

Abstract
The MRE11-RAD50-NBS1 (MRN) complex is essential for the detection of DNA double-strand breaks (DSBs) and initiation of DNA damage signaling. Here, we show that Rad17, a replication checkpoint protein, is required for the early recruitment of the MRN complex to the DSB site that is independent of MDC1 and contributes to ATM activation. Mechanistically, Rad17 is phosphorylated by ATM at a novel Thr622 site resulting in a direct interaction of Rad17 with NBS1, facilitating recruitment of the MRN complex and ATM to the DSB, thereby enhancing ATM signaling. Repetition of these events creates a positive feedback for Rad17-dependent activation of MRN/ATM signaling which appears to be a requisite for the activation of MDC1-dependent MRN complex recruitment. A point mutation of the Thr622 residue of Rad17 leads to a significant reduction in MRN/ATM signaling and homologous recombination repair, suggesting that Thr622 phosphorylation is important for regulation of the MRN/ATM signaling by Rad17. These findings suggest that Rad17 plays a critical role in the cellular response to DNA damage via regulation of the MRN/ATM pathway.
Keywords: ATM activation, MRE11-RAD50-NBS1 complex, Rad17
See also: TT Paull & J-H Lee (April 2014)
Introduction
Genome integrity is constantly challenged by generation of DNA damage, which can be induced by environmental agents as well as by metabolic byproducts generated during normal cellular activity. DNA double-strand breaks (DSBs) are among the most deleterious forms of DNA damage and, if not properly repaired, may cause genomic instability, cell death, and cancer (Jackson & Bartek, 2009; Ciccia & Elledge, 2010). A conserved and intricate signaling network of responses to DSB damage is critical for the maintenance of genomic integrity. Two distinct pathways, homologous recombination (HR) and non-homologous end joining (NHEJ), are responsible for repair of DSBs. It has been established that HR precisely repairs the broken DNA ends by utilizing an intact sister chromatid as a template, whereas NHEJ directly re-ligates the DNA ends resulting in a potentially error-prone repair (Lieber, 2008; San Filippo et al, 2008; Ciccia & Elledge, 2010).
MRE11-RAD50-NBS1 (MRN) complex is a multifaceted molecular machine which is implicated in multiple aspects of the DNA damage response (DDR), such as initial DSB detection, signal transduction, and promotion of DSB repair by HR and NHEJ (Williams et al, 2010). The role of the MRN complex in HR repair is largely dependent on resection of DSBs that occurs in an MRE11-, CtIP-, and BRCA1-dependent manner generating replication protein A (RPA)-coated single-stranded DNA (ssDNA) fragments. Rad51 is then loaded onto the ssDNA overhangs forming a nucleoprotein filament and catalyzing DNA strand invasion and exchange to facilitate HR repair (Daboussi et al, 2002). The MRN complex has also been suggested to be essential for ATM recruitment to DNA DSBs for optimal activation (Falck et al, 2005; You et al, 2005). Thus, the MRN complex has emerged as a critical regulator of the cellular response to DSBs (Williams et al, 2010). ATM kinase is another keystone in maintaining genomic stability as it activates a network of checkpoint and DNA repair proteins in response to DNA damage. Both the MRN complex and ATM are central components of the DDR. Although the mechanism of MRN complex accumulation and retention at chromatin surrounding the DSB that involves the interaction of NBS1 with phospho-MDC1 is well established (Chapman & Jackson, 2008; Melander et al, 2008; Spycher et al, 2008; Wu et al, 2008), the mechanism of MRN complex recruitment at an early stage of DDR is not fully understood since γ-H2AX that serves as a key anchor at sites of DSBs facilitating the recruitment of MDC1 is dispensable for the initial recognition of DNA breaks by the MRN complex (Celeste et al, 2003; Stucki et al, 2005; Yuan & Chen, 2010). Here, we found a specific mechanism by which Rad17 is required for early MRN complex recruitment to the DSB that is independent of MDC1.
Rad17 is a replication factor C (RFC)-like DNA damage sensor protein which has been demonstrated to be essential for ATR-dependent checkpoint signaling and the maintenance of genomic stability (Zou et al, 2002; Wang et al, 2003, 2006). Phosphorylation of Rad17 at Ser635 and Ser645 has been shown to play a role in loading Claspin and the 9-1-1 complex and activating the Chk1 kinase after DNA damage (Bao et al, 2001; Wang et al, 2006), indicating that phosphorylation of Rad17 at these two serine residues is involved in checkpoint activation after genotoxic stress. Interestingly, several studies showed that deletion of the N-terminus of Rad17 results in an impaired homologous recombination activity but does not affect the cell-cycle checkpoint activation in mouse cells, and that chicken DT40 cells lacking Rad17 were compromised in the process of targeting integration (Budzowska et al, 2004; Nishino et al, 2008), suggesting that Rad17 may be involved in DNA repair by HR.
Here, by exploring the mechanisms of the effect of Rad17 on HR repair, we show that Rad17 is required for early recruitment and retention of the MRN complex at DSB sites, which significantly affects activation of ATM signaling and HR repair. The early Rad17-mediated MRN complex recruitment is independent of MDC1 and may provide immediate and efficient cellular response to DNA damage. Moreover, the effect of Rad17 on MRN complex recruitment relies on a direct interaction of Rad17 with NBS1 that is enhanced by the phosphorylation of Rad17 at a novel Thr622 site by the ATM kinase. Our data suggest that Rad17 is involved in an ATM-Rad17-MRN-ATM amplification loop resulting in an efficient activation of MRN/ATM signaling. These findings demonstrate a novel ATM-mediated role for Rad17 in the cellular response to DNA damage that is distinct from its known function in ATR signaling pathway.
Results
Rad17 is involved in DNA repair by promoting HR in human cells
It was previously reported that C-terminal Ser635/Ser635 phosphorylation of Rad17 regulates induction of cell-cycle arrest upon DNA damage (Bao et al, 2001; Wang et al, 2006). Additionally, several studies have suggested that Rad17 plays a role in HR repair in chicken and mouse cells (Budzowska et al, 2004; Nishino et al, 2008). However, the molecular mechanism of regulation of HR repair by Rad17 is largely unexplored. To address this question, we initially utilized a well-established fluorescence-based assay assessing HR repair (Pierce et al, 1999). We observed that Rad17 knockdown by siRNA in U2OS cells resulted in a threefold reduction in the number of GFP-positive cells compared with control (Fig 1A), indicating a significant reduction of HR repair. Similar results were achieved with a second Rad17 siRNA and in A549 cells (Supplementary Fig S1A and B). Notably, cell viability and cell-cycle profile were marginally affected by transient siRNA-mediated knockdown of Rad17 (Supplementary Fig S1C and D). Consistent with these results, depletion of Rad17 by inducible shRNA resulted in defective DSB repair demonstrated by comet assay and sensitized cells to IR (Supplementary Fig S1E and F). Again, cell viability and cell-cycle profile were not affected by shRNA-mediated knockdown of Rad17 (Supplementary Fig S1G and H). These data indicate that Rad17 is involved in DNA repair by promoting HR in human cells.
Figure 1. Rad17 regulates HR repair in association with Rad51 recruitment to DSBs.

- Rad17 is required for HR repair in human cells. U2OS cells were transfected with the indicated siRNA for 2 days. HR assay was performed as previously described (Pierce et al, 1999). pGFP construct was transfected into cells to ensure similar transfection efficiency.
- Rad17 promotes Rad51 recruitment to DSBs. U2OS cells transfected with the indicated siRNA for 2 days were either left untreated or exposed to 5 Gy of IR. Immunostaining with anti-Rad17 and anti-Rad51 was performed. A cell containing 10 or more foci was considered as a foci-positive cell. The percentage of Rad51 foci-positive cells was plotted.
- Rad17 is required for γ-H2AX foci formation at DSB sites. U2OS cells transfected with the indicated siRNA for 2 days were either untreated or exposed to 5 Gy of IR. Immunofluorescence staining was performed with anti-Rad17 and anti-γ-H2AX. The intensity of γ-H2AX foci was analyzed using Image J software.
- Rad17 is required for γ-H2AX production. Inducible shRNA-mediated Rad17 knockdown cells (U2OS) treated with either Dox or DMSO for 2 days were either left untreated or exposed to 5 Gy of IR. Cell lysates were prepared at the indicated time points, and Western blots were performed as indicated. Dox, doxycycline; IR, ionizing radiation.
Data information: In (A–C): Error bars represent mean ± s.d. (n = 3).
Source data are available online for this figure.
We next investigated how Rad17 regulates HR repair. We first examined whether Rad51 is involved in the regulation of HR repair by Rad17 as Rad51 is known to facilitate DNA strand invasion and exchange, a key step in HR repair (Daboussi et al, 2002). We hypothesized that Rad17 may localize at the DSB and affect the recruitment of Rad51 to the break site. Indeed, we found that Rad17 co-localized with Rad51 in IR-induced nuclear foci, and observed a threefold decrease in the percentage of Rad51 foci-positive cells as a result of Rad17 knockdown (Fig 1B). BRCA1 is involved in HR repair by promoting DSB resection and acting as an upstream regulator of Rad51 (Greenberg et al, 2006; Huen et al, 2010). Interestingly, we found that down-regulation of Rad17 strongly attenuated BRCA1 foci formation after DSB damage (Supplementary Fig S2A–C), indicating that Rad17 promotes BRCA1 recruitment to sites of DSB damage.
It has been demonstrated that BRCA1 recruitment is largely dependent on γ-H2AX/MDC1-mediated histone ubiquitination (van Attikum & Gasser, 2009; Huen et al, 2010). Given that Rad17 is a DNA damage sensor protein that could act at the early stage of DDR (Niida & Nakanishi, 2006), we postulated that Rad17 might be involved in mediating γ-H2AX signaling, thus creating a platform for BRCA1 recruitment. Indeed, we observed a co-localization of Rad17 and γ-H2AX in IR-induced nuclear foci and a significant reduction in both the number and intensity of γ-H2AX foci due to down-regulation of Rad17 (Fig 1C). Moreover, Rad17 knockdown by inducible shRNA resulted in a significant decrease in γ-H2AX levels after IR (Fig 1D). Furthermore, we observed that recruitment of MDC1 that occurs downstream of γ-H2AX formation (Stucki et al, 2005) was significantly reduced when Rad17 was down-regulated (Supplementary Fig S2D). Collectively, these results suggest that Rad17 plays an important role in the early response to DSB damage by regulating γ-H2AX/MDC1 signaling pathway.
Rad17 directly interacts with NBS1
In order to understand how Rad17 regulates γ-H2AX formation, we sought to determine whether Rad17 is involved in the events upstream of H2AX histone phosphorylation. It has been shown that the formation and expansion of γ-H2AX at DSB sites depends on the MRN complex and ATM kinase (van Attikum & Gasser, 2009) and that the MRN complex is required for ATM recruitment and optimal activation of ATM (Falck et al, 2005; You et al, 2005). Therefore, we decided to determine whether there is a functional link between Rad17 and the MRN complex in the DDR. First, we examined the interaction between Rad17 and members of the MRN complex and found that endogenous Rad17 co-immunoprecipitates with NBS1, MRE11 and RAD50, and these interactions are significantly increased following DSB induction (Fig 2A–C). A reciprocal immunoprecipitation (IP) also showed an increased level of interaction between Rad17 and the MRN complex members after DSB induction (Fig 2D). Moreover, we noted that there was a basal constitutive interaction between Rad17 and NBS1 even in undamaged cells and investigated which regions of Rad17 and NBS1 are required for this interaction. GST pull-down assays using a series of GST-fused truncated mutants of Rad17 and NBS1 proteins showed that Rad17 can directly interact with NBS1, which requires the carboxyl terminus (residue 570–670) of Rad17 and the FHA domain of NBS1 (Fig 2E and F).
Figure 2. Rad17 interacts with the MRN complex by directly binding to NBS1.

A–D Rad17 interacts with the MRN complex members following DSB damage. Cell lysates prepared from U2OS cells either untreated or exposed to 5 Gy of IR were subjected to immunoprecipitation with control IgG, anti-NBS1 (A), anti-MRE11 (B), anti-RAD50 (C), or anti-Rad17 (D). The immunoprecipitates were blotted as indicated.
E, F Rad17 directly interacts with NBS1 through the FHA domain of NBS1 and the C-terminus of Rad17. U2OS cell lysate was incubated with either GST-alone beads, the indicated GST-Rad17 beads (E), or the indicated GST-NBS1 beads (F) at 4°C overnight. Proteins retained on beads were blotted as indicated. Arrows indicate the corresponding Rad17 fragments or NBS1 fragments.
Source data are available online for this figure.
Rad17 is required for early recruitment and retention of the MRN complex at DSBs
After identifying the biochemical interaction between Rad17 and the MRN complex, we investigated whether Rad17 is required for recruitment of the MRN complex to DSBs. Since NBS1 is a key component of the MRN complex, which acts as a bridging protein for the interaction of MRE11 and ATM (Williams et al, 2007), we first examined the effect of Rad17 on NBS1 recruitment to the DSB. As shown in Fig 3A, Rad17 co-localized with NBS1 and the percentage of NBS1 foci-positive cells was significantly reduced at time points assessed between 5 min and 3 h following IR in Rad17 siRNA-transfected cells compared with the control cells, indicating that Rad17 is required for early NBS1 recruitment and its retention at the DSB. A similar inhibitory effect of Rad17 knockdown on NBS1 recruitment was observed at least 15 min after laser micro-irradiation (Fig 3B), verifying that Rad17 is required for NBS1 recruitment in the early phase of DDR. Consistent with the knowledge that NBS1 is necessary for recruitment of MRE11 and RAD50 to the DSB site (Lee & Paull, 2007), we also found that the percentages of cells positive for MRE11 and RAD50 foci were significantly reduced in Rad17-depleted cells compared with the control cells (Supplementary Fig S3A and B), confirming that Rad17 is important for recruitment of the MRN complex to DSBs.
Figure 3. Rad17 is critical for recruitment and retention of the MRN complex at DSBs.

- Rad17 is required for NBS1 recruitment to the DSB sites. U2OS cells transfected with the indicated siRNA for 2 days were either untreated or exposed to 5 Gy of IR. Immunofluorescence staining was performed with anti-Rad17 and anti-NBS1. Error bars represent mean ± s.d. (n = 3).
- U2OS cells were transfected with the indicated siRNA for 2 days. Immunofluorescence staining was performed with anti-NBS1 following laser irradiation as indicated.
- Rad17 is required for NBS1 binding to specific DSBs. U2OS cells transfected with the indicated siRNA for 2 days were infected with HA-ER-I-PpoI retrovirus followed by addition of 4-hydroxytamoxifen (4-OHT). I-PpoI that causes specific DSB is induced just 2 h after induction, and ChIP assay was performed as previously described (Berkovich et al, 2007).
- Rad17 is important for the accumulation of MRN complex on chromatin. U2OS cells transfected with the indicated siRNA for 2 days were either untreated or exposed to 5 Gy of IR. Soluble nuclear and chromatin fractions were isolated as described in Materials and Methods. Western blot was performed as indicated.
Source data are available online for this figure.
To verify the role of Rad17 in NBS1 recruitment to DSBs, we employed an alternative method, in which DSBs at defined endogenous sites are induced by the homing endonuclease I-PpoI 2 h after 4-hydroxytamoxifen addition (Berkovich et al, 2007). We observed that NBS1 binding to the DSB induced at a unique cut site on chromosome 1 occurs quickly and is maintained for at least 10 h after I-PpoI induction, whereas Rad17 knockdown significantly decreased NBS1 binding to the DSB (Fig 3C), confirming that Rad17 is required for early recruitment of NBS1 and sustains its retention at specific DSBs. Moreover, subcellular fractionation analysis showed that following DSB induction, the abundance of MRN complex proteins in the chromatin fraction is markedly reduced as a result of Rad17 knockdown (Fig 3D). Together, these data demonstrate the importance of Rad17 for early recruitment and retention of the MRN complex at DSBs.
Previous studies showed that the MRN complex can be recruited to DSB surrounding regions by MDC1 due to an interaction between the N-terminal several acidic SDT motifs of MDC1 and the FHA domain of NBS1 (Chapman & Jackson, 2008; Melander et al, 2008; Spycher et al, 2008; Wu et al, 2008). Since MDC1 binds directly to γ-H2AX (Stucki et al, 2005) and γ-H2AX formation is affected by Rad17 (Fig 1C and D), but γ-H2AX is dispensable for the initial recognition of break sites (Celeste et al, 2003), we sought to determine how Rad17-dependent MRN complex recruitment is distinguished from MDC1-dependent MRN complex recruitment. We show that knockdown of Rad17 does not affect the interaction between MDC1 and NBS1 (Fig 4A and B) that was previously shown to be constitutive rather than DNA damage inducible, and that down-regulation of MDC1 does not affect the interaction of Rad17 and NBS1 after DSB induction (Fig 4C). In addition, we found no detectable interaction between Rad17 and MDC1 by reciprocal IP experiments (Supplementary Fig S3C and D). Furthermore, we observed that MDC1 knockdown has no effect on Rad17 foci formation following IR (Fig 4D). In contrast, Rad17 knockdown resulted in decreased MDC1 recruitment to DSB sites (Supplementary Fig S2D), suggesting that Rad17 might act upstream of MDC1 in DDR. Importantly, knockdown of MDC1 did not affect early NBS1 recruitment to sites of DNA damage after IR and laser micro-radiation as well as early NBS1 binding to specific DSBs following I-PpoI induction, but abolished the presence of NBS1 at the break site at later time points (Fig 4E–G, Supplementary Fig S3E). These data indicate that Rad17 regulates the early recruitment of the MRN complex to the DSB which is independent of MDC1, whereas a distinct γ-H2AX/MDC1-dependent pathway mediates MRN recruitment at a later stage of the DDR. Both pathways are likely important for the retention of the MRN complex at the DSB. Consistent with these results, γ-H2AX was shown to be dispensable for the initial recognition of DSB damage and the early phase of NBS1 recruitment to DSBs (Celeste et al, 2003; Yuan & Chen, 2010).
Figure 4. Rad17-mediated MRN complex recruitment is different from MDC1-mediated MRN complex recruitment.

A–C Rad17 depletion or MDC1 depletion does not affect MDC1-NBS1 or Rad17-NBS1 interaction. U2OS cells transfected with indicated siRNA for 2 days were either untreated or exposed to 5 Gy of IR. IP experiments and Western blots were performed as indicated.
D, E MDC1 knockdown has no effect on Rad17 recruitment and early NBS1 recruitment to DSBs. U2OS cells transfected with indicated siRNA for 2 days were either untreated or exposed to 5 Gy of IR. Immunofluorescence staining was performed as indicated. Error bars represent mean ± s.d. (n = 3).
F U2OS cells were transfected with the indicated siRNA for 2 days. Immunofluorescence staining was performed following laser irradiation as indicated.
G U2OS cells transfected with the indicated siRNA for 2 days were infected with HA-ER-I-PpoI retrovirus followed by addition of 4-hydroxytamoxifen (4-OHT). ChIP assay was performed as previously described.
Source data are available online for this figure.
Rad17 promotes ATM activation and DNA end resection
Having established an important role for Rad17 in MRN complex recruitment and retention at DSBs, we decided to assess the effect of Rad17 knockdown on the activation of ATM signaling. As shown in Fig 5A, upon IR treatment, depletion of Rad17 significantly impaired ATM phosphorylation at Ser1981 compared with control in U2OS cells. Reduction of ATM phosphorylation in Rad17-depleted cells upon IR correlated with the reduction of phosphorylation of NBS1 (Ser343), SMC1 (Ser957), and Chk2 (Thr68), three well-known substrates of ATM. A similar reduction in phosphorylation of ATM (Ser1981) and Chk2 (Thr68) was observed after IR due to depletion of Rad17 in both A549 cells and normal human foreskin fibroblasts (HFF) (Supplementary Fig S4A and B). Moreover, phospho-ATM foci formation following IR was significantly reduced in Rad17-depleted cells (Fig 5B; Supplementary Fig S4C). Together, these results indicate that Rad17 contributes to ATM activation after DSB induction. Given the role of Rad17 in MRN recruitment and ATM activation shown in this study, it appears likely that the decrease in IR-induced γ-H2AX formation due to Rad17 depletion is the result of decreased MRN-ATM signaling. Consistent with this conclusion, we (Supplementary Fig S4D) as well as another study (Zhang et al, 2005) show that NBS1 knockdown reduces the level of γ-H2AX after DNA damage induction. Notably, several PIKK kinases such as ATM, ATR, and DNA-PKcs are capable of phosphorylating H2AX (Fernandez-Capetillo et al, 2004; Stiff et al, 2004). As shown in Fig 5C, ATM knockdown resulted in a stronger reduction of γ-H2AX level following IR compared with DNA-PKcs and ATR knockdown. These data suggest that Rad17 affects γ-H2AX formation after DSB induction by regulating the MRN/ATM pathway activity.
Figure 5. Rad17 promotes ATM activation and DNA end resection.
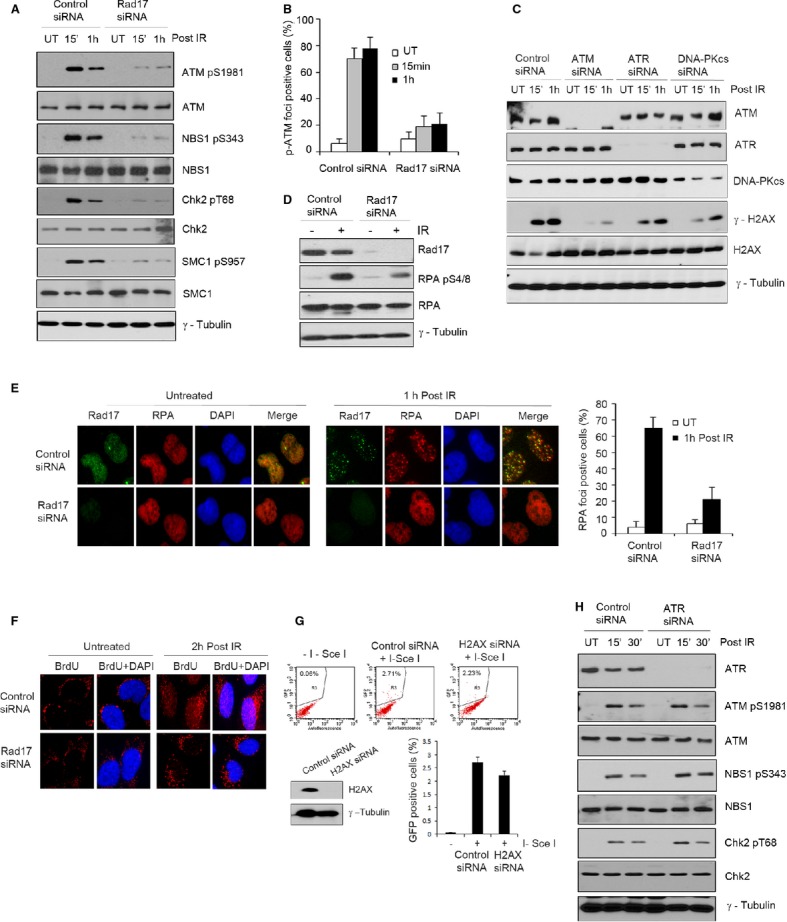
A, B Rad17 is required for activation of ATM signaling. U2OS cells transfected with the indicated siRNA for 2 days were either untreated or exposed to 5 Gy of IR. Western blot was performed as indicated (A). Immunofluorescence staining was performed, and the percentage of p-ATM foci-positive cells was plotted (mean ± s.d., n = 3) (B).
C U2OS cells transfected with indicated siRNAs were either untreated or exposed to 5 Gy of IR. Cell lysates were prepared at indicated times, and Western blotting was performed.
D–F Rad17 depletion significantly altered DNA end resection after IR. U2OS cells transfected with indicated siRNA were either untreated or exposed to 5 Gy of IR. Western blot was performed with anti-RPA pS4/8 as indicated (D). Immunostaining was performed as indicated, and the percentage of RPA foci-positive cells was plotted (mean ± s.d., n = 3) (E). Cells were grown in culture medium containing 10 μM BrdU for 24 h before IR (5 Gy) exposure. After IR, immunostaining was performed with anti-BrdU antibody as indicated according to the method as described (Hu et al, 2011) (F).
G U2OS cells were transfected with indicated siRNAs for 2 days. HR repair assay was performed as previously described.
H U2OS cells transfected with control siRNA or ATR siRNA were either left untreated or exposed to 5 Gy of IR. Western blot was performed as indicated.
Source data are available online for this figure.
It has been demonstrated that the MRN complex is essential for the initiation of DSB processing via the endonuclease activity of MRE11, resulting in formation of ssDNA overhangs coated by RPA (Jazayeri et al, 2006; Williams et al, 2010). Rad51 replaces RPA in a BRCA1- and BRCA2-dependent manner (Sugiyama & Kowalczykowski, 2002). Since Rad17 is critical for MRN complex recruitment to DSB sites, we hypothesized that the failure to recruit Rad51 to DSBs in the absence of Rad17 may also be due to inefficient resection of DSBs. To test this possibility, we examined RPA phosphorylation at Ser4 and Ser8 and RPA foci formation and found that knockdown of Rad17 resulted in significantly decreased RPA Ser4/8 phosphorylation and a decreased number of RPA foci-positive cells (Fig 5D and E; Supplementary Fig S4E). Furthermore, we examined BrdU incorporation under neutral conditions in order to directly monitor DNA resection (Hu et al, 2011). As shown in Fig 5F, ssDNA foci formation was significantly reduced in Rad17-depleted cells versus control cells. Together, these data indicate that Rad17 is critical for DSB resection activity of the MRN complex, thus affecting the initiation of HR repair. Although it has been demonstrated that γ-H2AX is required for stable accumulation of DNA repair proteins at sites of DNA breaks, H2AX depletion moderately affected HR repair (Fig 5G), which is consistent with multiple previous studies (Petersen et al, 2001; Bassing et al, 2002; Xie et al, 2004; Yuan & Chen, 2010). Moreover, IR-induced RPA foci were formed in the absence of H2AX (Supplementary Fig S4F), consistent with a previous report (Yuan & Chen, 2010). Collectively, these data suggest that while γ-H2AX may assist repair pathways, it unlikely plays a major role in HR repair and that Rad17 regulates HR repair most likely through regulating the MRN complex.
To determine whether alteration of MRN recruitment and ATM signaling as a result of Rad17 depletion is due to defective ATR signaling, we examined the effect of ATR depletion on NBS1 recruitment and ATM activation and found that there is no significant difference in NBS1 foci formation and phosphorylation of ATM (Ser1981), NBS1 (Ser343), and Chk2 (Thr68) between ATR-depleted cells and control cells (Fig 5H; Supplementary Fig S4G), which is consistent with previously reported data (Myers & Cortez, 2006). These results suggest that the effect of Rad17 on MRN/ATM signaling is independent of ATR activity in response to DSB induction.
ATM-dependent phosphorylation of Rad17 at a novel Thr622 phosphorylation site
Next, we investigated the molecular mechanism of Rad17-dependent regulation of MRN/ATM signaling during the DDR. Previous studies showed that serine phosphorylation is involved in the checkpoint function of Rad17 (Bao et al, 2001; Wang et al, 2006). Therefore, we hypothesized that phosphorylation of Rad17 might also play a role in regulating MRN/ATM signaling. To test this idea, we scanned the Rad17 amino acid sequence and found that Rad17 possesses several SQ/TQ motifs (Fig 6A). We focused on the Thr548 and Thr622 sites due to their proximity to the established Ser635 and Ser645 sites, as this area potentially constitutes an SQ/TQ “phosphorylation cluster” (Traven & Heierhorst, 2005). Since ATR is the major kinase phosphorylating Rad17 at Ser635 and Ser645 (Bao et al, 2001), we initially performed an in vitro ATR kinase assay which showed that mutation of threonine 622 to alanine (T622A) resulted in an additional reduction in phosphorylation of Rad17 S635A/S645A mutant, while mutation of threonine 548 to alanine (T548A) had no effect (Supplementary Fig S5A). A subsequent in vitro ATM kinase assay showed that T622A mutation resulted in a dramatic reduction in phosphorylation of Rad17 (Fig 6B), suggesting that ATM can directly phosphorylate Rad17 at Thr622, which may affect the function of Rad17 in regulating the DDR. We further observed that this Thr622 residue is phosphorylated as early as 5 min after IR in U2OS cells and other types of cells (Fig 6C; Supplementary Fig S5B). In contrast, UV and HU treatment led to weak Thr622 phosphorylation compared with IR treatment (Supplementary Fig S5C and D). Importantly, we found that knocking down ATM resulted in a significant decrease in Thr622 phosphorylation, while knocking down ATR marginally reduced this phosphorylation following IR (Fig 6D). Together, these results indicate that ATM is the major kinase responsible for Rad17 phosphorylation at Thr622 after DNA damage.
Figure 6. ATM-dependent phosphorylation of Rad17 at Thr622 is required for HR repair.
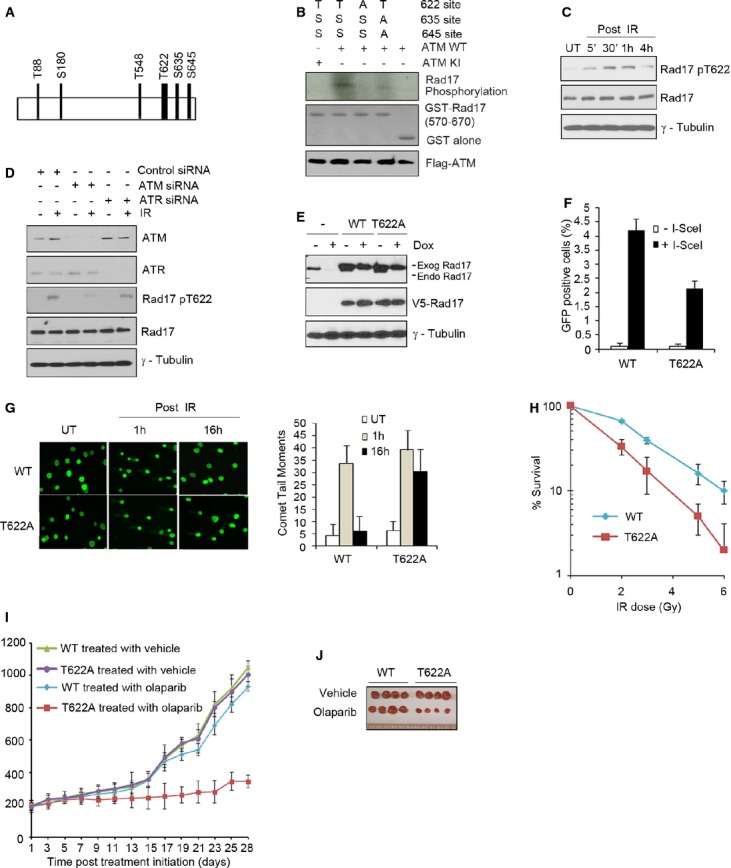
A Schematic presentation of potential phosphorylation TQ/SQ motifs in Rad17.
B ATM directly phosphorylates Rad17 at Thr622. An in vitro ATM kinase assay was performed as described in Supplementary Material and Methods.
C U2OS cells were either untreated or exposed to 5 Gy of IR. Western blot was performed as indicated.
D ATM but not ATR is responsible for Rad17 phosphorylation at Thr622 following DSB damage. U2OS cells transfected with the indicated siRNA were either untreated or exposed to 5 Gy of IR. Cell lysates were prepared 1 h after IR. Western blot was performed.
E Endogenous Rad17 is depleted by Dox induction and replaced by exogenous Rad17. Inducible Rad17 knockdown cells (U2OS) stably expressing shRNA-resistant Rad17 WT or Rad17 T622A mutant were treated with or without Dox for 2 days. Western blot was performed.
F–H Rad17 Thr622 phosphorylation is required for HR repair and cell survival. Inducible Rad17-knockdown cells reconstituted with shRNA-resistant Rad17 WT or Rad17 T622A mutant were used. HR repair assay was performed as previously described (F); comet assay was performed as previously described (G); clonogenic survival assay was performed using IR treatment (H).
I, J Rad17 Thr622 phosphorylation is necessary for tumor growth in the presence of PARP inhibitor. Mice bearing xenograft tumors of 200–220 mm3 were divided into two groups for each cell line as indicated: vehicle control group and Olaparib group. Each group contained four nude mice. Tumor size was measured and tumor growth curves plotted (I); tumors were harvested on day 28 after the start of Olaparib treatment (J).
Data information: Error bars represent mean ± s.d. (n = 4).
Source data are available online for this figure.
Thr622 phosphorylation affects Rad17-mediated regulation of HR repair
To investigate whether the effect of Rad17 on MRN/ATM signaling is dependent on Rad17 phosphorylation, we initially assessed whether Rad17 Ser635 and Ser645 phosphorylation affects Rad17-NBS1 interaction. We re-introduced shRNA-resistant Rad17 WT or Rad17 AA (S635A/S645A) mutant into U2OS cells depleted of endogenous Rad17 (Supplementary Fig S6A) and observed that the IR-induced interaction of Rad17 with NBS1 is not affected by Rad17 phosphorylation at these sites (Supplementary Fig S6B). We then examined the role of Rad17 Thr622 phosphorylation in MRN/ATM signaling and DNA repair. As shown in Fig 6E and F, cells reconstituted with Rad17 T622A mutant displayed a significant reduction in HR repair compared with cells reconstituted with WT Rad17. Consistent with this result, Rad17 T622A cells showed a marked decrease in overall DSB repair demonstrated by comet assay and a reduced cell survival following IR (Fig 6G and H).
Previous studies reported that the ATR-Chk1 pathway promotes HR repair (Hu et al, 2005; Sorensen et al, 2005). Given that Rad17 is an upstream regulator of the ATR-Chk1 signaling pathway (Zou et al, 2002; Wang et al, 2006) (Supplementary Fig S7A), it is possible that Rad17 may also contribute to HR repair via Chk1. We examined the Chk1 phosphorylation level and found that there was no significant difference in Chk1 Ser317 and Ser345 phosphorylation levels after UV and HU treatment and a moderate decrease after IR in Rad17 T622A cells versus Rad17 WT cells (Supplementary Fig S7B–D). Moreover, there were no significant changes in the percentage of Rad9 foci- and Hus1 foci-positive cells between Rad17 WT cells and T622A cells (Supplementary Fig S7E and F), suggesting that Thr622 phosphorylation has no effect on 9-1-1 complex loading at DNA damage sites. Taken together, these results indicate that Rad17 phosphorylation at Thr622 promotes HR in a manner that is likely independent of the role of Rad17 in Chk1 activation.
It has been demonstrated that deficient HR renders cells sensitive to selective killing by poly (ADP-ribose) polymerase (PARP) inhibition resulting in synthetic lethality (Bryant et al, 2005; Jackson & Bartek, 2009). We next investigate whether Rad17 Thr622 phosphorylation affects HR repair in vivo. We performed a subcutaneous xenograft of hypopharyngeal carcinoma (FaDu) cells reconstituted with Rad17 WT or Rad17 T622A mutant (Supplementary Fig S7G) in nude mice and found that the growth of tumors generated by Rad17 T622A cells was strongly reduced, whereas the growth of tumors initiated by Rad17 WT cells was not significantly altered by the PARP inhibitor Olaparib treatment (Fig 6I and J). These results suggest that Rad17 phosphorylation at Thr622 is necessary for HR repair that affects cellular survival following genotoxic stress in vivo, which provide the possibility that disruption of Thr622 phosphorylation might have potential therapeutic significance.
Rad17 Thr622 phosphorylation is important for MRN/ATM signaling
Since the Thr622 residue is within the region of Rad17 that is responsible for the interaction with NBS1 (Fig 2E), we reasoned that Thr622 phosphorylation is likely to be important for mediating the interaction of Rad17 with the MRN complex. To test this, we synthesized biotinylated phospho-T622 Rad17 peptide and control unphosphorylated peptide with the identical sequence. As shown in Fig 7A, a phospho-T622 Rad17 peptide is able to pull down the FHA domain of NBS1, but not the C-terminal region, with a higher efficiency than the non-phospho-T622 Rad17 peptide, indicating that Thr622 phosphorylation is directly involved in Rad17-NBS1 interaction. IP experiments showed that Rad17 T622A cells displayed a decreased interaction between Rad17 and NBS1 (Fig 7B). Additionally, inhibition of ATM activity by ATM inhibitor (KU55933) attenuated the interaction of Rad17 with NBS1 upon IR treatment (Fig 7C). Collectively, these results suggest that ATM-dependent Thr622 phosphorylation is required for Rad17-NBS1 interaction.
Figure 7. Thr622 phosphorylation is required for Rad17-NBS1 interaction.
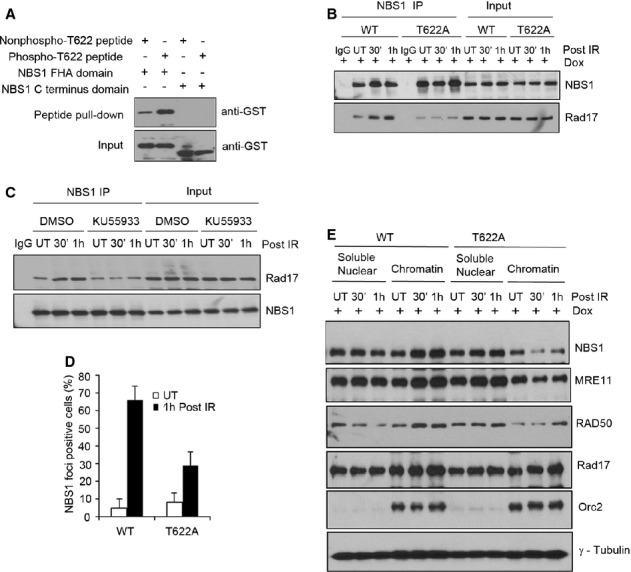
A Rad17 Thr622 phosphorylation is required for the interaction between Rad17 and the FHA domain of NBS1. Streptavidin-Sepharose beads coupled with the indicated peptides were incubated with purified GST-NBS1 fragments (FHA domain or C-terminus domain containing residues 682–754). Western blot was performed using anti-GST.
B, D–E The mutation of Thr622 phosphorylation attenuated the Rad17-NBS1 interaction and the recruitment of NBS1 to damaged chromatin. Inducible Rad17 knockdown cells (U2OS) stably expressing shRNA-resistant Rad17 WT or Rad17 T622A mutant treated with Dox for 2 days were either untreated or exposed to 5 Gy of IR. Cell lysates were subjected to immunoprecipitation with anti-NBS1 as indicated (B); Immunostaining was performed, and the percentage of NBS1 foci-positive cells was plotted (mean ± s.d., n = 3) (D); soluble nuclear and chromatin fractions were isolated and Western blot was performed (E).
C Inhibition of ATM kinase activity reduced the interaction of Rad17 with NBS1. U2OS cells were either untreated or treated with KU55933 (ATM inhibitor, 10 μM) for 2 h. IP experiment was performed as indicated.
Source data are available online for this figure.
Next we investigated whether MRN/ATM signaling is affected by Thr622 phosphorylation. Immunofluorescence analysis showed that reconstitution of Rad17-depleted cells with Rad17 WT, but not the Rad17 T622A mutant, restores NBS1 recruitment to DSBs (Fig 7D; supplementary Fig 8A). Further, we found that T622A mutation attenuates the enrichment of MRN complex members in the chromatin fraction after DSB induction (Fig 7E). These data suggest that Thr622 phosphorylation is important for MRN complex recruitment to the DSB. Importantly, we determined whether ATM signaling is affected by Thr622 phosphorylation and found that reconstitution of Rad17-depleted cells with Rad17 WT, but not the Rad17 T622A mutant, restored ATM Ser1981 phosphorylation and phosphorylation of its targets H2AX (Ser139), Chk2 (Thr68), and NBS1 (Ser343), as well as phospho-ATM and γ-H2AX foci formation following IR (Fig 8A–C; Supplementary Fig S8B and C). Phosphorylation of ATM and its targets peaked between 5 and 30 min in Rad17 WT cells following IR (Fig 8A), suggesting that there is amplification of ATM signaling. These results indicate that Rad17 Thr622 phosphorylation represents an important mechanism by which Rad17 contributes to activation of ATM during the phase of signal amplification in response to DSBs.
Figure 8. Rad17 phosphorylation at Thr622 is important for MRN/ATM signaling.

A–C Rad17 Thr622 phosphorylation is important for the activation of ATM signaling. Inducible Rad17-knockdown cells reconstituted with shRNA-resistant Rad17 WT or Rad17 T622A mutant were either untreated or exposed to 5 Gy of IR. Western blot was performed as indicated (A). Immunostaining was performed with anti-Rad17 and anti-p-ATM (B) or anti-γ-H2AX (C). ImageJ software was used to analyze the intensity of γ-H2AX foci.
D, E NBS1 regulates Rad17 foci formation and Thr622 phosphorylation level after IR. U2OS cells transfected with control siRNA or NBS1 siRNA were either untreated or exposed to 5 Gy of IR. Immunostaining (D) and Western blot (E) were performed as indicated. Error bars represent mean ± s.d. (n = 3).
Source data are available online for this figure.
Taken together, the findings reported here led us to propose how MRN/ATM signaling is regulated: Upon DSB induction, a fraction of ATM is initially activated and phosphorylates Rad17 at Thr622. This phosphorylation event facilitates a direct interaction between Rad17 and NBS1 resulting in the recruitment of MRN complex to DSBs, promoting ATM recruitment and activation and additional Rad17 phosphorylation. These events occur quickly at the early stage of DDR, and repetition of these events creates a positive feedback leading to the amplification of MRN/ATM signaling. Subsequently, γ-H2AX mediated by ATM results in MDC1 recruitment to DSB sites, in turn recruiting MRN complex to DSB flanking chromatin at a later stage of DDR (Stucki et al, 2005; van Attikum & Gasser, 2009). These two different mechanisms may together contribute to DDR. In support of Rad17-mediated feedback loop of MRN/ATM signaling, down-regulation of NBS1 resulted in decreased Rad17 foci formation and Thr622 phosphorylation level after IR treatment (Fig 8D and E), which also suggest that Rad17 might bind to NBS1 and is recruited to the DSB in complex with MRN. This model also explains why Rad17 knockdown leads to a marked decrease in ATM activation after IR (Fig 5A; Supplementary Fig S4A and B), although ATM appears to be required for Rad17 phosphorylation at Thr622. Collectively, the findings in this study indicate that Rad17 plays a critical role in regulating MRN/ATM signaling that is distinct from its known function in ATR signaling pathway.
Discussion
Rad17 was shown to be essential for the cell-cycle checkpoint response following exposure to genotoxic stress (Bao et al, 2001; Wang et al, 2006). The majority of Rad17 studies have focused on its ability to regulate checkpoint activation via the ATR signaling pathway. However, the potential connections of Rad17 with ATM signaling are not well characterized. In this study, we initially observed the requirement of Rad17 for HR repair in human cells, and through stepwise exploring the molecular mechanism by which Rad17 regulates HR repair, we demonstrate a critical role for Rad17 in early recruitment and retention of the MRN complex at the DSB sites and amplification of MRN/ATM signaling, which promotes the HR repair. These findings indicate that Rad17 plays an important role in the ATM signaling in addition to its role in the ATR signaling pathway.
In the present study, we propose the existence of two distinct mechanisms that promote MRN complex recruitment to DSBs. While Rad17-dependent mechanism facilitates early MRN recruitment to the DSB, a distinct γ-H2AX/MDC1-dependent pathway is responsible for accumulation of MRN at the break at a later stage of DDR. Accordingly, early MRN recruitment by Rad17 does not require MDC1. Thus, Rad17-mediated MRN complex recruitment and ATM activation appears to be a requisite for the activation of the γ-H2AX/MDC1-mediated pathway. Strikingly, while Rad17 is critical for DSB processing and HR following DSB induction, H2AX knockdown moderately affects HR activity. Given that γ-H2AX, which recruits MDC1 to the break site, is only present in DSB surrounding regions (Bekker-Jensen et al, 2006; Berkovich et al, 2007), we hypothesize that Rad17 facilitates MRN recruitment in the proximity of the DSB at an early DDR stage where it promotes DNA end resection and HR, while the subsequently activated MDC1-dependent pathway recruits MRN complex in the DSB surrounding regions amplifying DDR signaling and checkpoint activation.
Several studies showed that the MRN complex is capable of binding DNA ends directly in vitro (De Jager et al, 2001; Williams et al, 2008). However, it is important to point out that the circumstances of protein recruitment in vivo can differ from those in vitro due to the complex structure of chromatin. For example, while MRE11 is able to bind DNA ends in vitro (Williams et al, 2008), multiple in vivo studies have shown that in cells the whole MRN complex needs to be intact in order to form foci at DNA damage sites (Carney et al, 1998; Desai-Mehta et al, 2001). The fact that MDC1 is required for accumulation and maintenance of MRN complex also provides evidence that additional proteins are required for effective association of MRN complex with DSBs besides direct binding to DNA ends. Noteworthily, a study performed in yeast suggested that Rad24 (Rad17-homologue in yeast) is dispensable for MRE11 recruitment to the DSB (Lisby et al, 2004). However, there are significant differences with regard to MRN complex recruitment in yeast versus mammalian cells. For instance, a knockdown of Sae2 (CtIP-homologue in yeast) resulted in increased MRE11 foci formation in yeast (Lisby et al, 2004), while in human cells, CtIP and MRN form foci that are independent of each other, and CtIP depletion does not affect MRE11 foci formation after IR (Chen et al, 2008; Yuan & Chen, 2010).
It has been proposed that following DSB induction, MRN/ATM-dependent DNA end resection results in formation of ssDNA regions leading to ATR activation and subsequent Chk1 phosphorylation by ATR (Shiotani & Zou, 2009). This is called ATM-to-ATR switch. Our data showed that Rad17 knockdown results in a significant decrease in Chk1 phosphorylation after IR (Supplementary Fig S7A). Further, we observed a moderate decrease in Chk1 phosphorylation at Ser317 in Rad17 T622A cells following IR (Supplementary Fig S7D). We think that the residual Chk1 phosphorylation in the absence of WT Rad17 may result from an incomplete abrogation of DSB processing as evidenced by a residual presence of phospho-RPA (Fig 5D). Further, Rad17 T622A mutant protein still binds to NBS1 albeit less efficiently than WT protein after DSB induction (Fig 7B), suggesting that Thr622 phosphorylation is important but not essential for MRN/ATM activation. On the other hand, several studies show that Chk1 can also be phosphorylated in ATM deficient cells after IR or virus infection (Tomimatsu et al, 2009; Jiang et al, 2012), suggesting that activation of ATR at the DSB is not exclusively dependent on ATM activity. This can be due to ATR activation by DNA single-strand breaks that are also induced by IR other than DSBs. Rad17 is also known to promote Chk1 activation by loading 9-1-1 complex (Bao et al, 2001; Zou et al, 2002), which can explain why Rad17 knockdown results in a more pronounced decrease in Chk1 phosphorylation than observed in Rad17 T622A cells. Recent studies showed that the MRN complex is involved in recruitment of TopBP1 to stalled replication forks, thus contributing to ATR activation (Duursma et al, 2013; Kobayashi et al, 2013; Lee & Dunphy, 2013; Shiotani et al, 2013). While this function of MRN seems to be Rad17 independent (Kobayashi et al, 2013), it also differs from MRN function at the DSB since ATR activation may occur independent of ATM and DNA resection at stalled replication forks (Kobayashi et al, 2013; Shiotani et al, 2013), suggesting that the function of MRN complex in ATR activation might be different in response to different types of genotoxic stress.
It has been reported that constitutively di-phosphorylated pSer-Asp-pThr-Asp (SDTD)-like motifs in the N-terminus of MDC1 mediated by caseine kinase 2 (CK2) are important for the interaction of MDC1 with the N-terminal FHA domain of NBS1 (Chapman & Jackson, 2008; Melander et al, 2008; Spycher et al, 2008; Wu et al, 2008). However, although Rad17 lacks the SDTD-motif, we observed a solid interaction between the C-terminus of Rad17 and the FHA domain of NBS1 (Fig 2E and F; Fig 7A). The mechanism of this Rad17-NBS1 interaction is not clear yet. It appears possible that different proteins may bind to FHA domain of NBS1 through different mechanisms.
Since most traditional chemotherapies work by inducing DNA damage to kill cancer cells, DNA repair pathways can be targeted to slow cancer growth (Helleday et al, 2008). Our data suggest that the function of Rad17 in DNA repair makes it a possible therapeutic target. Rad17 phosphorylation at Thr622 is required for hypopharyngeal tumor growth in the presence of PARP inhibition (Fig 6I and J). Overexpression of Rad17 has been reported in human lung, colon and breast cancer, whereby Rad17 is frequently affected by genomic mutational events, potentially affecting the Thr622 residue (Bao et al, 1999; Wang et al, 2001). Therefore, blocking either this phosphorylation event through ATM inhibition or disrupting the Rad17-NBS1 interaction might increase the efficacy of radiation and certain chemotherapeutic agents or induce synthetic lethality if combined with PARP inhibition as demonstrated in this study using mouse xenografts.
In summary, our findings indicate that Rad17 is a pivotal protein that mediates early MRN recruitment to DSBs leading to amplification of MRN/ATM signaling via its Thr622 phosphorylation, which is important for an immediate and active cellular response to DNA damage. It is likely that both Rad17- and MDC1-dependent recruitment of MRN complex contribute to an efficient DDR via ATM signaling to maintain genome integrity. The connection between Rad17 and ATM signaling demonstrated in this study identifies a new function of Rad17 in the DDR and provides new details regarding the mechanism of ATM signaling activation.
Materials and Methods
Cell culture
U2OS cells were cultured in McCoy's 5A medium, HeLa, 293T, FaDu and HFF cells were maintained in Dulbecco's modified Eagle's medium, and A549 cells were cultured in F-12K medium. All media were supplemented with 10% fetal bovine serum, 100 Unit/ml penicillin, and 100 μg/ml streptomycin. All cell lines were obtained from the Duke Cell Culture Facility.
Immunoblotting, immunoprecipitation and immunofluorescent staining
Detailed methods and reagents used are described in Supplementary Materials and Methods.
HR repair assay
The fluorescence-based HR repair was performed as previously described (Pierce et al, 1999). To determine the effect of Rad17 on HR repair, a U2OS or A549 clone with the integrated HR reporter DR-GFP was generated as previously described (Pierce et al, 1999). The stable pDR-GFP cells transfected with either control siRNA or Rad17 siRNA for 2 days were further transfected with either I-SceI-expressing vector or a control vector. pGFP construct was transfected into these cells to ensure similar transfection efficiency. Forty-eight hours later, the cells were subjected to the analysis of GFP-positive cells using FACSCAN flow cytometer (Becton Dickinson) with CellQuest software. To determine the effects of Rad17 phosphorylation on HR repair, the stable pDR-GFP U2OS cells expressing shRNA-resistant Rad17 WT, Rad17 T622A mutant were treated with doxycycline for 2 days to deplete the endogenous Rad17. Cells were further transfected with either I-SceI-expressing vector or a control vector. Forty-eight hours later, the cells were analyzed by flow cytometry as described above.
In vitro GST pull-down and peptide pull-down assay
For GST pull-down assays, GST-fused Rad17 or NBS1 proteins were bacterially expressed using standard protocol and further immobilized on glutathione Sepharose 4B beads. The beads were incubated with U2OS cell lysates for 2 h or overnight at 4°C. Beads were then washed with NETN buffer (50 mM Tris, 150 mM NaCl, 1 mM EDTA, 1% NP40, 10% glycerol, 1 mM Na3SO4 and 10 mMNaF) four times, and proteins bound to beads were eluted with SDS sample buffer (100°C, 5 min) and separated by SDS–PAGE followed by Western blotting. For peptide pull-down assays, the biotinylated peptides (non-phospho-T622 peptide sequences: biotin-ESLGEPTQATVP, phospho-T622 peptide sequences: biotin-ESLGEP(p)TQATVP) were synthesized by Sigma. The peptides were coupled to streptavidin-Sepharose beads by mixing peptide with beads in coupling buffer (50 mM Tris, 5 mM EDTA, pH 8.5) for 30 min at room temperature. The resulting beads were incubated with purified GST-NBS1 proteins (FHA domain or C-terminus domain containing residues 682–754) 3 h at 4°C. Beads were washed four times, and proteins retained on the beads were eluted and subjected to Western blotting with anti-GST.
Subcellular fractionation
Isolation of nuclear soluble fractions and chromatin fractions was performed as previously described (Mendez & Stillman, 2000). Briefly, cells were lysed in hypotonic buffer to isolate nuclei. Nuclei were re-suspended, and insoluble chromatin was separated from soluble nuclear proteins and then sheared by sonication. A detail protocol is provided in Supplementary Materials and Methods.
Tumor xenograft
Mouse tumor xenograft experiments were carried out as previously described with some modifications (Weston et al, 2010). Stable FaDu cells containing inducible shRNA-resistant Rad17 WT or Rad17 T622A mutant were then inoculated into the right flank of nude mice by subcutaneous injection of 4 × 106 cells. Doxycycline was delivered daily into every mouse with 20 mg/kg by I.P. injection. Mice bearing tumors of 200–220 mm3 were divided into two groups for each cell line: vehicle control group (10% DMSO with 10% 2-hydroxypropyl-beta-cyclodextrin daily by oral gavage) and Olaparib group (100 mg/kg daily by oral gavage). Each group contained four nude mice. Tumor volume was subsequently measured every other day using calipers. Mice were sacrificed for tumor dissection on day 28 after the start of Olaparib treatment.
Acknowledgments
We thank Kuntian Luo for providing the NBS1 construct and technical advice and members of the Wang laboratory for helpful suggestions during the execution of the experiments. This research was supported by NIH Grants CA154151 and CA154586 (X.F.W.).
Author contributions
QW led the project and performed all experiments. MG reviewed and revised the manuscript. PA purified GST-Rad17 and GST-NBS1 recombinant proteins and performed qPCR experiments. TPW and JF contributed to the initial phase of the project. TS provided support with tissue culture maintenance and stable cell generation. ZL and MBK reviewed the manuscript and made helpful suggestions. QW and XFW conceived the study and wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information for this article is available online: http://emboj.embopress.org
References
- van Attikum H, Gasser SM. Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol. 2009;19:207–217. doi: 10.1016/j.tcb.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Bao S, Chang MS, Auclair D, Sun Y, Wang Y, Wong WK, Zhang J, Liu Y, Qian X, Sutherland R, Magi-Galluzi C, Weisberg E, Cheng EY, Hao L, Sasaki H, Campbell MS, Kraeft SK, Loda M, Lo KM, Chen LB. HRad17, a human homologue of the Schizosaccharomyces pombe checkpoint gene Rad17, is overexpressed in colon carcinoma. Cancer Res. 1999;59:2023–2028. [PubMed] [Google Scholar]
- Bao S, Tibbetts RS, Brumbaugh KM, Fang Y, Richardson DA, Ali A, Chen SM, Abraham RT, Wang XF. ATR/ATM-mediated phosphorylation of human Rad17 is required for genotoxic stress responses. Nature. 2001;411:969–974. doi: 10.1038/35082110. [DOI] [PubMed] [Google Scholar]
- Bassing CH, Chua KF, Sekiguchi J, Suh H, Whitlow SR, Fleming JC, Monroe BC, Ciccone DN, Yan C, Vlasakova K, Livingston DM, Ferguson DO, Scully R, Alt FW. Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proc Natl Acad Sci USA. 2002;99:8173–8178. doi: 10.1073/pnas.122228699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekker-Jensen S, Lukas C, Kitagawa R, Melander F, Kastan MB, Bartek J, Lukas J. Spatial organization of the mammalian genome surveillance machinery in response to DNA strand breaks. J Cell Biol. 2006;173:195–206. doi: 10.1083/jcb.200510130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkovich E, Monnat RJ, Kastan MB. Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat Cell Biol. 2007;9:683–690. doi: 10.1038/ncb1599. [DOI] [PubMed] [Google Scholar]
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- Budzowska M, Jaspers I, Essers J, de Waard H, van Drunen E, Hanada K, Beverloo B, Hendriks RW, de Klein A, Kanaar R, Hoeijmakers JH, Maas A. Mutation of the mouse Rad17 gene leads to embryonic lethality and reveals a role in DNA damage-dependent recombination. EMBO J. 2004;23:3548–3558. doi: 10.1038/sj.emboj.7600353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carney JP, Maser RS, Olivares H, Davis EM, Le Beau M, Yates JR, 3rd, Hays L, Morgan WF, Petrini JH. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double-strand break repair to the cellular DNA damage response. Cell. 1998;93:477–486. doi: 10.1016/s0092-8674(00)81175-7. [DOI] [PubMed] [Google Scholar]
- Celeste A, Fernandez-Capetillo O, Kruhlak MJ, Pilch DR, Staudt DW, Lee A, Bonner RF, Bonner WM, Nussenzweig A. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat Cell Biol. 2003;5:675–679. doi: 10.1038/ncb1004. [DOI] [PubMed] [Google Scholar]
- Chapman JR, Jackson SP. Phospho-dependent interactions between NBS1 and MDC1 mediate chromatin retention of the MRN complex at sites of DNA damage. EMBO Rep. 2008;9:795–801. doi: 10.1038/embor.2008.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Nievera CJ, Lee AY, Wu X. Cell cycle-dependent complex formation of BRCA1.CtIP.MRN is important for DNA double-strand break repair. J Biol Chem. 2008;283:7713–7720. doi: 10.1074/jbc.M710245200. [DOI] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daboussi F, Dumay A, Delacote F, Lopez BS. DNA double-strand break repair signaling: the case of RAD51 post-translational regulation. Cell Signal. 2002;14:969–975. doi: 10.1016/s0898-6568(02)00052-9. [DOI] [PubMed] [Google Scholar]
- De Jager M, van Noort J, van Gent DC, Dekker C, Kanaar R, Wyman C. Human Rad50/MRE11 is a flexible complex that can tether DNA ends. Mol Cell. 2001;8:1129–1135. doi: 10.1016/s1097-2765(01)00381-1. [DOI] [PubMed] [Google Scholar]
- Desai-Mehta A, Cerosaletti KM, Concannon P. Distinct functional domains of nibrin mediate Mre11 binding, focus formation, and nuclear localization. Mol Cell Biol. 2001;21:2184–2191. doi: 10.1128/MCB.21.6.2184-2191.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duursma AM, Driscoll R, Elias JE, Cimprich KA. A role for the MRN complex in ATR activation via TOPBP1 recruitment. Mol Cell. 2013;50:116–122. doi: 10.1016/j.molcel.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005;434:605–611. doi: 10.1038/nature03442. [DOI] [PubMed] [Google Scholar]
- Fernandez-Capetillo O, Lee A, Nussenzweig M, Nussenzweig A. H2AX: the histone guardian of the genome. DNA Repair. 2004;3:959–967. doi: 10.1016/j.dnarep.2004.03.024. [DOI] [PubMed] [Google Scholar]
- Greenberg RA, Sobhian B, Pathania S, Cantor SB, Nakatani Y, Livingston DM. Multifactorial contributions to an acute DNA damage response by BRCA1/BARD1-containing complexes. Genes Dev. 2006;20:34–46. doi: 10.1101/gad.1381306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helleday T, Petermann E, Lundin C, Hodgson B, Sharma R. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8:193–204. doi: 10.1038/nrc2342. [DOI] [PubMed] [Google Scholar]
- Hu B, Wang H, Wang X, Lu HR, Huang C, Powell SN, Huebner K, Wang Y. Fhit and CHK1 have opposing effects on homologous recombination repair. Cancer Res. 2005;65:8613–8616. doi: 10.1158/0008-5472.CAN-05-1966. [DOI] [PubMed] [Google Scholar]
- Hu Y, Scully R, Sobhian B, Xie A, Shestakova E, Livingston DM. RAP80-directed tuning of BRCA1 homologous recombination function at ionizing radiation-induced nuclear foci. Genes Dev. 2011;25:685–700. doi: 10.1101/gad.2011011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huen MS, Sy SM, Chen J. BRCA1 and its toolbox for the maintenance of genome integrity. Nat Rev Mol Cell Biol. 2010;11:138–148. doi: 10.1038/nrm2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J, Jackson SP. ATM-and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol. 2006;8:37–45. doi: 10.1038/ncb1337. [DOI] [PubMed] [Google Scholar]
- Jiang M, Zhao L, Gamez M, Imperiale MJ. Roles of ATM and ATR-mediated DNA damage responses during lytic BK polyomavirus infection. PLoS Pathog. 2012;8:e1002898. doi: 10.1371/journal.ppat.1002898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Hayashi N, Takata M, Yamamoto K. NBS1 directly activates ATR independently of MRE11 and TOPBP1. Genes Cells. 2013;18:238–246. doi: 10.1111/gtc.12031. [DOI] [PubMed] [Google Scholar]
- Lee J, Dunphy WG. The MRE11-Rad50-NBS1 (MRN) complex has a specific role in the activation of Chk1 in response to stalled replication forks. Mol Biol Cell. 2013;24:1343–1353. doi: 10.1091/mbc.E13-01-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Paull TT. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene. 2007;26:7741–7748. doi: 10.1038/sj.onc.1210872. [DOI] [PubMed] [Google Scholar]
- Lieber MR. The mechanism of human nonhomologous DNA end joining. J Biol Chem. 2008;283:1–5. doi: 10.1074/jbc.R700039200. [DOI] [PubMed] [Google Scholar]
- Lisby M, Barlow JH, Burgess RC, Rothstein R. Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell. 2004;118:699–713. doi: 10.1016/j.cell.2004.08.015. [DOI] [PubMed] [Google Scholar]
- Melander F, Bekker-Jensen S, Falck J, Bartek J, Mailand N, Lukas J. Phosphorylation of SDT repeats in the MDC1 N terminus triggers retention of NBS1 at the DNA –damage-modified chromatin. J Cell Biol. 2008;181:213–226. doi: 10.1083/jcb.200708210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez J, Stillman B. Chromatin association of human origin recognition complex, cdc6 and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol Cell Biol. 2000;20:8602–8612. doi: 10.1128/mcb.20.22.8602-8612.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers JS, Cortez D. Rapid activation of ATR by ionizing radiation requires ATM and MRE11. J Biol Chem. 2006;281:9346–9350. doi: 10.1074/jbc.M513265200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niida H, Nakanishi M. DNA damage checkpoints in mammals. Mutagenesis. 2006;21:3–9. doi: 10.1093/mutage/gei063. [DOI] [PubMed] [Google Scholar]
- Nishino K, Inoue E, Takada S, Abe T, Akita M, Yoshimura A, Tada S, Kobayashi M, Yamamoto K, Seki M, Enomoto T. A novel role for Rad17 in homologous recombination. Genes Genet Syst. 2008;83:427–431. doi: 10.1266/ggs.83.427. [DOI] [PubMed] [Google Scholar]
- Petersen S, Casellas R, Reina-San-Martin B, Chen HT, Difilippantonio MJ, Wilson PC, Hanitsch L, Celeste A, Muramatsu M, Pilch DR, Redon C, Ried T, Bonner WM, Honjo T, Nussenzweig MC, Nussenzweig A. AID is required to initiate NBS1/gamma-H2AX focus formation and mutations at sites of class switching. Nature. 2001;414:660–665. doi: 10.1038/414660a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999;13:2633–2638. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu Rev Biochem. 2008;77:229–257. doi: 10.1146/annurev.biochem.77.061306.125255. [DOI] [PubMed] [Google Scholar]
- Shiotani B, Nguyen HD, Håkansson P, Maréchal A, Tse A, Tahara H, Zou L. Two distinct modes of ATR activation orchestrated by Rad17 and Nbs1. Cell Rep. 2013;3:1651–1662. doi: 10.1016/j.celrep.2013.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiotani B, Zou L. Single-stranded DNA orchestrates an ATM-to-ATR switch at DNA breaks. Mol Cell. 2009;33:547–558. doi: 10.1016/j.molcel.2009.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen CS, Hansen LT, Dziegielewski J, Syljuasen RG, Lundin C, Bartek J, Helleday T. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat Cell Biol. 2005;7:195–201. doi: 10.1038/ncb1212. [DOI] [PubMed] [Google Scholar]
- Spycher C, Miller ES, Townsend K, Pavic L, Morrice NA, Janscak P, Stewart GS, Stucki M. Constitutive phosphorylation of MDC1 physically links the MRE11-RAD50-NBS1 complex to damaged chromatin. J Cell Biol. 2008;181:227–240. doi: 10.1083/jcb.200709008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiff T, O'Driscoll M, Rief N, Iwabuchi K, Löbrich M, Jeggo PA. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 2004;64:2390–2396. doi: 10.1158/0008-5472.can-03-3207. [DOI] [PubMed] [Google Scholar]
- Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell. 2005;123:1213–1226. doi: 10.1016/j.cell.2005.09.038. [DOI] [PubMed] [Google Scholar]
- Sugiyama T, Kowalczykowski SC. Rad52 protein associates with replication protein A (RPA)-single-stranded DNA to accelerate Rad51-mediated displacement of RPA and presynaptic complex formation. J Biol Chem. 2002;277:31663–31672. doi: 10.1074/jbc.M203494200. [DOI] [PubMed] [Google Scholar]
- Tomimatsu N, Mukherjee B, Burma S. Distinct roles of ATR and DNA-PKcs in triggering DNA damage responses in ATM-deficient cells. EMBO Rep. 2009;10:629–635. doi: 10.1038/embor.2009.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traven A, Heierhorst J. SQ/TQ cluster domains: concentrated ATM/ATR kinase phosphorylation site regions in DNA-damage-response proteins. BioEssays. 2005;27:397–407. doi: 10.1002/bies.20204. [DOI] [PubMed] [Google Scholar]
- Wang X, Wang L, Callister MD, Putnam JB, Mao L, Li L. Human Rad17 Is Phosphorylated upon DNA Damage and also Overexpressed in Primary Non-Small Cell Lung Cancer Tissues. Cancer Res. 2001;61:7417–7421. [PubMed] [Google Scholar]
- Wang X, Zou L, Lu T, Bao S, Hurov KE, Hittelman WN, Elledge SJ, Li L. Rad17 phosphorylation is required for claspin recruitment and Chk1 activation in response to replication stress. Mol Cell. 2006;23:331–341. doi: 10.1016/j.molcel.2006.06.022. [DOI] [PubMed] [Google Scholar]
- Wang X, Zou L, Zheng H, Wei Q, Elledge SJ, Li L. Genomic instability and endoreduplication triggered by Rad17 deletion. Genes Dev. 2003;17:965–970. doi: 10.1101/gad.1065103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston VJ, Oldreive CE, Skowronska A, Oscier DG, Pratt G, Dyer MJ, Smith G, Powell JE, Rudzki Z, Kearns P, Moss PA, Taylor AM, Stankovic T. The PARP inhibitor olaparib induces significant killing of ATM-deficient lymphoid tumor cells in vitro and in vivo. Blood. 2010;116:4578–4587. doi: 10.1182/blood-2010-01-265769. [DOI] [PubMed] [Google Scholar]
- Williams GJ, Lees-Miller SP, Tainer JA. MRE11-Rad50-NBS1 conformations and the control of sensing, signaling, and effector responses at DNA double-strand breaks. DNA Repair (Amst) 2010;9:1299–1306. doi: 10.1016/j.dnarep.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RS, Moncalian G, Williams JS, Yamada Y, Limbo O, Shin DS, Groocock LM, Cahill D, Hitomi C, Guenther G, Moiani D, Carney JP, Russell P, Tainer JA. MRE11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell. 2008;135:97–109. doi: 10.1016/j.cell.2008.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RS, Williams JS, Tainer JA. MRE11-Rad50-NBS1 is a keystone complex connecting DNA repair machinery, double-strand break signaling, and the chromatin template. Biochem Cell Biol. 2007;85:509–520. doi: 10.1139/O07-069. [DOI] [PubMed] [Google Scholar]
- Wu L, Luo K, Lou Z, Chen J. MDC1 regulates intra-S-phase checkpoint by targeting NBS1 to DNA double-strand breaks. Proc Natl Acad Sci. 2008;105:11200–11205. doi: 10.1073/pnas.0802885105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie A, Puget N, Shim I, Odate S, Jarzyna I, Bassing CH, Alt FW, Scully R. Control of sister chromatid recombination by histone H2AX. Mol Cell. 2004;16:1017–1025. doi: 10.1016/j.molcel.2004.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Z, Chahwan C, Bailis J, Hunter T, Russell P. ATM activation and its recruitment to damaged DNA require binding to the C terminus of NBS1. Mol Cell Biol. 2005;25:5363–5379. doi: 10.1128/MCB.25.13.5363-5379.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Chen J. MRE11-RAD50-NBS1 complex dictates DNA repair independent of H2AX. J Biol Chem. 2010;285:1097–1104. doi: 10.1074/jbc.M109.078436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Lim CU, Williams ES, Zhou J, Zhang Q, Fox MH, Bailey SM, Liber HL. NBS1 knockdown by small interfering RNA increases ionizing radiation mutagenesis and telomere association in human cells. Cancer Res. 2005;65:5544–5553. doi: 10.1158/0008-5472.CAN-04-4368. [DOI] [PubMed] [Google Scholar]
- Zou L, Cortez D, Elledge EJ. Regulation of ATR substrate selection by Rad17-dependent loading of Rad9 complexes onto chromatin. Genes Dev. 2002;16:198–208. doi: 10.1101/gad.950302. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.