

Abstract
Genetic variation in the multidrug resistance gene ABCB1, which encodes the efflux transporter P-glycoprotein (P-gp), has been associated with Parkinson disease. Our goal was to investigate P-gp transport of paraquat, a Parkinson-associated neurotoxicant. We used in vitro transport models of ATPase activity, xenobiotic-induced cytotoxicity, transepithelial permeability, and rhodamine-123 inhibition. We also measured paraquat pharmacokinetics and brain distribution in Friend leukemia virus B-type (FVB) wild-type and P-gp-deficient (mdr1a−/−/mdr1b−/−) mice following 10, 25, 50, and 100 mg/kg oral doses. In vitro data showed that: 1) paraquat failed to stimulate ATPase activity; 2) resistance to paraquat-induced cytotoxicity was unchanged in P-gp-expressing cells in the absence or presence of P-gp inhibitors GF120918 [N-(4-[2-(1,2,3,4-tetrahydro-6,7-dimethoxy-2-isoquinolinyl)ethyl]-phenyl)-9,10-dihydro-5-methoxy-9-oxo-4-acridine carboxamide] and verapamil—37.0 [95% confidence interval (CI): 33.2–41.4], 46.2 (42.5–50.2), and 34.1 µM (31.2–37.2)—respectively; 3) transepithelial permeability ratios of paraquat were the same in P-gp-expressing and nonexpressing cells (1.55 ± 0.39 and 1.39 ± 0.43, respectively); and 4) paraquat did not inhibit rhodamine-123 transport. Population pharmacokinetic modeling revealed minor differences between FVB wild-type and mdr1a−/−/mdr1b−/− mice: clearances of 0.47 [95% confidence interval (CI): 0.42–0.52] and 0.78 l/h (0.58–0.98), respectively, and volume of distributions of 1.77 (95% CI: 1.50–2.04) and 3.36 liters (2.39–4.33), respectively; however, the change in clearance was in the opposite direction of what would be expected. It is noteworthy that paraquat brain-to-plasma partitioning ratios and total brain accumulation were the same across doses between FVB wild-type and mdr1a−/−/mdr1b−/− mice. These studies indicate that paraquat is not a P-gp substrate. Therefore, the association between ABCB1 pharmacogenomics and Parkinson disease is not attributed to alterations in paraquat transport.
Introduction
P-glycoprotein (P-gp) is an efflux drug transporter encoded by the multidrug resistance gene ABCB1 (also known as MDR1). P-gp is a member of the ATP-binding cassette (ABC) superfamily of transporters and mediates the efflux of a variety of structurally diverse xenobiotics in healthy tissues throughout the body, including the intestine, liver, kidney, and blood-brain barrier (Giacomini, 1997; Cascorbi et al., 2001; Lin and Yamazaki, 2003a.b; Giacomini et al., 2010; Sharom, 2011). Although P-gp plays an important role in the pharmacokinetics and disposition of drugs from many therapeutic classes, there is less evidence for the role of P-gp in toxicant disposition. P-gp has also been associated with a number of diseases, including Parkinson disease (Le Couteur et al., 2001; Furuno et al., 2002; Drozdzik et al., 2003; Lee et al., 2004; Lee and Bendayan, 2004; Tan et al., 2004, 2005; Kortekaas et al., 2005; Bartels et al., 2008, 2009; Westerlund et al., 2008, 2009; Vautier and Fernandez, 2009; Dutheil et al., 2010). Although exposure to neurotoxic xenobiotics is a well-known risk factor in Parkinson disease, it is unknown whether P-gp mediates the transport of these compounds (Langston et al., 1984; Semchuk et al., 1992; Bonnet and Houeto, 1999; Gatto et al., 2009; Wirdefeldt et al., 2011; Kamel, 2013; Mostafalou and Abdollahi, 2013; Pezzoli and Cereda, 2013).
Paraquat dichloride, or methyl viologen, is an herbicide that has been highly implicated as a risk factor for Parkinson disease based on epidemiologic data (Wirdefeldt et al., 2011; Kamel, 2013; Mostafalou and Abdollahi, 2013; Pezzoli and Cereda, 2013). Although production and commercial sale of paraquat has been banned in several countries due to toxicity, paraquat remains one of the most commonly used herbicides worldwide, including in the United States. Some studies have suggested a link between P-gp activity and paraquat exposure. Specifically, induction of P-gp was protective against paraquat-induced toxicity in Caco-2 cells and rat lungs, although the inducers used, dexamethasone and doxorubicin, are not specific inducers of P-gp (Dinis-Oliveira et al., 2006a,b; Silva et al., 2011, 2013). Therefore, it remains unclear if P-gp transports paraquat and whether P-gp-mediated disposition of paraquat could play a role in Parkinson disease. One hypothesis is that if paraquat were a P-gp substrate, decreased P-gp activity at the blood-brain barrier would lead to increased brain accumulation of paraquat and an increased risk of Parkinson disease. One mechanism for decreased P-gp activity at the blood-brain barrier might be genetic variations in the ABCB1 gene that alter P-gp function (as reviewed in Woodahl and Ho, 2004; Wang and Sadee, 2006; Chinn and Kroetz, 2007; Cascorbi, 2011).
The goal of this study was to evaluate P-gp-mediated transport of paraquat in a combination of in vitro and in vivo models to evaluate a potential mechanism for the role of P-gp in Parkinson disease. We characterized P-gp transport of paraquat in vitro using cell- and membrane-based models. We also used an animal model to determine paraquat pharmacokinetics and brain accumulation in Friend leukemia virus B-type (FVB) wild-type mice and in P-gp deficient mice on an FVB background (mdr1a−/−/1b−/−) to evaluate the role of P-gp in paraquat disposition in vivo. This is the first comprehensive study to evaluate P-gp-mediated disposition of paraquat.
Materials and Methods
Chemicals
Paraquat dichloride, doxorubicin, verapamil, rhodamine-123 (R123), cyclosporine, and 4,6-diamidino-2-phenylindole (DAPI) were purchased from Sigma-Aldrich (St. Louis, MO). N-(4-[2-(1,2,3,4-Tetrahydro-6,7-dimethoxy-2-isoquinolinyl)ethyl]-phenyl)-9,10-dihydro-5-methoxy-9-oxo-4-acridine carboxamide (GF120918) was kindly provided by GlaxoSmithKline (Research Triangle Park, NC). [14C]Paraquat was purchased from American Radiolabeled Chemicals (St. Louis, MO).
ATPase Activity
Stimulation of ATPase activity was measured using SB MDR1/P-gp Sf9 ATPase membranes (SOLVO Biotechnology, Budaors, Hungary) according to the manufacturer’s instructions. ATPase activity in the presence of xenobiotics was estimated by measuring the release of inorganic phosphate (Pi) in a colorimetric reaction. P-gp membranes were incubated with either paraquat or the known P-gp substrate verapamil over a concentration range of 0.05–100 μM. ATPase activity was determined as the difference in Pi liberation measured in the absence or presence of 1.2 mM sodium orthovanadate, a nonspecific ATPase inhibitor. The reactions were incubated at 37°C for 20 minutes and stopped by the addition of 10% SDS. The detection reagent containing ammonium molybdate was added and incubated for 25 minutes at 37°C, and absorbance was read at 690 nm using a SpectraMax Gemini XS microplate reader (Molecular Devices, Sunnyvale, CA). The amount of Pi liberation was calculated from a phosphate standard curve. ATPase activity was reported as nanomolars of Pi liberated per minute of incubation time per milligram of total protein (nmol Pi/min/mg protein). Compounds were evaluated in duplicate, and the assay was performed twice. Michaelis-Menten parameters (Vmax and Km) were estimated using a nonlinear regression least squares model fit on Prism 5.0 software (GraphPad Software, Inc., San Diego, CA).
Cell Culture
LLC-PK1 vector cells (LLC-vector) and recombinant ABCB1/MDR1 cells (LLC-MDR1-WT), generously provided by Michael M. Gottesman (Laboratory of Cell Biology, National Cancer Institute, Bethesda, MD), were cultured in complete Media 199 (Mediatech, Manassas, VA) supplemented with 3% (v/v) fetal bovine serum (FBS) (Mediatech), 1% (v/v) l-glutamine (Mediatech), 1% (v/v) penicillin/streptomycin (Mediatech), and 1% (v/v) Geneticin (G418; Life Technologies, Carlsbad, CA) and grown at 37°C in the presence of 5% CO2.
Xenobiotic-Induced Cytotoxicity
Sensitivity to cytotoxic agents was evaluated in LLC-vector and LLC-MDR1-WT cells plated overnight at a density of 1,000 cells/well in 96-well plates (Thermo Fisher Scientific, Hampton, NH). Cells were treated with doxorubicin (0.02 nM–200 μM), colchicine (0.01–50,000 nM), or paraquat (1.95–500 µM) for 72 hours at 37°C. Cell viability was evaluated using the CellTiter-Glo Cell Viability Assay (Promega, Fitchburg, WI) according to the manufacturer’s instructions. Inhibition was performed at 0.5 µM GF120918 and 10 µM verapamil. Cell viability was measured by luminescence using a SynergyMX microplate reader (Biotek, Winooski, VT). Untreated cells were considered 100% viable. Compounds were evaluated in triplicate. Viability was estimated as the effective concentration necessary for 50% cell death (EC50) based on a nonlinear regression log (agonist) versus response variable slope least squares model fit using Prism 5.0.
Transepithelial Permeability
Transepithelial permeability assays were completed according to methods developed previously (Woodahl et al., 2004). LLC-vector and LLC-MDR1-WT recombinant cells were plated at a density of 2 × 106 cells/24-mm well on permeable supports (Transwell; 3.0-μm membrane pore size; Corning, Tewksbury, MA) and grown for 4 days before the initiation of the experiment. Transepithelial electrical resistance (TEER) values were measured with a Millicell-ERS volt-ohm meter (Millipore, Billerica, MA). All transport assays were performed at 37°C, and calculations were made accounting for the difference in volumes in the apical and basolateral compartments. Transport of the known P-gp substrate R123 (5 μM) or [14C]paraquat (450 nM) was performed in serum-free Media 199. Inhibition of transport was performed at 1 μM GF120918. Aliquots of 50 μl were taken from the apical and basal compartments at 0.5, 1, 2, 3, and 4 hours, time intervals within the linear range of permeability. Experiments were run in triplicate. R123 was quantified by measuring fluorescence with an excitation of 488 nm and an emission of 525 nm, using a Gemini XS Fluorescent microplate reader (Molecular Devices, Sunnyvale CA). [14C]Paraquat was quantified by liquid scintillation counting (Beckman LS 6500, Brea, CA). Apparent permeability (Papp) was calculated as Papp = 1/(A × C0) × (dQ/dt), where A is the surface area of the permeable support, C0 is the initial concentration in the donor compartment, and dQ/dt is the rate of transfer of compound into the acceptor compartment; Papp was estimated in both the apical-to-basolateral (Papp A→B) and basolateral-to-apical (Papp B→A) directions. The permeability efflux ratio of (Papp B→A)/(Papp A→B) was estimated to evaluate P-gp-mediated directional efflux. Permeability ratios > 2.0 are expected for P-gp substrates, and the ratio should reduce to approximately 1.0 in the presence of the P-gp inhibitor GF120918.
Inhibition of Intracellular Accumulation
Inhibition of R123 intracellular uptake in LLC-vector and LLC-MDR1-WT cells was performed based on a previously developed assay (Woodahl et al., 2004). Cells were plated overnight at a density of 1 × 106 cells/well in 6-well plates (Invitrogen, Carlsbad, CA). Cells were incubated in triplicate with 5 μM R123 with and without inhibitors in serum-free Media 199 at 37°C and then allowed to efflux in the presence of inhibitor in complete Media 199. Concentrations ranged from 0.1 to 500 µM for verapamil, 0.1 to 100 µM for cyclosporine, 1.56 to 1000 nM for GF120918, and 0.1 to 1000 µM for paraquat. After the efflux period, cells were washed in ice-cold phosphate-buffered saline, trypsinized, and resuspended in phosphate-buffered saline containing 5 mM EDTA, 25 mM HEPES, and 1% fetal bovine serum (pH 7.0). Immediately before flow cytometry, DAPI was added to the cells as a measure of cell viability. Cells were analyzed with a FACSAriaII flow cytometer (BD Biosciences, San Jose, CA) using FACSDiva software. Ten thousand cells from each sample were analyzed for forward scatter, side scatter, DAPI, and R123. Inhibition was estimated as the inhibitor concentration necessary for 50% inhibition (IC50) based on a nonlinear regression log (inhibitor) versus normalized response variable slope least squares model fit on Prism 5.0.
Animals
Male FVB wild-type and mdr1a−/−/1b−/− (FVB background) mice (Taconic Farms, Germantown, NY) were used in this study (ages 1.2–7.6 months). Mice were maintained on a 12-hour light-dark cycle, housed in microisolators, and were given food and deionized water ad libitum. All procedures were approved by the University of Montana Institutional Animal Care and Use Committee.
Paraquat Dosing and Sample Collection
Paraquat was prepared fresh in sterile water for each treatment. Paraquat was administered via oral gavage at doses of 10, 25, 50, or 100 mg/kg (n = 5–10 per dose group). Blood samples were collected via the saphenous vein at 1, 2, 4, and 8 hours following paraquat administration, and plasma was separated from whole blood via centrifugation. Paraquat was administered at approximately the same time of day for each treatment. Following the 8-hour blood collection, mice were killed via cervical dislocation. Whole brains were extracted and washed twice in phosphate-buffered saline. Plasma and brain samples were frozen at −80°C until analysis.
Quantitation of Paraquat in Plasma and Brain Samples
After thawing, sterile water was added to the brain samples at a 2:1 (w/v) ratio. The brain/water mixture was first homogenized for 1 minute and then sonicated for 5-second intervals for a total of 1 minute; samples were kept on ice throughout the entire homogenization and sonication periods. Ethyl viologen (100 μg/l) was used as an internal standard in plasma and brain samples before protein extraction. Standard curves were simultaneously prepared from brain and plasma samples from untreated mice at a range of 10–2500 μg/l paraquat and 100 μg/l ethyl viologen. Plasma and brain samples were extracted with 1:2 (v/v) acetonitrile (−20°C), vortexed, and centrifuged at 13,000 rpm for 10 minutes. The supernatant was collected, and extraction was repeated two more times for a total of three extractions. Supernatants were combined and filtered using Ultrafree-MC centrifugal filters (Millipore). Samples were stored at −80°C before quantitation.
Levels of paraquat in mouse plasma and brain samples were determined by liquid chromatography tandem mass spectroscopy (LC-MS/MS) using an Agilent 1200 Series HPLC and 6300 series MSD/XCT (Waldbronn, Germany), using a method adapted from previously published work (Ariffin and Anderson, 2006). Separations were accomplished using a Phenomenex Kinetex C18 column (Torrance, CA), 2.1 × 100 mm, 2.6-μm particles, and a flow rate of 0.1 ml/min. A gradient from 25% to 90% of methanol (mobile phase B) over 4 minutes was used with 20 mM ammonium formate and 15 mM heptafluorobutyric acid in water (mobile phase A; pH 3.2). MS detection was made from 5 to 10 minutes for plasma samples and 3.5 to 10 minutes for brain samples, with all other flow diverted to waste. Quantitation of paraquat in plasma or brain samples was determined using multiple reaction monitoring to characterize fragmentation of singly-charged analytes with transitions m/z 185→171 for paraquat and m/z 213→157 for ethyl viologen. Basic MS settings included nebulizer pressure 40 p.s.i., dry gas flow of 8 l/min, dry gas temperature of 350°C, isolation window of 4.0 amu, and fragmentation amplitude of 0.41 V. Plasma and brain concentrations were reported as microgram per liter and nanograms per gram brain tissue, respectively. The limit of detection (LOD) and limit of quantitation (LOQ) were 13.3 and 44.5 μg/l in plasma samples, respectively, and 22.2 and 74.7 ng/g in brain samples, respectively. Paraquat brain-to-plasma partitioning ratios were calculated using the paraquat concentrations in brain (nanograms per gram) and plasma (microgram per liter) at the 8-hour time point.
Pharmacokinetic Analysis
The concentration-time profiles of paraquat were explored graphically. Initial pharmacokinetic parameter estimates were made using noncompartmental methods (Phoenix WinNonlin 6.3; Pharsight, St. Louis, MO) (Gabrielsson and Weiner, 2012). Parameters that can be obtained from noncompartmental analysis include area under the plasma concentration–time curve (AUC), apparent oral clearance (CL/F), apparent volume of distribution (V/F), elimination half-life (t1/2), maximum observed concentration (Cmax), time of maximum observed concentration (tmax), and mean residence time (MRT) (Pharsight, 2005–2009). When possible, the linear trapezoidal method with the “partial area” option of 0–8 hours was used in each strain of mouse to estimate the elimination rate constant (λz) based on the modeled data. The AUC was calculated up to the last observation at 8 hours after the dose (AUC0–8). Initial estimates of apparent oral CL/F and V/F were calculated using following equations (Bauer, 2005):
Apparent Oral Clearance (CL/F)
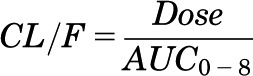 |
Apparent Volume of Distribution (V/F)
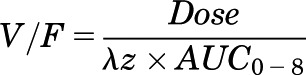 |
Final pharmacokinetic parameters were estimated using a population pharmacokinetic analysis of paraquat and nonlinear mixed effect modeling (NONMEM, version 7.2; Icon Development Solutions, Hanover, MD) with PDx-Pop (version 5; Icon Development Solutions) interfaced with Xpose (version 4.0, release 6, update 1; Uppsala, Sweden) (Yuh et al., 1994). The estimation method of first-order conditional estimation interaction (FOCE INTER) was used. The following models were tested: one-compartment linear model, one-compartment linear model with first-order absorption and lag, one-compartment linear model with first-order absorption, one-compartment linear model with first-order absorption with additive error model, one-compartment linear model with first-order absorption with proportional-error model, and one-compartment linear model with first-order absorption with combined additive and proportional error model.
Using the subroutines of NONMEM, the final model was ADVAN 2 (one-compartment linear model with first-order absorption) and TRANS 2 (CL/F and V/F). Population pharmacokinetic estimates of CL/F, V/F, and absorption rate constant (ka) were estimated by combining all dose groups in each mouse strain. The two groups of mice were simultaneously modeled, which allowed for estimation of the pharmacokinetics parameters in each group. Robust estimates for interindividual variability (IIV), which is the source of variability determined between mice, were predicted with exponential variability models. Estimates for residual unexplained variability (RUV), the coefficient of variance that accounts for the random and unexplained variability, were also determined. Residual variability, also called intraindividual variability, was estimated with a proportional error model.
Interindividual Variability
where CLi is paraquat clearance of the ith subject, θpop is the population value for paraquat clearance, and ηi is the interindividual random effect with mean zero and variance ω2.
Intraindividual Variability: Proportional Error Model
where Cij is the ith measured plasma concentration in the jth subject, Ci is the corresponding predicted plasma concentration, and εpropj is the residual variability term, representing independent identically distributed statistical errors with a mean of zero and variance σ2.
Pharmacokinetic Model Evaluation
The population pharmacokinetic models were compared using the Akaike and Schwarz information criterion to discriminate between nonhierarchical models in the selection of the structural model, which included one-compartment linear model, one-compartment linear model with first-order absorption and lag, and one-compartment linear model with first-order absorption. During model development, diagnostic plots of observed versus population predicted or individual predicted values were used to visually assess model fit (Sherwin et al., 2012). Plots of conditional weighted residuals versus time after dose or population predicted values were also examined. The population pharmacokinetic models were evaluated using a nonparametric resampling bootstrap method to assess model accuracy and stability. PDx-Pop (version 5; Icon Development Solutions) was used to generate bootstrap runs generated by random sampling using the original dataset. Standard errors for the estimated population parameters and random effects error models were also assessed. Relative standard error was estimated as the standard error divided by the mean and expressed as a percentage. Random error is considered to always be present and is unpredictable (Sheiner and Beal, 1981). Empirical Bayesian estimates for the predicted concentrations were obtained using the POSTHOC option in NONMEM. The performance of the final model was further evaluated by generating a visual predictive check, which compares statistics derived from the distribution of observations and the distribution of predictions (Bergstrand et al., 2011).
Statistical Analysis
Student’s two-tailed t test was used to analyze data from the transepithelial permeability assay. An extra sum of squares F-test was used to compare individual parameters for the cytotoxic sensitivity assay. The null hypothesis was that the EC50 estimates are the same within a cell type between xenobiotic treatment group alone and in the presence of a P-gp inhibitor, and the null hypothesis was only rejected if the inhibitor decreased cytotoxic resistance. A two-tailed Grubbs’ test for outliers was used prior to analysis of paraquat pharmacokinetics in plasma and brain samples. Levels of paraquat in brain samples were analyzed using an analysis of variance. For all analyses, P values < 0.05 were considered statistically significant.
Results
Membrane-Based Analysis of P-gp Transport of Paraquat.
ATPase activity in P-gp-expressing membranes was measured in the presence of paraquat and the P-gp substrate verapamil as a positive control (Fig. 1). Vmax and Km parameters for verapamil were estimated as 48.4 ± 2.6 nmol Pi/min/mg protein and 2.54 ± 0.68 μM, respectively. Paraquat did not stimulate ATPase activity over a wide concentration range above the level of the sodium orthovanadate–sensitive control (14.5 ± 1.0 nmol Pi/min/mg protein), a nonspecific ATPase inhibitor, indicating that paraquat is not a P-gp substrate.
Fig. 1.

ATPase stimulation in SB MDR1/P-gp Sf9 membranes. P-gp membranes were incubated with either paraquat (triangle, solid line) or the known P-gp substrate verapamil (circle, solid line). Orthovanadate-sensitive ATPase activity was also measured (square, dashed line). Insert displays the data on a log concentration scale. Compounds were measured in duplicate and tested twice (n = 4). Data are represented as mean ± S.D.
Cell-Based Analysis of P-gp Transport of Paraquat.
Xenobiotic-induced cytotoxicity was measured in LLC-vector and LLC-MDR1-WT cells. Dose-response curves were generated (Fig. 2) and EC50 values were estimated (Table 1) following exposure to paraquat or cytotoxic P-gp substrates, doxorubicin and colchicine, as positive controls. We also used P-gp inhibitors GF120918 and verapamil to confirm that changes in cellular sensitivities were due to P-gp. The number of cells used and the length of the cytotoxic exposure were optimized to ensure the experiments were conducted within the linear range of the assay. As expected, LLC-MDR1-WT cells exhibit significantly increased resistance to doxorubicin and colchicine compared with LLC-vector cells, a 61.4- and 48.5-fold increase in EC50 values, respectively. In addition, P-gp inhibitors, GF120918 and verapamil, reversed cellular resistance to doxorubicin and colchicine in LLC-MDR1-WT cells, confirming the increase in resistance was due to P-gp. Conversely, there was only a slight increase in resistance to paraquat toxicity in LLC-MDR1-WT cells compared with LLC-vector (2.9-fold increase in EC50 values). It is important to note, however, that the P-gp inhibitors GF120918 and verapamil did not reverse the resistance to paraquat in LLC-MDR1-WT cells, indicating that the modest increase in resistance to paraquat was not P-gp-mediated. The presence of GF120918 actually increased the resistance of the LLC-MDR1-WT to paraquat instead of reversing it. Additionally, the two different P-gp inhibitors did not have an effect on the maximum cell viability or maximum cell death observed in the dose-response curves, clearly showing that paraquat-induced cytotoxicity is not altered (Fig. 2, E and F). This provided further evidence that paraquat is not a P-gp substrate.
Fig. 2.

Xenobiotic-induced cytotoxicity in LLC-vector and LLC-MDR1-WT cells. LLC-vector and LLC-MDR-WT cells were treated with either doxorubicin (A and B), colchicine (C and D), or paraquat (E and F). Cell viability was tested with xenobiotic alone (square, solid line), or in the presence of P-gp inhibitors GF120918 (triangle, dashed line) or verapamil (circle, dotted line). Compounds were tested in triplicate (n = 3) at each concentration point; data are represented as mean ± S.D.
TABLE 1.
Xenobiotic-induced cytotoxicity in LLC-vector and LLC-MDR1-WT cells
| EC50 Values (95% Confidence Interval) |
||
|---|---|---|
| LLC-Vectora | LLC-MDR1-WTb | |
| Doxorubicin (nM) | 32.2 (13.7–85.6)*** | 1978 (1015–3850) |
| Doxorubicin (nM) + GF120918 | 24.8 (12.0–51.3) | 8.06 (1.32–49.2)*** |
| Doxorubicin (nM) + verapamil | 43.4 (12.0–155.9)** | 268 (90.7–98.6)*** |
| Colchicine (nM) | 13.3 (NDc) | 645 (467–892) |
| Colchicine (nM) + GF120918 | 12.6 (10.8–14.5)* | 17.5 (12.0–25.4)*** |
| Colchicine (nM) + verapamil | 12.8 (NDc) | 179 (117–273)*** |
| Paraquat (µM) | 12.8 (11.4–14.3)*** | 37.0 (33.2–41.4) |
| Paraquat (µM) + GF120918 | 13.6 (12.4–15.0)*** | 46.2 (42.5–50.2)** |
| Paraquat (µM) + verapamil | 12.9 (10.9–15.4)*** | 34.1 (31.2–37.2) |
ND, not determined. *P < 0.05; **P < 0.01; ***P < 0.001.
Comparisons address differences between cell types.
Comparisons address differences within an individual cell type (i.e., the xenobiotic treatment group alone versus in the presence of a P-gp inhibitor).
Nonlinear regression did not allow for estimation of confidence intervals.
P-gp-mediated directional transport of paraquat was evaluated by estimating transepithelial permeability ratios in LLC-vector or LLC-MDR1-WT cells using the P-gp substrate R123 as a positive control (Table 2). Transepithelial electrical resistance values were measured as 676 ± 157 and 782 ± 174 Ω*cm2 in LLC-vector and LLC-MDR1-WT cells, respectively, before the start of experiments confirming the integrity of the monolayers. Experiments were performed up to 4 hours to ensure that permeability rates were estimated in the linear range of transepithelial flux. As expected, we observed a significant increase in the R123 permeability ratios in LLC-MDR1-WT compared with LLC-vector cells, and the efflux was inhibited by GF120918. There was no directional transport of [14C]paraquat in LLC-vector or LLC-MDR1-WT cells and no effect of GF120918, confirming that paraquat is not a P-gp substrate.
TABLE 2.
Transepithelial permeability in LLC-vector and LLC-MDR1-WT cells
| Cell Type |
Papp B→A/Papp A→B ± S.D. |
|
|---|---|---|
| LLC-Vector | LLC-MDR1-WT | |
| R123 | 1.12 ± 0.40 | 5.10 ± 2.19* |
| R123 + GF120918 | 1.13 ± 0.29 | 1.33 ± 0.92 |
| Paraquat | 1.39 ± 0.43 | 1.55 ± 0.39 |
| Paraquat + GF120918 | 1.29 ± 0.35 | 1.58 ± 0.36 |
P < 0.05 for differences between cell types.
Finally, paraquat was also evaluated as an inhibitor of P-gp. The ability of paraquat to inhibit the P-gp efflux of R123 in LLC-vector and LLC-MDR1-WT cells was measured and compared with known P-gp inhibitors verapamil, cyclosporine, and GF120918 (Fig. 3). None of the test compounds altered R123 accumulation in LLC-vector cells (data not shown). Verapamil, cyclosporine, and GF120928 inhibited R123 efflux with IC50 values of 1.98 ± 0.12 μM, 1.39 ± 0.07 μM, and 22.9 ± 1.9 nM, respectively. Additionally, all three inhibitors produced 100% inhibition of R123 transport. Paraquat did not inhibit R123 accumulation in P-gp-expressing cells across a broad concentration range, even up to concentrations of 1 mM.
Fig. 3.

Inhibition of rhodamine-123 transport. Percent inhibition of rhodamine-123 transport was evaluated in LLC-vector and LLC-MDR-WT cells in the presence of verapamil (diamond), cyclosporine (triangle), GF120918 (circle), or paraquat (square). Compounds were tested in triplicate (n = 3) at each concentration point; data are presented as mean ± S.D.
Paraquat Pharmacokinetics and Brain Distribution.
The systematic screening of paraquat in vitro indicates that paraquat is not a substrate of P-gp, but to confirm this finding, we next investigated the role of P-gp in the disposition of paraquat in vivo. Paraquat pharmacokinetics and brain accumulation in FVB wild-type and mdr1a−/−/1b−/− mice were evaluated. Genotype of both FVB wild-type and mdr1a−/−/1b−/− mice was confirmed using methods obtained from Taconic Farms (data not shown). Both wild-type and knockout strains of mice have similar plasma concentration-time curves following oral administration of 10, 25, 50, or 100 mg/kg paraquat, and there was no evidence for nonlinearity across the range of doses (Fig. 4). Plasma concentrations were above the LOQ across all doses, although the 8-hour time point in the lowest dose group of 10 mg/kg was near the LOQ. Six samples were identified as outliers (three in each strain) and not included in further analyses. Noncompartmental analysis was used to generate initial parameter estimates (Supplemental Table 1).
Fig. 4.

Paraquat plasma concentration-time curves in FVB wild-type and mdr1a−/−/1b−/− mice. Plasma samples were collected at 1, 2, 4, and 8 hours after oral administration of paraquat. (A) FVB wild-type mice following doses of 10 (circles; n = 10), 25 (squares; n = 9), 50 (diamond; n = 5), or 100 mg/kg (triangle; n = 10). (B) mdr1a−/−/1b−/− mice following doses of 10 (circles; n = 6), 25 (squares; n = 6), 50 (diamond; n = 10), or 100 mg/kg (triangle; n = 6). Data are presented as mean ± S.D.
We used a one-compartment pharmacokinetic model to estimate final pharmacokinetic parameters (Table 3). Diagnostic plots were generated for observed paraquat concentrations versus population predicted concentrations (Supplemental Fig. 1) and population predicted concentrations versus time after the dose (Supplemental Fig. 2). Plots of conditional weighted residuals versus population predicted concentrations (Supplemental Fig. 3) and versus time after dose were also examined (Supplemental Fig. 4). The final covariate model generated reasonably stable and accurate estimates of the fixed and random effects. Simulations from the observed paraquat data were evaluated using a visual predictive check (Supplemental Fig. 5), with the median simulated value compared with the 5th, 10th, 90th, and 95th quantiles. Of the simulated observations, most fell within the 90% confidence interval, demonstrating model stability and reasonable agreement between the observed and simulated paraquat concentration data.
TABLE 3.
Population pharmacokinetic analysis of paraquat in FVB wild-type and mdr1a−/−/1b−/− mice following a single paraquat oral dose
| FVB Wild-Type (n = 34) |
mdr1a−/−/1b−/− (n = 28) |
|||||||
|---|---|---|---|---|---|---|---|---|
| Estimates | %RSE | 95% CI | CV% | Estimates | %RSE | 95% CI | CV% | |
| CL/F (l/h) | 0.473 | 5.18 | 0.425–0.521 | — | 0.777* | 13.3 | 0.575–0.979 | — |
| V/F (l) | 1.77 | 7.91 | 1.50–2.04 | — | 3.36* | 14.8 | 2.39–4.33 | — |
| ka (h−1) | 1.81 | 16.9 | 1.21–2.41 | — | 5.60a | — | — | — |
| IIV-CL/F | 0.056 | 35.1 | 0.018–0.095 | 23.7 | 0.344 | 32.0 | 0.128–0.560 | 58.7 |
| IIV-V/F | 0.072 | 49.6 | 0.002–0.141 | 26.8 | 0.514 | 24.7 | 0.265–0.763 | 71.7 |
| RUV | 0.118 | 23.3 | 0.064–0.172 | 34.4 | 0.133 | 22.5 | 0.074–0.192 | 36.5 |
IIV, interindividual variability; RUV, residual unexplained variability; RSE, relative standard error; CI, confidence interval.
Fixed pharmacokinetic estimate from one-compartmental model analysis. *P < 0.05 for parameter estimates between mouse strains.
Visual inspection of the plasma concentration time curves suggested that the majority of absorption had occurred prior to obtaining the first sample, making it difficult to estimate ka, particularly in the mdr1a−/−/1b−/− mice; therefore, the estimate for absorption in the mdr1a−/−/1b−/− mice from the one-compartment model is expressed as a fixed estimate. There were no differences between pharmacokinetic parameters as a function of age. There were modest increases in both CL/F and V/F between FVB wild-type and mdr1a−/−/1b−/− mice; the magnitude of the increases, however, were small (1.6- and 1.9-fold, respectively). Additionally, the difference in CL/F between the mice was in the opposite direction of what would be expected of a P-gp substrate (Polli et al., 1999; Choo et al., 2000).
We next measured brain accumulation in FVB wild-type and mdr1a−/−/1b−/− mice, which we would expect to be the parameter most altered if paraquat was a P-gp substrate. All brain samples were above the LOQ, with the exception of the 10 mg/kg dose group in the mdr1a−/−/1b−/− mice, although these samples were above the LOD. We observed no differences between FVB wild-type and mdr1a−/−/1b−/− mice in paraquat brain-to-plasma partitioning ratios calculated from combined dose groups (2.39 ± 1.57 and 1.92 ± 1.62, respectively) or total brain paraquat accumulation across dose groups (Fig. 5). The lack of a difference in brain distribution between FVB wild-type and mdr1a−/−/1b−/− mice confirms that paraquat is not a P-gp substrate.
Fig. 5.

Paraquat brain accumulation in FVB wild-type and mdr1a−/−/1b−/− mice. Brains were collected at 8 hours after oral administration of paraquat at doses of 10, 25, 50, or 100 mg/kg to FVB wild-type (closed squares) or mdr1a−/−/1b−/− mice (open squares). Data are presented as mean ± S.D. for each dose group—10 mg/kg: FVB, n = 10, mdr1a−/−/1b−/−, n = 6; 25 mg/kg: FVB, n = 9, mdr1a−/−/1b−/−, n = 6; 50 mg/kg: FVB, n = 5, mdr1a−/−/1b−/−, n = 10; and 100 mg/kg: FVB, n = 10; mdr1a−/−/1b−/−, n = 6.
DISCUSSION
We have systematically evaluated the P-gp-mediated transport of the herbicide paraquat using a combination of in vitro and in vivo models. We determined that paraquat is not a substrate or an inhibitor of P-gp using membrane- and cell-based assays. Additionally, pharmacokinetic and brain-accumulation studies in wild-type and P-gp-deficient mice showed that P-gp does not mediate the disposition of paraquat. Although there is evidence to show that P-gp is associated with the development of Parkinson disease, these data demonstrate that the causal relationship is not due to the P-gp-mediated transport of paraquat.
Paraquat was screened as a substrate for P-gp in three experimental models: ATPase activity, xenobiotic-induced cytotoxicity, and transepithelial permeability, all of which have been well characterized to study P-gp (Polli et al., 2001; Feng et al., 2008; Giacomini et al., 2010; Brouwer et al., 2013; Hillgren et al., 2013; Zamek-Gliszczynski et al., 2013). First, paraquat did not stimulate the hydrolysis of ATP in P-gp-expressing membranes. ATPase assays are commonly used screening tools to investigate potential substrates of P-gp; however, slowly transported substrates may not simulate ATPase activity (Giacomini et al., 2010; Bircsak et al., 2013). Therefore, we next assessed paraquat as a P-gp substrate in a cell-based model. LLC-PK1 cell lines are polarized cells that form tight junctions, have low endogenous expression levels of transporters, and are standard cell lines for predicting drug transport across the small intestine and blood-brain barrier (Giacomini et al., 2010; Brouwer et al., 2013; Hillgren et al., 2013; Zamek-Gliszczynski et al., 2013). We used recombinant LLC-vector and LLC-MDR1-WT cells as well as a combination of known P-gp substrates and inhibitors to ensure reliability of the transport models. In cytotoxicity assays, we observed a small increase in paraquat resistance between P-gp-expressing and nonexpressing cells, but importantly, observed no effect of P-gp inhibitors, GF120918 and verapamil, on the resistance. This indicates that P-gp was not contributing to the observed cellular resistance. As a comparison in our study, known P-gp substrates doxorubicin and colchicine exhibited marked increases in resistance, 61- and 48-fold, respectively, between P-gp-expressing and nonexpressing cells, and resistance was reversed in the presence of P-gp inhibitors. We also observed no directional transepithelial transport of paraquat mediated by P-gp when cells were grown on permeable supports, which also confirms that paraquat is not a P-gp substrate. Finally, paraquat also did not inhibit R123 efflux in P-gp-expressing cells. Therefore, we can conclude from the in vitro studies that paraquat is not a P-gp substrate or inhibitor.
We next evaluated paraquat pharmacokinetics and brain accumulation between FVB wild-type and mdr1a−/−/1b−/− mice in an oral dose-escalation study to confirm in vitro studies. We observed subtle differences in the primary pharmacokinetic parameters between wild-type and knockout mice. For instance, there was a significant increase in CL/F in mdr1a−/−/1b−/− mice when compared with FVB wild-type. This observation, however, is opposite of what would be expected if paraquat were a P-gp substrate, that CL/F of paraquat would be decreased in mdr1a−/−/1b−/− mice (Polli et al., 1999; Choo et al., 2000). Therefore, there may be other compensatory mechanisms altered in the mdr1a−/−/1b−/− mice that could account for the differences in paraquat pharmacokinetics. Alterations in drug-metabolizing enzymes and drug transporters have been observed in mdr1a−/−/1b−/− mice (Schuetz et al., 2000). Most notably, we did not observe differences in brain-to-plasma partitioning or total brain accumulation of paraquat between FVB wild-type and mdr1a−/−/1b−/− mice. Drugs that are P-gp substrates, such as amprenavir, ivermectin, and vinblastine, display approximately 20–80-fold increase in brain accumulation in knockout mice relative to wild-type mice due to loss of P-gp activity at the blood-brain barrier (Schinkel et al., 1994; van Asperen et al., 1996; Polli et al., 1999). Therefore, it is clear from the lack of differential brain distribution of paraquat that P-gp is not playing a role in paraquat disposition. The animal studies confirm our in vitro results that paraquat is not a P-gp substrate.
Our study is the first comprehensive study to measure P-gp transport of paraquat both in vitro and in vivo. Previous studies in rats have found that induction of P-gp expression was protective against paraquat-induced toxicity, which would suggest that paraquat is a P-gp substrate (Dinis-Oliveira et al., 2006a,b). These studies, however, used dexamethasone and doxorubicin as inducers, which are not P-gp specific and are known to be broad-spectrum inducers of not only other drug transporters but drug-metabolizing enzymes as well. Therefore, several mechanisms other than P-gp may have been involved in the protection against paraquat toxicity. This same group also examined P-gp transport of paraquat in Caco-2 cells following induction of P-gp by doxorubicin using paraquat concentrations ranging from 10–5000 μM. The researchers found less than a 2-fold increase in the reported EC50 values as a result of doxorubicin treatment, indicating a minimal protective effect by P-gp (Silva et al., 2011). Finally, another study evaluating paraquat as a P-gp substrate in Caco-2 cells observed differential paraquat cytotoxicity between untreated and doxorubicin-induced cells, but only at paraquat concentrations of 1 mM and above (Silva et al., 2013). These concentrations far exceed reported paraquat concentrations in humans. Blood samples from patients with acute paraquat poisoning had measured paraquat plasma concentrations that did not exceed 50,000 ng/ml (194 μM), and most patients’ concentrations were considerably lower (Shi et al., 2012). Thus, evaluating P-gp transport of paraquat in the millimolar concentration range is not relevant to systemic paraquat exposures in humans.
Although this is the first study to evaluate the role of P-gp in paraquat pharmacokinetics, there have been other studies to measure paraquat pharmacokinetics in chronic studies (Prasad et al., 2007, 2009; Breckenridge et al., 2013). The paraquat plasma concentrations we observed following an oral dose in FVB mice were approximately a magnitude less than what has been observed following an intraperitoneal dose to C57BlJ/6 mice; correspondingly, the paraquat brain concentrations we observed were also lower (Prasad et al., 2007, 2009; Breckenridge et al., 2013). Paraquat has highly variable bioavailability depending upon the route of administration, with the majority of an oral dose found in the feces, which may explain some of the differences in observed plasma concentrations in the different mouse strains (Daniel and Gage, 1966; Chui et al., 1988).
Several studies have described an association between ABCB1 pharmacogenomics and susceptibility to Parkinson disease (Le Couteur et al., 2001; Furuno et al., 2002; Drozdzik et al., 2003; Lee et al., 2004; Lee and Bendayan, 2004; Tan et al., 2004, 2005; Kortekaas et al., 2005; Bartels et al., 2008, 2009; Westerlund et al., 2008, 2009; Vautier and Fernandez, 2009; Dutheil et al., 2010). One mechanism may be that ABCB1 genetic variation decreases P-gp activity at the blood-brain barrier, leading to increased brain accumulation of neurotoxicants and an increased incidence of Parkinson disease. In European populations, the 1236C > T and 3435C > T single nucleotide polymorphisms have been shown to have an increased frequency in Parkinson disease, while another study evaluating the 2677G > T/A and 3435C > T single nucleotide polymorphisms found no association between allele frequencies and Parkinson disease (Furuno et al., 2002; Tan et al., 2004). Studies in Asian patients showed that the 2677T/3435T or 1236T/2677T/3435T haplotypes were significantly associated with a reduced risk of Parkinson disease (Lee et al., 2004; Lee and Bendayan, 2004; Tan et al., 2005). Finally, a recent study in Europeans observed an association between ABCB1 variation and Parkinson disease risk, but only after including occupational exposure to organochlorine pesticides in the analysis; the researchers observed a higher risk of Parkinson disease in patients with 2677TT or TA genotypes who were also exposed to organochlorines (Dutheil et al., 2010). Some of these conflicting associations with ABCB1 pharmacogenomics and Parkinson disease could be a result of small sample sizes as well as genetic heterogeneity within the study populations; however, there does appear to be a link between ABCB1 genetic variation and Parkinson disease risk. In addition to pharmacogenetic evidence, another link to P-gp activity in Parkinson disease was indicated by studies that showed increased brain accumulation of verapamil in Parkinson-disease patients compared with control subjects, indicating that P-gp activity at the blood-brain barrier may be decreased (Kortekaas et al., 2005; Bartels et al., 2009). While there is evidence for a role of P-gp in the development of Parkinson disease, it is clear from our data that altered P-gp transport of paraquat is not the cause.
In summary, we have demonstrated in both in vitro and in vivo models that paraquat is not a substrate or inhibitor of P-gp. Notably, P-gp expression did not alter the brain distribution of paraquat in mdr1a−/−/1b−/− mice. Therefore, the association of ABCB1 genetic variation and the increased risk of developing Parkinson disease is not due to alterations in P-gp efflux of the herbicide paraquat. Further research is needed to identify neurotoxicants that may play a role in the ABCB1 pharmacogenetic associations with Parkinson disease.
Supplementary Material
Acknowledgments
The authors thank the Molecular Computational Core Facility and Pam Shaw at the Fluorescence Cytometry Core at the University of Montana.
Abbreviations
- ABC
ATP-binding cassette
- AUC
area under the curve
- CL/F
apparent oral clearance
- DAPI
4,6-diamidino-2-phenylindole
- CI
confidence interval
- CL
confidence limit
- FVB
Friend leukemia virus B-type
- GF120918
N-(4-[2-(1,2,3,4-tetrahydro-6,7-dimethoxy-2-isoquinolinyl)ethyl]-phenyl)-9,10-dihydro-5-methoxy-9-oxo-4-acridine carboxamide
- ka
absorption rate constant
- λz
elimination rate constant
- LC-MS/MS
liquid chromatography tandem mass spectroscopy
- LLC-MDR1-WT
recombinant ABCB1/MDR1
- LLC-vector
LLC-PK1 vector
- LOD
limit of detection
- LOQ
limit of quantitation
- MDR
multidrug resistance
- MRT
mean residence time
- Papp
apparent permeability
- Papp A→B
apical-to-basolateral apparent permeability
- Papp B→A
basolateral-to-apical apparent permeability
- P-gp
P-glycoprotein
- Pi
inorganic phosphate
- t1/2
elimination half-life
- tmax
time of maximum observed concentration
- V/F
apparent volume of distribution
Authorship Contributions
Participated in research design: Lacher, Cardozo-Pelaez, Woodahl.
Conducted experiments: Lacher, Gremaud, Skagen, Steed, Dalton.
Contributed new reagents or analytic tools: Gremaud, Sugden, Sherwin.
Performed data analysis: Lacher, Gremaud, Skagen, Steed, Dalton, Sherwin, Woodahl.
Wrote or contributed to the writing of the manuscript: Lacher, Gremaud, Skagen, Dalton, Sherwin, Woodahl.
Footnotes
The authors report no conflict of interest.
This work was supported by the National Institutes of Health Centers of Biomedical Research Excellence grants that support the Center for Biomolecular Structure and Dynamics [Grant P20GM103546], the Center for Environmental Health Sciences [Grant P20RR017670], and the Center for Structural and Functional Neuroscience [Grant P20RR015583]; the American Foundation for Pharmaceutical Education, American Association of College of Pharmacy, New Investigators Program (E.L.W.); the Institute of Translational Health Sciences [Grant UL1TR000423] (E.L.W.]; and the American Foundation for Pharmacy Education Predoctoral Fellowship (S.E.L.).
 This article has supplemental material available at jpet.aspetjournals.org
This article has supplemental material available at jpet.aspetjournals.org
REFERENCES
- Ariffin MM, Anderson RA. (2006) LC/MS/MS analysis of quaternary ammonium drugs and herbicides in whole blood. J Chromatogr B Analyt Technol Biomed Life Sci 842:91–97 [DOI] [PubMed] [Google Scholar]
- Bartels AL, Kortekaas R, Bart J, Willemsen AT, de Klerk OL, de Vries JJ, van Oostrom JC, Leenders KL. (2009) Blood-brain barrier P-glycoprotein function decreases in specific brain regions with aging: a possible role in progressive neurodegeneration. Neurobiol Aging 30:1818–1824 [DOI] [PubMed] [Google Scholar]
- Bartels AL, Willemsen AT, Kortekaas R, de Jong BM, de Vries R, de Klerk O, van Oostrom JC, Portman A, Leenders KL. (2008) Decreased blood-brain barrier P-glycoprotein function in the progression of Parkinson’s disease, PSP and MSA. J Neural Transm 115:1001–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer LA. (2005) Clinical Pharmacokinetics Handbook, McGraw-Hill Companies, New York [Google Scholar]
- Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. (2011) Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J 13:143–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bircsak KM, Richardson JR, Aleksunes LM. (2013) Inhibition of human MDR1 and BCRP transporter ATPase activity by organochlorine and pyrethroid insecticides. J Biochem Mol Toxicol 27:157–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet AM, Houeto JL. (1999) Pathophysiology of Parkinson’s disease. Biomed Pharmacother 53:117–121 [DOI] [PubMed] [Google Scholar]
- Breckenridge CB, Sturgess NC, Butt M, Wolf JC, Zadory D, Beck M, Mathews JM, Tisdel MO, Minnema D, Travis KZ, et al. (2013) Pharmacokinetic, neurochemical, stereological and neuropathological studies on the potential effects of paraquat in the substantia nigra pars compacta and striatum of male C57BL/6J mice. Neurotoxicology 37:1–14 [DOI] [PubMed] [Google Scholar]
- Brouwer KL, Keppler D, Hoffmaster KA, Bow DA, Cheng Y, Lai Y, Palm JE, Stieger B, Evers R, International Transporter Consortium (2013) In vitro methods to support transporter evaluation in drug discovery and development. Clin Pharmacol Ther 94:95–112 [DOI] [PubMed] [Google Scholar]
- Cascorbi I. (2011) P-glycoprotein: tissue distribution, substrates, and functional consequences of genetic variations. Handb Exp Pharmacol 261–283 [DOI] [PubMed]
- Cascorbi I, Gerloff T, Johne A, Meisel C, Hoffmeyer S, Schwab M, Schaeffeler E, Eichelbaum M, Brinkmann U, Roots I. (2001) Frequency of single nucleotide polymorphisms in the P-glycoprotein drug transporter MDR1 gene in white subjects. Clin Pharmacol Ther 69:169–174 [DOI] [PubMed] [Google Scholar]
- Chinn LW, Kroetz DL. (2007) ABCB1 pharmacogenetics: progress, pitfalls, and promise. Clin Pharmacol Ther 81:265–269 [DOI] [PubMed] [Google Scholar]
- Choo EF, Leake B, Wandel C, Imamura H, Wood AJ, Wilkinson GR, Kim RB. (2000) Pharmacological inhibition of P-glycoprotein transport enhances the distribution of HIV-1 protease inhibitors into brain and testes. Drug Metab Dispos 28:655–660 [PubMed] [Google Scholar]
- Chui YC, Poon G, Law F. (1988) Toxicokinetics and bioavailability of paraquat in rats following different routes of administration. Toxicol Ind Health 4:203–219 [DOI] [PubMed] [Google Scholar]
- Daniel JW, Gage JC. (1966) Absorption and excretion of diquat and paraquat in rats. Br J Ind Med 23:133–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinis-Oliveira RJ, Duarte JA, Remião F, Sánchez-Navarro A, Bastos ML, Carvalho F. (2006a) Single high dose dexamethasone treatment decreases the pathological score and increases the survival rate of paraquat-intoxicated rats. Toxicology 227:73–85 [DOI] [PubMed] [Google Scholar]
- Dinis-Oliveira RJ, Remião F, Duarte JA, Ferreira R, Sánchez Navarro A, Bastos ML, Carvalho F. (2006b) P-glycoprotein induction: an antidotal pathway for paraquat-induced lung toxicity. Free Radic Biol Med 41:1213–1224 [DOI] [PubMed] [Google Scholar]
- Droździk M, Białecka M, Myśliwiec K, Honczarenko K, Stankiewicz J, Sych Z. (2003) Polymorphism in the P-glycoprotein drug transporter MDR1 gene: a possible link between environmental and genetic factors in Parkinson’s disease. Pharmacogenetics 13:259–263 [DOI] [PubMed] [Google Scholar]
- Dutheil F, Beaune P, Tzourio C, Loriot MA, Elbaz A. (2010) Interaction between ABCB1 and professional exposure to organochlorine insecticides in Parkinson disease. Arch Neurol 67:739–745 [DOI] [PubMed] [Google Scholar]
- Feng B, Mills JB, Davidson RE, Mireles RJ, Janiszewski JS, Troutman MD, de Morais SM. (2008) In vitro P-glycoprotein assays to predict the in vivo interactions of P-glycoprotein with drugs in the central nervous system. Drug Metab Dispos 36:268–275 [DOI] [PubMed] [Google Scholar]
- Furuno T, Landi MT, Ceroni M, Caporaso N, Bernucci I, Nappi G, Martignoni E, Schaeffeler E, Eichelbaum M, Schwab M, et al. (2002) Expression polymorphism of the blood-brain barrier component P-glycoprotein (MDR1) in relation to Parkinson’s disease. Pharmacogenetics 12:529–534 [DOI] [PubMed] [Google Scholar]
- Gabrielsson J, Weiner D. (2012) Non-compartmental analysis. Methods Mol Biol 929:377–389 [DOI] [PubMed] [Google Scholar]
- Gatto NM, Cockburn M, Bronstein J, Manthripragada AD, Ritz B. (2009) Well-water consumption and Parkinson’s disease in rural California. Environ Health Perspect 117:1912–1918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacomini KM. (1997) Membrane transporters in drug disposition. J Pharmacokinet Biopharm 25:731–741 [DOI] [PubMed] [Google Scholar]
- Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, Chu X, Dahlin A, Evers R, Fischer V, Hillgren KM, et al. International Transporter Consortium (2010) Membrane transporters in drug development. Nat Rev Drug Discov 9:215–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillgren KM, Keppler D, Zur AA, Giacomini KM, Stieger B, Cass CE, Zhang L, International Transporter Consortium (2013) Emerging transporters of clinical importance: an update from the International Transporter Consortium. Clin Pharmacol Ther 94:52–63 [DOI] [PubMed] [Google Scholar]
- Kamel F. (2013) Epidemiology. Paths from pesticides to Parkinson’s. Science 341:722–723 [DOI] [PubMed] [Google Scholar]
- Kortekaas R, Leenders KL, van Oostrom JC, Vaalburg W, Bart J, Willemsen AT, Hendrikse NH. (2005) Blood-brain barrier dysfunction in parkinsonian midbrain in vivo. Ann Neurol 57:176–179 [DOI] [PubMed] [Google Scholar]
- Langston JW, Irwin I, Langston EB, Forno LS. (1984) 1-Methyl-4-phenylpyridinium ion (MPP+): identification of a metabolite of MPTP, a toxin selective to the substantia nigra. Neurosci Lett 48:87–92 [DOI] [PubMed] [Google Scholar]
- Le Couteur DG, Davis MW, Webb M, Board PG. (2001) P-glycoprotein, multidrug-resistance-associated protein and Parkinson’s disease. Eur Neurol 45:289–290 [DOI] [PubMed] [Google Scholar]
- Lee CG, Tang K, Cheung YB, Wong LP, Tan C, Shen H, Zhao Y, Pavanni R, Lee EJ, Wong MC, et al. (2004) MDR1, the blood-brain barrier transporter, is associated with Parkinson’s disease in ethnic Chinese. J Med Genet 41:e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G, Bendayan R. (2004) Functional expression and localization of P-glycoprotein in the central nervous system: relevance to the pathogenesis and treatment of neurological disorders. Pharm Res 21:1313–1330 [DOI] [PubMed] [Google Scholar]
- Lin JH, Yamazaki M. (2003a) Clinical relevance of P-glycoprotein in drug therapy. Drug Metab Rev 35:417–454 [DOI] [PubMed] [Google Scholar]
- Lin JH, Yamazaki M. (2003b) Role of P-glycoprotein in pharmacokinetics: clinical implications. Clin Pharmacokinet 42:59–98 [DOI] [PubMed] [Google Scholar]
- Mostafalou S, Abdollahi M. (2013) Pesticides and human chronic diseases: evidences, mechanisms, and perspectives. Toxicol Appl Pharmacol 268:157–177 [DOI] [PubMed] [Google Scholar]
- Pezzoli G, Cereda E. (2013) Exposure to pesticides or solvents and risk of Parkinson disease. Neurology 80:2035–2041 [DOI] [PubMed] [Google Scholar]
- Pharsight (2005–2009). Getting Started Guide [PDF file]. Available from http://www.pharsight.com/library/Phoenix%201.1%20Getting%20Started%20Guide.pdf
- Polli JW, Jarrett JL, Studenberg SD, Humphreys JE, Dennis SW, Brouwer KR, Woolley JL. (1999) Role of P-glycoprotein on the CNS disposition of amprenavir (141W94), an HIV protease inhibitor. Pharm Res 16:1206–1212 [DOI] [PubMed] [Google Scholar]
- Polli JW, Wring SA, Humphreys JE, Huang L, Morgan JB, Webster LO, Serabjit-Singh CS. (2001) Rational use of in vitro P-glycoprotein assays in drug discovery. J Pharmacol Exp Ther 299:620–628 [PubMed] [Google Scholar]
- Prasad K, Tarasewicz E, Mathew J, Strickland PA, Buckley B, Richardson JR, Richfield EK. (2009) Toxicokinetics and toxicodynamics of paraquat accumulation in mouse brain. Exp Neurol 215:358–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad K, Winnik B, Thiruchelvam MJ, Buckley B, Mirochnitchenko O, Richfield EK. (2007) Prolonged toxicokinetics and toxicodynamics of paraquat in mouse brain. Environ Health Perspect 115:1448–1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinkel AH, Smit JJ, van Tellingen O, Beijnen JH, Wagenaar E, van Deemter L, Mol CA, van der Valk MA, Robanus-Maandag EC, te Riele HP, et al. (1994) Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell 77:491–502 [DOI] [PubMed] [Google Scholar]
- Schuetz EG, Umbenhauer DR, Yasuda K, Brimer C, Nguyen L, Relling MV, Schuetz JD, Schinkel AH. (2000) Altered expression of hepatic cytochromes P-450 in mice deficient in one or more mdr1 genes. Mol Pharmacol 57:188–197 [PubMed] [Google Scholar]
- Semchuk KM, Love EJ, Lee RG. (1992) Parkinson’s disease and exposure to agricultural work and pesticide chemicals. Neurology 42:1328–1335 [DOI] [PubMed] [Google Scholar]
- Sharom FJ. (2011) The P-glycoprotein multidrug transporter. Essays Biochem 50:161–178 [DOI] [PubMed] [Google Scholar]
- Sheiner LB, Beal SL. (1981) Evaluation of methods for estimating population pharmacokinetic parameters. II. Biexponential model and experimental pharmacokinetic data. J Pharmacokinet Biopharm 9:635–651 [DOI] [PubMed] [Google Scholar]
- Sherwin CM, Kiang TK, Spigarelli MG, Ensom MH. (2012) Fundamentals of population pharmacokinetic modelling: validation methods. Clin Pharmacokinet 51:573–590 [DOI] [PubMed] [Google Scholar]
- Shi Y, Bai Y, Zou Y, Cai B, Liu F, Fu P, Wang L. (2012) The value of plasma paraquat concentration in predicting therapeutic effects of haemoperfusion in patients with acute paraquat poisoning. PLoS ONE 7:e40911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva R, Carmo H, Dinis-Oliveira R, Cordeiro-da-Silva A, Lima SC, Carvalho F, Bastos MdeL, Remião F. (2011) In vitro study of P-glycoprotein induction as an antidotal pathway to prevent cytotoxicity in Caco-2 cells. Arch Toxicol 85:315–326 [DOI] [PubMed] [Google Scholar]
- Silva R, Carmo H, Vilas-Boas V, de Pinho PG, Dinis-Oliveira RJ, Carvalho F, Silva I, Correia-de-Sá P, Bastos MdeL, Remião F. (2013) Doxorubicin decreases paraquat accumulation and toxicity in Caco-2 cells. Toxicol Lett 217:34–41 [DOI] [PubMed] [Google Scholar]
- Tan EK, Chan DK, Ng PW, Woo J, Teo YY, Tang K, Wong LP, Chong SS, Tan C, Shen H, et al. (2005) Effect of MDR1 haplotype on risk of Parkinson disease. Arch Neurol 62:460–464 [DOI] [PubMed] [Google Scholar]
- Tan EK, Drozdzik M, Bialecka M, Honczarenko K, Klodowska-Duda G, Teo YY, Tang K, Wong LP, Chong SS, Tan C, et al. (2004) Analysis of MDR1 haplotypes in Parkinson’s disease in a white population. Neurosci Lett 372:240–244 [DOI] [PubMed] [Google Scholar]
- van Asperen J, Schinkel AH, Beijnen JH, Nooijen WJ, Borst P, van Tellingen O. (1996) Altered pharmacokinetics of vinblastine in Mdr1a P-glycoprotein-deficient Mice. J Natl Cancer Inst 88:994–999 [DOI] [PubMed] [Google Scholar]
- Vautier S, Fernandez C. (2009) ABCB1: the role in Parkinson’s disease and pharmacokinetics of antiparkinsonian drugs. Expert Opin Drug Metab Toxicol 5:1349–1358 [DOI] [PubMed] [Google Scholar]
- Wang D, Sadée W. (2006) Searching for polymorphisms that affect gene expression and mRNA processing: example ABCB1 (MDR1). AAPS J 8:E515–E520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerlund M, Belin AC, Anvret A, Håkansson A, Nissbrandt H, Lind C, Sydow O, Olson L, Galter D. (2009) Association of a polymorphism in the ABCB1 gene with Parkinson’s disease. Parkinsonism Relat Disord 15:422–424 [DOI] [PubMed] [Google Scholar]
- Westerlund M, Belin AC, Olson L, Galter D. (2008) Expression of multi-drug resistance 1 mRNA in human and rodent tissues: reduced levels in Parkinson patients. Cell Tissue Res 334:179–185 [DOI] [PubMed] [Google Scholar]
- Wirdefeldt K, Adami HO, Cole P, Trichopoulos D, Mandel J. (2011) Epidemiology and etiology of Parkinson’s disease: a review of the evidence. Eur J Epidemiol 26 (Suppl 1):S1–S58 [DOI] [PubMed] [Google Scholar]
- Woodahl EL, Ho RJ. (2004) The role of MDR1 genetic polymorphisms in interindividual variability in P-glycoprotein expression and function. Curr Drug Metab 5:11–19 [DOI] [PubMed] [Google Scholar]
- Woodahl EL, Yang Z, Bui T, Shen DD, Ho RJ. (2004) Multidrug resistance gene G1199A polymorphism alters efflux transport activity of P-glycoprotein. J Pharmacol Exp Ther 310:1199–1207 [DOI] [PubMed] [Google Scholar]
- Yuh L, Beal S, Davidian M, Harrison F, Hester A, Kowalski K, Vonesh E, Wolfinger R. (1994) Population pharmacokinetic/pharmacodynamic methodology and applications: a bibliography. Biometrics 50:566–575 [PubMed] [Google Scholar]
- Zamek-Gliszczynski MJ, Lee CA, Poirier A, Bentz J, Chu X, Ellens H, Ishikawa T, Jamei M, Kalvass JC, Nagar S, et al. International Transporter Consortium (2013) ITC recommendations for transporter kinetic parameter estimation and translational modeling of transport-mediated PK and DDIs in humans. Clin Pharmacol Ther 94:64–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.