

Abstract
Malaria is one of the strongest selective pressures in recent human evolution. African populations have been and continue to be at risk for malarial infections. However, few studies have re-sequenced malaria susceptibility loci across geographically and genetically diverse groups in Africa. We examined nucleotide diversity at Intercellular adhesion molecule-1 (ICAM-1), a malaria susceptibility candidate locus, in a number of human populations with a specific focus on diverse African ethnic groups. We used tests of neutrality to assess whether natural selection has impacted this locus and tested whether SNP variation at ICAM-1 is correlated with malaria endemicity. We observe differing patterns of nucleotide and haplotype variation in global populations and higher levels of diversity in Africa. Although we do not observe a deviation from neutrality based on the allele frequency distribution, we do observe several alleles at ICAM-1, including the ICAM-1Kilifi allele, that are correlated with malaria endemicity. We show that the ICAM-1Kilifi allele, which is common in Africa and Asia, exists on distinct haplotype backgrounds and is likely to have arisen more recently in Asia. Our results suggest that correlation analyses of allele frequencies and malaria endemicity may be useful for identifying candidate functional variants that play a role in malaria resistance and susceptibility.
Introduction
Selection due to infectious disease has had a major impact on patterns of genetic variation within and between modern human populations (Ayodo et al. 2007; Cook and Hill 2001; Fortin et al. 2002; Hamblin and Di Rienzo 2000; Hamblin et al. 2002; Ko et al. 2011; Verrelli et al. 2002). Malaria, in particular, has been an important selective force in recent human evolution (Clegg and Weatherall 1999; Fortin et al. 2002; Kwiatkowski 2000, 2005; Livingstone 1958, 1971; Verra et al. 2009). Characterizing patterns of nucleotide diversity at malaria susceptibility candidate loci and testing for signatures of natural selection may be informative for identifying functional loci that are related to malaria susceptibility (Ayodo et al. 2007; Ko et al. 2011; Tishkoff et al. 2001).
Intercellular adhesion molecule-1 (ICAM-1) is expressed on the surface of vascular endothelium cells and is thought to play a role in the pathogenesis of severe malaria (Chakraborty and Craig 2004; Chattopadhyay et al. 2004; Chen et al. 2000). During the erythrocytic phase of a Plasmodium falciparum infection, infected erythrocytes (IE) adhere to ICAM-1 and a number of other endothelial cell receptors (e.g. CD36, VCAM-1, and PECAM). The adherence of IEs to ICAM-1 is mediated through PfEMP-1 (P. falciparum erythrocyte membrane protein-1), which is a P. falciparum protein that is exported to the red blood cell membrane surface during malarial infections. The adherence of IEs to vascular endothelium is a critical component of a malaria infection because it is associated with complications of severe malaria, including cerebral malaria, which is a major cause of mortality, especially in Africa (Miller et al. 2002; Murphy and Breman 2001; Turner et al. 1994).
Because ICAM-1 has been implicated in the pathogenesis of severe malaria, a number of studies have investigated how genetic variation at ICAM-1 is related to malaria susceptibility and malarial disease phenotypes (Adams et al. 2000; Ayodo et al. 2007; Bellamy et al. 1998; Craig et al. 2000; Fry et al. 2008; Kun et al. 1999). In 1997 Fernandez-Reyes et al. (1997) identified a high-frequency non-synonymous single-nucleotide polymorphism (SNP) in ICAM-1 (rs5491). The derived allele of this SNP, designated ICAM-1Kilifi, causes a lysine to methionine change at amino acid position 29, which is located in the domain of ICAM-1 involved in IE adherence. When Fernandez-Reyes et al. (1997) examined the frequency of this allele among severe malaria cases and age-, time- and location-matched controls, they found that the ICAM-1Kilifi allele was associated with increased susceptibility to severe malaria, rather than protection. Subsequent studies have tested for evidence of an association between ICAM-1Kilifi and susceptibility to malarial disease. These studies have reported contradictory results. For example, Kun et al. (1999) suggest that ICAM-1Kilifi may be protective against severe malaria, whereas other studies suggest that no association exists between variation at ICAM-1 and malarial disease (Fry et al. 2008; Ndiaye et al. 2005; Ohashi et al. 2001). The ICAM-1Kilifi allele is also largely restricted to Africa and Asia, where the prevalence of malaria is high. This has caused some (Fry et al. 2008; Newbold et al. 1999) to propose that the restricted geographic distribution of this allele is the result of a selective event related to protection from malaria.
African populations have been and continue to be at risk for malarial disease. In 2007, approximately 345 million Africans lived in high malaria transmission areas (Hay et al. 2009) and in 2009, 91 % of the global malaria deaths occurred in Africa (WHO 2010). Because African populations have high levels of genetic sub-structure (Tishkoff et al. 2009) and live in diverse environments with differing exposure to P. falciparum, they may have population or regionally specific variants that play a role in malaria susceptibility. Therefore, we have re-sequenced approximately 6 kb of ICAM-1, located at 19p13.3–p13.2, in a sample of 416 unrelated, geographically and ethnically diverse, African and non-African individuals. We applied tests of neutrality to these data to identify signatures of natural selection and have tested whether patterns of genetic diversity at ICAM-1 are correlated with levels of malaria endemicity. The results of our study show that the ICAM-1Kilifi allele and SNPs in linkage disequilibrium (LD) with this site are correlated with malaria endemicity. This result implies that the ICAM-1Kilifi variant is indeed a candidate site for protection against malaria and deserves further study. Our results also suggest that correlation analyses may be useful to detect functional malaria resistance alleles that may not show simple signatures of natural selection.
Materials and methods
Populations sampled
We sequenced ICAM-1 in 318 individuals from 17 ethnically and geographically diverse African populations and 98 non-African individuals from four populations (Table 1). The Africans individuals included in this study come from diverse ethnic groups located in Nigeria, Cameroon, Tanzania, Kenya, Mozambique, São Tomé, Chad, and Sudan. In some cases, the specific ethnic group of a sampled individual was not known and these individuals are listed by geographic region of origin (see Table 1). In addition to the geographic and ethnic diversity represented by the African individuals included in this study, these individuals also live in different climates, practice different subsistence strategies, and live in areas of Africa where malaria endemicity differs (Snow et al. 2005; Tishkoff et al. 2009). All samples were collected and analyzed after obtaining approval from the institutional review boards (IRBs) of the University of Maryland and the University of Pennsylvania. Research and ethics approval and permits were obtained from the following institutions prior to sample collection: Commission for Science and Technology and the National Institute for Medical Research in Dar es Salaam, Tanzania; The Kenya Medical Research Institute in Nairobi, Kenya; The University of Khartoum in Sudan; The Nigerian Institute for Research and Pharmacological Development, Abuja, Nigeria; The Ministry of Health and National Committee of Ethics, Cameroon; The Eduardo Mondlane University, Maputo, Mozambique; The São Tomé Ministry of Health; The University of Porto, Portugal; The College of Public Health Sciences, Chulalongkorn University, Bangkok, Thailand. Written informed or oral consent was received from all study participants. The HapMap European (CEU) and Chinese (CHB) DNA samples were obtained from Coriell Cell Repositories in Camden, NJ.
Table 1.
Population diversity summary statistics
| Populations | 2N | S | Ssy | Snsy | ICAM-1Kilifi | π (× 10−3) | θw (× 10−3) | D |
|---|---|---|---|---|---|---|---|---|
| All samples pooled | 832 | 87 | 9 | 21 | 0.14 | 0.69 | 1.92 | –1.79* |
| Africans | 636 | 73 | 7 | 19 | 0.16 | 0.69 | 1.67 | −1.65 |
| Non-Africans | 196 | 27 | 2 | 5 | 0.07 | 0.56 | 0.75 | −0.72 |
| West Africa | ||||||||
| São Tomé (ST) | 40 | 14 | 1 | 2 | 0.17 | 0.62 | 0.70 | −0.06 |
| Cameroon | ||||||||
| Bakola Pygmy (PL)1 | 38 | 21 | 1 | 3 | 0.21 | 0.81 | 0.64 | −0.71 |
| Fulani (FU)1 | 38 | 18 | 1 | 3 | 0.16 | 0.62 | 0.70 | −0.38 |
| Mada (MD)1 | 38 | 18 | 0 | 5 | 0.11 | 0.50 | 0.70 | −0.91 |
| Lemande (LM)1 | 38 | 19 | 2 | 4 | 0.34 | 0.74 | 0.74 | 0.00 |
| Nigeria | ||||||||
| Yoruba (YR)1 | 36 | 17 | 1 | 4 | 0.25 | 0.58 | 0.67 | −0.41 |
| Chad | ||||||||
| Bulala (BU)1 | 26 | 16 | 1 | 4 | 0.08 | 0.60 | 0.68 | −0.44 |
| East Africa | ||||||||
| Mozambique (MO) | 40 | 14 | 2 | 2 | 0.25 | 0.58 | 0.54 | 0.28 |
| Sudan | ||||||||
| Beja (BJ)1 | 38 | 21 | 0 | 7 | 0.02 | 0.72 | 0.81 | −0.39 |
| Tanzania | ||||||||
| Hadza (HZ)1 | 48 | 17 | 1 | 5 | 0.13 | 0.55 | 0.62 | −0.39 |
| Sandawe (SW)1 | 46 | 30 | 1 | 7 | 0.22 | 0.64 | 1.10 | −1.44 |
| Datog (DT)1 | 38 | 26 | 3 | 8 | 0.11 | 0.75 | 1.01 | −0.88 |
| Iraqw (IQ)1 | 38 | 26 | 2 | 7 | 0.11 | 0.78 | 1.01 | −0.77 |
| Kenya | ||||||||
| Borana (BR)1 | 24 | 21 | 2 | 5 | 0.13 | 0.79 | 0.92 | −0.60 |
| Sengwer (SN)1 | 34 | 17 | 2 | 4 | 0.15 | 0.63 | 0.69 | 0.26 |
| Boni (BN)1 | 38 | 24 | 2 | 6 | 0.13 | 0.74 | 0.93 | −0.68 |
| Luo (LO)1 | 38 | 22 | 2 | 4 | 0.13 | 0.56 | 0.85 | −1.15 |
| Europe | ||||||||
| HapMap Europe (CEU) | 60 | 18 | 1 | 3 | 0.00 | 0.60 | 0.63 | −0.16 |
| Portugal (PO) | 44 | 11 | 0 | 2 | 0.00 | 0.57 | 0.41 | 1.13 |
| Asia | ||||||||
| HapMap China (CHB) | 58 | 18 | 1 | 3 | 0.13 | 0.52 | 0.63 | −0.83 |
| Thailand (THL) | 34 | 10 | 1 | 2 | 0.21 | 0.47 | 0.40 | 0.52 |
2N number of chromosomes in a sample; S number of segregating sites; Ssy number of segregating synonymous sites; Snsy number of segregating non-synonymous sites; π average pairwise sequence differences per site (× 10−3); θw Watterson’s estimator of theta (× 10−3) (Watterson 1975); D Tajima’s D statistic (Tajima 1989). ICAM-1Kilifi ICAM-1Kilifi allele frequency in a given population; The asterisk (*) indicates P < 0.05
a population that was included in the correlation analysis
PCR and sequencing methods
We re-sequenced 6,134 bp of the ICAM-1 gene, including all seven exons, adjacent intronic regions, 5′ and 3′ UTRs, as well as 1,198 bp upstream of the 5′ UTR and 199 bp downstream of the 3′UTR. PCR was used to amplify three regions (region I = 2,304 bp; region II = 527 bp; region III = 3,301 bp) of this locus (see Fig. 1). PCR and sequencing primers were designed using the PRIMER3 (www.frodo.wi.mit.edu) and Primer Select (Lasergene) software (PCR primers are listed in Supplemental Table 5; sequencing primers available upon request). PCR fragments were amplified using Platinum HiFi Taq DNA polymerase (Invitrogen). Each PCR reaction mixture contained 1 × of PCR buffer, 2 mM of MgSO4, 1 unit of Platinum HiFi Taq DNA polymerase (Invitrogen), 200 mM of dNTP (Promega), 0.2 mM of each primer, and 100 ng of genomic DNA (final volume, 25 μl). PCR cycles consisted of one cycle of pre-incubation (94 °C for 1 min), 30 cycles of amplification (94 °C for 50 s, 65 °C—region II; 62 °C—region I and III for 50 s, and 68 °C for 2–4 min—depending on product size), and one additional cycle of extension (72 °C 5 min). PCR amplicons were prepared for sequencing using shrimp alkaline phosphatase and exonuclease I (U.S. Biochemicals) to eliminate unincorporated dNTPs. Sequencing reactions were performed using Big Dye Terminator chemistry (Applied Biosystems) and run on an ABI 3730 XL automated capillary sequencer. The Sequencher program (Gene Codes) was used to assemble and edit all sequence trace files. All singletons were confirmed by re-sequencing a new PCR amplicon from the genomic DNA template.
Fig. 1.
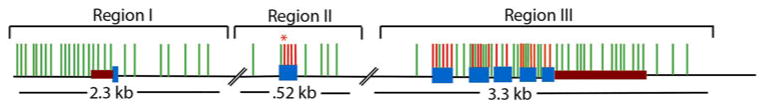
Representation of the ICAM-1 locus spanning ~15 kb. Exons are shown in blue boxes. The 5′ and 3′ UTRs are shown in maroon boxes. ICAM-1 was sequenced in three regions (I–III). The green lines represent silent (synonymous or non-coding) SNPs identified in this study, and the red lines represent non-synonymous SNPs identified in this study. The red asterisk (*) is placed over rs5491/ICAM-1Kilifi
Sequence alignment
Sequences were aligned using CLUSTAL X v.2.0 (Larkin et al. 2007) and adjustments were made manually to the alignment using the editing features of Bioedit (http://www.mbio.ncsu.edu/bioedit/bioedit.html) and Jalview (Waterhouse et al. 2009). Position one corresponds to the “A” in the ATG transcription start site, which corresponds to position 10,381,836 on chromosome 19 in GRCh37.p5/hg19 (Ref seq: NT_011295). The first sequenced nucleotide corresponds to −1520 from the “A” in the ATG transcription start site, which corresponds to position 10,380,316 on chromosome 19 in GRCh37.p5/hg19 (see Supplemental Table 1). The derived allele for polymorphic sites was determined using parsimony by comparing the human allele to the P. troglodytes sequence (NC006486.2). When the P. troglodytes data were unavailable the ancestral allele was determined using the ancestral allele determinations available in dbSNP (Sherry et al. 2001).
Nucleotide diversity, haplotype phase, and linkage disequilibrium analyses
We examined nucleotide diversity at ICAM-1 using two different measures of nucleotide diversity: average pairwise nucleotide difference (π) (Nei 1987) and Watterson’s θw (Watterson 1975) We used PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/), which is a tool that predicts the impact of non-synonymous SNPs on the structure and function of human proteins, to investigate potential consequences of the non-synonymous polymorphisms identified in this study (Table 2).
Table 2.
Summary of PolyPhen results at ICAM-1
| Amino acid change | PolyPhen2 score | Predicted impact | rsID | GRCh37.p5 coordinate | |
|---|---|---|---|---|---|
| Exon 2 | |||||
| +3704 | K56M (ICAM-1Kilifi) | 0.000 | Benign | 5491 | 10385540 |
| +3763 | R76W | 0.000 | Benign | N/a | 10385599 |
| +3827 | P97L | 0.771 | Possibly damaging | N/a | 10385663 |
| +3829 | D98N | 0.043 | Benign | N/a | 10385665 |
| Exon 3 | |||||
| +12332 | R115W | 0.998 | Probably damaging | N/a | 10394168 |
| +12333 | R115Q | 0.780 | Possibly damaging | N/a | 10394169 |
| +12444 | R152H | 0.999 | Probably damaging | N/a | 10394280 |
| +12454 | K155N | 0.002 | Benign | 5492 | 10394290 |
| +12602 | A205S | 0.135 | Benign | N/a | 10394438 |
| Exon 4 | |||||
| +12908 | R225W | 0.014 | Benign | N/a | 10394744 |
| +12948 | S238Y | 0.673 | Possibly damaging | N/a | 10394784 |
| +12956 | G241R | 0.995 | Probably damaging | 1799969 | 10394792 |
| +13082 | E283K | 0.342 | Possibly damaging | N/a | 10394918 |
| Exon 5 | |||||
| +13260 | V315M | 0.104 | Benign | 5495 | 10395096 |
| +13416 | R367C | 1.000 | Probably damaging | N/a | 10395252 |
| Exon 6 | |||||
| +13632 | R397Q | 0.624 | Possibly damaging | 5497 | 10395468 |
| +13653 | P404L | 0.851 | Probably damaging | N/a | 10395489 |
| +13814 | R458W | 0.001 | Benign | N/a | 10395650 |
| +13847 | K469E | 0.000 | Benign | 5498 | 10395683 |
| Exon 7 | |||||
| +13960 | R478W | 0.873 | Probably damaging | 5030400 | 10395796 |
| +14005 | M493V | 0.000 | Benign | N/a | 10395841 |
Predicted consequences of the non-synonymous SNPs identified in this study. Each site is numbered using the “A” in the ATG transcription as number 1. 48 % of SNPs are predicted to be benign, 24 % are predicted to be “possibly damaging”, and 29 % predicted to be “probably damaging”
Haplotype phase was inferred in individuals pooled by major geographic region (i.e. Africa, Asia, and Europe) using the program PHASE 2.1.1 (Stephens et al. 2001). Patterns of LD at ICAM-1 were examined using Haploview (Barrett et al. 2005). To maximize our power to detect significant patterns of LD, we examined pairwise LD between all SNPs that had a minor allele frequency ≥2 % in each continental sample (i.e. LD was examined separately in African, Asian, and European populations). The correlation between sites was measured using r2. The significance of each pairwise comparison was calculated using a Fisher’s exact test and a Bonferroni correction in DnaSP v. 5.10.01 (Librado and Rozas 2009). Because the frequency of some alleles differed between the African, Asian, and European populations, the SNPs that were included in analysis of LD in these populations differed slightly. In the African sample 18 SNPs met our criteria for inclusion into our study of LD. In the Asian populations 13 SNPs met the inclusion criteria, and in the European sample 11 SNPs were included in our analysis of LD (Fig. 2).
Fig. 2.

Pairwise linkage disequilibrium (LD) in African (a), Asian (b), and European (c) populations. LD was measured using r2. Gray shading represents values of 0 < r2 < 1; r2 = 0 is shown in white; r2 = 1 is shown in black. The number in each square is the observed r2value (%). When r2 = 1 no value is shown
Tests of neutrality
We used Tajima’s D statistic to examine the site frequency spectrum of ICAM-1 in each population and to test for deviations from expectations of neutral evolution. Tajima’s D is a comparison of the statistics π and Watterson’s θw which are both estimators of the population parameter θ. Because π and Watterson’s θw are estimates of the same parameter, they are expected to be equivalent when a locus is evolving neutrally in a population under a model of mutation/drift equilibrium (Tajima 1989). Nucleotide diversity and Tajima’s D were calculated using DnaSP (Librado and Rozas 2009). The significance of Tajima’s D values was determined using coalescent simulations of the null distribution of Tajima’s D generated in DnaSP using 10,000 simulations for given number of segregating sites under the assumption of constant population size.
We also used the McDonald Kreitman test (McDonald and Kreitman 1991) to compare the ratio of non-synonymous to synonymous polymorphisms within humans to the ratio of fixed non-synonymous to synonymous polymorphisms between the chimpanzee reference and humans. This test was used to detect whether natural selection has produced an excess of fixed or polymorphic non-synonymous changes. This test was implemented in DnaSP and significance was assessed using a Fisher’s exact test.
Population differentiation
We used FST to examine allele frequency differences between the populations within major continental groups (i.e. Africa, Asia, and Europe). FST was estimated following Akey et al. (2002), which includes a correction for differences in population sample size. When FST estimates were negative these values were set to zero. To determine whether our estimates of FST are similar to what is typically found across the genome, we compared our FST values with genome-wide FST estimates for the HapMap Phase III SNP data found in the SNP@Evolution database (Cheng et al. 2009). Although the populations included in the HapMap Phase III database are not identical to ours, comparison of FST values in our dataset, pooled by geographic region, with those from the Hap-Map III database provide a conservative comparison. This comparison is conservative because our study includes a more genetically diverse group of Africans, which are expected to have higher FST values than what is observed in the African HapMap III populations (Tishkoff et al. 2009).
Correlation between allele frequency at ICAM-1 and malaria endemicity
To examine correlations between the allele frequency of SNPs at ICAM-1 and malaria endemicity, we calculated Spearman’s rank correlation coefficient (r2), using the R statistical environment (R 2011), between the derived allele frequency of all non-singleton SNPs at ICAM-1 and the level of malaria endemicity in a subset of the sampled African populations (Table 1; Supplemental Table 1; Supplemental Table 4). Significance was determined by testing whether the observed value of r2 is significantly different from zero. Malaria endemicity for each population was determined by locating the latitude and longitude coordinates of a sampling site onto the country-level maps of the spatial distribution of P. falciparum malaria endemicity, measured by PfPR (P. falciparum parasite rate) and PfAPI (P. falciparum annual parasite incidence), created by the Malaria Atlas Project (Hay et al. 2009). PfAPI is a broad measure used to assess the spatial limits of P. falciparum malaria transmission using nationally reported incidence data, medical intelligence, and environmental factors that affect the locally dominant Anopheles vectors, including temperature and aridity (Hay et al. 2009). PfPR, a slightly different measure, is a commonly used index of malaria transmission intensity. It is measured as the proportion of the population that is found to carry asexual red blood cell-stage parasites (Smith et al. 2007). Here, we classified each population into one of three classes of malaria endemicity: the low class of endemicity corresponds to a PfAPI ≤0.1 per 1,000 people per annum (pa), the intermediate class of endemicity corresponds to 5 % > PfPR < 40 %, and the high class of endemicity corresponds to PfPR >40 % (Supplemental Table 1).
To assess whether any of our observed correlations are different than what is observed at other loci across the genome, we compared the observed ICAM-1 r2 values with empirical distributions of r2 calculated between the derived allele frequency (as indicated by dbSNP) of randomly chosen SNPs across the genome with malaria endemicity. The empirical distributions were built using allele frequencies of 260 genic and 370 intergenic SNPs (as defined by dbSNP) randomly selected from SNPs typed on an Illumina 1 M SNP array. The Illumina 1 M SNP array was assayed in the same populations used to assess the correlation between ICAM-1 allele frequencies and malaria endemicity (Table 1). The value of r2 used to build each distribution was calculated using the same procedure described above. We noted if any ICAM-1 r2 values occurred in the top 5 % of either the genic or intergenic distribution.
Results
Patterns of genetic diversity
We identified a total of 87 SNPs. Of the SNPs we identified, 55 (63 %) are not in dbSNP (Build 135) (Supplemental Table 1). Nucleotide diversity statistics (π and θw) are shown in Table 1. Overall, African populations had higher levels of diversity than non-Africans populations (Table 1). We identified 30 SNPs within the coding region of ICAM-1, 21 of which are non-synonymous (nine are synonymous), including the previously identified non-synonymous ICAM-1Kilifi allele (rs5491) (Table 1). Our PolyPhen results indicate that the majority of the non-synonymous SNPs are predicted to be benign (47 %); 23 % are predicted to be “possibly damaging,” and 28 % are predicted to be “probably damaging.” It is interesting to note that two SNPs (rs5491-ICAM-1Kilifi and rs5498) that are thought to be related to malaria susceptibility (Amodu et al. 2005; Fernandez-Reyes et al. 1997) are predicted to be benign. Of the non-synonymous SNPs with a minor allele frequency >3 % in the global sample (i.e. non-rare SNPs) only two, at positions +13416 and rs5497, are predicted to be “probably” or “possibly” damaging (Table 2).
The ICAM-1Kilifi allele was found in all African and Asian (Thailand and CHB) populations but was absent in our sampled European populations (Portuguese and CEU). The frequency of the ICAM-1Kilifi allele ranged from 0.08 to 0.34 in Africa. In the CHB and Thai populations the frequency of ICAM-1Kilifi allele was 0.13 and 0.21, respectively (Table 1). We calculated pairwise population FST for rs5491 (ICAM-1Kilifi) in Africans, Asians (Thailand and CHB), and Europeans (CEU and Portuguese) to determine continental levels of sub-structure at this site. The pairwise FST values for rs5491 are: FST Africa/Asia = 0, FST Africa/Europe = 0.095, FST Asia/Europe = 0.17. When compared with an empirical genome-wide distribution of FST values derived from HapMap Phase III SNP data, the FST Africa/Asia value is in the lowest 0.9 % of the empirical distribution. These results indicate that allele frequencies at rs5491 in Africa and Asia are more similar than what is generally observed across the genome.
Patterns of linkage disequilibrium (LD) and haplotype variation
Because of the different demographic histories among African, European, and Asian populations, LD was examined separately within major continental groups (Fig. 2). Generally, we observed low levels of LD in most populations (Fig. 2). In Africa, we observed significant LD between rs5491 (ICAM-1Kilifi) and two non-coding upstream SNPs, rs5030351 (r2 = 0.90; P < 0.001), and rs5490 (r2 = 0.56; P < 0.001). In the Asian populations we observed significant LD between rs5491 (ICAM-1Kilifi) and rs13306429 (r2 = 0.91; P < 0.001), which is a synonymous SNP in exon five.
In total, we inferred 157 haplotypes using all 87 identified SNPs (Supplemental Fig. 1). Because many of these haplotypes occurred only once we also inferred haplotypes using SNPs that showed a minor allele frequency ≥2 % in each continental region (i.e. the SNPs used in the LD analysis). The ≥2 % threshold was chosen because it allowed the greatest inclusion of polymorphic sites, while also eliminating singleton SNPs. Using this threshold, we observed a total of 76 haplotypes (Supplemental Table 2; Supplemental Fig. 2). Of these haplotypes, 45 are specific to African populations, seven are specific to Asian populations, and five are specific to European populations (Supplemental Table 2). We observed a total of 17 haplotypes that contain the ICAM-1Kilifi allele, 14 of which are observed only in African populations. Of the three ICAM-1Kilifi haplotypes that were observed in Asian populations (H40, H42 and H43) only one was shared between Asian and African populations (H40). Haplotype H40 has an observed frequency of 1 % in the Asian populations and a frequency of 0.4 % in the African populations. Haplotype H42, which was only found in the Thai population, has an observed frequency of 20 % in the Thai population, and H43, which was only observed in the CHB population, has an observed frequency of 10 % in the CHB population.
We used the program NETWORK 4.5 (www.fluxus-engineering.com) to build a median joining network (Bandelt et al. 1999) of the haplotypes that were inferred using SNPs with a minor allele frequency ≥2 % in each continental region. The network of all non-singleton haplotypes is shown in Fig. 3. Our results show that all of the African ICAM-1Kilifi haplotypes cluster together in the network. The shared Asian and African ICAM-1Kilifi halotype (H40) clusters with the other African ICAM-1Kilifi haplotypes. However, the uniquely Asian ICAM-1Kilifi haplotypes (H42 and H43) do not cluster with the other African ICAM-1Kilifi haplotypes. There are several median vectors that separate these haplotypes from H40 and the exclusively African ICAM-1Kilifi haplotypes.
Fig. 3.

Median-joining haplotype network consisting of SNPs with a frequency ≥2 % in each continental region spanning the entire region that was sequenced. Haplotypes that were observed at least two times are shown. The proportion of each haplotype that was observed in a major geographic region is shown in orange (West Africa), gray (East Africa), blue (Europe), and green (Asia). Note that the African ICAM-1Kilifi haplotypes cluster together in one part of the network (indicated by red circle). The arrows indicate the Asian specific ICAM-1Kilifi haplotypes. Median vectors are indicated by red circles. This figure is not drawn to scale. The number on each branch indicates the mutation that separates each haplotype and corresponds to the numbers presented in Supplemental Table 2
Tests of neutrality
To test whether the observed patterns of nucleotide diversity at ICAM-1 are consistent with neutrality we used Tajima’s D statistic (Tajima 1989) and the McDonald-Kreitman test (McDonald and Kreitman 1991). The results of Tajima’s D test are presented in Table 1. Generally, our values for Tajima’s D are negative and non-significant. We did observe a significant negative value for Tajima’s D when all African and non-African samples were pooled. However, this observation may be the result of pooling sub-structured populations (Ptak and Przeworski 2002; Wall et al. 2008). The results of our McDonald-Kreitman test (McDonald and Kreitman 1991) do not show a significant deviation from neutral expectations (P > 0.5, Fisher’s exact test; Supplemental Table 3).
Correlations between genetic diversity and malaria endemicity
To test the hypothesis that the distribution of SNPs at ICAM-1 is related to malaria endemicity, we assessed the correlation of the derived allele frequency of non-singleton SNPs at ICAM-1 with malaria endemicity in Africa using Spearman’s rank (r2) correlation test. Three SNPs (rs5491, rs5490, rs5030351) have derived alleles with an r2 value that is >0.55 (P < 0.001) and one SNP (rs5494) had r2 = 0.52 (P < 0.002) (Supplemental Table 4). To test whether the correlations we observed at ICAM-1 are due to demographic factors, which would affect all regions of the genome, we calculated the correlation between malaria endemicity and allele frequencies from 260 randomly chosen genic SNPs and 370 intergenic SNPs from across the genome, excluding the X and Y chromosomes, assayed on an Illumina 1 M SNP array in the same populations included in the correlation analysis. These correlation values were used to build a comparative empirical distribution of r2 values. We then compared the observed correlations we found at ICAM-1 with the empirical distribution of correlation values (Fig. 4). Of the SNPs we identified, the derived allele of four ICAM-1 SNPs (rs5494, rs5491, rs5490, rs5030351) were in the top 5 % of the empirical distribution of correlations. This result suggests that the high degree of correlation observed at SNPs at ICAM-1 is unique compared with the level of correlation with malaria endemicity that was observed at >600 SNPs from across the genome.
Fig. 4.

Frequency distribution of correlation coefficients (r2) calculated between a randomly chosen sample of genic (a) and intergenic (b) SNPs across the genome and malaria endemicity (see Supplemental Table 1 for population malaria endemicity values). Dashed line represents the top 5 % of each distribution. The four SNPs in the tails of both distributions include rs5491 (ICAM-1Kilifi), the two SNPs that are linked to it (rs5030351 and rs5490), and rs5494
Discussion
In the current study we characterized nucleotide variation through targeted re-sequencing of ICAM-1 in diverse African, European, and Asian populations. Because African populations are highly diverse it is important to examine genetic variation in many populations to understand the range of nucleotide variation and the patterns of LD at a given locus (Ko et al. 2011; Mortensen et al. 2011; Tarazona-Santos and Tishkoff 2005; Tishkoff et al. 2009). The populations included in this study were selected to maximize ethnic, linguistic, and genetic diversity (Tishkoff et al. 2009) and to encompass a range of malaria endemicities (Hay et al. 2009). Our study identified 87 SNPs, 55 of which are novel, in both the coding and non-coding regions of ICAM-1. The results of our study suggest that ICAM-1 is more diverse in African populations than non-African populations, consistent with other studies of genetic variation (Campbell and Tishkoff 2008).
We observed the ICAM-1Kilifi allele in all African populations on a number of different haplotype backgrounds and analyses of LD indicate that rs5490 and rs5030351 are in LD with rs5491 (ICAM-1Kilifi), which is consistent with previous studies (Fry et al. 2008). The ICAM-1Kilifi allele was also present in our sampled Asian populations, but was not identified in the populations of European descent, also consistent with previous reports in the literature (Ohashi et al. 2001; Vijgen et al. 2003). Because the ICAM-1Kilifi allele is found on several different haplotype backgrounds that are present in multiple African groups, it is likely that this SNP arose in Africa prior to the structuring of these groups.
In Asian populations, we observed the ICAM-1Kilifi allele on three different haplotype backgrounds (H40, H42, and H43), two of which were found exclusively in the Asian populations (H42 and H43). The presence of H40 in both Africa and Asia suggests that this haplotype was either present during early “Out-of-Africa” migrations or is present in Asia due to recent gene flow. Our phylogenetic network analysis shows that the exclusively Asian ICAM-1Kilifi haplotypes are separated from the other ICAM-1Kilifi haplotypes by several median vectors. Bandelt et al. (1999) suggests that median vectors can be biologically interpreted as possible extant un-sampled haplotypes or extinct ancestral haplotypes. Additionally, in the case of autosomal loci the presence of median vectors can result from recombination. The observation that several median vectors separate the exclusively Asian ICAM-1Kilifi haplotypes from the rest of the network indicates that Asian-specific ICAM-1Kilifi haplotypes are not closely related to African ICAM-1Kilifi haplotypes. Thus, it is possible that the exclusively Asian ICAM-1Kilifi haplotypes arose independently in Asia. Alternatively, the Asian ICAM-1Kilifi haplotypes could have been introduced during a migration of modern humans out of Africa and were lost due to genetic drift in most African populations. Under either hypothesis, the observation that the ICAM-1Kilifi allele is at intermediate frequency in Asia (Thai and CHB pooled allele frequency = 0.15) and has little associated haplotype diversity, indicates that the ICAM-1Kilifi allele rose to high frequency relatively recently in Asian populations, possibly due to natural selection resulting from malaria infection. The results of this study warrant further examination of ICAM-1 nucleotide variation in diverse Asian populations to better understand the evolutionary history of ICAM-1 in that region.
We used Tajima’s D and the MK test to test for signatures of natural selection. We failed to reject the null hypothesis in both cases (Table 1; Supplemental Table 3). However, it is possible that we do not have enough statistical power to observe a significant signature of natural selection using these tests (Simonsen et al. 1995; Wooding et al. 2004). In particular, Tajima’s D statistic requires a very strong and recent selection signal to be present to reject the null hypothesis (Simonsen et al. 1995; Wooding et al. 2004). In previous studies of genetic diversity at G6PD, a locus that is known to contain alleles that confer protection against malaria, a significant value of Tajima’s D was also not observed (Tishkoff et al. 2001). However, signatures of selection were detected using tests based on long-range LD (Sabeti et al. 2002; Tishkoff et al. 2001). Because ICAM-1Kilifi is quite old in Africa, as suggested by high levels of haplotype diversity in Africa, if the frequency of this allele has been shaped by recent natural selection it is possible that the selective event occurred on standing genetic variation. Thus, even if this allele is causally associated with malaria resistance in Africa any signatures of extended LD would be substantially weakened due to the age of the allele (Novembre and Di Rienzo 2009). Therefore, we would have limited power to detect a significant signature of positive natural selection using methods based on extended patterns of LD (Barrett and Schluter 2008; Novembre and Di Rienzo 2009; Przeworski et al. 2005). Indeed, genomic scans for recent positive selection (Pickrell et al. 2009; Voight et al. 2006) have not identified evidence of extended haplotype homozygosity (EHH) at ICAM-1.
In 2006, Ryan et al. (2006) showed, using publicly available re-sequencing data of African and European Americans, that the ICAM-1Kilifi allele has an unusually high FST value when compared with SNPs in genes that encode cytokines, adhesion molecules, cytokine receptors, and Toll-like receptors. Our results indicate that the FST value at rs5491 (ICAM-1Kilifi) between African and European populations (FST = 0.095) is similar to what has been observed at other loci across the genome (FST = 0.10–0.15) (Barreiro et al. 2008; Coop et al. 2009; Keinan et al. 2007). However, we observed an exceptionally low FST value (FST = 0) between African and Asian populations at rs5491. The overall similarity in allele frequencies at rs5491 between African and Asian populations could indicate that these populations have high frequencies of the ICAM-1Kilifi allele due to similar environmental selective pressures, like malaria. Malaria has been an important selective pressure in African populations (Sabeti et al. 2002; Tishkoff et al. 2001) and it has also been an important selective pressure in Asia as well (Ohashi et al. 2004). Hay et al. (2009) estimated that in 2007, 0.69 billion people in central and Southeast Asia were at risk for malaria caused by P. falciparum. These data suggest that malaria is a common selective pressure in both Southeast Asia and Africa and could explain the overall similar allele frequencies of the ICAM-1Kilifi allele in populations from those regions.
At ICAM-1 we observed four SNPs (rs5494, rs5491, rs5490, and rs5030351) whose derived allele frequencies are correlated with malaria endemicity in Africa. This includes the ICAM-1Kilifi allele (rs5491) and two sites in LD with the ICAM-1Kilifi (Fig. 4; Supplemental Table 4). The fourth allele correlated with malaria endemicity (rs5494) is a synonymous site in exon 4 that has not been shown to have clinical significance (Sherry et al. 2001). The correlation of polymorphic sites at ICAM-1 with malaria endemicity is similar to previous observations that variants that are associated with malaria resistance are correlated with malaria endemicity (e.g. G6PD and HBB) (Cook and Hill 2001). The results reported here suggest that one or more of the sites highly correlated with malaria endemicity could play a role in resistance to malaria.
The ICAM-1Kilifi allele causes a non-synonymous amino acid change in the IG-like domain of ICAM-1 that interacts with PfEMP1. Substantial research has been done to understand the functional consequences of this amino acid change (Adams et al. 2000; Bertonati and Tramontano 2007; Craig et al. 2000). For example, in vitro adhesion experiments have shown that a laboratory strain of P. falciparum called A4 shows reduced binding to ICAM-1Kilifi proteins when compared with ICAM-1Ref proteins (Adams et al. 2000; Craig et al. 2000; Tse et al. 2004). These data suggest the possible adaptive function conferred by the ICAM-1Kilifi allele. However, case–control association studies in Africa have produced conflicting results (Fernandez-Reyes et al. 1997; Fry et al. 2008; Kun et al. 1999; Ndiaye et al. 2005) and, indeed, other in vitro studies (Adams et al. 2000; Tse et al. 2004) indicate that although the ICAM-1Kilifi allele effects cytoadhesion to several laboratory strains of P. falciparum, these effects vary among laboratory strains. Therefore, the ultimate functional relevance of the ICAM-1Kilifi allele remains an open area of study. However, despite the conflicting epidemiological and experimental results, the data presented here show that the ICAM-1Kilifi allele is correlated with malaria endemicity in Africa, and has a similar intermediate frequency in Southeast Asia where malaria is also prevalent. Our data suggest that it is possible to use correlation analyses to identify functional variants that may not be identified using tests of neutrality designed to detect recent strong selective sweeps of new mutations and add strength to the biological/functional data that suggest the ICAM-1Kilifi allele may play a role in malaria resistance.
Supplementary Material
Acknowledgments
We are grateful to all of the individuals who contributed their DNA samples to this study. We would also like to thank Floyd Reed for his assistance in writing scripts and useful discussion. We are also grateful to Joseph Lachance, Charla Lambert, Joseph Jarvis, Laura Scheinfeldt, Simon Thompson, and Michael Campbell for their insightful comments during data analysis and manuscript preparation. We thank William Beggs and Karuna Panchapakesan for their technical assistance. This research is funded by Human Frontiers in Science grant BCS-0827436, and National Institutes of Health grants R01GM076637 and DP1-OD-006445-01 and NSF Hominid grant (BCS0827436) to S.A.T. An FCT grant (PPCDT/BIA-BDE/56654/2004) awarded to JR also supported this research. A Doctoral Dissertation Improvement Grant from the US National Science Foundation (NSF) (BCS0925802) given to F.G. and an NSF IGERT grant (9987590) to F.G. and S.A.T supported this research. F.G. was also supported by a Ford Foundation Pre-doctoral fellowship, a Cosmos Club research award, a Sigma Xi (GWU) Grant-in-Aid of Research (GIAR), and an American Anthropological Association Minority Dissertation Writing Fellowship.
Footnotes
The SNPs identified in this study are currently in submission to dbSNP (NCBI). The corresponding accession number is pending.
Electronic supplementary material The online version of this article (doi:10.1007/s00439-013-1284-5) contains supplementary material, which is available to authorized users.
Conflict of interest The authors have no conflicts of interest.
Contributor Information
Felicia Gomez, Email: fgomez@mail.med.upenn.edu, Department of Genetics and Biology, School of Medicine and School of Arts and Sciences, University of Pennsylvania, Philadelphia, PA 19104, USA. Hominid Paleobiology Doctoral Program, Department of Anthropology, The George Washington University, Washington, DC 20052, USA. Center for the Advanced Study of Hominid Paleobiology, Department of Anthropology, The George Washington University, Washington, DC 20052, USA.
Gil Tomas, IPATIMUP-Instituto de Patologia e Imunologia Molecular da Universidade do Porto, Porto, Portugal.
Wen-Ya Ko, Department of Genetics and Biology, School of Medicine and School of Arts and Sciences, University of Pennsylvania, Philadelphia, PA 19104, USA. CIBIO-Centro de Investigação em Biodiversidade e Recursos Genéticos, Universidade do Porto, Vairão, Portugal.
Alessia Ranciaro, Department of Genetics and Biology, School of Medicine and School of Arts and Sciences, University of Pennsylvania, Philadelphia, PA 19104, USA.
Alain Froment, UMR 208, Institut de Recherche pour le de’veloppement, Muse’um National d’Histoire Naturelle, Muse’e de l’Homme, 75116 Paris, France.
Muntaser Ibrahim, Department of Molecular Biology, Institute of Endemic Diseases, University of Khartoum, 15-Khartoum, Sudan.
Godfrey Lema, Department of Biochemistry, Muhimbili University of Health and Allied Sciences, Dar es Salaam, Tanzania.
Thomas B. Nyambo, Department of Biochemistry, Muhimbili University of Health and Allied Sciences, Dar es Salaam, Tanzania
Sabah A. Omar, Kenya Medical Research Institute, Center for Biotechnology Research and Development, 54840-00200 Nairobi, Kenya
Charles Wambebe, International Biomedical Research in Africa, Abuja, Nigeria.
Jibril B. Hirbo, Department of Genetics and Biology, School of Medicine and School of Arts and Sciences, University of Pennsylvania, Philadelphia, PA 19104, USA
Jorge Rocha, IPATIMUP-Instituto de Patologia e Imunologia Molecular da Universidade do Porto, Porto, Portugal. CIBIO-Centro de Investigação em Biodiversidade e Recursos Genéticos, Universidade do Porto, Vairão, Portugal. Departamento de Biologia, Faculdade de Ciências, Universidade do Porto, Porto, Portugal.
Sarah A. Tishkoff, Email: tishkoff@mail.med.upenn.edu, Department of Genetics and Biology, School of Medicine and School of Arts and Sciences, University of Pennsylvania, Philadelphia, PA 19104, USA
References
- Adams S, Turner GD, Nash GB, Micklem K, Newbold CI, Craig AG. Differential binding of clonal variants of Plasmodium falciparum to allelic forms of intracellular adhesion molecule 1 determined by flow adhesion assay. Infect Immun. 2000;68:264–269. doi: 10.1128/iai.68.1.264-269.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akey JM, Zhang G, Zhang K, Jin L, Shriver MD. Interrogating a high-density SNP map for signatures of natural selection. Genome Res. 2002;12:1805–1814. doi: 10.1101/gr.631202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amodu OK, Gbadegesin RA, Ralph SA, Adeyemo AA, Brenchley PE, Ayoola OO, Orimadegun AE, Akinsola AK, Olumese PE, Omotade OO. Plasmodium falciparum malaria in southwest Nigerian children: is the polymorphism of ICAM-1 and E-selectin genes contributing to the clinical severity of malaria? Acta Trop. 2005;95:248–255. doi: 10.1016/j.actatropica.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Ayodo G, Price AL, Keinan A, Ajwang A, Otieno MF, Orago AS, Patterson N, Reich D. Combining evidence of natural selection with association analysis increases power to detect malaria-resistance variants. Am J Hum Genet. 2007;81:234–242. doi: 10.1086/519221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandelt HJ, Forster P, Rohl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16:37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
- Barreiro LB, Laval G, Quach H, Patin E, Quintana-Murci L. Natural selection has driven population differentiation in modern humans. Nat Genet. 2008;40:340–345. doi: 10.1038/ng.78. [DOI] [PubMed] [Google Scholar]
- Barrett RD, Schluter D. Adaptation from standing genetic variation. Trends Ecol Evol. 2008;23:38–44. doi: 10.1016/j.tree.2007.09.008. [DOI] [PubMed] [Google Scholar]
- Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- Bellamy R, Kwiatkowski D, Hill AV. Absence of an association between intercellular adhesion molecule 1, complement receptor 1 and interleukin 1 receptor antagonist gene polymorphisms and severe malaria in a West African population. Trans R Soc Trop Med Hyg. 1998;92:312–316. doi: 10.1016/s0035-9203(98)91026-4. [DOI] [PubMed] [Google Scholar]
- Bertonati C, Tramontano A. A model of the complex between the PfEMP1 malaria protein and the human ICAM-1 receptor. Proteins. 2007;69:215–222. doi: 10.1002/prot.21691. [DOI] [PubMed] [Google Scholar]
- Campbell MC, Tishkoff SA. African genetic diversity: implications for human demographic history, modern human origins, and complex disease mapping. Annu Rev Genomics Hum Genet. 2008;9:403–433. doi: 10.1146/annurev.genom.9.081307.164258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S, Craig AG. The Role of ICAM-1 in Plasmodium falciparum cytoadherence. Eur J Cell Biol. 2004;84:15–27. doi: 10.1016/j.ejcb.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay R, Taneja T, Chakrabarti K, Pillai CR, Chitnis CE. Molecular analysis of the cytoadherence phenotype of a Plasmodium falciparum field isolate that binds intercellular adhesion molecule-1. Mol Biochem Parasitol. 2004;133:255–265. doi: 10.1016/j.molbiopara.2003.08.014. [DOI] [PubMed] [Google Scholar]
- Chen Q, Schlichtherle M, Wahlgren M. Molecular aspects of severe malaria. Clin Microbiol Rev. 2000;13:439–450. doi: 10.1128/cmr.13.3.439-450.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng F, Chen W, Richards E, Deng L, Zeng C. SNP@Evolution: a hierarchical database of positive selection on the human genome. BMC Evol Biol. 2009;9:221. doi: 10.1186/1471-2148-9-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clegg JB, Weatherall DJ. Thalassemia and malaria: new insights into an old problem. Proc Assoc Am Physicians. 1999;111:278–282. doi: 10.1046/j.1525-1381.1999.99235.x. [DOI] [PubMed] [Google Scholar]
- Cook G, Hill A. Genetics of Susceptibility to Human Disease. Nat Rev Genet. 2001;2:967–977. doi: 10.1038/35103577. [DOI] [PubMed] [Google Scholar]
- Coop G, Pickrell JK, Novembre J, Kudaravalli S, Li J, Absher D, Myers RM, Cavalli-Sforza LL, Feldman MW, Pritchard JK. The role of geography in human adaptation. PLoS Genet. 2009;5:e1000500. doi: 10.1371/journal.pgen.1000500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig A, Fernandez-Reyes D, Mesri M, McDowall A, Altieri DC, Hogg N, Newbold C. A functional analysis of a natural variant of intercellular adhesion molecule-1 (ICAM-1Kilifi) Hum Mol Genet. 2000;9:525–530. doi: 10.1093/hmg/9.4.525. [DOI] [PubMed] [Google Scholar]
- Fernandez-Reyes D, Craig AG, Kyes SA, Peshu N, Snow RW, Berendt AR, Marsh K, Newbold CI. A high frequency African coding polymorphism in the N-terminal domain of ICAM-1 predisposing to cerebral malaria in Kenya. Hum Mol Genet. 1997;6:1357–1360. doi: 10.1093/hmg/6.8.1357. [DOI] [PubMed] [Google Scholar]
- Fortin A, Stevenson MM, Gros P. Susceptibility to malaria as a complex trait: big pressure from a tiny creature. Hum Mol Genet. 2002;11:2469–2478. doi: 10.1093/hmg/11.20.2469. [DOI] [PubMed] [Google Scholar]
- Fry AE, Auburn S, Diakite M, Green A, Richardson A, Wilson J, Jallow M, Sisay-Joof F, Pinder M, Griffiths MJ, Peshu N, Williams TN, Marsh K, Molyneux ME, Taylor TE, Rockett KA, Kwiatkowski DP. Variation in the ICAM1 gene is not associated with severe malaria phenotypes. Genes Immun. 2008;9:462–469. doi: 10.1038/gene.2008.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamblin MT, Di Rienzo A. Detection of the signature of natural selection in humans: evidence from the Duffy blood group locus. Am J Hum Genet. 2000;66:1669–1679. doi: 10.1086/302879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamblin MT, Thompson EE, Di Rienzo A. Complex signatures of natural selection at the Duffy blood group locus. Am J Hum Genet. 2002;70:369–383. doi: 10.1086/338628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay SI, Guerra CA, Gething PW, Patil AP, Tatem AJ, Noor AM, Kabaria CW, Manh BH, Elyazar IR, Brooker S, Smith DL, Moyeed RA, Snow RW. A world malaria map: plasmodium falciparum endemicity in 2007. PLoS Med. 2009;6:e1000048. doi: 10.1371/journal.pmed.1000048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keinan A, Mullikin JC, Patterson N, Reich D. Measurement of the human allele frequency spectrum demonstrates greater genetic drift in East Asians than in Europeans. Nat Genet. 2007;39:1251–1255. doi: 10.1038/ng2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko WY, Kaercher KA, Giombini E, Marcatili P, Froment A, Ibrahim M, Lema G, Nyambo TB, Omar SA, Wambebe C, Ranciaro A, Hirbo JB, Tishkoff SA. Effects of natural selection and gene conversion on the evolution of human glycophorins coding for MNS blood polymorphisms in malaria-endemic African populations. Am J Hum Genet. 2011;88:741–754. doi: 10.1016/j.ajhg.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kun JF, Klabunde J, Lell B, Luckner D, Alpers M, May J, Meyer C, Kremsner PG. Association of the ICAM-1Kilifi mutation with protection against severe malaria in Lambarene, Gabon. Am J Trop Med Hyg. 1999;61:776–779. doi: 10.4269/ajtmh.1999.61.776. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski D. Genetic susceptibility to malaria getting complex. Curr Opin Genet Dev. 2000;10:320–324. doi: 10.1016/s0959-437x(00)00087-3. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski DP. How malaria has affected the human genome and what human genetics can teach us about malaria. Am J Hum Genet. 2005;77:171–192. doi: 10.1086/432519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin M, Blackshields G, Brown N, Chenna R, Mc Gettigan P, Mc William H, Valentin F, Wallace I, Wilm A, Lopez R, Thompson J, Gibson T, Higgins D. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- Livingstone F. Anthropological implications of sickle cell gene distribution in West Africa. Am Anthropol. 1958;60:533–562. [Google Scholar]
- Livingstone F. Malaria and human polymorphisms. Annu Rev Genet. 1971;5:33–64. doi: 10.1146/annurev.ge.05.120171.000341. [DOI] [PubMed] [Google Scholar]
- McDonald JH, Kreitman M. Adaptive protein evolution at the Adh locus in Drosophila. Nature. 1991;351:652–654. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- Miller LH, Baruch DI, Marsh K, Doumbo OK. The pathogenic basis of malaria. Nature. 2002;415:673–679. doi: 10.1038/415673a. [DOI] [PubMed] [Google Scholar]
- Mortensen HM, Froment A, Lema G, Bodo JM, Ibrahim M, Nyambo TB, Omar SA, Tishkoff SA. Characterization of genetic variation and natural selection at the arylamine N-acetyltransferase genes in global human populations. Pharmacogenomics. 2011;12:1545–1558. doi: 10.2217/pgs.11.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy SC, Breman JG. Gaps in the childhood malaria burden in Africa: cerebral malaria, neurological sequelae, anemia, respiratory distress, hypoglycemia, and complications of pregnancy. Am J Trop Med Hyg. 2001;64:57–67. doi: 10.4269/ajtmh.2001.64.57. [DOI] [PubMed] [Google Scholar]
- Ndiaye R, Sakuntabhai A, Casademont I, Rogier C, Tall A, Trape JF, Spiegel A, Dieye A, Julier C. Genetic study of ICAM1 in clinical malaria in Senegal. Tissue Antigens. 2005;65:474–480. doi: 10.1111/j.1399-0039.2005.00388.x. [DOI] [PubMed] [Google Scholar]
- Nei M. Molecular evolutionary genetics. Columbia University Press; New York: 1987. [Google Scholar]
- Newbold C, Craig A, Kyes S, Rowe A, Fernandez-Reyes D, Fagan T. Cytoadherence, pathogenesis and the infected red cell surface in Plasmodium falciparum. Int J Parasitol. 1999;29:927–937. doi: 10.1016/s0020-7519(99)00049-1. [DOI] [PubMed] [Google Scholar]
- Novembre J, Di Rienzo A. Spatial patterns of variation due to natural selection in humans. Nat Rev Genet. 2009;10:745–755. doi: 10.1038/nrg2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi J, Naka I, Patarapotikul J, Hananantachai H, Looareesuwan S, Tokunaga K. Absence of association between the allele coding methionine at position 29 in the N-terminal domain of ICAM-1 (ICAM-1(Kilifi)) and severe malaria in the northwest of Thailand. Jpn J Infect Dis. 2001;54:114–116. [PubMed] [Google Scholar]
- Ohashi J, Naka I, Patarapotikul J, Hananantachai H, Brittenham G, Looareesuwan S, Clark AG, Tokunaga K. Extended linkage disequilibrium surrounding the hemoglobin E variant due to malarial selection. Am J Hum Genet. 2004;74:1198–1208. doi: 10.1086/421330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK, Coop G, Novembre J, Kudaravalli S, Li JZ, Absher D, Srinivasan BS, Barsh GS, Myers RM, Feldman MW, Pritchard JK. Signals of recent positive selection in a worldwide sample of human populations. Genome Res. 2009;19:826–837. doi: 10.1101/gr.087577.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przeworski M, Coop G, Wall JD. The signature of positive selection on standing genetic variation. Evol Int J Org Evol. 2005;59:2312–2323. [PubMed] [Google Scholar]
- Ptak SE, Przeworski M. Evidence for population growth in humans is confounded by fine-scale population structure. Trends Genet. 2002;18:559–563. doi: 10.1016/s0168-9525(02)02781-6. [DOI] [PubMed] [Google Scholar]
- R. R: a language and environment for statistical computing. Austria, Vienna: 2011. [Google Scholar]
- Ryan AW, Mapp J, Moyna S, Mattiangeli V, Kelleher D, Bradley DG, McManus R. Levels of interpopulation differentiation among different functional classes of immunologically important genes. Genes Immun. 2006;7:179–183. doi: 10.1038/sj.gene.6364266. [DOI] [PubMed] [Google Scholar]
- Sabeti PC, Reich DE, Higgins JM, Levine HZ, Richter DJ, Schaffner SF, Gabriel SB, Platko JV, Patterson NJ, McDonald GJ, Ackerman HC, Campbell SJ, Altshuler D, Cooper R, Kwiat-kowski D, Ward R, Lander ES. Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002;419:832–837. doi: 10.1038/nature01140. [DOI] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonsen KL, Churchill GA, Aquadro CF. Properties of statistical tests of neutrality for DNA polymorphism data. Genetics. 1995;141:413–429. doi: 10.1093/genetics/141.1.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DL, Guerra CA, Snow RW, Hay SI. Standardizing estimates of the Plasmodium falciparum parasite rate. Malar J. 2007;6:131. doi: 10.1186/1475-2875-6-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow RW, Guerra CA, Noor AM, Myint HY, Hay SI. The global distribution of clinical episodes of Plasmodium falciparum malaria. Nature. 2005;434:214–217. doi: 10.1038/nature03342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarazona-Santos E, Tishkoff SA. Divergent patterns of linkage disequilibrium and haplotype structure across global populations at the interleukin-13 (IL13) locus. Genes Immun. 2005;6:53–65. doi: 10.1038/sj.gene.6364149. [DOI] [PubMed] [Google Scholar]
- Tishkoff SA, Varkonyi R, Cahinhinan N, Abbes S, Argyropoulos G, Destro-Bisol G, Drousiotou A, Dangerfield B, Lefranc G, Loiselet J, Piro A, Stoneking M, Tagarelli A, Tagarelli G, Touma EH, Williams SM, Clark AG. Haplotype diversity and linkage disequilibrium at human G6PD: recent origin of alleles that confer malarial resistance. Science. 2001;293:455–462. doi: 10.1126/science.1061573. [DOI] [PubMed] [Google Scholar]
- Tishkoff SA, Reed FA, Friedlaender FR, Ehret C, Ranciaro A, Froment A, Hirbo JB, Awomoyi AA, Bodo JM, Doumbo O, Ibrahim M, Juma AT, Kotze MJ, Lema G, Moore JH, Mortensen H, Nyambo TB, Omar SA, Powell K, Pretorius GS, Smith MW, Thera MA, Wambebe C, Weber JL, Williams SM. The genetic structure and history of Africans and African Americans. Science. 2009;324:1035–1044. doi: 10.1126/science.1172257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse MT, Chakrabarti K, Gray C, Chitnis CE, Craig A. Divergent binding sites on intercellular adhesion molecule-1 (ICAM-1) for variant Plasmodium falciparum isolates. Mol Microbiol. 2004;51:1039–1049. doi: 10.1046/j.1365-2958.2003.03895.x. [DOI] [PubMed] [Google Scholar]
- Turner GD, Morrison H, Jones M, Davis TM, Looareesuwan S, Buley ID, Gatter KC, Newbold CI, Pukritayakamee S, Nagachinta B, et al. An immunohistochemical study of the pathology of fatal malaria. Evidence for widespread endothelial activation and a potential role for intercellular adhesion molecule-1 in cerebral sequestration. Am J Pathol. 1994;145:1057–1069. [PMC free article] [PubMed] [Google Scholar]
- Verra F, Mangano VD, Modiano D. Genetics of susceptibility to Plasmodium falciparum: from classical malaria resistance genes towards genome-wide association studies. Parasite Immunol. 2009;31:234–253. doi: 10.1111/j.1365-3024.2009.01106.x. [DOI] [PubMed] [Google Scholar]
- Verrelli BC, McDonald JH, Argyropoulos G, Destro-Bisol G, Froment A, Drousiotou A, Lefranc G, Helal AN, Loiselet J, Tishkoff SA. Evidence for balancing selection from nucleotide sequence analyses of human G6PD. Am J Hum Genet. 2002;71:1112–1128. doi: 10.1086/344345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijgen L, Van Essche M, Van Ranst M. Absence of the Kilifi mutation in the rhinovirus-binding domain of ICAM-1 in a Caucasian population. Genet Test. 2003;7:159–161. doi: 10.1089/109065703322146885. [DOI] [PubMed] [Google Scholar]
- Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall JD, Cox MP, Mendez FL, Woerner A, Severson T, Hammer MF. A novel DNA sequence database for analyzing human demographic history. Genome Res. 2008;18:1354–1361. doi: 10.1101/gr.075630.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. Jalview Version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson GA. On the number of segregating sites in genetical models without recombination. Theor Pop Biol. 1975;7:256–276. doi: 10.1016/0040-5809(75)90020-9. [DOI] [PubMed] [Google Scholar]
- WHO. World malaria report 2010. WHO Press; Geneva, Switzerland: 2010. [Google Scholar]
- Wooding S, Kim UK, Bamshad MJ, Larsen J, Jorde LB, Drayna D. Natural selection and molecular evolution in PTC, a bitter-taste receptor gene. Am J Hum Genet. 2004;74:637–646. doi: 10.1086/383092. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.