

Abstract
Vibrio cholerae is a Gram-negative bacterium that persists in aquatic reservoirs and causes the diarrheal disease cholera upon entry into a human host. V. cholerae employs the second messenger molecule 3′,5′-cyclic diguanylic acid (c-di-GMP) to transition between these two distinct lifestyles. c-di-GMP is synthesized by diguanylate cyclase (DGC) enzymes and hydrolyzed by phosphodiesterase (PDE) enzymes. Bacteria typically encode many different DGCs and PDEs within their genomes. Presumably, each enzyme senses and responds to cognate environmental cues by alteration of enzymatic activity. c-di-GMP represses the expression of virulence factors in V. cholerae, and it is predicted that the intracellular concentration of c-di-GMP is low during infection. Contrary to this model, we found that bile acids, a prevalent constituent of the human proximal small intestine, increase intracellular c-di-GMP in V. cholerae. We identified four c-di-GMP turnover enzymes that contribute to increased intracellular c-di-GMP in the presence of bile acids, and deletion of these enzymes eliminates the bile induction of c-di-GMP and biofilm formation. Furthermore, this bile-mediated increase in c-di-GMP is quenched by bicarbonate, the intestinal pH buffer secreted by intestinal epithelial cells. Our results lead us to propose that V. cholerae senses distinct microenvironments within the small intestine using bile and bicarbonate as chemical cues and responds by modulating the intracellular concentration of c-di-GMP.
INTRODUCTION
Vibrio cholerae is a Gram-negative bacterium responsible for the human diarrheal disease cholera. This bacterium primarily resides in marine reservoirs and associates with aquatic organisms by preferentially forming biofilms on chitinous surfaces (1–3). V. cholerae multiplies in the small intestine and upregulates two key virulence factors, toxin-coregulated pilus (TCP) and cholera toxin (CT), to cause severe and acute diarrhea. Modulation of the second messenger 3′,5′-cyclic diguanylic acid (c-di-GMP) by V. cholerae is thought to be one mechanism by which V. cholerae mediates the transition from life in aquatic environments to a virulent state in the human host. It is well documented that c-di-GMP regulates many bacterial phenotypes, including biofilm formation, motility, expression of virulence genes, and cell cycle progression (4–9). It has been proposed that the intracellular c-di-GMP concentration in V. cholerae is relatively high in aquatic environments, leading to a sessile biofilm-forming lifestyle, whereas in the human host, reduced c-di-GMP concentrations stimulate virulence factor expression (10). Due to the technical limitations of measuring intracellular c-di-GMP, this model has not been directly tested, and the specific environmental cues that regulate the transition between these two niches remain unknown.
c-di-GMP is synthesized by diguanylate cyclase (DGC) enzymes and hydrolyzed by c-di-GMP-specific phosphodiesterase (PDE) enzymes. All DGCs contain a conserved C-terminal GGDEF domain with a GG[D/E]EF active site motif and a highly variable N terminus that often carries conserved signal recognition domains such as GAF, PAS, or receiver domains (11, 12). Conversely, c-di-GMP PDEs contain either a conserved EAL or a HD-GYP domain in their C terminus (13, 14). Bacteria typically encode many DGCs and PDEs within their genome; V. cholerae possesses 61 predicted c-di-GMP turnover enzymes (15). It is not well understood why V. cholerae encodes so many DGCs and PDEs, and the functions of the variable N-terminal domains for most of these enzymes have not been characterized. One hypothesis is that each DGC or PDE senses and responds to a specific environmental signal by altering c-di-GMP synthesis or degradation activity in the C-terminal domain. While a few environmental cues, including light, oxygen, zinc, arginine, quorum sensing (QS) autoinducers, and norspermidine, have been shown to directly regulate DGCs or PDEs in various bacteria (14, 16–24), the environmental signals recognized by the vast majority of DGCs and PDEs remain unidentified.
Upon entering the human host, it is imperative that V. cholerae recognize environmental signals to mediate the transition from an aquatic bacterium to a human pathogen. One signal prevalent in the small intestine is bile. Bile is an antimicrobial substance secreted from the liver into the proximal small intestine that aids in digestion by emulsifying lipids. The composition of secreted bile is heterogeneous and includes inorganic salts, cholesterol, phospholipids, pigments, and bile acids (25). Bile acids are derived from cholesterol in the liver and are processed by hepatic cells to produce a mixture dominated by taurine and glycine conjugates (25, 26). Bile acids have detergent properties that enable the interaction between bile and digestive lipids. For a complete review on the properties of bile, refer to the work of Begley et al. (27).
The interplay between enteric bacteria and bile is extensive. In bacteria, there is evidence that bile causes oxidative stress and DNA damage and perturbs the cell membrane (28–30). To counteract this stress, V. cholerae upregulates the porin protein ompU in a toxR-dependent manner to increase bile resistance (31). V. cholerae also employs six resistance-nodulation-division family efflux pumps that have been implicated in bile resistance (32–34). Additionally, V. cholerae increases biofilm formation in a vpsR-dependent manner in the presence of bile acids, presumably to increase resistance to the deleterious effects of bile (35). It is clear that the sensing of bile by V. cholerae leads to distinct physiological changes, but a role for c-di-GMP in this process has not been described.
We hypothesized that bile is an environmental signal sensed by V. cholerae to recognize and adapt to growth in the human environment by modulating c-di-GMP signaling pathways. Based on current models postulating that V. cholerae reduces intracellular c-di-GMP in the human host, we predicted that bile acids would reduce global c-di-GMP concentrations. Surprisingly, we discovered that bile acids increase intracellular c-di-GMP. A screen of the activity of all 61 V. cholerae c-di-GMP turnover enzymes in the presence and absence of bile acids identified three DGCs that showed increased c-di-GMP synthesis in the presence of bile acids. Furthermore, a screen on the expression of all 61 V. cholerae c-di-GMP turnover enzymes revealed that bile acids inhibited the expression of one PDE. Deletion of these four enzymes abolished the induction of c-di-GMP by bile and negated the ability of V. cholerae to form biofilms in the presence of bile. Bicarbonate, a biological pH buffer secreted by intestinal epithelial cells in the small intestine, suppressed the bile-induced increases in intracellular c-di-GMP. We propose that bile and bicarbonate inversely control c-di-GMP levels in V. cholerae, allowing this bacterium to sense and adapt to local environmental niches within the small intestine.
MATERIALS AND METHODS
Growth conditions and molecular methods.
The V. cholerae El Tor biotype strain C6706str2 was used for all experiments (36), and Escherichia coli strains DH10B (Invitrogen) and S17-λpir (37) were used to harbor and conjugate plasmid DNA into V. cholerae. The construction of the ΔvpsL, ΔluxO, and ΔhapR strains has been described elsewhere (38–40). For all experiments, unless otherwise specified, cultures were grown in Luria-Bertani (LB) medium at 35°C with shaking at 220 rpm. When necessary, medium was supplemented with kanamycin (Sigma) at 100 μg/ml or chloramphenicol (Sigma) at 10 μg/ml. The inducer isopropyl-β-d-thiogalactopyranoside (IPTG) was added at 0.1 mM when required.
Synthetic human bile (SHB) is a mixture of six purified conjugated bile acids added to LB medium at physiologically relevant concentrations to mimic the human small intestine (25, 26, 41, 42). All bile acids were purchased from Sigma. The conjugated bile acids added were taurocholate (0.46 mM), glycocholate (0.93 mM), taurochenodeoxycholate (0.46 mM), glycochenodeoxycholate (0.93 mM), taurodeoxycholate (0.32 mM), and glycodeoxycholate (0.64 mM). Bovine bile (BV; Sigma) was added as a supplement at 0.4% (wt/vol). 3-[(3-Cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) was added as a supplement at 0.4% (wt/vol), and sodium dodecyl sulfate (SDS) was added as a supplement at 0.01% (wt/vol) due to its potent bactericidal activity. Taurine and glycine were each added as a supplement at 4 mM. Sodium bicarbonate (BiC) was added as a supplement when indicated at 0.3% (wt/vol) (49.2 mM), consistent with cholera toxin-inducing conditions (43), while Tris was added as a supplement when indicated at 0.6% (wt/vol) (49.5 mM).
All of the protocols used in this study for DNA manipulation and plasmid construction were performed as previously described (44), and all strains, plasmids, and primers are listed in Table S1 in the supplemental material. The DNA polymerase Phusion (Thermo Scientific) was used for all PCRs. The expression plasmids for the DGCs and PDEs were constructed as described elsewhere (45). Briefly, these plasmids allow controlled expression of each DGC encoded by V. cholerae via induction of the Ptac promoter with IPTG. To construct the HD-GYP expression plasmids, each HD-GYP gene was amplified from the V. cholerae chromosome and then inserted into the pEVS143 vector using the EcoRI and BamHI insertion sites, as previously described (38). The identification of the c-di-GMP reporter plasmid 6:C9-lux is described elsewhere (46). The DGC mutant allele plasmids were generated using the Lightning site-directed mutagenesis kit (Agilent) with the DGC expression plasmid as the template using the ASM primers listed in Table S1. The VC2497-lux reporter was constructed by amplifying the promoter of VC2497 from the V. cholerae chromosome by PCR and then inserting it into the pBBRlux vector using the SacI and BamHI insertion sites.
To generate the DGC and PDE deletion strains, natural transformation and homologous recombination were used. A PCR product was generated that contained a chloramphenicol resistance cassette (cat) bordered by FLP recombination target (FRT) sites from the plasmid pKD3 (47), flanked by 500 bp upstream and downstream of the targeted gene using the primers KO1/KO2 and KO3/KO4. The PCR products generated with these promoters were fused to the cat gene using zipper PCR. Natural competence was induced by ectopically expressing tfoX (VC1153) (48) from the Ptac promoter using the plasmid pANDA2. pANDA2 was constructed by amplification of VC1153 with the primers CMW464 and CMW465 and insertion of this product into the EcoRI/BamHI sites of the plasmid pEVS143. Homologous recombination events were selected by growing the culture on LB medium supplemented with chloramphenicol at 1 μg/ml. The cat gene was then removed by ectopically expressing a FLP recombinase on the vector pTL17 (49).
Quantifying the intracellular concentration of c-di-GMP.
All c-di-GMP quantifications were analyzed using liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS). Unless otherwise specified, cultures were grown to an optical density at 600 nm (OD600) between 0.6 and 0.9, and a 1.5-ml aliquot of this culture was removed and centrifuged for 30 s at maximum speed. The supernatant was immediately removed, and the pellet was resuspended in 100 μl of cold extraction buffer (40% acetonitrile, 40% methanol, 0.1 N formic acid) and incubated for 20 min at −20°C. The insoluble fraction was pelleted in a benchtop centrifuge at 4°C for 5 min, and the supernatant was collected and stored at −80°C. Prior to mass spectrometry, the extraction buffer was evaporated using a vacuum manifold. The pellet was then resuspended in 100 μl water. Ten microliters of each sample was then analyzed on a Quattro Premier XE mass spectrometer (Waters) coupled with an Acquity Ultra Performance LC system (Waters) as previously described (50). The intracellular concentration of c-di-GMP was calculated by dividing the intracellular c-di-GMP of the sample by the total volume of the extracted bacteria, which was estimated by multiplying the number of bacterial cells in the extract by the average volume of the bacterial cell. The average cellular volume was determined for each strain analyzed under each growth condition by measuring individual cell dimensions using differential image contrast microscopy and assuming the cells to be cylindrical.
Systematic screen of DGC and PDE activity.
To determine the in vivo activity of each V. cholerae DGC, each of the 40 DGC expression vectors was conjugated into a ΔvpsL strain of V. cholerae harboring a separate reporter vector encoding a luciferase-transcriptional fusion of a c-di-GMP-inducible promoter located within the open reading frame (ORF) of VC1673 (6:C9-lux [46]). These strains were then grown in solid white 96-well clear-bottom plates (Costar 3903) in 150 μl of LB medium or LB medium with SHB in the presence of IPTG inoculated as a 1/100 dilution from an overnight culture. Luminescence and OD600 values were recorded in a SpectraMax M5 plate reader (Molecular Devices) after 8 h of growth and were reported as relative luminescence units (RLU). The screen for PDE activity was performed in the same manner as the DGC assay with the addition of a third vector, pBRP02, which carries the qrgB allele of DGC under Ptac control. pBRP02 has the Vibrio harveyi qrgB allele cloned into the vector pMMB67eh (51), allowing it to coexist with the PDE expression vectors and the 6:C9-lux reporter plasmid.
Systematic screen of DGC and PDE expression.
DGC-gfp (GGDEF domain proteins) and PDE-gfp (GGDEF + EAL and EAL domain proteins) transcriptional fusion plasmids were constructed as previously described (38). Strains containing the plasmids were grown in triplicate overnight and then inoculated 1/100 in 150 μl LB medium or LB medium with SHB in a Costar black, clear-bottom 96-well plate (catalog no. 3904). Cultures were grown for 8 h, and then fluorescence was quantified (excitation, 475 nm; emission, 510 nm) using an M5 SpectraMax plate reader (Molecular Devices). Relative fluorescence was quantified by dividing the fluorescence by the OD600 reading of the culture. PDE-lux transcriptional fusion plasmids were constructed as previously described (52). Strains containing the plasmids were grown in triplicate overnight and then inoculated 1/100 in 150 μl LB medium or LB medium with SHB in a Costar white, clear-bottom 96-well plate (catalog no. 3903). Cultures were grown for 6 h, and then luminescence was quantified using an Envision plate reader (PerkinElmer). Relative luminescence was quantified by dividing the luminescence by the OD600 reading of the culture.
Biofilm quantification.
Static biofilm formation was determined using a protocol modified from that in reference 35. Overnight planktonic cultures of V. cholerae and V. cholerae DGC mutants were grown in LB medium, and cultures were diluted 1:1,000 into 1 ml LB medium or LB medium with BV as specified in 17- by 100-mm polystyrene test tubes (BD Falcon). These tubes were incubated at 35°C for 24 h without shaking. The supernatant was then removed, an absorbance reading of the supernatant was taken (OD600), and the biofilm was gently washed with approximately 2 ml of phosphate-buffered saline (PBS). The biofilm was then stained with 0.41% crystal violet in 12% ethanol for 3 min, followed by three washes with PBS, elution of the crystal violet in 10 ml 95% ethanol, and OD570 measurement for each biofilm. Each OD570 biofilm measurement was normalized to the OD600 measurement of the planktonic culture to account for differences in growth.
RESULTS
Bile increases the intracellular concentration of c-di-GMP in V. cholerae.
It has been proposed that V. cholerae has a high intracellular c-di-GMP concentration in environmental reservoirs where it predominantly exists as surface-associated biofilm communities and reduces c-di-GMP upon entrance into the human host, leading to the activation of virulence factor expression (53). As bile is a prevalent constituent of the human small intestine, we hypothesized that V. cholerae would reduce c-di-GMP levels in response to bile. To test this hypothesis, we grew wild-type (WT) V. cholerae in the presence of a crude bovine bile extract (BV) or synthetic human bile (SHB) and quantified the intracellular c-di-GMP concentration using LC-MS/MS. BV was added as a supplement to the medium at 0.4% (wt/vol), a concentration which is physiologically relevant to that found in the small intestine (27, 54). SHB is a mixture of six purified bile acids that replicate the physiological concentrations of bile acids found in the human small intestine (25, 26, 41, 42). To our surprise, growth in both BV- and SHB-containing media significantly increased the intracellular concentration of c-di-GMP, 4.4- and 3.6-fold, respectively (Fig. 1).
FIG 1.
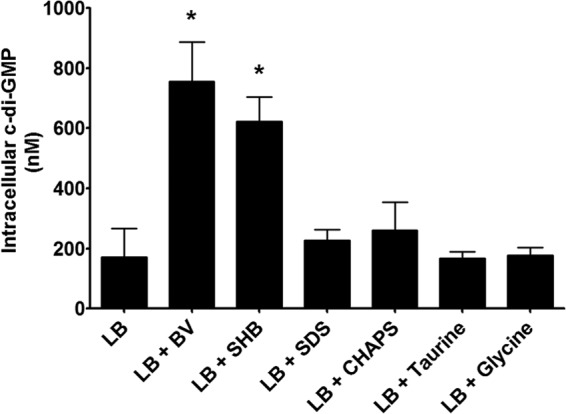
V. cholerae was grown in LB medium and LB medium with bovine bile (BV), synthetic human bile (SHB), the detergents SDS and CHAPS, and the amino acids taurine and glycine. Intracellular c-di-GMP was measured using LC-MS/MS. The reported values indicate the means. Error bars indicate standard deviations, and asterisks indicate statistical significance compared to LB medium as determined by a Student two-tailed t test (n = 3, P < 0.05).
To determine if the increase in intracellular c-di-GMP was due to the detergent activity of the bile acids, we grew V. cholerae in the presence of either the anionic detergent SDS (0.01%, wt/vol) or the zwitterionic detergent CHAPS (0.4%, wt/vol). We observed no significant difference in intracellular c-di-GMP upon addition of either detergent, suggesting that this increase in intracellular c-di-GMP is specific to bile acids. As the SHB mixture of bile acids contains bile acids conjugated to either taurine or a glycine, it was possible that these moieties were causing the increase in intracellular c-di-GMP. However, there was no significant difference in the intracellular c-di-GMP concentration of V. cholerae grown with the addition of either taurine or glycine at equivalent concentrations (Fig. 1). Therefore, we conclude that bile acids increase the intracellular concentration of c-di-GMP in V. cholerae.
The bile-mediated increase of intracellular c-di-GMP is growth phase dependent.
The experiments described thus far analyzed cultures grown to late exponential growth. We therefore examined the temporal dynamics of c-di-GMP induction by bile at various points during bacterial growth. When the bacterium is grown under standard culture conditions with shaking, there is no significant difference in the doubling time of V. cholerae in the presence or absence of SHB (LB medium, 22.3 ± 0.3 min; LB medium with SHB, 21.6 ± 0.6 min). We measured intracellular c-di-GMP in three growth phases in the presence and absence of SHB: early exponential growth (OD600, 0.2 to 0.3), late exponential growth (OD600, 0.6 to 0.8), and stationary phase (OD600, 1.0 to 1.2). When V. cholerae was grown in LB medium alone, we observed elevated c-di-GMP in early exponential growth, and the concentrations of c-di-GMP decreased as the cell density increased. These observations are consistent with previous studies showing that c-di-GMP is elevated in the low-cell-density quorum sensing (QS) state (38, 52). We observed significant increases of the intracellular c-di-GMP concentration in the presence of bile in early exponential and late exponential growth phases (Fig. 2). Alternatively, no significant induction of c-di-GMP was observed in stationary phase. These results indicate that bile induction of c-di-GMP is growth phase dependent. We speculated that QS might be responsible for density-dependent bile induction of c-di-GMP. However, bile induction of c-di-GMP was unchanged in V. cholerae mutants locked in either the low- or high-cell-density QS state, showing that QS does not drive growth-phase-dependent bile induction of c-di-GMP in V. cholerae (data not shown).
FIG 2.
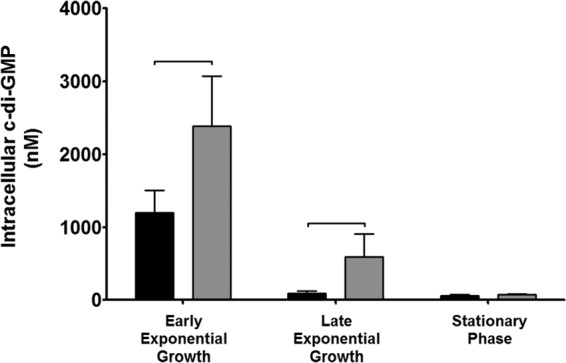
The intracellular c-di-GMP concentration of wild-type V. cholerae was quantified over the course of growth. All intracellular c-di-GMP concentrations were determined using LC-MS/MS. Black bars indicate strains grown in LB medium, and gray bars indicate strains grown in LB medium with SHB. The reported values indicate the means, and the error bars indicate the standard deviations. Brackets indicate statistical significance as determined by a one-tailed Student t test (n = 3, P > 0.05).
Three DGCs are more active in the presence of bile.
As SHB increases intracellular c-di-GMP, we hypothesized that specific DGCs in V. cholerae would increase c-di-GMP synthesis in the presence of SHB. To identify these DGCs, we developed a high-throughput screen where the in vivo activity of each of the 40 V. cholerae DGCs could be determined in the presence and absence of SHB. This screen utilized a series of expression vectors carrying every V. cholerae DGC under the control of the IPTG-inducible Ptac promoter and a common ribosome binding site (45). Therefore, we infer in this system that differences in c-di-GMP synthesis activity are not due to changes in gene expression. In addition to the DGC expression vector, each strain contained a separate reporter plasmid that carried a transcriptional fusion of a c-di-GMP-inducible promoter located within the ORF of VC1673 with the luciferase operon (named 6:C9-lux [46]). These vectors were introduced into a ΔvpsL strain of V. cholerae which abrogates biofilm formation and eliminates interference of aggregate formation in the analysis of reporter gene expression. In the absence of additional c-di-GMP generated by exogenous expression of DGCs, the reporter plasmid does not produce significant luminescence in the presence or absence of SHB (Fig. 3, vector control). c-di-GMP production by active DGCs leads to increased luciferase production, as can be seen by induction of luminescence by expression of the constitutively active DGC qrgB from V. harveyi (Fig. 3, QrgB). Induction of this transcriptional reporter is dependent on c-di-GMP synthesis as expression of qrgB*, an allele of qrgB harboring a mutation in the active site of the protein, does not induce luminescence (Fig. 3, qrgB*).
FIG 3.

V. cholerae strains containing a DGC expression plasmid and the 6:C9-lux reporter plasmid were measured for luminescence in LB medium (black bars) and LB medium with SHB (gray bars). Expression plasmids carrying qrgB and its mutant allele counterpart, qrgB*, were included as positive and negative controls, respectively. The vector control indicates expression of 6:C9-lux in the absence of protein induction. The error bars indicate standard deviations. Statistical significance (*) was determined for cultures exhibiting a positive fold change (LB medium + SHB/LB medium) greater than 2 as determined by a Student one-tailed t test (n = 3, P < 0.01).
6:C9-lux expression was determined upon induction of each DGC in the presence of LB medium and LB medium with SHB. Seven DGCs showed reduced luminescence in the presence of SHB, although the extent of this reduction was not large (<3.5-fold). It is not currently known if this reduction of expression is related to bile inhibition of the DGC activity of these proteins, unrelated transcriptional regulation of 6:C9-lux, or a nonspecific effect of SHB on luminescence. Due to the large number of DGCs that are negatively affected (seven, P < 0.01), the relatively low reduction, and the fact that bile increases the total intracellular c-di-GMP concentration, we favor the latter two possibilities. Nevertheless, three DGCs demonstrated more than a 5-fold increase in luminescence in the presence of SHB (P < 0.01; Fig. 3); these DGCs are VC1067 (7.0-fold increase), VC1372 (47.6-fold increase), and VC1376 (6.7-fold increase).
The activity of PDEs in V. cholerae is not altered by bile.
The hydrolysis of c-di-GMP is driven by c-di-GMP-specific PDE enzymes, which contain either a C-terminal EAL or an HD-GYP domain. Like DGC enzymes, these proteins are also modular, and the variable N-terminal domain is thought to respond to environmental stimuli. We hypothesized that an increase in intracellular c-di-GMP in response to bile acids could be caused by a repression of PDE activity. To examine this possibility, we developed a second high-throughput screen to analyze the enzymatic activity of the 29 predicted PDEs of V. cholerae. The sequence of one EAL protein, VC0515, was highly divergent from the sequenced N16961 strain upon amplification from the genome and was thus excluded from analysis; this is consistent with other studies reporting sequence variance (38). Similar to the screen above, each V. cholerae PDE is expressed from an inducible expression vector that has been introduced into a ΔvpsL strain of V. cholerae containing the 6:C9-lux reporter vector. A third vector (pBRP02) containing the constitutively active DGC qrgB was introduced, effectively increasing the baseline c-di-GMP concentration and subsequently the baseline luminescence values. Robust reporter gene expression can be seen in a strain expressing qrgB with a vector control that has no exogenous PDE expression (Fig. 4, vector control).
FIG 4.

V. cholerae strains containing a PDE expression plasmid, the 6:C9-lux reporter plasmid, and the qrgB expression plasmid were measured for luminescence in LB medium (black bars) and LB medium with SHB (gray bars). The vector control indicates expression of 6:C9-lux with qrgB induction and without PDE expression. The error bars indicate standard deviations (n = 3).
Strains of V. cholerae expressing each PDE alongside the DGC qrgB were grown in LB medium and LB medium with SHB, and the expression of 6:C9-lux was quantified. Twenty-two of the 29 PDEs showed appreciable decreases (>2-fold, P < 0.01) in luminescence when grown in LB medium compared to the vector control (Fig. 4), indicating that these PDEs are actively hydrolyzing c-di-GMP. However, none of the PDEs demonstrated any appreciable difference when grown in LB medium with SHB compared to LB medium alone (Fig. 4). Thus, we conclude that SHB does not affect the c-di-GMP hydrolysis of PDEs under the conditions that we examined here.
The three bile-inducible DGCs are inner membrane proteins.
We analyzed the predicted domain structure and subcellular localization of the three bile-inducible DGCs to search for commonalities. An analysis of the relative hydrophobicity of the amino acid sequence of these three DGCs using the program toppred predicts that all contain transmembrane spanning domains (Fig. 5) (55–57). The DGC VC1067 (GI 15641080), which has also been referred to as cdgH (58), is predicted to contain two sequential periplasmic substrate-binding domains (PBPb) in the N terminus while VC1376 (GI 15641388), which has also been referred to as cdgM (59), is predicted to contain a conserved CHASE domain. VC1372 (GI 15641384) does not contain any conserved protein domains in the N terminus; however, analysis of the relative hydrophobicity of the amino acid sequence using toppred predicts 6 sequential intramembrane spanning domains.
FIG 5.

The predicted hydrophobicity of the amino acid sequence of VC1067 (A), VC1372 (B), and VC1376 (C) was used to predict transmembrane domains using the toppred program (left). The dotted line indicates the cutoff value for a potential transmembrane domain. A depiction of the potential N-terminal signaling domains and transmembrane domains (black bars) and the C-terminal GGDEF domain of each DGC is shown (right). The images were created using the SMART database (57).
Three DGCs increase c-di-GMP synthesis in the presence of bile acids.
To directly examine if SHB increases the DGC activity of these three enzymes, we quantified the intracellular c-di-GMP concentration of strains ectopically expressing VC1067, VC1372, or VC1376 grown in the presence and absence of SHB. We similarly analyzed expression of active site mutant alleles of these genes (GG[D/E]EF→AA[D/E]EF) that render them incapable of c-di-GMP synthesis. As addition of SHB increases the intracellular c-di-GMP concentration of V. cholerae (Fig. 1), we hypothesized that the strains expressing each AA[D/E]EF mutant would show increased intracellular c-di-GMP in the presence of bile, similar to that of the wild-type (WT) strain. Moreover, induction of the bile-activated DGCs would lead to more c-di-GMP synthesis in the presence of bile than in the presence of LB medium alone.
When grown in LB medium, there was no notable difference between strains expressing any of the AA[D/E]EF alleles and the vector control in LB medium. As expected, growth of the vector control and that of DGC mutants in bile showed similar increases in c-di-GMP. Induction of the DGCs VC1067 and VC1376 produced significantly increased amounts of c-di-GMP in LB medium alone, leading to intracellular c-di-GMP concentrations of 48.4 ± 7.0 μM and 19.3 ± 10.0 μM, respectively (Fig. 6). Alternatively, the intracellular c-di-GMP of the VC1372 expression strain in the absence of bile (116.9 ± 17.4 nM) was not significantly altered compared to that of the vector control (263.7 ± 119.7 nM), indicating that this DGC does not produce c-di-GMP in LB medium alone. Importantly, the intracellular concentration of c-di-GMP was increased significantly upon ectopic expression of VC1067, VC1372, and VC1376 in the presence of LB medium with SHB over that in the presence of LB medium alone, resulting in intracellular concentrations of 89.2 ± 31.5 μM, 4.3 ± 0.4 μM, and 67.4 ± 20.1 μM c-di-GMP, respectively (Fig. 6). These data together indicate that VC1067, VC1372, and VC1376 synthesize more c-di-GMP in the presence of bile acids.
FIG 6.
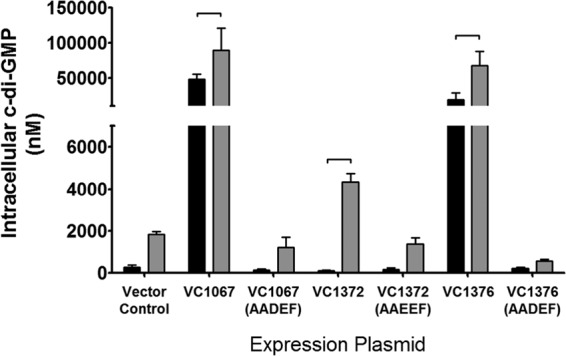
Intracellular levels of c-di-GMP in V. cholerae expressing an empty vector or DGC VC1067, VC1372, or VC1376 quantified with LC-MS/MS. The intracellular c-di-GMP levels of strains expressing alleles containing mutations in the active site motif of each DGC were also quantified. The black bars indicate strains grown in LB medium, while the gray bars indicate strains grown in LB medium with SHB. Error bars indicate standard deviations. Brackets indicate statistical significance, which was determined using a Student one-tailed t test (n = 3, P < 0.05).
Transcription of VC1295 is inhibited by bile acids.
We hypothesized that the presence of bile could also regulate the transcription of DGCs or PDEs to control intracellular c-di-GMP concentrations. Specifically, bile could increase the expression of DGCs or decrease the expression of PDEs to increase intracellular c-di-GMP. To examine this possibility, we measured the relative transcription of 51 GGDEF, GGDEF + EAL, and EAL proteins in LB medium and LB medium with SHB by growing strains of ΔvpsL V. cholerae containing transcriptional fusions of approximately 500 bp of DNA upstream of each gene to gfp. After 8 h of growth, we found that there was less than a 2-fold difference in fluorescence between all GGDEF, GGDEF + EAL, and EAL strains grown in the presence and absence of bile (see Fig. S1 in the supplemental material), suggesting that under the conditions examined here, regulation of GGDEF and EAL gene transcription by bile does not play a significant role in the bile-mediated increase of intracellular c-di-GMP.
We also examined the relative transcription of the 8 HD-GYP proteins in LB medium and LB medium with SHB using transcriptional fusions of each promoter, defined as approximately 500 bp upstream of each HD-GYP gene, to the lux operon. The HD-GYP gene VC1087 is carried in a putative operon with the EAL gene VC1086, and thus, the expression is presumed to be synonymous with VC1086 (see Fig. S1 in the supplemental material). The luminescence of each reporter strain was determined after 6 h of growth in either LB medium or LB medium with SHB. The expression of six of the genes encoding HD-GYP proteins was not significantly changed in LB medium with SHB from that in LB medium alone (>2-fold), whereas the expression of VC2497 was modestly increased in the presence of SHB (2.2-fold, P < 0.05). Importantly, the expression of VC1295 (GI 15641308) was decreased 2.8-fold when grown in the presence of SHB (Fig. 7; P < 0.05). VC1295 appears to be an active PDE when ectopically expressed in V. cholerae (Fig. 4). This result indicates that bile acids decrease the expression of VC1295, possibly resulting in decreased c-di-GMP hydrolysis contributing to increased intracellular c-di-GMP.
FIG 7.
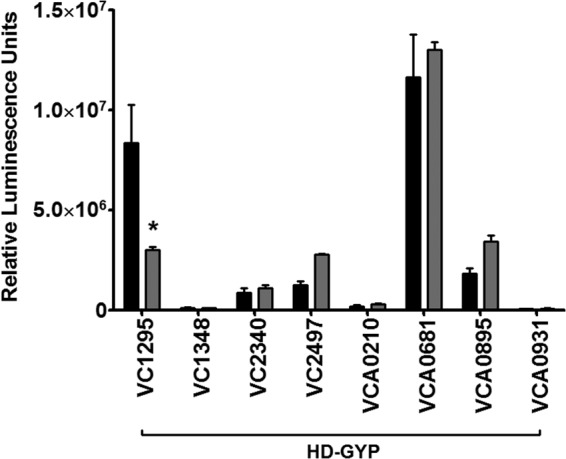
V. cholerae strains containing a transcriptional fusion of each HD-GYP promoter to luciferase were grown in LB medium (black) or LB medium with SHB (gray). Luminescence was quantified after 6 h of growth (n = 4) under each environmental condition. Each culture was normalized to an OD600 reading. Error bars indicate standard deviations. The asterisk indicates a statistically significant differences from the LB medium condition, determined by a one-tailed Student t test (P < 0.05).
Three DGCs and one HD-GYP account for bile-mediated c-di-GMP induction.
To determine if the DGCs VC1067, VC1372, and VC1376 contribute to the bile-mediated increase of intracellular c-di-GMP in V. cholerae, we constructed unmarked V. cholerae DGC deletion mutants and examined SHB-mediated induction of c-di-GMP (Fig. 8). Similar to our previous findings, the intracellular c-di-GMP concentration of V. cholerae increased 4.1-fold in the presence of SHB in the WT strain. Both the ΔVC1067 and ΔVC1376 single mutants showed a modest but significant reduction of c-di-GMP when grown in bile compared to the WT strain, having 29.6% and 24.9% less c-di-GMP, respectively. The ΔVC1372 single mutant and ΔVC1372 Δ1376 double mutant were not statistically different from the WT strain. Importantly, the ΔVC1067 ΔVC1372 ΔVC1376 triple mutant strain exhibited the greatest reduction of intracellular c-di-GMP in the presence of bile, losing 46.8% of the intracellular c-di-GMP compared to the WT strain in LB medium with SHB (P < 0.05). These results suggest that VC1067, VC1372, and VC1376 function redundantly in the bile-mediated c-di-GMP induction.
FIG 8.
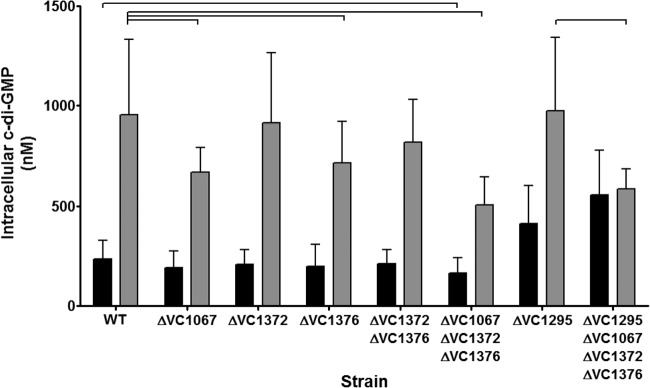
The intracellular c-di-GMP concentrations of wild-type V. cholerae and the DGC and PDE mutant strains were quantified after growth in LB medium (black bars) or LB medium with SHB (gray bars) using LC-MS/MS. The reported values indicate the means, and the error bars indicate standard deviations. Brackets indicate statistical significance as determined by a one-tailed Student t test (n = 9 to 10, P < 0.05).
As the expression of the HD-GYP VC1295 was inhibited by bile acids, we constructed an unmarked V. cholerae VC1295 deletion mutant and quantified intracellular c-di-GMP in the presence and absence of SHB. In LB medium alone, the intracellular c-di-GMP was modestly increased 1.8-fold in the ΔVC1295 mutant compared to the WT (Fig. 8; P < 0.05). This result is expected, as deletion of an active PDE will increase intracellular c-di-GMP. However, the intracellular c-di-GMP concentrations of the ΔVC1295 strain grown in the presence of SHB were indistinguishable from those of the WT. We hypothesized that both activation of DGC activity and transcriptional regulation of VC1295 contribute to bile induction of c-di-GMP. To test this, we created a quadruple ΔVC1295 ΔVC1067 ΔVC1372 ΔVC1376 mutant and measured intracellular c-di-GMP in the presence and absence of SHB. Similar to the ΔVC1295 single mutant, when grown in LB medium alone, the quadruple mutant had elevated intracellular c-di-GMP compared to the WT strain (2.4-fold, P < 0.05). Importantly, this strain showed no change in intracellular c-di-GMP in the presence of SHB. This indicates that these four proteins are responsible for the bile-mediated changes in intracellular c-di-GMP.
Deletion of the bile-responsive DGCs and PDE reduces bile induction of V. cholerae biofilm formation.
It has been previously reported that BV (i.e., bovine bile) increases biofilm formation of V. cholerae, and this induction is dependent on the transcriptional regulator vpsR (35). VpsR binds c-di-GMP to regulate the transcription of biofilm genes (46). We wondered if the levels of c-di-GMP measured in the various DGC and PDE mutant strains with and without bile would correlate with biofilm formation. To test this, we performed a static biofilm assay where the wild-type, ΔvpsL mutant, and the DGC and PDE V. cholerae mutant strains were grown in polystyrene test tubes containing LB medium or LB medium with BV without shaking followed by crystal violet staining of the resulting attached biofilm. The ΔvpsL mutant cannot produce exopolysaccharide and thus does not form biofilms. BV was used to induce biofilm formation to remain consistent with prior studies (35, 60) and because it induced more robust biofilm formation in this assay than did SHB (data not shown).
We observed that all cultures of V. cholerae grew to a significantly lower optical density, as measured by OD600, after static growth in the presence of BV. To account for these growth differences, the biofilm formation of each culture was normalized to the OD600 of the planktonic culture. Consistent with previous reports (35), BV increased biofilm formation in the WT strain 2.7-fold, and this response was eliminated in the ΔvpsL strain (Fig. 9). While the ΔVC1067, ΔVC1376, and ΔVC1372 ΔVC1376 mutants all showed modest to no loss of bile induced c-di-GMP (Fig. 8), these mutants no longer exhibited substantial bile-induced biofilm formation. Only the ΔVC1372 mutant induced biofilm formation in response to bile similar to that of the WT, indicating that this DGC contributes less to biofilm formation. As expected, the triple DGC mutant showed the lowest level of biofilm formation and was not responsive to bile addition. Although the ΔVC1295 strain had modestly elevated c-di-GMP in LB medium, deletion of VC1295 had no noticeable impact on biofilm formation. Like the triple mutant, the DGC/PDE quadruple mutant exhibited low biofilm formation that was unresponsive to bile addition. These results demonstrate that the bile-responsive DGCs are required for c-di-GMP-dependent biofilm formation in response to bile acids. However, the HD-GYP VC1295 did not contribute to biofilm formation in this assay.
FIG 9.

The biofilm formation of wild-type V. cholerae and V. cholerae DGC and PDE mutants was quantified in test tubes using crystal violet (CV). A strain containing a mutation in the vpsL gene was included as a negative control. The CV value was normalized by the OD600 value of the planktonic culture to account for differences in growth. The reported values indicate the means, and error bars indicate standard deviations from the means. Brackets indicate statistical significance as determined by a Student one-tailed t test (n = 3, P < 0.05).
Bicarbonate inhibits the bile-mediated increase of c-di-GMP in V. cholerae.
As c-di-GMP has been shown to repress virulence gene expression in V. cholerae, it remained puzzling why bile would increase c-di-GMP (53). We hypothesized that additional signals in the small intestine could override this induction. Another major component of the human small intestine is bicarbonate, which is secreted by the pancreas as well as the small intestinal epithelial cells. Bicarbonate acts as a pH buffer to neutralize acids secreted by the stomach (61, 62). Bicarbonate has also been implicated in virulence gene regulation in V. cholerae (63). As bicarbonate is abundant in the small intestine, we hypothesized that it may contribute to the regulation of intracellular c-di-GMP in V. cholerae. To examine if bicarbonate impacted bile induction of c-di-GMP, we measured intracellular c-di-GMP of V. cholerae when grown in the presence and absence of SHB and bicarbonate. Bicarbonate was added as a supplement at 0.3% (wt/vol), consistent with toxin-inducing conditions (43). We observed no significant difference in intracellular c-di-GMP of V. cholerae in the presence of bicarbonate alone (Fig. 10). However, growth of V. cholerae in the presence of both SHB and bicarbonate completely abolished bile induction of c-di-GMP (Fig. 10).
FIG 10.
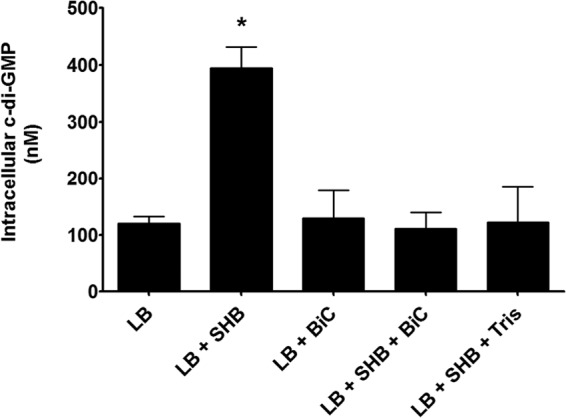
The intracellular c-di-GMP concentration of wild-type V. cholerae was quantified in LB medium, LB medium with bicarbonate (BiC), or LB medium with SHB and different supplements using LC-MS/MS. The reported values indicate the means, and the error bars indicate standard deviations. The asterisk indicates statistical significance compared to LB medium as determined by a one-tailed Student t test (n = 3, P > 0.05).
As bicarbonate acts as a buffer in the small intestine, we hypothesized that bicarbonate suppression of the bile induction of c-di-GMP was due to pH changes. We tested this idea by growing V. cholerae in the presence of SHB with the pH buffer Tris. The pHs of LB medium and LB medium with SHB were approximately the same and did not change significantly over the course of growth (pH of LB medium, 7.3 pregrowth, 7.0 postgrowth; pH of LB medium with SHB, 7.3 pregrowth, 7.1 postgrowth). Upon addition of bicarbonate, the pH increased substantially (pH of LB medium with SHB and BiC, 8.1 pregrowth, 8.7 postgrowth). The addition of Tris caused a similar increase in pH (pH of LB medium with SHB and Tris, 8.7 pregrowth, 8.6 postgrowth). Analogously to bicarbonate, the addition of Tris to LB medium with SHB inhibited the normal induction of c-di-GMP by bile (Fig. 10), indicating that this inhibition of bile-induced c-di-GMP is pH dependent.
DISCUSSION
Bile is an abundant component of the human small intestine and thus a probable physiological cue for V. cholerae to recognize upon entry into this environment. A number of lines of evidence suggest that bile is an important signal in the transition of V. cholerae between environmental and infectious lifestyles. Bile acids increase the expression of ompU in a toxR-dependent manner to increase bile resistance, indicating that the classical biotype of V. cholerae is capable of sensing the presence of bile (64). There are conflicting reports regarding bile control of virulence factor expression. Bile acids were reported to negatively regulate the expression of the toxin-coregulated pilus (TCP) and cholera toxin (CT) in a toxT-dependent manner in a classical biotype, as well as positively regulating motility, demonstrating that there is a link between bile and virulence (65, 66). Specifically, it has been shown that unsaturated fatty acids in bile inhibit the expression of virulence factors (67). Contrary to these findings, bile acids were reported to induce CT and TCP in a toxR-dependent manner in a classical V. cholerae biotype and a tcpP-dependent manner in the El Tor biotype used in this study (60, 68). From these studies, it is clear that V. cholerae responds to bile to induce a number of physiological changes. In this work, we explore the connections between bile and c-di-GMP.
Based on the prevailing V. cholerae disease model hypothesizing that c-di-GMP levels are decreased upon infection, we predicted that bile acids would decrease intracellular c-di-GMP concentrations. Contrary to this prediction, we found that bile acids increase the intracellular c-di-GMP concentration of V. cholerae (Fig. 1). This finding suggests that the dynamics of the c-di-GMP signaling system in the human host are more complex than previously appreciated. Furthermore, we have found that the difference in the bile-mediated change of c-di-GMP is largest during exponential growth and that at stationary phase this difference was negated. Consistent with these findings, it has been shown that intracellular c-di-GMP is depleted at high cell density and that these changes are due in part to quorum sensing (38, 52, 69). Our results indicate that this regulation is dominant over the increase in intracellular c-di-GMP caused by bile.
To begin to understand how V. cholerae modulates its intracellular c-di-GMP in response to bile, we determined if the activity of any DGCs or PDEs was affected by bile using a novel high-throughput in vivo assay. This assay is easily adaptable to examine the response of DGCs and PDEs to any environmental cue, and it can be modified to explore other bacteria if a suitable in vivo reporter of c-di-GMP levels is available. Three DGCs (VC1067, VC1372, and VC1376) exhibited increased c-di-GMP synthesis activity in the presence of SHB (Fig. 3 and 6). The DGC activity of VC1372 appeared to be absolutely dependent on bile whereas bile modulated the basal activities of VC1067 and VC1376. All of these DGCs are important for the bile-induced increase of intracellular c-di-GMP and biofilm formation of V. cholerae (Fig. 8 and 9).
The mechanisms by which these three DGCs respond to bile acids are currently unknown. All three DGCs are predicted to be associated with the inner membrane. VC1067 has been implicated in biofilm formation as it induces rugosity-associated phenotypes in V. cholerae (58). Furthermore, it has been shown that both VC1067 and VC1376 actively produce c-di-GMP and stimulate biofilm formation (45, 58), and a VC1376 mutant strain also demonstrates lower vpsL expression (59). VC1376 has also been implicated in the indole-induced increase in biofilm formation (70). Furthermore, all three DGCs also have been shown to repress motility when ectopically expressed in V. cholerae; this is particularly interesting as the levels of motility repression of the VC1372 and VC1376 expression strains do not seem to correlate with the intracellular c-di-GMP levels reported here (Fig. 6) (71). Analysis by BLAST revealed that sequences homologous to VC1372 are found only in a few Vibrio species, dominated by strains of V. cholerae. A previous study has also noted that VC1372 is unique to V. cholerae among members of the genus Vibrio (72).
The phylogenetic link of VC1372 with an enteric human pathogen, the domain structure of VC1372, and our observation that the activity of VC1372 is absolutely dependent on the presence of bile suggest that the physiological cue which controls VC1372 is bile. Bile acids are known to interact with the cell membrane due to their detergent activity (29). Moreover, we observed bile-mediated activation of VC1372 in E. coli (data not shown), an orthologous system with no clear homolog to VC1372. Alternatively, as both VC1067 and VC1376 maintain robust activity upon exogenous expression even in the absence of bile, we postulate that these DGCs might be controlled indirectly by bile through sensing perturbations in the membrane.
Additionally, the expression of the PDE VC1295 was inhibited by bile acids (Fig. 7). This HD-GYP actively hydrolyzes c-di-GMP in both LB medium and LB medium with SHB, but bile acids do not affect this activity (Fig. 4). VC1295 is predicted to be composed of 492 amino acids and contains 6 predicted N-terminal transmembrane domains preceding a HAMP domain linked to the HD-GYP domain (55–57). The mechanism governing this transcriptional regulation of VC1295 by bile remains unknown. Analysis of the VC1295 promoter region reveals motifs resembling the ToxR binding site (−127 bp, TCAAA-N11-TTAAA [73]). While this gene is not listed among the known genes regulated by ToxR (74), there is evidence that the activity of ToxR is altered by bile (60), suggesting that the ToxR regulon could be altered when bile acids are present. We are currently investigating the connection between the transcriptional regulation of VC1295, bile, and ToxR. Although VC1295 contributed to bile-induced c-di-GMP, we did not observe any effect of VC1295 on biofilm formation. We speculate that this result is due to the distinct experimental growth conditions under which c-di-GMP and biofilm formation were measured.
Another important host-derived cue is bicarbonate, a biological pH buffer that is highly abundant in the human small intestine (62). Bicarbonate has been shown to be important for virulence, as bicarbonate is critical for in vitro production of CT (43). Bicarbonate is capable of activating V. cholerae virulence gene expression via the transcriptional regulator toxT (63). We have shown that bicarbonate is able to suppress the bile-mediated induction of c-di-GMP in V. cholerae. Furthermore, this regulation is driven by changes in pH, as the bile-mediated induction is similarly repressed by Tris (Fig. 10). It is possible that the change in pH alters the structure of bile so that it no longer triggers the c-di-GMP synthesis activity of the DGCs. Alternatively, the bicarbonate could directly interact with DGCs or PDEs to competitively alter intracellular c-di-GMP.
As bile has strong antimicrobial properties (75), it may be physiologically advantageous for V. cholerae to increase c-di-GMP to promote biofilm formation in order to grant elevated tolerance to bile acids and other stresses associated with the intestinal environment. Other studies have indicated that biofilm formation is important for increased acid shock tolerance and protection from bile acids (35, 76), and biofilms increase infectivity and intestinal colonization in a mouse infection model, which has implications for transmission (77). Indeed, we confirmed that BV induces biofilm formation in V. cholerae and showed that all three bile-responsive DGCs were required for bile induction of biofilm formation.
We propose that bile stimulates a high intracellular c-di-GMP concentration in the intestinal lumen (Fig. 11). Upon penetration of the mucosal layer where the bicarbonate concentration and thus the pH are elevated (78), the response to bile is abrogated, leading to a corresponding decrease in intracellular c-di-GMP. The physiological consequences of spatial alteration of c-di-GMP within the small intestine remain to be determined, although we speculate that c-di-GMP could be modulating biofilm formation, motility, and virulence gene expression. Our model predicting high c-di-GMP concentrations in the lumen and decreased c-di-GMP proximal to the intestinal epithelial cells is consistent with previously described virulence gene regulatory models for V. cholerae and Salmonella enterica (63, 79, 80).
FIG 11.
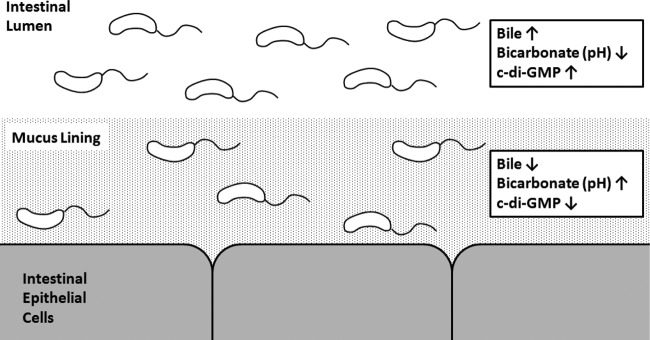
Proposed model of V. cholerae c-di-GMP regulation in the human small intestine. c-di-GMP is elevated in the lumen, where the concentration of bile is elevated and the concentrations of bicarbonate and pH are low. Upon entry into the mucosal layer, where bile is low and the bicarbonate concentration and pH are elevated, c-di-GMP is repressed.
Our findings indicate that both bile and bicarbonate are environmental cues that modulate c-di-GMP signaling in V. cholerae and facilitate the transition from aquatic environments to the human host. They suggest that modulation of c-di-GMP levels by V. cholerae upon entry into the human host is more complex than previously appreciated and that both bile and bicarbonate act together to inversely regulate the intracellular concentration of c-di-GMP to presumably enable the bacteria to adapt and thrive in the diverse intestinal environment.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH grants K22AI080937 and U19AI090872, membership within and support from the Region V “Great Lakes” RCE (NIH award 2U54AI057153), NSF grant MCB1253684, the NSF-sponsored BEACON Science and Technology Center (cooperative agreement no. DBI-0939454), and Michigan State University to C.M.W. We also acknowledge the Rudolph Hugh Fellowship and the Russell B. DuVall Scholarship to B.J.K.
We thank the MSU Mass Spectrometry Facility for assistance in measuring c-di-GMP. We also thank Robert Britton and Kristi Whitehead for providing the mixture for SHB; Benjamin Pursley and Carolyn Chan for the construction of pBRP2 and pANDA2, respectively; and Brian Hammer for generous sharing of the HD-GYP lux transcriptional fusions.
Footnotes
Published ahead of print 5 May 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01664-14.
REFERENCES
- 1.Halpern M, Broza YB, Mittler S, Arakawa E, Broza M. 2004. Chironomid egg masses as a natural reservoir of Vibrio cholerae non-O1 and non-O139 in freshwater habitats. Microb. Ecol. 47:341–349. 10.1007/s00248-003-2007-6 [DOI] [PubMed] [Google Scholar]
- 2.Huq A, Small EB, West PA, Huq MI, Rahman R, Colwell RR. 1983. Ecological relationships between Vibrio cholerae and planktonic crustacean copepods. Appl. Environ. Microbiol. 45:275–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tamplin ML, Gauzens AL, Huq A, Sack DA, Colwell RR. 1990. Attachment of Vibrio cholerae serogroup-O1 to zooplankton and phytoplankton of Bangladesh waters. Appl. Environ. Microbiol. 56:1977–1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tischler AD, Camilli A. 2004. Cyclic diguanylate (c-di-GMP) regulates Vibrio cholerae biofilm formation. Mol. Microbiol. 53:857–869. 10.1111/j.1365-2958.2004.04155.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simm R, Morr M, Kader A, Nimtz M, Romling U. 2004. GGDEF and EAL domains inversely regulate cyclic di-GMP levels and transition from sessility to motility. Mol. Microbiol. 53:1123–1134. 10.1111/j.1365-2958.2004.04206.x [DOI] [PubMed] [Google Scholar]
- 6.Boehm A, Kaiser M, Li H, Spangler C, Kasper CA, Ackermann M, Kaever V, Sourjik V, Roth V, Jenal U. 2010. Second messenger-mediated adjustment of bacterial swimming velocity. Cell 141:107–116. 10.1016/j.cell.2010.01.018 [DOI] [PubMed] [Google Scholar]
- 7.Kulasakara H, Lee V, Brencic A, Liberati N, Urbach J, Miyata S, Lee DG, Neely AN, Hyodo M, Hayakawa Y, Ausubel FM, Lory S. 2006. Analysis of Pseudomonas aeruginosa diguanylate cyclases and phosphodiesterases reveals a role for bis-(3′-5′)-cyclic-GMP in virulence. Proc. Natl. Acad. Sci. U. S. A. 103:2839–2844. 10.1073/pnas.0511090103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hecht GB, Newton A. 1995. Identification of a novel response regulator required for the swarmer-to-stalked-cell transition in Caulobacter crescentus. J. Bacteriol. 177:6223–6229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aldridge P, Paul R, Goymer P, Rainey P, Jenal U. 2003. Role of the GGDEF regulator PleD in polar development of Caulobacter crescentus. Mol. Microbiol. 47:1695–1708. 10.1046/j.1365-2958.2003.03401.x [DOI] [PubMed] [Google Scholar]
- 10.Tischler AD, Camilli A. 2005. Cyclic diguanylate regulates Vibrio cholerae virulence gene expression. Infect. Immun. 73:5873–5882. 10.1128/IAI.73.9.5873-5882.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galperin MY. 2004. Bacterial signal transduction network in a genomic perspective. Environ. Microbiol. 6:552–567. 10.1111/j.1462-2920.2004.00633.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ryan RP, Fouhy Y, Lucey JF, Dow JM. 2006. Cyclic di-GMP signaling in bacteria: recent advances and new puzzles. J. Bacteriol. 188:8327–8334. 10.1128/JB.01079-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmidt AJ, Ryjenkov DA, Gomelsky M. 2005. The ubiquitous protein domain EAL is a cyclic diguanylate-specific phosphodiesterase: enzymatically active and inactive EAL domains. J. Bacteriol. 187:4774–4781. 10.1128/JB.187.14.4774-4781.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ryan RP, Fouhy Y, Lucey JF, Crossman LC, Spiro S, He YW, Zhang LH, Heeb S, Camara M, Williams P, Dow JM. 2006. Cell-cell signaling in Xanthomonas campestris involves an HD-GYP domain protein that functions in cyclic di-GMP turnover. Proc. Natl. Acad. Sci. U. S. A. 103:6712–6717. 10.1073/pnas.0600345103 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15.Galperin MY, Nikolskaya AN, Koonin EV. 2001. Novel domains of the prokaryotic two-component signal transduction systems. FEMS Microbiol. Lett. 203:11–21. 10.1111/j.1574-6968.2001.tb10814.x [DOI] [PubMed] [Google Scholar]
- 16.Bernier SP, Ha DG, Khan W, Merritt JH, O'Toole GA. 2011. Modulation of Pseudomonas aeruginosa surface-associated group behaviors by individual amino acids through c-di-GMP signaling. Res. Microbiol. 162:680–688. 10.1016/j.resmic.2011.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carlson HK, Vance RE, Marletta MA. 2010. H-NOX regulation of c-di-GMP metabolism and biofilm formation in Legionella pneumophila. Mol. Microbiol. 77:930–942. 10.1111/j.1365-2958.2010.07259.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zähringer F, Lacanna E, Jenal U, Schirmer T, Boehm A. 13 June 2013. Structure and signaling mechanism of a zinc-sensory diguanylate cyclase. Structure 10.1016/j.str.2013.04.026 [DOI] [PubMed] [Google Scholar]
- 19.Kanazawa T, Ren S, Maekawa M, Hasegawa K, Arisaka F, Hyodo M, Hayakawa Y, Ohta H, Masuda S. 2010. Biochemical and physiological characterization of a BLUF protein-EAL protein complex involved in blue light-dependent degradation of cyclic diguanylate in the purple bacterium Rhodopseudomonas palustris. Biochemistry 49:10647–10655. 10.1021/bi101448t [DOI] [PubMed] [Google Scholar]
- 20.Ryan RP, McCarthy Y, Andrade M, Farah CS, Armitage JP, Dow JM. 2010. Cell-cell signal-dependent dynamic interactions between HD-GYP and GGDEF domain proteins mediate virulence in Xanthomonas campestris. Proc. Natl. Acad. Sci. U. S. A. 107:5989–5994. 10.1073/pnas.0912839107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trimble MJ, McCarter LL. 2011. Bis-(3′-5′)-cyclic dimeric GMP-linked quorum sensing controls swarming in Vibrio parahaemolyticus. Proc. Natl. Acad. Sci. U. S. A. 108:18079–18084. 10.1073/pnas.1113790108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tuckerman JR, Gonzalez G, Sousa EHS, Wan XH, Saito JA, Alam M, Gilles-Gonzalez MA. 2009. An oxygen-sensing diguanylate cyclase and phosphodiesterase couple for c-di-GMP control. Biochemistry 48:9764–9774. 10.1021/bi901409g [DOI] [PubMed] [Google Scholar]
- 23.Wan XH, Tuckerman JR, Saito JA, Freitas TAK, Newhouse JS, Denery JR, Galperin MY, Gonzalez G, Gilles-Gonzalez MA, Alam M. 2009. Globins synthesize the second messenger bis-(3′-5′)-cyclic diguanosine monophosphate in bacteria. J. Mol. Biol. 388:262–270. 10.1016/j.jmb.2009.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karatan E, Duncan TR, Watnick PI. 2005. NspS, a predicted polyamine sensor, mediates activation of Vibrio cholerae biofilm formation by norspermidine. J. Bacteriol. 187:7434–7443. 10.1128/JB.187.21.7434-7443.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dawson PA. 2010. Bile secretion and the enterohepatic circulation, p 1075–1088 In Feldman M, Friedman LS, Brandt LJ. (ed), Sleisenger and Fordtran's gastrointestinal and liver disease (9th), vol 1 WB Saunders, Philadelphia, PA [Google Scholar]
- 26.O'Connor CJ, Wallace RG. 1985. Physicochemical behavior of bile-salts. Adv. Colloid Interface Sci. 22:1–111. 10.1016/0001-8686(85)80002-6 [DOI] [Google Scholar]
- 27.Begley M, Gahan CGM, Hill C. 2005. The interaction between bacteria and bile. FEMS Microbiol. Rev. 29:625–651. 10.1016/j.femsre.2004.09.003 [DOI] [PubMed] [Google Scholar]
- 28.Bernstein C, Bernstein H, Payne CM, Beard SE, Schneider J. 1999. Bile salt activation of stress response promoters in Escherichia coli. Curr. Microbiol. 39:68–72. 10.1007/s002849900420 [DOI] [PubMed] [Google Scholar]
- 29.Schubert R, Schmidt KH. 1988. Structural changes in vesicle membranes and mixed micelles of various lipid compositions after binding of different bile salts. Biochemistry 27:8787–8794. 10.1021/bi00424a015 [DOI] [PubMed] [Google Scholar]
- 30.Cabral DJ, Small DM, Lilly HS, Hamilton JA. 1987. Transbilayer movement of bile-acids in model membranes. Biochemistry 26:1801–1804. 10.1021/bi00381a002 [DOI] [PubMed] [Google Scholar]
- 31.Provenzano D, Schuhmacher DA, Barker JL, Klose KE. 2000. The virulence regulatory protein ToxR mediates enhanced bile resistance in Vibrio cholerae and other pathogenic Vibrio species. Infect. Immun. 68:1491–1497. 10.1128/IAI.68.3.1491-1497.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Colmer JA, Fralick JA, Hamood AN. 1998. Isolation and characterization of a putative multidrug resistance pump from Vibrio cholerae. Mol. Microbiol. 27:63–72. 10.1046/j.1365-2958.1998.00657.x [DOI] [PubMed] [Google Scholar]
- 33.Bina JE, Mekalanos JJ. 2001. Vibrio cholerae tolC is required for bile resistance and colonization. Infect. Immun. 69:4681–4685. 10.1128/IAI.69.7.4681-4685.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bina XR, Provenzano D, Nguyen N, Bina JE. 2008. Vibrio cholerae RND family efflux systems are required for antimicrobial resistance, optimal virulence factor production, and colonization of the infant mouse small intestine. Infect. Immun. 76:3595–3605. 10.1128/IAI.01620-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hung DT, Zhu J, Sturtevant D, Mekalanos JJ. 2006. Bile acids stimulate biofilm formation in Vibrio cholerae. Mol. Microbiol. 59:193–201. 10.1111/j.1365-2958.2005.04846.x [DOI] [PubMed] [Google Scholar]
- 36.Thelin KH, Taylor RK. 1996. Toxin-coregulated pilus, but not mannose-sensitive hemagglutinin, is required for colonization by Vibrio cholerae O1 El Tor biotype and O139 strains. Infect. Immun. 64:2853–2856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Lorenzo V, Timmis KN. 1994. Analysis and construction of stable phenotypes in Gram-negative bacteria with Tn5-derived and Tn10-derived minitransposons. Methods Enzymol. 235:386–405. 10.1016/0076-6879(94)35157-0 [DOI] [PubMed] [Google Scholar]
- 38.Waters CM, Lu WY, Rabinowitz JD, Bassler BL. 2008. Quorum sensing controls biofilm formation in Vibrio cholerae through modulation of cyclic di-GMP levels and repression of vpsT. J. Bacteriol. 190:2527–2536. 10.1128/JB.01756-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miller MB, Skorupski K, Lenz DH, Taylor RK, Bassler BL. 2002. Parallel quorum sensing systems converge to regulate virulence in Vibrio cholerae. Cell 110:303–314. 10.1016/S0092-8674(02)00829-2 [DOI] [PubMed] [Google Scholar]
- 40.Zhu J, Miller MB, Vance RE, Dziejman M, Bassler BL, Mekalanos JJ. 2002. Quorum-sensing regulators control virulence gene expression in Vibrio cholerae. Proc. Natl. Acad. Sci. U. S. A. 99:3129–3134. 10.1073/pnas.052694299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Graham DY, Osato MS. 2000. H. pylori in the pathogenesis of duodenal ulcer: interaction between duodenal acid load, bile, and H. pylori. Am. J. Gastroenterol. 95:87–91. 10.1111/j.1572-0241.2000.01704.x [DOI] [PubMed] [Google Scholar]
- 42.Northfield TC, McColl I. 1973. Postprandial concentrations of free and conjugated bile acids down the length of the normal human small intestine. Gut 14:513–518. 10.1136/gut.14.7.513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iwanaga M, Yamamoto K. 1985. New medium for the production of cholera toxin by Vibrio cholerae 01 biotype El Tor. J. Clin. Microbiol. 22:405–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 45.Massie JP, Reynolds EL, Koestler BJ, Cong J-P, Agostoni M, Waters CM. 2012. Quantification of high-specificity cyclic diguanylate signaling. Proc. Natl. Acad. Sci. U. S. A. 109:12746–12751. 10.1073/pnas.1115663109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Srivastava D, Harris RC, Waters CM. 2011. Integration of cyclic di-GMP and quorum sensing in the control of vpsT and aphA in Vibrio cholerae. J. Bacteriol. 193:6331–6341. 10.1128/JB.05167-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645. 10.1073/pnas.120163297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meibom KL, Blokesch M, Dolganov NA, Wu CY, Schoolnik GK. 2005. Chitin induces natural competence in Vibrio cholerae. Science 310:1824–1827. 10.1126/science.1120096 [DOI] [PubMed] [Google Scholar]
- 49.Long T, Tu KC, Wang YF, Mehta P, Ong NP, Bassler BL, Wingreen NS. 2009. Quantifying the integration of quorum-sensing signals with single-cell resolution. PLoS Biol. 7:e68. 10.1371/journal.pbio.1000068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bobrov AG, Kirillina O, Ryjenkov DA, Waters CM, Price PA, Fetherston JD, Mack D, Goldman WE, Gomelsky M, Perry RD. 2011. Systematic analysis of cyclic di-GMP signalling enzymes and their role in biofilm formation and virulence in Yersinia pestis. Mol. Microbiol. 79:533–551. 10.1111/j.1365-2958.2010.07470.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fürste JP, Pansegrau W, Frank R, Blocker H, Scholz P, Bagdasarian M, Lanka E. 1986. Molecular cloning of the plasmid RP4 primase region in a multi-host-range tacP expression vector. Gene 48:119–131. 10.1016/0378-1119(86)90358-6 [DOI] [PubMed] [Google Scholar]
- 52.Hammer BK, Bassler BL. 2009. Distinct sensory pathways in Vibrio cholerae El Tor and classical biotypes modulate cyclic dimeric GMP levels to control biofilm formation. J. Bacteriol. 191:169–177. 10.1128/JB.01307-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tamayo R, Schild S, Pratt JT, Camilli A. 2008. Role of cyclic di-GMP during el tor biotype Vibrio cholerae infection: characterization of the in vivo-induced cyclic di-GMP phosphodiesterase CdpA. Infect. Immun. 76:1617–1627. 10.1128/IAI.01337-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hofmann AF. 1999. The continuing importance of bile acids in liver and intestinal disease. Arch. Intern. Med. 159:2647–2658. 10.1001/archinte.159.22.2647 [DOI] [PubMed] [Google Scholar]
- 55.Claros MG, Vonheijne G. 1994. TOPPRED-II—an improved software for membrane-protein structure predictions. Comput. Appl. Biosci. 10:685–686 [DOI] [PubMed] [Google Scholar]
- 56.Neron B, Menager H, Maufrais C, Joly N, Maupetit J, Letort S, Carrere S, Tuffery P, Letondal C. 2009. Mobyle: a new full web bioinformatics framework. Bioinformatics 25:3005–3011. 10.1093/bioinformatics/btp493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schultz J, Copley RR, Doerks T, Ponting CP, Bork P. 2000. SMART: a web-based tool for the study of genetically mobile domains. Nucleic Acids Res. 28:231–234. 10.1093/nar/28.1.231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Beyhan S, Odell LS, Yildiz FH. 2008. Identification and characterization of cyclic diguanylate signaling systems controlling rugosity in Vibrio cholerae. J. Bacteriol. 190:7392–7405. 10.1128/JB.00564-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shikuma NJ, Fong JCN, Yildiz FH. 2012. Cellular levels and binding of c-di-GMP control subcellular localization and activity of the Vibrio cholerae transcriptional regulator VpsT. PLoS Pathog. 8:e1002719. 10.1371/journal.ppat.1002719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hung DT, Mekalanos JJ. 2005. Bile acids induce cholera toxin expression in Vibrio cholerae in a ToxT-independent manner. Proc. Natl. Acad. Sci. U. S. A. 102:3028–3033. 10.1073/pnas.0409559102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Seidler U, Sjöblom M. 2012. Gastroduodenal bicarbonate secretion, p 1311–1339 In Johnson LR. (ed), Physiology of the gastrointestinal tract, 5th ed, vol 2 Elsevier, San Diego, CA [Google Scholar]
- 62.Hogan DL, Ainsworth MA, Isenberg JI. 1994. Gastroduodenal bicarbonate secretion. Aliment. Pharmacol. Ther. 8:475–488 [DOI] [PubMed] [Google Scholar]
- 63.Abuaita BH, Withey JH. 2009. Bicarbonate induces Vibrio cholerae virulence gene expression by enhancing ToxT activity. Infect. Immun. 77:4111–4120. 10.1128/IAI.00409-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Provenzano D, Klose KE. 2000. Altered expression of the ToxR-regulated porins OmpU and OmpT diminishes Vibrio cholerae bile resistance, virulence factor expression, and intestinal colonization. Proc. Natl. Acad. Sci. U. S. A. 97:10220–10224. 10.1073/pnas.170219997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schuhmacher DA, Klose KE. 1999. Environmental signals modulate ToxT-dependent virulence factor expression in Vibrio cholerae. J. Bacteriol. 181:1508–1514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gupta S, Chowdhury R. 1997. Bile affects production of virulence factors and motility of Vibrio cholerae. Infect. Immun. 65:1131–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chatterjee A, Dutta PK, Chowdhury R. 2007. Effect of fatty acids and cholesterol present in bile on expression of virulence factors and motility of Vibrio cholerae. Infect. Immun. 75:1946–1953. 10.1128/IAI.01435-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang MH, Liu Z, Hughes C, Stern AM, Wang H, Zhong ZT, Kan B, Fenical W, Zhu J. 2013. Bile salt-induced intermolecular disulfide bond formation activates Vibrio cholerae virulence. Proc. Natl. Acad. Sci. U. S. A. 110:2348–2353. 10.1073/pnas.1218039110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Koestler BJ, Waters CM. 2013. Exploring environmental control of cyclic di-GMP signaling in Vibrio cholerae by using the ex vivo lysate cyclic di-GMP assay (TELCA). Appl. Environ. Microbiol. 79:5233–5241. 10.1128/AEM.01596-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mueller RS, Beyhan S, Saini SG, Yildiz FH, Bartlett DH. 2009. Indole acts as an extracellular cue regulating gene expression in Vibrio cholerae. J. Bacteriol. 191:3504–3516. 10.1128/JB.01240-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hunter JL, Severin GB, Koestler BJ, Waters CM. 2014. The Vibrio cholerae diguanylate cyclase VCA0965 has an AGDEF active site and synthesizes cyclic di-GMP. BMC Microbiol. 14:22. 10.1186/1471-2180-14-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gu JY, Neary J, Cai H, Moshfeghian A, Rodriguez SA, Lilburn TG, Wang YF. 2009. Genomic and systems evolution in Vibrionaceae species. BMC Genomics 10(Suppl 1):S11. 10.1186/1471-2164-10-S1-S11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Goss TJ, Morgan SJ, French EL, Krukonis ES. 2013. ToxR recognizes a direct repeat element in the toxT, ompU, ompT and ctxA promoters of Vibrio cholerae to regulate transcription. Infect. Immun. 81:884–895. 10.1128/IAI.00889-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bina J, Zhu J, Dziejman M, Faruque S, Calderwood S, Mekalanos J. 2003. ToxR regulon of Vibrio cholerae and its expression in vibrios shed by cholera patients. Proc. Natl. Acad. Sci. U. S. A. 100:2801–2806. 10.1073/pnas.2628026100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Begley M, Gahan CGM, Hill C. 2002. Bile stress response in Listeria monocytogenes LO28: adaptation, cross-protection, and identification of genetic loci involved in bile resistance. Appl. Environ. Microbiol. 68:6005–6012. 10.1128/AEM.68.12.6005-6012.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhu J, Mekalanos JJ. 2003. Quorum sensing-dependent biofilms enhance colonization in Vibrio cholerae. Dev. Cell 5:647–656. 10.1016/S1534-5807(03)00295-8 [DOI] [PubMed] [Google Scholar]
- 77.Tamayo R, Patimalla B, Camilli A. 2010. Growth in a biofilm induces a hyperinfectious phenotype in Vibrio cholerae. Infect. Immun. 78:3560–3569. 10.1128/IAI.00048-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Quigley EMM, Turnberg LA. 1987. pH of the microclimate lining human gastric and duodenal mucosa in vivo. Gastroenterology 92:1876–1884 [DOI] [PubMed] [Google Scholar]
- 79.Prouty AM, Gunn JS. 2000. Salmonella enterica serovar Typhimurium invasion is repressed in the presence of bile. Infect. Immun. 68:6763–6769. 10.1128/IAI.68.12.6763-6769.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Krukonis ES, DiRita VJ. 2003. From motility to virulence: sensing and responding to environmental signals in Vibrio cholerae. Curr. Opin. Microbiol. 6:186–190. 10.1016/S1369-5274(03)00032-8 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.