

Abstract
Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) are the main therapeutic agents used to treat non–small-cell lung cancer patients harboring EGFR-activating mutations. However, most of these patients will eventually develop resistance, 50% of which are due to a secondary mutation at T790M in the EGFR. In this paper, we describe the development of an allele-specific DNAzyme, DzT, that can specifically silence EGFR T790M mutant messenger RNA while leaving wild-type EGFR intact. Allele-specific silencing of EGFR T790M expression and downstream signaling by DzT triggered apoptosis in non–small-cell lung cancer cells harboring this mutant. Adding a cholesterol-triethylene glycol group on the 3′-end of DzT (cDzT) improved drug efficacy, increasing inhibitory effect on cell viability from 46 to 79% in T790M/L858R-harboring H1975TM/LR non–small-cell lung cancer cells, without loss of allele specificity. Combined treatment with cDzT and BIBW-2992, a second-generation EGFR-tyrosine kinase inhibitor, synergistically inhibited EGFR downstream signaling and suppressed the growth of xenograft tumors derived from H1975TM/LR cells. Collectively, these results indicate that the allele-specific DNAzyme, DzT, may provide an alternative treatment for non–small-cell lung cancer that is capable of overcoming EGFR T790M mutant-based tyrosine kinase inhibitor resistance.
Keywords: DNAzyme, EGFR T790M, NSCLC, TKI
Introduction
Non–small-cell lung cancer (NSCLC) is the leading cause of cancer-related death.1,2 In one unique subset of NSCLC patients, lung cancer cells harbor activating mutations in the epidermal growth factor receptor (EGFR) and become addicted to aberrant EGFR signaling for their survival.3,4 Among activating mutations, L858R and exon 19 deletion in EGFR account for over 90% of drug-sensitive mutations and show increased binding affinity toward tyrosine kinase inhibitors (TKIs) compared to the wild-type EGFR.5,6 The administration of TKIs successfully induces intrinsic apoptosis pathways in EGFR-mutant lung cancer cells; however, dose-limiting side effects are also unavoidably triggered by the concurrent inhibition of wild-type EGFR signaling in normal cells.7,8 Moreover, despite the success of TKIs at the beginning of NSCLC treatment, the acquired secondary mutation at the gatekeeper residue 790 of EGFR (T790M), which is found in 50% of drug-resistant patients, weakens the interaction between TKIs and EGFR.9,10,11 Dose-limiting toxicity and EGFR T790M-based drug resistance are the main issues in NSCLC treatment that still remain to be solved.
DNAzymes—DNA oligonucleotides with catalytic activity toward specific target RNA sequences—have been comprehensively studied as a means for silencing various genes and have shown promise for use as therapeutic agents.12,13,14 The DNAzymes 10–23 and 8–17 have been most actively studied. DNAzymes consist of a conserved catalytic core that is essential for catalytic activity, flanked by two substrate-binding arms with sequences complementary to the targeted messenger RNA (mRNA) sequence.15 A single mismatch in the binding arm can impede the enzymatic activity of the DNAzyme; therefore, with rational design, DNAzymes are capable of distinguishing between mutant and wild-type mRNAs and achieving specific cleavage.16,17 It has been shown that a DNAzyme against the K-Ras (G12V) mutant form specifically cleaves K-Ras mutant mRNA and leaves wild-type K-Ras mRNA intact.18 Also, a c-Jun mRNA-targeting DNAzyme was shown to suppress the growth of skin cancers (both basal cell and squamous cell carcinomas) in mouse xenograft models.19,20,21 In preclinical studies, DNAzymes were shown to be safe and well tolerated, and exhibited acceptable off-target effects.21
In this study, we developed an allele-specific DNAzyme (DzT) against EGFR T790M and showed that it effectively inhibited EGFR T790M expression and suppressed xenograft tumor growth. Combined therapy with DzT and BIBW-2992 synergistically inhibited the growth of xenograft tumors derived from cells harboring the EGFR T790M mutant, suggesting that DzT may overcome T790M-based TKI resistance in NSCLC.
Results
Design of DNAzymes
DzT (Figure 1) was designed according to the prototype of the 10–23 DNAzyme, which comprises a 15-deoxyribonucleotide catalytic core22 and flanking side arms, I and II, with sequences complementary to its RNA target. The gene encoding EGFR T790M has a C to U substitution at the T790M mutation site.10 Several DNAzymes against EGFR T790M were tested for their efficacies in discriminating T790M from wild-type EGFR mRNA and specifically downregulating EGFR T790M mRNA, including a DNAzyme which is cleaved at a higher rate against mRNA with the EGFR point mutation A”U” positioned at the cleavage junction than one containing A”C” under simulated physiological conditions.23 DzT, which exhibited the best performance (unpublished data), was selected from these DNAzymes for subsequent experiments. Arm I of DzT contains a 10-nucleotide complementary sequence spanning the mutation site of EGFR T790M mRNA, with the cleavage site located four nucleotides away from the mutation. A one-nucleotide mismatch of DzT arm I with EGFR wild-type mRNA weakens DzT mRNA degradation activity, allowing DzT to discriminate the mutant variant from wild-type EGFR mRNA.
Figure 1.

Secondary structure of DzT in complex with EGFR T790M mRNA and wild-type mRNA. The catalytic core of 10–23 DzT (in blue) is flanked by two binding arms with sequences complimentary to EGFR T790M mRNA (in black). The sequences of wild-type EGFR mRNA (upper panel) and T790M mRNA (lower panel), RY cleavage site (arrow), and mutation site (in red) are indicated. EGFR, epidermal growth factor receptor; mRNA, messenger RNA.
DzT overcomes TKI resistance in EGFR T790M lung cancer cells
CL1-5 harbors wild-type EGFR (designated CL1-5wt). PC9 has a deletion in EGFR exon 19 (designated PC9d19). H1975 contains EGFR T790M and L858R mutations (designated H1975TM/LR) and T790M mutation in both alleles was confirmed by genomic DNA sequencing analysis (Supplementary Figure S1). EGF-activated EGFR in CL1-5wt cells or constitutively activated EGFR in PC9d19 cells was inactivated by treatment with Gefitinib, a potent EGFR inhibitor (Figure 2a). However, the phosphorylation level of EGFR in EGFR T790M mutant cells (H1975TM/LR) was not suppressed by Gefitinib treatment. The result suggested that H1975TM/LR was more resistant to Gefitinib treatment. Transfection of H1975TM/LR cells with DzT resulted in significant repression of EGFR protein expression compared to treatment with control DzC, a control DNAzyme with no complementarity to any known human mRNA (Figure 2b). DzT treatment caused no such repression in CL1-5wt or PC9d19 cells containing wild-type and del19 EGFR, respectively, demonstrating the allele-specific selectivity of the DNAzyme. The level of the activated form of EGFR (pEGFR) was downregulated in parallel with that of total EGFR. These results suggest that DzT can circumvent the EGFR-TKI resistance attributable to the T790M mutant.
Figure 2.

DzT overcomes TKI resistance in EGFR T790M mutant cells. Gefitinib (1 μmol/l; a) or DzT- (100 nmol/l; b) treated cells were analyzed by immunoblotting with the indicated primary antibodies listed on the left. EGF (100 ng/ml) was added to CL1-5wt 15 min before cell lysis. EGFR, epidermal growth factor receptor; TKI, tyrosine kinase inhibitors.
DzT selectively attenuates EGFR T790M expression and downstream signaling
A549 harbors wild-type EGFR (designated A549wt). CL97 contains EGFR T790M and G719A mutations (designated CL97TM/GA) and T790M mutation in both alleles was confirmed by genomic DNA sequencing analysis (Supplementary Figure S1). To examine the allele selectivity of DzT, we evaluated EGFR mRNA-knockdown efficiency in two cell lines harboring wild-type EGFR (A549wt and CL1-5wt) and two cell lines harboring T790M (H1975TM/LR and CL97TM/GA). Total RNA extracted from cells transfected with DzT was subjected to quantitative reverse transcription polymerase chain reaction; DzC transfections served as controls. As shown in Figure 3a, treatment of H1975TM/LR and CL97TM/GA cells with DzT significantly suppressed EGFR expression, decreasing EGFR mRNA levels to 32 and 23%, respectively, of those in DzC-treated controls. In contrast, EGFR mRNA expression was only slightly inhibited in A549wt (99% of control) and CL1-5wt (81% of control) cells. Thus, DzT exhibited at least a 2.5-fold increase in knockdown efficiency toward EGFR T790M mRNA compared with its wild-type counterpart, demonstrating the high allele-discrimination ability of this DNAzyme.
Figure 3.

Specific downregulation of EGFR T790M expression and downstream signaling by DzT. (a) RT-qPCR analysis of EGFR mRNA in cells treated with DzT (n = 3). Cells were harvested 48 hours after transfection with DzC or DzT (100 nmol/l). The relative amount of EGFR mRNA was normalized to ACTB mRNA. The data are presented as means ± SD and were analyzed by Student's t-test. Asterisks denote statistical significant differences (P ≤ 0.005). (b) Immunoblot analysis of EGFR and its downstream signaling pathways. Cells were harvested 72 hours after transfecting with 100 nmol/l DzC or DzT. EGFR in wild-type cells was activated by adding 100 ng/ml EGF 15 minutes before cell lysates were harvested. EGFR, epidermal growth factor receptor; mRNA, messenger RNA; RT-qPCR, quantitative reverse transcription polymerase chain reaction.
Like other members of the receptor tyrosine kinases family, EGFR binding to its extracellular ligands triggers receptor dimerization, tyrosine phosphorylation of downstream target molecules, and activation of various signaling pathways, including signal transducer and activator of transcription 3 (STAT3), AKT, extracellular signal-regulated kinase (ERK), and others.24 To examine the inhibitory effects of DzT on EGFR protein expression and downstream signaling, we performed immunoblot analysis. Control DzC did not affect phosphorylated EGFR, total EGFR, and its downstream substrates, including phosphorylated form of STAT3, AKT, and ERK when compared to untreated group in all four cell line examined (Supplementary Figure S2). Thus, DzC treatment was used as a reference control for the following experiments. On the other hand, DzT inhibited EGFR protein expression in both EGFR T790M mutant cell lines (H1975TM/LR and CL97TM/GA), with a concurrent decrease in the phosphorylated form of EGFR (Figure 3b, two panels at the right). DzT also inhibited the downstream activation of STAT3, AKT, and ERK without affecting the total amount of each individual protein. After EGF treatment, DzT remained its suppression effect on EGFR protein expression and downstream signaling including EGFR, STAT3, and ERK but not AKT (Supplementary Figure S3). In contrast, EGFR protein levels in DzT-treated groups did not differ from that of DzC-treated groups in A549wt and CL1-5wt cells (Figure 3b, two panels at the left); the phosphorylated form of EGFR and that of its downstream substrates were similarly unaffected by DzT treatment in A549wt and CL1-5wt.
DzT induces lung cancer cell apoptosis in an allele-specific manner
EGFR and its downstream signaling pathways regulate important cell functions, including cell proliferation and survival.3 To examine the effects of DzT on cell survival, we counted cell numbers after transfection of DzC or DzT. In A549wt and CL1-5wt cells, no differences in viable cell number were seen between DzC- and DzT-transfected groups (Figure 4a,b). In contrast, the viable cell number of EGFR T790M mutant cells (H1975TM/LR and CL97TM/GA) was significantly retarded by DzT transfection (Figure 4c,d). To determine whether DzT triggers apoptosis in EGFR T790M mutant cell lines, we immunoblotted for poly ADP-ribose polymerase (PARP) and performed flow cytometry analyses on annexin V (AV)- and propidium iodide (PI)-stained cells. The cleavage of PARP is caused by increased activity of caspase-3 and serves as a marker for apoptosis.25 Immunoblot analyses showed that the decrease in EGFR level induced by DzT treatment was accompanied by a concomitant increase in cleaved PARP in both EGFR T790M mutant cell lines (H1975TM/LR and CL97TM/GA) compared with that in DzC-treated groups (Figure 4e).
Figure 4.
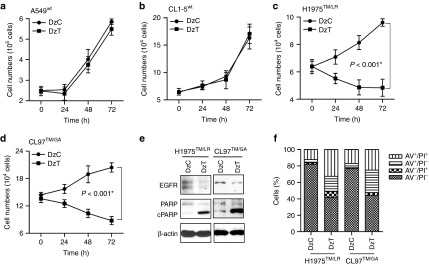
DzT induces apoptosis in an allele-specific manner. (a–d) A549wt (a), CL1-5wt (b), H1975TM/LR (c), and CL97TM/GA (d) cell numbers were determined at various times after DzT (100 nmol/l) transfection (n = 3). These data are presented as mean ± SD and were analyzed by Student's t-test. Asterisks denote statistical significant differences (P ≤ 0.001). (e, f) DzT induces apoptosis in cells harboring EGFR T790M. H1975TM/LR and CL97TM/GA cells were analyzed 72 hours after transfection with DzC or DzT by immunoblotting with the indicated antibodies (e) or flow cytometry stained with AV and PI (f). Counts are presented as percentages. AV, annexin V; PI, propidium iodide.
Dual staining with AV and PI in conjunction with flow cytometry is a commonly used method for evaluating cell viability and apoptosis status. AV-positive cells are generally regarded as apoptotic cells, and PI-positive cells as dead cells. A flow cytometric analysis of H1975TM/LR and CL97TM/GA cells stained with AV and PI showed 51 and 53% AV-positive in DzT-transfected groups; the corresponding values for DzC-transfected cells were only 16 and 22% (Figure 4f). A large proportion of PI-positive cells in the DzT-transfected groups were also AV positive (33 and 25% for H1975TM/LR and CL97TM/GA cells, respectively), indicating that the DzT-induced cell death was the result of cellular apoptosis.
Cholesterol modification enhances the inhibitory effect of DzT on cell viability
Cholesterol modification of anti-microRNAs (antagomirs) has been shown to increase their ability to escape endosomes and enhance their likelihood of making contact with their targets in the cytosol.26 Accordingly, we added a cholesterol-triethylene glycol (chol-TEG) group on the 3′-end of DNAzymes (cDzC and cDzT) and then tested their activity in cell viability assays. Modification of DzT with chol-TEG significantly increased the inhibitory effect on cell viability from 46 to 79% in H1975TM/LR cells with no loss of allele specificity (no inhibition in CL1-5wt cells). Neither modified nor primitive DzC inhibited cell viability (Figure 5a). Immunoblot showed that the phosphorylated form of EGFR and that of its downstream substrates were unaffected by cDzC treatment when compared to those in untreated group (Supplementary Figure S4). Thus, cDzC treatment was used as a reference control for the following experiments. On the other hand, cDzT inhibited expression of T790M but not wild-type EGFR (Figure 5b), demonstrating that the chol-TEG-modified DNAzyme retained the ability to specifically repress EGFR protein expression in an allele-specific manner. Downstream phosphorylation of STAT3, AKT, and ERK were also inhibited. Moreover, the cleaved form of PARP was elevated in H1975TM/LR cells, indicating that chol-TEG modification did not alter the ability of DzT to specifically trigger apoptosis in EGFR T790M mutant cells. Besides, after EGF treatment, cDzT remained its suppression effect on EGFR protein expression and downstream signaling including EGFR, STAT3, AKT, and ERK (Supplementary Figure S5).
Figure 5.

Cholesterol modification of DzT enhances the inhibitory effect on cell viability of DzT while retaining its allele specificity. (a) CL1-5wt and H1975TM/LR cells were treated with 100 nmol/l DzT with or without cholesterol modification. MTT assays were performed 72 hours after transfection to determine cell viability (n = 3). The data are presented as mean ± SD and were analyzed by Student's t-test. Asterisks denote statistical significant differences (P ≤ 0.001). (b) Cell lysates were analyzed by immunoblotting.
Synergistic antitumor effect of combined treatment with cDzT and BIBW-2992 in vitro and in vivo
cDzT could be used as single agent or as part of combination therapy with second generation TKIs to increase efficacy in the treatment of NSCLCs that harbor the EGFR T790M mutant.5 The efficacy of combined treatment with cDzT and the TKI, BIBW-2992, against EGFR T790M-based drug resistance was evaluated in vitro and in vivo. To evaluate the synergistic antitumor effects of BIBW-2992 and cDzT, we used lower, suboptimal concentrations of DzT (50 nmol/l) and BIBW-2992 (200 nmol/l) in these experiments. Immunoblot was performed to evaluate EGFR protein expression and trans-autophosphorylation status of its downstream signaling proteins (Figure 6a and Supplementary Figure S6). Both cDzT and BIBW-2992 alone inhibited growth and signaling in EGFR T790M mutant cells (H1975TM/LR and CL97TM/GA), albeit through different mechanisms. cDzT specifically degraded EGFR T790M mRNA and decreased EGFR protein levels, whereas BIBW-2992 inhibited autophosphorylation of EGFR without affecting EGFR protein level.
Figure 6.

Synergistic effects of cDzT and BIBW-2992. (a) Immunoblot analysis of H1975TM/LR cells treated with cDzC or cDzT (50 nmol/l) incubated with or without added BIBW-2992 (200 nmol/l). (b, c) MTT assay of H1975TM/LR cells treated with cDzC or cDzT (50 nmol/l) combined with 25 (●), 50 (■), 100 (▲), or 250 nmol/l (▼) BIBW-2992 (n = 3). The data are presented as the mean ± SD. The results were analyzed by Student's t-test and CI calculation. An asterisk denotes statistical significant difference (P ≤ 0.001). (d–f) Combined treatment silences EGFR signaling, triggers apoptosis, and suppresses xenograft tumor growth. (d) cDzT (500 pmoles) was intratumorally injected (twice per week) and BIBW-2992 (20 mg/kg) was orally administrated (three times per week) 10 days (arrow indicated) after inoculating xenograft mice with H1975TM/LR (n = 7). The data are presented as mean ± SD and were analyzed by Student's t-test. Asterisks denote statistical significant differences (P ≤ 0.005). (e) Xenograft tumor tissues were processed for immunostaining. Scale bars represent 200 μm in H&E images and 100 μm in EGFR L858R and caspase-3 images. (f) Xenograft tumor tissues were processed for immunoblotting. CI, combination index; EGFR, epidermal growth factor receptor.
At suboptimal concentrations, individual drugs did not efficiently suppress downstream signaling. In contrast, combined treatment significantly suppressed the phosphorylation of STAT3, AKT, and ERK (Figure 6a and Supplementary Figure S6).
Furthermore, BIBW-2992 enhanced the cell-killing effect of cDzT in a concentration-dependent manner on H1975TM/LR cells (50 nmol/l cDzT in Figure 6b and 25 nmol/l cDzT in Supplementary Figure S7a) and CL97TM/GA cells (25 nmol/l cDzT in Supplementary Figure S7c). Combination index (CI) values were calculated to evaluate the combined effect of cDzT and BIBW-2992. This analysis showed that combined treatment exerted a synergistic inhibitory effect (CI < 1) on the viability of H1975TM/LR cells (Figure 6c and Supplementary Figure S7b) and CL97TM/GA cells (Supplementary Figure S7d).
Synergistic effects of cDzT and BIBW-2992 were also seen in the xenograft animal model. Compared with the control group (cDzC), all three drug-treated groups (cDzC+BIBW-2992, cDzT, and cDzT+BIBW-2992) inhibited the growth of tumor originated from H1975TM/LR cells to different degrees (Figure 6d). Combined treatment with cDzT and BIBW-2992 showed the highest potency among all treatments in suppressing xenograft tumor growth. In this group, the average size of excised tumors was approximately fourfold smaller than that in the control group. An immunohistochemical analysis of tumor tissues showed severe necrosis in tumor tissues from the combined treatment group but not in tissues from the control group (Figure 6e, upper panel). EGFR in H1975TM/LR cells contains both L858R and T790M mutations, and thus can be detected using an antibody specific for the L858R mutant form. Tumor sections from the combined treatment group exhibited lower levels of EGFR L858R expression accompanied by higher caspase-3 protein expression levels compared with sections from the control group (Figure 6e, middle and lower panel). Tumor tissues were also evaluated for EGFR expression and downstream signaling. The results showed that the combination of cDzT and BIBW-2992 further suppressed total EGFR expression, the phosphorylation of EGFR in tumor tissues, and the levels of the phosphorylated forms of STAT3, AKT, and ERK compared with the control group (Figure 6f). The effect of cDzC treatment on EGFR expression and downstream signaling were evaluated by immunoblot regarding the tumor tissues from phosphate-buffered saline buffer group and cDzC-treated group. Immunoblot showed that the phosphorylated form of EGFR and that of its downstream substrates were unaffected by cDzC treatment when compared to those in phosphate-buffered saline buffer group (Supplementary Figure S8). Taken together, these results indicate that the combination of cDzT and BIBW-2992 synergistically inhibits EGFR protein expression and downstream signaling, triggering T790M-harboring cells to undergo apoptosis and suppressing xenograft tumor growth.
Discussion
In this study, we demonstrated that allele-specific DzT is capable of overcoming EGFR-TKI resistance attributable to a secondary EGFR T790M mutation in NSCLC. We also showed that modification of DzT with chol-TEG enhanced its efficacy in inhibiting cancer cell growth. Furthermore, we showed a synergistic effect of cDzT and BIBW-2992 on growth inhibition in vitro and in vivo in cancer cells harboring the EGFR T790M mutant. This study provides a successful example of allele-specific gene silencing therapy overcoming EGFR T790M-based drug resistance.
Since the first demonstration that synthetic small interfering RNAs (siRNAs) induced an RNA interference effect in mammalian cells by Thomas Tuschl,27 there has been a surge in interest in harnessing siRNA for biomedical research and drug development. Limitations in siRNA design has, at times, impeded the generation of siRNAs that are efficacious in degrading target mRNA, particularly with respect to allele-specific targeting. In addition, some mRNAs are inherently recalcitrant to perturbation by small RNAs.28 Given these limitations, DNAzymes provide a superb alternative for gene silencing, particularly for applications that require specific targeting of a mutant site. Unlike siRNA, which requires formation of an RNA-induced silencing complex with Dicer protein to cleave mRNA, divalent metal ions such as Mg2+, which are abundant in the cell cytosol, are sufficient for DNAzyme-mediated catalysis.29,30,31 The advantages of DNAzymes over siRNAs are more resistant to nuclease attack, cheaper to synthesize, and easier to modify.14 Modifications, such as introduction of nonstandard nucleotides32 and substitution of linkage bonds22 or functional groups,14 can be introduced into DNAzymes to enhance transport efficiency, pairing capacity, or enzymatic activity.
Owing to their highly negatively charged nature, oligonucleotides are difficult to transfer across cell membranes. Cholesterol modification has been demonstrated to enhance the ability of antagomirs to escape endosomes and increase the accessibility of their target RNA in the cytosol.26 We evaluated the efficacy of cholesterol-TEG modification of DzT in inhibiting cancer cell growth and found that this modification significantly enhanced the antiproliferative effect of DzT without affecting its allele specificity in distinguishing the EGFR T790M mutant form from its wild-type counterpart. Cholesterol provides a lipid moiety that allows cDzT to pass through cell membranes more efficiently, whereas both cholesterol and TEG may enhance resistance to degradation by exonucleases in biological systems.
We also demonstrated that cDzT and BIBW-2992 functioned in a synergistic manner against EGFR signaling pathways to suppress the growth of EGFR T790M-harboring NSCLC cells. The mechanisms of action of cDzT and TKIs are different. cDzT cleaves EGFR T790M mRNA, which leads to a decrease in EGFR protein levels, whereas BIBW-2992 is a second-line TKI that irreversibly binds to the intracellular kinase domain of EGFR and suppresses its downstream signaling. A recent report showed that EGFR support of cancer cell survival is independent of its kinase activity, suggesting that a TKI alone could not achieve maximum therapeutic efficacy.33 In addition to its pivotal role in signaling pathways, EGFR also interacts with and stabilizes the sodium/glucose cotransporter 1 to maintain intracellular glucose levels, which prevents against autophagic cell death. cDzT-induced inhibition of EGFR expression is advantageous compared with TKI effects in that it may also block the kinase-independent functions of EGFR. Combination therapies could allow the dosages of TKIs to be reduced and reduce unwanted toxicity toward normal cells. Thus, the allele-specific DNAzyme may solve the two main obstacles in targeted therapy of NSCLC: dose-limiting toxicity and T790M-based drug resistance.
To investigate the mechanisms by which EGFR knockdown inhibits cancer growth, we used suboptimal concentrations of cDzT in this study (higher concentrations of cDzT completely eradicated cancer cells, leaving no cells to study; unpublished data). As demonstrated by Cai et al.20 in a series of preclinical studies, DNAzymes are safe and well tolerated at dose as high as 100 μg for intratumor injection, an amount more than 20-fold greater than we used in our mouse studies (4.5 μg).
In this paper, we demonstrated that DzT is an allele-specific silencing agent against EGFR T790M mutant mRNA. In addition to T790M, prevalent EGFR mutants seen in NSCLC patients (with population percentages in parenthesis) include G719S in exon 18 (5%), E746-A750 deletion in exon 19 (45%), and L858R in exon 21 (40–45%).4 Like DzT, other DNAzymes can be designed to knock down the expression of each of the mutant forms of EGFR in an allele-specific manner without affecting wild-type EGFR expression. EGFR mutations vary among individual patients and can change within an individual due to genetic alterations that occur during the development and progression of cancer. Allele-specific DNAzymes could thus be used in combination to knock down all types of EGFR mRNA variants. They could also be used in combination with other drugs, such as EGFR-TKIs or EGFR-specific antibodies, to completely eradicate cancer cells.
Materials and methods
Cell lines and reagents. A549wt, CL1-5wt, H1975TM/LR, CL97TM/GA, and PC9d19 cells were cultivated at 37 °C with 5% CO2 in RPMI-1640 medium (Gibco BRL; Life Technologies, Grand Island, NY) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (Gibco BRL). A549wt, H1975TM/LR, and PC9d19 were purchased from American Type Culture Collection (Manassas, VA). CL1-5wt is a highly invasive human lung adenocarcinoma derived from parental CL1-0.34 Both CL1-5wt and CL97TM/GA35 were developed in our laboratory. Gefitinib and BIBW-2992 were kindly provided by Dr. James Chih-Hsin Yang (National Taiwan University). MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) solution was purchased from (Sigma-Aldrich, St Louis, MO).
Genomic DNA sequencing analysis. Genomic DNA was extracted from H1975TM/LR and CL97TM/GA cell lines using QIAGEN blood and cell culture DNA kit according to the manufacturer's protocol (Qiagen, Hilden, Germany). The primer pairs used for PCR amplification of EGFR exon 20 were: 5′-CCA TGA GTA CGT ATT TTG AAA CTC-3′ (forward) and 5′-CCA CAC TGA GCA CTC AAT AAA GAG-3′ (reverse). The purified PCR products were examined by sequencing analysis.
DNAzyme synthesis. All DNAzymes were synthesized by Integrated DNA Technologies (Coralville, IA). The sequence of the control DNAzyme (DzC) showed no complementarity to any known human mRNA. DzT was designed to hybridize specifically to EGFR T790M mutant mRNA. The DNA sequences of DzC and DzT are 5′-CAT CGG AGG CTA GCT ACA ACG AGA CAG CTG-3′ and 5′-AGC TGC ATG AGG CTA GCT ACA ACG AGA GC-3′ (Figure 1). Phosphorothioate bonds were introduced between underlined nucleotides at both ends of the DNAzymes. Chol-TEG was covalently attached to the 3′-end of DzC and DzT (cDzC and cDzT) at synthesis.
Cell culture and drug treatment. Cells were seeded onto six-well plates at 3 × 105 cells per well. After culturing overnight, cells were treated with 100 nmol/l DzC or DzT mixed with Lipofectamine 2000 (Invitrogen, Carlsbad, CA). In immunoblot experiments, for wild-type EGFR activation and inactivation, A549wt and CL1-5wt cells transfected with DNAzyme were serum-starved for 24 hours and incubated with or without 100 ng/ml EGF at 37 °C for 15 minutes immediately before cell lysis. To examine the potential EGF-mediated signaling in H1975TM/LR and CL97TM/GA cells, 100 ng/ml EGF was added 15 minutes before cell lysates were harvested. In TKI-resistance tests, 1 μmol/l Gefitinib was added into culture medium 6 hours before cell collection. Only CL1-5wt and H1975TM/LR cells were used in chol-TEG-modified DNAzyme studies. In order to leave enough surviving cells for analysis, a suboptimal concentration (50 nmol/l) of cDzC or cDzT was used for transfections. In the combined treatment study, H1975TM/LR and CL97TM/GA cells were treated with 50 nmol/l cDzC or cDzT together with 200 nmol/l BIBW-2992 or dimethyl sulfoxide (vehicle control), added to the culture medium.
Quantitative reverse transcription polymerase chain reaction. Forty-eight hours after transfection, total RNA was extracted using TRIzol, according to the manufacturer's protocol (Invitrogen). One-step reverse transcription polymerase chain reaction was performed using the LightCycler 480 system (Roche Applied Science, Mannheim, Germany). The relative amount of EGFR mRNA was normalized to β-actin (ACTB) mRNA. The sequences of PCR primer pairs were 5′-CAT CTC CGA AAG CCA ACA A-3′ (forward) and 5′-CTG CGT GAT GAG CTG CAC-3′ (reverse) for EGFR, and 5′-ATT GGC AAT GAG CGG TTC-3′ (forward) and 5′-GGA TGC CAC AGG ACT CCAT-3′ (reverse) for ACTB.
Immunoblot. Total protein was extracted 72 hours after transfection using radioimmunoprecipitation assay buffer containing complete protease inhibitor cocktail (Roche Applied Science). For animal studies, xenograft tumor tissues were also lysed with radioimmunoprecipitation assay buffer. Protein concentrations were determined using the Bio-Rad Protein Assay (Bio-Rad, Richmond, CA). Samples (50 μg protein) were resolved on 10% sodium dodecyl sulfate–polyacrylamide gels, transferred onto nitrocellulose membranes, and immunoblotted with primary antibodies. Primary antibodies against the following proteins were used in immunoblot analyses at a 1:1,000 dilution (the phosphorylation site is given in parenthesis): EGFR (Santa Cruz Biotechnology, Santa Cruz, CA), pEGFR (Y1068; Cell Signaling Technology, Beverly, MA), STAT3 (Cell Signaling Technology), pSTAT3 (Y705; Cell Signaling Technology), AKT (Santa Cruz Biotechnology), pAKT (S473; Cell Signaling Technology), ERK (Santa Cruz Biotechnology), pERK (T202/Y204; Cell Signaling), and cPARP (cleaved-PARP [Asp214]; Cell Signaling); anti-β-actin (Santa Cruz Biotechnology). Horseradish peroxidase–conjugated anti-rabbit or mouse IgG (Santa Cruz Biotechnology) was used as a secondary antibody at a 1:5,000 dilution. Protein bands on membranes were visualized using enhanced chemiluminescence reagents (Amersham Pharmacia, Piscataway, NJ).
Cell counting. Cells at exponential growth phase were obtained by seeding cells at different concentrations and cultivated in RPMI-1640 medium supplemented with 10% (v/v) heat-inactivated fetal bovine serum. Cells were trypsinized (Invitrogen) and counted with a hemocytometer 0, 24, 48, and 72 hours after transfection.
Apoptosis assay. Cells seeded onto 10-cm dishes were cultured overnight and treated with 100 nmol/l DzC or DzT mixed with Lipofectamine 2000. Cells were collected at 72 hours, double stained with AV and PI, and analyzed by flow cytometry (FACSCanto system; BD Biosciences, San Jose, CA) following the manufacturer's protocol (Alexa Fluor 488 Annexin V/Dead Cell Apoptosis Kit; Invitrogen).
Cell viability assay. Seventy-two hours after DzC or DzT transfection, cells were rinsed three times with phosphate-buffered saline prior to incubation with a 0.5 mg/ml MTT solution at 37 °C for 3 hours. MTT solution was then replaced with dimethyl sulfoxide, and cell proliferation was determined by measuring absorbance at 570 nm with a SpectraMax Plus384 Microplate Reader (Molecular Devices, Sunnyvale, CA).
Synergy. H1975TM/LR and CL97TM/GA cells were treated with 25, 50 nmol/l cDzC or cDzT together with 25, 50, 75, 100, 150, 250 nmol/l BIBW-2992 or dimethyl sulfoxide (vehicle control) added to the culture medium. Seventy-two hours after treatment, MTT assays were performed to monitor cell viability. CI values were calculated by CI equation of Chou–Talalay using CompuSyn version 3.0.1 software (ComboSyn, Paramus, NJ).36
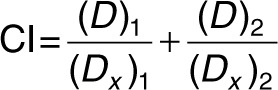 |
(Dx)1 was the dose of cDzT alone that inhibited x% of cell viability while (Dx)2 was the dose of BIBW-2992 alone that inhibited x% of cell viability. (D)1 was the portion of cDzT and (D)2 was the portion of BIBW-2992 that achieved x% of inhibition when combined treatment of cDzT and BIBW-2992. “Fraction affected (Fa)” on the x-axis of Fa-CI plot represented the fraction of cell viability inhibition on drug treated cells. CI values greater than 1, equal to 1, and less than 1 indicate antagonistic effects, additive effects, and synergistic effects, respectively.36
In vivo tumorigenesis assay. All animal studies were performed according to protocols approved by the Laboratory Animal Center, Academia Sinica. Eight-week-old Balb/c nude mice (BioLASCO, Taipei, Taiwan) were subcutaneously inoculated with 2 × 106 H1975TM/LR cells (day 0). In the combined-treatment study, mice were randomly divided into four groups on day 10 and administered the following drug or drug combinations: (i) cDzC, (ii) cDzC+BIBW-2992, (iii) cDzT, or (iv) cDzT+BIBW-2992. Chol-TEG-modified DNAzyme (500 pmoles) mixed with Lipofectamine 2000 was injected intratumorally twice per week. BIBW-2992 was suspended in phosphate-buffered saline and administered three times per week by oral gavage at 20 mg/kg. The length (L) and width (W) of tumors were measured with calipers every 3–4 days, and tumor volumes were calculated as (L × W2)/2. After mice were sacrificed, tumors were excised. Small sections of tumors were processed for immunoblot and the remaining tumor tissue was fixed with 10% formalin and embedded in paraffin. Xenograft tumor slides were stained with hematoxylin and eosin (H&E), anti-EGFR (L858R mutant specific; Cell Signaling), and anti-caspase 3 (Cell Signaling).
SUPPLEMENTARY MATERIAL Figure S1. The mutation status of EGFR exon 20 in H1975TM/LR and CL97TM/GA cell lines. Figure S2. EGFR expression and downstream signaling is unaffected by DzC treatment. Figure S3. DzT remains its suppression effect on EGFR T790M expression and downstream signaling after EGF treatment in T790M mutant cells. Figure S4. EGFR expression and downstream signaling is unaffected by cDzC treatment in CL1-5wt and H1975TM/LR. Figure S5. cDzT remains its suppression effect on EGFR T790M expression and downstream signaling after EGF treatment in H1975TM/LR. Figure S6. Combined treatment of cDzT and BIBW-2992 significantly suppresses the phosphorylation of STAT3, AKT, and ERK in CL97TM/GA cells. Figure S7. Combined treatment of cDzT and BIBW-2992 exerts a synergistic inhibitory effect on cell viability in cells harboring EGFR T790M mutants. Figure S8. EGFR expression and downstream signaling is unaffected in xenograft tissue after cDzC treatment.
Acknowledgments
This work was supported by grants from the National Science Council of the Republic of China (NSC100-2325-B001-016, NSC101-2325-B001-038, NSC 102-2911-I-002-303). The authors have declared that no conflicts of interest exist. The authors would like to dedicate this paper to the memory of Dr. Konan Peck, who passed away during the period of this research. Dr. Peck originated the rational design of DNAzymes; without his long-term devotion to lung cancer research, this paper could not have been completed. The authors thank Dr. James Chih-Hsin Yang (National Taiwan University) for providing BIBW-2992 and Dr. Yih-Leong Chang (National Taiwan University Hospital) for excellent guidance in immunohistochemistry staining procedures.
Supplementary Material
The mutation status of EGFR exon 20 in H1975TM/LR and CL97TM/GA cell lines.
EGFR expression and downstream signaling is unaffected by DzC treatment.
DzT remains its suppression effect on EGFR T790M expression and downstream signaling after EGF treatment in T790M mutant cells.
EGFR expression and downstream signaling is unaffected by cDzC treatment in CL1-5wt and H1975TM/LR.
cDzT remains its suppression effect on EGFR T790M expression and downstream signaling after EGF treatment in H1975TM/LR.
Combined treatment of cDzT and BIBW-2992 significantly suppresses the phosphorylation of STAT3, AKT, and ERK in CL97TM/GA cells.
Combined treatment of cDzT and BIBW-2992 exerts a synergistic inhibitory effect on cell viability in cells harboring EGFR T790M mutants.
EGFR expression and downstream signaling is unaffected in xenograft tissue after cDzC treatment.
References
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. 2008;359:1367–1380. doi: 10.1056/NEJMra0802714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med. 2008;358:1160–1174. doi: 10.1056/NEJMra0707704. [DOI] [PubMed] [Google Scholar]
- Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007;7:169–181. doi: 10.1038/nrc2088. [DOI] [PubMed] [Google Scholar]
- Imai K, Takaoka A. Comparing antibody and small-molecule therapies for cancer. Nat Rev Cancer. 2006;6:714–727. doi: 10.1038/nrc1913. [DOI] [PubMed] [Google Scholar]
- Linardou H, Dahabreh IJ, Bafaloukos D, Kosmidis P, Murray S. Somatic EGFR mutations and efficacy of tyrosine kinase inhibitors in NSCLC. Nat Rev Clin Oncol. 2009;6:352–366. doi: 10.1038/nrclinonc.2009.62. [DOI] [PubMed] [Google Scholar]
- Segaert S, Van Cutsem E. Clinical signs, pathophysiology and management of skin toxicity during therapy with epidermal growth factor receptor inhibitors. Ann Oncol. 2005;16:1425–1433. doi: 10.1093/annonc/mdi279. [DOI] [PubMed] [Google Scholar]
- Irmer D, Funk JO, Blaukat A. EGFR kinase domain mutations - functional impact and relevance for lung cancer therapy. Oncogene. 2007;26:5693–5701. doi: 10.1038/sj.onc.1210383. [DOI] [PubMed] [Google Scholar]
- Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong KK, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci USA. 2008;105:2070–2075. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao W, Chmielecki J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer. 2010;10:760–774. doi: 10.1038/nrc2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan ML, Choong PF, Dass CR. DNAzyme delivery systems: getting past first base. Expert Opin Drug Deliv. 2009;6:127–138. doi: 10.1517/17425240902751605. [DOI] [PubMed] [Google Scholar]
- Dass CR. Deoxyribozymes: cleaving a path to clinical trials. Trends Pharmacol Sci. 2004;25:395–397. doi: 10.1016/j.tips.2004.06.001. [DOI] [PubMed] [Google Scholar]
- Dass CR, Choong PF, Khachigian LM. DNAzyme technology and cancer therapy: cleave and let die. Mol Cancer Ther. 2008;7:243–251. doi: 10.1158/1535-7163.MCT-07-0510. [DOI] [PubMed] [Google Scholar]
- Schlosser K, Gu J, Lam JC, Li Y. In vitro selection of small RNA-cleaving deoxyribozymes that cleave pyrimidine-pyrimidine junctions. Nucleic Acids Res. 2008;36:4768–4777. doi: 10.1093/nar/gkn396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang TH, Li WT, Yu SH, Au LC. The use of 10-23 DNAzyme to selectively destroy the allele of mRNA with a unique purine-pyrimidine dinucleotide. Oligonucleotides. 2008;18:295–299. doi: 10.1089/oli.2008.0138. [DOI] [PubMed] [Google Scholar]
- Abdelgany A, Ealing J, Wood M, Beeson D. Selective DNAzyme-mediated cleavage of AChR mutant transcripts by targeting the mutation site or through mismatches in the binding arm. J RNAi Gene Silencing. 2005;1:32–37. [PMC free article] [PubMed] [Google Scholar]
- Yu SH, Wang TH, Au LC. Specific repression of mutant K-RAS by 10-23 DNAzyme: sensitizing cancer cell to anti-cancer therapies. Biochem Biophys Res Commun. 2009;378:230–234. doi: 10.1016/j.bbrc.2008.11.027. [DOI] [PubMed] [Google Scholar]
- Fahmy RG, Waldman A, Zhang G, Mitchell A, Tedla N, Cai H, et al. Suppression of vascular permeability and inflammation by targeting of the transcription factor c-Jun. Nat Biotechnol. 2006;24:856–863. doi: 10.1038/nbt1225. [DOI] [PubMed] [Google Scholar]
- Cai H, Santiago FS, Prado-Lourenco L, Wang B, Patrikakis M, Davenport MP, et al. DNAzyme targeting c-jun suppresses skin cancer growth. Sci Transl Med. 2012;4:139ra82. doi: 10.1126/scitranslmed.3003960. [DOI] [PubMed] [Google Scholar]
- Cho E, Moloney FJ, Cai H, Au-Yeung A, China C, et al. First-in-human trial of Dz13 DNAzyme targeting c-jun in patients with basal cell carcinoma. J Investig Dermatol. 2012;132:S66–S66. [Google Scholar]
- Schubert S, Gül DC, Grunert HP, Zeichhardt H, Erdmann VA, Kurreck J. RNA cleaving ‘10-23' DNAzymes with enhanced stability and activity. Nucleic Acids Res. 2003;31:5982–5992. doi: 10.1093/nar/gkg791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns MJ, King A, Sun LQ. Optimisation of the 10-23 DNAzyme-substrate pairing interactions enhanced RNA cleavage activity at purine-cytosine target sites. Nucleic Acids Res. 2003;31:2883–2889. doi: 10.1093/nar/gkg378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DL, Dunn EF, Harari PM. Understanding resistance to EGFR inhibitors-impact on future treatment strategies. Nat Rev Clin Oncol. 2010;7:493–507. doi: 10.1038/nrclinonc.2010.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Los M, Mozoluk M, Ferrari D, Stepczynska A, Stroh C, Renz A, et al. Activation and caspase-mediated inhibition of PARP: a molecular switch between fibroblast necrosis and apoptosis in death receptor signaling. Mol Biol Cell. 2002;13:978–988. doi: 10.1091/mbc.01-05-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenvang J, Petri A, Lindow M, Obad S, Kauppinen S. Inhibition of microRNA function by antimiR oligonucleotides. Silence. 2012;3:1. doi: 10.1186/1758-907X-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- Larsson E, Sander C, Marks D. mRNA turnover rate limits siRNA and microRNA efficacy. Mol Syst Biol. 2010;6:433. doi: 10.1038/msb.2010.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivas FV, Tolia NH, Song JJ, Aragon JP, Liu J, Hannon GJ, et al. Purified Argonaute2 and an siRNA form recombinant human RISC. Nat Struct Mol Biol. 2005;12:340–349. doi: 10.1038/nsmb918. [DOI] [PubMed] [Google Scholar]
- Rana TM. Illuminating the silence: understanding the structure and function of small RNAs. Nat Rev Mol Cell Biol. 2007;8:23–36. doi: 10.1038/nrm2085. [DOI] [PubMed] [Google Scholar]
- Khachigian LM. Catalytic DNAs as potential therapeutic agents and sequence-specific molecular tools to dissect biological function. J Clin Invest. 2000;106:1189–1195. doi: 10.1172/JCI11620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman SK. Catalytic DNA (deoxyribozymes) for synthetic applications-current abilities and future prospects. Chem Commun (Camb) 2008;30:3467–3485. doi: 10.1039/b807292m. [DOI] [PubMed] [Google Scholar]
- Weihua Z, Tsan R, Huang WC, Wu Q, Chiu CH, Fidler IJ, et al. Survival of cancer cells is maintained by EGFR independent of its kinase activity. Cancer Cell. 2008;13:385–393. doi: 10.1016/j.ccr.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu YW, Yang PC, Yang SC, Shyu YC, Hendrix MJ, Wu R, et al. Selection of invasive and metastatic subpopulations from a human lung adenocarcinoma cell line. Am J Respir Cell Mol Biol. 1997;17:353–360. doi: 10.1165/ajrcmb.17.3.2837. [DOI] [PubMed] [Google Scholar]
- Yeh CT, Wu AT, Chang PM, Chen KY, Yang CN, Yang SC, et al. Trifluoperazine, an antipsychotic agent, inhibits cancer stem cell growth and overcomes drug resistance of lung cancer. Am J Respir Crit Care Med. 2012;186:1180–1188. doi: 10.1164/rccm.201207-1180OC. [DOI] [PubMed] [Google Scholar]
- Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–446. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The mutation status of EGFR exon 20 in H1975TM/LR and CL97TM/GA cell lines.
EGFR expression and downstream signaling is unaffected by DzC treatment.
DzT remains its suppression effect on EGFR T790M expression and downstream signaling after EGF treatment in T790M mutant cells.
EGFR expression and downstream signaling is unaffected by cDzC treatment in CL1-5wt and H1975TM/LR.
cDzT remains its suppression effect on EGFR T790M expression and downstream signaling after EGF treatment in H1975TM/LR.
Combined treatment of cDzT and BIBW-2992 significantly suppresses the phosphorylation of STAT3, AKT, and ERK in CL97TM/GA cells.
Combined treatment of cDzT and BIBW-2992 exerts a synergistic inhibitory effect on cell viability in cells harboring EGFR T790M mutants.
EGFR expression and downstream signaling is unaffected in xenograft tissue after cDzC treatment.