

Abstract
The Set1 protein of Saccharomyces cerevisiae is a histone methyltransferase (HMTase) acting on lysine 4 of histone H3. Inactivation of the SET1 gene in a diploid leads to a sporulation defect. We have studied various processes that take place during meiotic differentiation in set1Δ diploid cells. The absence of Set1 leads to a delay of meiotic S-phase onset, which reflects a defect in DNA replication initiation. The timely induction of meiotic DNA replication does not require the Set1 HMTase activity, but depends on the SET domain. In addition, set1Δ displays a severe impairment of the DNA double-strand break formation, which is not only the consequence of the replication delay. Transcriptional profiling experiments show that the induction of middle meiotic genes, but not of early meiotic genes, is affected by the loss of Set1. In contrast to meiotic replication, the transcriptional induction of the middle meiotic genes appears to depend on the methylation of H3-K4. Our results unveil multiple roles of Set1 in meiotic differentiation and distinguish between HMTase-dependent and -independent Set1 functions.
Keywords: double-strand breaks, gene expression, meiosis, replication, Set1
Introduction
Sporulation in Saccharomyces cerevisiae is a process of cellular differentiation that is initiated in diploid a/α cells in response to nitrogen starvation, the presence of a non-fermentable carbon source, and the absence of glucose. Complex signal networks translate this combination of extracellular signals into an integrated cellular response that controls the switch from mitotic growth to meiotic differentiation (Honigberg and Purnapatre, 2003). The meiotic program can be divided into three major steps: (i) the meiotic prophase, during which after a single round of replication the homologous chromosomes pair and recombine, (ii) two nuclear divisions generating four haploid genomes, and (iii) spore formation and maturation (Mitchell, 1994).
One early landmark of the meiotic prophase is DNA replication, often called meiotic replication. Some important features distinguish meiotic from mitotic S phase. First, meiotic replication is strikingly longer (see the discussion in Cha et al, 2000). Second, the control of initiation displays some specific features. Whereas the S-phase function of the Clb5 and Clb6 cyclins can be fulfilled by the other B-type cyclins in mitotically growing cells, initiation of meiotic replication requires the activity of Clb5 and Clb6 (Stuart and Wittenberg, 1998). Meiosis also differs in the mechanism that controls the removal of the Cdk inhibitor Sic1 as it requires the action of the meiosis-specific kinase Ime2 (Dirick et al, 1998). Due to this specific genetic control, the onset of meiotic replication itself can be considered as a part of the sporulation differentiation process.
The formation of double-strand breaks (DSBs) constitutes the other meiotic-specific event experienced by the DNA. This process requires the meiotic replication as well as a meiosis-specific endonucleolytic activity (Baudat and Keeney, 2001). In the absence of S-phase cyclins (Stuart and Wittenberg, 1998), the meiotic replication does not occur and DSBs are not generated, in a manner independent of a replication checkpoint. Subsequent studies have led to the suggestion of a mechanistic coupling between meiotic replication and recombination initiation (Borde et al, 2000). The chromatin corresponding to future DSB sites exhibits an increased sensitivity to micrococcal nuclease (Ohta et al, 1994). These chromatin changes are temporally coupled to replication (Murakami et al, 2003) and do not occur in the absence of S-phase cyclins (Smith et al, 2001). Thus, the link between replication and DSB formation could involve chromatin reconfiguration. The meiosis-specific endonucleolytic cuts require several proteins, the majority of them being synthesized only during meiosis. One protein, Spo11, is suspected to have the endonuclease activity (Lichten, 2001).
A tightly coordinated transcriptional program accompanies the meiotic differentiation. Transcriptional analysis has led to group meiotic genes in various classes according to their timing of expression (Chu et al, 1998). The transcription factor Ime1 stimulates the expression of the early class of meiotic genes and thus is required for entry into the meiotic program (Vershon and Pierce, 2000). Early meiotic genes, which can be subdivided into subclasses (Chu et al, 1998), encode proteins that control the early steps of meiosis. Products of the middle genes are required for meiotic nuclear divisions and spore morphogenesis. Induction of the middle gene expression is controlled by the transcription factor Ndt80 (Chu and Herskowitz, 1998). Therefore, the normal progress of the transcriptional program constitutes the first control level for numerous processes during meiosis.
The budding yeast Set1 protein belongs to a complex of eight proteins, most of which are required to catalyze efficiently the methylation of lysine 4 on histone H3 (H3-K4) in vivo (Miller et al, 2001; Roguev et al, 2001; Krogan et al, 2002; Nagy et al, 2002). Each modified H3-K4 can be mono-, di-, or trimethylated and the state of methylation may be associated with specific biological responses. For example, it has been shown that trimethylation of H3-K4 was correlated with transcriptional activation (Santos-Rosa et al, 2002). Consistent with these results, Set1 has been found to be predominantly associated within the coding region of highly transcribed genes and the pattern of trimethylated H3-K4 correlates with Set1 occupancy (Ng et al, 2003). Together with the fact that Set1 interacts with the Paf1 complex involved in RNA polymerase II elongation (Krogan et al, 2003), it is thought that Set1 plays a role in transcription elongation. However, the functional consequence of the absence of Set1 on this process remains unknown. Set1 also regulates rDNA and telomeric silencing, telomere length, and DNA repair (Nislow et al, 1997; Corda et al, 1999; Roguev et al, 2001; Bryk et al, 2002).
The deletion of SET1 is associated with severe sporulation defects (Nislow et al, 1997). In this study, we have characterized the various defects in set1Δ diploid cells, by following different events that take place during the meiotic differentiation. Our results show that meiotic replication, meiotic DNA DSB formation, and gene expression are controlled by Set1.
Results
The loss of Set1 is associated with a delay of the meiotic S phase
The SET1 gene was deleted in the SK1 background. The sporulation of set1Δ diploid cells was found repeatedly to be less efficient and delayed compared to that of wild-type diploid cells. The sporulation delay was correlated to that of the progression through the first meiotic division, as assessed by DAPI staining (not shown). This led us to analyze meiotic replication, one of the earlier processes that occurs during the meiotic prophase.
As shown by the flow cytometry data presented in Figure 1, 4 h subsequent to the shift in sporulation medium, wild-type cells completed the meiotic S phase, whereas most set1Δ cells still have a 2n DNA content. In this case, the shift from 2n to 4n DNA content was detectable only after 4 h, with a large fraction of cells still in G1 at 10 h. Therefore, the onset of meiotic replication is largely delayed in set1Δ cells. Importantly, this delay was found to be meiosis-specific. If, after growth in presporulation medium, wild-type and set1Δ cells were returned to rich medium, they exhibited no difference of mitotic DNA replication kinetics (Figure 1). We concluded that Set1 is required for the efficient timing of meiotic DNA replication.
Figure 1.
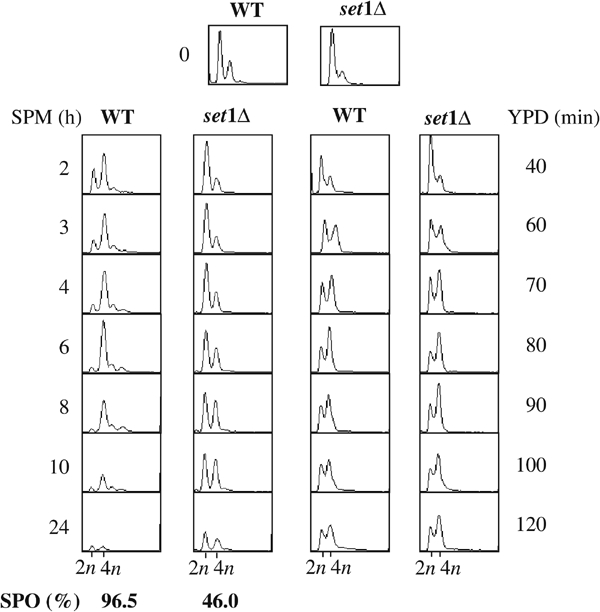
Meiotic DNA replication is delayed in set1Δ cells. Kinetics of DNA replication of SK1 WT and set1Δ diploid cells. After synchronization in YPA medium (time 0), cells were transferred either in sporulation medium (SPM, left) or rich medium (YPD, right) and replication was monitored by FACS analysis. Bottom left: sporulation rates were determined at 24 h.
The set1Δ meiotic S-phase delay reflects a defect in the initiation of DNA replication
Cells undergoing meiosis require Clb-dependent CDK activity. Two cyclins, Clb5 and to a lesser degree Clb6, play important roles in the initiation of meiotic replication (Stuart and Wittenberg, 1998). The activation of the Clb5/6-CDK complexes requires the degradation of the CDK inhibitor Sic1 (Dirick et al, 1998). To assess whether the delay of meiotic S-phase delay in set1Δ cells was due to a defect in DNA replication initiation, sporulation was induced in wild-type and set1Δ cells and samples collected at different times were analyzed for meiotic replication, Orc6 phosphorylation, and Clb5-associated kinase activity (Figure 2A). We considered Orc6 as a potential marker of meiotic DNA replication initiation because it has been shown that Orc6 is phosphorylated as cells initiate mitotic DNA synthesis, and dephosphorylated as cells exit mitosis and enter in G1 (Weinreich et al, 1999). Phosphorylation of Orc6, visualized by the mobility shift of the protein, was delayed in set1Δ by about 2 h (Figure 2A, 1 and 2). This correlates with the delay of entry in the S phase, and is consistent with the fact that deleting SET1 delays the initiation of meiotic DNA replication. The accumulation of Clb5myc and the Clb5-associated kinase activity was measured in the same cell extracts (Figure 2A, 3 and 4). We observed a slight defect in the accumulation of Clb5myc in set1Δ . However, the kinase activity (using histone H1 as a substrate) measured from Clb5-immunoprecipitates did not reveal a great difference between wild type and set1Δ (Figure 2A, 4). The results of Figure 2A indicate that the delayed Orc6 phosphorylation in set1Δ cells is not correlated to the Clb5 maximal activity. This suggests that the delay in meiotic replication might be independent of the Clb5 function.
Figure 2.
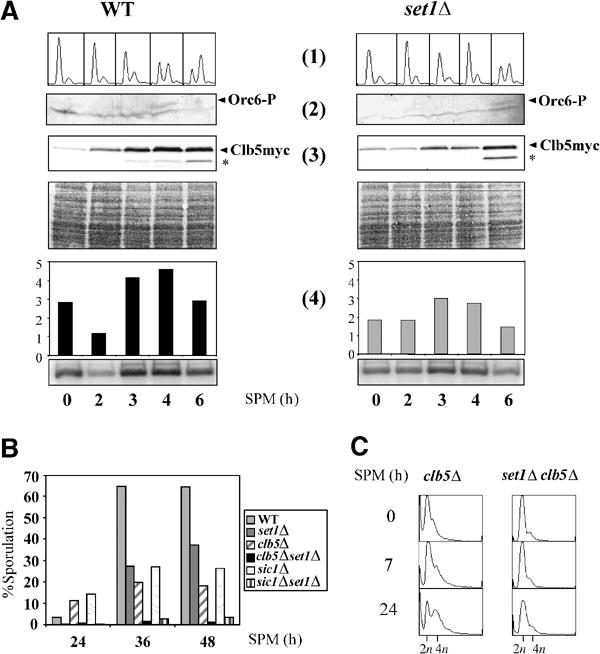
The initiation of meiotic DNA replication is delayed in set1Δ cells independently of the Clb5 function. (A) Sporulation was induced in WT CLB5myc and set1Δ CLB5myc cells. Samples were collected at the indicated times and used to analyze meiotic DNA replication (1), phosphorylation of Orc6 (2), Clb5myc accumulation (3), and kinase activity of immunoprecipitated Clb5myc (4). (1) Meiotic DNA replication was followed by FACS analysis; (2) kinetics of Orc6 phosphorylation was monitored by Western blot using anti-Orc6 antibodies (Weinreich et al, 1999); (3) Clb5myc accumulation was monitored by Western blot using anti-myc antibodies. Asterisk: degraded form of Clb5myc. Below: Ponceau red staining. (4) Kinase activity of the immunoprecipitated Clb5myc was assayed with histone H1 as a substrate. (B) Sporulation rates of the indicated strains were determined after 24, 36, and 48 h after plating on sporulation plates by measuring the ratio of sporulated versus non-sporulated diploid cells. (C) FACS analysis after induction of sporulation of clb5Δ and set1Δclb5Δ strains.
To further document this point, we next investigate the genetic interactions between Set1 and Clb5 in sporulation and meiotic replication. Sporulation rates of the set1Δ clb5Δ and set1Δ sic1Δ double mutants were clearly more affected than those of the single mutants, demonstrating a non-epistatic relationship (Figure 2B). This additivity of set1Δ and clb5Δ is also observed in meiotic replication, as inactivation of both, SET1 and CLB5, appears to block completely progression into the meiotic S phase (Figure 2C). Moreover, the limited replication seen with the set1Δ sic1Δ mutant never progressed further (not shown), a situation different from that described for the ime2Δ sic1Δ mutant (Dirick et al, 1998). Taken together, our analysis supports a model in which the meiotic defect caused by the absence of Set1 is likely to be independent of the Clb5 function.
The meiotic S-phase delay in set1Δ cells can be complemented by overproduction of the Set1 SET domain
We investigated the role of the Set1-dependent histone H3-K4 methylase activity in the meiotic replication. We introduced mutations within the SET domain of Set1 known to abolish the HMTase activity (Rea et al, 2000). The mutants carried a deletion of the conserved motifs NHCC and SEEL (residue 1016–1019 and 1047–1050, respectively). However, the corresponding Set1 mutant proteins were undetectable in SK1 (not shown). The same mutations only partially affected the Set1 protein level in an alternative strain background (DBY745) (not shown). We thus analyzed the behavior of H3-K4 methylase minus set1 mutants in DBY745. In this background, we also deleted the gene encoding Swd3, one component of the Set1 complex essential for the methylation of H3-K4 (Roguev et al, 2001). We failed to detect methylated H3-K4 in swd3Δ, set1ΔNHCC, and set1ΔSEEL mutants (Figure 3A); nevertheless, their meiotic DNA replication shows no set1Δ-like delay (Figure 3B). These data indicate that the HMTase activity of Set1 is not absolutely required for meiotic DNA replication. However, when we tested the substitution of the glycine 951 by a serine (G951S), which abolishes H3-K4 methylation (Figure 3A) without affecting the stability of Set1 (not shown), we observed a meiotic DNA replication defect similar to the one observed for set1Δ (Figure 3C). This result suggests that a function affected by the G951S mutation is involved in the control of DNA meiotic replication.
Figure 3.
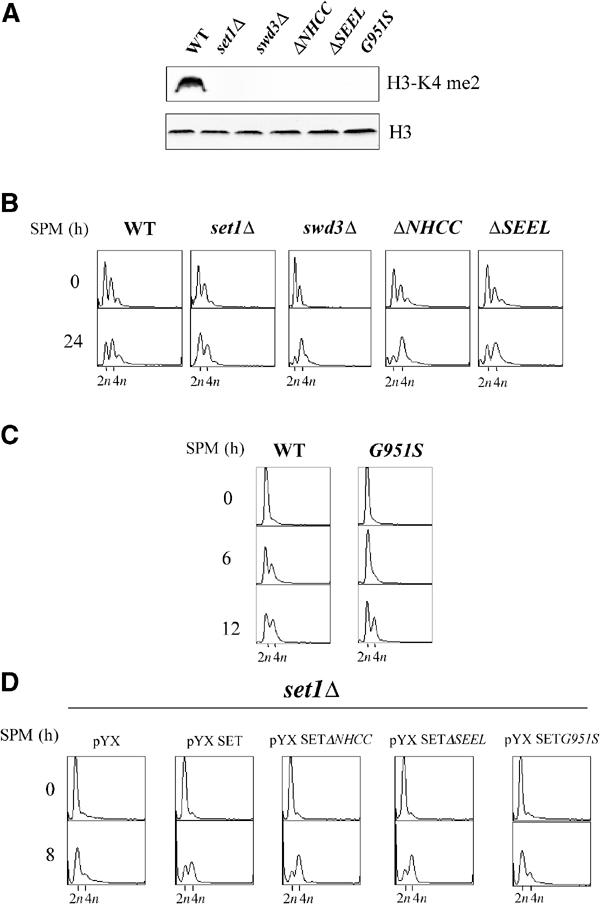
The role of Set1 in meiotic replication is independent of its HMTase activity but requires the presence of the SET domain. (A) Protein cell extracts from the indicated strains (DBY745 background) were subdivided and probed with dimethylated H3-K4 antiserum (top) or with C-term H3 antiserum (bottom). Strains carrying the deletion of the NHCC and SEEL motifs in the SET domain or the G951S substitution are denoted by set1ΔNHCC, set1ΔSEEL, and G951S, respectively. (B) Progression into meiotic DNA replication of WT, set1Δ, swd3Δ, set1ΔNHCC, and set1ΔSEEL was analyzed by FACS. (C) Progression into meiotic DNA replication of the G951S mutant. (D) The set1Δ diploid (SK1 background) was transformed by the vector pYX (pYX243), pYXSET900–1080, pYXSETΔNHCC, pYXSETΔSEEL, and pYXSETG951S directing the expression of the wild-type SET domain (900–1080) or of SET domains carrying the indicated mutations. Meiotic replication was analyzed by FACS analysis.
Despite the fact that swd3Δ, set1ΔNHCC, and set1ΔSEEL show no delayed meiotic DNA replication, the sporulation of all these mutants is affected. The appearance of spores is delayed and they accumulate at levels lower than wild-type levels, much like the set1Δ mutant (not shown). This suggests that, besides replication, some other set1Δ-induced defects in meiosis are due to the lack of H3-K4 methylation.
As we had previously observed that overexpression of the SET domain of Set1 rescued the set1Δ-induced telomeric silencing defect (Corda et al, 1999), we asked whether this could be the case for meiotic replication. We transformed a set1Δ SK1 diploid by a plasmid (pYX243-SET) that directs the synthesis of the isolated SET domain (residue 913–1080) under the control of the GAL promoter (Corda et al, 1999). As shown in Figure 3D, meiotic replication was restored by overexpression of SET domain. Overexpression of SET domain was without effect in wild-type cells (not shown). This result was confirmed using other constructs overexpressing Set1 fragments (Briggs et al, 2001). Indeed, overexpression of the C-terminal part of Set1 (Set1 900–1080) from the ADH1 promoter rescued the DNA replication defect, whereas overexpression of the N-terminal part (Set1 1–900) did not (Figure 4B, left). We concluded that this complementation was specific for the SET domain. Next, we introduced the set1ΔNHCC, set1ΔSEEL, and the G951S mutations into pYX243-SET. Consistent with the mutant phenotypes (see above), the set1ΔNHCC and set1ΔSEEL mutated SET domains complemented the DNA replication defect, whereas the G951S SET domain did not (Figure 3D). As shown previously (Briggs et al, 2001), neither the wild-type nor the mutated overexpressed SET domains produced from pYX243-SET were able to restore any H3-K4 methylation in set1Δ cells (not shown). This result shows that the SET domain, independently of its role in the HMTase activity of Set1, is sufficient for the meiotic replication function of Set1. This HMTase-independent activity of Set1 requires the presence of the glycine 951.
Figure 4.
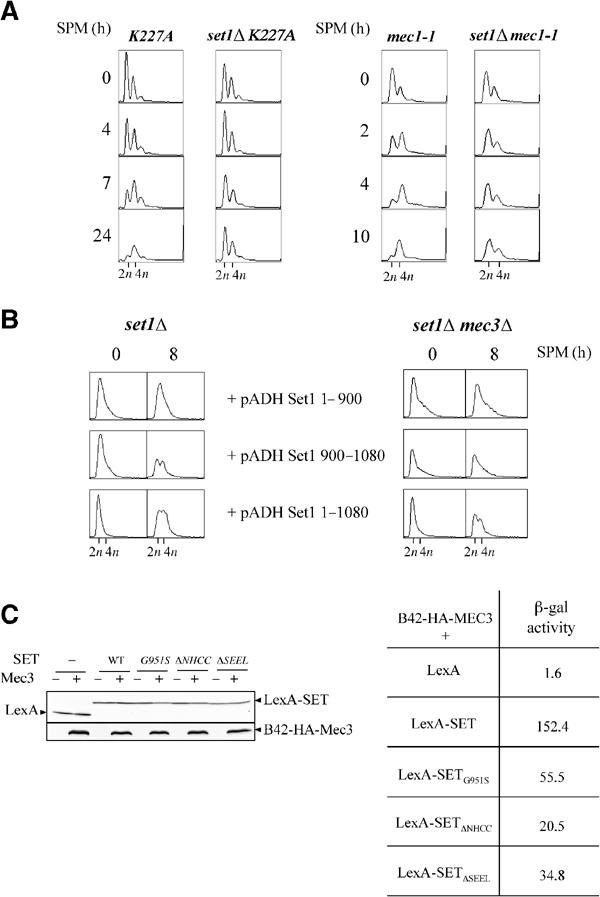
(A) Meiotic DNA replication is delayed in set1Δ cells independently of a checkpoint response. Meiotic DNA replication in strains with the K227A mutation (left, DBY745 background) and the mec1-1 mutation (right, SK1 background) was followed by FACS analysis. (B) Complementation by the SET domain requires Mec3. set1Δ and mec3Δset1Δ diploids (DBY745 background) were transformed by vectors overexpressing either the full-length Set1 (pADH Set1 1–1080) or fragments of Set1 extending from residue 1 to 900 (pADH Set1 1–900) or from 900 to 1080 (pADH Set1 900–1080) under the control of the ADH promoter (Bryk et al, 2002). Meiotic replication was analyzed by FACS analysis. (C) Interaction of wild-type and mutant SET domains with the C-terminal part of Mec3. Left: expression of the fusion proteins LexA-SET and B42-HA-Mec3 by Western blotting using an antibody against LexA or the HA epitope. Right: protein–protein interaction was monitored by measuring β-galactosidase activity (Miller units). The value represents the average of two measurements. Each measurement was made on five independent clones.
Delaying meiotic S phase in set1Δ does not depend on DNA-damage checkpoint proteins
In mitotic cells, the Mec1/Rad53 pathway is essential for checkpoint activation in response to DNA damage or replication block. In meiotic cells, different checkpoint pathways operate, which monitor DNA damage, incomplete replication, recombination, spindle formation, and chromosome segregation (Roeder and Bailis, 2000). The Mec1 protein kinase is one central component of the pachytene checkpoint response (Lydall et al, 1996), as well as of the meiotic S/M checkpoint (Stuart and Wittenberg, 1998). No role for the Rad53 kinase in a meiotic checkpoint has been described by now. A set1Δ-induced response was described in mitotic cells, which shares some components with the response produced by a DNA damage (Schramke et al, 2001). We tested whether activation of the Mec1/Rad53 pathway was responsible for delaying the meiotic DNA replication in set1Δ, either by introducing the rad53 K227A kinase-dead allele of RAD53 or by inactivating the MEC1 gene. The rad53 K227A and the mec1-1 diploids displayed normal meiotic replication kinetics. When introduced in the rad53 K227A or the mec1-1 mutant, the deletion of SET1 leads to a meiotic replication defect (Figure 4A). This defect appeared to be particularly severe with virtually no trace of replication at late times. This was correlated with an aggravation of the sporulation defect of the double mutants compared to set1Δ (not shown). These results show that the meiotic replication delay seen in set1Δ cells does not result from the activation of a Mec1/Rad53-dependent checkpoint.
It has been shown that Set1 interacts via its SET domain with the checkpoint protein Mec3 (Corda et al, 1999). We asked whether this interaction could have some functional implication during meiosis. Although meiotic replication occurs normally in the mec3Δ diploid, it is delayed in mec3Δ set1Δ (not shown). Interestingly, overexpression of the SET domain does not rescue the set1Δ-associated DNA replication defect in set1Δ mec3Δ cells, whereas it does in set1Δ cells (Figure 4B), indicating that the function of the SET domain depends on the presence of Mec3. We have investigated, via a two-hybrid approach, the effect of the mutations in the SET domain with respect to its interaction with Mec3. All the mutations lead to a decreased interaction of the SET domain with the C-terminal domain of Mec3, which is not related to differences in the amounts of the mutant SET domain fragments (Figure 4C). Importantly, the G951S mutation is not associated with a weaker interaction compared to the ΔNHCC and ΔSEEL mutations. Therefore, no correlation is evident between the capacity to sustain or not meiotic replication and the strength of interaction with Mec3. Thus, the only mutation that affects meiotic replication, that is, G951S, must affect a Set1 function independently of its interaction with Mec3.
Loss of Set1 affects meiotic DSB formation
The tight coupling existing between meiotic replication and meiotic DSB formation led us to analyze meiotic DSBs in set1Δ cells. In wild-type diploid SK1 cells, meiotic DSBs at the DEP1-CYS3 locus (Figure 5A) were detectable 4 h after transfer into sporulation medium (Figure 5B). In contrast, in set1Δ diploid cells, the DSB fragments were poorly detected, even after 24 h (Figure 5B). This was not only the consequence of an abnormal delay of meiotic replication as meiotic replication was completed between 6 and 8 h after transfer into sporulation medium (Figure 5C).
Figure 5.
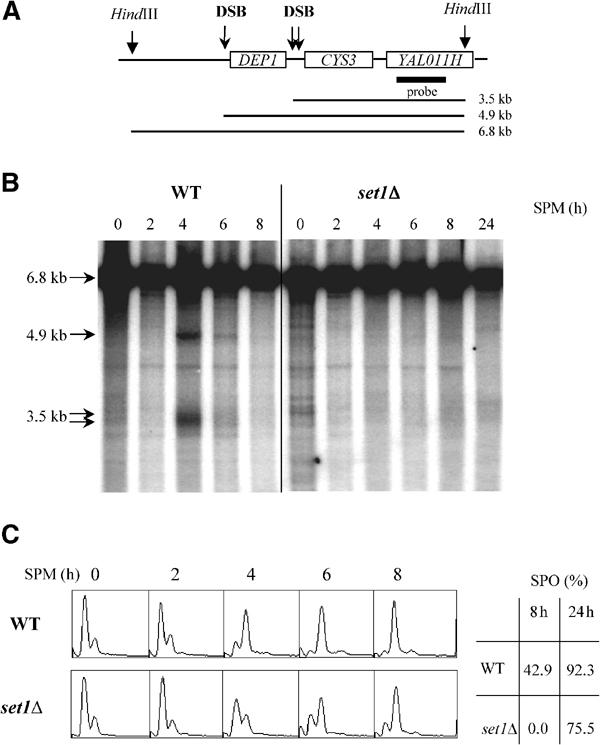
Reduced amounts of meiotic DSBs in set1Δ (SK1 background). (A) Map of the DEP1-CYS3 locus. The positions of the HindIII restriction sites and the sizes of the DNA fragments generated by the meiotic DSBs are indicated. The thick line represents the position of the DNA probe used for hybridization analysis. (B) Southern analysis of meiotic DSB kinetics. Genomic DNA from cells taken from sporulation medium (SPM) at the indicated times (hours) was extracted, digested with HindIII, and probed with a sequence specific to YAL011H. Arrows show the unbroken HindIII fragment and those corresponding to meiotic DSBs. (C) Left: FACS analysis were carried out to follow meiotic DNA replication in the same experiment as in (B) at the indicated times after shift in sporulation medium. Right: sporulation rates were determined after 8 and 24 h.
This very low steady-state level of DSBs in set1Δ could reflect either a decrease in the generation of DSBs or an acceleration of the repair of DSBs. To discriminate between the two alternatives, the formation of meiotic DSBs was further analyzed in the presence of the rad50S mutation (Figure 6). In this genetic context, DSBs are formed but not resected, and thus accumulate to high levels and persist throughout meiosis (Figure 6A, Cao et al, 1990). In contrast to rad50S, DSBs do not accumulate in set1Δ rad50S, showing that the set1Δ deletion is epistatic to the rad50S mutation (Figure 6A). This indicates that set1Δ affects the step of DSB formation. Whereas DSB formation was nearly absent, progression into meiotic program in set1Δ rad50S was only delayed, as shown by analysis of meiotic replication (Figure 6B) and of meiotic nuclear divisions (not shown). The set1Δ-induced defect in DSB formation was not restricted to the DEP1-CYS3 locus, as DSB fragments were poorly detectable at the YCR047c locus (not shown). Quantification of DSB levels at the two loci is shown in Figure 6C. We concluded that the loss of Set1 greatly affects DSB formation at different recombination hotspot loci. This defect in DSB formation does not appear to be the direct consequence of the delay in the onset of meiotic S phase.
Figure 6.
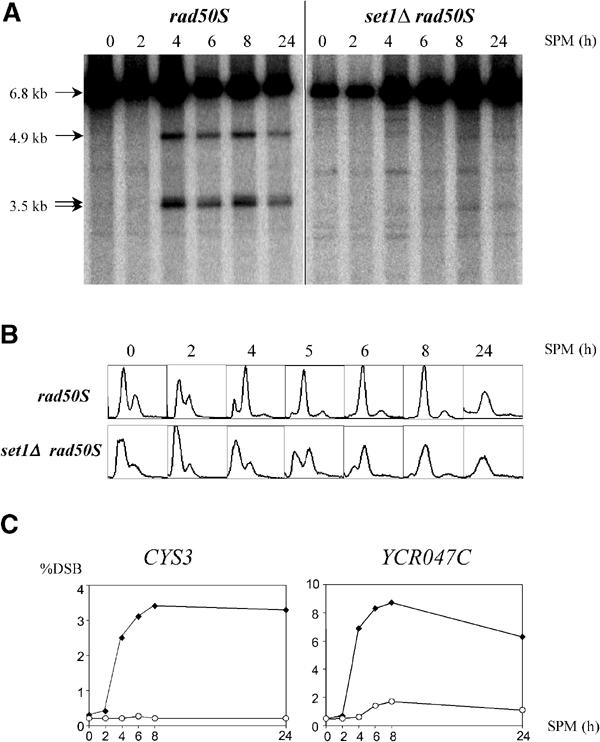
Levels of meiotic DSBs in the set1Δ rad50S mutant (SK1 background). (A) DSBs at the DEP1-CYS3 locus were analyzed as in Figure 5. (B) FACS analysis were carried out to follow meiotic DNA replication in the same experiment as in (A) at the indicated times after shift in sporulation medium. (C) Quantification of the DSB fragments to parental fragment at the DEP1-CYS3 locus and the YCR047c locus. Closed diamonds, rad50S; open circles, set1Δ rad50S.
H3-K4 methylation is required for the induction of middle meiotic gene expression
We have compared by DNA microarray analysis the temporal program of gene expression in wild-type, set1Δ, and swd3Δ SK1 cells at different times after the induction of sporulation. In the time courses used for transcription profiling, the meiotic replication kinetics of wild type and swd3Δ were identical, whereas that of set1Δ was delayed (Figure 7A). The expression level of various key genes of the meiotic program is presented for the 2 and 4 h time points (Figure 7B), that is, before meiotic S phase was evident in either strain and when it was completed in wild type and swd3Δ (Figure 7A). For each time point, the expression level corresponds to the ratio relative to the 0 h time point (see Materials and methods). IME2 is expressed very early after transfer to sporulation medium (Chu et al, 1998) and encodes a protein kinase specifically required for meiotic replication. The induction profile of this gene is completely similar in the three strains all along the time course (Figure 7B). Similarly, many genes involved in DNA replication are also induced early during sporulation (Chu et al, 1998). The average expression level of the corresponding genes (see REP on Figure 7B) was not affected by either swd3Δ or set1Δ. A small induction defect of the CLB5 gene was evident, but was similar for set1Δ and swd3Δ. Therefore, the meiotic replication delay in set1Δ cells could not be correlated to a specific transcriptional defect of any of the replication genes examined. In contrast, a delayed induction of transcription was evident for the early meiotic genes SPO11 and MEI4, whose products are required for the generation of DSBs (see DSB on Figure 7B). Such a delay could be involved, independently of or in addition to the replication delay, in the DSB formation defect seen in set1Δ (Figure 5).
Figure 7.
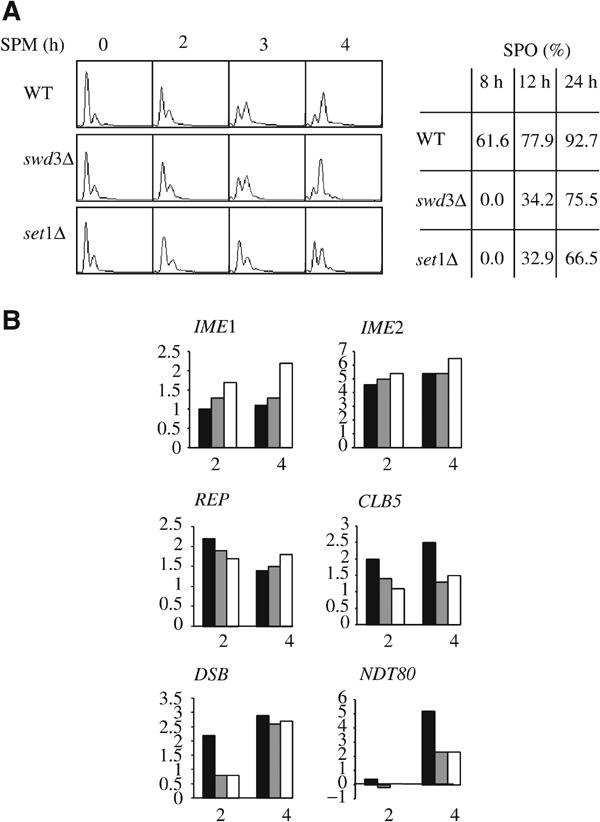
The delay of meiotic DNA replication is not correlated to a defect in gene expression (SK1 background). (A) Left: FACS analysis after induction of sporulation of WT, swd3Δ, and set1Δ strains. Right: sporulation rates at 8, 12, and 24 h. (B) Expression level of various key genes at 2 and 4 h after induction of sporulation. REP stands for many genes involved in DNA replication (i.e. RNR1, CDC21, RFA2, RFA3, POL1, POL30, MCM3, CDC47, and DBF4). DSB stands for SPO11 and MEI4, two genes involved in the generation of meiotic DSBs. Black bars: wild type; gray bars: swd3Δ; white bars: set1Δ.
Genes induced during meiosis have been previously classified into seven temporal patterns (Chu et al, 1998). The expression level of the genes belonging to these seven classes is presented for the 2 and 4 h time points (Figure 8). For commodity, genes in each class were ordered by their expression level in the wild type. At 2 h, with the exception of metabolic genes, the average expression level decreases according to the temporal class of genes (from early I to late). At this time point, the global transcription profiles of the three strains were quite similar. A minor difference was evident for early II, early—middle, and a fraction of middle genes that were slightly less expressed in set1Δ and swd3Δ than in the wild type. At 4 h, as expected, the expression level of genes belonging to the early–mid and middle classes was increased in the wild type. In set1Δ and swd3Δ, the expression level of metabolic and early genes was identical to that of the wild type. As reflected by SPO11 and MEI4 (see above), the defect in the induction of the early II genes in set1Δ and swd3Δ was transient, that is, visible at 2 h only. Importantly, the increase of early–mid and middle gene expression was very limited. Similar results were obtained by RT-PCR on individual genes in the DBY745 background (Supplementary Figure 1). Therefore, gene expression in set1Δ and swd3Δ appears to be differentially affected according to the class of genes considered.
Figure 8.
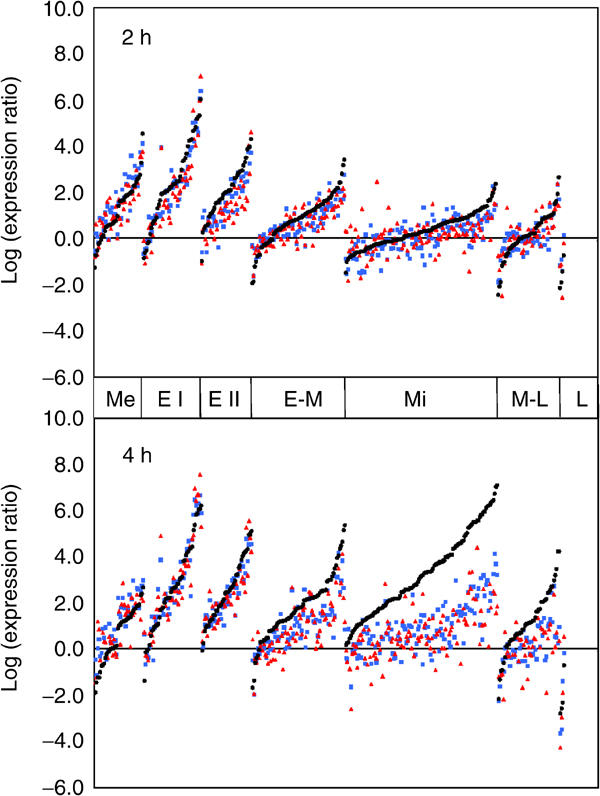
The absence of methylated H3-K4 is correlated to a defect in the induction of middle meiotic genes (SK1 background). The graph presents the expression level of the meiotic genes belonging to the different classes previously defined (Chu et al, 1998), 2 and 4 h after induction of sporulation of WT, swd3Δ, and set1Δ strains. Within each class and for each time point, the genes were ordered, from left to right, according to their expression level in the wild type. Log-expression levels of sporulation genes in the wild type, set1Δ, and swd3Δ are plotted as black circles, red triangles, and blue squares, respectively. Me: metabolic, E I: early I, E II: early II, E-M: early-mid, Mi: middle, M-L: mid-late, L: late (according to Chu et al, 1998).
Analysis of the genes corresponding to the key meiotic transcription factors revealed clear differences (Figure 7B). IME1, whose product is required for early meiotic gene expression, was expressed at higher levels in set1Δ and swd3Δ. In contrast, NDT80, which encodes a regulator of middle genes including itself, was expressed at lower levels in swd3Δ and set1Δ. This defect in NDT80 transcription could explain, at least in part, the reduced induction of middle gene transcription.
Discussion
Absence of Set1 is responsible for several meiotic defects
In the search for the molecular basis of the sporulation defect linked to the deletion of SET1, we have found that different aspects of the meiotic process are affected, that is, the meiotic replication, the formation of DSBs, and the expression of the middle meiotic genes. The meiotic phenotype of set1Δ differs from that associated with the deletion of SET3, which encodes another SET-domain-containing protein and for which a methyltransferase activity has not yet been demonstrated. Notably, contrary to Set1, Set3 is required for the repression of middle meiotic gene expression via its interaction with two potential histone deacetylases, Hos1 and Hst1, and the meiotic replication in set3Δ is not affected (Pijnappel et al, 2001).
Initiation of meiotic replication requires a non-canonical function of the SET domain
As suggested by several results, a defect in gene expression does not appear to be the leading cause of the meiotic replication delay. First, there is no defect in the induction of replication genes as well as IME2, encoding the meiosis-specific kinase essential for initiation of meiotic replication. Second, neither the ectopic expression of IME1 nor the deletion of RPD3, in order to induce or derepress early meiotic gene expression, can rescue the replication defect of set1Δ (not shown). Third, the meiotic replication phenotype of set1Δ is more severe in the absence of Mec1 or active Rad53. With regard to the function of Mec1 and Rad53 during replication (Lopes et al, 2001), it suggests that the loss of Set1 leads to a genuine replication defect. Fourth, the meiotic replication can be normal in the absence of H3-K4 methylation, whereas all the transcriptional effects of Set1 described to date are linked to the methylation of H3-K4 (Bernstein et al, 2002; Santos-Rosa et al, 2002). Finally, the swd3Δ and set1Δ transcriptional profiles are similar, whereas the meiotic replication kinetics of the two mutants differs.
As it is not primarily due to a defect in gene expression, what is the cause of the meiotic replication delay in set1Δ? The normal kinetics of Clb5-CDK activity induction in set1Δ contrasts with the delayed phosphorylation of Orc6. This suggests that a step just before the initiation of replication is affected. As the decision to enter meiotic S phase constitutes a major decision during the progression through meiosis, it is tempting to speculate that it requires an additional signal downstream Clb5-CDK activation. Interestingly, the complementation of set1Δ with the SET domain did not occur when the MEC3 gene is deleted. As the SET domain of Set1 interacts with the carboxy-terminal region of Mec3 (Corda et al, 1999), this interaction seems important for the meiotic replication function of Set1. Mec3 associates with two other proteins, Rad17 and Ddc1, to form a trimeric ring structurally and functionally related to PCNA. However, the possibility for a specific role of Mec3 in meiotic replication in a pathway that involves its interaction with the SET domain of Set1 is not supported by our data. First, the wild-type timing of mec3Δ meiotic replication indicates that Mec3 is dispensable. Second, double mutant combining the mec3Δ mutation with the set1ΔNHCC, the set1ΔSEEL or the swd3Δ mutations display a normal timing of meiotic replication, demonstrating that Mec3 is dispensable also in the absence of H3-K4 methylation (not shown). Third, no correlation is evident between the capacity of various mutant SET domains to sustain or not meiotic replication and the strength of their interaction with the C-terminal part of Mec3. Alternatively, the role of Mec3 could be aspecific. As Mec3 is dispensable as long as the full-length Set1 protein is present, one can imagine that Mec3 brings to the SET domain a function, such as the capacity to be imported into the nucleus, normally fulfilled by the N-terminal part of Set1.
Set1 is required for DSB formation
The question arises as to what extent the various phenotypic consequences associated to the deletion of SET1 are or not linked to one common primary cause. In fact, most of the processes affected in set1Δ are more or less interdependent. This is the case for meiotic replication and DSB formation. Thus, a delay in the onset of meiotic replication leads to a similar delay in DSB formation (Borde et al, 2000). Such a simple explanation is clearly not relevant in the case of set1Δ where no such kinetics relationship is evident. Although one cannot formally exclude a more complex relationship, this suggests that the loss of Set1 affects DSB formation independently of its effect on meiotic replication. In addition to replication, the formation of DSB requires the presence of meiotic-specific products, among which the Spo11 protein has a central role. In this respect, the delayed induction of SPO11 and MEI4 expression in set1Δ, as evidenced by global transcriptome analysis, may have some impact on the generation of DSBs. Strikingly, the good viability of set1Δ spores (not shown), which reflects an appropriate segregation of meiotic chromosomes, indicates that a sufficient number of DSBs are generated, and eventually repaired by homologous recombination to give full recombination products. Experiments are currently being performed to characterize more precisely how Set1 and H3-K4 methylation control the process of DSB formation.
Post-replication steps limit sporulation in set1Δ
Among the multiple consequences of the loss of Set1, some may contribute to the sporulation defect of set1Δ. The defect in DSB formation is probably not responsible for a block of progression into meiosis. The set1Δrad50Δ diploid shows the same sporulation defect as that of the set1Δ single mutant (not shown). As the rad50Δ mutation completely blocks the formation of DSBs without inhibiting the meiotic progression, this suggests that the sporulation defect of set1Δ is not the direct consequence of an anomaly in DSB metabolism. On the other hand, the sporulation of the HMTase mutants (set1ΔNHCC, set1ΔSEEL and swd3Δ) is much similar to that of set1Δ, that is, less spores are produced with a delayed kinetics, whereas only meiotic replication of set1Δ is delayed. Thus, it appears that a step following meiotic replication is limiting towards sporulation kinetics and rate. This step could be the expression of the middle, and perhaps late, genes whose normal induction requires methylated H3-K4.
Independent roles of Set1 during meiosis
Our study shows that the pleiotropic effects caused by the deletion of SET1 rely on different functions of Set1. The HMTase activity of Set1 is dispensable for meiotic DNA replication, but not for middle meiotic gene expression. Intriguingly, all the SET domain mutations that we have tested lead to an absence of detectable methylated H3-K4, but only the G951S substitution mimics the loss of Set1. Thus, this specific mutation may affect the HMTase-independent function of Set1 involved in meiotic replication, possibly by affecting the interaction with a protein. This protein does not appear to be Mec3. It has been shown that the Drosophila Trithorax SET domain was able to bind to the N-terminal tail of histone H3 in vitro and that this interaction was strongly affected by substituting the conserved glycine corresponding to G951 of the Set1 SET domain (Katsani et al, 2001). Assuming that these results can be transposed to Set1, this putative H3-binding activity of the Set1 SET domain could be involved in meiotic DNA replication. Unfortunately, we have not been able to detect a direct interaction between Set1 and H3 in vivo (Sollier et al, unpublished observations).
The similarity of the global meiotic transcriptomes of swd3Δ and set1Δ underlines the possible role of H3-K4 methylation in the transcription of middle genes that are expressed after meiotic replication. This brings a functional significance to the increase of SET1 expression during meiosis, the gene belonging to the early-middle class (Chu et al, 1998). In this context, methylated H3-K4 could be viewed as a mark of the transcriptional competence of the middle meiotic genes, that is, as a prerequisite for their full activation. However, the induction of this class of genes requires the meiosis-specific transcription factor Ndt80. Thus, the defect of middle gene induction in swd3Δ and set1Δ could be solely the consequence of the defect of NDT80 expression. As Ndt80 regulates its own expression, it is difficult to distinguish between the two possibilities.
An alternative explanation is conceivable, which involves Ndt80 as well. A checkpoint prevents cells from exiting from the pachytene stage of meiotic prophase until meiotic recombination or chromosome synapsis have been completed (Roeder and Bailis, 2000). Different observations suggest that the activity of Ndt80 is downregulated, most probably by post-translational modifications, in response to the activation of this pachytene checkpoint (Tung et al, 2000). Some anomaly in chromosome metabolism in set1Δ could activate, at least transiently, the pachytene checkpoint, which in turn would limit the induction of middle genes, including NDT80 itself. Interestingly, the Dot1 protein, which is required for the pachytene checkpoint (San-Segundo and Roeder, 2000), is also a H3 methyltransferase (van Leeuwen et al, 2002).
Materials and methods
Strains and plasmids
The genotypes of the yeast strains used in this study are listed in Table I. The set1Δ∷TRP1 and swd3Δ∷TRP1 null mutations were obtained after PCR amplifying a disruption cassette from plasmid pFA6a-TRP1. The set1Δ∷KAN and mec3Δ∷TRP1 null mutations were introduced as described (Corda et al, 1999). The RAD53K227A mutation was introduced using the EcoRI-linearized pCH8 plasmid (Pellicioli et al, 1999).
Table 1.
Yeast strains used in this study
| Strain | Relevant genotype | Source |
|---|---|---|
| SK1 background | ||
| ORT4601 | MATα ura3 lys2 ho∷LYS2 leu2-K arg4-nsp,bgl | A Nicolas |
| ORT4602 | MATa ura3 lys2 ho∷LYS2 leu2-R arg4-nsp,bgl | A Nicolas |
| ORT4607 | MATa ura3 lys2 ho∷LYS2 leu2∷hisG his4x∷LEU2∷URA3 arg4-nsp mec1-1 smlX | A Nicolas |
| ORT4608 | MATα ura3 lys2 ho∷LYS2 leu2∷hisG his4B∷LEU2 arg4-bgl mec1-1 smlX | A Nicolas |
| JSK1 | ORT4601 × ORT 4602 | This study |
| JSK2 | ORT4601 set1∷KAN × ORT 4602 set1∷KAN | This study |
| JSK3 | ORT4607 × ORT4608 | This study |
| JSK4 | ORT4607 set1∷KAN × ORT 4608 set1∷KAN | This study |
| JSK5 | ORT4601 swd3∷KAN × ORT 4602 swd3∷KAN | This study |
| JSK6 | ORT4601 CLB5-MYC × ORT 4602 CLB5-MYC | This study |
| JSK7 | ORT4601 set1∷KAN CLB5-MYC × ORT 4602 set1∷KAN CLB5-MYC | This study |
| 492 | MATa ura3 his4 ho∷LYS2 leu2∷hisG sic1∷URA3 | L Dirick |
| 494 | MATα ura3 his4 ho∷LYS2 leu2∷hisG sic1∷URA3 | L Dirick |
| 917 | MATa ura3 trp1 ho∷LYS2 leu2∷hisG clb5∷URA3 | L Dirick |
| 917α | MATα ura3 arg4 ho∷LYS2 leu2∷hisG clb5∷URA3 | This study |
| JSK8 | 492 × 494 | This study |
| JSK9 | 492 set1∷KAN × 494 set1∷KAN | This study |
| JSK10 | 917 × 917α | This study |
| JSK11 | 917 set1∷KAN × 917α set1∷KAN | This study |
| DBY745 background | ||
| JKM179 | MATα Δho Δhml∷ADE1 Δhmr∷ADE1 ade1-100 leu2-3,112 lys5 trp1∷hisG ura3-52 | J Haber |
| JKM139 | MATa Δho Δhml∷ADE1 Δhmr∷ADE1 ade1-100 leu2-3,112 lys5 trp1∷hisG ura3-52 | J Haber |
| JSJ1 | JKM179 × JKM139 | This study |
| JSJ2 | JKM179 set1∷TRP1 × JKM139 set1∷TRP1 | This study |
| JSJ3 | JKM179 mec3∷TRP1 × JKM139 mec3∷TRP1 | This study |
| JSJ4 | JKM179 set1∷KAN mec3∷TRP1 × JKM139 set1∷KAN mec3∷TRP1 | This study |
| JSJ5 | JKM179 RAD53K227A∷KAN × JKM139 RAD53K227A∷KAN | This study |
| JSJ6 | JKM179 set1∷TRP1 RAD53K227A∷KAN × JKM139 set1∷TRP1 RAD53K227A∷KAN | This study |
| JSJ7 | JKM179 URA3-TEL swd3∷TRP1 × JKM139 URA3-TEL swd3∷TRP1 | This study |
| JSJ8 | JKM179 URA3-TEL SET1-MYC × JKM139 URA3-TEL SET1-MYC | This study |
| JSJ9 | JSJ8 but homozygous set1Δ(NHCC)∷LEU2 | This study |
| JSJ10 | JSJ8 but homozygous set1Δ(SEEL)∷LEU2 | This study |
| JSJ11 | JSJ8 but homozygous set1G951S∷LEU2 | This study |
The G951S mutation and the ΔNHCC and ΔSEEL deletions were introduced in the SET1 gene by mutagenesis (QuickChange Site-directed Mutagenesis Kit of Stratagene). After sequencing, plasmids carrying the correct mutations were introduced in strains harboring an N-terminal myc-tagged version of SET1. The presence of the mutant proteins was confirmed by Western blot using a monoclonal anti-myc antibody (9E10).
A myc epitope was introduced into the C-terminal part of Clb5 using the plasmid pFA6a-13myc-KanMX6. Tagging was confirmed by PCR and immunoblotting using 9E10 antibody.
For complementation studies, JSK2 was transformed by pYX243, pYX243-SET1, pYX243-SET1Δ(NHCC), pYX243-SET1Δ(SEEL), or pYX243-SET1G951S, which directs the synthesis of the isolated SET domain (residues 914–1080) under the control of the GAL promoter. JSJ2 and JSJ4 were transformed by plasmids pRS416-SET1(1–1080), pRS416-SET1(900–1080), or pRS416-SET1(1–900), which directs the synthesis of the corresponding Set1 fragments under the control of the ADH promoter (Briggs et al, 2001).
Sporulation procedures
After growth in rich glucose medium (YPD), exponential phase cells were pregrown in rich acetate medium (YPA; 1% potassium acetate, 2% bacto-peptone, 1% bacto yeast extract supplemented with 25% amino acids) during 8 h, then diluted at 2 × 106 cells/ml and grown in YPA during 14 h. Cells were washed once with water and then inoculated into sporulation medium (1% potassium acetate supplemented with 25% amino acids) and incubated at 30°C with vigorous agitation.
For complementation studies, cells grown on SGal-leu (pYX243) or SD-ura (pRS416) plates during 3–4 days were directly resuspended in sporulation medium. Alternatively, cells were directly striked from YPD plates on sporulation medium plates (1% potassium acetate supplemented with 25% amino acids). Sporulation rate was determined by counting asci visualized by light microscopy.
Analysis of DNA content and of meiotic DSBs
Meiotic cells were fixed in 70% ethanol. After rehydration in PBS, the samples were incubated at least 2 h with RNase A (1 mg/ml) at 37°C. Cells were resuspended in 50 μg/ml propidium iodide in PBS for at least 15 min at room temperature. After a wash in PBS, cells were resuspended in 5 μg/ml propidium iodide, sonicated briefly to remove cell clumps if required, and the DNA content was determined by FACS with a Becton Dickinson FASCalibur. For meiotic DSBs analysis, DNA extracted from meiotic cells was digested, fractionated by electrophoresis, transferred to a nylon membrane, and probed with a radiolabelled specific DNA fragment. ImageQuant (Molecular Dynamics) was used to quantify the major DSB fragment at the DEP1-CYS3 locus (3.5 kb) and at the YCR047C locus (3.7 kb).
Immunoprecipitation and kinase assays
Cells (20 units) were harvested, rinsed with PBS, and resuspended in lysis buffer (Tris–HCl (pH 7.5) 50 mM, NaCl 250 mM, EDTA (pH 8) 5 mM, NP40 0.1%, NaVO4 1 mM, PMSF 1 mM, and protease inhibitors). Cells were then broken with glass beads, using a Vortex agitator at 4°C, for 30 min and microcentrifuged for 15 min. Supernatant protein concentration was assayed by the Bradford method. Clb5myc was immunoprecipitated from 1 mg of extract by addition of 9E10 beads (anti-myc monoclonal antibody covalently coupled to protein A-sepharose). After 3 h at 4°C, anti-myc beads were collected, washed three times with lysis buffer, and divided into two portions. One portion was assayed for H1 kinase activity in a 10 μl reaction volume composed of 2 μg of H1 histone, 10 μCi of ATP-γP32, 100 μM of cold ATP in MgCl2 buffer (Tris–HCl (pH 7.2) 50 mM, MgCl2 5 mM, KCl 25 mM). Samples were incubated for 30 min at 30°C and denaturated in loading buffer during 5 min at 100°C. The reaction mixture was then separated by SDS–polyacrylamide gel electrophoresis and revealed by autoradiography. Quantitation data were obtained using ImageQuant version 1.2 program. The other portion was analyzed by Western blot for the presence of Clb5myc with anti-myc antibody.
Analysis of H3-K4 methylation
NaOH 10 N (18.5 μl) was added to 1 ml of saturated culture. After 10 min at 4°C, 50 μl of TCA 100% was added. After 10 min at 4°C, extracts were centrifuged for 5 min at 13 000 rpm. The pellets were resuspended in loading buffer and denatured for 5 min at 100°C. Samples were separated by SDS–polyacrylamide gel electrophoresis (12.5%), transferred to a nitrocellulose membrane, and probed with polyclonal anti-dimethyl-histone H3 (Lys4) antibody from Upstate biotechnology (1:2500).
Two-hybrid assay
The EGY48Z strain was transformed by pEG202 alone or pEG202 expressing the wild-type and mutant SET domains (914–1080) fused to LexA (1–202), together with pJG4.5 alone or pJG4.5 expressing the HA-tagged C-terminal domain of Mec3 (324–474) fused to the B42 activation domain.
DNA microarrays
Yeast-coding regions were amplified by PCR. The resulting set containing more than 6200 PCR fragments (representing 93% of the ORFs) were printed on Cornings Gaps II slides. Strains JSK1, JSK2, and JSK5 were used for the meiotic time-course experiment. Samples were taken as the time 0 h from the presporulation culture just before centrifugation, then at 1, 2, 3, and 4 h after transfer into sporulation medium. Samples of saturated YPD cultures were taken as non-sporulating controls. For RNA extraction, 50 OD units of cells were harvested, washed with DEPC-treated H2O, and immediately frozen in liquid nitrogen. Total RNA, isolated by acid phenol extraction, was used for Poly(A)+RNA isolation with the Micro-Fast Track 2.0 kit (Invitrogen) following the manufacturer's protocol for yeast mRNA. A measure of 2 μg of mRNA for each time point was used for cDNA synthesis with incorporation of aminoallyl-dUTP (Sigma), then labeled with Cy5 dye (Amersham). A reference mRNA mix was prepared for each kinetics experiment by pooling equal amounts of mRNA from every time point of the considered time course, except for t=0 h, which was added four times the amount of the other samples. Probes were prepared from the pooled reference mix, labeled with Cy3 dye, aliquoted and hybridized against the Cy5-labeled probes from each time point. After hybridization, microarrays were washed and scanned with a GenePix 4000A scanner and data were quantified using GenePix Pro 3.0 software (Axon Instruments). Intensity ratios were normalized using the Lowess algorithm implemented in BASE (Saal et al, 2002). All the microarray data are presented at http://www.ebi.ac.uk/arrayexpr ess/ (Username: e-mexp-41, Password: sP73X5h).
Analysis of sporulation genes
Sporulation-specific genes were those previously defined in a whole-genome expression study (Chu et al, 1998). Genes overlapping with another open reading frame or with a BLAST hit to another gene within the yeast genome (as indicated in the data set from Chu et al) were discarded from the analysis. Genes that were not clearly identified by PCR for the microarray used in this study were also excluded. This left 371 genes that were first grouped according to their sporulation class (Chu et al, 1998) and then ordered by their log-expression level of the wild type (black circles in Figure 8).
Supplementary Material
Supplementary Figure 1
Acknowledgments
We thank B Stillman for the anti-Orc6 antibody, A Verreault for the C-term H3 antiserum, V Borde for the wild-type and mec1-1 SK1 haploid strains, L Dirick for the clb5Δ and the sic1Δ SK1 haploid strains, and D Allis for the Set1-expression pADH vectors. We thank F Fabre and E Schwob for helpful discussions. Work in the laboratory of VG was supported by l'Association pour la Recherche sur le Cancer and by le Ministère de la Recherche et des Nouvelles Technologies (Action Concertée Incitative ‘Biologie cellulaire, moléculaire et structurale'). JS was supported by la Ligue Nationale Contre le Cancer and l'Association pour la Recherche sur le Cancer. WL was supported by l'Association pour la Recherche sur le Cancer. CS was supported by a postdoctoral fellowship from CEA. All material requests should be addressed either to VG or CdLRSA.
References
- Baudat F, Keeney S (2001) Meiotic recombination: making and breaking go hand in hand. Curr Biol 23: R45–R48 [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Humphrey EL, Erlich RL, Schneider R, Bouman P, Liu JS, Kouzarides T, Schreiber SL (2002) Methylation of histone H3 Lys 4 in coding regions of active genes. Proc Natl Acad Sci USA 99: 8695–8700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borde V, Goldman AS, Lichten M (2000) Direct coupling between meiotic DNA replication and recombination initiation. Science 290: 806–809 [DOI] [PubMed] [Google Scholar]
- Briggs SD, Bryk M, Strahl BD, Cheung WL, Davie JK, Dent SY, Winston F, Allis CD (2001) Histone H3 lysine 4 methylation is mediated by Set1 and required for cell growth and rDNA silencing in Saccharomyces cerevisiae. Genes Dev 15: 3286–3295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryk M, Briggs SD, Strahl BD, Curcio MJ, Allis CD, Winston F (2002) Evidence that Set1, a factor required for methylation of histone H3, regulates rDNA silencing in S. cerevisiae by a Sir2-independent mechanism. Curr Biol 12: 165–170 [DOI] [PubMed] [Google Scholar]
- Cao L, Alani E, Kleckner N (1990) A pathway for generation and processing of double-strand breaks during meiotic recombination in S. cerevisiae. Cell 61: 1089–1101 [DOI] [PubMed] [Google Scholar]
- Cha RS, Weiner BM, Keeney S, Dekker J, Kleckner N (2000) Progression of meiotic DNA replication is modulated by interchromosomal interaction proteins, negatively by Spo11p and positively by Rec8p. Genes Dev 14: 493–503 [PMC free article] [PubMed] [Google Scholar]
- Chu S, DeRisi J, Eisen M, Mulholland J, Botstein D, Brown PO, Herskowitz I (1998) The transcriptional program of sporulation in budding yeast. Science 282: 699–705 [DOI] [PubMed] [Google Scholar]
- Chu S, Herskowitz I (1998) Gametogenesis in yeast is regulated by a transcriptional cascade dependent on Ndt80. Mol Cell 1: 685–696 [DOI] [PubMed] [Google Scholar]
- Corda Y, Schramke V, Longhese MP, Smokvina T, Paciotti V, Brevet V, Gilson E, Geli V (1999) Interaction between Set1p and checkpoint protein Mec3p in DNA repair and telomere functions. Nat Genet 21: 204–208 [DOI] [PubMed] [Google Scholar]
- Dirick L, Goetsch L, Ammerer G, Byers B (1998) Regulation of meiotic S phase by Ime2 and a Clb5,6-associated kinase in Saccharomyces cerevisiae. Science 281: 1854–1857 [DOI] [PubMed] [Google Scholar]
- Honigberg SM, Purnapatre K (2003) Signal pathway integration in the switch from the mitotic cell cycle to meiosis in yeast. J Cell Sci 116: 2137–2147 [DOI] [PubMed] [Google Scholar]
- Katsani KR, Arredondo JJ, Kal AJ, Verrijzer CP (2001) A homeotic mutation in the trithorax SET domain impedes histone binding. Genes Dev 15: 2197–2202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogan NJ, Dover J, Khorrami S, Greenblatt JF, Schneider J, Johnston M, Shilatifard A (2002) COMPASS, a histone H3 (Lysine 4) methyltransferase required for telomeric silencing of gene expression. J Biol Chem 277: 10753–10765 [DOI] [PubMed] [Google Scholar]
- Krogan NJ, Dover J, Wood A, Schneider J, Heidt J, Boateng MA, Dean K, Ryan OW, Golshani A, Johnston M, Greenblatt JF, Shilatifard A (2003) The Paf1 complex is required for histone H3 methylation by COMPASS and Dot1p: linking transcriptional elongation to histone methylation. Mol Cell 11: 721–729 [DOI] [PubMed] [Google Scholar]
- Lichten M (2001) Meiotic recombination: breaking the genome to save it. Curr Biol 11: R253–R256 [DOI] [PubMed] [Google Scholar]
- Lopes M, Cotta-Ramusino C, Pellicioli A, Liberi G, Plevani P, Muzi-Falconi M, Newlon CS, Foiani M (2001) The DNA replication checkpoint response stabilizes stalled replication forks. Nature 412: 557–561 [DOI] [PubMed] [Google Scholar]
- Lydall D, Nikolsky Y, Bishop DK, Weinert T (1996) A meiotic recombination checkpoint controlled by mitotic checkpoint genes. Nature 383: 840–843 [DOI] [PubMed] [Google Scholar]
- Miller T, Krogan NJ, Dover J, Erdjument-Bromage H, Tempst P, Johnston M, Greenblatt JF, Shilatifard A (2001) COMPASS: a complex of proteins associated with a trithorax-related SET domain protein. Proc Natl Acad Sci USA 98: 12902–12907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell AP (1994) Control of meiotic gene expression in Saccharomyces cerevisiae. Microbiol Rev 58: 56–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami H, Borde V, Shibata T, Lichten M, Ohta K (2003) Correlation between premeiotic DNA replication and chromatin transition at yeast recombination initiation sites. Nucleic Acids Res 31: 4085–4090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy PL, Griesenbeck J, Kornberg RD, Cleary ML (2002) A trithorax-group complex purified from Saccharomyces cerevisiae is required for methylation of histone H3. Proc Natl Acad Sci USA 99: 90–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng HH, Robert F, Young RA, Struhl K (2003) Targeted recruitment of Set1 histone methylase by elongating PolII provides a localized mark and memory of recent transcriptional activity. Mol Cell 11: 709–719 [DOI] [PubMed] [Google Scholar]
- Nislow C, Ray E, Pillus L (1997) SET1, a yeast member of the trithorax family, functions in transcriptional silencing and diverse cellular processes. Mol Biol Cell 8: 2421–2436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta K, Shibata T, Nicolas A (1994) Changes in chromatin structure at recombination initiation sites during yeast meiosis. EMBO J 13: 5754–5763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellicioli A, Lucca C, Liberi G, Marini F, Lopes M, Plevani P, Romano A, Di Fiore PP, Foiani M (1999) Activation of Rad53 kinase in response to DNA damage and its effect in modulating phosphorylation of the lagging strand DNA polymerase. EMBO J 18: 6561–6572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pijnappel WW, Schaft D, Roguev A, Shevchenko A, Tekotte H, Wilm M, Rigaut G, Seraphin B, Aasland R, Stewart AF (2001) The S. cerevisiae SET3 complex includes two histone deacetylases, Hos2 and Hst1, and is a meiotic-specific repressor of the sporulation gene program. Genes Dev 15: 2991–3004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea S, Eisenhaber F, O'Carroll D, Strahl BD, Sun ZW, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD, Jenuwein T (2000) Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406: 593–599 [DOI] [PubMed] [Google Scholar]
- Roeder GS, Bailis JM (2000) The pachytene checkpoint. Trends Genet 16: 395–403 [DOI] [PubMed] [Google Scholar]
- Roguev A, Schaft D, Shevchenko A, Pijnappel WW, Wilm M, Aasland R, Stewart AF (2001) The Saccharomyces cerevisiae Set1 complex includes an Ash2 homologue and methylates histone 3 lysine 4. EMBO J 20: 7137–7148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saal LH, Troein C, Vallon-Christersson J, Gruvberger S, Borg A, Peterson C (2002) BioArray Software Environment (BASE): a platform for comprehensive management and analysis of microarray data. Genome Biol 3, SOFTWARE0003: 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- San-Segundo PA, Roeder GS (2000) Role for the silencing protein Dot1 in meiotic checkpoint control. Mol Biol Cell 11: 3601–3615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NC, Schreiber SL, Mellor J, Kouzarides T (2002) Active genes are tri-methylated at K4 of histone H3. Nature 419: 407–411 [DOI] [PubMed] [Google Scholar]
- Schramke V, Neecke H, Brevet V, Corda Y, Lucchini G, Longhese MP, Gilson E, Géli V (2001) The set1Delta mutation unveils a novel signaling pathway relayed by the Rad53-dependent hyperphosphorylation of replication protein A that leads to transcriptional activation of repair genes. Genes Dev 15: 1845–1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KN, Penkner A, Ohta K, Klein F, Nicolas A (2001) B-type cyclins CLB5 and CLB6 control the initiation of recombination and synaptonemal complex formation in yeast meiosis. Curr Biol 11: 88–97 [DOI] [PubMed] [Google Scholar]
- Stuart D, Wittenberg C (1998) CLB5 and CLB6 are required for meiotic DNA replication and activation of the meiotic S/M checkpoint. Genes Dev 12: 2698–2710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung KS, Hong EJ, Roeder GS (2000) The pachytene checkpoint prevents accumulation and phosphorylation of the meiosis-specific transcription factor Ndt80. Proc Natl Acad Sci USA 97: 12187–12192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen F, Gafken PR, Gottschling DE (2002) Dot1p modulates silencing in yeast by methylation of the nucleosome core. Cell 109: 745–756 [DOI] [PubMed] [Google Scholar]
- Vershon AK, Pierce M (2000) Transcriptional regulation of meiosis in yeast. Curr Opin Cell Biol 12: 334–339 [DOI] [PubMed] [Google Scholar]
- Weinreich M, Liang C, Stillman B (1999) The Cdc6p nucleotide-binding motif is required for loading mcm proteins onto chromatin. Proc Natl Acad Sci USA 96: 441–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1