

Abstract
Krüppel-like factor 5 (KLF5) regulates multiple biologic processes. Its function in tumorigenesis appears contradictory though, showing both tumor suppressor and tumor promoting activities. In this study, we examined whether and how Klf5 functions in prostatic tumorigenesis using mice with prostate-specific deletion of Klf5 and phosphatase and tensin homolog (Pten), both of which are frequently inactivated in human prostate cancer. Histologic analysis demonstrated that when one Pten allele was deleted, which causes mouse prostatic intraepithelial neoplasia (mPIN), Klf5 deletion accelerated the emergence and progression of mPIN. When both Pten alleles were deleted, which causes prostate cancer, Klf5 deletion promoted tumor growth, increased cell proliferation, and caused more severe morphologic and molecular alterations. Homozygous deletion of Klf5 was more effective than hemizygous deletion. Unexpectedly, while Pten deletion alone expanded basal cell population in a tumor as reported, Klf5 deletion in the Pten-null background clearly reduced basal cell population while expanding luminal cell population. Global gene expression profiling, pathway analysis, and experimental validation indicate that multiple mechanisms could mediate the tumor-promoting effect of Klf5 deletion, including the up-regulation of epidermal growth factor and its downstream signaling molecules AKT and ERK and the inactivation of the p15 cell cycle inhibitor. KLF5 also appears to cooperate with several transcription factors, including CREB1, Sp1, Myc, ER and AR, to regulate gene expression. These findings validate the tumor suppressor function of KLF5. They also yield a mouse model that shares two common genetic alterations with human prostate cancer—mutation/deletion of Pten and deletion of Klf5.
Introduction
Prostate cancer is a common malignancy and a leading cause of cancer-related deaths. As in many other types of cancers, prostatic carcinogenesis is a multistep process that results from the accumulation of multiple genetic and epigenetic alterations. Identification and functional characterization of the cancer-causing alterations provides important molecular insights into how a cancer forms and improves cancer detection and treatment. While a number of genes have been identified with genetic/epigenetic alterations and causal roles in cancer, more such genes remain to be discovered and functionally examined. Chromosomal deletion is one of the most common genetic alterations in human cancer, and one common consequence of chromosomal deletion is the inactivation of tumor suppressor genes during carcinogenesis [1]. In prostate cancer, a number of commonly deleted chromosomal regions have been mapped, and one or more tumor suppressor genes have been identified for some of the commonly deleted regions, including phosphatase and tensin homolog (PTEN) from 10q22, NKX3-1 from 8p21, and ATBF1 from 16q22 [1], [2], [3], [4]. Key evidence for the tumor suppressor activity of a gene is the induction of neoplastic alteration after its deletion in mice, as seen for a number of tumor suppressor genes including Pten, Nkx3-1, p53, Rb, and Smad4 [5], [6], [7], [8].
Krüppel-like factor 5 (KLF5, also known as BTEB2), encoding a basic transcription factor and located at the q21 band of human chromosome 13 (13q21), centers the second most frequently deleted chromosomal region in human prostate cancer, as revealed by comparative genomic hybridization studies [1], [9], [10]. The deletion is exclusively hemizygous, causing the haploinsufficiency of KLF5 during cancer development [10], [11], [12]. In addition, KLF5 protein is degraded by the ubiquitin proteasome pathway, and one E3 ubiquitin ligase that degrades KLF5, WWP1, is amplified and overexpressed in human prostate and breast cancers, causing excessive protein degradation and functional insufficiency of KLF5 [13], [14], [15]. These findings indicate that KLF5 is frequently inactivated during human carcinogenesis and thus could be a tumor suppressor gene, and some functional studies indeed support a tumor suppressor function of KLF5. For example, KLF5 can inhibit the proliferation of epithelial cells including some cancer cell lines in culture [10], [11], [16], [17] and suppresses the tumorigenesis of human prostate cancer cell lines in nude mice [18]. However, KLF5 has also been demonstrated to promote the tumorigenesis of a bladder cancer cell line in a xenograft model [19] and mediate or be necessary for intestinal tumorigenesis induced by other oncogenic events in genetically modified mice [20], [21], [22], [23]. Our recent study indicates that KLF5 indeed can be both tumor suppressing and tumor promoting in the same cancer cell lines in xenograft models and that the acetylation status of KLF5 determines its function in tumorigenesis [24]. Nevertheless, whether frequent deletion of KLF5 in human prostate cancer has any functional consequences remains uncertain, because our previous study demonstrated that, in mouse prostates, hemizygous deletion of Klf5 alone increases cell proliferation and induces hyperplasia but does not cause neoplastic alterations such as mouse prostatic intraepithelial neoplasia (mPIN) or tumor [25], and homozygous deletion of Klf5 causes apoptosis rather than increases cell proliferation and does not induce any noticeable histologic alterations [25]. Therefore, a tumor suppressor function has not been established for KLF5 using genetically modified mice.
PTEN, a protein and lipid phosphatase that negatively regulates phosphoinositide 3-kinase (PI3K)/AKT oncogenic signaling, is a tumor suppressor that undergoes frequent mutations and deletions in human tumors including prostate cancer [26]. Activation of PI3K/AKT signaling by PTEN inactivation stimulates cell cycle progression and survival and, consequently, induces tumorigenesis [27], as demonstrated by the induction of prostate cancer after Pten homozygous deletion in mice [5]. Similar to KLF5, chromosomal deletion is also a common mechanism for the loss of PTEN function in different types of human cancers including prostate cancer [1], [26], [28]. As two of the most commonly deleted genetic loci in human prostate cancer, deletion of both PTEN and KLF5 can occur in the same tumors. For example, the commonly used human prostate cancer cell line LNCaP has the deletion of both PTEN and KLF5 [10], [29]. It is thus possible that simultaneous deletions of KLF5 and PTEN cooperate to initiate and promote tumorigenesis.
In this study, we tested whether and how simultaneous deletions of Klf5 and Pten interact to initiate and promote prostatic tumorigenesis in mice. We found that deletion of Klf5 accelerated the emergence and progression of mPIN induced by hemizygous deletion of Pten. In the Pten-null background, Klf5 deletion increased cell proliferation and promoted tumorigenesis, as indicated by more severe morphology. Interestingly, while Pten deletion alone increased the ratio of basal to luminal cells in a tumor as previously reported, Klf5 deletion in the Pten-null background increased luminal but diminished basal cell population in tumors. Mechanistically, Klf5 deletion dysregulated a large number of genes and signaling pathways, including the up-regulation of extracellular growth factor, epidermal growth factor (EGF), and its downstream signaling molecules AKT and extracellular signal-regulated kinases (ERK) and the inhibition of the p15 cell cycle inhibitor. Several transcription factors appear to cooperate with KLF5 to function in the prostate. These findings provide in vivo evidence for the tumor suppressor activity of KLF5. They also provide a mouse model of prostate cancer that shares two common genetic alterations with human prostate cancer.
Materials and Methods
Mouse Strains
The Klf5 floxed mouse strain, in which the genomic DNA from exon 2 to exon 3 of Klf5 is flanked by loxP sites, was generated as described in our previous study [25]. The PB-Cre4 transgenic mouse strain, where expression of Cre recombinase is driven by the prostatic epithelial specific probasin promoter [30], was purchased from the NCI Mouse Models of Human Cancers Consortium (MMHCC, Frederick, MD; Cat. No. 01XF5). Pten floxed mice were purchased from the Jackson Laboratory (Bar Harbor, ME; Cat. No. 004597). All mice were maintained in a mixed background of C57BL/6 J and 129Sv/J.
Mouse Breeding and Genotyping
The breeding strategy is shown in Figure 1A. Floxed-Klf5 mice (Klf5flox/flox) were crossed with floxed-Pten mice (Ptenflox/flox), and the F1 mice were then crossed with PB-Cre4 mice to generate the following six desired genotypes: Cre+/Ptenflox/wt/Klf5wt/wt, Cre+/Ptenflox/wt/Klf5flox/wt, Cre+/Ptenflox/wt/Klf5flox/flox, Cre+/Ptenflox/flox/Klf5wt/wt, Cre+/Ptenflox/flox/Klf5flox/wt, and Cre+/Ptenflox/flox/Klf5flox/flox.
Figure 1.
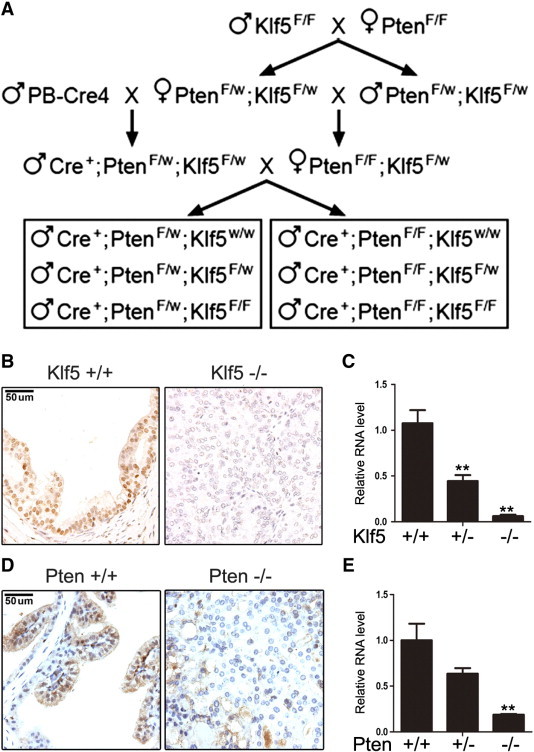
Breeding strategy and deletion confirmation for prostatic knockout of Klf5 and Pten. (A) Schematic of breeding design for the generation of mice with all six desired genotypes of Klf5 and Pten using mouse strains of floxed Klf5 (Klf5F/F), floxed Pten (PtenF/F) ,and PB-Cre4. F and W indicate floxed and wild-type alleles, respectively. (B and C) Detection of Klf5 protein expression by IHC staining in 6-month-old APs (B) and Klf5 mRNA expression by real-time RT-PCR in the whole prostate of 6-month-old mice (C) with different Klf5 deletion status and Pten homozygous deletion. (D and E) Detection of Pten protein expression by IHC staining in 6-month-old APs (D) and Pten mRNA expression by real-time RT-PCR in the whole prostate of 6-month-old mice (E) with different Pten deletion status. + and − indicate wild-type and deleted Klf5 or Pten alleles, respectively; * and ** indicate P < .05 and P < .01, respectively, based on Student's t test.
All mice were toe-clipped for identification and tail-clipped to collect tissues for genotyping at age 10 to 14 days. Freshly dissected tissues were washed in phosphate-buffered saline (PBS) to clean blood and incubated overnight at 56°C with tissue lysis buffer (100 mM Tris-HCl, 5 mM EDTA, 200 mM NaCl, 0.2% sodium dodecyl sulfate, 500 μg/ml proteinase K, pH 8.0) to release DNA. Diluted tissue lysates were used as templates for polymerase chain reaction (PCR)–based genotyping. PCR conditions were previously described [31]. Primer sequences and sizes of PCR products for genotyping the different genes are listed in Table S1.
RNA Extraction and Real-Time PCR Assay
For mouse tissues, freshly dissected prostate tissues were immediately immersed into the RNAlater solution (Qiagen, Valencia, CA) for at least 16 hours at 4°C. Total RNA was then isolated using the RNeasy Mini Kit (Qiagen), following the manufacturer’s instructions. For cultured cells, total RNA was isolated using the TRIzol reagent (Invitrogen, Carlsbad, CA) following the procedures established previously [32]. The first strand of cDNA was synthesized using the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA).
Real-time reverse transcription (RT)–PCR was performed using the SYBR Premix Ex Taq reagent (Takara Otsu, Shiga, Japan) with the ABI Prism 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA). The 2(− ΔΔCt) method was used to calculate the relative fold change for a gene’s expression, with the expression level of GAPDH as an internal control. Primers for real-time PCR assays are listed in Table S1.
Microarray-Based Expression Profiling and Signaling Pathway Analysis
The microarray experiment was performed at the Emory Integrated Genomics Core using standard procedures. Briefly, total RNA was extracted from dorsal prostates (DPs) of four pairs of 6-month-old mice with the genotype of Pten−/−;Klf5+/+ or Pten−/−;Klf5−/−. The quality of total RNA was confirmed using an Agilent Bioanalyzer (Agilent Technologies, Palo Alto, CA). After synthesis from the same amount of total RNA, the cDNA library for a tissue sample was hybridized to the Affymetrix Mouse Gene 1.0 ST Array, which covers 28,853 genes with approximately 27 probes per gene spread across the full length of each gene. Hybridization and washing and staining of probe arrays were performed according to Affymetrix’s protocol. GeneChips were scanned using the Affymetrix 3000 scanner and images were converted to digital data, which were then analyzed by using the GeneSpring GX program (Agilent Technologies). The initial list of genes was generated with the preliminary data using mean polish and robust multi-array average (RMA) with GC content adjustment.
Genes were sorted by their P values between the wild-type Klf5 and Klf5-null groups, and those with P < .05 were considered differentially expressed. After ranking by the fold change of expression, differentially expressed genes were categorized into the following seven groups: 1.5 to 2.0, 2.0 to 2.5, 2.5 to 3.0, 3.0 to 4.0, 4.0 to 5.0, 5.0 to 10, and > 10 fold change of expression. For each group, four to seven genes were chosen and subjected to real-time RT-PCR to validate their differential expression with four to eight RNA samples, including the four used for microarray hybridization. On the basis of published studies using the same gene arrays [33], [34], [35] and the validation of 31 of 33 genes, we set the threshold of fold change to 1.5 to establish the list of differentially expressed genes for pathway and ontology analyses. The original microarray data have been deposited in National Center for Biotechnology Information (NCBI)’s Gene Expression Omnibus database and are accessible through Gene Expression Omnibus Series Accession No. GSE58719.
Differentially expressed genes and their expression levels were uploaded into the web-based MetaCore program (http://thomsonreuters.com/metacore), and different algorithms were run with MetaCore database to identify or build signaling pathways, gene ontology (GO)–based functional processes, and molecular networks that are modulated by Klf5 deletion. For each of the 586 genes identified as prostate cancer–related genes by the Disease (by biomarkers) Ontology of MetaCore, a PubMed search with a gene’s name and “prostate cancer” as search terms indicated that 203 of the genes had been studied in prostate cancer. The publications were evaluated to identify those genes that have been shown to functionally impact prostate cancer cell behavior [proliferation, apoptosis, migration, epithelial-to-mesenchymal transition (EMT), metastasis, and so on], undergo promoter methylation, or have expression abnormalities that are associated with clinical or pathologic characteristics of prostate cancer. The resultant 172 genes and their normalized log2 values of intensity differences between Klf5 wild-type and Klf5-null groups were then subjected to the R software to generate a heat map.
Genes dysregulated by Klf5 deletion in this study were also compared to those identified in previous studies [24], [33], [34], [35] to identify the genes that are regulated by Klf5 in different tissues or experimental systems.
Histopathologic Analysis
Mice with different Klf5 and Pten deletion status were sacrificed at various time points (4, 6, 9, 12, 15, and 18 months for the Pten+/− group and 2, 4, and 6 months for the Pten−/− group), and prostates were freshly collected and dissected in cold PBS. Tissues for histopathologic analysis were fixed in 3.7% neutral buffered formaldehyde overnight. Fixed tissues were then embedded in paraffin, sectioned at 5-μm thickness, and stained with hematoxylin and eosin (H&E) following standard protocols.
Pathologic diagnosis was performed by pathologists led by Dr Robert D. Cardiff through a paid service at the Center for Comparative Medicine, Department of Pathology, University of California at Davis. Published guidelines were followed [36], [37].
Immunohistochemical and Immunofluorescence Staining
For immunohistochemical (IHC) staining, tissue sections on glass slides were deparaffinized in xylene, rehydrated in graded ethanol solutions, and washed in tap water. Antigen retrieval was performed by boiling the slides in a pressure cooker for 3 minutes in a citrate buffer (10 mM trisodium citrate, pH 6.0). After 10-minute treatment with 3% H2O2, tissue sections were then blocked with 5% normal goat serum in Tris-buffered saline with 0.1% Tween-20 for 1 hour at room temperature, incubated with primary antibodies at 4°C overnight, and then incubated with EnVision Polymer-HRP secondary antibodies (Dako, Glostrup, Denmark) for 30 minutes at room temperature. After the application of DAB chromogen, tissue sections were stained with hematoxylin, dehydrated, and mounted. The slides were then scanned with a Hamamatsu NanoZoomer scanner (Hamamatsu Corporation, Bridgewater, NJ). For some proteins, cell numbers were counted to determine the positive rate.
For immunofluorescence (IF) staining, the same procedures of deparaffinization, rehydration, and antigen retrieval were followed. Tissue sections were then incubated with the blocking solution (10% normal goat serum and 1% BSA in PBS) for 1 hour at room temperature, with primary antibodies at 4°C overnight, and with secondary antibodies (Alexa Fluor dyes; Invitrogen) for 30 minutes at room temperature. After 4',6-diamidino-2-phenylindole (DAPI) staining for nuclei, slides were mounted with anti-photobleaching mounting medium. Multichannel pictures were taken with a Zeiss Axioplan 2 Wide-Field Microscope (Carl Zeiss Microscopy, Thornwood, NY).
The primary antibodies used in this study (IHC or IF) and dilutions were given as follows: rabbit anti-KLF5 (IHC, 1:6000 dilution) [25], rabbit anti-PTEN (Cell Signaling Technology, Danvers, MA; IHC, 1:100), rabbit anti-Ki67 (Thermo Fisher Scientific, Waltham, MA; IHC, 1:200), rabbit anti–phospho-Akt (Cell Signaling Technology; IHC, 1:150), mouse anti–phospho-Erk1/2 (Cell Signaling Technology; IHC, 1:200), rabbit anti–phospho-mammalian target of rapamycin (mTOR) (Cell Signaling Technology; IHC, 1:100), rabbit anti–phospho-S6 (Cell Signaling Technology; IHC, 1:200), rabbit anti-p15/INK4B (Cell Signaling Technology; IHC, 1:1000), rabbit anti-CK5 (Covance, Princeton, NJ; IHC, 1:1000; IF, 1:200), mouse anti-Ck14 (Thermo Fisher Scientific; IF, 1:200), mouse anti-p63 (Santa Cruz Biotechnology, Santa Cruz, CA; IF, 1:100), rabbit anti-Ck18 (GeneTex, Irvine, CA; IHC, 1:1500; IF, 1:400), mouse anti–smooth muscle actin (Sma; Sigma-Aldrich, St Louis, MO; IHC, 1:5000), rabbit anti-EGF (Abcam, Cambridge, MA; IHC, 1:400), and rabbit anti–phospho–Egf receptor (Egfr) Y1068 (Cell Signaling Technology; IHC, 1:100).
Knockdown of KLF5 and PTEN by RNA Interference
The PNT2 human prostate luminal cell line, originated from SV40-immortalized normal prostate epithelium, was purchased from Sigma and cultured in RPMI 1640 supplemented with 10% FBS. PNT2 cells were transfected with siRNAs against KLF5, PTEN, or the control with the X-tremeGENE HP transfection reagent (Roche, Nutley, NJ) to knock down the expression of KLF5 or PTEN. Sequences of siRNAs were given as follows: siKLF5, AAGCUCACCUGAGGACUCATT [38]; siPTEN, AAGAUCUUGACCAAUGGCUAATT (Sigma); and siCtrl, AAUUCUCCGAACGUGUCACGUTT (Thermo Fisher Scientific).
Western Blot Analysis
Western blot analysis was performed following previously established procedures [32]. The antibody against human KLF5 was prepared as previously described [14], and those against other proteins are given as follow: rabbit anti-PTEN, rabbit anti–phospho-AKT, rabbit anti–phospho-ERK, rabbit anti-AKT, rabbit anti-ERK, and rabbit anti–phospho-S6 were from Cell Signaling Technology; and rabbit anti–β-actin, rabbit anti-p15, and rabbit anti-mouse Klf5 were from Sigma, Santa Cruz Biotechnology, and Millipore (Temecula, CA), respectively.
Detection of Apoptosis by the Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) Assay
The TumorTACS In Situ Apoptosis Detection Kit (Trevigen, Gaithersburg, MD) was used for the TUNEL assay following manufacturer’s instruction. Formalin-fixed prostate sections from 6-month-old mice were stained in triplicate for each genotype. Slides were then scanned, and the number of apoptotic cells and the number of total cells in the anterior and DPs were counted using ImageJ software to determine the percentage of apoptotic cells.
Statistical Analysis
Statistical analyses were performed using the GraphPad Instat statistical computer program (GraphPad Software, Inc, La Jolla, CA). One-way analysis of variance was used to determine statistical differences among groups in the analysis of Ki67-positive rate, while the Chi-square analysis was used for differences in phenotype changes among the three Klf5 genotypes in Table 1. P values less than .05 were considered statistically significant.
Table 1.
Numbers of Mice with Hyperplasia (HP) and mPIN in the AP, DP, LP, and VP at Different Ages (Months) with Hemizygous Deletion of Pten and Different Klf5 Deletion Status
| Klf5 Deletion | AP |
DP |
LP |
VP |
Total Mice | ||||
|---|---|---|---|---|---|---|---|---|---|
| HP | mPIN | HP | mPIN | HP | mPIN | HP | mPIN | ||
| 4 to 6 months | |||||||||
| +/+ | 2 | 1 | 3 | 0 | 2 | 1 | 3 | 0 | 3 |
| +/− | 8 | 0 | 3 | 5 | 1 | 7 | 8 | 0 | 8 |
| −/− | 5 | 2 | 3 | 4 | 3 | 4 | 7 | 0 | 7 |
| 9 to 12 months | |||||||||
| +/+ | 8 | 1 | 7 | 1 | 5 | 2 | 7 | 1 | 9 |
| +/− | 4 | 8 | 2 | 10** | 2 | 10* | 11 | 1 | 12 |
| −/− | 6 | 3 | 0 | 9** | 2 | 7 | 7 | 2 | 9 |
| 15 to 18 months | |||||||||
| +/+ | 4 | 7 | 0 | 11 | 4 | 6 | 11 | 0 | 11 |
| +/− | 2 | 13 | 0 | 15 | 4 | 11 | 13 | 2 | 15 |
| −/− | 3 | 7 | 1 | 9 | 1 | 8 | 8 | 1 | 10 |
| All ages | |||||||||
| +/+ | 14 | 9 | 10 | 12 | 11 | 9 | 21 | 1 | 23 |
| +/− | 14 | 21 | 5 | 30* | 7 | 28* | 32 | 3 | 35 |
| −/− | 14 | 12 | 4 | 22* | 6 | 19 | 22 | 3 | 26 |
Note: Chi-square analysis was used to determine the statistical significance of a phenotype caused by Klf5 deletion. + and − indicate the presence and absence, respectively, of a Klf5 allele in the prostate. *P < .05; **P < .01.
Results
Simultaneous Knockout of Pten and Klf5 in Mouse Prostates
Three strains of mice, including floxed Klf5, floxed Pten, and PB-Cre4, were used to generate the following six desired genotypes: Cre+/Ptenflox/wt/Klf5wt/wt, Cre+/Ptenflox/wt/Klf5flox/wt, Cre+/Ptenflox/wt/Klf5flox/flox, Cre+/Ptenflox/flox/Klf5wt/wt, Cre+/Ptenflox/flox/Klf5flox/wt, and Cre+/Ptenflox/flox/Klf5flox/flox (Figure 1A). These mice were divided into two groups according to Pten deletion status, i.e., hemizygous deletion (Pten+/−) and homozygous deletion (Pten−/−; Figure 1A). All three strains of mice have been successfully used in previous studies [5], [25], and efficient gene knockout in the prostate was confirmed by examining the expression of Pten and Klf5 at the mRNA and protein levels by real-time RT-PCR and IHC staining. The mRNA levels for both Klf5 and Pten were reduced by about half in tissues with hemizygous deletion and the reduction was dramatic in tissues with homozygous deletion (Figure 1, C and E). The protein expression of Klf5 or Pten was also significantly decreased on genomic deletion of the respective gene (Figures 1, B and D, and W1, and data not shown). These results validated the knockout system for further analyses.
Klf5 Deletion Accelerated the Development and Severity of mPIN Induced by Pten Hemizygous Deletion
Deletion of one Pten allele causes mPIN [5], so we first examined whether Klf5 deletion affects mPIN development induced by hemizygous deletion of Pten. At 4 to 6 months of age, prostates with Pten hemizygous deletion alone (Pten+/−/Klf5+/+) began to show scattered hyperplasia, but no mPIN was noticeable. Simultaneous deletion of Klf5, either of one allele (Pten+/−/Klf5+/−) or both alleles (Pten+/−/Klf5−/−), caused the appearance of low-grade mPIN in both DP and lateral prostate (LP), which was characterized by multiple layers of larger cells with abundant pale cytoplasm (Figure 2, panels at the left). At 9 to 12 months of age, while single allele deletion of Pten (Pten+/−/Klf5+/+) caused low-grade mPIN (grades I and II), simultaneous deletion of Klf5 (Pten+/−/Klf5+/− and Pten+/−/Klf5−/−) caused high-grade (HG) mPIN (grades III and IV), as indicated by the filling of the lumen with excess cells that had an increased nucleus-to-cytoplasm ratio and small intraepithelial blood vessels (Figure 2, panels in the middle). At 15 to 18 months of age, while HG mPIN developed in some but not all prostates with Pten hemizygous deletion alone (Pten+/−/Klf5+/+), inactivation of one or both Klf5 alleles (Pten+/−/Klf5+/− and Pten+/−/Klf5−/−) caused more severe HG mPIN with higher nucleus-to-cytoplasm ratios. The large cell pattern seen with deletions of both Pten and Klf5 was typical for AKT/PTEN-related tumors, further indicating a more severe cancer-related phenotype (Figure 2, panels at the right).
Figure 2.
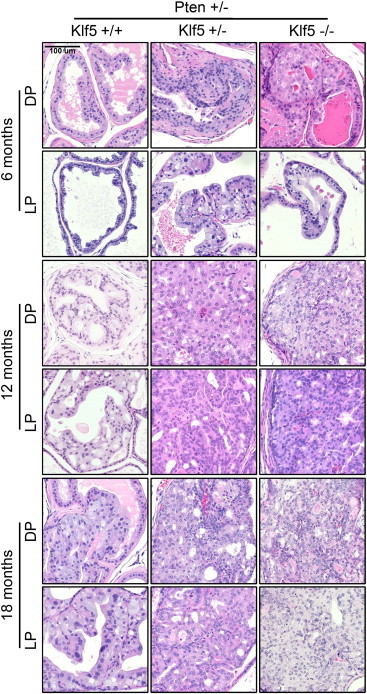
Deletion of Klf5 accelerates the development and severity of mPIN induced by hemizygous deletion of Pten. H&E-stained tissue section images of DPs and LPs at 6, 12, and 18 months with Klf5 and Pten deletions indicated. + and − indicate wild-type and deleted Klf5 or Pten alleles, respectively.
We compared the incidence of mPIN between prostates with and without the deletion of Klf5 and found that deletion of one or both Klf5 alleles caused a statistically significant increase in the incidence of mPIN in mice 9 to 12 months old (Table 1). When all age groups were combined, the difference was still statistically significant (Table 1). We also noticed that Klf5 deletion caused more severe abnormalities in the DP and LP (Figure 2) but less in the anterior prostate (AP) and ventral prostate (VP). In addition, few differences were noticeable between hemizygous and homozygous deletions of Klf5 in the development and progression of mPIN (Figure 2), although hemizygous deletion of Klf5 only decreased Klf5 expression in some cells (Figure S1), indicating that Klf5 is haploinsufficient and its haploinsufficiency is sufficient for the acceleration of Pten deletion–induced mPIN and related alterations.
Klf5 Deletion Promoted Pten Deletion–Initiated Prostatic Tumorigenesis and Disrupted the Luminal Architecture of Tumors
In the original study of Pten deletion in mouse prostates [5], Pten homozygous deletion caused invasive prostate cancer by the age of 9 weeks. In our study, prostate cancer was not detectable until the age of 16 weeks. We therefore focused on prostates at 6 months of age for analysis. Compared to prostates with Pten deletion alone at 6 months, those with both Klf5 and Pten deletions showed an increase in the size of prostate glands, as visualized in H&E-stained whole gland tissue section, and Klf5 homozygous deletion showed a more profound effect than Klf5 hemizygous deletion (Figure 3, upper row).
Figure 3.
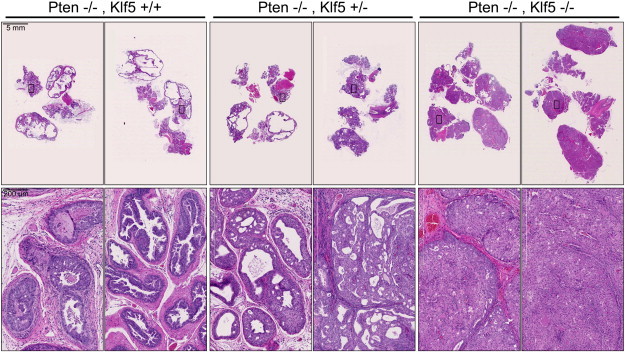
Deletion of Klf5 promotes the development and severity of prostate tumor induced by homozygous deletion of Pten. Upper row panels are images of H&E-stained tissue sections from the entire prostate gland at lower magnification, and lower row panels are magnified images of the boxed areas from the upper row panels. The prostates were from mice at 6 months of age. Note enlarged prostate glands and significant phenotypic alterations caused by Klf5 deletion. Prostates from two mice are shown for each genotype. + and − indicate wild-type and deleted Klf5 or Pten alleles, respectively.
Histologically, prostate tumors induced by Pten homozygous deletion alone maintained a luminal architecture (Figure 3), and necrosis was frequently observed in the center of the tumors, which is a common feature of HG mPIN and human prostate cancer due to insufficient nutrients. Interestingly, Klf5 hemizygous deletion resulted in a decreased luminal space in the prostate gland, and Klf5 homozygous deletion led to complete filling of the luminal space with tumor cells and thus the elimination of luminal architecture (Figure 3, lower panels of the Klf5−/− group). In addition, necrotic areas in the tumors were decreased by deletion of one Klf5 allele and abolished by deletion of both Klf5 alleles. In prostates with hemizygous Pten deletion, Klf5 deletion–enhanced mPIN mainly occurred in the DP and LP and the deletions of one or both Klf5 alleles showed similar effects. In contrast, Klf5 deletion caused morphologic abnormalities in Pten-null prostates in all four lobes, and deletion of both Klf5 alleles clearly showed a more profound effect than the deletion of one Klf5 allele (Figure 3).
Molecular Characterization of Architectural Abnormalities in Prostate Tumors with Deletions of Klf5 and Pten
Since the luminal structure was almost eliminated in prostate tumors with Klf5 and Pten deletions (Figure 3) and Pten deletion alone caused the expansion of the basal type of tumor cells, as indicated by positive staining for the Ck5 basal cell marker [39], we further evaluated whether Klf5 deletion altered the luminal architecture of Pten-null tumors. We examined the expression of molecular markers for major components of prostatic architecture, including those for luminal cells (Ck18), basal cells (Ck5 and Ck14), and the fibromuscular stroma (Sma), in 6-month-old prostate tumors with both Klf5 and Pten deletions. Without Klf5 deletion, cells in Pten-null prostate tumors were mostly positive for either a luminal marker (Ck18) or a basal marker (Ck5 or Ck14), and few cells were positive for both markers (Figure 4, A and B), which is consistent with the findings in a previous study [39]. When Klf5 was deleted, the number of basal cells decreased dramatically as indicated by the staining of Ck5 or Ck14 basal cell marker, while luminal cells, as indicated by positive staining for the Ck18 luminal cell marker, expanded and almost occupied the entire tumor (Figure 4, A and B). In Pten deletion–induced prostate tumors, expression of p63, a bona fide basal cell marker [40], was still restricted to basal layers, but Ck5 was expressed in both basal layers and cancerous acini [39], which was confirmed in our results (Figure 4C, left row). Interestingly, analysis of tumors with both Klf5 and Pten deletions demonstrated that, while cells in basal layers still expressed both p63 and Ck5, almost no tumor cells in acini expressed Ck5 (Figure 4C), which is different from the effect of Pten deletion alone. These results indicate that Klf5 deletion disrupts the ratio of Ck18-positive luminal cells to Ck5-positive basal cells in Pten deletion–induced prostate tumors.
Figure 4.
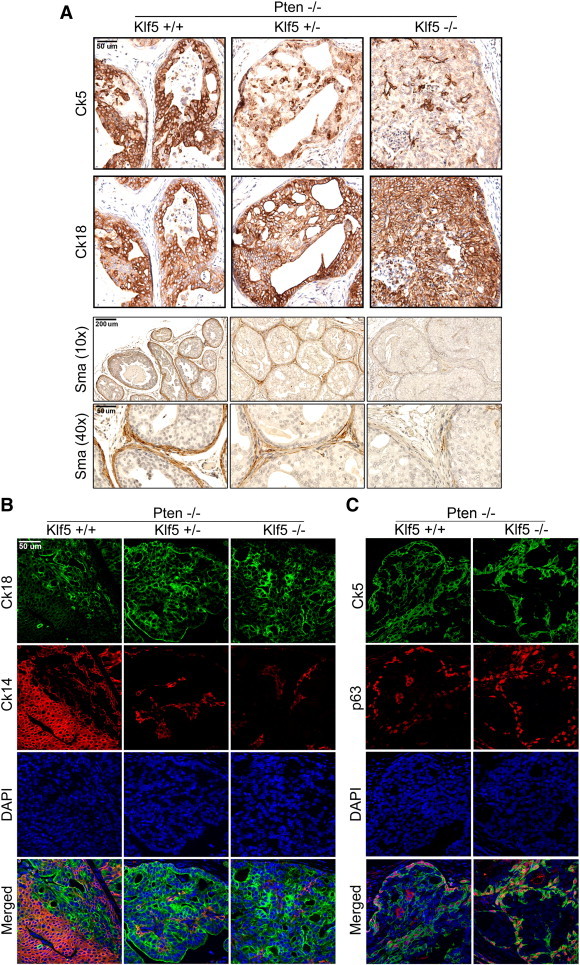
Knockout of Klf5 disrupts the architecture of prostate tumors induced by Pten deletion. Expression of basal cell markers Ck5, Ck14, and p63, luminal cell marker Ck18, and smooth muscle marker Sma was detected by IHC staining (A) or IF staining (B and C) in consecutive tissue sections (panel A only) of mouse prostates at 6 months of age. Pictures of basal and luminal markers were taken from the same area of tissue for better comparison (panel A only). DAPI staining was used to show nuclei (blue). Gene deletion status is indicated at the top and marker names at the left. The magnification for images of basal and luminal markers is × 200, and two magnifications are shown for Sma (× 10 in the upper and × 40 in the lower). + and − indicate the presence and absence of a Klf5 or a Pten allele.
We also analyzed the expression of Sma, a marker for the layer of fibromuscular stroma often used to evaluate tumor invasion to the stroma [17]. Whereas loss of Sma expression with Pten deletion alone was reported in a previous study [5], our analysis demonstrated that in prostate tumors with Pten deletion alone, Sma was still expressed, and the layer of Sma-positive fibromuscular stroma was still intact for an acinus (Figure 4A, the lower panel in the left two rows). When one Klf5 allele was deleted (Pten−/−/Klf5+/−), the surrounding smooth muscle layer became dramatically thinner, although it was still continuous. When both Klf5 alleles were deleted (Pten−/−/Klf5−/−), the smooth muscle layer became discontinuous or totally absent for most acini in tumors (Figure 4A, the lower panel in the right two rows).
Klf5 Deletion Dysregulates a Large Number of Genes
To understand how Klf5 deletion promotes prostate tumorigenesis initiated by Pten deletion, we performed microarray analysis using DPs at 6 months with wild-type or homozygous deletion of Klf5 in the Pten-null background. At 6 months, tumors were larger and had more severe phenotypic abnormalities (Figure 3). Using the Affymetrix mouse whole genome chips and associated computer programs, 7217 genes were identified on the basis of a significant (P < .05) expression difference between Klf5 wild-type and Klf5-null groups. In previous publications using the same expression chips, a > 1.5-fold change combined with P < .05 has been defined as the minimum meaningful fold change in the identification of differentially expressed genes [33], [34], [35]. Application of the same principle to our data resulted in a total of 1197 genes, 569 of which were upregulated and 628 were downregulated (Table S2).
To evaluate the quality of the microarray data, we selected 33 genes with different degrees of differential expression in the microarray analysis and performed real-time RT-PCR in four to eight wild-type or Klf5-null DPs. Differential expression was validated between the two groups for 31 of the 33 (94%) genes (Figure 5, A and B). In addition, almost all of the 33 genes showed similar fold changes between microarray and real-time RT-PCR analyses except for Klf5, which showed a much more dramatic decrease in the real-time RT-PCR assay (Figure 5B). These results indicate a good quality of the microarray data.
Figure 5.
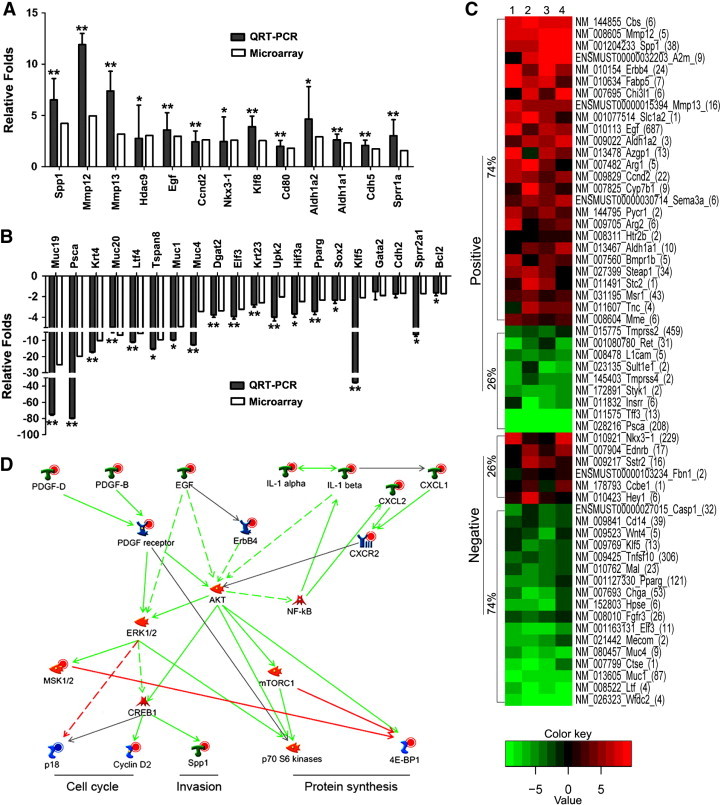
Bioinformatic identification and analysis of Klf5 deletion–dysregulated prostate cancer–related genes and molecular pathways in Pten-null mouse prostates. (A and B) Validation of differential expression for 13 upregulated (A) and 20 downregulated genes by real-time RT-PCR using the prostate samples including those used for microarray. White and black bars indicate microarray data and RT-PCR data, respectively. *P < .05; **P < .01. (C) Heat map of genes with > 2.0-fold expression change between wild-type and Klf5-null mouse prostates and associated with prostate cancer based on PubMed publications. The four wild-type prostates and the four Klf5-null prostates were randomly paired for map drawing. Genes are clustered on the basis of their association with prostate cancer, with those positively affecting prostate cancer cell behavior or upregulated in prostate cancer marked as “positive”, and those negatively affecting prostate cancer behavior or downregulated in prostate cancer marked as “negative”. The number in the parentheses after each gene name indicates the number of PubMed publications available on that gene. The percentage of upregulated or downregulated genes is shown. (D) Klf5 deletion activates canonical mitogenic signaling pathway involving AKT and ERK, as revealed by the analysis of Klf5 deletion–dysregulated genes using the MetaCore program. Up-regulation of signal initiating growth factors, cytokines, chemokines, and receptors activates downstream effectors involved in different processes through multiple canonical signaling pathways. Different shapes of the nodes represent functional classifications of genes. Red and blue circles in the nodes indicate up-regulation and down-regulation, respectively, of genes. Lines between every two nodes indicate their interactions, with red for inhibition, green for activation, gray for unspecified interaction, and dashed for indirect interaction. Arrow indicates the direction of a regulation.
As a basic transcription factor, KLF5 could regulate a common set of genes among different types of tissues. Therefore, we compared our gene list with published data from other mouse tissues. Among the genes dysregulated by Klf5 deletion during post-eyelid opening maturation of mouse corneas [41], 96 upregulated genes and 41 downregulated genes showed the same trends of changes, while 73 genes showed opposite changes compared to our data (Table S3). Among the available partial list of genes dysregulated by Klf5 deletion in intestine villus and bladder, a previous study identified 112 genes that are dysregulated (12 up and 100 down) by Klf5 deletion in both tissues [35]. Twenty of the downregulated genes but none of the upregulated genes showed the same pattern of dysregulation in the prostate in our study (Table S3). Two of the 20 genes, Reep6 and Dgat2, were downregulated in all four mouse models.
In the DU 145 human prostate cancer cell line, in which one PTEN allele has been lost [42] and KLF5 expression is rather low [10], RNA-Seq and data analysis showed that ectopic expression of KLF5 or the acetylation-deficient K369R mutant of KLF5 changed the expression of many genes [24]. Seven of the 65 KLF5-regulated genes (39 up and 26 down) and 54 K369R-regulated genes were also dysregulated by Klf5 deletion in mouse prostates (Table S4). The CHGA gene, which encodes a neuroendocrine differentiation marker associated with higher Gleason score in human prostate cancer [43], was the only gene that was downregulated by Klf5 deletion and K369R expression but upregulated by KLF5 expression, suggesting an up-regulation by acetylated KLF5.
Functional Processes and Molecular Pathways Modulated by Klf5 Deletion in Pten-Null Mouse Prostates
To explore the molecular mechanisms underlying Klf5 deletion–promoted tumorigenesis in Pten-null mouse prostates, we uploaded the 1197 differentially expressed genes to the MetaCore platform to enrich genes by diseases and identify significant GO processes, signaling pathways, and molecular networks that are dysregulated by Klf5 deletion.
Using the “Disease (by biomarker)” enrichment of the MetaCore program, we first collected all the genes that were associated with the disease of “prostate neoplasms”, which included 586 genes. Searching the PubMed database with a gene name and “prostate cancer” as keywords for these 586 genes revealed that 203 of them had been reported in at least one publication on prostate cancer. On the basis of the publications, 104 of the 203 genes were positively associated with prostate cancer, as indicated by a promoting effect on various behaviors and up-regulation or DNA hypomethylation in prostate cancer cells; 68 of the 203 genes were negatively associated with prostate cancer, as indicated by an inhibitory effect on cancer cell behavior and down-regulation or DNA hypermethylation in cancer cells (Table S5, Figure S2), and the remaining 31 of the 203 genes had inconclusive information. Deletion of Klf5 upregulated 70 of the 104 (67%) genes positively associated with prostate cancer and downregulated 37 of the 68 (54%) genes negatively associated with prostate cancer, which is consistent with the heat map generated by the R program using the 172 genes and their expression fold changes (Figure S2). The expression fold change was greater than 2 for 58 of the 172 (34%) genes. For these 58 genes, 35 and 23 were positively and negatively, respectively, associated with prostate cancer. Twenty-six of the 35 (74%) positively associated genes were upregulated, while 17 of the 23 (74%) negatively associated genes were downregulated by Klf5 deletion, which is obvious in the heat map for this group of genes (Figure 5C) and consistent with a tumor suppressor function of Klf5.
The GO analysis of the MetaCore program also identified a number of functional processes that were regulated by Klf5 deletion, including some that are associated with tumorigenesis: blood vessel morphogenesis, cell adhesion, ECM remodeling, EMT, chemotaxis, and inflammation (Table S6). Klf5 deletion also altered a number of canonical signaling pathways, as revealed by the pathway analysis function of the MetaCore program based on functional processes (Table S7). Consistent with a tumor suppressor function of Klf5, many of the pathways were activated by Klf5 deletion and their activation was shown to enhance multiple tumorigenesis-related processes such as MAPK cascades, EMT, G-protein signaling, and chemotaxis, including extracellular growth factors such as EGF, PDGF-B, and PDGF-D; cell membrane tyrosine kinase receptors such as ERBB4 and PDGFRβ; cytokine IL-1; and chemokines and chemokine receptor such as CXCL1, CXCL2, CXCR2, and so on (Table S7). These nine molecules are all located at the originating point of specific signaling pathways, and their expression changes could thus drive cascades of signaling changes. We applied the “batch search” program of MetaCore to these nine genes and used MetaCore’s “expand by one interaction” algorithm with all 1197 Klf5 deletion–dysregulated genes as “activated experiment” data to build networks and identify overconnected pathways that are most relevant to Klf5 deletion. Among the overconnected pathways identified, six were most overconnected, including AKT, ERK, NF-κB, protein kinase Cβ, p38, and PPARγ. Of all the pathways identified in Table S7, AKT and ERK appeared in 12 and 11 of them, respectively, whereas NF-κB, protein kinase Cβ, p38, and PPARγ appeared in 5, 4, 3, and 1 of them, respectively. We therefore focused on AKT and ERK to draw the pathway map of molecules that were dysregulated by Klf5 deletion, including the nine signal initiation molecules and some of their downstream effectors (p18, cyclin D2, Spp1, S6, and 4E-BP1). Molecules that were between an initiating molecule and an effector and not affected by Klf5 deletion were eliminated from the map, resulting a schematic pathway map that suggests that Klf5 deletion activates AKT and ERK signaling in the regulation of cell cycle, invasion, and protein synthesis (Figure 5D).
In addition to the canonical pathways affected by Klf5 deletion (Table S7 and Figure 5D), there could be novel networks affected by Klf5 deletion. We therefore built molecular networks involving Klf5 deletion–dysregulated genes with the network analysis algorithm of MetaCore. Networks with 50 total nodes and > 4 seed nodes (differentially expressed genes) were considered instructive and meaningful. Among the 25 networks built, 20 centered around CREB1, 2 centered around SP1 (ranked the first and third based on P values), and one each centered on Myc and RhoA (Figure S3). Similarly, 21 transcription factor–centered networks were also identified, of which CREB1 and SP1 were the first and second most significantly involved, while ER, Myc, and AR ranked third, fourth, and fifth, respectively (Table S8). These five molecules (CREB1, SP1, Myc, ER, and AR) and the Klf5 deletion–dysregulated genes that are directly regulated by these molecules are listed in Table 2.
Table 2.
Key Molecules Potentially Cooperating with Klf5 to Mediate Klf5 Deletion–Promoted Tumorigenesis, as Identified by the Network Analysis of MetaCore with Klf5 Deletion–Dysregulated Genes, and Those Differentially Expressed Genes that Are Also Direct Transcriptional Targets of These Key Molecules
| Key Molecules | Genes Regulated by Klf5 Deletion |
|---|---|
| CREB1 | Upregulated: Spp1, Tac1, Slc1a2, Egf, Ccnd2, Arg2, Msr1, Sstr2, Rgs2, Il1a, Gja1, Gzmb, Sfrp1, Selp, Vcam1, Cck, Tgm1, Il1b, Pfkfb3, Rgs1, Spry2, Mat2a, Pgf, and Map2. Downregulated: Ldlr, Bhlhe40, Zfp667, Avpi1, Pmaip1, Bcl2, Ndn, Cdo1, Ppp2r2b, Kcnh2, Kcne4, Cd14, Ppargc1b, Pparg, Agt, Chga, Muc4, Fam3b, Muc5b, and Ptges. |
| SP1 | Upregulated: Crabp1, Abo, Cbs, Spp1, Slc9a2, Bhlha15, Chil1, Ccl20, Ccnd2, Padi2, Nkx3-1, Cyp7b1, Klf8, Prlr, Trf, Ednrb, Raver2, Sstr2, Slc6a2, Ggt1, Tnc, Fbn1, Rgs2, Anxa6, Il1a, Serpina1a, Gja1, Bmp7, Serpina3n, Cd7, Pla2g7, Trem1, Crabp2, Selp, Vcam1, Cad, Pdgfd, Cck, Pdgfrb, Mgst1, Soat1, Tnnt2, Tgm1, Col1a1, Col1a1, Timp1, Kcnj2, Flt1, Col1a2, Col1a2, Aqp1, Slc5a8, Gstm7, Adm, Slc19a2, Apln, Col4a2, Angpt2, Spry2, Itgax, Mat2a, Ctns, Hdc, Padi3, Pdgfb, Fcgr4, Slc11a1, Aox1, Col4a1, Hhex, Nrp1, Cited2, Mgarp, Tfpi2, Phgdh, Mir365-1, Tlr13, and Eng. Downregulated: Stard3, Hcn2, Atp6ap2, Ldlr, Idh2, Mbp, Adam23, Cdkn2c, Bhlhe40, Cyp21a1, Zfp667, Ggh, Akap12, Cdk6, Noxo1, Cdh2, Atp4a, Efnb2, Bcl2, Ppl, Tnxb, Ppp2r2b, Ascl1, Kcnh2, S100a6, Smpd3, F3, Acacb, Mst1r, Cd14, Tmprss2, Klf5, Tnfsf10, Napepld, Upk1b, Chga, Cyp2f2, Ces1d, Clcn2, Hpse, Muc4, Kcnq1, Gpc3, Muc5b, Scnn1a, Krt7, Atp2a3, Ctse, Muc1, Ptges, Ltf, Hsd11b2, and Tff3. |
| MYC | Upregulated: Cbs, Spp1, Ccnd2, Fah, Ak4, Eif4ebp1, Bmp7, Sfrp1, Lox, Vcam1, Cad, Cck, Pdgfrb, Mgst1, Jag2, Adm, Rgs1, Itgax, Mt1, Cdh3, Sema6a, Angptl4, Slc11a1, Ndrg1, Cited2, Slc16a1, Bnip3, Spred2, and Alb. Downregulated: Mcm6, Abcd3, Vav2, Wnt5a, Ggh, Pmaip1, Rad51b, Cdk6, Nans, Bcl2, Mir200a, Peg10, Apol7a, Itpr2, Hmgcs2, Camk1d, Golm1, Rasgef1a, Gldc, Mir429, Casp1, Wnt4, Ppargc1b, Sestd1, Sox2, Pparg, Fa2h, Gpc3, and Hsd11b2. |
| ER | Upregulated: Ttr, Spp1, Ugt2b35, Erbb4, Slc1a2, Plod2, Cyp7b1, Prlr, Trf, Ccl28, Stc2, Sstr2, Mme, Mir181b-1, St3gal4, Gja1, Fam132a, Bmp7, Serpina3n, Thbs2, Cad, Rab31, Flt1, Aqp1, Adm, Pfkfb3, Mir181b-2, Cdh3, Adamts9, Padi3, Abtb2, Fcgr4, Mgarp, and Lamb1. Downregulated: Fabp6, Abcd3, Bcas1, Anxa3, Rap1gap, Cobl, Foxc1, Pmaip1, Mcf2l, Sytl2, Rad51b, Cdk6, Klf10, S100a6, Agr2, Mall, F3, Wnt4, Spire2, Por, Rapgefl1, Galnt4, Hsh2d, Agt, Ret, Sult1e1, Elf3, Gsta4, Krt7, Atp2a3, Muc1, Ptges, Ltf, Cyp2b10, Krt4, Tff3, and Psca. |
| AR | Upregulated: Spp1, Slc9a2, Ugt2b35, Azgp1, Ank, Nrn1, Nkx3-1, Pycr1, Mme, Eppin, Steap4, Cxcl2, Efna3, Pdgfrl, Slc19a2, Osbpl1a, Cxcl1, Gpr153, Cdh3, Angptl4, Ndrg1, Nrp1, and Cited2. Downregulated: AA986860, Pmaip1, Cdh2, Rab3b, Ppfibp2, Peg10, Agr2, F3, Mir31, Tmprss2, Klf5, Sox2, Pparg, Sult1e1, Scnn1a, Krt7, Muc1, and Psca. |
EGF Was Dramatically Upregulated by Klf5 Deletion in the Pten-Null Background
Whereas multiple growth factors and cytokines were significantly upregulated by Klf5 deletion in the microarray experiment, the Egf showed the highest fold change (2.97-fold). Considering the well-established role of EGF and its downstream signaling in cell proliferation and tumorigenesis, we performed additional experiments to test whether KLF5 regulates EGF transcription. Real-time RT-PCR was performed with DP samples (6 months old, eight samples for each of the three Klf5 genotypes, including the four with wild-type or Klf5-null genotype used in microarray hybridization), and up-regulation of Egf by homozygous deletion of Klf5 was confirmed in the Pten-null background (Figure 6A). Hemizygous deletion of Klf5 did not show an effect though (Figure 6A). We also detected Egf protein by IHC staining in the same samples used for RNA detection and obtained consistent results: Egf protein showed an evident increase in Klf5-null prostates but no change in prostates with hemizygous deletion of Klf5 (Figure 6B). We further tested the effect of KLF5 on EGF expression in the context of PTEN using the PNT2 human prostate luminal cell line, in which proliferation signaling is activated by SV40 [44]. RNA interference was applied to knock down KLF5 and/or PTEN, and the knockdown effect was confirmed by Western blot analysis (Figure 6C). Real-time RT-PCR demonstrated that, while knockdown of PTEN alone did not cause an apparent change in EGF expression, knockdown of KLF5 significantly increased EGF mRNA level (P < .01), and the increase was further enhanced by the knockdown of PTEN (P < .02) (Figure 6D). We then examined whether Egf up-regulation by Klf5 deletion in Pten-null mouse tumors has a functional effect by measuring the activity of Egfr. IHC staining demonstrated that the Y1068 phosphorylation of Egfr, an indication of Egfr activity [45], was clearly increased in Klf5-null prostates (Figure 6E), indicating that Klf5 deletion further enhances the EGF/EGFR/MAPK signaling activity.
Figure 6.
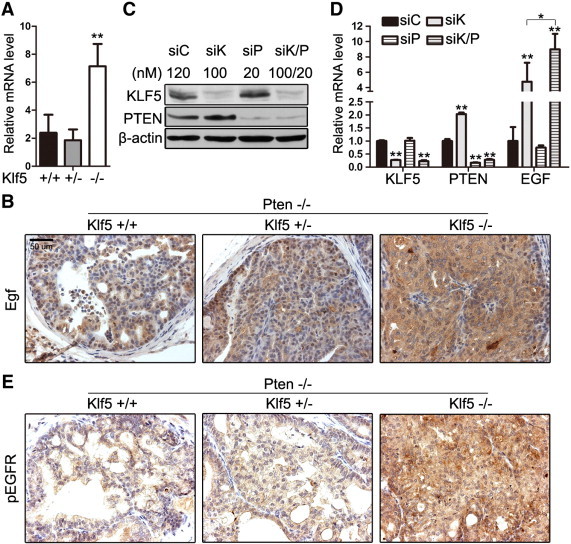
Klf5 deletion upregulates EGF in Pten-null mouse prostate tumors and human prostate epithelial cell line. (A, B, and E) Detection of EGF mRNA expression by real-time RT-PCR (A) and Egf and p-Egfr proteins by IHC staining (B and E) in 6-month-old Pten-null mouse DPs with indicated Klf5 deletion status. Data for each genotype in A was from eight mice. (C and D) Detection of protein expression of KLF5 and PTEN by Western blot analysis (C) and mRNA expression of KLF5, PTEN, and EGF by real-time RT-PCR (D) in the PNT2 immortalized human prostate luminal epithelial cell line with the knockdown of KLF5, PTEN, or both by RNA interference.
Deletion of Klf5 Increased Cell Proliferation with Consistent Changes in the Expression of Proliferation-Related Molecules
On the basis of the findings that Klf5 deletion significantly increased the size of Pten deletion–induced tumors (Figure 3), cell proliferation is increased by Pten deletion alone [5]. We determined whether Klf5 deletion causes more cells to proliferate. Expression of the Ki67 proliferation marker was determined by IHC staining in each of the prostate lobes, and the rate of Ki67 positivity in tumor cells was calculated. While Klf5 hemizygous deletion did not cause an obvious change in the rate of Ki67-positive cells, Klf5 homozygous deletion significantly increased the rate (Figure 7A), indicating that homozygous deletion of Klf5 increases cell proliferation in Pten deletion–induced prostate tumors.
Figure 7.
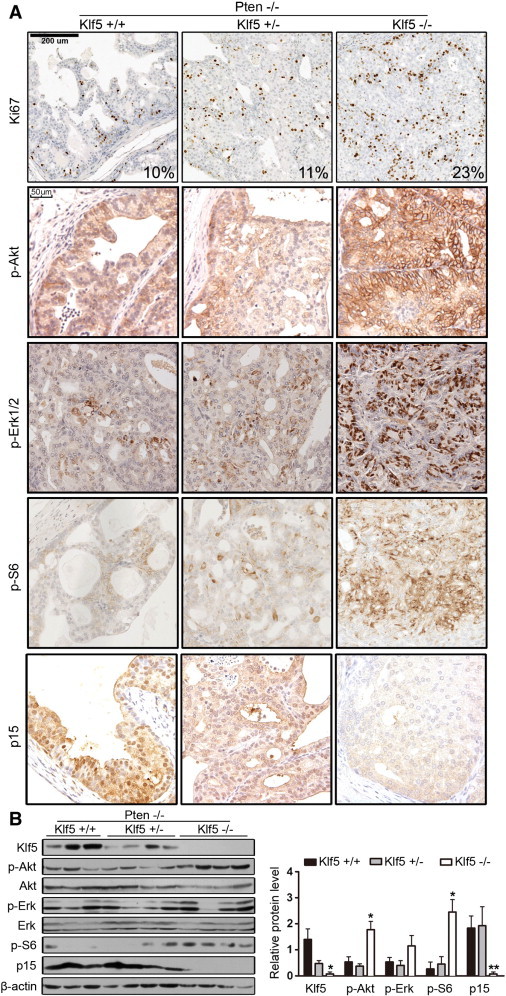
Molecular characterization of prostate tumors induced by Klf5 and Pten deletion. (A) IHC staining of the Ki67 proliferation marker, oncogenic kinases p-Akt, p-Erk1/2, and p-S6, and the p15 cell cycle inhibitor in tissue sections of APs at 6 months of age with different deletion status of Klf5 and Pten. Marker names are indicated at the left, and the deletion status of genes is indicated at the top. The ratio (%) of Ki67-positive cells, as determined by cell counting, is shown at the lower right corner of each image of the Ki67 panels. (B) Detection of protein expression for Klf5, p-Akt, Akt, p-Erk1/2, Erk1/2, p-S6, p15, and β-actin by Western blot analysis in tissue lysates from anterior mouse prostates with the same genotype and age used in A. Relative protein level was determined by using the ImageJ software, and statistical significance was evaluated by using Student’s t test. Marker names are indicated at the left, and the deletion status of genes is indicated at the top. + and − indicate the presence and absence of an allele for Klf5 and Pten. *P < .05; **P < .01.
Our microarray analyses indicated that Klf5 deletion increased the expression of genes positively associated with prostate cancer behaviors, activated mitogenic signaling pathways including AKT and ERK signaling pathways, and enhanced the EGF/EGFR signaling (Figures 5, C and D, and 6, A, B, and E). Loss of PTEN enhances PI3K/AKT signaling [46] to increase cell proliferation, KLF5 has been linked to Ras/MAPK proliferation signaling [20], [22], and loss of KLF5 downregulates the p15 cell cycle inhibitor in the context of transforming growth factor–β (TGF-β) [38], [47]. We therefore evaluated the expression of key players in the PI3K/AKT signaling pathway including phosphorylated Akt (p-Akt), phosphorylated Erk1/2 (p-Erk1/2), phosphorylated mTor (p-mTor), phosphorylated S6 (p-S6), and p15 cell cycle inhibitor by IHC staining and Western blot analysis in Pten-null prostate tumors at 6 months of age. While hemizygous deletion of Klf5 mildly increased the expression of p-Akt and p-S6, homozygous deletion of Klf5 led to much more dramatic increases in their expression (Figure 7, A and B), indicating that Klf5 deletion significantly enhances the effect of Pten deletion on PI3K/Akt signaling. Expression of p-Erk1/2 showed a similar pattern, with a much greater increase by Klf5 homozygous deletion than that by Klf5 hemizygous deletion (Figure 7, A and B). These results support the findings from MetaCore’s pathway analysis. For p15, hemizygous deletion of Klf5 decreased nuclear staining, and homozygous deletion of Klf5 led to a more significant decrease in p15 expression (Figure 7, A and B). Although mTor is one of the crucial players in PI3K/Akt signaling, we were unable to detect p-mTor by Western blot analysis and detected few cells that were positive in IHC staining for p-mTor in Klf5-null tumors (Figure S4), suggesting that p-mTor is less relevant in Klf5-null tumors.
Discussion
Klf5 Deletion Promotes Pten Deletion–Induced Prostatic Tumorigenesis
The current literature is somewhat inconsistent about the function of KLF5 in tumor development. On the one hand, KLF5 promotes tumor growth in xenograft models [19], [48], is necessary for intestinal tumorigenesis in genetically modified mice [49], and mediates Ras activation–induced intestinal tumorigenesis [22], [23]. On the other hand, KLF5 can also suppress tumorigenesis in xenograft models [18], [50]. Using a tissue-specific knockout mouse model, which provides more definitive evidence for the function of a given tumor suppressor gene, we were able to prove the tumor suppressor activity of KLF5. For example, in mouse prostates with hemizygous deletion of Pten, which develop mPIN [5], simultaneous deletion of Klf5 shortened the latency of mPIN development and caused more severe phenotypic alterations in the mPIN (Figure 2). When both Pten alleles were deleted, which alone causes prostate cancer [5], Klf5 deletion caused more rapid tumor growth and more severe phenotypic and molecular alterations in cancer (Figure 3, Figure 4, Figure 5, Figure 6, Figure 7).
In various human cancers including prostate cancer, deletion of the KLF5 locus is the second most frequent chromosomal deletion, occurring in about 42% of tumors [9], [51]. KLF5 is also inactivated in more than 20% of human cancers through excess protein degradation mediated by the amplification and overexpression of the WWP1 oncogenic ubiquitin E3 ligase [15], [52], [53], [54]. The establishment of a tumor-promoting effect of Klf5 deletion in this study suggests that KLF5 inactivation could impact more than half of human cancers.
While Klf5 deletion clearly promoted the development of prostate cancer initiated by Pten deletion (Figure 2, Figure 3), our previous study indicated that deletion of Klf5 alone in prostatic epithelial cells does not have a pro-tumorigenic effect [25], as hemizygous deletion of Klf5 caused hyperplasia with increased cell proliferation but did not cause neoplasms, and homozygous deletion of Klf5 induced apoptosis without causing detectable cell proliferation [25]. Multiple genetic events are usually necessary for a tumor to form and progress, and it appears that at least another oncogenic event, such as Pten deletion, is necessary to release the pro-tumorigenic effect of Klf5 deletion.
In human prostate cancer, a number of chromosomal regions are deleted or amplified, and deletions of NKX3-1 at 8p21, KLF5 at 13q21, and PTEN at 10q23 are among the most common deletions [1], and at least some prostate cancers possess the deletions of both KLF5 and PTEN loci. For example, the LNCaP prostate cancer cell line has a 2-bp homozygous deletion of PTEN [29] and a hemizygous deletion of KLF5 [10], and prostate cancer xenografts PC82 and LuCaP 58 have loss of both PTEN and KLF5 loci [10], [55], [56], [57]. Our findings in this study thus establish a positive interaction between KLF5 and PTEN deletions in prostatic carcinogenesis. They also provide a mouse model of prostate cancer involving two common genetic alterations in human prostate cancer.
In addition to the additive effect between Pten and Klf5 deletions described above, Pten deletion also interacts with Nkx3-1 deletion in prostatic carcinogenesis [58], [59]. Deletions of Klf5 and Nkx3-1, however, do not appear to have an additive effect [60]. PTEN is an inhibitor of the PI3K/AKT signaling pathway, and PTEN loss leads to increased PI3K/AKT signaling activities that enhance both cell proliferation and survival [27], [61].
While homozygous deletion of Klf5 alone increases cell death at least at age 18 months, as demonstrated in our previous study [25], it did not show a similar effect in Pten-null prostate tumors at 6 months of age (Figure S5); it rather enhanced cell proliferation significantly in these tumors (Figure 7A). It is well established that Pten loss leads to the activation of the PI3K/AKT signaling, which is pro-survival [62], [63]. It is thus possible that enhanced survival by PI3K/AKT activation overcomes cell death induced by Klf5 homozygous deletion, thus unmasking the tumor-promoting effect of Klf5 homozygous deletion by activating multiple oncogenic signaling pathways including those described in our recent study [24]. The same mechanism could apply to other tumor suppressors that are part of the TGF-β signaling pathway and whose deletions have a positive interaction with Pten deletion in tumor development, including Smad4 and p27 [8], [64]. Furthermore, it is possible that other signaling pathways that prevent cell death could also unmask pro-tumorigenic functions of Klf5 deletion, which remains unexplored.
Klf5 Deletion Promotes the Luminal but Attenuates the Basal Type of Pten Deletion–Induced Prostate Tumor Cells
Most human prostate cancers have a luminal phenotype, and very few (< 1%) human prostate cancer cells express basal cell markers [65]. In addition, it appears that cells in both the basal and luminal layers can be transformed to give rise to prostate cancer [66]. In Pten deletion–induced mouse prostate tumors, the basal cell population is expanded to the acinus with an increased expression of the PSCA transient amplifying cell marker [5], [39], [67], indicating a more profound promoting effect of Pten deletion on basal cells during tumorigenesis. Unexpectedly, Klf5 deletion reversed the effect of Pten deletion on the ratio of basal to luminal cells in prostate tumors. While Klf5 deletion significantly increased the number of luminal tumor cells, which could be due to increased proliferation of this type of cells as indicated by the percentage of Ki67-expressing cells (Figure 7A), it actually diminished the number of basal tumor cells, as indicated by Ck5 expression (p63 −), in the cancerous acini (Figure 4B). Basal cells in the basal layers, as identified by p63 expression, were not affected (Figure 4C). Therefore, the tumor-promoting effect of Klf5 deletion appears to be restricted to luminal tumor cells. Whereas the increase in the luminal to basal ratio by Klf5 deletion could result from the differentiation of Pten deletion–induced Ck5+/p63− TA cells, it is more likely that Klf5 deletion causes the death or prevents the proliferation of Ck5+/p63− TA cells, because these cells were almost absent in cancerous acini (Figure 4C) and the expression of the PSCA marker for such cells was dramatically reduced (more than 80-fold) on Klf5 deletion (Figure 5, B and C, and Table S2). Another possibility is that Klf5 deletion prevented the differentiation of p63+/Ck5+ cells in the basal layer into Ck5+/p63− TA cells. To fully address whether Klf5 deletion has different effects on basal and luminal cells, it is necessary to perform additional studies using approaches such as lineage tracing and luminal or basal cell–specific Cre mice.
In normal prostates, our previous study suggests that Klf5 could also have different or even opposing functions in basal and luminal cells [25]. For example, whereas Klf5 is expressed in both luminal and basal cells, the protein is typically acetylated in luminal cells (AcKlf5) while unacetylated in basal cells (unAcKlf5) [25], and AcKLF5 and unAcKLF5 suppress and promotes, respectively, cell proliferation and tumor growth [24], [38]. Taken together with the different effects of Klf5 deletion on luminal and basal tumor cells, we propose that unAcKLF5 is necessary for basal cell proliferation and AcKLF5 is necessary for luminal differentiation. We further propose that Klf5 deletion compromises the pro-differentiation function of AcKLF5, leading to partial dedifferentiation and subsequent proliferation of luminal cells while attenuating the proliferation or survival of basal cells in Pten-null prostates. We are currently testing these predictions by developing a mouse model in which acetylation-deficient Klf5 replaces wild-type Klf5 in the prostate.
Klf5 Deletion Promotes Cancer Cell Proliferation by Multiple Mechanisms
In understanding how Klf5 deletion promotes Pten deletion–initiated prostatic tumorigenesis, microarray-based expression profiling and molecular pathway/network analysis indicated that a large number of genes involved in a number of molecular signaling pathways are dysregulated by Klf5 deletion. One significant group of upregulated molecules included extracellular growth factors EGF and PDGFs, cytokine IL-1, chemokines CXCL and CCL, and tyrosine kinase and chemokine receptors ERBB4, PDGFR, and CXCR2 (Table S2, Table S7 and Figure 5). Egf, in particular, is downregulated by Pten deletion alone [5] but was upregulated by Klf5 deletion in the Pten-null background (Figures 5, A and C, and 6A). Up-regulation of Egf protein and activation of Egfr by Klf5 deletion were also detected by IHC staining in mouse prostates (Figure 6, B and E). Consistently, in the PNT2 human prostate epithelial cell line, knockdown of PTEN alone slightly downregulated but knockdown of KLF5 alone or in combination with PTEN knockdown significantly regulated EGF mRNA expression (Figure 6D). EGF is a potent growth factor that activates AKT and ERK signaling [68], [69], further implicating AKT and ERK in Klf5 deletion–promoted tumorigenesis. Up-regulation of Egf by Klf5 deletion could therefore be one of the driving factors underlying Klf5 deletion–promoted cell proliferation and tumorigenesis. This possibility remains to be tested.
Taken together with previous observations that Pten deletion–initiated mouse prostate tumors have an increased rate of cell proliferation due to the activation of PI3K/AKT and MAPK signaling [5], [27], [61], [70], [71], we determined the effects of Klf5 deletion on cell proliferation and AKT and ERK activities in Pten-null prostate tumors. When Klf5 was homozygously deleted, the cell proliferation rate more than doubled (Figure 7A, Ki67 panel), the expression of p-Akt and Erk were obviously increased (Figure 7A), and the rate of cell death was unaffected (Figure S5). Expression of activated Akt and Erk and their downstream molecule p-S6 [27], [72] was significantly increased, while p-mTor was slightly upregulated. These results indicate that Klf5 deletion further promotes the proliferation of tumor cells, and an enhanced PI3K/AKT mitogenic signaling pathway is involved. KLF5 itself directly regulates the p15 cell cycle inhibitor in cell proliferation [38], [47], and this mechanism also appeared to function in Pten and Klf5-null prostate tumors, where Klf5 deletion led to decreased p15 expression in a dose-dependent manner (Figure 7, A and B). Therefore, Klf5 deletion likely mediates more rapid tumor growth by multiple pro-proliferative molecular pathways.
In a previous study, homozygous deletion of Pten in mouse prostates induced invasive adenocarcinoma and subsequent metastasis to the lymph node and lung at 4 months of age [5]. In our study, however, no tumor invasion, based on Sma staining, or metastasis, based on the dissection of lungs and livers, was evident in mice carrying Pten- and Klf5-null tumors even at 6 months of age. However, Klf5 deletion could still promote tumor invasion and metastasis. For example, Klf5 deletion led to a thinner or discontinuous smooth muscle layer (Figure 4) and the up-regulation of multiple matrix metalloproteinases, including MMP-12, MMP-13, and MMP-8 (Figure 5A and Table S2). In addition, Klf5 deletion also upregulated Spp1, one of four signature molecules predictive of prostate cancer metastasis [8], and KLF5 inactivation promoted EMT, a strong indicator of tumor invasion and metastasis, by downregulating miR-200 micro-RNAs [73]. Klf5 deletion also downregulated miR-200 genes in mouse prostates (Table S2, Table S6, Table S7). Therefore Klf5 deletion could still promote tumor invasion and metastasis, and the lack of metastasis in this study could be caused by multiple reasons, including insufficient time for tumor progression and variation in genetic background among different mouse strains.
Multiple Molecules Could Cooperate with Klf5 in Gene Regulation
Despite the fact that KLF5 functions in a variety of pathophysiological processes in a context-dependent manner in various tissues [24], [38], [51], a large number of the same genes dysregulated by Klf5 deletion in mouse prostate tumors were also regulated by KLF5 or its acetylation-deficient mutant K369R in xenograft tumors of the DU 145 human prostate cancer cell line (Table S4). A large number of the same genes were also dysregulated by Klf5 deletion across multiple types of tissues including cornea, villus, and bladder (Table S3), suggesting that there is a common set of genes that is regulated by Klf5 in different tissues or under different contexts.
In identifying novel pathways and networks affected by Klf5 deletion using the Klf5 deletion–dysregulated genes, we found that CREB1, SP1, and Myc were the most significantly affected (Figure S3 and Table 2), all of which are transcription factors. Network analysis for transcription factors identified five transcription factors that most significantly related to Klf5 deletion–dysregulated genes, and CREB1, SP1, and Myc were again among the five. The other two transcription factors among the top 5 were ER and AR (Tables 2 and S8). Interestingly, the same five transcription factors were also identified as the most relevant molecules to the function of KLF5 and K369R in the DU 145 human prostate cancer cell [24]. It is thus possible that, as a transcription factor, KLF5 cooperates with CREB1, SP1, Myc, ER, and AR, e.g., in a transcription complex, to maintain the normal homeostasis of prostate epithelium, and disruption of this complex could promote tumorigenesis.
Some published studies support this possibility. For example, in human prostate cancer cells, KLF5 has been demonstrated to suppress tumor growth by forming a transcriptional complex with CREB1 and ERβ to upregulate the FOXO1 tumor suppressor gene [18]. Phosphorylation of CREB1 is a classic mechanism of CREB1 activation [74], so it is worth testing whether CREB1 phosphorylation affects KLF5 function. ERα has also been shown to physically interact with KLF5 in gene regulation and cell proliferation in ERα-positive breast cancer cells [75]. Sp1 and KLF5 belong to the same family of Krüppel-like factors that bind to GC-rich promoter elements [76], so they may both bind to promoters of the same genes to cooperate, although this idea has not been tested. For Myc, not only is its gene transcription directly regulated by KLF5 [77], it also directly interacts with KLF5 in a TGF-β–dependent manner to regulate cell proliferation [47]. AR is essential for both the normal development and function of the prostate gland and the growth of prostate cancer [78]. Although it is unknown whether AR and KLF5 form a complex, some molecules including Smad3 and p300 have been demonstrated to physically interact with both AR [79], [80] and KLF5 [47]. It is worth testing whether and how KLF5 cooperates with CREB1, Sp1, Myc, ER, and AR in gene regulation in epithelial homeostasis.
In summary, we examined the effects of Klf5 deletion on Pten deletion–initiated prostatic tumorigenesis in genetically modified mice and found that Klf5 deletion promoted the development and increased the severity of Pten deletion–induced mPIN and prostate tumor. Interestingly, Klf5 deletion increased the ratio of Ck18-positive luminal tumor cells to Ck5-positive basal tumor cells in tumors, suggesting a restricted promoting effect of Klf5 deletion on luminal tumor cells. Klf5 deletion in Pten-null prostate tumors increased the cell proliferation rate, which involved multiple signaling pathways including the up-regulation of growth factor EGF, activation of PI3K/AKT and MAPK, and the inactivation of the p15 cell cycle inhibitor. Further pathway analysis suggests that KLF5 cooperates with other transcription factors including CREB1, Sp1, Myc, ER, and AR to regulate gene expression in the prostate. These findings validate the tumor suppressor function of KLF5 in genetically modified mice and also provide a mouse model that shares two common genetic alterations with human prostate cancer, deletions of PTEN and KLF5.
The following are the supplementary data related to this article.
Primer Sequences Used in This Study
Differentially Expressed Genes between Klf5 Wild-Type and Klf5-Null Mouse DPs in the Pten-Null Background, as Identified by Microarray Analysis. Fold Change Was the Mean Value of Four Pairs of Samples
Genes Upregulated or Downregulated by Klf5 Deletion in More than One Type of Tissue. Up-regulation in a Tissue Is Indicated by “Up” or a Positive Fold Change Number, while Down-Regulation Is Indicated by “Down” or a Negative Fold Change Number. Data for Cornea, Villus, and Bladder Were from Published Studies [41], [35], [33]
Genes Dysregulated by Both Klf5 Deletion in Pten-Null Mouse Prostates and Ectopic Expression of KLF5 or Its Acetylation-Deficient Mutant K369R in Xenograft Tumors of the DU 145 Human Prostate Cancer Cell Line (DU 145-KLF5 or DU 145-K369R). Data for DU 145 Xenografts Were from a Previous Study [24]
Differentially Expressed Genes that Were Prostate Cancer Related, as Identified by the MetaCore Program, and Had Appeared in at Least One PubMed Publication on Prostate Cancer. Fold Change between Klf5 Wild-Type and Klf5-Null Groups and the Number of PubMed Publications Available Are Also Shown for Each of the Genes. Positive (P) or Negative (N) Refers to a Gene’s Association with the Development or Progression of Prostate Cancer Based on PubMed Publications, with P Indicating a Gene’s Promoting Role in Prostate Tumorigenesis or Up-Regulation in Prostate Tumors and N indicating the Opposite. N/A, Information Is Not Clear
Functional Processes Enriched by MetaCore Analysis Based on the Differentially Expressed Genes between Klf5 Wild-Type and Klf5-Null Mouse DPs. Different Processes with the Same Function Are Grouped Together and Ranked Based on P Values (from Low to High, P Value < .01). Genes Contributing to a Process Are Listed
Significant Pathways Identified by the MetaCore Analysis Based on the Differentially Expressed Genes between Klf5 Wild-Type and Klf5-Null Mouse DPs. All Pathways Are Ranked Based on Their P Values (from Low to High, P Value < .01)
Key Transcription Factors Significantly Associated with Differentially Expressed Genes between Klf5 Wild-Type and Klf5-Null Mouse DPs, as Defined by MetaCore’s Interactome Analysis for Transcription Factors. Related Biologic Processes Are Also Shown. The Ranking Is Based on P Values. Total Nodes Refer to All Nodes in a Transcription Factor Network, while Seed Nodes Refer to the Genes Differentially Expressed between the Experimental Groups
Confirmation of Klf5 deletion in mouse prostates with Pten hemizygous knockout. Klf5 protein was detected by IHC staining in 12-month-old APs with indicated Klf5 deletion status; + and − indicate wild-type and deleted Klf5 alleles, respectively.
Heat map illustrating gene expression changes caused by Klf5 deletion in Pten-null prostate tumors and associated with prostate cancer in the literature. A total of 173 differentially expressed genes with > 1.5-fold expression change and literature-based prostate cancer association was used in the heat map drawing. Each lane represents one pair of wild-type and Klf5-null DPs at 6 months old. Clustering in the heat map is based on the relation of genes with prostate cancer, with those reported to promote prostate cancer or be upregulated in prostate cancer as “positive”, and those reported to inhibit prostate cancer or be downregulated in prostate cancer as “negative”. The percentages of upregulated or downregulated genes in each group are shown. The number in the parentheses after each gene name indicates the number of publications on that gene in prostate cancer. Heat map colors reflect normalized log2 fold-change values of the ratio in each pair.
Molecular networks constructed for differentially expressed genes between Klf5 knockout and wild-type by the network analysis algorithm in the MetaCore platform. The 25 networks that had more than four seed nodes with P < .05 are shown. Different shapes of the nodes represent functional classifications of genes. Red and blue in the nodes indicate up-regulation and down-regulation, respectively, of genes. Lines between every two nodes indicate their interactions, with red for inhibition, green for activation, and gray for unspecified interaction. Arrow indicates the direction of a regulation. Colored thicker lines mark canonical pathways.
Klf5 deletion upregulates p-mTor in Pten-null mouse prostate tumors. IHC staining was used to detect p-mTor in tissue sections of APs and DPs at 6 months of age. Deletion status of Klf5 is indicated at the top. + and − indicate the presence and absence, respectively, of a Klf5 or Pten allele.
Klf5 deletion has no effect on apoptosis in Pten-null mouse prostate tumors. Apoptotic cells (indicated by arrows) were detected by the TUNEL assay in 6-month-old prostates. Deletion status of Klf5 is indicated at the left and that of Pten indicated at the top. The ratio (%) of apoptotic to total cells for each group is shown at the lower left corner of each image. + and − indicate the presence and absence, respectively, of a Klf5 or Pten allele. No statistical significance was detected between Klf5+/+ and Klf5−/−.
Acknowledgements
The authors thank Robert Cardiff of the University of California at Davis for pathologic diagnosis, Rini Pauly and Gregory Doho of the Biostatistics and Bioinformatics Shared Resource at Emory Winship Cancer Institute for assistance in microarray data analysis, Yudong Xia of E-GENE (Shenzhen, China) for assistance in the generation of heat map, Jenny Jianping Ni and Baotong Zhang of Emory for technical assistance, and Anthea Hammond of Emory for manuscript editing.
Footnotes
This article refers to supplementary materials, which are designated by Tables S1 to S8 and Figures S1 to S5 and are available online at www.neoplasia.com.
X.C. was supported by the graduate student program of the China Scholarship Council (No. 201206200049). This work was supported in part by grants R01CA87921 and R01CA171189 from the National Cancer Institute (NCI), National Institutes of Health (NIH) and grant 81130044 from the National Natural Science Foundation of China. The research reported in this publication was supported in part by the Integrated Cellular Imaging Shared Resource, the Emory Integrated Genomics Core, the Biostatistics and Bioinformatics Shared Resource of Winship Cancer Institute of Emory University, and NIH/NCI under award number P30CA138292. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
References
- 1.Dong J.T. Chromosomal deletions and tumor suppressor genes in prostate cancer. Cancer Metastasis Rev. 2001;20:173–193. doi: 10.1023/a:1015575125780. [DOI] [PubMed] [Google Scholar]
- 2.Sun X., Frierson H.F., Chen C., Li C., Ran Q., Otto K.B., Cantarel B.L., Vessella R.L., Gao A.C., Petros J. Frequent somatic mutations of the transcription factor ATBF1 in human prostate cancer. Nat Genet. 2005;37:407–412. doi: 10.1038/ng1528. [DOI] [PubMed] [Google Scholar]
- 3.Grasso C.S., Wu Y.M., Robinson D.R., Cao X., Dhanasekaran S.M., Khan A.P., Quist M.J., Jing X., Lonigro R.J., Brenner J.C. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sun X., Fu X., Li J., Xing C., Frierson H.F.J., Wu H., Ding X., Ju T., Cummings R.D., Dong J.T. Deletion of Atbf1/Zfhx3 in mouse prostate causes neoplastic lesions, likely by attenuation of membrane and secretory proteins and multiple signaling pathways. Neoplasia. 2014;16:377–389. doi: 10.1016/j.neo.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang S., Gao J., Lei Q., Rozengurt N., Pritchard C., Jiao J., Thomas G.V., Li G., Roy-Burman P., Nelson P.S. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell. 2003;4:209–221. doi: 10.1016/s1535-6108(03)00215-0. [DOI] [PubMed] [Google Scholar]
- 6.Kim M.J., Bhatia-Gaur R., Banach-Petrosky W.A., Desai N., Wang Y., Hayward S.W., Cunha G.R., Cardiff R.D., Shen M.M., Abate-Shen C. Nkx3.1 mutant mice recapitulate early stages of prostate carcinogenesis. Cancer Res. 2002;62:2999–3004. [PubMed] [Google Scholar]
- 7.Zhou Z., Flesken-Nikitin A., Corney D.C., Wang W., Goodrich D.W., Roy-Burman P., Nikitin A.Y. Synergy of p53 and Rb deficiency in a conditional mouse model for metastatic prostate cancer. Cancer Res. 2006;66:7889–7898. doi: 10.1158/0008-5472.CAN-06-0486. [DOI] [PubMed] [Google Scholar]
- 8.Ding Z., Wu C.J., Chu G.C., Xiao Y., Ho D., Zhang J., Perry S.R., Labrot E.S., Wu X., Lis R. SMAD4-dependent barrier constrains prostate cancer growth and metastatic progression. Nature. 2011;470:269–273. doi: 10.1038/nature09677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Knuutila S., Aalto Y., Autio K., Bjorkqvist A.M., El-Rifai W., Hemmer S., Huhta T., Kettunen E., Kiuru-Kuhlefelt S., Larramendy M.L. DNA copy number losses in human neoplasms. Am J Pathol. 1999;155:683–694. doi: 10.1016/S0002-9440(10)65166-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen C., Bhalala H.V., Vessella R.L., Dong J.T. KLF5 is frequently deleted and down-regulated but rarely mutated in prostate cancer. Prostate. 2003;55:81–88. doi: 10.1002/pros.10205. [DOI] [PubMed] [Google Scholar]
- 11.Chen C., Bhalala H.V., Qiao H., Dong J.T. A possible tumor suppressor role of the KLF5 transcription factor in human breast cancer. Oncogene. 2002;21:6567–6572. doi: 10.1038/sj.onc.1205817. [DOI] [PubMed] [Google Scholar]
- 12.Shindo T., Manabe I., Fukushima Y., Tobe K., Aizawa K., Miyamoto S., Kawai-Kowase K., Moriyama N., Imai Y., Kawakami H. Krüppel-like zinc-finger transcription factor KLF5/BTEB2 is a target for angiotensin II signaling and an essential regulator of cardiovascular remodeling. Nat Med. 2002;8:856–863. doi: 10.1038/nm738. [DOI] [PubMed] [Google Scholar]
- 13.Chen C., Sun X., Ran Q., Wilkinson K.D., Murphy T.J., Simons J.W., Dong J.T. Ubiquitin-proteasome degradation of KLF5 transcription factor in cancer and untransformed epithelial cells. Oncogene. 2005;24:3319–3327. doi: 10.1038/sj.onc.1208497. [DOI] [PubMed] [Google Scholar]
- 14.Chen C., Sun X., Guo P., Dong X.Y., Sethi P., Zhou W., Petros J., Frierson H.F.J., Vessella R.L., Atfi A., Dong J.T. Ubiquitin E3 ligase WWP1 as an oncogenic factor in human prostate cancer. Oncogene. 2007;26:2386–2394. doi: 10.1038/sj.onc.1210021. [DOI] [PubMed] [Google Scholar]
- 15.Chen C., Zhou Z., Ross J.S., Zhou W., Dong J.T. The amplified WWP1 gene is a potential molecular target in breast cancer. Int J Cancer. 2007;121:80–87. doi: 10.1002/ijc.22653. [DOI] [PubMed] [Google Scholar]
- 16.Bateman N.W., Tan D., Pestell R.G., Black J.D., Black A.R. Intestinal tumor progression is associated with altered function of KLF5. J Biol Chem. 2004;279:12093–12101. doi: 10.1074/jbc.M311532200. [DOI] [PubMed] [Google Scholar]
- 17.Yang Y., Goldstein B.G., Chao H.H., Katz J.P. KLF4 and KLF5 regulate proliferation, apoptosis and invasion in esophageal cancer cells. Cancer Biol Ther. 2005;4:1216–1221. doi: 10.4161/cbt.4.11.2090. [DOI] [PubMed] [Google Scholar]
- 18.Nakajima Y., Akaogi K., Suzuki T., Osakabe A., Yamaguchi C., Sunahara N., Ishida J., Kako K., Ogawa S., Fujimura T. Estrogen regulates tumor growth through a nonclassical pathway that includes the transcription factors ERβ and KLF5. Sci Signal. 2011;4:ra22. doi: 10.1126/scisignal.2001551. [DOI] [PubMed] [Google Scholar]
- 19.Chen C., Benjamin M.S., Sun X., Otto K.B., Guo P., Dong X.Y., Bao Y., Zhou Z., Cheng X., Simons J.W. KLF5 promotes cell proliferation and tumorigenesis through gene regulation in the TSU-Pr1 human bladder cancer cell line. Int J Cancer. 2006;118:1346–1355. doi: 10.1002/ijc.21533. [DOI] [PubMed] [Google Scholar]
- 20.Nandan M.O., Yoon H.S., Zhao W., Ouko L.A., Chanchevalap S., Yang V.W. Krüppel-like factor 5 mediates the transforming activity of oncogenic H-Ras. Oncogene. 2004;23:3404–3413. doi: 10.1038/sj.onc.1207397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nandan M.O., Chanchevalap S., Dalton W.B., Yang V.W. Krüppel-like factor 5 promotes mitosis by activating the cyclin B1/Cdc2 complex during oncogenic Ras-mediated transformation. FEBS Lett. 2005;579:4757–4762. doi: 10.1016/j.febslet.2005.07.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nandan M.O., McConnell B.B., Ghaleb A.M., Bialkowska A.B., Sheng H., Shao J., Babbin B.A., Robine S., Yang V.W. Krüppel-like factor 5 mediates cellular transformation during oncogenic KRAS-induced intestinal tumorigenesis. Gastroenterology. 2008;134:120–130. doi: 10.1053/j.gastro.2007.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nandan M.O., Ghaleb A.M., McConnell B.B., Patel N.V., Robine S., Yang V.W. Krüppel-like factor 5 is a crucial mediator of intestinal tumorigenesis in mice harboring combined ApcMin and KRASV12 mutations. Mol Cancer. 2010;9:63. doi: 10.1186/1476-4598-9-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li X., Zhang B., Wu Q., Ci X., Zhao R., Zhang Z., Xia S., Su D., Chen J., Ma G. Interruption of KLF5 acetylation converts its function from tumor suppressor to tumor promoter in prostate cancer cells. Int J Cancer. 2014 doi: 10.1002/ijc.29028. [in press] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xing C., Fu X., Sun X., Guo P., Li M., Dong J.T. Different expression patterns and functions of acetylated and unacetylated Klf5 in the proliferation and differentiation of prostatic epithelial cells. PLoS One. 2013;8:e65538. doi: 10.1371/journal.pone.0065538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hollander M.C., Blumenthal G.M., Dennis P.A. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat Rev Cancer. 2011;11:289–301. doi: 10.1038/nrc3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blanco-Aparicio C., Renner O., Leal J.F., Carnero A. PTEN, more than the AKT pathway. Carcinogenesis. 2007;28:1379–1386. doi: 10.1093/carcin/bgm052. [DOI] [PubMed] [Google Scholar]
- 28.Di Cristofano A., Pandolfi P.P. The multiple roles of PTEN in tumor suppression. Cell. 2000;100:387–390. doi: 10.1016/s0092-8674(00)80674-1. [DOI] [PubMed] [Google Scholar]
- 29.Li J., Yen C., Liaw D., Podsypanina K., Bose S., Wang S.I., Puc J., Miliaresis C., Rodgers L., McCombie R. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 30.Wu X., Wu J., Huang J., Powell W.C., Zhang J., Matusik R.J., Sangiorgi F.O., Maxson R.E., Sucov H.M., Roy-Burman P. Generation of a prostate epithelial cell-specific Cre transgenic mouse model for tissue-specific gene ablation. Mech Dev. 2001;101:61–69. doi: 10.1016/s0925-4773(00)00551-7. [DOI] [PubMed] [Google Scholar]
- 31.Sun X., Fu X., Li J., Xing C., Martin D.W., Zhang H.H., Chen Z., Dong J.T. Heterozygous deletion of Atbf1 by the Cre-loxP system in mice causes preweaning mortality. Genesis. 2012;50:819–827. doi: 10.1002/dvg.22041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu X., Zhu Z., Li W., Fu X., Su D., Fu L., Zhang Z., Luo A., Sun X., Dong J.T. Chromodomain helicase DNA binding protein 5 plays a tumor suppressor role in human breast cancer. Breast Cancer Res. 2012;14:R73. doi: 10.1186/bcr3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bell S.M., Zhang L., Mendell A., Xu Y., Haitchi H.M., Lessard J.L., Whitsett J.A. Kruppel-like factor 5 is required for formation and differentiation of the bladder urothelium. Dev Biol. 2011;358:79–90. doi: 10.1016/j.ydbio.2011.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kenchegowda D., Swamynathan S., Gupta D., Wan H., Whitsett J., Swamynathan S.K. Conditional disruption of mouse Klf5 results in defective eyelids with malformed meibomian glands, abnormal cornea and loss of conjunctival goblet cells. Dev Biol. 2011;356:5–18. doi: 10.1016/j.ydbio.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bell S.M., Zhang L., Xu Y., Besnard V., Wert S.E., Shroyer N., Whitsett J.A. Kruppel-like factor 5 controls villus formation and initiation of cytodifferentiation in the embryonic intestinal epithelium. Dev Biol. 2013;375:128–139. doi: 10.1016/j.ydbio.2012.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park J.H., Walls J.E., Galvez J.J., Kim M., Abate-Shen C., Shen M.M., Cardiff R.D. Prostatic intraepithelial neoplasia in genetically engineered mice. Am J Pathol. 2002;161:727–735. doi: 10.1016/S0002-9440(10)64228-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ittmann M., Huang J., Radaelli E., Martin P., Signoretti S., Sullivan R., Simons B.W., Ward J.M., Robinson B.D., Chu G.C. Animal models of human prostate cancer: the consensus report of the New York meeting of the Mouse Models of Human Cancers Consortium Prostate Pathology Committee. Cancer Res. 2013;73:2718–2736. doi: 10.1158/0008-5472.CAN-12-4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo P., Dong X.Y., Zhang X., Zhao K.W., Sun X., Li Q., Dong J.T. Pro-proliferative factor KLF5 becomes anti-proliferative in epithelial homeostasis upon signaling-mediated modification. J Biol Chem. 2009;284:6071–6078. doi: 10.1074/jbc.M806270200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang S., Garcia A.J., Wu M., Lawson D.A., Witte O.N., Wu H. Pten deletion leads to the expansion of a prostatic stem/progenitor cell subpopulation and tumor initiation. Proc Natl Acad Sci U S A. 2006;103:1480–1485. doi: 10.1073/pnas.0510652103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choi N., Zhang B., Zhang L., Ittmann M., Xin L. Adult murine prostate basal and luminal cells are self-sustained lineages that can both serve as targets for prostate cancer initiation. Cancer Cell. 2012;21:253–265. doi: 10.1016/j.ccr.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kenchegowda D., Harvey S.A., Swamynathan S., Lathrop K.L., Swamynathan S.K. Critical role of Klf5 in regulating gene expression during post-eyelid opening maturation of mouse corneas. PLoS One. 2012;7:e44771. doi: 10.1371/journal.pone.0044771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bastola D.R., Pahwa G.S., Lin M.F., Cheng P.W. Downregulation of PTEN/MMAC/TEP1 expression in human prostate cancer cell line DU145 by growth stimuli. Mol Cell Biochem. 2002;236:75–81. doi: 10.1023/a:1016191913274. [DOI] [PubMed] [Google Scholar]
- 43.Ishida E., Nakamura M., Shimada K., Tasaki M., Konishi N. Immunohistochemical analysis of neuroendocrine differentiation in prostate cancer. Pathobiology. 2009;76:30–38. doi: 10.1159/000178153. [DOI] [PubMed] [Google Scholar]
- 44.Berthon P., Cussenot O., Hopwood L., Leduc A., Maitland N. Functional expression of SV40 in normal human prostatic epithelial and fibroblastic cells—differentiation pattern of nontumorigenic cell-lines. Int J Oncol. 1995;6:333–343. doi: 10.3892/ijo.6.2.333. [DOI] [PubMed] [Google Scholar]
- 45.Okutani T., Okabayashi Y., Kido Y., Sugimoto Y., Sakaguchi K., Matuoka K., Takenawa T., Kasuga M. Grb2/Ash binds directly to tyrosines 1068 and 1086 and indirectly to tyrosine 1148 of activated human epidermal growth factor receptors in intact cells. J Biol Chem. 1994;269:31310–31314. [PubMed] [Google Scholar]
- 46.Cantley L.C., Neel B.G. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci U S A. 1999;96:4240–4245. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guo P., Zhao K.W., Dong X.Y., Sun X., Dong J.T. Acetylation of KLF5 alters the assembly of p15 transcription factors in transforming growth factor-β-mediated induction in epithelial cells. J Biol Chem. 2009;284:18184–18193. doi: 10.1074/jbc.M109.007096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zheng H.Q., Zhou Z., Huang J., Chaudhury L., Dong J.T., Chen C. Krüppel-like factor 5 promotes breast cell proliferation partially through upregulating the transcription of fibroblast growth factor binding protein 1. Oncogene. 2009;28:3702–3713. doi: 10.1038/onc.2009.235. [DOI] [PubMed] [Google Scholar]
- 49.McConnell B.B., Bialkowska A.B., Nandan M.O., Ghaleb A.M., Gordon F.J., Yang V.W. Haploinsufficiency of Krüppel-like factor 5 rescues the tumor-initiating effect of the ApcMin mutation in the intestine. Cancer Res. 2009;69:4125–4133. doi: 10.1158/0008-5472.CAN-08-4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang Y., Nakagawa H., Tetreault M.P., Billig J., Victor N., Goyal A., Sepulveda A.R., Katz J.P. Loss of transcription factor KLF5 in the context of p53 ablation drives invasive progression of human squamous cell cancer. Cancer Res. 2011;71:6475–6484. doi: 10.1158/0008-5472.CAN-11-1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dong J.T., Chen C. Essential role of KLF5 transcription factor in cell proliferation and differentiation and its implications for human diseases. Cell Mol Life Sci. 2009;66:2691–2706. doi: 10.1007/s00018-009-0045-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen C., Sun X., Guo P., Dong X.Y., Sethi P., Zhou W., Zhou Z., Petros J., Frierson H.F., Vessella R.L. Ubiquitin E3 ligase WWP1 as an oncogenic factor in human prostate cancer. Oncogene. 2007;26:2386–2394. doi: 10.1038/sj.onc.1210021. [DOI] [PubMed] [Google Scholar]
- 53.Byrne J.A., Chen Y., Martin La Rotta N., Peters G.B. Challenges in identifying candidate amplification targets in human cancers: chromosome 8q21 as a case study. Genes Cancer. 2012;3:87–101. doi: 10.1177/1947601912456287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ishkanian A.S., Mallof C.A., Ho J., Meng A., Albert M., Syed A., van der Kwast T., Milosevic M., Yoshimoto M., Squire J.A. High-resolution array CGH identifies novel regions of genomic alteration in intermediate-risk prostate cancer. Prostate. 2009;69:1091–1100. doi: 10.1002/pros.20959. [DOI] [PubMed] [Google Scholar]
- 55.Vlietstra R.J., van Alewijk D.C., Hermans K.G., van Steenbrugge G.J., Trapman J. Frequent inactivation of PTEN in prostate cancer cell lines and xenografts. Cancer Res. 1998;58:2720–2723. [PubMed] [Google Scholar]
- 56.Hermans K.G., van Alewijk D.C., Veltman J.A., van Weerden W., van Kessel A.G., Trapman J. Loss of a small region around the PTEN locus is a major chromosome 10 alteration in prostate cancer xenografts and cell lines. Genes Chromosomes Cancer. 2004;39:171–184. doi: 10.1002/gcc.10311. [DOI] [PubMed] [Google Scholar]
- 57.Saramaki O.R., Porkka K.P., Vessella R.L., Visakorpi T. Genetic aberrations in prostate cancer by microarray analysis. Int J Cancer. 2006;119:1322–1329. doi: 10.1002/ijc.21976. [DOI] [PubMed] [Google Scholar]
- 58.Kim M.J., Cardiff R.D., Desai N., Banach-Petrosky W.A., Parsons R., Shen M.M., Abate-Shen C. Cooperativity of Nkx3.1 and Pten loss of function in a mouse model of prostate carcinogenesis. Proc Natl Acad Sci U S A. 2002;99:2884–2889. doi: 10.1073/pnas.042688999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Abate-Shen C., Banach-Petrosky W.A., Sun X., Economides K.D., Desai N., Gregg J.P., Borowsky A.D., Cardiff R.D., Shen M.M. Nkx3.1; Pten mutant mice develop invasive prostate adenocarcinoma and lymph node metastases. Cancer Res. 2003;63:3886–3890. [PubMed] [Google Scholar]
- 60.Xing C., Fu X., Sun X., Dong J.T. Lack of an additive effect between the deletions of Klf5 and Nkx3-1 in mouse prostatic tumorigenesis. J Genet Genomics. 2013;40:315–318. doi: 10.1016/j.jgg.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Simpson L., Parsons R. PTEN: life as a tumor suppressor. Exp Cell Res. 2001;264:29–41. doi: 10.1006/excr.2000.5130. [DOI] [PubMed] [Google Scholar]
- 62.Downward J. PI 3-kinase, Akt and cell survival. Semin Cell Dev Biol. 2004;15:177–182. doi: 10.1016/j.semcdb.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 63.Plas D.R., Thompson C.B. Akt-dependent transformation: there is more to growth than just surviving. Oncogene. 2005;24:7435–7442. doi: 10.1038/sj.onc.1209097. [DOI] [PubMed] [Google Scholar]
- 64.Di Cristofano A., De Acetis M., Koff A., Cordon-Cardo C., Pandolfi P.P. Pten and p27KIP1 cooperate in prostate cancer tumor suppression in the mouse. Nat Genet. 2001;27:222–224. doi: 10.1038/84879. [DOI] [PubMed] [Google Scholar]
- 65.Maitland N.J., Collins A.T. Prostate cancer stem cells: a new target for therapy. J Clin Oncol. 2008;26:2862–2870. doi: 10.1200/JCO.2007.15.1472. [DOI] [PubMed] [Google Scholar]
- 66.Goldstein A.S., Witte O.N. Does the microenvironment influence the cell types of origin for prostate cancer? Genes Dev. 2013;27:1539–1544. doi: 10.1101/gad.222380.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lu T.L., Huang Y.F., You L.R., Chao N.C., Su F.Y., Chang J.L., Chen C.M. Conditionally ablated Pten in prostate basal cells promotes basal-to-luminal differentiation and causes invasive prostate cancer in mice. Am J Pathol. 2013;182:975–991. doi: 10.1016/j.ajpath.2012.11.025. [DOI] [PubMed] [Google Scholar]
- 68.Galbaugh T., Cerrito M.G., Jose C.C., Cutler M.L. EGF-induced activation of Akt results in mTOR-dependent p70S6 kinase phosphorylation and inhibition of HC11 cell lactogenic differentiation. BMC Cell Biol. 2006;7:34. doi: 10.1186/1471-2121-7-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cantley L.C. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 70.Yoon S., Seger R. The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors. 2006;24:21–44. doi: 10.1080/02699050500284218. [DOI] [PubMed] [Google Scholar]
- 71.Chetram M.A., Hinton C.V. PTEN regulation of ERK1/2 signaling in cancer. J Recept Signal Transduct Res. 2012;32:190–195. doi: 10.3109/10799893.2012.695798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hay N. The Akt-mTOR tango and its relevance to cancer. Cancer Cell. 2005;8:179–183. doi: 10.1016/j.ccr.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 73.Zhang B., Zhang Z., Xia S., Xing C., Ci X., Li X., Zhao R., Tian S., Ma G., Zhu Z. KLF5 activates microRNA 200 transcription to maintain epithelial characteristics and prevent induced epithelial-mesenchymal transition in epithelial cells. Mol Cell Biol. 2013;33:4919–4935. doi: 10.1128/MCB.00787-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Naqvi S., Martin K.J., Arthur J.S. CREB phosphorylation at Ser133 regulates transcription via distinct mechanisms downstream of cAMP and MAPK signalling. Biochem J. 2014;458:469–479. doi: 10.1042/BJ20131115. [DOI] [PubMed] [Google Scholar]
- 75.Guo P., Dong X.Y., Zhao K.W., Sun X., Li Q., Dong J.T. Estrogen-induced interaction between KLF5 and estrogen receptor (ER) suppresses the function of ER in ER-positive breast cancer cells. Int J Cancer. 2010;126:81–89. doi: 10.1002/ijc.24696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dang D.T., Pevsner J., Yang V.W. The biology of the mammalian Krüppel-like family of transcription factors. Int J Biochem Cell Biol. 2000;32:1103–1121. doi: 10.1016/s1357-2725(00)00059-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Guo P., Dong X.Y., Zhao K.W., Sun X., Li Q., Dong J.T. Opposing effects of KLF5 on the transcription of MYC in epithelial proliferation in the context of transforming growth factor β. J Biol Chem. 2009;284:28243–28252. doi: 10.1074/jbc.M109.036160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zegarra-Moro O.L., Schmidt L.J., Huang H., Tindall D.J. Disruption of androgen receptor function inhibits proliferation of androgen-refractory prostate cancer cells. Cancer Res. 2002;62:1008–1013. [PubMed] [Google Scholar]
- 79.Hayes S.A., Zarnegar M., Sharma M., Yang F., Peehl D.M., ten Dijke P., Sun Z. SMAD3 represses androgen receptor-mediated transcription. Cancer Res. 2001;61:2112–2118. [PubMed] [Google Scholar]
- 80.Culig Z., Bartsch G. Androgen axis in prostate cancer. J Cell Biochem. 2006;99:373–381. doi: 10.1002/jcb.20898. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primer Sequences Used in This Study
Differentially Expressed Genes between Klf5 Wild-Type and Klf5-Null Mouse DPs in the Pten-Null Background, as Identified by Microarray Analysis. Fold Change Was the Mean Value of Four Pairs of Samples
Genes Upregulated or Downregulated by Klf5 Deletion in More than One Type of Tissue. Up-regulation in a Tissue Is Indicated by “Up” or a Positive Fold Change Number, while Down-Regulation Is Indicated by “Down” or a Negative Fold Change Number. Data for Cornea, Villus, and Bladder Were from Published Studies [41], [35], [33]
Genes Dysregulated by Both Klf5 Deletion in Pten-Null Mouse Prostates and Ectopic Expression of KLF5 or Its Acetylation-Deficient Mutant K369R in Xenograft Tumors of the DU 145 Human Prostate Cancer Cell Line (DU 145-KLF5 or DU 145-K369R). Data for DU 145 Xenografts Were from a Previous Study [24]
Differentially Expressed Genes that Were Prostate Cancer Related, as Identified by the MetaCore Program, and Had Appeared in at Least One PubMed Publication on Prostate Cancer. Fold Change between Klf5 Wild-Type and Klf5-Null Groups and the Number of PubMed Publications Available Are Also Shown for Each of the Genes. Positive (P) or Negative (N) Refers to a Gene’s Association with the Development or Progression of Prostate Cancer Based on PubMed Publications, with P Indicating a Gene’s Promoting Role in Prostate Tumorigenesis or Up-Regulation in Prostate Tumors and N indicating the Opposite. N/A, Information Is Not Clear
Functional Processes Enriched by MetaCore Analysis Based on the Differentially Expressed Genes between Klf5 Wild-Type and Klf5-Null Mouse DPs. Different Processes with the Same Function Are Grouped Together and Ranked Based on P Values (from Low to High, P Value < .01). Genes Contributing to a Process Are Listed
Significant Pathways Identified by the MetaCore Analysis Based on the Differentially Expressed Genes between Klf5 Wild-Type and Klf5-Null Mouse DPs. All Pathways Are Ranked Based on Their P Values (from Low to High, P Value < .01)
Key Transcription Factors Significantly Associated with Differentially Expressed Genes between Klf5 Wild-Type and Klf5-Null Mouse DPs, as Defined by MetaCore’s Interactome Analysis for Transcription Factors. Related Biologic Processes Are Also Shown. The Ranking Is Based on P Values. Total Nodes Refer to All Nodes in a Transcription Factor Network, while Seed Nodes Refer to the Genes Differentially Expressed between the Experimental Groups
Confirmation of Klf5 deletion in mouse prostates with Pten hemizygous knockout. Klf5 protein was detected by IHC staining in 12-month-old APs with indicated Klf5 deletion status; + and − indicate wild-type and deleted Klf5 alleles, respectively.
Heat map illustrating gene expression changes caused by Klf5 deletion in Pten-null prostate tumors and associated with prostate cancer in the literature. A total of 173 differentially expressed genes with > 1.5-fold expression change and literature-based prostate cancer association was used in the heat map drawing. Each lane represents one pair of wild-type and Klf5-null DPs at 6 months old. Clustering in the heat map is based on the relation of genes with prostate cancer, with those reported to promote prostate cancer or be upregulated in prostate cancer as “positive”, and those reported to inhibit prostate cancer or be downregulated in prostate cancer as “negative”. The percentages of upregulated or downregulated genes in each group are shown. The number in the parentheses after each gene name indicates the number of publications on that gene in prostate cancer. Heat map colors reflect normalized log2 fold-change values of the ratio in each pair.
Molecular networks constructed for differentially expressed genes between Klf5 knockout and wild-type by the network analysis algorithm in the MetaCore platform. The 25 networks that had more than four seed nodes with P < .05 are shown. Different shapes of the nodes represent functional classifications of genes. Red and blue in the nodes indicate up-regulation and down-regulation, respectively, of genes. Lines between every two nodes indicate their interactions, with red for inhibition, green for activation, and gray for unspecified interaction. Arrow indicates the direction of a regulation. Colored thicker lines mark canonical pathways.
Klf5 deletion upregulates p-mTor in Pten-null mouse prostate tumors. IHC staining was used to detect p-mTor in tissue sections of APs and DPs at 6 months of age. Deletion status of Klf5 is indicated at the top. + and − indicate the presence and absence, respectively, of a Klf5 or Pten allele.
Klf5 deletion has no effect on apoptosis in Pten-null mouse prostate tumors. Apoptotic cells (indicated by arrows) were detected by the TUNEL assay in 6-month-old prostates. Deletion status of Klf5 is indicated at the left and that of Pten indicated at the top. The ratio (%) of apoptotic to total cells for each group is shown at the lower left corner of each image. + and − indicate the presence and absence, respectively, of a Klf5 or Pten allele. No statistical significance was detected between Klf5+/+ and Klf5−/−.