

Abstract
Platinum compounds represent one of the great success stories of metals in medicine. Following the serendipitous discovery of the anticancer activity of cisplatin by Rosenberg, a large number of cisplatin variants have been prepared and tested for their ability to kill cancer cells and inhibit tumor growth. These efforts continue today with increased realization that new strategies are needed to overcome issues of toxicity and resistance inherent to treatment by the approved platinum anticancer agents. One approach has been the use of so-called “non-traditional” platinum(II) and platinum(IV) compounds that violate the structure-activity relationships that governed platinum drug-development research for many years. Another is the use of specialized drug delivery strategies. Here we describe recent developments from our laboratory involving monofunctional platinum(II) complexes together with an historical account of the manner by which we came to investigate these compounds and their relationship to previously studied molecules. We also discuss work carried out using platinum(IV) prodrugs and the development of nanoconstructs designed to deliver them in vivo.
Introduction
The discovery in 1965 that soluble electrolytic oxidative decomposition products of a platinum electrode in ammonia-containing buffer, including cis-diamminedichloroplatinum(II) or cisplatin (Chart 1), inhibit cell division in Escherichia coli1 was the key step in the discovery of an important class of anticancer agents.2 Subsequent Food and Drug Administration (FDA) approval of cisplatin in 1978 had a profound and far-reaching impact on the field of chemotherapy, substantially increasing survival for many cancer patients. Most notably, long-term survival rates of testicular cancer patients improved from less than 10% to greater than 90% following the introduction of cisplatin to the treatment regimens.3 Subsequently, two related platinum-based drugs, carboplatin and oxaliplatin, were approved for clinical use in the United States. Three others, nedaplatin, lobaplatin, and heptaplatin, are widely employed in Asia (Chart 1).4
Chart 1.
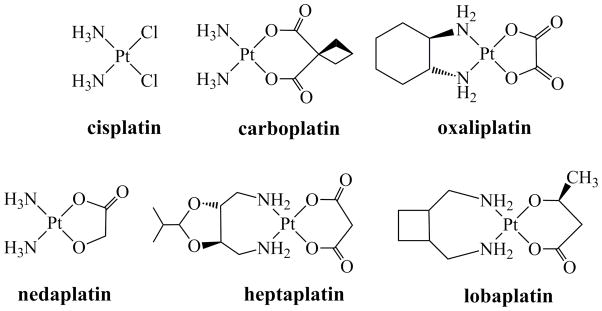
Chemical Structures of Platinum-Based Anticancer Drugs in Clinical Use Worldwide
Apart from its wonderful clinical value, cisplatin, one of few approved transition metal-based drugs, inspired generations of inorganic chemists to pursue applications of their research in the medical sciences. A portion of this every-growing body of work on the creative use of inorganic chemistry in medicine is showcased in this Inorganic Chemistry Forum, as well as in recently published books.5,6 Here, we provide an overview of developments in the field of platinum anticancer agents. Although cisplatin has been in use for over four decades, new and more effective platinum-based therapeutics are finally on the horizon.
This article is organized in the following manner. First, we present the canonical mechanism of action of cisplatin for readers who are generally unfamiliar with the topic. Next, monofunctional platinum(II) complexes are discussed. Although now a flourishing research program, investigations of monofunctional compounds in our laboratory were initiated three decades ago, as will be described. The sequence of events that led to renewed interest in monofunctional platinum(II) compounds and the discovery of very promising new clinical candidates will then be relayed. The final section of this article covers the use of octahedral platinum(IV) complexes as small-molecule prodrugs and their incorporation into various nanodelivery devices. The focus is primarily on work from our laboratory with limited discussion of that from other research groups. For a more comprehensive survey of the larger body of work carried out in this field, the reader is referred to recent review articles.7–10
Mechanism of Action of Cisplatin and Related Platinum-Based Drugs
Cisplatin and the related FDA-approved platinum-based drugs (Chart 1) operate by similar mechanisms of action. These agents kill cancer cells by binding to nuclear DNA.11 The process by which such compounds do so involves several steps (Figure 1), of which cellular uptake is the first. The efficacy of a platinum-anticancer agent depends upon its ability to enter the cell and penetrate the nucleus where the critical target, DNA, resides. Much research has been devoted to elucidating the pathways by which cisplatin is internalized by cells.12 Both passive and active transport pathways have been implicated.12 Of particular interest is the reported uptake of cisplatin by the copper transporter, CTR1,13 which may link cisplatin efficacy to intracellular copper trafficking.
Figure 1.

Different pathways of cisplatin before and after it enters the cell.
Once inside the cell, cisplatin becomes activated by aquation, the substitution of chloride ligands by water. This reaction gives rise to the potent electrophilic cations cis-[Pt(NH3)2Cl(OH2)]+ and cis-[Pt(NH3)2(OH2)2]2+, which readily bind DNA (Scheme 1). The aquation reaction is reversible, and thus the diminished chloride ion concentration (10–20 mM) in the cytoplasm compared to blood (0.1 M) favors the formation of the platinum aqua complexes. The preferred DNA-binding sites of cisplatin are the N7 positions of the nucleobases guanine and adenine. Intrastrand cross-links, formed by binding to two adjacent guanosine residues, comprise the majority of the cisplatin-DNA cross-links. In addition to DNA, soft sulfur-donor nucleophiles such as glutathione and sulfur-containing amino acids, also readily interact with cisplatin. These latter binding events can prevent substantial quantities of cisplatin from reaching DNA in the nucleus and ultimately limit its efficacy. The clinical relevance of off-target platinum-protein interactions is not yet fully understood.14
Scheme 1.
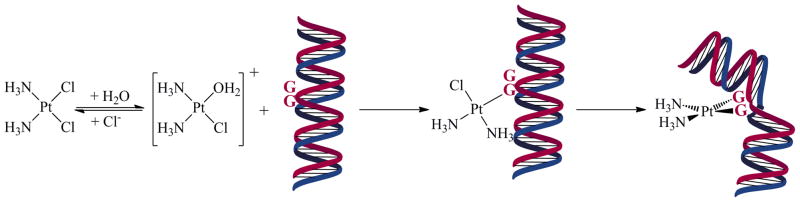
DNA-Binding Mechanism of Cisplatin where “G” Represents a Guanine Base
After aquation and evasion of deactivating endogenous ligands, cisplatin binds to DNA. The bifunctional platinum-DNA cross-links that are formed induce significant structural distortion in the double-helix and impede transcription and replication, triggering cell death pathways.15 The unique structural distortion of cisplatin-DNA intrastrand cross-links is also recognized by a variety of nuclear components.16–19 Among these are proteins and protein complexes comprising the cellular DNA repair machinery, including nucleotide excision repair (NER) proteins, which preferentially remove cisplatin intrastrand cross-links and enable cells to recover.20–22 Alternatively, other proteins that bind to platinated DNA17,18 can potentiate the activity of cisplatin.23 HMG-domain proteins, for example, selectively bind to cisplatin intrastrand d(GpG) cross-links in DNA,24–27 and the resulting complex increases the efficacy of these cytotoxic platinum lesions by blocking the access of proteins required for NER.28,29 Hence, high expression levels of HMG-domain proteins may increase cellular sensitivity to cisplatin.30 This phenomenon is commonly referred to as “repair-shielding.” The finding that HMGB4, a recently discovered HMG-domain protein that is preferentially expressed in the testis over other tissues,31 binds more strongly to platinated DNA than HMGB1, the protein first discovered to bind platinated DNA,24 may be relevant with respect to the hypersensitivity of testicular cancer to cisplatin.32
An additional degree of complexity is that, in eukaryotic cells, genomic DNA wraps around an octamer of four histone proteins, forming nucleosomes, which are further condensed into chromatin. Our investigations reveal that cisplatin damage decreases the mobility and alters the geometry of the DNA-nucleosome complex.33–35 Despite the decrease in mobility of platinated DNA in the nucleosomes, RNA polymerase can still transcribe until it reaches the sites of platinum damage.35 This property argues against the hypothesis15 that cisplatin inhibits transcription by preventing chromatin remodeling.
Monofunctional Platinum(II) Complexes
A complex of special importance to research in our laboratory is monofunctional phenanthriplatin, cis-[Pt(NH3)2(phenanthridine)Cl](NO3) (Chart 2, phenanthridine N-donor),36 where the term monofunctional refers to its ability to bind to DNA through only one coordination site, that of the chloride ligand. Although a large number of monofunctional compounds of the form cis-[Pt(NH3)2(L)Cl]+ have been prepared in which L is an N-heterocycle,36–49 phenanthriplatin is one of only a few that display in vitro cytotoxicity greater than that of cisplatin across a broad range of cancer cell types. We describe here the sequence of experiments and discoveries by which other laboratories and we came to focus on monofunctional platinum complexes. The line of research that culminated in our interest in the potential of such compounds for treating cancer, and the discovery and investigation of phenanthriplatin as a lead candidate for clinical development, are highlighted.
Chart 2.
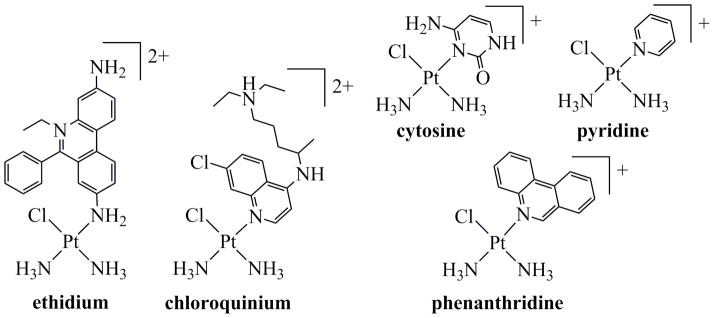
Monofunctional Platinum Complexesa
aDesignations refer to the names of the N-donor ligands.
The monofunctional platinum complexes in which we are currently interested typically contain two am(m)ine ligands, one leaving group ligand, and a heterocyclic N-donor. Before our recent work on these compounds, however, we had investigated a number of monofunctional complexes containing exocyclic N-donor atoms on the heterocycle. The following discussion relates the manner by which we came to study these compounds and the eventual progression towards analogues containing endocyclic N-donor heterocyclic ligands. Chart 2 depicts several structures that mark key developments in the course of this research.
Early Work on Monofunctional Compounds Containing Exocyclic N-Donor Ligands
Early contributions to the field of platinum anticancer drug research from our laboratory improved our understanding of the mechanism of action of cisplatin and related compounds,50–53 a line of research continued to the present day.15,54 It is now generally accepted that nuclear DNA is a target of cisplatin and that the drug primarily exerts its biological effects through formation of bifunctional DNA adducts.51,55 Cisplatin is typically administered in combination with other anticancer drugs,56 however, including DNA intercalators57 such as doxorubicin. In the 1980’s, in an attempt to probe the manner by which platinum compounds and intercalators may work in concert, we studied the platination of DNA containing the classical intercalator ethidium.58 When DNA was treated with cisplatin in the presence of ethidium, the presence of this cationic intercalator altered the pattern of platination on the duplex. In particular, it was proposed that this alteration might arise from a switch in the DNA binding mode from bifunctional to monofunctional.58
Subsequent work confirmed that the binding of cisplatin to DNA containing intercalated ethidium resulted in the formation of monofunctional platinum-DNA adducts.59 When the monofunctionally platinated DNA containing ethidium was extensively dialyzed, the intercalator could be removed, and the platinum proceeded to form bifunctional adducts. The ability to remove the ethidium by dialysis, and the lack of a reaction between cisplatin and ethidium when solutions of the two were mixed, led to the initial conclusion that no covalent bonds formed between them. As described below, subsequent experiments resulted in a re-evaluation of this conclusion.
Renewed interest in the area was sparked by a study confirming that DNA platination in the presence of intercalators removed the preference for binding to poly(dG) stretches.60 A difference was noted, however, between experiments carried out with acridine versus either ethidium or proflavin as intercalators. Acridine could be readily removed from DNA that had subsequently been platinated by tert-butanol extraction, filtration at acidic pH, or thin layer chromatography at basic pH. Neither ethidium nor proflavine could be removed in this manner and were therefore referred to as “tightly-bound.” These tightly bound intercalators underwent slow exchange at 37 °C, consistent with prior work.59 The strength of the interaction, however, led to the proposal that a ternary complex formed involving the intercalator, DNA, and the cis-{Pt(NH3)2}2+ unit. Further studies provided supporting evidence for ternary complex formation.61 In particular, fluorescence measurements revealed that, after platination, the intercalated ethidium no longer emitted, consistent with the presence of an interaction with platinum, which quenched the luminescence by the heavy atom effect.
Prompted by these results, we prepared, isolated, and characterized cis-[Pt(NH3)2(Etd)Cl]2+, where Etd is the ethidium cation (Chart 2).62 The characteristics of the optical absorption spectrum of this compound match those of DNA platinated in the presence of ethidium, confirming coordination of Etd to platinum in the latter instance. Moreover, DNA adducts formed by cis-[Pt(NH3)2(Etd)Cl]2+ released ethidium following incubation at 37 °C, as observed for the postulated ternary complexes in the earlier studies. Because cisplatin and ethidium do not interact in the absence of DNA, it was proposed that DNA acts to promote ligand substitution of chloride for ethidium following preliminary platinum aquation and binding to the duplex. Molecular mechanics computations were used to model a DNA hexamer containing an intercalated ethidium ion, platinated at the N7 position of a deoxyguanosine residue adjacent to the intercalator. The resulting energy minimized structure illustrates the manner in which the DNA preorganizes the two molecules for ligand substitution chemistry (Figure 2). In summary, cisplatin effectively cross-linked the intercalator and an adjacent deoxy-guanosine residue, similar to the manner by which it typically cross-links adjacent deoxyguanosines.
Figure 2.
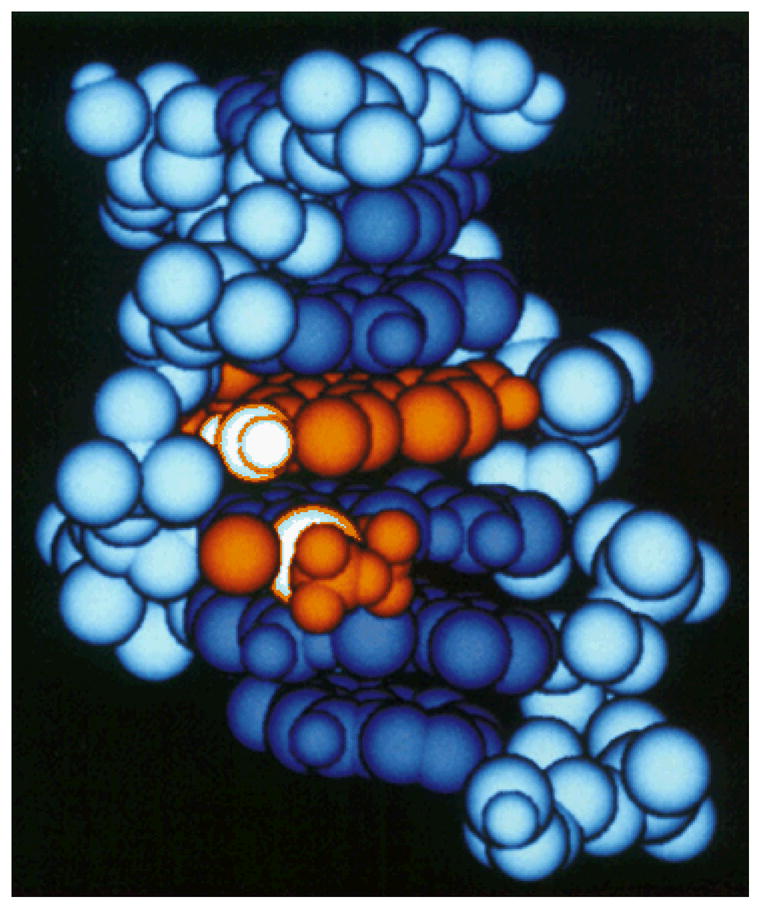
Molecular mechanics energy minimized structure of ethidium intercalated into platinated DNA.62 Copyright American Chemical Society, 1988.
An examination of the platinum ethidium complex in Chart 2, reveals that the ethidium can bind to the platinum through either of its two exocyclic amines, formally N3 and N8. Owing to the similar spectroscopic features of the N3 and N8 linkage isomers, firm experimental evidence as to which, if either, linkage isomer was favored in the presence of DNA had not been obtained. This situation was rectified by our discovery of the differential thermochromic behavior of the two isomers of cis-[Pt(NH3)2(Eth)Cl]2+.44 These studies indicated that DNA favors formation of the N8 regioisomer, the one depicted in Chart 2.
Monofunctional Compounds Containing Endocyclic N-Donor Ligands
Structure-activity relationships (SARs) established in the drug development community stated that monofunctional platinum compounds were not likely to be of clinical significance.63 Most of the compounds to enter into clinical trials in the early years of platinum anticancer drug research obeyed these traditional SARs, which required for activity charge neutrality, a square-planar coordination geometry, the presence of a pair of inert ligands cis to one another in the coordination sphere, and a pair of labile ligands in the remaining two sites.4,64
How did these SARs arise? Following the initial discovery of cisplatin as an anticancer drug, many platinum compounds were evaluated, and those that inhibited tumor growth in animals were used to establish the rules for activity.1,2 Monofunctional complexes such as [Pt(NH3)3Cl]+ and [Pt(dien)Cl]+ were inactive both in vivo and in vitro, consistent with the SARs.63,65–69 Although these particular complexes are inactive, it was also recognized that cisplatin and its analogues are not effective against all types of cancers, and this deficiency spurred the search for platinum complexes that deviated significantly in structure from cisplatin in order to find complexes that might overcome inherent or acquired drug resistance.70
One such program, active at about the time when the aforementioned studies of the ternary Pt-DNA-ethidium complex were being carried out, was being undertaken at Engelhard Industries. In a seminal paper by Stern, Hollis, and Amundsen, complexes of the type cis-[Pt(NH3)2(Am)Cl]+, where Am is an N-donor ligand derived from pyridine, purine, pyrimidine, or aniline, were described that displayed significant tumor cell growth inhibition in vitro and in L1210 and P388 mouse leukemia models.37 Although charged and carrying only one labile ligand, features incommensurate with the traditional SARs, these compounds had significant activity.
In a parallel set of experiments, we had prepared a pair of platinum(II) complexes of general formula cis-[Pt(NH3)2(Int)Cl]+, where Int is an intercalating moiety.43 In this study, the intercalators used were 9-aminoacridine and chloroquine. The former coordinates to platinum through its exocyclic amine and the latter through the endocyclic nitrogen atom. For purification purposes, the chloroquine ligand was protonated, resulting in the isolation of a complex of the chloroquinium ion (HCQ+). These complexes belonged to the cis-[Pt(NH3)2(Am)Cl]+ family of complexes found to be active by the Engelhard group and so preliminary animal studies were carried out. The levels of toxicity were considered to be prohibitive, however, and so their biological activity was not explored further. 43
We briefly highlight here that the compound cis-[Pt(NH3)2(N1-HCQ)Cl](NO3)2 (Chart 2) was the first complex of the form cis-[Pt(NH3)2(Am)Cl]n+ from our laboratory, where Am is an N-heterocyclic ligand, with the exception of the α-pyridone complex cis-[Pt(NH3)2(C5H4NOH)Cl](NO3).71,72 Studies of the latter, performed by Hollis while he was a graduate student in our group, were part of an extensive program to understand the chemistry of platinum blues,73,74 which did not include investigations of their biological activity.
In collaboration with the researchers at Engelhard Corporation we next investigated the nature of the interaction of cis-[Pt(NH3)2(Am)Cl]+ complexes with DNA.75,76 It was of interest, inter alia, to examine the possibility that the Am ligand was lost upon DNA binding, facilitating bifunctional coordination. It was not easy to let go of the prevailing SARs! Select members of the series were able to block replication in vitro, however. With the use of “replication mapping,” a technique previously devised by us to investigate bifunctional adducts formed by cisplatin,66 we quickly learned that the Engelhard compounds inhibited DNA polymerases at individual guanine residues. This result stood in contrast to that obtained with cisplatin, whereby DNA replication typically halted at d(Gp)n sequences, where n ≥ 2. The implication was that this new class of compounds did indeed form monofunctional adducts on DNA and that these compounds were functionally quite different from the simpler analogues, like [Pt(dien)Cl]+, which are unable to inhibit the progression of a DNA polymerase.66
The hypothesis that the biological activity of these complexes arises from monofunctional adducts was corroborated by the failure of antibodies, raised against DNA containing bifunctional platinum adducts, to recognize the lesions formed by cis-[Pt(NH3)2(Am)Cl]+. Formation of a bifunctional adduct from this complex would require the loss of either an Am or NH3 ligand, and it was observed that, only under conditions where cis-[Pt(NH3)2(4-Br-pyridine)Cl]+ was heated to 37 °C for 14 days in phosphate buffered saline (PBS), was NH4+ released, forming trans-[Pt(NH3)(4-Br-pyridine)Cl2]. Such harsh conditions are physiologically irrelevant, and trans-[Pt(NH3)(4-Br-pyridine)Cl2] displayed no anticancer properties, ruling out its potential role as an obligatory intermediate in this activity. Finally, NMR spectroscopic and high performance liquid chromatographic characterization of the products of the reaction of cis-[Pt(NH3)2(N3-cytosine)Cl]+ (Chart 2) with one molar equivalent of d(GpG) revealed the platinum complex to bind to only one of the guanine bases.75,76
An interesting observation was that binding of cis-[Pt(NH3)2(N3-cytosine)Cl]+ to d(GpG), or even to deoxyguanosine, produced species that displayed fluxionality as revealed by variable temperature NMR studies. The 1H NMR signals of the product were split but coalesced with increasing temperature to 70 °C. This result was interpreted to denote the presence of two interconverting rotational isomers arising from the asymmetry of the cytosine ring with respect to the coordination plane. cis-[Pt(NH3)2(4-methylpyridine)(dG)]2+, in which the 4-methylpyridine is symmetric about the platinum coordination plane, exhibited a single set of sharp NMR signals.
These results, indicating that these complexes did indeed bind DNA and exert their biological effects through monofunctional adduct formation, were confirmed by work carried out in the Reedijk group.77 They used NMR spectroscopy to investigate the interaction of cis-[Pt(NH3)2(4-methylpyridine)Cl]Cl with d(GpG) and found that the platinum complex binds to the N7 position of only one guanine residue. They also found no evidence for release of ammonium ion, which would be required if a bifunctional adduct were to form. The differential occurrence of mutations in bacteria treated with cisplatin versus cis-[Pt(NH3)2(4-methylpyridine)Cl]Cl was also investigated, and it was confirmed that distinct types of DNA adducts are formed by these species.77
We next continued our studies of monofunctional platinum complexes by interrogating whether DNA that was site-specifically modified by cis-[Pt(NH3)2(N3-cytosine)Cl]+ was bent.78 DNA bending induced by bifunctional binding of cisplatin was believed to be intimately tied to the manner by which it exerted its anticancer activity. Using an electrophoretic mobility shift assay (EMSA), we determined that DNA oligomers platinated with cis-[Pt(NH3)2(N3-cytosine)Cl]+ remain rigid and rod-like, in contrast to oligomers platinated with cisplatin, which are bent by 32–34°. At the same time, we reported the discovery of SSRP1, a DNA damage-recognition protein from mammalian cells that bound specifically to intrastrand d(GpG) and d(ApG) cross-links formed by cisplatin.79 This protein, later identified to be a high mobility group box (HMGB)-containing protein,24 did not, however, bind to the adduct formed by cis-[Pt(NH3)2(N3-cytosine)Cl]+. The lack of DNA bending by the monofunctional adduct was believed to be the reason for its different recognition by SSRP1, pointing to a differing mechanism of action. A further EMSA study employing supercoiled plasmid DNA80 revealed that, not does cis-[Pt(NH3)2(4-bromopyridine)Cl]+ bend DNA less, it also unwinds it less than bifunctional compounds like cisplatin.
More Recent Developments
Over the next 16 years, platinum research conducted in our laboratory proceeded in other directions, with no further work being carried out on a monofunctional compound except for the occasional inclusion of complexes like [Pt(dien)Cl]+ as controls in studies of cisplatin and other bifunctional complexes. In 2006, however, during the course of a collaboration with the Giacomini group at UCSF on the role that the organic cation transporters (OCTs) play in the uptake of oxaliplatin,81 we were inspired to re-examine monofunctional platinum complexes. The Giacomini laboratory were pursuing the hypothesis that overexpression of OCTs in colon cancer cells might explain the efficacy of oxaliplatin in the treatment of this disease. During the collaboration, we wondered how oxaliplatin, itself not a cation, might be recognized by OCTs and were curious to examine actual cationic platinum complexes containing ligands with organic character for uptake by OCTs. Many such candidates were readily available in our laboratory, and one of these, cis-[Pt(NH3)2(pyridine)Cl]+ or CDPCP for cis-diamminepyridinechloro-platinum(II), was especially remarkable in this regard (Chart 2). Its reinvestigation marks the beginning of our renewed interest in monofunctional platinum compounds.82
CDPCP was one of the cis-[Pt(NH3)2(Am)Cl]+ species initially reported by Hollis and coworkers in 1989 and bore structural similarities to many of the monofunctional compounds that we had previously studied. With its positive charge and organic pyridine ligand, this complex served as an excellent substrate for OCTs 1 and 2. Cells that overexpressed these transporters were more sensitive to treatment with CDPCP, or pyriplatin as we subsequently referred to the complex, than cells that did not. The differential in cell killing (87 to 137-fold) was greater than that of oxaliplatin (12 to 53-fold), which is taken up by the same transporters.
An EMSA analysis of the interaction of pyriplatin with closed-circular supercoiled plasmid DNA revealed that its monofunctional adducts did not significantly unwind the duplex. An X-ray structure analysis of a DNA dodecamer duplex site-specifically platinated at the N7 position of a central guanosine residue (Figure 3) revealed the presence of B-form DNA with the platinated guanine fully base- paired with its cytosine complement in the opposite strand. A study of the effect of this adduct on transcription revealed that pyriplatin placed on the template strand blocks progression of RNA polymerase II (pol II). We additionally found that, although the lesion is repaired by the nucleotide excision repair (NER) machinery of the cell, the pathway is much less effective than in the repair of bifunctional cisplatin adducts.82
Figure 3.
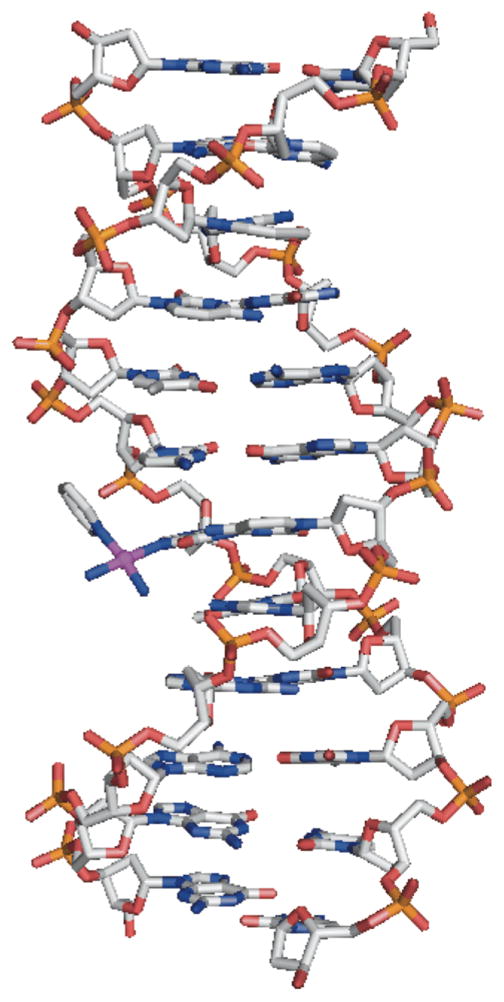
Diagram of a DNA dodecamer platinated with pyriplatin as revealed by X-ray crystallography. PDB code: 3CO3.82
More information about the mechanism by which pyriplatin disrupts cell function was obtained through an analysis of the crystal structure of transcribing RNA pol II stalled at the site of a pyriplatin monofunctional adduct (Figure 4A).83 The structure revealed that the platinated nucleotide captures pol II in a post-translational state, where the addition site is empty and ready to be loaded and the platinated guanosine residue sits directly above the bridge helix of the enzyme. The cis-[Pt(NH3)2(pyridine)]2+ unit is rotated around the Pt–Npy bond with respect to that observed in the structure of the dodecamer duplex described above so (Figure 3) as to avoid unfavorable steric interactions. Another structure was solved with CMP present in the addition site (Figure 4B). Although Watson-Crick base-pairing can occur between CMP and the platinated guanosine, the complex remained stalled in a pre-translocation site, potentially due to steric hindrance involving the pyridine ring. An interesting feature is that, in both the dodecamer structure and the structure of the platinated DNA stalled pol II with an empty addition site, the pyridine ring points towards the 5′ end of the platinated strand, but in the structure with CMP filling the active site, the pyridine ring is directed toward the 3′ end of the platinated strand.
Figure 4.
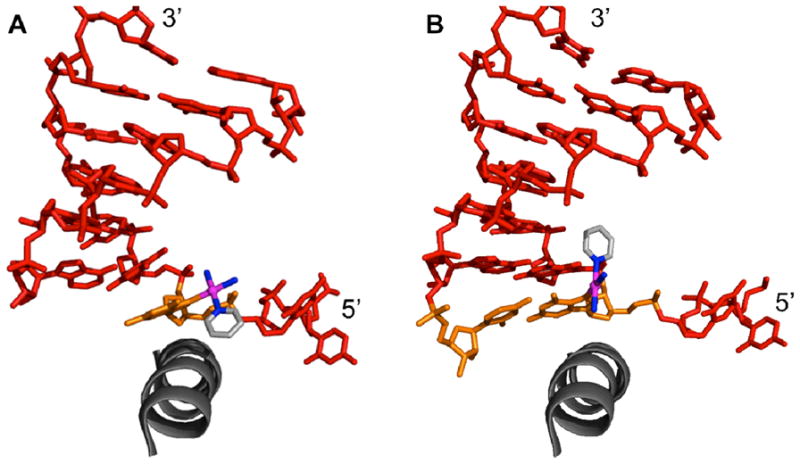
Diagrams of duplex DNA from crystal structures of transcribing RNA pol II stalled at the site of pyriplatin binding. A) Addition site opposite platinated G (orange) is empty and the pyridine ring is directed toward the 5′ end of the platinated strand. PDB code: 3M4O B) Addition site occupied by CMP (orange) with pyridine now directed more toward the 3′ end of the platinated strand. PDB code: 3M3Y.
With this information in hand, detailed studies were performed to determine the potential of pyriplatin as an anticancer agent.84 Its cytotoxicity was evaluated in the NCI-60 panel of human cancer cell lines, and a subsequent analysis by the online COMPARE algorithm revealed that the spectrum of activity of pyriplatin is unrelated to that of either cisplatin/carboplatin or oxaliplatin. However, the overall degree of toxicity, or potency, was an order of magnitude less for pyriplatin. A flow cytometry study showed that pyriplatin, like cisplatin and oxaliplatin, induces a cell cycle block at G2-M and induces apoptosis. This result was further confirmed by Western blotting for phosphorylated H2AX and Chk2 following treatment of HOP-62 cells with pyriplatin.84 The genetic effects induced by pyriplatin were compared to those for cisplatin and oxaliplatin. Among the differences was a decrease in the levels of ERCC1 mRNA in cells treated with pyriplatin. The protein encoded by this mRNA plays a role in NER, and lower levels are consistent with earlier results showing repair of pyriplatin adducts to be less efficient than that of cisplatin adducts. Finally, a synergistic effect was observed when cells were treated with both cisplatin and pyriplatin, strongly supporting the conclusion that these compounds operate by different mechanisms. A more detailed study revealed that the main pathway for repair of monofunctional pyriplatin adducts is NER and not mismatch repair or double-strand break repair.85 The ability of pyriplatin to efficiently inhibit transcription in a variety of cancer cell lines was also evaluated using both globally and site-specifically platinated plasmids.
The results of these studies indicated that pyriplatin operates by a mechanism of action distinct from that of cisplatin, albeit with less potency. Pyriplatin itself is therefore not likely to be taken forward, but it did serve as a lead compound in a project designed to capitalize on and maintain its differentiated spectrum of activity but to improve its potency. In pursuit of this idea, we recently generated a small library of compounds with various N-heterocyclic Am ligands in cis-[Pt(NH3)2(Am)Cl]+.36 The choice of ligands was guided by the crystallographic results described above and involved increasing the steric bulk of the Am ligand to further increase transcription inhibition and consequential cytotoxicity. Of the compounds generated, the phenanthridine complex, cis-[Pt(NH3)2(phenanthridine)Cl](NO3) or phenanthriplatin (Chart 2), was most potent. It exhibited IC50 values, the concentration required to inhibit cell growth by 50%, 4 to 40 times lower than those of cisplatin or oxaliplatin. Evaluation in the NCI-60 cancer cell line panel confirmed that the spectrum of activity of phenanthriplatin differs from those of other platinum anticancer agents. Its cellular uptake is enhanced over that of either cisplatin or phenanthriplatin, and the bulky ligand provides the metal center with a measure of protection from deactivating thiols, a feature similar to that designed to occur with picoplatin.86 Phenanthriplatin also inhibits transcription as effectively as cisplatin. Work is currently underway in our lab to further investigate the mechanism of action of phenanthriplatin and to devise improved analogs.
Platinum(IV) Anticancer Complexes
Properties and Mechanism of Action
The exploration of platinum(IV) complexes as potential anticancer agents is an active field of research, summarized in several recent reviews on the topic.87–91 Although the clinical potential of such complexes has only been realized within the last decade, Pt(IV) compounds were identified as having anticancer activity in the early days of cisplatin research.2 The properties of platinum(IV) complexes differ substantially from those of platinum(II). In contrast to the four-coordinate square-planar geometries of platinum(II) complexes, platinum(IV) complexes are typically six-coordinate with octahedral geometries. The saturated coordination sphere of low-spin d6 platinum(IV) complexes renders them kinetically more inert than those of platinum(II). These differences have been exploited in the pursuit of more effective platinum anticancer drug candidates. The kinetic inertness helps to prevent diversion to off-target biological nucleophiles before the platinum complex can reach the purine bases in nuclear DNA. The presence of two additional ligands enables modification of the chemical properties and provides attachment points for cancer-targeting units and conjugation to nanoparticles and other carriers.
Because most platinum(IV) complexes do not directly engage in ligand substitution reactions on a therapeutically relevant time scale (hours), reduction to platinum(II), accompanied by the loss of two ligands, is typically required for binding to biological targets.92,93 Once reduced, the mechanism of action of platinum(IV) anticancer agents resembles that of platinum(II) complexes, with binding to genomic DNA and inhibition of transcription and replication. Important features of uncertainty regarding this process are the timescale and location of the reduction event, the identity of the reducing agent(s), and the chemical nature of the reduction products. These factors have a great influence on the ultimate efficacy of a platinum(IV) complex compared to that of its platinum(II) reduction products.
Platinum(IV) complexes are typically potent oxidizing agents.94,95 As a consequence, the hypoxic environments of cancer cells and tissues, which contain a high concentration of reducing agents, enables the facile reduction of Pt(IV). Because of the significant structural change from octahedral to square planar geometry that occurs upon reduction, kinetic barriers most likely limit this process. Cyclic voltammograms of such complexes display only irreversible reductions,96 presumably due to a combination of slow heterogeneous reduction kinetics and ligand dissociation. The reduction of platinum(IV) complexes by small-molecule agents, like ascorbic acid and glutathione, can occur by both inner- and outer-sphere electron transfer mechanisms.97–101 The operative mechanism depends on the stereochemical arrangement and nature of ligands within the platinum(IV) coordination sphere. Hydroxide and halide ligands can serve as bridges to facilitate inner-sphere electron transfer chemistry. An additional requirement for inner-sphere electron transfer is that the bridging ligand be trans to a good leaving group.99 Thus satraplatin, which contains two chloride ligands trans to strongly bound am(m)ines, undergoes reduction by ascorbate and glutathione by a slow, outer-sphere mechanism.99
Although small-molecule reducing agents can reduce and activate platinum(IV), the identity of in vivo reductants remains to be defined conclusively. Some studies investigating reduction of platinum(IV) complexes in cancer cell extracts revealed that high molecular weight intracellular components, mainly proteins, are responsible for reduction.102 Another factor is that the reduction products often comprise a mixture of species, depending on ligands available to be lost.103 For complexes that undergo inner-sphere reductions, however, elimination of two trans ligands appears to occur exclusively.99,104
Synthetic Chemistry of Platinum(IV) Complexes
Oxidation of platinum(II) complexes with aqueous hydrogen peroxide affords trans dihydroxo platinum(IV) complexes.105 These complexes are useful for preparing a number of other derivatives, because the hydroxide ligands are nucleophilic and react with electrophiles, providing access to new compounds without the need for ligand substitution chemistry.106 The use of alcohols and carboxylic acids as solvents for the oxidation of platinum(II) complexes by hydrogen peroxide usually affords mixed trans hydroxo-alkoxo or hydroxo-carboxylato complexes (Scheme 2).107–112
Scheme 2.
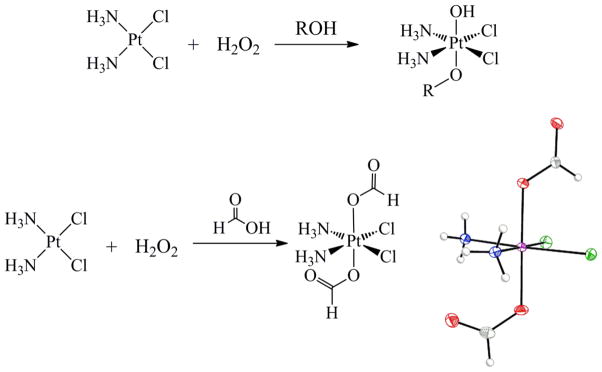
Oxidation Reactions of Cisplatin with Hydrogen Peroxide in Protic Solvents*
*The molecular diagram of cis,cis,trans-[Pt(NH3)2Cl2(O2CH)2] is depicted with thermal ellipsoids drawn at the 50% probability level. Full crystallographic details are provided in Supporting Information in CIF format.
If a sufficiently acidic carboxylic acid is used, however, dicarboxylato species form, presumably with protonation of the second hydroxo ligand, which leaves as water.113 The oxidation of cisplatin in formic acid, for example, cleanly generates the diformate complex with no evidence of monoformate formation. Stirring the dihydroxo complex, cis,cis,trans-[Pt(NH3)2Cl2(OH)2] in formic acid also produces this product, albeit of lower purity. This observation is consistent with the relatively low pKa (3.77) of formic acid. An optimal synthesis and characterization of the diformate complex cis,cis,trans-[Pt(NH3)2Cl2(O2CH)2] is given in the Experimental Section. Full crystallographic characterization is provided in the Supporting Information (SI).
Carboxylic acid anhydrides, isocyanates, acid chlorides, and pyrocarbonates are additional electrophiles that react with platinum(IV) hydroxides (Scheme 3).106,114,115 The products of such reactions are platinum(IV) carboxylates, carbamates, or carbonates. Another reaction of importance is that of platinum(IV) hydroxides with cyclic anhydrides.116–118 The resulting platinum(IV) dicarboxylates contain terminal, uncoordinated carboxylic acid functional groups that can be readily modified by standard amide-bond-forming chemistry (Scheme 3).118,119
Scheme 3.

Nucleophilic Reactivity of cis,cis,trans-[Pt(NH3)2Cl2(OH)2]
Because platinum(IV) complexes are kinetically inert to direct ligand substitution, these strategies, which use outer-sphere functionalization, are the preferred methods for preparing novel platinum(IV) complexes with potentially biologically compatible ligands.
Platinum(IV) Complexes with Biologically Active Axial Ligands
By converting a molecule with a carboxylic acid functional group to an acid chloride or anhydride, it can readily be attached to a platinum(IV) complex by the chemistry described above. Thus any compound that contains a carboxylic acid can in principle be attached to platinum(IV). Such modular reactivity is of particular interest for the design of dual-threat pharmaceutical agents, which combine two biologically active components into a single molecule. Ideally, the two compounds will have different intracellular targets so that resistance mechanisms protecting against one agent would not affect the activity of the other. Ethacrynic, valproic, and dichloroacetic acids are examples of biologically active carboxylates that have been coordinated to a platinum(IV) prodrug for this purpose (Chart 3). Ethacrynic acid is an inhibitor of the enzyme glutathione-S-transferase (GST), which catalyzes the conjugation of glutathione with xenobiotics and confers drug resistance to many cell types.120 Hence, a complex with two ethacrynic acid ligands (ethacraplatin) increased the efficacy of the released cisplatin by simultaneously inhibiting GST.121–123 Valproic acid inhibits histone deacetylase (HDAC), an emerging target for cancer therapy.122 The platinum(IV)-divalproate complex (VAAP) displayed effective in vivo anticancer activity, possibly due to a synergistic effect of the synchronized delivery of the HDAC inhibitor and cisplatin.124 The rationale for the design of mitaplatin, discovered and first investigated in our laboratory, is discussed next.
Chart 3.

Platinum(IV) Complexes with Biologically Active Carboxylate Ligands
Cancer cells operate by a unique cytosolic metabolic pathway known as “aerobic glycolysis,” a phenomenon commonly referred to as the Warburg effect.125 Non-malignant cells generate ATP primarily from the citric acid (TCA) cycle and oxidative phosphorylation, both of which operate in the mitochondria and rely on oxygen. In non-malignant cells, a small amount of ATP is also produced directly from glycolysis, but the main purpose of this pathway is to provide pyruvate. The pyruvate is then converted to acetyl-CoA in mitochondria and enters the citric acid cycle. Under hypoxic conditions, cells cannot produce ATP by the efficient oxidative phosphorylation pathway and instead must obtain their energy predominantly by glycolysis. The large amounts of pyruvate that accumulate as a result are transformed into lactic acid. Warburg discovered that, even in the presence of sufficient quantities of oxygen to produce ATP via the citric acid cycle and oxidative phosphorylation, cancer cells continue to employ glycolysis, giving rise to the term aerobic glycolysis.125 The distinctive operation of the glycolytic pathway in cancer cells provides a means of selectively targeting them for cancer therapy.126
Dichloroacetate (DCA) is a commercially available inhibitor of pyruvate dehydrogenase kinase (PDK).127 The ultimate effect of inhibiting PDK is an increase in the production of acetyl-CoA from pyruvate, which in turn initiates the citric acid cycle and oxidative phosphorylation. DCA thereby inhibits aerobic glycolysis, restores normal mitochondrial function, and decreases the mitochondrial membrane potential (Δψm).128,129 These effects act in concert to increase the ability of cancer cells to undergo apoptosis by facilitating release of apoptosis-inducing factor (AIF) from mitochondria.128,129 The platinum(IV) prodrug mitaplatin, cis,cis,trans-[Pt(NH3)2Cl2(DCA)2], was designed in our laboratory to simultaneously deliver cisplatin as a cytotoxic agent and two equivalents of DCA as apoptosis inducers.130
Against a panel of seven cancer cell lines, mitaplatin exhibited comparable or better cytotoxicity than cisplatin. Further studies revealed that, as planned, mitaplatin also depolarized the mitochondrial membrane and increased the release of AIF. By treating a co-culture of normal lung fibroblasts (MRC5) and lung carcinoma cells (A549) with mitaplatin or cisplatin, the selectivity of the former for cancer cells and tissues was modeled. These studies revealed that mitaplatin effectively killed the cancer cells, while leaving the normal cells untouched; cisplatin killed substantial quantities of both cell types. The DCA, arising from the axial ligands of mitaplatin, appears to selectively sensitize cancer cells to the platinum cytotoxic agent. Later studies by another research group investigated the efficacy of mitaplatin in cisplatin-resistant cell lines.131 Mitaplatin was more effective than cisplatin in resistant cell lines. The expected effects of DCA, such as decreased glucose uptake and inhibition of PDK, were observed.131 Synergism of DCA with commonly used platinum(II) anticancer drugs has also been noted.132–134 The concentrations of DCA required to see such effects, however, is between 2 to 10 mM, substantially higher than the low μM concentrations provided by mitaplatin. Thus mitaplatin may also act to increase cellular uptake of DCA and deliver it more selectively to its intracellular targets.
As discussed briefly above, there are a large number of DNA-binding proteins that selectively recognize Pt-DNA. One of these proteins, HMGB1, which contains an HMG-domain, can inhibit repair of cisplatin-DNA 1,2-d(GpG) intrastrand cross-links by effectively blocking DNA repair proteins from accessing the adduct.28 Overexpression of HMGB1 is thereby expected to increase the sensitivity of cells to cisplatin through such repair-shielding. There are conflicting data in the literature regarding the validity of this hypothesis, however.30,135 Recent studies from our group suggest that the ability of HMGB1 to sensitize cells to cisplatin can be modulated by the redox environment in the cell.136 In estrogen-receptor positive, ER(+) MCF-7 breast cancer cell lines, the addition of estrogen increases expression levels of HMGB1, which translates into an increase in the sensitivity of these cells to cisplatin, presumably because of repair shielding.30 With these results in mind, we rationally designed a platinum(IV) complex bearing axial ligands with appended estrogen moieties, using the strategy described above (Chart 4).118 The estrogen ligands upregulated HMGB1 expression levels and potentiated the action of cisplatin. The cisplatin-estrogen conjugates increase HMGB1 levels in the ER(+) (MCF-7) but not the ER(−) (HCC-1937) breast cancer cell lines.118 The increased expression level also correlated with lower IC50 values in the ER(+) cell lines.
Chart 4.

Platinum(IV) Complexes with Biologically Active Ligands Attached by Amide-Bond-Forming Reactions
The use of peptides to selectively target cancerous tissue has arisen as a viable strategy for generating less toxic, more effective chemotherapeutics.137,138 Recent reports describe the preparation and anticancer activity of platinum(IV) complexes tethered to bioactive peptides via their axial ligands.139–143 Our group has prepared several platinum(IV) complexes containing tri- and pentapetides containing either the RGD (Arg-Gly-Asp) or NGR (Asn-Gly-Arg) sequences.139 Peptides containing the RGD sequence are recognized by αvβ3 and αvβ5 integrins, and those containing the NGR sequence bind selectively to aminopeptidase N (APN).144 The αvβ3 and αvβ5 integrins and APN are overexpressed in the angiogenic vasculature characteristic of tumor tissue.145 The RGD and NGR peptides can therefore be used to selectively target angiogenic tumors over healthy tissue. Platinum(IV)-RGD and –NGR conjugates exhibited cytotoxicity close to that of cisplatin in cell lines having high expression levels of the αvβ3 and αvβ5 integrins.139
A larger, 36-residue peptide, chlorotoxin (CTX), was attached to a platinum(IV) prodrug in a separate set of experiments.142 CTX, a component of the venom of the Israeli desert scorpion, binds to a specific chloride channel protein (CLC-3) that is highly expressed in gliomas146 and also to annexin 2A, which is present on the surfaces of many cancer cell types.147 Like the RGD and NGR sequences described above, the use of CTX provides a method for selective delivery of a therapeutic to cancer cells. Although the Pt(IV)-CTX conjugate was less cytotoxic than cisplatin in three cell lines tested, it did exhibit enhanced cytotoxicity relative to its platinum(IV) disuccinate precursor.142 The greatest enhancement occurred in HeLa (human cervical cancer) cells, where it was approximately 200-fold more effective than the platinum(IV) disuccinate complex.142 This increased cytotoxicity was attributed to the targeting effect of the CTX peptide, because HeLa cells express both CLC-3148 and annexin 2A on their surface.149
Nanoparticle Delivery of Platinum(IV) Prodrugs
Nanotechnology for delivery of anticancer drugs is a rapidly expanding field with many benefits over traditional chemotherapeutic regimens.150 Nanodelivery devices take advantage of the leaky vasculature and poor lymphatic drainage of angiogenic tumor tissue. These features, common to most solid tumors, enable nanoparticles in the size range of 10–500 nm to selectively accumulate in the interstitial space of tumors by entering through pores of the leaky vasculature. Ineffective lymphatic drainage in tumors cannot remove nanoparticles, and the interstitial fluid in which they are suspended, at an appreciable rate. The tendency for nanoparticles to localize in tumors owing to these physiological deficiencies is commonly known as the enhanced permeability and retention, or EPR, effect. 151
Because of their widespread clinical use, platinum anticancer agents have been explored in conjunction with a variety of nanodelivery devices to exploit the EPR effect and provide more effective and less toxic formulations.152–166 Although several of these constructs utilize platinum complexes in the 2+ oxidation state,152,153,155,157,158,160,163–165 the field is trending toward the development of systems using platinum(IV) prodrugs. The two additional ligand binding sites available in platinum(IV) complexes provide covalent bond attachment points for fine tuning chemical and physical properties. Several examples of this strategy from our laboratory are discussed here.
Although most commonly known for applications in physical sciences, carbon nanotubes have gained increasing importance as selective drug delivery agents.167,168 In their unmodified forms, carbon nanotubes are poorly soluble and toxic,169 not useful for drug delivery. When functionalized with the appropriate ligands, however, carbon nanotubes can exhibit good solubility and biocompatibility with low apparent toxicity.170 Such carbon nanotubes are able to enter cells effectively, predominately through a mechanism of clathrin-dependent endocytosis.171 Platinum(IV) prodrugs were attached to single-walled carbon nanotubes (SWCNT) solubilized by PEGylated amines.111,172 Using either the mono- or disuccinate-platinum(IV) complexes, amide-bond forming chemistry enabled covalent attachment of the platinum payload onto the nanotube (Scheme 4).
Scheme 4.

Design and Preparation of Single Walled Carbon Nanotubes to Deliver a Platinum(IV) Prodrug*
*The molecular diagram of cis,cis,trans-[Pt(NH3)2Cl2(OCH2CH3)(OH)] is depicted with thermal ellipsoids drawn at the 50% probability level. Full crystallographic details are provided in Supporting Information in CIF format.
In a study using human testicular cancer cells (N-Tera2), Pt(IV)-nanotube constructs were able to deliver higher amounts of platinum to the cell and exhibited cytotoxicity greater than that of cisplatin, when measured on a per platinum basis.111 A Pt(IV)-SWCNT conjugate was designed that selectively targeted cancer cells. To accomplish this goal, we exploited the fact that many different cancer types overexpress the folate receptor (FR) by incorporating a folate molecule into the construct. Aided by both the high cellular uptake properties of the carbon nanotube and the targeting properties of the folate molecule, this second generation analogue was able to induce cell death in FR(+) cell lines more effectively than cisplatin.172 Encapsulation of a hydrophobic platinum(IV) complex within the interior of multi-walled carbon nanotubes (MWCNT) was recently described.173 Although the in vitro cytotoxicity of this material was not reported, it demonstrates the feasibility of full encapsulation of platinum(IV) complexes in nanotubes, providing a novel strategy for drug delivery.
Gold nanoparticles (AuNP) are another class of materials that have recently attracted attention for their use in medicine.174 Several PEG-functionalized AuNPs and gold nanorods have been derivatized with platinum(II)160,164 and platinum(IV) anticancer agents,162,166 respectively. Our group reported the attachment of a platinum(IV) prodrug to polyvalent DNA-functionalized gold nanoparticles (DNA-AuNP) (Scheme 5).175 DNA-AuNPs, pioneered by the research group of Mirkin, are advantageous over other AuNPs with different surface-passivating ligands.176 For example, DNA-AuNPs are taken up effectively by all cell lines tested thus far and induce no toxicity. The Pt(IV)-DNA-AuNP were prepared with standard amide-bond-forming reactions and were tested against several different cancer cell lines, revealing a cytotoxic potential greater than that of cisplatin.175 Because the oligonucleotide sequences of DNA-AuNPs can be used to regulate protein expression levels by binding to complementary mRNA sequences,177 future Pt(IV)-DNA-AuNP constructs may be designed to deliver cytotoxic platinum prodrugs while simultaneously knocking down the expression of proteins that confer cellular resistance to these drugs.
Scheme 5.

Design and Preparation of Gold Nanoparticles to Deliver a Platinum(IV) Prodrug*
*Molecular diagram of cis,cis,trans-[Pt(NH3)2Cl2(O2CH2CH2COOH)(OH)] is depicted with thermal ellipsoids drawn at the 50% probability level. Full crystallographic details are provided in Supporting Information in CIF format.
The final category of nanodelivery devices investigated by our laboratory is comprised of polymeric nanoparticles. Although any material composed of polymers and exhibiting length scales less than 1 μm qualifies as a polymeric nanoparticle, the term is typically reserved for structures that are composed of self-assembled polymer chains.178 The properties of the resulting nanoparticle can be tuned by altering the chemical structure of the polymer.179 The particles first employed by our group were based on the self-assembly of amphiphilic block copolymers to generate polymeric micelles.180 The block copolymer was a carboxy-terminated poly(D,L-lactic-co-glycolic acid)-block-poly(ethylene glycol) (PLGA-PEG-COOH).181 The PLGA portion of the polymer was chosen for its biodegradability and lack of toxicity.182 The hydrophobic PLGA core of nanoparticles formed by PLGA-PEG-COOH can also serve as a reservoir for the encapsulation and controlled release of hydrophobic molecules.183 The PEG portion of the PLGA-PEG-COOH provides a hydrophilic shell to aid in water solubility and biocompatibility. PEG coatings reduce rates of opsonization, thereby increasing blood circulation time.184 Finally, a pendant carboxylate was incorporated to provide a chemical handle for attaching active targeting agents.181
The first example from our laboratory employed PLGA-PEG-COOH nanoparticles, employed a construct targeted to the prostate specific membrane antigen (PSMA) and capable of encapsulating and releasing the complex cis,cis,trans-[Pt(NH3)2Cl2(hexanoate)2].185 This particular platinum(IV) prodrug was chosen because its axial ligands are sufficiently hydrophobic to allow it to be encapsulated within the PLGA core (Scheme 6). The pendant carboxylates on the surfaces of the nanoparticles were coupled to RNA aptamers selected to bind to PSMA, thereby actively targeting prostate cancer cells that overexpress this protein on their cell surface.186,187 The endocytotic uptake of the nanoparticles by the PSMA(+) LNCaP prostate cancer cells was confirmed microscopically by observing colocalization of fluorescently labeled nanoparticles and the early endosomal marker EEA-1. Subsequent formation of platinum-induced 1,2-intrastrand d(GpG) cross-links on nuclear DNA was confirmed with the use of monoclonal antibodies raised against this adduct.188 This nanoparticle construct was also evaluated in vivo.189 Pharmacokinetic studies revealed persistent platinum circulation in the blood of rats treated with the nanoparticle construct compared to treatment with an equivalent dose of cis,cis,trans-[Pt(NH3)2Cl2(hexanoate)2] alone. Moreover, encapsulation diverted platinum accumulation from the kidneys, a traditional site of platinum-based toxicity.190 Treatment with the targeted nanoparticle inhibited growth of a subcutaneous LNCaP tumor xenograft at 1/3 of the concentration of cisplatin needed to obtain an equivalent effect.
Scheme 6.
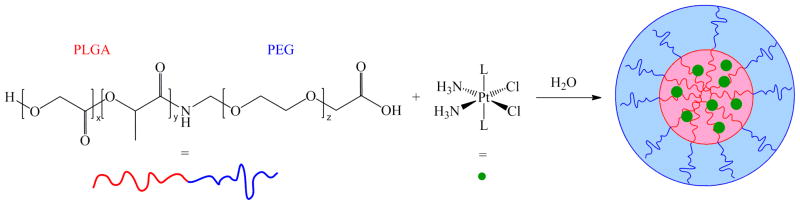
Design and Preparation of Polymer Nanoparticles to Deliver a Hydrophobic Platinum(IV) Prodrug
A similar approach was used to deliver cis,cis,trans-[Pt(NH3)2Cl2(hexanoate)2] to an orthotopic human breast cancer xenograft model in vivo with a c(RGDfK) targeted PLGA-PEG-COOH nanoparticle.191 The cyclic pentapeptide c(RGDfK) binds to αvβ3 integrins overexpressed on the surfaces of angiogenic epithelial cells and many cancers, as described above. The in vitro cytotoxicity of the construct was not only higher than that of cisplatin but also than that of the small molecule Pt(IV)-RGD conjugate. In the animal tumor model, treatment with cisplatin or the nanoparticle construct at equivalent doses of platinum resulted in equivalent degrees of inhibition of tumor growth. There was again, however, significantly reduced accumulation of platinum in the kidneys of the animals treated with the nanoparticle construct.
Although the platinum(IV) prodrug cis,cis,trans-[Pt(NH3)2Cl2(hexanoate)2] has proved to be satisfactory for these studies, we were interested to study the effect of systematically varying ligand hydrophobicity on the degree of NP encapsulation. A series of complexes cis,cis,trans-[Pt(NH3)2Cl2L2] was prepared in which L = CnH2n+1CO2−, n = 1–9.192 The lipophilicities of these compounds were evaluated by reversed-phase HPLC. The degree of encapsulation varied linearly with the lipophilicity of the complex, but solubility placed limitations on the amount of platinum that could be successfully loaded into PLGA-PEG-COOH polymeric micelles.
The encapsulation approach described above relies on the hydrophobicity of the platinum compound to be delivered. We have, however, developed a number of water-soluble platinum complexes, and we were interested to investigate the effect of nanoparticle encapsulation on the properties of these complexes. Mitaplatin, for instance, cannot be encapsulated in the manner described above. To prepare a PLGA-PEG-COOH nanoparticle containing this complex a different strategy was employed utilizing double emulsion.193 This nanoparticle construct provided mitaplatin with prolonged circulation time in the bloodstream and, as in the previous instances, diverted platinum accumulation from the kidneys. The efficacy of the nanoparticle construct was compared with that of un-encapsulated mitaplatin in a mouse xenograft model of triple-negative breast cancer. Although the free agent has a greater acute effect on tumor growth, in the long term, equivalent doses of platinum, either encapsulated or free, result in equivalent degrees of tumor growth inhibition.
The nanoparticle constructs described above employ non-covalent interactions to encapsulate the platinum compounds to be delivered. It is possible, however, to covalently conjugate a platinum complex to a polymer backbone. To investigate this alternative delivery strategy, the platinum(IV) prodrug cis,cis,trans-[Pt(NH3)2Cl2(O2CCH2CH2COOH)(OH)] was attached through the succinate to a poly(lactic acid) modified with pendant hydroxyl groups (PLA-OH).194 The resulting platinated polymer, PLA-Pt(IV), was then encapsulated within the core of a PLGA-PEG-COOH nanoparticle (Scheme 7). The core of this construct can more appropriately be viewed as a polymer blend. The pendant carboxylates on the surface of this nanoparticle were functionalized with RNA aptamers to target PSMA. In vitro studies showed that these nanoparticles are taken up by endocytosis and capable of releasing the prodrug, which forms 1,2-intrastrand d(GpG) cross-links on nuclear DNA.
Scheme 7.

Design and Preparation of Polymer Nanoparticles to Deliver Polymer-Conjugated Platinum(IV) Prodrugs
Conclusions and Future Directions
The widespread success of cisplatin as the first clinically approved transition metal-based anticancer drug significantly altered the paradigm of traditional medicinal chemistry, which previously focused primarily on organic drugs. We hope that the present survey of studies, mainly from our laboratory, have convinced the reader that, despite the fact that cisplatin has been in use for over 40 years, rational improvements to existing platinum-based anticancer agents can be made. Many other groups have similarly worked to design and implement platinum and other transition metal constructs as novel drug candidates. These advances have been aided by both a careful analysis of older literature, in the case of the monofunctional complexes, as well as utilization of new scientific discoveries in the field of nanoscience. A viable strategy for further advancement of platinum anticancer agents lies in both a careful reevaluation of compounds previously considered to be inactive, and the merging of these agents with drug delivery devices. Given the vibrant research activity in this field, and the pressing need for better cancer therapies, many new discoveries, and potentially the next FDA-approved platinum-based drug, are on the horizon. What are greatly needed are mechanisms to move such compounds past the stage of pre-clinical development into clinical trials. We look forward to future progress in this regard.
Experimental
Synthesis of cis,cis,trans-[Pt(NH3)2Cl2(O2CH)2]
Cisplatin (100 mg, 0.33 mmol) was suspended in 4 mL of formic acid and 220 μL (3.3 mmol) of 50% aqueous H2O2 were added. The mixture was heated to 60 °C. Within the first 0.5 h, the yellow cisplatin suspension became a greenish-grey mixture. Upon continued stirring at 60 °C for a total of 3.5 h, the mixture converted to a suspension of an off-white solid. The solid was collected by filtration, washed with 3 × 2 mL of diethyl ether, dissolved in 40 mL of hot (~90 °C) water, and the resulting solution was stored at 4 °C for 16 h. Colorless crystals formed and were collected and washed sequentially with 2 × 5 mL water, 2 × 5 mL ethanol, and 2 × 5 mL diethyl ether, prior to drying under vacuum. Yield: 55 mg (43%). Crystals suitable for X-ray diffraction studies were grown by vapor diffusion of diethyl ether into an N,N-dimethylacetamide (DMA) solution of the complex. Mp: 124–131 °C (dec). 1H NMR (DMSO-d6, 400 MHz): δ 7.94 (s, 2H, 3JPtH = 34 Hz), 6.31 (t, 6H, 1JNH = 50 Hz). 13C NMR (DMSO-d6, 100 MHz): δ 169.7 (2JPtC = 15 Hz). 195Pt NMR (DMSO-d6, 108 MHz): δ 1068. IR (KBr, cm−1): 3254 s, 3184 s, 3108 m, 1662 vs, 1618 vs, 1562 s, 1373 m, 1329 m, 1221 s, 896 w, 814 m, 789 w, 528 w, 465. ESI-MS (neg. ion mode, MeOH): m/z 388.8 [M-H]− (calcd: 388.9). Anal. Calcd. for C2H8Cl2N2O4Pt: C, 6.16; H, 2.07; N, 7.18. Found: C, 6.27; H, 2.09; N, 7.31.
X-ray Crystallography
Crystals of cis,cis,trans-[Pt(NH3)2Cl2(O2CH)2]·DMA (1·DMA) were grown as described above. The complexes cis,cis,trans-[Pt(NH3)2Cl2(OCH2CH3)(OH)] (2) and cis,cis,trans- [Pt(NH3)2Cl2(O2CH2CH2COOH)(OH)] (3) were prepared as described previously.111,175 Crystals of the ethanol solvate of the former were grown by cooling a concentrated ethanolic solution of the complex to − 40 °C. Crystals of the DMSO solvate of the latter were grown by cooling a concentrated DMSO/DMF (50:50) solution of the complex to 4 °C. Suitable crystals were selected by microscopic examination through crossed polarizers and each was mounted on a nylon cryoloop in Paratone oil and cooled to 100 K under a stream of nitrogen. A Bruker APEX CCD X-ray diffractometer controlled by the APEX2 software195 was used to collect the diffraction of graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) from the crystal. The data were integrated with SAINT196 and absorption, Lorentz, and polarization corrections were calculated by SADABS.197 The space groups were determined by analyzing the systematic absences of the diffraction pattern with XPREP.198 Using the SHELXTL-97 software package the structures were solved by direct methods and refined against F2 using standard procedures.199 All non-hydrogen atoms were located on difference Fourier maps and refined anisotropically. Unless otherwise indicated in the note below or the appropriate CIF, hydrogen atoms were placed at calculated positions and refined with their isotropic displacement parameters (Uiso) set equal to 1.2 times the Uiso of the atom to which they were attached. A multiplicative factor of 1.5 was used for terminal methyl groups. CIF data are provided in the Supporting Information. The structures, deposited in the Cambridge Structural Database, were checked for missed higher symmetry and twinning with PLATON200,201 and were further validated using CheckCIF. Selected crystallographic parameters are presented in Table 1.
Table 1.
Crystallographic Parameters
| 1·DMA | 2·EtOH | 3·DMSO | |
|---|---|---|---|
| Formula | C6H17Cl2N3O5Pt | C4H18Cl2N2O3Pt | C6H18Cl2N2O6PtS |
| Formula weight | 477.22 | 408.19 | 512.27 |
| Crystal system | triclinic | monoclinic | orthorhombic |
| Space group | P1̄ | P21/c | Pbca |
| Color | Colorless | Yellow | Colorless |
| a, Å | 7.6011(8) | 11.2499(9) | 9.6918(10) |
| b, Å | 8.9779(10) | 12.7069(10) | 13.2067(14) |
| c, Å | 10.4662(11) | 8.0645(6) | 23.235(2) |
| α, ° | 94.369(2) | ||
| β, ° | 96.246(2) | 95.8720(10) | |
| γ, ° | 107.314(2) | ||
| V, Å3 | 673.37(13) | 1146.78(15) | 2974.0(5) |
| Z | 2 | 4 | 8 |
| T, K | 100(2) | 100(2) | 100(2) |
| R1a (%) | 1.70 | 2.66 | 2.05 |
| wR2b (%) | 3.51 | 6.27 | 4.11 |
| GOFc | 1.055 | 1.066 | 1.037 |
R1=Σ||Fo| − |Fc||/ Σ|Fo|.
wR2= {Σ[w(Fo2−Fc2)2]/Σ[w(Fo2)2]}1/2.
GOF= {Σ[w(Fo2−Fc2)2]/(n−p)}1/2, where n is the number of data and p is the number of refined parameters.
Note on the crystal structure of cis,cis,trans-[Pt(NH3)2Cl2(O2CH2CH2COOH)(OH)] ·DMSO
The positions of the hydrogen atoms on the hydroxide ligand and the pendant carboxylate were refined semi-freely with the O–H distances restrained to 0.84(2) Å. The final structural model shows the positions of the hydrogen atoms to be consistent with the hydrogen-bonding network holding the crystal together. One of these hydrogen bonds forms between the pendant carboxylate of one molecule (OH donor) and the hydroxide of a symmetry generated molecule (OH acceptor). The O···O distance of this donor-acceptor pair is 2.524(4) Å, placing it within the category of a low-barrier hydrogen bond.202,203 The donor-acceptor distance approaches the commonly accepted 2.5 Å limit of the short-strong hydrogen bond (SSHB), but the hydrogen atom is not symmetrically located between the two oxygen atoms. Moreover, there is no evidence of positional disorder of the hydrogen atoms in this structure. Although the barrier to proton transfer between the two oxygen atoms is likely low, consistent with the expected nucleophilicity of the coordinated hydroxide, a single-welled potential, characteristic of a SSHB, does not appear to be present.
Supplementary Material
Acknowledgments
This work was supported by grant CA034992 from the National Cancer Institute. Spectroscopic instrumentation at the MIT DCIF is maintained with funding from NIH Grant 1S10RR13886-01. T.C.J. received partial funding from the Harvard/MIT CCNE, NIH grant 5-U54-CA151884. J.J.W. is the grateful recipient of David H. Koch Graduate Fellowship. A. D. Liang is thanked for her help in preparing Figure 4.
Footnotes
Potential Conflict of Interest. SJL declares a financial interest in Blend Therapeutics.
Supporting Information. X-ray crystallographic data in CIF file format for cis,cis,trans-[Pt(NH3)2Cl2(O2CH)2], cis,cis,trans-[Pt(NH3)2Cl2(OCH2CH3)(OH)], and cis,cis,trans-[Pt(NH3)2Cl2(O2CH2CH2COOH)(OH)]. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Rosenberg B, Van Camp L, Krigas T. Nature. 1965;205:698–699. doi: 10.1038/205698a0. [DOI] [PubMed] [Google Scholar]
- 2.Rosenberg B, Van Camp L, Trosko JE, Mansour VH. Nature. 1969;222:385–386. doi: 10.1038/222385a0. [DOI] [PubMed] [Google Scholar]
- 3.O’Dwyer PJ, Stevenson JP, Johnson SW. Clinical Status of Cisplatin, Carboplatin, and Other Platinum-Based Antitumor Drugs. In: Lippert B, editor. Cisplatin - Chemistry and Biochemistry of a Leading Anticancer Drug. Verlag Helvetica Chimica Acta; Zürich, Switzerland: 1999. pp. 31–69. [Google Scholar]
- 4.Kelland L. Nat Rev Cancer. 2007;7:573–584. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- 5.Dabrowiak JC. Metals in Medicine. John Wiley & Sons; Chichester, U.K: 2009. [Google Scholar]
- 6.Alessio E, editor. Bioinorganic Medicinal Chemistry. Wiley-VCH Verlag & Co; Weinheim, Germany: 2011. [Google Scholar]
- 7.Gabano E, Ravera M, Osella D. Curr Med Chem. 2009;16:4544–4580. doi: 10.2174/092986709789760661. [DOI] [PubMed] [Google Scholar]
- 8.Lovejoy KS, Lippard SJ. Dalton Trans. 2009:10651–10659. doi: 10.1039/b913896j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harper BW, Krause-Heuer AM, Grant MP, Manohar M, Garbutcheon-Singh KB, Aldrich-Wright JR. Chem–Eur J. 2010;16:7064–7077. doi: 10.1002/chem.201000148. [DOI] [PubMed] [Google Scholar]
- 10.Wang X, Guo Z. Chem Soc Rev. 2013;42:202–224. doi: 10.1039/c2cs35259a. [DOI] [PubMed] [Google Scholar]
- 11.Roberts JJ, Thomson AJ. Prog Nucleic Acid Res Mol Biol. 1979;22:71–133. doi: 10.1016/s0079-6603(08)60799-0. [DOI] [PubMed] [Google Scholar]
- 12.Hall MD, Okabe M, Shen DW, Liang XJ, Gottesman MM. Annu Rev Pharmacol Toxicol. 2008;48:495–535. doi: 10.1146/annurev.pharmtox.48.080907.180426. [DOI] [PubMed] [Google Scholar]
- 13.Ishida S, Lee J, Thiele DJ, Herskowitz I. Proc Natl Acad Sci U S A. 2002;99:14298–14302. doi: 10.1073/pnas.162491399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Casini A, Reedijk J. Chem Sci. 2012;3:3135–3144. [Google Scholar]
- 15.Todd RC, Lippard SJ. Metallomics. 2009;1:280–291. doi: 10.1039/b907567d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chu G. J Biol Chem. 1994;269:787–790. [PubMed] [Google Scholar]
- 17.Zhang CX, Chang PV, Lippard SJ. J Am Chem Soc. 2004;126:6536–6537. doi: 10.1021/ja049533o. [DOI] [PubMed] [Google Scholar]
- 18.Guggenheim ER, Xu D, Zhang CX, Chang PV, Lippard SJ. ChemBioChem. 2009;10:141–157. doi: 10.1002/cbic.200800471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu G, Lippard SJ. Biochemistry. 2009;48:4916–4925. doi: 10.1021/bi900389b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Selvakumaran M, Pisarcik DA, Bao R, Yeung AT, Hamilton TC. Cancer Res. 2003;63:1311–1316. [PubMed] [Google Scholar]
- 21.Ferry KV, Hamilton TC, Johnson SW. Biochem Pharmacol. 2000;60:1305–1313. doi: 10.1016/s0006-2952(00)00441-x. [DOI] [PubMed] [Google Scholar]
- 22.Johnson SW, Perez RP, Godwin AK, Yeung AT, Handel LM, Ozols RF, Hamilton TC. Biochem Pharmacol. 1994;47:689–697. doi: 10.1016/0006-2952(94)90132-5. [DOI] [PubMed] [Google Scholar]
- 23.Zlatanova J, Yaneva J, Leuba SH. FASEB J. 1998;12:791–799. doi: 10.1096/fasebj.12.10.791. [DOI] [PubMed] [Google Scholar]
- 24.Pil PM, Lippard SJ. Science. 1992;256:234–237. doi: 10.1126/science.1566071. [DOI] [PubMed] [Google Scholar]
- 25.Chow CS, Barnes CM, Lippard SJ. Biochemistry. 1995;34:2956–2964. doi: 10.1021/bi00009a027. [DOI] [PubMed] [Google Scholar]
- 26.Dunham SU, Lippard SJ. Biochemistry. 1997;36:11428–11436. doi: 10.1021/bi9709452. [DOI] [PubMed] [Google Scholar]
- 27.Ohndorf UM, Rould MA, He Q, Pabo CO, Lippard SJ. Nature. 1999;399:708–712. doi: 10.1038/21460. [DOI] [PubMed] [Google Scholar]
- 28.Huang JC, Zamble DB, Reardon JT, Lippard SJ, Sancar A. Proc Natl Acad Sci U S A. 1994;91:10394–10398. doi: 10.1073/pnas.91.22.10394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McA’Nulty MM, Lippard SJ. Mutat Res, DNA Repair. 1996;362:75–86. doi: 10.1016/0921-8777(95)00037-2. [DOI] [PubMed] [Google Scholar]
- 30.He Q, Liang CH, Lippard SJ. Proc Natl Acad Sci U S A. 2000;97:5768–5772. doi: 10.1073/pnas.100108697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Catena R, Escoffier E, Caron C, Khochbin S, Martianov I, Davidson I. Biol Reprod. 2009;80:358–366. doi: 10.1095/biolreprod.108.070243. [DOI] [PubMed] [Google Scholar]
- 32.Park S, Lippard SJ. Biochemistry. 2012;51:6728–6737. doi: 10.1021/bi300649v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ober M, Lippard SJ. J Am Chem Soc. 2007;129:6278–6286. doi: 10.1021/ja0706145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ober M, Lippard SJ. J Am Chem Soc. 2008;130:2851–2861. doi: 10.1021/ja710220x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Todd RC, Lippard SJ. Chem Biol. 2010;17:1334–1343. doi: 10.1016/j.chembiol.2010.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park GY, Wilson JJ, Song Y, Lippard SJ. Proc Natl Acad Sci U S A. 2012;109:11987–11992. doi: 10.1073/pnas.1207670109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hollis LS, Amundsen AR, Stern EW. J Med Chem. 1989;32:128–136. doi: 10.1021/jm00121a024. [DOI] [PubMed] [Google Scholar]
- 38.Lippert B, Lock CJL, Speranzini RA. Inorg Chem. 1981;20:335–342. [Google Scholar]
- 39.Wu S, Wang X, Zhu C, Song Y, Wang J, Li Y, Guo Z. Dalton Trans. 2011;40:10376–10382. doi: 10.1039/c1dt10555h. [DOI] [PubMed] [Google Scholar]
- 40.Huxley M, Sanchez-Cano C, Browning MJ, Navarro-Ranninger C, Quiroga AG, Rodger A, Hannon MJ. Dalton Trans. 2010;39:11353–11364. doi: 10.1039/c0dt00838a. [DOI] [PubMed] [Google Scholar]
- 41.Sanchez-Cano C, Huxley M, Ducani C, Hamad AE, Browning MJ, Navarro-Ranninger C, Quiroga AG, Rodger A, Hannon MJ. Dalton Trans. 2010;39:11365–11374. doi: 10.1039/c0dt00839g. [DOI] [PubMed] [Google Scholar]
- 42.Baird CL, Griffitts AE, Baffic S, Bryant P, Wolf B, Lutton J, Berardini M, Arvanitis GM. Inorg Chim Acta. 1997;256:253–262. [Google Scholar]
- 43.Sundquist WI, Bancroft DP, Lippard SJ. J Am Chem Soc. 1990;112:1590–1596. [Google Scholar]
- 44.Ren T, Bancroft DP, Sundquist WI, Masschelein A, Keck MV, Lippard SJ. J Am Chem Soc. 1993;115:11341–11352. [Google Scholar]
- 45.Wang X, Lin J, Zhang X, Liu Q, Xu Q, Tan RX, Guo Z. J Inorg Biochem. 2003;94:186–192. doi: 10.1016/s0162-0134(02)00618-9. [DOI] [PubMed] [Google Scholar]
- 46.Cavallo L, Cini R, Kobe J, Marzilli LG, Natile G. J Chem Soc, Dalton Trans. 1991:1867–1873. [Google Scholar]
- 47.Cerasino L, Intini FP, Kobe J, de Clercq E, Natile G. Inorg Chim Acta. 2003;344:174–182. [Google Scholar]
- 48.Malinge JM, Sip M, Blacker AJ, Lehn JM, Leng M. Nucleic Acids Res. 1990;18:3887–3891. doi: 10.1093/nar/18.13.3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gaucheron F, Malinge JM, Blacker AJ, Lehn JM, Leng M. Proc Natl Acad Sci U S A. 1991;88:3516–3519. doi: 10.1073/pnas.88.9.3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lippard SJ. Acc Chem Res. 1978;11:211–217. [Google Scholar]
- 51.Lippard SJ. Pure Appl Chem. 1987;59:731–742. [Google Scholar]
- 52.Sherman SE, Lippard SJ. Chem Rev. 1987;87:1153–1181. [Google Scholar]
- 53.Jamieson ER, Lippard SJ. Chem Rev. 1999;99:2467–2498. doi: 10.1021/cr980421n. [DOI] [PubMed] [Google Scholar]
- 54.Wang D, Lippard SJ. Nat Rev Drug Discov. 2005;4:307–320. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- 55.Siddik ZH. Oncogene. 2003;22:7265–7279. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- 56.Prestayko AW, Crooke ST, Carter SK University of Alabama in Birmingham. Comprehensive Cancer Center., Bristol Laboratories. . Cisplatin, current status and new developments. Academic Press; New York: 1980. p. xv.p. 527. [Google Scholar]
- 57.Berman HM, Young PR. Annu Rev Biophys Bioeng. 1981;10:87–114. doi: 10.1146/annurev.bb.10.060181.000511. [DOI] [PubMed] [Google Scholar]
- 58.Tullius TD, Lippard SJ. Proc Natl Acad Sci U S A. 1982;79:3489–3492. doi: 10.1073/pnas.79.11.3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Merkel CM, Lippard SJ. Cold Spring Harbor Symp Quant Biol. 1983;47:355–360. doi: 10.1101/sqb.1983.047.01.041. [DOI] [PubMed] [Google Scholar]
- 60.Malinge JM, Leng M. Proc Natl Acad Sci U S A. 1986;83:6317–6321. doi: 10.1073/pnas.83.17.6317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Malinge JM, Schwartz A, Leng M. Nucleic Acids Res. 1987;15:1779–1797. doi: 10.1093/nar/15.4.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sundquist WI, Bancroft DP, Chassot L, Lippard SJ. J Am Chem Soc. 1988;110:8559–8560. [Google Scholar]
- 63.Cleare MJ, Hoeschele JD. Bioinorg Chem. 1973;2:187–210. [Google Scholar]
- 64.Wheate NJ, Walker S, Craig GE, Oun R. Dalton Trans. 2010;39:8113–8127. doi: 10.1039/c0dt00292e. [DOI] [PubMed] [Google Scholar]
- 65.Macquet JP, Butour JL. J Natl Cancer Inst. 1983;70:899–905. [PubMed] [Google Scholar]
- 66.Pinto AL, Lippard SJ. Proc Natl Acad Sci U S A. 1985;82:4616–4619. doi: 10.1073/pnas.82.14.4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brabec V, Boudny V. Met-Based Drugs. 1994;1:195–200. doi: 10.1155/MBD.1994.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brabec V, Reedijk J, Leng M. Biochemistry. 1992;31:12397–12402. doi: 10.1021/bi00164a014. [DOI] [PubMed] [Google Scholar]
- 69.Bursova V, Kasparkova J, Hofr C, Brabec V. Biophys J. 2005;88:1207–1214. doi: 10.1529/biophysj.104.051771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kelland LR. Crit Rev Oncol Hemat. 1993;15:191–219. doi: 10.1016/1040-8428(93)90042-3. [DOI] [PubMed] [Google Scholar]
- 71.Hollis LS, Lippard SJ. J Am Chem Soc. 1981;103:1230–1232. [Google Scholar]
- 72.Hollis LS, Lippard SJ. Inorg Chem. 1983;22:2708–2713. [Google Scholar]
- 73.Barton JK, Lippard SJ. Ann N Y Acad Sci. 1978;313:686–700. doi: 10.1111/j.1749-6632.1978.tb39455.x. [DOI] [PubMed] [Google Scholar]
- 74.Barton JK, Rabinowitz HN, Szalda DJ, Lippard SJ. J Am Chem Soc. 1977;99:2827–2829. [Google Scholar]
- 75.Hollis LS, Sundquist WI, Burstyn JN, Heiger-Bernays WJ, Bellon SF, Ahmed KJ, Amundsen AR, Stern EW, Lippard SJ. Cancer Res. 1991;51:1866–1875. [PubMed] [Google Scholar]
- 76.Hollis LS, Miller AV, Amundsen AR, Stern EW, Sundquist WI, Toni J, Burstyn JN, Heiger W, Lippard SJ. J Inorg Biochem. 1989;36:153. [Google Scholar]
- 77.Lempers ELM, Bloemink MJ, Brouwer J, Kidani Y, Reedijk J. J Inorg Biochem. 1990;40:23–35. doi: 10.1016/0162-0134(90)80037-x. [DOI] [PubMed] [Google Scholar]
- 78.Bellon SF, Lippard SJ. Biophys Chem. 1990;35:179–188. doi: 10.1016/0301-4622(90)80007-t. [DOI] [PubMed] [Google Scholar]
- 79.Donahue BA, Augot M, Bellon SF, Treiber DK, Toney JH, Lippard SJ, Essigmann JM. Biochemistry. 1990;29:5872–5880. doi: 10.1021/bi00476a032. [DOI] [PubMed] [Google Scholar]
- 80.Keck MV, Lippard SJ. J Am Chem Soc. 1992;114:3386–3390. [Google Scholar]
- 81.Zhang S, Lovejoy KS, Shima JE, Lagpacan LL, Shu Y, Lapuk A, Chen Y, Komori T, Gray JW, Chen X, Lippard SJ, Giacomini KM. Cancer Res. 2006;66:8847–8857. doi: 10.1158/0008-5472.CAN-06-0769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lovejoy KS, Todd RC, Zhang S, McCormick MS, D’Aquino JA, Reardon JT, Sancar A, Giacomini KM, Lippard SJ. Proc Natl Acad Sci U S A. 2008;105:8902–8907. doi: 10.1073/pnas.0803441105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang D, Zhu G, Huang X, Lippard SJ. Proc Natl Acad Sci U S A. 2010;107:9584–9589. doi: 10.1073/pnas.1002565107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lovejoy KS, Serova M, Bieche I, Emami S, D’Incalci M, Broggini M, Erba E, Gespach C, Cvitkovic E, Faivre S, Raymond E, Lippard SJ. Mol Cancer Ther. 2011;10:1709–1719. doi: 10.1158/1535-7163.MCT-11-0250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhu G, Myint M, Ang WH, Song L, Lippard SJ. Cancer Res. 2012;72:790–800. doi: 10.1158/0008-5472.CAN-11-3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chen Y, Guo Z, Parsons S, Sadler PJ. Chem–Eur J. 1998;4:672–676. [Google Scholar]
- 87.Hall MD, Hambley TW. Coord Chem Rev. 2002;232:49–67. [Google Scholar]
- 88.Hall MD, Dolman RC, Hambley TW. Met Ions Biol Syst. 2004;42:297–322. [PubMed] [Google Scholar]
- 89.Hall MD, Mellor HR, Callaghan R, Hambley TW. J Med Chem. 2007;50:3403–3411. doi: 10.1021/jm070280u. [DOI] [PubMed] [Google Scholar]
- 90.Galanski M, Jakupec MA, Keppler BK. Curr Med Chem. 2005;12:2075–2094. doi: 10.2174/0929867054637626. [DOI] [PubMed] [Google Scholar]
- 91.Reisner E, Arion VB, Keppler BK, Pombeiro AJL. Inorg Chim Acta. 2008;361:1569–1583. [Google Scholar]
- 92.Battle AR, Deacon GB, Dolman RC, Hambley TW. Aust J Chem. 2002;55:699–704. [Google Scholar]
- 93.Dolman RC, Deacon GB, Hambley TW. J Inorg Biochem. 2002;88:260–267. doi: 10.1016/s0162-0134(01)00360-9. [DOI] [PubMed] [Google Scholar]
- 94.Connelly NG, Geiger WE. Chem Rev. 1996;96:877–910. doi: 10.1021/cr940053x. [DOI] [PubMed] [Google Scholar]
- 95.Harris DC. Quantitative Chemical Analysis. 5. W. H. Freeman and Company; New York, NY: 1999. [Google Scholar]
- 96.Ellis LT, Er HM, Hambley TW. Aust J Chem. 1995;48:793–806. [Google Scholar]
- 97.Hindmarsh K, House DA, van Eldik R. Inorg Chim Acta. 1998;278:32–42. [Google Scholar]
- 98.Lemma K, Berglund J, Farrell N, Elding LI. J Biol Inorg Chem. 2000;5:300–306. doi: 10.1007/pl00010658. [DOI] [PubMed] [Google Scholar]
- 99.Lemma K, Sargeson AM, Elding LI. J Chem Soc, Dalton Trans. 2000:1167–1172. [Google Scholar]
- 100.Lemma K, Shi T, Elding LI. Inorg Chem. 2000;39:1728–1734. doi: 10.1021/ic991351l. [DOI] [PubMed] [Google Scholar]
- 101.Lemma K, House DA, Retta N, Elding LI. Inorg Chim Acta. 2002;331:98–108. [Google Scholar]
- 102.Nemirovski A, Kasherman Y, Tzaraf Y, Gibson D. J Med Chem. 2007;50:5554–5556. doi: 10.1021/jm070740j. [DOI] [PubMed] [Google Scholar]
- 103.Nemirovski A, Vinograd I, Takrouri K, Mijovilovich A, Rompel A, Gibson D. Chem Commun. 2010;46:1842–1844. doi: 10.1039/b925721g. [DOI] [PubMed] [Google Scholar]
- 104.Sinisi M, Intini FP, Natile G. Inorg Chem. 2012;51:9694–9704. doi: 10.1021/ic300957v. [DOI] [PubMed] [Google Scholar]
- 105.Tschugájeff L, Chlopin W, Fritzmann E. Z Anorg Allg Chem. 1926;151:253–268. [Google Scholar]
- 106.Giandomenico CM, Abrams MJ, Murrer BA, Vollano JF, Rheinheimer MI, Wyer SB, Bossard GE, Higgins JD., III Inorg Chem. 1995;34:1015–1021. doi: 10.1021/ic00109a004. [DOI] [PubMed] [Google Scholar]
- 107.Dunham SO, Larsen RD, Abbott EH. Inorg Chem. 1993;32:2049–2055. [Google Scholar]
- 108.Lee YA, Jung OS. Bull Chem Soc Jpn. 2002;75:1533–1537. [Google Scholar]
- 109.Lee YA, Yoo KH, Jung OS. Inorg Chem Commun. 2003;6:249–251. [Google Scholar]
- 110.Chung TS, Na YM, Kang SW, Jung OS, Lee YA. Transition Met Chem. 2005;30:541–545. [Google Scholar]
- 111.Feazell RP, Nakayama-Ratchford N, Dai H, Lippard SJ. J Am Chem Soc. 2007;129:8438–8439. doi: 10.1021/ja073231f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pichler V, Heffeter P, Valiahdi SM, Kowol CR, Egger A, Berger W, Jakupec MA, Galanski M, Keppler BK. J Med Chem. 2012;55:11052–11061. doi: 10.1021/jm301645g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhang JZ, Bonnitcha P, Wexselblatt E, Klein AV, Najajreh Y, Gibson D, Hambley TW. Chem–Eur J. 2013;19:1672–1676. doi: 10.1002/chem.201203159. [DOI] [PubMed] [Google Scholar]
- 114.Wilson JJ, Lippard SJ. Inorg Chem. 2011;50:3103–3115. doi: 10.1021/ic2000816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Galanski M, Keppler BK. Inorg Chem. 1996;35:1709–1711. doi: 10.1021/ic9509490. [DOI] [PubMed] [Google Scholar]
- 116.Perez JM, Camazón M, Alvarez-Valdes A, Quiroga AG, Kelland LR, Alonso C, Navarro-Ranninger MC. Chem-Biol Interact. 1999;117:99–115. doi: 10.1016/s0009-2797(98)00100-8. [DOI] [PubMed] [Google Scholar]
- 117.Alvarez-Valdés A, Pérez JM, López-Solera I, Lannegrand R, Continente JM, Amo-Ochoa P, Camazón MJ, Solans X, Font-Bardía M, Navarro-Ranninger C. J Med Chem. 2002;45:1835–1844. doi: 10.1021/jm010968l. [DOI] [PubMed] [Google Scholar]
- 118.Barnes KR, Kutikov A, Lippard SJ. Chem Biol. 2004;11:557–564. doi: 10.1016/j.chembiol.2004.03.024. [DOI] [PubMed] [Google Scholar]
- 119.Reithofer M, Galanski M, Roller A, Keppler BK. Eur J Inorg Chem. 2006:2612–2617. [Google Scholar]
- 120.Townsend DM, Tew KD. Oncogene. 2003;22:7369–7375. doi: 10.1038/sj.onc.1206940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ang WH, Khalaila I, Allardyce CS, Juillerat-Jeanneret L, Dyson PJ. J Am Chem Soc. 2005;127:1382–1383. doi: 10.1021/ja0432618. [DOI] [PubMed] [Google Scholar]
- 122.Johnstone RW. Nat Rev Drug Discov. 2002;1:287–299. doi: 10.1038/nrd772. [DOI] [PubMed] [Google Scholar]
- 123.Parker LJ, Italiano LC, Morton CJ, Hancock NC, Ascher DB, Aitken JB, Harris HH, Campomanes P, Rothlisberger U, De Luca A, Lo Bello M, Ang WH, Dyson PJ, Parker MW. Chem–Eur J. 2011;17:7806–7816. doi: 10.1002/chem.201100586. [DOI] [PubMed] [Google Scholar]
- 124.Yang J, Sun X, Mao W, Sui M, Tang J, Shen Y. Mol Pharmaceutics. 2012;9:2793–2800. doi: 10.1021/mp200597r. [DOI] [PubMed] [Google Scholar]
- 125.Kim J-w, Dang CV. Cancer Res. 2006;66:8927–8930. doi: 10.1158/0008-5472.CAN-06-1501. [DOI] [PubMed] [Google Scholar]
- 126.Chen Z, Lu W, Garcia-Prieto C, Huang P. J Bioenerg Biomembr. 2007;39:267–274. doi: 10.1007/s10863-007-9086-x. [DOI] [PubMed] [Google Scholar]
- 127.Berendzen K, Theriaque DW, Shuster J, Stacpoole PW. Mitochondrion. 2006;6:126–135. doi: 10.1016/j.mito.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 128.Michelakis ED, Webster L, Mackey JR. Br J Cancer. 2008;99:989–994. doi: 10.1038/sj.bjc.6604554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Papandreou I, Goliasova T, Denko NC. Int J Cancer. 2011;128:1001–1008. doi: 10.1002/ijc.25728. [DOI] [PubMed] [Google Scholar]
- 130.Dhar S, Lippard SJ. Proc Natl Acad Sci U S A. 2009;106:22199–22204. doi: 10.1073/pnas.0912276106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Xue X, You S, Zhang Q, Wu Y, Zou G-z, Wang PC, Zhao Y-l, Xu Y, Jia L, Zhang X, Liang X-J. Mol Pharmaceutics. 2012;9:634–644. doi: 10.1021/mp200571k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Olszewski U, Poulsen TT, Ulsperger E, Poulsen HS, Geissler K, Hamilton G. Clin Pharmacol: Adv Appl. 2010;2:177–183. doi: 10.2147/CPAA.S11795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Fiebiger W, Olszewski U, Ulsperger E, Geissler K, Hamilton G. Clin Transl Oncol. 2011;13:43–49. doi: 10.1007/s12094-011-0615-z. [DOI] [PubMed] [Google Scholar]
- 134.Xie J, Wang BS, Yu DH, Lu Q, Ma J, Qi H, Fang C, Chen HZ. Int J Oncol. 2011;38:409–417. doi: 10.3892/ijo.2010.851. [DOI] [PubMed] [Google Scholar]
- 135.Wei M, Burenkova O, Lippard SJ. J Biol Chem. 2003;278:1769–1773. doi: 10.1074/jbc.M210562200. [DOI] [PubMed] [Google Scholar]
- 136.Park S, Lippard SJ. Biochemistry. 2011;50:2567–2574. doi: 10.1021/bi2000214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Aina OH, Sroka TC, Chen ML, Lam KS. Pept Sci. 2002;66:184–199. doi: 10.1002/bip.10257. [DOI] [PubMed] [Google Scholar]
- 138.Boohaker RJ, Lee MW, Vishnubhotla P, Perez JM, Khaled AR. Curr Med Chem. 2012;19:3794–3804. doi: 10.2174/092986712801661004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Mukhopadhyay S, Barnés CM, Haskel A, Short SM, Barnes KR, Lippard SJ. Bioconjugate Chem. 2008;19:39–49. doi: 10.1021/bc070031k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Abramkin S, Valiahdi SM, Jakupec MA, Galanski M, Metzler-Nolte N, Keppler BK. Dalton Trans. 2012;41:3001–3005. doi: 10.1039/c2dt12024k. [DOI] [PubMed] [Google Scholar]
- 141.Gaviglio L, Gross A, Metzler-Nolte N, Ravera M. Metallomics. 2012;4:260–266. doi: 10.1039/c2mt00171c. [DOI] [PubMed] [Google Scholar]
- 142.Graf N, Mokhtari TE, Papayannopoulos IA, Lippard SJ. J Inorg Biochem. 2012;110:58–63. doi: 10.1016/j.jinorgbio.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wong DYQ, Lau JY, Ang WH. Dalton Trans. 2012;41:6104–6111. doi: 10.1039/c2dt30264k. [DOI] [PubMed] [Google Scholar]
- 144.Ruoslahti E. Annu Rev Cell Dev Biol. 1996;12:697–715. doi: 10.1146/annurev.cellbio.12.1.697. [DOI] [PubMed] [Google Scholar]
- 145.Strömblad S, Cheresh DA. Chem Biol. 1996;3:881–885. doi: 10.1016/s1074-5521(96)90176-3. [DOI] [PubMed] [Google Scholar]
- 146.Soroceanu L, Gillespie Y, Khazaeli MB, Sontheimer H. Cancer Res. 1998;58:4871–4879. [PubMed] [Google Scholar]
- 147.Kesavan K, Ratliff J, Johnson EW, Dahlberg W, Asara JM, Misra P, Frangioni JV, Jacoby DB. J Biol Chem. 2010;285:4366–4374. doi: 10.1074/jbc.M109.066092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hermoso M, Satterwhite CM, Andrade YN, Hidalgo J, Wilson SM, Horowitz B, Hume JR. J Biol Chem. 2002;277:40066–40074. doi: 10.1074/jbc.M205132200. [DOI] [PubMed] [Google Scholar]
- 149.Filipenko NR, MacLeod TJ, Yoon CS, Waisman DM. J Biol Chem. 2004;279:8723–8731. doi: 10.1074/jbc.M311951200. [DOI] [PubMed] [Google Scholar]
- 150.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nat Nanotechnol. 2007;2:751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 151.Torchilin V. Adv Drug Delivery Rev. 2011;63:131–135. doi: 10.1016/j.addr.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 152.Sood P, Thurmond KB, II, Jacob JE, Waller LK, Silva GO, Stewart DR, Nowotnik DP. Bioconjugate Chem. 2006;17:1270–1279. doi: 10.1021/bc0600517. [DOI] [PubMed] [Google Scholar]
- 153.Yang Z, Wang X, Diao H, Zhang J, Li H, Sun H, Guo Z. Chem Commun. 2007:3453–3455. doi: 10.1039/b705326f. [DOI] [PubMed] [Google Scholar]
- 154.Rieter WJ, Pott KM, Taylor KML, Lin W. J Am Chem Soc. 2008;130:11584–11585. doi: 10.1021/ja803383k. [DOI] [PubMed] [Google Scholar]
- 155.Bhirde AA, Patel V, Gavard J, Zhang G, Sousa AA, Masedunskas A, Leapman RD, Weigert R, Gutkind JS, Rusling JF. ACS Nano. 2009;3:307–316. doi: 10.1021/nn800551s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Taylor-Pashow KML, Rocca JD, Xie Z, Tran S, Lin W. J Am Chem Soc. 2009;131:14261–14263. doi: 10.1021/ja906198y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Xing R, Wang X, Zhang C, Zhang Y, Wang Q, Yang Z, Guo Z. J Inorg Biochem. 2009;103:1039–1044. doi: 10.1016/j.jinorgbio.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 158.Xu C, Wang B, Sun S. J Am Chem Soc. 2009;131:4216–4217. doi: 10.1021/ja900790v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Aryal S, Hu CMJ, Zhang L. ACS Nano. 2010;4:251–258. doi: 10.1021/nn9014032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Brown SD, Nativo P, Smith JA, Stirling D, Edwards PR, Venugopal B, Flint DJ, Plumb JA, Graham D, Wheate NJ. J Am Chem Soc. 2010;132:4678–4684. doi: 10.1021/ja908117a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Duong HTT, Huynh VT, de Souza P, Stenzel MH. Biomacromolecules. 2010;11:2290–2299. doi: 10.1021/bm100396s. [DOI] [PubMed] [Google Scholar]
- 162.Min Y, Mao C, Xu D, Wang J, Liu Y. Chem Commun. 2010;46:8424–8426. doi: 10.1039/c0cc03108a. [DOI] [PubMed] [Google Scholar]
- 163.Xing R, Wang X, Zhang C, Wang J, Zhang Y, Song Y, Guo Z. J Mater Chem. 2011;21:11142–11149. [Google Scholar]
- 164.Craig GE, Brown SD, Lamprou DA, Graham D, Wheate NJ. Inorg Chem. 2012;51:3490–3497. doi: 10.1021/ic202197g. [DOI] [PubMed] [Google Scholar]
- 165.Li J, Yap SQ, Yoong SL, Nayak TR, Chandra GW, Ang WH, Panczyk T, Ramaprabhu S, Vashist SK, Sheu FS, Tan A, Pastorin G. Carbon. 2012;50:1625–1634. doi: 10.1016/j.carbon.2011.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Min Y, Mao CQ, Chen S, Ma G, Wang J, Liu Y. Angew Chem, Int Ed. 2012;51:6742–6747. doi: 10.1002/anie.201201562. [DOI] [PubMed] [Google Scholar]
- 167.Prato M, Kostarelos K, Bianco A. Acc Chem Res. 2008;41:60–68. doi: 10.1021/ar700089b. [DOI] [PubMed] [Google Scholar]
- 168.Liu Z, Tabakman S, Welsher K, Dai H. Nano Res. 2009;2:85–120. doi: 10.1007/s12274-009-9009-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Lam C-w, James JT, McCluskey R, Arepalli S, Hunter RL. Crit Rev Toxicol. 2006;36:189–217. doi: 10.1080/10408440600570233. [DOI] [PubMed] [Google Scholar]
- 170.Liu Z, Chen K, Davis C, Sherlock S, Cao Q, Chen X, Dai H. Cancer Res. 2008;68:6652–6660. doi: 10.1158/0008-5472.CAN-08-1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kam NWS, Liu Z, Dai H. Angew Chem, Int Ed. 2006;45:577–581. doi: 10.1002/anie.200503389. [DOI] [PubMed] [Google Scholar]
- 172.Dhar S, Liu Z, Thomale J, Dai H, Lippard SJ. J Am Chem Soc. 2008;130:11467–11476. doi: 10.1021/ja803036e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Li J, Yap SQ, Chin CF, Tian Q, Yoong SL, Pastorin G, Ang WH. Chem Sci. 2012;3:2083–2087. [Google Scholar]
- 174.Tiwari PM, Vig K, Dennis VA, Singh SR. Nanomaterials. 2011;1:31–63. doi: 10.3390/nano1010031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Dhar S, Daniel WL, Giljohann DA, Mirkin CA, Lippard SJ. J Am Chem Soc. 2009;131:14652–14653. doi: 10.1021/ja9071282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Giljohann DA, Seferos DS, Daniel WL, Massich MD, Patel PC, Mirkin CA. Angew Chem, Int Ed. 2010;49:3280–3294. doi: 10.1002/anie.200904359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Rosi NL, Giljohann DA, Thaxton CS, Lytton-Jean AKR, Han MS, Mirkin CA. Science. 2006;312:1027–1030. doi: 10.1126/science.1125559. [DOI] [PubMed] [Google Scholar]
- 178.Soppimath KS, Aminabhavi TM, Kulkarni AR, Rudzinski WE. J Controlled Release. 2001;70:1–20. doi: 10.1016/s0168-3659(00)00339-4. [DOI] [PubMed] [Google Scholar]
- 179.Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Mol Pharmaceutics. 2008;5:505–515. doi: 10.1021/mp800051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Cheng J, Teply BA, Sherifi I, Sung J, Luther G, Gu FX, Levy-Nissenbaum E, Radovic-Moreno AF, Langer R, Farokhzad OC. Biomaterials. 2007;28:869–876. doi: 10.1016/j.biomaterials.2006.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Farokhzad OC, Cheng J, Teply BA, Sherifi I, Jon S, Kantoff PW, Richie JP, Langer R. Proc Natl Acad Sci U S A. 2006;103:6315–6320. doi: 10.1073/pnas.0601755103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Jain RA. Biomaterials. 2000;21:2475–2490. doi: 10.1016/s0142-9612(00)00115-0. [DOI] [PubMed] [Google Scholar]
- 183.Langer R. Science. 1990;249:1527–1533. doi: 10.1126/science.2218494. [DOI] [PubMed] [Google Scholar]
- 184.Owens DE, III, Peppas NA. Int J Pharm. 2006;307:93–102. doi: 10.1016/j.ijpharm.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 185.Dhar S, Gu FX, Langer R, Farokhzad OC, Lippard SJ. Proc Natl Acad Sci U S A. 2008;105:17356–17361. doi: 10.1073/pnas.0809154105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Lupold SE, Hicke BJ, Lin Y, Coffey DS. Cancer Res. 2002;62:4029–4033. [PubMed] [Google Scholar]
- 187.Farokhzad OC, Jon S, Khademhosseini A, Tran TNT, LaVan DA, Langer R. Cancer Res. 2004;64:7668–7672. doi: 10.1158/0008-5472.CAN-04-2550. [DOI] [PubMed] [Google Scholar]
- 188.Liedert B, Pluim D, Schellens J, Thomale J. Nucleic Acids Res. 2006;34:e47. doi: 10.1093/nar/gkl051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Dhar S, Kolishetti N, Lippard SJ, Farokhzad OC. Proc Natl Acad Sci U S A. 2011;108:1850–1855. doi: 10.1073/pnas.1011379108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Miller RP, Tadagavadi RK, Ramesh G, Reeves WB. Toxins. 2010;2:2490–2518. doi: 10.3390/toxins2112490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Graf N, Bielenberg DR, Kolishetti N, Muus C, Banyard J, Farokhzad OC, Lippard SJ. ACS Nano. 2012;6:4530–4539. doi: 10.1021/nn301148e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Johnstone TC, Lippard SJ. Manuscript in preparation. [Google Scholar]
- 193.Johnstone TC, Kulak N, Pridgen EM, Farokhzad OC, Langer R, Lippard SJ. doi: 10.1021/nn401905g. Manuscript in preparation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Kolishetti N, Dhar S, Valencia PM, Lin LQ, Karnik R, Lippard SJ, Langer R, Farokhzad OC. Proc Natl Acad Sci U S A. 2010;107:17939–17944. doi: 10.1073/pnas.1011368107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.APEX2, Version 2008-40. Bruker AXS, Inc; Madison, WI: 2008. [Google Scholar]
- 196.SAINT: SAX Area-Detector Integration Program, Version 768. Bruker AXS, Inc; Madison, WI: 2009. [Google Scholar]
- 197.Sheldrick GM. SADABS: Area-Detector Absorption Correction, Version 2008/1. University of Göttingen; Göttingen, Germany: 2008. [Google Scholar]
- 198.XPREP, Version 2008/2. Bruker AXS, Inc; Madison, WI: 2008. [Google Scholar]
- 199.Müller P. Crystallogr Rev. 2009;15:57–83. [Google Scholar]
- 200.Spek AL. J Appl Crystallogr. 2003;36:7–13. [Google Scholar]
- 201.Spek AL. PLATON, A Multipurpose Crystallographic Tool, Version 160312. Utrecht University; Utrecht, The Netherlands: 2012. [Google Scholar]
- 202.Gilli P, Bertolasi V, Ferretti V, Gilli G. J Am Chem Soc. 1994;116:909–915. [Google Scholar]
- 203.Warshel A, Papazyan A. Proc Natl Acad Sci U S A. 1996;93:13665–13670. doi: 10.1073/pnas.93.24.13665. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.