

Abstract
Vascular calcification is an actively regulated process that culminates in organized extracellular matrix mineral deposition by osteoblast-like cells. The origins of the osteoblastic cells involved in this process and the underlying mechanisms remain to be defined. We previously revealed that active transforming growth factor (TGFβ) released from the injured arteries mobilizes mesenchymal stem cells (MSCs) to the blood stream and recruits the cells to the injured vessels for neointima formation. In this study, we used a low-density lipoprotein receptor (LDLR)-deficient mouse model (ldlr−/−), which develop progressive arterial calcification after having fed high-fat western diets (HFD), to examine whether TGFβ is involved in the mobilization of MSCs during vascular calcification. Nestin+/Sca1+ cells were recruited to the diseased aorta at earlier time points, and osteocalcin+ osteoblasts and the aortic calcification were seen at later time point in these mice. Importantly, we generated parabiotic pairs with shared blood circulation by crossing ldlr−/−mice fed HFD with transgenic mice, in which all the MSC-derived cells were fluorescently labeled. The labeled cells were detected not only in the peripheral blood but also in the arterial lesions in ldlr−/− mouse partners, and these blood circulation-originated cells gave rise to Ocn+ osteoblastic cells at the arterial lesions. Both active TGFβ1 levels and MSCs in circulating blood were upregulated at the same time points when these cells appeared at the aortic tissue. Further, conditioned medium prepared by incubating the aortae from ldlr−/−mice fed HFD stimulated the migration of MSCs in the ex vivo transwell assays, and either TGFβ neutralizing antibody or the inhibitor of TGFβ Receptor I kinase (TβRI) antagonized this effect. Importantly, treatment of the mice with TβRI inhibitor blocked elevated blood MSC numbers and their recruitment to the arterial lesions. These findings suggest that TGFβ-recruited MSCs to the diseased vasculature contribute to the development of osteogenic vascular calcification.
Introduction
Vascular calcification is prevalent in cardiovascular disease, diabetes, and chronic kidney disease [1–3]. The health impact of vascular calcification is also striking: increased morbidity of hypertension, left ventricular hypertrophy, and heart failure caused by aortic stiffness in addition to increased frequencies of myocardial infarction, stroke, amputation caused by plaque instability, and rupture in small arteries [4–8]. The mechanisms involved in the pathogenesis of vascular calcification remain largely unknown, and no therapy is currently available to prevent and reverse the calcification of vasculature. Early theories considered vascular calcification to be a passive process that occurred as a nonspecific response to tissue injury or necrosis; however, evidences over the past two decades suggest that it is an actively and tightly regulated process as recapitulation of bone formation through deposition of minerals in extracellular matrix by osteoblast-like cells [9–11].
The origins of the osteoblastic cells in arteries are controversial and remain to be defined. It has been proposed that most of the osteochondrogenic cells in media layer may be transdifferentiated from medial smooth muscle cells in situ [12]. Other studies have demonstrated the mesenchymal-derived progenitor cells in the vessel wall, including microvascular pericytes, retain an osteochondrogenic potential [13,14] and are most likely involved in vascular calcification. Moreover, a recent “Circulating Cell Theory” suggested that bone marrow-derived circulating stem cells/osteoprogenitors home to diseased arteries and contribute to vascular calcification [15–18]. Mesenchymal stem cells (MSCs), in particular, have the ability to differentiate into many cell types such as osteoblastic- and chondrogenic-lineage of cells. Both human and animal studies demonstrated that MSCs/osteoprogenitors can be mobilized into blood circulation [19–24]. Supporting this notion, our recent study demonstrated that MSCs were mobilized into the blood stream and further migrated to the injured vessel wall [25], where they commit and differentiate into either endothelial cells to repair the injured vessels or fibroblasts/myofibroblasts to form a pathological intima hyperplasia [25]. Therefore, it is most likely that the migration of remote MSCs from their original niche with subsequent activation toward osteoblastic cells at the diseased vessels also plays a role in the vascular calcification process.
It is believed that pro-migratory factor(s) released from damaged/inflammatory tissue or cells stimulate the directed migration of stem cells from peripheral blood or surrounding tissue to the target sites [26–29]. These migratory factor(s) may also diffuse into circulation and lead to the mobilization of the cells from their original niche into peripheral blood. We recently revealed that active transforming growth factor 1 (TGFβ1) released from tissue matrix in response to tissue remodeling or injury acts as a pro-migratory factor to induce MSC migration to participate in normal or pathological tissue repair/remodeling [25,30]. Moreover, active TGFβ also regulates the mobilization of these cells in the process [25]. Thus, unlike hematopoitic stem cells, the mobilization of which is regulated by granulocyte colony-stimulating factor (G-CSF) through disruption of CXCR4-CXCL12 axis in bone marrow, MSCs are mainly mobilized by active TGFβ. Several studies showing that G-CSF failed to stimulate MSC mobilization [31–33] support the view that the mobilization of discrete subsets of stem cells is regulated through different mechanisms.
On the basis of these findings, in the present study we asked whether active TGFβ is also a key factor that regulates the mobilization of MSCs and their migration to participate in osteogenic vascular calcification. Importantly, elevated TGFβ production or activation has been implicated in the development of many vascular disorders including vascular calcification [34,35]. Specifically, elevated TGFβ level and TGFβ bioavailability to activate downstream signaling were observed in calcified aortas [34–37]. However, the mechanisms by which TGFβ contributes to the pathogenesis of vascular calcification are largely unknown. Here, we took advantage of a low-density lipoprotein receptor (LDLR)-deficient mouse model (ldlr−/−) to determine the involvement of MSCs in the development of arterial calcification and how TGFβ regulates this process. When ldlr−/−mice are fed high-fat western diet (HFD), male animals develop obesity, diabetes, hyperlipidemia, and atherosclerosis, with progressively severe arterial calcification [38]. We found that nestin+/Sca1+MSCs were elevated in circulating blood at earlier time points (1 and 2 weeks) after the ldlr−/−mice were fed HFD, and these cells were recruited to arterial lesions where the arterial calcification was eventually formed at later time point (12 weeks) after the mice fed HFD. Further, chimerism and lesion formation were investigated using a mouse parabiosis model in which two mice were conjoined subcutaneously to build up cross-circulation. Observation revealed that circulating cells were exchanged between the two joined mice. Partner-derived circulating MSCs were recruited and differentiated into osteoblasts at the arterial lesions after the ldlr−/−mouse partners were fed HFD. Both the elevated MSC numbers in circulating blood and their recruitment to the arterial lesions were abolished when mice were treated with the inhibitor of TGFβ Receptor I kinase (TβRI). In addition, in an ex vivo aorta conditioned medium (CM)-based cell migration assay, TGFβ1 released from the diseased aorta stimulated the migration of MSCs to the aortic lesions, and this effect is mediated through Smad signaling activation.
Materials and Methods
Animals
ldlr−/− mice in the C57BL/6J background were purchased from the Jackson Laboratory. The genotype was confirmed in animals upon arrival and periodically in offspring using multiplex polymerase chain reaction (PCR). Male ldlr−/− mice were fed chew diet (CHD, Tekland Global 18% protein diet) after birth. At 2 months of age, mice were either challenged with HFD (Harlan Teklad diet TD88137; 42% fat calories, 0.2% cholesterol) or remained on CHD for different periods of time (1, 2, 4, or 12 weeks). Nestin-GFP mice were provided by Dr. Grigori Enikolopov at Cold Spring Harbor Laboratory [39]. Nes-cre and C57BL/6J-Gtrosa26 tm1EYFP (R26-EYFP) mice were purchased from Jackson Lab. All animals were maintained in the animal facility of the Johns Hopkins University School of Medicine. The experimental protocol was reviewed and approved by the Institutional Animal Care and Use Committee of the Johns Hopkins University (Baltimore, MD).
Parabiosis
Parabiotic surgery was performed as described previously [40–42]. A male Nes-Cre; R26-EYFP or Nes-Cre (control) mouse was surgically joined to a male ldlr−/− mouse. Briefly, at 2 months of age, the mice were anesthetized, and the hair of the dorsal and lateral aspects were removed, and matching skin incisions were made from the shoulder to the knee joint of each mouse. Approximate 1-cm incisions in the peritoneum were made in each mouse, and the mice were attached using 3-0 coated Vicryl (Ethicon). The dorsal and ventral skin was stitched through continuous suture. Individual parabiotic mouse pairs were placed in clean cages, and food pellets were provided on the floor to minimize the strain of reaching for food while adjusting to parabiotic existence. Established shared blood circulation was confirmed by injection of Evans blue dye (Sigma-Aldrich). A total of 200 mL 0.5% Evans blue dye in saline was intravenously injected into the sides of Nes-Cre; R26-EYFP mice. After the cross-circulation of the two mice was confirmed, the mice were fed either CHD or HFD for 2 or 10 weeks before the mice were sacrificed. Blood and aorta tissue from the ldlr−/− mouse of each parabiotic pair were collected for analysis.
Ex vivo aorta CM-based cell migration assays
To prepare vascular CM, mice were perfused with the 200 mL 0.9% saline and their ascending thoracic aortae were isolated from the peri-adventitial tissue under a dissection microscope. The isolated aortae were flushed with sterilized phosphate-buffered saline (PBS). Each aorta were cultured in 24-well tissue culture plates with 500 μL serum-free DMEM at 37°C. Two days later, the conditioned media were collected and stored at −80°C.
To collect bone marrow MSCs from Nestin-GFP mice, the bone marrow from the femora and tibia was flushed in FACS buffer containing 1 mg·mL−1 bovine serum albumin (BSA; Sigma), 10 mM HEPES (Sigma) pH 7.4, and 1% penicillin-streptomycin (Invitrogen). After erythrocyte lysis, CD45−GFP+ cells were further purified using an automated cell sorter. GFP+ cells were cultured in α-MEM medium (Mediatech, Inc.) supplemented with 10% fetal bovine serum (FBS; Atlanta Biologicals), 5% donor equine serum (Thermo Scientific), 100 U/mL penicillin, and 100 μg/mL streptomycin (Mediatech, Inc.) to expand.
Cell migration was assessed in 96-well Transwells (Corning, Inc.) as described previously [30]. The 8 μm pore membrane between the upper and lower chambers was precoated with 0.5 μg/mL type I collagen (BD Biosciences). 1×104 GFP+ MSCs collected as described above in 100 μL serum-free MEM were plated in the upper chambers and 150 μL undiluted conditioned media from the cultured aortae were added to the lower chambers. After 10 h-incubation, cells were fixed with 10% formaldehyde for 4 h, and then the MSCs remained on the upper chamber membrane were removed with cotton swabs. The cells that had migrated through the pores to the bottom surface of the membrane were stained with crystal violet (Sigma). Five fields at 200× magnification were selected. Micrographic images were obtained and the cell number on each image was counted.
Analysis of aortic calcium content
Aortic calcium content was measured as described [43]. Briefly, aortic segments were resected from ascending aorta, weighed, heated, and speed evaporated. Subsequently, 10% formic acid was added to extract the aortic calcium overnight at 37°C. The samples were then centrifuged for 5 min at 13,200 rpm, and the supernatant was added to deproteinization buffer (0.3 mL of glacial acetic acid and 3.8 mL of 1 N KOH diluted to 50 mL with deionized water, pH 5.2). After heating for 5 min at 95°C, samples were centrifuged 5 min at 13,200 rpm. The supernatant was mixed with freshly prepared ortho-cresolphthalein complexone (OCPC) color reagent (Sigma) as described [44]. The purple OCPC calcium complex is spectrophotometrically determined by absorbance at 570 nm using a uQuant BIO-TEK plate reader.
Western blot analysis and ELISA of TGFβ1 in plasma
Western blot analysis of cell lysates were performed as described previously [45]. All blots were developed by the enhanced chemiluminescence technique (Amersham). Levels of active TGFβ1 in plasma of mice were determined using a DuoSet ELISA Development kit (R&D Systems) as described previously [30].
Flow cytometry analysis
Flow cytometry analysis of MSCs in peripheral blood was performed as described previously [25]. Blood samples were collected from mice by cardiac puncture. After the process of red blood cell lysis with commercial ACK lysis buffer (Quality Biological, Inc.), cells were washed with 0.5% BSA in PBS and then counted. 1×106/mL cells were incubated with PE-conjugated anti-Sca1, PE/Cy7-conjugated anti-CD29, PerCP- conjugated anti-CD45, and APC-conjugated anti-CD11b antibodies or isotype control for 1 h at 37°C in dark room, and then washed twice with 0.1% BSA in PBS. Probes were analyzed using a FACSC alibur flow cytometer and CellQuest software (Becton Dickinson).
Colony forming unit-fibroblast assays
Colony forming unit-fibroblast assays of bone marrow cells were performed as described [46]. In brief, 1×106 murine marrow cells were plated into six-well plates in 3 mL of α-MEM supplemented with glutamine (2 mM), penicillin (100 U/mL), streptomycin sulfate (100 μg/mL), and 20% lot-selected FBS. Duplicate cultures were established. After 2 to 3 h of adhesion, unattached cells were removed, and 2.5×106 irradiated guinea pig feeder cells (provided by Dr. Brendan J. Canning) were added to cultivation medium of adherent cultures just after washing. On day 14, cultures were fixed and stained with crystal violet. The colonies containing 50 or more cells were counted.
Histological and immunohistochemical analysis of the aortic and bone sections
The mice were exsanguinated under anesthesia. To study the morphology of the arteries, vessels were perfused with PBS followed by 4% paraformaldehyde by cannulating the left ventricle. The aortic arch and the proximal descending thoracic aorta were harvested and cut into three segments of near equal lengths (∼2–3 mm long). The distal ends of the segments were embedded in paraffin. Serial cross sections of 5 μm were prepared from the proximal ends of each segment. Mineral deposition of the aortic tissue was visualized with Alizarin Red (calcium) and von Kossa (phosphate) stains as described previously [47]. Immunohistochemistry analysis of aortic and bone tissue was performed using standard protocol as the manufacturer recommended (EnVision™ System; Dako). The sections were processed for antigen retrieval by digestion in 0.05% trypsin (pH 7.8) for 15 min at 37°C, and then incubated with primary antibodies against osteocalcin (Ser463/465) (diluted 1:100; Takara), osterix (diluted 1:400; Abcam), nestin (diluted 1:500; Aves Labs), Sca1 (dilute 1:40; R&D), and active TGFβ1 (LC(1–30) [48–50], kindly provided by Dr. Kathy Flanders at NIH/NCI) overnight at 4°C. An HRP-streptavidin detection system (Dako) was subsequently used to detect the immunoactivity followed by counterstaining with hematoxylin (Sigma). Sections incubated with 1% nonimmune serum PBS solution served as negative controls.
Quantitative real-time PCR
Quantitative real-time PCR (qRT-PCR) was conducted as described previously [51]. Briefly, total RNA for qRT-PCR was extracted from aortic tissue using TRIzol reagent (Invitrogen) according to the manufacturer's protocol. cDNA was prepared with random primers using the SuperScript First-Strand Synthesis System (Invitrogen) and analyzed with SYBR GreenMaster Mix (QIAGEN) in the thermal cycler with two sets of primers specific for each targeted gene. Relative expression was calculated for each gene by 2−CT method with GAPDH for normalization. Primers used for qRT-PCR were as follows: Col1A (NM_007742.3, 5′-GCGAAGGCAACAGTCGCT-3′) and (5′-CTTGGTGGTTTTGTATTCGATGAC-3′); Osteocalcin (NM_001032298.2, 5′-CTGACCTCACAGATCCCAAGC-3′) and (5′-TGGTCTGATAGCTCGTCACAAG-3′); Runx2 (NM_001145920.1, 5’-CACTGCCACCTCTGACTTCT-3′) and (5′- GCTCTCAGTGAGGGATGAAA-3′); and GAPDH (NM_008084.2, 5′-AGGTCGGTGTGAACGGATTTG-3′) and (5′-GGGGTCGTTGATGGCAACA-3′).
Statistics
Statistical analysis was done using SAS software. The unpaired two-tailed Student's t-test and χ2test were used to calculate P values.
Results
Nestin+/Sca1+ cells are recruited to the arterial lesions in ldlr−/−mice fed HFD for 1 or 2 weeks
It is known that HFD induces arterial calcification in male ldlr−/− mice [38]. We examined whether ldlr−/− mice fed HFD develop osteogenic calcification and if such mineralization is MSC-involved. Calcification/ossification was observed in aortic tissue from mice fed HFD for 12 weeks but not in mice fed CHD detected by Alizarin Red (Fig. 1A) and von kossa staining (Fig. 1B). Calcification/ossification was not found at earlier time points, that is, 1 and 2 weeks after HFD. Consistently, calcium content in aorta tissue of ldlr−/− mice fed HFD dramatically increased relative to that of mice fed CHD (Fig. 1E). To determine whether the mineralization of the aortic tissue is mediated by osteoblast-like cells, we assayed the expressions of genes encoding osteoblast differentiation markers including osteocalcin (Ocn), ColA1, and Runx2 in aortic tissue. Ocn-positive cells were only detected in aortic tissue of ldlr−/− mice fed HFD for 12 weeks but not detected in those of ldlr−/−mice fed CHD or mice fed HFD for shorter periods of time (1 or 2 weeks) (Fig. 1C, D). Further, mRNA expression levels of Ocn, ColA1, and Runx2 were all significantly increased in aortic tissue of ldlr−/− mice fed HFD for 12 weeks (Fig. 1F). Thus, the formation of osteoblastic cells and the development of calcification/ossification in aortic tissue showed a same tempo-spatial pattern in ldlr−/−mice fed HFD.
FIG. 1.

Osteogenic calcification was formed in aortic tissue of ldlr−/− mice fed HFD for 12 weeks. Representative images of Alizarin Red stain (A), von Kossa stain (B), and osteocalcin immunostain (Ocn, C) in aortae of ldlr−/− mice fed chew diet (CHD) or high-fat western diets (HFD) for 1, 2, and 12 weeks. Arrows, Ocn+ cells. Scale bars: 100 μm. (D) Number of osteocalcin (Ocn)+ cells were counted from four random high-power fields in 1 mm2 tissue area per aortic cross section. A total of three aortic sections from each mouse, and five mice per treatment group were analyzed. *P<0.001 versus CHD group. (E) Calculation of the calcium content in the aortae of ldlr−/− mice fed CHD or HFD for 12 weeks. n=5. *P<0.001 versus CHD group. (F) Quantitative real-time PCR analysis of osteoblast marker genes (Col1A, collagen 1A; Ocn, osteocalcin; and Runx2) in aortas of ldlr−/− mice fed CHD or HFD for 12 weeks. n=5; *P<0.01. PCR, polymerase chain reaction.
We previously demonstrated that nestin+ cells were recruited to the injured arteries to participate in vascular repair/remodeling [25]. We then examined whether nestin+ cells were also recruited to the diseased arteries in ldlr−/−H mice after HFD by immunohistochemical analysis of the aortic tissue using antibodies against nestin and Sca1. Nestin+ cells (Fig. 2A, C) and Sca1+ cells (Fig. 2B, D) were detected in aortic tissue of ldlr−/− mice fed HFD for 1 or 2 weeks but not in mice fed HFD for 12 weeks and in mice fed CHD for all periods of time. Interestingly, nestin+ cells and Sca1+ cells showed the same pattern, that is, they were localized near the intra-luminal side of the intimal layer of the vessels in mice fed HFD for 1 week, but became closer to the media layer in mice fed HFD for 2 weeks, indicating a movement of these cells over time before vascular calcification is formed. The earlier appearance of nestin+/Sca1+cells relative to Ocn+ osteoblastic cells imply that these cells may differentiate into osteoblastic lineage of cells to participate in the calcification of the aorta in ldlr−/− mice fed HFD.
FIG. 2.

Nestin+/Sca1+ cells are recruited to the arterial lesions in ldlr−/− mice fed HFD for 1 or 2 weeks. Immunohistochemical staining of the aortae from of ldlr−/− mice fed CHD or HFD for 1, 2, and 12 weeks with antibody against nestin (A) or Sca1 (B) as indicated. Arrows, positive cells. Scale bars: 100 μm. Number of nestin+ cells (C) or Sca1+ cells (D) were counted from four random high-power fields in 1 mm2 tissue area per aortic cross section. A total of three aortic sections from each mouse, and five mice per treatment group were analyzed. *P<0.001 versus CHD group. (E, F) Nestin+/Sca1+ cells numbers are increased in circulating blood at early time points in ldlr−/− mice fed HFD. Flow cytometry measured percentages of Sca1+CD29+CD11b−CD45− cells (E) and nestin+ cells (F) in peripheral blood of ldlr−/− mice fed CHD or HFD for 1, 2, 4, and 12 weeks. n=4, *P<0.05, **P<0.01, versus CHD group.
We previously demonstrated that MSCs recruited from the blood stream to the injured arteries contribute to arterial repair/remodeling [25]. To examine whether blood-derived MSCs may also participate in the development of osteogenic calcification, we measured the numbers of the nestin+/Sca1+ cells in blood at different time points after ldlr−/− mice fed HFD by flow cytometric analysis. Sca1+CD29+CD11b−CD45− are commonly used cell surface markers for isolation of MSC population. We found that the percentage of Sca1+CD29+CD11b−CD45− MSCs in blood were increased almost three-fold at 1 week, two-fold at 2 and 4 weeks, being unchanged at 12 weeks in HFD-treated mice relative to that of the CHD-treated mice (Fig. 2E). Same pattern was observed for the percentages of nestin+ cells in blood in HFD-treated mice (Fig. 2F).
Circulating nestin+ MSCs are recruited to the arterial lesions and differentiate to osteoblastic cells in ldlr−/−mice fed HFD
To provide direct in vivo evidence for the migration of nestin+ cells from blood circulation into the aortic wall and their differentiation into osteogenic cells, we generated parabiotic pairs, in which male Nes-Cre; R26-EYFP mice were surgically joined to male ldlr−/− mice (Fig. 3A). Nes-Cre; R26-EYFP mice were generated by crossing mice carrying a nestin promoter/enhancer-driven cre-recombinase (Nes-cre) with C57BL/6J-Gtrosa26 tm1EYFP (R26-EYFP) mice. Therefore, both nestin+ cells and their descendants can be marked by green fluorescence detected by immunofluorescence staining of the tissue with GFP antibody. The parabiotic mouse pairs were fed with either CHD (Para-1) or HFD (Para-3) for 2 or 10 weeks. To avoid the nonspecific autofluorescence staining of the tissue, another group of control parabiotic pairs (Para-2, Nes-Cre mice joined to ldlr−/− mice and fed with HFD) were also used. GFP+ cells were found within the aortic lesions of ldlr−/− mouse partners in Para-3 at both 2 and 10 weeks after the mice fed HFD (Fig. 3B, C), but they were undetectable in ldlr−/− mouse partners in either Para-1 or Para-2. GFP+ cells in circulating blood were also significantly elevated at 2 weeks but not at 10 weeks after the mice fed HFD in ldlr−/− mouse partners in Para-3 relative to Para-1 and Para-2 control groups (Fig. 3D), indicating the mobilization of GFP+ cells from the Nes-Cre; R26-EYFP mouse partners to ldlr−/− mouse partners mainly occur at earlier time point, which is consistent with dynamic pattern of nestin+ cells in peripheral blood (Fig. 2F). Majority of these GFP+ cells (>95%) are nestin+, Sca1+, and CD45− (Fig. 3E), and therefore are considered as MSCs. Of note, Ocn+ cells were also detected in the aortic lesions of ldlr−/− mouse partners in Para-2 and Para-3 at 10 weeks after the mice fed HFD (Fig. 3B, F). However, Ocn+GFP+ double positive cells, which are descendants of nestin+ cells derived from Nes-Cre; R26-EYFP mouse partners through circulation, were detected only in the aortic lesions of ldlr−/− mouse partners in Para-3 but not in Para-2 (Fig. 3B, G). These results clearly suggest that GFP+ osteoblastic cells originating from circulating nestin+ MSCs were localized at the arterial lesions and participate in active arterial calcification.
FIG. 3.

Circulating nestin+ MSCs are recruited to the arterial lesions and differentiate to osteoblastic cells in ldlr−/−mice fed HFD. (A) Schematic showing parabiotic pairings. (B) Representative fields of double immunofluorescence staining of the aorta tissue sections from ldlr−/− mouse partners in Para-1 (left panels), Para-2 (middle panels), and Para-3 (right panels) at 2 weeks (upper panels) or 10 weeks (bottom panels) with anti-GFP and anti-Ocn antibodies. (C) Quantification of the number of GFP+ cells per tissue area. (D) Flow cytometry measured percentages of GFP+ cells in peripheral blood of ldlr−/− mouse partners at 2 or 10 weeks after CHD or HFD. (E) Flow cytometry analysis of the percentage of nestin+, Sca1+, and CD45+ cells in the sorted GFP+ cell population. (F) Quantification of the number of Ocn+ cells per tissue area. (G) Calculation of the percentage of GFP+Ocn+ double positive cells out of total Ocn+ cells. Data from five Para-1, five Para-2, and six Para-3. *P<0.001 versus control (Para-2 group). MSCs, mesenchymal stem cells.
Bone marrow is a primary reservoir for nestin+ MSCs [39], which give rise to osteoprogenitors, preosteoblasts, and mature osteoblasts for bone formation during bone growth and bone remodeling. Accumulated studies have demonstrated that low bone mineral density is associated with increases in the risk of vascular calcification. As we detected nestin+ MSCs in aorta of ldlr−/−mice fed HFD, we examined whether MSCs in bone marrow and their osteogenic function in bone also altered. Osterix (Osx)-positive osteoprogenitors and Ocn-positive osteoblasts were arrayed on bone surface of the trabecular bone area of femur tissue from ldlr−/− mice fed CHD. These cells were almost undetectable in those from mice fed HFD (Supplementary Fig. S1A; Supplementary Data are available online at www.liebertpub.com/scd). Quantitative data confirmed the dramatic reduction of Osx+ and Ocn+ cells in bone (Supplementary Fig. S1B, C), indicating decreased osteogenesis during bone remodeling in these mice. To determine the changes of MSCs in ldlr−/− mice fed HFD, we measured the frequencies of bone marrow Sca1+CD29+CD11b−CD45− Cells by flow cytometry analysis. Much reduced frequencies of Sca1+CD29+CD11b−CD45− cells were detected in bone marrow suspension from ldlr−/− mice fed HFD for 12 weeks relative to those in mice fed CHD (Supplementary Fig. S1D). Further, colony-forming capacity of the cells from HFD-treated mice was also significantly decreased (Supplementary Fig. S1E). Therefore, while the MSCs appeared at the arterial lesions, the number and osteogenic potential of these cells in bone marrow were decreased.
CM prepared by incubating the aortae of the ldlr−/− mice fed HFD stimulates the migration of MSCs
To examine the direct migration of MSCs in response to the factor(s) released from the diseased arteries from ldlr−/− mice, we took advantage of an ex vivo Aorta CM-based cell migration assay previously established [25] and modified slightly. In this assay, bone marrow CD45−GFP+ MSCs sorted from Nestin-GFP mice were placed in the upper chamber and the Aorta CM, prepared by incubating aortae isolated from ldlr−/− mice fed CHD or HFD for 2 weeks (Fig. 4A), was placed in the lower chamber of a transwell chamber (Fig. 4B). Aorta CM prepared from the aorta of HFD-treated mice significantly enhanced MSCs migration compared with CM prepared from the aorta of CHD-treated mice (Fig. 4C), indicating early changes of vascular tissue such as inflammation and subsequent tissue damage after HFD stimulated the released of pro-migratory factor(s) from the aortic tissue, which induced the migration of MSCs to the target sites. We also examined the possibility that factor(s) in Aorta CM may stimulate the proliferation of MSCs. Cell proliferation measured by population doubling time in cells incubated in Aorta CM prepared from ldlr−/− mice fed CHD and HFD, respectively, had no significant difference (Fig. 4D).
FIG. 4.
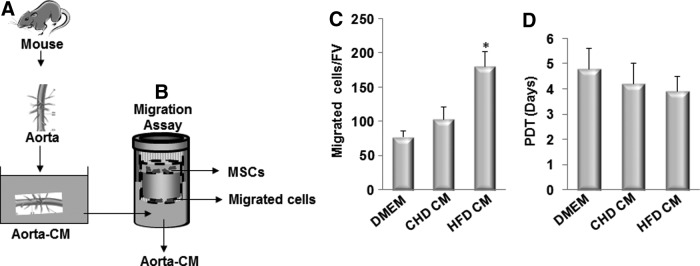
Conditioned medium prepared by incubating the aorta from ldlr−/− mice fed HFD stimulates the migration of MSCs. (A) Diagrams show the preparation of aorta conditioned medium (Aorta CM). Ascending thoracic aortae were isolated from ldlr−/− mice fed CHD or HFD for 2 week. The aortae were cultured in serum-free DMEM for 48 h, and culture media were collected as CHD CM or HFD CM. (B) Diagrams show the Aorta CM-based migration assays. CMs collected from (A) were placed in the lower chamber of a transwell, and MSCs were placed in the upper chamber. (C) Migrated cells were stained with crystal violet and counted. Numbers of migrated cells in Aorta CM-based migration assays using control DMEM, CHD CM, or HFD CM were counted per field of view (FV,×200 magnification). n=4. *P<0.001 versus CHD group. (D) Population doubling time (PDT) of the cells incubated with control DMEM, CHD CM, or HFD CM was calculated.
TGFβs released from the diseased aorta induce the migration of MSCs to the aortic lesions
To examine whether active TGFβs are the pro-migratory factor released from the aortic tissue in response to HFD, we measured the levels of active TGFβ1 in the aortic tissue from CHD- or HFD-treated mice. Immunohistostaining analysis showed that increased active TGFβ1 level was seen throughout the wall of the aorta in LDLR−/− mice fed HFD for 1 and 2 weeks compared with that in CHD-treated mice (Fig. 5A). We then performed the Aorta CM-based migration assay, in which we added either a neutralizing antibody against TGFβs (1D11) [52] or an isotype-matched murine IgG1 monoclonal control antibody (13C4) (kindly provided by Genzyme, Inc.) in the CM prepared from the aorta of HFD-treated mice. The migration of MSCs stimulated by the CM was inhibited by 1D11 (Fig. 5B). We also examined the key TGFβ signaling pathway(s) that mediates this effect. In canonical TGFβ signaling pathway, TβRI/II complex phosphorylates receptor-activated Smad2/3, which leads to recruitment of Smad4 and the transcription of downstream genes [53–55]. TGFβs also induce other (noncanonical) pathways, including the RhoA and MAPK cascades, the latter of which includes ERK, JNK, and p38 [56–58], which ultimately activate transcription of different downstream genes. The roles of canonical and noncanonical TGFβ signaling pathways in mediating MSCs migration stimulated by diseased Aorta CM were detected. Addition of SB505124 (TβRI/Smads inhibitor) in the CM prepared from the aorta of HFD-treated mice significantly suppressed the migration of MSCs to the level of that with the CM prepared from the aorta of ldlr−/−C mice, whereas Y27632 (RhoA inhibitor), U0126 (ERK inhibitor), SB202190 (p38 inhibitor), or SP600125 (JNK inhibitor) had marginal effect (Fig. 5C). Consistently, CM prepared from the aorta of HFD-treated mice stimulated the phosphorylation of Smad2/3 (p-Smad2/3), a key event in canonical signaling activation (Fig. 5D). Addition of either TGFβ neutralizing antibody (1D11) or SB-505124 alone significantly reduced the p-Smad2/3 level. In addition to the signaling elicited by the three isoforms of TGFβ, TGFβ receptor kinase inhibitor SB-505124 also interferes the signaling of other ligands such as activins and inhibins. We found that the level of p-Smad2/3 was not further reduced with combination treatment of 1D11 and SB-505124 relative to that of individual reagent-treated cells (Fig. 5D). The results suggest that TGFβs are primary factors released by the diseased arteries in inducing the migration of MSCs, and other ligands such as activins and inhibins may be involved in this event. CM prepared from the aorta of ldlr−/− mice fed HFD stimulated osteogenic differentiation detected by Alizarin Red staining. Neutralizing antibody of TGFβs, however, did not inhibit the effect (Fig. 5E). The results suggest that growth factor(s)/cytokine(s) other than TGFβ released from the diseased aorta induced the differentiation of the cells toward osteoblast lineage.
FIG. 5.

TGFβ1 released from the diseased aorta induces the migration of MSCs to the aortic lesions. (A) Immunohistochemical staining of active TGFβ1 in aorta of ldlr−/− mice fed CHD or HFD for 1 or 2 weeks. (B) Numbers of migrated MSCs in Aorta CM-based migration assays using CHD CM or HFD CM with 10 μg/mL neutralizing TGFβ antibody (1D11) or same concentration of control antibody (13C4) were counted per field of view (FV,×20 magnification). n=4. *P<0.01. (C) Numbers of migrated cells in Aorta CM-based migration assays using HFD CM with addition of individual inhibitors (2 μM SB505124, 10 μM Y27632, 10 μM SB202190, 10 μM U0126, and 10 μM SP600125). Numbers of migrated cells and differentiated cells were counted per field of view. n=4. *P<0.001, versus vehicle control. (D) MSCs were incubated with CHD CM or HFD CM with addition of 10 μg/mL control IgG antibody (13C4) or TGFβ neutralizing antibody (1D11) together with vehicle or 2 μM SB505124 as indicated for 2 h. Protein was extracted from MSCs, and western blot analysis was conducted using antibodies against phosphorylated Smad2/3 (p-Smad2/3) or total Smad2/3, respectively. (E) Alizarin Red staining of the MSCs incubated with control DMEM, CHD CM, or HFD CM with addition of 10 μg/mL neutralizing TGFβ antibody (1D11) or same concentration of control antibody (13C4). TGFβ1, transforming growth factor.
Active TGFβ1 is essential to induce the mobilization of MSCs to the circulating blood and their migration to the aortic lesions in ldlr−/− mice fed HFD
We have shown that the active TGFβ1 stimulates the mobilization of MSCs into circulating blood in response to vascular injury [25]. We therefore investigated whether TGFβ1 exerts similar effect on these cells in ldlr−/− mice fed HFD, eventually leading to osteogenic vascular calcification. We measured the levels of TGFβ1 in peripheral blood. While the total TGFβ1 levels in blood were not changed at 1, 2, and 12 weeks after the mice fed HFDs (Fig. 6B), active TGFβ1 levels in blood were elevated at 1 and 2 weeks but decreased at 12 weeks after the mice fed HFDs, as detected by ELISA (Fig. 6A). The ratios of active to total TGFβ1 levels were in a similar pattern (Fig. 6C). We then examined whether TGFβ is required for the mobilization of the cells. TβRI inhibitor SB-505124 (SB) was injected daily into ldlr−/− mice fed HFDs for 2 weeks. Blocking of TβRI signaling almost abolished the increase in Sca1+CD29+CD11b−CD45− cells in blood circulation (Fig. 6D, E). We also examined whether TGFβ is required for the recruitment of nestin+/Sca1+cells to the arterial lesions. Consistent with Fig. 2, nestin+/Sca1+ cells were detected in the aortic tissue from ldlr−/− mice fed HFD for 2 weeks. Notably, in TβRI inhibitor-treated mice, both nestin+ and Sca1+ cells were undetectable on the vessel wall of the ldlr−/− mice fed HFD (Fig. 6F–H).
FIG. 6.

Active TGFβ1 is required to induce the mobilization of MSCs to the circulating blood and their migration to the aortic lesions of ldlr−/− mice fed HFD. ELISA analysis of active TGFβ1 (A) and total TGFβ1 (B) levels in plasma of ldlr−/− mice fed HFD for 1, 2, and 12 weeks. C, Ratio of active/total TGFβ1 is calculated. n=6, *P<0.05, **P<0.01, versus CHD group. Injection of TβRI blocker SB-505124 (SB, 1 mg/kg-b.wt., i.p. daily) blocked mobilization of MSCs following arterial injury. Flow cytometrical quantitation of the numbers of Sca1+CD11b−CD45− cells (% of total cell counted) in blood of ldlr−/− mice fed HFD for 2 weeks with either SB or vehicle treatment. Representative flow cytometry analysis images are shown in (D). Quantified results are shown in (E). Results are mean±SD, n=3 mice per group per time point, *P<.001 versus respective vehicle-treated mice. (F) Immunohistochemical staining of the aortae from ldlr−/− mice fed HFD for 2 weeks with either TβRI blocker SB-505124 (SB, 1 mg/kg-b.wt., i.p. daily) or vehicle treatment using antibody against Sca1 (upper panels) and nestin (bottom panel) as indicated. Scale bars: 100 μm. Number of Sca1 cells (G) or nestin+ (H) per mm2 tissue area (N.nestin+ cells/T.Ar). n=5. *P<0.001 versus ldlr−/−C group. (I) Hypothetical model indicating the recruitment of MSCs in arterial calcification. Active TGFβ, released from the diseased arteries, stimulates the migration of bone marrow/circulating-derived MSCs to the vascular lesions, where they differentiate into osteoblast-like cells (OB) in response to other local osteogenic growth factor(s) to participate in vascular calcification. TβRI, TGFβ Receptor I kinase.
Discussion
Recent animal and human studies highlight the potentiality of blood circulation-derived stem cells/progenitor cells to engraft into the pathological tissue to participate in the calcification processes. However, direct evidence showing the contribution of MSCs to vascular calcification is still lacking. The present study, for the first time, demonstrates that MSCs were mobilized into circulating blood and recruited to the aortic tissue at earlier time points when vascular calcification was not developed, whereas osteocalcin+ osteoblastic cells and the formation of calcification appeared at later time point after HFD in ldlr−/− mice. Importantly, the parabiosis mouse study provides direct evidence to support the concept that blood circulation-derived nestin+ MSCs participate in osteogenic arterial calcification. Importantly, both circulating nestin+/Sca1+ cell numbers and nestin+/Sca1+ cells in the arterial lesions were dramatically reduced after TβRI inhibitor treatment. Further, at the earlier time point after HFD, CM prepared by incubating the aorta from ldlr−/− mice stimulated the migration of MSCs in the ex vivo assays. These findings suggest that nestin+/Sca1+ cells mobilized to the circulating blood from the remote sites (bone marrow or other tissues/organs) is one of the sources of the osteoprogenitors at the arterial lesions that cause osteogenic calcification of the arteries. We consider that most of the circulating nestin+ cells are MSCs as they are highly overlapped with CD45− nonhematopoitic Sca1+ cell population and exhibited same dynamic pattern with the Sca1+CD29+CD11b−CD45− MSCs in ldlr−/− mice fed HFD and in mice treated with TβRI inhibitor. We realize that circulating blood-derived MSCs may not be the only source contributing to arterial calcification. It is possible that the resident stem/progenitor cells and/or medial smooth muscle cells (SMCs) within the vasculature may directly migrate to the vascular lesions to participate in the calcification process. Whether the vascular resident cells are involved in this process and the underlying mechanisms are interesting topics for future study.
Our previous studies revealed that TGFβ1 is activated in bone [30] and vascular tissue [25] in response to normal bone remodeling and mechanical injury, respectively. Active TGFβ1 stimulates the mobilization of MSCs from bone marrow to blood circulation and their homing to the injured sites for vascular repair and remodeling [25]. In the present study, active TGFβs also stimulates the mobilization and recruitment of MSCs to the diseased sites for vascular calcification, further supporting the concept that TGFβs are stress/damage-activated messenger for the recruitment of MSCs to participate in both physiological and pathological tissue repair/remodeling. The TGFβ has three isoforms: TGFβ1, 2, and 3. Even though we detected increased active TGFβ1 level throughout the aorta in ldlr−/− mice fed HFD, we cannot exclude the involvement of TGFβ2 and 3. Since the distribution of the predominant isoform of TGFβs in different types of vessels varies, TGFβ isoform(s) that is activated in different types of vasculatures may also be different. Nevertheless, all three TGFβ isoforms share the same activation mechanism and common signaling pathway, and therefore their roles in recruiting MSCs during vascular calcification should be the same. The lineage commitment/differentiation of the recruited MSCs seems different at distinct environments even in the same types of tissue. The cells give rise to either SMC/myofibroblast-like cells or endothelial cells after they were recruited to the injured vessels [25], but they differentiate into osteoblastic cells during the development of vascular calcification. The possibility that TGFβ also induces the differentiation of MSCs into osteoblastic cells may not exist as we demonstrate that TGFβ-neutralizing antibody was unable to block the Aorta CM-induced osteoblast differentiation of MSCs. Thus, the primary function of TGFβ in this process is to drive the migration of MSCs to the right sites and impose the cells to other growth factors/cytokines produced in the specific environments for cell differentiation (Fig. 6I). A typical example is that IGF-I produced in bone microenvironment, stimulates osteoblast differentiation of MSCs recruited by TGFβ during bone remodeling [59,60]. The local growth factor(s)/cytokine(s) responsible for the differentiation of MSCs into myofibroblasts in neointima formation following mechanical injury and the differentiation into osteoblastic cells during vascular calcification following vascular inflammation are most likely different.
Of note, active TGFβ in peripheral circulation was increased at earlier time points in ldlr−/− mice following HFDs. One of the sources of active TGFβ in blood may be the injured vascular matrix. TGFβ is normally maintained in a sequestered state in extracellular matrix as a latent form, and the active TGFβ is released in response to the perturbations of the extracellular matrix at the situations of mechanical stress, wound repair, tissue injury, and inflammation [61]. Indeed, increased active TGFβ1 was observed throughout the aorta in the ldlr−/− mice fed HFD. The activation of TGFβs within the aortic tissue is most likely caused by the initial vascular damages due to inflammation and oxidative response. Supporting this view, Aorta CM prepared from ldlr−/− mice with 2 week-HFD treatment stimulated the migration of MSCs, and an addition of a neutralizing antibody of TGFβ in the Aorta CM significantly inhibited the effects. In addition, inflammatory cells such as white blood cells and macrophages, major players in the initial response, may also release active TGFβ1 into blood circulation.
There is increasing evidence for a link between bone metabolism and the vasculature, that is, so called “bone-vascular axis” [18,62,63]. Cross-sectional and longitudinal studies have demonstrated that low bone mineral density is associated with increases in the risk of cardiovascular events in prospective studies. In addition, high bone turnover itself is associated with increased cardiovascular mortality in elderly subjects independent of age, sex, overall health, serum parathyroid hormone levels, and hip fracture status. Our results show that numbers of MSCs in bone marrow and osteoblastogenesis in bone are decreased in ldlr−/− mice fed HFD. As nestin+/Sca1+ MSCs are osteoblasts-forming stem cells, the reduction of Osx+ osteoprogenitor cells and Ocn+ mature osteoblasts in bone suggests a decline in osteogenic potential of these MSCs in ldlr−/− mice fed HFD, which most likely is caused by the diminishing migratory ability of these cells to bone surface and/or their declined osteoblastic differentiation ability. Interestingly, the changes of MSCs and bone defects occurred at the same time point at which the vascular calcification develops. Further clarification of whether bone marrow is of primary origin from which MSCs are mobilized to participate in arterial calcification is needed for a better understanding on the pathogenesis of this disorder and the pathology of bone-vascular axis.
Supplementary Material
Acknowledgments
We gratefully thank Dr. Kathleen Flanders for providing us the active TGFβ1 antibody LC(1–30). This work was supported by the National Institutes of Health DK083350 (to M.W.) and the American Heart Association #13GRNT17050013 (to M.W.)
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Demer LL. and Tintut Y. (2008). Vascular calcification: pathobiology of a multifaceted disease. Circulation 117:2938–2948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hamirani YS, Pandey S, Rivera JJ, Ndumele C, Budoff MJ, Blumenthal RS. and Nasir K. (2008). Markers of inflammation and coronary artery calcification: a systematic review. Atherosclerosis 201:1–7 [DOI] [PubMed] [Google Scholar]
- 3.Sage A, Tintut Y, Garfinkel A. and Demer L. (2009). Systems biology of vascular calcification. Trends Cardiovasc Med 19:118–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blacher J, Guerin AP, Pannier B, Marchais SJ. and London GM. (2001). Arterial calcifications, arterial stiffness, and cardiovascular risk in end-stage renal disease. Hypertension 38:938–942 [DOI] [PubMed] [Google Scholar]
- 5.Lutgens E, van Suylen RJ, Faber BC, Gijbels MJ, Eurlings PM, Bijnens AP, Cleutjens KB, Heeneman S. and Daemen MJ. (2003). Atherosclerotic plaque rupture: local or systemic process?. Arterioscler Thromb Vasc Biol 23:2123–2130 [DOI] [PubMed] [Google Scholar]
- 6.Abedin M, Tintut Y. and Demer LL. (2004). Vascular calcification: mechanisms and clinical ramifications. Arterioscler Thromb Vasc Biol 24:1161–1170 [DOI] [PubMed] [Google Scholar]
- 7.Sage AP, Tintut Y. and Demer LL. (2010). Regulatory mechanisms in vascular calcification. Nat Rev Cardiol 7:528–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shao JS, Cheng SL, Sadhu J. and Towler DA. (2010). Inflammation and the osteogenic regulation of vascular calcification: a review and perspective. Hypertension 55:579–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giachelli CM. (2004). Vascular calcification mechanisms. J Am Soc Nephrol 15:2959–2964 [DOI] [PubMed] [Google Scholar]
- 10.Guzman RJ. (2007). Clinical, cellular, and molecular aspects of arterial calcification. J Vasc Surg 45Suppl A:A57–A63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson RC, Leopold JA. and Loscalzo J. (2006). Vascular calcification: pathobiological mechanisms and clinical implications. Circ Res 99:1044–1059 [DOI] [PubMed] [Google Scholar]
- 12.Speer MY, Yang HY, Brabb T, Leaf E, Look A, Lin WL, Frutkin A, Dichek D. and Giachelli CM. (2009). Smooth muscle cells give rise to osteochondrogenic precursors and chondrocytes in calcifying arteries. Circ Res 104:733–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tintut Y, Alfonso Z, Saini T, Radcliff K, Watson K, Bostrom K. and Demer LL. (2003). Multilineage potential of cells from the artery wall. Circulation 108:2505–2510 [DOI] [PubMed] [Google Scholar]
- 14.Farrington-Rock C, Crofts NJ, Doherty MJ, Ashton BA, Griffin-Jones C. and Canfield AE. (2004). Chondrogenic and adipogenic potential of microvascular pericytes. Circulation 110:2226–2232 [DOI] [PubMed] [Google Scholar]
- 15.Pal SN, Rush C, Parr A, Van CA. and Golledge J. (2010). Osteocalcin positive mononuclear cells are associated with the severity of aortic calcification. Atherosclerosis 210:88–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pal SN, Clancy P. and Golledge J. (2011). Circulating concentrations of stem-cell-mobilizing cytokines are associated with levels of osteoprogenitor cells and aortic calcification severity. Circ J 75:1227–1234 [DOI] [PubMed] [Google Scholar]
- 17.Pal SN. and Golledge J. (2011). Osteo-progenitors in vascular calcification: a circulating cell theory. J Atheroscler Thromb 18:551–559 [DOI] [PubMed] [Google Scholar]
- 18.Fadini GP, Rattazzi M, Matsumoto T, Asahara T. and Khosla S. (2012). Emerging role of circulating calcifying cells in the bone-vascular axis. Circulation 125:2772–2781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fernandez M, Simon V, Herrera G, Cao C, Del FH. and Minguell JJ. (1997). Detection of stromal cells in peripheral blood progenitor cell collections from breast cancer patients. Bone Marrow Transplant 20:265–271 [DOI] [PubMed] [Google Scholar]
- 20.Zvaifler NJ, Marinova-Mutafchieva L, Adams G, Edwards CJ, Moss J, Burger JA. and Maini RN. (2000). Mesenchymal precursor cells in the blood of normal individuals. Arthritis Res 2:477–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuznetsov SA, Mankani MH, Gronthos S, Satomura K, Bianco P. and Robey PG. (2001). Circulating skeletal stem cells. J Cell Biol 153:1133–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huss R, Lange C, Weissinger EM, Kolb HJ. and Thalmeier K. (2000). Evidence of peripheral blood-derived, plastic-adherent CD34(−/low) hematopoietic stem cell clones with mesenchymal stem cell characteristics. Stem Cells 18:252–260 [DOI] [PubMed] [Google Scholar]
- 23.Rochefort GY, Delorme B, Lopez A, Herault O, Bonnet P, Charbord P, Eder V. and Domenech J. (2006). Multipotential mesenchymal stem cells are mobilized into peripheral blood by hypoxia. Stem Cells 24:2202–2208 [DOI] [PubMed] [Google Scholar]
- 24.Otsuru S, Tamai K, Yamazaki T, Yoshikawa H. and Kaneda Y. (2008). Circulating bone marrow-derived osteoblast progenitor cells are recruited to the bone-forming site by the CXCR4/stromal cell-derived factor-1 pathway. Stem Cells 26:223–234 [DOI] [PubMed] [Google Scholar]
- 25.Wan M, Li C, Zhen G, Jiao K, He W, Jia X, Wang W, Shi C, Xing Q, et al. (2012). Injury-activated TGFbeta controls mobilization of MSCs for tissue remodeling. Stem Cells 30:2498–2511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wojakowski W, Landmesser U, Bachowski R, Jadczyk T. and Tendera M. (2011). Mobilization of stem and progenitor cells in cardiovascular diseases. Leukemia 26:23–33 [DOI] [PubMed] [Google Scholar]
- 27.Krankel N, Spinetti G, Amadesi S. and Madeddu P. (2011). Targeting stem cell niches and trafficking for cardiovascular therapy. Pharmacol Ther 129:62–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schraufstatter IU, Discipio RG. and Khaldoyanidi S. (2011). Mesenchymal stem cells and their microenvironment. Front Biosci 17:2271–2288 [DOI] [PubMed] [Google Scholar]
- 29.Caplan AI. and Correa D. (2011). The MSC: an injury drugstore. Cell Stem Cell 9:11–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tang Y, Wu X, Lei W, Pang L, Wan C, Shi Z, Zhao L, Nagy TR, Peng X, et al. (2009). TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat Med 15:757–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones CP, Pitchford SC, Lloyd CM. and Rankin SM. (2009). CXCR2 mediates the recruitment of endothelial progenitor cells during allergic airways remodeling. Stem Cells 27:3074–3081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lazarus HM, Haynesworth SE, Gerson SL. and Caplan AI. (1997). Human bone marrow-derived mesenchymal (stromal) progenitor cells (MPCs) cannot be recovered from peripheral blood progenitor cell collections. J Hematother 6:447–455 [DOI] [PubMed] [Google Scholar]
- 33.Brouard N, Driessen R, Short B. and Simmons PJ. (2010). G-CSF increases mesenchymal precursor cell numbers in the bone marrow via an indirect mechanism involving osteoclast-mediated bone resorption. Stem Cell Res 5:65–75 [DOI] [PubMed] [Google Scholar]
- 34.Jian B, Narula N, Li QY, Mohler ER, III, and Levy RJ. (2003). Progression of aortic valve stenosis: TGF-beta1 is present in calcified aortic valve cusps and promotes aortic valve interstitial cell calcification via apoptosis. Ann Thorac Surg 75:457–465 [DOI] [PubMed] [Google Scholar]
- 35.Trion A. and van der LA. (2004). Vascular smooth muscle cells and calcification in atherosclerosis. Am Heart J 147:808–814 [DOI] [PubMed] [Google Scholar]
- 36.Watson KE, Bostrom K, Ravindranath R, Lam T, Norton B. and Demer LL. (1994). TGF-beta 1 and 25-hydroxycholesterol stimulate osteoblast-like vascular cells to calcify. J Clin Invest 93:2106–2113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grainger DJ, Metcalfe JC, Grace AA. and Mosedale DE. (1998). Transforming growth factor-beta dynamically regulates vascular smooth muscle differentiation in vivo. J Cell Sci 111(Pt 19):2977–2988 [DOI] [PubMed] [Google Scholar]
- 38.Towler DA, Bidder M, Latifi T, Coleman T. and Semenkovich CF. (1998). Diet-induced diabetes activates an osteogenic gene regulatory program in the aortas of low density lipoprotein receptor-deficient mice. J Biol Chem 273:30427–30434 [DOI] [PubMed] [Google Scholar]
- 39.Mendez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, Scadden DT, Ma'ayan A, Enikolopov GN. and Frenette PS. (2010). Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 466:829–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tanaka K, Sata M, Natori T, Kim-Kaneyama JR, Nose K, Shibanuma M, Hirata Y. and Nagai R. (2008). Circulating progenitor cells contribute to neointimal formation in nonirradiated chimeric mice. FASEB J 22:428–436 [DOI] [PubMed] [Google Scholar]
- 41.Jiang Q, Oldenburg R, Otsuru S, Grand-Pierre AE, Horwitz EM. and Uitto J. (2010). Parabiotic heterogenetic pairing of Abcc6−/−/Rag1−/− mice and their wild-type counterparts halts ectopic mineralization in a murine model of pseudoxanthoma elasticum. Am J Pathol 176:1855–1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kotani M, Kikuta J, Klauschen F, Chino T, Kobayashi Y, Yasuda H, Tamai K, Miyawaki A, Kanagawa O, Tomura M. and Ishii M. (2013). Systemic circulation and bone recruitment of osteoclast precursors tracked by using fluorescent imaging techniques. J Immunol 190:605–612 [DOI] [PubMed] [Google Scholar]
- 43.Cheng SL, Shao JS, Halstead LR, Distelhorst K, Sierra O. and Towler DA. (2010). Activation of vascular smooth muscle parathyroid hormone receptor inhibits Wnt/beta-catenin signaling and aortic fibrosis in diabetic arteriosclerosis. Circ Res 107:271–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shao JS, Cheng SL, Pingsterhaus JM, Charlton-Kachigian N, Loewy AP. and Towler DA. (2005). Msx2 promotes cardiovascular calcification by activating paracrine Wnt signals. J Clin Invest 115:1210–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wan M, Yang C, Li J, Wu X, Yuan H, Ma H, He X, Nie S, Chang C. and Cao X. (2008). Parathyroid hormone signaling through low-density lipoprotein-related protein 6. Genes Dev 22:2968–2979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu X, Pang L, Lei W, Lu W, Li J, Li Z, Frassica FJ, Chen X, Wan M. and Cao X. (2010). Inhibition of Sca-1-positive skeletal stem cell recruitment by alendronate blunts the anabolic effects of parathyroid hormone on bone remodeling. Cell Stem Cell 7:571–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu B, Zhao X, Yang C, Crane J, Xian L, Lu W, Wan M. and Cao X. (2012). Parathyroid hormone induces differentiation of mesenchymal stromal/stem cells by enhancing bone morphogenetic protein signaling. J Bone Miner Res 27:2001–2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Flanders KC, Thompson NL, Cissel DS, Van Obberghen-Schilling E, Baker CC, Kass ME, Ellingsworth LR, Roberts AB. and Sporn MB. (1989). Transforming growth factor-beta 1: histochemical localization with antibodies to different epitopes. J Cell Biol 108:653–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Williams AO, Flanders KC. and Saffiotti U. (1993). Immunohistochemical localization of transforming growth factor-beta 1 in rats with experimental silicosis, alveolar type II hyperplasia, and lung cancer. Am J Pathol 142:1831–1840 [PMC free article] [PubMed] [Google Scholar]
- 50.Barcellos-Hoff MH, Ehrhart EJ, Kalia M, Jirtle R, Flanders K. and Tsang ML. (1995). Immunohistochemical detection of active transforming growth factor-beta in situ using engineered tissue. Am J Pathol 147:1228–1237 [PMC free article] [PubMed] [Google Scholar]
- 51.Li C, Xing Q, Yu B, Xie H, Wang W, Shi C, Crane JL, Cao X. and Wan M. (2013). Disruption of LRP6 in osteoblasts blunts the bone anabolic activity of PTH. J Bone Miner Res 28:2094–2108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dasch JR, Pace DR, Waegell W, Inenaga D. and Ellingsworth L. (1989). Monoclonal antibodies recognizing transforming growth factor-beta. Bioactivity neutralization and transforming growth factor beta 2 affinity purification. J Immunol 142:1536–1541 [PubMed] [Google Scholar]
- 53.Kretzschmar M. and Massague J. (1998). SMADs: mediators and regulators of TGF-beta signaling. Curr Opin Genet Dev 8:103–111 [DOI] [PubMed] [Google Scholar]
- 54.Massague J. and Chen YG. (2000). Controlling TGF-beta signaling. Genes Dev 14:627–644 [PubMed] [Google Scholar]
- 55.Feng XH. and Derynck R. (2005). Specificity and versatility in tgf-beta signaling through Smads. Annu Rev Cell Dev Biol 21:659–693 [DOI] [PubMed] [Google Scholar]
- 56.Derynck R. (1994). TGF-beta-receptor-mediated signaling. Trends Biochem Sci 19:548–553 [DOI] [PubMed] [Google Scholar]
- 57.Lee MK, Pardoux C, Hall MC, Lee PS, Warburton D, Qing J, Smith SM. and Derynck R. (2007). TGF-beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J 26:3957–3967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yamashita M, Fatyol K, Jin C, Wang X, Liu Z. and Zhang YE. (2008). TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-beta. Mol Cell 31:918–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xian L, Wu X, Pang L, Lou M, Rosen CJ, Qiu T, Crane J, Frassica F, Zhang L, et al. (2012). Matrix IGF-1 maintains bone mass by activation of mTOR in mesenchymal stem cells. Nat Med 18:1095–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Crane JL. and Cao X. (2014). Function of matrix IGF-1 in coupling bone resorption and formation. J Mol Med (Berl) 92:107–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Annes JP, Munger JS. and Rifkin DB. (2003). Making sense of latent TGFbeta activation. J Cell Sci 116:217–224 [DOI] [PubMed] [Google Scholar]
- 62.Thompson B. and Towler DA. (2012). Arterial calcification and bone physiology: role of the bone-vascular axis. Nat Rev Endocrinol 8:529–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.London GM. (2012). Bone-vascular cross-talk. J Nephrol 25:619–625 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.