

Abstract
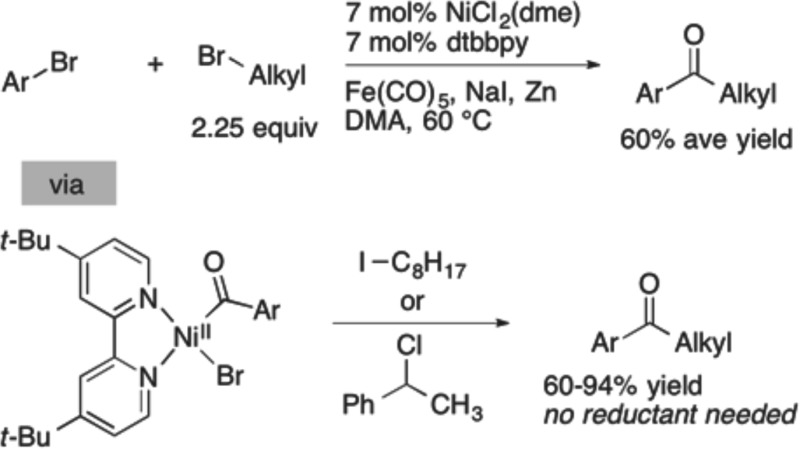
Acylnickel(II) complexes feature prominently in cross-electrophile coupling (XEC) reactions that form ketones, yet their reactivity has not been systematically investigated. We present here our studies on the reactivity of acylnickel(II) complexes with a series of carbon electrophiles. Bromobenzene, α-chloroethylbenzene, bromooctane, and iodooctane were reacted with (dtbbpy)NiII(C(O)C5H11)(Br) (1b) and (dtbbpy)NiII(C(O)tolyl)(Br) (1c) to form a variety of organic products. While reactions with bromobenzene formed complex mixtures of ketones, reactions with α-chloroethylbenzene were highly selective for the cross-ketone product. Reactions with iodooctane and bromooctane also produced the cross-ketone product, but in intermediate yield and selectivity. In most cases the presence or absence of a chemical reductant (zinc) had only a small effect on the selectivity of the reaction. The coupling of 1c with iodooctane (60% yield) was translated into a catalytic reaction, the carbonylative coupling of bromoarenes with primary bromoalkanes (six examples, 60% average yield).
Introduction
The ketone is ubiquitous in organic synthesis because it is a synthetically useful functional group and a common motif in biologically active molecules. Among the many synthetic methods known, carbonylative cross-coupling of a carbon nucleophile with a carbon electrophile is attractive because it is convergent (eq 1).1 A common challenge for all of these carbonylative cross-coupling reactions is the availability and stability of the carbon nucleophile component, which has led to the development of alternative methods, such as C–H activation,1b and milder organometallic reagents.2
 |
1 |
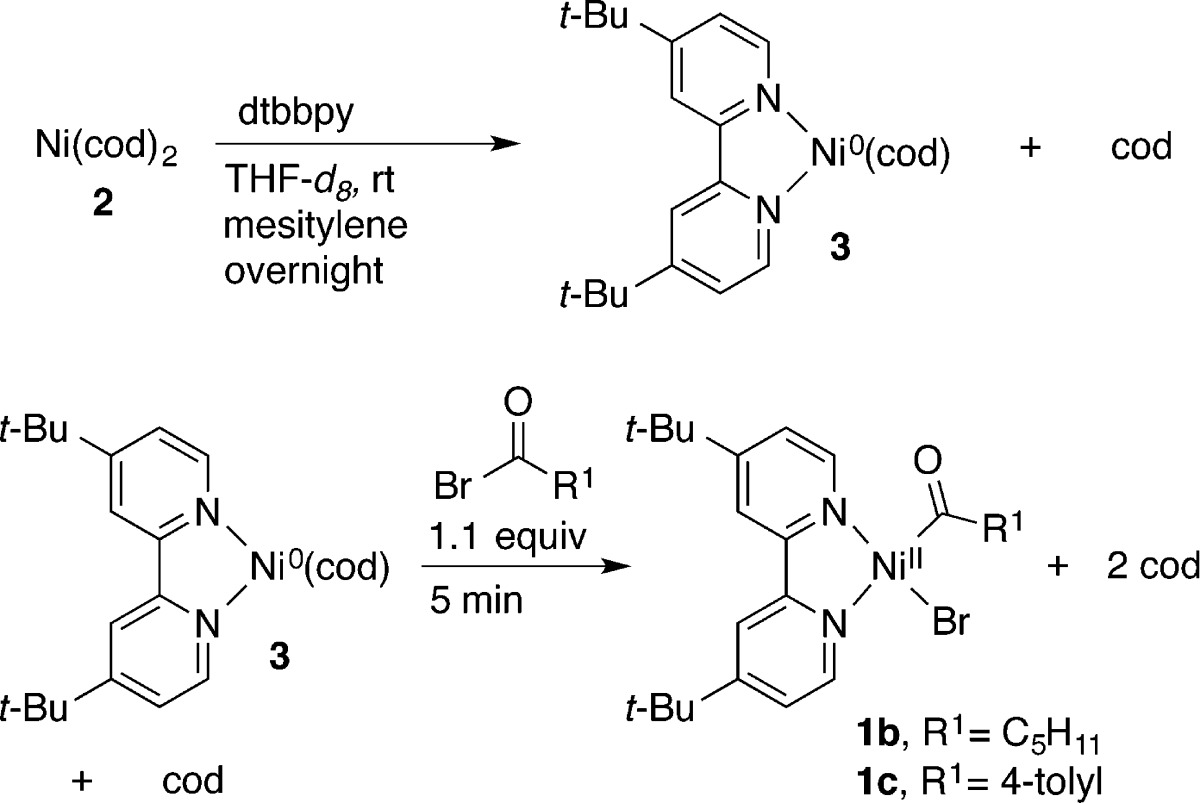
An alternative strategy to avoid the challenges of nucleophilic carbon reagents is the coupling of two different carbon electrophiles (Figure 1). Cross-electrophile coupling (XEC) has been much less studied than the cross-coupling of electrophiles with nucleophiles, but recent reports have demonstrated the generality of this approach3−6 and have shed light on the origins of selectivity.7 Two different nickel-catalyzed XEC approaches to ketones have been developed: the coupling of acid chlorides with alkyl and benzyl halides4 and the carbonylative coupling of aryl halides with alkyl halides.6 The carbonylative coupling approach has only been demonstrated with electrochemical reduction (Ni/Fe sacrificial anode), and high yields were only obtained with aryl iodides and benzyl bromides.6f
Figure 1.
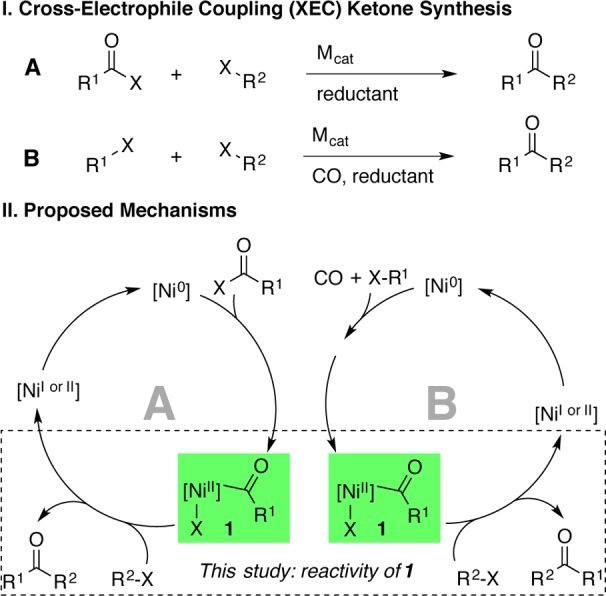
Acylnickel(II) intermediates 1 important to both XEC ketone synthesis methods A and B. Their reactivity with organic halides (R2X) has not been systematically studied.
A common feature of many4d−4f,6 of these approaches (A and B, Figure 1) is the intermediacy of an acylnickel(II) intermediate (1) that is proposed to react with a carbon electrophile (R2X) to form the ketone product (R1C(O)R2). Studies of the generation of acylnickel(II) intermediates from nickel(0) have clearly delineated its formation, but no such systematic studies on the reactivity of 1 with carbon electrophiles have been reported.
Despite our recent success in developing and understanding the mechanism of nickel-catalyzed cross-electrophile coupling of aryl halides with alkyl halides, we foresaw several challenges for the carbonylative reaction. First, the coupling of two electrophiles with a third component (CO) results in a large number of potential products in comparison to two-component couplings.8 Second, the reactivity of the proposed acylnickel intermediates is relatively unknown, limiting our ability to troubleshoot reactions.
We report here our progress toward understanding the reactivity of acylnickel(II) complexes toward aryl, alkyl, and benzylic electrophiles and a cross-selective carbonylative coupling of unactivated primary alkyl bromides with aryl halides to form aryl alkyl ketones.
Background
Stoichiometric reactions with Ni(CO)4 with electrophiles to form ketones are well-known to proceed via intermediate acylnickel(II) complexes (eq 2),9 but these reactions are not often used today due to toxicity concerns10 and limited availability.11,12 Catalytic versions of these stoichiometric reactions have proven to be difficult to develop.
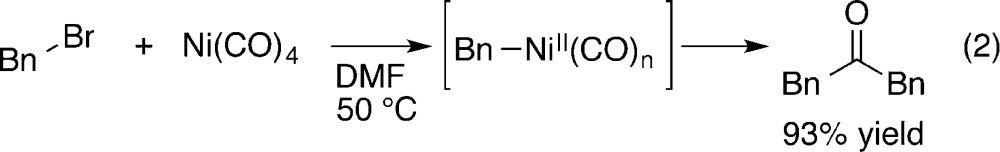 |
2 |
The catalytic, carbonylative coupling of two organic halides has primarily been developed by the group of Troupel. Early work revealed that Fe(CO)5 was a more convenient and reliable CO source1,13,14 for these reactions than CO gas because the reaction was strongly inhibited by the formation of unreactive (L)Ni0(CO)2 at higher CO concentrations.6a,6b
To date, the electrolysis conditions of Troupel are the best conditions for carbonylative XEC (Scheme 1).6f With 30 mol % of (bpy)NiBr2, benzyl chlorides could be coupled with aryl iodides (2 equiv) to form aryl benzyl ketones in 43–88% yield, but yields were lower with aryl bromides (32–60%) and an unactivated alkyl iodide, iodooctane (35–42%). Functional group compatibility was promising, with free phenol and aniline groups coupling in good yield.
Scheme 1. Electroreductive Synthesis of Ketones6f.

Troupel’s electrochemical mechanistic studies on this process implicated an acylnickel(II) intermediate, (bpy)Ni(COR1)X (1), and examined the rate and mechanism of its formation under catalytically relevant conditions. Further reaction of 1 with another organic halide (R2X) to form ketone (R1COR2) was not studied, but it was proposed that it could occur through a number of pathways, including intermediate reduction of 1 from nickel(II) to nickel(I).
Acylnickel complexes may be generated by addition of CO to the corresponding organonickel(II) complex15 or by the reaction of nickel(0) with an activated ester.16 Phosphine-ligated acylnickel(II) halide complexes are reported to lose carbon monoxide over time16 or to reversibly eliminate CO and exchange it with other nickel complexes.15a Only one study has reported on the reactivity of an acylnickel(II) species with a bidentate N–N ligand (bpy).15c All of the reported ketone-forming XEC reactions call for N–N ligands of this type.4,6
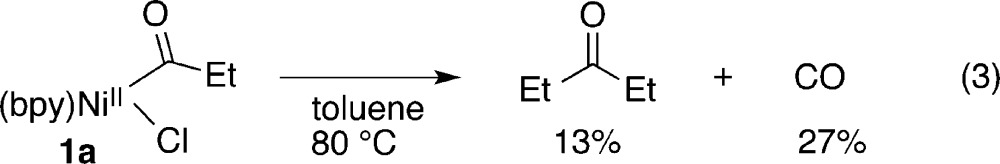
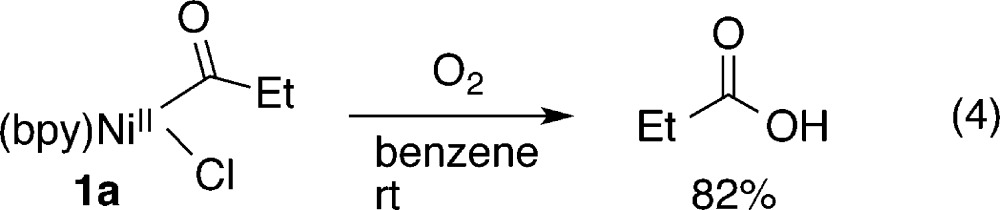
Yamamoto and co-workers explored the reactivity of 1 under thermolysis (eq 3) and with dioxygen (eq 4),15c but the stoichiometric reactivity of 1 with organic electrophiles has never been systematically examined. Amatore and Jutand generated 1 under electrochemical conditions in their study on the acylation of benzyl bromides and showed it can easily be reduced to an acylnickel(I) species but did not study the reactivity of either species with organic electrophiles.7a
 |
3 |
 |
4 |
In studies related to the acylation of alkyl halides, Gong reacted (dmbpy)Ni0 (dmbpy = 4,4′-dimethyl-2,2′-bipyridine) with a mixture of benzoic anhydride and a secondary alkyl bromide and reported that only the anhydride was consumed (Scheme 2).4e No ketone formation was observed from the resulting uncharacterized mixture unless a reductant (Zn0) was added. A similar reaction with a mixture of anhydride and iodoalkane resulted in partial consumption of both electrophiles and no ketone formation. From these studies it appeared that the initial acylnickel(II) (1) had to be reduced to an acylnickel(I)7a before it could form ketone products from the reaction with secondary bromoalkanes.
Scheme 2. Stoichiometric Studies on Acylation of RX4e.

An improved fundamental understanding of how (L)NiII(C(O)R)X (1) reacts with electrophiles would be generally helpful to developing XEC reactions for ketone synthesis, especially for an improved method for the carbonylative XEC of aryl halides with alkyl halides to form aryl alkyl ketones. We also sought to improve upon the results of Troupel in the carbonylative coupling of aryl halides with unactivated alkyl halides (Scheme 1).
Results and Discussion
Stoichiometric Studies
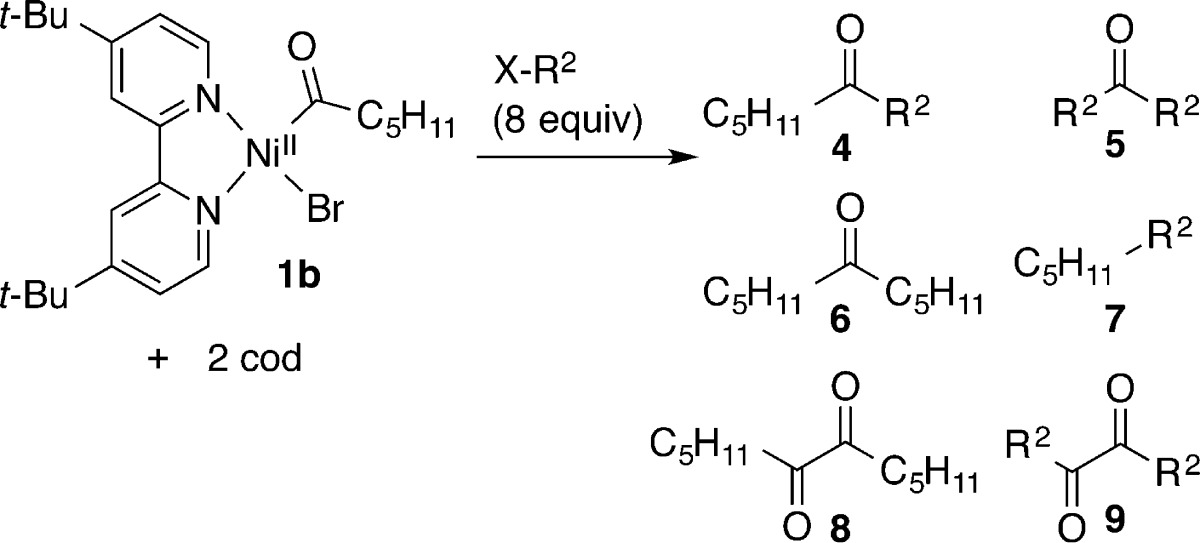
In order to study the key proposed bond-forming step, we synthesized the dark red (dtbbpy)NiII(C(O)C5H11)Br (1b) from the reaction of deep purple (dtbbpy)Ni0(cod)3g (3) with hexanoyl bromide in THF, in analogy to several literature reports (Table 1).16 The resulting acylnickel(II) intermediate proved difficult to isolate; therefore, it was characterized in solution by 1H NMR, IR, and UV–vis. The yield was determined to be 74% by NMR (average of two runs).
Table 1. Synthesis of (dtbbpy)NiII(COR1)Br and Selected Characterization Dataa.
| entryb | [Ni] | yield (%)c | IR (cm–1)d | NMR (ppm)e | UV–vis (cm–1) (103 M–1 cm–1)f |
|---|---|---|---|---|---|
| 1 | 1b | 74 | 1612 | 3.12 (t) | 20325 (5) |
| 2 | 1c | 89 | 1617 | N/A | 20576 (7) |
dtbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine.
See the Supporting Information for full characterization data and procedures.
1H NMR yield vs liberated cyclooctadiene; average of two runs.
C=O stretches reported that are characteristic of M–C(O)R complexes.15e
1H NMR chemical shift and multiplicity for α proton of M–C(O)CH2R.15c N/A = not applicable.
UV–vis absorbances reported as wavenumbers (ε values are given in parentheses).
The results compared well to those of Yamamoto.15c Distinctive features were as follows: (1) the Ni–C(O)CH2CH2CH2CH3 triplet (3.12 ppm) agrees well with the 3.19 ppm triplet reported for (bpy)NiII(C(O)Et)(Cl); (2) the IR spectrum revealed a CO stretch at 1612 cm–1, in good agreement with (bpy)NiII(C(O)Et)(Cl) at 1655 cm–1 and consistent with other acylnickel complexes (1600–1640 cm–1);15b,15d,15e (3) the UV–vis spectrum showed a strong MLCT at 20325 cm–1, in good agreement with Yamamoto’s report for (bpy)NiII(C(O)Et)(Cl) (19880 cm–1).15c
A similar reaction with 4-methylbenzoyl bromide afforded (dtbbpy)NiII(C(O)tolyl)Br (1c). This complex was similarly characterized in solution by 1H NMR, IR, and UV–vis. 1H NMR analysis clearly showed the formation of a single, new diamagnetic complex in 89% yield. The IR stretch was in good agreement with that reported for (dppe)NiII(C(O)Ph)Cl: 1617 vs 1630 cm–1.16b
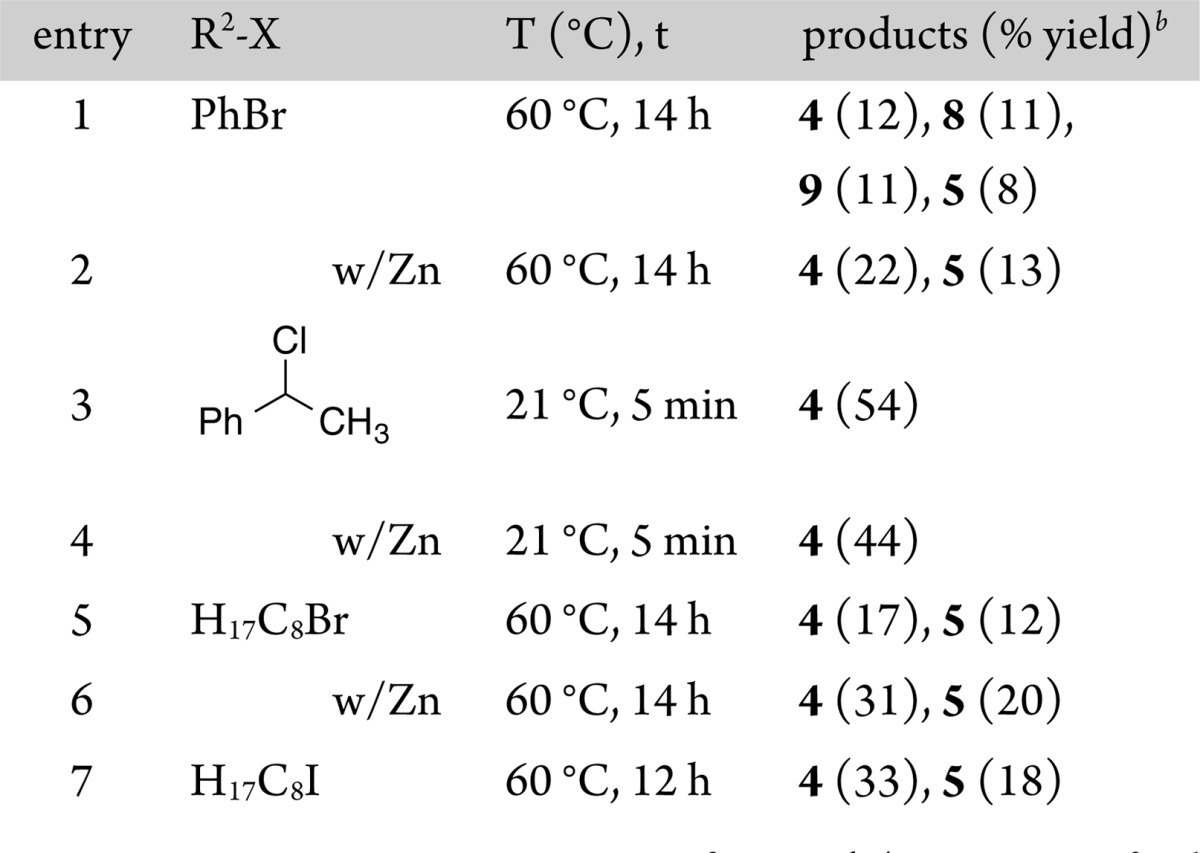
Reactions of hexanoylnickel 1b with four different organic halides (bromobenzene, α-chloroethylbenzene, bromooctane, and iodooctane) were performed, and the results are summarized in Table 2.17 Because 1b decomposes over about 2 h in solution, we generated it fresh for each experiment in THF-d8 and then diluted it with DMA to a 1/1 mixture for reactions with electrophiles. Finally, because it has been proposed that intermediate reduction of an acylnickel complex is needed before it can react with organic halides, we also ran reactions with an added reductant, zinc flake.
Table 2. Reaction of (dtbbpy)NiII(COC5H11)Br with RXa.
Reactions run in a 1/1 THF-d8/DMA mixture using freshly prepared 1b. See the Supporting Information for full experimental details and tables with all products formed.
Yields are uncorrected GC yields of products formed in >7% yield.
The reaction of 1b with bromobenzene produced similar amounts of cross-ketone (4) and symmetric ketone or diketone products (8 and 9; Table 2, entry 1). The addition of zinc flake eliminated the symmetric diketone products (8 and 9), but the reaction remains only marginally selective (entry 2).
Hexanoyl complex 1b reacts with α-chloroethylbenzene rapidly (under 5 min at room temperature) to exclusively form cross-ketone product 4 (Table 2, entry 3) in a yield that compares well with those reported for the XEC of acid chlorides with α-chloroethylbenzene (49–52% for similar conditions).4c,4f The addition of zinc flake did not improve the reaction yield or selectivity (entry 4), suggesting that reduction of 1b is not required.
Finally, the reaction of 1b with bromooctane or iodooctane produced nearly equal amounts of cross-ketone product 4 and dioctyl ketone 5 (Table 2, entries 5–7). The addition of zinc flake improved the yield of ketone products (entry 6) but did not improve the selectivity for cross-ketone over symmetric ketone significantly (1.4/1 to 1.6/1). XEC of acid chlorides with alkyl halides is reported to be sensitive to the reductant and solvent, perhaps explaining the difference between these modest yields and those reported by Gong4d and ourselves.4c
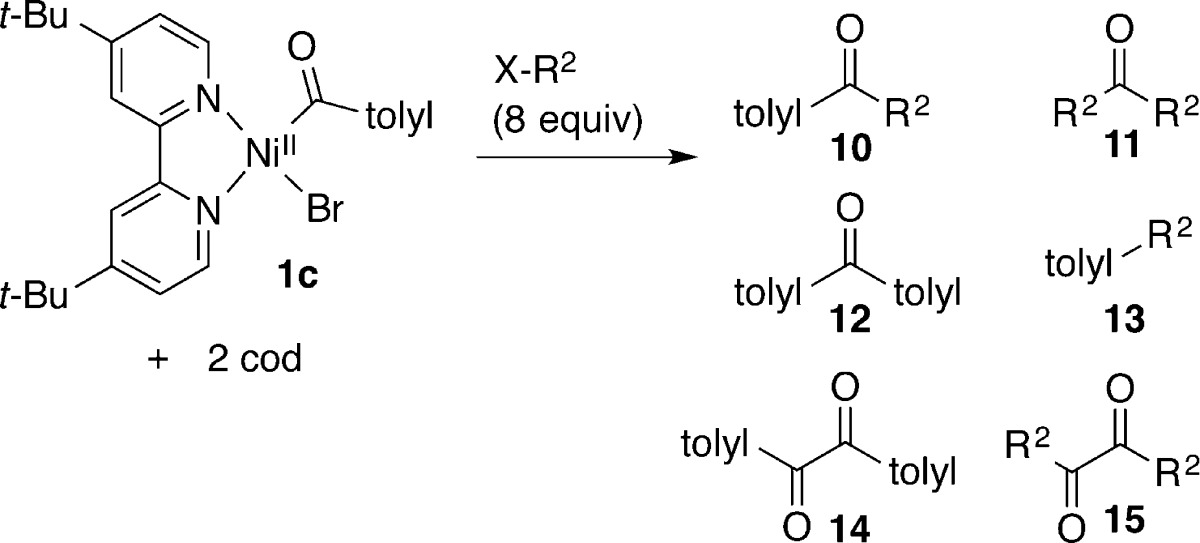
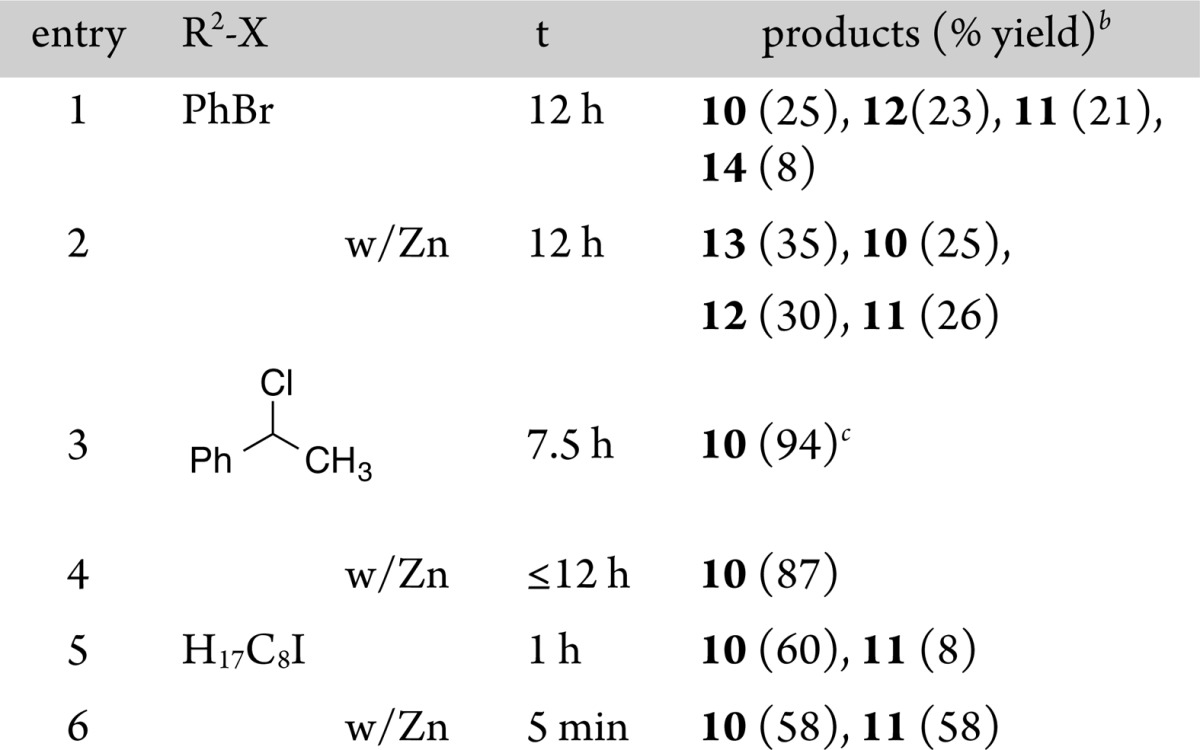
The reactivity of aroylnickel 1c was examined in the same manner as for 1b, and the results are shown in Table 3. Complex 1c was more stable than 1b, but we again prepared it fresh for each experiment.
Table 3. Reaction of (dtbbpy)NiII(COC6H4CH3)Br with RXa.
Reactions were run in a 1/1 mixture of THF-d8/DMA using freshly prepared 1c. See the Supporting Information for full experimental details and tables with all products formed.
Yields are uncorrected GC yields of products formed in >7% yield.
76% 1H NMR yield after workup and filtration through silica gel.
Reaction of 1c with bromobenzene again produced a mixture of ketone products (Table 3, entry 1). The addition of zinc as a reductant improved the total yield of all coupled products from 1c (nearly quantitative from 70%) but also resulted in the formation of significant amounts of noncarbonylated cross-aryl product 7 (entry 2).
The reaction of aroylnickel 1c with α-chloroethylbenzene was again rapid and clean, affording a 94% GC yield of the cross-ketone product 10 (Table 3, entry 3). In this case, we filtered the product mixture and obtained a 76% NMR yield of the cross-ketone product. There are no reports of a catalytic version of this reaction, but Gong reported that benzyl bromide and methallyl chloride couple with benzoyl chloride in high yield (70 and 76%).4d This result suggests that, as long as the aroyl activated ester reacts with nickel(0) first, such a cross-coupling will be high-yielding. Again, the addition of a reductant (zinc flake) did not improve the yield or selectivity (entry 4).
The reaction of iodooctane with aroylnickel 1c formed cross-ketone in 60% yield along with some dioctyl ketone 11 (Table 3, entry 5). If zinc flake was added, the amount of dioctyl ketone increased significantly (entry 6), suggesting decarbonylation of 1c, loss of the tolyl group, and coupling with iodooctane. Dioctyl ketone is not a major side product in the carbonylative coupling of aryl bromides with alkyl bromides, but the isolated yields of cross-ketone 10 are similar between the two methods (vide infra; Table 4, entry 17). Here, the addition of zinc increased the rate of the reaction (entry 6) but not the selectivity.
Table 4. Optimization and Controlsa.
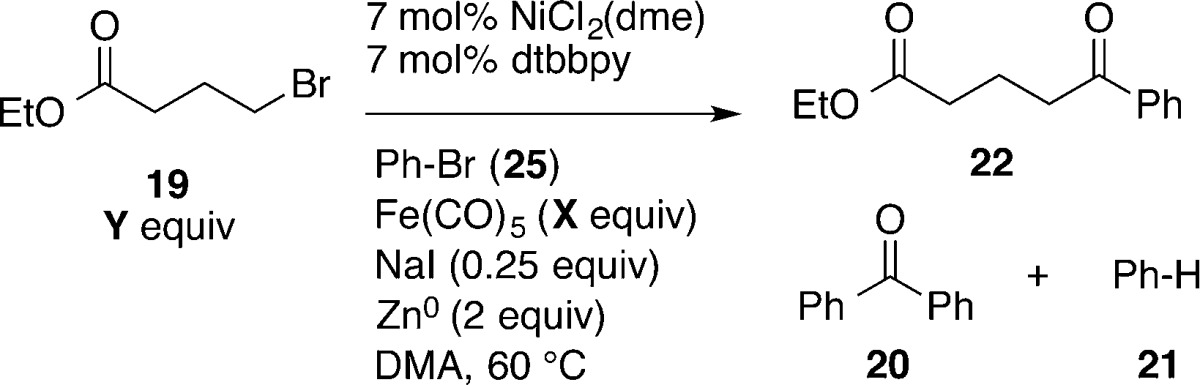
| entry | notes | X | Y | 22 | 20 | 21 |
|---|---|---|---|---|---|---|
| 1 | 0.35 | 2.50 | 69 | 5 | 4 | |
| 2 | 0.35 | 2.25 | 72 (60) | 6 | 6 | |
| 3 | 0.35 | 2.0 | 56 | 7 | 8 | |
| 4 | 0.35 | 1.5 | 50 | 9 | 15 | |
| 5b | 0.40 | 2.0 | 51 | 7 | 8 | |
| 6c | 0.45 | 2.0 | 42 | 3 | 5 | |
| 7c | 0.50 | 2.0 | 32 | 3 | 4 | |
| 8d | no NaI | 0.35 | 2.0 | 0 | 0 | 0 |
| 9 | LiI for NaI | 0.35 | 2.0 | 49 | 7 | 7 |
| 10 | KI for NaI | 0.35 | 2.0 | 48 | 4 | 8 |
| 11 | 5 mol % catalyst | 0.35 | 2.0 | 42 | 6 | 9 |
| 12e | no dtbbpy | 0.35 | 2.25 | 0 | 0 | 2 |
| 13e,f | no Ni or Zn | 0.35 | 2.0 | 0 | 0 | 1 |
| 14e,g | no Ni | 0.35 | 2.0 | <1 | <1 | 0 |
| 15c,g | no Zn | 0.35 | 2.25 | 25 | <1 | 4 |
| 16 | 6/1 DMA/THF | 0.35 | 2.25 | 76 | 7 | 0 |
| 17 | 1/1 DMA/THF | 0.35 | 2.25 | 52 | 7 | 3 |
| 18e,g | 1/2 DMA/THF | 0.35 | 2.25 | <1 | 0 | 1 |
Reactions were run on a 0.75 mmol scale in 3 mL of DMA. Yields are uncorrected GC yields vs dodecane. The yield in parentheses is an isolated yield of purified product.
PhBr (8%) remained.
PhBr (27%–47%) remained.
Starting materials were unchanged after 40 h, except for a 13% yield of hydrodehalogenated 19.
PhBr (75%) remained.
Alkyl-Br 19 not consumed.
Dialkyl ketone (12–23%) was formed.
A comparison of the reaction of hexanoylnickel 1b with bromobenzene (Table 2, entries 1 and 2) and the reaction of aroylnickel 1c with iodooctane (Table 3, entries 5 and 6) strongly suggests that the carbonylative coupling of aryl halides with alkyl halides will work best if the aryl halide reacts first with the nickel catalyst. Our recent work on the reactivity of (dtbbpy)Ni0(cod) demonstrated that aryl halides do react more quickly than alkyl halides under similar conditions.7b
Catalytic Studies
We started our catalytic studies by adding carbon monoxide sources to our published conditions for the coupling of aryl bromides with alkyl bromides.3d Initial results with 20 mol % of Fe(CO)5 were modest (26% yield). Examination of several metal carbonyls (Mn2CO10, W(CO)6, Mo(CO)6, Cr(CO)6) showed that iron carbonyl was best and that the reaction could be run in DMA instead of DMPU, but no significant improvements in yield were forthcoming.
The major reaction products were our desired aryl alkyl ketone, hydrodehalogenated aryl bromide, diaryl ketone, and noncarbonylated product. We envisioned that both the diaryl ketone and the aryl alkyl ketone product could arise from the aroyl nickel (1) formed by CO insertion into the aryl–nickel bond of 17 (Scheme 3). Aroylnickel(II) complexes are well-known to decompose into diaryl ketones by disproportionation upon heating.15,16 The noncarbonylated product presumably arose when not enough CO was available in solution, allowing for the formation of 18.7b
Scheme 3. Proposed Source of Aryl Side Products.

Consistent with these hypotheses, the addition of more alkyl bromide (2.25 equiv from 1 equiv) resulted in less diaryl ketone, although little effect was observed above 2.25 equiv (Table 4, entries 1–4). Increasing the amount of iron carbonyl eliminated noncarbonylated products, but too much iron carbonyl diminished yields (Table 4, entry 3 vs entries 5–7).
The addition of iodide was essential for reactivity. A reaction with no NaI gave no ketone product (Table 4, entry 8), but substitution of NaI with LiI and KI provided results similar to those for NaI (Table 4, entry 3 vs entries 9 and 10). Lowering the catalyst loading from 7 to 5 mol % resulted in a 14% lower yield (entry 11). A reaction run without added reductant produced a small amount of 22. In this case, the Fe(CO)5 likely served as the reductant (entry 15). Finally, 6/1 and 1/1 mixtures of DMA to THF formed products in 76% and 52% yields, respectively, but a 1/2 mixture did not consume most of the starting material (entries 16–18).
The highest yields were obtained when water was strictly excluded. For example, if starting materials were left on the bench for extended periods of time, the yield of product would drop (up to 20% difference in yield). Reactions also worked best when the mixtures were heated immediately after assembly. Stirring a reaction mixture at room temperature for 30 min before heating to 60 °C diminished the yield by about 10%.
In order to examine the scope of this reaction, several aryl bromides were coupled with primary alkyl bromides (Table 5). Yields were similar to those for the optimized reaction, but the coupling of an aryl bromide containing an electron-withdrawing group, −CF3, resulted in a lower yield (Table 5, entries 1 and 5), due to competitive formation of both diaryl ketone and biaryl.18 Aryl chlorides were not reactive (entry 2), and both protected alcohols and fluorinated substrates were also well tolerated (entries 4–6). Although α-chloroethylbenzene formed high yields of ketone product from reactions with acylnickel complexes (Tables 2 and 3), the catalytic reactions were plagued by rapid dimerization of this reactive substrate.
Table 5. Scope of Reaction with Aryl-Xa.
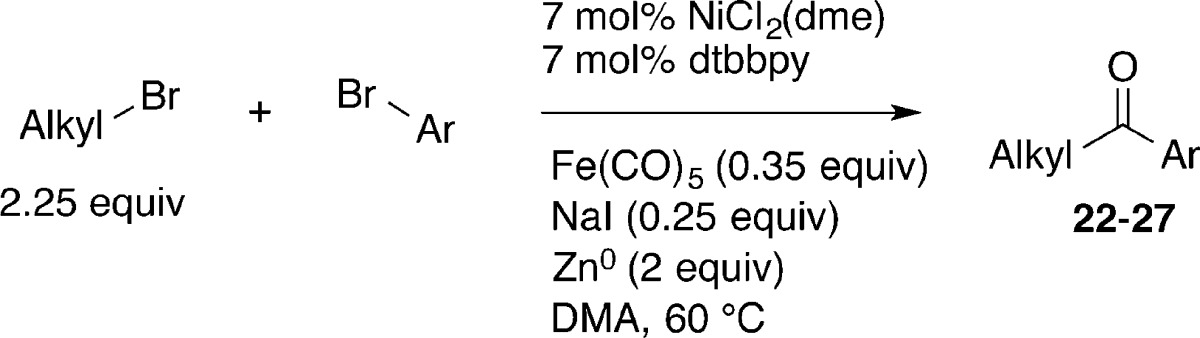
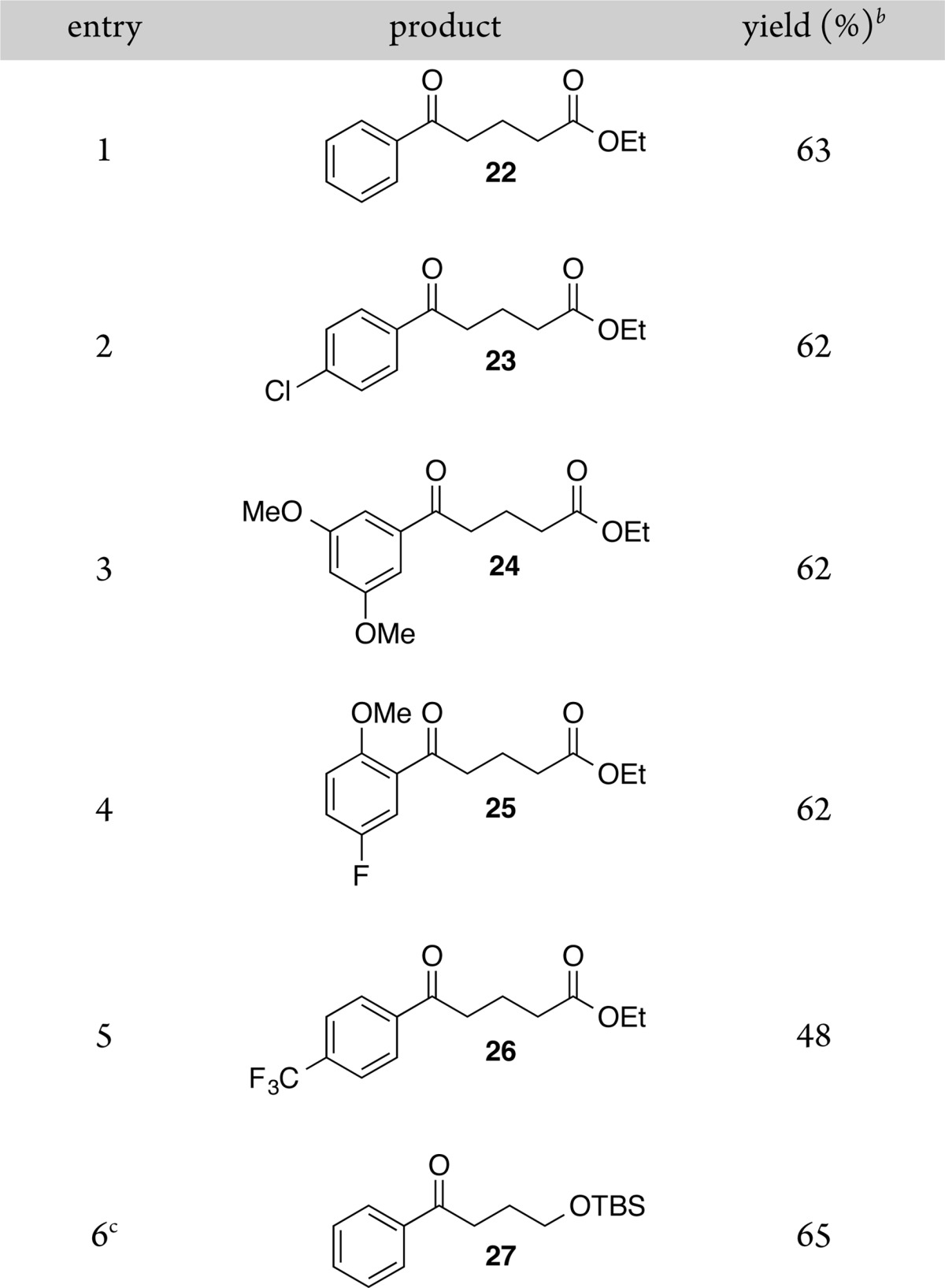
Reactions were performed on a 0.75 mmol scale on the benchtop in 3 mL of DMA.
Isolated yield of purified product.
Product contains an additional 3% of benzophenone.
Conclusions
In conclusion, our studies on acylnickel complexes have revealed differential reactivity with respect to electrophiles. Both aroyl- and alkanoylnickel complexes react with secondary benzylic chlorides to form product without added reductant, but the aroylnickel complex reacts with alkyl iodides to produce more cross-ketone than the alkanoylnickel complex. Neither complex reacts with bromobenzene to selectively produce cross-ketone product. The addition of an external reductant (zinc flake) did not improve the selectivity of reactions with alkyl or benzyl halides but did improve the rate in one case, suggesting that reduction of acylnickel(II) to acylnickel(I)4d−4f is not a necessary step in XEC ketone synthesis reactions and that zinc might facilitate a radical initiation step.7 Gong reported a reductant effect more dramatic than the effect we observed, but under significantly different conditions. Finally, the observed reactivity of 1c with alkyl halides can be translated into the first carbonylative cross-coupling of bromoarenes with bromoalkanes. Further studies on the reactivity of acylnickel complexes and translation to new reactions is ongoing.
Experimental Section
Materials and Methods
NiCl2(1,2-dimethoxyethane) (NiCl2(dme)) was purchased from Strem or synthesized according to the literature procedure.19 Ni(cod)2 was purchased from Strem. 4,4′-Di-tert-butyl-2,2′-bipyridine (dtbbpy) was purchased from Aldrich or synthesized according to the literature procedures.20,21 Zinc flake (−325 mesh; Alfa) was used as received. Anhydrous N,N-dimethylacetamide (DMA; Aldrich) was stored over activated 4 Å molecular sieves. DMA used for stoichiometric reactions was sequentially dried over 4 Å MS, degassed, and then vacuum-transferred. The water content was routinely measured using Karl Fischer titration (Metrohm) and was less than 70 ppm. THF-d8 was purchased from Cambridge Isotopes, dried sequentially over MS 4 Å, degassed, and then vacuum-transferred. Hexanoyl bromide and p-toluoyl bromide were synthesized using the literature procedures. 1-Iodooctane (Aldrich), 1-bromooctane (Aldrich), bromobenzene, and ethyl 4-bromobutanoate (Aldrich) were filtered through a dry, activated, basic alumina pad (1.5 cm) under nitrogen until they were colorless before use.
1H and 13C NMR spectra were acquired on 500 or 400 MHz (proton) Bruker NMR instruments, and data analysis was performed using the iNMR software package (version 5.3.5, http://www.inmr.net). NMR chemical shifts are reported in ppm and referenced to the residual solvent peak as an internal standard (for CDCl3 δ 7.260 ppm (1H), δ 77.160 ppm (13C); for THF-d8 δ 1.720, 3.573 ppm (1H)).
GC analyses were performed on an Agilent 7890A GC instrument equipped with dual DB-5 columns (20 m × 180 μm × 0.18 μm) and dual FID detectors and with hydrogen as the carrier gas. The analysis method used in all cases was a 1 μL injection of sample, an injection temperature of 300 °C, and a 100:1 split ratio; the initial inlet pressure was 20.3 psi but was varied as the column flow was held constant at 1.8 mL/min for the duration of the run. The initial oven temperature of 50 °C was held for 0.46 min, followed by a temperature ramp up to 300 °C at 65 °C/min; finally the temperature was held at 300 °C for 0.69 min. The total run time was ∼5 min. The FID temperature was 325 °C. Dodecane (Aldrich) was used as an internal standard for GC analysis of catalytic reactions. Mesitylene (Aldrich) was used as an internal standard for GC analysis of stoichiometric reactions. GC analysis of reaction mixtures was accomplished by removal of a 25 μL aliquot with a 100 μL gastight syringe which was quenched with 50 μL of 1 M aqueous NaHSO4, diluted with ethyl ether or TBME (1 mL), and filtered through a short silica pad (2 cm) in a pipet packed with glass wool. The filtrate was analyzed by GC.
Thin-layer chromatography was performed on EMD Chemicals TLC silica gel 60 F254 plates. Visualization was accomplished with p-anisaldehyde stain after inspection under UV light. Flash chromatography was performed using EMD silica gel 60, particle size 0.040–0.063 mm, using standard flash techniques. Some compounds were purified using a Combiflash Rf 200 (Teledyne Isco) with Redisep Rf Gold normal-phase silica columns.
4,4′-Di-tert-butyl-2,2′-bipyridine (dtbbpy)
1H NMR (400 MHz; THF-d8): δ 8.56 (s, 2H), 8.51 (d, J = 5.1 Hz, 2H), 7.31 (d, J = 5.1 Hz, 2H), 1.37 (s, 18 H).
Ni(cod)2
1H NMR (400 MHz; THF-d8): δ 4.30 (br s, 4H), 2.11 (br s, 8H).
1,5-Cyclooctadiene (cod)
1H NMR (400 MHz; THF-d8): δ 5.49 (s, 4H), 2.32 (s, 8H).
(dtbbpy)Ni0(cod) (2)
In a nitrogen-filled glovebox, an oven-dried 1 dram vial was charged with Ni(cod)2 (10.0 mg, 0.0364 mmol), 4,4′-di-tert-butyl-2,2′-bipyridine (9.8 mg, 0.036 mmol), and dry, degassed THF-d8 (0.75 mL). The resulting mixture was swirled by hand to give a deep purple solution. The mixture was then quantitatively transferred (2 × 0.125 mL of THF-d8 wash) to a screw-capped NMR tube with a PTFE-coated septum and sealed. The NMR tube was shaken until the mixture was homogeneous and was allowed to sit overnight at 22 °C (room temperature). The solution of (dtbbpy)Ni0(cod) and free cod was characterized by 1H NMR, paramagnetic 1H NMR, and 13C NMR. Data for the complex matched those in our previous literature report.7b
(dtbbpy)NiII(C(O)R)Br
The NMR tube was returned to the glovebox, and a carboxylic acid bromide (0.0382 mmol, 1.05 equiv) was added. The color immediately changed to deep red in both cases, and the resulting mixture was characterized by 1H NMR, paramagnetic 1H NMR, IR, and UV–vis spectroscopy.22
(dtbbpy)Ni(C(O)C5H11)(Br) (1b)
The general procedure was followed with hexanoyl bromide23 (6.8 mg, 0.038 mmol). The yield was determined by 1H NMR: 72% (vs mesitylene, average of two runs), 74% (vs cyclooctadiene, average of two runs). 1H NMR (400 MHz; THF-d8): δ 9.09 (d, J = 4.1 Hz, 1H), 8.11 (m, 3H), 7.53–7.49 (m, 2H), 3.12 (t, J = 7.3 Hz, 2H), 1.64 (dt, J = 13.0, 6.3 Hz, 2H), 1.38 (s, 18H), 1.29–1.25 (m, 10H), 0.84 (t, J = 6.7 Hz, 3H). IR (cm–1): 1612 (C=O). UV–vis (THF) 1/λ (ε): 20325 cm–1 (5 × 103).
(dtbbpy)Ni(C(O)C6H4Me)(Br) (1c)
The general procedure was followed using 4-methylbenzoyl bromide24 (7.6 mg, 0.038 mmol). The yield was determined by 1H NMR: 95% (vs mesitylene, average of two runs), 89% (vs cyclooctadiene, average of two runs). The product mixture contained 7 mol % of toluoyl bromide, as evidenced by the characteristic doublets at 7.95 and 7.37 ppm. 1H NMR (400 MHz; THF-d8): δ 9.14 (d, J = 5.7 Hz, 1H), 8.37 (d, J = 8.0 Hz, 2H), 8.17 (d, J = 7.3 Hz, 2H), 7.74 (d, J = 6.0 Hz, 1H), 7.58 (d, J = 5.3 Hz, 1H), 7.29 (d, J = 4.3 Hz, 1H), 7.12 (d, J = 7.8 Hz, 2H), 2.32 (s, 3H), 1.41 (s, 9H), 1.34 (s, 9H). IR (cm–1): 1617 (C=O). UV–vis (THF) 1/λ (ε): 20576 cm–1 (7 × 103).
Reactivity Studies (Tables 2 and 3)
The NMR tube was returned to the glovebox, and dry, degassed DMA (1 mL, <16 ppm of H2O by KF titration) was added to the red solution, resulting in a small change to dark red-orange. The appropriate electrophile (0.291 mmol, 8 equiv) was added, and the NMR tube was again sealed, removed from the glovebox, and heated to 60 °C (oil bath). The reaction was judged complete when the vivid red-orange color of the nickel acyl halide complex had disappeared. The resulting reaction was quenched with diethyl ether (1 mL) and aqueous NaHSO4 (1 M, 0.5 μL), and the resulting mixture was filtered through a pad of silica gel before being analyzed by GC, GC/MS, and/or 1H NMR. See the Supporting Information for tables of all products produced in these reactions. Attempts to visualize the nickel products of these reactions were unsuccessful, and in some cases precipitation of unknown products occurred.
General Procedure for Aryl Bromides
A flame-dried 10 mL round-bottom flask (14/20 joint) containing a Teflon-coated magnetic stir bar was charged with NiCl2(dme) (11.5 mg, 0.053 mmol), 4,4′-di-tert-butyl-2,2′-bipyridine (14.5 mg, 0.054 mmol), anhydrous NaI (28.1 mg, 0.1875 mmol), and zinc flake (98.0 mg, 2 equiv). The round-bottom flask was then fitted with a septum (14/20) and sealed tightly with a double strand of 20 gauge copper wire. The round-bottom flask was purged with nitrogen (1 min), and 2 mL of anhydrous DMA (<70 ppm of H2O by KF titration) was added by syringe. The resulting mixture was stirred for 1 h at room temperature (∼21 °C) until the supernatant had turned dark gray. Alkyl bromide (1.69 mmol, 2.25 equiv), a solution of aryl halide (0.75 mmol in 1 mL of anhydrous DMA), and Fe(CO)5 (35 μL, 52 mg, 0.27 mmol) were added sequentially by syringe. The reaction mixture was stirred (1000 rpm) at 60 °C (oil bath) until judged complete by GC analysis (less than 1% aryl bromide remaining, 17–36 h). The resulting mixture was filtered through a 1 cm silica gel pad and the pad rinsed with diethyl ether (∼100 mL). The collected organic layer was then washed with 60 mL of water, and the aqueous layer was extracted with diethyl ether (3 × 15 mL). The combined organic extracts were dried over MgSO4, filtered, and concentrated in vacuo. The resulting residue was purified by flash chromatography (SiO2) to yield the desired ketone products.
Ethyl 5-(Phenyl)-5-oxopentanoate (22) [73172-56-2].25
The general procedure was followed with bromobenzene (80 μL, 0.75 mmol) and ethyl 4-bromobutyrate (240 μL, 1.69 mmol). The reaction was judged complete after 18 h. Following chromatography (1/10 ethyl acetate/hexanes, Rf = 0.25, UV and p-anisaldehyde visualization), the product was isolated as a light yellow oil (103.6 mg, 63% yield). 1H NMR (400 MHz; CDCl3): δ 7.97–7.95 (m, 2H), 7.55 (tt, J = 7.4, 1.6 Hz, 1H), 7.47–7.43 (m, 2H), 4.13 (q, J = 7.1 Hz, 2H), 3.05 (t, J = 7.2 Hz, 2H), 2.43 (t, J = 7.2 Hz, 2H), 2.07 (quintet, J = 7.2 Hz, 2H), 1.25 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz; CDCl3): δ 199.6, 173.4, 136.9, 133.2, 128.7, 128.2, 60.5, 37.6, 33.5, 19.5, 14.4. IR (cm–1): 1728 (s, C=O, ester), 1682 (s, C=O, aryl ketone). EIMS (70 eV): m/z 220 (M+).
Ethyl 5-(4-Chlorophenyl)-5-oxopentanoate (23) [54029-03-7].26
The general procedure was followed using 4-chlorobromobenzene (143.6 mg, 0.75 mmol) and ethyl 4-bromobutyrate (240 μL, 1.68 mmol). The reaction was judged complete after 17 h. Following chromatography (6/1 hexanes/ethyl acetate, Rf = 0.42, stained purple with p-anisaldehyde stain), the product was isolated as a white crystalline solid (118.3 mg, 62%). Mp: 52–55 °C (lit.26 mp 34–36 °C). 1H NMR (500 MHz; CDCl3): δ 7.90 (d, J = 8.6 Hz, 2H), 7.43 (d, J = 8.5 Hz, 2H), 4.14 (q, J = 7.1 Hz, 2H), 3.02 (t, J = 7.2 Hz, 2H), 2.43 (t, J = 7.1 Hz, 2H), 2.09–2.03 (m, 2H), 1.25 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz; CDCl3): δ 198.4, 173.3, 139.7, 135.3, 129.6, 129.1, 60.6, 37.6, 33.4, 19.5, 14.4. IR (cm–1): 1724 (s, C=O, ester), 1686 (s, C=O, aryl ketone). EIMS (70 eV): m/z 254 (M+).
Ethyl 5-(3,5-Dimethoxyphenyl)-5-oxopentanoate (24) [898758-62-8]
The general procedure was followed using 1-bromo-3,5-dimethoxybenzene (162.8 mg, 0.75 mmol) and ethyl 4-bromobutyrate (240 μL, 1.68 mmol). The reaction was judged complete after 36 h. Following chromatography (23% ethyl acetate in hexanes, Rf = 0.40, stained purple with p-anisaldehyde stain), the product was isolated as a light pink amorphous solid (130.8 mg, 62%). Mp: 40.5–45 °C. 1H NMR (400 MHz; CDCl3): δ 7.09 (d, J = 2.3 Hz, 2H), 6.64 (t, J = 2.2 Hz, 1H), 4.14 (q, J = 7.1 Hz, 2H), 3.83 (s, 6H), 3.00 (t, J = 7.2 Hz, 2H), 2.42 (t, J = 7.2 Hz, 2H), 2.05 (quintet, J = 7.2 Hz, 2H), 1.25 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz; CDCl3): δ 198.5, 172.8, 160.5, 138.5, 105.8, 105.3, 60.6, 55.8, 38.0, 33.8, 20.0, 14.7. IR (cm–1): 1728 (s, C=O, ester), 1670 (s, C=O, aryl ketone). EIMS (70 eV): m/z 280 (M+).
Ethyl 5-(2-Methoxy-5-fluorophenyl)-5-oxopentanoate (25) [951889-85-3]
The general procedure was followed using 2-bromo-5-fluoroanisole (97 μL, 0.75 mmol) and ethyl 4-bromobutyrate (240 μL, 1.68 mmol). The reaction was judged complete after 22 h. Following chromatography (8/1 hexanes/ethyl acetate, Rf = 0.20, stained purple with p-anisaldehyde stain), the product was isolated as a white amorphous solid (132.1 mg, 62%). Mp: 30–33 °C. 1H NMR (400 MHz; CDCl3): δ 7.40 (dd, J = 7.1, 2.6 Hz, 1H), 7.14 (ddd, J = 7.2, 5.9, 2.6 Hz, 1H), 6.90 (dd, J = 7.3, 3.2 Hz, 1H), 4.12 (q, J = 5.7 Hz, 2H), 3.87 (s, 3H), 3.02 (t, J = 5.7 Hz, 2H), 2.37 (t, J = 5.9 Hz, 2H), 2.00 (quintet, J = 5.8 Hz, 2H), 1.23 (t, J = 5.7 Hz, 3H). 13C NMR (126 MHz; CDCl3): δ 200.3, 173.3, 156.9 (d, J = 240 Hz), 154.9 (d, J = 2.3 Hz), 129.2 (d, J = 5.7 Hz), 119.7 (d, J = 23.6 Hz), 116.4 (d, J = 23.8 Hz), 113.1 (d, J = 7.1 Hz), 60.3, 56.2, 42.7, 33.6, 19.6, 14.3. 19F NMR (376 MHz; CDCl3): δ −60.66. IR (cm–1): 1728 (s, C=O, ester), 1674 (s, C=O, aryl ketone). EIMS (70 eV): m/z 268 (M+).
Ethyl 5-(4-Trifluoromethylphenyl)-5-oxopentanoate (26) [898777-81-6].27
The general procedure was followed using 4-bromobenzotrifluoride (105 μL, 0.75 mmol) and ethyl 4-bromobutyrate (240 μL, 1.68 mmol). The reaction was judged complete after 24 h. Following chromatography (15% ethyl acetate in hexanes, Rf = 0.40, stained purple with p-anisaldehyde stain), the product was isolated as a white amorphous solid (102.9 mg, 48%). Mp: 29–32.5 °C. 1H NMR (500 MHz; CDCl3): δ 8.07 (d, J = 8.0 Hz, 2H), 7.73 (d, J = 8.1 Hz, 2H), 4.14 (q, J = 7.1 Hz, 2H), 3.08 (t, J = 7.1 Hz, 2H), 2.44 (t, J = 7.1 Hz, 2H), 2.08 (quintet, J = 7.1 Hz, 2H), 1.26 (t, J = 7.1 Hz, 4H). 13C NMR (126 MHz; CDCl3): δ 198.5, 173.3, 139.5, 134.5 (q, J = 32.7 Hz), 128.5, 125.8 (q, J = 3.7 Hz), 123.7 (q, J = 273 Hz), 60.6, 37.9, 33.3, 19.3, 14.3. 19F NMR (376 MHz; CDCl3): δ −0.38. IR (cm–1): 1728 (s, C=O, ester), 1674 (s, C=O, aryl ketone). EIMS (70 eV): m/z 288 (M+).
1-Phenyl-4-(tert-butyldimethylsilyloxy)-1-butanone (27) [143878-47-1]
The general procedure was followed using bromobenzene (80 μL, 0.75 mmol) and 3-(tert-butyldimethylsilyloxy)-1-bromopropane (427 mg, 1.68 mmol). The reaction was judged complete after 18 h. Following chromatography (4% diethyl ether in pentane, Rf = 0.22, stained yellow with p-anisaldehyde stain), the product was isolated with 3% benzophenone impurity as a clear, colorless oil (135.7 mg, 65%). 1H NMR (500 MHz; CDCl3): δ 7.96 (d, J = 7.7 Hz, 2H), 7.54 (t, J = 7.4 Hz, 1H), 7.44 (t, J = 7.7 Hz, 2H), 3.70 (t, J = 6.0 Hz, 2H), 3.05 (t, J = 7.2 Hz, 2H), 1.95 (quintet, J = 6.7 Hz, 2H), 0.88 (s, 9H), 0.03 (s, 6H). 13C NMR (126 MHz; CDCl3): δ 200.4, 137.3, 133.0, 128.7, 128.2, 62.4, 35.0, 27.6, 26.1, 18.5, −5.2. IR (cm–1): 1686 (C=O aryl ketone). EIMS (70 eV): m/z 221 (M+ – t-Bu).
Acknowledgments
This work was supported by the NIH (R01 GM097243). A.C.W. thanks the University of Rochester for a Weisburger Fellowship. R.D.R. was an NSF REU summer student at the University of Rochester (CHE-1156340). D.J.W. is an Alfred P. Sloan Research Fellow. We are grateful to Jill Caputo (University of Rochester) for experimental assistance with Table 4.
Supporting Information Available
Tables S1 and S2, giving the full product distribution for Tables 2 and 3, respectively, pictures of the the reaction of 1c with electrophiles, and figures giving NMR spectra for 1b,c, 3, and 22–27. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Reviews:; a Colquhoun H. M.; Thompson D. J.; Twigg M. V.. Carbonylation, Direct Synthesis of Carbonyl Compounds; Plenum Press: New York, 1991. [Google Scholar]; b Wu X.-F.; Neumann H.; Beller M. Chem. Soc. Rev. 2011, 40, 4986–5009. [DOI] [PubMed] [Google Scholar]
- Knochel P.Handbook of Functionalized Organometallics: Applications in Synthesis; Wiley-VCH: Weinheim,Germany, 2005; p 653. [Google Scholar]
- For C(sp2)–C(sp3) bond formation:; a Everson D. A.; Shrestha R.; Weix D. J. J. Am. Chem. Soc. 2010, 132, 920–921. [DOI] [PubMed] [Google Scholar]; b Amatore M.; Gosmini C. Chem. Eur. J. 2010, 16, 5848–5852. [DOI] [PubMed] [Google Scholar]; c Anka-Lufford L. L.; Prinsell M. R.; Weix D. J. J. Org. Chem. 2012, 77, 9989–10000. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Everson D. A.; Jones B. A.; Weix D. J. J. Am. Chem. Soc. 2012, 134, 6146–6159. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Wang S.; Qian Q.; Gong H. Org. Lett. 2012, 14, 3352–3355. [DOI] [PubMed] [Google Scholar]; f Yan C.-S.; Peng Y.; Xu X.-B.; Wang Y.-W. Chem. Eur. J. 2012, 18, 6039–6048. [DOI] [PubMed] [Google Scholar]; g Biswas S.; Weix D. J. J. Am. Chem. Soc. 2013, 135, 16192–16197. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Buonomo J. A.; Everson D. A.; Weix D. J. Synthesis 2013, 45, 3099–3102. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Everson D. A.; George D. T.; Weix D. J.; Buergler J. F.; Wood J. L. Org. Synth. 2013, 90, 200–214. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Zhao Y.; Weix D. J. J. Am. Chem. Soc. 2014, 136, 48–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Marzouk H.; Rollin Y.; Folest J. C.; Nédélec J. Y.; Périchon J. J. Organomet. Chem. 1989, 369, C47–C50. [Google Scholar]; b Amatore C.; Jutand A.; Périchon J.; Rollin Y. Monatsh. Chem. 2000, 131, 1293–1304. [Google Scholar]; c Wotal A. C.; Weix D. J. Org. Lett. 2012, 14, 1476–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Wu F.; Lu W.; Qian Q.; Ren Q.; Gong H. Org. Lett. 2012, 14, 3044–3047. [DOI] [PubMed] [Google Scholar]; e Yin H.; Zhao C.; You H.; Lin K.; Gong H. Chem. Commun. 2012, 48, 7034–7036. [DOI] [PubMed] [Google Scholar]; f Cherney A. H.; Kadunce N. T.; Reisman S. E. J. Am. Chem. Soc. 2013, 135, 7442–7445. [DOI] [PubMed] [Google Scholar]
- Other disconnections include the following. Conjugate addition:; a Shrestha R.; Weix D. J. Org. Lett. 2011, 13, 2766–2769. [DOI] [PubMed] [Google Scholar]; b Millan A.; Martin-Lasanta A.; Miguel D.; Cienfuegos L. A. d.; Cuerva J. M. Chem. Commun. 2011, 47, 10470–10472. [DOI] [PubMed] [Google Scholar]; c Shrestha R.; Dorn S. C. M.; Weix D. J. J. Am. Chem. Soc. 2013, 135, 751–762. [DOI] [PMC free article] [PubMed] [Google Scholar]; Cross-Wurtz coupling:; d Yu X.; Yang T.; Wang S.; Xu H.; Gong H. Org. Lett. 2011, 13, 2138–2141. [DOI] [PubMed] [Google Scholar]; e Xu H.; Zhao C.; Qian Q.; Deng W.; Gong H. Chem. Sci. 2013, 4, 4022–4029. [Google Scholar]; Coupling of RX with CO2:; f Troupel M.; Rollin Y.; Perichon J.; Fauvarque J. F. Nouv. J. Chim. 1981, 5, 621–625. [Google Scholar]; g Fauvarque J. F.; Chevrot C.; Jutand A.; François M.; Perichon J. J. Organomet. Chem. 1984, 264, 273–281. [Google Scholar]; h Correa A.; Martín R. J. Am. Chem. Soc. 2009, 131, 15974–15975. [DOI] [PubMed] [Google Scholar]; i Fujihara T.; Nogi K.; Xu T.; Terao J.; Tsuji Y. J. Am. Chem. Soc. 2012, 134, 9106–9109. [DOI] [PubMed] [Google Scholar]
- a Ocafrain M.; Devaud M.; Troupel M.; Perichon J. J. Chem. Soc., Chem. Commun. 1995, 2331–2332. [Google Scholar]; b Dolhem E.; Oçafrain M.; Nédélec J. Y.; Troupel M. Tetrahedron 1997, 53, 17089–17096. [Google Scholar]; c Oçafrain M.; Dolhem E.; Nedelec J.; Troupel M. J. Organomet. Chem. 1998, 571, 37–42. [Google Scholar]; d Troupel M.; Oçafrain M.; Dolhem E.; Folest J.-C.; Barhdadi R. Can. J. Chem. Eng. 1998, 76, 1013–1019. [Google Scholar]; e Oçafrain M.; Devaud M.; Nédelec J. Y.; Troupel M. J. Organomet. Chem. 1998, 560, 103–107. [Google Scholar]; f Dolhem E.; Barhdadi R.; Folest J.; Nedelec J.; Troupel M. Tetrahedron 2001, 57, 525–529. [Google Scholar]
- a Amatore C.; Jutand A.; Périchon J.; Rollin Y. Monatsh. Chem. 2000, 131, 1293–1304. [Google Scholar]; b Biswas S.; Weix D. J. J. Am. Chem. Soc. 2013, 135, 16192–16197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potential coupling products: three dicarbonyls (R1(CO)2R1, R2(CO)2R1, R2(CO)2R2), three ketone products (R1C(O)R1, R1(CO)R2, R2(CO)R2), three non-carbonylated products (R1R1, R1R2, R2R2). Other side products: one β-hydride elimination product (olefin from R2X), two hydrodehalogenation products (R1H, R2H), two carboxylic acids (R1CO2H, R2CO2H).
- a Bauld N. L. Tetrahedron Lett. 1963, 4, 1841–1845. [Google Scholar]; b Chiusoli G. P.; Cassar L. Angew. Chem., Int. Ed. Engl. 1967, 6, 124–133. [Google Scholar]; c Yoshisato E.; Tsutsumi S. J. Org. Chem. 1968, 33, 869–871. [Google Scholar]; d Hirota Y.; Ryang M.; Tsutsumi S. Tetrahedron Lett. 1971, 12, 1531–1534. [Google Scholar]; e Rhee I.; Ryang M.; Watanabe T.; Omura H.; Murai S.; Sonoda N. Synthesis 1977, 1977, 776–777. [Google Scholar]; f .
- Semmelhack M. F. Org. React. 1972, 19, 115–198. [Google Scholar]
- Only two common US research chemical suppliers stock Ni(CO)4: Strem, Inc. ($1661/mol, 450 g) and Pfaltz & Bauer ($3434/mol, 450 g). It is supplied in a lecture bottle (bp 42 °C).
- Reike developed a system of activated nickel and oxaylyl chloride that avoided preformed nickel carbonyl:Inaba S.-i.; Rieke R. D. Chem. Lett. 1984, 13, 25–28. [Google Scholar]
- Although iron pentacarbonyl should also be handled with care in a well-ventilated fume hood, it is sold by most large chemical suppliers in quantity (∼$34–69/mol, 1 kg) and is a relatively stable liquid (bp 103 °C).
- For a review of alternative CO sources, see:Morimoto T.; Kakiuchi K. Angew. Chem., Int. Ed. 2004, 43, 5580–5588. [DOI] [PubMed] [Google Scholar]
- a Klein H.-F. Angew. Chem., Int. Ed. Engl. 1973, 12, 402–402. [Google Scholar]; b Carmona E.; González F.; Poveda M. L.; Atwood J. L.; Rogers R. D. Dalton Trans. 1980, 2108–2116. [Google Scholar]; c Yamamoto T.; Kohara T.; Yamamoto A. Bull. Chem. Soc. Jpn. 1981, 54, 2161–2168. [Google Scholar]; d Carmona E.; Marin J. M.; Paneque M.; Poveda M. L. Organometallics 1987, 6, 1757–1765. [Google Scholar]; e Carmona E.; Paneque M.; Poveda M. L. Polyhedron 1989, 8, 285–291. [Google Scholar]; f Belderrain T. R.; Knight D. A.; Irvine D. J.; Paneque M.; Poveda M. L.; Carmona E. J. Chem. Soc., Dalton Trans. 1992, 1491–1495. [Google Scholar]
- a Otsuka S.; Nakamura A.; Yoshida T. J. Am. Chem. Soc. 1969, 91, 7196–7198. [Google Scholar]; b Corain B.; Mattinelli M. Inorg. Nucl. Chem. Lett. 1972, 8, 39–43. [Google Scholar]; c Otsuka S.; Nakamura A.; Yoshida T.; Naruto M.; Ataka K. J. Am. Chem. Soc. 1973, 95, 3180–3188. [Google Scholar]; d Corain B.; Favero G. J. Chem. Soc., Dalton Trans. 1975, 283–285. [Google Scholar]; e Fahey D. R.; Mahan J. E. J. Am. Chem. Soc. 1977, 99, 2501–2508. [Google Scholar]; f Bochmann M.; Hawkins I.; Hursthouse M. B.; Short R. L. J. Chem. Soc., Dalton Trans. 1990, 1213–1219. [Google Scholar]; g Zell T.; Radius U. Z. Chem. 2013, 639, 334–339. [Google Scholar]
- Complete tables of all products formed in Tables 2 and 3 can be found in the Supporting Information.
- Aryl bromides with stronger electron-withdrawing groups provided still lower yields of cross-coupled ketone under these conditions. Expansion of the synthetic utility of this coupling chemistry is ongoing.
- Ward L. G. L.; Pipal J. R. Inorg. Synth. 1971, 13, 154–164. [Google Scholar]
- Hadda T. B.; Le Bozec H. Polyhedron 1988, 7, 575–577. [Google Scholar]
- Buonomo J. A.; Everson D. A.; Weix D. J. Synthesis 2013, 45, 3099–3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attempts to obtain clean 13C NMR spectra were unsuccessful.
- Bernady K. F.; Floyd M. B.; Poletto J. F.; Schaub R. E.; Weiss M. J.. Novel 11-hydroxy-9-keto-5,6-cis-13,14-cis-prostadienoic acid derivatives. US 4123456 A, 1978.
- Coulson E. A. Synthesis 1973, 10, 612–613. [Google Scholar]
- Yasuda M.; Chiba K.; Ohigashi N.; Katoh Y.; Baba A. J. Am. Chem. Soc. 2003, 125, 7291–7300. [DOI] [PubMed] [Google Scholar]
- Yang X.-H.; Xie J.-H.; Liu W.-P.; Zhou Q.-l. Angew. Chem., Int. Ed. 2013, 52, 7833–7836. [DOI] [PubMed] [Google Scholar]
- Ebel H.; Frattini S.; Giovannini R.; Hoenke C.; Scheuerer S.. Ccr2 receptor antagonists. WO2011147772 A1, 2011.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.