

Abstract
Protein folding is a spontaneous process that is essential for life, yet the concentrated and complex interior of a cell is an inherently hostile environment for the efficient folding of many proteins. Some proteins—constrained by sequence, topology, size, and function—simply cannot fold by themselves and are instead prone to misfolding and aggregation. This problem is so deeply entrenched that a specialized family of proteins, known as molecular chaperones, evolved to assist in protein folding. Here we examine one essential class of molecular chaperones, the large, oligomeric, and energy utilizing chaperonins or Hsp60s. The bacterial chaperonin GroEL, along with its co-chaperonin GroES, is probably the best-studied example of this family of protein-folding machine. In this review, we examine some of the general properties of proteins that do not fold well in the absence of GroEL and then consider how folding of these proteins is enhanced by GroEL and GroES. Recent experimental and theoretical studies suggest that chaperonins like GroEL and GroES employ a combination of protein isolation, unfolding, and conformational restriction to drive protein folding under conditions where it is otherwise not possible.
Keywords: protein folding, molecular chaperone, chaperonin, GroEL, GroES, Hsp60
INTRODUCTION
Over 40 years ago, Christian Anfinsen demonstrated that all the information necessary to encode the three-dimensional structure of a protein is contained in its linear sequence of amino acids (Anfinsen, 1973). The correct translation of this information, from stored nucleic acid code into well folded and functional protein, depends upon a cell’s ability to carry out a complex, multi-step process at high speed while making very few mistakes. Each and every stage of protein production, therefore, requires high-fidelity error correction. Despite the simplifying observation that thermodynamics, in the end, specifiy protein structure, the folding of many essential proteins is not error free. For some proteins, the barriers to efficient folding are so significant that spontaneous folding is essentially impossible, at least under conditions and on time scales relevant for life. To solve this problem, cells have developed a number of specialized systems that monitor and correct protein folding mistakes. As a group, these accessory proteins are known as molecular chaperones (Ellis and van der Vies, 1991). A number of different molecular chaperone classes have been identified, each specialized for dealing with a different aspect of the cellular folding problem. Years of effort have uncovered some of the mechanisms by which chaperones accomplish this action and, further, have shown that interlocking networks of multiple molecular chaperones govern many essential aspects of protein biogenesis and homeostasis (Fenton and Horwich, 1997; Bukau and Horwich, 1998; Frydman, 2001; Deuerling and Bukau, 2004; Young et al., 2004; Mayer and Bukau, 2005). All molecular chaperones share in common the basic functions of recognizing, binding, and releasing other proteins. For some proteins, the assistance of molecular chaperones is necessary at virtually every stage of protein folding and assembly, involving multiple classes of chaperones along the way. Other proteins, however, seem to require little if any folding assistance, bypassing much of the molecular chaperone machinery altogether and folding rapidly and efficiently on their own. In this review, we will examine what is known about one essential class of molecular chaperones, the so-called chaperonins or Hsp60s, and how they drive the folding of proteins that cannot fold on their own.
PROTEIN-FOLDING CONSTRAINTS
Small, globular proteins (<15 kDa) tend to fold quickly and efficiently (Jackson, 1998; Radford, 2000; Grantcharova et al., 2001; Myers and Oas, 2002). Many small proteins display two-state folding behavior, in which the rapid, highly cooperative coalescence of native three-dimensional structure occurs without the population of stable intermediate states. Even in cases where small proteins populate intermediate states, they are generally short-lived and do not dramatically inhibit folding (Jackson, 1998). In general, the free energy barriers that separate intermediate states of small proteins from productive routes to their native state are not large and are readily overcome by thermal motion. While such simple folding can be visualized as a conversion between discreet conformational states, protein folding is actually a rather more complex problem of polymer dynamics. A more realistic representation of protein folding involves relating a protein’s accessible conformational space to the free energy of each polypeptide conformation (Dill and Chan, 1997; Onuchic et al., 1997). The resulting picture of protein folding is one of trajectories across an energy hypersurface or landscape that globally slopes toward the native state and becomes dramatically narrower as a protein approaches its native conformation (a folding funnel). Rapid and efficient folding of a small protein, then, is explained by a folding landscape where various routes to the native state are available, with no particularly large energy barriers to inhibit the efficient downward slide (Figure 1).
Figure 1.
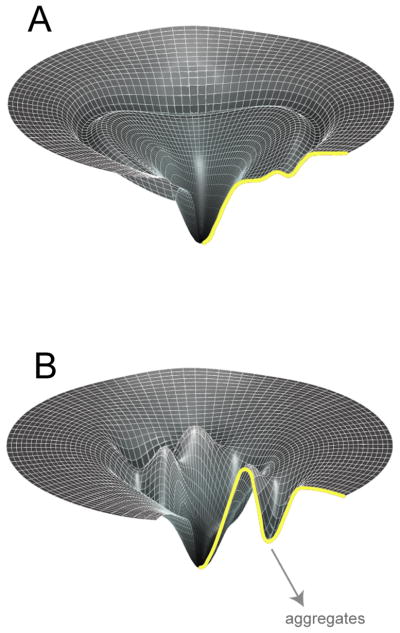
The folding landscapes of large and topologically complex proteins are filled with kinetic traps. (A) Folding landscape of a small, rapidly folding protein with no requirement for folding assistance from a molecular chaperone. The vertical axis of the landscape captures the internal free energy of each protein conformation, while the width of the landscape is a measure of the configurational entropy of the protein at a particular energy level (Dill and Chan, 1997; Onuchic et al., 1997). (B) Folding landscape of a large and complex protein that depends upon molecular chaperones for productive folding. Spontaneous folding is inefficient or impossible, due to the prominent and deep local energy minima that easily trap the protein and lead to aggregation.
Larger and more topologically complex proteins, however, often possess slower and more complicated folding reactions (Garel, 1992; Dobson, 2001; Grantcharova et al., 2001). In general, large proteins do not display two-state folding behavior, are found to populate a range of meta-stable intermediate states when their folding is triggered from a fully denatured state, and are highly prone to aggregation during folding. Additionally, large proteins are likely to have a greater probability of populating intermediate states that contain significant amounts of low free energy, non-native contacts. In other words, large proteins are more prone to misfolding by virtue of having many more possible contacts than smaller proteins, some of which are sufficiently stable that they get a large protein into serious trouble (Dobson, 2001; Grantcharova et al., 2001). The folding landscape of such a protein is thus imagined to be replete with kinetic traps, conformational wells (either native-like or not) of sufficient depth that thermal motion only rarely manages to drive the protein back onto a productive folding pathway (Figure 1; Todd et al., 1994; Thirumalai and Lorimer, 2001).
The increase in folding difficulty faced by large and topologically complex proteins can result in the highly efficient trapping of such proteins in either misfolded or aggregated states (Dobson, 2001; Horwich, 2002). Non-native proteins possess unsatisfied and exposed interaction sites, commonly unburied hydrophobic surface. These unsatisfied contact sites can lead to interactions with other proteins that have complementary surfaces, typically other molecules of the same non-native protein. If a given non-native protein contains multiple such sites, and if the sites are exposed for too long because of slow folding or misfolding, a rapid, multi-molecular aggregation reaction generally takes place (Garel, 1992; King et al., 1996; Jaenicke and Lilie, 2000). In the earliest stages, aggregation can often be reversed, but if allowed to proceed unchecked, aggregation is, in most cases, eventually irreversible. Irreversible aggregation of an important protein not only deprives a cell of a critical structural or chemical activity, but protein aggregates themselves seem to cause additional problems. Work on a variety of human diseases has shown that the incorrect folding and/or aggregation of important cellular proteins can be involved in serious pathologies (Thomas et al., 1995; Koo et al., 1999; Sanders and Nagy, 2000; Dobson, 2001; Horwich, 2002; Selkoe, 2002). Examples include cystic fibro-sis, thalassemias, alpha1-antitrypsin deficiency, and a variety of amyloid neuropathies such as Alzheimer’s and Huntington’s diseases. Misfolded and aggregated proteins can even generate infectious particles that can, in the case of prion proteins, propagate their own formation upon spreading from cell to cell (Prusiner et al., 1998; McKintosh et al., 2003; Weissmann, 2005). One critical role of molecular chaperones, then, is to ensure that essential cellular proteins, which fold poorly on their own, efficiently find their native state, instead of misfolding and aggregating.
CHAPERONINS
In the network of molecular chaperones that fold, monitor, and maintain cellular proteins, the large, barrel-shaped oligomers known as chaperonins play a central and essential role (Cheng et al., 1989; Fayet et al., 1989; Kerner et al., 2005). These remarkable molecular machines employ the energy of ATP hydrolysis to power a facilitated protein-folding reaction. GroEL, the chaperonin of the bacterium Escherichia coli, is the archetypal member of this ubiquitous family of protein folding engines. Over the last two decades, a large body of experimental work has established that GroEL and its co-chaperonin GroES are directly involved in driving protein folding under conditions where the spontaneous folding reaction simply does not proceed (Fenton and Horwich, 1997; Sigler et al., 1998; Thirumalai and Lorimer, 2001; Hartl and Hayer-Hartl, 2002; Saibil and Ranson, 2002; Fenton and Horwich, 2003). While we now possess a detailed understanding of the basic structure and reaction cycle of GroEL, the precise way in which GroEL and GroES accomplish facilitated protein folding is not well understood. Before proceeding on to this key issue, we will first briefly review some of the structural, allosteric and enzymatic properties of the GroEL-GroES system. We examine the oligomeric structures of GroEL and GroES, present the basic ATPase cycle that constitutes the heart of the functional chaperonin reaction, and then touch on some of the common physical characteristics of substrate proteins recognized by GroEL. We do not consider in this review the related chaperonins of the archeabacteria and eukaryotic cytoplasm, the so-called Type II chaperonins. For more complete treatments of these subjects, the reader is referred to other recent reviews (Sigler et al., 1998; Horovitz et al., 2001; Saibil and Ranson, 2002; Fenton and Horwich, 2003; Gomez-Puertas et al., 2004; Spiess et al., 2004).
GroEL and GroES Structure
The ability of GroEL and GroES to drive protein folding is rooted in the molecular structures of these proteins. Both GroEL and GroES are seven-fold, rotationally symmetric, ring shaped oligomers (Figure 2). GroES is composed of seven, identical 10 kDa subunits arranged as a domed disk roughly 80 Å in diameter (Hunt et al., 1996; Mande et al., 1996). GroEL is a much larger oligomeric complex, composed of fourteen identical 57kDa subunits (Braig et al., 1994; Braig et al., 1995). The GroEL subunits are arranged in two, seven-membered rings that are stacked back to back to form a double doughnut-like cylindrical structure that is approximately 147 Å in length and 137 Å in diameter. Each of the GroEL subunits can be divided into three domains: apical, intermediate, and equatorial (Figure 2). The equatorial domains of each ring abut each other, with the apical domains of each ring positioned at the outer ends of the cylinder. The apical domains contain the binding sites for non-native proteins and GroES. The equatorial domain contains the ATP binding site and the apical and intermediate domains are connected to one another through the slender intermediate domain. A GroEL ring contains a large central cavity that is roughly 45 Å in diameter and a GroEL tetradecamer, then, possesses two cavities that are isolated from one another by the equatorial domains and the C-terminal tails of each subunit. The inner apical surface of each ring is lined with non-polar amino acids (Braig et al., 1994; Braig et al., 1995) and this hydrophobic surface is used to capture and tightly bind protein folding intermediates (Fenton et al., 1994; Farr et al., 2000).
Figure 2.

GroES binding to a GroEL ring creates an enclosed and enlarged cavity. (A) The molecular structure of unliganded GroEL (Braig et al., 1994; Braig et al., 1995) is illustrated as a space-filling model, with three subunits highlighted. The secondary structure of one subunit is shown, and for clarity this subunit is also illustrated alone, with the apical (‘a’), intermediate (‘i’) and equatorial (‘e’) domains highlighted. A cross-section of the unliganded GroEL barrel, illustrating the central cavity in each ring, is shown to the right. The substrate-binding apical surface is highlighted in the upper ring. Note that the apparent connection between the two cavities is an artifact of unresolved polypeptide density in the crystal structure and the two GroEL ring cavities are actually isolated from one another. (B) The molecular structure of the GroEL-ADP-GroES complex (Xu et al., 1997) as a space-filling model is illustrated. The secondary structure of one GroEL subunit from the cis ring is shown, along with a cross-section of the GroEL-GroES structure to the right. Note that the substrate-binding apical residues are again highlighted, but have been rotated away and are much less exposed to the cavity. Molecular structure images were created with PyMOL (DeLano, 2002).
The GroEL ATPase Cycle
In order to function properly, GroEL must bind GroES (Tilly et al., 1981; Chandrasekhar et al., 1986; Saibil et al., 1991; Langer et al., 1992; Ishii et al., 1994). The hydrophobic apical surface of a GroEL ring provides the critical binding site for a flexible loop at the base of each GroES subunit (Landry et al., 1993; Fenton et al., 1994; Xu et al., 1997). The GroES binding sites on a GroEL ring therefore substantially overlap the regions of the GroEL ring that bind non-native substrate proteins. However, GroES binding requires a set of key structural rearrangements of the GroEL ring, driven by adenine nucleotide binding, that reposition the GroEL apical domain surface (Figure 2; Chen et al., 1994; Roseman et al., 1996; Xu et al., 1997). These nucleotide-driven structural rearrangements constitute the kinematic heart of the functional chaperonin protein folding cycle.
Stable binding of GroES to a GroEL ring necessitates that the nucleotide binding sites of the ring be filled with either ADP or ATP (Todd et al., 1994; Burston et al., 1995). However, ATP is required to power the specific structural transitions of a GroEL ring that lead to productive folding of the most GroEL-dependent substrate proteins (Rye et al., 1997; Chaudhry, et al., 2003; Motojima et al., 2004). In a typical reaction cycle (Figure 3), ATP binds to a GroEL ring in a highly cooperative fashion, followed by GroES binding to the same ring (Gray and Fersht, 1991; Bochkareva et al., 1992; Jackson et al., 1993; Burston et al., 1995). Occupancy of one ring by ATP then inhibits binding of ATP to the other ring by inducing strong negative cooperativity across the rings, creating an asymmetric GroEL-GroES complex (a cis ternary complex; Yifrach and Horovitz, 1994; Burston et al., 1995; Yifrach and Horovitz, 1995). These ATP-driven allosteric transitions are central to GroEL’s ability to function as a folding machine (Burston et al., 1995; Yifrach and Horovitz, 1995; Kad et al., 1998; Rye et al., 1999; Yifrach and Horovitz, 2000). Using both steady-state and pre-steady-state kinetic analysis, Horovitz and colleagues (2001) have suggested that the ATP-induced allosteric transitions of a GroEL tetradecamer are best described by a nested allosteric model, involving symmetry-driven (MWC-type) positive cooperativity within a ring, nested within a sequential (KNF-type) negatively cooperative transition between the rings (Yifrach and Horovitz, 1994; Yifrach and Horovitz, 1995; for review see Horovitz et al., 2001). Once seven molecules of ATP and GroES are bound to one GroEL ring to form an ATP-bound cis complex, the bound ATP is committed to hydrolyze to ADP. Hydrolysis of the cis-bound ATP then generates an ADP-bound cis complex that is stable until a disassembly signal is delivered from the opposite or trans ring (Todd et al., 1994; Rye et al., 1997).
Figure 3.

The GroEL-GroES reaction cycle. The full GroEL reaction involves two half-cycles (a–d and e–h), where the assembly a GroEL-GroES cis complex on one ring is directly coupled to disassembly of a cis complex on the other ring. In (a), the open trans ring on a GroEL-ADP-GroES complex binds non-native substrate protein and ATP, triggering disassembly of the cis complex on the other ring (b). Rapid binding of GroES to the ATP and substrate occupied ring (c) encloses the substrate and releases the protein into the cis cavity. Subsequent hydrolysis of ATP within the cis complex to generate the GroEL-ADP-GroES complex primes the cis complex for disassembly and activates the trans ring for another round of ATP, substrate and GroES binding (d–f). The last two steps (g–h) then return the cycle to the starting point (shown dim). With each round of cis complex disassembly, the contents of the cavity are ejected into solution, including both folded and unfolded protein intermediates. A protein would typically remain within the enclosed cis cavity for 6–10 sec at 25°C.
As shown by both electron cryomicroscopy (Chen et al., 1994; Roseman et al., 1996) and X-ray crystallography (Xu et al., 1997), nucleotide binding to a given GroEL ring results in large-scale, rigid body rearrangements of the GroEL subunits that are mediated by bending and rotational movements around hinge points that connect the intermediate domain with the apical and equatorial domains (Figure 2). These movements close the nucleotide into its binding site and result in an elevation and rotation of the apical domains. The rearrangements have a number of profound structural consequences. First, movement of the apical domains rotates the substrate binding sites away from the cavity while apparently optimizing the positioning of the GroES binding sites. This results in the tight association of GroES to form an enclosed chamber. Second, the surface of the GroEL oligomer facing the cavity is switched in character from hydrophobic to hydrophilic through the rearrangement of the amino acids that line the cavity (Figure 2).
The formation and stability of the GroEL-GroES complex is governed by the rate at which the ATP used to build the cis complex is hydrolyzed. Using a hydrolysis-deficient mutant of GroEL, it has been shown that the GroEL-GroES cis complex does not dissociate until the ATP in the cis complex hydrolyzes to ADP (Rye et al., 1997). Once this turnover takes place, the cis complex is “primed” for disassembly, which is driven by a round of ATP binding to the trans ring (Todd et al., 1994; Rye et al., 1997). The ATP-driven release trigger supplied by the trans-ring is prevented from firing, however, until the ATP present in the cis ring is hydrolyzed (Rye et al., 1997; Rye et al., 1999). The open trans ring of the GroEL-GroES complex is also used to capture non-native substrate proteins (Rye et al., 1999). Substrate protein binding to the trans ring dramatically accelerates cis complex disassembly but cannot occur until hydrolysis of ATP within the cis complex takes place. In essence, ATP hydrolysis by GroEL is used to power a dynamic cycle of substrate protein binding and release (Figure 3).
Recognition of Substrate Proteins by GroEL
GroEL binds to non-native substrate proteins but does not interact with the folded, native states of the same proteins (Viitanen et al., 1992). A common property of non-native, incompletely folded proteins is their tendency to expose hydrophobic surfaces that are normally buried in the native state (Radford, 2000; Horwich, 2002). Several studies have highlighted the importance of these hydrophobic surfaces in the recognition of substrate proteins by GroEL. Both mutational studies with a model substrate protein, where hydrophobic amino acids were systematically changed (Itzhaki et al., 1995), as well as direct calorimetric measurements on substrate protein binding to GroEL (Lin et al., 1995), support a central role for hydrophobicity in substrate protein binding by GroEL. In another study, limited proteolysis of a non-native protein was employed to probe the structure of the protein while it was bound to GroEL (Hlodan et al., 1995). Two regions of the substrate protein were identified that were well protected from proteolysis, presumably by virtue of being tightly associated with GroEL and therefore protected from the protease. These regions were highly populated with hydrophobic amino acids, forming both hydrophobic and amphipathic α-helices in the native state. More recent studies with small model peptides also support a key role for large stretches of hydrophobic surface in substrate protein recognition by GroEL (Brazil et al., 1997; Chen and Sigler, 1999; Preuss et al., 1999; Wang et al., 1999). Complementary observations on the role of hydrophobic interactions have also been made with GroEL itself. Mutations that disrupt the hydrophobicity of the GroEL inner apical surface dramatically reduce or eliminate substrate protein binding (Fenton et al., 1994). Non-native substrate proteins also appear to make multiple contacts with the hydrophobic apical domain ring, and this multi-valent binding appears to be essential for stable substrate protein capture and productive folding (Farr et al., 2000).
The binding of substrate proteins to GroEL does not appear, however, to be generally dependent on a particular amino acid sequence or specific structural motif. Comparisons of model peptides that show strong binding to GroEL demonstrate no significant sequence commonality, beyond their dramatic enrichment in hydrophobic amino acids (Coyle et al., 1997). Biophysical and structural studies have demonstrated that the GroEL apical surface can recognize a range of local structures, including α-helices (Landry and Gierasch, 1991; Landry et al., 1992; Preuss et al., 1999; Wang et al., 1999), extended strands (Buckle et al., 1997; Wang et al., 1999) and β-hairpins (Chen and Sigler, 1999). In all cases, the most important contacts appear to be hydrophobic in nature although, GroEL does demonstrate a preference for hydrophobic peptides with an overall positive charge (Hutchinson et al., 1997). An ionic contribution to the binding of full-sized, non-native proteins to GroEL has also been observed (Katsumata et al., 1996; Sparrer et al., 1996; Aoki et al., 1997; Perrett et al., 1997). Crystal structures of model peptides bound to both intact GroEL and to an isolated apical domain fragment have suggested that substrate proteins make a specific contact with the GroEL apical face, in a groove between two central helices (H and I) that constitute a significant part of the GroEL apical surface (Buckle et al., 1997; Chen and Sigler, 1999). This binding mode is highly reminiscent of the way in which the GroES mobile loop binds to the GroEL apical face (Xu et al., 1997). Interestingly, a competition experiment between a model peptide derived from the GroES mobile loop and another peptide derived from the GroEL substrate protein rhodanese were found to bind simultaneously to isolated GroEL apical domains (Ashcroft et al., 2002). This result implies that the simple binding of peptides to the groove between helices H and I, while suggestive, is not a complete picture of hydrophobic surface recognition by GroEL. Indeed, mutational studies indicate that important substrate binding contacts are also present in a loop beneath the H and I helices (Fenton et al., 1994). In the end, it remains unclear precisely how full-sized substrate proteins, with their more complex conformational dynamics and capacity to bind across multiple apical domains, interact with the apical surface of GroEL.
In general, GroEL-bound proteins appear to be collapsed and loosely structured states, possessing conformations somewhere between fully unfolded and fully native (Martin et al., 1991; Mendoza et al., 1992; Hayer-Hartl et al., 1994; Weissman et al., 1994; Zahn and Pluckthun, 1994). In some cases, GroEL has been observed to bind conformations reminiscent of molten globules (Martin et al., 1991), while in other cases GroEL appears to prefer poorly structured intermediates formed at very early stages of folding, and in still other cases GroEL binds more structured intermediates populated at relatively late stages of folding (Badcoe et al., 1991; Staniforth et al., 1994; Lilie and Buchner, 1995; Clark and Frieden, 1997). While we will consider the nature of GroEL-bound proteins in more detail below, we note that highly detailed structural information on non-native proteins bound to GroEL has been difficult to come by, due to their inherently unstable and poorly organized structure. However, a low-resolution picture of the gross morphology of a few non-native proteins bound to a GroEL ring has been developed through low angle neutron scattering and cryo-electron microscopy (Chen et al., 1994; Thiyagarajan et al., 1996; Falke et al., 2005). These studies are consistent with a poorly structured but collapsed conformation of the GroEL-bound protein, with a significant amount of polypeptide mass extending out of the mouth of the GroEL cavity.
HOW DOES GroEL FACILITATE PROTEIN FOLDING?
Passive versus Active Models of GroEL Action
GroEL, and its close relatives in other organisms, have been shown to assist the folding of a wide range of proteins (Cheng et al., 1989; Horwich et al., 1993; Ewalt et al., 1997; Dubaquie et al., 1998; Houry et al., 1999; Kerner et al., 2005; Goloubinoff et al., 1989b). The early recognition that GroEL assists the folding of many different proteins strongly implied that GroEL does not facilitate protein folding by providing a structural template for protein native states. GroEL must, therefore, target a common, folding-inhibitory property of the substrate proteins upon which it works. Proteins that require GroEL are typically large (>20 kDa), slow folding, and aggregation prone (Kerner et al., 2005). Thus, GroEL-dependent proteins are those for which folding kinetics have trumped the sequence-encoded thermodynamic drive to the native state. In the most general terms then, GroEL must function to alter the kinetic balance between productive folding and misfolding or aggregation. As an example, consider a GroEL-dependent protein that folds through the population of a single, aggregation-prone intermediate state (Figure 4). The intermediate state could be either an inherently productive intermediate with direct access to the native state (an on-pathway intermediate) or a non-productive, misfolded state (an off-pathway intermediate). In either case, the intermediate is assumed to form an irreversible aggregate at some fixed rate. In general, GroEL could promote the productive folding, and prevent the irreversible loss of such a protein, by either (1) blocking aggregation or (2) accelerating productive folding. Several of possible mechanisms have been proposed to explain how GroEL might accomplish these tasks. For the purposes of this review, we will classify these mechanisms as either passive or active. In a passive folding mechanism, GroEL has no direct effect on the conformation of a non-native protein, serving simply to block inhibitory aggregation. An active model, by contrast, involves the direct modification of a substrate protein’s structure or accessible conformational space by GroEL.
Figure 4.

GroEL can facilitate protein folding by either passive or active mechanisms. The dominant folding routes for an idealized, GroEL-dependent protein are shown. In the absence of GroEL, the protein can proceed to the native state via a productive, on-pathway intermediate state (Ion) or can form a misfolded and kinetically trapped off-pathway state (Ioff) with no direct access to the native state. Both intermediates, as well as the unfolded ensemble (U), are highly prone to irreversible aggregation. GroEL could passively rescue a protein by binding Ion and U and blocking aggregation. Alternately, GroEL could actively enhance the folding of this protein by (1) binding and unfolding Ioff, (2) binding U and Ion and preventing the formation of Ioff, or (3) binding U and Ion and increasing the rate that N is produced.
Passive Models of GroEL-Mediated Folding
Early models imagined that molecular chaperones functioned by passively suppressing protein aggregation (Pelham, 1986; Ellis and van der Vies, 1991; Agard, 1993). By binding to the exposed hydrophobic surfaces that get protein folding intermediates into trouble, a molecular chaperone like GroEL could directly block aggregation. For example, specific recognition and binding of an on-pathway intermediate state like Ion in Figure 4 would shift the overall folding equilibrium away from aggregation and provide a pool of folding-competent monomers that could proceed on to the native state. In a purely passive folding model, the primary event blocking folding is the formation of multi-molecular agglomerations that form faster than monomers can fold and bury hydrophobic surfaces. The high concentrations of macromolecules in cytoplasm would be expected to dramatically amplify this effect, both because high concentrations of an aggregating molecule non-linearly accelerate the rate of aggregation and because non-specific excluded volume effects in the “molecularly crowded” environment of living cytoplasm should also increase aggregation (van den Berg et al., 1999; Ellis, 2001). To prevent this aggregation process from progressing, GroEL could simply block the non-productive intermolecular associations that lead to aggregation. For such a passive, aggregation-dominated folding model, the conformation of the non-native substrate protein would not, however, need to be altered by GroEL.
Consistent with a direct involvement of GroEL in preventing aggregation, early in vivo studies in bacteria and yeast demonstrated that depletion of either GroEL or mitochondrial Hsp60, by temperature inactivation of conditional alleles of these proteins, resulted in whole-scale aggregation of a large number of newly translated proteins (Cheng et al., 1989; Horwich et al., 1993). Additionally, a range of in vitro studies directly demonstrated that GroEL can rapidly and efficiently bind to non-native states of several proteins and arrest their aggregation, including malate dehydrogenase (MDH; Ranson et al., 1995), ribulose-1,5-bisphosphate oxygenase-carboxylase (RuBisCO; Goloubinoff et al., 1989b), rhodanese (Martin et al., 1991; Mendoza et al., 1991), citrate synthase (Buchner et al., 1991), α-glucosidase (Holl-Neugebauer et al., 1991), and glutamine synthase (Fisher, 1992).
While passive binding of an aggregation-prone intermediate by GroEL can block aggregation, captured proteins must eventually be released back into free solution in order to complete the final steps of folding or oligomer assembly. A purely passive folding model requires that the relative rates of binding, release, folding, and aggregation be balanced so as to provide a window of opportunity for productive folding to occur. In principle, GroEL could provide this key folding window by allowing some folding to take place while the substrate protein is associated with the chaperonin. In this case, folding would take place at infinite dilution, with each GroEL-assocated protein physically isolated from other, aggregation-prone proteins (Agard, 1993; Saibil et al., 1993; Ellis, 1994). Upon attaining a sufficiently native-like state, the protein would no longer associate with the chaperonin and would be released into free solution.
The Anfinsen Cage
Precisely how GroEL could bind a substrate protein with high affinity, and hold it tightly enough to prevent aggregation but not so tightly that folding was blocked, was not initially obvious. The observation that a GroEL oligomer is organized into a pair of heptameric rings with large internal cavities, and that non-native substrate proteins could be at least partially contained inside the open GroEL cavity, provided an intriguing possibility: the GroEL ring could provide an protected environment for at least part of a substrate protein (Langer et al., 1992; Braig et al., 1993; Saibil et al., 1993; Ishii et al., 1994). Despite the highly suggestive nature of these observations, it was not initially clear how partial protection of a folding intermediate would generally stimulate folding of different substrate proteins or how this binding mode was connected to the productive release of a substrate protein from GroEL by ATP and GroES. It was clear from the earliest in vitro experiments, however, that ATP hydrolysis and GroES were necessary for productive protein folding under environmental conditions most restrictive for folding (non-permissive conditions; Goloubinoff et al., 1989b; Viitanen et al., 1990; Buchner et al., 1991; Martin et al., 1991; Schmidt et al., 1994). Without GroES and ATP, the most dependent GroEL substrates do not fold and remain tightly associated with the chaperonin.
What is the essential connection between the GroEL cavity, substrate and GroES binding, and ATP hydrolysis? The answer to this central question was provided by a series of seminal investigations into the role played by GroES in the GroEL reaction cycle. The first key observation was the nucleotide-dependent binding of GroES to GroEL to form the enclosed GroEL-GroES cis cavity (see above). The second key observation was the demonstration that the productive folding of classically stringent GroEL substrates (e.g., RuBisCO, MDH and rhodanese) is initiated by the encapsulation and confinement of non-native proteins inside the enclosed GroEL-GroES cis cavity (Weissman et al., 1995; Mayhew et al., 1996; Weissman et al., 1996; Rye et al., 1997).
A rather simple and elegant model was developed to explain how GroEL could passively, but efficiently, stimulate protein folding (Agard, 1993; Ellis, 1994; Wang and Weissman, 1999). A GroEL ring was imagined to shift between high-and-low-affinity states for a non-native substrate protein, with this transition driven by ATP hydrolysis and GroES binding. While early biochemical studies strongly suggested that ATP binding and hydrolysis could drive GroEL though a shift in substrate affinity (Badcoe et al., 1991; Martin et al., 1991), the apical domain movements observed in structural studies provided a vivid molecular explanation of how ATP and GroES could drive substrate protein release into the cis cavity. The non-native protein binding sites on the inner face of the GroEL apical domains, those that initially face the open cavity and capture folding intermediates, elevate and shift away from the cavity lumen to be sequestered by GroES. Thus, capture of a folding intermediate on a high-affinity GroEL ring initially prevents protein aggregation. ATP-driven enclosure of non-native proteins within the GroEL-GroES cis cavity then provides an efficient, coordinated release mechanism. More importantly, substrate enclosure within the isolated, hydrophilic GroEL-GroES cavity was an obvious method for infinitely diluting the substrate protein. This model of GroEL-mediated folding came to be known to as the Anfinsen cage model (Saibil et al., 1993; Ellis, 1994). Essentially, the enclosed cis cavity was envisioned to be a near ideal, isolated environment where folding could proceed unhindered, propelled only by the intrinsic thermodynamic drive encoded by the protein amino acid sequence.
Kinetic Partitioning
Perhaps the most efficient way that the GroEL-GroES cage could promote protein folding would be for the chaperonin to prevent a folding intermediate from escaping into solution until it had reached its native state (or at least a state “committed” to reach the native state). However, GroEL appears to act in exactly the opposite manner. Several studies have demonstrated that GroEL releases non-native proteins during the course of each reaction cycle, with multiple cycles needed to fold a population of stringent, non-native proteins (Todd et al., 1994; Weissman et al., 1994; Ranson et al., 1995; Smith and Fisher, 1995; Taguchi and Yoshida, 1995). Additional observations demonstrated that non-native forms emerge as GroES is discharged and the cis chamber is opened (Weissman et al., 1995; Burston et al., 1996). These and other results (Martin et al., 1993; Mayhew et al., 1996; Weissman et al., 1996; Ranson et al., 1997; Sparrer and Buchner, 1997; Rye et al., 1999) have lead to the idea that a normal GroEL reaction is a highly dynamic and repeating cycle of protein binding, encapsulation and release. In essence, a substrate protein has a short window of opportunity to fold with each round of capture and confinement during the course of the GroEL reaction cycle. If a non-native intermediate follows a productive contour of the folding landscape then folding completes. Those that do not make it must either aggregate or return to the chaperonin for another round of capture and confinement inside the GroEL-GroES cavity. In other words, a population of protein molecules upon which GroEL operates is subjected to cycles of kinetic partitioning (Todd et al., 1994; Weissman et al., 1994; Burston et al., 1996). Proteins that have reached a point of commitment to the native state can proceed on to their native structures and their prescribed cellular roles. Molecules that fall into kinetic traps are either rebound by another GroEL molecule or are captured by other components of the cellular chaperone network, including protease components that can degrade damaged proteins (Schroder et al., 1993; Buchberger et al., 1996; Horwich et al., 1999; Wickner et al., 1999; Hohfeld et al., 2001; Dougan et al., 2002). Full release of the cis complex with each turn of the GroEL cycle, in combination with kinetic partitioning, effectively explains why, in a cell, GroEL does not appear to get clogged with irretrievably damaged proteins.
The demonstration that non-native proteins are ejected from the GroEL-GroES cage with each turn of the chaperonin cycle at first seems at odds with a passive, Anfinsen cage model for GroEL-mediated folding. However, the highly dynamic GroEL cycle need not directly alter the conformational properties of a captured substrate protein in order to facilitate productive folding. While the overall process is actively driven by ATP hydrolysis, it could still qualify as a passive mechanism, if (1) folding is limited only by the aggregation of otherwise productive folding intermediates and (2) the free energy of ATP hydrolysis is used only to assemble and disassemble an isolation chamber. Provided that the rate of GroEL binding is significantly faster than the rate of aggregation, and that conformations with easy access to the native state are formed at a rate (the committed rate) that is at least comparable to the folding cavity lifetime, such a mechanism is theoretically capable of driving productive folding by repeated cycles of capture, isolation and release.
Active Models of GroEL-Mediated Folding
While the Anfinsen cage model is simple, general and consistent with the GroEL-GroES structure, it does not explain several theoretical and experimental observations. The demonstration of non-native substrate release and kinetic partitioning suggests that many GroEL-dependent substrate proteins complete a substantial fraction of their folding in free solution, outside the enclosed cage. Indeed, the short lifetime of the GroEL-GroES cavity (~ 8–10 sec at 25°C) provides precious little time for the infinitely dilute folding of stringent proteins that have intrinsic folding half-times of several minutes. In addition, recent experiments with RuBisCO and MDH suggest that GroEL can partially rescue the folding of these highly dependent substrate proteins in the complete absence of encapsulation by GroES, employing cycles of protein binding and release from the GroEL-GroES complex trans ring (Farr et al., 2003; see discussion below). While these observations are not necessarily inconsistent with a passive model of GroEL action, the strongest formulation of the passive Anfinsen cage model assumes that the folding intermediates bound by GroEL are inherently productive. In essence, purely passive models ignore the possibility that dominant kinetic traps could be misfolded states that have little or no possibility of accessing the native state.
The active modification of protein structure by GroEL provides an alternate strategy for facilitating protein folding, one that potentially addresses the limitations of purely passive models. How might GroEL-induced changes in substrate protein structure lead to enhanced folding? If a folding intermediate were to accumulate a sufficiently large number of stable, non-native interactions, the thermal fluctuations that normally help drive folding forward might not be adequate to rearrange the misfolded structure and permit folding to continue. A key action provided by GroEL, then, might be the transient injection of additional energy into a protein folding reaction through the unfolding of non-native intermediate states. Alternately, GroEL might bypass kinetic traps altogether by changing the physical constraints of the system (non-native protein plus surrounding solvent) so that the folding landscape itself is modified. In other words, the GroEL-GroES machine could be designed so that, while a substrate protein is associated with the chaperonin, the local free energy minima in the protein’s energy landscape are reduced, disallowed, or avoided, resulting in either faster or more efficient folding.
Substrate Protein Unfolding by GroEL
The idea that molecular chaperones might rearrange the structure of their substrate proteins dates to some of the earliest speculations on the mechanisms of molecular chaperone action (Pelham, 1986; Rothman, 1989; Hubbard and Sander, 1991). Whether or not substrate unfolding by GroEL serves an important role in driving folding, however, rests with three fundamental questions. First, can GroEL induce sig-nificant unfolding in non-native substrate proteins? If so, to what extent does unfolding occur? Finally, is unfolding necessary for stimulating productive folding or is it simply a byproduct of stable, non-native protein binding to the hydrophobic GroEL ring?
The evidence that GroEL can, in fact, unfold substrate proteins is substantial. Two general mechanisms, one thermodynamic and one catalytic, have been suggested to explain how GroEL could induce unfolding of substrate proteins (Figure 5; for additional reviews see Fenton and Horwich, 1997; Fenton and Horwich, 2003). In the thermodynamic partitioning model (Zahn et al., 1994a; Zahn and Pluckthun, 1994), GroEL preferentially binds less folded conformers within an ensemble of non-native states and thereby shifts a pre-existing, intrinsic equilibrium toward less folded states without altering the rate of unfolding. The kinetic partitioning model (Itzhaki et al., 1995), by contrast, posits that GroEL catalytically drives unfolding by lowering the free energy barriers that separate different folded states from one another, actively accelerating the rate of protein unfolding.
Figure 5.

Possible mechanisms of GroEL-induced protein unfolding. (A) Protein unfolding accomplished by thermodynamic coupling. A kinetically trapped folding intermediate that cannot bind to GroEL is in equilibrium with a less folded state that can bind to GroEL. In the presence of GroEL, mass action pulls the entire protein population into a GroEL-bound, unfolded state without affecting the intrinsic rate of unfolding. (B) Protein unfolding accomplished by catalytic unfolding. A kinetically trapped folding intermediate does not readily spontaneously unfold, but does bind to GroEL. The interaction of the folding intermediate with the hydrophobic GroEL ring results in a significant increase in the rate of unfolding. (C) Protein unfolding caused by forced unfolding. A kinetically trapped protein first binds to an open GroEL ring. Upon binding of ATP and GroES, the GroEL apical domains rearrange, resulting in significant stretching and unfolding of the bound substrate protein.
Evidence for Thermodynamically Coupled Unfolding
Unfolding a protein through the thermodynamic capture of poorly folded conformations is a simple method of exploiting the law of mass-action (Figure 5). Using such a mechanism, GroEL should be capable of unfolding any protein that populates, even transiently, non-native states that can be recognized and bound. Early studies with both DHFR (Viitanen et al., 1991) and pre-β-lactamase (Laminet et al., 1990; Zahn et al., 1994a) suggested that GroEL could indeed unfold proteins by capturing non-native conformers. Both DHFR, in the absence of its substrates, and unprocessed pre-β-lactamase are folded, but are not fully stable. When GroEL was added to these folded proteins, both were found to bind to GroEL in a non-native, less folded state. Thermally destabilized mature β-lactamase was found to behave in a similar fashion (Zahn and Pluckthun, 1994). A subsequent study by Schmid and colleagues put the thermodynamic partitioning model to a direct test, by asking whether GroEL could alter the rate of protein unfolding (Walter et al., 1996). These investigators employed a version RNAse T1 whose Cys residues were chemically blocked with carbamidomethyl groups, preventing the formation of key native state disulfide bonds. Without its disulfide bonds, RNAse T1 cannot fold to a fully native state and forms a metastable intermediate that does not bind to GroEL. The metastable intermediate is in rapid equilibrium with less folded states, however, and the rate that the metastable intermediate unfolds could be monitored by fluorescence spectroscopy upon shifts in the ionic strength of the solution or upon the addition of GroEL. As with DHFR and pre-β-lactamase, addition of GroEL resulted in the unfolding of the RNAse T1 intermediate, with the unfolded protein fully bound to GroEL. However, the rate of RNAse T1 unfolding was unaffected by the presence of GroEL. These observations demonstrated that unfolding of proteins like RNAse T1 by GroEL can be explained by thermodynamic coupling, with GroEL simply exploiting a facile, intrinsic equilibrium between more and less folded states of the protein (Walter et al., 1996). The GroEL-enhanced folding of two other small, non-stringent proteins, barstar and thioredoxin, was also shown to be consistent with simple thermodynamic coupling (Bhutani and Udgaonkar, 2000; Bhutani and Udgaonkar, 2001).
Evidence for Catalytic Unfolding
In contrast to thermodynamic partitioning, a catalytic unfolding mechanism requires that GroEL increase the rate of protein unfolding (Figure 5). Several lines of evidence suggest that GroEL can catalyze the disruption of protein structure, though the extent to which this catalytic effect is directed toward quaternary, tertiary or secondary structure (or some combination of all three) is not clear. In one early study, unfolded MDH was shown to populate a kinetically trapped intermediate state that could not fold spontaneously and only slowly formed irreversible aggregates (Peralta et al., 1994). Upon the addition of GroEL, GroES and ATP, the kinetically trapped MDH intermediate state was rapidly converted to native enzyme. Subsequent studies by Clarke and colleagues suggested that the kinetically trapped MDH intermediate state was a low-order, reversible aggregate (Ranson et al., 1995). The low-order MDH aggregate was formed rapidly and efficiently, essentially depopulating the productive folding pathway. Consistent with earlier observations, the low-order MDH aggregate only slowly converted to an irretrievable, high-order aggregate. Kinetic analysis of MDH folding in the presence of GroEL suggested that GroEL was capable of catalytically disrupting the low-order aggregate structure, essentially pumping the productive folding pathway with monomeric MDH subunits (Ranson et al., 1995). These observations, therefore, suggested that GroEL can accelerate productive protein folding by catalyzing the disruption of inhibitory protein quaternary structure.
Monomeric states held together by non-native tertiary or secondary structure are also likely to block productive folding. In this case, the disruption of a misfolded monomer would be the essential first step in facilitating productive folding. Several studies have suggested that GroEL can catalytically disrupt the tertiary and/or secondary structure of captured proteins. Fersht and colleagues have shown that GroEL can induce extensive unfolding in a small RNAse from B. amyloliquefaciens known as barnase (Zahn et al., 1996a; Zahn et al., 1996b). While barnase does not form a stable complex with GroEL, its spontaneous folding is dramatically slowed in the presence of GroEL as a result of transient interactions between non-native states of the barnase protein and open GroEL rings (Gray and Fersht, 1993; Corrales and Fersht, 1995). Remarkably, in the presence of GroEL, the exchange rate of the barnase backbone amide protons was found to increase significantly 4- to 25-fold. This enhancement in proton exchange rates was most dramatic for highly buried segments of the barnase polypeptide chain, suggesting that the interactions between barnase and GroEL results in transient, catalyzed global unfolding of the protein (Zahn et al., 1996a; Zahn et al., 1996b). GroEL was suggested to increase the overall rate of barnase unfolding by several orders of magnitude. While highly suggestive, the exact significance of complete (or near complete) substrate unfolding observed in these studies remained unclear, given that GroEL-driven unfolding is not required for barnase folding.
A direct connection between stimulated folding and the disruption of intra-molecular structure in a non-native and stringent protein has been more difficult to establish. Recently, we demonstrated that the GroEL-mediated folding of the stringent substrate protein RuBisCO involves the unfolding of a kinetically trapped, misfolded monomer (Lin and Rye, 2004). RuBisCO from Rhodospirillum ruburm was first shown to be a highly dependent GroEL substrate protein by Lorimer and colleagues (Goloubinoff et al., 1989a; Goloubinoff et al., 1989b). Non-native RuBisCO forms a metastable intermediate state that, like MDH, can be rescued by GroEL, GroES and ATP and that only forms irreversible, high-order aggregates at a slow rate (Goloubinoff et al., 1989a). Additionally, at low temperatures and high ionic strength (particularly in the presence of high Cl− ion concentrations), RuBisCO can fold spontaneously, albeit slowly and inefficiently (Viitanen et al., 1990; van der Vies et al., 1992). We recently demonstrated that at very low protein concentrations, low ionic strength, and low temperature, the RuBisCO metastable intermediate is not a low order aggregate, but is in fact a non-native, misfolded monomer (Lin and Rye, 2004). Using small fluorescent probes, fluorescence resonance energy transfer (FRET), and fluorescence anisotropy decay, we were able to establish that the kinetically trapped monomer is ensnared in a non-native conformation (or ensemble of conformations).
When the kinetically trapped RuBisCO monomer was mixed with GroEL, GroES and ATP, the enzyme was found to efficiently refold. Remarkably, upon binding to a GroEL ring, the average distance between the ends of the kinetically trapped RuBisCO monomer rapidly expanded (Lin and Rye, 2004). This observation strongly suggested that the RuBisCO monomer is unfolded or stretched apart upon binding to a GroEL ring. Overall, the total, through-space expansion between the ends of the RuBisCO monomer was extensive, increasing by 20 to 40 Å from the relative distance between these positions in the kinetically trapped intermediate. The observation that the initial unfolding rate of the RuBisCO monomer was dependent upon the GroEL concentration in a classically bimolecular fashion is consistent with a kinetic partitioning model for GroEL-driven unfolding of RuBisCO. However, because the intrinsic rate of the RuBisCO expansion in the absence of GroEL could not be measured, such a mechanism could not be unequivocally assigned.
How can simple binding of a non-native protein to a GroEL ring result in catalyzed unfolding? One possibility is suggested by the demonstration that binding of proteins like RuBisCO and MDH to GroEL requires multiple contacts between the non-native protein and the multi-valent GroEL ring (Farr et al., 2000). The extremely high local concentration of hydrophobic binding sites around an open GroEL ring might stabilize less folded, high-energy conformational transition states of a non-native protein. Subsequent on-ring rearrangements of a folding intermediate (e.g., Lin and Rye, 2004) would then result in highly stable and simultaneous binding of several regions of a folding intermediate to different segments of a GroEL ring. The overall effect might be similar to surface-dependent protein denaturation (Sharp et al., 2002; Swain and Gierasch, 2005).
Forced Unfolding
An alternate mechanism for unfolding was proposed by Lorimer and colleagues who suggested that substrate protein unfolding could be directly linked to the ATP-driven structural rearrangements of the GroEL ring itself (Figure 5; Shtilerman et al., 1999). Termed “forced unfolding,” this model suggested that the ATP-driven elevation and rotation of the GroEL apical domains that are necessary for GroES binding might apply a mechanical strain to a non-native protein bound across multiple GroEL apical domains, resulting in protein unfolding. In fact, the application of modest pulling forces using atomic force microscopes and laser tweezers has been shown to be sufficient to cause dramatic unfolding in single protein molecules (e.g., Rief et al., 1997; Tskhovrebova et al., 1997; Cecconi et al., 2005). Hydrogen-tritium exchange experiments with RuBisCO and GroEL suggested that a partially stable core of structure within a GroEL-bound RuBisCO intermediate (detected by a set of well protected backbone amide tritiums) was rapidly and completely disrupted upon the addition of GroES and ATP (Shtilerman et al., 1999). These observations were highly consistent with forced unfolding, as were recent molecular dynamics simulations (van der Vaart et al., 2004) and earlier observations of rapid fluorescence anisotropy shifts of the RuBisCO Trp residues upon encapsulation beneath GroES (Rye et al., 1997).
Experiments from Horwich and colleagues, however, have called the forced unfolding model into question (Chen et al., 2001; Park et al., 2005). In a set of experiments employing pulsed hydrogen-deuterium exchange, rapid on-column protease fragmentation and mass spectrometry, these investigators found that an MDH folding intermediate bound to a GroEL ring possessed a core of low stability, but detectable, structure (Chen et al., 2001). In these experiments, the non-native MDH was bound to a single ring version of GroEL known as SR1. SR1 was created by introducing a set of mutations into the GroEL equatorial domain that prevent the two GroEL rings from interacting with one another (Weissman et al., 1995; Weissman et al., 1996). SR1 thus exists as a single, seven subunit ring that possess normal ATP binding, hydrolysis and GroES binding activities. However, because the trans ring signals that drive cis cavity disassembly are missing, SR1 can only complete a single round of substrate binding and encapsulation, essentially a half-cycle of a normal GroEL reaction. Remarkably, SR1 is fully capable of driving productive folding of stringent substrate proteins (Weissman et al., 1996; Rye et al., 1997). Because SR1 is composed of a single GroEL ring, when it binds non-native substrate, ATP and GroES, all of the substrate protein is encapsulated within an SR1-GroES chamber that is essentially a cis ring complex. However, a similar addition of non-native substrate, GroES and ATP to wild-type, double-ring GroEL results in a mixture, with substrate protein both encapsulated within the cis cavity and bound to the trans ring (Weissman et al., 1995). Such a mixture of bound states could seriously complicate the interpretation of any observed changes in amide protection, and therefore compromise any conclusions about how structural changes are induced in the substrate protein. By using SR1 to force the population of only cis complexes, this problem can be avoided and only changes that result from substrate encapsulation can be observed.
Upon the addition of GroES and ATP to the SR1-MDH binary complex, the distribution and magnitude of backbone amide protections of the partially structured MDH intermediate changed only slightly (Chen et al., 2001). While a set of less well protected positions did appear to become rapidly less protected upon GroES and ATP binding, the most highly protected structural elements of the non-native MDH intermediate appeared to be essentially unaffected by the movements of the SR1 apical domains and GroES encapsulation. In a more recent set of experiments, Horwich and colleagues reexamined the RuBisCO-GroEL complex, again using hydrogen-tritium exchange (Park et al., 2005). Strikingly, they were unable to detect any significant protection of the tritium-labeled backbone amides in the RuBisCO intermediate, suggesting that GroEL-bound RuBisCO does not, in fact, contain any structure of sufficient stability to provide significant amide protection. This result precluded a re-test of the forced unfolding model with RuBisCO, as no structural probes were found to exist whose forced unfolding could be observed. Additionally, our FRET experiments with RuBisCO detected no evidence for forced unfolding upon GroES and ATP binding to a RuBisCO-SR1 binary complex (Lin and Rye, 2004). Based upon these studies, then, there is currently no good experimental evidence that ATP-driven structural changes in the GroEL ring drive extensive unfolding of substrate proteins. However, encapsulation of substrate proteins within the GroEL-GroES cavity clearly can induce structural changes in substrate proteins prior to the commencement of folding (e.g., Rye et al., 1997; Lin and Rye, 2004). While these changes could simply reflect the process of protein release into the cis cavity, some level of forced conformational change in substrate proteins upon ATP and GroES binding remains a possibility.
In summary, GroEL appears capable of causing substrate protein unfolding. Data supporting both thermodynamically and kinetically coupled mechanisms of protein unfolding have accumulated, suggesting that the exact mechanism employed depends substantially on the energetic and dynamic properties of the substrate protein itself. Indeed, thermodynamic and kinetic unfolding mechanisms are not mutually exclusive. For example, with a protein that readily populates non-native states well matched to the geometric and energetic constraints of a GroEL ring, thermodynamic partitioning is likely sufficient and further unfolding upon binding to GroEL would not be necessary, but might still occur. For other substrate proteins, the equilibrium between more and less folded states might be less facile, and overcoming critical free energy barriers that prevent productive folding could require catalytic unfolding by GroEL.
On the Magnitude of Substrate Protein Unfolding by GroEL
How extensive is the structural disruption induced by GroEL in the substrate proteins that it captures? Some evidence suggests that GroEL can cause substantial unfolding in a captured protein, completely destabilizing both tertiary and secondary contacts. As noted above, hydrogen-deuterium exchange studies on barnase suggested that GroEL could catalyze the global unfolding of this small protein (Gray and Fersht, 1993; Corrales and Fersht, 1995; Zahn et al., 1996b). An earlier study with another small protein, cyclophilin, reached a similar conclusion (Zahn et al., 1994b). While cyclophilin does not depend upon GroEL for folding, thermal destabilization of cyclophilin leads to stable GroEL binding. The pattern and kinetics of hydrogen-deuterium exchange for cyclophilin bound to GroEL were examined by NMR and suggested that GroEL binding caused extensive destabilization of cyclophilin’s secondary structure. Complementary results were obtained in yet another study, in which GroEL was found to induce extensive unfolding in rhodanese (Reid and Flynn, 1996). In this case, a folded and stable domain of rhodanese was generated by in vitro translation using an mRNA without a stop codon. The rhodanese remained associated with the stalled ribosome and the amino terminal 17 kDa domain was found to fold to a stable and protease resistant conformation. When the rhodanese was released from the ribosome with puromycin, the protein was found to efficiently bind to GroEL and the previously well-folded amino terminal domain was observed to be highly sensitive to protease, suggesting that rhodanese binding to GroEL caused the amino terminal domain to unfold (Reid and Flynn, 1996).
By contrast, GroEL does not appear to cause extensive unfolding in other proteins. Thermally unfolded β-lactamase binds to GroEL, but appears to retain a significant level of native-like secondary structure, based upon high-sensitivity hydrogen-deuterium exchange and mass spectrometry experiments (Gervasoni et al., 1996). Similar experiments with non-native, disulfide scrambled α-lactalbumin (Robinson et al., 1994) and with MDH (Chen et al., 2001) found significant, if unstable, residual structure in the GroEL-bound states of these proteins. While the observed protection factors were low (between 2 and 100), the methods employed were sensitive enough to detect some low stability, native-like structure in the GroEL-bound proteins. Several detailed studies of DHFR bound to GroEL have reached similar conclusions. As noted above, GroEL can drive DHFR unfolding by binding and stabilizing non-native states of the enzyme in the absence of its substrates (Viitanen et al., 1991). GroEL was subsequently found to bind a wide range of non-native conformations of DHFR, at both early and late stages of folding (Goldberg et al., 1997). A detailed study using hydrogen-deuterium exchange and NMR found that DHFR retains low, but significant, protection of backbone amides in the central β-sheet of the protein core while bound to GroEL (Goldberg et al., 1997). These results suggested that at least part of the DHFR core possess a native-like topology with some correct secondary structure, similar to the results obtained with MDH (Chen et al., 2001). Again, the low protection factors observed (5 to 50) suggested that the residual DHFR structure was unstable and probably only rarely sampled in the GroEL-bound state (Goldberg et al., 1997). Another hydrogen-deuterium exchange study of DHFR bound to GroEL by mass spectroscopy reached a similar conclusion, although higher protection factors (~1000) were observed for a core of approximately 20 residues (Gross et al., 1996). Finally, a very recent, state-of-the-art NMR study has examined the structure of DHFR bound to SR1 with a set of novel relaxation methods that permit direct examination of the conformational state of DHFR while bound to a GroEL ring (Horst et al., 2005). Strikingly, little well organized structure is observed in SR1-bound DHFR. In fact, the non-native DHFR appears to be highly dynamic, exchanging between an apparently random collection of mostly unfolded conformers while bound to the SR1 ring.
In the end, the real target of GroEL’s unfolding activity might be non-native tertiary contacts, rather than the detailed local structure of substrate proteins. In some cases, long-range and non-specific prying apart of tertiary contacts could result in collateral global unfolding, while in other cases, nascent secondary structure might remain mostly undisturbed. This conjecture is consistent with several observations, including studies on the folding of lysozyme in the presence of GroEL (Coyle et al., 1999). While lysozyme does not require GroEL for folding, it does possess a well characterized folding pathway that involves two general routes to the native state, one of which requires a rate-limiting disruption of non-native tertiary contacts. Using both fluorescence spectroscopy and hydrogen-deuterium exchange, GroEL was shown to stimulate the folding of lysozyme to a small (~2-fold) but significant extent by accelerating the disruption of the inhibitory tertiary contacts between the two sub-domains of a lysozyme folding intermediate (Coyle et al., 1999). At the same time, the nascent secondary structure present in the sub-domains appeared to remain intact. Finally, our observations on the long-range unfolding of a kinetically trapped RuBisCO monomer by GroEL are also consistent with this idea (Lin and Rye, 2004). GroEL was observed to directly disrupt the structure of a mis-folded RuBisCO folding intermediate by prying apart its large-scale structure. More recent measurements with additional FRET pairs suggest that this expansion takes place along multiple axes within the non-native RuBisCO structure (Lin, unpublished observations). While our experiments with RuBisCO could not access the consequences of large-range structural expansion on the detailed secondary and tertiary structure of the RuBisCO monomer, recent hydrogen-deuterium experiments suggest that the GroEL-bound RuBisCO monomer possesses no stable, well-organized structure (Park et al., 2005). Finally, the observation of a structural expansion in non-native carbonic anhydrase II bound to GroEL is also consistent with GroEL’s prying apart of long-range protein structure (Persson et al., 1999; Hammarstrom et al., 2001).
Folding by Unfolding: Annealing
While substrate protein unfolding by GroEL does occur and can be extensive, ultimately the key question remains: is unfolding necessary for productive folding? Despite experimental observations suggesting that unfolding may be important for some substrate proteins, it nonetheless seems fair to ask: what can protein unfolding possibly achieve, other than the opportunity to get stuck again? As noted above, the extremely rugged folding landscape of some proteins dramatically reduces the chance that these proteins can spontaneously locate their native state. There is, however, a relatively general method for locating the global minimum of such highly frustrated systems, at least in principle. Known as annealing, this process involves stimulating the acquisition of a global energetic minimum, in both physical (e.g., crystallization or metal cooling) and computational systems, by first increasing the internal energy of the system, followed by slow cooling (Kirkpatrick et al., 1983; Kirkpatrick, 1984; Todd et al., 1996). This process, often applied repetitively, can greatly reduce the overall frustration of the system by destabilizing local energetic minima relative to a global minimum. For a protein-folding reaction, increasing the effective internal energy would most simply involve unfolding.
Based upon observations on the unfolding of barnase, Fersht and colleagues suggested that GroEL might function as an annealing machine (Corrales and Fersht, 1996; Zahn et al., 1996a). GroEL was observed to not only catalyze the unfolding of native barnase to a less folded intermediate state, but to also suppress the rate of barnase unfolding to a fully unfolded state. At the same time, GroEL dramatically reduced the rate that the fully unfolded state collapsed back to the intermediate state. From these results, it was proposed that the interaction between the hydrophobic walls of the GroEL cavity and barnase resulted in transient global unfolding through a continuous annealing process. A repeated process of unfolding, rearrangement, and re-binding was imagined to continue until the native state was obtained (Corrales and Fersht, 1996; Zahn et al., 1996a).
Folding by continuous annealing should result in the progressive maturation of a non-native protein population, with the average conformational properties of the non-native molecules moving closer and closer to the native state. By contrast, GroEL-driven folding appears to occur in an all-or-nothing fashion. The experimental observation that GroEL-dependent proteins fold through cycles of non-native protein ejection and kinetic partitioning (Todd et al., 1994; Weissman et al., 1994; Ranson et al., 1995; Smith and Fisher, 1995; Taguchi and Yoshida, 1995) strongly suggests that most folding takes place either in the cis cavity or in free solution, so the opportunity for continuous annealing at the surface of GroEL appears to be quite limited (although some continuous annealing might take place in the cis cavity—see below). In addition, non-native proteins that bind to GroEL, even after repeated cycles of binding and release, appear to return to a GroEL bound state in about the same conformation in which they were originally captured. For example, the pattern of protease protection for GroEL-bound rhodanese is the same at early and late times in a folding reaction, suggesting that non-native rhodanese intermediates that do not commit to the native state are always bound by GroEL in roughly the same loosely folded state (Weissman et al., 1994). Another study on the structure of GroEL-bound DHFR reached a similar conclusion (Gross et al., 1996). Using hydrogen-deuterium exchange and mass spectrometry, these investigators examined the amount of low-stability structure in GroEL-bound DHFR, both before and several minutes after the addition of ATP. The extent and kinetics of hydrogen-deuterium exchange in each case was the same. This observation suggested that the gross structure of DHFR molecules that returned to a GroEL-bound state following a non-productive attempt at folding was always about the same. Thus, no evidence currently exists for the extensive and steady conformational progression predicted by continuous annealing. However, it remains possible that progressive conformational changes in a substrate protein may, in fact, take place on a small scale. In the same studies on GroEL-bound DHFR, significant differences before and after the addition of ATP were observed in both iodide quenching of internal Trp fluorescence and in the binding of the hydrophobic dye ANS to the GroEL-bound, non-native DHFR (Gross et al., 1996). These results suggest that some level of small-scale or localized progressive change can occur in a GroEL-bound protein, even though each time a protein is captured by GroEL it appears to return to roughly the same poorly structured, non-native state.
Thirumalai, Lorimer, and colleagues developed a more extensive annealing theory for GroEL, incorporating an explicit role for the experimental observation of cyclical binding and release of non-native proteins by GroEL (Todd et al., 1996; Thirumalai and Lorimer, 2001). Termed “iterative annealing,” this theory also assumes that substrate unfolding is a central feature of chaperonin action, and that it is required to reverse misfolded, kinetically trapped states (Figure 6). Upon release from GroEL into free solution, the unfolded protein is provided another chance to fold from a higher energy point on its folding landscape. Because of the extreme frustration in the folding landscape of GroEL-dependent proteins, the vast majority of these unfolding and release events fail to generate native protein. However, even if the probability of a successful run through the energy landscape to the global minimum is small on any given attempt, simple repetition could eventually push an entire population into the global minimum. In essence, the extensive frustration of a GroEL-dependent substrate protein is bypassed through repetition of a simple, if inefficient, process of unfolding and release. The iterative annealing model is similar to an earlier model proposed by Wolynes and colleagues, which postulated that molecular chaperones facilitate productive folding via kinetic proofreading (Gulukota and Wolynes, 1994). However, the iterative annealing model incorporates an explicit unfolding step as a key feature in stimulated folding by GroEL (Todd et al., 1996; Thirumalai and Lorimer, 2001). Based upon theoretical studies with computational 2-D lattice models, Dill and colleagues made a similar suggestion (Chan and Dill, 1996).
Figure 6.

Stimulation of protein folding by repetitive unfolding and release: iterative annealing. A kinetically trapped protein is located in a deep energetic well that it cannot spontaneously escape (a). Upon binding to GroEL, the structure of the trapped intermediate is unfolded, inducing a displacement of the protein up the energy gradient of its folding landscape (b). Upon release from GroEL, the protein is afforded another chance to fold from a higher point on its energy landscape. Most of the protein falls back into the kinetic trap, though a fraction follows a productive route to the native state (c). GroEL rebinds the trapped protein and repeats the process until most of the population partitions to the native state.
Stimulated Folding Inside the cis Cavity
While iterative annealing and related models are consistent with several key experimental observations, this family of models does possess one weakness. In these models, repetitive binding, unfolding and release are imagined to be the central and essential assistance that GroEL provides to a recalcitrant protein. Iterative annealing thus cannot easily explain how a single round of protein encapsulation within the GroEL-GroES cavity is sufficient for full and efficient folding. This experiment was first conducted using the single ring variant of GroEL, SR1 (Weissman et al., 1996). Because the cycle timer is essentially stuck in SR1, the substrate protein remains confined inside an SR1-GroES cis cavity following a single round of binding-induced unfolding. Remarkably, several stringent GroEL substrates not only initiate folding inside the SR1-GroES cavity, but also proceed all the way to fully committed states (Hayer-Hartl et al., 1996; Weissman et al., 1996; Rye et al., 1997; Chen et al., 2001). By contrast, only the smallest fraction (1% to 5%) of the same proteins manage to fold in free solution upon ejection from a wild-type GroEL ring following a normal encapsulation lifetime of 6 to 10 sec. Thus, if repetitive unfolding was obligatory for the folding of these proteins, complete folding within the SR1-GroES cavity should not work.
Confinement of a protein folding intermediate within the GroEL-GroES cavity, however, appears to directly influence how certain proteins fold. In one recent study, Hartl and colleagues developed a strategy for examining the folding of substrate proteins upon their release into free solution during a GroEL reaction cycle (Brinker et al., 2001). Normally, non-native proteins not committed to their native state rapidly rebind to GroEL, making an examination of their behavior immediately after release from the chaperonin difficult. To prevent this, Hartl and colleagues designed a GroEL variant carrying an apical domain Cys mutation that could be chemically biotinylated. While this modification had no apparent effect on the ability of the altered GroEL to drive protein folding, the addition of streptavidin resulted in very rapid occlusion of the GroEL cavity upon biotin binding by streptavidin, which completely blocked non-native substrate protein binding to the modified GroEL. Under non-permissive conditions, where aggregation of RuBisCO and rhodanese are significant, the addition of streptavidin to a folding reaction containing the modified GroEL resulted in an inhibition of folding for both proteins. The inhibition of folding for RuBisCO was particularly rapid and complete, suggesting that the vast majority of non-native RuBisCO released from GroEL under these conditions was incapable of folding to a committed state in free solution before succumbing to aggregation.
Hartl and colleagues (2001) next examined the folding of RuBisCO and rhodanese upon release from GroEL under permissive conditions, where aggregation does not prevent folding (Brinker et al., 2001). By shifting to much lower proteins concentrations, and in the case of RuBisCO, lower temperature and high ionic strength, the spontaneous folding rate of both proteins could be examined relative to the GroEL-mediated rate under the same conditions. For rhodanese, the presence of GroEL, GroES, and ATP appeared to have little effect, suggesting that GroEL primarily assisted rhodanese folding under non-permissive conditions by preventing aggregation. Strikingly, however, the refolding of RuBisCO in the presence of GroEL, GroES, and ATP under permissive conditions was several times (3- to 4-fold) faster than spontaneous folding. Upon the addition of streptavidin to a folding reaction, the rate of RuBisCO folding was found to shift immediately from the enhanced rate to the slower, spontaneous rate. These observations suggested that confinement of the RuBisCO intermediate within the GroEL-GroES cavity was responsible for the acceleration in folding. This conclusion was supported by experiments conducted under the same conditions with the single ring GroEL variant SR1, where similar folding rate enhancements were observed when the RuBisCO monomer was confined within the SR1-GroES cavity (Brinker et al., 2001).
Energy Landscape Smoothing
What properties of the enclosed GroEL-GroES cavity could account for the experimental observation of accelerated folding? Notably, the GroEL-GroES cavity defines a restricted space of finite volume. While the dimensions of the GroEL cavity expand by approximately 2-fold upon the binding of GroES and ATP (from ~85,000 Å3 to ~175,000 Å3; Xu et al., 1997), the distance between the surface of a non-native, expanded protein folding intermediate and the cavity wall would most likely accommodate no more than a thin hydration layer around the exterior of the substrate protein. Indeed, even for native and well folded proteins of modest size like GFP, confinement within the GroEL-GroES cavity appears to impose serious constraints on the rotational freedom of the protein. Direct measurements on native, fluorescent GFP inside an enclosed SR1-GroES cavity found that the rotational correlation time of the protein was up to 4-fold slower inside the cavity than in free solution (Weissman et al., 1996). This suggests either significant, direct interactions between the native GFP protein and the walls of the GroEL cavity, or a considerable increase in the effective viscosity of the hydration layer between the GFP and SR1-GroES cavity wall. For a much larger substrate protein like RuBisCO, the extent of physical constriction is likely to be far more significant. Indeed, the average hydration layer between a marginally expanded and large folding intermediate and the cavity wall might be no thicker than a single water molecule (Thirumalai and Lorimer, 2001).
Confinement of a protein within a cavity might prevent formation of non-native conformations possessing radii of gyration larger than the radius of the enclosing cavity. If so, an enclosed protein should demonstrate a significantly higher stability compared to the same protein in free solution, because unfolded states with expanded volumes would be disallowed (Thirumalai et al., 2003). In support of this supposition, a range of computational studies (Zhou and Dill, 2001; Klimov et al., 2002; Takagi et al., 2003), as well as experimental observations on proteins confined within silica glass nanopores (Eggers and Valentine, 2001), have shown that spatial confinement of a protein can, in fact, lead to large increases in protein stability. More significantly, however, the spatial restriction of a non-native folding intermediate could have a profound affect on the kinetics and mechanism of folding. The experimental observation of accelerated RuBisCO folding inside the GroEL-GroES cavity suggests that the folding landscape of a protein can be fundamentally altered by encapsulation (Brinker et al., 2001). Several computational studies, using both simple lattice and more detailed off-lattice models, have also provided support for this conclusion (Betancourt and Thirumalai, 1999; Gorse, 2001; Zhou and Dill, 2001; Klimov et al., 2002; Baumketner et al., 2003; Takagi et al., 2003; Jewett et al., 2004; Xu et al., 2005).
Because expanded, non-native conformational states are not compatible with the limited volume of the GroEL-GroES cavity, spatial confinement can be thought of as destabilizing expanded states relative to the more compact native state. As a result, the free energy barriers that promote the formation of well populated, expanded conformations are reduced, leading to a smoothing of the protein’s free energy landscape (Thirumalai et al., 2003). In addition, the total entropy of the most expanded, non-native ensembles should be significantly reduced by confinement (Klimov et al., 2002; Baumketner et al., 2003; Takagi et al., 2003). In essence, spatial restriction of a non-native protein should narrow a given protein’s folding funnel, physically excluding large regions of conformational space and confining the subsequent search for the native state to a smaller range of states. The effect of both smoothing and narrowing the folding funnel should, in theory, lead to accelerated folding (Figure 7). Interestingly, one recent computational analysis suggested that the effects of simple confinement should be most significant for proteins that demonstrate the greatest level of topological complexity or contact order (Takagi et al., 2003). Another recent study examined a theoretical α/β sandwich protein with a tendency to fold via a non-native, misfolded intermediate state (Baumketner et al., 2003). Under certain conditions, confinement was observed to enhance the folding of this protein (~2-fold) by preventing the formation of kinetically trapped, misfolded conformations possessing radii incompatible with the dimensions of an enclosing cavity. In essence, significant kinetic traps that would otherwise have been readily populated in bulk solvent were bypassed upon confinement (Baumketner et al., 2003). Our recent studies of RuBisCO folding inside an SR1-GroES cavity suggest a similar mechanism (Lin and Rye, 2004). As the SR1-bound RuBisCO intermediate was encapsulated beneath GroES, we observed a rapid compaction of the RuBisCO monomer that preceded the initiation of productive folding. More importantly, upon confinement within the SR1-GroES cavity, the RuBisCO monomer appeared to avoid the kinetically trapped and misfolded state observed in free solution, permitting the protein to follow an apparently different and productive route to a committed conformation.
Figure 7.

Confinement of a non-native folding intermediate within the GroEL-GroES cavity results in smoothing of the protein’s energy landscape. The energy landscape of a kinetically trapped protein (a) is modified when the protein is enclosed in the GroEL-GroES cavity (b). Confinement of the non-native protein results in: (1) narrowing of the protein’s folding funnel by reducing the total number of allowed polypeptide conformation, (2) destabilization of expanded, kinetically trapped states that possess large radii of gyration incompatible with the volume of the GroEL-GroES cavity, (3) reduction in the height of transition state energy barriers that separate kinetically trapped states from productive folding pathways. The overall result of spatial confinement is a smoothing of the energy landscape of the non-native protein, opening productive folding routes that were not previously available (c).
The surface character of the GroEL-GroES chamber has also been suggested to play an important role in editing the energy landscape of an encapsulated protein. In one recent study, a screen for GroEL and GroES mutants that enhance the folding of the non-stringent substrate protein GFP was conducted using a combination of directed evolution and combinatorial mutagenesis (Wang et al., 2002). While some of the modifications identified altered the GroEL ATPase cycle, other mutations appeared to affect the surface character of the GroEL-GroES cavity. Interestingly, modifications that enhanced the assisted folding of GFP were found to be deleterious for the GroEL-mediated folding of other substrate proteins. Similar observations on the influence of cavity surface character have also been made using computational methods (Chan and Dill, 1996; Betancourt and Thirumalai, 1999; Jewett et al., 2004; Xu et al., 2005). While the binding of GroES dramatically reduces the hydrophobic character of the GroEL apical surface, the resulting GroEL-GroES cavity is not an ideal hydrophilic chamber, and it retains some weak hydrophobic character (Xu et al., 1997; Betancourt and Thirumalai, 1999). Dynamic interactions between non-native folding intermediates and a weakly hydrophobic cavity wall have been suggested to accelerate folding by lowering the free energy barriers between different states (Chan and Dill, 1996; Betancourt and Thirumalai, 1999). This enhancement could be due to stabilization of more hydrophobic and less folded conformational transition states, providing an additional smoothing of the free energy landscape. Alternately, the weakly hydrophobic character of the GroEL-GroES cavity has been suggested to increase the ruggedness of a broad and slowly crossed transition state ensemble, opening folding routes that are not significantly populated in bulk solution (Jewett et al., 2004).
On the Role of Water
The mechanics and energetics of protein folding are deeply rooted in the chemical and physical properties of water. With its high dielectric constant, its capacity to serve as an excellent hydrogen bond donor and acceptor, and its remarkable bulk response to the presence of non-polar surfaces, water plays a key role in the specification of protein structure and stability (Edsall and McKenzie, 1978; Timasheff, 1993). Indeed, a cornerstone of our understanding of protein folding is the thermodynamic role played by the dehydration of hydrophobic amino acids as they cluster together to form the tightly packed, hydrophobic core of well folded, globular proteins (Kauzmann, 1959; Kuntz and Kauzmann, 1974; Baldwin, 1986; Privalov, 1992).
The ejection of water molecules from the core of an otherwise correctly organized protein structure could, in principle, be a late and critical step in protein folding. A range of fast events in protein folding, including hydrophobic collapse, nucleation, and secondary structure formation might proceed faster than water molecules can be fully ejected from internal surfaces of a collapsed protein (Cheung et al., 2002; Guo et al., 2003). The final acquisition of the native state of a protein might then require structural rearrangements that squeeze out the last of the water from the core. For example, NMR studies of the N-terminal SH3 domain from the D. melanogaster protein drk indicate that this proteins folds via a loosely packed but structured intermediate state that shows significant hydration of residues near the protein core (Zhang and Forman-Kay, 1997; Mok et al., 1999). Recent computational studies suggest that the non-native ensemble of this SH3 folding intermediate possesses a near-native structure with a partially hydrated core, and that the final step in folding of this protein involves the cooperative expulsion of the internal water molecules (Cheung et al., 2002; Guo et al., 2003).
Could GroEL act to facilitate the dehydration of non-native folding intermediates? By enforcing the formation of collapsed states with limited volumes, the GroEL-GroES cavity could help to drive the final dehydration of the protein core. The majority of proteins that require GroEL and GroES for productive folding are large enough (30 to 55 kDa) that their expanded, non-native intermediate states are likely to nearly fill the GroEL-GroES cavity, with little room for hydrating water molecules. By enforcing the formation of collapsed states incompatible with significant core (and even exterior) hydration, enclosure of a non-native protein within the GroEL-GroES cavity might specifically stabilize more internally dehydrated and compacted conformational states of the non-native protein.
Importantly, the large-scale unfolding or expansion of a substrate protein upon capture by a GroEL ring might, at least initially, result in an increased hydration of the interior of a bound protein. As noted above, a significant body of evidence suggests that GroEL can either stabilize or induce the formation of open, less folded conformations of captured proteins. The resulting “percolation” of water molecules through the internal structure of the folding intermediate has been suggested to significantly increase the internal flexibility of the protein core, releasing or destabilizing conformations of the protein that lead to kinetic trapping (Csermely, 1999; Kovacs et al., 2005). Thus, by transiently increasing the hydration of a protein core, GroEL could facilitate significant changes in the conformational state of a non-native protein, without necessarily causing extensive, global unfolding. Subsequent compaction and dehydration of the folding intermediate within the GroEL-GroES cavity could then provide a driving force that aids efficient ejection of the “conformation-erasing” water molecules from the protein core.
Folding from the trans Ring
Encapsulation of a folding intermediate within the GroEL-GroES cavity is a defining step in the facilitated protein folding reaction carried out by GroEL. However, this observation leaves a key question unresolved: can direct release of substrate proteins from an open GroEL ring result in productive protein folding? The answer to this question bears directly on what role, if any, GroEL plays in the folding of very large substrate proteins that cannot be enclosed within the GroEL-GroES cavity.
The open trans ring of the asymmetric GroEL-GroES complex could, in theory, bind proteins much larger than those that can be encapsulated beneath GroES. A wide range of polypeptides, including a number of large proteins (~60 kDa) were found to stably associate with GroEL when total E. coli lysate proteins were denatured and mixed with GroEL (Viitanen et al., 1992). More recent immunoprecipitation (Ewalt et al., 1997; Houry et al., 1999) and proteomics (Kerner et al., 2005) studies also identified several large proteins that persistently interact with GroEL in E. coli cytoplasm. However, it has remained unclear whether these proteins directly rely upon GroEL to drive their folding, employ GroEL to a limited extent to passively block aggregation or simply bind to GroEL for trivial and essentially non-functional reasons. Indeed, detailed studies of one large protein, phage P22 tailspike protein, have demonstrated that while GroEL can bind and release non-native states of this very large (~70 kDa) protein, these interactions with GroEL appear to have no direct effect on the folding of the tailspike protein (Brunschier et al., 1993; Gordon et al., 1994). Furthermore, release of tailspike folding intermediates from GroEL does not require GroES, as expected for a protein too large to be accommodated within the GroEL-GroES cavity.
The role of direct substrate protein release into solution without encapsulation has also been examined with a number of small substrate proteins. Several studies have demonstrated that ATP alone is sufficient to trigger release of some non-native proteins from GroEL and elicit their productive folding (e.g., Laminet et al., 1990; Badcoe et al., 1991; Martin et al., 1991; Viitanen et al., 1991; Fisher, 1992; Mizobata et al., 1992; Jackson et al., 1993; Corrales and Fersht, 1995). However, proteins that demonstrate GroEL-enhanced folding without GroES, in general, fold reasonably well on their own, while stringent substrate proteins require the presence of both GroES and ATP for significant folding under most conditions (Schmidt et al., 1994; Kerner et al., 2005). It was therefore assumed that a key characteristic of highly dependent GroEL substrates was a need for full encapsulation within the GroEL-GroES cavity. This conclusion was directly supported by a study on the GroEL-mediated folding of ornithine transcarbamylase (OTC; Weissman et al., 1995). Chemically denatured OTC was found to refold efficiently when encapsulated inside the GroEL-GroES cavity. However, when non-native OTC was first bound to the trans ring of a stable GroEL-ADP-GroES complex, the kinetics of OTC refolding upon the addition of ATP suggested that productive folding required the OTC monomers to transit through an enclosed GroEL-GroES complex at least once. Furthermore, in single turnover experiments employing SR1 as a GroES trap, OTC was found to fold and release productively from inside a GroEL-GroES cis complex but displayed virtually no folding when the same experiment was attempted with OTC bound to the trans ring. Finally, recent studies have suggested that the E. coli proteins that gain the most benefit from the action of GroEL and GroES in vivo, possess molecular weights less than ~55 kDa (Houry et al., 1999; Kerner et al., 2005). A requirement for substrate encapsulation within the limited volume of the GroEL-GroES cavity provides a straightforward explanation for these observations.
It was with some surprise then, that very large and GroEL-dependent substrate proteins were identified that appear to possess a requirement for GroES. One of the first hints that large substrate proteins might require GroEL and GroES for productive folding in vivo came from a study by Rospert and colleagues examining the import of proteins into isolated yeast mitochondria (Dubaquie et al., 1998). These investigators employed an in vitro assay in which radiolabled proteins were imported into mitochondria isolated from either wild-type yeast or yeast strains carrying temperature sensitive alleles of the mitochondrial GroEL and GroES homologs (Hsp60 and Hsp10). Upon import at the restrictive temperature, a number of proteins were identified that required functional versions of both Hsp60 and Hsp10 be present in the mitochondria for correct folding following their import. Remarkably, among the identified proteins was a very large (~83 kDa), iron-sulfur enzyme of the citric acid cycle, aconitase. Because aconitase was likely too large to be accommodated within the GroEL-GroES cavity, but nonetheless required GroES for correct folding, these observations suggested a possible direct and essential role for the open, GroEL-GroES complex trans ring in the assisted folding of large proteins. Another study, employing a large fusion protein of the maltose binding protein spliced onto part of a human mitochondrial alpha-ketoacid dehydrogenase reached a similar conclusion (Huang and Chuang, 1999).
The observations on aconitase folding in yeast mitochondria led Horwich and colleagues (2001) to revisit the question of productive folding of large substrate proteins from the GroEL trans ring (Chaudhuri et al., 2001). Expression of native and enzymatically active aconitase in E. coli was shown to depend upon the presence of both GroEL and GroES. Furthermore, direct examination of the folding of acid denatured aconitase demonstrated that GroEL, GroES, and ATP were required for refolding of the enzyme in vitro. As expected, non-native aconitase bound by GroEL was much too large to be accommodated beneath GroES based upon protease sensitivity experiments. Strikingly, however, this large, iron-sulfur protein appeared to gain a significant folding benefit through exclusive binding and release from the trans ring of a GroEL-GroES complex. In the absence of GroES, the immature aconitase folding intermediate did not complete folding and could not dissociate from GroEL, even in the presence of ATP. By contrast, the binding and release of aconitase from the trans ring of a GroEL-GroES complex appeared to be directly coupled to the efficient production of the apo state of aconitase. Remarkably, apo aconitase remained associated with the trans ring of a GroEL-GroES complex until insertion of the iron-sulfer cluster into the enzyme active site generated active, holo aconitase (Chaudhuri et al., 2001).
How could folding be stimulated by trans ring binding alone? In the absence of substrate encapsulation within the GroEL-GroES complex, many of the mechanisms outlined above clearly cannot be employed to drive productive folding. At least one GroEL action, however, is likely to still occur upon binding of a non-native protein to the GroEL-GroES complex trans ring: repetitive unfolding. For both cis-cavity and trans ring folding, a non-native protein must first bind to a multivalent GroEL ring (Farr et al., 2000). This binding action alone appears capable of inducing a significant level of structural disruption, expansion or stretching even in stringent substrate protein folding intermediates (Lin and Rye, 2004). This structural alteration, mediated either by global unfolding, prying apart of non-native tertiary structure or increased core hydration, could be sufficient to enhance productive folding through the destabilization of kinetically trapped conformations of a folding intermediate. In support of this conjecture, a recent study using a novel trans-only GroEL-GroES complex, has demonstrated that even the classically stringent GroEL substrates RuBisCO and MDH can fold productively as a result of simple binding and release from the trans ring (Farr et al., 2003). This surprising set of observations strongly suggests that the conformational modifications imposed by binding to an open GroEL ring are sufficient to open a productive folding route to at least a fraction of a non-native protein ensemble upon release into free solution.
Conformational changes in the trans ring could also impose additional structural alterations in a captured folding intermediate. The trans ring apical domains of the GroEL-ATP-GroES complex are known to move through a set of distinct structural alterations that are essential for the normal sequencing of the GroEL reaction cycle and ejection of substrate proteins (Rye et al., 1999; Ranson et al., 2006) Structural changes in a bound substrate protein induced by these movements could be important for triggering both folding and release of proteins from the trans ring (Chaudhuri et al., 2001; Farr et al., 2003). Indeed, a requirement for trans ring apical domain movements in the GroEL-ATP-GroES complex might explain why earlier trans ring experiments with OTC failed (Weissman et al., 1995). In this case, the non-native OTC was bound to the trans ring of a GroEL-ADP-GroES complex, but did not pass through the ejection step of a trans ring GroEL-ATP-GroES complex. Finally, it is worth noting that, while exclusive trans ring binding and release did result in the folding of MDH and RuBisCO, this mechanism was significantly less efficient (2- to 4-fold) than folding with normally cycling GroEL and GroES (Farr et al., 2003). These observations suggest that, while repetitive structural disruption upon binding to a GroEL ring is sufficient to provide significant assistance, fully efficient folding of stringent substrates like MDH and RuBisCO requires the confinement of their non-native intermediates within the enclosed GroEL-GroES cavity.
CONCLUSIONS AND PERSPECTIVES
Is there a general and unifying principle behind GroEL-mediated folding? A much more detailed understanding of the conformational dynamics of substrate proteins as they transit through the GroEL-GroES folding cycle is clearly needed to address this question fully. While the inherent complexity of non-native protein ensembles makes this a challenging problem, advances in protein folding theory, computational modeling, single molecule spectroscopies and large-molecule NMR techniques hold enormous promise for studying the folding of GroEL-dependent proteins in greater detail (Riek et al., 2000; Daggett and Fersht, 2003; Thirumalai et al., 2003; Zhuang and Rief, 2003; Gillespie and Plaxco, 2004; Haustein and Schwille, 2004; Horst et al., 2005; Vendruscolo and Dobson, 2005; Wolynes, 2005). Though it is possible that a single, specific mechanism will eventually be shown to underpin how GroEL drives stimulated folding for most or all of its substrate proteins, in the end, the potential actions outlined above for GroEL are not mutually exclusive. Maximally efficient folding of highly recalcitrant substrate proteins like RuBisCO may require a combination of both unfolding and confinement (Brinker et al., 2001; Lin and Rye, 2004). Other proteins, like rhodanese, may require little more than simple confinement and infinite dilution (Brinker et al., 2001). In the most general terms, GroEL could be designed to provide a range of stimulatory effects that are more or less effective, depending upon the particular size, energetics, topology, and folding kinetics of a given protein. An important challenge will be to determine which of the potential folding mechanisms is the most critical for which substrate proteins and why. GroEL may well have evolved to provide an overall optimum for stimulated folding of a range of proteins, without necessarily being capable of maximally efficient folding of any single protein. This suggestion is supported by observations on GroEL mutants optimized for folding GFP that demonstrate a dramatically reduced ability to fold other substrate proteins (Wang et al., 2002). The fact that large substrate proteins, which cannot fit inside the GroEL-GroES cavity, nonetheless gain a significant folding enhancement upon interacting with the GroEL trans ring, further underlines the mechanistic flexibility of this remarkable molecular machine.
Finally, molecular chaperones have been recognized in recent years to be essential for regulating many aspects of basic cellular biology, in addition to their roles in protein folding. In a general sense, molecular chaperones like GroEL can be considered broad-spectrum sensors and regulators of protein three-dimensional structure. Using essentially the same basic mechanisms of protein binding and release employed to assist protein folding, molecular chaperones are involved in processes as varied as protein translocation and trafficking, protein degradation and steroid hormone signaling (Young et al., 2004). In many cases, we are just beginning to develop a detailed mechanistic picture of molecular chaperone involvement in these cellular events. In addition to providing a better understanding of basic cell biology, a more detailed picture of molecular chaperone action is likely to prove critical for the development of therapeutics for diseases that result from protein misfolding and aggregation.
Acknowledgments
We thank Drs. C. M. Carr and J. L. Puchalla for comments on the manuscript. We also thank Dr. Puchalla for assistance in preparing elements of several figures. This work was supported by a grant from the Nation Institutes of Health (GM065421).
References
- Agard DA. To fold or not to fold. Science. 1993;260:1903–1904. doi: 10.1126/science.8100365. [DOI] [PubMed] [Google Scholar]
- Anfinsen CB. Principles that govern the folding of protein chains. Science. 1973;181:223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- Aoki K, Taguchi H, et al. Calorimetric observation of a GroEL-protein binding reaction with little contribution of hydrophobic interaction. J Biol Chem. 1997;272:32158–32162. doi: 10.1074/jbc.272.51.32158. [DOI] [PubMed] [Google Scholar]
- Ashcroft AE, Brinker A, et al. Structural plasticity and noncovalent substrate binding in the GroEL apical domain. A study using electrospay ionization mass spectrometry and fluorescence binding studies. J Biol Chem. 2002;277:33115–33126. doi: 10.1074/jbc.M203398200. [DOI] [PubMed] [Google Scholar]
- Badcoe IG, Smith CJ, et al. Binding of a chaperonin to the folding intermediates of lactate dehydrogenase. Biochemistry. 1991;30:9195–9200. doi: 10.1021/bi00102a010. [DOI] [PubMed] [Google Scholar]
- Baldwin RL. Temperature dependence of the hydrophobic interaction in protein folding. Proc Natl Acad Sci U S A. 1986;83:8069–8072. doi: 10.1073/pnas.83.21.8069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumketner A, Jewett A, et al. Effects of confinement in chaperonin assisted protein folding: rate enhancement by decreasing the roughness of the folding energy landscape. J Mol Biol. 2003;332:701–713. doi: 10.1016/s0022-2836(03)00929-x. [DOI] [PubMed] [Google Scholar]
- Betancourt MR, Thirumalai D. Exploring the kinetic requirements for enhancement of protein folding rates in the GroEL cavity. J Mol Biol. 1999;287:627–644. doi: 10.1006/jmbi.1999.2591. [DOI] [PubMed] [Google Scholar]
- Bhutani N, Udgaonkar JB. A thermodynamic coupling mechanism can explain the GroEL-mediated acceleration of the folding of barstar. J Mol Biol. 2000;297:1037–1044. doi: 10.1006/jmbi.2000.3648. [DOI] [PubMed] [Google Scholar]
- Bhutani N, Udgaonkar JB. GroEL channels the folding of thioredoxin along one kinetic route. J Mol Biol. 2001;314:1167–1179. doi: 10.1006/jmbi.2000.5193. [DOI] [PubMed] [Google Scholar]
- Bochkareva ES, Lissin NM, et al. Positive cooperativity in the functioning of molecular chaperone GroEL. J Biol Chem. 1992;267:6796–6800. [PubMed] [Google Scholar]
- Braig K, Adams PD, et al. Conformational variability in the refined structure of the chaperonin GroEL at 2.8 A resolution [see comments] Nat Struct Biol. 1995;2:1083–1094. doi: 10.1038/nsb1295-1083. [DOI] [PubMed] [Google Scholar]
- Braig K, Otwinowski Z, et al. The crystal structure of the bacterial chaperonin GroEL at 2.8 A [see comments] Nature. 1994;371:578–586. doi: 10.1038/371578a0. [DOI] [PubMed] [Google Scholar]
- Braig K, Simon M, et al. A polypeptide bound by the chaperonin groEL is localized within a central cavity. Proc Natl Acad Sci U S A. 1993;90:3978–3982. doi: 10.1073/pnas.90.9.3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazil BT, Cleland JL, et al. Model peptide studies demonstrate that amphipathic secondary structures can be recognized by the chaperonin GroEL (cpn60) J Biol Chem. 1997;272:5105–5111. doi: 10.1074/jbc.272.8.5105. [DOI] [PubMed] [Google Scholar]
- Brinker A, Pfeifer G, et al. Dual function of protein confinement in chaperonin-assisted protein folding. Cell. 2001;107:223–233. doi: 10.1016/s0092-8674(01)00517-7. [DOI] [PubMed] [Google Scholar]
- Brunschier R, Danner M, et al. Interactions of phage P22 tailspike protein with GroE molecular chaperones during refolding in vitro. J Biol Chem. 1993;268:2767–2772. [PubMed] [Google Scholar]
- Buchberger A, Schroder H, et al. Substrate shuttling between the DnaK and GroEL systems indicates a chaperone network promoting protein folding. J Mol Biol. 1996;261:328–333. doi: 10.1006/jmbi.1996.0465. [DOI] [PubMed] [Google Scholar]
- Buchner J, Schmidt M, et al. GroE facilitates refolding of citrate synthase by suppressing aggregation. Biochemistry. 1991;30(6):1586–1591. doi: 10.1021/bi00220a020. [DOI] [PubMed] [Google Scholar]
- Buckle AM, Zahn R, et al. A structural model for GroEL-polypeptide recognition. Proc Natl Acad Sci U S A. 1997;94:3571–3575. doi: 10.1073/pnas.94.8.3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukau B, Horwich AL. The Hsp70 and Hsp60 chaperone machines. Cell. 1998;92:351–366. doi: 10.1016/s0092-8674(00)80928-9. [DOI] [PubMed] [Google Scholar]
- Burston SG, Ranson NA, et al. The origins and consequences of asymmetry in the chaperonin reaction cycle. J Mol Biol. 1995;249:138–152. doi: 10.1006/jmbi.1995.0285. [DOI] [PubMed] [Google Scholar]
- Burston SG, Weissman JS, et al. Release of both native and non-native proteins from a cis-only GroEL ternary complex. Nature. 1996;383:96–99. doi: 10.1038/383096a0. [DOI] [PubMed] [Google Scholar]
- Cecconi C, Shank EA, et al. Direct observation of the three-state folding of a single protein molecule. Science. 2005;309:2057–2060. doi: 10.1126/science.1116702. [DOI] [PubMed] [Google Scholar]
- Chan HS, Dill KA. A simple model of chaperonin-mediated protein folding. Proteins. 1996;24:345–351. doi: 10.1002/(SICI)1097-0134(199603)24:3<345::AID-PROT7>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Chandrasekhar GN, Tilly K, et al. Purification and properties of the groES morphogenetic protein of Escherichia coli. J Biol Chem. 1986;261:12414–12419. [PubMed] [Google Scholar]
- Chaudhry C, Farr GW, et al. Role of the gamma-phosphate of ATP in triggering protein folding by GroEL-GroES: function, structure and energetics. Embo J. 2003;22:4877–4887. doi: 10.1093/emboj/cdg477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri TK, Farr GW, et al. GroEL/GroES-mediated folding of a protein too large to be encapsulated. Cell. 2001;107:235–246. doi: 10.1016/s0092-8674(01)00523-2. [DOI] [PubMed] [Google Scholar]
- Chen J, Walter S, et al. Folding of malate dehydrogenase inside the GroEL-GroES cavity. Nat Struct Biol. 2001;8:721–728. doi: 10.1038/90443. [DOI] [PubMed] [Google Scholar]
- Chen L, Sigler PB. The crystal structure of a GroEL/peptide complex: plasticity as a basis for substrate diversity. Cell. 1999;99:757–768. doi: 10.1016/s0092-8674(00)81673-6. [DOI] [PubMed] [Google Scholar]
- Chen S, Roseman AM, et al. Location of a folding protein and shape changes in GroEL-GroES complexes imaged by cryo-electron microscopy. Nature. 1994;371:261–264. doi: 10.1038/371261a0. [DOI] [PubMed] [Google Scholar]
- Cheng MY, Hartl FU, et al. Mitochondrial heat-shock protein hsp60 is essential for assembly of proteins imported into yeast mitochondria. Nature. 1989;337:620–625. doi: 10.1038/337620a0. [DOI] [PubMed] [Google Scholar]
- Cheung MS, Garcia AE, et al. Protein folding mediated by solvation: water expulsion and formation of the hydrophobic core occur after the structural collapse. Proc Natl Acad Sci U S A. 2002;99:685–690. doi: 10.1073/pnas.022387699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AC, Frieden C. GroEL-mediated folding of structurally homologous dihydrofolate reductases. J Mol Biol. 1997;268:512–525. doi: 10.1006/jmbi.1997.0969. [DOI] [PubMed] [Google Scholar]
- Corrales FJ, Fersht AR. The folding of GroEL-bound barnase as a model for chaperonin-mediated protein folding. Proc Natl Acad Sci U S A. 1995;92:5326–5330. doi: 10.1073/pnas.92.12.5326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrales FJ, Fersht AR. Kinetic significance of GroEL14.(GroES7)2 complexes in molecular chaperone activity. Fold Des. 1996;1:265–273. doi: 10.1016/s1359-0278(96)00040-5. [DOI] [PubMed] [Google Scholar]
- Coyle JE, Jaeger J, et al. Structural and mechanistic consequences of polypeptide binding by GroEL. Fold Des. 1997;2:R93–104. doi: 10.1016/S1359-0278(97)00046-1. [DOI] [PubMed] [Google Scholar]
- Coyle JE, Texter FL, et al. GroEL accelerates the refolding of hen lysozyme without changing its folding mechanism. Nat Struct Biol. 1999;6:683–690. doi: 10.1038/10735. [DOI] [PubMed] [Google Scholar]
- Csermely P. Chaperone-percolator model: a possible molecular mechanism of Anfinsen-cage-type chaperones. Bioessays. 1999;21:959–965. doi: 10.1002/(SICI)1521-1878(199911)21:11<959::AID-BIES8>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Daggett V, Fersht AR. Is there a unifying mechanism for protein folding? Trends Biochem Sci. 2003;28:18–25. doi: 10.1016/s0968-0004(02)00012-9. [DOI] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL Molecular Graphics System. San Carlos, CA: DeLano Scientific; 2002. http://www.pymol.org. [Google Scholar]
- Deuerling E, Bukau B. Chaperone-assisted folding of newly synthesized proteins in the cytosol. Crit Rev Biochem Mol Biol. 2004;39:261–277. doi: 10.1080/10409230490892496. [DOI] [PubMed] [Google Scholar]
- Dill KA, Chan HS. From Levinthal to pathways to funnels. Nat Struct Biol. 1997;4:10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- Dobson CM. The structural basis of protein folding and its links with human disease. Philos Trans R Soc Lond B Biol Sci. 2001;356:133–145. doi: 10.1098/rstb.2000.0758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougan DA, Mogk A, et al. Protein folding and degradation in bacteria: to degrade or not to degrade? That is the question. Cell Mol Life Sci. 2002;59:1607–1616. doi: 10.1007/PL00012487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubaquie Y, Looser R, et al. Identification of in vivo substrates of the yeast mitochondrial chaperonins reveals overlapping but nonidentical requirement for hsp60 and hsp10. Embo J. 1998;17:5868–5876. doi: 10.1093/emboj/17.20.5868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edsall JT, McKenzie HA. Water and proteins. I. The significance and structure of water; its interaction with electrolytes and non-electrolytes. Adv Biophys. 1978;10:137–207. [PubMed] [Google Scholar]
- Eggers DK, Valentine JS. Crowding and hydration effects on protein conformation: a study with sol-gel encapsulated proteins. J Mol Biol. 2001;314:911–922. doi: 10.1006/jmbi.2001.5166. [DOI] [PubMed] [Google Scholar]
- Ellis RJ. Molecular chaperones. Opening and closing the Anfinsen cage. Curr Biol. 1994;4:633–635. doi: 10.1016/s0960-9822(00)00140-8. [DOI] [PubMed] [Google Scholar]
- Ellis RJ. Macromolecular crowding: obvious but underappreciated. Trends Biochem Sci. 2001;26:597–604. doi: 10.1016/s0968-0004(01)01938-7. [DOI] [PubMed] [Google Scholar]
- Ellis RJ, van der Vies SM. Molecular chaperones. Annu Rev Biochem. 1991;60:321–347. doi: 10.1146/annurev.bi.60.070191.001541. [DOI] [PubMed] [Google Scholar]
- Ewalt KL, Hendrick JP, et al. In vivo observation of polypeptide flux through the bacterial chaperonin system. Cell. 1997;90:491–500. doi: 10.1016/s0092-8674(00)80509-7. [DOI] [PubMed] [Google Scholar]
- Falke S, Tama F, et al. The 13 angstroms structure of a chaperonin GroEL-protein substrate complex by cryo-electron microscopy. J Mol Biol. 2005;348:219–230. doi: 10.1016/j.jmb.2005.02.027. [DOI] [PubMed] [Google Scholar]
- Farr GW, Fenton WA, et al. Folding with and without encapsulation by cis- and trans-only GroEL-GroES complexes. Embo J. 2003;22:3220–3230. doi: 10.1093/emboj/cdg313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farr GW, Furtak K, et al. Multivalent binding of nonnative substrate proteins by the chaperonin GroEL. Cell. 2000;100:561–573. doi: 10.1016/s0092-8674(00)80692-3. [DOI] [PubMed] [Google Scholar]
- Fayet O, Ziegelhoffer T, et al. The groES and groEL heat shock gene products of Escherichia coli are essential for bacterial growth at all temperatures. J Bacteriol. 1989;171:1379–1385. doi: 10.1128/jb.171.3.1379-1385.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton WA, Horwich AL. GroEL-mediated protein folding. Protein Sci. 1997;6:743–760. doi: 10.1002/pro.5560060401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton WA, Horwich AL. Chaperonin-mediated protein folding: fate of substrate polypeptide. Q Rev Biophys. 2003;36:229–256. doi: 10.1017/s0033583503003883. [DOI] [PubMed] [Google Scholar]
- Fenton WA, Kashi Y, et al. Residues in chaperonin GroEL required for polypeptide binding and release [see comments] Nature. 1994;371:614–619. doi: 10.1038/371614a0. [DOI] [PubMed] [Google Scholar]
- Fisher MT. Promotion of the in vitro renaturation of dodecameric glutamine synthetase from Escherichia coli in the presence of GroEL (chaperonin-60) and ATP. Biochemistry. 1992;31:3955–3963. doi: 10.1021/bi00131a010. [DOI] [PubMed] [Google Scholar]
- Frydman J. Folding of newly translated proteins in vivo: the role of molecular chaperones. Annu Rev Biochem. 2001;70:603–647. doi: 10.1146/annurev.biochem.70.1.603. [DOI] [PubMed] [Google Scholar]
- Garel J. Folding of large proteins: multidomain and multisubunit proteins. In: Creishton TE, editor. Protein Folding. New York: W. H. Freeman and Company; 1992. pp. 405–454. [Google Scholar]
- Gervasoni P, Staudenmann W, et al. beta-Lactamase binds to GroEL in a conformation highly protected against hydrogen/deuterium exchange. Proc Natl Acad Sci U S A. 1996;93:12189–12194. doi: 10.1073/pnas.93.22.12189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie B, Plaxco KW. Using protein folding rates to test protein folding theories. Annu Rev Biochem. 2004;73:837–859. doi: 10.1146/annurev.biochem.73.011303.073904. [DOI] [PubMed] [Google Scholar]
- Goldberg MS, Zhang J, et al. Native-like structure of a protein-folding intermediate bound to the chaperonin GroEL. Proc Natl Acad Sci U S A. 1997;94:1080–1085. doi: 10.1073/pnas.94.4.1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goloubinoff P, Christeller JT, et al. Reconstitution of active dimeric ribulose bisphosphate carboxylase from an unfoleded state depends on two chaperonin proteins and Mg-ATP. Nature. 1989a;342:884–889. doi: 10.1038/342884a0. [DOI] [PubMed] [Google Scholar]
- Goloubinoff P, Gatenby AA, et al. GroE heat-shock proteins promote assembly of foreign prokaryotic ribulose bisphosphate carboxylase oligomers in Escherichia coli. Nature. 1989b;337:44–47. doi: 10.1038/337044a0. [DOI] [PubMed] [Google Scholar]
- Gomez-Puertas P, Martin-Benito J, et al. The substrate recognition mechanisms in chaperonins. J Mol Recognit. 2004;17:85–94. doi: 10.1002/jmr.654. [DOI] [PubMed] [Google Scholar]
- Gordon CL, Sather SK, et al. Selective in vivo rescue by GroEL/ES of thermolabile folding intermediates to phage P22 structural proteins. J Biol Chem. 1994;269:27941–27951. [PubMed] [Google Scholar]
- Gorse D. Global minimization of an off-lattice potential energy function using a chaperone-based refolding method. Biopolymers. 2001;59:411–426. doi: 10.1002/1097-0282(200111)59:6<411::AID-BIP1046>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Grantcharova V, Alm EJ, et al. Mechanisms of protein folding. Curr Opin Struct Biol. 2001;11(1):70–82. doi: 10.1016/s0959-440x(00)00176-7. [DOI] [PubMed] [Google Scholar]
- Gray TE, Fersht AR. Cooperativity in ATP hydrolysis by GroEL is increased by GroES [published erratum appears in FEBS Lett 1992 Sep 21;310(1):99] FEBS Lett. 1991;292:254–258. doi: 10.1016/0014-5793(91)80878-7. [DOI] [PubMed] [Google Scholar]
- Gray TE, Fersht AR. Refolding of barnase in the presence of GroE. J Mol Biol. 1993;232:1197–1207. doi: 10.1006/jmbi.1993.1471. [DOI] [PubMed] [Google Scholar]
- Gross M, Robinson CV, et al. Significant hydrogen exchange protection in GroEL-bound DHFR is maintained during iterative rounds of substrate cycling. Protein Sci. 1996;5:2506–2513. doi: 10.1002/pro.5560051213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulukota K, Wolynes PG. Statistical mechanics of kinetic proofreading in protein folding in vivo. Proc Natl Acad Sci U S A. 1994;91:9292–9296. doi: 10.1073/pnas.91.20.9292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Lampoudi S, et al. Posttransition state desolvation of the hydrophobic core of the src-SH3 protein domain. Biophys J. 2003;85:61–69. doi: 10.1016/S0006-3495(03)74454-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarstrom P, Persson M, et al. Protein compactness measured by fluorescence resonance energy transfer. Human carbonic anhydrase ii is considerably expanded by the interaction of GroEL. J Biol Chem. 2001;276:21765–21775. doi: 10.1074/jbc.M010858200. [DOI] [PubMed] [Google Scholar]
- Hartl FU, Hayer-Hartl M. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science. 2002;295:1852–1858. doi: 10.1126/science.1068408. [DOI] [PubMed] [Google Scholar]
- Haustein E, Schwille P. Single-molecule spectroscopic methods. Curr Opin Struct Biol. 2004;14:531–540. doi: 10.1016/j.sbi.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Hayer-Hartl MK, Ewbank JJ, et al. Conformational specificity of the chaperonin GroEL for the compact folding intermediates of alpha-lactalbumin. Embo J. 1994;13:3192–3202. doi: 10.1002/j.1460-2075.1994.tb06618.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayer-Hartl MK, Weber F, et al. Mechanism of chaperonin action: GroES binding and release can drive GroEL-mediated protein folding in the absence of ATP hydrolysis. Embo J. 1996;15:6111–6121. [PMC free article] [PubMed] [Google Scholar]
- Hlodan R, Tempst P, et al. Binding of defined regions of a polypeptide to GroEL and its implications for chaperonin-mediated protein folding. Nat Struct Biol. 1995;2:587–595. doi: 10.1038/nsb0795-587. [DOI] [PubMed] [Google Scholar]
- Hohfeld J, Cyr DM, et al. From the cradle to the grave: molecular chaperones that may choose between folding and degradation. EMBO Rep. 2001;2:885–890. doi: 10.1093/embo-reports/kve206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holl-Neugebauer B, Rudolph R, et al. Reconstitution of a heat shock effect in vitro: influence of GroE on the thermal aggregation of alpha-glucosidase from yeast. Biochemistry. 1991;30:11609–11614. doi: 10.1021/bi00114a001. [DOI] [PubMed] [Google Scholar]
- Horovitz A, Fridmann Y, et al. Review: allostery in chaperonins. J Struct Biol. 2001;135:104–114. doi: 10.1006/jsbi.2001.4377. [DOI] [PubMed] [Google Scholar]
- Horst R, Bertelsen EB, et al. Direct NMR observation of a substrate protein bound to the chaperonin GroEL. Proc Natl Acad Sci U S A. 2005;102:12748–12753. doi: 10.1073/pnas.0505642102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwich A. Protein aggregation in disease: a role for folding intermediates forming specific multimeric interactions. J Clin Invest. 2002;110:1221–1232. doi: 10.1172/JCI16781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwich AL, Low KB, et al. Folding in vivo of bacterial cytoplasmic proteins: role of GroEL. Cell. 1993;74:909–917. doi: 10.1016/0092-8674(93)90470-b. [DOI] [PubMed] [Google Scholar]
- Horwich AL, Weber-Ban EU, et al. Chaperone rings in protein folding and degradation. Proc Natl Acad Sci U S A. 1999;96:11033–11040. doi: 10.1073/pnas.96.20.11033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houry WA, Frishman D, et al. Identification of in vivo substrates of the chaperonin GroEL. Nature. 1999;402:147–154. doi: 10.1038/45977. [DOI] [PubMed] [Google Scholar]
- Huang YS, Chuang DT. Mechanisms for GroEL/GroES-mediated folding of a large 86-kDa fusion polypeptide in vitro. J Biol Chem. 1999;274:10405–10412. doi: 10.1074/jbc.274.15.10405. [DOI] [PubMed] [Google Scholar]
- Hubbard TJ, Sander C. The role of heat-shock and chaperone proteins in protein folding: possible molecular mechanisms. Protein Eng. 1991;4:711–717. doi: 10.1093/protein/4.7.711. [DOI] [PubMed] [Google Scholar]
- Hunt JF, Weaver AJ, et al. The crystal structure of the GroES co-chaperonin at 2.8 A resolution. Nature. 1996;379:37–45. doi: 10.1038/379037a0. [DOI] [PubMed] [Google Scholar]
- Hutchinson JP, Oldham TC, et al. Electrostatic as well as hydrophobic interactions are important for the association of Cpn60 (groEL) with peptides. J Chem Soc, Perkin Trans. 1997;2:279–288. [Google Scholar]
- Ishii N, Taguchi H, et al. Folding intermediate binds to the bottom of bullet-shaped holo-chaperonin and is readily accessible to antibody. J Mol Biol. 1994;236:691–696. doi: 10.1006/jmbi.1994.1181. [DOI] [PubMed] [Google Scholar]
- Itzhaki LS, Otzen DE, et al. Nature and consequences of GroEL-protein interactions. Biochemistry. 1995;34:14581–14587. doi: 10.1021/bi00044a037. [DOI] [PubMed] [Google Scholar]
- Jackson GS, Staniforth RA, et al. Binding and hydrolysis of nucleotides in the chaperonin catalytic cycle: implications for the mechanism of assisted protein folding. Biochemistry. 1993;32:2554–2563. doi: 10.1021/bi00061a013. [DOI] [PubMed] [Google Scholar]
- Jackson SE. How do small single-domain proteins fold? Fold Des. 1998;3:R81–91. doi: 10.1016/S1359-0278(98)00033-9. [DOI] [PubMed] [Google Scholar]
- Jaenicke R, Lilie H. Folding and association of oligomeric and multimeric proteins. In: Mathews CR, editor. Protein Folding Mechansims. San Diego: Academic Press; 2000. pp. 329–401. [DOI] [PubMed] [Google Scholar]
- Jewett AI, Baumketner A, et al. Accelerated folding in the weak hydrophobic environment of a chaperonin cavity: creation of an alternate fast folding pathway. Proc Natl Acad Sci U S A. 2004;101:13192–13197. doi: 10.1073/pnas.0400720101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kad NM, Ranson NA, et al. Asymmetry, commitment and inhibition in the GroE ATPase cycle impose alternating functions on the two GroEL rings. J Mol Biol. 1998;278:267–278. doi: 10.1006/jmbi.1998.1704. [DOI] [PubMed] [Google Scholar]
- Katsumata K, Okazaki A, et al. Effect of GroEL on the re-folding kinetics of alpha-lactalbumin. J Mol Biol. 1996;258:827–838. doi: 10.1006/jmbi.1996.0290. [DOI] [PubMed] [Google Scholar]
- Kauzmann W. Some factors in the interpretation of protein denaturation. Adv Protein Chem. 1959;14:1–63. doi: 10.1016/s0065-3233(08)60608-7. [DOI] [PubMed] [Google Scholar]
- Kerner MJ, Naylor DJ, et al. Proteome-wide analysis of chaperonin-dependent protein folding in Escherichia coli. Cell. 2005;122:209–220. doi: 10.1016/j.cell.2005.05.028. [DOI] [PubMed] [Google Scholar]
- King J, Haase-Pettingell C, et al. Thermolabile folding intermediates: inclusion body precursors and chaperonin substrates. Faseb J. 1996;10:57–66. doi: 10.1096/fasebj.10.1.8566549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick S. Optimization by simulated annealing: quantitative studies. J Stat Phys. 1984;34:975–986. [Google Scholar]
- Kirkpatrick S, Gelatt CD, et al. Optimazation by Simulated Annealing. Science. 1983;220:671–680. doi: 10.1126/science.220.4598.671. [DOI] [PubMed] [Google Scholar]
- Klimov DK, Newfield D, et al. Simulations of beta-hairpin folding confined to spherical pores using distributed computing. Proc Natl Acad Sci U S A. 2002;99:8019–8024. doi: 10.1073/pnas.072220699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo EH, Lansbury PT, Jr, et al. Amyloid diseases: abnormal protein aggregation in neurodegeneration. Proc Natl Acad Sci U S A. 1999;96:9989–9990. doi: 10.1073/pnas.96.18.9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs IA, Szalay MS, et al. Water and molecular chaperones act as weak links of protein folding networks: energy landscape and punctuated equilibrium changes point towards a game theory of proteins. FEBS Lett. 2005;579:2254–2260. doi: 10.1016/j.febslet.2005.03.056. [DOI] [PubMed] [Google Scholar]
- Kuntz ID, Jr, Kauzmann W. Hydration of proteins and polypeptides. Adv Protein Chem. 1974;28:239–345. doi: 10.1016/s0065-3233(08)60232-6. [DOI] [PubMed] [Google Scholar]
- Laminet AA, Ziegelhoffer T, et al. The Escherichia coli heat shock proteins GroEL and GroES modulate the folding of the beta-lactamase precursor. Embo J. 1990;9:2315–2319. doi: 10.1002/j.1460-2075.1990.tb07403.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry SJ, Gierasch LM. The chaperonin GroEL binds a polypeptide in an alpha-helical conformation. Biochemistry. 1991;30:7359–7362. doi: 10.1021/bi00244a001. [DOI] [PubMed] [Google Scholar]
- Landry SJ, Jordan R, et al. Different conformations for the same polypeptide bound to chaperones DnaK and GroEL. Nature. 1992;355:455–457. doi: 10.1038/355455a0. [DOI] [PubMed] [Google Scholar]
- Landry SJ, Zeilstra-Ryalls J, et al. Characterization of a functionally important mobile domain of GroES. Nature. 1993;364:255–258. doi: 10.1038/364255a0. [DOI] [PubMed] [Google Scholar]
- Langer T, Pfeifer G, et al. Chaperonin-mediated protein folding: GroES binds to one end of the GroEL cylinder, which accommodates the protein substrate within its central cavity. Embo J. 1992;11:4757–4765. doi: 10.1002/j.1460-2075.1992.tb05581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilie H, Buchner J. Interaction of GroEL with a highly structured folding intermediate: iterative binding cycles do not involve unfolding. Proc Natl Acad Sci U S A. 1995;92:8100–8104. doi: 10.1073/pnas.92.18.8100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z, Rye HS. Expansion and compression of a protein folding intermediate by GroEL. Mol Cell. 2004;16:23–34. doi: 10.1016/j.molcel.2004.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z, Schwartz FP, et al. The hydrophobic nature of GroEL-substrate binding. J Biol Chem. 1995;270:1011–1014. doi: 10.1074/jbc.270.3.1011. [DOI] [PubMed] [Google Scholar]
- Mande SC, Mehra V, et al. Structure of the heat shock protein chaperonin-10 of Mycobacterium leprae. Science. 1996;271:203–207. doi: 10.1126/science.271.5246.203. [DOI] [PubMed] [Google Scholar]
- Martin J, Langer T, et al. Chaperonin-mediated protein folding at the surface of groEL through a ‘molten globule’-like intermediate [see comments] Nature. 1991;352:36–42. doi: 10.1038/352036a0. [DOI] [PubMed] [Google Scholar]
- Martin J, Mayhew M, et al. The reaction cycle of GroEL and GroES in chaperonin-assisted protein folding [see comments] Nature. 1993;366:228–233. doi: 10.1038/366228a0. [DOI] [PubMed] [Google Scholar]
- Mayer MP, Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cell Mol Life Sci. 2005;62:670–684. doi: 10.1007/s00018-004-4464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayhew MC, da Silva AC, et al. Protein folding in the central cavity of the GroEL-GroES chaperonin complex. Nature. 1996;379:420–426. doi: 10.1038/379420a0. [DOI] [PubMed] [Google Scholar]
- McKintosh E, Tabrizi SJ, et al. Prion diseases. J Neurovirol. 2003;9:183–193. doi: 10.1080/13550280390194082. [DOI] [PubMed] [Google Scholar]
- Mendoza JA, Butler MC, et al. Characterization of a stable, reactivatable complex between chaperonin 60 and mitochondrial rhodanese. J Biol Chem. 1992;267:24648–24654. [PubMed] [Google Scholar]
- Mendoza JA, Rogers E, et al. Chaperonins facilitate the in vitro folding of monomeric mitochondrial rhodanese. J Biol Chem. 1991;266:13044–13049. [PubMed] [Google Scholar]
- Mizobata T, Akiyama Y, et al. Effects of the chaperonin GroE on the refolding of tryptophanase from Escherichia coli. Refolding is enhanced in the presence of ADP. J Biol Chem. 1992;267:17773–17779. [PubMed] [Google Scholar]
- Motojima F, Chaudhry C, et al. Substrate polypeptide presents a load on the apical domains of the chaperonin GroEL. Proc Natl Acad Sci. 2004;101:15005–15012. doi: 10.1073/pnas.0406132101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok YK, Kay CM, et al. NOE data demonstrating a compact unfolded state for an SH3 domain under non-denaturing conditions. J Mol Biol. 1999;289:619–638. doi: 10.1006/jmbi.1999.2769. [DOI] [PubMed] [Google Scholar]
- Myers JK, Oas TG. Mechanism of fast protein folding. Annu Rev Biochem. 2002;71:783–815. doi: 10.1146/annurev.biochem.71.110601.135346. [DOI] [PubMed] [Google Scholar]
- Onuchic JN, Luthey-Schulten Z, et al. Theory of protein folding: the energy landscape perspective. Annu Rev Phys Chem. 1997;48:545–600. doi: 10.1146/annurev.physchem.48.1.545. [DOI] [PubMed] [Google Scholar]
- Park ES, Fenton WA, et al. No evidence for a forced-unfolding mechanism during ATP/GroES binding to substrate-bound GroEL: no observable protection of metastable Rubisco intermediate or GroEL-bound Rubisco from tritium exchange. FEBS Lett. 2005;579:1183–1186. doi: 10.1016/j.febslet.2005.01.013. [DOI] [PubMed] [Google Scholar]
- Pelham HR. Speculations on the functions of the major heat shock and glucose-regulated proteins. Cell. 1986;46:959–961. doi: 10.1016/0092-8674(86)90693-8. [DOI] [PubMed] [Google Scholar]
- Peralta D, Hartman DJ, et al. Generation of a stable folding intermediate which can be rescued by the chaperonins GroEL and GroES. FEBS Lett. 1994;339:45–49. doi: 10.1016/0014-5793(94)80381-1. [DOI] [PubMed] [Google Scholar]
- Perrett S, Zahn R, et al. Importance of electrostatic interactions in the rapid binding of polypeptides to GroEL. J Mol Biol. 1997;269:892–901. doi: 10.1006/jmbi.1997.1081. [DOI] [PubMed] [Google Scholar]
- Persson M, Hammarstrom P, et al. EPR mapping of interactions between spin-labeled variants of human carbonic anhydrase II and GroEL: evidence for increased flexibility of the hydrophobic core by the interaction. Biochemistry. 1999;38:432–441. doi: 10.1021/bi981442e. [DOI] [PubMed] [Google Scholar]
- Preuss M, Hutchinson JP, et al. Secondary structure forming propensity coupled with amphiphilicity is an optimal motif in a peptide or protein for association with chaperonin 60 (GroEL) Biochemistry. 1999;38:10272–10286. doi: 10.1021/bi990342l. [DOI] [PubMed] [Google Scholar]
- Privalov PL. Physical basis of the stability of the folded conformations of proteins. In: Creishton TE, editor. Protein Folding. New York: W. H. Freeman and Company; 1992. pp. 83–126. [Google Scholar]
- Prusiner SB, Scott MR, et al. Prion protein biology. Cell. 1998;93:337–348. doi: 10.1016/s0092-8674(00)81163-0. [DOI] [PubMed] [Google Scholar]
- Radford SE. Protein folding: progress made and promises ahead. Trends Biochem Sci. 2000;25:611–618. doi: 10.1016/s0968-0004(00)01707-2. [DOI] [PubMed] [Google Scholar]
- Ranson NA, Burston SG, et al. Binding, encapsulation and ejection: substrate dynamics during a chaperonin-assisted folding reaction. J Mol Biol. 1997;266:656–664. doi: 10.1006/jmbi.1996.0815. [DOI] [PubMed] [Google Scholar]
- Ranson NA, Clare DK, et al. Allosteric signaling of ATP hydrolysis in GroEL-GroES complexes. Nat Struct Mol Biol. 2006 doi: 10.1038/nsmb1046. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranson NA, Dunster NJ, et al. Chaperonins can catalyse the reversal of early aggregation steps when a protein misfolds. J Mol Biol. 1995;250:581–586. doi: 10.1006/jmbi.1995.0399. [DOI] [PubMed] [Google Scholar]
- Reid BG, Flynn GC. GroEL binds to and unfolds rhodanese posttranslationally. J Biol Chem. 1996;271:7212–7217. doi: 10.1074/jbc.271.12.7212. [DOI] [PubMed] [Google Scholar]
- Rief M, Gautel M, et al. Reversible unfolding of individual titin immunoglobulin domains by AFM. Science. 1997;276:1109–1112. doi: 10.1126/science.276.5315.1109. [DOI] [PubMed] [Google Scholar]
- Riek R, Pervushin K, et al. TROSY and CRINEPT: NMR with large molecular and supramolecular structures in solution. Trends Biochem Sci. 2000;25:462–468. doi: 10.1016/s0968-0004(00)01665-0. [DOI] [PubMed] [Google Scholar]
- Robinson CV, Gross M, et al. Conformation of GroEL-bound alpha-lactalbumin probed by mass spectrometry. Nature. 1994;372:646–651. doi: 10.1038/372646a0. [DOI] [PubMed] [Google Scholar]
- Roseman AM, Chen S, et al. The chaperonin ATPase cycle: mechanism of allosteric switching and movements of substrate-binding domains in GroEL. Cell. 1996;87:241–251. doi: 10.1016/s0092-8674(00)81342-2. [DOI] [PubMed] [Google Scholar]
- Rothman JE. Polypeptide chain binding proteins: catalysts of protein folding and related processes in cells. Cell. 1989;59:591–601. doi: 10.1016/0092-8674(89)90005-6. [DOI] [PubMed] [Google Scholar]
- Rye HS, Burston SG, et al. Distinct actions of cis and trans ATP within the double ring of the chaperonin GroEL [see comments] Nature. 1997;388:792–798. doi: 10.1038/42047. [DOI] [PubMed] [Google Scholar]
- Rye HS, Roseman AM, et al. GroEL-GroES cycling: ATP and nonnative polypeptide direct alternation of folding-active rings. Cell. 1999;97:325–338. doi: 10.1016/s0092-8674(00)80742-4. [DOI] [PubMed] [Google Scholar]
- Saibil H, Dong Z, et al. Binding of chaperonins. Nature. 1991;353:25–26. doi: 10.1038/353025b0. [DOI] [PubMed] [Google Scholar]
- Saibil HR, Ranson NA. The chaperonin folding machine. Trends Biochem Sci. 2002;27:627–632. doi: 10.1016/s0968-0004(02)02211-9. [DOI] [PubMed] [Google Scholar]
- Saibil HR, Zheng D, et al. ATP induces large quaternary rearrangements in a cage-like chaperonin structure. Curr Biol. 1993;3:265–273. doi: 10.1016/0960-9822(93)90176-o. [DOI] [PubMed] [Google Scholar]
- Sanders CR, Nagy JK. Misfolding of membrane proteins in health and disease: the lady or the tiger? Curr Opin Struct Biol. 2000;10:438–442. doi: 10.1016/s0959-440x(00)00112-3. [DOI] [PubMed] [Google Scholar]
- Schmidt M, Buchner J, et al. On the role of groES in the chaperonin-assisted folding reaction. Three case studies. J Biol Chem. 1994;269:10304–10311. [PubMed] [Google Scholar]
- Schroder H, Langer T, et al. DnaK, DnaJ and GrpE form a cellular chaperone machinery capable of repairing heat-induced protein damage. Embo J. 1993;12:4137–4144. doi: 10.1002/j.1460-2075.1993.tb06097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Deciphering the genesis and fate of amyloid beta-protein yields novel therapies for Alzheimer disease. J Clin Invest. 2002;110:1375–1381. doi: 10.1172/JCI16783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp JS, Forrest JA, et al. Surface denaturation and amyloid fibril formation of insulin at model lipid-water interfaces. Biochemistry. 2002;41:15810–15819. doi: 10.1021/bi020525z. [DOI] [PubMed] [Google Scholar]
- Shtilerman M, Lorimer GH, et al. Chaperonin function: folding by forced unfolding. Science. 1999;284:822–825. doi: 10.1126/science.284.5415.822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigler PB, Xu Z, et al. Structure and function in GroEL-mediated protein folding. Annu Rev Biochem. 1998;67:581–608. doi: 10.1146/annurev.biochem.67.1.581. [DOI] [PubMed] [Google Scholar]
- Smith KE, Fisher MT. Interactions between the GroE chaperonins and rhdanese. Multiple intermediates and release and rebinding. J Biol Chem. 1995;270:21517–21523. doi: 10.1074/jbc.270.37.21517. [DOI] [PubMed] [Google Scholar]
- Sparrer H, Buchner J. How GroES regulates binding of nonnative protein to GroEL. J Biol Chem. 1997;272:14080–14086. doi: 10.1074/jbc.272.22.14080. [DOI] [PubMed] [Google Scholar]
- Sparrer H, Lilie H, et al. Dynamics of the GroEL-protein complex: effects of nucleotides and folding mutants. J Mol Biol. 1996;258:74–87. doi: 10.1006/jmbi.1996.0235. [DOI] [PubMed] [Google Scholar]
- Spiess C, Meyer AS, et al. Mechanism of the eukaryotic chaperonin: protein folding in the chamber of secrets. Trends Cell Biol. 2004;14:598–604. doi: 10.1016/j.tcb.2004.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staniforth RA, Burston SG, et al. Affinity of chaperonin-60 for a protein substrate and its modulation by nucleotides and chaperonin-10. Biochem J. 1994;300:651–658. doi: 10.1042/bj3000651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swain JF, Gierasch LM. First glimpses of a chaperonin-bound folding intermediate. Proc Natl Acad Sci U S A. 2005;102:13715–13716. doi: 10.1073/pnas.0506510102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi H, Yoshida M. Chaperonin releases the substrate protein in a form with tendency to aggregate and ability to rebind to chaperonin. FEBS Lett. 1995;359:195–198. doi: 10.1016/0014-5793(95)00041-7. [DOI] [PubMed] [Google Scholar]
- Takagi F, Koga N, et al. How protein thermodynamics and folding mechanisms are altered by the chaperonin cage: molecular simulations. Proc Natl Acad Sci U S A. 2003;100:11367–11372. doi: 10.1073/pnas.1831920100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thirumalai D, Klimov DK, et al. Caging helps proteins fold. Proc Natl Acad Sci U S A. 2003;100:11195–11197. doi: 10.1073/pnas.2035072100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thirumalai D, Lorimer GH. Chaperonin-mediated protein folding. Annu Rev Biophys Biomol Struct. 2001;30:245–269. doi: 10.1146/annurev.biophys.30.1.245. [DOI] [PubMed] [Google Scholar]
- Thiyagarajan P, Henderson SJ, et al. Solution structures of GroEL and its complex with rhodanese from small- angle neutron scattering. Structure. 1996;4:79–88. doi: 10.1016/s0969-2126(96)00011-1. [DOI] [PubMed] [Google Scholar]
- Thomas PJ, Qu BH, et al. Defective protein folding as a basis of human disease. Trends Biochem Sci. 1995;20:456–459. doi: 10.1016/s0968-0004(00)89100-8. [DOI] [PubMed] [Google Scholar]
- Tilly K, Murialdo H, et al. Identification of a second Escherichia coli groE gene whose product is necessary for bacteriophage morphogenesis. Proc Natl Acad Sci U S A. 1981;78:1629–1633. doi: 10.1073/pnas.78.3.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timasheff SN. The control of protein stability and association by weak interactions with water: how do solvents affect these processes? Annu Rev Biophys Biomol Struct. 1993;22:67–97. doi: 10.1146/annurev.bb.22.060193.000435. [DOI] [PubMed] [Google Scholar]
- Todd MJ, Lorimer GH, et al. Chaperonin-facilitated protein folding: optimization of rate and yield by an iterative annealing mechanism. Proc Natl Acad Sci U S A. 1996;93:4030–4035. doi: 10.1073/pnas.93.9.4030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd MJ, Viitanen PV, et al. Dynamics of the chaperonin ATPase cycle: implications for facilitated protein folding. Science. 1994;265:659–666. doi: 10.1126/science.7913555. [DOI] [PubMed] [Google Scholar]
- Tskhovrebova L, Trinick J, et al. Elasticity and unfolding of single molecules of the giant muscle protein titin. Nature. 1997;387:308–312. doi: 10.1038/387308a0. [DOI] [PubMed] [Google Scholar]
- van den Berg B, Ellis RJ, et al. Effects of macromolecular crowding on protein folding and aggregation. Embo J. 1999;18:6927–6933. doi: 10.1093/emboj/18.24.6927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Vaart A, Ma J, et al. The unfolding action of GroEL on a protein substrate. Biophys J. 2004;87:562–573. doi: 10.1529/biophysj.103.037333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Vies SM, Viitanen PV, et al. Conformational states of ribulosebisphosphate carboxylase and their interaction with chaperonin 60. Biochemistry. 1992;31:3635–3644. doi: 10.1021/bi00129a012. [DOI] [PubMed] [Google Scholar]
- Vendruscolo M, Dobson CM. Towards complete descriptions of the free-energy landscapes of proteins. Philos Transact A Math Phys Eng Sci. 2005;363:433–450. doi: 10.1098/rsta.2004.1501. discussion 50–52. [DOI] [PubMed] [Google Scholar]
- Viitanen PV, Donaldson GK, et al. Complex interactions between the chaperonin 60 molecular chaperone and dihydrofolate reductase. Biochemistry. 1991;30:9716–9723. doi: 10.1021/bi00104a021. [DOI] [PubMed] [Google Scholar]
- Viitanen PV, Gatenby AA, et al. Purified chaperonin 60 (groEL) interacts with the nonnative states of a multitude of Escherichia coli proteins. Protein Sci. 1992;1:363–369. doi: 10.1002/pro.5560010308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viitanen PV, Lubben TH, et al. Chaperonin-facilitated refolding of ribulosebisphosphate carboxylase and ATP hydrolysis by chaperonin 60 (groEL) are K+ dependent. Biochemistry. 1990;29:5665–5671. doi: 10.1021/bi00476a003. [DOI] [PubMed] [Google Scholar]
- Walter S, Lorimer GH, et al. A thermodynamic coupling mechanism for GroEL-mediated unfolding. Proc Natl Acad Sci U S A. 1996;93:9425–9430. doi: 10.1073/pnas.93.18.9425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JD, Herman C, et al. Directed evolution of substrate-optimized GroEL/S chaperonins. Cell. 2002;111:1027–1039. doi: 10.1016/s0092-8674(02)01198-4. [DOI] [PubMed] [Google Scholar]
- Wang JD, Weissman JS. Thinking outside the box: new insights into the mechanism of GroEL- mediated protein folding. Nat Struct Biol. 1999;6:597–600. doi: 10.1038/10636. [DOI] [PubMed] [Google Scholar]
- Wang Z, Feng H, et al. Basis of substrate binding by the chaperonin GroEL. Biochemistry. 1999;38:12537–12546. doi: 10.1021/bi991070p. [DOI] [PubMed] [Google Scholar]
- Weissman JS, Hohl CM, et al. Mechanism of GroEL action: productive release of polypeptide from a sequestered position under GroES. Cell. 1995;83:577–587. doi: 10.1016/0092-8674(95)90098-5. [DOI] [PubMed] [Google Scholar]
- Weissman JS, Kashi Y, et al. GroEL-mediated protein folding proceeds by multiple rounds of binding and release of nonnative forms. Cell. 1994;78:693–702. doi: 10.1016/0092-8674(94)90533-9. [DOI] [PubMed] [Google Scholar]
- Weissman JS, Rye HS, et al. Characterization of the active intermediate of a GroEL-GroES-mediated protein folding reaction. Cell. 1996;84:481–490. doi: 10.1016/s0092-8674(00)81293-3. [DOI] [PubMed] [Google Scholar]
- Weissmann C. Birth of a prion: spontaneous generation revisited. Cell. 2005;122:165–168. doi: 10.1016/j.cell.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Wickner S, Maurizi MR, et al. Posttranslational quality control: folding, refolding, and degrading proteins. Science. 1999;286:1888–1893. doi: 10.1126/science.286.5446.1888. [DOI] [PubMed] [Google Scholar]
- Wolynes PG. Energy landscapes and solved protein-folding problems. Philos Transact A Math Phys Eng Sci. 2005;363:453–464. doi: 10.1098/rsta.2004.1502. discussion 64–67. [DOI] [PubMed] [Google Scholar]
- Xu WX, Wang J, et al. Folding behavior of chaperonin-mediated substrate protein. Proteins. 2005;61:777–794. doi: 10.1002/prot.20689. [DOI] [PubMed] [Google Scholar]
- Xu Z, Horwich AL, et al. The crystal structure of the asymmetric GroEL-GroES-(ADP)7 chaperonin complex [see comments] Nature. 1997;388:741–750. doi: 10.1038/41944. [DOI] [PubMed] [Google Scholar]
- Yifrach O, Horovitz A. Two lines of allosteric communication in the oligomeric chaperonin GroEL are revealed by the single mutation Arg196–>Ala. J Mol Biol. 1994;243:397–401. doi: 10.1006/jmbi.1994.1667. [DOI] [PubMed] [Google Scholar]
- Yifrach O, Horovitz A. Nested cooperativity in the ATPase activity of the oligomeric chaperonin GroEL. Biochemistry. 1995;34:5303–5308. doi: 10.1021/bi00016a001. [DOI] [PubMed] [Google Scholar]
- Yifrach O, Horovitz A. Coupling between protein folding and allostery in the GroE chaperonin system. Proc Natl Acad Sci U S A. 2000;97:1521–1524. doi: 10.1073/pnas.040449997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JC, Agashe VR, et al. Pathways of chaperone-mediated protein folding in the cytosol. Nat Rev Mol Cell Biol. 2004;5:781–791. doi: 10.1038/nrm1492. [DOI] [PubMed] [Google Scholar]
- Zahn R, Axmann SE, et al. Thermodynamic partitioning model for hydrophobic binding of polypeptides by GroEL. I. GroEL recognizes the signal sequences of beta-lactamase precursor. J Mol Biol. 1994a;242:150–164. doi: 10.1006/jmbi.1994.1566. [DOI] [PubMed] [Google Scholar]
- Zahn R, Perrett S, et al. Conformational states bound by the molecular chaperones GroEL and secB: a hidden unfolding (annealing) activity. J Mol Biol. 1996a;261:43–61. doi: 10.1006/jmbi.1996.0440. [DOI] [PubMed] [Google Scholar]
- Zahn R, Perrett S, et al. Catalysis of amide proton exchange by the molecular chaperones GroEL and SecB. Science. 1996b;271:642–645. doi: 10.1126/science.271.5249.642. [DOI] [PubMed] [Google Scholar]
- Zahn R, Pluckthun A. Thermodynamic partitioning model for hydrophobic binding of polypeptides by GroEL. II. GroEL recognizes thermally unfolded mature beta-lactamase. J Mol Biol. 1994;242:165–174. doi: 10.1006/jmbi.1994.1567. [DOI] [PubMed] [Google Scholar]
- Zahn R, Spitzfaden C, et al. Destabilization of the complete protein secondary structure on binding to the chaperone GroEL. Nature. 1994b;368:261–265. doi: 10.1038/368261a0. [DOI] [PubMed] [Google Scholar]
- Zhang O, Forman-Kay JD. NMR studies of unfolded states of an SH3 domain in aqueous solution and denaturing conditions. Biochemistry. 1997;36:3959–3970. doi: 10.1021/bi9627626. [DOI] [PubMed] [Google Scholar]
- Zhou HX, Dill KA. Stabilization of proteins in confined spaces. Biochemistry. 2001;40:11289–11293. doi: 10.1021/bi0155504. [DOI] [PubMed] [Google Scholar]
- Zhuang X, Rief M. Single-molecule folding. Curr Opin Struct Biol. 2003;13:88–97. doi: 10.1016/s0959-440x(03)00011-3. [DOI] [PubMed] [Google Scholar]