

Abstract
Stable integration in the host genome renders murine leukemia virus (MLV)-derived vectors attractive tools for gene therapy. Adverse events in otherwise successful clinical trials caused by proto-oncogene activation due to vector integration hamper their application. MLV and MLV-based vectors integrate near strong enhancers, active promoters, and transcription start sites (TSS) through specific interaction of MLV integrase (IN) with the bromodomain and extra-terminal (BET) family of proteins, accounting for insertional mutagenesis. We identified a BET-interaction motif in the C-terminal tail of MLV IN conserved among gammaretroviruses. By deletion of this motif or a single point mutation (INW390A), BET-independent MLV (BinMLV) were engineered. BinMLV vectors carrying INW390A integrate at wild-type efficiency, with an integration profile that no longer correlates with BET chromatin distribution nor with the traditional markers of MLV integration. In particular, BinMLV vector integration associated less with oncogene TSS compared to the MLV vectors currently used in clinical trials. Together, these findings open perspectives to increase the biosafety of gammaretroviral vectors for gene therapy.
Introduction
Gene transfer vectors based on retroviruses have been used successfully in several gene transfer trials to treat genetic disorders,1,2,3,4,5 with clear signs of efficacy for more than 90% of the patients in clinical trials for primary immunodeficiencies.6 However, in 10% of the patients, insertional mutagenesis resulted in uncontrolled clonal proliferation. The first reported adverse events originated from vector integration in the proximity of the LMO2 proto-oncogene promoter, resulting in aberrant LMO2 expression and deregulated premalignant cell proliferation.1,2 Similar events were reported for integrations deregulating CCDN2, BMl1, and EVI1.6 The fact that these events are reported in several clinical trials involving the transplantation of stem cells genetically-corrected with retroviral vectors, indicates that insertional mutagenesis is not a mere theoretical event and highlights the importance to identify the mechanisms underlying vector-induced genotoxicity.
Stable insertion of the viral DNA into the host-cell genome is a key feature of the retroviral life cycle and part of the evolutionary strategy by which retroviruses maximize survival and propagation, combining transmission of the viral genome to the host-cell progeny with persistent viral gene expression. Integration is a nonrandom process, with different integration site distribution patterns for each retroviral family. Lentiviruses (including the human immunodeficiency virus (HIV)-1) prefer integration into the body of actively transcribed genes,7 while gammaretroviruses, such as murine leukemia virus (MLV), predominantly integrate in the vicinity of strong enhancers, transcription start sites (TSS), CpG islands, and DNaseI-hypersensitive sites (DHS), accounting for their integration close to proto-oncogene promoters and increased risk for mutational oncogenesis.8,9,10,11 Retroviral integration preference is dictated by host proteins that tether the viral preintegration complex to the chromatin. For HIV-1, the lens epithelium-derived growth factor (LEDGF/p75) is the dominant cellular cofactor.12,13,14,15,16 Via direct interaction with the lentiviral integrase (IN), LEDGF/p75 tethers the preintegration complex to the body of active genes explaining the integration bias of lentiviruses.12,17,18,19,20,21 Recently, we and others reported that the bromodomain and extra-terminal (BET) family of proteins (BRD2, BRD3, and BRD4) interact with MLV integrase (IN) and target MLV integration.22,23,24
As chromatin readers, BET proteins bind acetylated histones via tandem bromodomains. In addition to a role in transcriptional elongation, cell cycle progression and cancer,25 we showed that BET proteins bind MLV IN and colocalize in the nucleus of the cell.23 MLV integration site distribution corresponds to the chromatin-binding profile of BET proteins,22,23 and inhibition of BET chromatin binding via bromodomain inhibitors or BET protein knockdown inhibited gammaretrovirus replication,22,23,24 targeting MLV integration away from TSS.22 Stable expression of an artificial fusion that links the BRD4 ET domain with the LEDGF/p75 chromatin binding domain resulted in an integration site profile reminiscent of that of HIV,23 underscoring BET proteins as main determinant for MLV integration site distribution. Apart from re-engineering the cellular cofactor,23 redesigning MLV-based vectors to render them BET-independent is more interesting from a translational point of view. In this manuscript, we report the development of BET-independent MLV (BinMLV) vectors with an altered integration profile, particularly showing diminished integration in the vicinity of markers for retroviral integration.
Results
The BET interaction domain locates to the unstructured C-terminal tail of MLV IN and is dictated by W390
To increase biosafety of retroviral vectors, we set out to develop cofactor-independent retroviral vectors. We previously pinpointed the interaction between BET proteins and MLV IN to the ET domain of BET proteins and the C-terminal tail of MLV IN (amino acids 381–408), respectively.23 In this study, we further dissected and characterized the BET-MLV IN interaction. Alignment of the C-terminal tail (Ct) of gammaretroviral integrases (amino acids 381–408, INCt) revealed a conserved sequence (390W-X(3)-R/K-S/T-X(2)-PLK-I/L-R-I/L-X-R405) exclusively found in gammaretroviruses (Figure 1a) but absent in all other retroviral genera, such as α-, β-, Δ-, and ɛ-retroviruses (data not shown). Of note, the INCt coding sequence overlaps with that of the envelope (Env) open reading frame (Supplementary Figure S1a,b). Contrary to INCt, alignment of the N-terminal tails of gammaretroviral Env proteins showed low conservation, underscoring that the sequence conservation in this region is due to selective pressure on this portion of the IN sequence.
Figure 1.

Characterization of the MLV IN-BET interface. (a) Schematic representation of MLV IN. The N-terminal HHCC zinc-binding domain, the catalytic core domain (CCD) and the C-terminal domain (CTD) are indicated. The three residues in the CCD analyzed in this manuscript are indicated. The C-terminal (Ct) tail (indicated in gray) is aligned from different gammaretroviruses. Note that a consensus BET-interaction motif (390W-X(3)-R/K-S/T-X(2)-PLK-I/L-R-I/L-X-R405) is conserved among all gammaretroviruses. Protein sequences were downloaded from the UniProt database, aligned using t-coffee and manually refined. * indicates amino acids mutated in this manuscript. (b–d) Interaction of 20 nmol/l His-tagged mBRD4ET with increasing amounts of GST-tagged MLV IN C-terminal tail (amino acids 381–408)(GST-INCt) or the indicated derived mutants as measured by AlphaScreen. (e) Interaction of increasing amounts of GST-tagged BRD4 ET with 80 nmol/l His-INWT or the indicated mutants as measured by AlphaScreen. Representative experiments are shown. Error bars indicate the standard deviations of triplicate data points. AKR-MLV, AKR murine leukemia virus (P03356); BaEV, Baboon endogenous virus (P10272); Cas-BR-E, Cas-Br-E murine leukemia virus (P08361); en-FeLV, endogenous feline leukemia virus (P10273); GaLV, Gibbon ape leukemia virus (P21414); KoRV, Koala retrovirus (Q9TTC1); MLV, murine leukemia virus (P03355); PERV, Porcine endogenous retrovirus (Q8UM96).
In a first step to further define the BET-interaction domain in MLV IN, we selectively mutated two patches of conserved amino acids into alanine in the MLV C-terminal tail peptide, fused N-terminally to a glutathione-S-transferase (GST) tag, generating GST-INCt_W390A/R391A/R394A and GST-INCt_K400A/R402A/R405A, respectively. Protein integrity was confirmed by SDS-PAGE (Supplementary Figure S1c). Interaction with His-tagged BRD4ET (His-BRD4ET) was evaluated in an AlphaScreen assay, using GST-INCt as a control (Figure 1b). While GST-INCt interacted with His-BRD4ET, as previously described,23 none of the triple alanine mutant C-terminal peptides interacted. Site-directed mutagenesis of each individual conserved amino acid showed that five out of six single mutants still interacted with His-BRD4ET (Figure 1c,d), while binding of GST-INCt_W390A was severely affected (Figure 1c). These data were corroborated in a similar AlphaScreen assay evaluating binding to maltose-binding protein (MBP)-tagged BRD2/3ET (Supplementary Figure S1d,e). Next, we introduced the W390A mutation into full-length MLV IN protein (INW390A) and in parallel, deleted the C-terminal tail (dCt, containing a 27 amino acid C-terminal truncation) of MLV IN (INdCt). Both INW390A and INdCt lost interaction with BRD4ET in an AlphaScreen assay (Figure 1e). These data were confirmed by coimmunoprecipitation of GFP-tagged BRD4 from 293T nuclear extracts transiently expressing flag-tagged INWT, INW390A, or INdCt (data not shown). Gupta et al.24 reported that residues in the MLV IN catalytic core domain (CCD; E266, L268, and Y269) were important for the BET interaction.24 However, E266A, L268A, or Y269A substitutions into full-length IN did not abolish the interaction with BRD4ET (Figure 1e). These results establish the C-terminal tail of MLV IN and more specifically INW390 as a critical hot spot for the interaction with BET proteins.
BET-independent MLV (BinMLV) vectors efficiently transduce cells
Inspired by older reports that viruses with C-terminal truncation of MLV IN are replication competent,26,27 we engineered MLV vector packaging plasmids carrying an IN with a truncated C-terminal tail (27 amino acids) or the single W390A mutation. VSV-G pseudotyped MLV-based vectors defective for BET-interaction and encoding an enhanced green fluorescent protein (eGFP) reporter, referred to as MLVIN_dCt and MLVIN_W390A, were produced in parallel with wild-type MLV vector (MLVIN_WT). Viral vector production efficiency was monitored by reverse transcriptase (RT) activity. While RT units for MLVIN_WT and MLVIN_W390A vectors were comparable (P > 0.05), a sixfold lower RT activity was detected for MLVIN_dCt (P < 0.001), indicating that fewer viral particles were produced of MLVIN_dCt (Figure 2a). Following normalization for RT activity, vector preparations were used to transduce SupT1 cells. Transduction efficiency (% gated cells) was evaluated 2 days post-transduction for different vector dilutions. MLVIN_W390A transduced SupT1 cells as efficient as MLVIN_WT (P > 0.01), while truncation of the C-terminal tail significantly reduced transduction efficiency of MLVIN_dCt (P < 0.001), when compared to MLVIN_WT and MLVIN_W390A (Figure 2b). These data were corroborated at 10 days post-transduction, underscoring stable vector integration and excluding expression from nonintegrated vector particles (Supplementary Figure S2a). Identical results were obtained upon transduction of primary human CD4+ T cells (Figure 2c). Vector integration in SupT1 cells was quantified by quantitative-polymerase chain reaction (PCR) demonstrating comparable integrated copies for MLVIN_W390A and MLVIN_WT, whereas integrated copies were twofold lower for MLVIN_dCt (P < 0.01 compared to MLVIN_WT), which is in line with the transduction efficiency (Figure 2d). Interestingly, although transduction efficiency and integrated copies were comparable for MLVIN_W390A and MLVIN_WT in SupT1 cells and CD4+ T cells, eGFP mean fluorescence intensity was 25% lower for MLVIN_W390A and MLVIN_dCt (Supplementary Figure S2b,c) (P < 0.01). This decrease of mean fluorescence intensity in both BET-independent conditions may be indicative of an altered integration site distribution resulting in lower gene expression.
Figure 2.
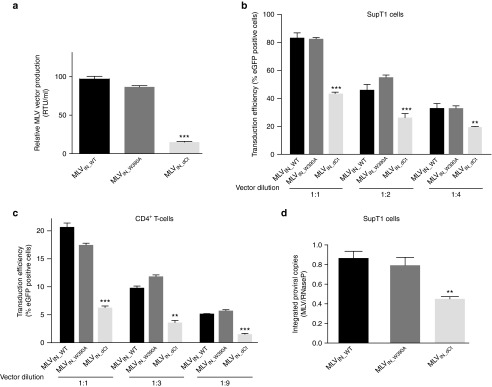
Transduction efficiency of BinMLV vectors. (a) Relative murine leukemia virus (MLV) vector production determined by reverse transcriptase activity per ml of BinMLV vectors as measured by SYBRGreen-I product-enhanced reverse transcriptase assay (SG-PERT). RTU, reverse transcriptase units. Average values and standard deviations of triplicate measurements are shown. (b,c) Transduction of SupT1 cells (b) and CD4+ T cells (c) with equal RT-units of the indicated BinMLV vectors expressing eGFP. After 48 hours, the percentage of eGFP-positive cells was determined for the indicated vector dilutions. Average values and standard deviations of triplicate measurements are shown. (d) Normalized integrated proviral copies (to RNaseP) as determined via quantitative polymerase chain reaction in transduced SupT1 cells at 10 days post-transduction. Representative experiments are shown. Error bars indicate the standard deviations of triplicate data points. Differences were determined using Student's t-test. **P < 0.01, ***P < 0.001.
Loss of BET interaction does not affect the local MLV integration site sequence
We next asked whether truncation of the C-terminal tail of MLV IN or the introduction of W390A, resulted in redistribution of integration sites. Integration sites were determined as described previously,19 yielding 4896, 5906, and 4129 unique vector integration sites in SupT1 cells for MLVIN_WT, MLVIN_W390A, and MLVIN_dCt, respectively. In addition, we determined integration sites in primary human CD4+ T cells for MLVIN_WT and MLVIN_W390A, yielding and 1,831 and 485 unique integrations, respectively. Random control sites were generated computationally and matched to experimental sites (matched random control (MRC)).
Retroviral INs show weak but discernable target sequence specificity at the local site of integration. In an effort to control whether truncation or mutation of MLV IN influences the consensus sequence flanking the integration site, we determined sequence logos (Supplementary Figure S3). The local integration site neighborhood remained unaffected, in agreement with the fact that IN binding to local target DNA is determined by IN-DNA interactions of the IN catalytic core.
Loss of the BET interaction uncouples MLV integration from BET hot spots and traditional markers of MLV integration
We previously showed that MLV integration sites and BET protein (BRD-2, -3, and -4) chromatin immunoprecipitation sequencing (ChIP-seq) tags in 293T cells tightly correlate and concentrate around RefGene TSS with a comparable bimodal distribution.23 We binned BET protein chromatin occupancy (BET protein ChIP-seq tag densities) in a 10 kb window around MLVIN_WT, MLVIN_W390A, and MLVIN_dCt integration sites in SupT1 cells (Figure 3). Whereas MLVIN_WT integration correlated with ChIP-seq read density for BRD-2, -3, and -4, densities around MLVIN_W390A and MLVIN_dCt sites were markedly lower. Similar results were obtained when assessing CD4+ T-cell BRD4 ChIP-seq tag frequencies28 (Supplementary Figure S4a). Together, these results indicate that both MLVIN_W390A and MLVIN_dCt no longer integrate via BET proteins.
Figure 3.
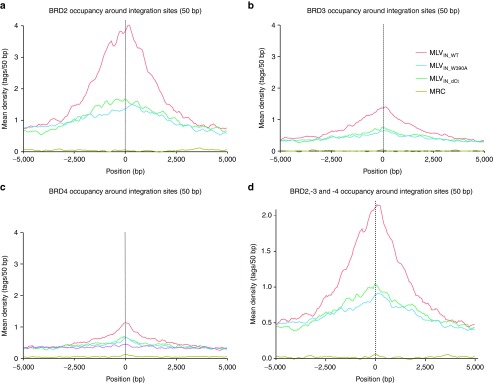
Loss of the BET-interaction uncouples BinMLV integration from BET hot spots. Mean background-subtracted ChIP-seq read density for BRD-2, -3, and -4 or the combination thereof in 50 bp bins in a 10 kb window around MLVIN_WT, MLVIN_W390A, and MLVIN_dCt integration sites.
In line with previous reports,8,29 MLVIN_WT integration in SupT1 cells was enriched within a 2 kb window around TSS (19.24%), CpG islands (18.67%), and DHS (43.53%) (P < 0.001 compared to MRC) (Figure 4a). Interestingly, for MLVIN_W390A and MLVIN_dCt integration associated significantly less with these features (TSS 9.81–10.17%, CpG islands 9.47–9.91%, and DHS 31.62–32.48%, for MLVIN_W390A and MLVIN_dCt, respectively; P < 0.001 compared to MLVIN_WT) (Figure 4a). Comparable data were obtained for larger window sizes (only 4 kb is shown) (Figure 4a). For comparison, data sets were juxtaposed to integration site sets of other retroviruses, such as foamy virus30,31 (FV) and HIV-1, indicating that uncoupling of BET-interaction for BinMLV vectors results in an integration site pattern that resembles that of FV for these features (Figure 4a). When integration sites were binned based on their distance to TSS, CpG islands, or DHS island midpoints (Figure 4b–d, respectively), the lack of BET interaction resulted in a shift of integration away from those features and toward a more random integration pattern (compare brown/green and red bars), phenocopying FV distribution (blue bars). Similar data were obtained when zooming in on oncogene TSS (Figure 4a,e). While 3.23% of MLVIN_WT integration sites landed within a 2 kb window around oncogene TSS, only 1.93 and 2.08% of MLVIN_W390A and MLVIN_dCt integrations occurred in this window, respectively (P < 0.001, compared to MLVIN_WT), much alike FV distribution (1.63%), whereas HIV disfavors TSS (0.75%), in line with previous reports.9
Figure 4.

BinMLV vectors integration is targeted away from transcription start sites. (a) Murine leukemia virus (MLV)-based vector integration sites obtained from SupT1 cells and their genomic distribution. Integration percentages in 2 and 4 kb windows around TSS, CpG island midpoints, DHS, and oncogene TSS are listed. For comparison, foamy virus (FV) and HIV-1 data sets are included. P values (*) show significant departures (***P < 0.001, pairwise Fishers test) from MRC (not shown) and MLVIN_WT. (b–d) Integration frequencies for MLVIN_WT, MLVIN_dCt, MLVIN_W390A, FV, and HIV-1 in 750 bp bins around TSS, CpG islands, and DHS in SupT1 cells. (e) Integration frequency (%) of the indicated vectors in a 2 kb window around TSS of oncogenes (***P < 0.001, pairwise Fishers test). (f,g) Heat maps summarizing the relation between vector integration site frequency and genomic (e) or epigenetic (f) features in SupT1 cells. Evaluated vectors are indicated above the columns. Features analyzed are shown to the left of the corresponding row of the heat map. Tile colors indicate whether a particular feature is favored or disfavored for integration of the respective data sets relative to their MRCs, as detailed in the colored ROC area scale at the bottom of the panel. P values (*) show significance of departures from MLVIN_WT integration sites in SupT1 cells (**P < 0.01; ***P < 0.001, Wald statistics referred to χ2 distribution). CpG, CpG-rich islands; DHS, DNase I-hypersensitive sites; TSS, transcription start sites.
In addition, we analyzed integration preferences for MLVIN_W390A and MLVIN_dCt in SupT1 cells relative to a wide range of genomic features. MLVIN_W390A and MLVIN_dCt showed a decreased frequency of integration in areas rich in CpG islands, DHS, and high in GC content, and overall distributed more randomly (toward MRC) compared to MLVIN_WT (Figure 4f). Nonetheless, integration frequencies were still significantly different from MRC (P < 0.001, data not shown). Likewise, we compared the density of integration sites with that of a set of histone modifications (acetylation/methylation) and three chromatin-bound proteins (Pol II, H2AZ, and CTCF), mapped using chromatin immunoprecipitation and Solexa sequencing (ChIP-Seq) (detailed information on these epigenetic marks and their roles can be found in ref. 32,33). Compared to MLVIN_WT, BinMLV integration occurred less frequent near sites marked for active transcription by epigenetic modifications acetylations, H3K4me1, H3K4me2, H3K9me1 and H4K20me1, bound RNA Pol II, or H2AZ (a histone variant associated with promoters; Figure 4g). Overall, BinMLV distributed more randomly (shifting toward MRC) compared to MLVIN_WT, whereas MLVIN_W390A and MLVIN_dCt integration site distributions were identical (Supplementary Figure S4b,c; no statistical difference between both integration site data sets).
Similar results were obtained when assessing MLVIN_WT and MLVIN_W390A integration site distributions in primary human CD4+ T-cells (Supplementary Figure S5) and HeLa cells (data not shown). Whereas CD4+ T-cell BRD4 ChIP-seq tag frequencies28 correlated with MLVIN_WT, this association was lost for MLVIN_W390A (Supplementary Figure S5a). In line with the data obtained in SupT1 cells, MLVIN_W390A associated significantly less with TSS, CpG islands and DHS compared to MLVIN_WT (p<0.001, Supplementary Figure S4b–e). Likewise, MLVIN_W390A integration shifted more toward random compared to MLVIN_WT for a range of genomic features (Supplementary Figure S5f), and histone modifications32,33 (Supplementary Figure S5g).
Together, these findings demonstrate that BinMLV vectors do no longer integrate via BET proteins, leading to more random integration pattern, detargeted from traditional markers of MLV integration, suggesting that BinMLV vectors display a safer integration site profile, which opens perspectives for future translation of BinMLV vectors to gene therapeutic applications.
Discussion
Different animal models and several clinical trials underscored the potential of γ-retroviral vectors for gene marking and gene therapy. Unfortunately, further progress was thwarted with the advent of severe side effects, such as induced clonal dominance and malignant transformation, referred to as insertional mutagenesis.1,2 The main determinants of retroviral insertional oncogenesis are the integration site profile and transactivation of neighboring genes by strong promoter/enhancer elements in the U3 region of retroviral LTRs.1,2 To reduce the potential of clonal proliferation, self-inactivating (SIN) vectors were developed34,35,36 showing a lower tumorigenic potential in preclinical assays and gene-marking studies.35,36,37 However, even SIN vectors are mutagenic, given that they contain a sufficiently strong internal promoter, albeit at decreased incidences.34 Likewise, SIN lentiviral vectors harboring physiological promoter/enhancers caused insertional dysregulation of cellular genes in erythroid cells at high frequencies.38 In addition, it should be taken into account that SIN architecture is potentially genotoxic, because integration may disrupt or impede open-reading frames or regulatory regions, since their integration profile is not different from traditional retroviral vectors.39,40
Design of safer viral vectors for gene therapy requires mechanistic insight in the molecular mechanism of integration site selection. Retroviral integration is a nonrandom process, with different patterns of favored and disfavored target sites for each retroviral family. Interestingly, cluster analyses of different retroviruses based on their integration preferences mirrors phylogenetic trees based on the sequence similarity of their INs, and both are in good agreement with traditional trees based on genomic sequences.41 This strongly suggests a link between integration site selection and evolution, and puts forward integration site selection as part of the strategy by which retroviruses maximize their fitness.
The integration pattern of retroviruses or retrotransposons varies among different genera. While some members of the Ty3 retrotransposon lineage acquired a chromodomain at their integrase C-terminal end,42 other retrotransposons interact with cellular proteins to establish chromatin targeting. In this regard, the involvement of the TFIIIB component of the PolIII transcription apparatus in Ty3 retrotransposon targeting and Sir4p in Ty5 targeting were studied before.43,44 Lentiviruses are targeted to active transcription units via their interaction with LEDGF/p75.18,19,21,45 The latter protein does not bind the C-terminal end of lentiviral IN, but a cleft that is formed by the IN core dimer.46 Current retargeting strategies are based on (transient) expression of alternative tethers by linking the C-terminal domain of LEDGF/p75 to heterologous chromatin binding domains such as CBX.19,47 Although these methods allow retargeting, they are not readily applicable in a clinical gene therapy setting.
In this manuscript, we developed BET-independent MLV (BinMLV) vectors with wild-type transduction efficiency. The MLV IN-BET interaction is mediated by the BET ET domain and the unstructured IN C-terminal tail.23 We identified a motif (termed BET-interaction motif) in the C-terminal tail that is conserved in all gammaretroviral integrase proteins. Specific searches in protein databases did not reveal other proteins harboring this full motif. Our in vitro analysis revealed that deletion of the C-terminal tail or introduction of W390A mutation in BET-interaction motif is sufficient to abrogate the BET interaction. In contrast, Gupta et al.24 reported residues E266, L268, and Y269 in the IN core domain to be critical for BET interaction. Although our in vitro analysis did not reveal a significant effect of these residues in the direct interaction with BET proteins, we cannot exclude a role for the MLV IN core domain in BET binding in vivo.
Analysis of the integration profile of MLVIN_W390A and MLVIN_dCt revealed that integration site distribution of BinMLV vectors is less associated with TSS, CpG islands and DHS as compared to MLVIN_WT. Of note, the integration profile of MLVIN_W390A and MLVIN_dCt are similar, indicating that the W390A mutant is as potent as the complete deletion of the C-terminal tail to abrogate the BET interaction in vivo. Although BinMLV integration is still significantly different from MRC for the large majority of genomic features (P < 0.001), the distribution shifts more toward random (Figure 4f). Possibly, the open chromatin surrounding TSS is more accessible to BinMLV vectors and other cellular or viral determinants.
Earlier studies showed that a virus lacking the IN C-terminal tail is viable, however this deletion resulted in a two- to fourfold decrease in IN catalytic activity in vitro.26,27 Our results show that deletion of the C-terminal tail in the context of a vector results in a sixfold production and a twofold integration defect. On the contrary, transduction efficiency of MLVIN_W390A was in line with that of MLVIN_WT, opening perspectives for the use of BinMLVIN_W390A vectors in a clinical gene therapy setting. The decreased expression levels (mean fluorescence intensity, Supplementary Figure S2c,d) can be attributed to the more random integration pattern.
Comparison of MLVIN_W390A and MLVIN_dCt Bin vector integration sites to a set of epigenetic modifications revealed that overall integration is distributed more randomly and as such, is less associated with markers of active chromatin. Interestingly, we observed significantly less association with oncogene TSS, a primary determinant for insertional mutagenesis. These results suggest that BinMLV vectors have superior properties with respect to the current MLV-derived vectors used in clinical trials in regard to their oncogenic potential. In a next step, the biosafety of these vectors should be determined.
Material and methods
Plasmids. All oligonucleotides used are listed in Supplementary Table S1. All enzymes were purchased from Fermentas (Thermo Scientific, St Leon-Rot, Germany). To generate the recombinant protein expression constructs for GST-INCt and derived mutants, oligos 1 to 18 were annealed and ligated in BamHI/XhoI digested pGEX-6P-2 (GE Healthcare, Diegem, Belgium). The plasmid encoding full-length His6-tagged recombinant MLV IN (pKB-IN6H-MLV-IN) was kindly provided by C. Johnson (Picathaway, NJ). To generate protein expression constructs for GST-tagged INW390A and INdCt, oligos 19–20 and oligos 21–22 were annealed and ligated into XmaI/SalI digested pKB-IN6H-MLV-IN, respectively. For clarity, W390A is the actual position in MLV IN that interacts with ET domain of BET proteins. However, when using recombinant proteins, an additional start codon (ATG, methionine) is included, shifting all IN amino acids one position. In our previous paper, only using recombinant proteins, we therefore referred to this position as W391A.23 For simplicity, we employ W390A throughout this manuscript. MLV IN mutations E266A, L268A, and Y269A in full-length MLV IN were introduced via site-directed, ligase-independent mutagenesis (SLIM) in pKB-IN6H-MLV-IN using oligos 23–30 as previously described.23 To create the His6-Brd4ET expression construct, BRD4 ET was recombined from the pDONR221-BRD4ET plasmid into pHXGWA using the Gateway system (Invitrogen, Merelbeke, Belgium).23 Expression constructs for MBP-tagged BRD-2 and -3ET were described earlier.23 The MLV vector packaging plasmid (pCgp 608) was kindly provided by F.D. Busman (Philadelphia, NJ).
The Gagpol reading frame from pCgp 608 was PCR amplified with oligos 31 and 32, digested with EcoRI/NheI and subcloned into EcoRI/NheI digested peGFP-C1 (pC1_608). The MLV IN W390A mutation and dCt truncation in pC1_608 were first introduced in a shuttle plasmid containing part of MLV Gagpol by SLIM mutagenesis using oligos 33–36 and 37–40, respectively. The resulting shuttle plasmids were digested with BbvcI/EcoRI and the released fragments containing the mutation were cloned back into BbvcI/EcoRI digested pC1_608 to create MLVIN_W390A and MLVIN_dCt, respectively. The integrity of all plasmids was verified by DNA sequencing.
Cell culture. SupT1 cells and CD4+ T-cells (ATCC CRL-1942) were cultured in Roswell Park Memorial Institutes medium (RPMI-1640, Gibco-BRL, Merelbeke, Belgium) supplemented with 10% heat inactivated fetal calf serum (Sigma-Aldrich, Bornem, Belgium) and gentamicin (50 µg/ml, Gibco-BRL). HeLa cells and 293T cells were cultured in Dulbecco's modified Eagle medium (Gibco-BRL) supplemented with 8% heat inactivated fetal calf serum and gentamicin. All cells are grown in a humidified atmosphere with 5% CO2 at 37 °C.
T-cell purification. Peripheral blood mononuclear cells were purified from a buffy coat using density-gradient centrifugation (Lymphoprep; Axis-Shield PoC AS, Oslo, Norway). Primary CD4+ T cells were isolated using negative selection (MACS; Miltenyi Biotec, Leiden, the Netherlands) and stimulated with CD2, CD3, CD28 beads (MACS).
Retroviral vector production. Viral vectors were produced as previously described.23 Briefly, MLV-based vectors were produced by a triple PEI-based transfection of 293T cells with pVSV-G envelope, pC1_608 packaging plasmid or its derived mutants (see above) and p450-GFP transfer plasmid (kindly provided by F.D. Bushman). Vector titer, represented as reverse transcriptase units, was determined by the SYBRGreen-I product-enhanced reverse transcriptase assay.
Retroviral vector transduction. SupT1 cells (12 × 104/well), CD4+ T-cells (20 × 104/well) and NIH3T3 or HeLa cells (2 × 104/well) were seeded in 96-well plates and subsequently transduced with a dilution series of the respective vectors. Forty-eight hours post-transduction, 50% of the cells were harvested for fluorescence-activated cell sorting analysis, while the remaining 50% were cultured for 10 days for fluorescence-activated cell sorting analysis, to determine integrated copies and to perform integration site analysis.
gDNA isolation and quantitative PCR. Two million cells were pelleted and genomic DNA was extracted using a mammalian genomic DNA miniprep kit (Sigma-Aldrich). Genomic DNA concentrations were determined using standard spectrophotometric methods. Samples corresponding to 700 ng genomic DNA were used for analysis. Each reaction contained 12.5 µl iQ Supermix (Biorad, Nazareth, Belgium), 40 nmol/l forward and reverse primer (oligo 41 and 42 respectively) and 40 nmol/l of GFP probe (oligo 43) in a final volume of 25 µl. RNaseP was quantified as an endogenous control (TaqMan RNaseP control reagent, Applied Biosystems, The Netherlands). Samples were run in triplicate for 3 minutes at 95 °C followed by 50 cycles of 10 seconds at 95 °C and 30 seconds at 55 °C in a LightCycler 480 (Roche-applied-science, Vilvoorde, Belgium). Analysis was performed using the LightCycler 480 software supplied by the manufacturer.
Protein purification. Escherichia coli BL21 chemically competent cells were transformed with prokaryotic expression constructs. Cultures were grown at 37 °C to OD 0.6 and induced with 1 mmol/l isopropyl β-D-l-thiogalactopyranoside at 30 °C for 3 hours for induction of the C-terminal tail and at 16 °C for 1 hour for full-length MLV IN. Cell pellets were lysed in a lysis buffer (50 mmol/l Tris/HCl pH 7.3, 250 mmol/l NaCl, 1 mmol/l, PMSF, 5 mmol/l DTT, 10IU recombinant DNAse/10 ml lysate) and sonicated. The lysates were cleared by centrifugation at 15,000g for 30 minutes. Proteins were purified on a column containing an appropriate affinity resin for the specific tag. Glutathione Sepharose (GE Life Sciences, Diegem, Belgium) and amylose resin (New England Biolabs, Leiden, Netherlands) were used for GST and MBP purifications, respectively. Ni+-resin (Invitrogen) was used for His6-tagged proteins. The columns were washed in wash buffer (50 mmol/l Tris/HCl pH 7.3, 250 mmol/l NaCl, and 5 mmol/l DTT). Subsequently, proteins were eluted in wash buffer supplemented with 50 mmol/l reduced glutathione for GST-purifications, 20 mmol/l maltose for MBP purifications or 250 mmol/l imidazole for Ni+-purifications, respectively. Eluted proteins were dialyzed overnight with wash buffer containing 10% glycerol and stored at −80 °C.
AlphaScreen binding assay. AlphaScreen measurements were performed in a total volume of 25 µl in 384-well Optiwell microtiter plates (PerkinElmer, Zaventem, Belgium). All components were diluted to the desired concentrations in assay buffer (25 mmol/l Tris/HCl pH 7.4, 150 mmol/l NaCl, 1 mmol/l MgCl2, 0.1% Tween-20, 5 mmol/l DTT, and 0.1% bovine serum albumin). The affinities of GST-INCt and its respective mutants were determined against a fixed concentration of His-BRD4ET (20 nmol/l) or MBP-BRD4ET (2 nmol/l) while for His6-tagged full-length MLV-IN, its respective mutants or INdCt, a fixed concentration of 80 nmol/l was tested against dilution series of GST-BRD4ET. After addition of the proteins, the plate was incubated for 1 hour at 4 °C. Subsequently, 20 µg/ml anti-GST or anti-MBP donor and Ni2+-chelate acceptor beads (PerkinElmer) were added, bringing the final volume to 25 µl. After 1 hour incubation at 30 °C in the dark, the plate was read on an EnVision Multilabel Reader in AlphaScreen mode (PerkinElmer). Results were analyzed with Prism5.0 (GraphPad software) after nonlinear regression with the appropriate equations (one-site specific binding).
Recovery of integration sites and analysis of integration site distributions. Recovery of integration sites was performed as previously described.23 Briefly, linkers were ligated to restriction enzyme-digested (MseI) genomic DNA isolated from transduced cells and virus-host DNA junctions were amplified by nested PCR. Samples were individually barcoded with the second pair of PCR primers to generate 454 libraries. PCR products were purified and sequenced using 454/Roche pyrosequencing (Titanium Technology, Roche). Reads were quality-filtered by requiring perfect matches to the LTR linker, barcode, and flanking LTR and subsequently mapped to the human/mouse genome. All sites were required to align to the reference genome within 3 bp of the LTR edge. In order to control for possible biases in the datasets due to the choice of the MseI restriction endonuclease in cloning integration sites, MRC sites were generated in silico. To do so, each experimental integration site was paired with three sites in the genome, locating at the same distance from a randomly selected MseI site in the genome.
Analyses were carried out as described.18 A detailed account of the statistical methods used and the methods for forming and analyzing heat maps using ROC curves can be found in 48. Consensus sequence analysis at the point of integration was performed using WebLogo (http://weblogo.Berkeley.edu/logo.cgi). For association with specific genomic features, the distance of each integration site (in kb) to the respective genomic feature was calculated (midpoint of the CpG island or DHS, and X5-end of genes as a measure for TSS). Integration sites left of the genomic feature were given negative kb values, while integration sites toward the right were calculated as positive. Subsequently, the integration site data were pooled in bins ranging from 0 to 750 bp and increasing or decreasing with steps of 750 bp distance (−7,500 till 7500 bp window size around the respective genomic features). The percentage of integration sites occurring at a certain distance from the feature was plotted versus the distance. For heat maps, comparisons were carried out over three different interval sizes surrounding each integration site (5, 10, and 50 kb), since previous studies have shown that the interval sizes chosen for comparison can influence the conclusions. In this study, results were similar for each interval size examined (data not shown), so only the data for 10 kb intervals are shown. Results of statistical tests comparing the distributions of integration sites to the reference dataset are summarized as asterisks on each tile of the heat map. Type I error was accounted for by subjecting all P values to Bonferroni correction.
BET protein ChIP-seq data obtained in HEK293T cells and CD4+ T cells were retrieved from the Gene Expression Omnibus (accession codes GSM971946-8 for BRD-2, -3, and -4 in HEK293T cells, respectively and GSE33281 for BRD4 in CD4+ T cells).28,49 Extended sequence read densities were determined in 10 kb windows around MLVIN_WT, MLVIN_dC, MLVIN_W390A integration, or MRC sites respectively. Densities were normalized for total sequencing depth; input control (GSM971951) was subtracted and results were plotted using R 3.0.1.
SUPPLEMENTARY MATERIAL Figure S1. Characterization of the MLV-BET interface. Figure S2. Transduction efficiency and mean fluorescence intensity for BinMLV vectors. Figure S3. Loss of the BET interaction does not affect MLV integration site neighborhood. Figure S4. Loss of the BET interaction uncouples MLV integration from BET hot spots. Figure S5. Integration site distribution analysis for MLVIN_WT and MLVIN_W390A in human primary CD4+ T cells. Table S1. Oligonucleotides used in this study.
Acknowledgments
We thank Paulien Van de Velde, NamJoo Van der Veeken, and Martine Michiels for excellent technical assistance, Nirav Malani and Frederick Bushman (University of Pennsylvania, School of Medicine) for helpful discussions and support on integration site analysis. Viral vector production was performed at the Leuven Viral Vector Core. Research at KU Leuven received support from the FWO, the KU Leuven Research Council (OT), the KU Leuven IDO program and the Belgian IAP Belvir (IDO/12/008, ZKB9996 SB/0881057 and FWO ZKC0523, ZKC3378). S.E.A. is a doctoral fellow supported by Interdisciplinary Research Programmes (IDO; Interdisciplinaire onderzoeksprogramma's). J.D. and K.C. are doctoral fellows for the FWO Flanders. S.V. is a doctoral fellow supported by the Agency for Innovation by Science and Technology in Flanders (IWT). P.M. is a postdoctoral fellow for Belspo (Belgian Science Policy), cofunded by the Marie Curie Actions of the European Commission.
Supplementary Material
Characterization of the MLV-BET interface.
Transduction efficiency and mean fluorescence intensity for BinMLV vectors.
Loss of the BET interaction does not affect MLV integration site neighborhood.
Loss of the BET interaction uncouples MLV integration from BET hot spots.
Integration site distribution analysis for MLVIN_WT and MLVIN_W390A in human primary CD4+ T cells.
Oligonucleotides used in this study.
References
- Hacein-Bey-Abina S, Garrigue A, Wang GP, Soulier J, Lim A, Morillon E, et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. 2008;118:3132–3142. doi: 10.1172/JCI35700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe SJ, Mansour MR, Schwarzwaelder K, Bartholomae C, Hubank M, Kempski H, et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Invest. 2008;118:3143–3150. doi: 10.1172/JCI35798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott MG, Schmidt M, Schwarzwaelder K, Stein S, Siler U, Koehl U, et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat Med. 2006;12:401–409. doi: 10.1038/nm1393. [DOI] [PubMed] [Google Scholar]
- Avedillo Díez I, Zychlinski D, Coci EG, Galla M, Modlich U, Dewey RA, et al. Development of novel efficient SIN vectors with improved safety features for Wiskott-Aldrich syndrome stem cell based gene therapy. Mol Pharm. 2011;8:1525–1537. doi: 10.1021/mp200132u. [DOI] [PubMed] [Google Scholar]
- Cavazzana-Calvo M, Hacein-Bey S, de Saint Basile G, Gross F, Yvon E, Nusbaum P, et al. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science. 2000;288:669–672. doi: 10.1126/science.288.5466.669. [DOI] [PubMed] [Google Scholar]
- Mukherjee S, Thrasher AJ. Gene therapy for PIDs: progress, pitfalls and prospects. Gene. 2013;525:174–181. doi: 10.1016/j.gene.2013.03.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder AR, Shinn P, Chen H, Berry C, Ecker JR, Bushman F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell. 2002;110:521–529. doi: 10.1016/s0092-8674(02)00864-4. [DOI] [PubMed] [Google Scholar]
- Wu X, Li Y, Crise B, Burgess SM. Transcription start regions in the human genome are favored targets for MLV integration. Science. 2003;300:1749–1751. doi: 10.1126/science.1083413. [DOI] [PubMed] [Google Scholar]
- Mitchell RS, Beitzel BF, Schroder AR, Shinn P, Chen H, Berry CC, et al. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. 2004;2:E234. doi: 10.1371/journal.pbio.0020234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Ravin SS, Su L, Theobald N, Choi U, Macpherson JL, Poidinger M, et al. Enhancers are major targets for murine leukemia virus vector integration. J Virol. 2014;88:4504–4513. doi: 10.1128/JVI.00011-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFave MC, Varshney GK, Gildea DE, Wolfsberg TG, Baxevanis AD, Burgess SM. MLV integration site selection is driven by strong enhancers and active promoters. Nucleic Acids Res. 2014;42:4257–4269. doi: 10.1093/nar/gkt1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherepanov P, Maertens G, Proost P, Devreese B, Van Beeumen J, Engelborghs Y, et al. HIV-1 integrase forms stable tetramers and associates with LEDGF/p75 protein in human cells. J Biol Chem. 2003;278:372–381. doi: 10.1074/jbc.M209278200. [DOI] [PubMed] [Google Scholar]
- Llano M, Saenz DT, Meehan A, Wongthida P, Peretz M, Walker WH, et al. An essential role for LEDGF/p75 in HIV integration. Science. 2006;314:461–464. doi: 10.1126/science.1132319. [DOI] [PubMed] [Google Scholar]
- Hombrouck A, De Rijck J, Hendrix J, Vandekerckhove L, Voet A, De Maeyer M, et al. Virus evolution reveals an exclusive role for LEDGF/p75 in chromosomal tethering of HIV. PLoS Pathog. 2007;3:e47. doi: 10.1371/journal.ppat.0030047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shun MC, Raghavendra NK, Vandegraaff N, Daigle JE, Hughes S, Kellam P, et al. LEDGF/p75 functions downstream from preintegration complex formation to effect gene-specific HIV-1 integration. Genes Dev. 2007;21:1767–1778. doi: 10.1101/gad.1565107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rijck J, Vandekerckhove L, Gijsbers R, Hombrouck A, Hendrix J, Vercammen J, et al. Overexpression of the lens epithelium-derived growth factor/p75 integrase binding domain inhibits human immunodeficiency virus replication. J Virol. 2006;80:11498–11509. doi: 10.1128/JVI.00801-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciuffi A, Ronen K, Brady T, Malani N, Wang G, Berry CC, et al. Methods for integration site distribution analyses in animal cell genomes. Methods. 2009;47:261–268. doi: 10.1016/j.ymeth.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall HM, Ronen K, Berry C, Llano M, Sutherland H, Saenz D, et al. Role of PSIP1/LEDGF/p75 in lentiviral infectivity and integration targeting. PLoS One. 2007;2:e1340. doi: 10.1371/journal.pone.0001340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gijsbers R, Ronen K, Vets S, Malani N, De Rijck J, McNeely M, et al. LEDGF hybrids efficiently retarget lentiviral integration into heterochromatin. Mol Ther. 2010;18:552–560. doi: 10.1038/mt.2010.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rijck J, Bartholomeeusen K, Ceulemans H, Debyser Z, Gijsbers R. High-resolution profiling of the LEDGF/p75 chromatin interaction in the ENCODE region. Nucleic Acids Res. 2010;38:6135–6147. doi: 10.1093/nar/gkq410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrijvers R, De Rijck J, Demeulemeester J, Adachi N, Vets S, Ronen K, et al. LEDGF/p75-independent HIV-1 replication demonstrates a role for HRP-2 and remains sensitive to inhibition by LEDGINs. PLoS Pathog. 2012;8:e1002558. doi: 10.1371/journal.ppat.1002558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Larue RC, Plumb MR, Malani N, Male F, Slaughter A, et al. BET proteins promote efficient murine leukemia virus integration at transcription start sites. Proc Natl Acad Sci USA. 2013;110:12036–12041. doi: 10.1073/pnas.1307157110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rijck J, de Kogel C, Demeulemeester J, Vets S, El Ashkar S, Malani N, et al. The BET family of proteins targets moloney murine leukemia virus integration near transcription start sites. Cell Rep. 2013;5:886–894. doi: 10.1016/j.celrep.2013.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta SS, Maetzig T, Maertens GN, Sharif A, Rothe M, Weidner-Glunde M, et al. Bromo- and extraterminal domain chromatin regulators serve as cofactors for murine leukemia virus integration. J Virol. 2013;87:12721–12736. doi: 10.1128/JVI.01942-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaiah BN, Singer DS. Two faces of brd4: mitotic bookmark and transcriptional lynchpin. Transcription. 2013;4:13–17. doi: 10.4161/trns.22542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth MJ. Mutational analysis of the carboxyl terminus of the Moloney murine leukemia virus integration protein. J Virol. 1991;65:2141–2145. doi: 10.1128/jvi.65.4.2141-2145.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson CB, Donzella GA, Gaucan E, Smith CM, Roth MJ. Functional domains of Moloney murine leukemia virus integrase defined by mutation and complementation analysis. J Virol. 1996;70:4585–4597. doi: 10.1128/jvi.70.7.4585-4597.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Prakash C, Sum C, Gong Y, Li Y, Kwok JJ, et al. Bromodomain-containing protein 4 (BRD4) regulates RNA polymerase II serine 2 phosphorylation in human CD4+ T cells. J Biol Chem. 2012;287:43137–43155. doi: 10.1074/jbc.M112.413047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felice B, Cattoglio C, Cittaro D, Testa A, Miccio A, Ferrari G, et al. Transcription factor binding sites are genetic determinants of retroviral integration in the human genome. PLoS One. 2009;4:e4571. doi: 10.1371/journal.pone.0004571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trobridge GD, Miller DG, Jacobs MA, Allen JM, Kiem HP, Kaul R, et al. Foamy virus vector integration sites in normal human cells. Proc Natl Acad Sci USA. 2006;103:1498–1503. doi: 10.1073/pnas.0510046103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowrouzi A, Dittrich M, Klanke C, Heinkelein M, Rammling M, Dandekar T, et al. Genome-wide mapping of foamy virus vector integrations into a human cell line. J Gen Virol. 2006;87 Pt 5:1339–1347. doi: 10.1099/vir.0.81554-0. [DOI] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol. 2007;14:1025–1040. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zychlinski D, Schambach A, Modlich U, Maetzig T, Meyer J, Grassman E, et al. Physiological promoters reduce the genotoxic risk of integrating gene vectors. Mol Ther. 2008;16:718–725. doi: 10.1038/mt.2008.5. [DOI] [PubMed] [Google Scholar]
- Montini E, Cesana D, Schmidt M, Sanvito F, Bartholomae CC, Ranzani M, et al. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. J Clin Invest. 2009;119:964–975. doi: 10.1172/JCI37630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modlich U, Navarro S, Zychlinski D, Maetzig T, Knoess S, Brugman MH, et al. Insertional transformation of hematopoietic cells by self-inactivating lentiviral and gammaretroviral vectors. Mol Ther. 2009;17:1919–1928. doi: 10.1038/mt.2009.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newrzela S, Cornils K, Li Z, Baum C, Brugman MH, Hartmann M, et al. Resistance of mature T cells to oncogene transformation. Blood. 2008;112:2278–2286. doi: 10.1182/blood-2007-12-128751. [DOI] [PubMed] [Google Scholar]
- Hargrove PW, Kepes S, Hanawa H, Obenauer JC, Pei D, Cheng C, et al. Globin lentiviral vector insertions can perturb the expression of endogenous genes in beta-thalassemic hematopoietic cells. Mol Ther. 2008;16:525–533. doi: 10.1038/sj.mt.6300394. [DOI] [PubMed] [Google Scholar]
- Moiani A, Miccio A, Rizzi E, Severgnini M, Pellin D, Suerth JD, et al. Deletion of the LTR enhancer/promoter has no impact on the integration profile of MLV vectors in human hematopoietic progenitors. PLoS One. 2013;8:e55721. doi: 10.1371/journal.pone.0055721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modlich U, Schambach A, Brugman MH, Wicke DC, Knoess S, Li Z, et al. Leukemia induction after a single retroviral vector insertion in Evi1 or Prdm16. Leukemia. 2008;22:1519–1528. doi: 10.1038/leu.2008.118. [DOI] [PubMed] [Google Scholar]
- Derse D, Crise B, Li Y, Princler G, Lum N, Stewart C, et al. Human T-cell leukemia virus type 1 integration target sites in the human genome: comparison with those of other retroviruses. J Virol. 2007;81:6731–6741. doi: 10.1128/JVI.02752-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik HS, Burke WD, Eickbush TH. The age and evolution of non-LTR retrotransposable elements. Mol Biol Evol. 1999;16:793–805. doi: 10.1093/oxfordjournals.molbev.a026164. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Zou S, Wright DA, Voytas DF. Tagging chromatin with retrotransposons: target specificity of the Saccharomyces Ty5 retrotransposon changes with the chromosomal localization of Sir3p and Sir4p. Genes Dev. 1999;13:2738–2749. doi: 10.1101/gad.13.20.2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchner J, Connolly CM, Sandmeyer SB. Requirement of RNA polymerase III transcription factors for in vitro position-specific integration of a retroviruslike element. Science. 1995;267:1488–1491. doi: 10.1126/science.7878467. [DOI] [PubMed] [Google Scholar]
- Ciuffi A, Llano M, Poeschla E, Hoffmann C, Leipzig J, Shinn P, et al. A role for LEDGF/p75 in targeting HIV DNA integration. Nat Med. 2005;11:1287–1289. doi: 10.1038/nm1329. [DOI] [PubMed] [Google Scholar]
- Cherepanov P, Ambrosio AL, Rahman S, Ellenberger T, Engelman A. Structural basis for the recognition between HIV-1 integrase and transcriptional coactivator p75. Proc Natl Acad Sci USA. 2005;102:17308–17313. doi: 10.1073/pnas.0506924102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vets S, De Rijck J, Brendel C, Grez M, Bushman F, Debyser Z, et al. Transient Expression of an LEDGF/p75 Chimera Retargets Lentivector Integration and Functionally Rescues in a Model for X-CGD. Mol Ther Nucleic Acids. 2013;2:e77. doi: 10.1038/mtna.2013.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady T, Lee YN, Ronen K, Malani N, Berry CC, Bieniasz PD, et al. Integration target site selection by a resurrected human endogenous retrovirus. Genes Dev. 2009;23:633–642. doi: 10.1101/gad.1762309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy G, Chepelev I, Dimaggio PA, Blanco MA, Zee BM, Zhao K, et al. Proteogenomic characterization and mapping of nucleosomes decoded by Brd and HP1 proteins. Genome Biol. 2012;13:R68. doi: 10.1186/gb-2012-13-8-r68. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Characterization of the MLV-BET interface.
Transduction efficiency and mean fluorescence intensity for BinMLV vectors.
Loss of the BET interaction does not affect MLV integration site neighborhood.
Loss of the BET interaction uncouples MLV integration from BET hot spots.
Integration site distribution analysis for MLVIN_WT and MLVIN_W390A in human primary CD4+ T cells.
Oligonucleotides used in this study.