

Abstract
Background
Paracoccus aminophilus JCM 7686 is a methylotrophic α-Proteobacterium capable of utilizing reduced one-carbon compounds as sole carbon and energy source for growth, including toxic N,N-dimethylformamide, formamide, methanol, and methylamines, which are widely used in the industry. P. aminophilus JCM 7686, as many other Paracoccus spp., possesses a genome representing a multipartite structure, in which the genomic information is split between various replicons, including chromids, essential plasmid-like replicons, with properties of both chromosomes and plasmids. In this study, whole-genome sequencing and functional genomics approaches were applied to investigate P. aminophilus genome information.
Results
The P. aminophilus JCM 7686 genome has a multipartite structure, composed of a single circular chromosome and eight additional replicons ranging in size between 5.6 and 438.1 kb. Functional analyses revealed that two of the replicons, pAMI5 and pAMI6, are essential for host viability, therefore they should be considered as chromids. Both replicons carry housekeeping genes, e.g. responsible for de novo NAD biosynthesis and ammonium transport. Other mobile genetic elements have also been identified, including 20 insertion sequences, 4 transposons and 10 prophage regions, one of which represents a novel, functional serine recombinase-encoding bacteriophage, ϕPam-6. Moreover, in silico analyses allowed us to predict the transcription regulatory network of the JCM 7686 strain, as well as components of the stress response, recombination, repair and methylation machineries. Finally, comparative genomic analyses revealed that P. aminophilus JCM 7686 has a relatively distant relationship to other representatives of the genus Paracoccus.
Conclusions
P. aminophilus genome exploration provided insights into the overall structure and functions of the genome, with a special focus on the chromids. Based on the obtained results we propose the classification of bacterial chromids into two types: “primary” chromids, which are indispensable for host viability and “secondary” chromids, which are essential, but only under some environmental conditions and which were probably formed quite recently in the course of evolution. Detailed genome investigation and its functional analysis, makes P. aminophilus JCM 7686 a suitable reference strain for the genus Paracoccus. Moreover, this study has increased knowledge on overall genome structure and composition of members within the class Alphaproteobacteria.
Keywords: Paracoccus aminophilus JCM 7686, Genome, Chromid, Plasmid, Mobile genetic element, Bacteriophage
Background
The genus Paracoccus (Alphaproteobacteria) currently comprises 40 recognized and validly named species, isolated from different environments in various geographical locations. Members of this genus exhibit a broad range of metabolic flexibility, especially in respiratory processes, e.g. employing nitrate, nitrite, nitrous oxide and nitric oxide as alternative electron acceptors in denitrification and the ability to use one-carbon (C1) compounds (e.g. methanol, methylamine) as electron donors to respiratory chains [1]. Moreover, Paracoccus spp. as facultative chemolithoautotrophs may utilize reduced sulfur compounds (e.g. thiocyanate, thiosulfate or elemental sulfur), molecular hydrogen and Fe(II) as energy sources [2-4]. Paracoccus spp. are also able to use a broad range of organic compounds as their sole source of carbon and energy, including pollutants such as acetone, dichloromethane, formamide, N,N-dimethylformamide (DMF) and methylamine [3,5]. Another common feature of Paracoccus spp. is methylotrophy, defined as the ability to utilize reduced C1 carbon substrates containing no carbon-carbon bonds (including methane, methanol, methylated amines, halogenated methanes and methylated sulfur species) as their sole source of carbon and energy for growth.
Having a versatile metabolism, Paracoccus spp. play an important role in biogeochemical cycles and they have also been successfully employed in the biotreatment of contaminated environments, e.g. bioremediation of soils contaminated with polycyclic aromatic hydrocarbons (PAHs) using Paracoccus sp. HPD-2 [6].
Although Paracoccus spp. constitute an interesting and metabolically versatile group of bacteria with substantial biotechnological potential, little is known about the content and organization of their genomes. Only one complete genome of Paracoccus denitrificans PD1222 has been deposited in the NCBI database ([GenBank:CP000489], [GenBank:CP000490] and [GenBank:CP000491]). This genome is composed of two chromosomes (ChI – 2.9 Mb and ChII – 1.7 Mb) and a single megaplasmid (plasmid 1) of 653 kb. Four Paracoccus spp. genome sequencing projects (Paracoccus sp. TRP [7], Paracoccus denitrificans SD1 [8], Paracoccus sp. N5 and P. zeaxanthinifaciens ATCC 21588) are currently in progress.
Much more is known about mobile genetic elements (MGEs) of Paracoccus spp. Baj and colleagues (2000) [9] demonstrated that bacteria belonging to this genus usually harbor at least one plasmid (in most cases a mega-sized replicon, exceeding 100 kb). Several multireplicon strains carrying 4 or more plasmids were identified, including P. aminophilus JCM 7686. Our group has already obtained the sequences of 18 plasmids, ranging in size from 2.7 to 40 kb, that originate from different Paracoccus spp.: P. methylutens DM12 (2 plasmids) [10], P. pantotrophus DSM 11072 (1 plasmid) [11] and four carotenoid producers, P. aestuarii DSM 19484 (5 plasmids) ([GenBank:JQ041633], [GenBank:JQ065021], [GenBank:JQ066766], [GenBank:JQ684025], [GenBank:JQ796370]), P. haeundaensis LMG P-21903 (2 plasmids) ([GenBank:JQ066767], [GenBank:JQ684024]), P. marcusii DSM 11574 (5 plasmids) ([GenBank: KC542384], [GenBank:KC561053], [GenBank:KC561054], [GenBank:KC561055], [GenBank:JQ796371) and P. marcusii OS22 (3 plasmids) ([GenBank:JQ664550], [GenBank:JQ678602], [GenBank:JQ684023]). We have also sequenced the basic replicons of three other plasmids from P. alcaliphilus JCM 7364 (pALC1) and P. versutus UW1 (pTAV1 and pTAV3) [12-14]. Moreover, complex analyses of 25 Paracoccus spp. strains using trap plasmid systems have led to the identification and characterization of (i) 48 insertion sequences (ISs), (ii) a composite transposon Tn6097 carrying genetic modules involved in the arginine deiminase pathway and daunorubicin/doxorubicin resistance, (iii) 3 non-composite transposons of the Tn3 family, (iv) a transposable genomic island TnPpa1 (45 kb) and (v) several transposable modules (TMos) generated by a single copy of the IS1380 family insertion sequence [10,15-20]. The findings outlined above suggest that horizontal gene transfer (HGT) events occur frequently in Paracoccus spp. genomes, which may explain their metabolic flexibility.
Among Paracoccus species, P. aminophilus JCM 7686 is of particular interest since it is a methylotrophic bacterium capable of utilizing several toxic C1 compounds, including N,N-dimethylformamide, formamide as well as tri-, di- and monomethylamine, which are widely used in the chemical industry [21]. P. aminophilus JCM 7686 carries eight indigenous, extrachromosomal replicons (pAMI1 to pAMI8) ranging in size from 5.6 kb to approximately 440 kb. Our previous analyses, focused on the three smallest plasmids, pAMI3 (5.6 kb), pAMI2 (18.6 kb) and pAMI7 (20.5 kb), revealed the presence of (i) novel types of plasmid-encoded maintenance systems [22,23], (ii) a type II restriction-modification module with NcoI specificity [24] and (iii) genes crucial for the first step in the degradation of DMF [25].
In the present study, the genome of P. aminophilus JCM 7686 was completely sequenced and analyzed. An in-depth exploration of this genome sequence, followed by functional analyses, provided considerable insights into its overall architecture, as well as the functions of particular replicons. Moreover, in this study, the first inducible Paracoccus phage was identified.
Results and discussion
Sequencing and general features of the P. aminophilus JCM 7686 genome
A 454-pyrosequencing run for the P. aminophilus JCM7686 genomic DNA yielded 598348 shotgun and 8-kb-long paired-end reads with a total number of 225,950,536 bp that were assembled into 17 scaffolds. The scaffolds consisted of 429 large (> 500 nucleotides) and 129 small (100–500 nucleotides) contigs. The gaps in the chromosome and plasmids were closed by a PCR-based approach followed by sequencing of the corresponding amplicons. Subsequently, the genome was re-sequenced (for the quality check) applying the Illumina HiScanSQ Genome Analyzer.
The genome of P. aminophilus JCM 7686 is composed of a single circular chromosome of 3,613,807 bp and eight circular plasmids: pAMI1 (118,164 bp), pAMI2 (18,563 bp), pAMI3 (5575 bp), pAMI4 (438,126 bp), pAMI5 (294,017 bp), pAMI6 (206,583 bp), pAMI7 (20,542 bp) and pAMI8 (202,421 bp) (Figure 1). Thus, the total size of the genome is 4,917,798 bp. The overall GC content of the chromosome is 63.4%, which is consistent with other sequenced Paracoccus genomes. The GC contents of the JCM 7686 plasmids ranges between 57.4% (pAMI7) and 64.2% (pAMI4) (Table 1).
Figure 1.

Circular representations of the P. aminophilus JCM 7686 genome. Circles displayed (from the outside): (i) predicted CDSs transcribed in the clockwise direction, (ii) predicted CDSs transcribed in the counterclockwise direction, (iii) the position of tRNA genes, (iv) the position of rRNA genes, (v) the GC percent deviation, (vi) GC skew (G + C/G-C). The tRNA and rRNA genes are present only within the chromosome and plasmid pAMI1. Yellow arrow heads indicate transposable elements. Violet lines represents the prophage-like regions. Circles are drawn not in scale.
Table 1.
General features of the P. aminophilus JCM 7686 genome
| General features | Chromosome | pAMI1 | pAMI2 | pAMI3 | pAMI4 | pAMI5 | pAMI6 | pAMI7 | pAMI8 |
|---|---|---|---|---|---|---|---|---|---|
| Size (bp) |
3,613,807 |
118,164 |
18,563 |
5575 |
438,126 |
294,017 |
206,583 |
20,542 |
202,421 |
| GC content (%) |
63.4 |
63.3 |
62 |
60.8 |
64.2 |
62.8 |
63.9 |
57.4 |
62.3 |
| Coding density (%) |
90.2 |
87.5 |
79.1 |
72 |
88.1 |
88.5 |
92.8 |
83.6 |
89.2 |
| Number of ORFs |
3416 |
99 |
19 |
7 |
375 |
264 |
171 |
19 |
203 |
| Number of tRNA genes |
56 |
3 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
| Number of 16S-23S-5S rRNA operons |
3 |
1 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
| Phage regions |
9 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
| GTA regions |
1 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
| Transposases (including truncated) |
25 |
1 |
4 |
0 |
20 |
4 |
0 |
1 |
17 |
| Complete non-composite transposons |
0 |
1 |
0 |
0 |
0 |
0 |
0 |
1 |
2 |
| Complete insertion sequences | 5 | 0 | 2 | 0 | 6 | 1 | 0 | 0 | 6 |
The genome of JCM 7686 possesses 59 tRNAs and four clusters of 5S, 16S and 23S rRNA genes, which are located within the chromosome and plasmid pAMI1 (Table 1). This strain can produce tRNAs for all 20 amino acids, and genes encoding all the aminoacyl-tRNA synthetases are present within its chromosome. Moreover, a single tRNA for selenocyteine (tRNA-SeC) was also identified.
The JCM 7686 genome contains 4573 putative coding sequences (CDSs): 3416 in the chromosome and 1157 in the plasmids (Table 1). The coding density within the chromosome is 90.2%, while within the plasmids it varies from 72% (pAMI3) to 92.8% (pAMI6) (Table 1). We could assign putative biological functions to 3506 CDSs (76.6%), while 1069 CDSs (23.4%) were annotated as encoding hypothetical proteins of unknown function.
Predicted P. aminophilus proteins were functionally categorized and the proportions in each COG category were calculated. A total of 1550 (33.9%) predicted proteins were described as involved in the overall cellular metabolism, being assigned to COG functional categories C, G, E, F, H, I or Q. It appeared that more than 40% of the genes encoding proteins involved in metabolic processes are located within extrachromosomal elements, mainly pAMI1, pAMI4, pAMI5 and pAMI6 (426 genes in total). This finding suggests that these four replicons perform an important role (Additional file 1).
The most abundant class of proteins comprises those involved in amino acid transport and metabolism (COG category E). A total of 576 (12.6%) CDSs were assigned to this category, of which 191 occur within the aforementioned replicons: pAMI1 (41 genes), pAMI4 (65), pAMI5 (52) and pAMI6 (31) (Additional file 1).
Transcription regulatory network
Within the JCM 7686 genome, we identified 12 genes (7 in the chromosome, 3 in pAMI4, 1 in pAMI1 and 1 in pAMI5) encoding predicted Sigma factors: (i) FecI-like σ19 – required for the uptake of iron (5 copies, JCM7686_1690, JCM7686_1812, JCM7686_pAMI1p074, JCM7686_pAMI4p059 and JCM7686_pAMI5p016), (ii) RpoE-like σ24 – enabling the expression of genes involved in the heat shock response, as well as extracytoplasmic transport (4 copies, JCM7686_2748, JCM7686_2865, JCM7686_3466 and JCM7686_pAMI4p292), (iii) RpoH-like σ32 – involved in the heat shock and other stress responses (JCM7686_2477), (iv) RpoN-like σ54 – involved in nitrogen metabolism (JCM7686_pAMI4p216) and (v) RpoD-like σ70 – regulating gene expression during exponential growth (JCM7686_2204). Genes equivalent to rpoF (σ28) and rpoS (σ38), responsible for the expression of flagellar genes and genes in the stationary phase of growth, respectively, were not detected in this genome. In addition, we identified anti-sigma (JCM7686_3151) and anti-anti-sigma (JCM7686_3150) factors within the JCM 7686 chromosome.
The presence of eleven alternative sigma factors (of four types – σ19, σ24, σ32 and σ54), enables global changes in gene expression and permits alteration of the overall cellular metabolism in response to changing environmental conditions [26]. Thus, this feature indirectly reflects the metabolic flexibility and adaptive abilities of the strain JCM 7686. It is noteworthy that three of five P. aminophilus fecI (σ19) genes were identified within plasmids, which suggests an important role for these replicons in iron transport and metabolism. Moreover, the only rpoN gene copy was carried by a plasmid, pAMI4. It was previously demonstrated that RpoN, besides regulating genes involved in nitrogen metabolism, enables the transcription of a wide range of other genes encoding proteins participating in the regulation of virulence-related factors, as well as in amino acid, carbohydrate and organic acid synthesis, utilization and transport [26]. This indicates that pAMI4 may be highly significant in overall gene regulation in the strain JCM 7686.
Global regulators, which have the ability to regulate operons that belong to various metabolic pathways, are another basic component of transcription regulatory networks in bacteria. It has been shown that the expression of 51% of the E. coli genes is under the control of only seven regulatory proteins: CRP, FNR, IHF, FIS, ArcA, NarL and Lrp [27]. Within the JCM 7686 genome we identified genes encoding 5 types of putative global gene regulators: (i) three CRP/FNR family transcriptional regulators (JCM7686_1106, JCM7686_3086, JCM7686_pAMI8p144), (ii) three FIS family transcriptional regulators (JCM7686_3395, JCM7686_pAMI4p202, JCM7686_pAMI5p202), (iii) a H-NS proteins (JCM7686_3426, JCM7686_pAMI8p115), (iv) a Lrp regulator (JCM7686_0212) and (v) integration host factor (IHF) subunits A (IhfA; JCM7686_1093) and B (IhfB; JCM7686_1288).
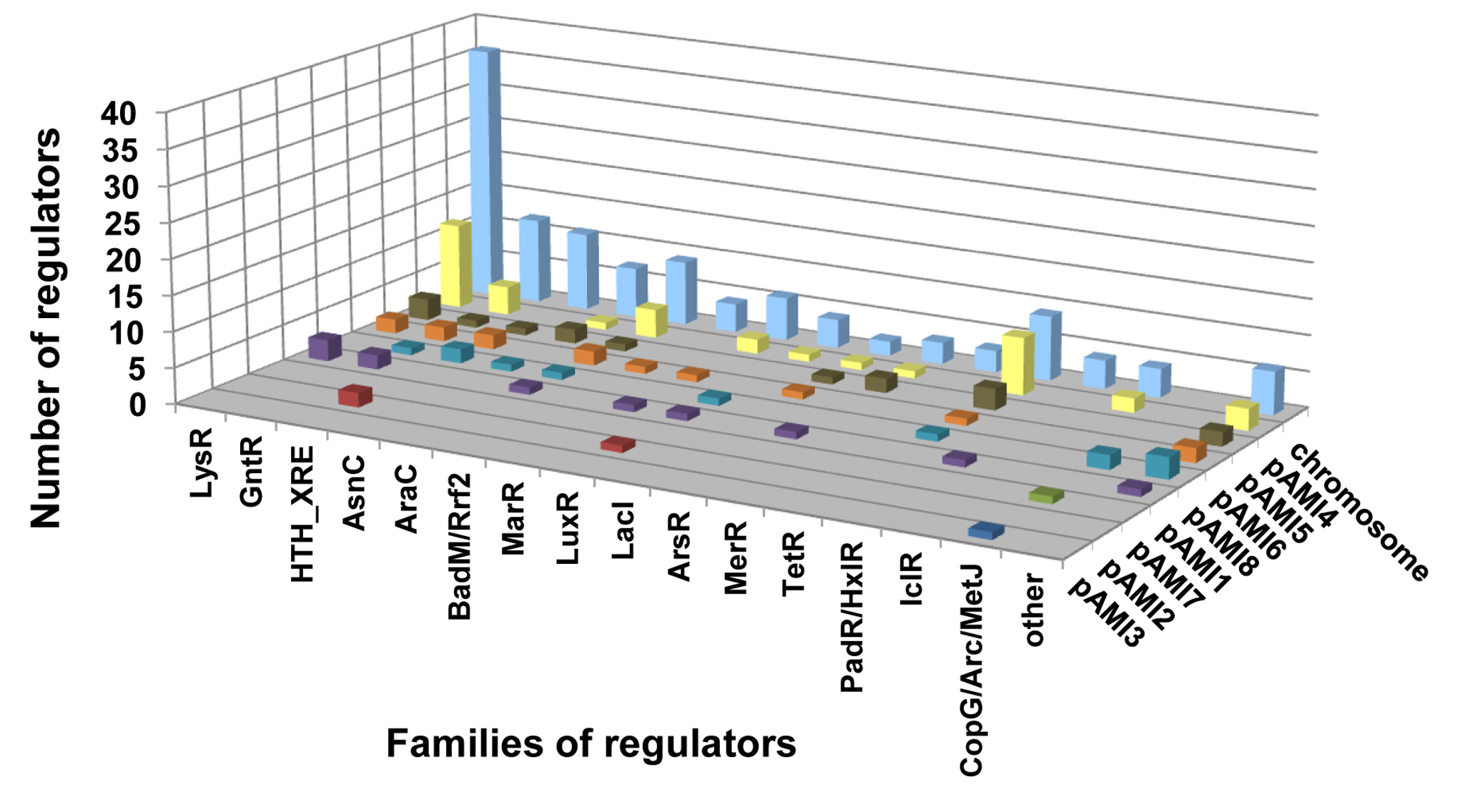
We also identified 217 genes encoding transcriptional regulators with predicted local specificity of action, probably limited to a single gene/gene cluster (Additional file 2). A quarter of the identified regulators were assigned to the LysR family – the most abundant type of transcriptional regulators in bacteria [28]. Interestingly, we identified genes encoding 8 regulators of the LuxR family, which are located within the chromosome and plasmids pAMI1, pAMI2, pAMI4 and pAMI8. The majority of LuxR-type proteins represents transcription activators, which specifically bind to N-acyl homoserine lactones (AHL; synthesized by a LuxI protein) that are secreted signaling molecules involved in quorum sensing in a variety of Gram-negative bacteria (e.g. [29]). We also identified CDSs for 3 putative N-acyl-L-homoserine lactone synthetases (LuxI-like proteins): two localized within the chromosome (JCM7686_2124, JCM7686_3181) and one in plasmid pAMI1 (JCM7686_pAMI1p026). The presence of luxR and luxI genes strongly suggests that quorum sensing plays a role in regulation of P. aminophilus gene expression.
Two-component systems are another important part of bacterial transcription regulatory networks present in P. aminophilus. Such systems are directly involved in sensing a cell’s external environment and signal transduction (e.g. [30]). Within the JCM 7686 genome we identified 23 pairs of genes encoding histidine protein kinases (HPKs) and phospho-aspartyl response regulators: 15 of them within the chromosome and others located on plasmids pAMI4, pAMI5, pAMI6 and pAMI8 (Additional file 3). Moreover, we identified 6 HPKs with unknown partner response regulators in the chromosome and pAMI6 (Additional file 3).
Among the P. aminophilus HPKs we found homologs of enzymes of well-described two-component phosphorelay systems involved in regulating (i) nitrogen assimilation – NtrB (JCM7686_0575 and NtrX (JCM7686_0577) [31], (ii) chemotaxis – CheA (JCM7686_1281) [32], (iii) phosphate homeostasis – PhoR (JCM7686_2063) [33], (iv) differentiation and cell cycle progression – CckA (JCM7686_2539) [34], (v) C4-dicarboxylate metabolism – DctB (JCM7686_2824) [35], (vi) expression of virulence factors – QseC (JCM7686_3369; JCM7686_pAMI5p117) [36], (vii) methanol and formaldehyde oxidation – FlhS (JCM7686_3383), (viii) several anaerobic processes and assimilation of CO2 and N2 – RegB (JCM7686_3423) [37], (ix) high affinity potassium-uptake – KdpD (JCM7686_pAMI4p337) [38] and (x) expression of the tor structural operon encoding the trimethylamine N-oxide reductase respiratory system in response to substrate availability – TorS (JCM7686_pAMI5p018) [39].
Stress response
In the majority of cases, the processes underlying the global stress response of bacteria are dependent on alterations in gene expression, usually controlled at the transcriptional level by various sigma factors [40]. However, there are also several more specific stress response mechanisms.
A fundamental trigger of the cellular stress response is DNA damage. In this scenario, the bacterial defense mechanism relies on a conserved inducible pathway – the SOS response (e.g. [41]). The primary components of this pathway, RecA recombinase and the LexA repressor, are encoded within the JCM 7686 chromosome (JCM7686_2538 and JCM7686_0753, respectively).
Moreover, the SpoT/RelA, (p)ppGpp synthetase I (JCM7686_1948), involved in the bacterial stringent response (triggered by nutritional deprivation) was also identified. During the stringent response process, an accumulation of the signaling nucleotides pppGpp and ppGpp occurs, which causes dramatic alterations in gene expression [42]. An important role in this process is also played by the chromosomally-encoded RelE toxin of the relBE systems, which, as mRNA-cleaving enzymes, globally inhibits translation during amino acid starvation [43]. Within the P. aminophilus genome we identified 11 toxin-antitoxin (TA) pairs representing the families relBE/parDE (5 TA systems), phd-doc (1), ccdAB (1), hipAB (2), and 2 hybrid systems (vapC-phd). The majority of the identified TA modules were localized within the plasmids (Additional file 4), and they most probably perform replicon stabilization functions. Only a single TA system of the hipAB family was located on the chromosome.
Among the most important factors in the maintenance of cellular fitness under changing environmental conditions are molecular chaperones. They are involved in various processes in bacterial cells, such as assisting the folding of newly synthesized proteins, protein secretion, preventing the aggregation of proteins under heat shock conditions, and repairing proteins that have been damaged or misfolded due to stress conditions (e.g. [44]). Within the P. aminophilus genome we identified 35 putative molecular chaperones, the majority of which (32) are encoded by the chromosome. These include heat and cold shock proteins, proteases, disulfide bond chaperones and protein-export chaperones (Additional file 5). We also identified a major RNA chaperone, Hfq, which is a key player in small RNA (sRNA)-mediated regulation of target mRNAs, that promotes sRNA-mRNA base pairing to permit rapid adaptive responses [45]. Surprisingly, we did not identify homologs of the bacterial HtpG (Hsp90) protein, which is responsible for heat or chemical shock responses. This is unusual because genes encoding HtpG proteins are present in the majority of bacterial genomes [46].
DNA recombination and repair
Since the integrity of the genomic DNA is fundamental to bacterial persistence, it is extremely important that the cell is equipped with sufficient DNA repair proteins to protect it against genetic damage [47]. Within the JCM 7686 genome we identified 59 genes encoding proteins predicted to be involved in various DNA repair pathways. The vast majority of them (47) are located within the chromosome. We performed a detailed characterization and classification of all genes encoding DNA repair-related proteins (summarized in Additional file 6). Comparison of the DNA repair genes of P. aminophilus with those of two well defined species, Escherichia coli (Gammaproteobacteria) and Caulobacter crescentus (Alphaproteobacteria), revealed the presence of 53 and 58 orthologous genes, respectively (Additional file 6).
As in C. crescentus[47], the JCM 7686 genome contains no mutH and dam homologs, which implies that both strains use different proteins to recognize and repair DNA replication errors. P. aminophilus, like many other Alphaproteobacteria (including C. crescentus) also lacks the RecBCD module, but possesses the related system AddAB instead, which recognizes a 5-base Chi site (5′-AGCGG-3′; 17,218 such recognition sequences were identified within the JCM 7686 genome) [48]. Curiously, we also found that P. aminophilus does not encode a typical deoxyribodipyrimidine photolyase (Phr-like). These enzymes employ visible light as the energy source to monomerize pyrimidine dimers induced by UV irradiation [49], and orthologs are found within the C. crescentus genome. Instead of Phr, JCM 7686 encodes a non-related SplB-like DNA repair photolyase. It was shown that the SplB protein is a DNA repair enzyme responsible for the process of reversion of the thymine dimer, 5-thyminyl-5,6-dihydrothymine (spore photoproduct), formed during UV irradiation of Bacillus subtilis spores [50]. We hypothesize that this protein may function as the major photolyase of P. aminophilus, responsible for the photo-reactivation process.
Although the majority of the DNA repair proteins are found within the JCM 7686 chromosome, 12 genes encoding such proteins are present in three plasmids: pAMI4 (3 genes), pAMI5 (3) and pAMI8 (6). Among them is an alkB gene (carried by pAMI5), which encodes a highly conserved and usually chromosomally-encoded protein (AlkB), responsible for the repair of alkylation damage in DNA via an oxidative demethylation pathway [51]. The presence of this alkB gene within pAMI5 suggests that it may be an essential replicon.
DNA methylation
Genes encoding predicted DNA methyltransferases (MTases), which may play important roles in the regulation of replication, gene expression and mismatch repair systems (e.g. [52]) were detected in the JCM 7686 genome. In addition to the previously described M.PamI [24], that is part of the type II restriction-modification system encoded by plasmid pAMI7, we identified seven chromosomally-encoded putative MTase genes. All of them seem to be orphan MTases, since they lack associated partner endonucleases.
The gene JCM7685_3079 encodes a CcrM (cell-cycle regulating MTase) homolog. Its ability to modify adenine residues in GANTC sequences was confirmed in vivo and in vitro (Additional file 7). The CcrM methyltransferases were shown to be essential for the viability of various Alphaproteobacteria (including Caulobacter crescentus, Sinorhizobium meliloti, Agrobacterium tumefaciens and Brucella abortus) and to play a crucial role in the regulation of bacterial cell division (e.g. [53]). To confirm the pivotal role of the JCM7686_3079 gene product in the P. aminophilus cell cycle, we attempted to disrupt this gene. However, this proved to be impossible, unless a wild-type copy was provided in trans, demonstrating that this CcrM homolog is essential for the viability of the host strain.
The other six genes encoding putative MTases are located within predicted prophage regions. Based on automatic methyltransferase prediction algorithms we allocated the following genes to different MTase classes: m6A MTases – JCM7686_1231, JCM7686_2255 and JCM7686_2934; m4C MTase – JCM7686_0815; m5C MTases – JCM7686_0772 and JCM7686_2655. Each of these predicted methylase genes was cloned into an expression vector and expressed in E. coli. The activity of the recombinant MTases was assessed and their sequence specificity determined using the endonuclease protection assay (according to [54]) (Additional files 8, 9, 10).
This analysis revealed that both m5C MTases have relaxed substrate specificity [partial protection of sequences CCNGG, CCWGG, AGCT, CCGG, GCN7GC and others (Additional file 8)], but they do not protect DNA against cleavage by R.PamI, encoded by the restriction-modification system of pAMI7. The m4C MTase of JCM 7686 recognizes and methylates YGGCCR sequences (Additional file 9). The three m6A MTases are highly similar (at least 95% aa sequence identity) and they methylate the sequence GANTC (Additional file 10). This is the same sequence specificity as assigned to the CcrM methylase (the main cell cycle regulator).
Multireplicon structure of the JCM 7686 genome
As mentioned above, the genome of P. aminophilus JCM 7686 is composed of a single chromosome and eight smaller replicons (pAMI1-pAMI8) (Figure 1). Predicted plasmids constitute 26.5% of the genome and they carry 1158 CDSs (about 25% of all JCM 7686 CDSs), which means that a huge amount of genetic information is stored within these replicons. Among the JCM 7686 plasmids we distinguished 5 mega-sized replicons (118–438 kb), pAMI1, pAMI4, pAMI5, pAMI6 and pAMI8, all carrying genes conserved in bacterial chromosomes, including chromosomes I and II of P. denitrificans PD1222 (Additional file 11). Since size is not an infallible criterion for distinguishing plasmids from secondary chromosomes, functional analyses of the JCM 7686 replicons was undertaken.
To characterize the function of the mega-sized replicons in the cellular metabolism we constructed mini-derivatives, which were used to remove the native plasmids from the host cell by incompatibility (such analyses of the smallest plasmids pAMI2, pAMI3 and pAMI7 were performed previously [22,23,25]. The mini-derivatives were constructed by cloning DNA fragments containing the plasmid maintenance modules (including plasmid replication initiation modules), into the E. coli-specific, mobilizable, narrow-host-range vector pABW1 (ColE1-type ori of pMB1) [55]. Using the obtained mini-derivatives containing incompatibility determinants, we were able to remove plasmids pAMI1, pAMI4, pAMI6 and pAMI8 from JCM 7686 (Additional file 12). Interestingly, curing this strain of pAMI1 resulted in a change in colony morphology, which became irregular, flat and dry. We were unable to introduce the mini-derivative of pAMI5 into JCM 7686 cells. It was therefore impossible to remove pAMI5, which strongly suggests that this replicon might be essential to host viability.
Growth rate analysis of strains deprived of the individual megaplasmids was performed in rich LB medium, as well as in AC minimal salt medium. This revealed that the removal of plasmids pAMI1, pAMI4 and pAMI8 had no effect on the growth rate (Figure 2). Interestingly, the pAMI1-less derivative grew in the form of irregular flocks (Additional file 13), which were in fact the aggregations of bacterial cells. In the case of the pAMI6-less strain, growth in LB medium was significantly decreased, and it was completely inhibited in AC medium (Figure 2). This indicates that the presence of plasmid pAMI6 enables growth of this P. aminophilus strain in minimal medium.
Figure 2.
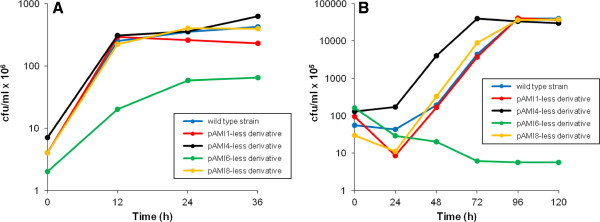
The growth rates of pAMI1-, pAMI4-, pAMI6- and pAMI8-less derivatives of P. aminophilus JCM 7686. Growth rates of the bacterial cultures were determined by viable cell counts (cfu).The strains [wild-type strain, pAMI1-less derivative, pAMI4-less derivative, pAMI6-less derivative and pAMI8-less derivative] were grown in 20 ml of liquid LB (A) or AC medium (B) with arabinose added as the sole source of carbon and energy. Samples were taken every 12 h for 36 h (in the case of LB medium) and every 24 h for 120 h (in the case of AC medium). The values plotted are the means of three replicates.
Based on the obtained results we have classified the pAMI-replicons into two groups: (i) essential (pAMI5, pAMI6) and (ii) non-essential (pAMI1, pAMI2, pAMI3, pAMI4, pAMI7 and pAMI8) genetic elements.
Additionally, to extend the functional analyses, the host ranges of pAMI1-pAMI8 replicons were examined using their plasmid mini-derivatives cloned within plasmid pABW1. The tested strains of Alphaproteobacteria were classified into two orders: (i) Rhodobacterales, represented by Paracoccus versutus UW225 and Paracoccus pantotrophus KL100, and (ii) Rhizobiales, represented by the families Brucellaceae (Ochrobactrum sp. LM19R) and Rhizobiaceae, including members of the Rhizobium/Agrobacterium group (Rhizobium etli CE3 and Agrobacterium tumefaciens LBA288) and the Sinorhizobium/Ensifer group (Sinorhizobium sp. LM21R). Replication abilities of pAMI1-pAMI8 were also tested in Alcaligenes sp. LM16R (Betaproteobacteria) and E. coli BR825 (Gammaproteobacteria).
This analysis revealed that mini-derivatives of pAMI1, pAMI2, pAMI3, pAMI4, pAMI7 and pAMI8 could replicate in all analyzed strains of Alphaproteobacteria, while the introduction of pAMI5 and pAMI6 into Sinorhizobium sp. and Ochrobactrum sp. was impossible, which may be due to strong incompatibility with their native replicons. We were also unable to introduce any of the analyzed plasmids into representatives of the Beta- or Gammaproteobacteria (Additional file 14). These findings indicate the relatively narrow host range of the analyzed replicons, being limited to members of the Alphaproteobacteria class.
Essential genetic elements – chromids
The curing experiments showed that the pAMI5 replicon could not be removed from JCM 7686 cells. Close inspection of the genetic content of this plasmid revealed that 62 (23.5%) of its genes are conserved in the chromosomes of P. denitrificans PD1222 (Additional file 11), and that 45 of them are singletons in the JCM 7686 genome. These genes encode several proteins of unknown function, but also proteins that are probably involved in the host metabolism, including (i) iron transport, (ii) D-pantothenate synthesis, (iii) motility, (iv) folate one-carbon metabolism and (v) de novo NAD biosynthesis. Interestingly, the nadABC (JCM7686_pAMI5p135-137) genes are the only ones encoding components of the de novo NAD biosynthetic pathway (Additional file 15) identified within the JCM 7686 genome, which may explain the essential nature of the pAMI5 replicon. It is important to mention that the loss of nadABC genes most probably cannot be compensated via the NAD salvage pathway, since the predicted NAD pyrophosphatase (an enzyme involved in the process) is also encoded by pAMI5 replicon (JCM7686_pAMI5p033 gene) (the remaining components of this pathway were identified in the JCM 7686 chromosome). Moreover, pAMI5 contains the only copies of (i) the alkB gene (JCM7686_pAMI5p075) induced during adaptive responses and involved in the direct reversal of alkylation damage [56] and (ii) the hslJ gene (JCM7686_pAMI5p111) encoding a heat shock protein [57].
All the aforementioned genes may play an important role in the JCM 7686 metabolism and we speculate that some of them may be responsible for the pAMI5 essentiality. However this needs to be experimentally confirmed.
Essential element status can also be assigned to the pAMI6 replicon. Although, we were able to remove it from JCM 7686 cells cultivated in LB medium, the growth of the pAMI6-less strain was completely inhibited in minimal salt medium (Figure 2). Analysis of the genetic content of pAMI6 revealed the presence of several modules that are typically encoded on chromosomes. Corresponding genes are involved in histidine, folate, glycerophospholipid, purine, selenoamino acid, sulfur, propanoate, glyoxylate and dicarboxylate metabolism. A surprising finding was the amtB gene (JCM7686_pAMI6p067, the only copy of this gene in the JCM 7686 genome), which encodes a NH4+ transporter of the Amt/Rh family [58]. Presence of this gene on pAMI6 may explain why this plasmid is essential for growth in minimal salt medium, where the only nitrogen source is NH4Cl, which cannot be transported into the cell in the absence of an appropriate transporter. This finding was confirmed by the analysis of the strain growth in minimal medium with casamino acids as a nitrogen source instead of ammonium chloride. pAMI6-less strain was able to growth in such medium, but its growth rate was significantly decreased. This suggests that there are also some additional (to amtB) genes, which are responsible for the essentiality of this replicon.
A growing number of studies in the field of genomics have produced data suggesting that the structure of many bacterial genomes is more complex than previously assumed. Many bacteria bear additional, large, autonomous replicons, which (like chromosomes) carry a pool of housekeeping genes [59-62]. Unlike typical plasmids, such replicons are necessary for the viability of their hosts, and for this reason they were initially defined as “secondary chromosomes”. However, this name is inadequate since these replicons possess many characteristics typical of plasmids: they contain plasmid-like replication systems and other genetic modules of plasmid origin. Bioinformatic analyses of these replicon sequences indicated that they were generated by the transfer of genetic information from chromosomes to plasmids co-residing in the cell. Due to their dualistic properties, they have been reclassified into a separate, newly distinguished group with properties of both chromosomes and plasmids: the chromids [60]. Our analyses indicate that pAMI5 and pAMI6 can be classified into this group of elements as well.
Non-essential genetic elements – plasmids
Six of the predicted JCM 7686 plasmids (including the previously described plasmids pAMI2, pAMI3 and pAMI7 [23,25]) were readily removed from the host cells. Among these dispensable replicons are the two repABC-type megaplasmids, pAMI4 and pAMI8 (Figure 1). Interestingly, pAMI4 and pAMI8 carry almost as many putative transposase genes (20 and 17, respectively; complete and truncated) as the whole chromosome (25 genes), and many more than the other replicons (0–4 genes) (Table 1). Within pAMI4 and pAMI8 we identified 101 genes encoding predicted transporters involved in the transport of (i) amino acids/dipeptides (dpp-like gene clusters), (ii) sn-glycerol-3-phosphate (ugp-like gene clusters), (iii) taurine (tau-like gene cluster) and (iv) inorganic ions [mainly iron (fec-like gene clusters), but also potassium, sodium and various heavy metals]. Genes encoding predicted transporters constitute 21 and 10% of the genetic information carried by plasmids pAMI4 and pAMI8, respectively. Moreover, within pAMI8 we identified a vir-gene cluster of a type IV secretion system, which is frequently found in Alphaproteobacteria megaplasmids [63], and 5 putative dsb-like genes the products of which may be involved in introducing disulfide bonds into diverse substrate proteins [64].
As mentioned above, the removal of pAMI1 from JCM 7686 cells influenced colony morphology and affected the growth mode in rich LB medium (but not in minimal-salt AC medium) (Additional file 13). Analysis of the pAMI1 sequence revealed that 40% of its genes encode proteins involved in amino acid transport and metabolism. Since amino acids serve as the main nitrogen source in LB medium, while nitrogen is provided by ammonium ions in AC medium, this may explain the observed growth differences.
Prophages and other prophage-related regions
In the P. aminophilus chromosome, we identified 10 regions encoding phage-related proteins (Figure 1). These constitute 8.2% of the host chromosome. Close inspection revealed that only 6 of them encode a full set of proteins crucial for the phage “life cycle”. Thus only these regions were considered to represent putative prophages and designated ϕPam-1 to ϕPam-6, respectively (Figure 3A). The predicted sizes of the identified prophages range from 32.9 to 43.9 kb and they comprise between 41 and 49 phage-related genes coding for proteins involved in integration, replication, packaging, capsid and tail assembly, and lysis. For four of the prophages we could identify putative integration sites, which were tRNA sequences (tRNA-Pro for ϕPam-1, tRNA-Arg for ϕPam-3, tRNA-Gly for ϕPam-5 and tRNA-Met for ϕPam-6).
Figure 3.

Prophage-like regions and transposable elements within the P. aminophilus JCM 7686 genome. The putative CDSs, colored according to their category, are shown by arrows representing their direction of transcription. (A) Genetic organization and comparison of the prophage-like regions within the P. aminophilus genome. Predicted gene clusters contain lysogeny, replication, structural and lysis-associated phage genes. The gray-shaded areas connect regions of prophages sharing ≥95% nucleotide sequence identity. (B) Genetic organization of the putative gene transfer agent (GTA) region. (C) Genetic organization of the insertion sequences and non-composite transposon Tn3434a of P. aminophilus. The families and names of the identified TEs are shown in the table. Inverted repeats (IRL – left IR; IRR – right IR) flanking ISs are marked by black vertical bars.
The vast majority of genes annotated within the prophage regions encode hypothetical proteins of unknown function. However, since prophages may encode fitness factors (e.g. virulence genes or metabolic modules) for their lysogenic hosts (e.g. [65]) we performed in-depth homology searches and found that genes JCM7686_0776 and JCM7686_1223 (within ϕPam-1 and ϕPam-2) encode tellurite resistance proteins (TerB). Moreover, we found that all but one (ϕPam-3) of the prophages encode methyltransferases (Figure 3A).
The JCM 7686 prophages exhibit high reciprocal nucleotide sequence similarity (Figure 3A), but they share only limited and localized homology with other prophages. This finding suggests their uniqueness among so far identified bacteriophages. Comparative analyses of the phage structural proteins revealed that five of the JCM 7686 prophages (ϕPam-2-ϕPam-6) encode major capsid proteins similar to those of HK97-like phages (Siphoviridae), which may suggest some evolutionary relationship.
To identify functional phages we applied the method of aggressive induction with mitomycin C. Using this procedure, only one of the JCM 7686 prophages, ϕPam-6, was induced. The ϕPam-6 prophage is flanked in the genome by 17-bp direct repeats (5′-CCCTCCTCCGCTACCAT-3′) which were recognized as the phage attachment site. In addition, ϕPam-6 encodes a serine family recombinase, rather than a tyrosine family integrase that is typical for phages (including the other JCM 7686 prophages). Following induction, bacteriophage particles were visualized by transmission electron microscopy using negative staining with uranyl acetate (Additional file 16) and it was confirmed by the restriction analysis that they contain ϕPam-6 DNA. Hence, ϕPam-6 is the first functional phage identified in Paracoccus spp.
Among the predicted prophage-related regions we also identified one gene transfer agent (GTA) cluster of 14.7 kb, which contains 18 putative genes (Figure 3B). This GTA cluster is located upstream of the cysE gene, encoding serine O-acetyltransferase (involved in cysteine biosynthesis), which is a common location for GTAs in bacterial genomes [66]. The identified region shares synteny and exhibits 80% nucleotide sequence identity with a GTA cluster found within chromosome II of Paracoccus denitrificans PD1222 [GenBank:CP000490]. Moreover, the P. aminophilus GTA exhibits homology to other RcGTA-like gene clusters (the archetype of this group is Rhodobacter capsulatus GTA) identified in various representatives of Alphaproteobacteria[66].
Transposable elements (TEs)
Analysis of the JCM 7686 genome identified 72 predicted transposase genes (Table 1); however, 32 of them are truncated (Additional file 17). The encoded transposases were classified and particular TEs distinguished (Additional file 17).
We identified 11 types of complete insertion sequences representing 5 IS families [IS3 (IS407, IS51 groups), IS5 (IS427, IS903 groups), IS66, IS110 and IS1182] (Additional file 17). Most of the identified ISs are present as a single copy in the JCM 7686 genome. The exceptions are ISPam1 and ISPam2 (both have 2 copies), as well as ISPam5 (8 copies). As shown in Figure 3C, the majority of the identified ISs contain a single ORF encoding a transposase (members of the IS903 group of the IS5 family, IS110 and IS1182 families) or carry two overlapping ORFs and possess a conserved frame-shift motif (members of the IS407 group of the IS3 family and IS427 group of the IS5 family). The frame-shift sequences are most probably responsible for the generation of a fusion protein (ORF1 + ORF2) as a result of programmed translational frame-shifting [67]. A putative trans-frame transposase may also be potentially produced by ISPam8 of the IS66 family. ISPam8 also contains two additional ORFs (Figure 3C), which may be involved in the regulation of transposition.
Within the plasmid component of the JCM 7686 genome we also identified a non-composite, cryptic transposon Tn3434a of the Tn3 family. It is present in 4 copies: in pAMI1, pAMI7 and pAMI8 (2 copies). The presence of two identical divergently oriented copies of Tn3434a within pAMI8 resulted in inversion of an approx. 15-kb DNA region due to site-specific resolvase-mediated recombination.
In a previous study, using a trap plasmid strategy, we showed that ISPam1, ISPam2, ISPam3 and Tn3434a are fully functional elements [17]. However, such analyses have some limitations because, although it may identify most active elements, other functional TEs can be missed due to their low transposition frequency.
Comparative genomics of Paracoccus spp
Until now, the annotated genome sequences of only four Paracoccus spp. strains are available in public databases: P. denitrificans PD1222 ([GenBank:CP000489], [GenBank:CP000490] and [GenBank:CP000491]), Paracoccus sp. strain TRP [7], P. denitrificans SD1 [8] and Paracoccus sp. N5 ([GenBank:NZ_AQUO01000001], [GenBank:NZ_AQUO01000002] and [GenBank:NZ_AQUO01000003]). In this study we add the genome of P. aminophilus JCM 7686 and comparative analysis of five Paracoccus spp. genomes is now possible.
A total of 1001 genes (20 - 27% of all genes) can be considered as core Paracoccus genes, since they are present in all five analyzed genomes (Figure 4). Functional classification of the proteins encoded by these genes showed that, in a vast majority of cases, they belong to five COG groups (J, O, C, F, H) that represent basic housekeeping functions, expected to be encoded on chromosomes, and therefore conserved among bacterial genomes.
Figure 4.
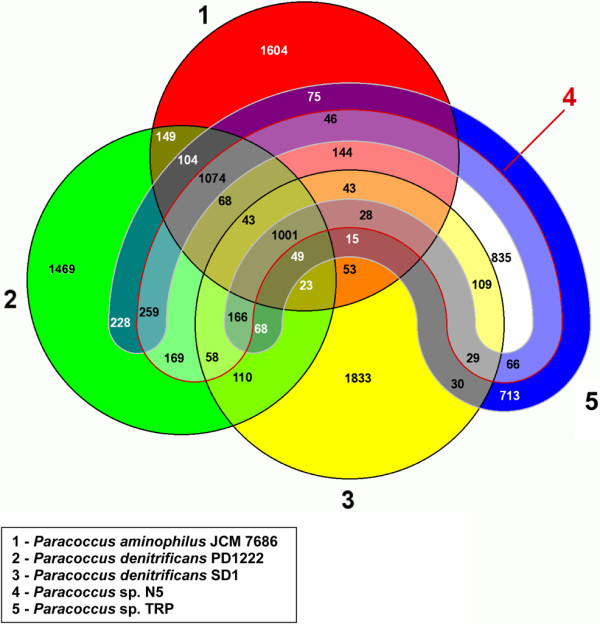
Venn diagram of the comparison of five sequenced and annotated Paracoccus genomes. The differently colored areas represent the compared strains. The central area describes the core genome consisting of 1001 genes.
Comparative genomic analyses disclosed some specific features of the P. aminophilus genome. The chromosome of strain JCM 7686 contains 757 singletons (22% of all genes) that are mainly located within prophages, although other gene clusters are also included, e.g. a type III secretion system with 19 unique genes. The remaining singleton genes are encoded by the plasmids (46% of all plasmid genes). A relatively large portion of singletons within the genome is a feature of all the analyzed Paracoccus genomes, which suggests that these strains are not that closely related.
We also performed a complex phylogenetic analysis of P. aminophilus JCM 7686 in relation to other fully sequenced members of the class Alphaproteobacteria. A phylogenetic tree based on 453 core genes was computed (Figure 5). P. aminophilus JCM 7686 is clustered together with the other completely sequenced Paracoccus spp. strains, but it forms an outgroup, which indicates its “distant” relationship to representatives of other species (Figure 5).
Figure 5.

Phylogenetic tree based on 453 core genes of the selected strains of Alphaproteobacteria. Multiple sequence alignments of concatenated core gene sequences were calculated using EDGAR tool. Plasmid sequences were included.
Conclusions
Chromids have been found in bacteria belonging to many different phyla, including Actinobacteria, Bacteroidetes, Chloroflexi, Cyanobacteria, Deinococcus-Thermus, Firmicutes, Proteobacteria and Spirochaetes[60,61]. It was shown that chromids of bacteria classified to separate phylogenetic groups carry replication systems of different types, which indicates that these replicons were derived from plasmid precursors typical for particular groups of hosts, and were formed independently in the course of evolution [60,61].
Chromids share a number of major characteristics: (i) considerable size (they are usually the second largest replicons in the cell, being larger than the chromosomes of some bacteria), (ii) the presence of plasmid-type replication systems, (iii) G + C nucleotide contents comparable to that of chromosomes, (iv) codon usage similar to that of chromosomes (points iii and iv indicate long-term co-evolution of chromosomes and chromids), (v) the presence of housekeeping genes, typical for chromosomes (removal of chromids from the bacterial cell causes a lethal effect), and (vi) the presence of adaptive genes, typical for plasmids, enabling adaptation of bacteria to a given ecological niche [60,68,69]. A typical feature of chromids is their high variability, resulting mainly from the low density of housekeeping genes. Therefore, they are considered to be specific “training sites” in which different evolutionary variants are “tested” [68,70,71], which leads to structural variability of multireplicon genomes in different strains of the same genus [72].
The formation of multi-replicon genomes, in which the basic genomic information is split between various replicons, seems to be beneficial to bacteria. The presence of additional replicons enables more rapid duplication of the genetic information, and permits the maintenance of a larger genome while keeping a high rate of cell division. Moreover, it was shown that the frequency of dimer generation by bacterial chromosomes increases exponentially in relation to their size; therefore, the reduction of chromosome size (by dividing the genetic information essential for host viability, into two or more replicons) allows minimization of this phenomenon [73].
Many Paracoccus spp. genomes also possess a multi-replicon structure. A good example of such a genome architecture is P. aminophilus JCM 7686, which carries one chromosome and eight plasmid-like replicons. Our in silico analyses revealed that pAMI1, pAMI4, pAMI5, pAMI6 and pAMI8 carry many genes (also predicted core genes) showing homology to ORFs within P. denitrificans PD1222 chromosomes (Additional file 11), thus they may be considered as putative chromids. However, relying only on the bioinformatic studies, the concept of essentiality of particular replicons is highly speculative, thus to define their nature we applied additional analyses.
In this study, acknowledging the importance of functional analyses, we have shown that only pAMI5 and pAMI6 should be classified as chromids, since their presence in the host cells is essential for their proper functioning. pAMI5 could not be removed from the host cells at all, and while it was possible to obtain a pAMI6-less derivative, this strain was unable to grow in minimal media, i.e. in conditions similar to those in the natural environment. Based on this observation we propose the classification of bacterial chromids into two types: “primary” chromids (e.g. pAMI5), which are indispensable for host viability and carry genes of the core genome, thus their elimination from the host cells is impossible under any environmental conditions, and “secondary” chromids (e.g. pAMI6) which were probably formed quite recently from an evolutionary point of view (e.g. they contain different REP systems, and have more characteristics typical for plasmids). Moreover, secondary chromids seems to carry a species (or genera) -specific pool of genes that are crucial for survival in the natural environment, but are not essential under “safe” laboratory conditions, thus the replicons are only “facultatively” essential [74].
According to this definition, the chromosome II of P. denitrificans PD1222 should be reclassified as a primary chromid. It is noteworthy that chromosome II of PD1222 and pAMI5 carry related dnaA-like replication systems, which may be a typical feature of the primary chromids of Paracoccus spp. This may facilitate further classifications of such replicons in other representative of this genus.
Methods
Strains, plasmids and culture conditions
The strains used in this study are presented in Additional file 18. They were grown in Luria-Bertani (LB) medium [75], TY medium (Rhizobium etli CE3) [76] and the minimal salts medium AC [77] at 37°C (E. coli) and 30°C (other strains). Where necessary, the media were supplemented with arabinose (0.2%), glucose (1%), kanamycin (50–1000 μg/ml) and rifampicin (50 μg/ml). The plasmids used and constructed in this study are described in Additional file 19.
DNA isolation, standard genetic manipulations and introduction of plasmid DNA into bacterial cells
The isolation of total DNA and plasmids, as well as common DNA manipulation methods were performed as described by Sambrook and Russell (2001) [75]. The visualization of mega-sized replicons was achieved by in-gel lysis and DNA electrophoresis according to a method described by Wheatcroft et al. (1990) [78]. PCR was performed in a Mastercycler using HiFi polymerase (Qiagen; with supplied buffer), dNTP mixture, total DNA (or plasmid DNA) of P. aminophilus as the template and appropriate oligonucleotide primers (Additional file 20). Transformation of E. coli strains was performed according to the method of Kushner [79]. Triparental mating was performed as previously described [80].
Determination of the sequence specificity of methyltransferases using the endonuclease protection assay
Putative MTase genes were PCR-amplified from P. aminophilus genomic DNA using specific primer pairs (Additional file 20). The amplified genes were cloned into expression vectors pET-28a or pET-30a (Additional file 19). Obtained plasmids were used to transform E. coli ER2566 and the strains were propagated for expression. To repress T7 RNA polymerase expression in the ER2566 strains before induction, glucose was added to cultures at a final concentration of 1%. The DNAs of recombinant plasmids isolated from IPTG-induced and non-induced bacterial cultures were used as substrates for cleavage by selected restriction enzymes.
Phage excision and transmission electron microscopy
Mitomycin C (0.5 mg/ml) was added to a logarithmic phase culture of P. aminophilus in 20 ml LB medium. Following incubation for a further 8 h, the culture was centrifuged at 11,000 g for 10 min to pellet the cells and the supernatant fraction was passed through a membrane filter (0.22 μm pore size). Phage particles were collected by centrifugation (2 h at 100,000 g) and resuspended in 50 μl of SM buffer (50 mM Tris–HCl, pH 7.5, 100 mM NaCl, 8 mM MgSO4 × 7H2O, 0.01% gelatin). One drop of the suspension was applied to the surface of a Formvar-coated grid, negatively stained with 2% uranyl acetate [81], and then examined using a LEO 912AB transmission electron microscope.
DNA sequencing
Total genomic DNA was isolated from an overnight culture of P. aminophilus JCM 7686 using the DNeasy Blood & Tissue Kit (Qiagen). This was used for the construction of NGS (Next Generation Sequencing) libraries: (i) GS FLX + shotgun library [using the GS FLX + library preparation kit (Roche)], (ii) an 8-kb-long paired-end library [using the GS FLX Paired-End kit (Roche)] and (iii) Illumina Paired-End library [using the Illumina TruSeq2.0 kit (Illumina Inc.)]. The genomic libraries were sequenced using a Genome Sequencer FLX + System (Roche) and Illumina HiScanSQ Genome Analyzer (Illumina Inc.) in the DNA Sequencing and Oligonucleotide Synthesis Laboratory (oligo.pl) at the Institute of Biochemistry and Biophysics, Polish Academy of Sciences. The sequence data was assembled into contigs and scaffolds using Newbler De Novo Assembler (454 Sequencing System Software, Roche). Any remaining gaps were closed using the Expand Long Template PCR System (Roche), followed by Sanger sequencing with an ABI3500 Genetic Analyzer (Life Technologies) using BigDye Terminator Mix v. 3.1 chemistry (Life Technologies).
Bioinformatics
Automatic gene prediction and annotation of the JCM 7686 genome were performed using GenDB 2.0 [82]. Automatic annotations and intergenic regions were analyzed and corrected manually by means of BLAST programs [83] and the PRIAM tool [84]. Putative tRNA genes were identified with the tRNAscan-SE program [85]. Each gene was functionally classified by assigning a Cluster of Orthologous Groups (COG) number and its corresponding COG category [86]. Comparison searches for insertion sequences were performed with ISfinder [87]. Finally, the Artemis software was used to visualize the genome [88].
Comparisons between the JCM 7686 genome and the genomes of other representatives of the Alphaproteobacteria (i.e. synteny analyses, identification of orthologous genes and classification of genes as core genes or singletons) were performed using the EDGAR tool [89]. It was also used for the creation of the phylogenetic tree. To construct the tree, 453 core genes from 27 genomes of the Alphaproteobacteria were computed. The multiple alignments for all core genes were created using MUSCLE [90]. Non matching parts of the alignments were masked using GBLOCKS [91] and removed subsequently. The remaining parts of all alignments were concatenated to one multiple alignment, which then was used to generate the phylogenetic tree applying PHYLIP [92]. Other comparative analyses were performed with the GeneOrder4.0 tool [93].
Nucleotide sequence accession numbers
The nucleotide sequences of P. aminophilus JCM 7686 chromosome and extrachromosomal replicons pAMI1-8 have been annotated and deposited in GenBank (NCBI) with respective accession numbers: CP006650, CP006651, GQ410978, GQ468939, CP006652, CP006653, CP006654, GQ468938, CP006655.
Abbreviations
CDS: Coding sequence; COG: Cluster of Orthologous Group; DMF: N,N-dimethylformamide; GTA: Gene transfer agent; HPK: Histidine protein kinase; IR: Inverted repeat; IS: Insertion sequence; LB: Luria-bertani; MGE: Mobile genetic elements; MTase: methyltransferase; NAD: Nicotinamide adenine dinucleotide; NaMN: Nicotinic acid mononucleotide; NCBI: National Center for Biotechnology Information; ORF: Open reading frame; PCR: Polymerase chain reaction; TE: Transposable element; TMo: Transposable module; Tn: Transposon.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
LD and DB conceived and designed the experiments; LD coordinated the project; LD, JC, DW, MR, PM and MS performed the experiments; LD, JC, MR and DW analyzed the data; LD, DB, AS and AP contributed reagents/materials/analysis tools; DB, AS and AP supervised the work; LD, DB and DW wrote the paper. All authors read and approved the final manuscript.
Supplementary Material
COG categories of the proteins of P. aminophilus JCM 7686.
Diversity and distribution of 217 transcriptional regulators of P. aminophilus JCM 7686.
{kind=link}
Two-component systems and histidine kinases encoded by the P. aminophilus JCM 7686 genome.
Toxin-antitoxin systems encoded by the P. aminophilus JCM 7686 genome.
Chaperones and co-chaperonins encoded by the P. aminophilus JCM 7686 genome.
Genes encoding DNA repair related proteins within the P. aminophilus JCM 7686 genome.
Restriction patterns of P. aminophilus JCM 7686 genomic DNA cleaved with selected restriction endonucleases showing the protection of GANTC sites by the CcrM methylase (JCM7685_3079). ND - undigested DNA. M – GeneRuler 100–10,000 bp size marker.
Summary of the sensitivity of various restriction endonucleases to DNA modifications introduced by the JCM7686_0772 and JCM7686_2655 proteins (m 5 C MTases).
Summary of the sensitivity of various restriction endonucleases to DNA modifications introduced by the JCM7686_0815 protein (m 4 C MTase).
Summary of the sensitivity of various restriction endonucleases to DNA modifications introduced by the JCM7686_1231, JCM7686_2255 and JCM7686_2934 proteins (m 6 A MTases).
The number of homologous proteins encoded by JCM 7686 plasmids and P. denitrificans PD1222 chromosomes. The analysis was performed using the GeneOrder 4.0 tool. Proteins were considered homologous only if the BLAT threshold scores were >200. The results were verified manually by BLAST comparisons.
{kind=link}
Plasmid profiles of the JCM 7686 wild-type strain and its derivatives deprived of particular megasized-replicons.
The growth mode of wild-type JCM 7686 and the pAMI1-less derivative in liquid LB medium.
{kind=link}
Host ranges of P. aminophilus JCM 7686 pAMI1-8 replicons.
Schematic diagram of de novo NAD biosynthesis from aspartate to nicotinic acid mononucleotide (NaMN).
Transmission electron micrograph of tailed bacteriophage ϕPam-6 of P. aminophilus JCM 7686.
{kind=link}
Transposase genes within the P. aminophilus JCM 7686 genome.
Bacterial strains used in this study.
Plasmids used and constructed in this study.
Oligonucleotide primers used in this study.
Contributor Information
Lukasz Dziewit, Email: ldziewit@biol.uw.edu.pl.
Jakub Czarnecki, Email: jczarnecki@biol.uw.edu.pl.
Daniel Wibberg, Email: dwibberg@cebitec.uni-bielefeld.de.
Monika Radlinska, Email: m.radlinska@biol.uw.edu.pl.
Paulina Mrozek, Email: mrozek.paulina@gmail.com.
Michal Szymczak, Email: panszymczak@gmail.com.
Andreas Schlüter, Email: aschluet@cebitec.uni-bielefeld.de.
Alfred Pühler, Email: puehler@cebitec.uni-bielefeld.de.
Dariusz Bartosik, Email: bartosik@biol.uw.edu.pl.
Acknowledgements
We would like to thank J. Baj for scientific advice, as well as L. Kowalski, K. Kuzmicz, K. Romaniuk and A. Szych for technical assistance. We are also grateful to J. Gawor from the DNA Sequencing and Oligonucleotide Synthesis Laboratory (oligo.pl; Institute of Biochemistry and Biophysics, Polish Academy of Sciences) for his excellent work during the genome sequencing. The work was supported by the Ministry of Science and Higher Education, Poland (grants: IP2010 008670 and IP2011 011471).
References
- Baker SC, Ferguson SJ, Ludwig B, Page MD, Richter OM, van Spanning RJ. Molecular genetics of the genus Paracoccus: metabolically versatile bacteria with bioenergetic flexibility. Microbiol Mol Biol Rev. 1998;62(4):1046–1078. doi: 10.1128/mmbr.62.4.1046-1078.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama Y, Hiraishi A, Kuraishi H. Paracoccus thiocyanatus sp. nov., a new species of thiocyanate-utilizing facultative chemolithotroph, and transfer of Thiobacillus versutus to the genus Paracoccus as Paracoccus versutus comb. nov. with emendation of the genus. Microbiology. 1995;141:1469–1477. doi: 10.1099/13500872-141-6-1469. [DOI] [PubMed] [Google Scholar]
- Kelly DP, Euzeby JP, Goodhew CF, Wood AP. Redefining Paracoccus denitrificans and Paracoccus pantotrophus and the case for a reassessment of the strains held by international culture collections. Int J Syst Evol Microbiol. 2006;56:2495–2500. doi: 10.1099/ijs.0.64401-0. [DOI] [PubMed] [Google Scholar]
- Kumaraswamy R, Sjollema K, Kuenen G, van Loosdrecht M, Muyzer G. Nitrate-dependent [Fe(II)EDTA]2- oxidation by Paracoccus ferrooxidans sp. nov., isolated from a denitrifying bioreactor. Syst Appl Microbiol. 2006;29:276–286. doi: 10.1016/j.syapm.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Doronina NV, Trotsenko YA, Krausova VI, Suzina NE. Paracoccus methylutens sp. nov. - a new aerobic facultatively methylotrophic bacterium utilizing dichloromethane. Syst Appl Microbiol. 1998;21:230–236. doi: 10.1016/S0723-2020(98)80027-1. [DOI] [Google Scholar]
- Sun M, Luo Y, Teng Y, Christie P, Jia Z, Li Z. Tenax TA extraction to understand the rate-limiting factors in methyl-beta-cyclodextrin-enhanced bioremediation of PAH-contaminated soil. Biodegradation. 2013;24:365–375. doi: 10.1007/s10532-012-9593-2. [DOI] [PubMed] [Google Scholar]
- Li K, Wang S, Shi Y, Qu J, Zhai Y, Xu L, Xu Y, Song J, Liu L, Rahman MA. et al. Genome sequence of Paracoccus sp. Strain TRP, a chlorpyrifos biodegrader. J Bacteriol. 2011;193(7):1786–1787. doi: 10.1128/JB.00014-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddavattam D, Karegoudar TB, Mudde SK, Kumar N, Baddam R, Avasthi TS, Ahmed N. Genome of a novel isolate of Paracoccus denitrificans capable of degrading N,N-dimethylformamide. J Bacteriol. 2011;193(19):5598–5599. doi: 10.1128/JB.05667-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baj J, Piechucka E, Bartosik D, Wlodarczyk M. Plasmid occurrence and diversity in the genus Paracoccus. Acta Microbiol Pol. 2000;49(3–4):265–270. [PubMed] [Google Scholar]
- Bartosik D, Putyrski M, Dziewit L, Malewska E, Szymanik M, Jagiello E, Lukasik J, Baj J. Transposable modules generated by a single copy of insertion sequence ISPme1 and their influence on structure and evolution of natural plasmids of Paracoccus methylutens DM12. J Bacteriol. 2008;190(9):3306–3313. doi: 10.1128/JB.01878-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartosik D, Baj J, Sochacka M, Piechucka E, Wlodarczyk M. Molecular characterization of functional modules of plasmid pWKS1 of Paracoccus pantotrophus DSM 11072. Microbiology. 2002;148(Pt 9):2847–2856. doi: 10.1099/00221287-148-9-2847. [DOI] [PubMed] [Google Scholar]
- Bartosik D, Baj J, Wlodarczyk M. Molecular and functional analysis of pTAV320, a repABC-type replicon of the Paracoccus versutus composite plasmid pTAV1. Microbiology. 1998;144:3149–3157. doi: 10.1099/00221287-144-11-3149. [DOI] [PubMed] [Google Scholar]
- Bartosik D, Witkowska M, Baj J, Wlodarczyk M. Characterization and sequence analysis of the replicator region of the novel plasmid pALC1 from Paracoccus alcaliphilus. Plasmid. 2001;45(3):222–226. doi: 10.1006/plas.2000.1505. [DOI] [PubMed] [Google Scholar]
- Bartosik D, Baj J, Bartosik AA, Wlodarczyk M. Characterization of the replicator region of megaplasmid pTAV3 of Paracoccus versutus and search for plasmid-encoded traits. Microbiology. 2002;148(Pt 3):871–881. doi: 10.1099/00221287-148-3-871. [DOI] [PubMed] [Google Scholar]
- Bartosik D, Sochacka M, Baj J. Identification and characterization of transposable elements of Paracoccus pantotrophus. J Bacteriol. 2003;185(13):3753–3763. doi: 10.1128/JB.185.13.3753-3763.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartosik D, Szymanik M, Baj J. Identification and distribution of insertion sequences of Paracoccus solventivorans. Appl Environ Microbiol. 2003;69(12):7002–7008. doi: 10.1128/AEM.69.12.7002-7008.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziewit L, Baj J, Szuplewska M, Maj A, Tabin M, Czyzkowska A, Skrzypczyk G, Adamczuk M, Sitarek T, Stawinski P. et al. Insights into the transposable mobilome of Paracoccus spp. (Alphaproteobacteria) PLoS One. 2012;7(2):e32277. doi: 10.1371/journal.pone.0032277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikosa M, Sochacka-Pietal M, Baj J, Bartosik D. Identification of a transposable genomic island of Paracoccus pantotrophus DSM 11072 by its transposition to a novel entrapment vector pMMB2. Microbiology. 2006;152(Pt 4):1063–1073. doi: 10.1099/mic.0.28603-0. [DOI] [PubMed] [Google Scholar]
- Szuplewska M, Bartosik D. Identification of a mosaic transposable element of Paracoccus marcusii composed of insertion sequence ISPmar4 (ISAs1 family) and an IS1247a-driven transposable module (TMo) FEMS Microbiol Lett. 2009;292(2):216–221. doi: 10.1111/j.1574-6968.2009.01495.x. [DOI] [PubMed] [Google Scholar]
- van Spanning RJ, de Boer AP, Slotboom DJ, Reijnders WN, Stouthamer AH. Isolation and characterization of a novel insertion sequence element, IS1248, in Paracoccus denitrificans. Plasmid. 1995;34(1):11–21. doi: 10.1006/plas.1995.1029. [DOI] [PubMed] [Google Scholar]
- Urakami T, Araki H, Oyanagi H, Suzuki K, Komagata K. Paracoccus aminophilus sp. nov. and Paracoccus aminovorans sp. nov., which utilize N, N-dimethylformamide. Int J Syst Bacteriol. 1990;40(3):287–291. doi: 10.1099/00207713-40-3-287. [DOI] [PubMed] [Google Scholar]
- Dziewit L, Jazurek M, Drewniak L, Baj J, Bartosik D. The SXT conjugative element and linear prophage N15 encode toxin-antitoxin-stabilizing systems homologous to the tad-ata module of the Paracoccus aminophilus plasmid pAMI2. J Bacteriol. 2007;189(5):1983–1997. doi: 10.1128/JB.01610-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziewit L, Adamczuk M, Szuplewska M, Bartosik D. DIY series of genetic cassettes useful in construction of versatile vectors specific for Alphaproteobacteria. J Microbiol Methods. 2011;86(2):166–174. doi: 10.1016/j.mimet.2011.04.016. [DOI] [PubMed] [Google Scholar]
- Dziewit L, Kuczkowska K, Adamczuk M, Radlinska M, Bartosik D. Functional characterization of the type II PamI restriction-modification system derived from plasmid pAMI7 of Paracoccus aminophilus JCM 7686. FEMS Microbiol Lett. 2011;324(1):56–63. doi: 10.1111/j.1574-6968.2011.02388.x. [DOI] [PubMed] [Google Scholar]
- Dziewit L, Dmowski M, Baj J, Bartosik D. Plasmid pAMI2 of Paracoccus aminophilus JCM 7686 carries N, N-dimethylformamide degradation-related genes whose expression is activated by a LuxR family regulator. Appl Environ Microbiol. 2010;76(6):1861–1869. doi: 10.1128/AEM.01926-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studholme DJ, Buck M. The biology of enhancer-dependent transcriptional regulation in bacteria: insights from genome sequences. FEMS Microbiol Lett. 2000;186:1–9. doi: 10.1111/j.1574-6968.2000.tb09074.x. [DOI] [PubMed] [Google Scholar]
- Martinez-Antonio A, Collado-Vides J. Identifying global regulators in transcriptional regulatory networks in bacteria. Curr Opin Microbiol. 2003;6:482–489. doi: 10.1016/j.mib.2003.09.002. [DOI] [PubMed] [Google Scholar]
- Maddocks SE, Oyston PC. Structure and function of the LysR-type transcriptional regulator (LTTR) family proteins. Microbiology. 2008;154:3609–3623. doi: 10.1099/mic.0.2008/022772-0. [DOI] [PubMed] [Google Scholar]
- Subramoni S, Venturi V. LuxR-family ‘solos’: bachelor sensors/regulators of signalling molecules. Microbiology. 2009;155(Pt 5):1377–1385. doi: 10.1099/mic.0.026849-0. [DOI] [PubMed] [Google Scholar]
- Wolanin PM, Thomason PA, Stock JB. Histidine protein kinases: key signal transducers outside the animal kingdom. Genome Biol. 2002;3(10) doi: 10.1186/gb-2002-3-10-reviews3013. REVIEWS3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drepper T, Wiethaus J, Giaourakis D, Gross S, Schubert B, Vogt M, Wiencek Y, McEwan AG, Masepohl B. Cross-talk towards the response regulator NtrC controlling nitrogen metabolism in Rhodobacter capsulatus. FEMS Microbiol Lett. 2006;258:250–256. doi: 10.1111/j.1574-6968.2006.00228.x. [DOI] [PubMed] [Google Scholar]
- Wang X, Vu A, Lee K, Dahlquist FW. CheA-receptor interaction sites in bacterial chemotaxis. J Mol Biol. 2012;422:282–290. doi: 10.1016/j.jmb.2012.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamarche MG, Wanner BL, Crepin S, Harel J. The phosphate regulon and bacterial virulence: a regulatory network connecting phosphate homeostasis and pathogenesis. FEMS Microbiol Rev. 2008;32:461–473. doi: 10.1111/j.1574-6976.2008.00101.x. [DOI] [PubMed] [Google Scholar]
- Ausmees N, Jacobs-Wagner C. Spatial and temporal control of differentiation and cell cycle progression in Caulobacter crescentus. Annu Rev Microbiol. 2003;57:225–247. doi: 10.1146/annurev.micro.57.030502.091006. [DOI] [PubMed] [Google Scholar]
- Janausch IG, Zientz E, Tran QH, Kroger A, Unden G. C4-dicarboxylate carriers and sensors in bacteria. Biochim Biophys Acta. 2002;1553:39–56. doi: 10.1016/S0005-2728(01)00233-X. [DOI] [PubMed] [Google Scholar]
- Hughes DT, Clarke MB, Yamamoto K, Rasko DA, Sperandio V. The QseC adrenergic signaling cascade in Enterohemorrhagic E. coli (EHEC) PLoS Pathog. 2009;5:e1000553. doi: 10.1371/journal.ppat.1000553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird TH, Du S, Bauer CE. Autophosphorylation, phosphotransfer, and DNA-binding properties of the RegB/RegA two-component regulatory system in Rhodobacter capsulatus. J Biol Chem. 1999;274:16343–16348. doi: 10.1074/jbc.274.23.16343. [DOI] [PubMed] [Google Scholar]
- Jung K, Veen M, Altendorf K. K + and ionic strength directly influence the autophosphorylation activity of the putative turgor sensor KdpD of Escherichia coli. J Biol Chem. 2000;275:40142–40147. doi: 10.1074/jbc.M008917200. [DOI] [PubMed] [Google Scholar]
- Jourlin C, Bengrine A, Chippaux M, Mejean V. An unorthodox sensor protein (TorS) mediates the induction of the tor structural genes in response to trimethylamine N-oxide in Escherichia coli. Mol Microbiol. 1996;20:1297–1306. doi: 10.1111/j.1365-2958.1996.tb02648.x. [DOI] [PubMed] [Google Scholar]
- Boor KJ. Bacterial stress responses: what doesn’t kill them can make then stronger. PLoS Biol. 2006;4:e23. doi: 10.1371/journal.pbio.0040023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butala M, Zgur-Bertok D, Busby SJ. The bacterial LexA transcriptional repressor. Cell Mol Life Sci. 2009;66:82–93. doi: 10.1007/s00018-008-8378-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferullo DJ, Lovett ST. The stringent response and cell cycle arrest in Escherichia coli. PLoS Genet. 2008;4:e1000300. doi: 10.1371/journal.pgen.1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen SK, Mikkelsen M, Pedersen K, Gerdes K. RelE, a global inhibitor of translation, is activated during nutritional stress. Proc Natl Acad Sci U S A. 2001;98:14328–14333. doi: 10.1073/pnas.251327898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund PA. Microbial molecular chaperones. Adv Microb Physiol. 2001;44:93–140. doi: 10.1016/s0065-2911(01)44012-4. [DOI] [PubMed] [Google Scholar]
- Wagner EG. Cycling of RNAs on Hfq. RNA Biol. 2013;10:619–626. doi: 10.4161/rna.24044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Zhong D, Monteiro A. Comparative genomics and evolution of the HSP90 family of genes across all kingdoms of organisms. BMC Genomics. 2006;7:156. doi: 10.1186/1471-2164-7-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins-Pinheiro M, Marques RC, Menck CF. Genome analysis of DNA repair genes in the alpha proteobacterium Caulobacter crescentus. BMC Microbiol. 2007;7:17. doi: 10.1186/1471-2180-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigley DB. Bacterial DNA repair: recent insights into the mechanism of RecBCD, AddAB and AdnAB. Nat Rev Microbiol. 2013;11:9–13. doi: 10.1038/nrmicro2917. [DOI] [PubMed] [Google Scholar]
- Menck CF. Shining a light on photolyases. Nat Genet. 2002;32:338–339. doi: 10.1038/ng1102-338. [DOI] [PubMed] [Google Scholar]
- Rebeil R, Nicholson WL. The subunit structure and catalytic mechanism of the Bacillus subtilis DNA repair enzyme spore photoproduct lyase. Proc Natl Acad Sci U S A. 2001;98:9038–9043. doi: 10.1073/pnas.161278998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falnes PO, Johansen RF, Seeberg E. AlkB-mediated oxidative demethylation reverses DNA damage in Escherichia coli. Nature. 2002;419:178–182. doi: 10.1038/nature01048. [DOI] [PubMed] [Google Scholar]
- Marinus MG, Casadesus J. Roles of DNA adenine methylation in host-pathogen interactions: mismatch repair, transcriptional regulation, and more. FEMS Microbiol Rev. 2009;33:488–503. doi: 10.1111/j.1574-6976.2008.00159.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier J. Epigenetic regulation of the bacterial cell cycle. Curr Opin Microbiol. 2009;12:722–729. doi: 10.1016/j.mib.2009.08.005. [DOI] [PubMed] [Google Scholar]
- Drozdz M, Piekarowicz A, Bujnicki JM, Radlinska M. Novel non-specific DNA adenine methyltransferases. Nucleic Acids Res. 2012;40(5):2119–2130. doi: 10.1093/nar/gkr1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartosik D, Bialkowska A, Baj J, Wlodarczyk M. Construction of mobilizable cloning vectors derived from pBGS18 and their application for analysis of replicator region of a pTAV202 mini-derivative of Paracoccus versutus pTAV1 plasmid. Acta Microbiol Pol. 1997;46(4):387–392. [PubMed] [Google Scholar]
- Falnes PO, Rognes T. DNA repair by bacterial AlkB proteins. Res Microbiol. 2003;154:531–538. doi: 10.1016/S0923-2508(03)00150-5. [DOI] [PubMed] [Google Scholar]
- Lilic M, Jovanovic M, Jovanovic G, Savic DJ. Identification of the CysB-regulated gene, hslJ, related to the Escherichia coli novobiocin resistance phenotype. FEMS Microbiol Lett. 2003;224:239–246. doi: 10.1016/S0378-1097(03)00441-5. [DOI] [PubMed] [Google Scholar]
- Tremblay PL, Hallenbeck PC. Ammonia-induced formation of an AmtB-GlnK complex is not sufficient for nitrogenase regulation in the photosynthetic bacterium Rhodobacter capsulatus. J Bacteriol. 2008;190:1588–1594. doi: 10.1128/JB.01643-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acosta-Cruz E, Wisniewski-Dye F, Rouy Z, Barbe V, Valdes M, Mavingui P. Insights into the 1.59-Mbp largest plasmid of Azospirillum brasilense CBG497. Arch Microbiol. 2012;194:725–736. doi: 10.1007/s00203-012-0805-2. [DOI] [PubMed] [Google Scholar]
- Harrison PW, Lower RP, Kim NK, Young JP. Introducing the bacterial ‘chromid’: not a chromosome, not a plasmid. Trends Microbiol. 2010;18:141–148. doi: 10.1016/j.tim.2009.12.010. [DOI] [PubMed] [Google Scholar]
- Villasenor T, Brom S, Davalos A, Lozano L, Romero D, Los Santos AG. Housekeeping genes essential for pantothenate biosynthesis are plasmid-encoded in Rhizobium etli and Rhizobium leguminosarum. BMC Microbiol. 2011;11:66. doi: 10.1186/1471-2180-11-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landeta C, Davalos A, Cevallos MA, Geiger O, Brom S, Romero D. Plasmids with a chromosome-like role in rhizobia. J Bacteriol. 2011;193:1317–1326. doi: 10.1128/JB.01184-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie PJ. Type IV secretion: the Agrobacterium VirB/D4 and related conjugation systems. Biochim Biophys Acta. 2004;1694:219–234. doi: 10.1016/j.bbamcr.2004.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depuydt M, Messens J, Collet JF. How proteins form disulfide bonds. Antioxid Redox Signal. 2011;15:49–66. doi: 10.1089/ars.2010.3575. [DOI] [PubMed] [Google Scholar]
- Tinsley CR, Bille E, Nassif X. Bacteriophages and pathogenicity: more than just providing a toxin? Microbes Infect. 2006;8:1365–1371. doi: 10.1016/j.micinf.2005.12.013. [DOI] [PubMed] [Google Scholar]
- Lang AS, Beatty JT. Importance of widespread gene transfer agent genes in alpha-proteobacteria. Trends Microbiol. 2007;15:54–62. doi: 10.1016/j.tim.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Sekine Y, Eisaki N, Ohtsubo E. Translational control in production of transposase and in transposition of insertion sequence IS3. J Mol Biol. 1994;235(5):1406–1420. doi: 10.1006/jmbi.1994.1097. [DOI] [PubMed] [Google Scholar]
- Galardini M, Pini F, Bazzicalupo M, Biondi EG, Mengoni A. Replicon-dependent bacterial genome evolution: the case of Sinorhizobium meliloti. Genome Biol Evol. 2013;5:542–558. doi: 10.1093/gbe/evt027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazur A, Stasiak G, Wielbo J, Koper P, Kubik-Komar A, Skorupska A. Phenotype profiling of Rhizobium leguminosarum bv. trifolii clover nodule isolates reveal their both versatile and specialized metabolic capabilities. Arch Microbiol. 2013;195:255–267. doi: 10.1007/s00203-013-0874-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper VS, Vohr SH, Wrocklage SC, Hatcher PJ. Why genes evolve faster on secondary chromosomes in bacteria. PLoS Comput Biol. 2010;6:e1000732. doi: 10.1371/journal.pcbi.1000732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow JD, Cooper VS. Evolutionary effects of translocations in bacterial genomes. Genome Biol Evol. 2012;4:1256–1262. doi: 10.1093/gbe/evs099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazur A, Stasiak G, Wielbo J, Kubik-Komar A, Marek-Kozaczuk M, Skorupska A. Intragenomic diversity of Rhizobium leguminosarum bv. trifolii clover nodule isolates. BMC Microbiol. 2011;11:123. doi: 10.1186/1471-2180-11-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Val ME, Skovgaard O, Ducos-Galand M, Bland MJ, Mazel D. Genome engineering in Vibrio cholerae: a feasible approach to address biological issues. PLoS Genet. 2012;8(1):e1002472. doi: 10.1371/journal.pgen.1002472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen J, Frank O, Goker M, Pradella S. Extrachromosomal, extraordinary and essential–the plasmids of the Roseobacter clade. Appl Microbiol Biotechnol. 2013;97:2805–2815. doi: 10.1007/s00253-013-4746-8. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Russell DW. Molecular Cloning: a laboratory manual. New York: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- Noel KD, Sanchez A, Fernandez L, Leemans J, Cevallos MA. Rhizobium phaseoli symbiotic mutants with transposon Tn5 insertions. J Bacteriol. 1984;158(1):148–155. doi: 10.1128/jb.158.1.148-155.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood AP, Kelly DP. Heterotrophic growth of Thiobacillus A2 on sugars and organic acids. Arch Microbiol. 1977;113(3):257–264. doi: 10.1007/BF00492033. [DOI] [PubMed] [Google Scholar]
- Wheatcroft R, McRae GD, Miller RW. Changes in the Rhizobium meliloti genome and the ability to detect supercoiled plasmids during bacteroid development. Mol Plant Microbe Interact. 1990;3:9–17. doi: 10.1094/MPMI-3-009. [DOI] [Google Scholar]
- Kushner SR. In: Genetic Engineering. Boyer HB, Nicosia S, editor. Amsterdam: Elsevier/North-Holland; 1978. An improved method for transformation of E. coli with ColE1 derived plasmids; pp. 17–23. [Google Scholar]
- Bartosik D, Szymanik M, Wysocka E. Identification of the partitioning site within the repABC-type replicon of the composite Paracoccus versutus plasmid pTAV1. J Bacteriol. 2001;183(21):6234–6243. doi: 10.1128/JB.183.21.6234-6243.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackermann HW. Basic phage electron microscopy. Methods Mol Biol. 2009;501:113–126. doi: 10.1007/978-1-60327-164-6_12. [DOI] [PubMed] [Google Scholar]
- Meyer F, Goesmann A, McHardy AC, Bartels D, Bekel T, Clausen J, Kalinowski J, Linke B, Rupp O, Giegerich R. et al. GenDB - an open source genome annotation system for prokaryote genomes. Nucleic Acids Res. 2003;31:2187–2195. doi: 10.1093/nar/gkg312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claudel-Renard C, Chevalet C, Faraut T, Kahn D. Enzyme-specific profiles for genome annotation: PRIAM. Nucleic Acids Res. 2003;31:6633–6639. doi: 10.1093/nar/gkg847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN. et al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 2003;4:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siguier P, Perochon J, Lestrade L, Mahillon J, Chandler M. ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006;34:D32–D36. doi: 10.1093/nar/gkj014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver T, Berriman M, Tivey A, Patel C, Bohme U, Barrell BG, Parkhill J, Rajandream MA. Artemis and ACT: viewing, annotating and comparing sequences stored in a relational database. Bioinformatics. 2008;24:2672–2676. doi: 10.1093/bioinformatics/btn529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blom J, Albaum SP, Doppmeier D, Puhler A, Vorholter FJ, Zakrzewski M, Goesmann A. EDGAR: a software framework for the comparative analysis of prokaryotic genomes. BMC Bioinformatics. 2009;10:154. doi: 10.1186/1471-2105-10-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talavera G, Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 2007;56(4):564–577. doi: 10.1080/10635150701472164. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. PHYLIP - phylogeny inference package (version 3.2) Cladistics. 1989;5:164–166. [Google Scholar]
- Mahadevan P, Seto D. Rapid pair-wise synteny analysis of large bacterial genomes using web-based GeneOrder4.0. BMC Res Notes. 2010;3:41. doi: 10.1186/1756-0500-3-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
COG categories of the proteins of P. aminophilus JCM 7686.
Diversity and distribution of 217 transcriptional regulators of P. aminophilus JCM 7686.
Two-component systems and histidine kinases encoded by the P. aminophilus JCM 7686 genome.
Toxin-antitoxin systems encoded by the P. aminophilus JCM 7686 genome.
Chaperones and co-chaperonins encoded by the P. aminophilus JCM 7686 genome.
Genes encoding DNA repair related proteins within the P. aminophilus JCM 7686 genome.
Restriction patterns of P. aminophilus JCM 7686 genomic DNA cleaved with selected restriction endonucleases showing the protection of GANTC sites by the CcrM methylase (JCM7685_3079). ND - undigested DNA. M – GeneRuler 100–10,000 bp size marker.
Summary of the sensitivity of various restriction endonucleases to DNA modifications introduced by the JCM7686_0772 and JCM7686_2655 proteins (m 5 C MTases).
Summary of the sensitivity of various restriction endonucleases to DNA modifications introduced by the JCM7686_0815 protein (m 4 C MTase).
Summary of the sensitivity of various restriction endonucleases to DNA modifications introduced by the JCM7686_1231, JCM7686_2255 and JCM7686_2934 proteins (m 6 A MTases).
The number of homologous proteins encoded by JCM 7686 plasmids and P. denitrificans PD1222 chromosomes. The analysis was performed using the GeneOrder 4.0 tool. Proteins were considered homologous only if the BLAT threshold scores were >200. The results were verified manually by BLAST comparisons.
Plasmid profiles of the JCM 7686 wild-type strain and its derivatives deprived of particular megasized-replicons.
The growth mode of wild-type JCM 7686 and the pAMI1-less derivative in liquid LB medium.
Host ranges of P. aminophilus JCM 7686 pAMI1-8 replicons.
Schematic diagram of de novo NAD biosynthesis from aspartate to nicotinic acid mononucleotide (NaMN).
Transmission electron micrograph of tailed bacteriophage ϕPam-6 of P. aminophilus JCM 7686.
Transposase genes within the P. aminophilus JCM 7686 genome.
Bacterial strains used in this study.
Plasmids used and constructed in this study.
Oligonucleotide primers used in this study.