

Background: The function of the CXXXC motifs in P3H1 and CRTAP has not been previously characterized.
Results: The model GCRALCG peptide and the P3H1·CRTAP·CypB complex show disulfide isomerase activity in vitro.
Conclusion: Our results suggest that this complex could function as a disulfide isomerase.
Significance: This indicates an additional function of the P3H1 complex in the rER.
Keywords: Collagen, Disulfide, Endoplasmic Reticulum (ER), Oxidation-Reduction, Protein Isomerase, Collagen Biosynthesis, Prolyl 3-Hydroxylase
Abstract
Collagen biosynthesis occurs in the rough endoplasmic reticulum, and many molecular chaperones and folding enzymes are involved in this process. The folding mechanism of type I procollagen has been well characterized, and protein disulfide isomerase (PDI) has been suggested as a key player in the formation of the correct disulfide bonds in the noncollagenous carboxyl-terminal and amino-terminal propeptides. Prolyl 3-hydroxylase 1 (P3H1) forms a hetero-trimeric complex with cartilage-associated protein and cyclophilin B (CypB). This complex is a multifunctional complex acting as a prolyl 3-hydroxylase, a peptidyl prolyl cis-trans isomerase, and a molecular chaperone. Two major domains are predicted from the primary sequence of P3H1: an amino-terminal domain and a carboxyl-terminal domain corresponding to the 2-oxoglutarate- and iron-dependent dioxygenase domains similar to the α-subunit of prolyl 4-hydroxylase and lysyl hydroxylases. The amino-terminal domain contains four CXXXC sequence repeats. The primary sequence of cartilage-associated protein is homologous to the amino-terminal domain of P3H1 and also contains four CXXXC sequence repeats. However, the function of the CXXXC sequence repeats is not known. Several publications have reported that short peptides containing a CXC or a CXXC sequence show oxido-reductase activity similar to PDI in vitro. We hypothesize that CXXXC motifs have oxido-reductase activity similar to the CXXC motif in PDI. We have tested the enzyme activities on model substrates in vitro using a GCRALCG peptide and the P3H1 complex. Our results suggest that this complex could function as a disulfide isomerase in the rough endoplasmic reticulum.
Introduction
Secreted proteins such as extracellular matrix proteins are synthesized in the rough endoplasmic reticulum (rER).2 Collagen is the most abundant extracellular matrix protein in humans and has important roles in building the structural framework in tissues like bone, skin, tendon, and cartilage (1, 2). Fibrillar collagens are the major components in these tissues. The biosynthesis of type I collagen has been well characterized compared with other types of collagens. Many molecular chaperones, modifying enzymes, and folding catalysts are involved in this process in the rER (3). This molecular ensemble is required for proper post-translational modifications, folding, and secretion of type I collagen. Disruption of this ensemble results in connective tissue disorders such as osteogenesis imperfecta (3, 4).
Hydroxylation of proline residues is one of the important post-translational modification during collagen biosynthesis. Two different types of hydroxylation of proline residues occur, prolyl 4-hydroxylation and prolyl 3-hydroxylation, and these modifications are performed by distinct enzyme complexes. The prolyl 4-hydroxylase (P4H)·protein disulfide isomerase (PDI) and prolyl 3-hydroxylase 1 (P3H1)·cartilage-associated protein (CRTAP)·cyclophilin B (CypB) complexes are responsible for prolyl 4-hydroxylation and prolyl 3-hydroxylation, respectively (3). Prolyl 4-hydroxylation increases the thermal stability of the collagen molecule, and almost all prolines at position Yaa are 4-hydroxylated in collagen Gly-Xaa-Yaa sequences (5–7). In contrast, the function of prolyl 3-hydroxylation still unknown, and prolyl 3-hydroxylation is a much less frequent event in collagenous sequences. Type IV collagen contains the greatest number of 3(S)-hydroxyproline residues among all collagen types studied. However, the contribution of 3-hydroxylation to the amino acid composition of type IV is <1%. By contrast, 4-hydroxyproline represents close to 10% of the amino acids in collagens (8–10). This suggests a more subtle role for 3-hydroxyproline residues.
The tetrameric P4H complex has a 2:2 stoichiometry of the α-subunit (P4H) to PDI, which is called the β-subunit in this tetramer (11, 12). PDI is a rER resident oxido-reductase that contains two thioredoxin motifs each with an active site CGHC sequence (13, 14). In humans, >20 PDI homologues are reported to exist in the rER (15). Most PDIs share the thioredoxin-like domain containing a CXXC motif, which is responsible for their oxido-reductase activity. The number of -CXXC- motifs and the identity of -CXXC- sequences differ among these homologues. PDI has been suggested to be a key player in the formation of native disulfide bonds in the noncollagenous carboxyl-terminal propeptide of type I collagen (16–18). The α-subunit requires the β-subunit for solubility and for retention inside the rER because it does not have a rER retention signal (19). The P4H complex hydroxylates proline residues in the Yaa position of collagen Gly-Xaa-Yaa sequences to 4(R)-hydroxyproline using its 2-oxoglutarate- and iron-dependent dioxygenase domains (20, 21). P3H1 has been well studied compared with the other two isoforms, P3H2 and P3H3. The lack of prolyl 3-hydroxylase 1 has been implicated in severe recessive osteogenesis imperfecta, and P3H1 knock-out mice showed abnormalities in the collagen fibril ultrastructure in many tissues (22–26). P3H1 forms a hetero-trimeric complex with CRTAP and CypB, and this complex has been shown to be a multifunctional complex that acts as prolyl 3-hydroxylase, peptidyl prolyl cis-trans isomerase, and molecular chaperone (27, 28). This complex hydroxylates proline residues in collagen Gly-Pro-4(R)-hydroxyproline sequences to 3(S)-hydroxyproline (28). The primary structure of P3H1 suggests two domains: a carboxyl-terminal domain that corresponds to the 2-oxoglutarate- and iron-dependent dioxygenase domains similar to the α-subunit in P4H and lysyl hydroxylases and an amino-terminal domain (Fig. 1). Two CXXXC sequences are located between two tetratricopeptide repeat motifs, and this subdomain occurs twice in the amino-terminal domain (29). However, the function of this domain is not known. CRTAP, which is a part of the P3H1 complex, also contains the same arrangement of CXXXC sequence repeats. The primary structure of CRTAP shares a high similarity to the amino-terminal domain of P3H1 (Fig. 1). The lack of CRTAP also resulted in a recessive form of osteogenesis imperfecta, and CRTAP-null mice showed abnormalities in the collagen fibril ultrastructure in many tissues (30). Besides being part of the P3H1·CRTAP·CypB complex no other biological function of CRTAP has been described. SC65 shows a size and primary sequence similar to those of CRTAP. It was reported to be a nucleolar protein (31); however, the function of this protein is still unclear. Understanding the role of CXXXC motifs may aid in the understanding of the function of CRTAP and SC65.
FIGURE 1.
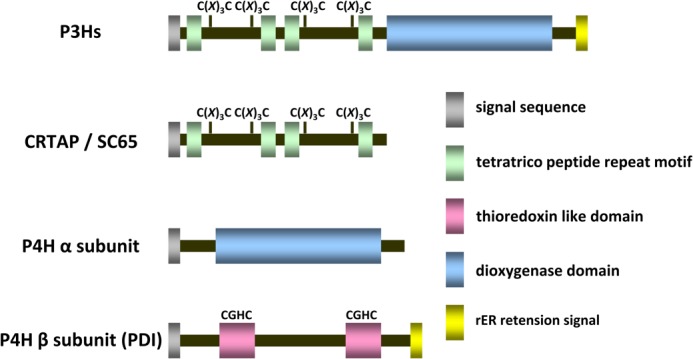
Schematic representation of the domain structures. The domain structures of P3Hs, CRTAP/SC65 and the α- and β-subunits of P4H are shown.
Several publications have reported that short peptides containing a CXC or a CXXC sequence show oxido-reductase activity similar to PDI in vitro (32–35). Here we test whether the CXXXC sequence has oxido-reductase activity similar to the CXXC motif in PDI. We synthesized a model peptide GCRALCG, whose sequence is located in the third position of the amino-terminal domain of P3H1 and which is well conserved in various species (Table 1). This peptide was used to test whether the CXXXC sequence behaved in a similar way to the CXXC sequence. The pKa value and reduction potential of the GCRALCG peptide were determined and compared with reported values of peptides with a CXC or CXXC sequences as well as PDI family proteins. The enzyme activity of this peptide was also measured by classical oxido-reductase assays. Finally, the purified P3H1 complex, which is a biologically functional protein complex in the rER, was used in these enzyme assays to measure its enzyme activities in vitro.
TABLE 1.
The sequence alignment of -CXXXC- in prolyl 3-hydroxylases, CRTAP, and SC65 from human, mouse, and chicken
NCBI Protein Databases are as follows: human P3H1 (NCBI accession NP_071751.3), P3H2 (NCBI accession NP_060662.2), P3H3 (NCBI accession NP_055077.2), CRTAP (NCBI accession NP_006362.1), and SC65 (NCBI accession NP_006446.1); mouse P3H1 (NCBI accession NP_062756.2), P3H2 (NCBI accession NP_775555.1), P3H3 (NCBI accession NP_038562.2), CRTAP (NCBI accession NP_064306.2), and SC65 (NCBI accession NP_789800.1); chicken P3H1 (NCBI accession NP_001001529.1), P3H2 (NCBI accession NP_001001530.1), and CRTAP (NCBI accession AAS45240.1). Ensemble Protein Databases are as follows: chicken P3H3 (Ensemble accession ENSGALP00000023347) and SC65 (Ensemble accession ENSGALP00000041385).
| Molecule and species | -CXXXC- sequences from the amino terminus |
|||
|---|---|---|---|---|
| 1st | 2nd | 3rd | 4th | |
| Prolyl 3-hydroxylases 1 | ||||
| Human | -CRTQC- | -CLRRC- | -CRALC- | -CKQNC- |
| Mouse | -CRTRC- | -CLRRC- | -CRALC- | -CKQNC- |
| Chicken | -CRLRC- | -CLRGC- | -CRALC- | -CKQGC- |
| Prolyl 3-hydroxylases 2 | ||||
| Human | -CARHC- | -CYRSC- | -CRTLC- | -CQHEC- |
| Mouse | -CARHC- | -CSRSC- | -CRALC- | -CQHEC- |
| Chicken | -CRRRC- | -CLRSC- | -CQIMC- | -CKHDC- |
| Prolyl 3-hydroxylases 3 | ||||
| Human | -CGASC- | -CLTQC- | -CRADC- | -CRQRC- |
| Mouse | -CGASC- | -CLTQC- | -CRAAC- | -CRQHC- |
| Chicken | -CRDAC- | -CLQHC- | -CRALC- | -CRQQC- |
| CRTAP | ||||
| Human | -CHRNC- | -CLKRC- | -CLAAC- | -CKIQC- |
| Mouse | -CHRNC- | -CLKRC- | -CLAAC- | -CKIRC- |
| Chicken | -CHHNC- | -CLRRC- | -CIAAC- | -CKVQC- |
| SC65 | ||||
| Human | -CHANC- | -CLRRC- | -CLAGC- | -CKVDC- |
| Mouse | -CHANC- | -CLRRC- | -CLAGC- | -CKVDC- |
| Chicken | -CHRRC- | -CLRAC- | -CLAGC- | -CKVGC- |
EXPERIMENTAL PROCEDURES
Synthesis of GCRALCG Peptide
The peptide GCRALCG was synthesized on an ABI 433A synthesizer using Fmoc (N-(9-fluorenyl)methoxycarbonyl) chemistry and HATU O-(7-azabenzotriazol-1-yl)-1.1.3.3-tetramethyluronium hexafluorophosphate (Perseptive Biosystems) (4.0 eq)/diisopropylethylamine-mediated peptide couplings. The peptide was cleaved from the resin and purified by preparative HPLC (Vydac® C18, 5 μm, 300 Å, 250 × 50 mm; W. R. Grace). The synthesized peptide was characterized by mass spectrometry.
Determination of Thiol pKa
The thiol pKa value was determined according to the method described by Woycechowsky et al. (34, 35). Thiol titration curves were obtained by measuring the absorbance at 238 nm as a function of pH. The GCRALCG peptide (63 μm) was mixed into 0.10 m potassium phosphate buffers with various pH values. Data were analyzed using a double titration model as described previously (34).
Preparation of Reduced and Oxidized GCRALCG Peptide
Reduced GCRALCG peptide was generated by incubating 5 mg of peptide in 1 ml of 0.1 m Tris acetate, pH 8.0, containing 2 mm of EDTA, 6 m guanidine hydrochloride, and 0.14 m DTT overnight. Oxidized peptide was formed by dialysis into 0.1 m Tris acetate, pH 8.0, mixed with 10 mm reduced glutathione (GSH), and 1 mm oxidized glutathione (GSSG) for a week. Peptide concentrations were 5 mg/ml and 0.4 mg/ml for reduced and oxidized forms, respectively. After the reactions were quenched by HCl, these peptides were purified by HPLC and lyophilized. These peptides were characterized by mass spectrometry.
Determination of Reduction Potential on the GCRALCG Peptide
The stability of the cyclic disulfide formed by the GCRALCG peptide was evaluated as described previously (34, 35), with minor modifications. The thiol-disulfide interchange equilibria were established between the reduced GCRALCG peptide and β-hydroxyethyl disulfide (Sigma-Aldrich) or the oxidized GCRALCG peptide and β-mercaptoethanol (Sigma-Aldrich) in various ratios at 25 °C for 24 h. After the reaction was quenched by trifluoroacetic acid, an aliquot of the mixture was loaded onto a Vydac analytical C4 reverse phase column using a HPLC system, and the separated peaks were detected by absorbance at 205 nm. The areas of each peak were converted to concentrations of compound using calibration curves that were obtained by peak areas as a function of concentrations of these compounds (data not shown). The reduction potential of the GCRALCG disulfide was calculated using the Nernst equation and Eβ-hydroxyethyl disulfide = −260 mV.
Purification of the Chicken P3H1 Complex and Bovine PDI
The chicken P3H1 complex and bovine PDI were extracted from 17-day-old chick embryos and bovine liver and purified as described (27, 36).
Insulin Disulfide Bond Reductase Activity
The insulin reductase activity was measured by the method of Lambert and Freedman et al. (37). Enzyme assays were performed in 20 mm sodium phosphate buffer, pH 7.5, containing 5 mm EDTA, 8 mm reduced glutathione, 0.12 mm NADPH, 1 units/ml glutathione reductase, and 30 μm insulin at 25 °C. All substrates were purchased from Sigma-Aldrich. Enzyme activity reduces the disulfide bond of insulin via the oxidation of reduced glutathione. The oxidized glutathione was reduced by glutathione reductase using NADPH. Activities were monitored continuously at 340 nm by the oxidation of NADPH in a Cary 4 spectrophotometer (Varian Inc.). All enzyme and peptide concentrations were determined by amino acid analysis.
Determination of Disulfide Bond Oxidase and Isomerase Activity Using Reduced and Scrambled RNase A
Disulfide bond oxidase and isomerase activity on reduced and scrambled RNase A were measured with a modified procedure published by Lyles and Gilbert (38). Reduced RNase A was prepared by incubating the protein in denaturation buffer (0.1 m Tris-HCl, pH 8.0, containing 6 m guanidine hydrochloride and 0.15 m DTT) overnight at room temperature. Scrambled RNase was purchased from Sigma-Aldrich and dissolved in 10 mm acetic acid. Enzyme assays were performed in 50 mm Tris-HCl, pH 7.8, containing 0.15 m NaCl, a 1:5 ratio of oxidized and reduced glutathione, and cCMP. The concentration of cCMP was 4.5 mm and 0.9 mm for disulfide bond oxidase and isomerase assay, respectively. Enzyme activities were monitored continuously at 296 nm by hydrolysis of cCMP in a Cary4 spectrophotometer. Temperature was controlled at 25 and 30 °C for disulfide bond oxidase and isomerase assay, respectively. Both assays were initiated by the addition of RNase A to the reaction buffer. Reduced RNase A was added into the reaction mixture immediately after spinning down in a ZebaTM spin desalting column (Thermo Scientific) to remove the denaturant and the reducing agent. All enzyme and peptide concentrations were determined by amino acid analysis.
RESULTS
Characterization of the GCRALCG Peptide
First, we determined whether the synthetic GCRALCG peptide could act as an active site of oxido-reductase activity. The thiol pKa value reflects the ability for a proton to dissociate from the side chain of cysteine residues at a particular pH thus forming a thiolate anion. The equilibrium between among, thiol-thiolate, and dithiolate in the reduced GCRALCG peptide was monitored by measuring the absorbance at 238 nm in solutions of varying pH. The values were titrated over a pH range (Fig. 2). The pKa values were calculated to be 8.19 and 9.29 by a double titration model.
FIGURE 2.

Effect of pH on the absorbance at 238 nm of the GCRALCG peptide. The titration was performed in 0.10 m potassium phosphate buffer. Fitting the data to an equation (see “Experimental Procedures”) gives pKa values of 8.19 and 9.29.
The reduction potential was determined by the thiol-disulfide interchange equilibrium between the reduced GCRALCG peptide and β-hydroxyethyl disulfide or between the oxidized GCRALCG peptide and β-mercaptoethanol using HPLC separation. This value indicates the tendency for the side chain of the cysteine residue to either gain or lose a proton. A lower potential suggests that the cysteine residue has a tendency to reduce or be oxidized by the substrate. The reduction potential of the GCRALCG peptide was determined to be E°′ = −197 ± 19 mV.
Table 2 shows the thiol pKa values and reduction potential of other C(X)nC peptides and PDI family proteins compared with that of GCRALCG peptide. Our results are comparable with earlier reported values.
TABLE 2.
Values of the reduction potential and pKa for peptides and enzymes containing -C(X)nC- motifs
| Molecule | Sequence | pKa | Reduction potential E°′ |
|---|---|---|---|
| mV | |||
| Escherichia coli thioredoxin | -CGPC- | 7.5 (77) | −270 (78) |
| Rat protein disulfide isomerase | -CGHC- | 6.7 (79) | −180 (80) |
| Human ERdj5 | -CXXC- | Not determined | −190 (40) |
| CXC peptide | CGC- | 8.7 (35) | −167 (35) |
| Active site peptide of PDI | -WCGHCKAL- | Not determined | −205 (32) |
| GCXXXCG peptide | -GCRALCG- | 8.19 | −197 ± 19 |
Oxido-reductase Activities of the GCRALCG Peptide and the P3H1 Complex
To build on our initial results with the GCRALCG peptide, three classical oxido-reductase assays were performed. Previous studies showed that small peptides containing a CXC or a CXXC sequence had oxido-reductase activity in vitro (32–35). The purified P3H1 complex was also used in these assays to test whether the CXXXC motifs were biologically active. The disulfide oxido-reductase activities of the GCRALCG peptide were measured at millimolar concentrations, whereas that of P3H1 complex was measured at a concentration similar to PDI, which was always used as a positive control for these assays.
First, we tested the disulfide oxidase activity using reduced RNase A. Oxidation of cysteines to form disulfide bonds is an important process for protein folding, and PDI family proteins assist with this process in the rER. Neither the GCRALCG peptide nor the P3H1 complex catalyzed the oxidation of reduced RNase A, and these curves were indistinguishable from a curve in the absence of any enzymes (Fig. 3). PDI-treated reduced RNase A and native RNase A showed reasonable enzyme activities.
FIGURE 3.
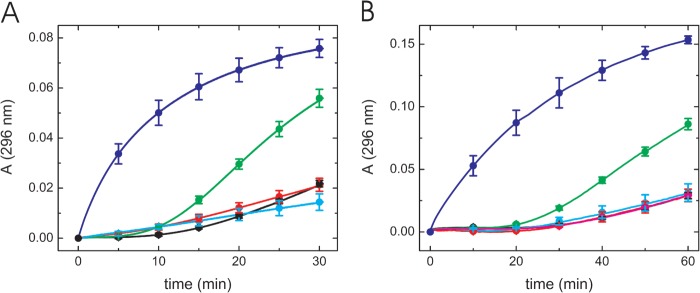
Disulfide bond oxidase activity of the GCRALCG peptide and the P3H1 complex using reduced RNase A. Disulfide bond oxidase activity of the GCRALCG peptide (A) and the P3H1 complex (B) were monitored continuously at 296 nm. A, native (blue) and reduced (black) RNase A by itself were used as controls. Both RNase A concentrations were 8.4 μm. The activities to reduced RNase A are shown as 0.7 5 mm (red) and 1.5 mm (cyan) GCRALCG peptide, and 0.5 μm PDI (green). B, native (blue) and reduced (black) RNase A by itself were used as controls. Both RNase A concentrations were 8.4 μm. The activities to reduced RNase A are shown as 0.2 μm (red) and 0.3 μm (cyan) P3H1 complex. The curves of 0.2 μm PDI (green) and 0.2 μm BSA (magenta) in presence of reduced RNase A are shown as positive and negative controls, respectively. The curves of black, red, magenta, and cyan are completely overlapped and difficult to distinguish from each other.
Next, disulfide isomerase activity was assayed using scrambled RNase A which contains nonnative disulfide bonds and little enzyme activity. PDI activity contributes to a reshuffling of the nonnative disulfide bonds and regenerates RNase activity. Native RNase A and PDI were used as positive controls for these experiments. The GCRALCG peptide showed disulfide isomerase activity and a lag phase for the initial 5 min of the experiment. This observation was likely caused by the altered redox conditions in the presence of a high concentration of peptide (millimolar level). Nonnative disulfide bonds in scrambled RNase A may be sensitive to shuffling in this redox buffer. 0.1 mm GCRALCG peptide showed a similar activity compared with 0.05 μm PDI at approximately 30 min (Fig. 4A). This peptide also exhibited disulfide isomerase activity in a concentration-dependent manner (Fig. 4B). The same tendency was observed using the P3H1 complex in this experiment. A solution of 0.02 μm P3H1 complex had almost the same catalytic efficiency as 0.04 μm PDI (Fig. 4C). This complex also showed a concentration dependence of its disulfide isomerase activity (Fig. 4D).
FIGURE 4.

Disulfide bond isomerase activity of the GCRALCG peptide and the P3H1 complex using scrambled RNase A. Disulfide bond isomerase activity of the GCRALCG peptide (A and B) and the P3H1 complex (C and D) were monitored continuously at 296 nm. Individual curves were averaged by at least three measurements. Error bars (S.D.) were included at every 5 min and 10 min for the GCRALCG peptide and the P3H1 complex, respectively. A, comparison of disulfide bond isomerase activity toward 1.2 μm scrambled RNase A between the GCRALCG peptide and PDI. The activities of 0.12 μm native RNase A are shown as a blue curve, in the absence (black) and presence of 0.1 mm GCRALCG peptide (green) and 0.05 μm PDI (red). B, concentration dependence of disulfide bond isomerase activity toward 1.2 μm scrambled RNase A in presence of the GCRALCG peptide, in the absence (black) and presence of 0.1 mm (red), 0.2 mm (blue), and 2 mm (green) GCRALCG peptide. C, comparison of disulfide bond isomerase activity toward 1.2 μm scrambled RNase A between the P3H1 complex and PDI. The activities of 0.12 μm native RNase A are shown as a blue curve, in the absence (black) and presence of 0.02 μm P3H1 complex (green), 0.04 μm PDI (red). D, concentration dependence of disulfide bond isomerase activity toward 1.2 μm scrambled RNase A in the presence of various concentrations of P3H1 complex, in the absence (black) and presence of 0.005 μm (red), 0.01 μm (blue), and 0.02 μm (green) P3H1 complex. Error bars indicate standard deviations.
To confirm that both the GCRALCG peptide and the P3H1 complex had disulfide isomerase activity, we examined enzyme activity with a different substrate. Insulin has a single disulfide bond and has been used in the past to assay disulfide isomerase/reductase activity (37, 39, 40). Disulfide isomerase activity of the GCRALCG peptide was shown to behave in a concentration-dependent manner (Fig. 5A). A similar level of activity was observed with 0.1 μm PDI and 0.3 mm peptide (Fig. 5B). The P3H1 complex also showed concentration-dependent activity similar to the activity of PDI. The isomerase activity of 0.09 μm P3H1 complex was almost equal to the activity of 0.1 μm PDI in this assay (Fig. 5, C and D).
FIGURE 5.

Insulin disulfide bond reductase activity using the GCRALCG peptide and the P3H1 complex. Disulfide bond reductase activity of the GCRALCG peptide (A and B) and the P3H1 complex (C and D) were monitored continuously at 340 nm. Individual curves were averaged by at least three measurements. Error bars (S.D.) were included at every 40-s interval. A, concentration dependence of disulfide bond reductase activity in the presence of various concentrations of the GCRALCG peptide, in the absence (black) and presence of 0.1 mm (red), 0.3 mm (green), and 0.5 mm (blue) GCRALCG peptide. B, comparison of disulfide bond reductase activity between the GCRALCG peptide and PDI, in the absence (black) and presence of 0.1 mm (red) and 0.3 mm (blue) GCRALCG peptide. The activities of PDI are shown as 0.05 μm (green) and 0.1 μm (cyan). C, concentration dependence of disulfide bond reductase activity in the presence of various concentrations of P3H1 complex, in the absence (black) and presence of 0.03 μm (red), 0.06 μm (green), and 0.09 μm (blue) P3H1 complex. D, comparison of disulfide bond reductase activity between the P3H1 complex and PDI, in the absence (black) and presence of 0.06 μm (red) and 0.09 μm (blue) P3H1 complex. The activities of PDI are shown as 0.05 μm (green) and 0.1 μm (cyan). Error bars indicate standard deviations.
DISCUSSION
The P3H1 Complex Likely Has Disulfide Isomerase Activity in the rER
The characterization of the GCRALCG peptide indicates that this peptide possesses a similar pKa value and reduction potential to the previously reported values for CXC or CXXC peptides or PDI homologues. It has been reported that the pH in the rER is close to neutral (41) and the reduction potential in the rER is assumed to be around −180 mV (42). Our results suggest that this peptide could act as a disulfide reductase. We further examined its activity using classical oxido-reductase assays in vitro. The peptide did not assist in the folding of reduced RNase A; however, it showed concentration-dependent contributions to disulfide isomerization of scrambled RNase A and reduction of insulin. The much higher concentration of the peptide required for activity comparable with PDI is likely due to the lack of structure, as the peptide is studied out of the context of the folded P3H1 or CRTAP. Nevertheless, these data suggest that the CXXXC sequences in P3Hs, CRTAP, and SC65 potentially also have disulfide isomerase activity. Indeed, the P3H1 complex showed activity similar to that of the peptide toward these model substrates, with nearly equal activities compared with PDI. We hypothesize that the P3H1 complex behaves as a disulfide isomerase in the rER, similar to the PDI homologues. In contrast to PDI, the P3H1 complex and the GCRALCG peptide do not show disulfide oxidase activity. Fig. 6 shows a schematic diagram of the reactions catalyzed by PDI and the P3H1 complex. Whereas PDI is active in the oxidation of cysteines, the GCRALCG peptide and the P3H1 complex only are involved in the reduction/isomerization reaction.
FIGURE 6.

Schematic diagram of the reactions catalyzed by the P3H1 complex and PDI in the rER. The oxidation and reduction/isomerization reactions are shown for a hypothetical substrate with two disulfide bonds.
Which Functions in the P3H1 Complex Are Crucial during Collagen Biosynthesis?
The disruption of any component of the P3H1 complex affects type I collagen biosynthesis leading to osteogenesis imperfecta (22–26, 30, 43–48). It has been reported that these disruption are caused by the absence of 3-hydroxylation at Pro-986 of the α1 chain. The absence of P3H1 and CRTAP results in similar defects to the type I collagen molecule. The collagen becomes overmodified by increased glycosylation of hydroxylysine residues in both collagen α1 and α2 chains and exhibits a higher melting temperature. Additionally a slower rate of folding and secretion is observed (23–26, 46, 47).
CypB forms multiple protein complexes in the rER (27, 49–52). It was reported that a mutation in the molecular interaction site of horse CypB affects collagen folding, secretion, and post-translational modifications. This mutation did not disrupt the PPIase activity but altered the interaction with lysyl hydroxylase 1 (50) and possibly calnexin and calreticulin (51). This study indicated that collagen biosynthesis is both influenced by the PPIase activity and other molecular interactions.
The P3H1 complex was shown to be a multifunctional complex, acting as a general molecular chaperone, prolyl 3-hydroxylase, and PPIase during collagen biosynthesis (27, 28). Our results suggest that this complex also plays a role as a disulfide isomerase. The PPIase activity is attributed to CypB (27), but CypB exhibits poor molecular chaperone activity toward model substrates (27, 49). P3H1 includes the 2-oxoglutarate- and iron-dependent dioxygenase domain, which is responsible for the prolyl 3-hydroxylase activity (28). This suggests that the amino-terminal domain of P3H1 and/or CRTAP is functioning as a molecular chaperone and a disulfide isomerase.
Several reports indicated that the absence of any individual component of the P3H1·CRTAP·CypB complex could alter the complex formation (43, 44, 48). This could result in losing four different functions during collagen biosynthesis. The function of the 3-hydroxylation of proline residues is still unknown. The molecular chaperone activity of the complex has been proposed to stabilize the transient junction between folded and unfolded chains during triple helix formation (27). Proline residues are frequently found in collagenous sequence, and the rate-limiting step in triple helix formation is the cis-trans isomerization of prolyl peptide bonds (53–56). PPIase activity contributes to the conformational change from cis to trans prolyl peptide bonds. The biological relevance of the additional function, disulfide isomerase activity, in the P3H1 complex is unclear. The carboxyl- and amino-terminal propeptide regions of type I collagen contain many cysteine residues that form interchain and intrachain disulfide bonds. PDI has been suggested as a key player in the formation of the correct disulfide bonds in the carboxyl-terminal propeptide (16–18). The P3H1 complex may also be involved in disulfide bond formation either directly or indirectly. Another possibility is that the complex is involved in establishing the redox environment in the rER.
The Consensus Sequence and Functions of CXXXC Motifs
Many PDI homologues share the thioredoxin-like domain with a CXXC sequence, and CGHC is the most common sequence. Mutagenesis studies have shown that neither the number of CXXC motifs nor their sequence can account for the strength of PDI activity (39, 57–59). This indicates that there are different contributions of individual CXXC sequences in PDI homologues.
We chose the GCRALCG sequence because it is well conserved in many species in P3H1 (Table 1). P3Hs, CRTAP, and SC65 show greater sequence variations in CXXXC than PDI homologues do in CXXC. Our peptide acted as a disulfide isomerase for model substrates in vitro, and the P3H1 complex also showed disulfide isomerase activity at a concentration range similar to that of PDI. The P3H1 complex contains eight CXXXC sequences that are derived from P3H1 and CRTAP. Despite its activity similar to that of the P3H1 complex, PDI contains only two CGHC sequences. Further studies are required to identify the most active CXXXC sequences and their level of activity in the P3H1 complex.
Mitochondria also have a well developed redox network system, and there are many protein-protein disulfide linkages in these compartments (60, 61). While in the rER the CXXC-based PDI proteins play an important role, mitochondrial CXXXC-containing proteins function to import proteins into the mitochondria intermembrane space (62, 63). Small translocase of innermembrane proteins contain two highly conserved CXXXC sequences which are seemingly unique to this protein family. These twin CXXXC sequences were proposed to coordinate Zn2+ by forming a zinc-finger-like structure (61, 64). They form a 70-kDa complex that acts as a chaperone in the intermembrane space, and the CXXXC motifs are essential for assembly of these complexes (61–64). A metal binding domain called the TRASH domain found in both prokaryote and eukaryote proteins also contains distinct conserved cysteines (CX19–22CXXXC) in its primary sequence (65). So far, no biologically functional correlation between the CXXXC sequences in the P3H1 complex, in small translocase of innermembrane proteins and in TRASH domains, has been identified.
A Comparison between the P3H1 Complex and the PDI·P4H tetramer
The P4H α-subunit shows poor solubility in the rER and does not possess an rER retention signal. PDI solves both issues through its molecular chaperone activity and the rER retention signal. Similarly, CRTAP does not have a rER retention signal. This protein was initially characterized as a secreted protein after it was identified in the extracellular matrix in cartilage by immunohistochemistry (66). CRTAP also shows poor solubility in various in vitro expression systems in our laboratory (data not shown). This suggests that neither the α-subunit of P4H nor CRTAP exists as free proteins in the rER. In contrast, free PDI and CypB are present in extracts of chicken rER (30). Association of the α-subunit of P4H with PDI and association of CRTAP with P3H1 and CypB are essential for these proteins to function properly in the rER.
PDI consists of four thioredoxin domains, the a, b, b′, and a′ domain and an acidic carboxyl-terminal extension with the rER retention signal KDEL (13, 67). Both the a and a′ domains contain the catalytic site CGHC, whereas the b′ domain principally recognizes the substrates of PDI. Studies of tetramer formation using mutant or truncated domains of PDI showed that b′ and a′ domains fulfill the minimum requirement to form the active tetramer with P4H (68, 69). In the case of the P3H1 complex, it is predicted that tetratricopeptide repeat motifs are present and surround the CXXXC sequences (29). This motif mediates protein-protein interactions (70, 71). It is possible that these motifs contribute to complex formation and/or substrate recognition in the rER. The P3H1 complex also has molecular chaperone activity (27), and this activity may rely on the amino-terminal domains including the CXXXC sequences.
The redox environment in the rER is oxidative compared with that of the cytosol (72). Redox networks and/or cascades exist in the rER via multiple and multistep electron transfer pathways to enhance or maintain oxidation, isomerization, and reduction of disulfide bonds during protein folding. PDI homologues are involved in maintaining this redox environment in the rER (13, 14, 73, 74). A recent study suggested that this environment not only affected protein folding, but also contributed to maintaining the presence of ascorbic acid in the rER (75). A loss-of-function study in mice of the three thiol oxidases, ERO1α, ERO1β, and PRDX4, showed defects in intracellular procollagen maturation and an abnormal connective tissue with a lower content of 4-hydroxyl proline (75). The authors concluded that these defects were caused by a depletion of ascorbic acid and a noncanonical form of scurvy due to thiol oxidase deficiency (75). Ascorbic acid is an important co-factor in the reduction of the inactive iron(III) state to the active iron(II) state in the dioxygenase domains of P4H (21) and probably also in P3Hs and lysyl hydroxylases. As previously described, prolyl 4-hydroxylation is the most frequent post-translational modification in collagenous sequences involved in the increase of the thermal stability of the triple helix. Knock-out mice of P4H show embryonic lethality and basement membrane defects caused by the loss of type IV collagen assembly (76).
Thus, the redox environment in the rER indirectly affects the function of the PDI·P4H tetramer. We speculate that the functions of P3H1 complex can also be influenced by the redox environment in the rER directly or indirectly.
Acknowledgments
We thank the Analytical Core facility of Shriners Hospitals for Children Portland for peptide synthesis, mass spectrometry analysis, and amino acid analysis.
This work was supported by grants from Shriners Hospitals for Children (to H. P. B.).
- rER
- rough endoplasmic reticulum
- CRTAP
- cartilage-associated protein
- CypB
- cyclophilin B
- P3H1
- prolyl 3-hydroxylase 1
- P4H
- prolyl 4-hydroxylase
- PDI
- protein disulfide isomerase
- PPIase
- peptidylprolyl cis-trans isomerase.
REFERENCES
- 1. Bächinger H. P., Mizuno K., Vranka J., Boudko S. (2010) in Comprehensive Natural Products II: Chemistry and Biology (Mander L., Liu H.-W. eds) pp. 469–530, Elsevier, New York [Google Scholar]
- 2. Bateman J. F., Boot-Handford R. P., Lamandé S. R. (2009) Genetic diseases of connective tissues: cellular and extracellular effects of ECM mutations. Nat. Rev. Genet. 10, 173–183 [DOI] [PubMed] [Google Scholar]
- 3. Ishikawa Y., Bächinger H. P. (2013) A molecular ensemble in the rER for procollagen maturation. Biochim. Biophys. Acta 1833, 2479–2491 [DOI] [PubMed] [Google Scholar]
- 4. Eyre D., Weis M. (2013) Calcified Tissue Int. 10.1007/s00223-013-9723-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Berg R. A., Prockop D. J. (1973) The thermal transition of a non-hydroxylated form of collagen: evidence for a role for hydroxyproline in stabilizing the triple-helix of collagen. Biochem. Biophys. Res. Commun. 52, 115–120 [DOI] [PubMed] [Google Scholar]
- 6. Jimenez S., Harsch M., Rosenbloom J. (1973) Hydroxyproline stabilizes the triple helix of chick tendon collagen. Biochem. Biophys. Res. Commun. 52, 106–114 [DOI] [PubMed] [Google Scholar]
- 7. Rosenbloom J., Harsch M., Jimenez S. (1973) Hydroxyproline content determines the denaturation temperature of chick tendon collagen. Arch. Biochem. Biophys. 158, 478–484 [DOI] [PubMed] [Google Scholar]
- 8. Kresina T. F., Miller E. J. (1979) Isolation and characterization of basement membrane collagen from human placental tissue: evidence for the presence of two genetically distinct collagen chains. Biochemistry 18, 3089–3097 [DOI] [PubMed] [Google Scholar]
- 9. Kefalides N. A. (1975) Basement membranes: structural and biosynthetic considerations. J. Invest. Dermatol. 65, 85–92 [DOI] [PubMed] [Google Scholar]
- 10. Gryder R. M., Lamon M., Adams E. (1975) Sequence position of 3-hydroxyproline in basement membrane collagen: isolation of glycyl-3-hydroxyprolyl 4-hydroxyproline from swine kidney. J. Biol. Chem. 250, 2470–2474 [PubMed] [Google Scholar]
- 11. Kivirikko K. I., Myllylä R. (1982) Posttranslational enzymes in the biosynthesis of collagen: intracellular enzymes. Methods Enzymol. 82, 245–304 [DOI] [PubMed] [Google Scholar]
- 12. Myllyharju J., Kivirikko K. I. (2004) Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet. 20, 33–43 [DOI] [PubMed] [Google Scholar]
- 13. Wilkinson B., Gilbert H. F. (2004) Protein disulfide isomerase. Biochim. Biophys. Acta 1699, 35–44 [DOI] [PubMed] [Google Scholar]
- 14. Hatahet F., Ruddock L. W. (2009) Protein disulfide isomerase: a critical evaluation of its function in disulfide bond formation. Antioxid. Redox Signal. 11, 2807–2850 [DOI] [PubMed] [Google Scholar]
- 15. Ellgaard L., Ruddock L. W. (2005) The human protein disulphide isomerase family: substrate interactions and functional properties. EMBO Rep. 6, 28–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wilson R., Lees J. F., Bulleid N. J. (1998) Protein disulfide isomerase acts as a molecular chaperone during the assembly of procollagen. J. Biol. Chem. 273, 9637–9643 [DOI] [PubMed] [Google Scholar]
- 17. Bottomley M. J., Batten M. R., Lumb R. A., Bulleid N. J. (2001) Quality control in the endoplasmic reticulum: PDI mediates the ER retention of unassembled procollagen C-propeptides. Curr. Biol. 11, 1114–1118 [DOI] [PubMed] [Google Scholar]
- 18. Koide T., Nagata K. (2005) Collagen Biosynthesis (Brinckmann J., Notbohm H., Müller P. K., eds), pp. 85–114, Springer, Berlin [Google Scholar]
- 19. John D. C., Grant M. E., Bulleid N. J. (1993) Cell-free synthesis and assembly of prolyl 4-hydroxylase: the role of the β-subunit (PDI) in preventing misfolding and aggregation of the α-subunit. EMBO J. 12, 1587–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Myllyharju J. (2003) Prolyl 4-hydroxylases, the key enzymes of collagen biosynthesis. Matrix Biol. 22, 15–24 [DOI] [PubMed] [Google Scholar]
- 21. Gorres K. L., Raines R. T. (2010) Prolyl 4-hydroxylase. Crit. Rev. Biochem. Mol. Biol. 45, 106–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vranka J. A., Pokidysheva E., Hayashi L., Zientek K., Mizuno K., Ishikawa Y., Maddox K., Tufa S., Keene D. R., Klein R., Bächinger H. P. (2010) Prolyl 3-hydroxylase 1-null mice display abnormalities in fibrillar collagen-rich tissues such as tendons, skin, and bones. J. Biol. Chem. 285, 17253–17262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cabral W. A., Chang W., Barnes A. M., Weis M., Scott M. A., Leikin S., Makareeva E., Kuznetsova N. V., Rosenbaum K. N., Tifft C. J., Bulas D. I., Kozma C., Smith P. A., Eyre D. R., Marini J. C. (2007) Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta. Nat. Genet. 39, 359–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Baldridge D., Schwarze U., Morello R., Lennington J., Bertin T. K., Pace J. M., Pepin M. G., Weis M., Eyre D. R., Walsh J., Lambert D., Green A., Robinson H., Michelson M., Houge G., Lindman C., Martin J., Ward J., Lemyre E., Mitchell J. J., Krakow D., Rimoin D. L., Cohn D. H., Byers P. H., Lee B. (2008) CRTAP and LEPRE1 mutations in recessive osteogenesis imperfecta. Hum. Mutat. 29, 1435–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Willaert A., Malfait F., Symoens S., Gevaert K., Kayserili H., Megarbane A., Mortier G., Leroy J. G., Coucke P. J., De Paepe A. (2009) Recessive osteogenesis imperfecta caused by LEPRE1 mutations: clinical documentation and identification of the splice form responsible for prolyl 3-hydroxylation. J. Med. Genet. 46, 233–241 [DOI] [PubMed] [Google Scholar]
- 26. Takagi M., Ishii T., Barnes A. M., Weis M., Amano N., Tanaka M., Fukuzawa R., Nishimura G., Eyre D. R., Marini J. C., Hasegawa T. (2012) A novel mutation in LEPRE1 that eliminates only the KDEL ER-retrieval sequence causes non-lethal osteogenesis imperfecta. PLoS One 7, e36809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ishikawa Y., Wirz J., Vranka J. A., Nagata K., Bächinger H. P. (2009) Biochemical characterization of the prolyl 3-hydroxylase 1·cartilage-associated protein·cyclophilin B complex. J. Biol. Chem. 284, 17641–17647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vranka J. A., Sakai L. Y., Bächinger H. P. (2004) Prolyl 3-hydroxylase 1, enzyme characterization and identification of a novel family of enzymes. J. Biol. Chem. 279, 23615–23621 [DOI] [PubMed] [Google Scholar]
- 29. Marini J. C., Cabral W. A., Barnes A. M., Chang W. (2007) Components of the collagen prolyl 3-hydroxylation complex are crucial for normal bone development. Cell Cycle 6, 1675–1681 [DOI] [PubMed] [Google Scholar]
- 30. Morello R., Bertin T. K., Chen Y., Hicks J., Tonachini L., Monticone M., Castagnola P., Rauch F., Glorieux F. H., Vranka J., Bächinger H. P., Pace J. M., Schwarze U., Byers P. H., Weis M., Fernandes R. J., Eyre D. R., Yao Z., Boyce B. F., Lee B. (2006) CRTAP is required for prolyl 3- hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell 127, 291–304 [DOI] [PubMed] [Google Scholar]
- 31. Ochs R. L., Stein T. W., Jr., Chan E. K., Ruutu M., Tan E. M. (1996) cDNA cloning and characterization of a novel nucleolar protein. Mol. Biol. Cell 7, 1015–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Siedler F., Rudolph-Böhner S., Doi M., Musiol H. J., Moroder L. (1993) Redox potentials of active-site bis(cysteinyl) fragments of thiol-protein oxidoreductases. Biochemistry 32, 7488–7495 [DOI] [PubMed] [Google Scholar]
- 33. Cabrele C., Fiori S., Pegoraro S., Moroder L. (2002) Redox-active cyclic bis(cysteinyl)peptides as catalysts for in vitro oxidative protein folding. Chem. Biol. 9, 731–740 [DOI] [PubMed] [Google Scholar]
- 34. Woycechowsky K. J., Wittrup K. D., Raines R. T. (1999) A small-molecule catalyst of protein folding in vitro and in vivo. Chem. Biol. 6, 871–879 [DOI] [PubMed] [Google Scholar]
- 35. Woycechowsky K. J., Raines R. T. (2003) The CXC motif: a functional mimic of protein disulfide isomerase. Biochemistry 42, 5387–5394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lambert N., Freedman R. B. (1983) Structural properties of homogeneous protein disulphide-isomerase from bovine liver purified by a rapid high-yielding procedure. Biochem. J. 213, 225–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lambert N., Freedman R. B. (1983) Kinetics and specificity of homogeneous protein disulphide-isomerase in protein disulphide isomerization and in thiol-protein-disulphide oxidoreduction. Biochem. J. 213, 235–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lyles M. M., Gilbert H. F. (1991) Catalysis of the oxidative folding of ribonuclease A by protein disulfide isomerase: dependence of the rate on the composition of the redox buffer. Biochemistry 30, 613–619 [DOI] [PubMed] [Google Scholar]
- 39. Kikuchi M., Doi E., Tsujimoto I., Horibe T., Tsujimoto Y. (2002) Functional analysis of human P5, a protein disulfide isomerase homologue. J. Biochem. 132, 451–455 [DOI] [PubMed] [Google Scholar]
- 40. Ushioda R., Hoseki J., Araki K., Jansen G., Thomas D. Y., Nagata K. (2008) ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science 321, 569–572 [DOI] [PubMed] [Google Scholar]
- 41. Kim J. H., Johannes L., Goud B., Antony C., Lingwood C. A., Daneman R., Grinstein S. (1998) Noninvasive measurement of the pH of the endoplasmic reticulum at rest and during calcium release. Proc. Natl. Acad. Sci. U.S.A. 95, 2997–3002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hwang C., Sinskey A. J., Lodish H. F. (1992) Oxidized redox state of glutathione in the endoplasmic reticulum. Science 257, 1496–1502 [DOI] [PubMed] [Google Scholar]
- 43. Barnes A. M., Carter E. M., Cabral W. A., Weis M., Chang W., Makareeva E., Leikin S., Rotimi C. N., Eyre D. R., Raggio C. L., Marini J. C. (2010) Lack of cyclophilin B in osteogenesis imperfecta with normal collagen folding. N. Engl. J. Med. 362, 521–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Choi J. W., Sutor S. L., Lindquist L., Evans G. L., Madden B. J., Bergen H. R., 3rd, Hefferan T. E., Yaszemski M. J., Bram R. J. (2009) Severe osteogenesis imperfecta in cyclophilin B-deficient mice. PLoS Genet. 5, e1000750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pyott S. M., Schwarze U., Christiansen H. E., Pepin M. G., Leistritz D. F., Dineen R., Harris C., Burton B. K., Angle B., Kim K., Sussman M. D., Weis M., Eyre D. R., Russell D. W., McCarthy K. J., Steiner R. D., Byers P. H. (2011) Mutations in PPIB (cyclophilin B) delay type I procollagen chain association and result in perinatal lethal to moderate osteogenesis imperfecta phenotypes. Hum. Mol. Genet. 20, 1595–1609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Valli M., Barnes A. M., Gallanti A., Cabral W. A., Viglio S., Weis M. A., Makareeva E., Eyre D., Leikin S., Antoniazzi F., Marini J. C., Mottes M. (2012) Deficiency of CRTAP in non-lethal recessive osteogenesis imperfecta reduces collagen deposition into matrix. Clin. Genet. 82, 453–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Van Dijk F. S., Nesbitt I. M., Nikkels P. G., Dalton A., Bongers E. M., van de Kamp J. M., Hilhorst-Hofstee Y., Den Hollander N. S., Lachmeijer A. M., Marcelis C. L., Tan-Sindhunata G. M., van Rijn R. R., Meijers-Heijboer H., Cobben J. M., Pals G. (2009) CRTAP mutations in lethal and severe osteogenesis imperfecta: the importance of combining biochemical and molecular genetic analysis. Eur. J. Hum. Genet. 17, 1560–1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. van Dijk F. S., Nesbitt I. M., Zwikstra E. H., Nikkels P. G., Piersma S. R., Fratantoni S. A., Jimenez C. R., Huizer M., Morsman A. C., Cobben J. M., van Roij M. H., Elting M. W., Verbeke J. I., Wijnaendts L. C., Shaw N. J., Högler W., McKeown C., Sistermans E. A., Dalton A., Meijers-Heijboer H., Pals G. (2009) PPIB mutations cause severe osteogenesis imperfecta. Am. J. Hum. Genet. 85, 521–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Horibe T., Yosho C., Okada S., Tsukamoto M., Nagai H., Hagiwara Y., Tujimoto Y., Kikuchi M. (2002) The chaperone activity of protein disulfide isomerase is affected by cyclophilin B and cyclosporin A in vitro. J. Biochem. 132, 401–407 [DOI] [PubMed] [Google Scholar]
- 50. Ishikawa Y., Vranka J. A., Boudko S. P., Pokidysheva E., Mizuno K., Zientek K., Keene D. R., Rashmir-Raven A. M., Nagata K., Winand N. J., Bächinger H. P. (2012) Mutation in cyclophilin B that causes hyperelastosis cutis in American Quarter Horse does not affect peptidylprolyl cis-trans isomerase activity but shows altered cyclophilin B-protein interactions and affects collagen folding. J. Biol. Chem. 287, 22253–22265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kozlov G., Bastos-Aristizabal S., Määttänen P., Rosenauer A., Zheng F., Killikelly A., Trempe J. F., Thomas D. Y., Gehring K. (2010) Structural basis of cyclophilin B binding by the calnexin/calreticulin P-domain. J. Biol. Chem. 285, 35551–35557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Smith T., Ferreira L. R., Hebert C., Norris K., Sauk J. J. (1995) Hsp47 and cyclophilin B traverse the endoplasmic reticulum with procollagen into pre-Golgi intermediate vesicles: a role for Hsp47 and cyclophilin B in the export of procollagen from the endoplasmic reticulum. J. Biol. Chem. 270, 18323–18328 [DOI] [PubMed] [Google Scholar]
- 53. Bächinger H. P., Bruckner P., Timpl R., Engel J. (1978) The role of cis-trans isomerization of peptide bonds in the coil leads to and comes from triple helix conversion of collagen. Eur. J. Biochem. 90, 605–613 [DOI] [PubMed] [Google Scholar]
- 54. Bächinger H. P., Bruckner P., Timpl R., Prockop D. J., Engel J. (1980) Folding mechanism of the triple helix in type-III collagen and type-III pN-collagen: role of disulfide bridges and peptide bond isomerization. Eur. J. Biochem. 106, 619–632 [DOI] [PubMed] [Google Scholar]
- 55. Bächinger H. P., Morris N. P., Davis J. M. (1993) Thermal stability and folding of the collagen triple helix and the effects of mutations in osteogenesis imperfecta on the triple helix of type I collagen. Am. J. Med. Genet. 45, 152–162 [DOI] [PubMed] [Google Scholar]
- 56. Steinmann B., Bruckner P., Superti-Furga A. (1991) Cyclosporin A slows collagen triple-helix formation in vivo: indirect evidence for a physiologic role of peptidyl-prolyl cis-trans-isomerase. J. Biol. Chem. 266, 1299–1303 [PubMed] [Google Scholar]
- 57. Hagiwara M., Maegawa K., Suzuki M., Ushioda R., Araki K., Matsumoto Y., Hoseki J., Nagata K., Inaba K. (2011) Structural basis of an ERAD pathway mediated by the ER-resident protein disulfide reductase ERdj5. Mol. Cell 41, 432–444 [DOI] [PubMed] [Google Scholar]
- 58. Horibe T., Gomi M., Iguchi D., Ito H., Kitamura Y., Masuoka T., Tsujimoto I., Kimura T., Kikuchi M. (2004) Different contributions of the three CXXC motifs of human protein-disulfide isomerase-related protein to isomerase activity and oxidative refolding. J. Biol. Chem. 279, 4604–4611 [DOI] [PubMed] [Google Scholar]
- 59. Satoh M., Shimada A., Kashiwai A., Saga S., Hosokawa M. (2005) Differential cooperative enzymatic activities of protein disulfide isomerase family in protein folding. Cell Stress Chaperones 10, 211–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mesecke N., Terziyska N., Kozany C., Baumann F., Neupert W., Hell K., Herrmann J. M. (2005) A disulfide relay system in the intermembrane space of mitochondria that mediates protein import. Cell 121, 1059–1069 [DOI] [PubMed] [Google Scholar]
- 61. Koehler C. M., Beverly K. N., Leverich E. P. (2006) Redox pathways of the mitochondrion. Antioxid. Redox Signal. 8, 813–822 [DOI] [PubMed] [Google Scholar]
- 62. Mokranjac D., Neupert W. (2009) Thirty years of protein translocation into mitochondria: unexpectedly complex and still puzzling. Biochim. Biophys. Acta 1793, 33–41 [DOI] [PubMed] [Google Scholar]
- 63. Wagner K., Mick D. U., Rehling P. (2009) Protein transport machineries for precursor translocation across the inner mitochondrial membrane. Biochim. Biophys. Acta 1793, 52–59 [DOI] [PubMed] [Google Scholar]
- 64. Koehler C. M. (2004) The small Tim proteins and the twin CX3C motif. Trends Biochem. Sci. 29, 1–4 [DOI] [PubMed] [Google Scholar]
- 65. Ettema T. J., Huynen M. A., de Vos W. M., van der Oost J. (2003) TRASH: a novel metal-binding domain predicted to be involved in heavy-metal sensing, trafficking and resistance. Trends Biochem. Sci. 28, 170–173 [DOI] [PubMed] [Google Scholar]
- 66. Castagnola P., Gennari M., Morello R., Tonachini L., Marin O., Gaggero A., Cancedda R. (1997) Cartilage-associated protein (CASP) is a novel developmentally regulated chick embryo protein. J. Cell Sci. 110, 1351–1359 [DOI] [PubMed] [Google Scholar]
- 67. Tian G., Xiang S., Noiva R., Lennarz W. J., Schindelin H. (2006) The crystal structure of yeast protein disulfide isomerase suggests cooperativity between its active sites. Cell 124, 61–73 [DOI] [PubMed] [Google Scholar]
- 68. Klappa P., Koivunen P., Pirneskoski A., Karvonen P., Ruddock L. W., Kivirikko K. I., Freedman R. B. (2000) Mutations that destabilize the a′-domain of human protein-disulfide isomerase indirectly affect peptide binding. J. Biol. Chem. 275, 13213–13218 [DOI] [PubMed] [Google Scholar]
- 69. Pirneskoski A., Ruddock L. W., Klappa P., Freedman R. B., Kivirikko K. I., Koivunen P. (2001) Domains b′ and a′ of protein disulfide isomerase fulfill the minimum requirement for function as a subunit of prolyl 4-hydroxylase: the N-terminal domains a and b enhance this function and can be substituted in part by those of ERp57. J. Biol. Chem. 276, 11287–11293 [DOI] [PubMed] [Google Scholar]
- 70. Blatch G. L., Lässle M. (1999) The tetratricopeptide repeat: a structural motif mediating protein-protein interactions. Bioessays 21, 932–939 [DOI] [PubMed] [Google Scholar]
- 71. D'Andrea L. D., Regan L. (2003) TPR proteins: the versatile helix. Trends Biochem. Sci. 28, 655–662 [DOI] [PubMed] [Google Scholar]
- 72. Wouters M. A., Fan S. W., Haworth N. L. (2010) Disulfides as redox switches: from molecular mechanisms to functional significance. Antioxid. Redox Signal. 12, 53–91 [DOI] [PubMed] [Google Scholar]
- 73. Araki K., Nagata K. (2011) Functional in vitro analysis of the ERO1 protein and protein-disulfide isomerase pathway. J. Biol. Chem. 286, 32705–32712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sato Y., Inaba K. (2012) Disulfide bond formation network in the three biological kingdoms, bacteria, fungi and mammals. FEBS J. 279, 2262–2271 [DOI] [PubMed] [Google Scholar]
- 75. Zito E., Hansen H. G., Yeo G. S., Fujii J., Ron D. (2012) Endoplasmic reticulum thiol oxidase deficiency leads to ascorbic acid depletion and noncanonical scurvy in mice. Mol. Cell 48, 39–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Holster T., Pakkanen O., Soininen R., Sormunen R., Nokelainen M., Kivirikko K. I., Myllyharju J. (2007) Loss of assembly of the main basement membrane collagen, type IV, but not fibril-forming collagens and embryonic death in collagen prolyl 4-hydroxylase I-null mice. J. Biol. Chem. 282, 2512–2519 [DOI] [PubMed] [Google Scholar]
- 77. Chivers P. T., Prehoda K. E., Volkman B. F., Kim B. M., Markley J. L., Raines R. T. (1997) Microscopic pKa values of Escherichia coli thioredoxin. Biochemistry 36, 14985–14991 [DOI] [PubMed] [Google Scholar]
- 78. Moore E. C., Reichard P., Thelander L. (1964) Enzymatic synthesis of deoxyribonucleotides. V. Purification and properties of thioredoxin reductase from Escherichia coli B. J. Biol. Chem. 239, 3445–3452 [PubMed] [Google Scholar]
- 79. Hawkins H. C., Freedman R. B. (1991) The reactivities and ionization properties of the active-site dithiol groups of mammalian protein disulphide-isomerase. Biochem. J. 275, 335–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lundström J., Holmgren A. (1993) Determination of the reduction-oxidation potential of the thioredoxin-like domains of protein disulfide-isomerase from the equilibrium with glutathione and thioredoxin. Biochemistry 32, 6649–6655 [DOI] [PubMed] [Google Scholar]