

Abstract
Mitogen-activated protein kinases (MAPKs) are highly conserved protein kinase modules, and they control fundamental cellular processes. While the activation of MAPKs has been well studied, little is known on the mechanisms driving their inactivation. Here we uncover a role for ubiquitination in the inactivation of a MAPK module. Extracellular-signal-regulated kinase 5 (ERK5) is a unique, conserved member of the MAPK family and is activated in response to various stimuli through a three-tier cascade constituting MEK5 and MEKK2/3. We reveal an unexpected role for Inhibitors of Apoptosis Proteins (IAPs) in the inactivation of ERK5 pathway in a bimodal manner involving direct interaction and ubiquitination. XIAP directly interacts with MEKK2/3 and competes with PB1 domain-mediated binding to MEK5. XIAP and cIAP1 conjugate predominantly K63-linked ubiquitin chains to MEKK2 and MEKK3 which directly impede MEK5–ERK5 interaction in a trimeric complex leading to ERK5 inactivation. Consistently, loss of XIAP or cIAP1 by various strategies leads to hyperactivation of ERK5 in normal and tumorigenic cells. Loss of XIAP promotes differentiation of human primary skeletal myoblasts to myocytes in a MEKK2/3-ERK5-dependent manner. Our results reveal a novel, obligatory role for IAPs and ubiquitination in the physical and functional disassembly of ERK5-MAPK module and human muscle cell differentiation.
Keywords: ERK5, IAP, MAPK, myoblasts, ubiquitin
See also: AM Klein & MH Cobb (August 2014)
Introduction
Mitogen-activated protein kinases (MAPKs) are highly conserved, ubiquitously expressed enzymes that are activated in response to a plethora of extracellular stimuli to regulate a variety of cellular processes (Chang & Karin, 2001; Coulombe & Meloche, 2007; Raman et al, 2007). Typically, MAPKs are activated by dual phosphorylation in the Thr-X-Tyr motif in their activation loop by a MAPKK. MAPKKs, on the other hand, are activated by phosphorylation of Ser-Thr residues by upstream MAPKKKs, thus constituting a three-tier kinase cascade (Zhang & Dong, 2007). RAF-MEK1/2-ERK1/2 is the first discovered, well-studied classical MAPK cascade. In mammalian cells, four distinct classical MAPK cascades are recognized: ERK1/2, JNK1/2/3, p38 MAPK α/β/γ/δ, and the ERK5 pathways (Chang & Karin, 2001). ERK5, also called the Big MAPK 1 (BMK1), was cloned by two groups in 1995. It is a unique member of this family as it is double the size of ERK1/2 with a kinase domain in the N-terminus and a long C-terminus (Nithianandarajah-Jones et al, 2012). The N-terminal kinase domain is 66% identical to the kinase domain of ERK2, and the C-terminus encompasses a nuclear localization signal (NLS), two proline-rich domains, a myocyte enhancer factor 2 (MEF2)-interacting region, and a transcriptional activation domain (Nithianandarajah-Jones et al, 2012). MEK5 was identified as an upstream MAPKK through yeast two-hybrid analysis, phosphorylating the activation loop of ERK5 at Thr218 and Tyr220 leading to its activation (Zhou et al, 1995). Activated ERK5 undergoes autophosphorylation at numerous residues in its C-terminus, which causes conformational change so that the NLS is exposed, facilitating its nuclear translocation (Mody et al, 2003; Nithianandarajah-Jones et al, 2012). MEKK2 and MEKK3 kinase domains are 90% identical, and they form heteromers with MEK5 through their PB1 (Phox, Bemp1) domains, an evolutionarily conserved protein–protein interaction motif (Seyfried et al, 2005; Moscat et al, 2006; Sumimoto et al, 2007). MEKK2/3 and MEK5 interact through their PB1 domains in a front-to-back orientation where the basic residues in the front end of MEKK2/3 PB1 domain mediate electrostatic interactions with the acidic residues in the back end of MEK5 PB1 domain (Nakamura & Johnson, 2003, 2007). ERK5, though lacks the PB1 domain, interacts through its N-terminus (a.a. 78–139) with the PB1 domain and the following C-terminal stretch (a.a. 98–131) of MEK5 (Nithianandarajah-Jones et al, 2012). Like many MAPKs, ERK5 possesses a common docking domain (a.a. 350–358) with negatively charged amino acids, with which it interacts with its substrates and MEK5 consequently maintaining the signaling fidelity and efficiency (Tanoue & Nishida, 2002). The ERK5 cascade is activated in response to various growth factors (EGF, PDGF, VEGF, FGF-2, NGF, and BDNF), cytokines (like IL-6), and other stimuli like oxidative stress and ischemia. Gene knockout studies have revealed a crucial role of this cascade in the development of cardiovascular system (Wang & Tournier, 2006). There is mounting evidence for the role of ERK5/MAPK pathway in regulating cancer as ERK5 amplifications (17p11) are detected in hepatocellular carcinoma and high MEK5 and ERK5 levels are associated with bone and lymph node metastases in prostate cancer and oral squamous cell carcinoma, respectively (Lochhead et al, 2012). Despite the physiological and pathophysiological importance of the ERK5 signaling cascade, relatively less is known on the upstream signaling machinery as well as the factors contributing to the activation/inactivation of this pathway. Here we identify Inhibitors of Apoptosis Proteins (IAPs) as negative regulators of ERK5 activation.
IAPs are a class of evolutionarily conserved, multifunctional proteins (Srinivasula & Ashwell, 2008). In mammals, eight IAPs exist distinguished by the presence of their signature BIR (Baculoviral IAP Repeat) domain, a conserved protein–protein interaction motif. Many of them possess a RING domain with E3 ubiquitin ligase activity (Srinivasula & Ashwell, 2008). X-linked inhibitor of apoptosis protein (XIAP, also called BIRC4) is considered as a legitimate inhibitor of caspases, and cellular IAPs (cIAPs) were shown to control both canonical and non-canonical pathways of NF-κB activation (O'Riordan et al, 2008). Apart from their well-established role in apoptosis, inflammation and innate immunity, emerging evidence revealed a role for IAPs in the regulation of cell shape, cell migration, and tumor metastases (Gyrd-Hansen & Meier, 2010). XIAP and cIAPs regulate classical MAPK activation as they can promote the proteasomal degradation of C-RAF through the Hsp90 protein quality-control machinery (Dogan et al, 2008). Further XIAP and cIAP1 can function as the direct E3 ubiquitin ligase of RhoGTPase Rac1, thus controlling cell shape and migration (Oberoi et al, 2011). Here we uncover that XIAP and cIAP1 function as negative regulators of ERK5 signaling cascade by physically interacting with and ubiquitinating MEKK2 and MEKK3. As a consequence, loss of XIAP enhances ERK5 activation and MEF2C activity. This in turn promotes human myoblast differentiation in a MEKK2/3-ERK5-dependent manner. Our results reveal a thus far unknown role of IAPs and ubiquitination in the functional disassembly of a classical MAPK module and human myocyte generation.
Results
Loss of IAPs leads to hyperactivation of ERK5 in normal and tumor cells
We have recently shown that XIAP and cIAPs can modulate ERK1/2 activation by directly regulating the stability of C-RAF kinase (Dogan et al, 2008). To test whether XIAP can influence other related classical MAPKs, we checked for the activation of ERK5 (pERK5) in IAP-depleted cells. Interestingly, depletion of XIAP with different siRNAs led to a striking increase in the phosphorylation of ERK5 at steady state in HeLa cells (Fig1A and B). Loss of XIAP did not lead to any significant alterations in the protein levels of ERK5 (Fig1A and B). Similarly, depletion of cIAP1 but not cIAP2 led to an increase in ERK5 phosphorylation in these cells (Supplementary Fig S1A and B). These effects were evident in other tumor cell lines as well as in human primary tracheal epithelial cells (Supplementary Fig S1C and D and data not shown). To exclude any potential off target effects, we performed complementation experiments. Expression of XIAP in trans-reduced pERK5 levels in HeLa cells transfected with 3′UTR-XIAP siRNA (Supplementary Fig S1E), thus confirming the specificity of the observed phenotype. To corroborate these observations, we checked for MEF2 transcriptional activity in HeLa cells stably expressing MEF2 luciferase reporter constructs. Consistent with the previous observations, depletion of XIAP led to a significant increase in MEF2 activity in an ERK5-dependent manner (Fig1C). Further, we checked for the kinetics of ERK5 activation in HeLa cells and MEFs. Loss of XIAP increased ERK5 activation in HeLa cells and MEFs in response to FCS and FGF-2 stimulation, respectively (Fig1B and D). This suggests that loss of XIAP influences the intensity of ERK5 activation. As XIAP loss can lead to stabilization of C-RAF and Rac1 protein levels, we checked for their possible role in the activation of ERK5. Co-knockdown experiments in HeLa cells revealed that depletion of XIAP led to an increase in pERK5 levels despite the absence of C-RAF and Rac1 (Supplementary Fig S1F–H). As MEKK2/3 and MEK5 are the cognate upstream MAPK members, we checked for their role in activating ERK5 in XIAP-depleted cells. As expected, co-knockdown of MEKK2, MEKK3, or MEK5 prevented activation of ERK5 in XIAP-depleted cells (Fig1E–G). These results confirmed that XIAP and cIAP1 negatively regulate ERK5 activation. Further, depletion of both MEKK2 and MEKK3 led to reduced ERK5 activation unveiling functional non-redundancy between these two MAP3Ks in activating MEK5–ERK5 pathway (Fig1E and F).
Figure 1. Depletion of XIAP leads to enhanced activation of the MEKK2/3-MEK5-ERK5 pathway.
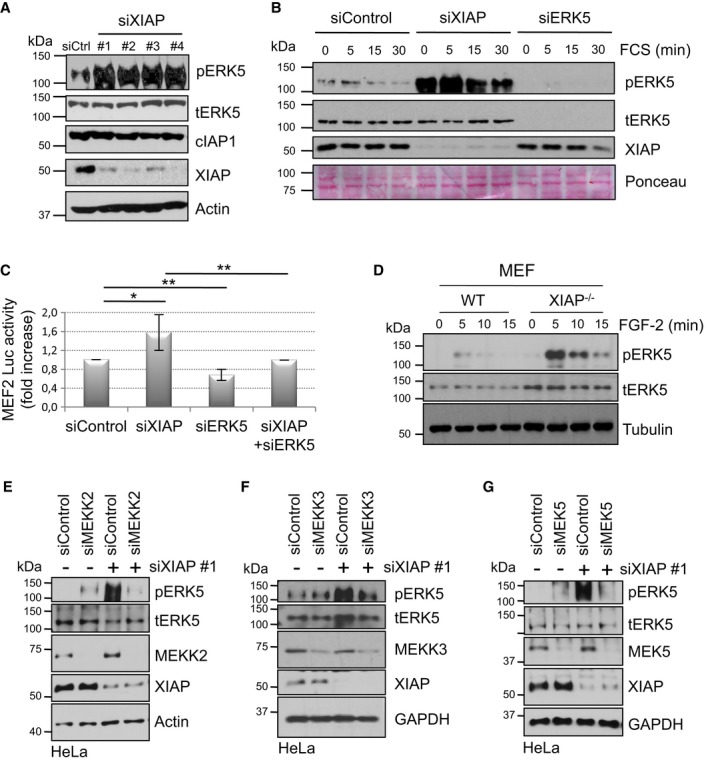
A Depletion of XIAP increases the basal phosphorylation of ERK5. HeLa cells were transiently transfected with Control or XIAP siRNAs. Total lysates were analyzed by Western blotting with various antibodies.
B Depletion of XIAP enhances the strength of ERK5 activation. HeLa-MEF2 cells were transiently transfected with siControl or siXIAP or siERK5 and then stimulated with 10% FCS after serum starvation. Total lysates were analyzed by Western blotting.
C Depletion of XIAP enhances MEF2 transcriptional activity. HeLa cells stably expressing the MEF2-luciferase (luc) reporter gene were transiently transfected with siRNAs against XIAP, ERK5, or both. Cells were then lysed and the luciferase activity was measured and normalized to the Control activity as mentioned in the methods section (shown is the quantification of five independent experiments with *P < 0.05 and **P < 0.01, Student's t-test).
D Loss of XIAP in MEFs also enhances ERK5 phosphorylation. WT, and XIAP−/− MEFs were stimulated with 25 ng/ml of FGF-2. Total lysates were analyzed by Western blotting.
E–G XIAP-mediated effect on ERK5 phosphorylation is dependent of MEKK2, MEKK3, and MEK5. HeLa cells were co-transfected with siRNAs against XIAP and/or MEKK2 (E), MEKK3 (F), or MEK5 (G). Total lysates were analyzed by Western blotting as indicated.
Source data are available online for this figure.
XIAP directly binds to MEKK2/3 and competes with MEK5 for PB1-mediated interaction
As XIAP has been shown to interact with MEKK2 (Winsauer et al, 2008), we checked whether this is a direct interaction. In vitro interaction experiments with purified recombinant proteins revealed a direct interaction between XIAP and MEKK2 or MEKK3 (Fig2A). Consistently, we could detect constitutive interaction between XIAP and MEKK2 at endogenous levels in HeLa cells. However, we failed to detect MEKK2 or MEKK3 in XIAP immunoprecipitates (Fig2B and data not shown). We then checked for the role of various domains of XIAP in mediating the interaction with MEKK2. In vitro interaction experiments with purified proteins encompassing various domains of XIAP (Supplementary Fig S2A) revealed that the RING domain of XIAP is dispensable for binding to MEKK2 and that this interaction can possibly be mediated through BIR1 and BIR2 domains (Supplementary Fig S2B). Next, we investigated the role of PB1 domain of MEKK2 in mediating the interaction with XIAP, although XIAP did not possess any PB1 domain. Interestingly, mutating the conserved basic lysine residue in the PB1 domain (K47A) severely impaired the direct interaction between XIAP and MEKK2 (Fig2C). As XIAP could bind to the PB1 domain of MEKK2, we expected a potential competition between XIAP and MEK5 in binding to MEKK2. XIAP fails to bind directly to MEK5 (Fig2D). In vitro competition experiments with recombinant full-length proteins revealed that XIAP could directly compete with MEK5 in binding to MEKK2 (Fig2E). Consistent with these observations, we could detect increasing amounts (˜1.5-fold) of MEK5 co-precipitating with MEKK2 at endogenous levels in XIAP-depleted cells (Fig2F and Supplementary Fig S2C). These results revealed that XIAP could directly bind to MEKK2/3 and compete with MEK5 interaction.
Figure 2. Characterizing the mode of interaction between XIAP and MEKK2/3-MEK5.

- XIAP binds to MEKK2 and MEKK3 in vitro. Purified XIAP protein was incubated with MEKK2-GST or MEKK3-GST, and GST pull down was performed as described in Materials and Methods.
- XIAP binds to MEKK2 in vivo. Endogenous XIAP and MEKK2 were immunoprecipitated from HeLa cell lysates, and IP fractions were analyzed by Western blotting for co-precipitation of complex components.
- Mutation in the PB1 domain of MEKK2 prevents XIAP binding. XIAP-GST protein was incubated with in vitro translated MEKK2-wt, MEKK2-K47A and GST pull down were performed. Total lysates and GST fraction were analyzed by Western blotting.
- XIAP does not bind to MEK5. MEK5-GST protein was incubated with either in vitro translated MEKK2-wt or XIAP-wt protein, and GST pull down was performed. Total lysates and GST fraction were analyzed by Western blotting.
- XIAP competes for MEKK2 binding to MEK5. MEK5-GST protein was incubated with in vitro translated MEKK2-wt in the presence or absence of increasing amounts of in vitro translated XIAP-wt and subjected to GST pull down. The amount of XIAP in the supernatant (SN) was also tested. The relative amounts of co-precipitated MEKK2 were quantified, and the mean of three independent experiments is shown as a graph with **P < 0.01 and ***P < 0.001, Student's t-test.
- Depletion of XIAP improves MEK5 binding to MEKK2 in vivo. A431 cells were transiently transfected with siControl or siXIAP, stimulated with 10% FCS after serum starvation and endogenous MEKK2 were immunoprecipitated. The relative fold increase in the amount of MEK5 band co-precipitating with MEKK2 was quantified and presented below the blots.
Source data are available online for this figure.
XIAP regulates ERK5 activation in a RING-dependent manner
As XIAP possesses a RING domain with E3 ubiquitin ligase activity, we tested for the role of RING domain in regulating ERK5 activation. To check for potential ubiquitination of MEKK2, we immunoprecipitated endogenous MEKK2 from control and XIAP-deficient MEFs stimulated with FGF-2. Upon FGF-2 stimulation, ERK5 phosphorylation increased and was inactivated at 15 min post-induction in control cells. Interestingly, XIAP-deficient cells exhibited pronounced ERK5 phosphorylation at 15 min post-induction (Fig3A). Intriguingly, the MEKK2 antibody detected a smear at 15 min post-induction in control cells, which was clearly absent in the XIAP-deficient cells (Fig3A). Furthermore, the appearance of MEKK2 smear correlates with the inactivation phase of ERK5. We then checked for direct ubiquitination of MEKK2/3 by XIAP or cIAP1. Ubiquitination experiments revealed that XIAP and cIAP1 can directly ubiquitinate MEKK2 and MEKK3 in vitro (Fig3B, Supplementary Fig S3A–C). In addition, we have also detected autoubiquitination of the respective IAPs in these reactions (Supplementary Fig S3C). As the ubiquitin smears were detected in the absence of any proteasomal inhibitors (Fig3A), we suspected that XIAP might conjugate non-degradative ubiquitin chains on MEKK2 and MEKK3. Recent studies revealed that several kind of ubiquitin chains (K-63, K-11, M0, K27/29, and K6) are involved in signaling and in the assemblage of protein complexes (Fulda et al, 2012). By employing linkage-specific antibodies and ubiquitin mutants, we detected that XIAP and cIAP1 conjugate predominantly K63-linked ubiquitin chains to MEKK2 and MEKK3 both in vitro and in vivo (Fig3C–E and Supplementary Fig S3D–F). To confirm these observations, we employed K-63 ubiquitin-specific DUB AMSH [associated molecule with the Src homology 3 domain of signal transducing adaptor molecule (STAM)] (Huang et al, 2013). As expected, AMSH treatment led to deubiquitination of MEKK2 similar to Usp2, suggesting that the chains synthesized on MEKK2 by XIAP are predominantly K-63 linked chains (Supplementary Fig S3G). Consistently, MEF2 luciferase activity induced by MEKK2 is impaired by co-expression of XIAP (Supplementary Fig S3H). Subsequently, we examined the role of ubiquitination in regulating the activation dynamics of ERK5. To pursue these experiments, we employed MEFs derived from XIAP ΔRING knock-in mice or MEFs derived from XIAP knockout mice complemented with either wild-type or XIAP-H467A, a RING mutant of XIAP. Interestingly, the activation of ERK5 is sustained in XIAP ΔRING MEFs as loss of the RING domain of XIAP enhanced the basal as well as FGF-2-mediated activation of ERK5 (Fig3F). Similarly, stable expression of wild-type XIAP, but not XIAP-H467A, rescued FGF-induced ERK5 overactivation in the XIAP-deficient MEFs (Fig3G). Finally, we attempted mass spectrometry-based approach to identify the ubiquitination sites on MEKK2 and MEKK3 (Supplementary Fig S4). Interestingly, most of the ubiquitination sites identified in this screen were primarily localized in the kinase domains of MEKK2 or MEKK3. Mutation of these individual lysine residues failed to prevent direct ubiquitination of MEKK2 or MEKK3 by XIAP, suggesting that these sites are probably redundant (KR, TKOK unpublished observations). However, transfection of MEKK2-K450R mutant increased pERK5 levels at steady state, suggesting that ubiquitination at this site is possibly involved in the activation dynamics of ERK5 (Supplementary Fig S4C).
Figure 3. XIAP promotes K63-linked ubiquitin chain formation on MEKK2 and MEKK3 in a RING-dependent manner.

A XIAP mediates ubiquitination of MEKK2 after ERK5 activation in vivo. WT and XIAP−/− MEFs were stimulated with 25 ng/ml of FGF-2 for the indicated time points and MEKK2 was immunoprecipitated. Total lysates and IP fraction were analyzed by Western blotting.
B XIAP promotes MEKK2 ubiquitination in vitro. In vitro ubiquitination of purified MEKK2 by XIAP was performed and analyzed by Western blotting using MEKK2 antibody.
C K63-ubiquitination of MEKK2 is dependent of XIAP-E3 ligase activity. Flag-MEKK2 and HA-Ubiquitin were transfected in HEK293T cells with myc-EV, XIAP, or XIAP-H467A, an XIAP RING mutant. MEKK2 was immunoprecipitated using Flag antibody, and ubiquitination was checked using Western blot analysis with HA (ubiquitin) and K63-linkage-specific antibodies. *denotes overexpressed XIAP.
D, E XIAP promotes MEKK2 and MEKK3 K63-specific ubiquitination in vitro. MEKK2 (D) and MEKK3 (E) were subjected to in vitro ubiquitination, and MEKK2 or MEKK3, respectively, was immunoprecipitated from the samples. Western blot analysis was performed using K63-linkage-specific antibody.
F ERK5 phosphorylation is dependent on XIAP RING domain. WT and XIAP ΔRING knock-in MEFs were stimulated with 25 ng/ml of FGF-2 for indicated time points, and phosphorylation of ERK5 was detected by immunoblots.
G XIAP−/− MEFs were stably reconstituted with pEGZ-Flag EV, pEGZ-Flag-XIAP wt, or pEGZ-Flag-XIAP H467A were stimulated with 25 ng/ml of FGF-2 for the indicated time points. The activation of ERK5 was monitored by immunoblots.
Source data are available online for this figure.
Ubiquitination of MEKK2 and MEKK3 does not impair their kinase activity but impedes ERK5 activation
As IAPs directly ubiquitinate the kinase domain of MEKK2 and MEKK3, we then tested whether ubiquitination of MEKK2 or MEKK3 directly alters its kinase activity or its interaction with the downstream MAP2K, MEK5. We conducted in vitro ubiquitination-coupled phosphorylation assays to measure the activity of ubiquitin-conjugated or non-ubiquitinated MEKK2. Ubiquitination of MEKK2 did not impair the direct phosphorylation of MEK5 (Fig4A). Similar results were obtained in in vitro kinase assays employing 32P with MEKK3 using myeline basic protein (MyBP), a common kinase substrate (Fig4B). As XIAP-mediated ubiquitination fails to impair the kinase activity of MEKK2/3 directly, we performed in vitro reconstituted kinase assays with the entire MEKK2/3-MEK5-ERK5 kinase module components. Mutationally, inactivated ERK5, ERK5_KI D182A, was used in all kinase experiments. Consistent with the previous observations, we detected efficient ERK5 phosphorylation with non-ubiquitinated MEKK2 or MEKK3 but not with ubiquitinated kinases, suggesting that ubiquitination of MEKK2 or MEKK3 by IAPs has a direct influence on the activation of ERK5 in a trimeric complex (Fig4C). We employed concentrations of XIAP that will not compete with the interaction between MEKK2/3 and MEK5 in these experiments. Further, in the absence of E1 and E2 (or ubiquitination), ERK5 phosphorylation is not inhibited by the presence of XIAP (Supplementary Fig S5A). In addition, we detected that phosphorylation of ERK5 but not MEK5 is impaired in these coupled assays, suggesting that ubiquitination of MEKK2/3 has a direct effect on the activation of ERK5 (Fig4D). Further, XIAP autoubiquitination fails to prevent ERK5 phosphorylation in the presence of constitutively active MEK5 (MEK5DD) and thus XIAP requires ubiquitination of MEKK2/3 for impairing ERK5 phosphorylation (Fig4E). MEKK2/3 kinases also activate the JNK pathway by directly phosphorylating and activating MKK7. To confirm whether the observed effect of ubiquitination is specific for the ERK5 kinase module, we conducted similar experiments with purified MEKK2/3, MKK7, and JNK1. Interestingly, ubiquitination of MEKK2/3 by XIAP failed to prevent JNK1 phosphorylation in these coupled assays (Supplementary Fig S5B). These results revealed a direct, specific role for ubiquitinated MEKK2 and MEKK3 in negatively regulating the activation of ERK5 but not JNK1.
Figure 4. XIAP-mediated MEKK2/3 ubiquitination directly impedes ERK5 activation.
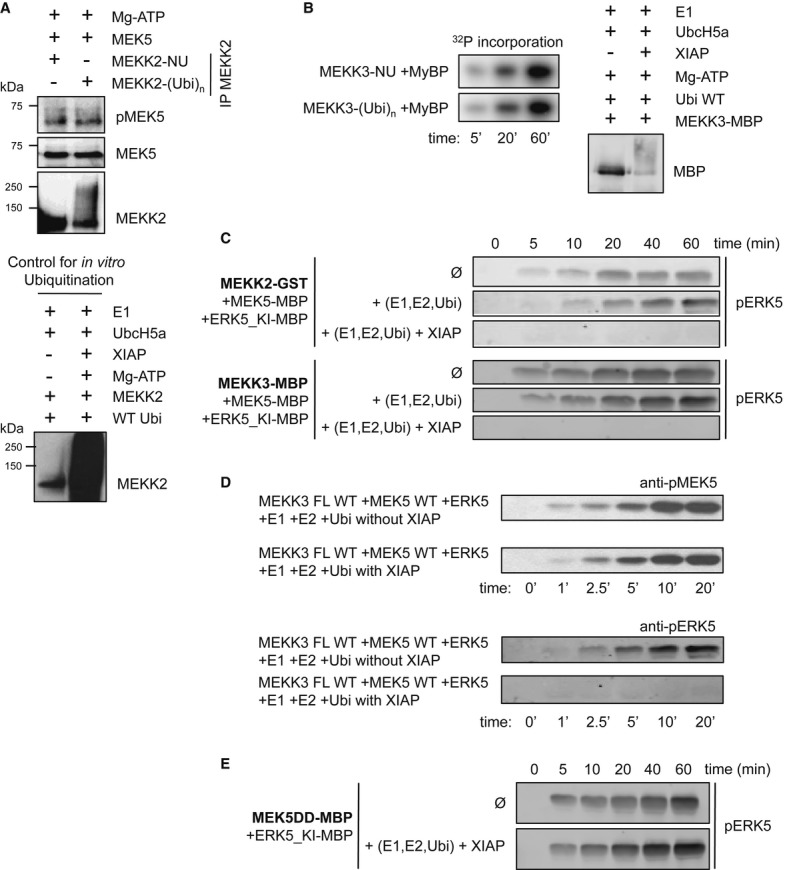
- MEKK2 activity on MEK5 is not affected by ubiquitination. In vitro ubiquitination of MEKK2 was performed (lower panel) and then was immunoprecipitated out from the ubiquitination mix. Beads with non-ubiquitinated (-NU) and ubiquitinated (-(Ubi)n) MEKK2 were employed for kinase assay with recombinant MEK5 as substrate (higher panel). Western blotting with phospho-specific MEK5 antibody was used as readout for the experiment.
- MEKK3 activity on a general kinase substrate (myelin basic protein, MyBP) is not affected by ubiquitination. In vitro ubiquitination of MEKK3-MBP was performed as described in Materials and Methods and checked by Western blot (right panel). Non-ubiquitinated (-NU) and ubiquitinated (-(Ubi)n) MEKK3 was incubated with 20 μM MyBP in the presence of ATP(γ-32P) and MyBP phosphorylation was monitored by phosphorimaging (left panel).
- XIAP-mediated ubiquitination interferes with ERK5 phosphorylation in the reconstituted MEKK2-MEK5-ERK5 MAPK module (higher panel). Similar results were found in MEKK3-MEK5-ERK5 reconstitution experiments (lower panel). ERK5 phosphorylation by MEK5 was monitored by Western blots (mutationally inactivated ERK5 was used in all experiments, ERK5_KI D182A). MEKK2 and MEKK3 were ubiquitinated as described for (B) and then an in vitro kinase assay was started by adding additional 0.5 mM ATP and recombinantly expressed and purified MEK5 and ERK5 (1 μM and 5 μM, respectively).
- XIAP-mediated ubiquitination interferes with ERK5 phosphorylation in the reconstituted MEKK3-MEK5-ERK5 MAPK module without any effect on MEK5 activation. MEK5 and ERK5 phosphorylation were monitored by Western blots (mutationally inactivated ERK5 was used in all kinase assays, ERK5_KI D182A). MEKK3 was ubiquitinated and then an in vitro kinase assay was performed as described for (C).
- Ubiquitination does not interfere with MEK5 mediated phosphorylation of ERK5, as the constitutively activated form of MEK5 (MEK5DD) activates ERK5 independent of the presence of XIAP (in all assays, the following concentrations were employed: MEKK2/3: 0.2 μM, MKK5: 1 μM, ERK5: 5 μM).
Source data are available online for this figure.
Ubiquitination promotes MEKK2/3 dimerization but directly impairs MEK5–ERK5 interaction
As ubiquitination is not influencing the kinase activity of MEKK2 and MEKK3, we checked for the complex formation between the members of this kinase cascade. MEKK2 and MEKK3, like many kinases, are activated by dimerization via their catalytic domain, and they form weak homodimers. Consistent with the kinase activity of ubiquitin-conjugated MEKK2, ubiquitination directly promoted homo- as well as heterodimerization of MEKK2 with MEKK3 (Fig5A and B). We then checked whether ubiquitination impairs the interaction between MEK5 and MEKK2/3. Ubiquitination of MEKK2 or MEKK3 by XIAP did not impair the interaction with MEK5 (Fig5C and D). The PB1 domain of MEK5 coordinates the interaction between the PB1 domain of MEKK2/3 and the N-terminus of ERK5 to form a kinase competent complex (Fig5E) (Glatz et al, 2013). While the PB1-mediated interaction between MEKK2/3 and MEK5 is mediated via electrostatic interactions, ERK5 interaction with MEK5 is non-canonical as ERK5 lacks a PB1 domain (Nithianandarajah-Jones et al, 2012). We envisaged that ubiquitination of MEKK2/3 might compete with MEK5–ERK5 interaction as PB1 domain also presents an ubiquitin-like β-grasp fold (Sumimoto et al, 2007). We then repeated the assays with all three kinases and checked for the formation of the entire kinase module. As expected, ubiquitinated MEKK2/3 prevented the direct interaction between MEK5 and ERK5 in a trimeric complex (Fig5F and G and Supplementary Fig S6A). To confirm these observations in cells, we have attempted to immunoprecipitate the entire kinase module from cells stably expressing ERK5 and MEK5. Interestingly, increasing amounts of MEK5 were co-precipitating with ERK5 in the absence of XIAP (Supplementary Fig S6B). Taken together, these data suggest that ubiquitin chains conjugated to MEKK2/3 directly prevented the non-canonical interaction between MEK5 and ERK5, thus inactivating ERK5-MAPK module (Fig5G). This effect is specific for ERK5 module as ubiquitination of MEKK2/3 kinases failed to prevent MKK7 or JNK1 activation under identical settings (Supplementary Fig S5B).
Figure 5. Functional significance of MEKK2/3 ubiquitination.

- Ubiquitination of MEKK2 promotes its homodimerization. Purified GST-tagged MEKK2 was subjected to in vitro ubiquitination. In vitro transcribed and translated MEKK2 was added to the non-ubiquitinated (-NU) and ubiquitinated (-(Ubi)n) MEKK2. In vitro ubiquitination mix and GST pull down were performed as stated in Materials and Methods. Homodimerization of MEKK2 was analyzed by Western blotting.
- Ubiquitination of MEKK2 promotes its heterodimerization with MEKK3. Purified MEKK2 protein was subjected to in vitro ubiquitination. Purified MEKK3 was added to the non-ubiquitinated (-NU) and ubiquitinated (-(Ubi)n) MEKK2 samples, and MEKK2 was pulled down. Co-precipitated MEKK3 was detected using Western blotting.
- Ubiquitination of MEKK2 does not impair MEK5 binding. In vitro ubiquitination of MEKK2 by XIAP was performed as stated above. His-tagged ubiquitin was pulled down from non-ubiquitinated (-NU) and ubiquitinated (-(Ubi)n) MEKK2 samples using Ni-NTA beads as detailed in Materials and Methods section. Co-precipitated MEK5 was analyzed using Western blotting.
- Ubiquitination of MEKK3 does not impair MEK5 binding. Ubiquitination of MBP-MEKK3 was carried out after the protein was bound to amylose resin and non-ubiquitinated (-NU), and ubiquitinated (-(Ubi)n) MBP-MEKK3 was used for pulling down GST-MEK5. Samples were subjected to SDS–PAGE and stained with Coomassie.
- MEKK2/3 and MEK5 binding is mediated through their PB1 domains in the MEKK2/3-MEK5-ERK5 ternary complex. The structural model of the ternary complex is shown in surface representation (Glatz et al, 2013).
- The trimeric complex formation between MEKK2-MEK5-ERK5 was analyzed using non-ubiquitinated and ubiquitinated MEKK2. GST-tagged MEK5 was used for immunoprecipitating GST-MEKK2 and GST-ERK5 using rabbit monoclonal antibody against MEK5. Difference between binding of ERK5 to MEK5 in the presence of ubiquitinated and non-ubiquitinated MEKK2 was analyzed by Western blotting.
- Schematic representation of the MEKK2/3-MEK5-ERK5 module. Ubiquitin conjugated to MEKK2/3 competes with the non-canonical interaction between the MEK5 PB1 domain and ERK5. The XIAP–cIAP1 complex promotes MEKK2/3 K63-ubiquitination, which, in turn, interferes with the recruitment of ERK5 to the MEKK2/3-MEK5 binary complex. (Kinases are depicted schematically with their classical N- and C-terminal bipartite lobe structure where “P” indicates activation loop phosphorylation sites, * marks the position of the kinase active site, and the rectangle at the back of the ERK5 kinase domain shows the MEK5-docking motif binding to the MAPK-docking groove (Glatz et al, 2013).
Source data are available online for this figure.
Loss of XIAP promotes MEF2 activity and human myoblast differentiation
As XIAP negatively regulates ERK5 activation by direct interaction and ubiquitination, we investigated the physiological significance of these observations. ERK5 regulates neuronal and muscle differentiation through the activation of MEF2 transcription factors (Dinev et al, 2001; Hindi et al, 2013). Consequently, we explored the possible role of XIAP in the regulation of human primary skeletal myoblasts (HSM) differentiation. Human primary skeletal myoblasts differentiate to myocytes in a span of 6–7 days as evidenced by the formation of polynucleated myotubes and expression of differentiation markers like Myosin Heavy Chain (MHC), MEF2C, MyoD, and p21. We initially checked whether depletion of XIAP modulates ERK5 activation in HSM cells. As expected, depletion of XIAP led to an increase in pERK5 signals in the immunoblots and MEF2 activity in HSM cells (Fig6A and B). Consistently, loss of XIAP strongly promoted (˜10- to 12-fold increase) the differentiation of myoblasts as evidenced by the presence of polynucleated muscle fibers with a strong staining of Myosin Heavy Chain (Fig6C and D). Immunoblot analysis revealed that loss of XIAP led to a consistent increase in pERK5 levels with a concomitant increase in the appearance of various differentiation markers including MHC, p21, Myogenin, and MEF2C (Fig6E and F). As cIAP1 loss also leads to ERK5 activation in tumor cells, we initially tested for the role of cIAP1 in regulating human myoblast differentiation. However, single depletion of cIAP1 failed to enhance myocyte formation possibly due to cross-stabilization of XIAP in these cells (ANT, KR unpublished observations). These results confirmed that loss of XIAP promotes ERK5 activation and myogenesis in human cells.
Figure 6. Depletion of XIAP enhances myoblasts differentiation.

A Depletion of XIAP leads to an increase in ERK5 phosphorylation in undifferentiated myoblasts. Undifferentiated HSM were transiently transfected with siControl or siXIAP.
B Depletion of XIAP leads to an increase in ERK5 activation in undifferentiated myoblasts. Undifferentiated HSM cells stably expressing the MEF2-luciferase (Luc) reporter gene were transiently transfected with siControl or siXIAP. Part of the lysates was used for Western blot detection and another part to measure the luciferase activity (shown is the fold increase of MEF2 activity upon XIAP knockdown).
C, D Depletion of XIAP leads to an increase of myoblasts differentiation. HSM were transiently transfected with siControl or siXIAP, and differentiation was started the next day. Transfection was performed every 2 days, and media were changed every following day. Coverslips were fixed and stained everyday as described in methods (shown in (C) are data from a representative experiment from day 0, day 4, and day 7). Polynucleation of MHC positive cells was quantified from four independent experiments, and the average fold increase is shown in (D).
E, F Depletion of XIAP leads to an increase in ERK5 phosphorylation and expression of differentiation markers in myoblasts. HSM were transfected and differentiated as described for (C). Cells were lysed, run on SDS–PAGE, and blotted for various differentiation markers (E). Increase in ERK5 phosphorylation was quantified from three independent experiments, and the average fold increase is shown in (F) (Student's t-test, *P < 0.05, **P < 0.01).
Source data are available online for this figure.
XIAP regulates human myogenic differentiation in MEKK2/3-ERK5-dependent manner
We then investigated whether the enhanced differentiation observed in XIAP-depleted HSM cells was indeed due to the hyperactivation of ERK5 signaling. To address this issue, we performed several co-knockdown experiments. Interestingly, knockdown of ERK5, MEKK2, and MEKK3 completely prevented basal as well as siXIAP-mediated differentiation of myoblasts (Fig7A–G and Supplementary Fig S7A and B). To corroborate these observations, we employed ERK5-specific kinase inhibitor XMD 8–92. Consistent with the observations made with ERK5 siRNAs, treatment of HSM cells with XMD 8–92 prevented both normal and XIAP-depletion-induced differentiation as evidenced by the lack of polynucleation or expression of differentiation markers (Fig7C and D and Supplementary Fig S7C). These results confirmed the physiological role of XIAP–ERK5 interface in human myogenic differentiation.
Figure 7. Depletion of MEKK2, MEKK3 or ERK5 inhibits the enhanced differentiation triggered by XIAP depletion.

A HSM cells were transiently co-transfected with siRNAs against XIAP and MEKK2 or ERK5, and differentiation was initiated with the appropriate media the next day. Coverslips were fixed and stained everyday as described (Shown are data from day 6 of differentiation).
B The cells were lysed and the expression of various differentiation markers was tested.
C Pharmacological inhibition of ERK5 prevents the enhanced differentiation provoked by XIAP depletion. HSM were transiently transfected with siXIAP and treated with DMSO or 5 μM of XMD 8–92, and differentiation was started the day after. Transfection was performed every 2 days, and media and drug treatment were renewed every following day. The polynucleation index was calculated as mentioned in (A).
D Experiments were performed as mentioned in (C) and cell lysates were prepared. The expression of various differentiation markers was tested by Western blots.
E HSMs were transiently co-transfected with siRNAs against XIAP and MEKK3 and differentiated as described for (A). Polynucleation of MHC positive cells at day 6 was quantified, and the average fold increase is shown in the right panel (n = 3, Student's t-test, *P < 0.05 and ***P < 0.001).
F, G HSMs were transfected with different siRNAs and differentiated as described in (A). Cells were lysed, run on SDS–PAGE, and blotted for various differentiation markers.
H Schematic model of MEKK2 ubiquitination by XIAP and its effect on the MEKK2/3-MEK5-ERK5 pathway in response to various stimuli. XIAP regulates this cascade by direct interaction and ubiquitination. At steady state, XIAP competes with MEK5 in binding to MEKK2. Upon stimulation with growth factors, XIAP–cIAP1 complex ubiquitinates MEKK2/3, thus leading to the inactivation of ERK5. Loss of XIAP promotes human myogenic differentiation in a MEKK2/3-ERK5-dependent manner. Depending on the stimuli, MEKK2/3 might bind and activate other effectors like MKK7 or p62, thus leading to JNK or NF-κB activation respectively.
Source data are available online for this figure.
Discussion
MAPKs are activated in response to various stimuli to perform fundamental cellular processes (Raman et al, 2007). The signaling fidelity and efficiency of MAPK modules are controlled by scaffolding proteins and docking interactions occurring in a spatiotemporal manner (Tanoue & Nishida, 2002). Dephosphorylation by phosphatases is one of the major means of inactivating MAPKs (Raman et al, 2007). The cross-talk between phosphorylation and ubiquitination is multilayered, and there is a lot of evidence for the role of ubiquitination in the regulation of kinase activity (Hunter, 2007). To the best of our knowledge, this is the first evidence where non-degradative ubiquitination (predominantly K63-linked ubiquitin) is involved in the physical and functional disassembly of a “classical” MAPK module, thus adding another layer in the inactivation of MAPKs. ERK5 is a relatively less studied MAPK despite its importance in vertebrate development and in pathogenic conditions such as cardiac hypertrophy and cancer (Wang & Tournier, 2006). The MEKK2/3-MEK5-ERK5 cascade is activated by numerous stimuli and assembled by distinct PB1 domain-mediated protein–protein interactions, though a role for scaffold proteins such as Lad has been suggested (Sun et al, 2001; Drew et al, 2012). This study reveals a novel role for IAP-mediated ubiquitination events in directly uncoupling a MAPK from its cognate MAPKK.
XIAP and cIAP1 are highly conserved RING domain-containing E3 ubiquitin ligases, and they can catalyze the conjugation of ubiquitin chains of various kinds to their growing list of substrates. We have previously shown that XIAP can regulate Rac1 protein stability (Oberoi et al, 2011). While we fail to detect a significant role for Rac1 in ERK5 activation (Supplementary Fig S1F and G), an upstream, cell type-specific role in activating MEKK2/3 cannot be ruled out as chemical inhibition of Rac1 has been shown to reduce MEKK3 activation in T cells (Wang et al, 2011). XIAP and cIAP1 negatively regulate ERK5 activation, and as both IAPs are required, XIAP–cIAP1 complex is possibly functional in vivo in regulating the ubiquitination of MEKK2/3.
In the same lines, both MAP3Ks are functionally non-redundant as depletion of either MEKK2 or MEKK3 led to an identical reduction in pERK5 levels and inhibition of myocyte formation. These data suggest that MEKK2 and MEKK3 might function as heterodimers (like B-RAF and C-RAF kinases) and that this interaction could be stabilized by ubiquitination. In fact, ubiquitination of MEKK2 by XIAP promotes homodimerization and heteromerization with MEKK3 (Fig5A and B). IAPs directly conjugate ubiquitin predominantly to the kinase domains of MEKK2 and MEKK3, and ubiquitination at Lys450 in MEKK2 contributes to the dynamics of ERK5 activation (Supplementary Fig S4). Mutation of two or more lysine sites severely compromises the stability and expression of MEKK2, thus limiting further studies. We have however characterized the kind of ubiquitin chains and their functional significance. Our mass spectrometry-based screen detects traces of K-11, K27/29, and K-48 chains apart from K-63 linked chains. However, by employing ubiquitin mutants, linkage-specific antibodies and DUBs, we confirm that they are predominantly K-63 linked chains. Further quantification of other kind of chains and the ubiquitination sites is clearly warranted.
As expected, K63-linked ubiquitination of MEKK2/3 did not impair their kinase activity as they still can directly phosphorylate MEK5 or MKK7. On the other hand, in vitro reconstitution experiments revealed that ubiquitination of MEKK2 by XIAP decreases the MEK5–ERK5 interaction. At stoichiometric concentrations in vitro, ubiquitination of MEKK2/3 directly and completely inhibits the activation of ERK5 but not JNK1 (Fig4 and Supplementary Fig S5). Ubiquitination of MEKK2 in vivo directly correlates to ERK5 dephosphorylation in cells in a XIAP-dependent manner (Fig3A), and consistently, loss of RING activity or the domain itself led to an increased ERK5 activation (Fig3F and G), confirming the role of ubiquitination in this process. Ubiquitination of MEKK2 in vivo “at endogenous levels” seems to be partial (Fig3A), but this could be due to either (i) the sensitivity of the antibody toward ubiquitinated MEKK2 in cells or (ii) that only a part of MEKK2 which is in complex with MEK5-ERK5 gets ubiquitinated. Further, interaction between the members of this kinase module can also be regulated by compartmentalization within the cells. Thus, in vitro reconstitution at stoichiometric concentrations is much needed to confirm the direct role of ubiquitin in the disassembly of this kinase module (Fig5G). As PB1 domain and ubiquitin are structurally similar (Sumimoto et al, 2007), it is quite likely that K63-ubiquitin competes directly with the interaction between MEK5 and ERK5 but not MKK7 and JNK1. In these lines, Seyfried et al (2005) have previously shown that MEKK2/3 and ERK5 can compete for binding to MEK5 PB1 domain. However, recent structure-based modeling studies confirmed that MEK5 PB1 domain can indeed function as an adaptor as it involves two distinct surfaces for binding to MEKK2/3 and ERK5 (Glatz et al, 2013). Our data favor an ubiquitin-PB1 competition model, where K63-ubiquitin chains linked to MEKK2/3 prevent ERK5 from binding to MEK5, thereby preventing ERK5 activation (Figs5G and 7H).
Further, XIAP binds to the PB1 domain of MEKK2 as mutation of a conserved lysine in this domain prevents this interaction (Fig2C) and loss of XIAP enhances MEK5–MEKK2 interaction in vivo (Fig2F and Supplementary Fig S2C). We detect XIAP-dependent ubiquitination of MEKK2 during the inactivation phase of ERK5 (Fig3A) and thereof hypothesize that XIAP regulates ERK5 activation in a two-step process. While XIAP limits MEKK2–MEK5 interaction at steady state levels, stimulation of cells with FCS or growth factor triggers the ubiquitin ligase activity of XIAP–cIAP1 complex that leads to the conjugation of K63 polyubiquitin to MEKK2/3. This excludes ERK5 from interacting with MEK5, in turn leading to ERK5 inactivation (Fig7H). Thus, loss of XIAP led to increased ERK5 phosphorylation both at steady state and at stimulated conditions. It is currently unclear whether the kinase activity of MEKK2 or MEKK3 is required for interaction with XIAP. Currently, we lack sensitive tools to detect the dynamics of interaction between MEKK2/3 and XIAP in these cells at endogenous levels. Further, the upstream regulators of MEKK2/3 are largely unknown though WNK1 has been suggested to function as an upstream kinase (Xu et al, 2004).
Apart from the negatively regulatory role in ERK5 activation, what could be the significance of IAP-mediated MEKK2/3 ubiquitination? Previous studies have shown that XIAP can also regulate the second wave of TNF-α-dependent NF-κB activation by binding to MEKK2 (Winsauer et al, 2008). Further, MEKK2 binds to MEK5 at steady state levels and to MKK7 upon sorbitol treatment to mediate JNK activation (Nakamura & Johnson, 2007). Ubiquitination of MEKK2/3 by IAPs did not inactivate these kinases. This suggests that the ubiquitinated MEKK2/3 dimer might bind to other effectors like MKK7. In addition, we uncovered that MEKK2 is a ubiquitin binding protein (KR, TKOK unpublished observations). Thus, XIAP-mediated ubiquitination of MEKK2 might result in different outcomes depending on the external stimuli, that is TNF-α triggering late-phase of NF-κB activation, sorbitol stimulating JNK, and growth factors leading to ERK5 inactivation and regulation of cellular differentiation (Fig7H). In this context, it is interesting to point out that p62 can bind to MEKK3 to mediate NF-κB activation (Nakamura et al, 2010). However, XIAP was reported not to control the early phase of NF-κB activation which is mediated by MEKK3 (Winsauer et al, 2008). Further, ubiquitination of MEKK2/3 failed to prevent the activation of JNK1 directly unlike ERK5 (Supplementary Fig S5).
Consistent with the negative regulatory role of XIAP in ERK5 activation, we uncover that depletion of XIAP augments activation of ERK5 and MEF2, thus promoting the formation of human myotubes. Further, loss of MEKK2, MEKK3, and ERK5 led to a complete block in myogenic differentiation in control and XIAP-depleted cells. These data reveal the physiological significance of XIAP–ERK5–cascade interactions in regulating human myogenic differentiation.
While the role of IAPs in regulation of apoptosis, inflammation, and immune signaling is well established, their role in regulation of cell migration and differentiation is just emerging (Kenneth & Duckett, 2012). cIAP1 has been previously shown to regulate monocyte to macrophage differentiation and NAIP, to regulate neuronal differentiation. TWEAK and cIAP1 have been shown to regulate myoblast fusion through non-canonical pathway of NF-κB (Enwere et al, 2012). While cIAP1 functions as a direct E3 ligase of NIK, loss of XIAP does not lead to constitutive NF-κB activation, negating the possible involvement of this pathway in XIAP-regulated human myogenic differentiation. In this context, XIAP antagonists might be valuable in treating muscular degenerative diseases and for promoting muscle regeneration. Taken together, our data reveal a novel role of IAPs and IAP-mediated non-degradative ubiquitination in the regulation of ERK5-MAPK module and myogenic differentiation (Fig7H). It would be indeed interesting to investigate whether the assemblage and the activation dynamics of other MAPK modules are also directly influenced by non-degradative ubiquitination.
Materials and Methods
Cell culture
HeLa and A549 cells were cultured in RPMI-1640 medium (Gibco BRL) supplemented with 10% FCS (Gibco BRL) and 0.2% penicillin (100 U/ml)/streptomycin (100 μg/ml) (Gibco BRL) at 37°C in 5% CO2. HEK293T, A431, and BT474 were cultured in DMEM medium supplemented with 10% FCS (Gibco BRL) and 0.2% penicillin (100 U/ml)/streptomycin (100 μg/ml) (Gibco BRL) at 37°C in 5% CO2. HTEpC were cultured in Airway Epithelial Cell Growth Medium from Promocell (C-21060). MEFs wt, MEFs XIAP−/−, and MEFs XIAP-ΔRING were obtained from John Silke and Hermann Steller, and were cultured in DMEM medium supplemented with 10% FCS (Gibco BRL), 0.2% penicillin (100 U/ml)/streptomycin (100 μg/ml) (Gibco BRL), 1 mM non-essential amino acids, 1 mM sodium pyruvate, and 1 mM β-mercaptoethanol at 37°C in 5% CO2. SKB are Human Skeletal Muscle Myoblast obtained from ZenBio (Cat #: SKB-F (Lot 1 #: SK052009, Lot 2 #: SK051810)). SKB were cultured in the Skeletal Muscle Cell Growth Medium (Cat #: SKM-M, ZenBio) and were differentiated with the Skeletal Muscle Cell Differentiation Medium (Cat #: SKM-D, ZenBio) at 37°C in 5% CO2.
In order to obtain the HeLa MEKK2-wt, K450R, and ERK5/MEK5 stable cell lines, we first transfected HEK293T with pHAGE-CMV-N-Flag-HA-GAW-IRES-Puro-MEKK2-wt or pHAGE-CMV-N-Flag-HA-GAW-IRES-Puro-MEKK2-K450R or pHAGE-CMV-N-Flag-HA-GAW-IRES-PURO-ERK5, and pLenti4TO/V5-DEST-MEK5 together with the pLenti package (HDM-VSV-G; HDM-tatlb; HDM-Hgprn2 (gag-pol); RC-CMV-Rev1b). The media containing the virus were sterile filtrated and then added to Hela cells in presence of 8 μg/μl of polybrene. After 24 h, cells were selected with 2.5 μg/ml puromycin or in the case of ERK5/MEK5 overexpression with 2.5 μg/ml puromycin and 100 μg/ml Zeocin. pHAGE plasmid was a kind gift from Dr. Behrends.
Wherever appropriate, cells were stimulated with FGF-2 (human FGFb147 from eBioscience, Cat. No. 14-8986-80) in presence of serum for the indicated times at a final concentration of 25 ng/ml. In case of FCS stimulation, cells were starved for 15 h in FCS-free medium and then stimulated with media containing 10% FCS.
Transfection of siRNAs
In order to silence XIAP, cIAP1, cIAP2, MEKK2, MEKK3, MEK5, ERK5, and C-RAF expression by siRNA interference, approximately 75,000 cells/well were seeded in a 12-well plate at least 20 h prior to transfection. siRNAs directed against various genes and scrambled control siRNA as negative control were transfected at a final concentration of 60 nM using Lipofectamine™ RNAiMAX (Invitrogen, Cat. No. 13778) transfection reagent. For complementation experiments, siRNA and plasmid were co-transfected using HiPerFect (QIAGEN, Cat. No. 301705) transfection reagent. Unless otherwise mentioned, cells were lysed at 48 h post-transfection. The following siRNAs and shRNAs were employed in this study (sense strand sequence):
siControl: 5′-UUCUCCGAACGUGUCACGU-3′ (QIAGEN, Cat. No. 1027310)
siXIAP #1 (3′utr): 5′-GACUGAUCUAAUUGUAUUATT-3′ (QIAGEN)
siXIAP #2: 5′-AAGTGCTTTCACTGTGGAGGA-3′ (QIAGEN, Hs_BIRC4_5_HP Validated siRNA, Cat. No. SI00299446)
siXIAP #3: 5′-ACACUGGCACGAGCAGGGUUUCUUU-3′ (Invitrogen Stealth, XIAPHSS100564)
siXIAP #4: 5′-GAAGGAGAUACCGUGCGGUGCUUUA-3′ (Invitrogen Stealth, XIAPHSS100565)
sicIAP1 #1: 5′-GAAUGAAAGGCCAAGAGUU-3′ (Thermo Scientific, ON-TARGET plus siRNA human BIRC2 (329), Cat No. J-004390-13-0020)
sicIAP1 #2: 5′-CAGAAAGCUUUGAAUACUATT-3′ (QIAGEN, Hs_BIRC2_12, Cat No. SI05067258)
sicIAP1 #3: 5′-CAUAGUAGCUUGUUCAGUGTT-3′ (QIAGEN, Hs_BIRC2_7, Cat No. SI02654435)
sicIAP2 #1: 5′-UUGGGAACCGAAGGAUAAUTT-3′ (QIAGEN, Hs_BIRC3_5_HP, Cat No. SI00299439)
sicIAP2 #2: 5′-GCUGCAGAUUCGUUCAGAGUCUAAA-3′ (Invitrogen Stealth, BIRC3HSS100561)
siMEKK2: 5′-GCUACCCAGAUAAUCAUCAGGAAUU-3′ (Invitrogen Stealth, MAP3K2HSS116574)
siMEKK3: 5′-CUGACAACAGACAGGAAUA-3′ (SIGMA-ALDRICH, SASI_Hs01_00047591)
siMEK5: 5′-AGGCCAGCACCUGAAGAAUTT-3′ (QIAGEN, Hs_MAP2K5_10_HP validated siRNA, Cat. No. SI00300713)
siERK5: 5′-GACCCACCUUUCAGCCUUATT-3′ (QIAGEN, Hs_MAPK7_9 validated siRNA, Cat. No. SI00606039)
siC-RAF (3′utr): GTGGATGTTGATGGTAGTACA (QIAGEN, target sequence)
siRac1: 5′-AUGAAAGUGUCACGGGUAA-3′ (Thermo Scientific, siGENOME siRNA human Rac1 (5879), D-003560-30-0020)
shC-RAF: 5′-CCGGCAAGCAAAGAACAGTGGTCAACTCGAGTTGACCACTG TTCTTTGCTTGTTTTT-3′ (TRCN0000001067, Sigma-Aldrich)
shRac1: 5′-CCGGCCCTACTGTCTTTGACAATTACTCGAGTAATTGTCAAAGA CAGTAGGGTTTTT (TRCN0000004872, Sigma-Aldrich)
Plasmids and constructs
The plasmids employed for the expression of indicated proteins are listed here:
pGEX6p1-GST-XIAP wt, pGEX6p1-GST-XIAP-BIR1, pGEX6p1-GST-XIAP-BIR2, pGEX6p1-XIAP-BIR3, pGEX6p1-GST-XIAP-BIR1+2+3, pGEX6p1-GST-XIAP-ΔRING, pGEX6p1-GST-AMSH, pcDNA3.1B-myc/his-XIAPwt, pcDNA3.1B-myc/his-MEKK2-wt, pcDNA3-flag-MEKK2-wt, pLenti4TO/V5-DEST-MEKK2-wt, pHAGE-CMV-N-Flag-HA-GAW-IRES-Puro-MEKK2-wt, pcDNA3.1B-myc/his-XIAP wt, pEGZ-flag-XIAP wt, pRK5-HA-Ubiquitin wt.
The various point mutations in MEKK2 (K47A, K384M, K360R, K450R), XIAP (H467A), and cIAP1 (H588A) were generated with a Site-Directed Mutagenesis Kit (Stratagene) following manufacturer's instructions.
SDS–PAGE and Western blot
For SDS–PAGE, cells were lysed in 5× Laemmli buffer and boiled at 100°C for 5 min before loading onto polyacrylamide gels. The proteins were then transferred to nitrocellulose membranes by Western blotting. After transfer, membranes were stained with Ponceau (AppliChem). For immunoblot analysis, membranes were blocked with 5% low-fat milk in phosphate-buffered saline for 1 h at room temperature and then incubated with various primary antibodies overnight at 4°C. For K63 linkage-specific antibody, the nitrocellulose membrane was incubated for 2 h at room temperature in the primary antibody and then proceeded as stated above. Antigen–antibody complexes were detected by horseradish peroxidase-coupled secondary antibodies followed by enhanced chemiluminescence (Amersham Biosciences, Millipore, Thermo Scientific). Quantification of Western blots was performed by densitometry (ImageJ software, NIH). The following antibodies have been employed in this study:
Phospho-ERK5 (Thr218/Tyr220; Cat. No. 3371) and total-ERK1/2 (p44/42 MAPKinase; Cat. No. 9102) rabbit polyclonal from Cell Signaling; total-ERK5 (C-term, Cat. No. 1719-1), MEKK2 (Cat. No. 1662-1), MEKK3 (N-term, Cat. No. 1672-1) rabbit monoclonal and MEF2c (T300, Cat. No. T1324), MyoD (Cat. No. S0549) rabbit polyclonal antibodies from Epitomics; XIAP (Cat. No. 610763), total-MEK5 (Cat. No. 610956) and Rac1 (Cat. No. 610650) mouse monoclonal antibodies from BD Biosciences; Actin rabbit polyclonal (Cat. No. A2066) and Tubulin (Cat. No. T9026), Flag (M2, Cat. No. F3165), GAPDH (clone GAPDH-71.1, Cat. No. G8795) mouse monoclonal antibodies from Sigma-Aldrich; cIAP1 (1E1-1-10, Cat. No. ALX-803-335) and cIAP2 (16E-6-3, Cat. No. ALX-803-341) rat antibody from ENZO; normal mouse IgG (Cat. No. sc-3877), normal rabbit IgG (Cat. No. sc-3888), phospho-MEK5 (Ser311/Thr315; Cat. No. sc-135702), C-RAF (Raf-1, C-12, Cat. No. sc-133), p21 (C-19, Cat. No. sc-397) rabbit polyclonal, GST (B-14, Cat. No. sc-138) and HA (12CA5; Cat. No. sc-57592) mouse monoclonal antibodies from Santa Cruz Biotechnology; Ubiquitin mouse monoclonal antibody from Invitrogen (Cat. No. 13-1600); Goat anti-human IgG-HRP from Invitrogen (Cat. No. AHI0704); MHC (MF20) mouse monoclonal antibody from DSHB; Myogenin mouse monoclonal antibody from Millipore (Cat. No. MAB3876); Cy™3-conjugated AffiniPure Goat Anti-Mouse IgG (Jackson ImmunoResearch, Code No. 115-165-146). The antibody against K63-linked polyubiquitin was a kind gift from Genentech (San Fransisco, CA, USA).
Luciferase assay
HeLa cells were transduced with the Cignal Lenti Pathway Reporter lentiviral particles using Cignal Lenti MEF2 Reporter (luc) Kit (Qiagen, Cat. No. CLS-4024L). Following transduction, the cells were cultured under puromycin selection to generate a homogenous population of transduced cells. Briefly, transduced and selected HeLa cells were seeded on a 12-well plate and transfected with XIAP, ERK5, and/or MEKK2 siRNAs using Lipofectamine RNAimax.
HEK293T cells were transiently transfected with the Cignal MEF2 Reporter (luc) Kit (QIAGEN, Cat. No. CCS-7024L). Cells were transfected using GeneJuice (MERCK Millipore, Cat. No. 70967) with 500 ng of each of the following plasmids: the luciferase reporter gene, MEKK2-myc/his, and XIAP-flag. As negative and positive controls, we used the control plasmids provided by the Kit.
In both case, the cells were harvested in Passive Lysis Buffer 48 h after transfection. The MEF2 luciferase assay was performed using the Luciferase® Reporter Assay System (Promega, Mannheim, Germany) in accordance with manufacturer's protocol.
Ubiquitination experiments
For detecting the ubiquitination of MEKK2 in vivo, 293T cells were transfected with flag-MEKK2 in combination with HA-Ubiquitin wt, with or without myc/his-XIAP and XIAP-H467A, using GeneJuice® transfection reagent (Novagen®, Merck Millipore, Cat. No. 70967) at a final concentration of 1 μg/ml for 48 h. The cells were lysed and immunoprecipitated with Flag antibody for 15 h. The bound proteins were analyzed by SDS–PAGE and immunoblotting.
In vitro ubiquitylation of MEKK2 or MEKK3 was performed in the presence of ubiquitylation buffer (50 mM Tris–HCl, pH 7.5, 100 mM NaCl, 2.5 mM MgCl2, 1 mM DTT), 100 nM XIAP or 100 nM c-IAP1 (R&D Systems), 100 nM E1 (Boston Biochem), 150 μM UbcH5a (Boston Biochem), 107 μM His-Ubiquitin (Boston Biochem), 50 mM EDTA, 1× Mg-ATP (Enzo Life Sciences), 1 U inorganic pyrophosphatase (Fluka), and 50 mM DTT. Wherever specified, in vitro ubiquitination was performed using K48-only, K63-only, or K29-only ubiquitin (Boston Biochem). The reaction was incubated at 37°C for 1 h. For Western blot analyses, the reaction was stopped using Laemmli buffer and MEKK2 was visualized using MEKK2 rabbit monoclonal antibody (Epitomics).
For GST and His-ubi pull down after in vitro ubiquitination, 300 μl of GST pull-down buffer (GPB) was added to the in vitro ubiquitination reaction mix with 50 μl of either equilibrated glutathione sepharose or Ni-NTA beads. We then proceeded for GST pull down. For immunoprecipitation after in vitro ubiquitination, 300 μl of lysis buffer (constituents described above) was added to the in vitro ubiquitination reaction mix with 2 μg of MEKK2 antibody or MEKK3 antibody and immunoprecipitated. For the deubiquitinase experiments, we treated in vitro ubiquitinated MEKK2-GST (SignalChem) with 5 μM Amsh (K63 linkage specific) or 5 μM Usp2 (linkage unspecific) for 60 min at 30°C. The incubation was terminated by denaturation in Laemmli Buffer.
Kinase assay
For kinase assay, in vitro ubiquination of MEKK2 was performed as described above, with or without the addition of E3 ligase, XIAP. MEKK2 is immunoprecipitated from these samples using MEKK2 antibody and sepharose-coupled protein A/G beads according to procedure described above. Beads were washed three times with lysis buffer and then the lysis buffer was removed using insulin syringe. To the beads, added 4 μl of 10× kinase buffer (100 mM MgCl2, 250 mM β-glycerophosphate, 250 mM HEPES pH 7.5, 50 mM benzamidine, 5 mM DTT, 10 mM sodiumorthovanadate), 2 μl of 20× Mg-ATP (Enzo Lifesciences), 400 ng of GST-MEK5 (Abcam), and distilled water up to 40 μl of reaction mix. The reaction was incubated at 30°C for 30 min and then stopped by adding 8 μl of 5× Laemmli. The entire reaction mix was loaded on the SDS–PAGE gel for immunoblot analysis.
Proteins for the MEKK2/3-MEK5-ERK5 and MEKK2/3-MKK7-JNK1 MAPK module reconstitution experiments were produced in E. coli or in SF9 insect cells (with the baculoviral expression system) as N-terminal GST or MBP fusions which contained a C-terminal His6 tag (apart from JNK1 that contained an N-terminal His6-tag only). Samples were normally purified by double-affinity chromatography or by ion exchange close to homogeneity checked by SDS–PAGE. In vitro ubiquitination and phosphorylation reactions were carried out in 50 mM HEPES pH 7.5, 100 mM NaCl, 5 mM MgCl2, 0.05% IGEPAL, 5% glycerol, 2 mM DTT. MEKK3 was expressed with the Bac-to-Bac baculoviral expression system with a C-terminal His6-tag and an N-terminal maltose binding protein (MBP) tag and purified by double-affinity chromatography (using Ni-NTA and amylose resin, New England Biolabs). GST-MEKK2 was purchased from Signalchem. MAPKKKs (0.2 μM) were first ubiquitinylated in the presence of ubiquitin (25 μM, Boston Biochem) at room temperature for 30′ using E1, E2 (0.1 μM, Boston Biochem) and GST-XIAP (0.2 μM, expressed in E. coli and purified) similarly as described in the ubiquitination experiments section. Phosphorylation assays were started by adding MAPKKs (1 μM) and MAPKs (5 μM) and additional 0.5 mM fresh Mg-ATP. For non-ubi reactions, samples were treated exactly the same, but XIAP was not added to the reaction mix. Reactions were stopped with protein-loading sample buffer complemented with 20 mM EDTA, boiled and then subjected to SDS–PAGE. Gels were dried before analysis by phosphorimaging or by Western blots on a Typhoon Trio+ scanner (GE Healthcare). Western blots were done by using anti-phospho ERK5 antibody, anti-phospho MEK5, anti-phospho JNK1 antibody (9251S, Cell Signaling), and anti-phospho MKK7 antibody (4171S, Cell Signaling). The constitutive active form of MEK5 (MEK5DD: S311D, T315D) and the kinase inactive version of ERK5 (ERK5_KI: D182A)—that was used as substrate in the in vitro kinase assays—were generated by QuickChange Site-Directed Mutagenesis or by PCR, and proteins were expressed and purified as described earlier (Glatz et al, 2013). JNK1 kinase_dead (KD) was expressed with N-terminal His6-tag in E. coli Rosetta (DE3) pLsyS (Novagen), and purification was done with nickel affinity and ion-exchange chromatography (ResourseQ 1 ml column, GE Healthcare). Dephosphorylated MKK7 (with N-terminal GST- and C-terminal His6-tag) was co-expressed with λ phage phosphatase in E. coli (MG950 vector) and purified by double-affinity chromatography (on Ni-NTA and glutathione sepharose). For the myeline basic protein (MyBP) phosphorylation experiments, the reaction was started by adding 0.5 mM ATP with ˜5 μCi of ATP(γ-32P).
Expression of GST-tagged proteins and their purification
XIAP-GST (M1-S497), XIAP-BIR1-GST (M1-S123), XIAP-BIR2-GST (L121-S261), XIAP-BIR3-GST (S261-E350), XIAP-BIR1+2+3-GST (M1-E350), XIAP-ΔRING-GST (M1-L449), or GST alone were transformed into BL21-CodonPlus competent cells and transformation, and protein purification was done according to manufacturer's instructions. Briefly, the transformed cells were grown in a liquid culture at 37°C overnight with chloramphenicol and ampicillin with constant shaking. Part of this overnight culture was then added to fresh LB media and incubated at 37°C till it reached OD600 0.6. Transformed bacteria were then induced with 1 mM IPTG for 4 h at 37°C. The cells were pelleted and lysed in GST lysis buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 1 mM EDTA, 5% glycerol, 0.1% NP-40), 1 mM DTT and 1 mM protease inhibitor cocktail (Calbiochem). The GST proteins were bound to glutathione–sepharose beads (GE Healthcare). MEKK2-GST and MEKK3-GST were obtained from SignalChem (active MEKK2, Cat. No. M10-10G-20; active MEKK3, Cat. No. 11-10G-20) and MEK5-GST from Abnova (Cat. No. H00005607-P01).
In vitro translation
In vitro translation was performed using the TNT® T7 Coupled Reticulocyte Lysate Systems from Promega following the manufacturer's protocol (Cat. No. L4610). Briefly, 1 μg of template DNA was mixed in the following reaction: 25 μl TNT® lysates, 2 μl TNT® Reaction Buffer, 1 μl TNT® T7 RNA Polymerase, 1 mM Amino Acids Mixture, minus Leucine, 1 mM Amino Acids Mixture, minus Methionine and 40 units of RNasin® Ribonuclease Inhibitor, and incubated at 30°C for 90 min. pcDNA3.1B-myc/his-MEKK2-wt, pcDNA3.1B-myc/his-MEKK2-K47A, pcDNA3.1B-myc/his-MEKK2-K384M, pcDNA3.1B-myc/his-XIAP plasmids were used as template.
Pull-down experiments
Glutathione Sepharose™ (GE Healthcare, Cat. No. 17-0756-05) was washed and equilibrated in the so-called “GST Pull-down Buffer” (GPB): 50 mM Tris pH 7.5, 150 mM NaCl, 1% NP-40, 1 mM DTT. For each condition, 50 μl of beads were resuspended in 300 μl of GPB and incubated on rotator for 2 h at 4°C with 1 μg of GST protein. The beads were then washed three times with GPB and incubated on rotator for 1 h at 4°C with 100 mg/ml BSA-GPB solution. After washing three times, beads were finally incubated on rotator for 2 h at 4°C with purified XIAP protein or in vitro translated protein. The final washing was performed with GPB containing either 150 mM or 250 mM NaCl. Buffer was completely removed using an insulin syringe, and samples were then prepared for SDS–PAGE.
For MBP pull-down experiments, first the amylose resin (New England BioLabs) was equilibrated with binding buffer (20 mM Tris, 100 mM NaCl, 0.1% IGEPAL, 2 mM β-mercaptoethanol) and 10 μg immobilized MBP-fusion protein (and MBP protein as negative control) was incubated in the presence of 10 μM prey in 200 μl binding buffer for 30 min at room temperature. Binding reactions typically contained 10–20 μl resin saturated with baits. Amylose beads were pelleted with centrifugation and washed three times. Retained proteins were eluted from the resin with SDS-loading buffer. Samples were subjected to SDS–PAGE and stained with Coomassie staining.
Immunoprecipitation
To immunoprecipitate endogenous proteins, HeLa or MEFs cells were seeded on 10-cm dishes and if required, transfected after 24 h and then lysed 48 h post-transfection. Next day, 80% confluent cells were lysed with lysis buffer containing 20 mM Tris–HCl pH 7.5, 150 mM NaCl, 0.5% NP-40, 0.5% Triton X-100, 1 mM NaVO3, 10 mM Na-pyrophosphate, 1 mM NaF, 0.5 mM EDTA, 0.5 mM EGTA, 1 mM DTT, 1 μg/ml aprotinin, 0,5 μg/ml leupeptin, 10 μM pepstatin for 30 min on ice. Lysates were cleared by centrifugation for 15 min at 14,000 rpm. Endogenous MEKK2, MEKK3, or XIAP was then immunoprecipitated overnight. The antigen–antibody complexes were precipitated by sepharose-coupled protein A/G beads (Roche, Cat. No. 11-134-515-001 and 11-243-233-001). The beads were washed with the lysis buffer, and bound proteins were analyzed by SDS–PAGE and immunoblotting.
For immunoprecipitation of co-expressed proteins in 293T cells, we have transfected 293T cells with various plasmids using GeneJuice® transfection reagent (Novagen®, Merck Millipore, Cat. No. 70967). The cells were lysed at 48 h post-transfection, and proteins were immunoprecipitated as mentioned above. Control IPs were performed with IgG isotype control antibodies (Santa Cruz).
For immunoprecipitation of the entire protein complex (MEKK2–MEK5-ERK5), HeLa cells stably overexpressing ERK5-FLAG and MEK5-V5 were employed. Two days post-transfection with siXIAP 3′UTR using RNAiMAX, the cells were lysed in RIPA buffer (250 mM NaCl, 50 mM Tris pH 7.5, 10% glycerine, 1% Triton X-100, 1 mM DTT, 1% of Protease Inhibitor Cocktail Set I (Calbiochem)) for 30 min at 4°C. Lysates were cleared by centrifugation for 15 min at 14,000 rpm. FLAG-ERK5 fusion protein was isolated using ANTI-FLAG M2 Magnetic beads (Sigma). For each condition, 10 μl magnetic beads were used and incubated with the lysate for 4 h at 4°C. The immunoprecipitation and subsequent elution with FLAG peptides were performed according to the manufacturer's instructions (Sigma).
Mass spectrometry
Samples from the in vitro ubiquitination assay were cleaned and fractionated using SDS–PAGE and digested in-gel using trypsin as described by earlier with minor modifications. The sample was alkylated with chloroacetamide to reduce formation of lysine modifications with the same atomic composition as the di-glycine modification (Nielsen et al, 2008). Before performing LC-MS analysis, peptides were desalted using C18 StageTips (Rappsilber et al, 2002). Peptides were loaded with Solvent A (0.5% acetic acid) to an Easy-nLC II coupled to an Orbitrap Elite (Thermo Fisher Scientific) and separated using a C18 fused silica emitter packed in-house. Elution was performed using a segmented gradient of 5–50–90% of solvent B (80% ACN in 0.5% acetic acid) with a constant flow of 200 nl/min over 87 min. Full-scan MS spectra were acquired in the Orbitrap mass analyser in the range of 300–2,000 Th at a resolution of 120,000. The 20 most intense ions were successively isolated for CID fragmentation in the linear ion trap at a target value of 5,000 charges. Ions selected for fragmentation were included in a dynamic exclusion list for 60 s.
The acquired data were processed using the MaxQuant software suite (Cox & Mann, 2008) which contains the Andromeda search engine (Cox et al, 2011). Mass spectra data was searched against a human database containing 73,929 entries (http://www.uniprot.org, Protein data set in FASTA format, downloaded on the 18.01.2012) and 248 frequently, seen laboratory contaminants. Carbamidomethylation (Cys) was defined as fixed, and protein N-terminal acetylation, oxidation (Met), and GlyGly (Lys) were defined as variable modifications. Initial mass tolerance was set to 7 ppm for precursor ions, and to 0.5 Da for fragment ions. False discovery rate was set to 1% at the protein, peptide and modification levels.
Myoblast differentiation
Human Skeletal Muscle Myoblast (HSM) was seeded in a 12-well plate at 10,000 cells per well. When immunofluorescence analysis was required, cells were seeded out on glass coverslips, previously coated with purified bovine collagen solution from Nutragen® (Advanced BioMatrix, Cat. No. 5010-D) for at least 2 h. At 70–80% confluency, cells were transfected with siRNAs as explained above. The following day differentiation was started by first rinsing residual growth medium with PBS and then by adding differentiation medium (ZenBio, see above). Transfection of siRNA was performed every 2 days to ensure the knockdown of the genes of interest. In order to inhibit ERK5 activation, cells were treated with 5 μM of XMD 8–92 (R&D Systems, Cat. No. 4132) or with the equivalent amount of DMSO, every 2 days while renewing the differentiation medium. Cells were lysed at various time points by adding 5× Laemmli and prepared for SDS–PAGE and immunoblotting. For the immunofluorescence, cells were fixed with 2% PFA for 20 min at room temperature (RT). After washing three times with PBS, cells were permeabilized 7 min at RT with 0.1% Triton X-100 diluted in PBS. Cells were then blocked in 5% BSA-PBS overnight at 4°C. The first antibody against Myosine Heavy Chain (MHC) was added at a dilution of 1/200 overnight at 4°C and was followed by an overnight incubation with the secondary anti-mouse-Cy3 antibody at 4°C. The DAPI staining was obtained by incubating the fixed cells in 1/5,000 Hoechst 33342 solution for 10 min at RT. Finally, cover slips were fixed on slides using Mowiol 4-88 reagent (Calbiochem, Cat. No. 475904). The images were acquired using a Leica fluorescent microscope (DMIRE2) fitted with a digital camera with both high NA and low NA 10× objectives.
Acknowledgments
We would like to thank Vishva Dixit and Genentech-Roche for kindly providing ubiquitin-linkage-specific antibodies. We thank Carrie Anderson for excellent technical assistance, Conny Pyko for purification of XIAP mutants, Anita Alexa for the production of MBP-MEKK3, Koraljka Husnjak for providing recombinant ubiquitin chains and the deubiquitinases AMSH and USP2, John Silke for providing IAP deficient MEFs, and Hermann Stellar for XIAP delta RING MEFs. This work was partially supported by an Emmy Noether Programme grant RA1739/1-1, Lowe Ubiquitin-Network Grant as well as CRC-128 grants from the DFG to KR. KR is a PLUS3 fellow of the Boehringer Ingelheim Foundation and a Heisenberg professor of the DFG (RA1739/4-1). AR is an International Senior Research Fellow of the Wellcome Trust. The work was also supported by the “Lendület” Grant (LP2013-57/2013) from the Hungarian Academy of Sciences (to AR).
Author contributions
ANT and TKO performed most of the experiments, analyzed and interpreted data, and prepared figures. GG, KS, and RPS performed additional experiments, analyzed and interpreted data, and prepared figures. AC and BM performed mass spectrometric analysis. AR designed experiments, and analyzed and interpreted data. KR conceived and designed the project, analyzed and interpreted data, coordinated the study, and wrote the paper with input from all authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information for this article is available online: http://emboj.embopress.org
References
- Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- Coulombe P, Meloche S. Atypical mitogen-activated protein kinases: structure, regulation and functions. Biochim Biophys Acta. 2007;1773:1376–1387. doi: 10.1016/j.bbamcr.2006.11.001. [DOI] [PubMed] [Google Scholar]
- Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Cox JR, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M. Andromeda: a Peptide Search Engine Integrated into the MaxQuant Environment. J Proteome Res. 2011;10:1794–1805. doi: 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- Dinev D, Jordan BW, Neufeld B, Lee JD, Lindemann D, Rapp UR, Ludwig S. Extracellular signal regulated kinase 5 (ERK5) is required for the differentiation of muscle cells. EMBO Rep. 2001;2:829–834. doi: 10.1093/embo-reports/kve177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogan T, Harms GS, Hekman M, Karreman C, Oberoi TK, Alnemri ES, Rapp UR, Rajalingam K. X-linked and cellular IAPs modulate the stability of C-RAF kinase and cell motility. Nat Cell Biol. 2008;10:1447–1455. doi: 10.1038/ncb1804. [DOI] [PubMed] [Google Scholar]
- Drew BA, Burow ME, Beckman BS. MEK5/ERK5 pathway: the first fifteen years. Biochim Biophys Acta. 2012;1825:37–48. doi: 10.1016/j.bbcan.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enwere EK, Holbrook J, Lejmi-Mrad R, Vineham J, Timusk K, Sivaraj B, Isaac M, Uehling D, Al-awar R, LaCasse E, Korneluk RG. TWEAK and cIAP1 regulate myoblast fusion through the noncanonical NF-kappaB signaling pathway. Sci Signal. 2012;5:ra75. doi: 10.1126/scisignal.2003086. [DOI] [PubMed] [Google Scholar]
- Fulda S, Rajalingam K, Dikic I. Ubiquitylation in immune disorders and cancer: from molecular mechanisms to therapeutic implications. EMBO Mol Med. 2012;4:545–556. doi: 10.1002/emmm.201100707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glatz G, Gogl G, Alexa A, Remenyi A. Structural mechanism for the specific assembly and activation of the extracellular signal regulated kinase 5 (ERK5) module. J Biol Chem. 2013;288:8596–8609. doi: 10.1074/jbc.M113.452235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyrd-Hansen M, Meier P. IAPs: from caspase inhibitors to modulators of NF-kappaB, inflammation and cancer. Nat Rev Cancer. 2010;10:561–574. doi: 10.1038/nrc2889. [DOI] [PubMed] [Google Scholar]
- Hindi SM, Tajrishi MM, Kumar A. Signaling mechanisms in Mammalian myoblast fusion. Sci Signal. 2013;6:re2. doi: 10.1126/scisignal.2003832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F, Zeng X, Kim W, Balasubramani M, Fortian A, Gygi SP, Yates NA, Sorkin A. Lysine 63-linked polyubiquitination is required for EGF receptor degradation. Proc Natl Acad Sci USA. 2013;110:15722–15727. doi: 10.1073/pnas.1308014110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter T. The age of crosstalk: phosphorylation, ubiquitination, and beyond. Mol Cell. 2007;28:730–738. doi: 10.1016/j.molcel.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Kenneth NS, Duckett CS. IAP proteins: regulators of cell migration and development. Curr Opin Cell Biol. 2012;24:871–875. doi: 10.1016/j.ceb.2012.11.004. [DOI] [PubMed] [Google Scholar]
- Lochhead PA, Gilley R, Cook SJ. ERK5 and its role in tumour development. Biochem Soc Trans. 2012;40:251–256. doi: 10.1042/BST20110663. [DOI] [PubMed] [Google Scholar]
- Mody N, Campbell DG, Morrice N, Peggie M, Cohen P. An analysis of the phosphorylation and activation of extracellular-signal-regulated protein kinase 5 (ERK5) by mitogen-activated protein kinase kinase 5 (MKK5) in vitro. Biochem J. 2003;372:567–575. doi: 10.1042/BJ20030193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscat J, Diaz-Meco MT, Albert A, Campuzano S. Cell signaling and function organized by PB1 domain interactions. Mol Cell. 2006;23:631–640. doi: 10.1016/j.molcel.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Johnson GL. PB1 domains of MEKK2 and MEKK3 interact with the MEK5 PB1 domain for activation of the ERK5 pathway. J Biol Chem. 2003;278:36989–36992. doi: 10.1074/jbc.C300313200. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Johnson GL. Noncanonical function of MEKK2 and MEK5 PB1 domains for coordinated extracellular signal-regulated kinase 5 and c-Jun N-terminal kinase signaling. Mol Cell Biol. 2007;27:4566–4577. doi: 10.1128/MCB.00125-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Kimple AJ, Siderovski DP, Johnson GL. PB1 domain interaction of p62/sequestosome 1 and MEKK3 regulates NF-kappaB activation. J Biol Chem. 2010;285:2077–2089. doi: 10.1074/jbc.M109.065102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen ML, Vermeulen M, Bonaldi T, Cox J, Moroder L, Mann M. Iodoacetamide-induced artifact mimics ubiquitination in mass spectrometry. Nat Methods. 2008;5:459–460. doi: 10.1038/nmeth0608-459. [DOI] [PubMed] [Google Scholar]
- Nithianandarajah-Jones GN, Wilm B, Goldring CE, Muller J, Cross MJ. ERK5: structure, regulation and function. Cell Signal. 2012;24:2187–2196. doi: 10.1016/j.cellsig.2012.07.007. [DOI] [PubMed] [Google Scholar]
- Oberoi TK, Dogan T, Hocking JC, Scholz RP, Mooz J, Anderson CL, Karreman C, Meyer Z, Heringdorf D, Schmidt G, Ruonala M, Namikawa K, Harms GS, Carpy A, Macek B, Koster RW, Rajalingam K. IAPs regulate the plasticity of cell migration by directly targeting Rac1 for degradation. EMBO J. 2011;31:14–28. doi: 10.1038/emboj.2011.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Riordan MX, Bauler LD, Scott FL, Duckett CS. Inhibitor of apoptosis proteins in eukaryotic evolution and development: a model of thematic conservation. Dev Cell. 2008;15:497–508. doi: 10.1016/j.devcel.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman M, Chen W, Cobb MH. Differential regulation and properties of MAPKs. Oncogene. 2007;26:3100–3112. doi: 10.1038/sj.onc.1210392. [DOI] [PubMed] [Google Scholar]
- Rappsilber J, Ishihama Y, Mann M. Stop and Go Extraction Tips for Matrix-Assisted Laser Desorption/Ionization, Nanoelectrospray, and LC/MS Sample Pretreatment in Proteomics. Anal Chem. 2002;75:663–670. doi: 10.1021/ac026117i. [DOI] [PubMed] [Google Scholar]
- Seyfried J, Wang X, Kharebava G, Tournier C. A novel mitogen-activated protein kinase docking site in the N terminus of MEK5alpha organizes the components of the extracellular signal-regulated kinase 5 signaling pathway. Mol Cell Biol. 2005;25:9820–9828. doi: 10.1128/MCB.25.22.9820-9828.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasula SM, Ashwell JD. IAPs: what's in a name? Mol Cell. 2008;30:123–135. doi: 10.1016/j.molcel.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumimoto H, Kamakura S, Ito T. Structure and function of the PB1 domain, a protein interaction module conserved in animals, fungi, amoebas, and plants. Sci STKE. 2007;2007:re6. doi: 10.1126/stke.4012007re6. [DOI] [PubMed] [Google Scholar]
- Sun W, Kesavan K, Schaefer BC, Garrington TP, Ware M, Johnson NL, Gelfand EW, Johnson GL. MEKK2 associates with the adapter protein Lad/RIBP and regulates the MEK5-BMK1/ERK5 pathway. J Biol Chem. 2001;276:5093–5100. doi: 10.1074/jbc.M003719200. [DOI] [PubMed] [Google Scholar]
- Tanoue T, Nishida E. Docking interactions in the mitogen-activated protein kinase cascades. Pharmacol Ther. 2002;93:193–202. doi: 10.1016/s0163-7258(02)00188-2. [DOI] [PubMed] [Google Scholar]
- Wang X, Tournier C. Regulation of cellular functions by the ERK5 signalling pathway. Cell Signal. 2006;18:753–760. doi: 10.1016/j.cellsig.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Wang X, Zhang F, Chen F, Liu D, Zheng Y, Zhang Y, Dong C, Su B. MEKK3 regulates IFN-gamma production in T cells through the Rac1/2-dependent MAPK cascades. J Immunol. 2011;186:5791–5800. doi: 10.4049/jimmunol.1002127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winsauer G, Resch U, Hofer-Warbinek R, Schichl YM, de Martin R. XIAP regulates bi-phasic NF-kappaB induction involving physical interaction and ubiquitination of MEKK2. Cell Signal. 2008;20:2107–2112. doi: 10.1016/j.cellsig.2008.08.004. [DOI] [PubMed] [Google Scholar]
- Xu BE, Stippec S, Lenertz L, Lee BH, Zhang W, Lee YK, Cobb MH. WNK1 activates ERK5 by an MEKK2/3-dependent mechanism. J Biol Chem. 2004;279:7826–7831. doi: 10.1074/jbc.M313465200. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Dong C. Regulatory mechanisms of mitogen-activated kinase signaling. Cell Mol Life Sci. 2007;64:2771–2789. doi: 10.1007/s00018-007-7012-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G, Bao ZQ, Dixon JE. Components of a new human protein kinase signal transduction pathway. J Biol Chem. 1995;270:12665–12669. doi: 10.1074/jbc.270.21.12665. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.