

Abstract
A major challenge in current systems biology is the combination and integrative analysis of large data sets obtained from different high-throughput omics platforms, such as mass spectrometry based Metabolomics and Proteomics or DNA microarray or RNA-seq-based Transcriptomics. Especially in the case of non-targeted Metabolomics experiments, where it is often impossible to unambiguously map ion features from mass spectrometry analysis to metabolites, the integration of more reliable omics technologies is highly desirable. A popular method for the knowledge-based interpretation of single data sets is the (Gene) Set Enrichment Analysis. In order to combine the results from different analyses, we introduce a methodical framework for the meta-analysis of p-values obtained from Pathway Enrichment Analysis (Set Enrichment Analysis based on pathways) of multiple dependent or independent data sets from different omics platforms. For dependent data sets, e.g. obtained from the same biological samples, the framework utilizes a covariance estimation procedure based on the nonsignificant pathways in single data set enrichment analysis. The framework is evaluated and applied in the joint analysis of Metabolomics mass spectrometry and Transcriptomics DNA microarray data in the context of plant wounding. In extensive studies of simulated data set dependence, the introduced correlation could be fully reconstructed by means of the covariance estimation based on pathway enrichment. By restricting the range of p-values of pathways considered in the estimation, the overestimation of correlation, which is introduced by the significant pathways, could be reduced. When applying the proposed methods to the real data sets, the meta-analysis was shown not only to be a powerful tool to investigate the correlation between different data sets and summarize the results of multiple analyses but also to distinguish experiment-specific key pathways.
Introduction
High-throughput omics platforms, such as mass spectrometry (MS) based Metabolomics and Proteomics or DNA microarray or RNA-seq-based Transcriptomics, allow the comprehensive analysis of an organism's reaction under different experimental conditions [1]–[5]. A current major challenge in systems biology is the combination and integrative analysis of the large data sets obtained from these platforms [6]–[8]. A single data set usually contains the intensity/expression profiles (intensities for all measured samples) of thousands of features, such as different ion species in MS or spots in DNA microarray analysis. After individual preprocessing of each data set, which includes the statistical analysis, ranking, or filtering of features according to the relevance of their profiles [9]–[11], the features have to be assigned to known biological entities [12], such as metabolites, genes, or proteins. Especially in MS-based Metabolomics, a major bottleneck is the identification of metabolites in non-targeted experiments [13]. In many applications, the putative monoisotopic masses of measured ion species cannot unambiguously mapped to metabolite entries in public databases. The integration of data from other omics platforms which provide a more reliable mapping, such as DNA microarrays, can significantly support the metabolite identification in this case. After annotation, the results are usually interpreted in the context of current knowledge, e.g. known biochemical pathways or processes [14]–[16]. A popular method for this knowledge-based interpretation of single data sets is the Gene Set Enrichment Analysis [17] or Overrepresentation Analysis [18], [19]. Many similar approaches have been developend and the methodology was transferred to other omics platforms [20]–[23]. In general, the enrichment analysis is based on sets of entities, e.g. pathways with associated metabolites, and results in a list of relevant sets which are enriched in high-ranking features (in comparison to all features in the data set). In most methods, the enrichment level of a single set is expressed as p-value. Modelling metabolic pathways as well-defined sets of biological entities, e.g. metabolites, enzymes, and corresponding genes, has shown to be a powerful approach to interpreting complex omics data sets. Furthermore, the concept of pathways associated with different types of biological entities facilitates the joint analysis of different data sets [24].
The combination of results from different studies sharing the same experimental design in terms of null and alternative hypothesis (meta-analysis) is a central task in various statistical applications [25]–[27]. In case of the combination of independent p-values, Fisher's method [28] or Stouffer's method [29], also known as normal, Z-method, or Z-transform test, are often applied. For dependent p-values and known covariances, in [30] an extended version of Fisher's method was proposed (Brown's method). In order to increase statistical power, meta-analysis has been applied to Pathway Enrichment Analysis (Set Enrichment Analysis utilizing pathways as sets) in the context of cancer studies [31]. The proposed methods were focused on the combination of independent p-values based on DNA microarray data. In contrast, we introduce a general methodical framework for the meta-analysis of multiple dependent or independent data sets resulting from different omics platforms applied to Pathway Enrichment Analysis. In order to cope with dependent data sets, such as obtained from the same biological samples analyzed by MS in negative and positive ionization mode, the framework utilizes a covariance estimation procedure based on the nonsignificant pathways in single data set enrichment analysis. The framework is applied and evaluated on two Metabolomics MS data sets [32] and two Transcriptomics DNA microarray studies [11] in the context of wounding of Arabidopsis thaliana. The main focus of this exemplary meta-analysis lies on the enhancement of MS based Metabolomics results by means of the microarray studies.
Materials and Methods
Data sets and preprocessing
For application and evaluation of the meta-analysis, two Metabolomics MS data sets (M1 and M2) [11] and two Transcriptomics DNA microarray data sets (T1 and T2) [32] were used (see Table 1 and Dataset S1 for details). All studies investigate the wounding of Arabidopsis thaliana wild type and the jasmonate-deficient dde 2-2 mutant plants [33], the experimental designs comprise conditions for control plants as well as plants harvested at different times after wounding (see Table 1). The two Metabolomics data sets derive from an Ultra Performance Liquid Chromatography (UPLC) analysis coupled to a Time-Of-Flight (TOF) MS detection. With this method, the non-polar extraction phase of one set of samples was analyzed in positive and negative ionization mode. Since some metabolites may have been measured in both ionization modes following different (partially unknown) ionization rules [34], the level of dependence between both data sets is not clear. In case of the MS data sets, a single feature corresponds to a particular ion species, which is characterized by an exact mass-to-charge ratio and a retention time. A single metabolite may be represented by multiple features, e.g. corresponding to different adduct formations and isotopologues. The features in the microarray data sets correspond to different spots on the array containing DNA probes that match a particular sequence. Also in this case, a single transcript may be represented by multiple features corresponding to particular sequences of the respective gene. The feature profiles of all data sets were ranked separately utilizing a signal-to-noise ratio (similar to the method described in [9], see TechnicalDescription S1).
Table 1. Overview on data sets.
| Label | Number of features | Times | Platform | Ionization mode | Reference |
| M1 | 24796 | 0.5 h, 2 h, 5 h | Mass spectrometry | negative | [11] |
| M2 | 23325 | 0.5 h, 2 h, 5 h | Mass spectrometry | positive | [11] |
| T1 | 25392 | 1 h | DNA microarray | - | [32], E-ATMX-9 |
| T2 | 25392 | 3 h | DNA microarray | - | E-MEXP-1475 |
The table gives an overview on the four data sets used for evaluation and application. The third column (Times) summarizes the different points in time when the wounded plants were harvested in the respective experiment. The T1 and T2 data sets can be obtained from the ArrayExpress [44] website.
Pathway enrichment analysis
The ranked features were mapped to the pathway entries in AraCyc [35] and the Arabidopsis-specific pathways in the Kyoto Encyclopedia of Genes and Genomes (KEGG) database [14] (see TechnicalDescription S1). In case of the Metabolomics MS data sets, all potential monoisotopic masses were calculated per feature based on the ionization rules and number of isotopes used in [11] and mapped to the metabolite masses in the databases. In case of the Transcriptomics DNA microarray data, the features were mapped to the A. thaliana genes utilizing their CATMA IDs [36]. Based on the mappings, a set of feature ranks was extracted for each pathway and data set. In order to test for an over-representation of high-ranked features, a p-value was calculated for each set of ranks (pathway) utilizing a one-sided Kolmogorov-Smirnov (KS) or Wilcoxon rank-sum test (also known as Mann-Whitney U test) [21]. In case of the KS test, the empirical distribution of ranks in a given set is compared to the distribution of ranks in the respective data set. In case of the rank-sum test, the sum of feature ranks within a given set is evaluated. Especially for Gene Set Enrichment Analysis of DNA microarrays, many methods have been published [20]. Most of these methods are based on KS-like or average gene-specific statistics. For a general meta-analysis and in order to combine the Metabolomics and Transcriptomics data sets in a robust way, we decided to utilize the rank-based KS and rank-sum test. However, more specialized methods for the pathway-specific p-value calculation may be employed as well. The resulting p-values for the dependent Metabolomics data sets were used for the covariance estimation (see corresponding section). The covariances between both Transcriptomics data sets and between the Metabolomics and Transcriptomics data sets, which were obtained from independent biological samples, were set to zero.
Meta-analysis of p-values
In statistical meta-analysis, the most common methods for combining independent p-values from related tests are Fisher's [28] and Stouffer's method [29]. In Fisher's method, the meta-p-value is calculated based on a chi-squared distribution (see TechnicalDescription S1). In Stouffer's method, the test statistic is the sum of p-values transformed into normally distributed random variables (standard normal deviates). For dependent p-values, a powerful approach is Brown's method [30], which is an extension of Fisher's method based on a scaled chi-squared distribution and modified degrees of freedom utilizing a known covariance matrix for standard normal deviates. The given p-values can be transformed into standard normal deviates by means of the inverse cumulative distribution function of the standard normal distribution. The covariance matrix of the standard normal deviates can also be utilized in order to extend Stouffer's method to dependent p-values.
Estimation of covariances
In most applications with dependent data sets, the covariance matrix is not known and has to be estimated. In our proposed procedure, the pairwise covariance between two data sets is estimated based on the standard normal deviates of the pathway-specific p-values, which were obtained for each single data set in Pathway Enrichment Analysis. This estimation is expected to be biased by the alternative hypothesis since the similar or same experimental setup of the data sets imposes a certain dependence and significant pathways associated with very low p-values will strongly influence the results. In order to minimize this bias in the estimation of the pairwise covariance between two data sets, a parameter  is introduced and only pathways with p-values in the range
is introduced and only pathways with p-values in the range 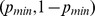 are considered. This procedure leaves out significant pathways for which the null hypothesis is likely to be rejected for at least one of the data sets. Instead of directly estimating the sample covariance of the transformed p-values in this range (which would again be biased because of the range restriction), Pearson's correlation coefficient is used.
are considered. This procedure leaves out significant pathways for which the null hypothesis is likely to be rejected for at least one of the data sets. Instead of directly estimating the sample covariance of the transformed p-values in this range (which would again be biased because of the range restriction), Pearson's correlation coefficient is used.
Results
The Pathway Enrichment Analysis, the transformation of pathway-specific p-values into standard normal deviates, the estimation of covariances for dependent data sets, and the meta-analysis based on the previous results were applied and evaluated on the four Metabolomics/Transcriptomics data sets (see previous section). First, in order to check the distribution of transformed p-values, the histograms of the standard normal deviates were inspected. Because of significant pathways which are highly relevant in this context, the p-values are expected to be not fully uniformly distributed, which may result in a distribution of transformed p-values that deviates from the standard normal distribution. In this case, the p-values/normal deviates should be corrected for significance analysis. Second, the performance of the introduced method in reconstructing simulated data set correlations was evaluated for different 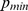 values. This performance was not clear, since the proposed correlation estimation includes several complex steps, such as the mapping of a proportion of feature ranks to pathways of different size, the calculation and restriction of p-values, and the transformation into normal deviates. Additionally, the
values. This performance was not clear, since the proposed correlation estimation includes several complex steps, such as the mapping of a proportion of feature ranks to pathways of different size, the calculation and restriction of p-values, and the transformation into normal deviates. Additionally, the  parameter might have a strong influence on the results. Therefore, another objective of the simulation studies was the identification of an appropriate parameter value for the real data sets. Third, the correlation estimation and meta-analysis were applied to all four real data sets. All data sets, containing the annotation information from the pathway mapping, and the results from Pathway Enrichment Analysis are available as comma-separated-values files (see Dataset S1 and Table S1). The source code of functions for the meta-analysis of p-values can be found in File S1.
parameter might have a strong influence on the results. Therefore, another objective of the simulation studies was the identification of an appropriate parameter value for the real data sets. Third, the correlation estimation and meta-analysis were applied to all four real data sets. All data sets, containing the annotation information from the pathway mapping, and the results from Pathway Enrichment Analysis are available as comma-separated-values files (see Dataset S1 and Table S1). The source code of functions for the meta-analysis of p-values can be found in File S1.
Distribution of standard normal deviates
Figure 1 shows the histograms of the transformed p-values (standard normal deviates) from Pathway Enrichment Analysis for the two Metabolomics and two Transcriptomics data sets within the p-value range 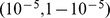 . The histograms for the KS and the rank-sum test are similar and confirm the normal-like distribution of deviates. In both cases however, the sample standard deviation is higher than the unit standard deviation used for the transformation. Additionally, the sample mean for the combined Transcriptomics data sets is smaller than zero. This difference may be caused by pathways which are directly or indirectly influenced by the experimental setup. Although the highly significant pathways with p-values below the threshold
. The histograms for the KS and the rank-sum test are similar and confirm the normal-like distribution of deviates. In both cases however, the sample standard deviation is higher than the unit standard deviation used for the transformation. Additionally, the sample mean for the combined Transcriptomics data sets is smaller than zero. This difference may be caused by pathways which are directly or indirectly influenced by the experimental setup. Although the highly significant pathways with p-values below the threshold 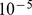 were left out, many other pathways are expected to be indirectly affected by the wounding process. Another explanation would be the dependence of feature ranks used for p-value calculation, e.g. introduced by the dependence of different microarray spots representing the same gene or by gene-gene correlations [17]. In order to eliminate the observed bias, the p-values were restandardized [37] for significance analysis by means of the sample mean and sample standard deviation of observed normal deviates per data set and retransforming of the standardized deviates into corrected p-values. This is a conservative correction because the observed bias also includes the pathways which are directly influenced by the wounding process.
were left out, many other pathways are expected to be indirectly affected by the wounding process. Another explanation would be the dependence of feature ranks used for p-value calculation, e.g. introduced by the dependence of different microarray spots representing the same gene or by gene-gene correlations [17]. In order to eliminate the observed bias, the p-values were restandardized [37] for significance analysis by means of the sample mean and sample standard deviation of observed normal deviates per data set and retransforming of the standardized deviates into corrected p-values. This is a conservative correction because the observed bias also includes the pathways which are directly influenced by the wounding process.
Figure 1. Histograms of standard normal deviates for the Metabolomics and Transcriptomics data sets.

For the p-value calculation, the Kolmogorov-Smirnov (KS) and rank-sum tests were utilized. The p-values were restricted to the range 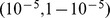 . The red graph represents the expected density assuming the standard normal distribution. The green graph shows the expected density assuming a normal distribution with the sample mean and standard deviation as parameters. The histograms for both tests are similar and confirm the normal-like distribution of deviates. In both cases however, the sample standard deviation is higher than the unit standard deviation used for the transformation. Additionally, the sample mean for the combined Transcriptomics data sets is smaller than zero.
. The red graph represents the expected density assuming the standard normal distribution. The green graph shows the expected density assuming a normal distribution with the sample mean and standard deviation as parameters. The histograms for both tests are similar and confirm the normal-like distribution of deviates. In both cases however, the sample standard deviation is higher than the unit standard deviation used for the transformation. Additionally, the sample mean for the combined Transcriptomics data sets is smaller than zero.
Estimation of data set correlation
In simulated studies (see TechnicalDescription S1 for details), the correlation estimation was evaluated by calculating the pairwise Pearson correlation coefficients between all four data sets and a copy of the respective data set with different percentages of feature ranks randomly permuted. For each original and permuted data set, the p-values were calculated for all pathways using the KS or rank-sum test. The correlation coefficient between each original and permuted data set was computed based on the respective standard normal deviates (not restandardized) and the restriction of p-values utilizing different parameter values 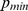 . As measurement of the introduced artificial correlation, the correlation coefficient between the feature ranks of each data set and the permuted ranks (feature rank correlation) was calculated and averaged, respectively. The whole procedure was repeated for negative correlation by randomly permuting a percentage of the inverted original feature ranks per data set.
. As measurement of the introduced artificial correlation, the correlation coefficient between the feature ranks of each data set and the permuted ranks (feature rank correlation) was calculated and averaged, respectively. The whole procedure was repeated for negative correlation by randomly permuting a percentage of the inverted original feature ranks per data set.
Table S2 shows the average results over all data sets in detail. Figure 2 and 3 summarize the differences between the reconstructed correlation coefficients from pathway enrichment and the introduced positive or negative feature rank correlation. In comparison to the average feature rank correlation coefficients (x-axis), the absolute correlation is overestimated for low 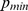 values and underestimated for high values. A
values and underestimated for high values. A 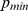 value of 0.01 results in the best reconstruction of data set correlation, the absolute difference between the correlation coefficients from pathway enrichment and the feature rank correlation is close to zero for both tests. In case of the observed overestimation for low
value of 0.01 results in the best reconstruction of data set correlation, the absolute difference between the correlation coefficients from pathway enrichment and the feature rank correlation is close to zero for both tests. In case of the observed overestimation for low 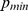 values, the relevant pathways, which are associated with many top-ranking features, are assigned a low p-value, even when randomly permuting some of the features, and have a high influence on the correlation estimation. In case of the underestimation for high
values, the relevant pathways, which are associated with many top-ranking features, are assigned a low p-value, even when randomly permuting some of the features, and have a high influence on the correlation estimation. In case of the underestimation for high 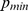 values, the introduced correlation over all features and pathways cannot be fully recovered when restricting the range of p-values and number of utilized pathways too much.
values, the introduced correlation over all features and pathways cannot be fully recovered when restricting the range of p-values and number of utilized pathways too much.
Figure 2. Differences between the reconstructed correlation coefficients from pathway enrichment and the introduced positive feature correlation.

The differences were calculated for different 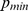 values and the Kolmogorov-Smirnov (KS) and rank-sum test. The best reconstruction, corresponding to differences near zero, can be observed for
values and the Kolmogorov-Smirnov (KS) and rank-sum test. The best reconstruction, corresponding to differences near zero, can be observed for 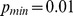 .
.
Figure 3. Differences between the reconstructed correlation coefficients from pathway enrichment and the introduced negative feature correlation.

The differences were calculated for different 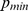 values and the Kolmogorov-Smirnov (KS) and rank-sum test. The best reconstruction, corresponding to differences near zero, can be observed for
values and the Kolmogorov-Smirnov (KS) and rank-sum test. The best reconstruction, corresponding to differences near zero, can be observed for 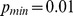 . The KS test is not able to fully reconstruct strong negative feature correlations.
. The KS test is not able to fully reconstruct strong negative feature correlations.
For the KS test and small negative feature rank correlations, the estimated coefficients from enrichment are considerably larger, e.g. showing a difference between 0.2 and 0.4 in case of a feature rank correlation of −1 (see Figure 3). This can be explained by the non-symmetric properties of the one-sided KS test. A set enriched in both high-ranking and low-ranking features would receive a low p-value when performing the one-sided KS test on the original as well as the inverted ranks. The rank-sum test, on the contrary, would result in an average p-value in both cases because the sum of ranks in the set is near the expected value. For a 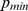 value of 0.01 and negative correlation, the KS test is still able to reconstruct feature rank correlation coefficients between 0 and −0.3 with a difference near zero.
value of 0.01 and negative correlation, the KS test is still able to reconstruct feature rank correlation coefficients between 0 and −0.3 with a difference near zero.
For the correlation estimation between the two dependent Metabolomics data sets, a 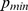 value of 0.01, which showed the best reconstruction in the simulations, was utilized. The estimation resulted in relatively small coefficients, 0.12 (KS test) and 0.08 (rank-sum test).
value of 0.01, which showed the best reconstruction in the simulations, was utilized. The estimation resulted in relatively small coefficients, 0.12 (KS test) and 0.08 (rank-sum test).
Meta-analysis of pathway enrichment
Tables 2 and 3 show the results from meta-analysis of pathway enrichment utilizing Brown's and Stouffer's extended method integrating the correlation estimation for the Metabolomics data sets. The pathways are sorted according to the False Discovery Rate (FDR) [38] calculated based on the meta-p-values. Pathways with more than 500 associated entries were left out in this analysis for better interpretability. For both methods, the top-ranked pathways are the “alpha-Linolenic acid metabolism” (KEGG, 214 feature hits), the “jasmonic acid biosynthesis” (AraCyc, 176 feature hits), and the “lycolipid desaturation”(AraCyc, 325 feature hits). These pathways specifically describe parts of the biosynthesis of the well-known wound hormone jasmonate [39]. The first two pathways cover all biosynthetic steps from the fatty acid alpha-linolenic acid to jasmonic acid. The first committed step is catalyzed by the allene oxide synthase (AOS), whose gene is mutated in the dde 2-2 mutant plants [33]. The glycolipid desaturation pathway describes the formation of the alpha-linolenic acid via sequential steps of glycolipid-linked desaturation. The FDRs for these key pathways are much lower compared to the following pathways. Tables 4, 5, 6, 7, 8, 9, 10, and 11 show the results from enrichment analysis of the four single data sets and selected mappings of top-ranked features which were assigned to entries in the three key pathways, respectively. The enrichment analysis of the M1 data set (negative ionization mode, see Table 4) provides a major contribution to the results from meta-analysis. The first two pathways are also top-ranked but associated with much higher FDRs. The high-ranked features associated with jasmonic acid and its precursor metabolites, such as OPDA and OPC-8:0, are mainly responsible for this ranking (see Table 5). However, the mapping of putative monoisotopic feature masses to metabolites is error-prone and ambiguous. For example, OPDA, EOTrE, and a couple of other metabolites provided by KEGG and AraCyc share the same sum formula and single ion features cannot be unambiguously assigned without further information. In contrast to the alpha-linolenic acid metabolism pathway (KEGG), the very similar jasmonic acid biosynthesis pathway (AraCyc) is associated with a much higher FDR. This can be explained by a number of additional entries found only in the AraCyc version of the pathway and representing general substrates, such as acetyl-CoA, intermediate products which could not be measured with a high signal-to-noise ratio, such as OPC6-3-hydroxyacyl-CoA, or other side products. The glycolipid desaturation pathway, which can be found at position seven, is associated with a very high FDR. Most of the glycolipid species show higher intensities and signal-to-noise ratios in positive compared to negative ionization mode, which results in a very low FDR in pathway enrichment analysis of the M2 data set (see Tables 6 and 7). In contrast, jasmonate and many direct precursor metabolites cannot be measured in positive ionization mode with sufficient intensity, which explains the less prominent ranking of the alpha-linolenic acid metabolism (rank 12) and jasmonic acid biosynthesis (rank 13). Nonetheless, metabolites such as OPDA can be measured in both ionization modes with high signal-to-noise ratio and these findings confirm the corresponding pathways in meta-analysis. Integrating the Transcriptomics data sets T1 and T2 results in a much more comprehensive data interpretation (see Tables 9 and 11). Figure 4 exemplarily shows the pathway map of the alpha-linolenic acid metabolism with marked entries matched by high-ranking features from all data sets. In this combination, the ambiguous mapping of the MS data is supported by unambiguously matching transcripts. Almost all of the transcripts corresponding to enzymes in the alpha-linolenic acid metabolism can be found in the T1 and T2 data sets with relatively high signal-to-noise ratios. This results in much lower FDRs for the jasmonate-specific pathways in meta-analysis compared to the results from single Metabolomics data set analysis. Also in the analysis of the single Transcriptomics data sets (see Tables 8 and 10), these two pathways are associated with relatively high FDRs. In case of the T1 data set, both pathways can be found at less prominent positions (rank 14 and 23, see Table 8). For both Transcriptomics data sets, the glycolipid desaturation is ranked in the middle of all pathways (rank 420 and 161). Only a small number of transcripts associated with fatty acid desaturase show a high signal-to-noise ratio (see Tables 9 and 11).
Table 2. Results from meta-analysis of pathway enrichment (Brown's method).
| Rank | DB | Pathway | Hits | KS | Rank-sum |
| 1 | KEGG | alpha-Linolenic acid metabolism | 214 | 0.0001321 | 2.383e-05 |
| 2 | AraCyc | jasmonic acid biosynthesis | 176 | 0.003676 | 0.0003101 |
| 3 | AraCyc | glycolipid desaturation | 325 | 0.0007046 | 0.0102 |
| 4 | KEGG | Linoleic acid metabolism | 147 | 0.5252 | 0.3968 |
| 5 | AraCyc | superpathway of phenylalanine, tyrosine and tryptophan biosynthesis | 86 | 0.5364 | 0.5253 |
| 6 | AraCyc | traumatin and (Z)-3-hexen-1-yl acetate biosynthesis | 131 | 0.4538 | 0.5557 |
| 7 | KEGG | 2-Oxocarboxylic acid metabolism | 578 | 0.5252 | 0.767 |
| 8 | AraCyc | glucosinolate biosynthesis from dihomomethionine | 153 | 0.5364 | 0.7857 |
| 9 | KEGG | Starch and sucrose metabolism | 335 | 0.5252 | 0.7857 |
| 10 | KEGG | Proteasome | 114 | 0.5252 | 0.7857 |
The table contains the high-ranking pathways from meta-analysis of pathway enrichment (Brown's method) based on the Kolmogorov-Smirnov (KS) and rank-sum test utilizing all data sets. The p-values per data set were restandardized. The pathways are sorted according to the meta-p-values derived from the rank-sum test. The second column (DB) contains the name of the source database, the fourth column (Hits) the number of feature assignments. The last two columns comprise the false discovery rates calculated from the meta-p-values.
Table 3. Results from meta-analysis of pathway enrichment (Stouffer's extended method).
| Rank | DB | Pathway | Hits | KS | Rank-sum |
| 1 | KEGG | alpha-Linolenic acid metabolism | 214 | 6.708e-05 | 1.127e-05 |
| 2 | AraCyc | jasmonic acid biosynthesis | 176 | 0.001774 | 7.328e-05 |
| 3 | AraCyc | glycolipid desaturation | 325 | 0.02043 | 0.2122 |
| 4 | AraCyc | traumatin and (Z)-3-hexen-1-yl acetate biosynthesis | 131 | 0.4545 | 0.4326 |
| 5 | AraCyc | superpathway of phenylalanine, tyrosine and tryptophan biosynthesis | 86 | 0.4545 | 0.4326 |
| 6 | KEGG | Linoleic acid metabolism | 147 | 0.7866 | 0.499 |
| 7 | AraCyc | glucosinolate biosynthesis from dihomomethionine | 153 | 0.4545 | 0.6282 |
| 8 | AraCyc | glucosinolate biosynthesis from tryptophan | 132 | 0.4545 | 0.6583 |
| 9 | AraCyc | glucosinolate biosynthesis from phenylalanine | 115 | 0.6522 | 0.6583 |
| 10 | AraCyc | glucosinolate biosynthesis from tetrahomomethionine | 114 | 0.4545 | 0.6583 |
The table contains the high-ranking pathways from meta-analysis of pathway enrichment (Stouffer's extended method) based on the Kolmogorov-Smirnov (KS) and rank-sum test utilizing all data sets. The last two columns comprise the false discovery rates calculated from the meta-p-values.
Table 4. Results from pathway enrichment analysis of data set M1.
| Rank | DB | Pathway | Hits | KS | Rank-sum |
| 1 | KEGG | alpha-Linolenic acid metabolism | 65 | 0.02084 | 0.03717 |
| 2 | AraCyc | jasmonic acid biosynthesis | 68 | 0.1531 | 0.1503 |
| 3 | KEGG | Linoleic acid metabolism | 43 | 0.4598 | 0.8524 |
| 4 | AraCyc | indole-3-acetyl-amino acid biosynthesis | 29 | 0.4598 | 0.8524 |
| 5 | AraCyc | traumatin and (Z)-3-hexen-1-yl acetate biosynthesis | 38 | 0.4598 | 0.8524 |
| 6 | AraCyc | galactosylcyclitol biosynthesis | 14 | 0.4598 | 0.8524 |
| 7 | AraCyc | glycolipid desaturation | 144 | 0.4598 | 0.8524 |
| 8 | KEGG | Porphyrin and chlorophyll metabolism | 222 | 0.8841 | 0.8524 |
| 9 | AraCyc | poly-hydroxy fatty acids biosynthesis | 59 | 0.9248 | 0.8524 |
| 10 | KEGG | Lysine degradation | 46 | 0.4598 | 0.8524 |
The table contains the high-ranking pathways from enrichment analysis of data set M1 based on the Kolmogorov-Smirnov (KS) and rank-sum test. The pathways are sorted according to the restandardized p-values derived from the rank-sum test. The last two columns comprise the false discovery rates calculated from the restandardized p-values.
Table 5. Selected feature mappings from data set M1.
| Rank | rt | m/z | Mappings |
| 1 | 0.73 | 255.1218 | Jasmonic acid |
| 3 | 0.73 | 209.1168 | Jasmonic acid |
| 7 | 0.73 | 256.1264 | Jasmonic acid |
| 8 | 2.08 | 337.1999 | OPDA, EOTrE |
| 11 | 2.08 | 338.2044 | OPDA, EOTrE |
| 321 | 5.66 | 986.6145 | 18:3/18:1-DGD, 18:2/18:2-DGD |
| 324 | 5.78 | 822.5428 | 18:2/16:0-MGD, 18:1/16:1-MGD |
| 410 | 5.53 | 820.5295 | 18:3/16:0-MGD, 18:2/16:1-MGD, 18:1/16:2-MGD |
| 447 | 2.33 | 339.2155 | OPC-8:0 |
| 540 | 5.64 | 960.5985 | 18:3/16:0-DGD |
| 542 | 6.02 | 823.5541 | 18:1/16:0-MGD, 18:0/16:1-MGD |
| 554 | 5.67 | 858.5064 | 18:3/18:3-MGD |
| 563 | 5.74 | 795.5232 | 18:3/18:1-MGD, 18:2/18:2-MGD |
| 650 | 6.18 | 939.5986 | 18:2/18:3-DGD |
| 846 | 5.89 | 962.613 | 18:2/16:0-DGD |
| 879 | 6.23 | 859.5155 | 18:2/18:3-MGD |
| 899 | 1.86 | 309.2055 | HpOTrE |
| 1445 | 6.17 | 964.6258 | 18:1/16:0-DGD |
| 1727 | 7.53 | 278.2245 | Linolenic acid |
| 2142 | 0.52 | 239.0895 | 9-Oxononanoic acid |
The table shows selected mappings of features from data set M1 (24796 features) to entries in the first three pathways in tables 2 and 3. The first column contains the feature rank. The second and third column show the corresponding retention times and mass-to-charge ratios. Multiple mappings correspond to different ionization rules or isotopologues.
Table 6. Results from pathway enrichment analysis of data set M2.
| Rank | DB | Pathway | Hits | KS | Rank-sum |
| 1 | AraCyc | glycolipid desaturation | 167 | 0.0009173 | 0.002862 |
| 2 | AraCyc | antheraxanthin and violaxanthin biosynthesis | 63 | 0.1033 | 0.2477 |
| 3 | KEGG | Carotenoid biosynthesis | 389 | 0.3608 | 0.8365 |
| 4 | AraCyc | zeaxanthin biosynthesis | 29 | 0.5251 | 0.8365 |
| 5 | AraCyc | lutein biosynthesis | 34 | 0.5251 | 0.8365 |
| 6 | AraCyc | capsanthin and capsorubin biosynthesis | 38 | 0.5251 | 0.8365 |
| 7 | AraCyc | brassinosteroids inactivation | 20 | 0.5251 | 0.8365 |
| 8 | KEGG | Porphyrin and chlorophyll metabolism | 236 | 0.8693 | 0.8365 |
| … | … | … | … | … | … |
| 12 | KEGG | alpha-Linolenic acid metabolism | 89 | 0.8693 | 0.8365 |
| 13 | AraCyc | jasmonic acid biosynthesis | 54 | 0.8693 | 0.8365 |
The table contains the high-ranking pathways from enrichment analysis of data set M2 based on the Kolmogorov-Smirnov (KS) and rank-sum test. The last two columns comprise the false discovery rates calculated from the restandardized p-values.
Table 7. Selected feature mappings from data set M2.
| Rank | rt | m/z | Mappings |
| 2 | 2.08 | 310.2377 | OPDA, EOTrE |
| 8 | 2.08 | 293.2117 | OPDA, EOTrE |
| 11 | 2.08 | 311.2422 | OPDA, EOTrE |
| 48 | 2.08 | 315.1932 | OPDA, EOTrE |
| 180 | 6.17 | 942.6175 | 18:1/16:0-DGD |
| 211 | 6.17 | 941.6124 | 18:2/18:3-DGD |
| 231 | 6.22 | 772.5912 | 18:2/16:0-MGD, 18:1/16:1-MGD |
| 248 | 5.51 | 776.5365 | 18:3/16:0-MGD, 18:2/16:1-MGD, 18:1/16:2-MGD |
| 295 | 6.00 | 960.6576 | 18:3/18:1-DGD, 18:2/18:2-DGD |
| 297 | 4.69 | 772.5034 | 18:3/16:2-MGD |
| 310 | 5.07 | 937.5843 | 18:3/18:3-DGD |
| 330 | 5.87 | 935.6452 | 18:2/16:0-DGD |
| 413 | 6.15 | 915.5996 | 16:0/18:1-DGD |
| 459 | 6.45 | 774.6054 | 18:1/16:0-MGD, 18:0/16:1-MGD |
| 507 | 5.72 | 768.56 | 18:3/16:1-MGD, 18:2/16:2-MGD, 18:1/16:3-MGD |
| 615 | 5.18 | 748.5052 | 18:3/16:3-MGD |
| 699 | 1.41 | 441.3184 | Volicitin |
Table 8. Results from pathway enrichment analysis of data set T1.
| Rank | DB | Pathway | Hits | KS | Rank-sum |
| 1 | KEGG | Glycolysis/Gluconeogenesis | 108 | 0.3527 | 0.2952 |
| 2 | KEGG | Proteasome | 57 | 0.2885 | 0.2952 |
| 3 | KEGG | Protein processing in endoplasmic reticulum | 176 | 0.2885 | 0.2952 |
| 4 | KEGG | Ribosome | 220 | 0.02489 | 0.2952 |
| 5 | KEGG | Oxidative phosphorylation | 118 | 0.2885 | 0.2952 |
| 6 | KEGG | Phenylalanine, tyrosine and tryptophan biosynthesis | 54 | 0.492 | 0.2952 |
| 7 | AraCyc | superpathway of phenylalanine, tyrosine and tryptophan biosynthesis | 43 | 0.4966 | 0.3302 |
| … | … | … | … | … | … |
| 14 | AraCyc | jasmonic acid biosynthesis | 27 | 0.8201 | 0.35 |
| … | … | … | … | … | … |
| 23 | KEGG | alpha-Linolenic acid metabolism | 30 | 0.6202 | 0.4782 |
| … | … | … | … | … | … |
| 420 | AraCyc | glycolipid desaturation | 7 | 0.976 | 0.9758 |
The table contains the high-ranking pathways from enrichment analysis of data set T1 based on the Kolmogorov-Smirnov (KS) and rank-sum test. The last two columns comprise the false discovery rates calculated from the restandardized p-values.
Table 9. Selected feature mappings from data set T1.
| Rank | ID | Mappings |
| 6 | AT2G06050 | 12-oxophytodienoate reductase 3 |
| 12 | AT3G11170 | fatty acid desaturase 7 |
| 16 | AT5G42650 | allene oxide synthase |
| 18 | AT1G17420 | lipoxygenase 3 |
| 82 | AT2G06050 | 12-oxophytodienoate reductase 3 |
| 120 | AT4G15440 | hydroperoxide lyase 1 |
| 226 | AT5G48880 | peroxisomal 3-keto-acyl-CoA thiolase 5 |
| 241 | AT2G44810 | phospholipase A1 |
| 316 | AT1G20510 | OPC-8:0 CoA ligase 1 |
| 436 | AT1G76680 | 12-oxophytodienoate reductase 1 |
| 638 | AT1G72520 | lipoxygenase 4 |
| 737 | AT4G16760 | peroxisomal acyl-coenzyme A oxidase 1 |
| 744 | AT1G17420 | lipoxygenase 3 |
| 1037 | AT3G45140 | lipoxygenase 2 |
| 1487 | AT1G13280 | allene oxide cyclase 4 |
| 2116 | AT2G06925 | phospholipase A2-ALPHA |
| 2788 | AT2G31360 | delta 9 acyl-lipid desaturase 2 |
| 3146 | AT3G15290 | 3-hydroxyacyl-CoA dehydrogenase |
| 4263 | AT5G04040 | triacylglycerol lipase SDP1 |
| 4276 | AT1G76150 | enoyl-CoA hydratase 2 |
Table 10. Results from pathway enrichment analysis of data set T2.
| Rank | DB | Pathway | Hits | KS | Rank-sum |
| 1 | KEGG | alpha-Linolenic acid metabolism | 30 | 0.5885 | 0.0794 |
| 2 | KEGG | Starch and sucrose metabolism | 142 | 0.6748 | 0.3277 |
| 3 | AraCyc | jasmonic acid biosynthesis | 27 | 0.7192 | 0.3277 |
| 4 | KEGG | Linoleic acid metabolism | 11 | 0.7192 | 0.7457 |
| 5 | AraCyc | glucosinolate biosynthesis from phenylalanine | 16 | 0.7192 | 0.7457 |
| 6 | AraCyc | glucosinolate biosynthesis from dihomomethionine | 19 | 0.7192 | 0.7457 |
| 7 | KEGG | Valine, leucine and isoleucine biosynthesis | 19 | 0.7192 | 0.7457 |
| 8 | AraCyc | glucosinolate biosynthesis from tryptophan | 21 | 0.7192 | 0.7457 |
| 9 | AraCyc | starch degradation I | 37 | 0.7192 | 0.7457 |
| … | … | … | … | … | … |
| 161 | AraCyc | glycolipid desaturation | 7 | 0.8851 | 0.9666 |
The table contains the high-ranking pathways from enrichment analysis of data set T2 based on the Kolmogorov-Smirnov (KS) and rank-sum test. The last two columns comprise the false discovery rates calculated from the restandardized p-values.
Table 11. Selected feature mappings from data set T2.
| Rank | ID | Mappings |
| 25 | AT5G42650 | allene oxide synthase |
| 104 | AT2G06050 | 12-oxophytodienoate reductase 3 |
| 355 | AT1G76680 | 12-oxophytodienoate reductase 1 |
| 376 | AT5G48880 | peroxisomal 3-keto-acyl-CoA thiolase 5 |
| 426 | AT1G17420 | lipoxygenase 3 |
| 484 | AT3G15870 | oxidoreductase |
| 631 | AT1G19640 | jasmonic acid carboxyl methyltransferase |
| 1019 | AT3G11170 | fatty acid desaturase 7 |
| 1263 | AT5G04040 | triacylglycerol lipase SDP1 |
| 1354 | AT4G16760 | peroxisomal acyl-coenzyme A oxidase 1 |
| 1371 | AT1G17420 | lipoxygenase 3 |
| 1544 | AT3G45140 | lipoxygenase 2 |
| 1812 | AT3G15850 | fatty acid desaturase 5 |
| 1940 | AT2G06925 | phospholipase A2-ALPHA |
| 2139 | AT4G30950 | fatty acid desaturase 6 |
| 2413 | AT2G06050 | 12-oxophytodienoate reductase 3 |
| 2653 | AT3G15290 | 3-hydroxyacyl-CoA dehydrogenase |
| 3022 | AT1G67560 | lipoxygenase 3 |
| 3297 | AT3G06860 | 3-hydroxyacyl-CoA dehydrogenase |
| 3383 | AT2G33150 | peroxisomal 3-keto-acyl-CoA thiolase 2 |
Figure 4. Pathway map of the alpha-linolenic acid metabolism (KEGG) with marked entries.

Entries mapped to features from all data sets are marked in gray, selected entries from Tables 5, 7, 9, and 11 are marked in red.
In case of both methods for meta-analysis, the pathways “Linoleic acid metabolism” and “traumatin and (Z)-3-hexen-1-yl acetate biosynthesis” can be found in the list of top-ten. These pathways are directly connected with the alpha-linolenic acid metabolism and affected by the AOS mutation as well [40]. However, it should be noted that the second pathway is only of limited relevance in this context because the used genotype Columbia is a natural mutant in its second enzymatic step, the fatty acid hydroperoxide lyase reaction [41]. The 2-Oxocarboxylic acid metabolism (Brown's method) and several pathways in the ranking based on Stouffer's extended method describe glucosinolate biosynthesis, the major chemical defense reaction of Arabidopsis plants upon wounding that is regulated by jasmonates [42]. Though, these pathways are associated with comparably high FDRs.
Comparing the results based on the KS and the rank-sum test, no clear trend towards lower FDRs can be observed. In case of Brown's method, the glycolipid desaturation pathway is associated with a much lower FDR for both tests. In case of Stouffer's extended method, both jasmonate-specific pathways are scored with lower FDRs.
Discussion
The meta-analysis of pathway enrichment was evaluated and applied on two Metabolomics and two Transcriptomics data sets in the context of plant wounding. The meta-analysis based on Brown's and Stouffer's extended method is able to incorporate information from different independent and dependent omics data sets and distinguish key pathways in the experimental context. The FDRs calculated based on the meta-p-values are much lower compared to the single data set analysis. Especially for the pathway analysis of non-targeted Metabolomics studies, where the identification of metabolites is a bottleneck, the integration of data from other omics platforms, such as DNA microarrays, increases the value and reliability of results. In this application, Brown's and Stouffer's extended method showed overall similar results. However, Brown's method seems to be more powerful in case of pathways which are associated with extreme p-values for only a proportion of the data sets. The glycolipid desaturation pathway for example is associated with very small p-values (KS and rank-sum test) for the M2, relatively small p-values for the M1, and much larger p-values for the T1 and T2 data sets (see Table S1). In case of Brown's method, this pathway is associated with smaller FDRs (0.0007 and 0.01) in comparison to Stouffer's method (0.02 and 0.21). In contrast, Stouffer's method seems to be more powerful in case a pathway is associated with comparably small p-values for all data sets (see alpha-linolenic acid metabolism and jasmonic acid biosynthesis pathways). The choice of method depends on the objective of the meta-analysis, e.g. focus on pathways which show a consensus for all data sets or also including pathways with significant p-values for only a single or small number of data sets [26], [43]. In the context of heterogeneous omics data sets, which contain entities that cannot be measured in all experiments, e.g. metabolites that can be ionized either in positive or negative ionization mode, and pathways that may be associated with only a small number of entries for a particular omics platform, Brown's (or Fisher's method in case of independent p-values) seems to be the better choice. In both meta-analyses, a couple of pathways related to the wounding process were detected with relatively large FDRs. In order to combine the Metabolomics and Transcriptomics data sets in a robust way, we utilized general rank-based tests and a conservative restandardization of p-values per data set. The introduced framework may also be combined with more powerful tests specialized on microarray data analysis [37]. The enrichment analysis of the single T1 and T2 data sets resulted in considerably different rankings. This is likely to be related to the different time points when the wounded plants have been harvested (one and three hours).
In the performed simulation studies, the introduced feature rank correlation could be fully reconstructed utilizing the correlation estimation from pathway enrichment. By restricting the range of p-values via the parameter 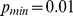 , leaving out significant pathways, the estimation bias could be reduced. The comparison of the two dependent Metabolomics data sets, which were obtained from the same biological samples analyzed in positive and negative ionization mode, resulted in relatively small positive correlation coefficients. This indicates that only a small proportion of metabolites could be detected in both ionization modes with comparable quality of intensity profiles and that data from both modes should be considered in a comprehensive analysis. In general, the statistical power of the meta-analysis increases with decreasing dependence of data sets. Therefore, nearly independent data sets are desirable.
, leaving out significant pathways, the estimation bias could be reduced. The comparison of the two dependent Metabolomics data sets, which were obtained from the same biological samples analyzed in positive and negative ionization mode, resulted in relatively small positive correlation coefficients. This indicates that only a small proportion of metabolites could be detected in both ionization modes with comparable quality of intensity profiles and that data from both modes should be considered in a comprehensive analysis. In general, the statistical power of the meta-analysis increases with decreasing dependence of data sets. Therefore, nearly independent data sets are desirable.
Comparing the one-sided KS and rank-sum test, both tests resulted in a similar distribution of normal deviates. In the simulation studies, the one-sided KS test was not able to fully reconstruct strong negative feature correlations. In most applications however, this type of data set correlation is not expected.
Supporting Information
Matlab source code for functions used in meta-analysis.
(GZ)
Data sets with database entry and pathway annotations. The archive file contains the data sets in comma separated values format. The first column contains the feature IDs, respectively. The rt and Former m/z columns (M1 and M2 data set) contain the retention times and mass-to-charge ratios from MS analysis. The raw intensities for each sample can be found in the following columns. The s/n column shows the feature-specific signal-to-noise ratios and the last columns contain the KEGG and AraCyc entries and pathways mapped to the corresponding features and separated by slash characters.
(ZIP)
Pathways with p-values and FDRs from Pathway Enrichment Analysis. The comma separated values file contains the p-values, restandardized p-values, meta-p-values, and corresponding FDRs for single data set and meta-analysis.
(CSV)
Supplementary tables for simulation studies.
(PDF)
Technical description of methods.
(PDF)
Acknowledgments
We would like to thank Helmut Grubmüller for fruitful discussions.
Funding Statement
Alexander Kaever and Manuel Landesfeind were funded by the German Federal Ministry of Education and Research (BMBF BioFung project 0315595A), and were supported by the Biomolecules program of the Göttingen Graduate School for Neurosciences, Biophysics, and Molecular Biosciences (GGNB). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Fiehn O, Kopka J, Dörmann P, Altmann T, Trethewey R, et al. (2000) Metabolite profiling for plant functional genomics. Nature Biotechnology 18: 1157–1161. [DOI] [PubMed] [Google Scholar]
- 2. Meinicke P, Lingner T, Kaever A, Feussner K, Göbel C, et al. (2008) Metabolite-based clustering and visualization of mass spectrometry data using one-dimensional self-organizing maps. Algorithms for Molecular Biology 3: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aebersold R, Mann M (2003) Mass spectrometry-based proteomics. Nature 422: 198–207. [DOI] [PubMed] [Google Scholar]
- 4. Brown PO, Botstein D (1999) Exploring the new world of the genome with DNA microarrays. Nature Genetics 21: 33–37. [DOI] [PubMed] [Google Scholar]
- 5. Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods 5: 621–628. [DOI] [PubMed] [Google Scholar]
- 6. Joyce AR, Palsson BØ (2006) The model organism as a system: integrating ‘omics’ data sets. Nature Reviews Molecular Cell Biology 7: 198–210. [DOI] [PubMed] [Google Scholar]
- 7. Weckwerth W, Wenzel K, Fiehn O (2004) Process for the integrated extraction, identification and quantification of metabolites, proteins and RNA to reveal their co-regulation in biochemical networks. Proteomics 4: 78–83. [DOI] [PubMed] [Google Scholar]
- 8. Gehlenborg N, O'Donoghue SI, Baliga NS, Goesmann A, Hibbs MA, et al. (2010) Visualization of omics data for systems biology. Nature Methods 7: S56–S68. [DOI] [PubMed] [Google Scholar]
- 9. Tusher VG, Tibshirani R, Chu G (2001) Significance analysis of microarrays applied to the ionizing radiation response. Proceedings of the National Academy of Sciences 98: 5116–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Smyth GK (2004) Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Statistical Applications in Genetics and Molecular Biology 3: 3. [DOI] [PubMed] [Google Scholar]
- 11.Kaever A, Landesfeind M, Possienke M, Feussner K, Feussner I, et al.. (2012) MarVis-Filter: Ranking, Filtering, Adduct and Isotope Correction of Mass Spectrometry Data. Journal of Biomedicine and Biotechnology 2012. [DOI] [PMC free article] [PubMed]
- 12. Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, et al. (2003) DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biology 4: P3. [PubMed] [Google Scholar]
- 13. Dunn WB, Erban A, Weber RJ, Creek DJ, Brown M, et al. (2013) Mass appeal: metabolite identification in mass spectrometry-focused untargeted metabolomics. Metabolomics 9: 44–66. [Google Scholar]
- 14. Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M (2012) KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Research 40: D109–D114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Caspi R, Altman T, Dreher K, Fulcher CA, Subhraveti P, et al. (2012) The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Research 40: D742–D753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, et al. (2000) Gene Ontology: tool for the unification of biology. Nature Genetics 25: 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, et al. (2005) Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. PNAS 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Draghici S, Khatri P, Martins RP, Ostermeier GC, Krawetz SA (2003) Global functional profiling of gene expression. Genomics 81: 98–104. [DOI] [PubMed] [Google Scholar]
- 19. Hosack DA, Dennis G Jr, Sherman BT, Lane HC, Lempicki RA, et al. (2003) Identifying biological themes within lists of genes with EASE. Genome Biology 4: R70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ackermann M, Strimmer K (2009) A general modular framework for gene set enrichment analysis. BMC Bioinformatics 10: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Barry WT, Nobel AB, Wright FA (2005) Significance analysis of functional categories in gene expression studies: a structured permutation approach. Bioinformatics 21: 1943–1949. [DOI] [PubMed] [Google Scholar]
- 22. Goeman JJ, Van De Geer SA, De Kort F, Van Houwelingen HC (2004) A global test for groups of genes: testing association with a clinical outcome. Bioinformatics 20: 93–99. [DOI] [PubMed] [Google Scholar]
- 23. Xia J, Wishart DS (2010) MSEA: a web-based tool to identify biologically meaningful patterns in quantitative metabolomic data. Nucleic Acids Research 38: W71–W77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wägele B, Witting M, Schmitt-Kopplin P, Suhre K (2012) MassTRIX Reloaded: Combined Analysis and Visualization of Transcriptome and Metabolome Data. PLoS ONE 7: e39860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hedges LV, Olkin I (1985) Statistical Methods for Meta-Analysis. San Diego: Academic Press.
- 26. Whitlock M (2005) Combining probability from independent tests: the weighted Z-method is superior to Fisher's approach. Journal of Evolutionary Biology 18: 1368–1373. [DOI] [PubMed] [Google Scholar]
- 27. Loughin TM (2004) A systematic comparison of methods for combining p-values from independent tests. Computational Statistics & Data Analysis 47: 467–485. [Google Scholar]
- 28.Fisher RA (1925) Statistical methods for research workers. Edinburgh: Oliver and Boyd.
- 29.Stouffer SA, Suchman EA, DeVinney LC, Star SA, Williams RM Jr (1949) The American soldier: adjustment during army life. Princeton: Princeton University Press.
- 30. Brown MB (1975) A method for combining non-independent, one-sided tests of significance. Biometrics 31: 987–992. [Google Scholar]
- 31. Shen K, Tseng GC (2010) Meta-analysis for pathway enrichment analysis when combining multiple genomic studies. Bioinformatics 26: 1316–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yan Y, Stolz S, Chételat A, Reymond P, Pagni M, et al. (2007) A downstream mediator in the growth repression limb of the jasmonate pathway. The Plant Cell 19: 2470–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. von Malek B, van der Graaff E, Schneitz K, Keller B (2002) The Arabidopsis male-sterile mutant dde2-2 is defective in the ALLENE OXIDE SYNTHASE gene encoding one of the key enzymes of the jasmonic acid biosynthesis pathway. Planta 216: 187–192. [DOI] [PubMed] [Google Scholar]
- 34. Draper J, Enot D, Parker D, Beckmann M, Snowdon S, et al. (2009) Metabolite signal identification in accurate mass metabolomics data with MZedDB, an interactive m/z annotation tool utilising predicted ionisation behaviour ‘rules’. BMC Bioinformatics 10: 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mueller LA, Zhang P, Rhee SY (2003) AraCyc: a biochemical pathway database for Arabidopsis. Plant Physiology 132: 453–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sclep G, Allemeersch J, Liechti R, De Meyer B, Beynon J, et al. (2007) CATMA, a comprehensive genome-scale resource for silencing and transcript profiling of Arabidopsis genes. BMC Bioinformatics 8: 400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Efron B, Tibshirani R (2007) On testing the significance of sets of genes. The Annals of Applied Statistics: 107–129.
- 38. Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B (Methodological) 57: 289–300. [Google Scholar]
- 39. Wasternack C, Hause B (2013) Jasmonates: biosynthesis, perception, signal transduction and action in plant stress response, growth and development. An update to the 2007 review in Annals of Botany. Annals of Botany 111: 1021–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stumpe M, Feussner I (2006) Formation of oxylipins by cyp74 enzymes. Phytochemistry Reviews 5: 347–357. [Google Scholar]
- 41. Duan H, Huang MY, Palacio K, Schuler MA (2005) Variations in cyp74b2 (hydroperoxide lyase) gene expression differentially affect hexenal signaling in the columbia and landsberg erecta ecotypes of arabidopsis. Plant Physiology 139: 1529–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sønderby IE, Geu-Flores F, Halkier BA (2010) Biosynthesis of glucosinolates–gene discovery and beyond. Trends in Plant Science 15: 283–290. [DOI] [PubMed] [Google Scholar]
- 43.Rice WR (1990) A consensus combined p-value test and the family-wide significance of component tests. Biometrics: 303–308.
- 44. Brazma A, Parkinson H, Sarkans U, Shojatalab M, Vilo J, et al. (2003) ArrayExpress-a public repository for microarray gene expression data at the EBI. Nucleic Acids Research 31: 68–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Matlab source code for functions used in meta-analysis.
(GZ)
Data sets with database entry and pathway annotations. The archive file contains the data sets in comma separated values format. The first column contains the feature IDs, respectively. The rt and Former m/z columns (M1 and M2 data set) contain the retention times and mass-to-charge ratios from MS analysis. The raw intensities for each sample can be found in the following columns. The s/n column shows the feature-specific signal-to-noise ratios and the last columns contain the KEGG and AraCyc entries and pathways mapped to the corresponding features and separated by slash characters.
(ZIP)
Pathways with p-values and FDRs from Pathway Enrichment Analysis. The comma separated values file contains the p-values, restandardized p-values, meta-p-values, and corresponding FDRs for single data set and meta-analysis.
(CSV)
Supplementary tables for simulation studies.
(PDF)
Technical description of methods.
(PDF)