

The methyltransferase Skb1 and the previously uncharacterized protein Slf1 physically interact and colocalize at a 1:1 molar ratio in cortical nodes. Skb1 and Slf1 are mutually dependent for node localization, and the Slf1 C-terminus binds to lipids for anchoring nodes at the plasma membrane.
Abstract
The plasma membrane contains both dynamic and static microdomains. Given the growing appreciation of cortical microdomains in cell biology, it is important to determine the organizational principles that underlie assembly of compartmentalized structures at the plasma membrane. The fission yeast plasma membrane is highly compartmentalized by distinct sets of cortical nodes, which control signaling for cell cycle progression and cytokinesis. The mitotic inhibitor Skb1 localizes to a set of cortical nodes that provide spatial control over signaling for entry into mitosis. However, it has been unclear whether these nodes contain other proteins and how they might be organized and tethered to the plasma membrane. Here we show that Skb1 forms nodes by interacting with the novel protein Slf1, which is a limiting factor for node formation in cells. Using quantitative fluorescence microscopy and in vitro assays, we demonstrate that Skb1-Slf1 nodes are megadalton structures that are anchored to the membrane by a lipid-binding region in the Slf1 C-terminus. We propose a mechanism for higher-order node formation by Skb1 and Slf1, with implications for macromolecular assemblies in diverse cell types.
INTRODUCTION
The spatial regulation of large macromolecular complexes can generate microdomains and compartments for distinct processes in cells. Large-scale protein–lipid complexes organize these domains at the plasma membrane and serve as signaling hubs for diverse cellular processes (Laude and Prior, 2004; Lingwood and Simons, 2010; Hartman and Groves, 2011). For example, the fidelity of Ras GTPase signaling requires dynamic multiprotein nanoclusters at the plasma membrane (Cebecauer et al., 2010). More-static microdomains such as caveolae form plasma membrane compartments that control diverse signaling networks, including Src family kinases, receptor tyrosine kinases, and nitric oxide (Laude and Prior, 2004). Given the growing appreciation of cortical microdomains in cell biology, it is important to determine the organizational principles that underlie assembly of compartmentalized structures at the plasma membrane.
The fission yeast Schizosaccharomyces pombe provides a striking example of plasma membrane compartmentalization, with both dynamic and static cortical microdomains. The growing tips of these cylindrical cells contain dynamic microdomains of polarity proteins that ensure spatial control of polarized growth (Dodgson et al., 2013). By contrast, the plasma membrane in the nongrowing middle of interphase cells contains at least three stable, nonoverlapping populations of cortical microdomains. First, a set of cortical filaments called eisosomes is generated by the BAR-domain protein Pil1 and functions to regulate phosphatidylinositol (4,5)-bisphosphate (PI(4,5)P2) levels (Kabeche et al., 2011, 2014; Snaith et al., 2011). Second, a set of 50–75 cortical nodes, organized by the mitotic inducer Cdr2, contains several well-studied cell cycle regulatory proteins (Morrell et al., 2004; Martin and Berthelot-Grosjean, 2009; Moseley et al., 2009). Third, the mitotic inhibitor Skb1, a PRMT5-like methyltransferase, localizes to a band of 50–100 cortical nodes that are distinct from eisosomes and Cdr2 nodes (Deng and Moseley, 2013). The number of Skb1 nodes scales with cell size, and these nodes ensure proper cell cycle progression by sequestering Skb1 from its inhibitory target Cdr1 in Cdr2 nodes.
The Skb1-containing nodes are the least understood of these three cortical structures. For example, it is unknown whether these nodes contain other proteins and how they attach to the plasma membrane. It is also interesting to note that Skb1 function and localization are remarkably divergent from those of its budding yeast orthologue Hsl7, which promotes mitotic entry and localizes to a continuous ring at the bud neck (Cid et al., 2001; Theesfeld et al., 2003; Sakchaisri et al., 2004). In this study, we investigated the components and mechanisms that promote Skb1 node formation. Our findings reveal a cellular strategy for the assembly of stable microdomains and also have implications for the role of specific ligands in the functional divergence of highly conserved proteins.
RESULTS
Identification of Slf1 as an Skb1 interaction protein
Skb1 forms node-like structures that are confined to the fission yeast plasma membrane (Deng and Moseley, 2013), but the Skb1 protein sequence contains no identifiable motifs for membrane anchoring. We hypothesized that Skb1 nodes might represent multiprotein structures with additional factors that dock Skb1 to the plasma membrane. As a first step toward testing this model, we identified proteins that physically interact with Skb1. We integrated a 3x hemagglutinin (3HA) epitope tag at the C-terminus of endogenous Skb1 and used anti-HA beads to purify protein complexes from both skb1-3HA cells and untagged control cells (Figure 1A). We then used liquid chromatography–tandem mass spectrometry (LC-MS/MS) to identify proteins that were purified specifically from skb1-3HA cells (Supplemental Figure S1). The most abundant Skb1-associated factor was the previously uncharacterized protein SPAC821.03c, which we designated Slf1 (Skb1 localization factor) for reasons to be described. We verified the Skb1-Slf1 interaction in vivo using reciprocal coimmunoprecipitation experiments. Endogenously tagged Slf1–monomeric enhanced green fluorescent protein (mEGFP) copurified with Skb1-3HA (Figure 1B), and Skb1-13myc coimmunoprecipitated with hexahistidine (6His)-Slf1 (Figure 1C). We additionally found that Skb1 and Slf1 interact by the yeast two-hybrid assay (Figure 1D), consistent with a direct physical interaction. Together these data indicate that Slf1 and Skb1 form a physical complex in fission yeast cells.
FIGURE 1:

Slf1 physically interacts with Skb1. (A) Silver-stained SDS-polyacrylamide gel of proteins isolated from skb1-3HA and untagged control strains. LC-MS/MS identified Slf1 specifically in the skb1-3HA sample. (B) Coimmunoprecipitation of endogenously tagged Skb1-3HA and Slf1-mEGFP from fission yeast cells using anti-HA beads. (C) Coimmunoprecipitation of 6His-Slf1 and Skb1-13myc. 6His-Slf1 was induced for 20 h using the pREP3X-6His-slf1 plasmid and isolated using nickel-agarose chromatography. (D) Skb1 and Slf1 physically interact in the yeast two-hybrid assay. Transformants were selected on double dropout (DDO) plates, and interactions were tested by growth on quadruple dropout plus aureobasidin A (QDO/A) plates.
The Skb1-Slf1 interaction suggested that Slf1 might be a component of Skb1 nodes at the plasma membrane. Therefore we tested the spatial distribution of Slf1 in cells. Similar to Skb1, endogenously tagged Slf1-mEGFP localized to a set of cortical nodes that were excluded from growing cell tips, which were marked by the cell wall dye Blankophor (Figure 2A). In cells expressing both Skb1-3GFP and mCherry-Slf1, we found that Skb1 and Slf1 colocalize in the same cortical nodes (Figure 2B). We encountered significant variation of mCherry-Slf1 signal during the acquisition of multiple focal planes, with the appearance of some Skb1-3GFP nodes having reduced or no signal from mCherry-Slf1. These differences in signal might represent dynamic changes in the abundance of these two proteins over the lifetime of a node or alternatively reflect loss of signal due to photobleaching. This technical limitation also prevented time-lapse experiments to monitor Skb1 and Slf1 levels at nodes over time. Nonetheless, our combined experiments indicate that these cortical nodes are multiprotein structures that contain the interacting proteins Skb1 and Slf1.
FIGURE 2:
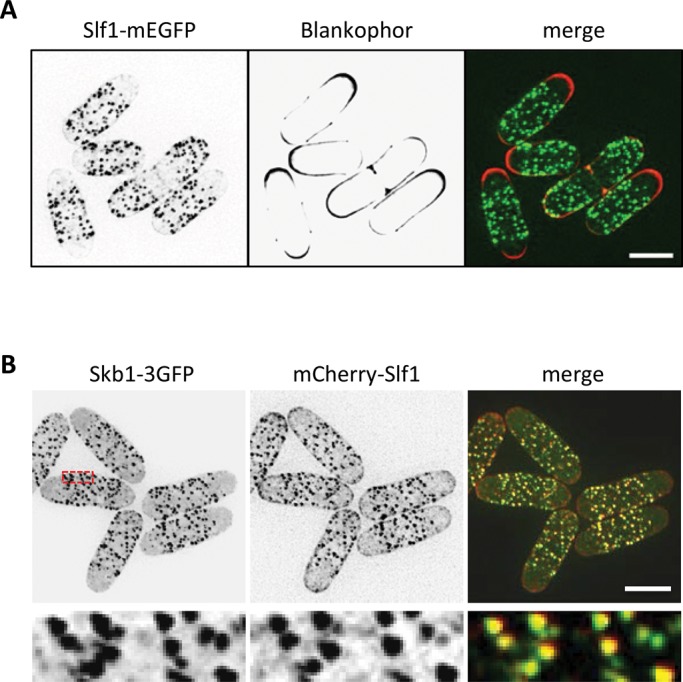
Slf1 colocalizes with Skb1 at cortical nodes. (A) Localization of endogenously tagged Slf-mEGFP. Cells were also stained by Blankophor. Images are inverted maximum projections from Z-planes in the top half of the cells. Note the exclusion of cortical nodes from Blankophor-stained regions. (B) Skb1 and Slf1 colocalize in cortical nodes. Images are inverted maximum projections from Z-planes in the top half of the cells. Region boxed in red is magnified in the bottom row. Scale bars, 5 μm.
Both Skb1 and Slf1 are required to assemble megadalton nodes
To test the roles of Skb1 and Slf1 in node formation, we examined the localization of each protein in the absence of the other. In wild-type cells, Skb1-3GFP localized to cortical nodes and in the cytoplasm. By contrast, Skb1 nodes were absent in slf1∆ cells, and all Skb1 protein was in the cytoplasm (Figure 3A). Similarly, Slf1 node localization was lost in skb1∆ cells (Figure 3B). Slf1-mEGFP remained largely cortical in skb1∆ cells but was not concentrated in nodes. Instead, Slf1-mEGFP localized in small cortical puncta at growing cell tips and the division septum in the absence of Skb1. These results indicate that Skb1 and Slf1 are mutually dependent for node formation and suggest that their interaction promotes the assembly of a higher-order structure in cells.
FIGURE 3:
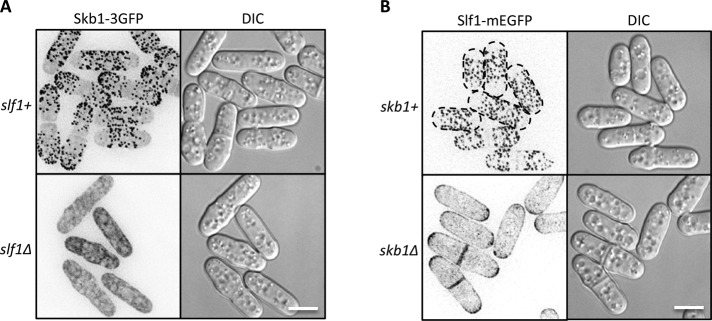
Skb1 and Slf1 are mutually dependent for node localization. (A) Slf1 is required for Skb1 cortical localization. Note the cytoplasmic localization of Skb1-3GFP in slf1∆ cells. (B) Skb1 is required for localization of Slf1 to cortical nodes. Note the absence of Slf1 nodes in skb1∆ cells. Images are inverted maximum projections. Scale bars, 5 μm.
We next sought to gain quantitative insight into the relationship between Skb1 and Slf1 at cortical nodes. Therefore we counted the number of Skb1 and Slf1 molecules both globally in cells and locally in nodes. We used mEGFP-tagged proteins of known cellular concentration (Cdc12, Mid1, Sad1, Rlc1, and Spn1; Wu and Pollard, 2005) to establish a linear relationship between fluorescence intensity and molecule numbers in a single cell (Figure 4A). We then quantified total cellular fluorescence from endogenously tagged slf1-mEGFP and skb1-mEGFP cells and counted 14,000 ± 2600 Skb1 molecules/cell and 8700 ± 1900 Slf1 molecules/cell (Figure 4A). Next we used local fluorescence in a node to count the number of Skb1-mEGFP and Slf1-mEGFP molecules per node. We quantified an average of 69 Skb1 molecules and 77 Slf1 molecules per single node (Figure 4B). This means that cortical nodes contain ∼1:1 ratio of Skb1 and Slf1 molecules, although the total cellular concentration of Skb1 is nearly twofold higher than that of Slf1. These numbers represent a “lifetime average” for a single node, and the ratio of Skb1 and Slf1 may vary during the initiation and disassembly of a node. Nonetheless, a cortical node containing 69 Skb1 molecules (73 kDa each) and 77 Slf1 molecules (54 kDa each) represents a 9.1-MDa structure. This is a conservative estimate because nodes may contain additional factors that have not yet been identified. For comparison, a 9-MDa cortical node is bigger than a ribosome (∼3 MDa) and smaller than a nuclear pore complex (∼50 MDa). We conclude that Skb1 and Slf1 interact in 1:1 stoichiometry to generate large intracellular structures at the plasma membrane.
FIGURE 4:

Global and local quantification of Skb1 and Slf1 concentrations. (A) Fluorescence intensity analyses for counting the numbers of Skb1 and Slf1 molecules per cell. Linear relationship between fluorescence intensity (mean ± SD) and molecule numbers per cell (Wu and Pollard, 2005) were established using mEGFP-tagged Spn1, Rlc1, Mid1, Sad1, and Cdc12 cells (n > 50 cells for each protein). Fluorescence intensities from Skb1-mEGFP and Slf1-mEGFP were measured (n > 50) to calculate their global abundance, shown in the graph. (B) Box plots showing the numbers of Slf1 and Skb1 molecules per individual nodes. Fluorescence intensities of Skb1-mEGFP and Slf1-mEGFP in the cortical nodes (n > 160) were measured and converted to number of molecules using the equation from A. (C) Slf1 overexpression promotes the formation of Skb1 nodes. 6His-Slf1 protein was induced at 32°C for 20 h. Numbers under the images show the number of Skb1 nodes per cell (mean ± SD; n > 100). Images are inverted maximum projections from deconvolved Z-series. Scale bar, 5 μm.
Several lines of evidence suggested that Slf1 might be a limiting component for node formation in cells. For example, the cellular concentration of Skb1 is nearly twofold higher than that of Slf1, but these proteins exist in a 1:1 ratio at nodes. This is consistent with the previously observed cytoplasmic pool of Skb1 (Deng and Moseley, 2013). If the number of Slf1 molecules in a cell limits node formation, then increased Slf1 protein levels should lead to more nodes. We found that Slf1 overexpression doubled the number of Skb1 nodes per cell (Figure 4C), indicating that Slf1 protein levels limit the number of nodes in cells. Because overexpressed Slf1 recruits cytoplasmic Skb1 to the plasma membrane, these results also suggest a role for Slf1 in promoting the cortical localization of nodes.
Slf1 is a dosage-dependent membrane anchor for Skb1
How are Skb1-Slf1 nodes anchored to the plasma membrane? Slf1-mEGFP remained cortical in skb1∆ cells that lack nodes, and we identified a lysine-rich region between amino acids 451 and 485 in the C-terminus of Slf1 (Supplemental Figure S2). Thus we hypothesized that the Slf1 C-terminus might bind to anionic lipids in the plasma membrane. To test this possibility, we first generated a truncated Slf1∆C-mEGFP construct that abolished localization to the cell cortex (Figure 5A). This change is not due to disruption of Skb1-Slf1 binding, as we found that Slf1(1-200) is sufficient for interaction with Skb1 (Figure 5B). Of interest, the truncated Slf1∆C-mEGFP localized to both the nucleus and cytoplasm, but Skb1 remained largely cytosolic in this mutant (Supplemental Figure S3A). These data mean that the Slf1 C-terminus promotes cortical localization, whereas the Slf1 N-terminus interacts with Skb1.
FIGURE 5:

Slf1 is a membrane anchor for cortical nodes. (A) Localization of Slf1-mEGFP and the truncated Slf1ΔC-mEGFP, which lacks amino acids 451–484. Images are inverted maximum projections. (B) Mapping of Skb1 interaction region on Slf1 by yeast two-hybrid assay. Bait and prey plasmids were cotransformed and selected on DDO plates; interactions were tested on QDO plates. (C) Localization of Slf1C-mEGFP and mEGFP. Slf1C includes amino acids 451–484. Note the membrane localization of Slf1C-mEGFP. Images are single focal planes. (D) Liposome pelleting assays for recombinant GST-Slf1C, which contains Slf1 amino acids 451–485. Note the increase of GST-Slf1C in the pellet fraction for reactions containing PC + phosphatidylethanolamine (PE) + phosphatidylserine (PS) + PI(4,5)P2 liposomes. GST was used as a negative control. (E) Localization of Skb1-mEGFP and Skb1-Slf1C-mEGFP in slf1+ or slf∆ cells. Images are inverted maximum projections. Scale bars, 5 μm.
We next tested the ability of the Slf1 C-terminus to interact with membranes in cells and in vitro. First, we fused the Slf1 C-terminus to GFP to generate the construct Slf1C-mEGFP. This fusion protein was recruited to the cell cortex, in contrast to the cytoplasmic localization of mEGFP alone (Figure 5C). We conclude that the Slf1 C-terminus is both necessary and sufficient for localization to the plasma membrane in cells. Next we purified the recombinant C-terminus of Slf1 fused to glutathione S-transferase (GST-Slf1C; Supplemental Figure S3B) and tested direct binding to lipids with liposome pelleting assays. GST-Slf1C pelleted with liposomes containing negatively charged lipids phosphatidylserine (PS) and PI(4,5)P2 but did not pellet with liposomes containing only neutral lipid phosphatidylcholine (PC) (Figure 5D). By contrast, GST alone did not pellet with liposomes under any conditions (Figure 5D). Thus the Slf1 C-terminus is both necessary and sufficient for cortical localization in cells and binds directly to lipids in vitro.
We further tested the cortical targeting activity of the Slf1 C-terminus by fusing it to Skb1, thereby generating an Skb1-Slf1C fusion protein. Compared to Skb1-mEGFP, the chimeric protein Skb1-Slf1C formed more cortical nodes and reduced cytoplasmic signal in slf1+ cells (Figure 5E). This indicates that the Slf1 C-terminus is sufficient for cortical tethering of Skb1 and that recruitment of Skb1 to the plasma membrane is a limiting step in the assembly of cortical nodes. However, cortical node assembly by the chimeric Skb1-Slf1C protein was lost in slf1∆ cells (Figure 5E). This means that Slf1 does not act as a simple membrane anchor. Instead, the Slf1 N-terminus, which interacts with Skb1, is required for higher-order assembly of node structures in cells. This requirement cannot be bypassed by artificial recruitment of Skb1 to the cortex.
Slf1 functions with Skb1
Because Slf1 and Skb1 interact to generate cortical nodes, we next tested whether these two proteins function in similar pathways and processes. Skb1 delays mitotic entry through regulation of Cdr1 and Wee1 (Gilbreth et al., 1998; Deng and Moseley, 2013) and has been suggested to promote cell polarity (Gilbreth et al., 1996; Wiley et al., 2003). We previously showed that cortical nodes sequester Skb1 from its regulatory targets Cdr1 and Wee1 (Deng and Moseley, 2013). Fission yeast cells divide reproducibly at 14 μm due to a cell size checkpoint that controls mitotic entry. We found that slf1∆ cells divide at a larger size than wild-type cells, indicating that Slf1 promotes mitotic entry (Figure 6A). By contrast, skb1∆ reduced cell size at division, consistent with mitotic inhibition by Skb1. To determine whether Skb1 and Slf1 regulate mitotic entry through the same pathway, we generated the double mutant skb1∆ slf1∆, which divided at the same size as the single mutant slf1∆ (Figure 6A). Thus slf1∆ is epistatic to skb1∆, and these proteins likely function in a linear pathway. In support of this conclusion, the slf1∆ size defect was additive with cdc25-22 but not with cdr1∆ (Figure 6A), similar to previous results for skb1∆ (Deng and Moseley, 2013). The mechanism of Slf1 function in regulating mitotic entry will require additional studies, but our genetic data indicate that Slf1 functions with Skb1 in the regulation of cell size at division through Cdr1.
FIGURE 6:

Slf1 is a novel mitotic inducer. (A) Length of dividing, septated cells of the indicated strains (mean ± SD; n > 50 for each value). (B) Phenotype of Slf1 overexpression. Images of Blankophor-stained cells are inverted single focal planes from deconvolved Z-series. The indicated plasmids were transformed and induced for 36 h at 25°C. Note the abnormal cell morphology in 6His-Slf1–overexpressed cells. Scale bar, 5 μm. (C) Slf1 overexpression is lethal. Cells transformed with pREP3X or pREP3X-6His-slf1 were spotted onto EMM-Leu plate or EMM-Leu plate supplemented with 10 μg/ml thiamine with 10× serial dilutions. Plates were incubated at 32°C for 4 d.
We next tested the effects of Slf1 overexpression by using the pREP3X multicopy plasmid with thiamine-repressible P3nmt1 promoter. At early time points of expression, increased levels of Slf1 protein reduced the size of dividing cells (11.7 ± 1.0 μm for pREP3X-6His-slf1 vs. 13.9 ± 0.9 μm for pREP3X alone), indicating that Slf1 can act as a dosage-dependent inducer of mitotic entry. At later time points, Slf1 overexpression led to defects in cell morphology and septation (Figure 6B). This suggests that Slf1 might regulate cell polarity, similar to previous models for its interacting partner Skb1 (Gilbreth et al., 1996; Wiley et al., 2003). Consistent with this severe phenotype, Slf1 overexpression in serial dilution growth assays was lethal in wild-type, cdr1∆, and skb1∆ strains (Figure 6C). We conclude that cortical node components Skb1 and Slf1 function together in the regulation of cell cycle progression and cell morphology.
Evolutionary conservation of Slf1 and Skb1
Finally, we considered that the evolutionary conservation of Slf1 and Skb1 might provide insight into the striking difference between Skb1-like molecules in the related budding and fission yeasts. Whereas fission yeast Skb1 inhibits mitotic entry and localizes to cortical nodes, the budding yeast Skb1-like protein Hsl7 promotes mitotic entry and localizes in a continuous ring at the bud neck (Ma et al., 1996; Cid et al., 2001; Theesfeld et al., 2003; Sakchaisri et al., 2004). Both Skb1 and Hsl7 are members of the PRMT5 family of arginine methyltransferase proteins, which have been highly conserved from yeast through humans (Bao et al., 2001; Sayegh and Clarke, 2008). Of interest, we identified Slf1 homologues in many Schizosaccharomyces species, including S. pombe, S. cryophilus, S. octosporus, and S. japonicus (Figure 7), but not in other fungi, such as Aspergillus niger, Neurospora crassa, and the budding yeast Saccharomyces cerevisiae. Thus Slf1 may have evolved in fission yeasts to drive the formation of a distinct localization pattern for Skb1. In addition, we found that sequence conservation of Skb1-related proteins correlates with the presence of Slf1-like proteins. All Skb1-related proteins contain a highly conserved PRMT5 methyltransferase domain that is flanked by regulatory N- and C-terminal regions. These Skb1 N- and C-terminal regulatory regions are well conserved in species that contain Slf1 but are divergent when compared with species that lack Slf1 (Figure 7). This raises the possibility that Skb1 and Slf1 coevolved under selective pressures for signaling by cortical nodes. The role of Slf1 also demonstrates how species-specific adaptor proteins can modulate the localization and function of highly conserved factors such as Skb1 and other PRMT5-like proteins.
FIGURE 7:
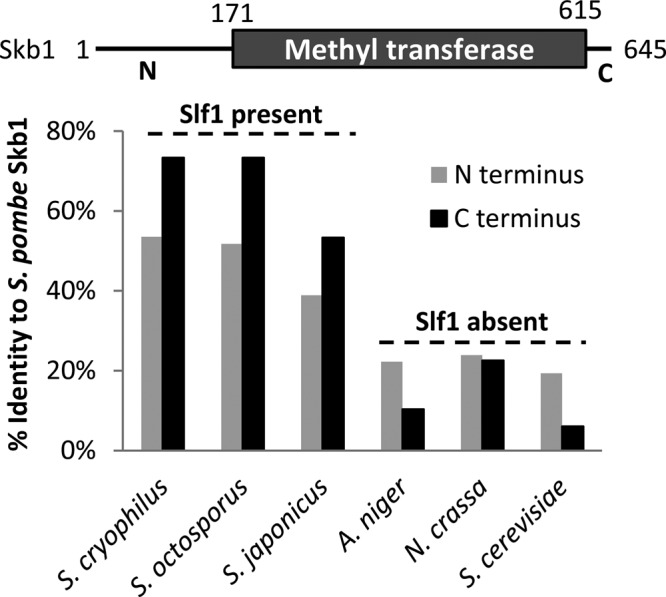
Conservation of Skb1-Slf1 interactions. Sequence identity scoring for the N- and C- termini of Skb1 orthologues in other fungi, as compared with S. pombe Skb1 protein sequence. The top scheme shows the structure of S. pombe Skb1. Note that species with Slf1-like proteins have higher conservation of Skb1 N- and C-termini. Residues included in this analysis are reported in Materials and Methods.
DISCUSSION
Macromolecular structures control a wide range of functions in cells, and a long-standing goal of cell biology is to understand organizational principles that guide assembly of such large structures. Our identification of Slf1 as a ligand of Skb1 led to the discovery of megadalton nodes at the fission yeast plasma membrane. These two proteins are present at a 1:1 stoichiometry in nodes, but Slf1 protein abundance limits the number of nodes per cell. The local concentrations of Skb1 and Slf1 at a node may change over time, so this 1:1 ratio represents a lifetime average. Indeed, we encountered variability in quantifying these local concentrations, and this might reflect dynamic changes in Skb1 and Slf1 localization during the lifetime of a node or heterogeneity between different nodes in a cell. Changes in the ratio of these two proteins during time might provide insights into the assembly and disassembly mechanism of nodes, and this represents an important future challenge.
On the basis of our findings, we propose a simple working model for these large, two-component macromolecular structures (Figure 8). Slf1 binds to both Skb1 and anionic lipids and thus represents the membrane anchor for this population of cortical nodes. These Slf1-Skb1 protein complexes may assemble into higher-order structures through the previously demonstrated multimerization of Skb1 (Gilbreth et al., 1996; Deng and Moseley, 2013). We note that artificial membrane tethering of Skb1 by the Skb1-Slf1C fusion protein is not sufficient for node assembly, indicating that both Slf1 N-terminus and lipids likely promote the assembly of these complexes into megadalton structures. One possibility is that Slf1 N-terminus, which interacts with Skb1, promotes Skb1 multimerization at nodes. Our model does not address what limits the size of an individual node or the organization of Skb1, Slf1, and lipid molecules in these structures. The molecular mechanisms that guide node assembly are likely to be conserved in cortical microdomains from other cell types and organisms. In this sense, our findings and the remaining open questions have implications for the assembly of macromolecular structures in a wide range of systems.
FIGURE 8:
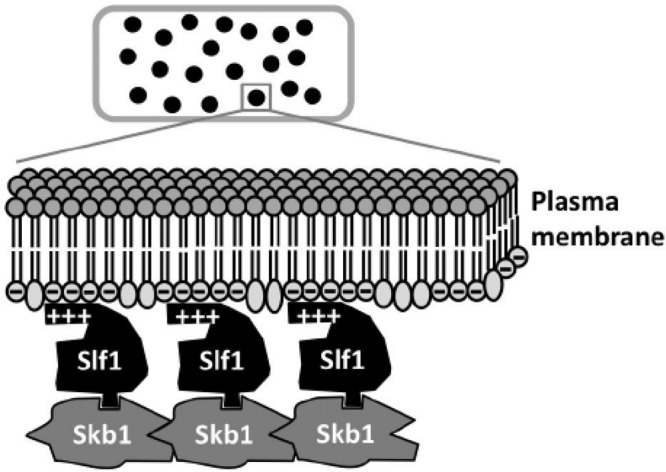
Working model for molecular interactions within a cortical node. See the text for details and discussion.
Our genetic data indicate that both Skb1 and Slf1 function in a linear pathway that regulates mitotic entry through Cdr1 and Wee1. We previously demonstrated that Skb1 prevents mitotic entry by inhibiting the Cdr1-Wee1 signaling pathway (Deng and Moseley, 2013). Here we found that slf1∆ cells are elongated at division, indicating that Slf1 promotes mitotic entry. This slf1∆ phenotype was epistatic to the skb1∆ cell size defect, which suggests that Slf1 functions downstream of Skb1 in signaling to Cdr1 and Wee1. The underlying mechanism is unknown, as Skb1 physically interacts with both Cdr1 and Wee1 (Deng and Moseley, 2013), but we have not detected physical interactions for Slf1 with Cdr1 and/or Wee1 (our unpublished results). Additional work is needed to reconcile these genetic and biochemical data, which suggest that other proteins or layers of regulation may contribute to these signaling pathways.
The assembly of cortical nodes represents a general organizing principle for signaling molecules at the fission yeast plasma membrane. During interphase, the central cortex of fission yeast cells contains both Skb1-Slf1 nodes and a second set of stable nodes that contain many interacting proteins (Cdr2, Cdr1, Wee1, Mid1, Blt1, Klp8, Gef2, and Nod1; Paoletti and Chang, 2000; Morrell et al., 2004; Martin and Berthelot-Grosjean, 2009; Moseley et al., 2009; Jourdain et al., 2013; Zhu et al., 2013). This second set of nodes has been further subdivided into type I and type II nodes, which assemble independently but then merge to form a single set of type I/II nodes with all components (Akamatsu et al., 2014). Skb1-Slf1 nodes and type I/II Cdr2 nodes exhibit no colocalization and do not depend on each other (Deng and Moseley, 2013), indicating that they are distinct cortical structures. The organization of these signaling molecules into distinct sets of nodes is in stark contrast to their budding yeast orthologues, which largely colocalize in a continuous ring at the bud neck. These different localization patterns have the potential to monitor distinct aspects of cell growth for downstream signaling events in these two types of cells.
Both Skb1-Slf1 nodes and “Cdr2 nodes” appear as stable, punctate structures, but they also exhibit important differences. For example, Skb1-Slf1 nodes may contain only these two proteins, whereas “Cdr2 nodes” contain many more components that likely assemble through complex physical interactions. Although additional proteins may be discovered in each set of nodes, this suggests that Skb1-Slf1 nodes have a simpler composition. The dependence of different proteins in recruitment to these nodes is another distinction between Cdr2 nodes and Skb1-Slf1 nodes. Cdr2 nodes have been proposed to follow a linear hierarchy, where Cdr2 itself is sufficient to assemble node structures, followed by the recruitment of other proteins (Moseley et al., 2009). This contrasts with our demonstration that both Skb1 and Slf1 are mutually required for node localization. Finally, we estimated that Skb1-Slf1 nodes are 9 MDa in size, whereas Cdr2 nodes have not yet been analyzed by quantitative fluorescence microscopy. Given the large number of proteins in these Cdr2 nodes, we suspect that they are likely larger than Skb1-Slf1 nodes. We anticipate that future biochemical isolation of these various node structures will provide a more thorough understanding of their composition, size, and organization.
The exclusion of stable nodes from growing cell tips provides another intriguing but mysterious finding. Both Skb1-Slf1 nodes and Cdr2 interphase nodes are excluded from sites of polarized cell growth, most notably the growing cell tip. We found that Slf1 is present at sites of cell growth but is not concentrated into nodes at these sites. Previously, we demonstrated that Skb1 makes transient, unstable interactions with sites of cell growth (Deng and Moseley, 2013). This suggests an active mechanism to prevent the stable interaction of Skb1 and Slf1 at growing cell tips. It is possible that the protein kinases Orb6 and Orb2 contribute to this inhibitory mechanism, as both kinases localize to cell tips and physically interact with Skb1 (Gilbreth et al., 1996; Wiley et al., 2003). We also note that the local concentration and/or dynamics of specific membrane lipids has the potential to affect node assembly. It will be important to understand the role of these lipid species in the formation of fission yeast node structures, as well as cortical microdomains in other cell types.
MATERIALS AND METHODS
Yeast strains and growth
Standard S. pombe media and methods were used (Moreno et al., 1991). Strains used in this study are listed in Supplemental Table S1, and plasmids are listed in Supplemental Table S2. One-step PCR-based homologous recombination was performed for chromosomal tagging and deletion (Bähler et al., 1998). For Slf1 N-terminal mCherry tagging, PCR product from pJM775 (pQE30-mCherry-Slf1-Tslf1::KanR) was inserted between slf1+ promoter and open reading frame. Double mutants were generated through tetrad dissections. To construct pREP3X-slf1C-mEGFP plasmid, the Slf1 C-terminus (amino acids 451–485) fused to mEGFP was PCR amplified from JM1359 (slf1-mEGFP::KanR) and then inserted into pREP3X plasmid. To assess cell length at division, >50 septated cells grown in EMM4S medium at 32°C were measured in exponential phase with Blankophor (MP Biomedicals, Illkirch, France) staining. For yeast two-hybrid assay, bait and prey plasmids were cotransformed into yeast strain Y2H-Gold (Clontech Laboratories, Mountain View, CA) and selected on double dropout (DDO) plates (SD-Leu-Trp). Interactions were tested on QDO/A plate (SD-Leu-Trp-Ade-His with 125 ng/ml aureobasidin A). For overexpression assays, plasmids were transformed into fission yeast strains and selected against leucine auxotroph. In Figure 6B, mid–log-phase cells in minimal medium with 10 μg/ml thiamine at 25°C were collected and washed into medium lacking thiamine for 36 h. In Figure 6C, fresh cells were resuspended in minimal medium and spotted by 10× serial dilutions on EMM-Leu plates supplemented or not with 10 μg/ml thiamine. Plates were incubated under at 32°C for 4 d before scanning.
Immunoprecipitation and protein identification by LC-MS/MS
Fission yeast cells were grown at least eight generations at 32°C in rich medium. For Skb1-3HA pull-down experiment, 1 l of log-phase (OD595 = 0.5) cells was harvested and lysed in lysis buffer (20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], pH 7.4, 1 mM EDTA, 150 mM NaCl, 0.2% Triton X-100, 1 mM phenylmethylsulfonyl fluoride [PMSF], complete EDTA-free protease inhibitor tablets [Roche, Indianapolis, IN]) with glass beads using a Mini-beadbeater-16 (Biospec, Bartlesville, OK) at 4°C. Lysates were incubated at 4°C for 2 h with 500 μl anti-HA magnetic beads (Thermo Scientific, Rockford, IL). Beads were then washed three times with lysis buffer and boiled in 80 μl of sample buffer at 95°C for 5 min. Proteins were separated by SDS–PAGE and visualized by silver staining or Coomassie staining. Proteins >50 kDa were extracted from Coomassie-stained gels and then cut into 1-mm2 cubes and further destained with three washes of 25 mM NH4HCO3 in 50% CH3CN with 10-min incubations. Each group of gel cubes was then dehydrated in CH3CN for 10 min and dried in a SpeedVac, followed by reduction with 3 μl of 300 mM dithiothreitol in 300 μl of mM NH4HCO3 for 1 h at 56°C and alkylation with 3 μl of 300 mM iodoacetamide in 300 μl of 100 mM NH4HCO3 for 30 min in the dark. The hydrated gel slice was covered with 60 μl of trypsin solution (10 ng/μl) and incubated overnight at 37°C. Then 6 μl of 10% formic acid was added to stop the digestion, followed by centrifugation at 2800 × g. The supernatant was transferred to a new vial. The tryptic peptides in the gel cubes were extracted again by 60 μl of 5% formic acid in 50% CH3CN. The two supernatant fractions were combined and dried by SpeedVac, then 20 μl of 2.5% formic acid in 3% CH3CN was added, and the samples were ready for LC-MS/MS.
For LC-MS/MS analysis, a fused silica microcapillary LC column (15 cm ∆ 100 μm inner diameter) packed with C18 reversed-phase resin (5-μm particle size; 20-nm pore size; Magic C18AQ; Michrom Bioresources) was used with nanospray electrospray ionization (ESI). The nanospray ESI was fitted onto a linear trap quadrupole (LTQ) mass spectrometer (Thermo Electron, San Jose, CA). A 6-μl amount of tryptic peptide samples was injected and separated by applying a gradient of 3–50% acetonitrile in 0.1% formic acid at a flow rate of 250 nl/min for 45 min. After acquisition of the full MS scan in the data-dependent acquisition mode, the 10 most abundant ions were subjected to MS/MS analysis.
After an LC-MS run was completed and spectra were obtained, the spectra were searched against the S. pombe protein sequence databases using Proteome Discoverer software (version 1.4; Thermo Electron). The search parameters permitted a 20-ppm precursor MS tolerance and a 1.0-Da MS/MS tolerance. Oxidation of methionine (M) and carboxymethylation of cysteines (C) were allowed as variable modifications. Up to two missed tryptic cleavages of peptides were considered. The cutoffs for SEQUEST assignments were cross-correlation (Xcorr) scores >1.9, 2.5, and 3.0 for peptide charge states of +1, +2, and +3, respectively, and peptide prophet false discovery rate (FDR) <0.5%.
Coimmunoprecipitations and Western blots
For coimmunoprecipitations, we modified the foregoing immunoprecipitation protocol to use a smaller scale, starting with 50 ml of log-phase (OD595 = 0.5) cells. For Figure 1C, nickel-nitriloacetic acid agarose (Qiagen) was used instead of anti-HA beads, and 10 mM imidazole was added in lysis buffer. Western blots were probed with anti-6His (SC-8036; Santa Cruz Biotechnology), anti-GFP (Moseley et al., 2009), and anti-myc (SC-40, Santa Cruz Biotechnology, Dallas, TX) antibodies.
Protein purification and liposome pelleting assays
Recombinant GST and GST-Slf1C (amino acids 451–485) were purified using the plasmids pJM209 and pJM1016, respectively. Transformed Escherichia coli BL21(DE3) cells were grown to OD600 = 0.3 at 37°C and then induced for 3 h at 37°C by addition of isopropyl-β-d-thiogalactoside to 0.4 mM. Cells were harvested by centrifugation, washed into lysis buffer (20 mM HEPES, pH 7.4, 1 mM EDTA, 1 mM dithiothreitol, 250 mM NaCl, 5% glycerol, Roche complete protease inhibitors, and PMSF), and lysed by French press. The lysate was clarified and incubated with glutathione-agarose (Sigma-Aldrich, St. Louis, MO) for 1.5 h at 4°C. GST and GST-Slf1C were eluted by 20 mM glutathione (pH 8.0) for 30 min, analyzed by SDS–PAGE with Coomassie staining, and dialyzed twice against 20 mM HEPES (pH 7.4), 100 mM NaCl, and 5% glycerol.
For liposome pelleting assays, liposomes were prepared as previously described (Kabeche et al., 2014). GST or GST-Slf1C at 3 μM was incubated in the presence or absence of 10 mM liposomes at room temperature for 20 min. Samples were centrifuged with an OptimaTXL ultracentrifuge (Beckman, Indianapolis, IN) using a TLA-100 rotor at 47,000 rpm at 4°C for 30 min. Supernatants and pellets were collected, adjusted to equal volumes, and analyzed by SDS–PAGE and Coomassie staining.
Sequence analysis
BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi) was performed using S. pombe Slf1 and Skb1 protein sequences as seeds. Sequence alignment for S. pombe Slf1 and its homologues (Supplemental Figure S2) was conducted using DNAMAN 6.0. To assess conservation of N-/C-terminus of Skb1, amino acids 1–150/596–625 of S. cryophilus Skb1, 1–150/596–625 of S. octosporus Skb1, 1–180/625–654 of S. japonicas Skb1, 1–191/665–719 of A. niger Skb1, 1–182/691–718 of N. crassa Skb1, and 1–186/680–827 of S. cerevisiae Hsl7 were individually compared with 1–170/616–645 of S. pombe Skb1 using DNAMAN 6.0. The resulting identity values from aligned regions are graphed in Figure 7.
Microscopy and image processing
Fission yeast cells were imaged at 25°C with a DeltaVision Imaging System (Applied Precision, Issaquah, WA), equipped with an Olympus IX-71 inverted wide-field microscope, a Photometrics CoolSNAP HQ2 camera, and Insight solid-state illumination unit. Stacks of Z-series were acquired with 0.5-μm step size and iteratively deconvolved in SoftWoRx software (Applied Precision). Images were rendered by two-dimensional maximum intensity projection by ImageJ 1.45 (National Institutes of Health, Bethesda, MD).
For molecule quantification, cells were grown in exponential phase in YE5S rich medium and washed into EMM5S medium before imaging to reduce background autofluorescence (Wu et al., 2006). To measure the fluorescence intensity, slf1-mEGFP (JM1359), skb1-mEGFP (JM2325), and the control strains were individually mixed with wild-type cells just before cell collection. Cells were placed on a thin layer of EMM5S medium with 20% gelatin and 5 μM n-propyl-gallate and imaged at 23°C. Images were collected using a 100×/1.4 numerical aperture Plan-Apo objective lens on a spinning disk confocal microscope (UltraVIEW Vox CSUX1 system; PerkinElmer Life and Analytical Sciences) with 488-nm solid-state laser and back-thinned electron-multiplying charge-coupled device camera (Hamamatsu C9100-13) without binning. Stacks of Z-series were acquired with 0.5-μm step size, similar to the full width at half-maximum of the point spread function along the Z-axis as tested previously (Wu et al., 2006; Laporte et al., 2011), and summed for measuring intensity. Uneven illumination was corrected using images of purified 6His–monomeric yellow fluorescent protein solution (Wu and Pollard, 2005). From 40 to 70 mEGFP-tagged cells and 20–30 wild-type cells were measured. The intensity of mEGFP-tagged cells subtracted by wild-type mean intensity was used for counting molecules. Standard curve was plotted using the control strains (Wu and Pollard, 2005). Numbers of Skb1 or Slf1 molecules were calculated using the equation obtained from the standard curve.
Supplementary Material
Acknowledgments
We thank members of the Moseley laboratory and the Department of Biochemistry for discussions; W. Wickner, C. Barlowe, and D. Madden for reagents and shared instruments; and J. Fields and B. Deng at the University of Vermont (Burlington, VT) for help with mass spectrometry. This work was supported by National Institutes of Health Grants GM099774 (J.B.M.) and GM086546 (J.-Q.W.), and an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences/National Institutes of Health under Grant P20GM103449. J.B.M. is a Pew Scholar in the Biomedical Sciences.
Abbreviations used:
- EDTA
ethylenediaminetetraacetic acid
- GFP
green fluorescent protein
- GST
glutathione S-transferase
- 3HA
3x hemagglutinin
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- 6His
hexahistidine
- LC-MS/MS
liquid chromatography–tandem mass spectrometry
- PC
phosphatidylcholine
- PI(4,5)P2
phosphatidylinositol (4,5)-bisphosphate
- PMSF
phenylmethylsulfonyl fluoride
- PRMT5
protein arginine methyltransferase 5
- PS
phosphatidylserine
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-04-0896) on July 9, 2014.
REFERENCES
- Akamatsu M, Berro J, Pu KM, Tebbs IR, Pollard TD. Cytokinetic nodes in fission yeast arise from two distinct types of nodes that merge during interphase. J Cell Biol. 2014;204:977–988. doi: 10.1083/jcb.201307174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bähler J, Wu JQ, Longtine MS, Shah NG, McKenzie A, 3rd, Steever AB, Wach A, Philippsen P, Pringle JR. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast. 1998;14:943–951. doi: 10.1002/(SICI)1097-0061(199807)14:10<943::AID-YEA292>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Bao S, Qyang Y, Yang P, Kim H, Du H, Bartholomeusz G, Henkel J, Pimental R, Verde F, Marcus S. The highly conserved protein methyltransferase, Skb1, is a mediator of hyperosmotic stress response in the fission yeast Schizosaccharomyces pombe. J Biol Chem. 2001;276:14549–14552. doi: 10.1074/jbc.C100096200. [DOI] [PubMed] [Google Scholar]
- Cebecauer M, Spitaler M, Serge A, Magee AI. Signalling complexes and clusters: functional advantages and methodological hurdles. J Cell Sci. 2010;123:309–320. doi: 10.1242/jcs.061739. [DOI] [PubMed] [Google Scholar]
- Cid VJ, Shulewitz MJ, McDonald KL, Thorner J. Dynamic localization of the Swe1 regulator Hsl7 during the Saccharomyces cerevisiae cell cycle. Mol Biol Cell. 2001;12:1645–1669. doi: 10.1091/mbc.12.6.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Moseley JB. Compartmentalized nodes control mitotic entry signaling in fission yeast. Mol Biol Cell. 2013;24:1872–1881. doi: 10.1091/mbc.E13-02-0104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodgson J, Chessel A, Yamamoto M, Vaggi F, Cox S, Rosten E, Albrecht D, Geymonat M, Csikasz-Nagy A, Sato M, et al. Spatial segregation of polarity factors into distinct cortical clusters is required for cell polarity control. Nat Commun. 2013;4:1834. doi: 10.1038/ncomms2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbreth M, Yang P, Bartholomeusz G, Pimental RA, Kansra S, Gadiraju R, Marcus S. Negative regulation of mitosis in fission yeast by the shk1 interacting protein skb1 and its human homolog, Skb1Hs. Proc Natl Acad Sci USA. 1998;95:14781–14786. doi: 10.1073/pnas.95.25.14781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbreth M, Yang P, Wang D, Frost J, Polverino A, Cobb MH, Marcus S. The highly conserved skb1 gene encodes a protein that interacts with Shk1, a fission yeast Ste20/PAK homolog. Proc Natl Acad Sci USA. 1996;93:13802–13807. doi: 10.1073/pnas.93.24.13802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman NC, Groves JT. Signaling clusters in the cell membrane. Curr Opin Cell Biol. 2011;23:370–376. doi: 10.1016/j.ceb.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jourdain I, Brzezinska EA, Toda T. Fission yeast Nod1 is a component of cortical nodes involved in cell size control and division site placement. PLoS One. 2013;8:e54142. doi: 10.1371/journal.pone.0054142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeche R, Baldissard S, Hammond J, Howard L, Moseley JB. The filament-forming protein Pil1 assembles linear eisosomes in fission yeast. Mol Biol Cell. 2011;22:4059–4067. doi: 10.1091/mbc.E11-07-0605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeche R, Roguev A, Krogan NJ, Moseley JB. A Pil1-Sle1-Syj1-Tax4 functional pathway links eisosomes with PI(4,5)P2 regulation. J Cell Sci. 2014;127:1318–1326. doi: 10.1242/jcs.143545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laporte D, Coffman VC, Lee IJ, Wu JQ. Assembly and architecture of precursor nodes during fission yeast cytokinesis. J Cell Biol. 2011;192:1005–1021. doi: 10.1083/jcb.201008171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laude AJ, Prior IA. Plasma membrane microdomains: organization, function and trafficking. Mol Membr Biol. 2004;21:193–205. doi: 10.1080/09687680410001700517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingwood D, Simons K. Lipid rafts as a membrane-organizing principle. Science. 2010;327:46–50. doi: 10.1126/science.1174621. [DOI] [PubMed] [Google Scholar]
- Ma XJ, Lu Q, Grunstein M. A search for proteins that interact genetically with histone H3 and H4 amino termini uncovers novel regulators of the Swe1 kinase in Saccharomyces cerevisiae. Genes Dev. 1996;10:1327–1340. doi: 10.1101/gad.10.11.1327. [DOI] [PubMed] [Google Scholar]
- Martin SG, Berthelot-Grosjean M. Polar gradients of the DYRK-family kinase Pom1 couple cell length with the cell cycle. Nature. 2009;459:852–856. doi: 10.1038/nature08054. [DOI] [PubMed] [Google Scholar]
- Moreno S, Klar A, Nurse P. Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol. 1991;194:795–823. doi: 10.1016/0076-6879(91)94059-l. [DOI] [PubMed] [Google Scholar]
- Morrell JL, Nichols CB, Gould KL. The GIN4 family kinase, Cdr2p, acts independently of septins in fission yeast. J Cell Sci. 2004;117:5293–5302. doi: 10.1242/jcs.01409. [DOI] [PubMed] [Google Scholar]
- Moseley JB, Mayeux A, Paoletti A, Nurse P. A spatial gradient coordinates cell size and mitotic entry in fission yeast. Nature. 2009;459:857–860. doi: 10.1038/nature08074. [DOI] [PubMed] [Google Scholar]
- Paoletti A, Chang F. Analysis of mid1p, a protein required for placement of the cell division site, reveals a link between the nucleus and the cell surface in fission yeast. Mol Biol Cell. 2000;11:2757–2773. doi: 10.1091/mbc.11.8.2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakchaisri K, Asano S, Yu LR, Shulewitz MJ, Park CJ, Park JE, Cho YW, Veenstra TD, Thorner J, Lee KS. Coupling morphogenesis to mitotic entry. Proc Natl Acad Sci USA. 2004;101:4124–4129. doi: 10.1073/pnas.0400641101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayegh J, Clarke SG. Hsl7 is a substrate-specific type II protein arginine methyltransferase in yeast. Biochem Biophys Res Commun. 2008;372:811–815. doi: 10.1016/j.bbrc.2008.05.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snaith HA, Thompson J, Yates JR, 3rd, Sawin KE. Characterization of Mug33 reveals complementary roles for actin cable-dependent transport and exocyst regulators in fission yeast exocytosis. J Cell Sci. 2011;124:2187–2199. doi: 10.1242/jcs.084038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theesfeld CL, Zyla TR, Bardes EG, Lew DJ. A monitor for bud emergence in the yeast morphogenesis checkpoint. Mol Biol Cell. 2003;14:3280–3291. doi: 10.1091/mbc.E03-03-0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley DJ, Marcus S, D'Urso G, Verde F. Control of cell polarity in fission yeast by association of Orb6p kinase with the highly conserved protein methyltransferase Skb1p. J Biol Chem. 2003;278:25256–25263. doi: 10.1074/jbc.M209703200. [DOI] [PubMed] [Google Scholar]
- Wu JQ, Pollard TD. Counting cytokinesis proteins globally and locally in fission yeast. Science. 2005;310:310–314. doi: 10.1126/science.1113230. [DOI] [PubMed] [Google Scholar]
- Wu JQ, Sirotkin V, Kovar DR, Lord M, Beltzner CC, Kuhn JR, Pollard TD. Assembly of the cytokinetic contractile ring from a broad band of nodes in fission yeast. J Cell Biol. 2006;174:391–402. doi: 10.1083/jcb.200602032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu YH, Ye Y, Wu Z, Wu JQ. Cooperation between Rho-GEF Gef2 and its binding partner Nod1 in the regulation of fission yeast cytokinesis. Mol Biol Cell. 2013;24:3187–3204. doi: 10.1091/mbc.E13-06-0301. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.