

1. Introduction
It has long been axiomatic that a protein’s structure determines its function. Intrinsically disordered proteins (IDPs) and disordered protein regions (IDRs) defy this structure–function paradigm. They do not exhibit stable secondary and/or tertiary structures and exist as dynamic ensembles of interconverting conformers with preferred, nonrandom orientations.1−4 The concept of IDPs and IDRs as functional biological units was initially met with skepticism. For a long time, disorder, intuitively implying chaos, had no place in our perception of orchestrated molecular events controlling cell biology.
Over the past years, however, this notion has changed. Aided by findings that structural disorder constitutes an ubiquitous and abundant biological phenomenon in organisms of all phyla,5−7 and that it is often synonymous with function,8−11 disorder has become an integral part of modern protein biochemistry. Disorder thrives in eukaryotic signaling pathways12 and functions as a prominent player in many regulatory processes.13−15 Disordered proteins and protein regions determine the underlying causes of many neurodegenerative disorders and constitute the main components of amyloid fibrils.16 They further contribute to many forms of cancer, diabetes and to cardiovascular and metabolic diseases.17,18
Research into disordered proteins produced significant findings and established important new concepts. On the structural side, novel experimental and computational approaches identified and described disordered protein ensembles3,19,20 and led to terms such as secondary structure propensities, residual structural features, and transient long-range contacts.1,21 The discovery of coupled folding-and-binding reactions defined the paradigm of disorder-to-order transitions22 and high-resolution insights into the architectures of amyloid fibrils were obtained.23,24 On the biological side, we learned about the unexpected intracellular stability of disordered proteins, their roles in integrating post-translational protein modifications in cell signaling and about their functions in regulatory processes ranging from transcription to cell fate decisions.15,25,26 One open question remaining to be addressed is how these in vitro structural insights relate to biological in vivo effects. How do complex intracellular environments modulate the in vivo properties of disordered proteins and what are the implications for their biological functions (Figure 1)?27−29
Figure 1.
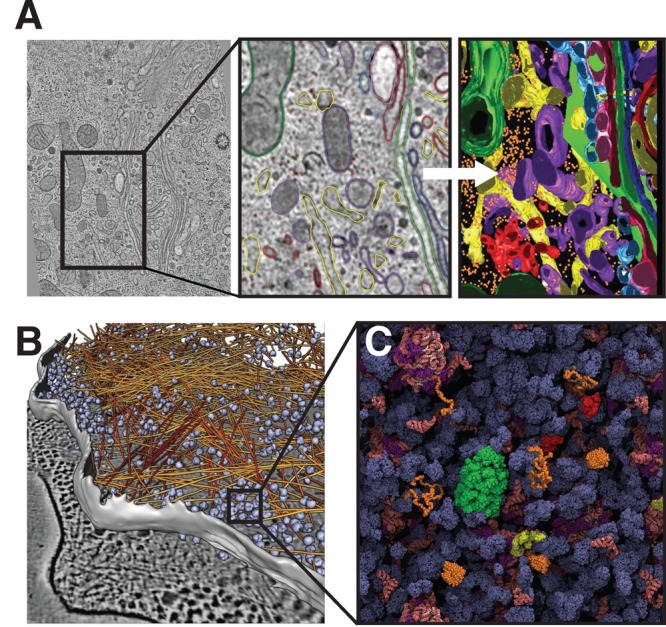
Intracellular complexity. (A) Left: Cryo-electron tomography slice of a mammalian cell. Middle: Close-up view of cellular structures colored according to their identities: Right: Three-dimensional surface representation of the same region. Yellow, endoplasmic reticulum; orange, free ribosomes; green, mitochondria; blue, dense core vesicles; red, clathrin-positive compartments and vesicles; purple, clathrin-negative compartments and vesicles. Reprinted with permission from ref (27). Copyright 2012 Public Library of Science. (B) Tomography image of the interior of a Dictyostelium cell with actin filaments shown in orange and ribosomes in blue. Reprinted with permission from ref (29). Copyright 2012 Rockefeller University Press. (C) Schematic representation of the E. coli cytosol. Ribosomes and tRNA are shown in pink, chaperones in green and red, disordered proteins in orange, and all other proteins in dark blue. Reprinted with permission from ref (28). Copyrigth 2011 Elsevier.
Here, we attempt to answer these questions by reviewing the physical and biological properties of intracellular environments in relation to structural and functional parameters of disordered proteins. Specifically, we discuss how IDPs may experience in vivo environments differently to ordered proteins. To this end, we provide a description of the compositional and physical parameters of the cellular milieu and their effects on ordered and disordered proteins (section 2). We evaluate how biological processes may act differently on ordered and disordered proteins (section 3) and discuss how combined physical and biological contributions modulate the intracellular aggregation behavior of IDPs (section 4). Finally, we review theoretical and experimental approaches to study the structural and functional properties of disordered proteins in cells (section 5).
2. Physicochemical Properties of the Intracellular Environment
To understand how proteins function inside cells, one needs to consider the particular physical properties of the intracellular environment and how they shape the cellular behaviors of ordered and intrinsically disordered proteins. In the following paragraphs, we discuss the composition of the prokaryotic and eukaryotic cytoplasm in terms of average ion and metabolite concentrations, dielectric properties, macromolecular crowding and how these parameters affect intracellular viscosity, rotational and translational diffusion, and macromolecular association events.
2.1. Composition of the Cytoplasm
We begin by reviewing cytosolic ion and metabolite compositions and concentrations, and delineate their effects on cellular dielectric constants, pH and viscosity. We do so by making use of the CyberCell database from David Wishart’s laboratory30 (http://ccdb.wishartlab.com/CCDB/) and of BioNumbers, and references therein, from the Systems Biology Department at Harvard Medical School (www.bionumbers.hms.harvard.edu).
2.1.1. Inorganic Ions
The total concentration of cytoplasmic inorganic ions in E. coli is ∼300 mM according to the CyberCell database. The concentration of K+, by far the most abundant inorganic ion, varies drastically with osmotic conditions.31 200 mM is reported to be physiologically relevant32 and CyberCell notes a concentration range of 200–250 mM. (For the remainder of this paragraph, concentrations reported by CyberCell, where available, are given in brackets when following concentrations provided by other sources). In separate studies of E. coli grown in McIlvaine’s medium,33 glucose,34 or LB,35 for example, the concentrations of K+ were determined to be ∼250 (free), ∼180–200, and ∼100 mM, respectively. Similarly, large variations in the total concentration of Mg2+ have been reported, with estimates ranging from 2035 to 100 mM,36 although the amount of free Mg2+ is estimated to be much smaller in comparison at 1–2 mM.37,38 Estimates for other common inorganic ions include Na+ at ∼5 mM,34 Ca2+ at ∼0.1 mM,35 and CyberCell reports concentrations of Cl– and total phosphate (H2PO4–/HPO42–/PO43–) at 6 and 5 mM,30 respectively. Although variations between tissues exist, average concentrations of inorganic ions in E. coli, yeast and mammalian cells are in the same range.30,34,35,38−41 For example, differences have been reported for some specialized tissues/organs such as resting frog muscle with 141 mM for K+, 9.0 mM for Na+, 2.2 mM for Cl–, and 1.4 mM for total inorganic phosphate, while the concentrations of free Ca2+ and Mg2+ were reported as 52 nM and 0.8 mM, respectively.42
Bacteria can store metal ions at concentrations much higher than those in the growth medium. The concentrations of several transition metals have indeed been measured, with Fe and Zn at 0.1 mM35,43 [18 and 4 mM, respectively],30 and Cu, Mn, Mo, and Se at ∼10 μM35,43 [4 mM for Cu, Mn, and Mo].30 In eukaryotic cells, intracellular concentrations of metal ions are more difficult to determine accurately, because large variations between different organelles exist (Figure 2A).44 In HeLa cells for example, the concentration of free Zn2+ in mitochondria is in the pM range but nM in the cytoplasm.45 In addition, many proteins bind metals, and their abundance and localization leads to dynamic changes in metal content.46−48 Specialization and metabolic activities further modulate intracellular metal concentrations. The total iron load in red blood cells is ∼20 mM for example, while it is 1 mM in neurons.47,49,50 Despite these differences, global intracellular concentrations of free metals are similar in prokaryotic and eukaryotic cells.51 Concentrations of free Fe2+/Fe3+, Zn2+ and Cu+/Cu2+ are usually low because of their poor solubility at neutral pH and strong affinities toward intracellular metal-binding proteins.
Figure 2.
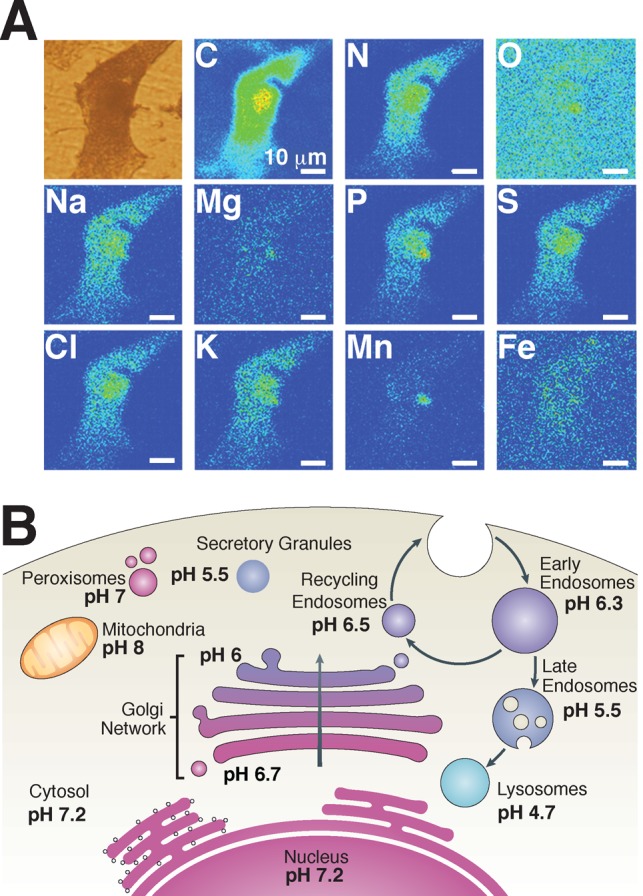
Intracellular composition. (A) Localization of various elements in a mammalian PC12 cell (exposed to MnCl2). Reprinted with permission from ref (44). Copyright 2009 Royal Society Publishing. (B) Average pH values of organelles and compartments in mammalian cells. Adapted with permission from ref (92). Copyright 2010 Nature Publishing Group.
Changes in intracellular ion levels can have pronounced effects on the conformational properties of disordered proteins as seen in the enhanced aggregation of α-synuclein at high salt concentrations.52 Because charged amino acids are selectively enriched in IDPs53 and play important roles in compaction,54,55 their capacities to mediate, as well as to shield electrostatic interactions depend on their charge states and counterion coordination.56 In addition, many metal-binding proteins contain extended disordered regions (∼30–50 residues),57 and metal binding can affect their mean net charge to a degree that they become ordered. Zn2+ coordination by the disordered N-terminal domain of HIV-1 integrase, for example, results in α-helix formation.58 For several other IDPs such effects are more subtle. Zn2+ binding to ProThymosin α (ProTα) increases transient helicity from <1% to 12%59,60 and induces partial folding of its C-terminal Glu-rich region.60 Metal binding can also promote oligomerization. In the case of HIV-1 integrase, the Zn2+-bound protein tetramerizes readily.58 Deletion of the disordered C-terminal 27 residues of the scavenger protein cardiac calsequestrin results in the loss of Ca2+-dependent tetramerization.61 Growing evidence suggests that changes in metal homeostasis and altered IDP-metal interactions contribute to the pathogeneses of several neurodegenerative disorders.62−64 Indeed, many amyloidogenic IDPs such as α-synuclein, tau or amyloid beta (Aβ) peptides directly bind metals, and metal interactions modulate their in vitro aggregation behaviors (see section 4).
2.1.2. Metabolites
Recent advances in metabolomics technologies have allowed the concentrations of large numbers of metabolites to be measured in E. coli.65 In glucose-fed, exponentially growing E. coli cells the combined concentrations of metabolites have been estimated to be ∼300 mM, with glutamate (Glu–) being the most abundant metabolite by far (96 mM), followed by glutathione, fructose-1,6-bisphophate and adenosine triphosphate (ATP) at 17 mM, 15 mM and 9.6 mM, respectively.65 However, these concentrations depend on the culture medium. By changing the carbon source from glucose to glycerol or acetate, intracellular Glu– levels change from 96 to 149 to 45 mM, respectively. Similarly, intracellular glutathione concentrations change from 17 to 18 to 8 mM; Fructose-1,6-bisphophate from 15 to 6 to <0.15 mM; ATP from 9.6 to 9.0 to 4.1 mM.65 Significant variations in intracellular Glu– levels due to changes in glucose levels in the growth media or due to changes in osmotic conditions have also been seen in other studies.33,66 When E. coli cells were grown in McIlvaine’s medium at pH 6 and harvested at midexponential phase, the total concentration of all amino acids was determined to be ∼90 mM, of which Glu– comprises ∼60 mM.33 In the presence of 200 mM glucose, Glu– concentration increase to ∼117 mM. At 400 mM glucose, it is ∼160 mM. By contrast, CyberCell lists the combined E. coli concentrations of all “small organic molecules” as 40–50 mM (undefined growth-medium and -stage), concentrations of free amino acids total ∼15 mM, and ATP is indicated between 1.3 and 7.0 mM, depending on growth conditions and sugar sources.30
In eukaryotes, metabolite concentrations are subject both to variations between subcellular organelles, and to variations between cell and tissue types. In the yeast S. cerevisiae, total cellular concentrations of glutamate, glutathione, and ATP are ∼75, ∼15, and ∼2.5 mM, respectively.39,67,68 In human cells, according to the human metabolome database, HCO3– is the most abundant metabolite at ∼11 mM, followed by 2,3-diphosphoglyceric acid at 4.0–5.0 mM, glutathione at ∼2–5 mM, l-malic acid at ∼3–4 mM, ATP at ∼1–2 mM, and Glu– at ∼1–2 mM.69 Again, these concentrations are subject to cell- and tissue-type variations. One example is frog muscle, where phosphocreatine is the most abundant metabolite (∼50 mM), followed by carnosine, total free amino acids, creatine and total ATP at respective concentrations of 19.5 mM, 11.7 mM, 11.0 mM, and 6.2 mM.42
Not measured in the above studies are the important polyamine metabolites putrescine, spermidine, and spermine.70 These polycations, found in all eukaryotes and most prokaryotes, and have roles in cell growth and proliferation.71,72 Decreased levels are associated with aging and increased levels are associated with cancer.72 Putrescine and spermidine are generally found at high concentrations in Gram-negative bacteria such as E. coli, where spermine is absent. Studies in E. coli reported concentrations of ∼20–30 mM for putrescine and ∼6–7 mM for spermidine.73 Levels of putrescine and spermidine in mammalian cells are significantly lower. Spermidine was measured in bovine lymphocytes and rat liver cells at ∼1 mM.74 For spermine, the concentrations are ∼2 and ∼1 mM, respectively. Relative to these two, putrescine is present at ∼5-fold lower levels in rat liver and human promyelocytic leukemia HL-60 cells.70,71
Polyamines have been found to accelerate the in vitro aggregation and fibrillation of α-synuclein, an IDP implicated in Parkinson’s disease (PD), in vitro.75 The extent of these effects increases with polyamine charge, length and concentration, suggesting that they can also occur in vivo.75 Other metabolites such as glycerol, trehalose and zwitterions such as trimethylamine-N-oxide, proline, betaine, and ectoine, stabilize proteins at intracellular concentrations between 100 and 300 mM.76 These compounds may represent a special class of metabolites, because they also function as powerful stabilizing agents in vitro. Nevertheless, these data indicate that metabolite concentrations in the range of ∼300 mM are sufficient to alter the properties of individual proteins. While it is unlikely that metabolites generally induce folding of disordered proteins in cellular environments, they may modify the structural features of some of them.
2.1.3. Dielectric Properties
Ions and charged metabolites contribute to the dielectric properties of the intracellular environment. The static dielectric constant of pure water is 78.4 at 25 °C,77 but dissolved ions can decrease this value substantially due to the tendency of water molecules to align with local electric fields caused by nearby ions.78 This effect is concentration-dependent, but linear in the dilute regime, i.e., at ion concentrations below ∼2 M. Based on measurements by Hasted et al., we estimate that in physiological solutions the dielectric constant of water would be reduced by only ∼2 units (i.e., to ∼76) primarily because of K+; given the complexity of the intracellular environment it is questionable whether this estimate is accurate. In addition to affecting the dielectric behavior of the water component of a solution, charged ions and metabolites also contribute to a solution’s conductivity. On the other hand, many metabolites, such as free amino acids, carry neutral net charges and, therefore, do not contribute to conductivity but the overall dieletric properties of the environment.79−81 Free amino acids increase the dielectric constant by molar increments of 20–30 M–1. For example, at its physiological concentration of 0.8 mM in E. coli (CCDB), alanine is expected to increase the dielectric constant of the cytosol by a very modest ∼0.02 units.
The dielectric properties of many cell types have been investigated, including E. coli,82−84 murine lymphocytes and erythrocytes,85 murine erythroleukemia cells,86 and human breast- and colon-cancer cells,87,88 as well as red blood cells.87 It is difficult to draw firm conclusions from these studies because the reported values for the dielectric constants of the cytoplasm range from ∼50–150, while those of membranes range from ∼2–15.88 For eukaryotic systems, a further complication is the possibility of significant variations between organelles. Wang et al. have recently reported the use of a surface plasmon resonance (SPR)-based electrochemical impedance microscope (EIM) that enables intracellular resistance levels to be measured at submicrometer resolution;89 in combination with sophisticated mathematical modeling, these measurements could be used to calculate intraorganelle, approximative conductivities and dielectric constants. The authors obtained indicative values for intracellular dielectric constants in the range of ∼30 and ∼60.89 Dielectric constants of 15 and 60 were recently determined for the cytoplasm and cell nucleus of mammalian CHO cells, respectively.90
Variations in permittivity modify the strength of charge–charge interactions. Hence, intracellular dielectric constants can impact the conformational properties of disordered proteins, whose shapes are modulated by intramolecular charge–charge interactions.53−55 Intracellular electric susceptibility depends on ion and metabolite concentrations, and it is difficult to separate the effects of these two parameters. Having shown that different subcellular compartments exhibit different dielectric properties, it is moreover possible that ion and metabolite effects scale differently in different intracellular microenvironments (see section 2.4). In turn, this complexity may lead to spatially modulated effects on IDP structure and function.
2.1.4. pH
As a rule of thumb, the pH of the cytoplasm is 7.2,91 and it is critical to maintain this value for any given organism.92 Phosphate or bicarbonate ions and other weak acids and bases within the cell provide the intracellular buffering capacity, to which the side-chains and free amino- and carboxy-termini of amino acids and proteins contribute less than 1%.93,94 Indeed, only histidine (pKa ∼6.04) and other imidazoles affect buffering near neutral pH.94 The pKa’s of other amino acids are either too far from neutral92 or, as in the case of cysteine (pKa 8.3) have chemistries that are too complex to contribute to buffering.94
At optimal growth conditions, E. coli maintains an intracellular pH of 7.4–7.8, provided that the pH of the external environment is between 5.5 and 9.0.95−98 Using fluorescence imaging, approximate pH values of HeLa cytoplasm, mitochondria, endoplasmic reticulum, and Golgi were determined to be 7.4, 8.0, 7.5, and 6.6, respectively.99 In a recent review, the pH values of various eukaryotic compartments and organelles were reported to be 7.2 for the cytosol, 7.2 for the nucleus (due to permeability), 7.2 for the endoplasmic reticulum, 6.7 for cis-Golgi cisterns and 6.0 at trans-Golgi networks, 8.0 in mitochondria, 7.0 in peroxisomes, 5.5 in secretory granules, 6.3 in early endosomes, 6.5 in recycling endosomes, 5.5 in late endosomes, and 4.7 in lysosomes (Figure 2B).92
How do changes in intracellular pH affect disordered proteins? Ordered proteins are sensitive to pH, and unfolding is common at pH values <3.100 By contrast, disordered proteins withstand “denaturing” pH titrations, as judged by circular dichroism (CD), fluorescence- and NMR-spectroscopy, and resistance to pH changes serves as a common indicator for native “unfoldedness”.101 Some IDPs adopt more ordered structures in response to “drastic” pH changes. Examples include the gain of α-helical content in histones at pH 10,102 increased helicity of the calpastatin domain I at pH levels below 4,103 and an increase in transient helicity of ProTα at pH 2 (from <1% to 10.7%).104 Below pH 5.5, α-synuclein displays higher levels of compaction105 brought about by the neutralization of its acidic C-terminal residues and the concomitant abolishment of intramolecular contacts with positively charged amino acids at its N-terminus.106,107 Low pH also favors α-synuclein aggregation.105 However, given that most of these pH conditions are not compatible with physiological environments, it is questionable whether these results are biologically meaningful.
2.2. Viscosity
Intracellular viscosity is a bulk phenomenon that is manifested at multiple scales. Because large pools of molecules, macromolecules, and macromolecular assemblies are present in the cytoplasm, ranging from water to metabolites, from soluble monomeric proteins to the cytoskeleton, from isolated lipids and fatty acids to membranes and organelles, objects of different sizes, shapes and chemistries contribute to what we perceive as effective intracellular viscosity. Each of these components evokes different dissipative forces that oppose motions and affect different molecules to different degrees. Here, we limit our discussion to “solvent viscosity”, i.e., to local viscosity effects that act on the subnanometer scale.
Concerning water motions on the pico- (ps) to nano- (ns) second time scale, corresponding to the subnanometer (nm) length scale, ∼85% of all intracellular water displays translational (macroscopic) and rotational (microscopic) dynamics that are indistinguishable from pure water, both in prokaryotic and eukaryotic cells.108,109 This amount of bulk water is not coordinated in the first hydration shell of cellular macromolecules and only marginally affects the rotational dynamic properties of small fluorescent dyes.110 Moreover, ∼90% of the water molecules in the hydration layer of proteins, and other macromolecules, display an average 2-fold reduction in dynamic properties compared to bulk water,108,109 although 10-fold reductions may occur at macromolecule-to-solvent ratios above 30–50% (v/v), according to in vitro measurements.111 Therefore, and despite the astonishingly large intracellular concentrations of small solutes (see above), these molecules contribute little to intracellular viscosity. This observation is consistent with the measured effects of ions and metabolites, such as amino acids on water viscosity in vitro. With the exception of K+, other ions and amino acids generally increase viscosity by 5–15% at concentrations typical of intracellular conditions.112,113 As we outline next, macroscopic intracellular viscosity is primarily governed by the pool of much larger macromolecules, which collectively give rise to yet another important phenomenon in cellular biophysics: macromolecular crowding.
2.3. Macromolecular Crowding
A general property of the intracellular space is its high net content of large biological macromolecules. The typical E. coli cell contains ∼25% protein by volume, of which ∼10% are cytoskeletal filaments and ∼90% are soluble globular proteins, along with substantial amounts of RNA, DNA, and biopolymers such as lipids and glycans. In the E. coli cytoplasm, this corresponds to 200–320 g/L of protein (∼4 mM), 75–120 g/L of RNA, and 11–18 g/L of DNA.66,114 In mammalian cells, protein concentrations ranging from 50 to 250 g/L and nucleic acid concentrations of 20–50 g/L have been determined, which vary with cell types.115−117 Thus, macromolecules occupy 10–40% of any cell volume and make this space unavailable to other macromolecules. The resulting macromolecular crowding effect has several consequences.
It was realized early on that steric repulsion between individual molecules in highly volume-occupied solutions such as the cytoplasm decreases the volume available to other molecules. The resulting “excluded volume effect” stipulates that the effective concentration of a test solute is determined by the number of molecules of that solute per unit of available volume, rather than the total volume (Figure 3A). Therefore, the thermodynamic activity of a given solute in a crowded environment does not depend on its nominal concentration but on its effective concentration, which depends on the available volume.118,119 The thermodynamic activity of the test species might thus exceed its nominal concentration by several orders of magnitude, especially in cases of severe crowding. The term macromolecular crowding was coined in 1981 to connote the influence of mutual volume exclusion on the thermodynamic, kinetic and structural properties of macromolecules in crowded media.120 While the original use of term denoted effects of inert repulsive forces, it has since been updated to also include weak attractive interactions.121 In the first part of our discussion, we describe “classical” macromolecular crowding effects that result from steric repulsion and volume exclusion. In the second part, we outline additional attractive effects that contribute to crowding in cells.
Figure 3.
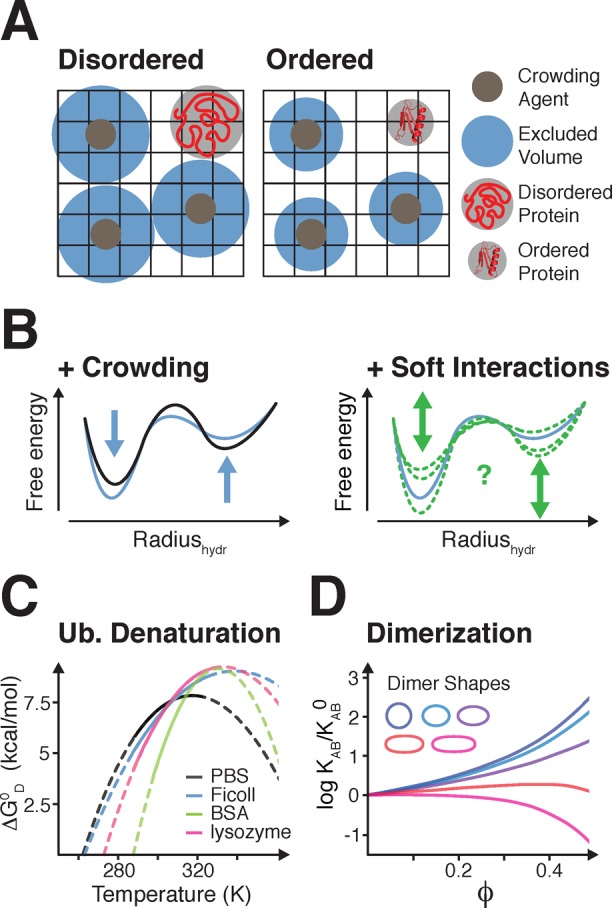
Excluded volume effects. (A) Schematic representation of excluded volume effects for disordered and folded proteins. (B) Left: IDP free energy as a function of compaction in noncrowded (black line) and crowded environments (blue line). Right: Representation of stabilizing or destabilizing effects via soft interactions (green lines). (C) Free energy of ubiquitin denaturation as a function of temperature in buffer pH 5.4 (black), 100 g/L Ficoll (blue), 100 g/L BSA (green), and 100 g/L lysozyme (pink). Solid lines are fits based on experimental measurements, dashed lines are extrapolations for different temperatures. Adapted with permission from ref (124). Copyright 2012 American Chemical Society. (D) Dimerization equilibrium constants for different dimer shapes (constant volumes) as a function of volume fraction ϕ of hard-sphere crowders. Adapted with permission from ref (123). Copyright 2008 Annual Reviews.
2.3.1. Macromolecular Crowding and Steric Repulsion
In a simplified model of macromolecular crowding, intracellular background molecules are considered inert hard-spheres that exert steric repulsions on other molecules and thereby increase the total free energy of the system. In turn, the system shifts equilibria toward states that maximize the available volume. One predicted outcome is that macromolecular crowding promotes protein compaction (Figure 3B), which may stabilize globular proteins (Figure 3C), or promote protein–protein associations (Figure 3D), which may, in turn, lead to conformational collapse and aggregation (see section 4).122−124 Mimicking crowding with high-pressure experiments for example, showed that poly alanine helices are more compact under such conditions.125 Macromolecular crowding can also increase protein hydration.126 Surface hydration generally favors protein folding by promoting the formation of hydrophobic cores, but also because water is a poor solvent for the polypeptide backbone.56,127 However, at high concentrations of macromolecules (>100 g/L), weak interactions between the crowding molecules induce cluster formation128 and crowder-solute interactions, which can destabilize globular proteins.124,129,130 Accordingly, in vivo studies of protein folding reported weak modulatory effects. Macromolecular crowding within the E. coli cytoplasm was found to not be sufficient to fold the slightly (1 kcal/mol) destabilized B1 domain of streptococcal protein L.131 In mammalian cells, the melting temperature of folded phosphoglycerate kinase (PGK) increases by ∼3 K compared to isolated in vitro conditions,132 whereas that of the surface antigen VlsE decreases by ∼4 K.133 These effects are cell type-specific and vary in different organelles,134,135 and at different stages of the cell cycle,136 which is largely due to modulations of intracellular crowding, and changes of specific and unspecfic interactions (see section 2.3.4).
How does intracellular macromolecular crowding affect the conformational properties of disordered proteins? IDPs are more flexible and less compact than ordered proteins;137,138 therefore, they may experience exacerbated crowding effects and respond with even greater degrees of compactions. Indeed, IDP compaction was reported for the disordered carboxyamidated ribonuclease T1 (TCAM) in the presence of 400 g/L of dextran139 and for the C-terminal domain of histone H1 in the presence of PEG and Ficoll.140 Similarly, the disordered FlgM protein displays structural alterations toward more folded conformations in intact E. coli cells and in glucose, BSA and ovalbumin-crowded solutions (see section 5.2). Urea denaturated CRABP, which normally folds into a β-rich structure, was also found to be more compact in the presence of Ficoll.141 Higher concentrations and larger sizes of PEG molecules lead to greater intramolecular FRET efficiencies, and enhanced compaction of N- and C-terminal fragments of human prothymosin-α, the binding domain of the activator for thyroid hormones and retinoid receptors (ACTR) and the N-terminal domain of HIV-1 integrase.142 By contrast, other disordered proteins, including the c-Fos transactivation domain, the p27 (Kip1, Cdkn1b) kinase-inhibition domain, the acidic extracellular α-casein, the basic cytoplasmic protein MAP2c, the nuclear kinase inhibitor p21 (Cdkn1a), the highly acidic protein ProTα, the basic protein TC-1, the repeat-in-toxin (RTX) motif (which folds upon binding to Ca2+), as well as the bacteriophage λ N protein failed to show compaction under similarly crowded in vitro conditions.143−147 In-cell IDP compaction or folding was not reported for α-synuclein or human tau.148−151 Together, these results show that cellular and in vitro crowding contributions on protein compaction do not follow the uniform trend predicted by “classical” theory and that intracellular “soft” interactions, often of unspecific nature, may indeed determine the net effects observed in intact cells (Figure 4B).136,147
Figure 4.
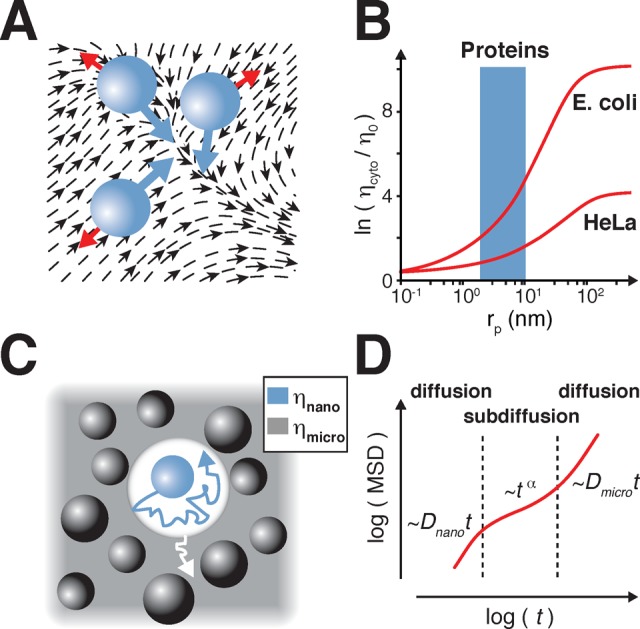
Macromolecular crowding and viscosity. (A) Schematic representation of water flows (black arrows) in response to protein movements (blue spheres and arrows). Forces resulting from water–protein friction are shown as red arrows. Translational diffusion is slowed down by hydrodynamic interactions with water flows caused by protein movements. (B) Different viscosity regimes experienced by probes of radii (rp) in the cytosol of E. coli and HeLa cells (red curves). Average size range of proteins is indicated in blue. Adapted with permission from references (157) and (170). Copyright 2011 American Chemical Society and Copyright 2012 Oxford University Press, respectively (C) Protein motions in crowded environments. Proteins experience Brownian motion in the depletion layer of apparent nanoviscosity (ηnano) (blue). The depletion layer moves according to the microviscosity (ηmicro) (gray) of its surrounding. (D) Particle mean square displacements (MSD) in different diffusion regimes as a function of time. Anomalous subdiffusion occurs at the transition between fast nano- (Dnano) and slow microdiffusion (Dmicro). Adapted with permission from ref (169). Copyright 2013 IOP Publishing.
2.3.2. Macromolecular Crowding and Viscosity
At identical (w/v) concentrations, macromolecular crowders cause greater macroscopic viscosity than small solutes. For example, PEG solutions are more viscous than ethylene glycol solutions. At a given concentration of PEG, viscosity further increases with increasing chain length.152,153 Similar observations were made for dextran154 and other crowding agents.155−158 To explore these viscosity anomalies, Lu et al. employed a theoretical capture-flow model, in which macromolecules exhibit size-dependent characteristics, i.e., capture capacities and weak interactions with other molecules that they drag along. These particles exert draining forces on noncaptured molecules, which result in passive flows. The combination of these effects determines the overall macroscopic viscosity.159 This description provides a valuable dynamic and scale-dependent view: Shape, size, and surface properties of crowders and solute molecules mutually influence each other by exerting dragging and draining forces.
As we discussed above, and consistent with the model by Lu et al., neither metabolites nor macromolecules alter the viscosity of bulk intracellular water on the nm scale.108,109 At this level, macromolecular crowding only influences water molecules in the first hydration layer. Because these properties are similarly displayed in pure protein-buffer solutions, additional contributions may affect the viscosity of intracellular water at larger time- and length-scales. Indeed, when intracellular water dynamics are measured by magnetic resonance imaging (MRI), i.e., in the ∼10–100 ms range or at 1–10 μm, water diffusion coefficients in intra- and extra-cellular brain sections are found to be reduced by factors of 2 to 10, which could reflect different spatial or compositional cell organizations or time-averages of movements of water molecules in and out of hydration layers.160 In general, however, these results indicate that intracellular solvent viscosity displays the same features as viscosity in aqueous protein solutions at the nm scale: unperturbed properties of bulk water, with increased solvent viscosity and reduced dynamics of hydration shell water.
Moving to larger objects, such as proteins, it is beneficial to also discuss viscosity effects with respect to different time- and length-scales. At the ns time scale, i.e., in the Å range, crowding induces local nanoviscosity via hydrodynamic interactions, not hard-sphere repulsions. Movements of solute molecules induce water flows, which, in turn, reduce the self-diffusion of other solute molecules (Figure 4A).161 In crowded environments, a transition from nano- to microviscosity is observed. Below the nm scale, normal diffusion is guided by Brownian motion and results in particle mean square displacements that increase linearly with time, while diffusion coefficients decrease with hydrodynamic forces. Above the nm scale, overall increases in apparent microviscosity are felt by solute particles. At this point, intermolecular hard-sphere repulsions between crowders and solute particles impair diffusion. The extent to which microviscosity is experienced by a particle depends on its radius, and the crowding agents’ dimensions and concentrations.157,162−169 Holyst et al. rationalized this effect in an advanced manner: (i) When a particle is much larger than the crowding agent, it experiences microviscosity, which increases exponentially with the concentration of the crowding agent. (ii) When a particle is much smaller than the crowding agent it experiences nanoviscosity, which corresponds to the viscosity of bulk water. (iii) Between these extremes, viscosity increases for particles that are larger than the average distance between the individual crowding molecules. This correlation length scales inversely with the concentration of the crowding agent. Using this formalism, the authors fitted data from previous studies of intracellular tracer molecules of different sizes and types (protein, DNA, dextran) and deduced average correlation lengths of ∼0.5 and ∼5 nm for prokaryotic and eukaryotic cells, respectively (Figure 4B).157,170
The hydrodynamic radii of most disordered proteins are in the range of these correlation lengths. 140 residue α-synuclein has a radius of ∼4 nm,171 whereas tau (441 residues) has a radius of ∼6 nm.172 According to the correlation length model, and provided that intracellular crowding does not significantly change the hydrodynamic radii of these IDPs, both proteins experience an intracellular microviscosity inside E. coli and mammalian cells that is 8- and 3-times greater than in buffer, respectively. Because both radii are representative for many disordered proteins, these approximate values are likely to be generally valid.173,174 Accordingly, IDPs succumb to an effective intracellular nanoviscosity that is ∼2-fold greater than in dilute solution, whereas microviscosity is ∼5-fold greater, on average. Holyst et al. conceptualized these differences in apparent viscosities by modeling proteins as spheres that move through a solution of polymers. These spheres create a solvent layer depleted in polymers due to macromolecular repulsion or low polymer entropy at the protein–solvent interface. The protein moves within the depletion layer according to its nanoviscosity, whereas the solvent layer diffuses independently and according to the microviscosity of the system (Figure 4C).168,175 This model conceptualizes many of the measured intracellular viscosity effects and also accounts for differences in diffusion behaviors in cells (although intracellular viscosity and diffusion are strongly related, we treat cellular diffusion processes separately in the next paragraph). This model also offers a compelling explanation for why rotational diffusion, governed by nanoviscosity, is less affected than translational diffusion, which is primarily influenced by microviscosity (see below).155,176
2.3.3. Macromolecular Crowding and Diffusion
Macromolecular crowding-induced nano- and microviscosity affect intracellular protein diffusion. In general, we distinguish between two main types of diffusion: Rotational diffusion, as a property that is felt at the subnano- to nanometer scale and that denotes the localized reorientations of solute molecules according to their internal mobilities. Translational diffusion, on the other hand, is felt at length scales above 10 nm and describes movements of solutes from one point in space to another.
2.3.3.1. Rotational Diffusion
In E. coli, rotational diffusion was found to be 2–3 times slower than in pure water.177 For eukaryotic intracellular compartments, the measured rotational correlation times in the cytoplasm are 10–20% larger than in dilute solution, 10–30% larger in mitochondria and 100% larger in the endoplasmic reticulum.178−180 These values directly reflect the local cellular viscosity on the subnano to nanometer scale, which influences the rotational correlation times of proteins, but also their internal motions. In the case of disordered proteins, backbone motions are difficult to dissect from rotational diffusion properties, because both occur on the same ns time scale. It is therefore advantageous to describe the dynamic properties of IDPs in a residue-resolved manner. In the absence of protein–protein interactions or global conformational rearrangements, IDPs display residue-specific correlation times that are directly proportional to the nanoviscosity of their environment, i.e., their internal dynamics scale inversely with nanoviscosity.181 Hence, disordered proteins may exhibit intracellular correlation times that correspond to low nanoviscosity environments (∼2-fold greater than in buffer), whereas their translational diffusion properties may be slowed 5-fold or more (see section 2.3.3.2). However, these numbers need to be considered with caution as they reflect ideal dynamic behaviors in the absence of intracellular, intermolecular interactions and proteins tend to constantly engage in short-lived, weak interactions with other intracellular biomolecules.121 As we discuss in section 2.3.4, Li et al. showed that, in E. coli, these interactions slowed the rotational diffusion properties of the small folded protein G B1 domain (GB1) and ubiquitin (Ub) by as much as viscosity increases of 5 and 22 cP, respectively.177 Given the average solvent viscosity of the E. coli cytoplasm (∼3 cP), this is in good agreement with the 8-fold increase in the rotational correlation time of GB1 that Wang et al. measured by in-cell NMR spectroscopy.182
Do transient intracellular interactions affect ordered and disordered proteins differently? In folded proteins, weak interactions often target localized structural entities such as hydrophobic or charged surface patches.182,183 In disordered proteins, transient contacts may predominantly occur at clustered charged or hydrophobic residues or at regions of transient secondary structure functioning as promiscuous binding interfaces (see section 3.7). Hence, weak IDP interactions may result in more discontinuous viscosity effects that target individual protein segments differently. In-cell NMR data on human tau, α-synuclein and other disordered proteins support this notion (see section 5.2).149,151,184,185
2.3.3.2. Translational Diffusion
Translational diffusion is defined as the mean-square displacement of a given particle with time (t), and according to Fick’s law, the diffusion coefficient (D) denotes the velocity with which a solvent unit cross section (μm2) is transversed in time (s–1). In a homogeneous solvent, where the solute size is comparable to or greater than that of the solvent, translational diffusion is primarily determined by the solute’s size and shape. We refer to this type of diffusion as normal diffusion, which is primarily governed by Brownian motion (i.e., Brownian diffusion). In inhomogeneous environments or where the solute is smaller than the solvent or where large fractions of the volume are occupied by other solutes (i.e., macromolecular crowding), translational diffusion can become complex and exhibit characteristics of anomalous diffusion (non Brownian diffusion). While normal diffusion scales linearly with time, anomalous diffusion does not. To describe non-Brownian diffusion, a tα term is introduced. α values between zero and one denote subdiffusion. α values greater than one denote superdiffusion.
A central assumption in describing normal diffusion is that the solute moves within a continuous hydrodynamic fluid, which is not the case in cells. Cellular interiors harbor vast amounts of metabolites and macromolecules, exhibit finite dimensions, spatial restrictions, confinements and intricate networks of organelles, vesicles and the cytoskeleton. Given that macromolecular crowding increases viscosity at different length scales, it is clear that normal diffusion is restricted in cells and that intracellular diffusion displays anomalous characteristics.168,169 In the following paragraph, we discuss experimentally determined diffusion behaviors of proteins on the micrometer scale in prokaryotic and eukaryotic cells. Methods to measure these properties are mostly based on fluorescence imaging in combination with single-particle tracking. We discuss these techniques in section 5.3.
As indicated above, prokaryotes exhibit much higher intracellular concentrations of metabolites and biological macromolecules than eukaryotes. At physiological osmotic concentrations (∼0.44 Osm), bacteria contain ∼200 g/L of protein. When shifted to 1.02 Osm, protein levels reach ∼320 g/L, which corresponds to a concentration typically observed in protein crystals. Translational diffusion of GFP [∼27 kDa, hydrodynamic radius (RH) 2.82 nm] in E. coli is ∼10 times slower (Dcyt = 3–8 μm2/s) than in water (D = 87 μm2/s).164 These diffusion properties are contrasted by the 3- to 9-fold higher intracellular diffusion coefficients inside Dictyostelium discoideum (Dcyt 24 μm2/s) and in mammalian NIH-3T3 fibroblasts (Dcyt = 27 μm2/s), which reinforces the notion that macromolecular crowding in mammalian cells exerts weaker effects on intracellular diffusion than in bacteria. Moreover, the intracellular translational diffusion coefficients of Ficoll and dextran scale similarly with size in mammalian cells and in pure water, up to molecular weights of ∼500 kDa (radius of gyration ∼17.5 nm) and their magnitudes are only ∼4 times smaller.163,186 The average hydrodynamic radii of disordered proteins (4–6 nm) are slightly larger than those of similarly sized, ordered proteins, which suggests that the reductions in intracellular GFP diffusion similarly apply to IDPs. Thus, translational protein diffusion in E. coli and mammalian cells is ∼8- and ∼3-times slower than in pure water in the supra-nanometer range (see section 2.3.2).
When bacteria are exposed to external osmotic upshifts, the passive loss of intracellular water (up to 70%) increases the molar concentrations of intracellular macromolecules. Under such conditions, most intracellular water is coordinated within the first hydration layer of the “suspended” biomolecules. At 1.02 Osm, corresponding to 400 mM of external NaCl, the intracellular diffusion coefficient of GFP is 0.014 μm2/s.164 Under similar osmotic conditions, van den Boogaart et al. reported a drastic reduction in intracellular GFP diffusion, which coincides with the formation of noncontinuous pools of intracellular GFP indicating that active diffusion barriers are formed.187 Interestingly, no such diffusion barriers or impairments on intracellular diffusion behaviors are detected for low molecular weight compounds, such as fluorescently labeled sugars, even at 14.7 Osm (2 M NaCl).164 These results imply that the bacterial cytoplasm displays sieve-like properties toward larger molecules that result in strong impairments of translational diffusion and local entrapments under conditions of severe macromolecular crowding. These properties, however, largely depend on the bacterial metabolic state and cytoplasmic fluidity can be drastically different in active or inactive cells.188
The Stokes–Einstein relationship states that diffusion of a particle scales inversely with its Stokes radius, also called hydrodynamic radius (RS or RH), which is determined by its size and shape. When Mika et al. plotted intracellular diffusion coefficients of different proteins in E. coli against their individual molecular weights they found that, while diffusion scales inversely with size as expected, cytosolic diffusion coefficients (Dcyt) are generally smaller than predicted by the Stokes–Einstein equation.164 This disparity increases with molecular weight, which indicates that larger proteins or protein complexes diffuse much slower than suggested by their size. Theoretical and experimental approaches in artificial crowded in vitro environments recapitulated some of these properties via excluded volume effects (see section 2.3.2).157,167,168 However, they were unable to explain the quasi-immobile nature of intracellular particles larger than 50 nm.186,189 Similar observations were made in E. coli for particles larger than 4.5 nm, which is equivalent to 4 covalently bound GFP molecules.166 These results suggest that organized intracellular structures, such as the cytoskeleton, form a restrictive sieve-like meshwork impeding the diffusion of macromolecular assemblies above certain sizes. These structures were estimated to exhibit pores of ∼5 and ∼50 nm diameters in E. coli and mammalian cells, respectively. In the case of mammalian cells, this size is above the average RH of folded and intrinsically disordered proteins in their monomeric states. However, when one considers the average hydrodynamic radii of IDP aggregates (∼90 nm for fibrillar tau, for instance), oligomeric IDP species may exhibit reduced intracellular diffusion (see section 2.3.2). In fact, IDP aggregates may constitute barriers to intracellular translational diffusion and increase intracellular viscosity or exacerbate the effects of existing sieve-like structures. In the case of α-synuclein for example, aggregates could hamper vesicle transport in neuronal cells and contribute to Parkinson’s disease. Similarly, fibrillar tau tangles may increase the diffusion-limiting effects of the microtubule network, with which the protein physiologically interacts, and thereby impair active transport along microtubules.
What are the biological consequences of slowed intracellular diffusion? Although the crowded cytoplasm of prokaryotic and eukaryotic cells restricts translational diffusion, most biological reactions occur faster than diffusion. Mika et al. calculated that inside E. coli (∼3 μm long) it takes 30 ms for fluorescently labeled glucose (MW ∼0.3 kDa; D = 50 μm2/s), 0.5 s for GFP (MW ∼27 kDa; D = 3 μm2/s), 2 s for 4xGFP-tagged β-galactosidase (MW ∼580 kDa; D = 0.8 μm2/s), and 75 s for 25–50 MDa ribosome-loaded mRNAs (D = 0.02 μm2/s) to transverse the cytoplasm.164 Given the average doubling time of E. coli (∼30 min), even the largest macromolecular assemblies can travel back and forth multiple times during one cell division cycle. Osmotic stress will increase these times and progressively affect biological reactions that rely on fast macromolecular diffusion.
Recent data suggest that even simple organisms such as bacteria employ forms of spatial organization that anchor large macromolecular assemblies at defined intracellular positions (see section 4.2).190,191 This organization may ensure that biomolecules, such as newly synthesized proteins are abundantly available in certain intracellular areas to enable fast interactions en route to supramolecular complex formation. In turn, such spatial restrictions may promote the establishment of functional compartments in the absence of organelles. In higher eukaryotes, translational diffusion in the lumen of intracellular organelles contributes to many cellular processes. Diffusion in mitochondria has been studied extensively, primarily because of the functional importance of this organelle and its high degree of macromolecular crowding, with protein concentrations ranging from 270 to 560 g/L.192 These concentrations prompted researchers to propose that “metabolite channeling” along spatially arranged, membrane-bound enzymes was the only way to ensure efficient mitochondrial activity.180,193 However, fluorescence studies of GFP targeted to the mitochondrial matrix established that its diffusion coefficient in this environment is 20–30 μm2/s, only 3–4 fold lower than in pure water.180 These measurements revealed that inside mitochondria effective contributions from macromolecular crowding are much smaller than expected based on absolute protein concentrations. Herrmann et al. later showed that membrane embedded polypeptides make up the largest portion of these mitochondrial proteins and do not contribute significantly to matrix lumen macromolecular crowding.194 When one subtracts the spatial restrictions that the inner mitochondrial membrane and its invaginations impose on translational diffusion, the effective viscosity felt by GFP is only ∼2 times higher than in water.195,196 Similarly, GFP diffusion in the lumen of the endoplasmic reticulum is only 9 to 18 times slower than in water, including reductions due to membrane intrusions.179 The viscosity in the nucleus is 1.2 to 1.4 times greater than in water.197 Together, these results establish that general intracellular- or organelle-macromolecular crowding exerts small effects on the diffusion properties of average-sized proteins at physiological concentrations, irrespective of whether they are ordered or intrinsically disordered.
2.3.4. Macromolecular Crowding and Weak Interactions
One additional observation from these, and other experiments concerns the notion that similarly sized, but chemically distinct particles or proteins, often exhibit marked differences in their intracellular diffusion behaviors. Extended DNA fragments for example, diffuse slower in the cytoplasm and nucleus of HeLa cells than dextran molecules of similar sizes.189 Clearly, up to now we adopted an overly simplified view of the cellular interior and neglected possible “biological” contributions to macromolecular crowding. It is therefore reasonable that size and shape alone do not determine a biomolecule’s intracellular diffusion. Especially weak transient interactions exert a strong influence on rotational and translational diffusion processes in cells. We collectively refer to these contributions as “soft” interactions, also to contrast their effects with “hard-sphere repulsions” according to classical macromolecular crowding theory.121 Soft interactions can be repulsive or attractive, and they can either exacerbate or counteract hard-sphere effects. Depending on their nature and magnitudes, soft interactions can enforce or diminish the previously outlined, purely physical crowding effects on protein diffusion.
Because “soft” interactions are omnipresent in biological systems, several studies have aimed at providing concise descriptions about their origins. Simple examples include positive or negative electrostatic contributions: Strongly charged surface properties, especially when they occur in a highly localized manner, can differentially modulate a protein’s diffusion behavior in positively or negatively charged environments.198,199 Similarly, hydrophobic surface patches or promiscuous binding sites can result in soft interactions.177,182,183,200 In contrast to ordered proteins, IDPs expose their residues to the solvent and, therefore, have much larger solvent accessible surface areas. In turn, this increases the likelihood for multivalent weak interactions, which may lead to complex subdiffusion.162
2.3.5. Macromolecular Crowding and Intermolecular Association
So far, we discussed how macromolecular crowding has the capacity to modulate the structures and diffusion behaviors of biological macromolecules, as well as how weak transient interactions can counteract or exacerbate these effects. In our introduction to classical macromolecular crowding theory, we also stated that greater effective concentrations and enhanced thermodynamic activities result in direct consequence of excluded volumes and crowding. Here, we outline how these combined crowding and interaction-effects influence protein–protein association. We separately discuss the impact of macromolecular crowding on aggregation in section 4.
2.3.5.1. Contributions to Equilibrium Thermodynamics
Using the same simplistic thermodynamic rationale employed to explain protein compaction, i.e., minimizing volume occupancy; macromolecular crowding can favor protein–protein association, because it also enables the system to decrease its volume. However, this statement cannot be generalized, because some associations result in complexes with dimensions that are greater than those of the individually interacting molecules.123 Therefore, macromolecular crowding can either stabilize or destabilize protein–protein interactions, depending on the size and shape of the reacting molecules and the resulting complexes (Figure 3D). Mildly stabilizing (∼2–5 fold) effects were observed in vitro for the formation of heterodimers between E. coli polymerase III theta-epsilon subunits in dextran and Ficoll-crowded solutions and for the interaction of superoxide dismutase and xanthine oxidase in PEG-, dextran-, and Ficoll-containing environments.201 Destabilizing effects (∼2–4 fold) were noted for the barnase-barstar interaction in the presence of PEG and for TEM1 and β-lactamase-BLIP inhibitor in PEG- and dextran-crowded solutions.201 In agreement with theory, stronger stabilizing effects were detected for complexes with more than two subunits.123,201 A 2-fold in vivo affinity increase between the GTPase Cdc42 and its various effector-proteins was reported in HeLa cells.201
2.3.5.2. Contributions to Kinetics
Viscosity-dependent diffusion of reacting molecules determines their interaction rates. Higher viscosity decreases association rates but also decreases dissociation rates. Because intracellular viscosity depends on different macromolecular crowding contributions and scales differently at different length scales (see sections 2.3.2 and 2.3.3), kinetic effects can have complex behaviors. On the subnm scale, translational diffusion in cells is much faster than on the μm scale, where anomalous diffusion prevails. According to theory, anomalous diffusion produces greater probabilities for protein–protein interactions in the vicinity of binding interfaces, i.e., at distances below 10 to 100-times the radii of interacting molecules.202 In other words, proteins succumb to macromolecular crowding-induced partial confinement in a concentration-dependent manner. Following this notion, one concludes that chances for interactions increase when two proteins are abundant and in close proximity. This behavior is exacerbated by steric repulsion, but unaffected by slower translation diffusion. When two proteins are present at low abundance and separated, their chances to interact decrease. This opposite behavior is equally affected by macromolecular crowding, but largely governed by slower translational diffusion. Such a view is nevertheless biased toward intracellular association rates and oblivious to dissociation kinetics. Because specifically interacting proteins tend to colocalize in cells, these assumptions may not accurately reflect physiological scenarios, and in vivo association kinetics may differ only marginally from in vitro rates. Some experimental results point in this direction. Association rates of the barnase-barstar interaction are not affected by polyvinylpyrrolidone (PVP) crowding and reduced 3-fold in the presence of 300 g/L PEG.201 Similarly, TEM1-BLIP binding is less than 4-fold slower in PEG-, dextran-, and Ficoll-solutions, and only 2-fold slower in HeLa cells.201
2.4. Cellular Interfaces and Environments
So far, we discussed the compositional and physical properties of intracellular environments as continuum parameters. However, in cells, these properties fluctuate in a spatial and temporal manner. Subcellular organization directly correlates with evolutionary advancement, and organismal complexity and compartmentalization are instrumental in many biological processes.203−206 In turn, different regions in prokaryotic and eukaryotic cells exhibit different microenvironments that affect biomolecules in their vicinity.
Organelles are separated by membranes that function as physical barriers in preserving environmental properties and counteracting exchange. Their interiors offer different environmental conditions, which are often determined by their biomolecular compositions. The cell nucleus for example, harbors vast amounts of DNA, a negatively charged polyelectrolyte with an overwhelming charge potential of −2e per base pair, which imposes considerable electrostatic constraints on architectural proteins such as histones or DNA-binding proteins such as transcription factors.207−209 Histones contain extended, disordered “tail” regions that are highly positively charged. In the context of nucleosomes, the basic packaging unit of nuclear DNA,210 histone tails experience electrostatic DNA contributions, that ultimately dictate their structural and functional behaviors.211 Post-translational histone modifications alter these electrostatic properties and, in turn, modulate interactions with DNA. Because these modifications partially establish the epigenetic histone code, thereby regulating the transcriptional states of entire genomes, they provide a compelling example of a context-regulated biological activity that relies on structural disorder as the main determinant for function.212
Enveloped by membranes, organelles offer additional lipid-solvent interfaces at their interiors and exteriors. Given the chemical and compositional diversity of biological membranes, and their differing physical properties including thickness, fluidity, curvature, lateral pressure, bilayer coupling and surface charge, they constitute highly specialized environments.213−215 All biological events at membranes occur with strong electrostatic contributions.216 One classical example of how structural disorder is functionally linked to these properties is the electrostatic myristoyl switch.217 Peripheral membrane proteins such as the myristoylated alanine-rich C kinase substrate (MARCKS), Src kinase and the transducin αt subunit contain N-myristoylated glycines, followed by long stretches of disordered residues with characteristic patterns of basic and hydrophobic amino acids, so-called basic effector domains. Membrane attachment is mediated by the respective myristoyl moieties. However, myristoyl-membrane binding is not strong enough to firmly anchor these proteins in the lipid bilayer. Complementary charge interactions between acidic phospholipids and the basic effector domains are required for high affinity binding. In turn, phosphorylation of conserved serines within the effector domain of MARCKS, for example, reduces its net charge and results in membrane dissociation.218 Thus, synergistic hydrophobic and electrostatic effects, conferred by disordered protein regions mediate the reversible membrane interaction.217 In fact, most signal sequences that target proteins to organelles are intrinsically disordered and exhibit high contents of hydrophobic and/or positively charged residues. Therefore, some of the mechanisms of membrane binding and membrane crossing rely on similar biophysical principles.219−222
In prokaryotes and eukaryotes, different types of lipids assemble into functional microdomains termed lipid rafts.223 Lipid rafts harbor specific sets of trans-membrane proteins and act as platforms for complex biological processes such as signal transduction, cytoskeletal organization, membrane transport and pathogen invasion.223−225 Different lipid rafts exhibit distinct lipid compositions and have characteristic permeabilities, fluidities, and overall electrostatic properties.226,227 In eukaryotes, they are characterized by high degrees of stiffness, which results from enrichment in cholesterol and sphingolipids.221,224 Microenvironments around these lipid rafts exacerbate or reduce electrostatic effects and mediate attractive or repulsive long-range interactions.228 Because all membranes exert hydrophobic and electrostatic effects, the latter predominantly through acidic phosphoplipids, disordered proteins with clustered positively charged residues experience attractive forces, which can be weak, but biologically meaningful in mediating membrane interactions, as we have seen above. By the same token, patches of negatively charged residues in disordered proteins are electrostatically repulsive, which may prevent unspecific membrane interactions.
Protein complexes spanning membranes and connecting cellular compartments, often mediate selective transport and exchange of biomolecules. To do so, they frequently form microenvironments of defined physical properties, especially with regard to permeability. One striking example is the nuclear pore complex. Bridging the cytoplasm and the nucleus, it contains an interior microenvironment that is made up entirely by disordered segments of distinct nucleoporins.229,230 Their extended, Phe-, Gly-rich (FG) regions form a meshwork of filaments that functions as a sieve-like hydrogel.231−233 Proteins, and other biomolecules, entering or exiting the nucleus, have to pass through this microenvironment to reach their respective destinations.234,235 Interestingly, FG-filaments form amyloid-like, β-rich structures and thus represent another cunning example of a functional IDP aggregate (see section 4.2).232
The cytoplasm harbors additional interfaces and microenvironments. As we discuss in section 2.3.3.2, the cytoskeleton establishes multiple physical barriers that spatially confine vesicles, organelles and large cellular machines such as ribosomes. The segregation of the cytoplasm into nanometer-sized compartments bounded by cytoskeleton filaments creates defined pools of biomolecules and metabolites. Building on early work by McConkey et al.236,237 and others,66,238,239 Spitzer and Poolman developed a comprehensive physicochemical model of the cytoplasm to explain these effects. Although originally delineated for prokaryotic systems, their concepts can be extended to eukaryotic cells.204,205,240 The Spitzer and Poolman model postulates the existence of intracellular metabolic zones that comprise electrolyte pools and levels of macromolecular crowding different from other regions, such as cell membranes and organelles.204 Formation of metabolic zones largely depends on asymmetric charge distributions, which are generated by electrostatic surface properties, especially anionic patches.65,241 Positively charged species are less abundant and only partially neutralize the entirety of cellular macro- and microanions.242 In their model, the cytoplasm is steeped in electrolyte pools that contain unique temporal compositions of metabolites, ions and freely diffusing macromolecules.205,240 The boundaries of these pools are defined by the charged surfaces of macromolecules, which further provide electrochemical gradients that drive the flow of charged metabolites. Overall, the surfaces of these structures remain negatively charged and repel each other, thereby preventing spontaneous aggregation. Cellular machines such as ribosomes reside at specific locations in these metabolic zones, where more or less defined compartments insulate concurrent biological processes and prevent mutual perturbations.205,237,243
The model by Spitzer and Poolman is a good description of the heterogeneous cytoplasm. Importantly, it emphasizes the notion that the physical and compositional characteristics outlined in this section only describe a basis set of general parameters influencing the structural and functional properties of disordered and ordered proteins in cells. To fully grasp in vivo contributions to cellular structural biology we must take spatiotemporal variations of these physicochemical parameters into account.
3. Biological Properties of IDPs Inside Cells
Proteomic analyses of numerous organisms show that intrinsic disorder scales linearly with evolutionary progression and that disorder contents are larger in proteomes of higher eukaryotes than in prokaryotes or archaea. Furthermore, proteomes of multicellular eukaryotic organisms contain larger proportions of disorder than unicellular ones.5,6 Approximately 30% of all mammalian proteins are disordered, whereas 75% of all signaling proteins contain extended disordered regions. Globular proteins make up the majority of cellular enzymes and transport proteins, which require stable three-dimensional structures to execute their respective biological functions.15 Proteins or protein domains that regulate and mediate adaptive and malleable biological responses require significant conformational flexibility and are predominantly disordered (see section 3.6).13,244 Accordingly, disorder is particularly prevalent in eukaryotic proteins and processes that rely on these functions, such as cell differentiation, cell–cell communication, cell-cycle progression, transcriptional regulation, apoptosis, etc..15 Comprehensive functional annotations of disorder can be found in several reviews.13,245
Given their prominence in many proteomes, the question arises as to how intracellular levels of disordered proteins, their intactness, stability and degradation are regulated, especially in comparison to ordered proteins. IDP homeostasis must be tightly controlled to avoid incidences of self- and nonself associations, which can directly result in several pathologies. Abnormal accumulations of α-synuclein, Huntingtin, tau, or the Aβ peptides for instance, promote the formation of toxic amyloid fibrils and oligomeric species implicated in Parkinson’s, Huntington’s and Alzheimer’s disease, respectively (see section 4).16 Failure of the proteasome to clear aggregated IDPs additionally contributes to the multifactorial etiology of these disorders.246−249 Besides neurodegenerative pathologies, misregulation of cellular disordered proteins is strongly associated with many forms of cancer. Overexpression of Stathmin and low levels of p27 for example, contribute to the metastatic phenotypes in human sarcomas.250,251 In the following paragraphs, we outline how cells regulate the synthesis, cellular diversity, stability and degradation of intrinsically disordered proteins.
3.1. Regulation of IDP Synthesis
Protein synthesis is generally controlled on the transcriptional (DNA), post-transcriptional (RNA), and translational (ribosome) levels. Because the primary characteristics of IDPs are manifested on the protein level, information about the regulation of their genes and transcripts is sparse. Nevertheless, insights into these processes exemplify how cellular diversity and abundance are regulated.
Two independent studies analyzed gene expression and mRNA levels in higher eukaryotes and compared the relative abundance of transcripts encoding intrinsically disordered-, versus ordered proteins.252,253 Results indicated that, on average, genes encoding disordered proteins are more intricately regulated than genes encoding folded proteins, while the mRNA levels of IDP genes are lower than those of ordered proteins. IDP transcripts were grouped into five categories based on their expression levels and protein abundance.253 Highly expressed mRNAs encode ribosomal proteins or disordered housekeeping proteins such as splicing factors and architectural chromatin components, protease inhibitors, or regulators of enzymatic activities. Intermediately expressed IDP transcripts encode general transcription factors, tissue specific regulators such as receptor ligands, or transcriptional cofactors. Low abundance mRNAs encode disordered proteins involved in organ development and differentiation.253 In another study in E. coli, the authors found a weak, positive correlation between intrinsic disorder, mRNA expression levels and predicted protein abundance.254 Although bacteria have fewer disordered proteins in their proteomes (∼5%) than higher eukaryotes (∼30-50%), their prominence in copius cellular factors such as ribosomal proteins, transcriptional regulators and chaperones253 renders IDP mRNAs more abundant than transcripts of folded proteins. The Babu group also investigated the synthesis and degradation rates of IDP transcripts and compared them to the rates of ordered proteins (Table 1).252 They found that the number of transcription factors regulating IDP gene expression is roughly the same as for folded proteins, and so are their transcriptional rates. Also, that the half-lives of mRNAs encoding highly disordered proteins are shorter compared to ordered proteins.252,253 These results indicated that IDP transcript abundance is primarily regulated by degradation. To validate this assumption, the authors analyzed the lengths of mRNA poly-A tails, which govern intracellular lifetimes and degradation rates.252 They found that IDP transcripts contain shorter poly-A tails than ordered proteins. When the occurrence of Pumilio repeat (PUF)-binding sites on both transcript types was analyzed, PUF proteins bind and down-regulate mRNAs either by signaling degradation or via translational repression by 3′ UTR binding,255,256 more sites were found in IDP mRNAs than in transcripts encoding ordered proteins.252 In agreement with these observations, the Jones group showed that human IDP mRNAs display a higher abundance of predicted miRNA binding sites and faster degradation rates than transcripts encoding ordered proteins.253 However, these values are statistical averages and their distributions are broad.
Table 1. Intracellular Regulation of IDPs and Ordered Proteinsa.
| cellular quantity/regulation mechanism | ordered proteins | disordered proteins |
|---|---|---|
| mRNA level | ||
| transcription factors/gene | 2.0 | 2.0 |
| transcriptional rate (mRNAs/hour) | 2.2 | 1.8 |
| transcript abundance (copies per cell) | 0.9 | 0.8 |
| transcript half-life (min.) | 23.0 | 19.0 |
| transcript degradation (% with short polyA tails) | 28 | 56 |
| miRNA targeting (% sequences)b | 15 | 30 |
| protein level | ||
| translational rate (ribosomes/ORF) | 0.5 | 0.37 |
| protein abundance (proteins/cell) | 2900 | 1860 |
| protein half-life (min.) | 45 | 37 |
| % PEST sequences | 19 | 42 |
| ubiquitination sitesb,c | 50 | 70 |
3.2. Cellular IDP Diversity
Several biological processes can regulate the cellular diversity of disordered proteins. In this section, we discuss the evolvability of IDPs and their greater resistance to mutational damage, as well as how alternative mRNA splicing generates functionally distinct proteins. Furthermore, we provide examples of proteolytic processing reactions that specifically target disordered proteins and describe how they extend IDP diversity in vivo.
3.2.1. Evolutionary Selection
Disordered proteins exhibit greater capacity for mutational evolvability and resistance to mutational damage. Evolutionary rates of ordered proteins are determined by their requirements to preserve structural integrity, which is not the case for disordered proteins. Therefore, IDPs should evolve faster than folded proteins. To investigate this notion, Dunker and Lin compared the genetic distances of ordered and disordered protein regions within 26 families.257,258 Their analysis showed that disorder-rich regions in otherwise ordered proteins evolve more rapidly. The authors propose that these differences primarily arise because globular proteins typically employ more residues to “construct” their three-dimensional structures, leaving them fewer degrees of freedom to accommodate substitutions. Because disordered proteins exhibit fewer intramolecular contacts, their primary amino acid structures tolerate mutational changes better. By the same token, new interfaces arise without deleterious structural consequences, which increases the functional repertoire of IDPs. Dunker et al. use this evidence to convincingly argue that the higher evolutionary rates of disordered proteins also lend strong support to the notion that they exist freely in cells.13 If they were always bound to interacting partners in vivo, probably in folded conformations, their evolutionary rates are expected to be comparable to those of ordered proteins, which is not the case. We discuss additional aspects of IDP evolvability and mutational tolerance in the section on post-translational IDP modifications (see section 3.6).
3.2.2. Alternative Splicing
Alternative splicing constitutes another process regulating IDP diversity.259 Interestingly, alternative splicing occurs more frequently in mRNAs encoding disordered than ordered proteins.260,261 Dunker and co-workers analyzed 46 alternatively spliced human genes and found that 81% of all alternative splice sites occur in fully (57%) or partially (24%) disordered protein domains. Only 19% occur in ordered regions.261 More recently, the Tompa group analyzed ∼500 isoforms of spliced human proteins and found that alternative splicing “avoids” folded domains and preferentially “targets” disordered protein segments.260 The authors propose that alternative splicing favors these regions to minimize deleterious effects of truncated proteins that lead to loss of function, misfolding, and aggregation. Alternative splice-site selection can also entail the use of different exon combinations to generate functionally distinct proteins or dual-coding regions that are alternatively assembled into different open reading frames. A sequence analysis of 62 dual-coding regions revealed that their protein products are particularly rich in disordered regions.262 For +1 frame shifts, the amount of disorder in the resulting protein is comparable with that of the original frame, whereas −1 frame shift proteins are more disordered. The authors showed that this mechanism results in novel functions and also protects mRNAs from nonsense-mediated decay.262
Alternative splicing is particularly versatile in controlling protein diversity and abundance. Splice-isoforms of disordered proteins can contain different sequences, different numbers of linear motifs, or novel post-translational modification sites that can affect functions by changing binding properties, stabilities and/or subcellular localizations. Such changes can rewire protein interaction networks and facilitate new phenotypes in cell-, tissue-, organ- and organism-specific manners.244 One example is p53 (TP53).263 Its central folded domain binds DNA, while the disordered N- and C-termini act as regulatory elements that interact with different effector proteins and harbor numerous modification sites (see section 3.6).264 In humans, 12 alternatively spliced p53 isoforms are known, and splicing mostly occurs within the disordered N- and C-terminal regions.265 In turn, these p53 isoforms display different functional activities with regard to triggering cell-cycle arrest, or apoptosis, they bind effector proteins such as Mdm2 differently, which affects their intracellular stability and degradation, and they localize to different subcellular compartments, i.e., the cytoplasm versus the cell nucleus.266
Similar to alternative splicing, isoforms of disordered proteins are also generated on the protein level by targeted proteolysis. We discuss examples for this regulatory mechanism in the following section.
3.2.3. Proteolytic Processing
Disordered proteins particularly amenable to functional and site-specific processing events, such as targeted proteolysis. While some of these cleavage reactions produce toxic fragments, including aggregation prone species (see section 4), others result in modified proteins with novel functions. Proteolytic processing of the Bcl-2 family of proteins is one example. These proteins contain extended (∼50 residues) disordered loop regions that control pro- and antiapoptotic cellular responses.267,268 Depending on the extent of cell damage, opposing phosphorylation- and site-specific caspase cleavage-reactions within these loops disrupt or enforce existing interactions, and enable new binding events that collectively determine whether cells commit to irreversible cell death or reversible cell-cycle arrest and senescence.268 In a similar fashion, site-specific cleavage within a disordered loop of plasma membrane-bound sterol regulatory element-binding protein (SREBP) by a subtilisin-related serine protease and a Zn(II)-metalloprotease promotes membrane dissociation, nuclear import and, in turn, transcriptional activity.269 Analogous endoproteolytic activation and translocation have been reported for casein kinase I epsilon (CKIε), the NF-κB associated factor Relish and other proteins.270,271
Other examples of productive proteolysis are the prion protein and the amyloid precursor protein. The nontoxic form of the prion protein (PrPc) is membrane-anchored and contains a disordered N-terminus and an α-helical C-terminal domain.272 Conversion of PrPc into toxic prion species (PrPsc) causes Creutzfeld-Jakob’s disease (see section 4). In cells, cleavage of PrPc by members of the desintegrin and metalloproteinase family of proteases dissociates the disordered N-terminus of PrPc from its membrane-bound C-terminus.273 Other proteases such as calpain and cathepsins target PrPsc at different sites, generating longer and shorter N- and C-terminal fragments, while autocatalytic PrPc cleavage also occurs in the presence of reactive oxygen species.273 Whether these proteolytic PrPc and PrPsc fragments execute biological functions remains unclear, although some data suggest they might be neuroprotective.274−276
The amyloid precursor protein (APP) is another ordered, membrane-bound protein with well-defined heparin and Cu/Zn binding domains.277 Along the amyloidogenic pathway, N-terminal portions of APP are cleaved by β- and γ-secretase releasing the disordered, aggregation-prone and highly neurotoxic Aβ peptides. Depositions of these peptides in the forms of amyloid plaques are the pathological hallmarks of Alzheimer’s disease (see section 4). By contrast, nonamyloidogenic processing by α-secretase produces nontoxic Aβ species that regulate gene expression and are considered neuroprotective.277 Together, these examples illustrate how site-specific protease reactions regulate the cellular diversity of intrinsically disordered proteins. Besides functional proteolytic processing, IDPs are also targeted by general cellular proteolysis as a means to maintain homeostasis. Next, we outline how the abundance, stability and degradation of disordered proteins are regulated by intracellular proteases.
3.3. Intracellular IDP Stability and Degradation
In vitro, disordered proteins are more susceptible to proteolysis than ordered proteins. Historically, this property was used to identify and characterize IDPs.278,279 The simplest explanation is that disordered proteins provide unrestricted access to proteases over their entire lengths. Similarly, denaturation of ordered proteins increases their susceptibility to cleavage280 and surveys of crystal structures of substrate-bound proteases show that their substrates are in extended conformations.281 Accordingly, conformational selection is thought to steer proteolytic degradation of ordered and disordered proteins.281 In prokaryotes and eukaryotes, broadly acting proteases degrade misfolded or otherwise damaged proteins. In addition, complex proteolytic systems and processes, such as the proteasome and autophagy regulate global protein homeostasis, acting on a wide range of proteins.26 70–90% of all eukaryotic proteins are cleared by the proteasome282,283 and 40% of all cellular proteasomes exist as 20S proteasomal core units, with poorly defined activities.284 Protein turnover is mainly achieved via ubiquitin-mediated degradation, although 20S particles act in an ubiquitin-independent manner.284 Both 20S and 26S holo-proteasomes require disordered regions for initiating proteolysis, however, catalytic activity is primarily governed by proteasomal targeting motifs (20S) and poly ubiquitination (26S).285−288
Some classes of cellular enzymes, such as the Lon family of serine-specific proteases are tailored to target disordered proteins. ATP-independent Lon isoforms exclusively degrade unfolded substrates, whereas ATP-dependent isoforms require ATP hydrolysis to degrade ordered substrates, but not disordered ones.289,290 The three-dimensional structures of Lon proteases reveal that individual degradation chamber entry channels are too small for ordered proteins,290 suggesting that disordered polypeptides are preferred Lon substrates, whereas ordered proteins require ATP-dependent disassembly to be degraded. Similar observations were made for the proteasome system. Disordered proteins such as α-synuclein, p21 or c-Jun, are efficiently degraded by the 20S core particle in vitro, while ordered proteins are not under the same conditions.26 Interestingly, proteinase K-sensitive, molten-globule proteins are also resistant to 20S degradation.279 As for Lon proteases, these observations suggest that extended, flexible IDP conformations facilitate proteasome entry. Support for this hypothesis comes from experiments measuring 20S proteolysis rates of disordered peptide chains coupled to bulky gold particles.291 Without the gold labels, peptides are efficiently degraded. When bound to gold particles, proteolytic cleavage is severely impaired, likely because the substrates are unable to enter the proteasome. In vitro, the 20S proteasome also degrades folded proteins that contain extended disordered regions at their termini, such as p53 for example. While terminal disorder is required to initiate proteolysis, once globular protein domains are encountered during the degradation process they are actively unfolded in an ATP-independent manner and further proteolyzed.292,293 Similarly, a priming disordered region of ∼30 amino acids is required for polyubiquitin-dependent proteolytic processing by the 26S proteasome.294,295 Indeed, the 26S proteasome degrades fully disordered proteins even in the absence of polyubiquitination,296 suggesting that ubiquitination is not absolutely required for degradation.296,297
So far, we discussed in vitro aspects of proteolytic processing. How do these considerations apply to the degradation of ordered and disordered proteins in vivo? The Babu group used different data sets reporting on protein half-life in a proteome-wide fashion to investigate differences in abundance, stability and degradation of ordered versus disordered proteins (Table 1).252 They concluded that, on average, the half-lives of IDPs are slightly shorter than those of folded proteins. They focused on two parameters known to modulate proteasome-dependent degradation, namely the frequency and abundance of disordered N-end residues and PEST motifs.298,299 The N-end rule states that the half-life of a protein is determined by the identity of its N-terminal residues. In E. coli bulky, hydrophobic amino acids, such as Leu, Phe, Trp, and Tyr are most destabilizing, followed by basic residues (Arg and Lys). In eukaryotes, hydrophobic and basic amino acids act independently in determining the stability of a protein and its properties as a proteolytic substrate.299 In eukaryotic proteins, PEST sequences contain between 10 and 50 amino acids rich in Pro, Glu, Ser, and Thr that cause rapid degradation. Indeed, PEST sequences are overrepresented in proteins with high intracellular turnover rates (<2 h).298,299 Although destabilizing N-end residues are not more prevalent in disordered proteins, PEST motifs are enriched.252 An independent study by Tompa and co-workers using a yeast data set,300 concluded that polypeptide length is the key determinant of in vivo degradation rates, with longer proteins being degraded faster than shorter ones, irrespective of whether they are ordered or intrinsically disordered. Interestingly, when the authors reanalyzed their data after normalizing to length, disordered proteins displayed a weak, although probably significant correlation with enhanced degradation rates.301 According to their study, PEST motifs and other degradation sequences such as the KEN box, only yielded weak correlations with the half-lives of disordered proteins.301
Because polyubiquitination plays a pivotal role in cellular protein degradation, Edwards et al. analyzed the occurrence of predicted ubiquitination sites in disordered and ordered proteins.253,302 They showed that ubiquitination sites are more abundant in disordered proteins. Hagai et al investigated a set of 482 in vivo ubiquitinated proteins and found a strong correlation between confirmed degradation sites and regions of structural disorder.303 In another study on protein turnover in human cells, Yen et al. did not find a correlation between disorder content and protein half-life.304 Given the technical challenges in many of these studies, especially with regard to annotating ubiquitination sites on a proteomic scale, it is difficult to establish a conclusive link between protein disorder and ubiquitin-mediated degradation.305 The Selbach group has measured absolute mRNA and protein levels in mammalian NIH3T3 cells, and their turnover rates and average stabilities, thus providing a superior data set for a genome wide comparison.307 This resource has not yet been interrogated with respect to disorder.
Collectively, many in vivo studies suggest that disordered proteins are not degraded appreciably faster than ordered ones, which is contrasted by their in vitro behaviors. What causes these discrepancies and do they accurately reflect high in vivo stabilities of disordered proteins? One experimental shortcoming in measuring a protein’s half-life with fluorescently labeled tags is the presence of bulky, folded entities at either the N-, or the C-terminus of the investigated polypeptide.300,304 As pointed out by Suskiewicz et al., given the importance of disorder at protein ends in steering degradation (see above), tagging is likely to affect these processes.26,306 In other cases, partial degradation may preserve the globular tags and result in incorrect scoring. Another issue may relate to the use of data sets from different cellular systems and conditions.252 These variations might skew correlations between individual transcript and protein levels.
While these considerations are valid and important, it is unlikely that methodological shortcomings alone account for the observed differences in in vitro and in vivo degradation behaviors. It is plausible that additional cellular factors and effectors synergistically modulate these processes.
3.4. Factors Affecting Intracellular IDP Degradation
Purely physical factors such as intracellular viscosity and macromolecuar crowding are expected to increase, rather than decrease, proteolytic activity. Limiting scenarios may be encountered in cases of high molecular weight aggregates or upon compaction, folding, and binding.308,309 In PEG-crowded solutions for example, the affinity of α-chymotrypsin for its substrates increases, whereas its proteolytic activity decreases. Verma et al. showed that this is caused by crowding-induced structural changes in the protease that favor an open conformation of its substrate entry site, while sterically restricting the exit path.310 A similar behavior was reported for the HIV-1 protease, where crowding stabilizes the closed conformation of the catalytic site.311 For SARS-CoV, protease activity depends on the oligomerization state of the protein. Under dilute in vitro conditions, the enzyme exists in a monomer–dimer equilibrium, with only the dimer being proteolytically active. In the presence of BSA or PEG, dimeric SARS-CoV is stabilized, which leads to a 3-fold increase in activity.312 Therefore, intracellular macromolecular crowding cannot explain the observed differences between in vitro and in vivo degradation behaviors of disordered proteins.
Another explanation may be afforded by intracellular compartmentalization, separating disordered proteins from the degradation machinery. Although organelles such as lyzosomes, function as general destruction units, there are no indications that disordered substrates are excluded from these organelles in vivo. Several other factors may tune the stability of disordered proteins in cells, especially because they interact with diverse sets of biomolecules (see sections 3.6 and 3.7). Accordingly, many disordered proteins are likely bound by other biomolecules in cells, modulating their degradation. α-synuclein for example, exists in a membrane-bound conformation that entails its first 100 residues.313,314 Hence, larger portions of cytoplasmic α-synuclein can potentially be proteolyzed when it is not bound to membranes. Indeed, when free α-synuclein is treated with proteases such as thermolysin, proteinase K, or the Glu-specific V8-protease it is efficiently degraded into small peptide fragments that span its entire sequence. When it is bound to SDS micelles, proteolysis preferentially targets its free C-terminus.315 Similarly, disordered regions in transcription factors may be protected from proteolysis upon DNA binding.26,316 Recent data suggest that many nonfunctional transcription factor-binding sites on genomic DNA serve as proteolytic safe havens protecting these proteins from degradation.317 Stabilizing effects can also be mediated by interactions with other proteins318 such as proteasome gatekeepers and nanny proteins.26 The proteasome gatekeeper NQO1 for example, binds and protects many disordered proteins,319,320 whereas nanny proteins interact with newly synthesized polypeptides and counteract sporadic degradation.293,321 Nanny proteins might also explain how newly synthesized, disordered proteins escape the general degradation machinery when they emerge from the ribosome. It has been suggested that nascent disordered protein regions transiently interact with ribosomal proteins until sequences emerge that interact with nanny proteins.26 In that sense, proteasome gatekeepers and nanny proteins function as protective chaperones preserving the integrity of disordered proteins (see section 3.5). Other cellular chaperones might also influence the fate of IDPs, but their individual roles are less clear. According to the prevalent view, disordered proteins are not preferred chaperone targets.322 In fact, recent data on tau and p53 suggest that IDP-chaperone interactions promote degradation, rather than protection, either via the 26S proteasome or the autophagy pathway.323,324
Post-translational modifications also determine intracellular stability of disordered proteins and their degradation. As we discuss in section 3.6, disordered regions are often post-translationally modified which, in turn, leads to altered structures and functions.252,302 In many cases, modifications regulate protein–protein interactions, which may either protect disordered proteins from degradation or signal their turnover. In this manner, modifications can directly modulate intracellular protein abundance and stability. One such example is p27. Phosphorylation of Tyr88 results in its release from Cdk2 and further phosphorylation of Thr187, which serves as the p27 degradation signal.251 One example of a protective modification effect is the MAP2 protein, which is rapidly degraded by calpain in its unmodified form, whereas hyper-phosphorylation prevents degradation.325 These results underscore the importance of post-translational modifications in regulating the intracellular stability of disordered proteins and their turnover. Thus, several mechanisms influence abundance, stability and degradation in cells, acting at the level of transcription, mRNA stability and decay, synthesis and homeostasis. Because intracellular protein clearance mainly occurs via ubiquitin-dependent proteasomal degradation and given that intrinsically disordered proteins are not targeted by ubiquitination to an appreciable greater extent than ordered proteins (see section 3.3), this may constitute the most convincing argument for the comparable in vivo stabilities of both classes of proteins.287,303,326
3.5. Molecular Chaperones
Protein quality control is an important aspect of cellular life. It is executed by proteins called molecular chaperones, which assist in protein folding, monitor and maintain structural protein integrity, refold misfolded proteins, disentangle aggregated proteins, and target proteins for degradation.327−329 For these reasons, interest in molecular chaperones and disordered proteins is closely aligned. In the early days of IDP research, disordered proteins were considered in vitro artifacts and an indication of the absence of appropriate chaperones to help them adopt their “native”, ordered conformations. We now know that, in vitro, disordered proteins are equally stable as ordered ones (see section 3.3), that they are not preferred targets of cellular proteases (see section 3.4) and that many of them maintain their disordered conformations inside the crowded cytoplasms of prokaryotic and eukaryotic cells (see section 2.3). When these notions became more widely accepted, interest in cellular chaperone functions shifted toward the prevention of spontaneous IDP aggregation. In fact, a large portion of recent IDP/chaperone literature addresses the functional roles of chaperones as cellular anti-aggregation factors (see section 4). Here, we discuss chaperone functions that target nonaggregated disordered proteins. We focus on intrinsic disorder as an inherent structural property of chaperones, before we review the role of disordered proteins as chaperone “substrates”.
3.5.1. Disordered Chaperones
As part of their surveillance function, chaperones facilitate protein folding and counteract interactions that result in misfolding.330 In response to cell stress, chaperones prevent and reverse protein aggregation and ameliorate refolding. The realization that disordered protein regions constitute integral parts of chaperones is largely based on in vitro studies of heat shock proteins (Hsp). Within the ATP-dependent chaperones, including the GroEL-GroES system, Hsp70 and Hsp90, conformational flexibility of their “active sites” confers the domain motions that comprise the chaperoning cycle.331−334 The disordered C-terminus of Hsp70 for example, functions as an auxiliary domain in binding misfolded proteins, which enhances chaperone activity in vitro and in vivo.335 In the presence of ATP, hydrophobic segments of the disordered linker domain dock onto the nucleotide-binding domain and confer interdomain allostery.333,336−338
Extended disordered regions in ATP-independent chaperones such as the small heat shock proteins (sHsp) mediate interactions with different substrates.339,340 These regions efficiently cross-link with many cellular proteins, suggesting that they continuously sample and transiently interact with folded and misfolded polypeptides in cells.339,340 HdeA and Hsp33 for instance, undergo order-to-disorder transitions in response to cellular stress, which concomitantly activates their chaperoning functions.341,342 Promiscuous chaperone interactions may further destabilize misfolded proteins and smoothen energy landscapes to enable escapes from local minima, thereby promoting refolding.343−345 These so-called entropy transfer reactions transmit conformational entropy to misfolded proteins.331,340,346−348 Indeed, weak chaperone activities were reported for many “classical” IDPs such as α-synuclein349,350 and the plant LEA proteins, members of the dehydrin family.340,348,351 Dehydrins such as ERD10 and ERD14 are entirely disordered yet prevent temperature-induced aggregation of several substrates.352 Other LEA proteins, such as nematode AavLEA1 and plant Em protect organisms from desiccation-induced aggregation of citrate synthase.351 Surprisingly, however, they fail to protect against temperature-induced aggregation. These disordered proteins do not bind their substrates as conventional chaperones, but, instead, act as physical barriers against unfavorable interactions and are, therefore, referred to as molecular shields.351,352 Hence, molecular shielding via disordered regions is another mechanism by which chaperones reduce unfavorable interactions.
Another long-standing question in cellular homeostasis is that of protein solubility. Molecular chaperones and disordered proteins may represent one part of the answer.28 Because IDPs constitute a significant proportion of the proteome, they might add to an overall solubility increase by creating extended hydrated and hydrophilic intracellular environments. A higher proteome-wide content of disorder may also help to decrease the load on cellular chaperone system(s), enabling cells to better cope with different stress conditions. In that sense, chaperones and IDPs may exert complementary functions and thereby control cellular homeostasis on multiple levels.
Other classes of “protective” proteins with high contents of disordered regions are cochaperones, i.e., proteins that interact with, and activate chaperones.331 Although the role of disorder in cochaperones is not yet fully understood, some data suggest that it expands the ability of chaperones to interact with substrate molecules.328 The chaperoning effect of disorder, however, is not limited to proteins but also extends to the realm of RNA and, in fact, is common in RNA chaperones.346,353 RNA chaperones such as the core proteins of flaviviruses resolve misfolded RNA species, which critically depends on their disordered regions.354 Upon binding to kinetically entrapped RNA molecules, many of these chaperones undergo disorder-to-order transitions. Some partially disordered ribosomal proteins, such as L16 and L18 exhibit dual protein and RNA chaperoning activities, earning them the name Janus chaperones.355
3.5.2. Disordered Chaperone Substrates
While the importance of disordered protein contributions to the functions of molecular chaperones is widely appreciated, considerably less is known about IDPs as chaperone substrates. Importantly, a computational study by Hegyi and Tompa concluded that they do not constitute preferred chaperone targets.322 It is therefore unlikely that disordered proteins spend more time in chaperone-bound states than ordered ones.
Several specific chaperone/IDP interactions have been reported. Aggregation-prone regions of human tau for example, interact with Hsp72 and Hsc70, two homologous variants of the Hsp70 family of proteins, as well as with Hsp90.356,357 These tau sites become available and accessible only when the protein dissociates from microtubules.358 In the presence of ATP, Hsp70 and Hsp90 bind tau with a dissociation-constant in the low micromolar range (∼5 μM for Hsp90).357,359 In the absence of ATP or ADP, the affinity between tau and Hsp70 is likely to be weaker,356 wheras Hsp90 binding is unaffected.357 Reports of chaperone interactions with α-synuclein are more controversial. Binding to αB-crystallin was not detected by NMR spectroscopy, but dissociation constants in the ∼60 nM range were delineated from surface plasmon resonance experiments.360−362 The Dobson group reported no binding of α-synuclein to Hsp70,363 although a later report postulated a weak interaction.364 α-Synuclein does not bind Hsp70 according to fluorescence anisotropy and pull-down affinity capture experiments,365 but chemical cross-linking was confirmed by mass-spectrometry.366 Weak binding to Hsp90365 and GroEL367 was reported in the absence of ATP or ADP. Overall, it is difficult to assess whether such isolated in vitro chaperone interactions with nonaggregated IDPs are physiologically relevant in vivo.
Another level of complexity may involve cochaperone interactions with both IDPs, and chaperones. The disordered C-terminal region of measles virus nucleoprotein for example, binds Hsp70 with ∼70 μM affinity.368 In the presence of stoichiometric amounts of the cochaperone Hsp40, the dissociation constant decreases to ∼50 nM. Thus, cochaperones can modulate IDP-chaperone affinities, which may be particularly important during translation, when multiple chaperones and cochaperones line the ribosomal exit tunnel.369 Recent findings suggest that short, aggregation-prone, hydrophobic and disordered polypeptides are preferred substrates for yeast ribosome-associated Hsp70,370 although cotranslational ubiquitination was not found to be statistically different in ordered and disordered proteins.371
3.6. IDPs and Post-Translational Modifications (PTMs)
The reversible nature of most PTMs enables eukaryotic proteins to rapidly adopt different structural and functional states in response to internal and external cues. These modifications greatly expand their repertoire of biological activities. Based on a proteome-wide PTM analysis it was suggested that, on average, all human proteins are modified on at least three sites (Figure 5A).372 This number does not include N-terminal acetylation, a constitutive chemical modification in most mammalian proteins, which occurs cotranslationally when nascent polypeptide chains exit the ribosome.373 Hence, eukaryotic PTMs represent the single most important means to dynamically regulate protein function in a reversible manner.374,375 We often think of PTMs as binary “switches” that toggle between “on” and “off” states. Such notions assume that PTMs control all or nothing decisions, which is not always the case, as we describe in the following paragraphs. Here, we review the functional and structural properties of post-translational modifications and show that disordered proteins and protein regions are particularly well suited to integrate cellular PTM activities.
Figure 5.
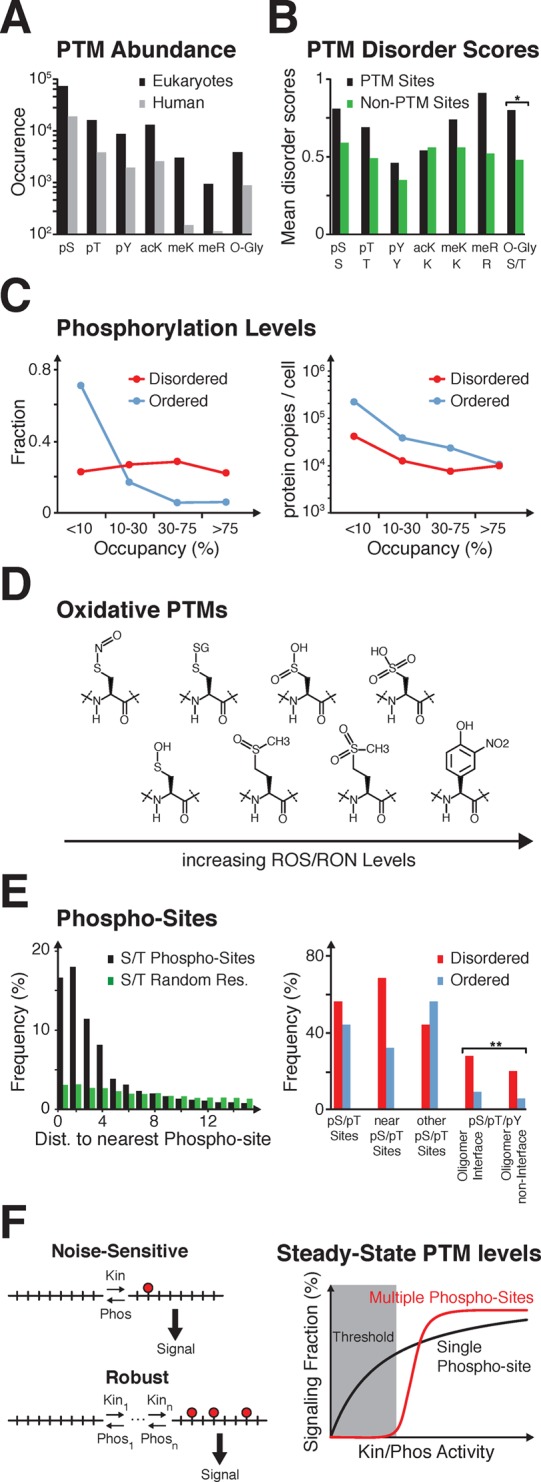
Post-translational modifications. (A) Experimental and predicted phospho-serines (pS), -threonines (pT), -tyrosines (pY), acetyl-lysines (acK), mono-, di-, or trimethyl-lysines (meK), symmetric and asymmetric dimethyl-arginines (meR), and O-glycosylated serines or threonines (O-Gly) according to ref (372). (B) Disorder scores for pS, pT, pY, acK, meK, and meR (black), and nonmodified residues (green), according to VSL2B, ref (382) and SwissProt, ref (380). Scores above or below 0.5 predict disorder or order, respectively. Scores for O-Gly (*) were calculated as the ratio of O-glycosylation sites in predicted IDRs over the total number of O-glycosylation sites in 190 proteins. For nonmodified sites, the ratio of serines/threonines in IDRs was determined over their numbers in the same set of proteins, ref (379). (C) Left: Fraction of pS/pT sites in disordered and ordered S. cerevisiae proteins and their relative phosphorylation levels (occupancy), according to ref (386). Right: Average number of proteins per S. cerevisiae cell and their relative phosphorylation levels, according to ref (394). (D) Chemical structures of post-translational amino acid modifications at increasing ROS/RNS levels. (E) Left: Distances between phosphorylated serine/threonine residues in eukaryotic proteins (black), versus average distances between random (modified and nonmodified) serine and threonine residues (green). Adapted with permission from ref (470). Copyright 2010 Biomed Central Ltd., Springer Science+Business media Right: Disorder and order at serine/threonine phosphorylation sites (pS/pT), at pS/pT sites with neighboring modification sites less than 4 residues apart (near pS/pT sites) and at pS/pT sites with no other modification site less than 4 residues away (other pS/pT sites). Adapted with permission from ref (470). Copyright 2010 Biomed Central Ltd., Springer Science+Business media. Average serine/threonine/tyrosine phosphorylation sites at oligomer interfaces and noninterface regions (**). Adapted with permission from ref (469). Copyright 2013 Royal Society of Chemistry. (F) Left: Sensitivity and robustness of single-, versus multiple-PTM signaling modes. Right: Switch-like responses result from multiple modifications in the presence of balanced kinase (Kin) and phosphatase (Phos) activities.
3.6.1. IDPs as PTM Targets
Most eukaryotic PTMs occur in disordered protein regions (Figure 5B), because disorder provides unrestricted access for modifying and demodifying enzymes and enables facile interactions with protein modules that specifically recognize and interact with modified residues.25,376−382 This property is reflected in the higher abundance of disordered regions in eukaryotic proteomes and correlates with the greater complexities of eukaryotic signaling networks and regulatory pathways. Disorder is overrepresented in signaling proteins and in proteins controlling gene activity, such as transcription factors.12,15,383−385 Indeed, the occurrence of PTMs in disordered proteins exceeds often the three to four modifications per protein mentioned above.372
Among 50 proteins phosphorylated at 20 or more sites, most are disordered or contain extended disordered regions.386 Prominent examples are the N-terminal “tails” of histones with more than ∼40 chemically distinct modifications over a narrow sequence range (∼35 residues for longest histone H3 tail, for example).387,388 Histone tail modifications directly affect the architecture of chromatin and regulate gene activity. Similarly, the human transcription factor and oncoprotein p53 contains ∼20 annotated PTM sites in its disordered N- and C-terminal transactivation domains.389,390 In p53, modifications modulate protein stability, DNA binding and transcription. The amyloidogenic tau protein represents another prominent example with multiple modification sites. Intrinsically disordered over its entire length (between ∼350 and 440 residues depending on the splice isoform), tau contains 75 verified and 85 predicted phosphorylation sites, 45 of which are serines, 35 are threonines and 5 are tyrosines.391,392 Strikingly, only 7 of these sites are annotated and known to affect microtubule binding. The function(s) of the other sites are unknown. Given the large number of tau phosphorylation sites, it remains to be determined whether all of them are physiologically relevant. Besides phosphorylation, 50 additional chemically distinct modifications of tau have been reported.391,392
3.6.2. IDP Diversity and PTMs
By assessing phospho-site conservation, a recent study suggested that 65% of these sites might lack unique functional roles.393 This observation raises an important question about the relative phosphorylation levels at the individual modification sites. Indeed, most modifications are present substoichiometrically (occupancy below 50%) (Figure 5C), meaning that roughly equal numbers of substrate molecules lack the modification.386,394 Given the high frequency of modifiable amino acids in disordered proteins,395 low “penetrance” can lead to substantial chemical heterogeneity, which may, in turn, have functional consequences. We divide the following discussion into two parts. First, we describe the functional and structural characteristics of modifications that require dedicated enzymatic activities for their establishment. Second, we outline nonenzymatic modifications.
3.6.2.1. Enzymatic PTMs
As mentioned above, modification sites in disordered protein regions are easily accessible by modifying and demodifying enzymes. Kinases and phosphatases regulate serine, threonine, tyrosine and sometimes histidine phosphorylation.77,396−398 Acetyltransferases and deacetylases control lysine acetylation399,400 and methyltransferases and demethylases mediate lysine and arginine methylation,401,402 to mention just a few enzymes.
While protein phosphorylation constitutes the most abundant modification in eukaryotes, the ∼500 kinases that establish this modification in humans display moderate substrate site specificities, typically governed by ∼9 residues flanking the phosphorylatable amino acid.403 Site selection is encoded and decoded by the kinase domains, whereas distant docking sites, often in separate protein modules as in the case of tyrosine kinases for example, create supplementary recognition features.397 Conversely, Thr-Pro motifs represent 30% of all threonine phosphorylation sites in eukaryotic proteins, whereas Ser-Pro constitutes ∼20% of all phosphorylated serine residues.404 Both motifs serve as minimal consensus sequences for numerous kinases, including the large families of cell cycle regulated, mitogen-activated protein kinases (MAPKs) and the cyclin-dependent protein kinases (CDKs).405,406 Dephosphorylating enzymes, i.e., protein phosphatases are ten-times less abundant than kinases,407 which is also reflected in their weaker substrate site specificities, compared to kinases. Importantly, however, kinase and phosphatase activities and specificities are often modulated by subunit interactions, which can either activate or deactivate these enzymes depending on cellular conditions.397,398,407 In cells, enzymes of both classes are further regulated by their spatial context397,407,408 and kinases are often tethered to subcellular structures such as membranes or occur as parts of immobile scaffolding complexes.397
Other enzymes that establish post-translational modifications, such as acetyltransferases and deacetylases, display similarly weak specificities. No consensus sequence has been determined for the broadly acting transcriptional regulator and acetyltransferase CBP/p300. Instead, in vitro studies suggest that it follows the classical Theorell-Chance “hit and run” model.409 In vivo, broad substrate specificity may be counteracted by localized enzyme action and dynamic subunit interactions in different holo-enzyme complexes.409,410 Weak substrate site specificities are also observed for many deacetylases411 and the methyltransferases G9a and SET7/9.412,413
Given these weak specificities, certain cellular modifications may occur transiently on secondary target sites. In line with this notion, abundant proteins, prone to random encounters with kinases and phosphatases, are more frequently phosphorylated, although their individual phospho-site “occupancies” are low.394 Within this group, 8% of all serines and threonines are phosphorylated in disordered proteins, whereas the number is only 2.5% in ordered ones. Hence, IDPs likely display greater modification heterogeneities than ordered proteins.
3.6.2.2. PTM Recognition
As discussed above, disordered proteins are frequently targeted by cellular modifications. Given their disordered nature, they are also more versatile in their capacities to interact with different modification recognition domains. These domains are typically small, folded protein motifs (∼120 or fewer amino acids) that bind, albeit weakly, to modified residues (dissociation constants 10 to 100 μM414), in a context independent manner,415 although some surrounding residues confer low-level binding specificity.416 While they specifically recognize specific modifications, their interactions are context independent,415 although residues surrounding the modification sites confer some specificity.416 Examples include recognition domains that interact with phosphorylated residues, such as WW-, 14–3–3-, and SH2-domains,417−419 as well as protein modules that interact with methylated lysines and arginines, i.e., chromo-, PHD- and Tudor-domains,420 or acetylated lysines such as bromo-domains.421
Many of the ∼100 human SH2 domains, for example, bind phospho-tyrosines with dissociation constants above 0.1 μM.422 Similarly, bromo-domains interact with acetylated lysines with dissociation constants above ∼10 μM. Analogously, the 7 human phospho-serine and phospho-threonine specific 14–3–3 domain isoforms (up to 15 in plants) are not specific and interact with target sites within multiple kinase consensus motifs, such as those of AuroraA/B/C, CaMKII, RSK, PKA and PKCG.423 Finally, Tudor-, PHD-, and chromo-domains bind methylated lysines irrespective of sequence context.424−426
3.6.2.3. Nonenzymatic PTMs
The intracellular milieu offers several ways to establish non enzyme-catalyzed modifications. Reactive oxygen- and reactive nitrogen-species (ROS and RNS, respectively), for example, create multiple chemically distinct modifications, such as oxidized cysteines, and methionines, or nitrated tyrosines (Figure 5D).427,428 ROS and RNS are natural, metabolic products of mitochondrial respiration.427−429 In healthy cells, surveillance mechanisms and scavenging systems control their cellular levels to protect proteins, and other biomolecules, from oxidative damage.429,430 ROS and RNS induced modifications accumulate during aging, as manifested by higher numbers of oxidized cysteines and methionines, and covalently cross-linked dityrosines.427,429,431−433 Cellular scavenging of ROS and RNS constitutes a process of fundamental biological importance, and misregulation is often implicated in human disorders, although it is difficult to determine whether oxidative modifications are a cause or a consequence of the disease.427,431,433 In other biological processes, inflammation for example, ROS and RNS function as signaling molecules, and modified proteins mediate cellular responses.430 Given the solvent-exposed nature of most side-chains in IDPs, one may assume that these proteins constitute preferred scavengers of cellular oxidative PTMs. However, recent data suggest that this is not always the case.
Cysteines are most sensitive to oxidation and oxidized cysteines are often found in folded proteins, especially in enzyme active sites.427,434 However, cysteine is the second least abundant amino acid type in disordered proteins (less than 1%)395 and cysteine oxidation is rarely detected in these proteins.376 The same is true for tyrosine nitration since the overall abundance of hydrophobic and aromatic residues in IDPs is low.395 Sequence proximity to charged residues, as well as enhanced solvent accessibility favors tyrosine nitration,435−437 which should render tyrosines in IDPs preferred nitration targets. Indeed, high levels of tyrosine nitration are found in Lewy body aggregates of α-synuclein438 and the same modification is detected in the soluble protein under conditions of cellular oxidative stress.439 Similarly, tyrosine-nitrated tau is found in brain plaques of Alzheimer’s disease patients and nitration inhibits tau binding to microtubules.440 Although nitrated tyrosines generally occur at low abundance and only 0.01% of all tyrosine are nitrated under inflammatory conditions,435,437 they may serve as important disease factors. Because postulated mechanisms of amyloid formation involve nucleation and aggregation-prone seed structures, even low quantities of potentially harmful IDP variants may trigger aggregation.437,441 Alternatively, nitrated tyrosines may function as immunogenic signals that activate inflammatory responses. Interestingly, many of the tyrosine sites in IDPs that are readily nitrated in vitro, do not correspond to residues modified in vivo, indicating that intracellular conditions affect substrate site selection and/or site-specific repair efficiencies.437,442
Little is known about methionine oxidation on a proteomic scale. Studies of independent replicates of H2O2 exposed mammalian Jurkat cells show that methionine oxidation does not occur randomly, although no particular class of proteins, nor subcellular compartment are preferentially affected.443 Methionine oxidation does not reach full penetrance and 85% of the characterized modification sites show oxidation levels of ∼30%. However, a clear sequence bias is evident. Oxidation does not occur on methionines that are flanked by aromatic or cysteine residues and it is enriched at sites in the vicinity of acidic-, basic- and glutamine residues,443 reflecting the bias in amino-acid composition of IDPs.53 Levels of cellular methionine oxidation appear to be balanced by endogenous methionine reductase activities. In vitro, H2O2 mediated methionine oxidation is less efficient when Lys, Arg, His, or Pro precede the modifiable amino acid, and solvent exposed methionines are modified faster than buried residues.443,444 Similarly, methionine sulfoxide reductases are less active when acidic, or proline residues, enriched in IDPs, precede the modified methionines and are more active in disordered regions.443,444 Together, these in vitro and proteomic data suggest that IDPs constitute preferred substrates for methionine oxidation. Complete oxidation of all four α-synuclein methionines results in a decompaction of the protein,445 inhibits its fibrillization and promotes the generation of off-pathway species. Given that methinione oxidation occurs with rather low penetrance under physiological conditions,443 these effects may be mitigated in cells.
Several other oxidative modifications, such as tyrosine halogenation, dityrosine formation, tryptophan nitration and carbonylation of various amino acids427 are mediated by reactive oxygen species that accumulate with aging, impairment of oxidative stress response pathways and inflammation. Because many neurodegenerative diseases are age-related and associated with neuro-inflammation,446 functional roles of oxidative modifications in these diseases were postulated early on.440 Open questions remain with regard to timing, abundance and effects of oxidative modifications and whether they are causative or symptomatic.
3.6.3. PTMs and IDP-Mediated Signaling
Given the complexity and heterogeneity of cellular modifications, especially with regard to chemical diversity, specificity and penetrance, the notion of global PTM “noise” emerges, postulating that cellular proteins continuously experience transient, chemically distinct modifications, many of which with no biological function. From an analytical point of view, this situation makes it difficult to discriminate between meaningful, regulated modifications and coincidental “bystanders”. In cells, differences in growth conditions and intracellular environments may further embellish the heterogeneity of physiological modification patterns. Here, we discuss the role of disordered proteins in processing PTM “noise”. We entertain the idea that these proteins may be particularly well suited to “buffer” noise and thereby regulate eukaryotic signaling.
Intrinsic disorder is generally viewed as favoring rapid association and dissociation,447 while maintaining a balance between ligand binding specificity and affinity,448,449 and greater binding partner promiscuity.450,451 Moreover, IDP mutations have weaker impacts on general protein–protein interactions.452 IDPs display higher mutation rates than folded proteins453 (see section 3.2) and several studies suggest that greater levels of genomic evolvability correlate with larger pools of mutable IDPs.450,454 These properties are equally important in cell signaling, which requires transient interactions with multiple partners, while maintaining high levels of tolerance to mutagenic alterations.12,383,448,455
A number of signaling pathways exhibit ultrasensitive responses, meaning that switch-like functional changes occur only above a threshold of stimulatory signals.456−458 Modification multiplicity on individual target proteins mediates robust, ultrasensitive responses and low interference from signaling noise (Figure 5F).459 Intramolecular substrate-site competition, cooperativity, or stepwise modifications of multiple substrate sites can amplify these responses.460−468 Protein disorder favors cooperative mechanisms not only because PTM sites are exposed and easily accessible to modifying and demodifying enzymes, but also because modified residues can rapidly engage in novel protein–protein or protein–membrane interactions and directly alter local or global IDP conformations. Therefore, cumulative modifications on a single protein can promote the establishment of additional modifications in a stepwise manner or ramp up binding affinities to new partners, once a certain threshold is surpassed. At the same time, such behaviors may ensure that non biologically relevant modifications (i.e., noise) do not trigger aberrant signaling activities. Phosphorylation sites in disordered proteins are often clustered, which can enhance multisite cooperativity (Figure 5E).469,470 Following this rationale, low-level PTM noise may actually improve signaling sensitivity and robustness by allowing faster responses, without the requirement for positional substrate site conservation. The higher mutational tolerance of IDPs may therefore provide an evolutionary advantage for such regulatory mechanisms.
Substrates of cyclin-dependent kinase 1 (Cdk1), for example, show low positional conservation of their substrate sites, but a high degree of net conservation that preserves overall PTM effects in fast evolving disordered regions.471 Among the many Cdk1 substrates, one well-studied system, Cdk1 inhibitor Sic1 (the yeast ortolog of human p27), serves as a good example to outline the concept of graded response behaviors upon cooperative, multisite phosphorylation events. Sic1 is intrinsically disordered and contains seven Cdk1 phosphorylation sites. Phosphorylation of these sites triggers binding to Cdc4, a member of the ubiquitin ligase complex. Upon Cdc4 binding, Sic1 is degraded and full Cdk1 activity required for entering S-phase is established. While Sic1 phosphorylation by Cdk1 occurs cooperatively,472 high affinity Cdc4 binding also requires double phosphorylation of at least one of three paired modification sites on Sic1.464,473−475 Therefore, some phosphorylation events might occur at low abundance in the absence of sustained Cdk1 activity, whereas full kinase activation overcomes the signaling threshold that triggers the robust biological response.
3.6.4. PTMs and IDP Structures
Another important aspect of post-translational protein modifications concerns their ability to modulate protein structure. Whereas PTM-induced structural changes in folded proteins have been analyzed,476 comparable studies for intrinsically disordered proteins are not available. Many IDPs undergo disorder-to-order transitions upon binding to cognate partners and for equilibrium thermodynamic reasons, binding conformations are often transiently sampled in their free states, resulting in residual secondary structure. These kinds of “motifs” have been termed short linear motifs (SLiMs), molecular recognition features (MoRFs), prestructured motifs (PreSMos) or anchor sites.477−480 Given the structural plasticity of disordered proteins, different modifications or sets of modifications may modulate their structural propensities in either a positive or negative way, similar to folded proteins, and thereby exert important regulatory functions in cells.
3.6.4.1. PTM Effects on Secondary Structure
Several studies investigated the influence of glycosylation and phosphorylation on peptide secondary structures. Glycans for example, can modulate conformations via the formation of new hydrogen bonds or by steric restriction. These effects depend on the nature of the glycan and the protein sequence context. N-glycosylation promotes β-turn conformations,481 whereas O-glycosylation can increase or decrease α-helix, β-strand, β-turn or poly proline type II helix propensities.482−489 Phosphorylation can also induce or stabilize α-helical structures via side-chain salt bridges in cases where phosphorylated serines or threonines at positions i are preceded or followed by lysines at positions i–4 or i+4, respectively.490 In another example, multiple phosphorylation of short peptides derived from the proline-rich regions of human tau induces poly proline type II helices,488,491 which is particularly intriguing given the large number of proline-directed kinases and kinase consensus sequences.492 Using short model peptides, Andrew et al. and Elbaum and Zondlo showed that serine and threonine phosphorylation at internal, or C-terminal helix positions destabilizes the secondary structure, whereas N-terminal phosphorylation stabilizes α-helices.489,493 These properties are often mentioned in relation to secondary structure changes in response to modifications occurring in vivo, although their structural impact in physiological environments is less clear.494−498 Phosphorylated Ser-Pro and Thr-Pro motifs are further targeted by peptidyl-prolyl isomerases such as Pin1, which collectively increase the rate of proline cis/trans interconversion.499 In the absence of these enzymes, serine/threonine phosphorylation N-terminal to prolines does not affect cis/trans equilibria.500−503 However, in cells, proline cis/trans isomers may be differently sequestered by binding partners, which may, in turn, change equilibrium distributions.504
Other types of modifications may also affect the structural properties of disordered proteins. N-terminal acetylation of α-synuclein for example, increases transient helicity within its first ten residues (from 10% to ∼30%).505−507 Although this modification is not strictly speaking a PTM because it mostly occurs cotranslationally,373 such findings confirm that cellular modifications may have structural-, and ultimately functional-consequences.505−508 Considering all possible PTM effects, it is expected that they also impact the aggregation behavior of disordered proteins (see section 4).392,509
3.6.4.2. PTM Effects on IDP Compaction
Besides altering the conformations of IDPs, modifications can also affect their global appearance. In general, levels of compaction decrease with net charge.54,55 Therefore, PTMs that introduce charged entities, such as phosphate groups or neutralize existing charges, such as lysine acetylation, are likely to modulate the shapes of disordered protein ensembles. Lower compaction has been reported for a mutant version of tau that contained six serine/threonine to glutamic acid substitutions, mimicking multisite phosphorylation,510 although increased local compaction was observed in the vicinity of the pseudophosphorylation sites.510,511 Similarly, multisite phosphorylation of Sic1 leads to compaction of this positively charged protein.512 A decrease of compaction was observed for α-synuclein upon methionine oxidation.513 Therefore, these effects often vary from protein to protein. Similarly, ubiquitination and sumoylation introduce new interaction surfaces that may mask, reveal, or modulate IDP structures.514
In summary, we presented an exemplary overview of possible PTM effects on disordered proteins in physiological environments. We showed that cellular modifications greatly expand the chemical heterogeneity of these proteins, which, in turn, may increase their structural and functional diversity. It is important to keep in mind that in cells all the scenarios discussed above occur in a much more complex environment that offers many additional possibilities for modulatory effects.
3.7. Coupled Folding and Binding
Energy landscapes of disordered proteins are shallow and contain many local minima,345 enabling them to adopt different structures when they interact with other macromolecules.515,516 Indeed, many IDPs undergo disorder-to-order transitions upon binding to proteins, nucleic acids, or lipids.53,347 Given the abundance of disordered proteins in cells, the physiological milieu offers ample opportunities for interactions that promote disorder-to-order transitions, prompting researchers to ask whether free disordered proteins ever exist in cells, or whether they only occur as complex-bound, ordered entities.517 If that were the case, interacting molecules triggering these transitions needed to be as abundant as disordered proteins, serving as the prerequisite for an in vivo world without disorder. For folding transitions that depend on cell membranes or other abundant structures, such as the cytoskeleton, this might indeed be the case. For folding transitions that require protein–protein interactions the situation is less clear and relative intracellular availabilities and concentrations come into play. Organelle transport, intracellular localization and spatial confinement may additionally impose restrictions onto intracellular binding events and these spatial parameters may change in response to different biological activities such as post-translational protein modifications. Therefore, mixed populations of disordered and ordered protein states likely reflect realistic in vivo situations.
In this section, we discuss how different intracellular environments may affect coupled folding and binding reactions. We begin by introducing two prevalent models to explain the thermodynamic and kinetic properties of these events, namely the induced fit mechanism and conformational selection (Figure 6A). In the following, we outline how disorder-to-order transitions can be of biological advantage, and discuss how coupled folding and binding may occur in cells.
Figure 6.
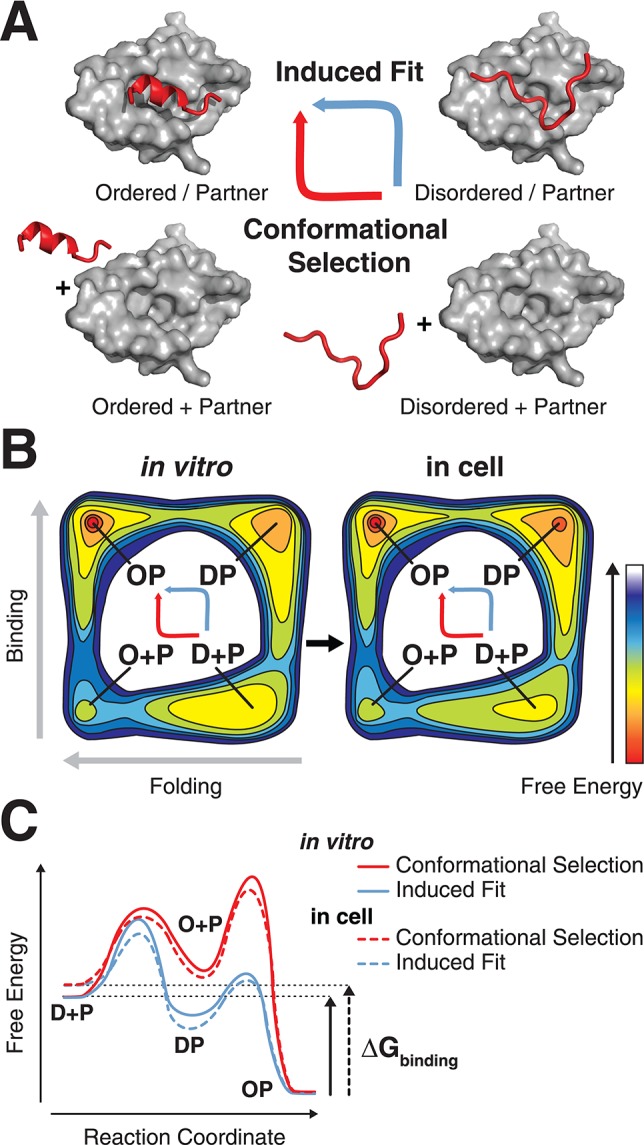
Coupled folding and binding. (A) Representations of induced fit and conformational selection mechanisms of coupled folding and binding reactions. The disordered and ordered ligand is shown in red (D and O, respectively), the folded binding partner (P) is shown in gray. (B) Schematic representations of possible differences between in vitro and in cell free energy landscapes of coupled folding and binding reactions. (C) Free energy/reaction coordinate profiles of coupled folding and binding systems and possible in vivo modulations. Resulting binding free energies (ΔGbinding) may be different.
3.7.1. Induced Fit
According to the induced fit model, disordered proteins engage in initial encounter complexes through weak interactions. Guided by these interactions, and the surface properties of their binding partners, they mold into their final conformations via disorder-to-order transitions establishing the strong intermolecular contacts of their bound states. One example to describe an induced fit interaction is the binding of intrinsically disordered pKID to the folded CBP KIX domain.518 In the initial encounter complex, the N-terminus of pKID forms an α-helix (αA), while its C-terminus remains flexible and disordered (30% transient helicity), and weakly samples different hydrophobic surface areas on KIX. Eventually, the C-terminal portion of pKID undergoes an induced fit disorder-to-order transition and docks onto the KIX domain in a second, α-helical conformation (αB) forming the final, high-affinity complex. Transient helicity of the αB region in the absence of KIX domain binding is only ∼10%, suggesting that αA formation and binding also induces a substantial increase in transient helicity of C-terminal pKID residues.
While the pKID-KIX interaction is often cited in reference to coupled folding-upon-binding reactions, it is not without bias. Strictly speaking, the helical conformations of pKID in the final KIX complex mirror its structural tendencies in the free state. pKID residues corresponding to αA exhibit ∼50% transient helicity in their free form, arguing that some secondary structure of the complex is already populated in the absence of KIX binding.519−521 In fact, the presence of transient αA features in free pKID may support the second reaction mechanism of disorder-to-order transitions: conformational selection.
3.7.2. Conformational Selection
Conformation selection postulates that free disordered proteins sample the structures of their bound states and that interacting partners scavenge these prestructured populations to form complexes.522 Based on this notion, and provided that interacting partners are abundant, levels of transient secondary structure determine the rates of disorder-to-order transitions, as well as the thermodynamic properties of the respective binding processes. One often cited example with regard to conformational selection is based on the work of the late Flemming Poulsen.523 His group established that the free nuclear coactivator binding domain (NCBD) of CBP exists in a conformation resembling its structure in the p160 coactivator (ACTR)-bound complex. They further determined that NCBD adopts this conformation, described as a molten globule, ∼90% of the time.524 To evoke an exclusive conformational selection mechanism is not without drawbacks. Dogan et al. showed that the NCBD-ACTR interaction involves a transient priming state, whereas most of the specific hydrophobic contacts are established after this rate-limiting encounter, reminiscent of the nucleation-condensation model in classical protein folding.525 Therefore, this interaction too requires at least 2 steps, with the first encounter probably driven by conformational selection, followed by a structural rearrangement that could easily pass as an induced fit mechanism.525,526
These two examples illustrate an important point in the ongoing induced fit versus conformational selection debate: The difficulty to clearly separate the two mechanisms. As we discuss next, the physical boundaries between both scenarios are diffuse and often depend on the experimental conditions employed to characterize them. These considerations also set the tone for the discussion about their physiological relevance in the context of live cells.
3.7.3. Induced Fit versus Conformational Selection
Cumulating experimental data conflict with mechanisms of disorder-to-order transitions solely based on conformational selection. Even for interactions between ordered proteins, conformational rearrangements, at least on the side-chain level are necessary to obtain high affinity complexes.527−529 In disordered proteins, side-chains display high degrees of flexibility and their time scales of motions (<10–8 s) are faster than most diffusion controlled binding processes (>10–7 s).530 The same is true for nucleation-dependent α-helix formations (<10–7 s).531 According to these considerations, binding via conformational selection alone is unlikely to result in the observed high affinity complexes and, as suggested by Zhou and co-workers, fast conformational sampling appears to favor interactions along the induced fit model.532 Although transient preformed secondary structures are frequently observed for regions that interact with cognate targets,477−479,533−540 IDP-ligand complexes often contain additional structural features that are not populated in their free states, exhibiting non uniform levels of bound IDP flexibility.347,541 Exclusive induced fit mechanisms have been proposed for some disorder-to-order transitions,525,532,542−545 whereas mixed mechanisms have been suggested for others.530,546−548 Given their high degree of conformational plasticity, disordered proteins may be particularly prone to mixed binding behaviors exhibiting features of both induced fit and conformational selection.522,537,542,543,545,549−556 As pointed out by Hammes et al., solution conditions and protein concentrations additionally affect the mechanistic properties of disorder-to-order transitions.557
3.7.4. Disorder-to-Order Transitions in Cells
We mentioned earlier that intracellular environments contain large numbers of biological interfaces that can potentially catalyze disorder-to-order transitions. At the same time, the physical properties of the intracellular space, including viscosity, macromolecular crowding, excluded volumes and soft interactions, contribute to the overall characteristics of cellular binding events (see section 2). We discussed flexibility and residual structural features as important determinants in disorder-to-order transitions and both properties are likely modulated in cells. Levels of transient secondary structure for example, may increase (i.e., stabilizing) or decrease (i.e., destabilizing), either enhancing or reducing contributions to interactions that involve conformational selection (Figure 6B). Perhaps more important, these effects may enhance or reduce binding free energies and thereby improve or weaken molecular recognition (Figure 6C). These considerations illustrate how intracellular contributions to disorder-to-order transitions can have different effects based on the types of interacting species, the nature of their interaction, and the physical and biological properties of their surrounding environments. Therefore, it is difficult to predict how these parameters modulate cellular disorder-to-order transitions.
It has been suggested that disordered proteins enable faster protein–protein interactions due to their extended hydrodynamic radii and enhanced ligand-capture efficiencies.558 However, this argument is contradicted by the fact that extended hydrodynamic radii also lead to slower translational diffusion.447 Moreover, such fly casting mechanisms may be counterbalanced by in-cell compaction.447,449 Kriwacki and co-workers provided important insights into how much disorder is needed for the cellular functions of the cyclin-dependent kinase (Cdk) regulators p21 and p27.559,560 These two disordered proteins bind Cdk-cyclins via interactions of their D1 and D2 subdomains. p27 inhibits Cdk2-cyclin A activity and establishes cell-cycle arrest in G1, whereas p21 broadly inhibits Cdk-cyclin complexes and elicits arrest at various stages.561 In both p21 and p27, the LH linker domain between D1 and D2 exhibits helical propensities in its free state559,560 and adopts a fully helical conformation in the p27:Cdk2-cyclin A complex.561 Mutants of p27 with reduced levels of transient LH secondary structure fail to elicit G1 cell-cycle arrest.562 Moreover, the structural flexibility of the p21 LH domain is essential to efficiently bind Cdk-cyclins and to initiate cell-cycle arrest. Restrained LH domain mutants elicit partial arrest at the G1-to-S transition, but no arrest at the G2-to-M transition. Mutants with shorter LH domains fail to induce cell-cycle arrest, in line with their inability to bind Cdks.561 Hence, the different degrees of p21 and p27 disorder specifically tune their binding affinities and specificities toward Cdk-cyclins in vitro and in vivo.
Another example of coupled folding and binding is the α-synuclein interaction with membranes.533 The protein is highly abundant in the brain and its physiological role is linked to synaptic vesicle trafficking and membrane fusion in dopaminergic neurons.314,563,564 In turn, lipid environments rather than protein–protein interactions mediate folding transitions of α-synuclein. In vitro, α-synuclein interacts with negatively charged lipid vesicles and artificial membranes, such as SDS micelles and unilamellar vesicles.313,533,565−572 These interactions induce an α-helical conformation of its first ∼100 residues.533 Different membrane environments induce different helical α-synuclein structures ranging from extended single helices, to broken double-helical horseshoe conformations.313,533,565−571 These findings led to the conclusion that membrane charge, composition and curvature determine the global structural features of folded α-synuclein. How are these disorder-to-order transitions mechanistically accomplished? We can rationalize these coupled folding and binding reactions by similar combinations of induced fit and conformational selection mechanisms. In its free monomeric state, the N-terminus of α-synuclein exhibits α-helical propensities (∼10%), which it fully adopts in the different membrane-bound states.573 Despite the low abundance of non membrane-bound, helical α-synuclein molecules, these species may initially interact with membranes via conformational selection and trigger additional helical transitions of more C-terminal α-synuclein residues in a second induced fit step. The observation that α-synuclein binds different membranes in different α-helical conformations supports this hypothesis.
In cells, α-synuclein is enriched at presynaptic terminals and colocalizes with synaptic vesicles, suggesting that the protein also exists in helical membrane-bound conformations in vivo.574−578 With respect to α-synuclein’s membrane-mediated disorder-to-order transition, a recent study by the George group is of particular interest. In their analysis the authors asked whether differences in α-helicity affected the colocalization of α-synuclein with physiological membranes.578 The authors studied the effects of N-terminal α-synuclein truncations and point mutations in rat primary hippocampal neurons and found that deletions of N-terminal α-helical residues disrupt presynaptic localization and result in diffuse axon staining. They also analyzed a prominent familial point mutation of α-synuclein, A30P, exhibiting reduced levels of transient helicity at residues surrounding the mutation site.573 In vitro, A30P displays severely impaired membrane binding.579−581 They found similarly reduced membrane colocalization of A30P α-synuclein in hippocampal neurons, whereas another mutant with unperturbed secondary structure, i.e., A53T, was not affected.578 In support of these conclusions, other groups also observed these effects in yeast cells.582,583
Recent evidence further suggests that N-terminal acetylation of α-synuclein increases transient helicity from ∼10% to ∼30%.505,506 In agreement with conformational selection driving the initial membrane encounter, N-terminally acetylated α-synuclein binds membranes with higher affinity than the nonacetylated form of the protein.505 Thus, arguing that this enzyme-mediated cotranslational protein modification enhances the intrinsic propensity of α-synuclein to interact with physiological membranes. Agreeing with this notion, α-synuclein overexpressed in N-terminal acetylation competent yeast cells, uniformly decorates the cytoplasmic side of the cell membrane.582,583 By contrast, disrupting the N-acetyltransferase gene in yeast abolishes α-synuclein membrane staining and results in diffuse cytoplasmic localization.583 Together, these results provide compelling evidence for the existence of membrane-bound pools of α-synuclein under physiological conditions. Whereas a direct proof of an α-helical in vivo conformation is missing, these indirect observations support the notion that membrane-bound α-synuclein exists in helical conformations in cells.
3.7.5. Fuzzy Complexes
Challenging the exclusiveness of coupled folding and binding reactions, Fuxreiter et al. proposed the concept of fuzzy complexes.584 In fuzzy complexes, IDPs exhibit different extents of disorder in their bound states, and display dynamic properties in between fully disordered, and ordered structures. One often cited example of a fuzzy complex is the interaction between the regulatory region and the nucleotide-binding domain (NBD) of the cystic fibrosis trans-membrane conductance regulator (CFTR).585 In this case, multiple sites of the regulatory region contribute to binding, each exhibiting transient helical features in their free states. Even upon binding, these regions remain dynamic and frequently exchange in the NBD binding site.
Forman-Kay and co-workers studied another example of a fuzzy complex, the interaction between phosphorylated Sic1, an inhibitor of Cdk kinases, and the ubiquitin ligase Cdc4.512 Sic1 is degraded in response to multisite phosphorylation, prompting binding to Cdc4.586 Cdc4 recognizes six different phosphorylation sites of Sic1 and engages the protein in transient, disordered interactions. The concept of fuzzy complexes also offers a means of describing modulatory cellular effects along a continuum of stabilizing and destabilizing interactions. In this sense, coupled folding and binding reactions, disorder-to-order transitions and folded conformations of disordered proteins and protein regions in complex-bound states likely exhibit higher degrees of fuzziness in vivo.
4. IDP Aggregation
Aggregation of ordered proteins requires initial unfolding.587,588 Because disordered proteins exist as dynamic ensembles of interconverting monomers, they do not have to unfold to aggregate.1−3 Therefore, IDPs have long been considered particularly prone to aggregation. However, not all IDPs aggregate, and those that do only represent a small fraction of the disordered proteome. Within this portion, some IDPs aggregate spontaneously, while others require additional factors, or defined solution conditions, highlighting the importance of contextual contributions from native in vivo environments. In the following paragraphs, we outline cellular aspects of IDP aggregation and discuss possible roles in physiology and pathology.
4.1. Aggregation and Disease
Early interest in IDPs was spurred by observations that post mortem brains of neurodegenerative disease patients contained high levels of intra- or extra-cellular IDP deposits that were later shown to be representative of the different pathologies.589,590 Intracellular aggregates of α-synuclein for example, are hallmarks of Parkinson’s disease (PD), extracellular deposits of Aβ peptides and intracellular inclusions of the tau protein are found in Alzheimer’s disease (AD). Insoluble aggregates of the prion protein are synonymous with Creutzfeld-Jacob’s disease (CJD), while insoluble fibrils of Huntingtin are characteristic of Huntington’s disease (HD). In their native states, these proteins are either fully or largely disordered, whereas they adopt extended β-sheet structures in their aggregated forms. Moreover, these aggregates, as well as their deposits in post mortem tissues, can be stained with iodine, which led to their initial, wrongful identification as starch deposits (amylum in Latin) and the creation of the term “amyloid”. Our knowledge about disordered proteins and amyloids has increased considerably since those days, but how ordered aggregates form in complex intra- and extra-cellular environments remains enigmatic. In the following, we describe how aggregation of disordered proteins results in cell damage and pathology. We further discuss how different intracellular environments may affect aggregation and thereby contribute to disease progression and prevalence.
Alzheimer’s disease is the most common neurodegenerative disorder. It is characterized by progressive memory loss and dementia that arises from tissue damage in the cortex and hippocampus.591 Cell death coincides with extracellular aggregation of Aβ peptides and intracellular deposition of the microtubule-binding protein tau into insoluble amyloid plaques and neurofibrillar tangles, respectively.592,593 Aβ peptides occur as 40- to 42-residue proteolytic fragments of the amyloid precursor protein, which functions in synaptic stabilization and plasticity. Tau is a 55–62 kDa protein with a role in microtubule-dependent transport in the nervous system. Both proteins readily form amyloid fibrils in vitro and in vivo, and several lines of evidence support their roles in AD.594
Parkinson’s disease is the second most common, age-related neurodegenerative disorder. It is accompanied by the progressive loss of dopaminergic neurons of the substantia nigra pars compacta, which translates into severe motor problems.595 Pathologically, the disease is characterized by proteinaceus cytoplasmic inclusions known as Lewy bodies,595,596 which primarily contain α-synuclein amyloid fibrils. Aggregation of α-synuclein is one of the underlying causes of PD,597 and other neurodegenerative disorders such as multiple system atrophy, and dementia with Lewy bodies, all of which are characterized by intracellular amyloid inclusions of α-synuclein. Accordingly, these diseases are called synucleinopathies.16
Creutzfeld-Jakob’s disease, also known as “mad cow” or prion disease, is a neurological disorder manifested by insoluble deposits of the prion protein (PrP).598 Prion aggregates are transmissible and can be passed on similar to an infectious agent.599 Native PrP (PrPC) is bound to the outer plasma membrane. The N-terminus of PrP constitutes the intrinsic disordered part of the protein, whereas the C-terminus is folded and adopts an α-helical structure.600,601 PrP aggregation constitutes the key determinant of CJD and a conformational rearrangement of its C-terminus into β-sheet structures is responsible for the pathological conversion of PrPc into PrPSc, the toxic form of the protein. PrPSc is self-propagating and catalyzes further conversions of PrPC into PrPSc. This process is strongly influenced by the N-terminal disordered region of PrPc.602
Expansions of poly glutamine repeats (polyQ) are encountered in many human disorders, including in Huntington’s disease. Huntington’s disease is characterized by the formation of granular and fibrous deposits of the Huntingtin protein in the nuclei and cytoplasm of several neuronal cell types, leading to pervasive cell death. Aggregating IDPs that cause this disorder contain different lengths of polyQ expansions with a direct correlation between numbers of polyQ repeats and aggregation propensities.16,603
Type II diabetes is another related amyloid disease that arises from the loss of pancreatic β-cells and results in an insufficient insulin response.604 Cell death is thought to occur due to aggregation of the 37-residue amylin peptide into amyloid fibrils.605
Many of these pathologies are of neuronal origin and it is likely that certain biological activities in brain cells contribute to the prevalence of amyloid phenotypes in this organ. Cellular oxidative stress and oxidative protein modifications for example, are risk factors in many neurodegenerative disorders.392,606,607 Indeed, the brain is particularly vulnerable to oxidative stress, because it consumes approximately one-fifth of the inspired oxygen and metabolizes ATP at high rates.608,609 Considering that 5% of all oxygen consumed by cells is converted into reactive oxygen species (ROS), levels in the brain are higher than in other tissues. In response, the brain has evolved several mechanisms to deal with elevated amounts of ROS, but whenever an imbalance between ROS production and clearance exists, oxidative damage may lead to protein oligomerization and/or the impairment of protein homeostasis. Many of the age-related characteristics of these diseases underscore the plausibility of such scenarios. Here, we discuss cellular aspects of disordered protein aggregation using α-synuclein as an example.
Whereas α-synuclein is abundantly expressed in various parts of the brain,610 intracellular aggregation and neuronal damage is primarily, and first manifested in dopaminergic neurons.597 These observations suggest that the biological makeup of these cells contributes to aggregation. Additionally, most sporadic cases of PD occur late in life, enforcing the notion that age, and age-related cellular processes constitute important disease factors. Abnormal accumulations of cytosolic dopamine for instance, produce highly reactive intracellular radicals and quinone derivatives with great oxidative potential that can readily oxidize α-synuclein in vitro and in vivo.611−614 At the same time, systems that protect cells against oxidative damage, such as glycogen synthase kinase 3β (Gsk3β), mitogen-activated protein kinases (MAPKs), superoxide dismutase (SOD) and catalases display age-related declines of their activities.615 In turn, co-occurrence of both effects may render an abundant, intrinsically disordered protein such as α-synuclein particularly prone to oxidative damage, and increase intracellular accumulations of toxic species forming amyloid fibrils. Experimental evidence supports such multifactorial scenarios for PD and many other neurodegenerative disorders.616
4.2. Functional IDP Aggregates
IDP aggregation and amyloid formation are usually linked to pathological conditions. However, recent evidence suggests that IDP amyloids can have physiological roles in prokaryotic and eukaryotic cells.617 The E. coli protein curlin forms amyloids in vivo, which enable bacteria to colonize epithelial surfaces and develop biofilms, two properties that provide survival benefits.618 In the yeast S. cerevisae, functional amyloid aggregates of the prion-like partially disordered proteins Ure2p and Sup35p have been reported.619 Ure2p binds Gln3p and prevents transcription of genes that regulate cell growth under low nitrogen conditions. Aggregation of Ure2p disrupts its interaction with Gln3p and enables cell growth and adaptation to nitrogen deprivation. The unrelated protein Sup35p acts as a release factor during translation termination, and its activity is lost upon amyloid formation.620 Once Sup35p aggregates, RNA polymerase reads through stop codons, which results in greater protein diversity and the generation of new protein activities that are beneficial for survival. Aggregation of Ure2p and Sup35p are mediated by their disordered, asparagine- and glutamine-rich N-termini.621 Related functional amyloids have also been reported in mammalian cells. Disordered Pmel17 forms amyloid-like fibrils in specialized organelles of epithelial cells called melanosomes. These structures act as polymerization seeds for small molecules forming melanin fibers that protect cells from UV and oxidative damage.622 Pmel17 aggregation also prevents melanin-associated toxicity by sequestering intermediate on-pathway species in the course of polymer formation. A disordered repeat domain that is rich in proline, threonine and glutamic acid residues drives aggregation,623,624 in a tightly controlled manner.622 These examples underscore the functional importance of disordered protein amlyoids in providing organisms with benefits for certain growth, and metabolic conditions.
So far, we outlined disease-related aggregation of disordered proteins and introduced the concept of functional amyloids. In the following paragraphs, we describe molecular mechanisms leading to aggregation in vitro, and outline factors that likely influence aggregation in cells. Specifically, we discuss how physical and biological properties such as macromolecular crowding, intracellular viscosity, post-translational protein modifications and ligand binding affect aggregation in vivo.
4.3. Mechanisms of IDP Aggregation
Despite their great divergence in sequence, size and native conformations, certain aggregation characteristics of disordered proteins are surprisingly similar. Most mechanisms entail initial steps of native ensemble perturbations that produce partially folded intermediates with higher aggregation propensities (Figure 7A).16 Intermolecular associations then lead to early oligomeric species, often rich in β-sheet structures that rapidly convert into proto-fibrils. Eventually, proto-fibrils assemble into elongated structures of mature amyloids.625−627 The rate-limiting step in many of these processes is the formation of early oligomeric species acting as aggregation seeds, out of which proto-fibrils and fibrils grow in a nucleation-dependent fashion.625,627 Equations 1 and 2 provide simplified representations of the molecular events that lead to amyloid formation:
| 1 |
| 2 |
Figure 7.
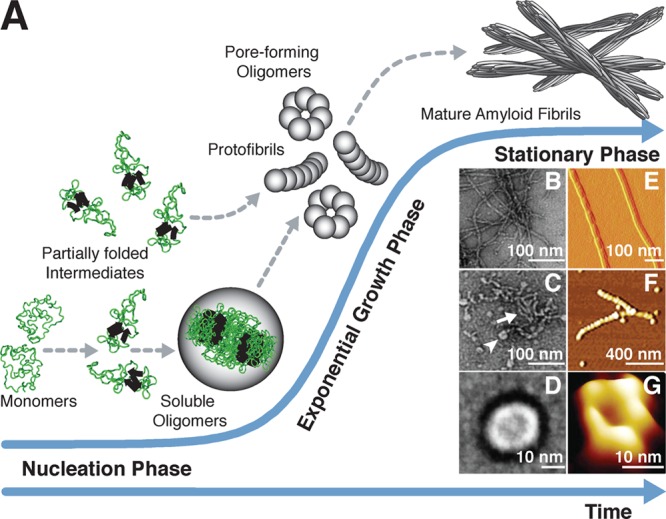
Amyloid cascade. (A) Stages of amyloid formation. In the nucleation phase, IDP monomers aggregate via partially folded intermediates into soluble oligomers. During the exponential growth phase, oligomers assemble into high-molecular weight proto-fibrils, into which monomers are incorporated. In the stationary phase, proto-fibrils mature into amyloids. (B–D) Transmission electron microscopy (TEM) and (E–G) atomic force microscopy (AFM) images of Aβ aggregates. Structures of mature amyloid fibrils (B and E) and coexistence of linear (yellow) and annular (white) proto-fibrils (C). AFM of linear and annular proto-fibrils (F and G) and TEM of annular oligomers (D). Panels B-G adapted with permission from refs (641−644). Panels B–D, Copyright 2003 Elsevier; Panel E, Copyright 2005 FASEB; Panel F, Copyright 2003 American Chemical Society and Panel G, Copyright 2005 US National Academy of Sciences.
P denotes the protein, i is the number of protein molecules that assemble in the aggregation seed and knuc, kpol and kdpol are the nucleation, polymerization and depolymerization rate constants, respectively. Accordingly, monomeric proteins are continuously consumed by initial oligomeric species that eventually extend into mature amyloid fibrils. More detailed mechanisms have been reported, some of which depend on the nature of the aggregating protein or on specific aggregation conditions.602,628−630 Several studies aimed at characterizing on-pathway species in vitro and the structural properties of early Aβ, α-synuclein and amylin oligomers have been analyzed to great detail.631−637 Typically, they display spherical shapes and contain 10–50 monomers that interact in chain-like proto-fibril conformations, often arranging into annular, pore-like structures (Figure 7A-G).637−644 Higher-order fibrils are insoluble and stain positive for dyes such as Congo red and Thioflavin T.645 They typically comprise 2 to 6 proto-filaments, with diameters of 2–5 nm and continue to form rope-like structures with diameters of 10–30 nm. Individual proto-filaments are rich in β-sheets that mostly align perpendicular to the extended fibril axis.646,647 Different factors increase or decrease the in vitro aggregation rates of disordered proteins, some by stabilizing early oligomeric or prefibrillar species. Interactions with binding partners, post-translational protein modifications and intracellular macromolecular crowding conditions exert additional effects. In the following paragraphs, we analyze these contributions with respect to aggregation and fibrillation in cells and tissues.
4.4. Aggregation and Macromolecular Crowding
As outlined in section 2.1, intracellular environments and extracellular fluids contain large amounts of macromolecules, reaching concentrations of up to 300–500 g/L, including proteins, nucleic acids, lipids, glycans, metabolites, and solvated ions.114,648 Together, these species occupy a defined fraction of the intracellular space and make it unavailable to other molecules.649−651 They also determine intracellular viscosity (see section 2.3).652−654 One general conclusion from several studies is that macromolecular crowding stabilizes compact protein structures.650,655 Because partially folded intermediates constitute key aggregation species,588,656−659 macromolecular crowding is thought to enhance aggregation. Viscosity, by contrast, is expected to exert the opposite effect, because it mainly interferes with diffusion-controlled processes, such as the addition of monomers to preexisting proto-fibrils.649−651,655,660−662 Accordingly, net in vivo effects of these opposing contributions are difficult to predict, because they depend on the geometries, concentrations, and surface properties of both the aggregating species and the macromolecules that define the crowded environment.
Several studies dissected the individual contributions of crowding and viscosity to IDP aggregation. Enhanced aggregation of α-synuclein for example, was observed under crowded in vitro conditions at PEG, BSA, lysozyme, Ficoll, and dextran concentrations ranging from 100 to 150 mg/mL (Table 2).660,661,663 In these studies, aggregation-enhancing effects were attributed to a selective decreases in the lag-phases of the respective aggregation processes, indicating that the formation of early oligomers is favored under crowded conditions. At higher concentrations of crowding agents, these effects were less pronounced, supporting the notion that higher viscosity slows down diffusion-controlled additions of monomers to preexisting oligomers and proto-fibrils. This hypothesis was independently confirmed in solutions of 40% glycerol, which severely impaired α-synuclein aggregation.661 Crowding has also been shown to modulate the aggregation behavior of Tau and the human prion protein.664,665 Increasing amounts of Ficoll and dextran enhanced aggregation in a concentration dependent manner (4-fold and higher at 200 g/L). In vitro, crowding further prompted fibrillation of GSK3-phosphorylated Tau, which does not normally occur under comparable dilute conditions. Similar aggregation-promoting effects in crowded in vitro solutions were also reported for Aβ peptides, amylin, tau and the prion protein,666,667 and for folded proteins under denaturing conditions, such as core histones, S-carboxymethyl-α-lactalbumin, β-lactoalbumin and human apolipoprotein CII.660,668,669 Crowding can also affect the morphologies of mature amyloid fibrils. In some intances, identical fibrils are formed under dilute and crowded conditions,664,665,670 whereas different shapes and structures such as shorter fibril lengths,664,665 or higher numbers of branching sites are observed in other cases.661 These results indicate that cellular conditions and activities, including post-translational protein modifications and biological interactions, likely modulate in vivo aggregation kinetics and aggregate morphologies.
Table 2. Modulations of α-Synuclein Aggregationa by Various Physiological/Pathological Factors and Effectors in Vitro.
| factors/effectors | effects on fibrillation kineticsb | main species | refs |
|---|---|---|---|
| macromolecular crowding and viscosity: | |||
| PEG (150 g/L) | enhancement (∼10-fold) | amyloid fibrils | (661, 663) |
| dextran (150 g/L) | enhancement (∼2-fold) | amyloid fibrils | (661, 663) |
| Ficoll 70 (150 g/L) | enhancement (∼5-fold) | amyloid fibrils | (661, 663) |
| Ficoll 400 (150 g/L) | enhancement (∼5-fold) | amyloid fibrils | (661, 663) |
| BSA (60 g/L) | enhancement (∼7-fold) | amyloid fibrils | (661, 663) |
| lysozyme (50 g/L) | enhancement (∼5-fold) | amyloid fibrils | (661, 663) |
| glycerol (40%) | enhancement (∼3-fold) | amyloid fibrils | (661, 663) |
| glycerol (50%) | enhancement (∼1-fold) | amyloid fibrils | (661, 663) |
| glycerol (60%) | no fibrillation | n.d.h | (661, 663) |
| post-translational modifications: | |||
| Ser87 phosphorylation | no fibrillation | monomers/amorphous aggregates | (675) |
| Ser129 phosphorylation | no fibrillation | monomers/amorphous aggregatesc | (678) |
| Tyr125 phosphorylation | no change | amyloid fibrils | (679) |
| Met-oxidation | no fibrillation | soluble oligomers | (700) |
| Tyr-nitration | no fibrillation | spherical aggregates | (696, 697) |
| monoubiquitination: | (694) | ||
| Lys10 | no change | amyloid fibrils | |
| Lys6, Lys12, Lys21, Lys23 | inhibition (>3-fold) | amyloid fibrils/proto-fibrils | |
| Lys32, Lys34, Lys43, Lys96 | no fibrillation | monomers/amorphous aggregates | |
| poly ubiquitination: | |||
| Tetra-Ub- Lys12 | no fibrillation | large, nonfibrillar aggregates | (692) |
| Sumoylationd | no fibrillation | amorphous aggregates | (693) |
| 4-hydroxy-2-nonenale | no fibrillation | monomers/soluble oligomers | (695) |
| N-terminal acetylation | no change | amyloid fibrils | (508) |
| truncation: | |||
| α-synuclein (aa1–108) | enhancement (∼10-fold) | shorter amyloid fibrils | (52) |
| α-synuclein (aa1–124) | enhancement (∼7-fold) | shorter amyloid fibrils | |
| ligand and protein interactions: | |||
| metal ionsf: | |||
| Cu(II) (1:1) | enhancement (∼2-fold) | amyloid fibrils | (687, 722) |
| Mn(II), Fe(II), Zn(II) (1:1) | no change | amyloid fibrils | (721) |
| Ca(II) (10:1) | n.d.h | spherical oligomers | (730) |
| Cu(II), Mn(II), Fe(II), Zn(II) (50:1) | enhancement (>5-fold) | amyloid fibrils | (659) |
| polyamines: | |||
| putrescine (3000:1) | enhancement (∼4-fold) | shorter amyloid fibrils | |
| spermidine (300:1) | enhancement (∼4-fold) | shorter amyloid fibrils | (75) |
| spermine (15:1) | enhancement (∼4-fold) | shorter amyloid fibrils | |
| polyphenols: | |||
| EGCG (1:1)g | no fibrillation | spherical aggregates | (746) |
| theaflavins | no fibrillation | spherical aggregates | (753) |
| dopamine | no fibrillation | amorphous aggregates | (734) |
| β-synuclein (2:1) | inhibition (∼2-fold) | amyloid fibrils | |
| γ-synuclein (2:1) | inhibition (>4-fold) | amyloid fibrils | (331) |
| chaperones: | |||
| Hsp20 (1:1) | inhibition (∼2-fold) | amyloid fibrils | (362) |
| Hsp27 (1:1) | inhibition (∼1.5-fold) | shorter amyloid fibrils | |
| HspB8 (1:1) | no fibrillation | spherical aggregates | |
| Hsp 70 (1:10) | no fibrillation | monomers | |
| Hsp104 (1:400 -ATP) | no change | amyloid fibrils | (758) |
| Hsp104 (1:400 +ATP) | inhibition (∼2-fold) | amyloid fibrils | |
| Hsp104 (1:40 + ATP) | no fibrillation | amorphous aggregates | (757) |
Aggregation is defined as the formation of Thioflavin-T (ThT) positive amyloid fibrils.
Quantification based on the half-time (t1/2) of in vitro aggregation. Each condition was compared to the aggregation rates of α-synuclein control samples.
Longer incubations produced ThT positive aggregates that were morphologically different from non phosphorylated control samples.
Complete sumoylation at Lys96 and Lys102.
Addition of up to six 4-hydroxy-nonenal molecules.
Metal:protein ratios are given in parentheses.
(−)-Epigallocatechingallate.
n.d. = not determined
4.5. PTMs and Aggregation
We outlined how unrelated disordered proteins can form amyloid fibrils of strikingly similar morphologies. However, not all IDPs aggregate into amyloids and even for those that do, their rates of fibrillation vary greatly. An IDP’s sequence, its amino acid composition and the positional clustering of individual amino acids affect its aggregation, because these factors influence the global and local physicochemical properties, including hydrophobicity, net charge, dynamic characteristics and the propensities to populate transient states of secondary structure.589,671 As we discuss in section 3.6, IDPs constitute preferred in vivo targets for post-translational modifications and these may, in turn, modulate aggregation. We use α-synuclein, Tau and other disease related IDPs as examples to discuss some of these aspects.
Early analyses of α-synuclein deposits in PD patients’ brains showed that ∼90% of the protein is phosphorylated at Ser129, whereas only 4% of cytosolic α-synuclein is modified.672−674 It was established later that phosphorylated Ser87 constitutes another pathological hallmark of intracellular α-synuclein inclusions.675 These results led to the hypothesis that phosphorylation promotes aggregation in vivo. In vitro studies of Ser129 and Ser87 phosphorylated α-synuclein revealed that the protein adopts a more extended structure, which supports the notion that phosphorylation may expose the aggregation-prone, hydrophobic nonamyloid component (NAC) region to intermolecular interactions.675−679 Surprisingly, in vitro aggregation of Ser129, and Ser87 phosphorylated α-synuclein is severely impaired (Table 2). The increased net charge and electrostatic repulsions between α-synuclein monomers were put forward as explanations for these inhibitory effects.675,678 In line with these arguments, a Ser129-to-alanine mutant of α-synuclein displays enhanced aggregation tendencies in vitro and in vivo.680,681 This example illustrates the difficulties to correlation with changes in in vitro aggregation behaviors to effects observed in vivo. Do higher levels of Ser129 phosphorylation in α-synuclein deposits reflect higher intracellular aggregation tendencies? Maybe aggregated α-synuclein constitutes a better kinase substrate than the monomeric protein and phosphorylation occurs after aggregation? Perhaps aggregated and nonaggregated species are equally good kinase substrates, but only nonaggregated α-synuclein is efficiently dephosphorylated by cellular phosphatases? Maybe phosphorylation of α-synuclein increases its affinity to Cu and Fe,682 which are known to promote aggregation?625,659,683
Unlike phosphorylation, truncations of α-synuclein correlate with enhanced aggregation in vitro and in vivo. C-terminal truncations are found in intracellular α-synuclein deposits672,684−686 and they aggregate more readily in vitro (Table 2).52,687−690 Indeed, removing C-terminal parts of the protein produces aggregation prone species with a decrease in net charge and an increase in hydrophobicity, as well as critical perturbations of autoinhibitory conformations that counteract aggregation of full-length α-synuclein.676,677 Aggregated α-synuclein isolated from post-mortem PD patients’ brains, exhibits a number of nonenzymatic and oxidative stress-associated modifications, such as tyrosine nitration, methionine oxidation and lipidation.689 Many of these modifications were carefully investigated given their prominence in the disease.607,609,691−695 Nitration of monomeric α-synuclein, for example, produces partially folded species that readily aggregate into off-pathway oligomers, inhibiting amyloid formation (Table 2).696,697 However, when nitrated protein is added to unmodified α-synuclein, it triggers aggregation and amyloid formation, even at substoichiometric concentrations.698 By contrast, higher amounts of nitrated α-synuclein inhibit fibrillation. Based on these results, Uversky et al. proposed that seed formation and growth comprises two independent events.696 Similarly, α-synuclein methionine oxidation induces soluble oligomers that do not aggregate into amyloid fibrils in vitro, acting as effective fibrillation inhibitors (Table 2).607,699,700 Similar to the reported phosphorylation effects, these in vitro studies must be considered with caution because different outcomes may be observed in vivo in the presence of other synergistically or antagonistically acting factors.701 Serine phosphorylation, tyrosine nitration and methionine oxidation also decrease the avidity of α-synuclein membrane interactions, which may result in abnormally high concentrations of cytosolic protein and enhanced aggregation.702−704
Tau hyperphosphorylation inhibits microtubule binding and thereby contributes to the formation of neurofibrillar tangles in the course of Alzheimer’s disease.705,706 In turn, overexpression of Asp/Glu-phosphomimetic Tau mutants in PC12 cells recapitulates some of the phosphorylation-dependent neurotoxic phenotypes.707 In vitro experiments also showed that phosphomimetics had opposing effects on Tau aggregation, which depended on the targeted protein region. While N-terminal Asp/Glu replacements suppressed aggregation, C-terminal substitutions enhanced aggregation.392,708,709 Other modifications, such as tyrosine nitration and proteolytic Tau processing, often observed in post mortem AD patients’ brains, also promote aggregation.392,710−713 Similarly, Aβ-oxidation enhances fibrillation.714
In the case of the Huntingtin protein, N-terminal Ser to Asp substitutions at positions 13 and 16 reduce aggregate formation in vitro and in vivo, and result in neuroprotective phenotypes in mouse models of HD.715,716 In line with these findings, in vitro experiments revealed diminished aggregation propensities of Thr3-, Ser13-, and Ser16-phosphorylated protein.716,717 In turn, Mishra et al. hypothesized that phosphorylated or mutant forms of Huntingtin failed to form α-helical oligomers, critically required as seed structures for aggregation to occur.509,716
Together, these results underscore the importance of cellular post-translational modifications in defining the aggregation behaviors of IDPs in cellular environments.
4.6. Ligand Interactions and Aggregation
Ligand interactions can similarly affect the aggregation properties of disordered proteins. A myriad of IDP-ligand interactions have been described that promote or inhibit aggregation. Among them, physiological IDP-metal interactions are of particular interest, because of the strong link between cellular metal homeostasis and neurodegenerative disorders.609,625,718−720 Cu for instance, was identified as a potent enhancer of α-synuclein, Aβ and tau oligomerization and/or fibrillation.718,721−724 Fe, Zn, Mn, and Ca are additional risk factors in several neurodegenerative disorders and they promote aggregation (Table 2).62,659,725−730 The mechanisms by which these metals act often involve alterations in the structures of disordered proteins, either by direct binding or via oxidation.625,724,731 Metal binding affects the net charges of IDPs, which may, in turn, promote structural changes that expose hydrophobic regions and lead to aggregation.659,724 Metals may also induce secondary modifications such as methionine oxidation or dityrosine cross-linking and thereby trigger oligomerization.625,699,724 Besides metals, other physiological ligands such as polyamines, dopamine, or other oxidized cathechols, glycosaminoglycanes, nucleic acids, lipids and membranes may additionally promote aggregation.75,732−743 On the other hand, ligands may also inhibit aggregation (Table 2).744−753 Many of these studies provided important insights into different aggregation processes. Collectively, they reemphasize the notion that exposed, hydrophobic IDP residues function as key aggregation determinants and that aromatic π–π stacking interactions are instrumental for fibril formation, thereby offering clues for therapeutic intervention.748,749,751,754,755
IDP interactions with proteins are worth noting. Cellular chaperones for example, often protect cells from deleterious effects of spontaneous aggregation (see section 3.5)331 and their roles in aggregation have been studied extensively. Chaperones such as the heat shock proteins Hsp20, Hsp70, Hsp90 and Hsp104, as well as TRAP1 prevent α-synuclein aggregation and exert neuro-protective effects in several cell and animal models of PD (Table 2).362,756−758 Hsp70 inhibits Tau aggregation in vitro and decreases neurotoxicity in live cells.759,760 Indeed, elevated levels of intracellular Hsp70 and Hsp90 diminish Tau deposits and enhance microtubule binding.761 Cellular Hsp40 and Hsp70 modulate the aggregation behavior of Huntingtin.762 Atomic force microscopy experiments revealed that these chaperones impair the formation of spherical and annular oligomers of expanded polyQ-tract Huntingtin, which correlates well with their generic roles in repressing intracellular potein aggregation and cytotoxicity.763 Besides ATP-dependent chaperones, members of ATP-independent chaperones, such as αB-Crystallin and clusterin, were shown to inhibit fibrillation of Aβ peptides.764,765 In most of these cases, chaperone-mediated antiaggregation activities result from direct interactions with target IDPs or via sequestration of early oligomeric species and the prevention of amyloid maturation.362,762,764,765 In cells, chaperones likely also function to clear aggregated species via chaperone-assisted autophagy or by proteasomal trafficking and degradation.761,762 These interactions exemplify the diversity of biological processes that affect protein aggregation in physiological environments.
4.7. Aggregation in Cells
As discussed above, macromolecular crowding, cellular post-translational modifications and ligand interactions affect aggregation in vitro. Given the numerous specific and unspecific interactions that disordered proteins experience in intra- and extra-cellular environments, aggregation processes are modulated in ways that in vitro experiments cannot fully recapitulate. Early transmission electron-microscopy studies on amyloids extracted from post-mortem tissues revealed that their overall β-rich structures are similar to fibrils grown in vitro.766−771 High-resolution methods such as electron paramagnetic resonance (EPR)-, and solid-state nuclear magnetic resonance (NMR)-spectroscopy later showed that despite these overall similarities, in vitro and in vivo aggregates display different morphologies.24,772,773 When recombinant Aβ (1–40) was seeded with proto-fibrils from AD patients, different structural arrangements of monomers in the amyloid core and novel side-chain interactions within the strand-bend-strand motifs emerged.774 The Tycko group further showed that amyloid fibrils grown in vitro from physiological Aβ seeds display polymorphisms that are AD patient-specific (Figure 8).775 These findings indicate that native Aβ amyloids have structural and/or biochemical features that modulate the outcomes of in vitro aggregation processes. Different fibril polymorphisms were also observed for Huntingtin aggregates.776−778 In situ Fourier-transform (FT)- and synchrotron infrared (FTIR) microspectroscopy experiments on mice and human brain slices confirmed the presence of alternate Huntingtin conformations containing different extents of secondary structure (see section 5.4). They also showed that aggregate morphologies depended on the lengths of polyQ-tract motifs, subcellular aggregate localization and the affected brain regions.776
Figure 8.
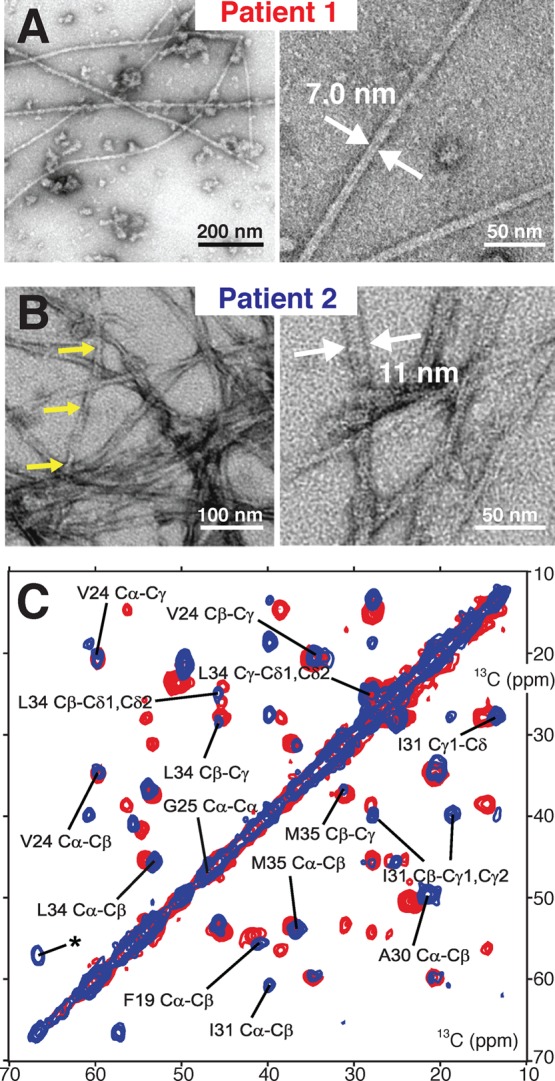
Amyloid polymorphisms. (A and B) TEM images of Aβ fibrils grown in vitro from pathological seeds of two Alzheimer’s disease patients. Yellow arrows point to fibrils with reduced diameters and periodic twists, not observed when grown from seeds of patient 1. (C) Superposition of two-dimensional, 13C–13C solid-state NMR spectra of patient 1 (red) and patient 2 fibrils (blue). Labels indicate cross-peak assignments for patient 2 fibrils. Reprinted with permission from ref (775). Copyright 2013 Elsevier.
Ignatova and Gierasch et al. investigated how different cellular conditions modulated the aggregation kinetics and amyloid structures of a Huntingtin-53 exon 1 fusion protein in E. coli (53 polyQ-repeats).779,780 Their data showed that nonamyloidogenic, amorphous aggregates formed in the presence of osmolytes, such as TMAO and proline. By contrast, glycerol and glycine-betaine prompted the formation of classical amyloid structures.779 In another study, Morimoto et al. followed the aggregation of a Huntingtin-78 exon 1 fusion construct in HeLa cells.781 Their results indicated that fibril formation occurred via compact polyQ-cores onto which monomers were progressively added, thus confirming previous models of in vitro aggregation.
Fluorescence imaging is often used to study cellular aggregation of disordered proteins (see section 5.3). A recent application analyzed the behaviors of Aβ (1–40) and Aβ (1–42) peptides in HeLa and SH-SY5Y cells (Figure 9).782 Although Aβ aggregates typically form in the extracellular space, new evidence suggests that intracellular deposits are also involved in pathogenesis.783,784 Indeed, exogenously added oligomeric and fibrillar Aβ (1–42) peptides were detected in the cytoplasm within 1 h of incubation.782 While most of these fibrils displayed in vitro-like lengths and diameters, ∼10% exhibited curvatures and distortion angles similar to amyloid fibrils from Alzheimer’s disease patients.774 In the case of Aβ (1–40) peptides, only small oligomeric species were detected, in agreement with the reduced aggregation propensities of these species.609
Figure 9.
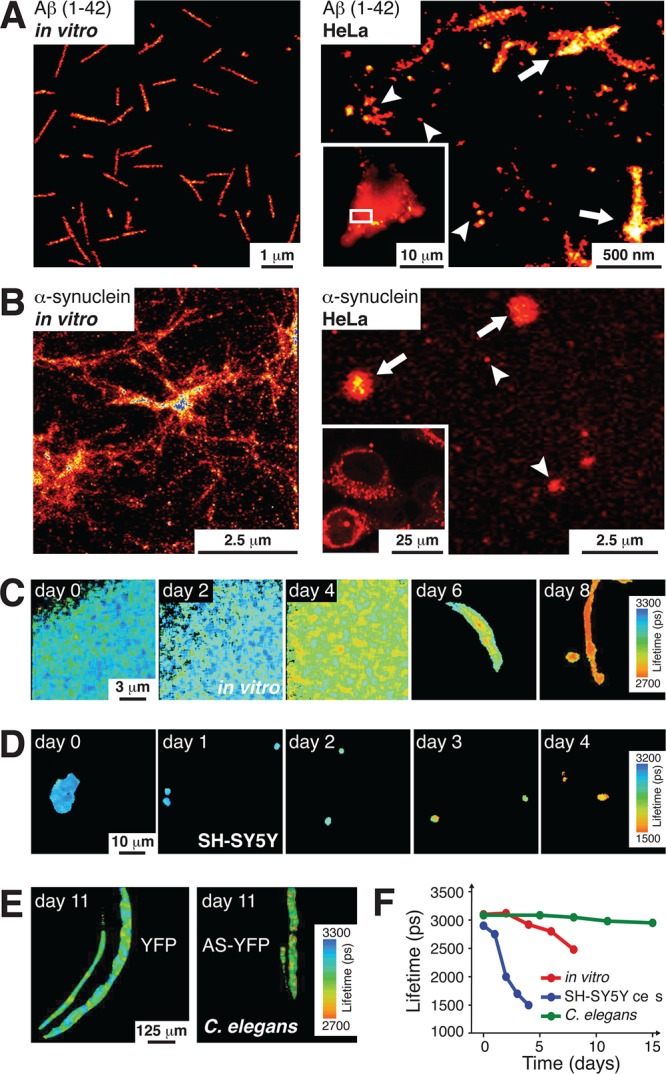
Intracellular morphology and aggregation kinetics of example IDPs. Super-resolution microscopy images of in vitro and in HeLa cell aggregates of Aβ peptides (A) and α-synuclein (B). Arrows and arrowheads denote fibrillar and small oligomeric species. Insets show intracellular peptide/protein localization. Aggregation kinetics of yellow-fluorescence protein (YFP)-coupled α-synuclein in vitro (C), in SH-SY5Y cells (D) and in C. elegans (E), as determined by single photon counting fluorescence lifetime microscopy. Shorter fluorescence lifetimes indicate more aggregated α-synuclein. (F) Time evolution of α-synuclein YFP fluorescence in the three systems. Reprinted with permissions from refs (782), (787), and (789). Panel A, Copyright 2011 American Chemical Society; panel B, Copyright 2012 Elsevier; panels C–E Copyright 2011 WILEY-VCH Verlag GmbH & Co.
Similar fluorescence imaging techniques were employed to study intracellular aggregation of α-synuclein.785 Biarsenic fluorescence labels were coupled onto the protein to enable spatially resolved, real-time aggregation monitoring in neuronal and non-neuronal SH-SY5Y and HeLa cells, respectively. In situ measurements revealed that α-synuclein distributed evenly throughout the cells at early time points, whereas it accumulated in cytoplasmic inclusions after 48 h. ThioS-staining subsequently confirmed that these deposits contained amyloid signatures. Förster resonance energy transfer (FRET) experiments further showed that aggregates displayed greater degrees of heterogeneity than in vitro samples and a more compact architecture, which was attributed to steric intracellular confinement. A detailed investigation of these cellular α-synuclein aggregates revealed that they contained diameters ranging from 40 to 200 nm, resembling those of large prefibrillar species observed in vitro (Figure 9B).786−788 Similar experiments in the presence of FeCl3 revealed larger numbers of intracellular deposits.785 In-cell aggregation of α-synuclein was also studied with novel FRET-based sensors that exploit the intrinsic fluorescence properties of amyloid-protein cores.789 Using such tools, these authors reported that α-synuclein aggregation occurred faster in SH-SY5Y cells than in vitro (Figure 9C,D),782 which they interpreted as an indication for cell type-specific contributions to aggregation.660,661,734,790,791 No intracellular aggregation of β-synuclein was observed in SH-SY5Y cells. In C. elegans, α-synuclein displays reduced aggregation propensities and forms oligomeric structures rather than characteristic amyloid fibrils (Figure 9E,F).789
Having discussed several lines of evidence supporting the notion that IDP aggregation in vitro and in vivo follows similar nucleation-dependent pathways that populate intermediate proto-fibrillar states, we note that the final outcomes of these reactions, especially with regard to their kinetics and the morphologies of the resulting species, may be strongly influenced by specific cellular contributions. Following, it is critically important to choose suitable cell and animal models to study these processes in physiologically relevant environments. The advent of technologies to generate such appropriate cellular systems for studying IDP aggregation, such as induced pluripotent stem cells (iPSC), offers exciting new possibilities in this direction.792−794
5. Theoretical and Experimental Methods to Study IDPs
In this section we discuss methods to study the structural and functional properties of disordered proteins in cellular environments. These include in silico molecular simulations (section 5.1), high-resolution in-cell NMR spectroscopy (section 5.2), fluorescence imaging methods (section 5.3) and Fourier-transform infrared (FTIR) microspectroscopy (section 5.4).
5.1. Molecular Simulations
Molecular simulations are an integral part of IDP investigations and they have been used extensively to simulate the structural properties of disordered proteins (for reviews see refs (20, 56, 544, and 795)). While molecular simulations are always subject to concerns about how well they reflect reality, the following features make them attractive vehicles both for exploring the behavior of disordered proteins and for modeling biomolecules in general. First, in simulations, all molecules are directly visualizable and there is no need to modify them to observe their behavior. Second, factors that might govern particular types of behaviors can often be adjusted directly without affecting other aspects of the system. For example, to unambiguously assess the contributions made by electrostatic interactions, a simulation can be carried out with all electrostatic interactions switched off (i.e., by setting all partial charges to zero) and compared with identical simulations in which electrostatic interactions are present. Moreover, since such adjustments can be made at the discretion of the simulator, i.e., switching some electrostatic interactions off while leaving others on, it becomes possible to dissect the contributions of individual molecules, residues, or atoms in a way that is not achievable in experiments. Third, and of direct relevance to modeling intracellular environments, the simulator can choose which molecules to include in the system and in what concentrations. Hence, there are no unknown components or quantities complicating the understanding of the resulting behaviors.
All of these advantages are to be weighted against two principal disadvantages of molecular simulations. The first one concerns the accuracy of the energy functions used to describe intra- and intermolecular interactions.544 When these are inaccurate or imbalanced, it is possible that simulated behaviors have little relation to reality. The second one is the problem of “sampling”. Since biomolecular systems are capable of assuming many different configurations it is critical to sample them in a meaningful way. In many cases, this issue can be addressed by “brute force”, i.e., by running a single simulation for a very long period of time or by running many short simulations in parallel. Good examples of the power of brute force approaches are provided by recent molecular dynamics simulations of protein folding carried out with the revolutionary Anton supercomputer developed by Shaw and co-workers.796−799 In other cases, more efficient ways of sampling might be required, i.e., replica-exchange techniques.800−802 Because we discuss cellular aspects of intrinsically disordered proteins, we focus on simulations of intracellular environments. Readers interested in simulations of IDPs alone are referred to recent reviews.20,56,544
5.1.1. Molecular Simulations of the Prokaryotic Cytoplasm
A number of groups have devised computational strategies to recapitulate the physical properties of the E. coli cytoplasm in silico. In 1996, Bicout and Field performed the first dynamic simulation of this kind.803 They assumed a mixture of three different types of macromolecules: ribosomes, tRNA, and proteins, all of which were modeled as spheres. Using Langevin dynamics simulations, coupled with simple descriptions of intermolecular interactions, they showed that translational diffusion is approximately 2-fold slower in their model than in dilute solution. In 2008, the Ellison group reported a conceptually similar, but substantially expanded model of the E. coli cytoplasm.804 Their model included a more diverse set of macromolecules and added concentrations according to quantitative proteomics studies.805 Although all molecules were modeled as spherical particles, and simplified approaches were used to calculate their interactions and diffusive behaviors, this work provided several additional insights into the effects of macromolecular crowding on translational diffusion and a prototypical protein–protein association reaction. Confirming the results by Bicout and Field,803 they determined that the average diffusion coefficients of GFP-sized molecules are decreased 2-fold relative to dilute solutions. By then, its was known that this decrease was smaller than what had been observed experimentally (see section 2.3.3.2).164,166,652,806 The authors duly noted that factors additional to steric repulsion needed to be considered to quantitatively reproduce macromolecular diffusion in vivo.
The next advance in dynamically modeling the bacterial cytoplasm was reported by McGuffee and Elcock.807 Their model was also based on quantitative proteomics data241 but included the atomically detailed models of 50 types of macromolecules. Interatomic interactions were treated as a combination of electrostatic and Lennard-Jones potentials, the latter to mimic hydrophobic interactions. The authors were able to adjust the simulated diffusion behavior of GFP until it corresponded to its measured in vivo properties by altering the strengths of these hydrophobic contributions. Hydrophobic intracellular encounters are consistent with “soft” interactions exerting important modulatory effects on translational diffusion (see section 2.3.4). Importantly, these soft interactions also recapitulated the destabilizing effects of the cytoplasm on the apparent folding thermodynamics of the CRABP protein, as determined experimentally by the Gierasch group.808,809
The Skolnick group reported the most recent dynamic simulation of the bacterial cytoplasm.810 In their study, hydrodynamic contributions to macromolecular translational diffusion were modeled for the first time. By using the sophisticated technique of Stokesian dynamics, together with spherical representations of all macromolecules, they showed that the inclusion of intermolecular hydrodynamic interactions explains the ∼10-fold decrease in intracellular GFP mobility observed in vivo, even in the absence of attractive hydrophobic interactions. Together, these combined modeling results suggest that macroscopic in vivo properties such as translational diffusion are shaped by macromolecular crowding, intermolecular hydrodynamic-, and weak attractive electrostatic-, and hydrophobic-interactions although their individual contributions remain to be determined quantitatively.
5.1.2. Outlook for IDP Simulations
At the time of writing, all of the published cytoplasm simulations treated intracellular macromolecules as rigid bodies. However, the only meaningful way to model IDPs is to include internal protein dynamics and conformational motions. One major challenge to simulating flexible proteins is that both inter- and intramolecular hydrodynamic interactions must be treated appropriately to derive meaningful in vivo diffusion behaviors. In particular, omitting the latter can lead to drastic underestimations of cellular diffusion coefficients.811 Owing to the expense involved in calculating hydrodynamic interactions, efforts are ongoing to develop alternative tools to treat them in simulations of large numbers of molecules.812,813 These methods will likely enable accurate modeling of intracellular IDP diffusion in the future.
5.2. In-Cell Nuclear Magnetic Resonance (NMR) Spectroscopy
One particularly appealing method to study the structural characteristics of disordered proteins inside live cells is high-resolution in-cell NMR spectroscopy. It exploits the atomic resolution properties of solution state NMR in the context of individual isotope-labeled proteins that are selectively enriched in non isotope-containing eukaryotic or prokaryotic cells.814,815 In bacteria, intracellular enrichment is achieved via the induction of recombinant protein expression in the presence of isotope-labeled metabolic precursors. Because most expression plasmids make use of strong promoters, recombinant protein production typically outperforms endogenous protein synthesis. By switching to isotope-labeled growth media during the induction process, labeled recombinant proteins are selectively enriched in the cytoplasm or periplasm of E. coli.816 Similar routines can be employed to produce in-cell NMR samples in yeast,817 insect,818 and mammalian cells.819,820
For the preparation of in-cell NMR samples in large eukaryotic cells, such as Xenopus laevis oocytes, isotope-labeled proteins can be directly delivered by microinjection.149,821,822 Mammalian cells can be targeted with isotope-labeled, cell penetrating peptide-tagged cargo proteins for active intracellular transport.823,824 Alternatively, cells can be permeabilized with pore-forming bacterial toxins to enable passive infusion of isotope-labeled proteins.824,825 By directly using such protein-loaded cells in in-cell NMR experiments, the isotope-effect is exploited as a selective visualization filter to detect the protein of interest against the backdrop of all other non isotope-labeled cellular components. In doing so, the structural and functional properties of individual disordered proteins can be analyzed in truly physiological environments that display native degrees of intracellular viscosity (see section 2.2), macromolecular crowding (see section 2.3), and biological activities, such as post-translational protein modifications (eukaryotic cells) (see section 3.6). Importantly, high-resolution in-cell NMR spectroscopy does not yield spatial information about intracellular protein localizations and must not be confused with magnetic resonance imaging (MRI). Instead, it provides ensemble descriptions of the combined effects that the cellular environment exerts on the isotope-labeled protein under investigation.
To obtain high-quality in-cell NMR signals of ordered and disordered proteins, the respective targets need to “tumble freely” in the cytoplasm (which determines their overall NMR relaxation behavior). In NMR terms, “tumbling” equates to rotational diffusion, i.e., the rates with which local reorientations occur and, hence, directly reports on intracellular viscosity and crowding in the absence of intracellular interactions (see section 2.3.3). “Tumbling” of ordered proteins is primarily governed by their molecular size. Small proteins tumble faster and their NMR signals relax slower, whereas large proteins tumble slower and their NMR signals relax faster. Slow relaxing NMR signals give rise to sharp resonance cross peaks, which is favorable, whereas fast relaxing NMR signals give rise to broad resonances, which is unfavorable. The “tumbling” behavior of disordered proteins is different to that of ordered ones. IDPs exhibit much higher intrinsic flexibilities on the per-residue scale and do not adopt compact globular shapes. Therefore, backbone and side-chain motions, rather than protein size determine their “tumbling” rates. On average, these are ∼2–3 times greater than for ordered proteins of the same size. High intracellular viscosity slows down rotational diffusion or “tumbling” and, accordingly, leads to faster relaxation characteristics and broadening of in-cell NMR signals of ordered and disordered proteins. Because of their favorable dynamic properties, disordered proteins often display superior in-cell NMR qualities. Nevertheless, given the nature of NMR as an ensemble method, IDP properties are similarly averaged over all intracellular molecules and it is not possible to dissect contributions from inhomogeneous intracellular microenvironments.
The validity of these statements critically depends on one sentence: “In the absence of intracellular interactions”. As we discuss in section 2.3.4, intracellular binding events are manifold and ubiquitous. However, they affect folded and disordered proteins differently. When folded proteins engage in transient intracellular interactions they often respond as single entities and display uniform degrees of signal attenuations.177,182,183,826 By contrast, transient interactions of disordered proteins often elicit line broadening of few residues only.151,184,185 In many instances, these effects identify weakly interacting proteins regions. It is important to note, however, that other scenarios can also lead to broadening of NMR signals. The intracellular environment often affects dynamic processes such as chemical reactions or conformational exchange on the μs to ms time scale, and exchange rates can experience modulations toward slower, more unfavorable NMR time-scales (i.e., intermediate exchange). Such effects are particularly abundant in disordered protein regions exhibiting features of transient secondary structure or for protein segments that adopt multiple conformations.
While most in-cell NMR studies investigated ordered proteins, some were geared toward deciphering the structural in vivo properties of disordered proteins.149,827−829 Out of these, some were primarily concerned with understanding post-translational protein modifications in intact cells,149,827,829 or cell extracts,830−834 whereas others were more specifically geared toward structural and dynamic analyses.835 Among the latter, in-cell NMR studies on α-synuclein feature most prominently.148,150,151,185,507,508,653,836−838 Two of the likely reasons for α-synuclein’s rise to fame in the in-cell NMR community are recent reports that relieved the protein of its disordered status by postulating a folded helical tetramer as representing the relevant in vivo structure in cells.839,840 Both papers stirred considerable interest in the α-synuclein and larger IDP community and prompted several follow up studies. Among them, proponents of the tetramer hypothesis argued that in vivo cross-linking in bacteria, primary neurons and human erythroleukemia cells confirmed the presence of tetrameric, although labile α-synuclein species,841 similar to in vitro experiments with N-terminally acetylated protein in the presence of membranes.842 Opponents contested the existence of a tetramer with results from similar experimental approaches.507,508,843 One argument that lay at the heart of the original discussion was in reference to a boiling step in the purification protocol of bacterially expressed, recombinant α-synuclein.844 In their papers, the groups of Selkoe and Petsko argue that the use of denaturing agents or boiling of bacterial cell lysates during initial purification steps of the recombinant protein destroys the alleged tetramer conformation and results in the accumulation of monomeric species.839,840 While reasonable in its own right, although oblivious to the original paper by Weinreb et al., both groups stated that this constitutes the main reason for why the α-synuclein tretramer evaded detection for so many years. However, these statements also ignore earlier in-cell NMR reports on α-synuclein showing that the protein is monomeric and disordered in intact E. coli cells.148,150,653,836 Several follow-up in-cell NMR studies reinvestigated the structural properties of α-synuclein in intact bacteria151,185,507,837,838 and concluded-, in agreement with earlier reports-, that α-synuclein is intrinsically disordered and monomeric when overexpressed in bacteria.
Another in-cell NMR analysis investigated the disordered bacterial protein FlgM.828 FlgM is a 97-residue polypeptide from Salmonella typhimurium, which regulates flagellar synthesis by binding to the transcription factor Δ28.845 Free FlgM is mostly unstructured in dilute solution, but its C-terminal half can form a transient α-helix.846 Upon binding to Δ28 in vitro this portion of FlgM undergoes a disorder-to-order transition, which is manifested by the disappearance of a set of C-terminal resonance signals.847 In their study, Dedmon et al. exploited this behavior to investigate the conformational properties of FlgM in different in vitro and in vivo environments. In E. coli, the same NMR resonances that disappear upon Δ28 binding are not detected. The authors reasoned that this indicates a similar structural rearrangement in bacteria. Binding to an endogenous homologue of Δ28 cannot account the observed effect, because neither Δ28, nor homologous proteins are present in E. coli. Further NMR analyses in glucose- (450g/L), BSA- (400 g/L), or ovalbumin-crowded (450 g/L) in vitro solutions revealed that the observed transition depends on the absolute amount of the respective crowding agent. This suggested that the intracellular environment modulates the structural in vivo properties of this disorderd protein, although transient weak interactions can similarly explain the disappearance of FlgM NMR signals.
With regard to eukaryotic in-cell IDP studies, Bodart et al. investigated the structural in vivo properties of tau in Xenopus laevis oocytes. Being one of the largest IDPs, with disordered features over its entire length (441 residues, ∼45 kDa), in-cell NMR experiments represented a veritable challenge. Tau exhibits only weak propensities for transient secondary structure and binds to microtubules, i.e., one of the largest macromolecular structures in cells, present at high natural abundance (∼10 μM). At intracellular concentrations of ∼5 μM, in-cell NMR spectra revealed that the predominantly unfolded conformation of tau was similarly populated in oocytes. Due to intracellular viscosity and sample inhomogeneity, line broadening and increased spectral overlap made it difficult to assess whether some portions of the protein adopted structural features not populated in vitro. No substantial conformational rearrangements or overall protein folding were observed. Additional characteristics of the in-cell NMR spectra indicated that tau existed in a microtubule-bound conformation, based on striking similarities with in vitro NMR spectra of tubulin-bound tau.848 Given the high endogenous abundance of microtubules in Xenopus oocytes, such scenarios are perfectly plausible. In-cell NMR spectra of tau also revealed several NMR signals indicative of phosphorylated amino acids. By comparing these signals with in vitro phosphorylated tau,849 the authors confirmed that in-cell phosphorylation by endogenous kinases occurred.
Together, these in-cell NMR results underscore several important points. Foremost, they indicate the overall feasibility of high-resolution IDP studies in prokaryotic and eukaryotic cells. They further prove that in vivo conformations of disordered proteins can be assessed with simple NMR experiments and that changes in protein structures can be readily detected. Given the abundance of cellular post-translational modifications, and their various effects on the structures and functions of disordered proteins (see section 3.6), paired with the unique ability of NMR spectroscopy to detect PTMs and their structural consequences,850 in-cell NMR methods are well-suited to report on the in vivo characteristics of IDPs in a fully integrated manner.
5.3. Fluorescence Microscopy and Spectroscopy
Fluorescence microscopy methods provide relatively simple ways to monitor localization, oligomerization, and diffusion of IDPs in cells and on cell surfaces thereby offering a window into IDP function and dysfunction in cells. For the simplest studies, the diffuse, dim fluorescence of fast moving monomers and oligomers can easily be distinguished from the punctate, bright fluorescence intensity of larger, less mobile aggregates. More complicated imaging and data analysis modalities primarily aimed at monitoring molecular motion include fluorescence recovery after photobleaching (FRAP), fluorescence loss in photobleaching (FLIP), fluorescence correlation and image correlation techniques, and single particle tracking. With the exception of FLIP,851 in-cell implementations of these techniques were recently reviewed.852 The basis of these and other fluorescence microscopy techniques that have been used to monitor mobility, function, conformations, oligomeric state(s), and/or localization of IDPs in cells are discussed briefly below.
FRAP and FLIP exploit photobleaching to monitor molecular diffusion. In FRAP, an intense laser pulse is used to bleach a small region of the cell and illumination is then returned to lower levels.852,853 If fluorescently labeled protein can diffuse into the bleached region within the experimental time scale fluorescence intensity in this region recovers, but little or no recovery is observed when the fluorescently labeled protein is immobile. FLIP uses a similar principle, but in this case the photobleaching beam is constantly on.851 In FLIP experiments, mobile fluorescent species move into the photobleaching region and are photobleached, leading to a loss of fluorescence intensity in the entire cell. By contrast, cells containing large proportions of immobile fluorescent species retain their fluorescence outside the volume illuminated by the photobleaching beam. Both FRAP and FLIP provide quantitative data on translational diffusion and whether IDP populations contain both mobile and immobile species.
Diffusion in cells can also be measured by fluorescence correlation spectroscopy (FCS) and related image correlation spectroscopy methods, recently reviewed by Digman and Gratton854 and by Fitzpatrick and Lillemeier.855 These take advantage of fluorescence intensity fluctuations, the time scales of which depend on how fast the fluorescent molecules diffuse in and out of the observation volume, as a function of molecular mass and shape. Correlating the fluorescence signal over time and/or space thus provides data on molecular mobility, oligomeric distributions, and binding of small fluorescent moieties to larger molecular assemblies or vesicles.854,855 Correlation methods also provide information on the cellular environment, including the local viscosity, barriers to diffusion and directed motion
If the probability of detecting multiple oligomers in the same region of an image (referred to as a region of interest) is low, photobleaching measurements can be employed to count the number of fluorescent subunits (i.e., monomers) in oligomers (for a recent review see ref (856)). This method is particularly powerful in cases where oligomers move relatively slow, i.e., when proteins bind to DNA857 or to cell membranes.858−860 In these experiments, the number of photobleaching steps (steps in fluorescence intensity before the fluorescence baseline is reached) report on the number of monomers in a single region of interest and thereby provide information on the stoichiometry of oligomers. Data from multiple regions of interest reveal the distribution of oligomeric species.
For molecules that diffuse quickly or as an adjunct to photobleaching experiments, fluorescence brightness analysis, such as fluorescence distribution analysis and photon counting histograms are used to determine oligomeric distributions.861,862 These techniques directly take advantage of the increase in fluorescence intensity that occurs when fluorescently labeled molecules oligomerize. Protein oligomerization can also be monitored using protein-fragment complementation methods where interacting proteins of interest are tagged with complementary pieces of a fluorescent protein, such as GFP or a luminescent protein, such as luciferase.863 Dimerization of the tagged proteins reconstitutes the full-length, folded fluorescent or luminescent entity and leads to an optical readout. See below for a more detailed discussion on labeling IDPs.
Distance sensitive Förster resonance energy transfer (FRET) spectroscopy provides a way to monitor protein–protein interactions or conformational changes, in cells. FRET utilizes the steep (1/r6) distance dependence of nonradiative dipole–dipole energy transfer from a donor molecule to an acceptor and is most sensitive for donor–acceptor distances between ∼0.5 and ∼1.5 times the Förster distance (Ro),864 where Ro, often ∼5 nm, is the donor–acceptor distance at which there is a 50% probability of energy transfer. The most common applications of FRET involve two labels with spectrally distinct fluorescence emissions, i.e., a donor that emits in the green and an acceptor that emits in the red. In vitro174 and in cells,134 FRET can be used to monitor conformational changes and conformational distributions when an IDP is labeled with both a donor and acceptor. FRET can also be used to monitor protein–protein interactions, oligomer formation and conformational changes within oligomers by labeling one pool of IDPs with a donor and another pool with an acceptor,865 or by energy transfer between labeled IDPs and dyes, such as thioflavin T, that bind to specific oligomeric species.215 Alternatively, energy migration Förster resonance energy transfer (emFRET, also called homoFRET) uses two identical fluorophores.866 In emFRET, energy transfer from the donor to the acceptor changes the polarization of the emitted fluorescence and reduces the rotational correlation time. Fluorescence anisotropy methods can thus report on oligomerization and, via emFRET, conformational changes of IDPs.867
Recent developments in super-resolution fluorescence imaging methods provide even more information on intracellular protein localization and motion, as highlighted in a number of recent reviews.868−870 In these techniques, single fluorophores are localized with a spatial resolution of tens of nm. Super-resolution methods have been used to probe oligomerization and localization of at least three different IDPs in cells providing valuable data on the formation and morphology of oligomers, inclusion bodies and fibrils.782,787,871 Advances in illumination procedures, such as light-sheet illumination that allows 3-D imaging of cells and tissues while reducing photodamage,872,873 will likely make super-resolution techniques even more powerful.
While steady-state methods provide valuable insights into intracellular behaviors of IDPs, the ability to quickly manipulate the cellular environment and monitor relaxation kinetics provides important additional information on protein stability and the conformations accessible to IDPs and folded proteins. Gruebele and co-workers have recently adapted in-cell temperature jump methods (up to 4 °C in milliseconds), which they call Fast Relaxation Imaging (FReI).874 For FReI, the protein of interest is tagged with a donor fluorescent protein, usually AcGFP, at the N-terminus and an acceptor fluorescent protein, usually mCherry, at the C-terminus. Two-color fluorescence is monitored with millisecond resolution, and FRET between the donor and acceptor reports on protein conformations, protein stability and protein folding/unfolding kinetics. In-cell FReI experiments on the globular protein phosphoglycerate kinase (PGK) have revealed that GFP-mCherry tagged PGK is more compact and more stable in vivo relative to dilute solutions.134,874 These studies also showed that PGK exhibited substantial differences in protein stability and folding within individual cells and in different cellular compartments.134 PGK was more stable, folding was faster and the PGK population was more homogeneous in the nucleus of U2OS bone cancer cells than in the endoplasmic reticulum or the cytoplasm. However, PGK folding displayed a two-state, rather than multistate, character in the ER. These results, attributed to differences in viscosity and macromolecular crowding effects in different microenvironments even within the same subcellular compartment, demonstrate the richness of these cellular data.
FReI has since been applied to α-synuclein875 and has the potential to reveal both the stability of the proteins in cells and the dynamics of conformational changes in response to temperature jumps. FReI may thus provide a measure of the conformational space accessible to IDPs in cells, the dynamics of in-cell conformational changes and how these properties vary between organelles in the same cell, and from cell to cell. Such new and improved technologies along with continuing advancements in fluorescence microscopy methods, including both single molecule fluorescence microscopy and ensemble methods such as fluorescence lifetime imaging,876 are likely to increase the utility of fluorescence microscopy for studying IDPs in cells.
5.3.1. Fluorescence Labeling
In-cell fluorescence microscopy and spectroscopy requires labeling of IDPs with fluorescent molecules. This is most easily accomplished genetically by fusing IDPs to a fluorescent protein (FP). FPs are quite large (∼27 kDa) compared to many IDPs and FP fusions can lead to experimental artifacts. Conjugation to FPs can result in mislocalization,877 and many FPs can dimerize or form higher order oligomers. Monomeric FP variants should be used for all experiments. For green fluorescent protein from the Aequorea victoria jellyfish and its variants, monomers can be obtained by mutating of Ala206 to Lys.878,879 But, there is no established method for ensuring that FPs derived from other organisms are monomeric, although an in-cell membrane protein based assay was recently suggested880 and FPs reported to be monomeric have, in some cases, been found to promote dimerization.881 Finally, FPs may affect the conformational behaviors of IDPs via weak intra- or intermolecular interactions. For a detailed description of how to choose an appropriate FP, see ref (878).
In addition to labeling with FPs, IDPs may be tagged with small organic fluorophores, such as the biarsenical dyes FlAsH and ReAsH that bind to tetra-Cys motifs not found in naturally occurring proteins.882,883 FlAsH and ReAsH are membrane permeable and allow facile in-cell labeling. They also show significant increases in fluorescence quantum yield when conjugated to specific sequence/structural motifs in proteins, enabling the detection of tagged proteins in the presence of unbound fluorophores.882 IDPs labeled with other fluorophores, such as rhodamines may be injected into cells or applied to cell surfaces to study IDP-membrane interactions and/or internalization. Even small organic fluorophores can be perturbing and care must be taken when implementing any of these labeling techniques. When possible, in vitro measurements of structures, i.e., by NMR,785 aggregation propensities, aggregate structures,867,884 as well as in-cell measurements of IDP behaviors and localizations should be performed with native and fluorescently labeled IDPs.
Pioneering studies by Ignatova and Gierasch demonstrated the utility of engineered tetra-Cys motifs and biarsenical dyes to study protein synthesis, stability and aggregation in E. coli cells (reviewed in ref (809)). In these experiments, the authors introduced a tetra-Cys motif into a loop region of the folded cellular retinoic acid binding protein (CRABP) and expressed it in E. coli. In the absence of tetra-Cys CRABP, FlAsH has a low quantum yield882 and the cells showed little fluorescence signal. FlAsH binding to tetra-Cys CRABP increased the FlAsH quantum yield and resulted in diffuse cellular fluorescence, as expected for a fast diffusing, monomeric species. By contrast, production of the aggregation prone Pro39Ala (P39A) tetra-Cys CRABP mutant resulted in bright fluorescent puncta due to inclusion body formation, which increased the total fluorescence intensity per cell. Similarly, denaturing tetra-Cys CRABP by incubating E. coli cells with increasing concentrations of urea led to aggregation and formation of fluorescence puncta at the cell poles. Inclusion body formation could be followed over time or as a function of urea concentration and revealed that tetra-Cys CRABP was destabilized in E. coli relative to in vitro experiments in dilute solutions. Further experiments on P39A tetra-Cys CRABP showed that the addition of the osmolyte proline to the cellular medium cleared inclusion bodies and led to the loss of fluorescence puncta and increases in diffuse fluorescence. Thus, tetra-Cys motifs and biarsenical probes provide a powerful tool for in-cell measurements of protein stability, protein aggregation and the screening of small molecules that prevent or break up aggregates.
Biarsenic dyes bind to the linear sequence Cys-Cys-X-Y-Cys-Cys where X and Y are usually Gly-Pro to yield the preferred beta turn motif.885 This sequence and structural specificity allowed the design of split-tetra-Cys motifs on adjacent beta strands that bind FlAsH only when the sheets are properly oriented, reporting on structure formation.886 Similarly, the tetra-Cys motif may be split between two proteins, allowing FlAsH binding to report on protein–protein interactions.887 Lee and co-workers used the split tetra-Cys motif to monitor Aβ (1–40) aggregation in vitro by appending Cys-Cys to the N-terminus of Aβ (1–40).215 FlAsH binding was reversible and the aggregation kinetics were not significantly altered by the N-terminal tetra-Cys motif or by FlAsH binding. Two processes with two different time scales were evident from increases in FlAsH fluorescence intensity: (i) an almost immediate fast increase that plateaued after 3 h and (ii) a second increase after ∼12 h that coincided with the increase in ThT fluorescence intensity, which reports on fibril formation. ThT binding also resulted in FRET from FlAsH to ThT. The early and swift increase in fluorescence intensity indicated that oligomers formed quickly, and the later, coincident increase in ThT and FlAsH fluorescence intensity as well as FRET suggested that oligomers were on-pathway for fibril formation, and that fibrils formed more slowly than smaller oligomers. Soluble oligomers are quite difficult to detect and these results suggested that split tetra-Cys motifs are suitable for monitoring IDP oligomer formation in cells.
5.3.2. Fluorescence Studies of IDP Aggregation
5.3.2.1. polyQ-Repeat Proteins
By appending polyglutamine (polyQ) repeats from the Huntingtin exon 1 to the C-terminus of tetra-Cys CRABP, Ignatova et al. investigated IDP aggregate formation in E. coli.809,888 As expected, in-cell aggregation of tetra-Cys CRABP-Huntingtin constructs depended on the length of the polyQ tracts. Tetra-Cys CRABP-Huntingtin-40 (where 40 is the length of the polyQ repeat) displayed both soluble and aggregated populations, while tetra-Cys CRABP constructs with longer polyQ tracts, Huntingtin-53 and Huntingtin-64, formed detergent insoluble aggregates over time. Long polyQ tracts also resulted in long, filamentous bacteria indicating that the detergent insoluble fibrils interfered with E. coli cell division.
The use of a flanking sequence can influence IDP aggregation. Both in vitro and in E. coli the aggregation prone P39A CRABP variant seeds early oligomer formation while polyQ tracts with lengths above the pathological threshold seeded the formation of later, detergent resistant aggregates.888,889 Thus, as observed for polyQs appended to other folded proteins,890−893 the context of the polyQ sequence is key. Aggregation of polyQ repeats tagged with an FP, (often cerulean fluorescent protein (CFP), GFP, or yellow fluorescent protein (YFP) at the C-terminal end) or complementary GFP fragments has been studied in yeast cells, in mammalian cells and in the transparent nematode C. elegans (for reviews and methods see refs (852, 894, and 895)). As expected, polyQ monomers and small oligomers in cells show dim, diffuse fluorescence throughout the cytoplasm while larger, less mobile inclusion bodies result in intense, punctate fluorescence.896 The translational diffusion of fluorescently labeled polyQ constructs directly report on how long it takes for inclusion bodies to form. In C. elegans, FRAP experiments revealed that short polyQ tracts containing 35, or fewer repeats were mobile in 3 to 4 day old nematodes, tracts with 40 repeats were polydisperse with mobile and immobile fractions, and constructs with 82 repeats were essentially immobile.897 Inclusion body formation was age dependent and polyQ constructs with 33 repeats, which were mobile in young nematodes displayed punctate fluorescence in 5 day, or older nematodes. Inclusion body formation by polyQ-FP constructs, in both mammalian cells and C. elegans, has proven to be an extremely effective sensor for monitoring changes in protein homeostasis due to aging,897,898 osmotic899 and other stresses, and for testing whether and how protein homeostasis can be restored.895,898,900,901 The high fluorescence intensity from inclusion bodies obscures lower intensity fluorescence signals from smaller, more mobile aggregates. By combining specific photobleaching of inclusion bodies with super-resolution microscopy, Frydman and co-workers were able to visualize smaller oligomers formed by Huntingtin polyQ-eYFP repeats in mammalian cells.871 Such super-resolution imaging is likely to provide more detailed data on the polyQ oligomeric distribution in cells, how larger oligomers form and how changes in the cellular environment affect the oligomeric distribution.
Specific fluorescent sensors for oligomerization can be constructed by combining labeling modalities. Onodera and co-workers have built an in-cell FRET sensor for oligomerization by expressing mixtures of polyQ constructs labeled with different FPs, i.e., polyQ-CFP and polyQ-YFP, where energy transfer only occurred if the polyQ-FP constructs adopted particular orientations in oligomers and large aggregates.865 FRET in mobile oligomers and large aggregates were only observed when the FPs were on the same termini of the polyQ construct (polyQ-CFP plus polyQ-YFP, or CFP-polyQ plus YFP-polyQ) and not on different termini (i.e., polyQ-CFP plus YFP-polyQ).865 Alternatively, Hatters and colleagues combined tetra-Cys tags and FPs in the same monomers.892,902 In these experiments, monomers displayed a high ratio of biarsenical dye fluorescence intensity to fluorescent protein intensity. Occlusion of the tetra-Cys binding sites due to inclusion body formation for example, reduced this ratio. When the polyQ repeat length was above the pathogenic threshold, the ratio was also reduced in the mobile population, but only for some locations of the tetra-Cys tag. While attention must be paid to how the location and presence of the fluorophores affects aggregation, the sensitivity of both of these methods to the location of the fluorescent tag should be helpful in modeling the molecular structure of in-cell oligomers. Already, the similarity of FRET signals for soluble oligomers and inclusion bodies provide limits on the conformational distributions of these species.
Identification of cytotoxic species is key to understanding how polyQ repeats and other aggregation prone species kill cells. Fluorescently labeled polyQ repeats can be monitored over time as cells or animals age and die providing data on cytotoxic species.894,895,903,904 Experiments in cells and in C. elegans showed that individual cells that accumulate inclusion bodies often live longer than cells with diffuse, mobile oligomers865,900,905,906 supporting models in which inclusion body formation is a cytoprotective mechanism.895,907−909 In addition, in C. elegans, some gene knockdowns that reduced inclusion body formation of a polyQ with 35 repeats (Q35) resulted in increased toxicity, again arguing that soluble oligomers can be cytotoxic.900 Identification of toxic oligomeric species is, however, more difficult and FCS experiments in which mobile Q35 was monitored in C. elegans cell lysates, or after being purified from C. elegans revealed a heterogeneous oligomeric population where the population shifted to slower moving species as the animals aged.910 Interestingly there was no correlation between changes in the oligomer distributions and gene knockdowns that enhanced cytotoxicity. While in-cell fluorescent techniques provide clues as to which polyQ species or sets of species may be toxic, truly deciphering the conformational distributions in cells will likely require creative applications of and innovations in fluorescence techniques combined with other techniques including NMR and ion-mobility mass spectrometry.
5.3.2.2. IDP Aggregation and Neurodegeneration
Research on other IDP containing proteins, or peptides associated with neurodegeneration have focused on the localization and function of native conformers, identifying and localizing cytotoxic species as well as how interactions with membranes alter IDP conformation and oligomerization. The techniques used are similar to those described above for polyQ repeats with an added emphasis on labeling with small fluorophores, particularly for the short Aβ peptides, and a number of studies using luciferase,911 or FP complementation912 to study oligomerization and aggregation. These experiments, discussed below, have mainly focused on the Parkinson’s disease associated protein α-synuclein and the Aβ peptides associated with Alzheimer’s disease. In cell culture, animal models and humans, conformational changes and oligomerization of proteins associated with neurodegenerative diseases appears to propagate from cell to cell (for recent reviews see refs (913 and 914)). Fluorescently labeled IDPs have been used to monitor cell–cell spread of oligomers and oligomerization in real time, to determine the conformational distributions of the species involved and to test interventions.784,915,916
To elucidate the cellular localization and function of α-synuclein, a number of groups have constructed α-synuclein-eGFP fusions with the enhanced GFP at the C-terminus.917−920 Imaging of neurons containing this construct has shown that α-synuclein is enriched at the synapse where it dynamically binds to synaptic vesicles.574,918,920 More recently, α-synuclein has been localized at the presynaptic terminals, in intraluminar vesicles of multivesicular bodies and in lysosomes in neuronal cell bodies by combining fluorescence imaging of α-synuclein-eGFP in cells with 3D electron tomography of cells containing α-synuclein-miniSOG constructs, where the miniSOG provides contrast.917 These studies suggest that α-synuclein remains bound to synaptic vesicles until they fuse with plasma membranes,918 and that overexpression of the protein may perturb intracellular membrane architecture.917
In C. elegans expressing human α-synuclein-YFP fusions, inclusion body formation increased as a function of age and this system has been used to test methods for ameliorating cytotoxicity.921 Like the polyQ model systems where molecular chaperones helped protect against cytotoxicity, overexpression of the molecular chaperone Hsp70 reduced cytotoxicity in C. elegans.(921) In neuronal cultures, overexpression of α-synuclein-FP constructs resulted in secretion into the extracellular medium and, as shown using α-synuclein fused to fragments of luciferase, the secreted protein was often oligomeric915 and associated with Hsp70 altering its size distribution. Nonetheless, RNA interference screens in C. elegans designed to identify proteins that reduced α-synuclein pathology did not pull up Hsp70, or other molecular chaperones, but rather identified a number of proteins involved in vesicular trafficking and lipid transport as key to reducing toxicity.921 These results are consistent with α-synuclein’s role in synaptic vesicle trafficking,918 lysosomal abnormalities observed in the nervous system of transgenic mice expressing human α-synuclein-eGFP,922 α-synuclein associated fragmentation of mitochondrial membranes in neuronal cells923 and membrane abnormalities in yeast.924 α-synuclein membrane interactions were reviewed by Lindquist and colleagues.924
While α-synuclein-FP fusions provided important information on cellular localization and function, FPs (∼27 kDa) are much larger than the protein and there is some evidence that α-synuclein-FP species can be mislocalized in cells.920 Therefore, a number of groups have pursued alternative labeling strategies using smaller organic fluorophores. The biarsenical dye, FlAsH, was employed to monitor the aggregation of α-synuclein modified with a C-terminal tetra-Cys tag in mammalian cells.785,788 Super-resolution fluorescence microscopy was used to monitor α-synuclein aggregates by labeling the protein with a rhodmaine spiroamide derivative, an organic fluorophore, in vitro and then microinjecting the protein into cells.787 Further super-resolution fluorescence microscopy experiments either using microinjected, or photoactivatable-, photoswitchable FP-tagged protein may help elucidate how α-synuclein is trafficked in cells providing more data on the physiological function and dysfunction of this important IDP.
Intrinsically disordered Aβ peptides derived from amyloid precursor protein are the main component of extracellular plaques in the brains of Alzheimer patients (for a recent review see ref (925)). The N-termini of Aβ peptides are easily labeled with small, organic fluorophores including fluorescein,926−930 rhodamine928,930−932 and HiLyte930,933,934 based dyes, and these N-terminal labels do not significantly perturb the peptides.931,933,935,936 Because these peptides are extracellular, incubation of cells with fluorescently labeled peptides can provide valuable, physiologically relevant data on how Aβ peptides interact with the extracellular leaflet of plasma membranes.
Renner et al. showed that preformed oligomers of fluorescently labeled Aβ peptides localize to synapses and cluster glutamate receptors when incubated with live hippocampal neurons.930 Similar synaptic localization of oligomers was observed by immunofluorescence for hippocampal neurons incubated with unlabeled oligomers937 and for Aβ oligomers found near plaques in a mouse model of Alzheimer disease.938 Fluorescently labeled oligomers, but not fibrils can also be internalized by cells.931 Cellular incubation with low concentrations (250–500 nM) of fluorescently labeled Aβ peptides results in internalization and formation of intracellular aggregates.782,928 Super-resolution imaging of such in-cell aggregates revealed fibril formation by internalized, fluorescence-labeled Aβ (see section 4.7).782
Physiological Aβ peptide concentrations range from sub-nM to low nM.939−941 At these concentrations in aqueous solution, fluorescent Aβ (1–40) and Aβ (1–42) peptides are mainly monomeric, with small populations of dimers and trimers.860,934,942 Single molecule fluorescence photobleaching,860,943 or multicolor934 experiments, as well as fluorescence correlation methods929,932 provide a way to monitor Aβ peptides-cell interactions at these physiologically relevant concentrations. In photobleaching experiments, both the fluorescence intensity and number of photobleaching steps were used to determine the number of labeled monomers in a single fluorescent spot.860,943 While in multicolor experiments, Aβ peptides labeled with two different color dyes were mixed at equimolar concentrations before incubation with cells and both fluorescence intensities and the colocalization of multiple colors were used to identify and characterize oligomers. The exact characteristics of cell bound species depended on which cell-types and Aβ peptides, i.e., Aβ (1–40) or Aβ (1–42)934 were used, or on the combination of peptides that was applied to cells.943 Nonetheless, in all of these experiments, as well as in experiments using higher peptide concentrations,926,927 the distribution of oligomeric states shifted from mainly monomeric in solution to dimers and higher-order oligomers on the surface of cells and a large portion of these oligomers showed restricted motions on cell membranes.860,929,934,943 Thus, these experiments demonstrate the utility of single molecule fluorescence and fluorescence correlation methods for determining the oligomeric states of membrane bound IDPs and their motions on cells. Taken together with the results from super-resolution fluorescence microscopy, these data suggest that by combining fluorescence microscopy modalities it may be possible to monitor Aβ on-cell aggregation on the single molecule level in real time.
Fluorescence labeling of IDPs and fluorescence microscopy in cells continues to provide facile methods for monitoring IDP conformations, interactions with other biomolecules including membranes, oligomerization, motion and localization in real time. With the continuing advances in fluorescence labeling modalities and the recent revolution in super-resolution methods, fluorescence microscopy in cells and whole organisms will likely continue to be important for understanding IDP localization, function and dysfunction.
5.4. Fourier Transform Infrared (FTIR) Microspectroscopy
Fourier transform infrared (FTIR) spectroscopy exploits bond vibrations in molecules to yield spectra with unique absorption patterns. Besides providing specific molecular fingerprints of investigated molecules, these patterns also contain structural information.944 Proteins, nucleic acids, lipids and carbohydrates have unique chemical features giving rise to distinct FTIR spectra. Protein FTIR spectra for example, contain different regions characteristic for the different modes of vibration. The amide I band in particular (∼1655 cm–1) is sensitive to protein secondary structure, because its spectral features are affected by the different hydrogen-bond patterns in α-helices, β-sheets, and disordered structures.944−947 FTIR additionally provides unique information about protein aggregates based on characteristic IR signatures of intermolecular β-sheets with absorption peaks at 1630–1620 cm–1.948,949 For these reasons, FTIR is extensively used to study the conformational properties of aggregated disordered proteins.950,951
Early FTIR studies were carried out on isolated, purified proteins, or aggregates. However, the recent combination of FTIR spectroscopy with light microscopy, i.e., FTIR microspectroscopy, made it possible to also study aggregated IDPs in intact cells and tissues, which is particularly attractive because is does not rely on exogenous protein labeling, or the use of contrast agents.777 Despite these advantages, FTIR microspectroscopy also bears several drawbacks, some of which are directly linked to the physical nature of the technique. First, IR wavelengths are long and, hence, achievable spatial resolution is restricted to the specific diffraction limit (∼2–10 μm for the mid-IR region, ∼4000–500 cm–1). In practice, however, spatial resolution is primarily limited by the low intensity of IR rays. In turn, large microscope apertures are needed, which further decreases spatial resolution. For these reasons, modern FTIR microspectrometers are installed at synchrotrons, providing a 100–1000 times brighter source of IR illumination.952 Synchrotron IR beams also offer the possibility to implement fast data collection and readout schemes, which enable time-resolved recordings of biological processes.952 Another impediment to FTIR microspectroscopy is water. Present at high endogenous concentrations in most biological samples, the water absorption band in the mid-IR region pollutes IR spectra of cells and tissues. Different strategies to reduce these water effects have been devised and are excellently reviewed in.944,952
IDP-based FTIR microspectroscopy applications in cells and tissues have focused almost entirely on intracellular aggregation of Aβ, Huntingtin and the Prion protein.776,953−955 In these approaches, IR spectra of different sample areas are spatially matched with immuno-electron, or -fluorescence microscopy. Thereby, intracellular regions of high secondary structure content, characteristic of β-rich amyloid aggregates for example, are correlated with areas of bright antibody-, or thioflavin S-fluorescence. In one such study, Andre et al. investigated the structures of Huntingtin fibrils in brain slices of control individuals and Huntington’s disease patients.776 By measuring IR signatures of different protein deposits that had been spatially localized by immuno-fluorescence microscopy, the authors established that the structural features of intracellular Huntingtin inclusions are nonuniform and display localized degrees of polymorphisms and β-sheet contents.
Similarly, FTIR microspectroscopy of amyloid fibrils of Alzheimer’s disease patients displays varying degrees of β-sheet content in different brain regions.953 However, IR detection of the amide I band at ∼1632 cm–1 conflicted with earlier reports of in vitro aggregated Aβ and IR absorption bands at 1620–1628 cm–1.956,957 The authors attributed this discrepancy to the presence of nonfibrillar amyloid-associated proteins, such as apolipoprotein E and ubiquitin, which are known components of native neuritic plaques.953 When Miller et al. investigated thioflavin S-positive inclusions in brain slices of AD patients they detected β-aggregate characteristic absorption bands at ∼1625 cm–1.958 However, they also noted that the β-sheet contents of these species are lower than those of in vitro aggregated Aβ fibrils. In a next step, they used synchroton X-ray fluorescence to probe for local accumulations of Cu and Zn and found that regions of high metal content correspond to areas with localized amyloid deposits. These results provided first-time experimental evidence for a spatial correlation between Aβ plaques and enhanced intracellular concentrations of Cu and Zn, in support of the postulated link between defects in metal homeostasis and the onset of AD.724
Diomede et al. used FTIR microspectroscopy to monitor Aβ aggregation in the presence of tetracyclines in C. elegans.954 Tetracyclines inhibit Aβ amyloid formation and are considered possible AD drugs.959 Induction of Aβ overexpression resulted in a time dependent increase of the 1623 cm–1 IR absorption signal. When Diomede et al. cultured worms in tetracycline-containing media they did not detect aggregate-indicative IR absorption signals and consistently found fewer intracellular amyloid aggregates.
Kneipp et al. used FTIR microspectroscopy to study cellular inclusions of the Prion protein in scrapie-infected hamster neurons.960 When the authors compared IR spectra of brain slices from control and infected animals, they noted prominent absorption bands in diseased animals only. Further PrP immuno-stainings revealed a strong colocalization with brain areas of high β-sheet contents. Wang et al. independently confirmed these results in a larger animal cohort and in different neuronal cells.961 They also probed for the accumulation of metals using X-ray fluorescence spectroscopy and found elevated levels of intracellular Fe in PrP-positive cells. When they prepared brain slices of infected hamsters at different postinfection time points, they discovered that local increases in β-sheet contents correlate with the abundance of insoluble PrP deposits. However, regions that were void of PrP deposits also featured high β-contents, which suggested that either β-aggregate formation precedes precipitation, or that other non-PrP aggregates are additionally present. Together, these FTIR microspectroscopy studies conveyed several important points. First, they indicate that intracellular IDP aggregates are structurally inhomogeneous, which contrasts with results obtained in vitro. Second, they show that IDP aggregation scavenges bystanders, either actively, or passively, including other intracellular proteins and/or metals. Third, they indicate that aggregate accumulation correlates with disease progression.
6. Summary and Outlook
With this work, we attempted to provide a comprehensive overview of factors influencing the structural and functional properties of intrinsically disordered proteins in different cellular environments. We discussed the elementary composition of prokaryotic and eukaryotic cells in terms of ions, metabolites and biological macromolecules such as proteins, RNA, DNA, lipids, and glycans and delineated how they contribute to general physical parameters such as viscosity and macromolecular crowding. We outlined how these properties affect micro- and macroscopic intracellular behaviors such as diffusion and association. We described biological activities encountered in cells, such as post-translational protein modifications, and how they influence in vivo properties of intrinsically disordered proteins, especially also with regard to protein association and aggregation, and vice versa. Finally, we presented methods to study IDPs in silico and in intact cells.
Some general conclusions can be drawn from these discussions. Ordered and disordered proteins experience the intracellular milieu in a similar manner and they are subject to the same types of physical and biological forces acting upon them. Given their unique structural features, disordered proteins may respond to these factors differently, especially in terms of compaction and aggregation. With regard to their cellular stability and lifetimes, ordered and disordered proteins behave similarly. IDPs are not preferred targets for proteolytic degradation or for chaperone interactions, and given their roughly equal abundance in proteomes of higher organisms, most of their cellular properties are indistinguishable from ordered proteins. IDPs are more prone to post-translational modifications and therefore likely to exist as chemically heterogeneous intracellular populations. They may interact with multiple physiological ligands and display ordered properties in their bound states.
By using complexity-reduced experimental setups such as artificially crowded in vitro solutions, or cell extracts, we can learn a great deal about the in vivo properties of disordered proteins. However, we must acknowledge that these environments only partially reflect the physical and biological contributions experienced in cells. Especially cell extracts fall short in recapitulating the effects exerted by cellular compartments and localized biological activities. Therefore, we must strive to develop and employ tools to analyze disordered proteins in intact physiological settings. While this presents a veritable challenge for the future, it also offers exciting new possibilities for IDP research.
Acknowledgments
We thank Dr. Carlos Bertoncini for providing original figure files for reproduction. F.X.T. acknowledges support from the Association pour la Recherche contre le Cancer (ARC). P.S. acknowledges funding by an Emmy Noether research grant (SE1794/1-1) from the Deutsche Forschungsgemeinschaft (DFG). The work of T.F.K. and A.H.E. is supported by NIH Grants R01 GM087290 and R01 GM099865. K.H. is supported, in part, by a fellowship from the University of Massachuessetts, Amherst, as part of the Chemistry-Biology Interface Program (funded by NIH Grant T32 GM08515). The laboratory of G.J.P. is supported by the National Science Foundation, Grant MCB 1051819. C.L. acknowledges funding from the Ministry of Science and Technology of China (2013CB910200), the 1000 Young Talents Program, and the National Sciences Foundation of China (Grants 21173258 and 21075134). P.B.C. and C.K. are supported by NUI Galway (Hardiman Research Scholarship to C.K.) and the Science Foundation of Ireland (Grants 10/RFP/BIC2807 and 11/W.1/B2067). L.G. is supported by NIH Grants NIH/NIGMS R01GM101644 and NIH/NIGMS R01GM027616. L.G. and A.G. are supported by NIH Grant NIH/NIGMS R01GM094848.
Biographies

François-Xavier Theillet studied Physics and Chemistry at the Ecole Normale Supérieure (ENS) de Lyon in France. For his Ph.D., he worked on the structure and antigenicity of bacterial polysaccharides in the laboratory of Muriel Delepierre at the Institute Pasteur in Paris (2005–2009). He obtained a doctorate in Biophysics and Biochemistry from the University Pierre et Marie Curie (Paris VI) in 2009. Since 2010 he is a postdoctoral fellow with Philipp Selenko at the FMP Berlin. His research covers cellular structural biology, with special emphasis on IDPs, post-translational protein modifications, and NMR method development.

Andrés Binolfi studied Biotechnology at the National University of Rosario, Argentina. He did his Diploma with Marisa Biasoli (2001–2004) and his Ph.D. with Claudio Fernandez at the Institute of Molecular and Cell Biology of Rosario (2005–2010). Since 2011, he is a postdoctoral fellow with Philipp Selenko at the FMP Berlin. His research focuses on α-synuclein and its structural and functional properties in different cellular environments. His expertise includes biophysical methods and NMR spectroscopy.

Tamara Frembgen-Kesner is a native Iowan. She obtained her B.A. in chemistry at the University of Iowa in 2002 and her Ph.D. in the group of Adrian H. Elcock at the University of Iowa in 2008. Her work focuses on using coarse-grained simulation methods to model the effects of hydrodynamic interactions on protein diffusion, folding and association.

Karan Hingorani received his Bachelor’s degree in Life Sciences and Biochemistry from St. Xavier’s college at the University of Mumbai in India. Subsequently, he worked for a year with Derek Wilson at York University in Toronto where he was introduced to microfluidics and mass spectrometry. In 2010 he joined the Molecular and Cellular Biology graduate program at the University of Massachusetts at Amherst where he works with Lila Gierasch. His graduate research seeks to understand the differences between protein folding inside cells compared to test tubes. In particular, he is developing a sensor for protein folding capable of reporting thermodynamic stability in vivo. Additionally, he is researching molecular chaperones and their influence on the folding landscape of proteins in cells.

Mohona Sarkar was born in India and received her B.Sc. and M.Sc. from the Indian Institute of Technology, Kharagpur. She earned her Ph.D. under the supervision of Gary Pielak at the University of North Carolina at Chapel Hill. Her research focused on the effects of biomolecular crowding on protein stability with emphasis on soft interactions. She is now a postdoctoral researcher in Patricia Clark’s laboratory at the University of Notre Dame in Indiana.

Ciara Kyne received her BSc (Chemistry) from NUI Galway in 2011 and is a Ph.D. candidate in the Crowley laboratory. She uses NMR spectroscopy and size exclusion chromatography to study protein interactions under native-like conditions. Recently, she was a trainee at the Centre to Advance Molecular Interaction Science, University of New Hampshire.

Conggang Li received his B.A. in Chemistry from Wuhan University and his Ph.D. in Chemistry and Biochemistry from Florida State University. He was a postdoctoral fellow in the laboratory of Gary J. Pielak at University of North Carolina—Chapel Hill. He is currently a principal investigator at the Wuhan Institute of Physics and Mathematics, Chinese Academy of Sciences. For his research he uses nuclear magnetic resonance spectroscopy to study protein dynamics and interactions. Currently, his laboratory focuses on understanding cellular crowding effects on protein structure, dynamics, interactions, and function.

Peter B. Crowley received his B.Sc. (Chemistry) from University College Dublin in 1998 and his Ph.D. in 2002 from Leiden University. After a Marie Curie Individual Fellowship at the New University of Lisbon, he returned to Ireland. Since 2008, he is a lecturer at NUI Galway. His research is focused on weak protein interactions and controlled protein assembly.

Lila Gierasch received her undergraduate education at Mount Holyoke College, graduating with an A.B. in Chemistry. Her interest in protein folding began with her undergraduate research on collagen refolding. She then obtained her Ph.D. at Harvard University in Biophysics, where she worked with Elkan Blout on cyclic peptide models for reverse turns. She held faculty positions at Amherst College, the University of Delaware, and the University of Texas Southwestern Medical School before joining the University of Massachusetts Amherst, where she is now Distinguished Professor of Biochemistry & Molecular Biology and Chemistry. Her research continues to focus on protein folding, with a current focus on how folding occurs in the challenging environment of the cell and how molecular chaperones facilitate this process.

Gary J. Pielak was born in Great Lakes, Illinois. He earned a B.A. in Chemistry from Bradley University in Peoria, Illinois and a Ph.D. in Biochemistry from Washington State University in Pullman, Washington. He was a postdoctoral fellow in the laboratory of Michael Smith at the University of British Columbia in Vancouver, Canada and in the laboratory of Robert J.P. Williams at the Univeristy of Oxford in England. Gary is currently the Glen H. Elder Jr., Distinguished Term Professor of Chemistry, Biochemistry and Biophysics at the University of North Carolina—Chapel Hill in the U.S. His research focuses on understanding protein chemistry in cells and under crowded conditions in vitro.

Adrian H. Elcock was born and raised in Sheffield, U.K. He obtained a first-class honors degree in chemical sciences from the University of East Anglia in 1989 and a D.Phil. in physical chemistry in the group of Prof. W. Graham Richards from the University of Oxford in 1994. Following postdoctoral research in the group of Prof. J. Andrew McCammon at UC San Diego he joined the faculty at the University of Iowa in 2000. His work focuses on the development and application of molecular simulation methods for modeling biological macromolecules in vitro and in vivo.

Anne Gershenson received her undergraduate education at Bryn Mawr College, graduating with an A.B. in Physics. As a graduate student in Physics at the University of Michigan, Ann Arbor with Ari Gafni and Duncan Steel she used triplet state spectroscopy to study protein folding. As a postdoctoral fellow she evolved thermostable proteins with Frances Arnold at Caltech and performed single molecule fluorescence experiments with Paul R. Selvin at the University of Illinois, Urbana–Champaign. She held a faculty position at Brandeis University before joining the University of Massachusetts Amherst, where she is now an Associate Research Professor in the Department of Biochemistry and Molecular Biology. She continues to study protein folding, primarily using single molecule fluorescence, with a focus on the folding of metastable proteins both in vitro and in cells.

Philipp Selenko studied Chemistry and Biochemistry at the University of Vienna, Austria. He did his Diploma at the Institute of Molecular Pathology (IMP) with Thomas Jenuwein (1998) and his Ph.D. with Michael Sattler at the European Molecular Biology Laboratory (EMBL) in Heidelberg (1998–2002). He was a postdoctoral fellow with Gerhard Wagner at Harvard Medical School (2003–2008) and is heading the in-cell NMR group at the FMP Berlin since 2008. His laboratory pursues cellular structural biology, with special emphasis on intrinsically disordered proteins.
Author Contributions
∇ Equal contribution.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Eliezer D. Curr. Opin. Struct. Biol. 2009, 19, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittag T.; Forman-Kay J. D. Curr. Opin. Struct. Biol. 2007, 17, 3. [DOI] [PubMed] [Google Scholar]
- Schneider R.; Huang J. R.; Yao M.; Communie G.; Ozenne V.; Mollica L.; Salmon L.; Jensen M. R.; Blackledge M. Mol. Biosyst. 2012, 8, 58. [DOI] [PubMed] [Google Scholar]
- Uversky V. N. Biochim.Biophy. Acta 2013, 1834, 932. [DOI] [PubMed] [Google Scholar]
- Dunker A. K.; Obradovic Z.; Romero P.; Garner E. C.; Brown C. J. Genome Inform. Ser. Workshop Genome Inform. 2000, 11, 161. [PubMed] [Google Scholar]
- Ward J. J.; Sodhi J. S.; McGuffin L. J.; Buxton B. F.; Jones D. T. J. Mol. Biol. 2004, 337, 635. [DOI] [PubMed] [Google Scholar]
- Xue B.; Dunker A. K.; Uversky V. N. J. Biomol. Struct. Dyn. 2012, 30, 137. [DOI] [PubMed] [Google Scholar]
- Dunker A. K.; Lawson J. D.; Brown C. J.; Williams R. M.; Romero P.; Oh J. S.; Oldfield C. J.; Campen A. M.; Ratliff C. R.; Hipps K. W.; Ausio J.; Nissen M. S.; Reeves R.; Kang C. H.; Kissinger C. R.; Bailey R. W.; Griswold M. D.; Chiu M.; Garner E. C.; Obradovic Z. J. Mol. Graph. Model. 2001, 19, 26. [DOI] [PubMed] [Google Scholar]
- Dunker A. K.; Obradovic Z. Nat. Biotechnol. 2001, 19, 805. [DOI] [PubMed] [Google Scholar]
- Tompa P. Trends Biochem. Sci. 2002, 27, 527. [DOI] [PubMed] [Google Scholar]
- Wright P. E.; Dyson H. J. J. Mol. Biol. 1999, 293, 321. [DOI] [PubMed] [Google Scholar]
- Tantos A.; Han K. H.; Tompa P. Mol. Cell. Endocrinol. 2012, 348, 457. [DOI] [PubMed] [Google Scholar]
- Dunker A. K.; Brown C. J.; Lawson J. D.; Iakoucheva L. M.; Obradovic Z. Biochemistry 2002, 41, 6573. [DOI] [PubMed] [Google Scholar]
- Dunker A. K.; Silman I.; Uversky V. N.; Sussman J. L. Curr. Opin. Struct. Biol. 2008, 18, 756. [DOI] [PubMed] [Google Scholar]
- Xie H.; Vucetic S.; Iakoucheva L. M.; Oldfield C. J.; Dunker A. K.; Uversky V. N.; Obradovic Z. J. Proteome Res. 2007, 6, 1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky V. N. Front. Biosci. 2009, 14, 5188. [DOI] [PubMed] [Google Scholar]
- Uversky V. N.; Oldfield C. J.; Dunker A. K. Annu. Rev. Biophys. 2008, 37, 215. [DOI] [PubMed] [Google Scholar]
- Vacic V.; Markwick P. R.; Oldfield C. J.; Zhao X.; Haynes C.; Uversky V. N.; Iakoucheva L. M. PLoS Comput. Biol. 2012, 8, e1002709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernado P.; Svergun D. I. Mol. Biosyst. 2012, 8, 151. [DOI] [PubMed] [Google Scholar]
- Jensen M. R.; Ruigrok R. W. H.; Blackledge M. Curr. Opin. Struct. Biol. 2013, 23, 426. [DOI] [PubMed] [Google Scholar]
- Tompa P. Curr. Opin. Struct. Biol. 2011, 21, 419. [DOI] [PubMed] [Google Scholar]
- Dyson H. J.; Wright P. E. Curr. Opin. Struct. Biol. 2002, 12, 54. [DOI] [PubMed] [Google Scholar]
- Tycko R. Annu. Rev. Phys. Chem. 2011, 62, 279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tycko R.; Wickner R. B. Acc. Chem. Res. 2013, 46, 1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iakoucheva L. M.; Radivojac P.; Brown C. J.; O’Connor T. R.; Sikes J. G.; Obradovic Z.; Dunker A. K. Nucleic Acids Res. 2004, 32, 1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suskiewicz M. J.; Sussman J. L.; Silman I.; Shaul Y. Protein Sci. 2011, 20, 1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali R. A.; Landsberg M. J.; Knauth E.; Morgan G. P.; Marsh B. J.; Hankamer B. PLoS One 2012, 7, e33697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershenson A.; Gierasch L. M. Curr. Opin. Struct. Biol. 2011, 21, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucic V.; Rigort A.; Baumeister W. J. Cell Biol. 2013, 202, 407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundararaj S.; Guo A.; Habibi-Nazhad B.; Rouani M.; Stothard P.; Ellison M.; Wishart D. S. Nucleic Acids Res. 2004, 32, D293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Record M. T.; Courtenay E. S.; Cayley D. S.; Guttman H. J. Trends Biochem. Sci. 1998, 23, 143. [DOI] [PubMed] [Google Scholar]
- Jewett M. C.; Swartz J. R. Biotechnol. Bioeng. 2004, 86, 19. [DOI] [PubMed] [Google Scholar]
- Roe A. J.; McLaggan D.; Davidson I.; O’Byrne C.; Booth I. R. J. Bacteriol. 1998, 180, 767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabala L.; Bowman J.; Brown J.; Ross T.; McMeekin T.; Shabala S. Environ. Microbiol. 2009, 11, 137. [DOI] [PubMed] [Google Scholar]
- Outten C. E.; O’Halloran T. V. Science 2001, 292, 2488. [DOI] [PubMed] [Google Scholar]
- Moncany M. L.; Kellenberger E. Experientia 1981, 37, 846. [DOI] [PubMed] [Google Scholar]
- Alatossava T.; Jutte H.; Kuhn A.; Kellenberger E. J. Bacteriol. 1985, 162, 413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyrrell J.; McGinnis J. L.; Weeks K. M.; Pielak G. J. Biochemistry 2013, 52, 8777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Eunen K.; Bouwman J.; Daran-Lapujade P.; Postmus J.; Canelas A. B.; Mensonides F. I. C.; Orij R.; Tuzun I.; van den Brink J.; Smits G. J.; van Gulik W. M.; Brul S.; Heijnen J. J.; de Winde J. H.; de Mattos M. J. T.; Kettner C.; Nielsen J.; Westerhoff H. V.; Bakker B. M. FEBS J. 2010, 277, 749. [DOI] [PubMed] [Google Scholar]
- Ingwall J. S.; Balschi J. A. J. Cardiovasc. Electrophysiol. 2006, 17Suppl 1S127. [DOI] [PubMed] [Google Scholar]
- Traut T. W. Mol. Cell. Biochem. 1994, 140, 1. [DOI] [PubMed] [Google Scholar]
- Godt R. E.; Maughan D. W. Am. J. Physiol. 1988, 254, C591. [DOI] [PubMed] [Google Scholar]
- Finney L. A.; O’Halloran T. V. Science 2003, 300, 931. [DOI] [PubMed] [Google Scholar]
- Ortega R.; Deves G.; Carmona A. J. R. Soc. Interface 2009, 6, S649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J. G.; Qin Y.; Galati D. F.; Palmer A. E. ACS Chem. Biol. 2012, 7, 1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banci L.; Bertini I.; McGreevy K. S.; Rosato A. Nat. Prod. Rep. 2010, 27, 695. [DOI] [PubMed] [Google Scholar]
- McRae R.; Bagchi P.; Sumalekshmy S.; Fahrni C. J. Chem. Rev. 2009, 109, 4780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRae R.; Lai B.; Fahrni C. J. Metallomics 2013, 5, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darbari D.; Loyevsky M.; Gordeuk V.; Kark J. A.; Castro O.; Rana S.; Apprey V.; Kurantsin-Mills J. Blood 2003, 102, 357. [DOI] [PubMed] [Google Scholar]
- Fiedler A.; Reinert T.; Morawski M.; Bruckner G.; Arendt T.; Butz T. Nucl. Instrum. Meth. B 2007, 260, 153. [Google Scholar]
- Lyons T. J.; Eide D. J. In Biological inorganic chemistry: structure and reactivity; Bertini I., Gray H., Stiefel E., Valentine J. S., Eds.; University Science Books: Mill Valley, CA, 2007. [Google Scholar]
- Hoyer W.; Cherny D.; Subramaniam V.; Jovin T. M. Biochemistry 2004, 43, 16233. [DOI] [PubMed] [Google Scholar]
- Uversky V. N.; Dunker A. K. Biochim. Biophys. Acta 2010, 1804, 1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao A. H.; Crick S. L.; Vitalis A.; Chicoine C. L.; Pappu R. V. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 8183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller-Spath S.; Soranno A.; Hirschfeld V.; Hofmann H.; Ruegger S.; Reymond L.; Nettels D.; Schuler B. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 14609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao A. H.; Lyle N.; Pappu R. V. Biochem. J. 2013, 449, 307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobley G. E.; Bremner D. M.; Holtrop G.; Johnstone A. M.; Maloney C. Br. J. Nutr. 2007, 97, 1099. [DOI] [PubMed] [Google Scholar]
- Zheng R. L.; Jenkins T. M.; Craigie R. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 13659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky V. N.; Gillespie J. R.; Millett I. S.; Khodyakova A. V.; Vasilenko R. N.; Vasiliev A. M.; Rodionov I. L.; Kozlovskaya G. D.; Dolgikh D. A.; Fink A. L.; Doniach S.; Permyakov E. A.; Abramov V. M. Biochem. Biophys. Res. Commun. 2000, 267, 663. [DOI] [PubMed] [Google Scholar]
- Yi S. L.; Boys B. L.; Brickenden A.; Konermann L.; Choy W. Y. Biochemistry 2007, 46, 13120. [DOI] [PubMed] [Google Scholar]
- Park H. J.; Park I. Y.; Kim E. J.; Youn B. Y.; Fields K.; Dunker A. K.; Kang C. H. J. Biol. Chem. 2004, 279, 18026. [DOI] [PubMed] [Google Scholar]
- Breydo L.; Uversky V. N. Metallomics 2011, 3, 1163. [DOI] [PubMed] [Google Scholar]
- Leal S. S.; Botelho H. M.; Gomes C. M. Coord. Chem. Rev. 2012, 256, 2253. [Google Scholar]
- Kepp K. P. Chem. Rev. 2012, 112, 5193. [DOI] [PubMed] [Google Scholar]
- Bennett B. D.; Kimball E. H.; Gao M.; Osterhout R.; Van Dien S. J.; Rabinowitz J. D. Nat. Chem. Biol. 2009, 5, 593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayley S.; Lewis B. A.; Guttman H. J.; Record M. T. Jr. J. Mol. Biol. 1991, 222, 281. [DOI] [PubMed] [Google Scholar]
- Ostergaard H.; Tachibana C.; Winther J. R. J. Cell Biol. 2004, 166, 337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ytting C. K.; Fuglsang A. T.; Hiltunen J. K.; Kastaniotis A. J.; Ozalp V. C.; Nielsen L. J.; Olsen L. F. Integr. Biol. 2012, 4, 99. [DOI] [PubMed] [Google Scholar]
- Wishart D. S.; Jewison T.; Guo A. C.; Wilson M.; Knox C.; Liu Y. F.; Djoumbou Y.; Mandal R.; Aziat F.; Dong E.; Bouatra S.; Sinelnikov I.; Arndt D.; Xia J. G.; Liu P.; Yallou F.; Bjorndahl T.; Perez-Pineiro R.; Eisner R.; Allen F.; Neveu V.; Greiner R.; Scalbert A. Nucleic Acids Res. 2013, 41, D801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegg A. E.; Mccann P. P. Am. J. Physiol. 1982, 243, C212. [DOI] [PubMed] [Google Scholar]
- Nakamura C.; Yasumoto E.; Nakano K.; Nakayachi T.; Hashimoto K.; Kusama K.; Fukuda M.; Sakashita H.; Shirahata A.; Sakagami H. Anticancer Res. 2003, 23, 4797. [PubMed] [Google Scholar]
- Minois N.; Carmona-Gutierrez D.; Madeo F. Aging-US 2011, 3, 716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabor C. W.; Tabor H. Mirobiol. Rev. 1985, 49, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S.; Kusamaeguchi K.; Kobayashi H.; Igarashi K. J. Biol. Chem. 1991, 266, 20803. [PubMed] [Google Scholar]
- Antony T.; Hoyer W.; Cherny D.; Heim G.; Jovin T. M.; Subramaniam V. J. Biol. Chem. 2003, 278, 3235. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay A.; Saxena K.; Kasturia N.; Dalal V.; Bhatt N.; Rajkumar A.; Maity S.; Sengupta S.; Chakraborty K. Nat. Chem. Biol. 2012, 8, 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endicott J. A.; Noble M. E.; Johnson L. N. Annu. Rev. Biochem. 2012, 81, 587. [DOI] [PubMed] [Google Scholar]
- Hasted J. B.; Ritson D. M.; Collie C. H. J. Chem. Phys. 1948, 16, 1. [Google Scholar]
- Aaron M. W.; Grant E. H. Trans. Faraday Soc. 1963, 59, 85. [Google Scholar]
- Aaron M. W.; Grant E. H. Trans. Faraday Soc. 1967, 63, 2177. [Google Scholar]
- Suzuki M.; Shigematsu J.; Fukunishi Y.; Kodama T. J. Phys. Chem. B 1997, 101, 3839. [Google Scholar]
- Asami K.; Hanai T.; Koizumi N. Biophys. J. 1980, 31, 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai W.; Zhao K. S.; Asami K. Biophys. Chem. 2006, 122, 136. [DOI] [PubMed] [Google Scholar]
- Castellarnau M.; Errachid A.; Madrid C.; Juarez A.; Samitier J. Biophys. J. 2006, 91, 3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asami K.; Takahashi Y.; Takashima S. Biochim. Biophys. Acta 1989, 1010, 49. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Wang X. B.; Holzel R.; Becker F. F.; Gascoyne P. R. C. Phys. Med. Biol. 1995, 40, 1789. [DOI] [PubMed] [Google Scholar]
- Becker F. F.; Wang X. B.; Huang Y.; Pethig R.; Vykoukal J.; Gascoyne P. R. C. Proc. Natl. Acad. Sci. U.S.A. 1995, 92, 860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L. Q.; Yung L. Y. L.; Lim K. M.. Biomicrofluidics 2012, 6. 14113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.; Foley K.; Shan X.; Wang S. P.; Eaton S.; Nagaraj V. J.; Wiktor P.; Patel U.; Tao N. J. Nat. Chem. 2011, 3, 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasmal D. K.; Ghosh S.; Das A. K.; Bhattacharyya K. Langmuir 2013, 29, 2289. [DOI] [PubMed] [Google Scholar]
- Pielak G. J.; Li C. G.; Miklos A. C.; Schlesinger A. P.; Slade K. M.; Wang G. F.; Zigoneanu I. G. Biochemistry 2009, 48, 226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey J. R.; Grinstein S.; Orlowski J. Nat. Rev. Mol. Cell Biol. 2010, 11, 50. [DOI] [PubMed] [Google Scholar]
- Roos A.; Boron W. F. Physiol. Rev. 1981, 61, 296. [DOI] [PubMed] [Google Scholar]
- Poznanski J.; Szczesny P.; Ruszczynska K.; Zielenkiewicz P.; Paczek L. Biochem. Bioph. Res. Co. 2013, 430, 741. [DOI] [PubMed] [Google Scholar]
- Slonczewski J. L.; Rosen B. P.; Alger J. R.; Macnab R. M. Proc. Natl. Acad. Sci. U.S.A. 1981, 78, 6271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilberstein D.; Agmon V.; Schuldiner S.; Padan E. J. Bacteriol. 1984, 158, 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilks J. C.; Slonczewski J. L. J. Bacteriol. 2007, 189, 5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slonczewski J. L.; Fujisawa M.; Dopson M.; Krulwich T. A. Adv. Microb. Physiol. 2009, 55, 1. [DOI] [PubMed] [Google Scholar]
- Llopis J.; McCaffery J. M.; Miyawaki A.; Farquhar M. G.; Tsien R. Y. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink A. L.; Calciano L. J.; Goto Y.; Kurotsu T.; Palleros D. R. Biochemistry 1994, 33, 12504. [DOI] [PubMed] [Google Scholar]
- Smith M. D.; Jelokhani-Niaraki M. Methods Mol. Biol. 2012, 896, 223. [DOI] [PubMed] [Google Scholar]
- Munishkina L. A.; Fink A. L.; Uversky V. N. J. Mol. Biol. 2004, 342, 1305. [DOI] [PubMed] [Google Scholar]
- Konno T.; Tanaka N.; Kataoka M.; Takano E.; Maki M. Biochim. Biophys. Acta 1997, 1342, 73. [DOI] [PubMed] [Google Scholar]
- Uversky V. N.; Gillespie J. R.; Millett I. S.; Khodyakova A. V.; Vasiliev A. M.; Chernovskaya T. V.; Vasilenko R. N.; Kozovskaya G. D.; Dolgikh D. A.; Fink A. L.; Doniach S.; Abramov V. M. Biochemistry 1999, 38, 15009. [DOI] [PubMed] [Google Scholar]
- Uversky V. N.; Eliezer D. Curr. Protein Pept. Sci. 2009, 10, 483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K. P.; Weinstock D. S.; Narayanan C.; Levy R. M.; Baum J. J. Mol. Biol. 2009, 391, 784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClendon S.; Rospigliosi C. C.; Eliezer D. Protein Sci. 2009, 18, 1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasnin M.; Stadler A.; Tehei M.; Zaccai G. Phys. Chem. Chem. Phys. 2010, 12, 10154. [DOI] [PubMed] [Google Scholar]
- Qvist J.; Persson E.; Mattea C.; Halle B. Faraday Discuss. 2009, 141, 131. [DOI] [PubMed] [Google Scholar]
- Kuimova M. K. Phys. Chem. Chem. Phys. 2012, 14, 12671. [DOI] [PubMed] [Google Scholar]
- King J. T.; Arthur E. J.; Brooks C. L.; Kubarych K. J. J. Am. Chem. Soc. 2014, 136, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins H. D. B.; Marcus Y. Chem. Rev. 1995, 95, 2695. [Google Scholar]
- Banipal T. S.; Kaur D.; Banipal P. K. J. Chem. Eng. Data 2004, 49, 1236. [Google Scholar]
- Zimmerman S. B.; Trach S. O. J. Mol. Biol. 1991, 222, 599. [DOI] [PubMed] [Google Scholar]
- Conlon I.; Raff M. J. Biol. 2003, 2, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeskind B. J.; Jordan C. D.; Timp W.; Trapani L.; Waller G.; Horodincu V.; Ehrlich D. J.; Matsudaira P. Nat. Methods 2007, 4, 567. [DOI] [PubMed] [Google Scholar]
- Cheung M. C.; LaCroix R.; McKenna B. K.; Liu L.; Winkelman J.; Ehrlich D. J. Cytom. Part A 2013, 83, 540. [DOI] [PubMed] [Google Scholar]
- Ellis R. J. Trends Biochem. Sci. 2001, 26, 597. [DOI] [PubMed] [Google Scholar]
- Minton A. P. J. Biol. Chem. 2001, 276, 10577. [DOI] [PubMed] [Google Scholar]
- Minton A. P.; Wilf J. Biochemistry 1981, 20, 4821. [DOI] [PubMed] [Google Scholar]
- Sarkar M.; Li C.; Pielak G. J. Biophys. Rev. 2013, 5, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall D.; Minton A. P. Biochim. Biophys. Acta 2003, 1649, 127. [DOI] [PubMed] [Google Scholar]
- Zhou H. X.; Rivas G. N.; Minton A. P. Annu. Rev. Biophys. 2008, 37, 375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Sarkar M.; Smith A. E.; Krois A. S.; Pielak G. J. J. Am. Chem. Soc. 2012, 134, 16614. [DOI] [PubMed] [Google Scholar]
- Neumaier S.; Büttner M.; Bachmann A.; Kiefhaber T. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 20988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timasheff S. N. Annu. Rev. Biophys. Biomol. Struct. 1993, 22, 67. [DOI] [PubMed] [Google Scholar]
- Teufel D. P.; Johnson C. M.; Lum J. K.; Neuweiler H. J. Mol. Biol. 2011, 409, 250. [DOI] [PubMed] [Google Scholar]
- Stradner A.; Sedgwick H.; Cardinaux F.; Poon W. C. K.; Egelhaaf S. U.; Schurtenberger P. Nature 2004, 432, 492. [DOI] [PubMed] [Google Scholar]
- McGuffee S. R.; Elcock A. H.. PLoS Comput. Biol. 2010, 6, e1000694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada R.; Tochio N.; Kigawa T.; Sugita Y.; Feig M. J. Am. Chem. Soc. 2013, 135, 3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlesinger A. P.; Wang Y. Q.; Tadeo X.; Millet O.; Pielak G. J. Am. Chem. Soc. 2011, 133, 8082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M.; Xu Y.; Gruebele M. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 17863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman I.; Gelman H.; Tai J.; Gruebele M. J. Mol. Biol. 2014, 426, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar A.; Girdhar K.; Singh D.; Gelman H.; Ebbinghaus S.; Gruebele M. Biophys. J. 2011, 101, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth A. J.; Gruebele M. Bioessays 2013, 35, 984. [DOI] [PubMed] [Google Scholar]
- Wirth A. J.; Platkov M.; Gruebele M. J. Am. Chem. Soc. 2013, 135, 19215. [DOI] [PubMed] [Google Scholar]
- Uversky V. N. Protein Sci. 2002, 11, 739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky V. N.; Santambrogio C.; Brocca S.; Grandori R. FEBS Lett. 2012, 586, 70. [DOI] [PubMed] [Google Scholar]
- Qu Y.; Bolen D. W. Biophys. Chem. 2002, 101–102, 155. [DOI] [PubMed] [Google Scholar]
- Roque A.; Ponte I.; Suau P. Biophys. J. 2007, 93, 2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong J. A.; Gierasch L. M. J. Am. Chem. Soc. 2010, 132, 10445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soranno A.; Koenig I.; Borgia M. B.; Hofmann H.; Zosel F.; Nettels D.; Schuler B. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 4874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaugh S. L.; Lumb K. J. Biomacromolecules 2001, 2, 538. [DOI] [PubMed] [Google Scholar]
- Szasz C. S.; Alexa A.; Toth K.; Rakacs M.; Langowski J.; Tompa P. Biochemistry 2011, 50, 5834. [DOI] [PubMed] [Google Scholar]
- Cino E. A.; Karttunen M.; Choy W. Y. PLoS One 2012, 7, e49876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotomayor-Perez A. C.; Subrini O.; Hessel A.; Ladant D.; Chenal A. J. Am. Chem. Soc. 2013, 135, 11929. [DOI] [PubMed] [Google Scholar]
- Goldenberg D. P.; Argyle B. Biophys. J. 2014, 106, 905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNulty B. C.; Young G. B.; Pielak G. J. J. Mol. Biol. 2006, 355, 893. [DOI] [PubMed] [Google Scholar]
- Bodart J. F.; Wieruszeski J. M.; Amniai L.; Leroy A.; Landrieu I.; Rousseau-Lescuyer A.; Vilain J. P.; Lippens G. J. Magn. Reson. 2008, 192, 252. [DOI] [PubMed] [Google Scholar]
- Bertini I.; Felli I. C.; Gonnelli L.; Kumar M. V. V; Pierattelli R. Angew. Chem., Int. Ed. 2011, 50, 2339. [DOI] [PubMed] [Google Scholar]
- Binolfi A.; Theillet F. X.; Selenko P. Biochem. Soc. Trans. 2012, 40, 950. [DOI] [PubMed] [Google Scholar]
- Kozer N.; Kuttner Y. Y.; Haran G.; Schreiber G. Biophys. J. 2007, 92, 2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holyst R.; Bielejewska A.; Szymanski J.; Wilk A.; Patkowski A.; Gapinski J.; Zywocinski A.; Kalwarczyk T.; Kalwarczyk E.; Tabaka M.; Ziebacz N.; Wieczorek S. A. Phys. Chem. Chem. Phys. 2009, 11, 9025. [DOI] [PubMed] [Google Scholar]
- Kasaai M. R. Carbohyd. Polym. 2012, 88, 373. [Google Scholar]
- Wang Y. Q.; Li C. G.; Pielak G. J. J. Am. Chem. Soc. 2010, 132, 9392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goins A. B.; Sanabria H.; Waxham M. N. Biophys. J. 2008, 95, 5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalwarczyk T.; Ziebacz N.; Bielejewska A.; Zaboklicka E.; Koynov K.; Szymanski J.; Wilk A.; Patkowski A.; Gapinski J.; Butt H. J.; Holyst R. Nano Lett. 2011, 11, 2157. [DOI] [PubMed] [Google Scholar]
- Grupi A.; Minton A. P. Anal. Chem. 2012, 84, 10732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y. Y.; An L. J.; Wang Z. G. Macromolecules 2013, 46, 5731. [Google Scholar]
- Zhong D. P.; Pal S. K.; Zewail A. H. Chem. Phys. Lett. 2011, 503, 1. [Google Scholar]
- Roosen-Runge F.; Hennig M.; Zhang F.; Jacobs R. M.; Sztucki M.; Schober H.; Seydel T.; Schreiber F. Proc. Natl. Acad. Sci. U.S.A. 2010, 108, 11815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss M.; Elsner M.; Kartberg F.; Nilsson T. Biophys. J. 2004, 87, 3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dix J. A.; Verkman A. S. Annu. Rev. Biophys. 2008, 37, 247. [DOI] [PubMed] [Google Scholar]
- Mika J. T.; Poolman B. Curr. Opin. Biotechnol. 2011, 22, 117. [DOI] [PubMed] [Google Scholar]
- Kumar M.; Mommer M. S.; Sourjik V. Biophys. J. 2010, 98, 552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nenninger A.; Mastroianni G.; Mullineaux C. W. J. Bacteriol. 2010, 192, 4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohli I.; Mukhopadhyay A. Macromolecules 2012, 45, 6143. [Google Scholar]
- Ochab-Marcinek A.; Wieczorek S. A.; Ziebacz N.; Holyst R. Soft Matter 2012, 8, 11173. [Google Scholar]
- Hofling F.; Franosch T. Rep. Prog. Phys. 2013, 76, 046602. [DOI] [PubMed] [Google Scholar]
- Kalwarczyk T.; Tabaka M.; Holyst R. Bioinformatics 2012, 28, 2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Uversky V. N.; Fink A. L. Biochemistry 2001, 40, 11604. [DOI] [PubMed] [Google Scholar]
- Mukrasch M. D.; Bibow S.; Korukottu J.; Jeganathan S.; Biernat J.; Griesinger C.; Mandelkow E.; Zweckstetter M. PLoS Biol. 2009, 7, 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn J. E.; Millett I. S.; Jacob J.; Zagrovic B.; Dillon T. M.; Cingel N.; Dothager R. S.; Seifert S.; Thiyagarajan P.; Sosnick T. R.; Hasan M. Z.; Pande V. S.; Ruczinski I.; Doniach S.; Plaxco K. W. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 12491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann H.; Soranno A.; Borgia A.; Gast K.; Nettels D.; Schuler B. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 16155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziebacz N.; Wieczorek S. A.; Kalwarczyk T.; Fiakowski M.; Holyst R. Soft Matter 2011, 7, 7181. [Google Scholar]
- Kuttner Y. Y.; Kozer N.; Segal E.; Schreiber G.; Haran G. J. Am. Chem. Soc. 2005, 127, 15138. [DOI] [PubMed] [Google Scholar]
- Ye Y.; Liu X.; Zhang Z.; Wu Q.; Jiang B.; Jiang L.; Zhang X.; Liu M.; Pielak G. J.; Li C. Chem.—Eur. J. 2013, 19, 12705. [DOI] [PubMed] [Google Scholar]
- Partikian A.; Olveczky B.; Swaminathan R.; Li Y. X.; Verkman A. S. J. Cell Biol. 1998, 140, 821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayel M. J.; Hom E. F. Y.; Verkman A. S. Biophys. J. 1999, 76, 2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkman A. S. Trends Biochem. Sci. 2002, 27, 27. [DOI] [PubMed] [Google Scholar]
- Neuweiler H.; Lollmann M.; Doose S.; Sauer M. J. Mol. Biol. 2007, 365, 856. [DOI] [PubMed] [Google Scholar]
- Wang Q. H.; Zhuravleva A.; Gierasch L. M. Biochemistry 2011, 50, 9225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley P. B.; Chow E.; Papkovskaia T. ChemBioChem 2011, 12, 1043. [DOI] [PubMed] [Google Scholar]
- Barnes C. O.; Monteith W. B.; Pielak G. J. ChemBioChem 2011, 12, 390. [DOI] [PubMed] [Google Scholar]
- Waudby C. A.; Camilloni C.; Fitzpatrick A. W.; Cabrita L. D.; Dobson C. M.; Vendruscolo M.; Christodoulou J. PLoS One 2013, 8, e72286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seksek O.; Biwersi J.; Verkman A. S. J. Cell Biol. 1997, 138, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bogaart G.; Hermans N.; Krasnikov V.; Poolman B. Mol. Microbiol. 2007, 64, 858. [DOI] [PubMed] [Google Scholar]
- Parry B. R.; Surovtsev I. V.; Cabeen M. T.; O’Hem C. S.; Dufresne E. R.; Jacobs-Wagner C. Cell 2014, 156, 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacs G. L.; Haggie P.; Seksek O.; Lechardeur D.; Freedman N.; Verkman A. S. J. Biol. Chem. 2000, 275, 1625. [DOI] [PubMed] [Google Scholar]
- Llopis P. M.; Jackson A. F.; Sliusarenko O.; Surovtsev I.; Heinritz J.; Emonet T.; Jacobs-Wagner C. Nature 2010, 466, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer J. Microbiol. Mol. Biol. Rev. 2011, 75, 491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srere P. A. Trends Biochem. Sci. 1980, 5, 120. [Google Scholar]
- Appelhans T.; Richter C. P.; Wilkens V.; Hess S. T.; Piehler J.; Busch K. B. Nano Lett. 2012, 12, 610. [DOI] [PubMed] [Google Scholar]
- Herrmann J. M.; Riemer J. Antioxid. Redox Sign. 2010, 13, 1341. [DOI] [PubMed] [Google Scholar]
- Dieteren C. E. J.; Gielen S. C. A. M.; Nijtmans L. G. J.; Smeitink J. A. M.; Swarts H. G.; Brock R.; Willems P. H. G. M.; Koopman W. J. H. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 8657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olveczky B. P.; Verkman A. S. Biophys. J. 1998, 74, 2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmo-Fonseca M.; Platani M.; Swedlow J. R. Trends Cell Biol. 2002, 12, 491. [DOI] [PubMed] [Google Scholar]
- Zustiak S. P.; Nossal R.; Sackett D. L. Biophys. J. 2011, 101, 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav S.; Laue T. M.; Kalonia D. S.; Singh S. N.; Shire S. J. Mol. Pharm. 2012, 9, 791. [DOI] [PubMed] [Google Scholar]
- Latham M. P.; Kay L. E. J. Biomol. NMR 2013, 55, 239. [DOI] [PubMed] [Google Scholar]
- Phillip Y.; Schreiber G. FEBS Lett. 2013, 587, 1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guigas G.; Weiss M. Biophys. J. 2008, 94, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters R. A. Trans. Faraday Soc. 1930, 26, 797. [Google Scholar]
- Spitzer J.; Poolman B. Microbiol. Mol. Biol. Rev. 2009, 73, 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer J.; Poolman B. FEBS Lett. 2013, 587, 2094. [DOI] [PubMed] [Google Scholar]
- Vale R. D. Cell 2003, 112, 467. [DOI] [PubMed] [Google Scholar]
- Boryskina O. P.; Tkachenko M. Y.; Shestopalova A. V. Biopolym. Cell 2010, 26, 360. [Google Scholar]
- Carrivain P.; Cournac A.; Lavelle C.; Lesne A.; Mozziconacci J.; Paillusson F.; Signon L.; Victor J.-M.; Barbi M. Soft Matter 2012, 8, 9285. [Google Scholar]
- Heddi B.; Foloppe N.; Hantz E.; Hartmann B. J. Mol. Biol. 2007, 368, 1403. [DOI] [PubMed] [Google Scholar]
- Luger K.; Mader A. W.; Richmond R. K.; Sargent D. F.; Richmond T. J. Nature 1997, 389, 251. [DOI] [PubMed] [Google Scholar]
- Pepenella S.; Murphy K. J.; Hayes J. J.. Chromosoma 2013, in press [DOI] [PMC free article] [PubMed]
- Kouzarides T. Cell 2007, 128, 693. [DOI] [PubMed] [Google Scholar]
- Bigay J.; Antonny B. Dev. Cell 2012, 23, 886. [DOI] [PubMed] [Google Scholar]
- Klose C.; Surma M. A.; Simons K. Curr. Opin. Cell Biol. 2013, 25, 406. [DOI] [PubMed] [Google Scholar]
- Lee J.; Culyba E. K.; Powers E. T.; Kelly J. W. Nat. Chem. Biol. 2011, 7, 602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivotto M.; Arcangeli A.; Carla M.; Wanke E. Bioessays 1996, 18, 495. [DOI] [PubMed] [Google Scholar]
- McLaughlin S.; Aderem A. Trends Biochem. Sci. 1995, 20, 272. [DOI] [PubMed] [Google Scholar]
- Thelen M.; Rosen A.; Nairn A. C.; Aderem A. Nature 1991, 351, 320. [DOI] [PubMed] [Google Scholar]
- Cho W. H.; Stahelin R. V. Annu. Rev. Biophys. Biomol. Struct. 2005, 34, 119. [DOI] [PubMed] [Google Scholar]
- Heymann J. B.; Zakharov S. D.; Zhang Y.-L.; Cramer W. A. Biochemistry 1996, 35, 2717. [DOI] [PubMed] [Google Scholar]
- Leventis P. A.; Grinstein S. Annu. Rev. Biophys 2010, 39, 407. [DOI] [PubMed] [Google Scholar]
- Mulgrewnesbitt A.; Diraviyam K.; Wang J.; Singh S.; Murray P.; Li Z.; Rogers L.; Mirkovic N.; Murray D. Biochim. Biophys. Acta 2006, 1761, 812. [DOI] [PubMed] [Google Scholar]
- Michel V.; Bakovic M. Biol. Cell 2007, 99, 129. [DOI] [PubMed] [Google Scholar]
- Lopez D.; Kolter R. Genes Dev. 2010, 24, 1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto K.; Kusaka J.; Nishibori A.; Hara H. Mol. Microbiol. 2006, 61, 1110. [DOI] [PubMed] [Google Scholar]
- van Meer G.; de Kroon A. I. P. M. J. Cell Sci. 2010, 124, 5. [DOI] [PubMed] [Google Scholar]
- Vanounou S.; Parola A. H.; Fishov I. Mol. Microbiol. 2003, 49, 1067. [DOI] [PubMed] [Google Scholar]
- Ben-Tal N.; Hoing B.; Miller C.; McLaughlin S. Biophys. J. 1997, 73, 1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Angelo M. A.; Hetzer M. W. Trends Cell Biol. 2008, 18, 456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raices M.; D’Angelo M. A. Nat. Rev. Mol. Cell Biol. 2012, 13, 687. [DOI] [PubMed] [Google Scholar]
- Frey S.; Gorlich D. Cell 2007, 130, 512. [DOI] [PubMed] [Google Scholar]
- Labokha A. A.; Gradmann S.; Frey S.; Hulsmann B. B.; Urlaub H.; Baldus M.; Gorlich D. EMBO J. 2013, 32, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denning D. P.; Patel S. S.; Uversky V.; Fink A. L.; Rexach M. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ader C.; Frey S.; Maas W.; Schmidt H. B.; Gorlich D.; Baldus M. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 6281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulsmann B. B.; Labokha A. A.; Gorlich D. Cell 2012, 150, 738. [DOI] [PubMed] [Google Scholar]
- McConkey E. H. Proc. Natl. Acad. Sci. U.S.A. 1982, 79, 3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srere P. A. Trends Biochem. Sci. 2000, 25, 150. [DOI] [PubMed] [Google Scholar]
- Cayley S.; Record M. T. Jr. J. Mol. Recognit. 2004, 17, 488. [DOI] [PubMed] [Google Scholar]
- Minton A. P. J. Cell Sci. 2006, 119, 2863. [DOI] [PubMed] [Google Scholar]
- Spitzer J. J.; Poolman B. Trends Biochem. Sci. 2005, 30, 536. [DOI] [PubMed] [Google Scholar]
- Link A. J.; Robison K.; Church G. M. Electrophoresis 1997, 18, 1259. [DOI] [PubMed] [Google Scholar]
- Laue T. M. J. Mol. Recognit. 2012, 3, 165. [DOI] [PubMed] [Google Scholar]
- Pastore A.; Temussi P. A. Curr. Opin. Struct. Biol. 2012, 22, 30. [DOI] [PubMed] [Google Scholar]
- Buljan M.; Chalancon G.; Dunker A. K.; Bateman A.; Balaji S.; Fuxreiter M.; Babu M. M. Curr. Opin. Struct. Biol. 2013, 23, 443. [DOI] [PubMed] [Google Scholar]
- Uversky V. N. Eur. J. Biochem. 2002, 269, 2. [DOI] [PubMed] [Google Scholar]
- Davies J. E.; Sarkar S.; Rubinsztein D. C. BMC Biochem 2007, 8Suppl 1S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lashuel H. A.; Overk C. R.; Oueslati A.; Masliah E. Nat. Rev. Neurosci. 2013, 14, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layfield R.; Cavey J. R.; Lowe J. Ageing Res. Rev. 2003, 2, 343. [DOI] [PubMed] [Google Scholar]
- Tseng B. P.; Green K. N.; Chan J. L.; Blurton-Jones M.; LaFerla F. M. Neurobiol. Aging 2008, 29, 1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldassarre G.; Belletti B.; Nicoloso M. S.; Schiappacassi M.; Vecchione A.; Spessotto P.; Morrione A.; Canzonieri V.; Colombatti A. Cancer Cell 2005, 7, 51. [DOI] [PubMed] [Google Scholar]
- Grimmler M.; Wang Y.; Mund T.; Cilensek Z.; Keidel E. M.; Waddell M. B.; Jakel H.; Kullmann M.; Kriwacki R. W.; Hengst L. Cell 2007, 128, 269. [DOI] [PubMed] [Google Scholar]
- Gsponer J.; Futschik M. E.; Teichmann S. A.; Babu M. M. Science 2008, 322, 1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards Y. J. K.; Lobley A. E.; Pentony M. M.; Jones D. T.. Genome Biol. 2009, 10, R50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paliy O.; Gargac S. M.; Cheng Y.; Uversky V. N.; Dunker A. K. J. Proteome Res. 2008, 7, 2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M. A.; Olivas W. M. Wiley Interdiscip. Rev.-RNA 2011, 2, 471. [DOI] [PubMed] [Google Scholar]
- Wickens M.; Bernstein D. S.; Kimble J.; Parker R. Trends Genet. 2002, 18, 150. [DOI] [PubMed] [Google Scholar]
- Brown C. J.; Takayama S.; Campen A. M.; Vise P.; Marshall T. W.; Oldfield C. J.; Williams C. J.; Dunker A. K. J. Mol. Evol. 2002, 55, 104. [DOI] [PubMed] [Google Scholar]
- Lin Y. S.; Hsu W. L.; Hwang J. K.; Li W. H. Mol. Biol. Evol. 2007, 24, 1005. [DOI] [PubMed] [Google Scholar]
- Nilsen T. W.; Graveley B. R. Nature 2010, 463, 457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegyi H.; Kalmar L.; Horvath T.; Tompa P. Nucleic Acids Res. 2011, 39, 1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero P. R.; Zaidi S.; Fang Y. Y.; Uversky V. N.; Radivojac P.; Oldfield C. J.; Cortese M. S.; Sickmeier M.; LeGall T.; Obradovic Z.; Dunker A. K. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 8390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs E.; Tompa P.; Liliom K.; Kalmar L. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joerger A. C.; Fersht A. R. Annu. Rev. Biochem. 2008, 77, 557. [DOI] [PubMed] [Google Scholar]
- Oldfield C. J.; Meng J.; Yang J. Y.; Yang M. Q.; Uversky V. N.; Dunker A. K.. BMC Genomics 2008, 9. [DOI] [PMC free article] [PubMed]
- Khoury M. P.; Bourdon J. C. Cold Spring Harbor Perspect. Med. 2011, 2, 453. [Google Scholar]
- Bourdon J. C.; Fernandes K.; Murray-Zmijewski F.; Liu G.; Diot A.; Xirodimas D. P.; Saville M. K.; Lane D. P. Genes Dev. 2005, 19, 2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng E. H. Y.; Kirsch D. G.; Clem R. J.; Ravi R.; Kastan M. B.; Bedi A.; Ueno K.; Hardwick J. M. Science 1997, 278, 1966. [DOI] [PubMed] [Google Scholar]
- Youle R. J.; Strasser A. Nat. Rev. Mol. Cell Biol. 2008, 9, 47. [DOI] [PubMed] [Google Scholar]
- Brown M. S.; Goldstein J. L. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 11041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cegielska A.; Gietzen K. F.; Rivers A.; Virshup D. M. J. Biol. Chem. 1998, 273, 1357. [DOI] [PubMed] [Google Scholar]
- Stoven S.; Ando I.; Kadalayil L.; Engstrom Y.; Hultmark D. EMBO Rep. 2000, 1, 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns C. S.; Aronoff-Spencer E.; Legname G.; Prusiner S. B.; Antholine W. E.; Gerfen G. J.; Peisach J.; Millhauser G. L. Biochemistry 2003, 42, 6794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J.; Kong Q. Prion 2012, 6, 453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillot-Sestier M. V.; Checler F. Neurodegener. Dis. 2012, 10, 294. [DOI] [PubMed] [Google Scholar]
- Guillot-Sestier M. V.; Sunyach C.; Druon C.; Scarzello S.; Checler F. J. Biol. Chem. 2009, 284, 35973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westergard L.; Turnbaugh J. A.; Harris D. A. J. Biol. Chem. 2011, 286, 44234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalivaeva N. N.; Turner A. J. FEBS Lett. 2013, 587, 2046. [DOI] [PubMed] [Google Scholar]
- Fontana A.; Polverino de Laureto P.; De Filippis V.; Scaramella E.; Zambonin M. Fold. Des. 1997, 2, R17. [DOI] [PubMed] [Google Scholar]
- Tsvetkov P.; Asher G.; Paz A.; Reuven N.; Sussman J. L.; Silman I.; Shaul Y. Proteins: Struct., Funct., Bioinf. 2008, 70, 1357. [DOI] [PubMed] [Google Scholar]
- Pace C. N.; Barrett A. J. Biochem. J. 1984, 219, 411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madala P. K.; Tyndall J. D.; Nall T.; Fairlie D. P. Chem. Rev. 2010, 110, PR1. [DOI] [PubMed] [Google Scholar]
- Lee D. H.; Goldberg A. L. Trends Cell Biol. 1998, 8, 397. [DOI] [PubMed] [Google Scholar]
- Rock K. L.; Gramm C.; Rothstein L.; Clark K.; Stein R.; Dick L.; Hwang D.; Goldberg A. L. Cell 1994, 78, 761. [DOI] [PubMed] [Google Scholar]
- Jung T.; Catalgol B.; Grune T. Mol. Aspects Med. 2009, 30, 191. [DOI] [PubMed] [Google Scholar]
- Schrader E. K.; Harstad K. G.; Matouschek A. Nat. Chem. Biol. 2009, 5, 815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inobe T.; Fishbain S.; Prakash S.; Matouschek A. Nat. Chem. Biol. 2011, 7, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erales J.; Coffino P.. Biochim. Biophys. Acta 2013, in press [DOI] [PMC free article] [PubMed]
- Ciechanover A.; Stanhill A.. Biochim. Biophys. Acta 2013, in press
- Fukui T.; Eguchi T.; Atomi H.; Imanaka T. J. Bacteriol. 2002, 184, 3689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao J. H.; Kuo C. I.; Huang Y. Y.; Lin Y. C.; Lin Y. C.; Yang C. Y.; Wu W. L.; Chang W. H.; Liaw Y. C.; Lin L. H.; Chang C. I.; Wu S. H. PLoS One 2012, 7, e40226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel T.; Baumeister W. Nat. Struct. Biol. 1995, 2, 199. [DOI] [PubMed] [Google Scholar]
- Asher G.; Reuven N.; Shaul Y. Bioessays 2006, 28, 844. [DOI] [PubMed] [Google Scholar]
- Tsvetkov P.; Reuven N.; Prives C.; Shaul Y. J. Biol. Chem. 2009, 284, 26234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash S.; Tian L.; Ratliff K. S.; Lehotzky R. E.; Matouschek A. Nat. Struct. Mol. Biol. 2004, 11, 830. [DOI] [PubMed] [Google Scholar]
- Takeuchi J.; Chen H.; Coffino P. EMBO J. 2007, 26, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarcsa E.; Szymanska G.; Lecker S.; O’Connor C. M.; Goldberg A. L. J. Biol. Chem. 2000, 275, 20295. [DOI] [PubMed] [Google Scholar]
- Hwang J.; Kalejta R. F. Virology 2007, 367, 334. [DOI] [PubMed] [Google Scholar]
- Dice J. F. FASEB J. 1987, 1, 349. [DOI] [PubMed] [Google Scholar]
- Mogk A.; Schmidt R.; Bukau B. Trends Cell Biol. 2007, 17, 165. [DOI] [PubMed] [Google Scholar]
- Belle A.; Tanay A.; Bitincka L.; Shamir R.; O’Shea E. K. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 13004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa P.; Prilusky J.; Silman I.; Sussman J. L. Proteins: Struct., Funct., Bioinf. 2008, 71, 903. [DOI] [PubMed] [Google Scholar]
- Babu M. M.; van der Lee R.; de Groot N. S.; Gsponer J. Curr. Opin. Struct. Biol. 2011, 21, 432. [DOI] [PubMed] [Google Scholar]
- Hagai T.; Azia A.; Toth-Petroczy A.; Levy Y. J. Mol. Biol. 2011, 412, 319. [DOI] [PubMed] [Google Scholar]
- Yen H. C.; Xu Q.; Chou D. M.; Zhao Z.; Elledge S. J. Science 2008, 322, 918. [DOI] [PubMed] [Google Scholar]
- Sylvestersen K. B.; Young C.; Nielsen M. L. Curr. Opin. Chem. Biol. 2013, 17, 49. [DOI] [PubMed] [Google Scholar]
- Schwanhausser B.; Busse D.; Li N.; Dittmar G.; Schuchhardt J.; Wolf J.; Chen W.; Selbach M. Nature 2011, 473, 337. [DOI] [PubMed] [Google Scholar]
- Liu C. W.; Corboy M. J.; DeMartino G. N.; Thomas P. J. Science 2003, 299, 408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivas G.; Fernandez J. A.; Minton A. P. Biochemistry 1999, 38, 9379. [DOI] [PubMed] [Google Scholar]
- Rivas G.; Fernandez J. A.; Minton A. P. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma P. K.; Rakshit S.; Mitra R. K.; Pal S. K. Biochimie 2011, 93, 1424. [DOI] [PubMed] [Google Scholar]
- Minh D. D.; Chang C. E.; Trylska J.; Tozzini V.; McCammon J. A. J. Am. Chem. Soc. 2006, 128, 6006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto D. N.; Oliveira L. C.; Kondo M. Y.; Cezari M. H.; Szeltner Z.; Juhasz T.; Juliano M. A.; Polgar L.; Juliano L.; Gouvea I. E. Biol. Chem. 2010, 391, 1461. [DOI] [PubMed] [Google Scholar]
- Chandra S.; Chen X.; Rizo J.; Jahn R.; Sudhof T. C. J. Biol. Chem. 2003, 278, 15313. [DOI] [PubMed] [Google Scholar]
- Chandra S.; Gallardo G.; Fernandez-Chacon R.; Schluter O. M.; Sudhof T. C. Cell 2005, 123, 383. [DOI] [PubMed] [Google Scholar]
- de Laureto P. P.; Tosatto L.; Frare E.; Marin O.; Uversky V. N.; Fontana A. Biochemistry 2006, 45, 11523. [DOI] [PubMed] [Google Scholar]
- Pariat M.; Carillo S.; Molinari M.; Salvat C.; Debussche L.; Bracco L.; Milner J.; Piechaczyk M. Mol. Cell. Biol. 1997, 17, 2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger A.; Walczak A. M.; Wolynes P. G. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irie M.; Hata Y.; Takeuchi M.; Ichtchenko K.; Toyoda A.; Hirao K.; Takai Y.; Rosahl T. W.; Sudhof T. C. Science 1997, 277, 1511. [DOI] [PubMed] [Google Scholar]
- Sollner S.; Schober M.; Wagner A.; Prem A.; Lorkova L.; Palfey B. A.; Groll M.; Macheroux P. EMBO Rep. 2009, 10, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsvetkov P.; Reuven N.; Shaul Y. Cell Death Differ. 2010, 17, 103. [DOI] [PubMed] [Google Scholar]
- Tsvetkov P.; Reuven N.; Shaul Y. Nat. Chem. Biol. 2009, 5, 778. [DOI] [PubMed] [Google Scholar]
- Hegyi H.; Tompa P. PLoS Comput. Biol. 2008, 4, e1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettern N.; Dreiseidler M.; Tawo R.; Hohfeld J. Biol. Chem. 2010, 391, 481. [DOI] [PubMed] [Google Scholar]
- Posokhova E.; Uversky V.; Martemyanov K. A. J. Proteome Res. 2010, 9, 1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez C.; Diaz-Nido J.; Avila J. Prog. Neurobiol. 2000, 61, 133. [DOI] [PubMed] [Google Scholar]
- Baugh J. M.; Viktorova E. G.; Pilipenko E. V. J. Mol. Biol. 2009, 386, 814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saibil H. Nat. Rev. Mol. Cell Biol. 2013, 13, 630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y. E.; Hipp M. S.; Bracher A.; Hayer-Hartl M.; Hartl F. U. Annu. Rev. Biochem. 2013, 82, 323. [DOI] [PubMed] [Google Scholar]
- Hilton G. R.; Lioe H.; Stengel F.; Baldwin A. J.; Benesch J. L. Top. Curr. Chem. 2013, 328, 69. [DOI] [PubMed] [Google Scholar]
- Hartl F. U. Nat. Med. 2011, 17, 1206. [DOI] [PubMed] [Google Scholar]
- Uversky V. N. Chem. Rev. 2011, 111, 1134. [DOI] [PubMed] [Google Scholar]
- Chen D. H.; Madan D.; Weaver J.; Lin Z.; Schroder G. F.; Chiu W.; Rye H. S. Cell 2013, 153, 1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuravleva A.; Clerico E. M.; Gierasch L. M. Cell 2012, 151, 1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi R.; Sarbeng E. B.; Liu Q.; Le K. Q.; Xu X.; Xu H.; Yang J.; Wong J. L.; Vorvis C.; Hendrickson W. A.; Zhou L.; Liu Q. Nat. Struct. Mol. Biol. 2013, 20, 900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smock R. G.; Blackburn M. E.; Gierasch L. M. J. Biol. Chem. 2011, 286, 31821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swain J. F.; Dinler G.; Sivendran R.; Montgomery D. L.; Stotz M.; Gierasch L. M. Mol. Cell 2007, 26, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel M.; Mayer M. P.; Bukau B. J. Biol. Chem. 2006, 281, 38705. [DOI] [PubMed] [Google Scholar]
- Zhuravleva A.; Gierasch L. M. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 6987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basha E.; O’Neill H.; Vierling E. Trends Biochem. Sci. 2012, 37, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardwell J. C.; Jakob U. Trends Biochem. Sci. 2012, 37, 517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichmann D.; Xu Y.; Cremers C. M.; Ilbert M.; Mittelman R.; Fitzgerald M. C.; Jakob U. Cell 2012, 148, 947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foit L.; George J. S.; Zhang B. W.; Brooks C. L. 3rd; Bardwell J. C. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, E1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y. J.; Inouye M. Curr. Opin. Struct. Biol. 2008, 18, 765. [DOI] [PubMed] [Google Scholar]
- Sharma A. K.; Ali A.; Gogna R.; Singh A. K.; Pati U. PLoS One 2009, 4, e7159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky V. N. FEBS Lett. 2013, 587, 1891. [DOI] [PubMed] [Google Scholar]
- Tompa P.; Csermely P. FASEB J. 2004, 18, 1169. [DOI] [PubMed] [Google Scholar]
- Kovacs D.; Szabo B.; Pancsa R.; Tompa P. Arch. Biochem. Biophys. 2013, 531, 80. [DOI] [PubMed] [Google Scholar]
- Kovacs D.; Tompa P. Biochem. Soc. Trans. 2013, 40, 963. [DOI] [PubMed] [Google Scholar]
- Souza J. M.; Giasson B. I.; Lee V. M.; Ischiropoulos H. FEBS Lett. 2000, 474, 116. [DOI] [PubMed] [Google Scholar]
- Rekas A.; Ahn K. J.; Kim J.; Carver J. A. Proteins: Struct., Funct., Bioinf. 2012, 80, 1316. [DOI] [PubMed] [Google Scholar]
- Chakrabortee S.; Tripathi R.; Watson M.; Schierle G. S.; Kurniawan D. P.; Kaminski C. F.; Wise M. J.; Tunnacliffe A. Mol. Biosyst. 2012, 8, 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs D.; Kalmar E.; Torok Z.; Tompa P. Plant Physiol. 2008, 147, 381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucera N. J.; Hodsdon M. E.; Wolin S. L. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanyi-Nagy R.; Lavergne J. P.; Gabus C.; Ficheux D.; Darlix J. L. Nucleic Acids Res. 2008, 36, 712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs D.; Rakacs M.; Agoston B.; Lenkey K.; Semrad K.; Schroeder R.; Tompa P. FEBS Lett. 2009, 583, 88. [DOI] [PubMed] [Google Scholar]
- Jinwal U. K.; Akoury E.; Abisambra J. F.; O’Leary J. C. 3rd; Thompson A. D.; Blair L. J.; Jin Y.; Bacon J.; Nordhues B. A.; Cockman M.; Zhang J.; Li P.; Zhang B.; Borysov S.; Uversky V. N.; Biernat J.; Mandelkow E.; Gestwicki J. E.; Zweckstetter M.; Dickey C. A. FASEB J. 2013, 27, 1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karagoz G. E.; Duarte A. M. S.; Akoury E.; Ippel H.; Biernat J.; Luengo T. M.; Radli M.; Didenko T.; Nordhues B. A.; Veprintsev D. B.; Dickey C. A.; Mandelkow E.; Zweckstetter M.; Boelens R.; Madl T.; Rudiger S. G. D. Cell 2014, 156, 963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelkow E. M.; Mandelkow E. Cold Spring Harbor Perspect. Med. 2012, 2, a006247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A. D.; Scaglione K. M.; Prensner J.; Gillies A. T.; Chinnaiyan A.; Paulson H. L.; Jinwal U. K.; Dickey C. A.; Gestwicki J. E. ACS Chem. Biol. 2012, 7, 1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rekas A.; Adda C. G.; Andrew Aquilina J.; Barnham K. J.; Sunde M.; Galatis D.; Williamson N. A.; Masters C. L.; Anders R. F.; Robinson C. V.; Cappai R.; Carver J. A. J. Mol. Biol. 2004, 340, 1167. [DOI] [PubMed] [Google Scholar]
- Waudby C. A.; Knowles T. P.; Devlin G. L.; Skepper J. N.; Ecroyd H.; Carver J. A.; Welland M. E.; Christodoulou J.; Dobson C. M.; Meehan S. Biophys. J. 2010, 98, 843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruinsma I. B.; Bruggink K. A.; Kinast K.; Versleijen A. A.; Segers-Nolten I. M.; Subramaniam V.; Kuiperij H. B.; Boelens W.; de Waal R. M.; Verbeek M. M. Proteins: Struct., Funct., Bioinf. 2011, 79, 2956. [Google Scholar]
- Dedmon M. M.; Christodoulou J.; Wilson M. R.; Dobson C. M. J. Biol. Chem. 2005, 280, 14733. [DOI] [PubMed] [Google Scholar]
- Roodveldt C.; Bertoncini C. W.; Andersson A.; van der Goot A. T.; Hsu S. T.; Fernandez-Montesinos R.; de Jong J.; van Ham T. J.; Nollen E. A.; Pozo D.; Christodoulou J.; Dobson C. M. EMBO J. 2009, 28, 3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falsone S. F.; Kungl A. J.; Rek A.; Cappai R.; Zangger K. J. Biol. Chem. 2009, 284, 31190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redeker V.; Pemberton S.; Bienvenut W.; Bousset L.; Melki R. J. Biol. Chem. 2012, 287, 32630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida N.; Yagi-Utsumi M.; Motojima F.; Yoshida M.; Shimada I.; Kato K. J. Biosci. Bioeng. 2013, 116, 160. [DOI] [PubMed] [Google Scholar]
- Couturier M.; Buccellato M.; Costanzo S.; Bourhis J. M.; Shu Y.; Nicaise M.; Desmadril M.; Flaudrops C.; Longhi S.; Oglesbee M. J. Mol. Recognit. 2009, 23, 301. [DOI] [PubMed] [Google Scholar]
- Pechmann S.; Willmund F.; Frydman J. Mol. Cell 2013, 49, 411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willmund F.; del Alamo M.; Pechmann S.; Chen T.; Albanese V.; Dammer E. B.; Peng J.; Frydman J. Cell 2013, 152, 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duttler S.; Pechmann S.; Frydman J. Mol. Cell 2013, 50, 379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury G. A.; Baliban R. C.; Floudas C. A. Sci. Rep. 2011, 1, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starheim K. K.; Gevaert K.; Arnesen T. Trends Biochem. Sci. 2012, 37, 152. [DOI] [PubMed] [Google Scholar]
- Deribe Y. L.; Pawson T.; Dikic I. Nat. Struct. Mol. Biol. 2010, 17, 666. [DOI] [PubMed] [Google Scholar]
- Walsh C. T.; Garneau-Tsodikova S.; Gatto G. J. Jr. Angew. Chem., Int. Ed. 2005, 44, 7342. [DOI] [PubMed] [Google Scholar]
- Xie H.; Vucetic S.; Iakoucheva L. M.; Oldfield C. J.; Dunker A. K.; Obradovic Z.; Uversky V. N. J. Proteome Res. 2007, 6, 1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L. L.; Wan S. B.; Niu S.; Shi X. H.; Li H. P.; Cai Y. D.; Chou K. C. Biochimie 2011, 93, 489. [DOI] [PubMed] [Google Scholar]
- Radivojac P.; Vacic V.; Haynes C.; Cocklin R. R.; Mohan A.; Heyen J. W.; Goebl M. G.; Iakoucheva L. M. Proteins: Struct., Funct., Bioinf. 2009, 78, 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa I.; Nakajima Y.; Ito M.; Fukuchi S.; Homma K.; Nishikawa K. Int. J. Mol. Sci. 2010, 11, 4991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J.; Xu D. Pac. Symp. Biocomput. 2012, 17, 94. [PMC free article] [PubMed] [Google Scholar]
- Song J.; Tan H.; Perry A. J.; Akutsu T.; Webb G. I.; Whisstock J. C.; Pike R. N. PLoS One 2012, 7, e50300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obradovic Z.; Peng K.; Vucetic S.; Radivojac P.; Dunker A. K. Proteins: Struct., Funct., Bioinf. 2005, 61, 176. [DOI] [PubMed] [Google Scholar]
- Galea C. A.; High A. A.; Obenauer J. C.; Mishra A.; Park C. G.; Punta M.; Schlessinger A.; Ma J.; Rost B.; Slaughter C. A.; Kriwacki R. W. J. Proteome Res. 2009, 8, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kragelund B. B.; Jensen M. K.; Skriver K. Trends Plant Sci. 2012, 17, 625. [DOI] [PubMed] [Google Scholar]
- Stavropoulos I.; Khaldi N.; Davey N. E.; O’Brien K.; Martin F.; Shields D. C. PLoS One 2012, 7, e44389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu R.; Haas W.; Dephoure N.; Huttlin E. L.; Zhai B.; Sowa M. E.; Gygi S. P. Nat. Methods 2011, 8, 677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latham J. A.; Dent S. Y. Nat. Struct. Mol. Biol. 2007, 14, 1017. [DOI] [PubMed] [Google Scholar]
- Tan M.; Luo H.; Lee S.; Jin F.; Yang J. S.; Montellier E.; Buchou T.; Cheng Z.; Rousseaux S.; Rajagopal N.; Lu Z.; Ye Z.; Zhu Q.; Wysocka J.; Ye Y.; Khochbin S.; Ren B.; Zhao Y. Cell 2011, 146, 1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse J. P.; Gu W. Cell 2009, 137, 609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meek D. W.; Anderson C. W. Cold Spring Harbor Perspect. Biol. 2009, 1, a000950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanger D. P.; Anderton B. H.; Noble W. Trends Mol. Med. 2009, 15, 112. [DOI] [PubMed] [Google Scholar]
- Martin L.; Latypova X.; Terro F. Neurochem. Int. 2011, 58, 458. [DOI] [PubMed] [Google Scholar]
- Landry C. R.; Levy E. D.; Michnick S. W. Trends Genet. 2009, 25, 193. [DOI] [PubMed] [Google Scholar]
- Levy E. D.; Michnick S. W.; Landry C. R. Philos. Trans. R. Soc. London B. Biol. Sci. 2012, 367, 2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campen A.; Williams R. M.; Brown C. J.; Meng J.; Uversky V. N.; Dunker A. K. Protein Pept. Lett. 2008, 15, 956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe M. B. Nat. Rev. Mol. Cell Biol. 2002, 3, 177. [DOI] [PubMed] [Google Scholar]
- Ubersax J. A.; Ferrell J. E. Jr. Nat. Rev. Mol. Cell Biol. 2007, 8, 530. [DOI] [PubMed] [Google Scholar]
- Roy J.; Cyert M. S. Sci. Signal 2009, 2, re9. [DOI] [PubMed] [Google Scholar]
- Yuan H.; Marmorstein R. Biopolymers 2012, 99, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H.; Marmorstein R. J. Biol. Chem. 2012, 287, 42428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan S.; Horowitz S.; Trievel R. C. ChemBioChem 2011, 12, 254. [DOI] [PubMed] [Google Scholar]
- Couture J. F.; Collazo E.; Ortiz-Tello P. A.; Brunzelle J. S.; Trievel R. C. Nat. Struct. Mol. Biol. 2007, 14, 689. [DOI] [PubMed] [Google Scholar]
- Mok J.; Kim P. M.; Lam H. Y. K.; Piccirillo S.; Zhou X. Q.; Jeschke G. R.; Sheridan D. L.; Parker S. A.; Desai V.; Jwa M.; Cameroni E.; Niu H. Y.; Good M.; Remenyi A.; Ma J. L. N.; Sheu Y. J.; Sassi H. E.; Sopko R.; Chan C. S. M.; De Virgilio C.; Hollingsworth N. M.; Lim W. A.; Stern D. F.; Stillman B.; Andrews B. J.; Gerstein M. B.; Snyder M.; Turk B. E. Sci. Signal 2010, 3, ra12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao R. S.; Møller I. M. Biochim. Biophys. Acta 2012, 1824, 405. [DOI] [PubMed] [Google Scholar]
- Cargnello M.; Roux P. P. Microbiol. Mol. Biol. Rev. 2011, 75, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echalier A.; Endicott J. A.; Noble M. E. M. Biochim. Biophys. Acta 2010, 1804, 511. [DOI] [PubMed] [Google Scholar]
- Shi Y. Cell 2009, 139, 468. [DOI] [PubMed] [Google Scholar]
- Sacco F.; Perfetto L.; Castagnoli L.; Cesareni G. FEBS Lett. 2012, 586, 2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berndsen C. E.; Denu J. M. Curr. Opin. Struct. Biol. 2008, 18, 682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H.; Marmorstein R. Biopolymers 2013, 99, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurard-Levin Z. A.; Kilian K. A.; Kim J.; Bahr K.; Mrksich M. ACS Chem. Biol. 2010, 5, 863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathert P.; Dhayalan A.; Murakami M.; Zhang X.; Tamas R.; Jurkowska R.; Komatsu Y.; Shinkai Y.; Cheng X.; Jeltsch A. Nat. Chem. Biol. 2008, 4, 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhayalan A.; Kudithipudi S.; Rathert P.; Jeltsch A. Chem. Biol. 2011, 18, 111. [DOI] [PubMed] [Google Scholar]
- Seet B. T.; Dikic I.; Zhou M. M.; Pawson T. Nat. Rev. Mol. Cell Biol. 2006, 7, 473. [DOI] [PubMed] [Google Scholar]
- Scott J. D.; Pawson T. Science 2009, 326, 1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B. A.; Engelmann B. W.; Nash P. D. Proteomics 2012, 12, 1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdecia M. A.; Bowman M. E.; Lu K. P.; Hunter T.; Noel J. P. Nat. Struct. Biol. 2000, 7, 639. [DOI] [PubMed] [Google Scholar]
- Bustos D. M. Mol. Biosyst. 2012, 8, 178. [DOI] [PubMed] [Google Scholar]
- Liu B. A.; Jablonowski K.; Shah E. E.; Engelmann B. W.; Jones R. B.; Nash P. D. Mol. Cell. Proteomics 2012, 9, 2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khorasanizadeh S. Curr. Opin. Struct. Biol. 2011, 21, 744. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P.; Picaud S.; Mangos M.; Keates T.; Lambert J. P.; Barsyte-Lovejoy D.; Felletar I.; Volkmer R.; Muller S.; Pawson T.; Gingras A. C.; Arrowsmith C. H.; Knapp S. Cell 2012, 149, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko T.; Joshi R.; Feller S. M.; Li S. S. Cell Commun. Signal. 2012, 10, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panni S.; Montecchi-Palazzi L.; Kiemer L.; Cabibbo A.; Paoluzi S.; Santonico E.; Landgraf C.; Volkmer-Engert R.; Bachi A.; Castagnoli L.; Cesareni G. Proteomics 2011, 11, 128. [DOI] [PubMed] [Google Scholar]
- Kim J.; Daniel J.; Espejo A.; Lake A.; Krishna M.; Xia L.; Zhang Y.; Bedford M. T. EMBO Rep. 2006, 7, 397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez R.; Zhou M. M. Trends Biochem. Sci. 2011, 36, 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolov M.; Fischle W. Mol. Biosyst. 2013, 9, 182. [DOI] [PubMed] [Google Scholar]
- Bachi A.; Dalle-Donne I.; Scaloni A. Chem. Rev. 2013, 113, 596. [DOI] [PubMed] [Google Scholar]
- Bindoli A.; Rigobello M. P. Antioxid. Redox Sign. 2013, 18, 1557. [DOI] [PubMed] [Google Scholar]
- Valko M.; Leibfritz D.; Moncol J.; Cronin M. T. D.; Mazur M.; Telser J. Int. J. Biochem. Cell Biol. 2007, 39, 44. [DOI] [PubMed] [Google Scholar]
- D’Autreaux B.; Toledano M. B. Nat. Rev. Mol. Cell Biol. 2007, 8, 813. [DOI] [PubMed] [Google Scholar]
- Butterfield D. A.; Perluigi M.; Reed T.; Muharib T.; Hughes C. P.; Robinson R. A. S.; Sultana R. Antioxid. Redox Sign. 2012, 17, 1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hekimi S.FASEB J. 2011, 25, 82.2. [Google Scholar]
- Sohal R. S.; Orr W. C. Free Radical Biol. Med. 2012, 52, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winterbourn C. C.; Hampton M. B. Free Radical Biol. Med. 2008, 45, 549. [DOI] [PubMed] [Google Scholar]
- Souza J. M.; Peluffo G.; Radi R. Free Radical Biol. Med. 2008, 45, 357. [DOI] [PubMed] [Google Scholar]
- Bayden A. S.; Yakovlev V. A.; Graves P. R.; Mikkelsen R. B.; Kellogg G. E. Free Radical Biol. Med. 2011, 50, 749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abello N.; Kerstjens H. A.; Postma D. S.; Bischoff R. J. Proteome Res. 2009, 8, 3222. [DOI] [PubMed] [Google Scholar]
- Danielson S. R.; Andersen J. K. Free Radical Biol. Med. 2008, 44, 1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielson S. R.; Held J. M.; Schilling B.; Oo M.; Gibson B. W.; Andersen J. K. Anal. Chem. 2009, 81, 7823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds M. R.; Berry R. W.; Binder L. I. Biochemistry 2007, 46, 7325. [DOI] [PubMed] [Google Scholar]
- Jones L. H. Chem. Biol. 2012, 19, 1086. [DOI] [PubMed] [Google Scholar]
- Vana L.; Kanaan N. M.; Hakala K.; Weintraub S. T.; Binder L. I. Biochemistry 2011, 50, 1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghesquiere B.; Jonckheere V.; Colaert N.; Van Durme J.; Timmerman E.; Goethals M.; Schymkowitz J.; Rousseau F.; Vandekerckhove J.; Gevaert K.. Mol. Cell. Proteomics 2011, 10, e16682. [DOI] [PMC free article] [PubMed]
- Tarrago L.; Kaya A.; Weerapana E.; Marino S. M.; Gladyshev V. N. J. Biol. Chem. 2012, 287, 24448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breydo L.; Wu J. W.; Uversky V. N. Biochim. Biophys. Acta 2012, 1822, 261. [DOI] [PubMed] [Google Scholar]
- Glass C. K.; Saijo K.; Winner B.; Marchetto M. C.; Gage F. H. Cell 2010, 140, 918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.; Liu Z. J. Mol. Biol. 2009, 393, 1143. [DOI] [PubMed] [Google Scholar]
- Liu J.; Faeder J. R.; Camacho C. J. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 19819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H. X. Trends Biochem. Sci. 2012, 37, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokuriki N.; Tawfik D. S. Science 2009, 324, 203. [DOI] [PubMed] [Google Scholar]
- Hazy E.; Tompa P. ChemPhysChem 2009, 10, 1415. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Liu Z. Chem.—Eur. J. 2013, 19, 4462. [DOI] [PubMed] [Google Scholar]
- Brown C. J.; Johnson A. K.; Dunker A. K.; Daughdrill G. W. Curr. Opin. Struct. Biol. 2011, 21, 441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosca R.; Pache R. A.; Aloy P. Mol. Cell. Proteomics 2012, 11, M111 014969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S.; Cox J.; Olsen J.; Mann M.; Frishman D. PLoS Comput. Biol. 2013, 9, e1002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerening J. R. Curr. Opin. Biotechnol. 2008, 19, 381. [DOI] [PubMed] [Google Scholar]
- Buchler N. E.; Louis M. J. Mol. Biol. 2008, 384, 1106. [DOI] [PubMed] [Google Scholar]
- Siegal-Gaskins D.; Mejia-Guerra M. K.; Smith G. D.; Grotewold E.. PLoS Comput. Biol. 2011, 7, e1002039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladbury J. E.; Arold S. T. Trends Biochem. Sci. 2012, 37, 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Nie Q.; Enciso G. Biophys. J. 2010, 99, L41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Bardwell L.; Nie Q. Biophys. J. 2010, 98, 1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dushek O.; van der Merwe P. A.; Shahrezaei V. Biophys. J. 2011, 100, 1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varedi K. S.; Ventura A. C.; Merajver S. D.; Lin X. N. PLoS One 2010, 5, e14029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borg M.; Mittag T.; Pawson T.; Tyers M.; Forman-Kay J. D.; Chan H. S. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 9650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar C.; Hofer T. FEBS J. 2009, 276, 3177. [DOI] [PubMed] [Google Scholar]
- Trunnell N. B.; Poon A. C.; Kim S. Y.; Ferrell J. E. Jr. Mol. Cell 2011, 41, 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickfaden S. C.; Winters M. J.; Ben-Ari G.; Lamson R. E.; Tyers M.; Pryciak P. M. Cell 2007, 128, 519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malleshaiah M. K.; Shahrezaei V.; Swain P. S.; Michnick S. W. Nature 2010, 465, 101. [DOI] [PubMed] [Google Scholar]
- Nishi H.; Fong J. H.; Chang C.; Teichmann S. A.; Panchenko A. R. Mol. Syst. 2013, 9, 1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweiger R.; Linial M. Biol. Direct 2010, 5, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt L. J.; Tuch B. B.; Villen J.; Johnson A. D.; Gygi S. P.; Morgan D. O. Science 2009, 325, 1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivomagi M.; Valk E.; Venta R.; Iofik A.; Lepiku M.; Balog E. R.; Rubin S. M.; Morgan D. O.; Loog M. Nature 2011, 480, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittag T.; Orlicky S.; Choy W. Y.; Tang X.; Lin H.; Sicheri F.; Kay L. E.; Tyers M.; Forman-Kay J. D. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 17772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X.; Orlicky S.; Mittag T.; Csizmok V.; Pawson T.; Forman-Kay J. D.; Sicheri F.; Tyers M. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao B.; Oehlmann S.; Sowa M. E.; Harper J. W.; Pavletich N. P. Mol. Cell 2007, 26, 131. [DOI] [PubMed] [Google Scholar]
- Xin F.; Radivojac P. Bioinformatics 2012, 28, 2905. [DOI] [PubMed] [Google Scholar]
- Vacic V.; Oldfield C. J.; Mohan A.; Radivojac P.; Cortese M. S.; Uversky V. N.; Dunker A. K. J. Proteome Res. 2007, 6, 2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey N. E.; Van Roey K.; Weatheritt R. J.; Toedt G.; Uyar B.; Altenberg B.; Budd A.; Diella F.; Dinkel H.; Gibson T. J. Mol. Biosyst. 2012, 8, 268. [DOI] [PubMed] [Google Scholar]
- Pancsa R.; Fuxreiter M. IUBMB Life 2012, 64, 513. [DOI] [PubMed] [Google Scholar]
- Lee S. H.; Kim D. H.; Han J. J.; Cha E. J.; Lim J. E.; Cho Y. J.; Lee C.; Han K. H. Curr. Protein Pept. Sci. 2012, 13, 34. [DOI] [PubMed] [Google Scholar]
- Meyer B.; M?ller H. Top. Curr. Chem. 2007, 267, 187. [Google Scholar]
- Tagashira M.; Iijima H.; Toma K. Glycoconjugate J. 2002, 19, 43. [DOI] [PubMed] [Google Scholar]
- Coltart D. M.; Royyuru A. K.; Williams L. J.; Glunz P. W.; Sames D.; Kuduk S. D.; Schwarz J. B.; Chen X. T.; Danishefsky S. J.; Live D. H. J. Am. Chem. Soc. 2002, 124, 9833. [DOI] [PubMed] [Google Scholar]
- Hashimoto R.; Fujitani N.; Takegawa Y.; Kurogochi M.; Matsushita T.; Naruchi K.; Ohyabu N.; Hinou H.; Gao X. D.; Manri N.; Satake H.; Kaneko A.; Sakamoto T.; Nishimura S. I. Chem.—Eur. J. 2011, 17, 2393. [DOI] [PubMed] [Google Scholar]
- Corzana F.; Busto J. H.; Engelsen S. B.; Jimenez-Barbero J.; Asensio J. L.; Peregrina J. M.; Avenoza A. Chem.—Eur. J. 2006, 12, 7864. [DOI] [PubMed] [Google Scholar]
- Simanek E. E.; Huang D. H.; Pasternack L.; Machajewski T. D.; Seitz O.; Millar D. S.; Dyson H. J.; Wong C. H. J. Am. Chem. Soc. 1998, 120, 11567. [Google Scholar]
- Wu W. G.; Pasternack L.; Huang D. H.; Koeller K. M.; Lin C. C.; Seitz O.; Wong C. H. J. Am. Chem. Soc. 1999, 121, 2409. [Google Scholar]
- Brister M. A.; Pandey A. K.; Bielska A. A.; Zondlo N. J. J. Am. Chem. Soc. 2014, 136, 3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbaum M. B.; Zondlo N. J.. Biochemistry 2014, in press [DOI] [PMC free article] [PubMed]
- Errington N.; Doig A. J. Biochemistry 2005, 44, 7553. [DOI] [PubMed] [Google Scholar]
- Bielska A. A.; Zondlo N. J. Biochemistry 2006, 45, 5527. [DOI] [PubMed] [Google Scholar]
- Songyang Z.; Lu K. P.; Kwon Y. T.; Tsai L. H.; Filhol O.; Cochet C.; Brickey D. A.; Soderling T. R.; Bartleson C.; Graves D. J.; DeMaggio A. J.; Hoekstra M. F.; Blenis J.; Hunter T.; Cantley L. C. Mol. Cell. Biol. 1996, 16, 6486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrew C. D.; Warwicker J.; Jones G. R.; Doig A. J. Biochemistry 2002, 41, 1897. [DOI] [PubMed] [Google Scholar]
- Dadarlat V. M.; Skeel R. D. Biophys. J. 2011, 100, 469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H. J.; Srinivasan D.; Coomber D.; Lane D. P.; Verma C. S. Cell Cycle 2007, 6, 2604. [DOI] [PubMed] [Google Scholar]
- Feng H.; Jenkins L. M.; Durell S. R.; Hayashi R.; Mazur S. J.; Cherry S.; Tropea J. E.; Miller M.; Wlodawer A.; Appella E.; Bai Y. Structure 2009, 17, 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez Y.; Gairi M.; Pons M.; Bernado P. J. Mol. Biol. 2009, 391, 136. [DOI] [PubMed] [Google Scholar]
- Tait S.; Dutta K.; Cowburn D.; Warwicker J.; Doig A. J.; McCarthy J. E. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 17627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou Y. C.; Zhou X. Z.; Lu K. P. Trends Biochem. Sci. 2011, 36, 501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutkowski M.; Bernhardt A.; Zhou X. Z.; Shen M.; Reimer U.; Rahfeld J. U.; Lu K. P.; Fischer G. Biochemistry 1998, 37, 5566. [DOI] [PubMed] [Google Scholar]
- Guo Y. T.; Li Y. M.; Zhu Z. T.; Zhao Y. F. Int. J. Pept. Res. Ther. 2005, 11, 159. [Google Scholar]
- Byun B. J.; Kang Y. K. Biopolymers 2009, 93, 330. [DOI] [PubMed] [Google Scholar]
- Velazquez H. A.; Hamelberg D. Biochemistry 2011, 50, 9605. [DOI] [PubMed] [Google Scholar]
- Theillet F. X.; Kalmar L.; Tompa P.; Han K. H.; Selenko P.; Dunker A. K.; Daughdrill G. W.; Uversky V. N. Intrinsically Disord. Proteins 2013, 1, e24360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltsev A. S.; Ying J. F.; Bax A. Biochemistry 2012, 51, 5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang L.; Moriarty G. M.; Woods L. A.; Ashcroft A. E.; Radford S. E.; Baum J. Protein Sci. 2012, 21, 911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauvet B.; Mbefo M. K.; Fares M. B.; Desobry C.; Michael S.; Ardah M. T.; Tsika E.; Coune P.; Prudent M.; Lion N.; Eliezer D.; Moore D. J.; Schneider B.; Aebischer P.; El-Agnaf O. M.; Masliah E.; Lashuel H. A. J. Biol. Chem. 2012, 287, 15345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauvet B.; Fares M. B.; Samuel F.; Dikiy I.; Tandon A.; Eliezer D.; Lashuel H. A. J. Biol. Chem. 2012, 287, 28243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra R.; Jayaraman M.; Roland B. P.; Landrum E.; Fullam T.; Kodali R.; Thakur A. K.; Arduini I.; Wetzel R. J. Mol. Biol. 2012, 415, 900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibow S.; Ozenne V.; Biernat J.; Blackledge M.; Mandelkow E.; Zweckstetter M. J. Am. Chem. Soc. 2011, 133, 15842. [DOI] [PubMed] [Google Scholar]
- Sibille N.; Huvent I.; Fauquant C.; Verdegem D.; Amniai L.; Leroy A.; Wieruszeski J. M.; Lippens G.; Landrieu I. Proteins: Struct., Funct., Bioinf. 2011, 80, 454. [DOI] [PubMed] [Google Scholar]
- Mittag T.; Marsh J.; Grishaev A.; Orlicky S.; Lin H.; Sicheri F.; Tyers M.; Forman-Kay J. D. Structure 2010, 18, 494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W. B.; Long C. M.; Reaney S. H.; Di Monte D. A.; Fink A. L.; Uversky V. N. Biochim. Biophys. Acta 2010, 1802, 322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotti M.; Longhi S. Mol. Biosyst. 2012, 8, 105. [DOI] [PubMed] [Google Scholar]
- Mohan A.; Oldfield C. J.; Radivojac P.; Vacic V.; Cortese M. S.; Dunker A. K.; Uversky V. N. J. Mol. Biol. 2006, 362, 1043. [DOI] [PubMed] [Google Scholar]
- Xue B.; Dunker A. K.; Uversky V. N. J. Biomol. Struct. Dyn. 2012, 29, 843. [DOI] [PubMed] [Google Scholar]
- Janin J.; Sternberg M. J. E.. F1000 Biol. Rep. 2013, 5, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugase K.; Dyson H. J.; Wright P. E. Nature 2007, 447, 1021. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan I.; Perez-Alvarado G. C.; Dyson H. J.; Wright P. E. FEBS Lett. 1998, 430, 317. [DOI] [PubMed] [Google Scholar]
- Ganguly D.; Chen J. H. J. Am. Chem. Soc. 2009, 131, 5214. [DOI] [PubMed] [Google Scholar]
- Espinoza-Fonseca L. M. Mol. Biosyst. 2012, 8, 237. [DOI] [PubMed] [Google Scholar]
- Espinoza-Fonseca L. M. Biochem. Biophys. Res. Commun. 2009, 382, 479. [DOI] [PubMed] [Google Scholar]
- Kjaergaard M.; Teilum K.; Poulsen F. M. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 12535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjaergaard M.; Andersen L.; Nielsen L. D.; Teilum K. Biochemistry 2013, 52, 1686. [DOI] [PubMed] [Google Scholar]
- Dogan J.; Mu X.; Engstrom A.; Jemth P. Sci. Rep. 2013, 3, 2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogan J.; Schmidt T.; Mu X.; Engstrom A.; Jemth P. J. Biol. Chem. 2012, 287, 34316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunberg R.; Leckner J.; Nilges M. Structure 2004, 12, 2125. [DOI] [PubMed] [Google Scholar]
- Wlodarski T.; Zagrovic B. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 19346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruvinsky A. M.; Kirys T.; Tuzikov A. V.; Vakser I. A. J. Mol. Biol. 2011, 408, 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teilum K.; Olsen J. G.; Kragelund B. B. Cell. Mol. Life Sci. 2009, 66, 2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Sancho D.; Best R. B. J. Am. Chem. Soc. 2011, 133, 6809. [DOI] [PubMed] [Google Scholar]
- Zhou H. X. Biophys. J. 2010, 98, L15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfefferkorn C. M.; Jiang Z.; Lee J. C. Biochim. Biophys. Acta 2012, 1818, 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells M.; Tidow H.; Rutherford T. J.; Markwick P.; Jensen M. R.; Mylonas E.; Svergun D. I.; Blackledge M.; Fersht A. R. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 5762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh J. A.; Dancheck B.; Ragusa M. J.; Allaire M.; Forman-Kay J. D.; Peti W. Structure 2010, 18, 1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen M. R.; Communie G.; Ribeiro E. A. Jr.; Martinez N.; Desfosses A.; Salmon L.; Mollica L.; Gabel F.; Jamin M.; Longhi S.; Ruigrok R. W.; Blackledge M. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 9839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fermani S.; Trivelli X.; Sparla F.; Thumiger A.; Calvaresi M.; Marri L.; Falini G.; Zerbetto F.; Trost P. J. Biol. Chem. 2012, 287, 21372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlap T. B.; Kirk J. M.; Pena E. A.; Yoder M. S.; Creamer T. P. Proteins: Struct., Funct., Bioinf. 2012, 81, 607. [DOI] [PubMed] [Google Scholar]
- Iesmantavicius V.; Jensen M. R.; Ozenne V.; Blackledge M.; Poulsen F. M.; Kjaergaard M. J. Am. Chem. Soc. 2013, 135, 10155. [DOI] [PubMed] [Google Scholar]
- Cino E. A.; Killoran R. C.; Karttunen M.; Choy W. Y. Sci. Rep. 2013, 3, 2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky V. N. Chem. Soc. Rev. 2010, 40, 1623. [DOI] [PubMed] [Google Scholar]
- Wright P. E.; Dyson H. J. Curr. Opin. Struct. Biol. 2009, 19, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiefhaber T.; Bachmann A.; Jensen K. S. Curr. Opin. Struct. Biol. 2012, 22, 21. [DOI] [PubMed] [Google Scholar]
- Chen J. Arch. Biochem. Biophys. 2012, 524, 123. [DOI] [PubMed] [Google Scholar]
- Rogers J. M.; Steward A.; Clarke J. J. Am. Chem. Soc. 2013, 135, 1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vertessy B. G.; Orosz F. Bioessays 2011, 33, 30. [DOI] [PubMed] [Google Scholar]
- Marsh J. A.; Teichmann S. A.; Forman-Kay J. D. Curr. Opin. Struct. Biol. 2012, 22, 643. [DOI] [PubMed] [Google Scholar]
- Zhou H. X.; Pang X. D.; Lu C. Phys. Chem. Chem. Phys. 2012, 14, 10466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Chu X.; Longhi S.; Roche P.; Han W.; Wang E.; Wang J. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, E3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann A.; Wildemann D.; Praetorius F.; Fischer G.; Kiefhaber T. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higo J.; Nishimura Y.; Nakamura H. J. Am. Chem. Soc. 2011, 133, 10448. [DOI] [PubMed] [Google Scholar]
- Karlsson O. A.; Chi C. N.; Engstrom A.; Jemth P. J. Mol. Biol. 2012, 417, 253. [DOI] [PubMed] [Google Scholar]
- Yu Q. F.; Ye W.; Wang W.; Chen H. F.. PLoS One 2013, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giri R.; Morrone A.; Toto A.; Brunori M.; Gianni S. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 14942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S.; Showalter S. A.; Noid W. G. J. Phys. Chem. B 2013, 117, 3074. [DOI] [PubMed] [Google Scholar]
- Drobnak I.; De Jonge N.; Haesaerts S.; Vesnaver G.; Loris R.; Lah J. J. Am. Chem. Soc. 2013, 135, 1288. [DOI] [PubMed] [Google Scholar]
- Hammes G. G.; Chang Y. C.; Oas T. G. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 13737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker B. A.; Portman J. J.; Wolynes P. G. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 8868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacy E. R.; Filippov I.; Lewis W. S.; Otieno S.; Xiao L.; Weiss S.; Hengst L.; Kriwacki R. W. Nat. Struct. Mol. Biol. 2004, 11, 358. [DOI] [PubMed] [Google Scholar]
- Otieno S.; Grace C. R.; Kriwacki R. W. Biophys. J. 2011, 100, 2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Fisher J. C.; Mathew R.; Ou L.; Otieno S.; Sublet J.; Xiao L.; Chen J.; Roussel M. F.; Kriwacki R. W. Nat. Chem. Biol. 2011, 7, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otieno S.; Kriwacki R. PLoS One 2012, 7, e47177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burre J.; Sharma M.; Sudhof T. C. J. Neurosci. 2012, 32, 15227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burre J.; Sharma M.; Tsetsenis T.; Buchman V.; Etherton M. R.; Sudhof T. C. Science 2010, 329, 1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussell R. Jr.; Ramlall T. F.; Eliezer D. Protein Sci. 2005, 14, 862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgieva E. R.; Ramlall T. F.; Borbat P. P.; Freed J. H.; Eliezer D. J. Am. Chem. Soc. 2008, 130, 12856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jao C. C.; Der-Sarkissian A.; Chen J.; Langen R. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 8331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jao C. C.; Hegde B. G.; Chen J.; Haworth I. S.; Langen R. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 19666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lokappa S. B.; Ulmer T. S. J. Biol. Chem. 2011, 286, 21450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shvadchak V. V.; Yushchenko D. A.; Pievo R.; Jovin T. M. FEBS Lett. 2011, 585, 3513. [DOI] [PubMed] [Google Scholar]
- Ulmer T. S.; Bax A.; Cole N. B.; Nussbaum R. L. J. Biol. Chem. 2005, 280, 9595. [DOI] [PubMed] [Google Scholar]
- Varkey J.; Mizuno N.; Hegde B. G.; Cheng N. Q.; Steven A. C.; Langen R. J. Biol. Chem. 2013, 288, 17620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussell R. Jr.; Eliezer D. J. Biol. Chem. 2001, 276, 45996. [DOI] [PubMed] [Google Scholar]
- Fortin D. L.; Nemani V. M.; Voglmaier S. M.; Anthony M. D.; Ryan T. A.; Edwards R. H. J. Neurosci. 2005, 25, 10913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen P. H.; Nielsen M. S.; Jakes R.; Dotti G.; Goedert M. J. Biol. Chem. 1998, 273, 26292. [DOI] [PubMed] [Google Scholar]
- Kahle P. J.; Neumann M.; Ozmen L.; Muller V.; Jacobsen H.; Schindzielorz A.; Okochi M.; Leimer U.; van der Putten H.; Probst A.; Kremmer E.; Kretzschmar H. A.; Haass C. J. Neurosci. 2000, 20, 6365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Withers G. S.; George J. M.; Banker G. A.; Clayton D. F. Dev. Brain Res. 1997, 99, 87. [DOI] [PubMed] [Google Scholar]
- Yang M. L.; Hasadsri L.; Woods W. S.; George J. M.. Mol. Neurodegener. 2010, 5, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodner C. R.; Maltsev A. S.; Dobson C. M.; Bax A. Biochemistry 2010, 49, 862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo E. J.; Fuller N.; Rand R. P.; St George-Hyslop P.; Fraser P. E. J. Mol. Biol. 2002, 315, 799. [DOI] [PubMed] [Google Scholar]
- Perrin R. J.; Woods W. S.; Clayton D. F.; George J. M. J. Biol. Chem. 2000, 275, 34393. [DOI] [PubMed] [Google Scholar]
- Outeiro T. F.; Lindquist S. Science 2003, 302, 1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabrocki P.; Bastiaens I.; Delay C.; Barnmens T.; Ghillebert R.; Pellens K.; De Virgilio C.; Van Leuven F.; Winderickx J. Biochim. Biophys. Acta 2008, 1783, 1767. [DOI] [PubMed] [Google Scholar]
- Fuxreiter M. Mol. Biosyst. 2012, 8, 168. [DOI] [PubMed] [Google Scholar]
- Baker J. M. R.; Hudson R. P.; Kanelis V.; Choy W. Y.; Thibodeau P. H.; Thomas P. J.; Forman-Kay J. D. Nat. Struct. Mol. Biol. 2007, 14, 738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash P.; Tang X. J.; Orlicky S.; Chen Q. H.; Gertler F. B.; Mendenhall M. D.; Sicheri F.; Pawson T.; Tyers M. Nature 2001, 414, 514. [DOI] [PubMed] [Google Scholar]
- Dobson C. M. Trends Biochem. Sci. 1999, 24, 329. [DOI] [PubMed] [Google Scholar]
- Uversky V. N.; Fink A. L. Biochim. Biophys. Acta 2004, 1698, 131. [DOI] [PubMed] [Google Scholar]
- Chiti F.; Dobson C. M. Annu. Rev. Biochem. 2006, 75, 333. [DOI] [PubMed] [Google Scholar]
- Dobson C. M. Semin. Cell Dev. Biol. 2004, 15, 3. [DOI] [PubMed] [Google Scholar]
- McKhann G.; Drachman D.; Folstein M.; Katzman R.; Price D.; Stadlan E. M. Neurology 1984, 34, 939. [DOI] [PubMed] [Google Scholar]
- Cavallucci V.; D’Amelio M.; Cecconi F. Mol. Neurobiol. 2012, 45, 366. [DOI] [PubMed] [Google Scholar]
- Farias G.; Cornejo A.; Jimenez J.; Guzman L.; Maccioni R. B. Curr. Alzheimer Res. 2011, 8, 608. [DOI] [PubMed] [Google Scholar]
- Jucker M.; Walker L. C. Ann. Neurol. 2011, 70, 532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M.; Spillantini M. G.; Del Tredici K.; Braak H. Nat. Rev. Neurol. 2012, 9, 13. [DOI] [PubMed] [Google Scholar]
- Forno L. S. J. Neuropathol. Exp. Neurol. 1996, 55, 259. [DOI] [PubMed] [Google Scholar]
- Lashuel H. A.; Overk C. R.; Oueslati A.; Masliah E. Nat. Rev. Neurosci. 2012, 14, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westaway D.; Carlson G. A.; Prusiner S. B. Trends Neurosci. 1989, 12, 221. [DOI] [PubMed] [Google Scholar]
- Asante E. A.; Linehan J. M.; Desbruslais M.; Joiner S.; Gowland I.; Wood A. L.; Welch J.; Hill A. F.; Lloyd S. E.; Wadsworth J. D.; Collinge J. EMBO J. 2002, 21, 6358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riek R.; Hornemann S.; Wider G.; Glockshuber R.; Wuthrich K. FEBS Lett. 1997, 413, 282. [DOI] [PubMed] [Google Scholar]
- Wuthrich K.; Riek R. Adv. Protein Chem. 2001, 57, 55. [DOI] [PubMed] [Google Scholar]
- Scott M.; Groth D.; Foster D.; Torchia M.; Yang S. L.; DeArmond S. J.; Prusiner S. B. Cell 1993, 73, 979. [DOI] [PubMed] [Google Scholar]
- Perutz M. F. Curr. Opin. Struct. Biol. 1996, 6, 848. [DOI] [PubMed] [Google Scholar]
- Hull R. L.; Westermark G. T.; Westermark P.; Kahn S. E. J. Clin. Endocrinol. Metab. 2004, 89, 3629. [DOI] [PubMed] [Google Scholar]
- Westermark G. T.; Westermark P. Exp. Diabetes Res. 2008, 2008, 528354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen J. K. Nat. Med. 2004, 10SupplS18. [DOI] [PubMed] [Google Scholar]
- Schildknecht S.; Gerding H. R.; Karreman C.; Drescher M.; Lashuel H. A.; Outeiro T. F.; Di Monte D. A.; Leist M. J. Neurochem. 2013, 125, 491. [DOI] [PubMed] [Google Scholar]
- Barnham K. J.; Masters C. L.; Bush A. I. Nat. Rev. Drug. Discovery 2004, 3, 205. [DOI] [PubMed] [Google Scholar]
- Gaggelli E.; Kozlowski H.; Valensin D.; Valensin G. Chem. Rev. 2006, 106, 1995. [DOI] [PubMed] [Google Scholar]
- Iwai A.; Masliah E.; Yoshimoto M.; Ge N.; Flanagan L.; de Silva H. A.; Kittel A.; Saitoh T. Neuron 1995, 14, 467. [DOI] [PubMed] [Google Scholar]
- Bisaglia M.; Greggio E.; Maric D.; Miller D. W.; Cookson M. R.; Bubacco L. BMC Neurosci. 2010, 11, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisaglia M.; Mammi S.; Bubacco L. J. Biol. Chem. 2007, 282, 15597. [DOI] [PubMed] [Google Scholar]
- Bisaglia M.; Tosatto L.; Munari F.; Tessari I.; de Laureto P. P.; Mammi S.; Bubacco L. Biochem. Biophys. Res. Commun. 2010, 394, 424. [DOI] [PubMed] [Google Scholar]
- Nakaso K.; Tajima N.; Ito S.; Teraoka M.; Yamashita A.; Horikoshi Y.; Kikuchi D.; Mochida S.; Nakashima K.; Matsura T. PLoS One 2013, 8, e55068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar H.; Lim H. W.; More S. V.; Kim B. W.; Koppula S.; Kim I. S.; Choi D. K. Int. J. Mol. Sci. 2012, 13, 10478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuner K.; Muller W. E.; Reichert A. S. Mol. Neurobiol. 2012, 46, 186. [DOI] [PubMed] [Google Scholar]
- Greenwald J.; Riek R. Structure 2010, 18, 1244. [DOI] [PubMed] [Google Scholar]
- Chapman M. R.; Robinson L. S.; Pinkner J. S.; Roth R.; Heuser J.; Hammar M.; Normark S.; Hultgren S. J. Science 2002, 295, 851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien P.; Weissman J. S.; DePace A. H. Annu. Rev. Biochem. 2004, 73, 617. [DOI] [PubMed] [Google Scholar]
- Serio T. R.; Lindquist S. L. Annu. Rev. Cell Dev. Biol. 1999, 15, 661. [DOI] [PubMed] [Google Scholar]
- Tuite M. F.; Cox B. S. Nat. Rev. Mol. Cell Biol. 2003, 4, 878. [DOI] [PubMed] [Google Scholar]
- Fowler D. M.; Koulov A. V.; Balch W. E.; Kelly J. W. Trends Biochem. Sci. 2007, 32, 217. [DOI] [PubMed] [Google Scholar]
- McGlinchey R. P.; Gruschus J. M.; Nagy A.; Lee J. C. Biochemistry 2011, 50, 10567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlinchey R. P.; Shewmaker F.; McPhie P.; Monterroso B.; Thurber K.; Wickner R. B. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 13731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binolfi A.; Fernandez C. O.. Interactions of alpha-synuclein with metal ions: new insights into the structural biology and bioinorganic chemistry of Parkinsońs disease; Pan Stanford Publishing: Singapore, 2012. [Google Scholar]
- Cohen S. I.; Vendruscolo M.; Dobson C. M.; Knowles T. P. J. Mol. Biol. 2012, 421, 160. [DOI] [PubMed] [Google Scholar]
- Liu T.; Bitan G. ChemMedChem. 2012, 7, 359. [DOI] [PubMed] [Google Scholar]
- Bemporad F.; Chiti F. Chem. Biol. 2012, 19, 315. [DOI] [PubMed] [Google Scholar]
- Morris A. M.; Watzky M. A.; Finke R. G. Biochim. Biophys. Acta 2009, 1794, 375. [DOI] [PubMed] [Google Scholar]
- Wetzel R. Acc. Chem. Res. 2006, 39, 671. [DOI] [PubMed] [Google Scholar]
- Chen M.; Zhang S.; Liu Q.; Liu P.; Busuttil K.; Wang C.; Besenbacher F.; Li Y. M.; Dong M. Chem.—Eur. J. 2012, 18, 2493. [DOI] [PubMed] [Google Scholar]
- Ding F.; Dokholyan N. V.; Buldyrev S. V.; Stanley H. E.; Shakhnovich E. I. J. Mol. Biol. 2002, 324, 851. [DOI] [PubMed] [Google Scholar]
- Diociaiuti M.; Gaudiano M. C.; Malchiodi-Albedi F. Int. J. Mol. Sci. 2011, 12, 9277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diociaiuti M.; Polzi L. Z.; Valvo L.; Malchiodi-Albedi F.; Bombelli C.; Gaudiano M. C. Biophys. J. 2006, 91, 2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper J. D.; Wong S. S.; Lieber C. M.; Lansbury P. T. Jr. Biochemistry 1999, 38, 8972. [DOI] [PubMed] [Google Scholar]
- Lashuel H. A.; Hartley D.; Petre B. M.; Walz T.; Lansbury P. T. Jr. Nature 2002, 418, 291. [DOI] [PubMed] [Google Scholar]
- Lashuel H. A.; Petre B. M.; Wall J.; Simon M.; Nowak R. J.; Walz T.; Lansbury P. T. Jr. J. Mol. Biol. 2002, 322, 1089. [DOI] [PubMed] [Google Scholar]
- Bitan G.; Vollers S. S.; Teplow D. B. J. Biol. Chem. 2003, 278, 34882. [DOI] [PubMed] [Google Scholar]
- Haass C.; Selkoe D. J. Nat. Rev. Mol. Cell Biol. 2007, 8, 101. [DOI] [PubMed] [Google Scholar]
- Ahmed M.; Davis J.; Aucoin D.; Sato T.; Ahuja S.; Aimoto S.; Elliott J. I.; Van Nostrand W. E.; Smith S. O. Nat. Struct. Mol. Biol. 2010, 17, 561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lashuel H. A.; Hartley D. M.; Petre B. M.; Wall J. S.; Simon M. N.; Walz T.; Lansbury P. T. J. Mol. Biol. 2003, 332, 795. [DOI] [PubMed] [Google Scholar]
- Arimon M.; Diez-Perez I.; Kogan M. J.; Durany N.; Giralt E.; Sanz F.; Fernandez-Busquets X. FASEB J. 2005, 19, 1344. [DOI] [PubMed] [Google Scholar]
- Chromy B. A.; Nowak R. J.; Lambert M. P.; Viola K. L.; Chang L.; Velasco P. T.; Jones B. W.; Fernandez S. J.; Lacor P. N.; Horowitz P.; Finch C. E.; Krafft G. A.; Klein W. L. Biochemistry 2003, 42, 12749. [DOI] [PubMed] [Google Scholar]
- Quist A.; Doudevski L.; Lin H.; Azimova R.; Ng D.; Frangione B.; Kagan B.; Ghiso J.; Lal R. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 10427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orcellet M. L.; Fernandez C. O. Curr. Protein Pept. Sci. 2011, 12, 188. [DOI] [PubMed] [Google Scholar]
- Serpell L. C.; Fraser P. E.; Sunde M. Methods Enzymol. 1999, 309, 526. [DOI] [PubMed] [Google Scholar]
- Serpell L. C.; Sunde M.; Benson M. D.; Tennent G. A.; Pepys M. B.; Fraser P. E. J. Mol. Biol. 2000, 300, 1033. [DOI] [PubMed] [Google Scholar]
- Zimmerman S. B.; Minton A. P. Annu. Rev. Biophys. Biomol. Struct. 1993, 22, 27. [DOI] [PubMed] [Google Scholar]
- Ellis R. J. Curr. Opin. Struct. Biol. 2001, 11, 114. [DOI] [PubMed] [Google Scholar]
- Ellis R. J.; Minton A. P. Biol. Chem. 2006, 387, 485. [DOI] [PubMed] [Google Scholar]
- Minton A. P. Biophys. J. 2005, 88, 971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elowitz M. B.; Surette M. G.; Wolf P. E.; Stock J. B.; Leibler S. J. Bacteriol. 1999, 181, 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C. G.; Charlton L. M.; Lakkavaram A.; Seagle C.; Wang G. F.; Young G. B.; Macdonald J. M.; Pielak G. J. J. Am. Chem. Soc. 2008, 130, 6310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullineaux C. W.; Nenninger A.; Ray N.; Robinson C. J. Bacteriol. 2006, 188, 3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minton A. P. Curr. Opin. Struct. Biol. 2000, 10, 34. [DOI] [PubMed] [Google Scholar]
- Uversky V. N.; Lee H. J.; Li J.; Fink A. L.; Lee S. J. J. Biol. Chem. 2001, 276, 43495. [DOI] [PubMed] [Google Scholar]
- Uversky V. N.; Li J.; Fink A. L. J. Biol. Chem. 2001, 276, 10737. [DOI] [PubMed] [Google Scholar]
- Wetzel R. Cell 1996, 86, 699. [DOI] [PubMed] [Google Scholar]
- Uversky V. N.; Li J.; Fink A. L. J. Biol. Chem. 2001, 276, 44284. [DOI] [PubMed] [Google Scholar]
- Munishkina L. A.; Ahmad A.; Fink A. L.; Uversky V. N. Biochemistry 2008, 47, 8993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munishkina L. A.; Cooper E. M.; Uversky V. N.; Fink A. L. J. Mol. Recognit. 2004, 17, 456. [DOI] [PubMed] [Google Scholar]
- Munishkina L. A.; Fink A. L.; Uversky V. N. Curr. Alzheimer Res. 2009, 6, 252. [DOI] [PubMed] [Google Scholar]
- Uversky V. N.; Cooper E. M.; Bower K. S.; Li J.; Fink A. L. FEBS Lett. 2002, 515, 99. [DOI] [PubMed] [Google Scholar]
- Ma Q.; Fan J. B.; Zhou Z.; Zhou B. R.; Meng S. R.; Hu J. Y.; Chen J.; Liang Y. PLoS One 2012, 7, e36288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z.; Fan J. B.; Zhu H. L.; Shewmaker F.; Yan X.; Chen X.; Chen J.; Xiao G. F.; Guo L.; Liang Y. J. Biol. Chem. 2009, 284, 30148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. F.; Bird S.; Shaw M.; Jean L.; Vaux D. J. J. Biol. Chem. 2012, 287, 38006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera E.; Straub J.; Thirumalai D. Biophys. J. 2009, 96, 4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatters D. M.; Minton A. P.; Howlett G. J. J. Biol. Chem. 2002, 277, 7824. [DOI] [PubMed] [Google Scholar]
- Ma B.; Xie J.; Wei L.; Li W. Int. J. Biol. Macromol. 2013, 53, 82. [DOI] [PubMed] [Google Scholar]
- Sukenik S.; Politi R.; Ziserman L.; Danino D.; Friedler A.; Harries D. PLoS One 2010, 6, e15608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monsellier E.; Chiti F. EMBO Rep. 2007, 8, 737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson J. P.; Walker D. E.; Goldstein J. M.; de Laat R.; Banducci K.; Caccavello R. J.; Barbour R.; Huang J.; Kling K.; Lee M.; Diep L.; Keim P. S.; Shen X.; Chataway T.; Schlossmacher M. G.; Seubert P.; Schenk D.; Sinha S.; Gai W. P.; Chilcote T. J. J. Biol. Chem. 2006, 281, 29739. [DOI] [PubMed] [Google Scholar]
- Fujiwara H.; Hasegawa M.; Dohmae N.; Kawashima A.; Masliah E.; Goldberg M. S.; Shen J.; Takio K.; Iwatsubo T. Nat. Cell Biol. 2002, 4, 160. [DOI] [PubMed] [Google Scholar]
- Takahashi M.; Kanuka H.; Fujiwara H.; Koyama A.; Hasegawa M.; Miura M.; Iwatsubo T. Neurosci. Lett. 2003, 336, 155. [DOI] [PubMed] [Google Scholar]
- Paleologou K. E.; Oueslati A.; Shakked G.; Rospigliosi C. C.; Kim H. Y.; Lamberto G. R.; Fernandez C. O.; Schmid A.; Chegini F.; Gai W. P.; Chiappe D.; Moniatte M.; Schneider B. L.; Aebischer P.; Eliezer D.; Zweckstetter M.; Masliah E.; Lashuel H. A. J. Neurosci. 2010, 30, 3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertoncini C. W.; Jung Y. S.; Fernandez C. O.; Hoyer W.; Griesinger C.; Jovin T. M.; Zweckstetter M. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedmon M. M.; Lindorff-Larsen K.; Christodoulou J.; Vendruscolo M.; Dobson C. M. J. Am. Chem. Soc. 2005, 127, 476. [DOI] [PubMed] [Google Scholar]
- Paleologou K. E.; Schmid A. W.; Rospigliosi C. C.; Kim H. Y.; Lamberto G. R.; Fredenburg R. A.; Lansbury P. T. Jr.; Fernandez C. O.; Eliezer D.; Zweckstetter M.; Lashuel H. A. J. Biol. Chem. 2008, 283, 16895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hejjaoui M.; Butterfield S.; Fauvet B.; Vercruysse F.; Cui J.; Dikiy I.; Prudent M.; Olschewski D.; Zhang Y.; Eliezer D.; Lashuel H. A. J. Am. Chem. Soc. 2012, 134, 5196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L.; Feany M. B. Nat. Neurosci. 2005, 8, 657. [DOI] [PubMed] [Google Scholar]
- Chen L.; Periquet M.; Wang X.; Negro A.; McLean P. J.; Hyman B. T.; Feany M. B. J. Clin. Invest. 2009, 119, 3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y.; Prudent M.; Fauvet B.; Lashuel H. A.; Girault H. H. ACS Chem. Neurosci. 2011, 2, 667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole N. B.; Murphy D. D.; Lebowitz J.; Di Noto L.; Levine R. L.; Nussbaum R. L. J. Biol. Chem. 2005, 280, 9678. [DOI] [PubMed] [Google Scholar]
- Baba M.; Nakajo S.; Tu P. H.; Tomita T.; Nakaya K.; Lee V. M.; Trojanowski J. Q.; Iwatsubo T. Am. J. Pathol. 1998, 152, 879. [PMC free article] [PubMed] [Google Scholar]
- Crowther R. A.; Jakes R.; Spillantini M. G.; Goedert M. FEBS Lett. 1998, 436, 309. [DOI] [PubMed] [Google Scholar]
- Spillantini M. G.; Crowther R. A.; Jakes R.; Hasegawa M.; Goedert M. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binolfi A.; Rodriguez E. E.; Valensin D.; D’Amelio N.; Ippoliti E.; Obal G.; Duran R.; Magistrato A.; Pritsch O.; Zweckstetter M.; Valensin G.; Carloni P.; Quintanar L.; Griesinger C.; Fernandez C. O. Inorg. Chem. 2010, 49, 10668. [DOI] [PubMed] [Google Scholar]
- Murray I. V.; Giasson B. I.; Quinn S. M.; Koppaka V.; Axelsen P. H.; Ischiropoulos H.; Trojanowski J. Q.; Lee V. M. Biochemistry 2003, 42, 8530. [DOI] [PubMed] [Google Scholar]
- Oueslati A.; Fournier M.; Lashuel H. A. Prog. Brain. Res. 2010, 183, 115. [DOI] [PubMed] [Google Scholar]
- Periquet M.; Fulga T.; Myllykangas L.; Schlossmacher M. G.; Feany M. B. J. Neurosci. 2007, 27, 3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson M. P.; Magnus T. Nat. Rev. Neurosci. 2006, 7, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haj-Yahya M.; Fauvet B.; Herman-Bachinsky Y.; Hejjaoui M.; Bavikar S. N.; Karthikeyan S. V.; Ciechanover A.; Lashuel H. A.; Brik A. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 17726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumova P.; Meulmeester E.; Garrido M.; Tirard M.; Hsiao H. H.; Bossis G.; Urlaub H.; Zweckstetter M.; Kugler S.; Melchior F.; Bahr M.; Weishaupt J. H. J. Cell Biol. 2011, 194, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier F.; Abeywardana T.; Dhall A.; Marotta N. P.; Varkey J.; Langen R.; Chatterjee C.; Pratt M. R. J. Am. Chem. Soc. 2012, 134, 5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Z. J.; Hu D. M.; Han S. B.; Reaney S. H.; Monte D. A.; Fink A. L. J. Biol. Chem. 2007, 282, 5862. [DOI] [PubMed] [Google Scholar]
- Uversky V. N.; Yamin G.; Munishkina L. A.; Karymov M. A.; Millett I. S.; Doniach S.; Lyubchenko Y. L.; Fink A. L. Mol. Brain Res. 2005, 134, 84. [DOI] [PubMed] [Google Scholar]
- Yamin G.; Uversky V. N.; Fink A. L. FEBS Lett. 2003, 542, 147. [DOI] [PubMed] [Google Scholar]
- Hodara R.; Norris E. H.; Giasson B. I.; Mishizen-Eberz A. J.; Lynch D. R.; Lee V. M.; Ischiropoulos H. J. Biol. Chem. 2004, 279, 47746. [DOI] [PubMed] [Google Scholar]
- Chavarria C.; Souza J. M. Arch. Biochem. Biophys. 2013, 533, 25. [DOI] [PubMed] [Google Scholar]
- Glaser C. B.; Yamin G.; Uversky V. N.; Fink A. L. Biochim. Biophys. Acta 2005, 1703, 157. [DOI] [PubMed] [Google Scholar]
- Yamin G.; Glaser C. B.; Uversky V. N.; Fink A. L. J. Biol. Chem. 2003, 278, 27630. [DOI] [PubMed] [Google Scholar]
- Maltsev A. S.; Chen J.; Levine R. L.; Bax A. J. Am. Chem. Soc. 2013, 135, 2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pronin A. N.; Morris A. J.; Surguchov A.; Benovic J. L. J. Biol. Chem. 2000, 275, 26515. [DOI] [PubMed] [Google Scholar]
- Sevcsik E.; Trexler A. J.; Dunn J. M.; Rhoades E. J. Am. Chem. Soc. 2011, 133, 7152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H.; Braak E. Neurobiol. Aging 1995, 16, 271. [DOI] [PubMed] [Google Scholar]
- Dickey C. A.; Kamal A.; Lundgren K.; Klosak N.; Bailey R. M.; Dunmore J.; Ash P.; Shoraka S.; Zlatkovic J.; Eckman C. B.; Patterson C.; Dickson D. W.; Nahman N. S. Jr.; Hutton M.; Burrows F.; Petrucelli L. J. Clin. Invest. 2007, 117, 648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fath T.; Eidenmuller J.; Brandt R. J. Neurosci. 2002, 22, 9733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraha A.; Ghoshal N.; Gamblin T. C.; Cryns V.; Berry R. W.; Kuret J.; Binder L. I. J. Cell Sci. 2000, 113, 3737. [DOI] [PubMed] [Google Scholar]
- Haase C.; Stieler J. T.; Arendt T.; Holzer M. J. Neurochem. 2004, 88, 1509. [DOI] [PubMed] [Google Scholar]
- Horowitz P. M.; Patterson K. R.; Guillozet-Bongaarts A. L.; Reynolds M. R.; Carroll C. A.; Weintraub S. T.; Bennett D. A.; Cryns V. L.; Berry R. W.; Binder L. I. J. Neurosci. 2004, 24, 7895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes J. F.; Reynolds M. R.; Horowitz P. M.; Fu Y.; Guillozet-Bongaarts A. L.; Berry R.; Binder L. I. Neurobiol Dis 2008, 31, 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds M. R.; Berry R. W.; Binder L. I. Biochemistry 2005, 44, 13997. [DOI] [PubMed] [Google Scholar]
- Reynolds M. R.; Berry R. W.; Binder L. I. Biochemistry 2005, 44, 1690. [DOI] [PubMed] [Google Scholar]
- Kummer M. P.; Hermes M.; Delekarte A.; Hammerschmidt T.; Kumar S.; Terwel D.; Walter J.; Pape H. C.; Konig S.; Roeber S.; Jessen F.; Klockgether T.; Korte M.; Heneka M. T. Neuron 2011, 71, 833. [DOI] [PubMed] [Google Scholar]
- Gu X.; Greiner E. R.; Mishra R.; Kodali R.; Osmand A.; Finkbeiner S.; Steffan J. S.; Thompson L. M.; Wetzel R.; Yang X. W. Neuron 2009, 64, 828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra R.; Hoop C. L.; Kodali R.; Sahoo B.; van der Wel P. C.; Wetzel R. J. Mol. Biol. 2012, 424, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansaloni A.; Wang Z. M.; Jeong J. S.; Ruggeri F. S.; Dietler G.; Lashuel H. A. Angew. Chem., Int. Ed. 2014, 53, 1928. [DOI] [PubMed] [Google Scholar]
- Binolfi A.; Quintanar L.; Bertoncini C. W.; Griesinger C.; Fernandez C. O. Coord. Chem. Rev. 2012, 256, 2188. [Google Scholar]
- Brown D. R.; Kozlowski H. Dalton T. 2004, 13, 1907. [DOI] [PubMed] [Google Scholar]
- Kozlowski H.; Luczkowski M.; Valensin D.; Valensin G.. Metal ion binding properties of protein related to neurodegeneration; Wiley: New York, 2006. [Google Scholar]
- Binolfi A.; Rasia R. M.; Bertoncini C. W.; Ceolin M.; Zweckstetter M.; Griesinger C.; Jovin T. M.; Fernandez C. O. J. Am. Chem. Soc. 2006, 128, 9893. [DOI] [PubMed] [Google Scholar]
- Rasia R. M.; Bertoncini C. W.; Marsh D.; Hoyer W.; Cherny D.; Zweckstetter M.; Griesinger C.; Jovin T. M.; Fernandez C. O. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 4294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soragni A.; Zambelli B.; Mukrasch M. D.; Biernat J.; Jeganathan S.; Griesinger C.; Ciurli S.; Mandelkow E.; Zweckstetter M. Biochemistry 2008, 47, 10841. [DOI] [PubMed] [Google Scholar]
- Viles J. H. Coord. Chem. Rev. 2012, 256, 2271. [Google Scholar]
- Bolognin S.; Messori L.; Zatta P. Neuromol. Med. 2009, 11, 223. [DOI] [PubMed] [Google Scholar]
- Brown D. R. Metallomics 2011, 3, 229. [DOI] [PubMed] [Google Scholar]
- Brown D. R. Metallomics 2011, 3, 226. [DOI] [PubMed] [Google Scholar]
- Faller P. ChemBioChem 2009, 10, 2837. [DOI] [PubMed] [Google Scholar]
- Kozlowski H.; Brown D.; Valensin G.. Metallochemistry of Neurodegeneration: Biological, Chemical, and Genetic Aspects; RSC Publishing: Cambridge, U.K., 2007. [Google Scholar]
- Lowe R.; Pountney D. L.; Jensen P. H.; Gai W. P.; Voelcker N. H. Protein Sci. 2004, 13, 3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binolfi A.; Valiente-Gabioud A. A.; Duran R.; Zweckstetter M.; Griesinger C.; Fernandez C. O. J. Am. Chem. Soc. 2011, 133, 194. [DOI] [PubMed] [Google Scholar]
- Ariga T.; Miyatake T.; Yu R. K. J. Neurosci. Res. 2010, 88, 2303. [DOI] [PubMed] [Google Scholar]
- Barrantes A.; Camero S.; Garcia-Lucas A.; Navarro P. J.; Benitez M. J.; Jimenez J. S. Curr. Alzheimer Res. 2012, 9, 924. [DOI] [PubMed] [Google Scholar]
- Cappai R.; Leck S. L.; Tew D. J.; Williamson N. A.; Smith D. P.; Galatis D.; Sharples R. A.; Curtain C. C.; Ali F. E.; Cherny R. A.; Culvenor J. G.; Bottomley S. P.; Masters C. L.; Barnham K. J.; Hill A. F. FASEB J. 2005, 19, 1377. [DOI] [PubMed] [Google Scholar]
- Fernandez C. O.; Hoyer W.; Zweckstetter M.; Jares-Erijman E. A.; Subramaniam V.; Griesinger C.; Jovin T. M. EMBO J. 2004, 23, 2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde M. L.; Vasudevaraju P.; Rao K. J. Front. Biosci. 2010, 15, 418. [DOI] [PubMed] [Google Scholar]
- Liu I. H.; Uversky V. N.; Munishkina L. A.; Fink A. L.; Halfter W.; Cole G. J. Glycobiology 2005, 15, 1320. [DOI] [PubMed] [Google Scholar]
- Mazzulli J. R.; Xu Y. H.; Sun Y.; Knight A. L.; McLean P. J.; Caldwell G. A.; Sidransky E.; Grabowski G. A.; Krainc D. Cell 2011, 146, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papy-Garcia D.; Christophe M.; Huynh M. B.; Fernando S.; Ludmilla S.; Sepulveda-Diaz J. E.; Raisman-Vozari R. Curr. Protein Pept. Sci. 2011, 12, 258. [DOI] [PubMed] [Google Scholar]
- Parekh-Olmedo H.; Wang J.; Gusella J. F.; Kmiec E. B. J. Mol. Neurosci. 2004, 24, 257. [DOI] [PubMed] [Google Scholar]
- Rabe M.; Soragni A.; Reynolds N. P.; Verdes D.; Liverani E.; Riek R.; Seeger S. ACS Chem. Neurosci. 2013, 4, 408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasahara K.; Morigaki K.; Okazaki T.; Hamada D. Biochemistry 2012, 51, 6908. [DOI] [PubMed] [Google Scholar]
- Zhou X.; Xu J. PLoS One 2012, 7, e46245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieschke J.; Herbst M.; Wiglenda T.; Friedrich R. P.; Boeddrich A.; Schiele F.; Kleckers D.; Lopez del Amo J. M.; Gruning B. A.; Wang Q.; Schmidt M. R.; Lurz R.; Anwyl R.; Schnoegl S.; Fandrich M.; Frank R. F.; Reif B.; Gunther S.; Walsh D. M.; Wanker E. E. Nat. Chem. Biol. 2011, 8, 93. [DOI] [PubMed] [Google Scholar]
- Bulic B.; Pickhardt M.; Mandelkow E. M.; Mandelkow E. Neuropharmacology 2010, 59, 276. [DOI] [PubMed] [Google Scholar]
- Ehrnhoefer D. E.; Bieschke J.; Boeddrich A.; Herbst M.; Masino L.; Lurz R.; Engemann S.; Pastore A.; Wanker E. E. Nat. Struct. Mol. Biol. 2008, 15, 558. [DOI] [PubMed] [Google Scholar]
- Hard T.; Lendel C. J. Mol. Biol. 2012, 421, 441. [DOI] [PubMed] [Google Scholar]
- Lamberto G. R.; Binolfi A.; Orcellet M. L.; Bertoncini C. W.; Zweckstetter M.; Griesinger C.; Fernandez C. O. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 21057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamberto G. R.; Torres-Monserrat V.; Bertoncini C. W.; Salvatella X.; Zweckstetter M.; Griesinger C.; Fernandez C. O. J. Biol. Chem. 2012, 286, 32036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono K.; Hasegawa K.; Naiki H.; Yamada M. Neurochem. Int. 2006, 48, 275. [DOI] [PubMed] [Google Scholar]
- Porat Y.; Abramowitz A.; Gazit E. Chem. Biol. Drug Des. 2006, 67, 27. [DOI] [PubMed] [Google Scholar]
- Shoval H.; Lichtenberg D.; Gazit E. Amyloid 2007, 14, 73. [DOI] [PubMed] [Google Scholar]
- Grelle G.; Otto A.; Lorenz M.; Frank R. F.; Wanker E. E.; Bieschke J. Biochemistry 2011, 50, 10624. [DOI] [PubMed] [Google Scholar]
- Azriel R.; Gazit E. J. Biol. Chem. 2001, 276, 34156. [DOI] [PubMed] [Google Scholar]
- Castelletto V.; Hamley I. W.; Cenker C.; Olsson U.; Adamcik J.; Mezzenga R.; Miravet J. F.; Escuder B.; Rodriguez-Llansola F. J. Phys. Chem. B 2011, 115, 2107. [DOI] [PubMed] [Google Scholar]
- Butler E. K.; Voigt A.; Lutz A. K.; Toegel J. P.; Gerhardt E.; Karsten P.; Falkenburger B.; Reinartz A.; Winklhofer K. F.; Schulz J. B. PLoS Genet. 2012, 8, e1002488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Bianco C.; Shorter J.; Regulier E.; Lashuel H.; Iwatsubo T.; Lindquist S.; Aebischer P. J. Clin. Invest. 2008, 118, 3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk K. C.; Mills I. P.; Trojanowski J. Q.; Lee V. M. Biochemistry 2008, 47, 12614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson K. R.; Ward S. M.; Combs B.; Voss K.; Kanaan N. M.; Morfini G.; Brady S. T.; Gamblin T. C.; Binder L. I. Biochemistry 2011, 50, 10300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss K.; Combs B.; Patterson K. R.; Binder L. I.; Gamblin T. C. Biochemistry 2012, 51, 888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou F.; Netzer W. J.; Tanemura K.; Li F.; Hartl F. U.; Takashima A.; Gouras G. K.; Greengard P.; Xu H. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker J. L.; Zareie M. H.; Fong H.; Sarikaya M.; Muchowski P. J. Nat. Struct. Mol. Biol. 2004, 11, 1215. [DOI] [PubMed] [Google Scholar]
- Jana N. R.; Tanaka M.; Wang G.; Nukina N. Hum. Mol. Genet. 2000, 9, 2009. [DOI] [PubMed] [Google Scholar]
- Narayan P.; Meehan S.; Carver J. A.; Wilson M. R.; Dobson C. M.; Klenerman D. Biochemistry 2012, 51, 9270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayan P.; Orte A.; Clarke R. W.; Bolognesi B.; Hook S.; Ganzinger K. A.; Meehan S.; Wilson M. R.; Dobson C. M.; Klenerman D. Nat. Struct. Mol. Biol. 2011, 19, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowther R. A.; Daniel S. E.; Goedert M. Neurosci. Lett. 2000, 292, 128. [DOI] [PubMed] [Google Scholar]
- Goedert M.; Jakes R.; Spillantini M. G.; Hasegawa M.; Smith M. J.; Crowther R. A. Nature 1996, 383, 550. [DOI] [PubMed] [Google Scholar]
- Inoue S.; Kisilevsky R. Methods Enzymol. 1999, 309, 496. [DOI] [PubMed] [Google Scholar]
- Inoue S.; Kuroiwa M.; Kisilevsky R. Brain Res. Rev. 1999, 29, 218. [DOI] [PubMed] [Google Scholar]
- Jaikaran E. T.; Clark A. Biochim. Biophys. Acta 2001, 1537, 179. [DOI] [PubMed] [Google Scholar]
- Spillantini M. G. Parkinsonism Relat. Disord. 1999, 5, 157. [DOI] [PubMed] [Google Scholar]
- Andrews M. E.; Inayathullah N. M.; Jayakumar R.; Malar E. J. J. Struct. Biol. 2009, 166, 116. [DOI] [PubMed] [Google Scholar]
- Heise H.; Hoyer W.; Becker S.; Andronesi O. C.; Riedel D.; Baldus M. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 15871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paravastu A. K.; Qahwash I.; Leapman R. D.; Meredith S. C.; Tycko R. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 7443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J. X.; Qiang W.; Yau W. M.; Schwieters C. D.; Meredith S. C.; Tycko R. Cell 2013, 154, 1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andre W.; Sandt C.; Dumas P.; Djian P.; Hoffner G. Anal. Chem. 2013, 85, 3765. [DOI] [PubMed] [Google Scholar]
- Miller L. M.; Bourassa M. W.; Smith R. J. Biochim. Biophys. Acta 2013, 1828, 2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nekooki-Machida Y.; Kurosawa M.; Nukina N.; Ito K.; Oda T.; Tanaka M. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 9679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borwankar T.; Rothlein C.; Zhang G.; Techen A.; Dosche C.; Ignatova Z. Biochemistry 2011, 50, 2048. [DOI] [PubMed] [Google Scholar]
- Ignatova Z.; Gierasch L. M. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 13357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto G.; Kim S.; Morimoto R. I. J. Biol. Chem. 2006, 281, 4477. [DOI] [PubMed] [Google Scholar]
- Kaminski Schierle G. S.; van de Linde S.; Erdelyi M.; Esbjorner E. K.; Klein T.; Rees E.; Bertoncini C. W.; Dobson C. M.; Sauer M.; Kaminski C. F. J. Am. Chem. Soc. 2011, 133, 12902. [DOI] [PubMed] [Google Scholar]
- Du H.; Guo L.; Fang F.; Chen D.; Sosunov A. A.; McKhann G. M.; Yan Y.; Wang C.; Zhang H.; Molkentin J. D.; Gunn-Moore F. J.; Vonsattel J. P.; Arancio O.; Chen J. X.; Yan S. D. Nat. Med. 2008, 14, 1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren P. H.; Lauckner J. E.; Kachirskaia I.; Heuser J. E.; Melki R.; Kopito R. R. Nat. Cell Biol. 2009, 11, 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberti M. J.; Bertoncini C. W.; Klement R.; Jares-Erijman E. A.; Jovin T. M. Nat. Methods 2007, 4, 345. [DOI] [PubMed] [Google Scholar]
- Fauerbach J. A.; Yushchenko D. A.; Shahmoradian S. H.; Chiu W.; Jovin T. M.; Jares-Erijman E. A. Biophys. J. 2012, 102, 1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberti M. J.; Folling J.; Celej M. S.; Bossi M.; Jovin T. M.; Jares-Erijman E. A. Biophys. J. 2012, 102, 1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberti M. J.; Jovin T. M.; Jares-Erijman E. PLoS One 2011, 6, e23338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminski Schierle G. S.; Bertoncini C. W.; Chan F. T.; van der Goot A. T.; Schwedler S.; Skepper J.; Schlachter S.; van Ham T.; Esposito A.; Kumita J. R.; Nollen E. A.; Dobson C. M.; Kaminski C. F. ChemPhysChem 2011, 12, 673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosharov E. V.; Larsen K. E.; Kanter E.; Phillips K. A.; Wilson K.; Schmitz Y.; Krantz D. E.; Kobayashi K.; Edwards R. H.; Sulzer D. Neuron 2009, 62, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham C. L.; Leong S. L.; Ali F. E.; Kenche V. B.; Hill A. F.; Gras S. L.; Barnham K. J.; Cappai R. J. Mol. Biol. 2009, 387, 771. [DOI] [PubMed] [Google Scholar]
- Devine M. J.; Ryten M.; Vodicka P.; Thomson A. J.; Burdon T.; Houlden H.; Cavaleri F.; Nagano M.; Drummond N. J.; Taanman J. W.; Schapira A. H.; Gwinn K.; Hardy J.; Lewis P. A.; Kunath T. Nat. Commun. 2011, 2, 440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grskovic M.; Javaherian A.; Strulovici B.; Daley G. Q. Nat. Rev. Drug. Discovery 2011, 10, 915. [DOI] [PubMed] [Google Scholar]
- Soldner F.; Hockemeyer D.; Beard C.; Gao Q.; Bell G. W.; Cook E. G.; Hargus G.; Blak A.; Cooper O.; Mitalipova M.; Isacson O.; Jaenisch R. Cell 2009, 136, 964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemkul J. A.; Bevan D. R. ACS Chem. Neurosci. 2012, 3, 845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen M. O.; Jogini V.; Borhani D. W.; Leffler A. E.; Dror R. O.; Shaw D. E. Science 2012, 336, 229. [DOI] [PubMed] [Google Scholar]
- Piana S.; Lindorff-Larsen K.; Shaw D. E. Proc. Natl. Acad. Sci. U.S.A. 2013, 109, 17845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piana S.; Lindorff-Larsen K.; Shaw D. E. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 5915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan Y.; Arkhipov A.; Kim E. T.; Pan A. C.; Shaw D. E. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 7270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee Y. M.; Pande V. S. Biophys. J. 2003, 84, 775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheraga H. A.; Khalili M.; Liwo A. Annu. Rev. Phys. Chem. 2007, 58, 57. [DOI] [PubMed] [Google Scholar]
- Sugita Y.; Okamoto Y. Chem. Phys. Lett. 1999, 314, 141. [Google Scholar]
- Bicout D. J.; Field M. J. J. Phys. Chem. 1996, 100, 2489. [Google Scholar]
- Ridgway D.; Broderick G.; Lopez-Campistrous A.; Ru’aini M.; Winter P.; Hamilton M.; Boulanger P.; Kovalenko A.; Ellison M. J. Biophys. J. 2008, 94, 3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Campistrous A.; Semchuk P.; Burke L.; Palmer-Stone T.; Brokx S. J.; Broderick G.; Bottorff D.; Bolch S.; Weiner J. H.; Ellison M. J. Mol. Cell. Proteomics 2005, 4, 1205. [DOI] [PubMed] [Google Scholar]
- Mika J. T.; Krasnikov V.; van den Bogaart G.; de Haan F.; Poolman B. PLoS One 2011, 6, e25664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuffee S. R.; Elcock A. H. J. Am. Chem. Soc. 2006, 128, 12098. [DOI] [PubMed] [Google Scholar]
- Ignatova Z.; Gierasch L. M. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignatova Z.; Krishnan B.; Bombardier J. P.; Marcelino A. M.; Hong J.; Gierasch L. M. Biopolymers 2007, 88, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando T.; Skolnick J. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 18457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frembgen-Kesner T.; Elcock A. H. J. Chem. Theory Comput. 2009, 5, 242. [DOI] [PubMed] [Google Scholar]
- Ando T.; Chow E.; Saad Y.; Skolnick J. J. Chem. Phys. 2012, 137, 064106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elcock A. H. J. Chem. Theory Comput. 2013, 9, 3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y.; Selenko P. Curr. Opin. Struct. Biol. 2010, 20, 640. [DOI] [PubMed] [Google Scholar]
- Maldonado A. Y.; Burz D. S.; Shekhtman A. Prog. Nucl. Magn. Reson. Spectrosc. 2011, 59, 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serber Z.; Corsini L.; Durst F.; Dotsch V. Methods Enzymol. 2005, 394, 17. [DOI] [PubMed] [Google Scholar]
- Bertrand K.; Reverdatto S.; Burz D. S.; Zitomer R.; Shekhtman A. J. Am. Chem. Soc. 2012, 134, 12798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamatsu J.; O’Donovan D.; Tanaka T.; Shirai T.; Hourai Y.; Mikawa T.; Ikeya T.; Mishima M.; Boucher W.; Smith B. O.; Laue E. D.; Shirakawa M.; Ito Y. J. Am. Chem. Soc. 2013, 135, 1688. [DOI] [PubMed] [Google Scholar]
- Banci L.; Barbieri L.; Luchinat E.; Secci E. Chem. Biol. 2013, 20, 747. [DOI] [PubMed] [Google Scholar]
- Banci L.; Barbieri L.; Bertini I.; Luchinat E.; Secci E.; Zhao Y.; Aricescu A. R. Nat. Chem. Biol. 2013, 9, 297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai T.; Tochio H.; Tenno T.; Ito Y.; Kokubo T.; Hiroaki H.; Shirakawa M. J. Biomol. NMR 2006, 36, 179. [DOI] [PubMed] [Google Scholar]
- Selenko P.; Serber Z.; Gadea B.; Ruderman J.; Wagner G. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 11904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielsson J.; Inomata K.; Murayama S.; Tochio H.; Lang L.; Shirakawa M.; Oliveberg M. J. Am. Chem. Soc. 2013, 135, 10266. [DOI] [PubMed] [Google Scholar]
- Ogino S.; Kubo S.; Umemoto R.; Huang S.; Nishida N.; Shimada I. J. Am. Chem. Soc. 2009, 131, 10834. [DOI] [PubMed] [Google Scholar]
- Kubo S.; Nishida N.; Udagawa Y.; Takarada O.; Ogino S.; Shimada I. Angew. Chem., Int. Ed. 2013, 52, 1208. [DOI] [PubMed] [Google Scholar]
- Luh L. M.; Hansel R.; Lohr F.; Kirchner D. K.; Krauskopf K.; Pitzius S.; Schafer B.; Tufar P.; Corbeski I.; Guntert P.; Dotsch V. J. Am. Chem. Soc. 2013, 135, 13796. [DOI] [PubMed] [Google Scholar]
- Amata I.; Maffei M.; Igea A.; Gay M.; Vilaseca M.; Nebreda A. R.; Pons M. ChemBioChem 2013, 14, 1820. [DOI] [PubMed] [Google Scholar]
- Dedmon M. M.; Patel C. N.; Young G. B.; Pielak G. J. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 12681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selenko P.; Frueh D. P.; Elsaesser S. J.; Haas W.; Gygi S. P.; Wagner G. Nat. Struct. Mol. Biol. 2008, 15, 321. [DOI] [PubMed] [Google Scholar]
- Cordier F.; Chaffotte A.; Terrien E.; Prehaud C.; Theillet F. X.; Delepierre M.; Lafon M.; Buc H.; Wolff N. J. Am. Chem. Soc. 2012, 134, 20533. [DOI] [PubMed] [Google Scholar]
- Dose A.; Liokatis S.; Theillet F. X.; Selenko P.; Schwarzer D. ACS Chem. Biol. 2011, 6, 419. [DOI] [PubMed] [Google Scholar]
- Liokatis S.; Dose A.; Schwarzer D.; Selenko P. J. Am. Chem. Soc. 2010, 132, 14704. [DOI] [PubMed] [Google Scholar]
- Theillet F. X.; Liokatis S.; Jost J. O.; Bekei B.; Rose H. M.; Binolfi A.; Schwarzer D.; Selenko P. J. Am. Chem. Soc. 2012, 134, 7616. [DOI] [PubMed] [Google Scholar]
- Theillet F.-X.; Rose H. M.; Liokatis S.; Binolfi A.; Thongwichian R.; Stuiver M.; Selenko P. Nat. Protoc. 2013, 8, 1416. [DOI] [PubMed] [Google Scholar]
- Li C.; Liu M. FEBS Lett. 2013, 587, 1008. [DOI] [PubMed] [Google Scholar]
- Croke R. L.; Sallum C. O.; Watson E.; Watt E. D.; Alexandrescu A. T. Protein Sci. 2008, 17, 1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waudby C. A.; Mantle M. D.; Cabrita L. D.; Gladden L. F.; Dobson C. M.; Christodoulou J. J. Am. Chem. Soc. 2012, 134, 11312. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Benton L. A.; Singh V.; Pielak G. J. J. Phys. Chem. Lett. 2012, 3, 2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartels T.; Choi J. G.; Selkoe D. J. Nature 2011, 477, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.; Perovic I.; Chittuluru J.; Kaganovich A.; Nguyen L. T.; Liao J.; Auclair J. R.; Johnson D.; Landeru A.; Simorellis A. K.; Ju S.; Cookson M. R.; Asturias F. J.; Agar J. N.; Webb B. N.; Kang C.; Ringe D.; Petsko G. A.; Pochapsky T. C.; Hoang Q. Q. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 17797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dettmer U.; Newman A. J.; Luth E. S.; Bartels T.; Selkoe D. J. Biol. Chem. 2013, 288, 6371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trexler A. J.; Rhoades E. Protein Sci. 2012, 21, 601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burre J.; Vivona S.; Diao J.; Sharma M.; Brunger A. T.; Sudhof T. C. Nature 2013, 498, E4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreb P. H.; Zhen W.; Poon A. W.; Conway K. A.; Lansbury P. T. Jr. Biochemistry 1996, 35, 13709. [DOI] [PubMed] [Google Scholar]
- Hughes K. T.; Gillen K. L.; Semon M. J.; Karlinsey J. E. Science 1993, 262, 1277. [DOI] [PubMed] [Google Scholar]
- Daughdrill G. W.; Hanely L. J.; Dahlquist F. W. Biochemistry 1998, 37, 1076. [DOI] [PubMed] [Google Scholar]
- Daughdrill G. W.; Chadsey M. S.; Karlinsey J. E.; Hughes K. T.; Dahlquist F. W. Nat. Struct. Biol. 1997, 4, 285. [DOI] [PubMed] [Google Scholar]
- Sillen A.; Barbier P.; Landrieu I.; Lefebvre S.; Wieruszeski J. M.; Leroy A.; Peyrot V.; Lippens G. Biochemistry 2007, 46, 3055. [DOI] [PubMed] [Google Scholar]
- Landrieu I.; Lacosse L.; Leroy A.; Wieruszeski J. M.; Trivelli X.; Sillen A.; Sibille N.; Schwalbe H.; Saxena K.; Langer T.; Lippens G. J. Am. Chem. Soc. 2006, 128, 3575. [DOI] [PubMed] [Google Scholar]
- Theillet F. X.; Smet-Nocca C.; Liokatis S.; Thongwichian R.; Kosten J.; Yoon M. K.; Kriwacki R. W.; Landrieu I.; Lippens G.; Selenko P. J. Biomol. NMR 2012, 54, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole N. B.; Smith C. L.; Sciaky N.; Terasaki M.; Edidin M.; Lippincott-Schwartz J. Science 1996, 273, 797. [DOI] [PubMed] [Google Scholar]
- Kim S. A.; Sanabria H.; Digman M. A.; Gratton E.; Schwille P.; Zipfel W. R.; Waxham M. N. J. Neurosci. 2010, 30, 16409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelrod D.; Koppel D. E.; Schlessinger J.; Elson E.; Webb W. W. Biophys. J. 1976, 16, 1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Digman M. A.; Gratton E. Annu. Rev. Phys. Chem. 2010, 62, 645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick J. A. J.; Lillemeier B. F. Curr. Opin. Struct. Biol. 2011, 21, 650. [DOI] [PubMed] [Google Scholar]
- Coffman V. C.; Wu J.-Q. Trends Biochem. Sci. 2012, 37, 499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Lamothe R.; Sherratt D. J.; Leake M. C. Science 2010, 328, 498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulbrich M. H.; Isacoff E. Y. Nat. Methods 2007, 4, 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dukes K. D.; Rodenberg C. F.; Lammi R. K. Anal. Biochem. 2008, 382, 29. [DOI] [PubMed] [Google Scholar]
- Johnson R. D.; Schauerte J. A.; Wisser K. C.; Gafni A.; Steel D. G. PLoS One 2011, 6, e23970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kask P.; Palo K.; Ullmann D.; Gall K. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 13756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald P.; Johnson J.; Smith E.; Chen Y.; Mueller J. D. Methods Enzymol. 2013, 518, 71. [DOI] [PubMed] [Google Scholar]
- Kerppola T. K. Chem. Soc. Rev. 2009, 38, 2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisamakis E.; Valeri A.; Kalinin S.; Rothwell P. J.; Seidel C. A. M. Methods Enzymol. 2010, 475, 455. [DOI] [PubMed] [Google Scholar]
- Takahashi T.; Kikuchi S.; Katada S.; Nagai Y.; Nishizawa M.; Onodera O. Hum. Mol. Genet. 2007, 17, 345. [DOI] [PubMed] [Google Scholar]
- Chan F. T. S.; Kaminski C. F.; Kaminski Schierle G. S. ChemPhysChem 2010, 12, 500. [DOI] [PubMed] [Google Scholar]
- van Ham T. J.; Esposito A.; Kumita J. R.; Hsu S.-T. D.; Schierle G. S. K.; Kaminski C. F.; Dobson C. M.; Nollen E. A. A.; Bertoncini C. W. J. Mol. Biol. 2010, 395, 627. [DOI] [PubMed] [Google Scholar]
- Gould T. J.; Hess S. T.; Bewersdorf J. Annu. Rev. Biomed. Eng. 2012, 14, 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B.; Babcock H.; Zhuang X. Cell 2010, 143, 1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta P.; Van Engelenburg S.; Lippincott-Schwartz J. Dev. Cell 2012, 23, 1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahl S. J.; Weiss L. E.; Duim W. C.; Frydman J.; Moerner W. E. Sci. Rep. 2012, 2, 895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L.; Shao L.; Higgins C. D.; Poulton J. S.; Peifer M.; Davidson M. W.; Wu X.; Goldstein B.; Betzig E. Cell 2012, 151, 1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höckendorf B.; Thumberger T.; Wittbrodt J. Dev. Cell 2012, 23, 1111. [DOI] [PubMed] [Google Scholar]
- Ebbinghaus S.; Dhar A.; McDonald D.; Gruebele M. Nat. Methods 2010, 7, 319. [DOI] [PubMed] [Google Scholar]
- Dhar A.; Prigozhin M.; Gelman H.; Gruebele M. Methods Mol. Biol. 2012, 895, 101. [DOI] [PubMed] [Google Scholar]
- Chen Y.-C.; Clegg R. M. Photosynth. Res. 2009, 102, 143. [DOI] [PubMed] [Google Scholar]
- Landgraf D.; Okumus B.; Chien P.; Baker T. A.; Paulsson J. Nat. Methods 2012, 9, 480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snapp E. L. Trends Cell Biol. 2009, 19, 649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacharias D. A.; Violin J. D.; Newton A. C.; Tsien R. Y. Science 2002, 296, 913. [DOI] [PubMed] [Google Scholar]
- Costantini L. M.; Fossati M.; Francolini M.; Snapp E. L. Traffic 2012, 13, 643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M.; Chang H.; Zhang Y.; Yu J.; Wu L.; Ji W.; Chen J.; Liu B.; Lu J.; Liu Y.; Zhang J.; Xu P.; Xu T. Nat. Methods 2012, 9, 727. [DOI] [PubMed] [Google Scholar]
- Griffin B. A.; Adams S. R.; Tsien R. Y. Science 1998, 281, 269. [DOI] [PubMed] [Google Scholar]
- Martin B. R.; Giepmans B. N. G.; Adams S. R.; Tsien R. Y. Nat. Biotechnol. 2005, 23, 1308. [DOI] [PubMed] [Google Scholar]
- Anderson V. L.; Webb W. W. BMC Biotechnol. 2011, 11, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams S. R.; Campbell R. E.; Gross L. A.; Martin B. R.; Walkup G. K.; Yao Y.; Llopis J.; Tsien R. Y. J. Am. Chem. Soc. 2002, 124, 6063. [DOI] [PubMed] [Google Scholar]
- Krishnan B.; Gierasch L. M. Chem. Biol. 2008, 15, 1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowder M. A.; Appelbaum J. S.; Hobert E. M.; Schepartz A. Curr. Opin. Chem. Biol. 2011, 15, 781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignatova Z.; Thakur A. K.; Wetzel R.; Gierasch L. M. J. Biol. Chem. 2007, 282, 36736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz J.; Gierasch L. M.; Ignatova Z. Biochemistry 2008, 47, 4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duennwald M. L.; Jagadish S.; Muchowski P. J.; Lindquist S. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 11045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellisdon A. M.; Thomas B.; Bottomley S. P. J. Biol. Chem. 2006, 281, 16888. [DOI] [PubMed] [Google Scholar]
- Ramdzan Y. M.; Nisbet R. M.; Miller J.; Finkbeiner S.; Hill A. F.; Hatters D. M. Chem. Biol. 2010, 17, 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetzel R. J. Mol. Biol. 2012, 421, 466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brignull H. R.; Morley J. F.; Garcia S. M.; Morimoto R. I. Methods Enzymol. 2006, 412, 256. [DOI] [PubMed] [Google Scholar]
- Finkbeiner S. Cold Spring Harbor Perspect. Biol. 2011, 3, a007476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igarashi S. S.; Koide R. R.; Shimohata T. T.; Yamada M. M.; Hayashi Y. Y.; Takano H. H.; Date H. H.; Oyake M. M.; Sato T. T.; Sato A. A.; Egawa S. S.; Ikeuchi T. T.; Tanaka H. H.; Nakano R. R.; Tanaka K. K.; Hozumi I. I.; Inuzuka T. T.; Takahashi H. H.; Tsuji S. S. Nat. Genet. 1998, 18, 111. [DOI] [PubMed] [Google Scholar]
- Morley J. F.; Brignull H. R.; Weyers J. J.; Morimoto R. I. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 10417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu A.-L.; Murphy C. T.; Kenyon C. Science 2003, 300, 1142. [DOI] [PubMed] [Google Scholar]
- Moronetti Mazzeo L. E.; Dersh D.; Boccitto M.; Kalb R. G.; Lamitina T. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 10587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva M. C.; Fox S.; Beam M.; Thakkar H.; Amaral M. D.; Morimoto R. I. PLoS Genetics 2011, 7, e1002438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsvetkov A. S.; Miller J.; Arrasate M.; Wong J. S.; Pleiss M. A.; Finkbeiner S. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 16982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramdzan Y. M.; Polling S.; Chia C. P. Z.; Ng I. H. W.; Ormsby A. R.; Croft N. P.; Purcell A. W.; Bogoyevitch M. A.; Ng D. C. H.; Gleeson P. A.; Hatters D. M. Nat. Methods 2012, 9, 467. [DOI] [PubMed] [Google Scholar]
- Brignull H. R.; Morley J. F.; Morimoto R. I. Adv. Exp. Med. Biol. 2007, 594, 167. [DOI] [PubMed] [Google Scholar]
- Miller J.; Arrasate M.; Shaby B. A.; Mitra S.; Masliah E.; Finkbeiner S. J. Neurosci. 2010, 30, 10541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrasate M.; Mitra S.; Schweitzer E. S.; Segal M. R.; Finkbeiner S. Nature 2004, 431, 805. [DOI] [PubMed] [Google Scholar]
- Taylor J. P.; Tanaka F.; Robitschek J.; Sandoval C. M.; Taye A.; Markovic-Plese S. S.; Fischbeck K. H. Hum. Mol. Genet. 2003, 12, 749. [DOI] [PubMed] [Google Scholar]
- Ross C. A.; Poirier M. A. Nat. Rev. Mol. Cell Biol. 2005, 6, 891. [DOI] [PubMed] [Google Scholar]
- Treusch S.; Cyr D. M.; Lindquist S. Cell Cycle 2009, 8, 1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi H. Y.; Orr H. T. Curr. Opin. Neurobiol. 1999, 9, 566. [DOI] [PubMed] [Google Scholar]
- Beam M.; Silva M. C.; Morimoto R. I. J. Biol. Chem. 2012, 287, 26136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Outeiro T. F.; Putcha P.; Tetzlaff J. E.; Spoelgen R.; Koker M.; Carvalho F.; Hyman B. T.; McLean P. J. PLoS One 2008, 3, e1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kothawala A.; Kilpatrick K.; Novoa J. A.; Segatori L. PLoS One 2012, 7, e43505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymenidou M.; Cleveland D. W. J. Exp. Med. 2012, 209, 889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker L. C.; LeVine H. J. Biol. Chem. 2012, 287, 33109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danzer K. M.; Ruf W. P.; Putcha P.; Joyner D.; Hashimoto T.; Glabe C.; Hyman B. T.; McLean P. J. FASEB J. 2011, 25, 326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desplats P.; Lee H.-J.; Bae E.-J.; Patrick C.; Rockenstein E.; Crews L.; Spencer B.; Masliah E.; Lee S.-J. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 13010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boassa D.; Berlanga M. L.; Yang M. A.; Terada M.; Hu J.; Bushong E. A.; Hwang M.; Masliah E.; George J. M.; Ellisman M. H. J. Neurosci. 2013, 33, 2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin D. L.; Troyer M. D.; Nakamura K.; Kubo S.-i.; Anthony M. D.; Edwards R. H. J. Neurosci. 2004, 24, 6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean P. J.; Kawamata H.; Hyman B. T. Neuroscience 2001, 104, 901. [DOI] [PubMed] [Google Scholar]
- Specht C. G.; Tigaret C. M.; Rast G. F.; Thalhammer A.; Rudhard Y.; Schoepfer R. Mol. Cell. Neurosci. 2005, 28, 326. [DOI] [PubMed] [Google Scholar]
- van Ham T. J.; Thijssen K. L.; Breitling R.; Hofstra R. M. W.; Plasterk R. H. A.; Nollen E. A. A. PLoS Genetics 2008, 4, e1000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockenstein E.; Schwach G.; Ingolic E.; Adame A.; Crews L.; Mante M.; Pfragner R.; Schreiner E.; Windisch M.; Masliah E. J. Neurosci. 2005, 80, 247. [DOI] [PubMed] [Google Scholar]
- Nakamura K.; Nemani V. M.; Azarbal F.; Skibinski G.; Levy J. M.; Egami K.; Munishkina L.; Zhang J.; Gardner B.; Wakabayashi J.; Sesaki H.; Cheng Y.; Finkbeiner S.; Nussbaum R. L.; Masliah E.; Edwards R. H. J. Biol. Chem. 2011, 286, 20710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auluck P. K.; Caraveo G.; Lindquist S. Annu. Rev. Cell Dev. Biol. 2010, 26, 211. [DOI] [PubMed] [Google Scholar]
- Masters C. L.; Selkoe D. J. Cold Spring Harbor Perspect. Med. 2012, 2, a006262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman D. A.; Chakrabartty A. Biophys. J. 2009, 96, 4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccotosto G. D.; Kozer N.; Chow T. T. Y.; Chon J. W. M.; Clayton A. H. A. Biophys. J. 2013, 104, 1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X.; Crick S. L.; Bu G.; Frieden C.; Pappu R. V.; Lee J. M. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 20324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nag S.; Chen J.; Irudayaraj J.; Maiti S. Biophys. J. 2010, 99, 1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner M.; Lacor P. N.; Velasco P. T.; Xu J.; Contractor A.; Klein W. L.; Triller A. Neuron 2010, 66, 739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chafekar S. M.; Baas F.; Scheper W. Biochim. Biophys. Acta 2008, 1782, 523. [DOI] [PubMed] [Google Scholar]
- Hossain S.; Grande M.; Ahmadkhanov G.; Pramanik A. Exp. Mol. Pathol. 2007, 82, 169. [DOI] [PubMed] [Google Scholar]
- Ding H.; Wong P. T.; Lee E. L.; Gafni A.; Steel D. G. Biophys. J. 2009, 97, 912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayan P.; Ganzinger K. A.; McColl J.; Weimann L.; Meehan S.; Qamar S.; Carver J. A.; Wilson M. R. St; George-Hyslop P.; Dobson C. M.; Klenerman D. J. Am. Chem. Soc. 2013, 135, 1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman D. A.; McLaurin J.; Chakrabartty A. BMC Neurosci. 2007, 8, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta P.; Garai K.; Sahoo B.; Shi Y.; Callaway D. J. E.; Maiti S. Biochemistry 2003, 42, 10506. [DOI] [PubMed] [Google Scholar]
- Lacor P. N.; Buniel M. C.; Chang L.; Fernandez S. J.; Gong Y.; Viola K. L.; Lambert M. P.; Velasco P. T.; Bigio E. H.; Finch C. E.; Krafft G. A.; Klein W. L. J. Neurosci. 2004, 24, 10191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koffie R. M.; Meyer-Luehmann M.; Hashimoto T.; Adams K. W.; Mielke M. L.; Garcia-Alloza M.; Micheva K. D.; Smith S. J.; Kim M. L.; Lee V. M.; Hyman B. T.; Spires-Jones T. L. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lue L. F.; Kuo Y. M.; Roher A. E.; Brachova L.; Shen Y.; Sue L.; Beach T.; Kurth J. H.; Rydel R. E.; Rogers J. Am. J. Pathol. 1999, 155, 853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean C. A.; Cherny R. A.; Fraser F. W.; Fuller S. J.; Smith M. J.; Beyreuther K.; Bush A. I.; Masters C. L. Ann. Neurol. 1999, 46, 860. [DOI] [PubMed] [Google Scholar]
- Seubert P.; Vigo-Pelfrey C.; Esch F.; Lee M.; Dovey H.; Davis D.; Sinha S.; Schlossmacher M.; Whaley J.; Swindlehurst C.; McCormack R.; Wolfert R.; Selkoe D.; Lieberburg I.; Schenk D. Nature 1992, 359, 325. [DOI] [PubMed] [Google Scholar]
- Nag S.; Sarkar B.; Bandyopadhyay A.; Sahoo B.; Sreenivasan V. K. A.; Kombrabail M.; Muralidharan C.; Maiti S. J. Biol. Chem. 2011, 286, 13827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson R. D.; Schauerte J. A.; Chang C.-C.; Wisser K. C.; Althaus J. C.; Carruthers C. J. L.; Sutton M. A.; Steel D. G.; Gafni A. Biophys. J. 2013, 104, 894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller L. M.; Dumas P. Curr. Opin. Struct. Biol. 2010, 20, 649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byler D. M.; Susi H. Biopolymers 1986, 25, 469. [DOI] [PubMed] [Google Scholar]
- Goormaghtigh E.; Ruysschaert J. M.; Raussens V. Biophys. J. 2006, 90, 2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthaus C.; Bird B.; Miljkovic M.; Chernenko T.; Romeo M.; Diem M. Methods Cell Biol. 2008, 89, 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ami D.; Natalello A.; Doglia S. M. Methods Mol. Biol. 2012, 895, 85. [DOI] [PubMed] [Google Scholar]
- Barth A.; Zscherp C. Q. Rev. Biophys. 2002, 35, 369. [DOI] [PubMed] [Google Scholar]
- Natalello A.; Doglia S.. Intrinsically disordered proteins and induced folding studied by fourier transform infrared spectroscopy; Wiley: Honoken, NJ, 2010. [Google Scholar]
- Natalello A.; Santarella R.; Doglia S. M.; de Marco A. Protein Expr. Purif. 2008, 58, 356. [DOI] [PubMed] [Google Scholar]
- Holman H. Y. N.; Bechtel H. A.; Hao Z.; Martin M. C. Anal. Chem. 2010, 82, 8757. [DOI] [PubMed] [Google Scholar]
- Choo L. P.; Wetzel D. L.; Halliday W. C.; Jackson M.; LeVine S. M.; Mantsch H. H. Biophys. J. 1996, 71, 1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diomede L.; Cassata G.; Fiordaliso F.; Salio M.; Ami D.; Natalello A.; Doglia S. M.; De Luigi A.; Salmona M. Neurobiol. Dis. 2010, 40, 424. [DOI] [PubMed] [Google Scholar]
- Kretlow A.; Wang Q.; Kneipp J.; Lasch P.; Beekes M.; Miller L.; Naumann D. Biochim. Biophys. Acta 2006, 1758, 948. [DOI] [PubMed] [Google Scholar]
- Kim Y. S.; Liu L.; Axelsen P. H.; Hochstrasser R. M. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 7720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M.; Sachse C.; Richter W.; Xu C.; Fandrich M.; Grigorieff N. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 19813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller L. M.; Dumas P. Biochim. Biophys. Acta 2006, 1758, 846. [DOI] [PubMed] [Google Scholar]
- Forloni G.; Colombo L.; Girola L.; Tagliavini F.; Salmona M. FEBS Lett. 2001, 487, 404. [DOI] [PubMed] [Google Scholar]
- Kneipp J.; Miller L. M.; Joncic M.; Kittel M.; Lasch P.; Beekes M.; Naumann D. Biochim. Biophys. Acta 2003, 1639, 152. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Kretlow A.; Beekes M.; Naumann D.; Miller L. Vib. Spectrosc. 2005, 38, 61. [Google Scholar]