respond to CCR5 and CXCR3 ligands (32) and enter tumor tissues. Our follow-up analyses will also allow us to determine whether the observed differences in the expression of chemokines and Teff markers in patients with metastatic colorectal cancer also translate into differences in clinical course of the disease and patient survival, as predicted by studies in primary colon cancer (1–4).
Supplementary Material
Acknowledgments
Grant Support: This work was supported by the NIH grants PO1CA132714, 1P50CA121973, and P30CA047904.
The authors thank Mark O’Malley for logistical support, Dr. James Schlesselman for his support with the statistical approaches and stimulating discussions, and Jeffrey Wong for critical reading of the manuscript.
Footnotes
Disclosure of Potential Conflicts of Interest: The methods of tumor-selective regulation of chemokine environment are the subject to a pending patent application by the University of Pittsburgh. None of the authors receives any form of remuneration related to these findings.
References
- 1.Galon J, Costes A, Sanchez-Cabo F, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313(5795):1960–4. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 2.Naito Y, Saito K, Shiiba K, et al. CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res. 1998;58(16):3491–4. [PubMed] [Google Scholar]
- 3.Pages F, Berger A, Camus M, et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med. 2005;353(25):2654–66. doi: 10.1056/NEJMoa051424. [DOI] [PubMed] [Google Scholar]
- 4.Ropponen KM, Eskelinen MJ, Lipponen PK, Alhava E, Kosma VM. Prognostic value of tumour-infiltrating lymphocytes (TILs) in colorectal cancer. J Pathol. 1997;182(3):318–24. doi: 10.1002/(SICI)1096-9896(199707)182:3<318::AID-PATH862>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 5.Clarke SL, Betts GJ, Plant A, et al. CD4+CD25+FOXP3+ regulatory T cells suppress anti-tumor immune responses in patients with colorectal cancer. PLoS ONE. 2006;1:e129. doi: 10.1371/journal.pone.0000129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ling KL, Dulphy N, Bahl P, et al. Modulation of CD103 expression on human colon carcinoma-specific CTL. J Immunol. 2007;178(5):2908–15. doi: 10.4049/jimmunol.178.5.2908. [DOI] [PubMed] [Google Scholar]
- 7.Michel S, Benner A, Tariverdian M, et al. High density of FOXP3-positive T cells infiltrating colorectal cancers with microsatellite instability. Br J Cancer. 2008;99(11):1867–73. doi: 10.1038/sj.bjc.6604756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chaput N, Louafi S, Bardier A, et al. Identification of CD8+CD25+Foxp3+ suppressive T cells in colorectal cancer tissue. Gut. 2009;58(4):520–9. doi: 10.1136/gut.2008.158824. [DOI] [PubMed] [Google Scholar]
- 9.Bonecchi R, Bianchi G, Bordignon PP, et al. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J Exp Med. 1998;187(1):129–34. doi: 10.1084/jem.187.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hirai H, Tanaka K, Yoshie O, et al. Prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophils, and basophils via seven-transmembrane receptor CRTH2. J Exp Med. 2001;193(2):255–61. doi: 10.1084/jem.193.2.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iellem A, Mariani M, Lang R, et al. Unique chemotactic response profile and specific expression of chemokine receptors CCR4 and CCR8 by CD4(+)CD25(+) regulatory T cells. J Exp Med. 2001;194(6):847–53. doi: 10.1084/jem.194.6.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loetscher P, Uguccioni M, Bordoli L, et al. CCR5 is characteristic of Th1 lymphocytes. Nature. 1998;391(6665):344–5. doi: 10.1038/34814. [DOI] [PubMed] [Google Scholar]
- 13.Sallusto F, Lenig D, Mackay CR, Lanzavecchia A. Flexible programs of chemokine receptor expression on human polarized T helper 1 and 2 lymphocytes. J Exp Med. 1998;187(6):875–83. doi: 10.1084/jem.187.6.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sallusto F, Mackay CR, Lanzavecchia A. Selective expression of the eotaxin receptor CCR3 by human T helper 2 cells. Science. 1997;277(5334):2005–7. doi: 10.1126/science.277.5334.2005. [DOI] [PubMed] [Google Scholar]
- 15.Musha H, Ohtani H, Mizoi T, et al. Selective infiltration of CCR5(+)CXCR3(+) T lymphocytes in human colorectal carcinoma. Int J Cancer. 2005;116(6):949–56. doi: 10.1002/ijc.21135. [DOI] [PubMed] [Google Scholar]
- 16.Kunz M, Toksoy A, Goebeler M, Engelhardt E, Brocker E, Gillitzer R. Strong expression of the lymphoattractant C-X-C chemokine Mig is associated with heavy infiltration of T cells in human malignant melanoma. J Pathol. 1999;189(4):552–8. doi: 10.1002/(SICI)1096-9896(199912)189:4<552::AID-PATH469>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 17.Ohtani H, Jin Z, Takegawa S, Nakayama T, Yoshie O. Abundant expression of CXCL9 (MIG) by stromal cells that include dendritic cells and accumulation of CXCR3+ T cells in lymphocyte-rich gastric carcinoma. J Pathol. 2009;217(1):21–31. doi: 10.1002/path.2448. [DOI] [PubMed] [Google Scholar]
- 18.Mlecnik B, Tosolini M, Charoentong P, et al. Biomolecular network reconstruction identifies T cell homing factors associated with survival in colorectal cancer. Gastroenterology. 2009 doi: 10.1053/j.gastro.2009.10.057. [DOI] [PubMed] [Google Scholar]
- 19.Curiel TJ, Coukos G, Zou L, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10(9):942–9. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 20.Soumaoro LT, Uetake H, Higuchi T, Takagi Y, Enomoto M, Sugihara K. Cyclooxygenase-2 expression: a significant prognostic indicator for patients with colorectal cancer. Clin Cancer Res. 2004;10(24):8465–71. doi: 10.1158/1078-0432.CCR-04-0653. [DOI] [PubMed] [Google Scholar]
- 21.Williams C, Shattuck-Brandt RL, DuBois RN. The role of COX-2 in intestinal cancer. Ann N Y Acad Sci. 1999;889:72–83. doi: 10.1111/j.1749-6632.1999.tb08725.x. [DOI] [PubMed] [Google Scholar]
- 22.Muthuswamy R, Urban J, Lee JJ, Reinhart TA, Bartlett D, Kalinski P. Ability of mature dendritic cells to interact with regulatory T cells is imprinted during maturation. Cancer Res. 2008;68(14):5972–8. doi: 10.1158/0008-5472.CAN-07-6818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lebre MC, Kalinski P, Das PK, Everts V. Inhibition of contact sensitizer-induced migration of human Langerhans cells by matrix metalloproteinase inhibitors. Arch Dermatol Res. 1999;291(7–8):447–52. doi: 10.1007/s004030050436. [DOI] [PubMed] [Google Scholar]
- 24.Greten FR, Eckmann L, Greten TF, et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118(3):285–96. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 25.Sakamoto K, Maeda S, Hikiba Y, et al. Constitutive NF-kappaB activation in colorectal carcinoma plays a key role in angiogenesis, promoting tumor growth. Clin Cancer Res. 2009;15(7):2248–58. doi: 10.1158/1078-0432.CCR-08-1383. [DOI] [PubMed] [Google Scholar]
- 26.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441(7092):431–6. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 27.Pikarsky E, Porat RM, Stein I, et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431(7007):461–6. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- 28.Hoffmann A, Levchenko A, Scott ML, Baltimore D. The IkappaB-NF-kappaB signaling module: temporal control and selective gene activation. Science. 2002;298(5596):1241–5. doi: 10.1126/science.1071914. [DOI] [PubMed] [Google Scholar]
- 29.Clarke DL, Clifford RL, Jindarat S, et al. TNFalpha and IFNgamma synergistically enhance transcriptional activation of CXCL10 in human airway smooth muscle cells via STAT- 1, NF-kappaB, and the transcriptional coactivator CREB-binding protein. J Biol Chem. 285(38):29101–10. doi: 10.1074/jbc.M109.0999952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu LY, Bates ME, Jarjour NN, Busse WW, Bertics PJ, Kelly EA. Generation of Th1 and Th2 chemokines by human eosinophils: evidence for a critical role of TNF-alpha. J Immunol. 2007;179(7):4840–8. doi: 10.4049/jimmunol.179.7.4840. [DOI] [PubMed] [Google Scholar]
- 31.Dudley ME, Wunderlich JR, Shelton TE, Even J, Rosenberg SA. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J Immunother. 2003;26(4):332–42. doi: 10.1097/00002371-200307000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Watchmaker PB, Berk E, Muthuswamy R, et al. Independent regulation of chemokine responsiveness and cytolytic function versus CD8+ T cell expansion by dendritic cells. J Immunol. 2010;184(2):591–7. doi: 10.4049/jimmunol.0902062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fallert BA, Reinhart TA. Improved detection of simian immunodeficiency virus RNA by in situ hybridization in fixed tissue sections: combined effects of temperatures for tissue fixation and probe hybridization. J Virol Methods. 2002;99(1–2):23–32. doi: 10.1016/s0166-0934(01)00378-0. [DOI] [PubMed] [Google Scholar]
- 34.Reinhart TA, Fallert BA, Pfeifer ME, et al. Increased expression of the inflammatory chemokine CXC chemokine ligand 9/monokine induced by interferon-gamma in lymphoid tissues of rhesus macaques during simian immunodeficiency virus infection and acquired immunodeficiency syndrome. Blood. 2002;99(9):3119–28. doi: 10.1182/blood.v99.9.3119. [DOI] [PubMed] [Google Scholar]
- 35.Ascierto ML, De Giorgi V, Liu Q, et al. An immunologic portrait of cancer. J Transl Med. 2011;9:146. doi: 10.1186/1479-5876-9-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spivey TL, Uccellini L, Ascierto ML, et al. Gene expression profiling in acute allograft rejection: challenging the immunologic constant of rejection hypothesis. J Transl Med. 2011;9:174. doi: 10.1186/1479-5876-9-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ugurel S, Schrama D, Keller G, et al. Impact of the CCR5 gene polymorphism on the survival of metastatic melanoma patients receiving immunotherapy. Cancer Immunol Immunother. 2008;57(5):685–91. doi: 10.1007/s00262-007-0407-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Inngjerdingen M, Damaj B, Maghazachi AA. Expression and regulation of chemokine receptors in human natural killer cells. Blood. 2001;97(2):367–75. doi: 10.1182/blood.v97.2.367. [DOI] [PubMed] [Google Scholar]
- 39.Martin-Fontecha A, Thomsen LL, Brett S, et al. Induced recruitment of NK cells to lymph nodes provides IFN-gamma for T(H)1 priming. Nat Immunol. 2004;5(12):1260–5. doi: 10.1038/ni1138. [DOI] [PubMed] [Google Scholar]
- 40.Mailliard RB, Wankowicz-Kalinska A, Cai Q, et al. alpha-type-1 polarized dendritic cells: a novel immunization tool with optimized CTL-inducing activity. Cancer Res. 2004;64(17):5934–7. doi: 10.1158/0008-5472.CAN-04-1261. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.